
Home » Posts tagged 'Aurigene'
Tag Archives: Aurigene
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TACACICLIB, AUR-102, AURIGENE
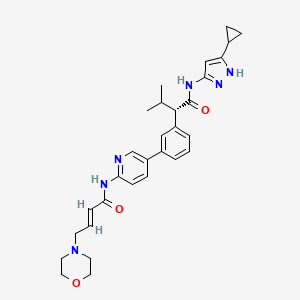
Tacaciclib
2768774-66-7
AUR-102
- Tacaciclib
- SCHEMBL24548621
- GTPL12880
- 528.6 g/mol
- C30H36N6O3
INN 12755
UNI D3G4JKK1MA
(2S)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-3-methyl-2-[3-[6-[[(E)-4-morpholin-4-ylbut-2-enoyl]amino]pyridin-3-yl]phenyl]butanamide
(αS)-N-(5-Cyclopropyl-1H-pyrazol-3-yl)-α-(1-methylethyl)-3-[6-[[(2E)-4-(4-morpholinyl)-1-oxo-2-buten-1-yl]amino]-3-pyridinyl]benzeneacetamide
Benzeneacetamide, N-(5-cyclopropyl-1H-pyrazol-3-yl)-α-(1-methylethyl)-3-[6-[[(2E)-4-(4-morpholinyl)-1-oxo-2-buten-1-yl]amino]-3-pyridinyl]-, (αS)-
Tacaciclib is a CDK inhibitor, antineoplastic effect.
The present invention is directed to methods of preparation of compound of formula (I) that is useful for inhibiting Cyclin-dependent kinase 7 (CDK7) and for treating diseases or disorders mediated thereby.
CDK7, which complexes with cyclin H and RING-finger protein MAT1, phosphorylates the cell cycle CDKs in the activation of T-loop, to promote their activities (Fisher et al., Cell., Aug 26;78(4):713- 24, 1994). As such, it has been proposed that inhibiting CDK7 would provide a potent means of inhibiting cell cycle progression, which may be especially relevant given that there is compelling evidence from gene knockout studies in mice for lack of an absolute requirement for CDK2, CDK4 and CDK6 for the cell cycle at least in most cell types (M alumbres et al., Nature Cell Biology, 11, 1275 – 1276, 2009), whilst different tumors appear to require some, but they are independent of other interphase CDKs (CDK2, CDK4 , CDK6). Recent genetic and biochemical studies have confirmed the importance of CDK7 for cell cycle progression (Larochelle. et al., Mol Cell., Mar 23;25(6):839-50. 2007; Ganuza et al., EM BO J., May 30; 31(11): 2498-510, 2012).
Cyclin-dependent kinase 7 (CDK7) activates cell cycle CDKs and is a member of the general Transcription factor II Human (TFIIH). CDK7 also plays a role in transcription and possibly in DNA repair. The trimeric Cak complex CDK7/CyclinH/MATl is also a component of TFIIH, the general transcription/DNA repair factor IIH (Morgan, DO., Annu.Rev. Cell Dev. Biol. 13, 261-91, 1997). As a TFIIH subunit, CDK7 phosphorylates the CTD (Carboxy-Terminal-Domain) of the largest subunit of RNA polymerase II (pol II). The CTD of mammalian pol (II) consists of 52 heptad repeats with the consensus sequence 1 YSPTSPS 7 and the phosphorylation status of the Ser residues at positions 2 and 5 has been shown to be
important in the activation of RNAP-II indicating that it is likely to have a crucial role in the function of the CTD. CDK7, which primarily phosphorylates Ser-5 (PSS) of RNAP-II at the promoter as part of transcriptional initiation (Gomes et ah, Genes Dev. 2006 Mar 1; 20(5):601-12, 2006), in contrast with CDK9, which phosphorylates both Ser-2 and Ser-5 of the CTD heptad (Pinhero et al., Eur. J. Biochem., 271, pp. 1004-1014, 2004).
In addition to CDK7, other CDKs have been reported to phosphorylate and regulate RNA pol (II) CTD. The other CDKs include, Cdk9/ Cyclin T1 or T2 that constitute the active form of the positive transcription elongation factor (P-TEFb) (Peterlin and Price, Mol Cell., Aug 4; 23(3): 297-305,2006) and Cdkl2/Cyclin K and Cdkl3/Cyclin K as the latest members of RNAPII CTD kinases (Bartkowiak et al., Genes Dev., Oct 1 5;24(20):2303-16, 2010; Blazek et al., Genes Dev .Oct 15;25(20):2158-72, 2011).
Disruption of RNAP II CTD phosphorylation has been shown to preferentially effect proteins with short half-lives, including those of the anti-apoptotic BCL-2 family. (Konig et al., Blood, 1, 4307-4312, 1997; The transcriptional non-selective cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1; (Gojoet al., Clin. Cancer Res. 8, 3527-3538, 2002).
This suggests that the CDK7 enzyme complexes are involved in multiple functions in the cell: cell cycle control, transcription regulation and DNA repair. It is surprising to find one kinase involved in such diverse cellular processes, some of which are even mutually exclusive. It also is puzzling that multiple attempts to find cell cycle dependent changes in CDK7 kinase activity remained unsuccessful. This is unexpected since activity and phosphorylation state of its substrate, CDC2, fluctuate during the cell cycle. In fact, it is shown that cdk7 activity is required for the activation of both Cdc2/Cyclin A and Cdc2/Cyclin B complexes, and for cell division. (Larochelle, S. et al. Genes Dev 12,370-81, 1998). Indeed, flavopiridol, a non-selective pan-CDK inhibitor that targets CTD kinases, has demonstrated efficacy for the treatment of chronic lymphocytic leukemia (CLL), but suffers from a poor toxicity profile (Lin et al.,). 27, 6012-6018, 2009; Christian et al., Clin. Lymphoma Myeloma, 9, Suppl.
3, S179-S185, 2009).
International publication WO2016193939, which is incorporated herein by reference for all purposes describes CDK7 inhibitors and processes for the preparation thereof. Inhibitors of CDK7 are currently being developed for the treatment of cancer. For drug development, it is typically advantageous to employ individual stereoisomers as they exhibit marked differences in pharmacodynamic, pharmacokinetic, and toxicological properties.
SYN
COUPLER

MAIN

Aurigene Discovery Technologies Ltd.
WO2022249141
WO2022130304
WO2022084930
WO2023224961
WO2023107861
WO2022249141
WO2022229835
WO2022130304
WO2022084930
PATENT
WO 2016/193939 COMPD 44
https://patents.google.com/patent/WO2016193939A1/en
InventorSusanta SamajdarRamulu PoddutooriChetan PanditSubhendu MUKHERJEERajeev Goswami
AURIGENE DISCOVERY TECHNOLOGIES LIMITED [IN]/[IN]
Inventors
- SAMAJDAR, Susanta
- PODDUTOORI, Ramulu
- PANDIT, Chetan
- MUKHERJEE, Subhendu
- GOSWAMI, Rajeev
PATENT
Applicants
- AURIGENE ONCOLOGY LIMITED [IN]/[IN]
Inventors
- PODDUTOORI, Ramulu
- VIJAYKUMAR BHAT, Uday
- THIMMASANDRA SEETHAPPA, Devaraja
WO2022229835
Example- 1: Preparation of compound of formula (I)
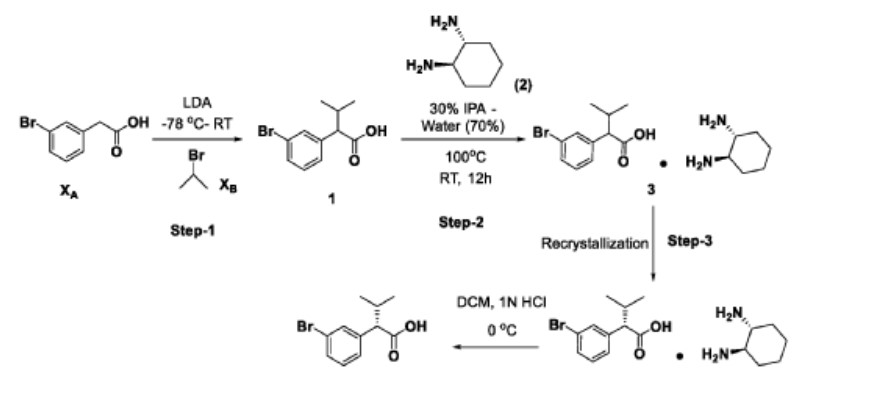
Scheme-1: Preparation of KRM-A

Step-4
KRM-A 4
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3-bromophenyl) acetic acid (XA, 150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by a drop wise addition of isopropyl bromide (X B , 255 g, 2.07 mol) over a period of 30 min. The reaction mixture was stirred at room temperature overnight. Then, the reaction mixture was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the crude compound which was purified by silica column by eluting with 0-10% ethyl acetate-hexane system to afford the title compound (150 g, 83% yield) , HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN 110590747.
Step-2: Preparation of Compound 3
2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IP A in water (10.2 L; 3.06 L of IPA-7.14 L of water) and ( 1L\ 2i ?)-cyclohexane-1,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then was heated to 100 °C until the solution became clear and stirred at the same temperature for another 30 min. The reaction mixture was allowed to slowly reach room temperature for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
Work up (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C until the clear solution was observed. The compound was extracted into DCM, dried over NaiSCL and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
The compound 3 (619.90 g) was taken in 30% of IP A in water (12.4 L), then the mixture was heated to 100 °C until the solution became clear and was stirred at the same temperature for another 30min. The reaction mixture was allowed to reach room temperature slowly for 8-12h.
The obtained solid was filtered and washed with 500mL 30% IPA-water and dried under vacuum to afford a desired compound (360g, wet).
Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C until the clear solution was observed and the compound was extracted to DCM, dried over NaiSCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
The recrystallization method was repeated for three more times by using 30% of IPA in water as per the aforesaid procedure to obtain the purity of greater than 98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated, washed with brine solution (500 mL) and dried over NaiSCL. The solvent was evaporated from the reaction mixture to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
*H NMR (400MHz, DMSO-de): d 12.5 (s, 1H), 7.50-7.44 (m, 2H), 7.34-7.26 (m, 2H), 3.16 (d, 1H), 2.23-2.11 (m, 1H), 0.98 (d, 3H), 0.63 (d, 3H); Chiral HPLC: 98.50% retention time: 4,588 min.
Scheme-2: Preparation of compound of formula (I)
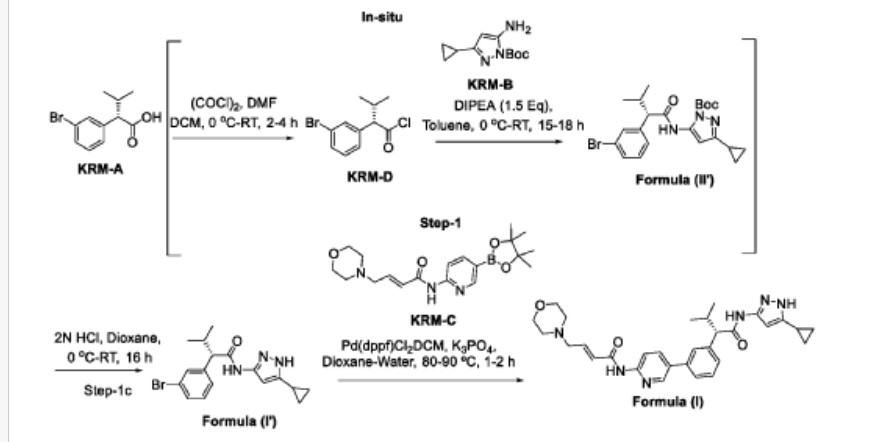

Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-3-methylbutanamide
Step-la: Preparation ofKRM-D
To a stirred solution of KRM-A (lOOg, O.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0°C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude (S)- 2-(3-bromophenyl)-3-methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step-lb: Preparation of compound of formula (II)
(5)-2-(3-bromophcnyl)-3-mcthylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of ieri-butyl 3-amino-5-cyclopropyl-1H-pyrazole- l-carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to reach RT and stirred overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HC1 (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL), brine solution (500 mL), dried over anhydrous NaiSCL , filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (S)-5-( 2-(3-bromophenyl)-3-methylbutanamido)-3-
cyclopropyl- lH-pyrazole- 1-carboxylate (compound of formula (IG)) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H) + , HPLC: 80.80%, retention time:15.89 min) . The crude product was taken as such for next step without further purification.
Step-1 c: Preparation of compound of formula (I)
To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-1H-pyrazole-1-carboxylate (180 g, 1,731 mol) in dioxane (360 mL ) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The obtained compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL), brine solution (300 mL), and dried over anhydrous NaiSCL . The organic layer was concentrated to obtain a crude (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula (G)) as a semi-solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The obtained solid was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (1 lOg, Yield: 78% over two steps). LCMS: m/z= 362 (M+H) + , HPLC: 97.66%, retention time: 24.10 min
Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2- yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound of formula (I))
To a degassed solution of (5)-2-(3-bromophcnyl)-N-(5-cyclopropyl-1 H-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (E)-4 -morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)Ch.DCM (3.38 g, 0.0042 mol, and 0.03eq) was added to the reaction mixture and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite ® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and
extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulfate. The organic layer was concentrated under vacuum at 35-40°C to obtain crude title compound (~80g).
The crude compound of formula (I), (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20% aqueous NaiCCL solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulfate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), then the reaction mixture was stirred overnight at room temperature and filtered through Celite ® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, until absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (70g, HPLC purity: 92.70%, retention time: 15.65 min).
Work-up for improved chiral purity: The above compound of formula (I) was dissolved in ethylacetate (~30 vol, 2L) and washed with aqueous citric acid (2 times, 400 mL x 1 and 200mL x 1), aqueous NaHCCL solution (2%, 500 mL x 1) and aqueous NaCl solution (10%, 500 mL x 1). The combined organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (~60g).
*H NMR (400MHz, DMSO-rfe): d: 10.79 (s, 1H), 10.46 (s, 1H), 8.61 (d, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.69 (s, 1H), 7.56 (d, 1H), 7.39 (m, 2H), 6.84-6.77 (m, 1H), 6.62 (s, 2H), 6.51 (d, 1H), 6.13 (s, 1H) , 3.62-3.59 (m, 4H), 3.35 (d, 1H), 3.15-3.13 (m, 2H), 2.42-2.39 (m, 5H), 1.80-1.77 (m, 1H), 0.98 (d, 3H) , 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H); LCMS: m/z= 529.25-free base (M+H) + , HPLC: 98.98%, retention time: 15.40 min.
Patent
PATENT
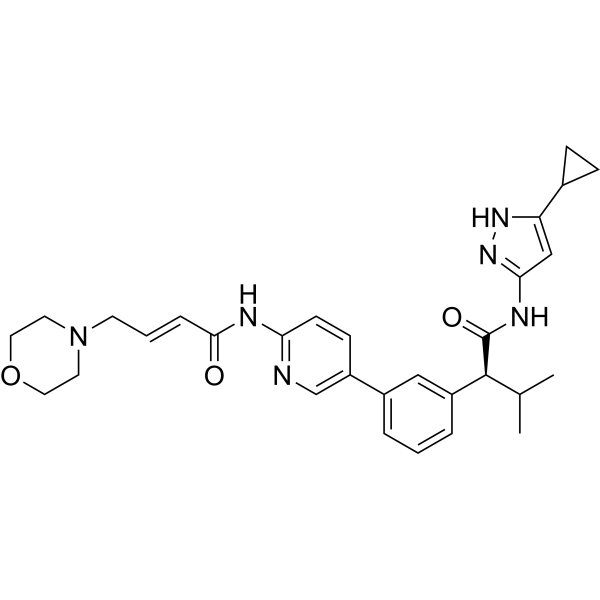
In some embodiments, the compound of formula (I) is (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide or a pharmaceutically acceptable salt or a stereoisomer thereof (Compound 44).
Compound 44 is disclosed in WO 2016/193939 Al, published December 8, 2016, entitled “Substituted heterocyclyl derivatives as cdk inhibitors,” the entire contents of which are incorporated herein by reference. Compound 44A can be in the form of a fumaric acid salt or cocrystal as described in WO 2022/130304 Al, published June 23, 2022, entitled “Cocrystal of a cdk inhibitor,” the entire contents of which are incorporated herein by reference.
Example 3: Synthesis of Compounds 44A & 44B via Chiral Separation
Scheme-1
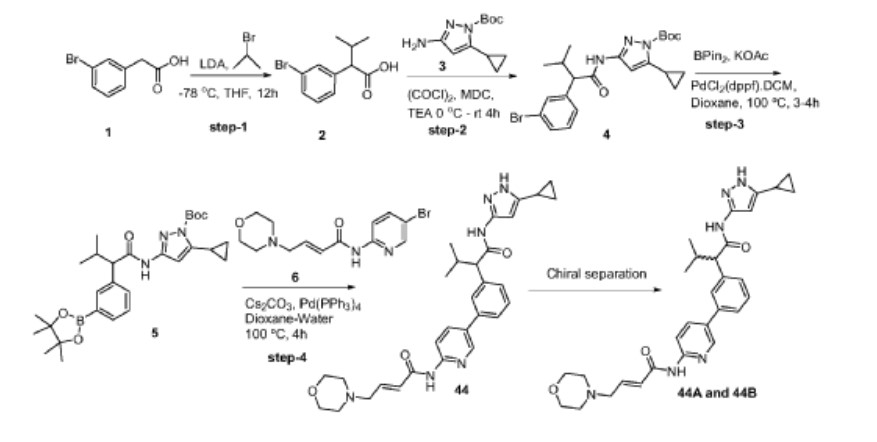

Step-1: Synthesis of 2-(3-bromophenyl)-3-methylbutanoic acid
[0352] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (reagent-1, 150g, 0.69mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mass was stirred for 2h at -78 °C followed by the drop wise addition of Isopropyl bromide (255 g, 2.07mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature for overnight. The reaction mass was quenched with IN HC1 (pH 2) and product extracted to ethyl acetate (500mL x 3). The combined organic layer washed with water followed by brine, dried and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound 2 (150g, 83% yield). LCMS: m/z = 254.80 (M-2H)’
Step-2: Synthesis of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate
[0353] 2-(3-bromophenyl)-3-methylbutanoic acid (intermediate-2, 70g, 0.0.27mol) was dissolved in dry DCM (500 mL) and added oxalyl chloride (68 mL, 0.78mol) dropwise at 0 °C followed by addition of catalytic amount of DMF (0.8mL) and maintained reaction mass at same temperature for 30min. The reaction mass was allowed to room temperature and stirred for 4h, distilled off the solvent and excess oxalyl chloride under vacuum. Re-dissolved the residue in DCM (250 mL) and added slowly to the cooled solution of tert-butyl 3 -amino-5 -cyclopropyl- 1H-pyrazole-1 -carboxylate (intermediate-3, 49g, 0.218mol) and TEA (55 mL, 0.546mol) in THF (250 mL) at 0 °C for 30min, The reaction was stirred at room temperature for 12h then the reaction mass was concentrated under reduced pressure and the residue was dissolved in DCM, washed with saturated NaHCO3 solution and brine. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure, the crude was purified by silica gel column chromatography by eluting with 15% ethyl acetate-hexane to afford the title compound 4 (90g, 71% ) LCMS: m/z = 363.80 (M-Boc+2).
Step-3: Synthesis of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)butanamido)-l H -pyr azole- 1 -carboxylate
[0354] To a degassed solution of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate (intermediate-4, 90g, 0.193mol) and 4, 4, 4′, 4′, 5, 5, 5′, 5′-octamethyl-2,2′-bi(l,3,2-dioxaborolane) (62g, 0.25 Imol) in 1,4-Dioxane (500 mL) was added potassium acetate (37.80g, 0.386mol). The reaction mass was allowed to stir for 10 min with degassing at RT and added PdC12(dppf).DCM complex (12.5g, 0.015mol). The reaction mass was heated for 3-4 h at 100 °C. Reaction mixture cooled to RT and filtered on celite bed, filtrate evaporated to get dark brown liquid. The crude material was purified by silica column chromatography by eluting with 20% ethyl acetate in hexane to afford the compound 5 (90g, 86%). LCMS: m/z = 410 (M-Boc+1)+.
Step-4: Synthesis of (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide
[0355] To a degassed solution of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4, 4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)phenyl)butanamido)- 1 H-pyrazole- 1 -carboxylate, 5 (10g, 0.019mol) and (E)-N-(5-bromopyridin-2-yl)-4-morpholinobut-2-enamide (7.7g, 0.023mol) in
1,4-Dioxane (lOOmL) and water (40mL) followed by Cs2CO3 (14.5g, 0.045mol) were added. The reaction mass was allowed to stir for 10 min with degassing and added Pd(PPh3)4 (1.1g, 0.00095mol), heated the reaction mass for 4 h at 100 °C in a sealed tube. The reaction mass was cooled and diluted with brine solution. The aqueous layer was separated and re-extracted with ethyl acetate. The combined organic layer was evaporated to dryness and crude material was purified by silica column chromatography by eluting with 10%-l 5 % methanol in DCM to get desired pure compound 44 (4.5g, 44%). LCMS: m/z = 529.15 (M+H)+; HPLC: 95.17%, rt: 6.34 min.
[0356] Racemic (E)-N-(5 -(3 -( 1 -((5 -cyclopropyl- 1 H-pyrazol-3 -yl)amino)-3 -methyl- 1 -oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide was separated by using chiral preparative HPLC column (Method: Column: Chiral Pak IA (20mm X 250 mm, 5 micron), Elution: isocratic (50:50), A=ACN, B= MeOH, Flow: 20mL/min ) to afford the pure Isomer- 1 and Isomer-2.
Isomer-1 (Compound 44-A):
[0357] 1HNMR (DMSO-d6, 400MHz): 5 12.02 (s, 1H), 10.78 (s, 1H), 10.44 (s, 1H), 8.61 (s, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.68 (s, 1H), 7.57 (d, 1H), 7.41-7.37 (m, 2H), 6.81-6.78 (m, 1H), 6.49 (d, 1H), 6.13 (s, 1H), 3.61-3.58 (m, 4H), 3.36-3.34 (m, 1H), 3.12 (d, 2H), 2.41-2.32 (m, 5H), 1.82-1.76 (m, 1H), 0.97 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.59 (m, 2H); LCMS: m/z = 529.15 (M+H)+; HPLC: 96.72%, rt: 6.39 min; Chiral HPLC: 97.68%, rt: 14.47.
Isomer-2 (Compound 44B):
[0358] 1HNMR (DMSO-d6, 400MHz): 5 12.02 (s, 1H), 10.78 (s, 1H), 10.44 (s, 1H), 8.61 (s, 1H), 8.28 (d, 1H), 8.07-8.04 (m, 1H), 7.68 (s, 1H), 7.57 (d, 1H), 7.41-7.37 (m, 2H), 6.81-6.78 (m, 1H), 6.50 (d, 1H), 6.14 (s, 1H), 3.61-3.58 (m, 4H), 3.36-3.34 (m, 1H), 3.12 (d, 2H), 2.40-2.39 (m, 5H), 1.82-1.76 (m, 1H), 0.97 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H); LCMS: m/z = 529.15 (M+H)+; HPLC: 96.24%, rt: 6.39 min; Chiral HPLC: 97.92%, rt: 8.80.
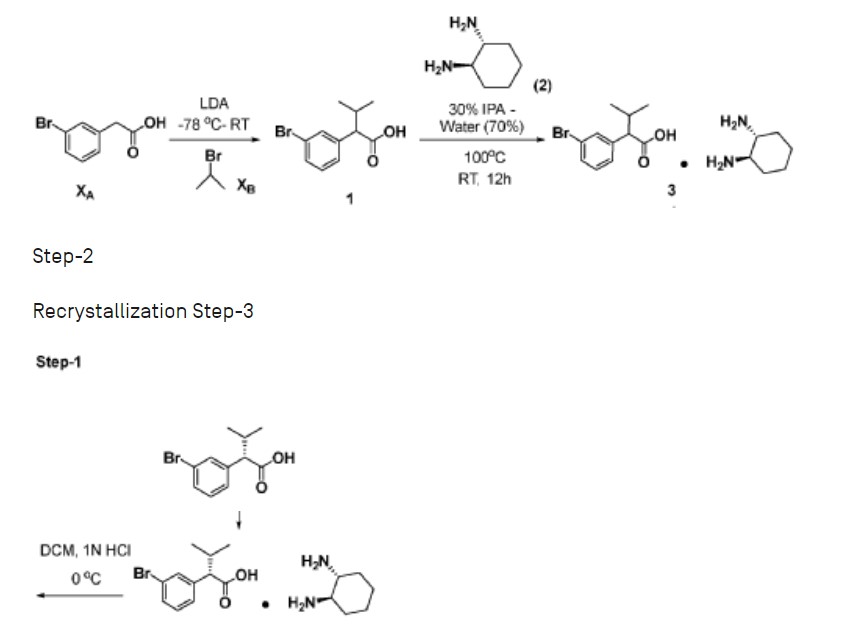
Example 4: Preparation of Compound 44-A via Chiral Synthesis
Preparation of KRM-A (chemical precursor to Compound 44-A)

Step-4
KRM-A
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
[0359] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by drop wise addition of isopropyl bromide (XB, 255 g, 2.07 mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature overnight. The reaction mass was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound (150 g, 83% yield), HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN110590747.
Step-2: Preparation of Compound 3
[0360] 2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IPA in water (10.2 L; 3.06 L of IPA-7.14 L of water) and (1R, 27?)-cyclohexane-l,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then heated to 100 °C till the solution becomes clear and was stirred at same temperature for another 30 min. The reaction mixture was allowed to attain room temperature slowly for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
[0361] Work up for analysis (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C till the clear solution was observed. The compound was extracted into DCM, dried over Na2SC>4 and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
[0362] In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
[0363] The compound 3 (619.90 g) was taken in 30% of IPA in water (12.4 L), then the mixture was heated to 100 °C till the solution becomes clear and stirred at same temperature for another 30min. The reaction mixture was allowed to attain room temperature slowly for 8-12h. The obtained solid was filtered and washed with 500mL 30% IP A- water and dried under vacuum to afford a desired compound (360g, wet).
[0364] Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C till the clear solution was observed and the compound was extracted to DCM, dried over Na2SCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
[0365] The recrystallization method was repeated for three more times by using 30% of IPA in water as described above to get the purity >98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
[0366] The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated and washed brine solution (500 mL) and dried over Na2SO4, the solvent was evaporated to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
[0367] ‘H NMR (400MHz, DMSO-d6): 8 12.5 (s, 1H), 7.50-7.44 (m, 2H), 7.34-7.26 (m, 2H), 3.16 (d, 1H), 2.23-2.11 (m, 1H), 0.98 (d, 3H), 0.63 (d, 3H); Chiral HPLC: 98.50% retention time: 4.588 min.
Preparation of Compound 44-A
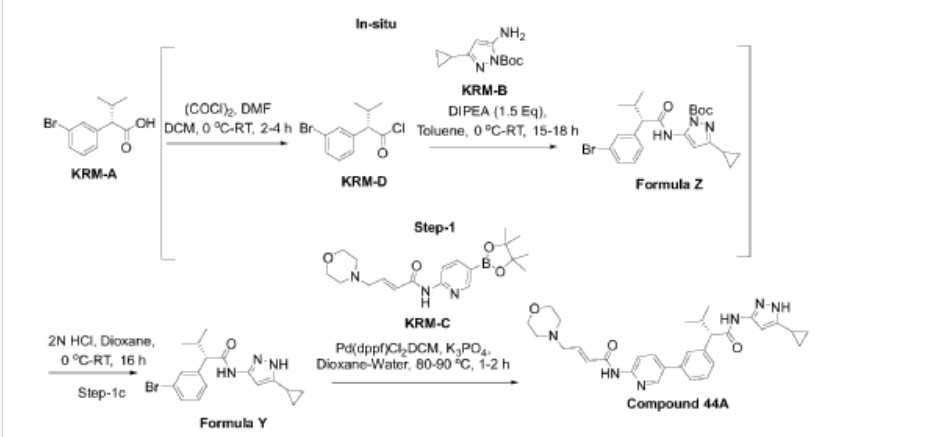

Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylhutanamide
Step- la: Preparation of KRM-D
[0368] To a stirred solution of KRM-A (100g, 0.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0 °C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude 2-(3-bromophenyl)-3 -methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step- lb: Preparation of compound of formula Z
[0369] (S)-2-(3-bromophenyl)-3 -methylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of te/7-butyl 3 -amino-5 -cyclopropyl- IH-pyrazole-l -carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to attain RT and stirred for overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HCI (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL),
brine solution (500 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (5)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (compound of formula Z) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H)+, HPLC: 80.80%, retention time: 15.89 min). The crude product was taken as such for next step without further purification.
Step-lc: Preparation of compound of formula Y
[0370] To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (180 g, 1.731 mol) in dioxane (360 mL) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature.
[0371] After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The resulted compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL) and brine solution (300 mL), and dried over anhydrous Na2SO4. The organic layer was concentrated to get a crude (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula Y) as a semi solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The solid formed was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (110g, Yield: 78% over two steps). LCMS: m/z= 362 (M+H)+, HPLC: 97.66%, retention time: 24.10 min
[0372] Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2-yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound 44A)
[0373] To a degassed solution of (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (£)-4-morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)C12.DCM [l,l’-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane] (3.38 g, 0.0042 mol, and 0.03eq) was added and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
[0374] After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulphate. The organic layer was concentrated under vacuum at 35-40°C to get crude title compound (~80g).
[0375] The crude compound 44A, (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20 % aqueous Na2COs solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulphate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), stirred overnight at room temperature and filtered through Celite® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, till absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound 44A (70g, HPLC purity: 92.70%, retention time: 15.65 min).
[0376] ‘ H NMR (400MHz, DMSO-^): <5: 10.79 (s, 1H), 10.46 (s, 1H), 8.61 (d, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.69 (s, 1H), 7.56 (d, 1H), 7.39 (m, 2H), 6.84-6.77 (m, 1H), 6.62 (s, 2H), 6.51 (d, 1H), 6.13 (s, 1H), 3.62-3.59 (m, 4H), 3.35 (d, 1H), 3.15-3.13 (m, 2H), 2.42-2.39 (m, 5H), 1.80-1.77 (m, 1H), 0.98 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H);
LCMS: m/z= 529.25-free base (M+H)+, HPLC: 98.98%, retention time: 15.40 min.


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
REF
POSTER SESSION: Molecular Targeted Agents| Volume 138, SUPPLEMENT 2, S47, October 01, 2020
//////Tacaciclib, GTPL12880, AUR-102
CC(C)C(C1=CC=CC(=C1)C2=CN=C(C=C2)NC(=O)C=CCN3CCOCC3)C(=O)NC4=NNC(=C4)C5CC5

NEW DRUG APPROVALS
ONE TIME
$10.00
AUPM 170, CA 170, PD-1-IN-1
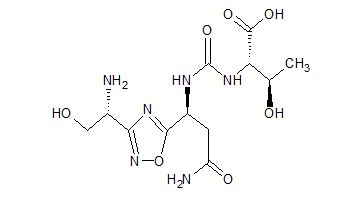



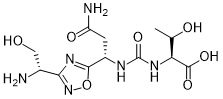
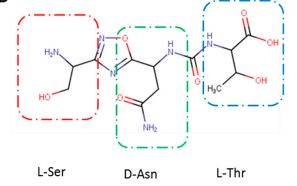
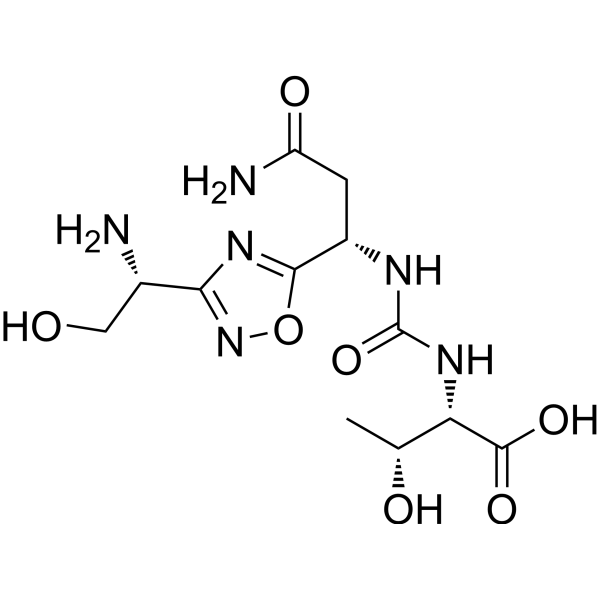
(2S,3R)-2-(3-((S)-3-amino-1-(3-((R)-1-amino-2-hydroxyethyl)-1,2,4-oxadiazol-5-yl)-3-oxopropyl)ureido)-3-hydroxybutanoic acid

| Molecular Weight (MW) | 360.33 |
|---|---|
| Formula | C12H20N6O7 |
| CAS No. | 1673534-76-3 |
N-[[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-L-threonine
L-Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-
AUPM 170, CA 170, AUPM-170, CA-170, PD-1-IN-1
Novel inhibitor of programmed cell dealth-1 (PD-1)
CA-170 (also known as AUPM170 or PD-1-IN-1) is a first-in-class, potent and orally available small molecule inhibitor of the immune checkpoint regulatory proteins PD-L1 (programmed cell death ligand-1), PD-L2 and VISTA (V-domain immunoglobulin (Ig) suppressor of T-cell activation (programmed death 1 homolog; PD-1H). CA-170 was discovered by Curis Inc. and has potential antineoplastic activities. CA-170 selectively targets PD-L1 and VISTA, both of which function as negative checkpoint regulators of immune activation. Curis is currently investigating CA-170 for the treatment of advanced solid tumours and lymphomas in patients in a Phase 1 trial (ClinicalTrials.gov Identifier: NCT02812875).
References: www.clinicaltrials.gov (NCT02812875); WO 2015033299 A1 20150312.
Aurigene Discovery Technologies Limited INNOVATOR
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
CURIS AND AURIGENE ANNOUNCE AMENDMENT OF COLLABORATION FOR THE DEVELOPMENT AND COMMERCIALIZATION OF CA-170
PRESS RELEASE
Curis and Aurigene Announce Amendment of Collaboration for the Development and Commercialization of CA-170
– Aurigene to fund and conduct a Phase 2b/3 randomized study of CA-170 in patients with non-squamous non-small cell lung cancer (nsNSCLC) –
– Aurigene to receive Asia rights for CA-170; Curis entitled to royalty payments in Asia –
LEXINGTON, Mass., February 5, 2020 /PRNewswire/ — Curis, Inc. (NASDAQ: CRIS), a biotechnology company focused on the development of innovative therapeutics for the treatment of cancer, today announced that it has entered into an amendment of its collaboration, license and option agreement with Aurigene Discovery Technologies, Ltd. (Aurigene). Under the terms of the amended agreement, Aurigene will fund and conduct a Phase 2b/3 randomized study evaluating CA-170, an orally available, dual
inhibitor of VISTA and PDL1, in combination with chemoradiation, in approximately 240 patients with nonsquamous
non-small cell lung cancer (nsNSCLC). In turn, Aurigene receives rights to develop and commercialize CA-170 in Asia, in addition to its existing rights in India and Russia, based on the terms of the original agreement. Curis retains U.S., E.U., and rest of world rights to CA-170, and is entitled to receive royalty payments on potential future sales of CA-170 in Asia.
In 2019, Aurigene presented clinical data from a Phase 2a basket study of CA-170 in patients with multiple tumor types, including those with nsNSCLC. In the study, CA-170 demonstrated promising signs of safety and efficacy in nsNSCLC patients compared to various anti-PD-1/PD-L1 antibodies.
“We are pleased to announce this amendment which leverages our partner Aurigene’s expertise and resources to support the clinical advancement of CA-170, as well as maintain our rights to CA-170 outside of Asia,” said James Dentzer, President and Chief Executive Officer of Curis. “Phase 2a data presented at the European Society for Medical Oncology (ESMO) conference last fall supported the potential for CA-170 to serve as a therapeutic option for patients with nsNSCLC. We look forward to working with our partner Aurigene to further explore this opportunity.”
“Despite recent advancements, patients with localized unresectable NSCLC struggle with high rates of recurrence and need for expensive intravenous biologics. The CA-170 data presented at ESMO 2019 from Aurigene’s Phase 2 ASIAD trial showed encouraging results in Clinical Benefit Rate and Prolonged PFS and support its potential to provide clinically meaningful benefit to Stage III and IVa nsNSCLC patients, in combination with chemoradiation and as oral maintenance” said Kumar Prabhash, MD, Professor of Medical Oncology at Tata Memorial Hospital, Mumbai, India.
Murali Ramachandra, PhD, Chief Executive Officer of Aurigene, commented, “Development of CA-170, with its unique dual inhibition of PD-L1 and VISTA, is the result of years of hard-work and commitment by many people, including the patients who participated in the trials, caregivers and physicians, along with the talented teams at Aurigene and Curis. We look forward to further developing CA-170 in nsNSCLC.”
About Curis, Inc.
Curis is a biotechnology company focused on the development of innovative therapeutics for the treatment of cancer, including fimepinostat, which is being investigated in combination with venetoclax in a Phase 1 clinical study in patients with DLBCL. In 2015, Curis entered into a collaboration with Aurigene in the areas of immuno-oncology and precision oncology. As part of this collaboration, Curis has exclusive licenses to oral small molecule antagonists of immune checkpoints including, the VISTA/PDL1 antagonist CA-170, and the TIM3/PDL1 antagonist CA-327, as well as the IRAK4 kinase inhibitor, CA- 4948. CA-4948 is currently undergoing testing in a Phase 1 trial in patients with non-Hodgkin lymphoma.
In addition, Curis is engaged in a collaboration with ImmuNext for development of CI-8993, a monoclonal anti-VISTA antibody. Curis is also party to a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are commercializing Erivedge® for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at http://www.curis.com.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision- oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene currently has several programs from its pipeline in clinical development. Aurigene’s ROR-gamma inverse agonist AUR-101 is currently in phase 2 clinical development under a US FDA IND. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has partnered with many large and mid-pharma companies in the United States and Europe and has 15 programs currently in clinical development. For more information, please visit Aurigene’s website at https://www.aurigene.com/
Curis with the option to exclusively license Aurigene’s orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field
Addressing immune checkpoint pathways is a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients.
Through its collaboration with Aurigene, Curis is now engaged in the discovery and development of the first ever orally bioavailable, small molecule antagonists that target immune checkpoint receptor-ligand interactions, including PD-1/PD-L1 interactions. In the first half of 2016, Curis expects to file an IND application with the U.S. FDA to initiate clinical testing of CA-170, the first small molecule immune checkpoint antagonist targeting PD-L1 and VISTA. The multi-year collaboration with Aurigene is focused on generation of small molecule antagonists targeting additional checkpoint receptor-ligand interactions and Curis expects to advance additional drug candidates for clinical testing in the coming years. The next immuno-oncology program in the collaboration is currently targeting the immune checkpoints PD-L1 and TIM3.
In November 2015, preclinical data were reported. Data demonstrated tha the drug rescued and sustained activation of T cells functions in culture. CA-170 resulted in anti-tumor activity in multiple syngeneic tumor models including melanoma and colon cancer. Similar data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
By August 2015, preclinical data had been reported. Preliminary data demonstrated that in in vitro studies, small molecule PD-L1 antagonists induced effective T cell proliferation and IFN-gamma production by T cells that were specifically suppressed by PD-L1 in culture. The compounds were found to have effects similar to anti-PD1 antibodies in in vivo tumor models
(Oral Small Molecule PD-L1/VISTAAntagonist)
Certain human cancers express a ligand on their cell surface referred to as Programmed-death Ligand 1, or PD-L1, which binds to its cognate receptor, Programmed-death 1, or PD-1, present on the surface of the immune system’s T cells. Cell surface interactions between tumor cells and T cells through PD-L1/PD-1 molecules result in T cell inactivation and hence the inability of the body to mount an effective immune response against the tumor. It has been previously shown that modulation of the PD-1 mediated inhibition of T cells by either anti-PD1 antibodies or anti-PD-L1 antibodies can lead to activation of T cells that result in the observed anti-tumor effects in the tumor tissues. Therapeutic monoclonal antibodies targeting the PD-1/PD-L1 interactions have now been approved by the U.S. FDA for the treatment of certain cancers, and multiple therapeutic monoclonal antibodies targeting PD-1 or PD-L1 are currently in development.
In addition to PD-1/PD-L1 immune regulators, there are several other checkpoint molecules that are involved in the modulation of immune responses to tumor cells1. One such regulator is V-domain Ig suppressor of T-cell activation or VISTA that shares structural homology with PD-L1 and is also a potent suppressor of T cell functions. However, the expression of VISTA is different from that of PD-L1, and appears to be limited to the hematopoietic compartment in tissues such as spleen, lymph nodes and blood as well as in myeloid hematopoietic cells within the tumor microenvironment. Recent animal studies have demonstrated that combined targeting/ blockade of PD-1/PD-L1 interactions and VISTA result in improved anti-tumor responses in certain tumor models, highlighting their distinct and non-redundant functions in regulating the immune response to tumors2.
As part of the collaboration with Aurigene, in October 2015 Curis licensed a first-in-class oral, small molecule antagonist designated as CA-170 that selectively targets PD-L1 and VISTA, both of which function as negative checkpoint regulators of immune activation. CA-170 was selected from the broad PD-1 pathway antagonist program that the companies have been engaged in since the collaboration was established in January 2015. Preclinical data demonstrate that CA-170 can induce effective proliferation and IFN-γ (Interferon-gamma) production (a cytokine that is produced by activated T cells and is a marker of T cell activation) by T cells that are specifically suppressed by PD-L1 or VISTA in culture. In addition, CA-170 also appears to have anti-tumor effects similar to anti-PD-1 or anti-VISTA antibodies in multiple in vivo tumor models and appears to have a good in vivo safety profile. Curis expects to file an IND and initiate clinical testing of CA-170 in patients with advanced tumors during the first half of 2016.
 Jan 21, 2015
Jan 21, 2015
Curis and Aurigene Announce Collaboration, License and Option Agreement to Discover, Develop and Commercialize Small Molecule Antagonists for Immuno-Oncology and Precision Oncology Targets
— Agreement Provides Curis with Option to Exclusively License Aurigene’s Antagonists for Immuno-Oncology, Including an Antagonist of PD-L1 and Selected Precision Oncology Targets, Including an IRAK4 Kinase Inhibitor —
— Investigational New Drug (IND) Application Filings for Both Initial Collaboration Programs Expected this Year —
— Curis to issue 17.1M shares of its Common Stock as Up-front Consideration —
— Management to Host Conference Call Today at 8:00 a.m. EST —
LEXINGTON, Mass. and BANGALORE, India, Jan. 21, 2015 (GLOBE NEWSWIRE) — Curis, Inc. (Nasdaq:CRIS), a biotechnology company focused on the development and commercialization of innovative drug candidates for the treatment of human cancers, and Aurigene Discovery Technologies Limited, a specialized, discovery stage biotechnology company developing novel therapies to treat cancer and inflammatory diseases, today announced that they have entered into an exclusive collaboration agreement focused on immuno-oncology and selected precision oncology targets. The collaboration provides for inclusion of multiple programs, with Curis having the option to exclusively license compounds once a development candidate is nominated within each respective program. The partnership draws from each company’s respective areas of expertise, with Aurigene having the responsibility for conducting all discovery and preclinical activities, including IND-enabling studies and providing Phase 1 clinical trial supply, and Curis having responsibility for all clinical development, regulatory and commercialization efforts worldwide, excluding India and Russia, for each program for which it exercises an option to obtain a license.
The first two programs under the collaboration are an orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field and an orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
“We are thrilled to partner with Aurigene in seeking to discover, develop and commercialize small molecule drug candidates generated from Aurigene’s novel technology and we believe that this collaboration represents a true transformation for Curis that positions the company for continued growth in the development and eventual commercialization of cancer drugs,” said Ali Fattaey, Ph.D., President and Chief Executive Officer of Curis. “The multi-year nature of our collaboration means that the parties have the potential to generate a steady pipeline of novel drug candidates in the coming years. Addressing immune checkpoint pathways is now a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients. Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers. We look forward to advancing these programs into clinical development later this year.”
Dr. Fattaey continued, “Aurigene has a long and well-established track record of generating targeted small molecule drug candidates with bio-pharmaceutical collaborators and we have significantly expanded our drug development capabilities as we advance our proprietary drug candidates in currently ongoing clinical studies. We believe that we are well-positioned to advance compounds from this collaboration into clinical development.”
CSN Murthy, Chief Executive Officer of Aurigene, said, “We are excited to enter into this exclusive collaboration with Curis under which we intend to discover and develop a number of drug candidates from our chemistry innovations in the most exciting fields of cancer therapy. This unique collaboration is an opportunity for Aurigene to participate in advancing our discoveries into clinical development and beyond, and mutually align interests as provided for in our agreement. Our scientists at Aurigene have established a novel strategy to address immune checkpoint targets using small molecule chemical approaches, and have discovered a number of candidates that modulate these checkpoint pathways, including PD-1/PD-L1. We have established a large panel of preclinical tumor models in immunocompetent mice and can show significant in vivo anti-tumor activity using our small molecule PD-L1 antagonists. We are also in the late stages of selecting a candidate that is a potent and selective inhibitor of the IRAK4 kinase, demonstrating excellent in vivo activity in preclinical tumor models.”
In connection with the transaction, Curis has issued to Aurigene approximately 17.1 million shares of its common stock, or 19.9% of its outstanding common stock immediately prior to the transaction, in partial consideration for the rights granted to Curis under the collaboration agreement. The shares issued to Aurigene are subject to a lock-up agreement until January 18, 2017, with a portion of the shares being released from the lock-up in four equal bi-annual installments between now and that date.
The agreement provides that the parties will collaborate exclusively in immuno-oncology for an initial period of approximately two years, with the option for Curis to extend the broad immuno-oncology exclusivity.
In addition Curis has agreed to make payments to Aurigene as follows:
- for the first two programs: up to $52.5 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any;
- for the third and fourth programs: up to $50 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any; and
- for any program thereafter: up to $140.5 million per program, including $87.5 million per program in approval and commercial milestones, plus specified approval milestone payments for additional indications, if any.
Curis has agreed to pay Aurigene royalties on any net sales ranging from high single digits to 10% in territories where it successfully commercializes products and will also share in amounts that it receives from sublicensees depending upon the stage of development of the respective molecule.
About Immune Checkpoint Modulation and Programmed Death 1 Pathway
Modulation of immune checkpoint pathways has emerged as a highly promising therapeutic approach in a wide range of human cancers. Immune checkpoints are critical for the maintenance of self-tolerance as well as for the protection of tissues from excessive immune response generated during infections. However, cancer cells have the ability to modulate certain immune checkpoint pathways as a mechanism to evade the immune system. Certain immune checkpoint receptors or ligands are expressed by various cancer cells, targeting of which may be an effective strategy for generating anti-tumor activity. Some immune-checkpoint modulators, such as programmed death 1 (PD-1) protein, specifically regulate immune cell effector functions within tissues. One of the mechanisms by which tumor cells block anti-tumor immune responses in the tumor microenvironment is by upregulating ligands for PD-1, such as PD-L1. Hence, targeting of PD-1 and/or PD-L1 has been shown to lead to the generation of effective anti-tumor responses.
About Curis, Inc.
Curis is a biotechnology company focused on the development and commercialization of novel drug candidates for the treatment of human cancers. Curis’ pipeline of drug candidates includes CUDC-907, a dual HDAC and PI3K inhibitor, CUDC-427, a small molecule antagonist of IAP proteins, and Debio 0932, an oral HSP90 inhibitor. Curis is also engaged in a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are developing and commercializing Erivedge®, the first and only FDA-approved medicine for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at www.curis.com.
About Aurigene
Aurigene is a specialized, discovery stage biotechnology company, developing novel and best-in-class therapies to treat cancer and inflammatory diseases. Aurigene’s Programmed Death pathway program is the first of several immune checkpoint programs that are at different stages of discovery and preclinical development. Aurigene has partnered with several large- and mid-pharma companies in the United States and Europe and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies. Aurigene is an independent, wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (NYSE:RDY). For more information, please visit Aurigene’s website at http://aurigene.com/.
POSTER


WO2011161699, WO2012/168944, WO2013144704 and WO2013132317 report peptides or peptidomimetic compounds which are capable of suppressing and/or inhibiting the programmed cell death 1 (PD1) signaling pathway.
PATENT

Inventors
- SASIKUMAR, Pottayil Govindan Nair
- RAMACHANDRA, Muralidhara
- NAREMADDEPALLI, Seetharamaiah Setty Sudarshan
Priority Data
| 4011/CHE/2013 | 06.09.2013 | IN |
Example 4: Synthesis of Co
The compound was synthesised using similar procedure as depicted in Example 2 for synthesising compound 2 using
instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.35 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.2 (M+H)+, HPLC: tR = 12.19 min.


REFERENCES
US20150073024
| WO2011161699A2 | 27 Jun 2011 | 29 Dec 2011 | Aurigene Discovery Technologies Limited | Immunosuppression modulating compounds |
| WO2012168944A1 | 21 Dec 2011 | 13 Dec 2012 | Aurigene Discovery Technologies Limited | Therapeutic compounds for immunomodulation |
| WO2013132317A1 | 4 Mar 2013 | 12 Sep 2013 | Aurigene Discovery Technologies Limited | Peptidomimetic compounds as immunomodulators |
| WO2013144704A1 | 28 Mar 2013 | 3 Oct 2013 | Aurigene Discovery Technologies Limited | Immunomodulating cyclic compounds from the bc loop of human pd1 |
http://www.curis.com/pipeline/immuno-oncology/pd-l1-antagonist
http://www.curis.com/images/stories/pdfs/posters/Aurigene_PD-L1_VISTA_AACR-NCI-EORTC_2015.pdf
References:
1) https://bmcimmunol.biomedcentral.com/articles/10.1186/s12865-021-00446-4
2) https://www.nature.com/articles/s42003-021-02191-1
3) https://www.esmoopen.com/article/S2059-7029(20)30108-3/fulltext
4) https://www.mdpi.com/1420-3049/24/15/2804
////////Curis, Aurigene, AUPM 170, CA 170, AUPM-170, CA-170, PD-L1, VISTA antagonist, PD-1-IN-1, phase 2, CANCER
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)[C@@H](C)O)CC(N)=O

NEW DRUG APPROVALS
ONE TIME
$9.00
AUR 101
AUR 101
AUR101-201
ANTIINNFLAMATORY
AUR-101, a ROR gamma inverse agonist for autoimmune disorders like psoriasis
AUR-101 is an ROR-gammaT inverse agonist in phase II clinical development at Aurigene for the treatment of patients with moderate-to-severe chronic plaque-type psoriasis.
- DrugsAUR 101 (Primary)
- IndicationsPlaque psoriasis
- FocusAdverse reactions; First in man
- AcronymsINDUS
- SponsorsAurigene Discovery Technologies
- OriginatorAurigene Discovery Technologies
- ClassAntipsoriatics; Small molecules
- Mechanism of ActionNuclear receptor subfamily 1 group F member 3 inverse agonists
- Phase IIPsoriasis
- 28 Aug 2021No recent reports of development identified for phase-I development in Psoriasis(In volunteers) in Australia (PO, Tablet)
- 23 Apr 2021Aurigene Discovery Technologies plans a phase II INDUS-3 trial for Psoriasis in USA (PO) in May 2021 (NCT04855721)
- 15 Apr 2021Aurigene Discovery Technologies completes a phase II trial in Psoriasis in India (PO) (NCT04207801)
- CDSCO
- https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/Aurigene20.pdf
- NCT04207801
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
AURIGENE ANNOUNCES FIRST PATIENT DOSED WITH AUR101 IN PHASE II STUDY IN PATIENTS WITH MODERATE TO SEVERE PSORIASIS
PRESS RELEASE
Aurigene Announces First Patient Dosed with AUR101 in Phase II Study in Patients with Moderate to Severe Psoriasis
Bangalore, February 17, 2020 — Aurigene, a development stage biotechnology company, today announced dose administration for the first patient in INDUS-2, a Phase II double blind placebo-controlled three-arm study of AUR101 in patients with moderate to severe psoriasis. AUR101 is an oral small molecule inverse agonist of RORγ and has shown desirable pharmacodynamic modulation of IL-17 and acceptable safety in a completed Phase I human study conducted in Australia.
“The initiation of this Phase II study under a US FDA IND represents a significant milestone for Aurigene, as it marks the first program which Aurigene has led from the bench side to the clinic all by itself,” said Murali Ramachandra, PhD, Chief Executive Officer of Aurigene. “We look forward to producing important clinical data by the end of 2020 to guide our future development plans and demonstrating Aurigene’s unique expertise in conducting Proof-of-Concept studies in a quality and fast-paced manner.”
About AUR101-201 and the Phase II Study of AUR101 in Patients with Moderate to Severe Psoriasis
The purpose of the Phase II multi-center, blinded, placebo-controlled, three-arm study is to evaluate the clinical activity of AUR101 in patients with moderate to severe psoriasis. In two of the arms, AUR101 will be administered twice daily, at 400 mg PO BID and 600 mg PO BID, for 12 weeks. Patients in the third arm will receive matched blinded placebo in a double dummy fashion. The trial is listed at clinicaltrials.gov with identifier NCT04207801.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY,NYSE: RDY). Aurigene is focused on precision- oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene currently has several programs from its pipeline in clinical development. Aurigene has also submitted an IND to DCGI, India for a Phase IIb/III trial of CA-170, a dual inhibitor of PD-L1 and VISTA, in non-squamous NSCLC. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has partnered with many large and mid-pharma companies in the United States and Europe and has 15 programs currently in clinical development. For more information, please visit Aurigene’s website at https://www.aurigene.com/.
CLIP
Signalling of multiple interleukin (IL)-17 family cytokines via IL-17 receptor A drives psoriasis-related inflammatory pathways
https://onlinelibrary.wiley.com/doi/10.1111/bjd.20090
M.A.X. Tollenaere,J. Hebsgaard,D.A. Ewald,P. Lovato,S. Garcet,X. Li,S.D. Pilger,M.L. Tiirikainen,M. Bertelsen,J.G. Krueger,H. Norsgaard,First published: 01 April 2021 https://doi.org/10.1111/bjd.20090Citations: 2Funding sources LEO Pharma A/S funded this study.Conflicts of interest M.A.X.T., J.H., D.A.E., P.L., S.D.P., M.L.T., M.B. and H.N. are employees of LEO Pharma. J.G.K. received grants paid to his institution from Novartis, Pfizer, Amgen, Lilly, Boehringer, Innovaderm, BMS, Janssen, AbbVie, Paraxel, LEO Pharma, Vitae, Akros, Regeneron, Allergan, Novan, Biogen MA, Sienna, UCB, Celgene, Botanix, Incyte, Avillion and Exicure; and personal fees from Novartis, Pfizer, Amgen, Lilly, Boehringer, Biogen Idec, AbbVie, LEO Pharma, Escalier, Valeant, Aurigene, Allergan, Asana, UCB, Sienna, Celgene, Nimbus, Menlo, Aristea, Sanofi, Sun Pharma, Almirall, Arena and BMS.Data Availability Statement The gene array dataset described in this publication has been deposited in NCBI’s Gene Expression Omnibus and is accessible through GEO Series accession number GSE158448 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158448).
CLOP
https://www.drugdiscoverychemistry.com/Anti-Inflammatories/16
10:35 Small Molecule Inhibitors of RORgamma and IRAK4 for the Treatment of Autoimmune Disorders
 Susanta Samajdar, Ph.D., Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Susanta Samajdar, Ph.D., Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Although biologics such as anti-TNFα antibody are fairly successful in the treatment of autoimmune disorders, there is significant unmet need due to heterogeneity in diseases and lack of response to established therapies in some patients. While biologics typically target one cytokine signaling pathway, small molecule therapeutics directed towards intracellular target(s) can interfere in the signaling from multiple cytokines potentially leading to improved response. Development of small molecule oral inhibitors of IRAK4 and RORgamma to target TLR/IL-R and Th17 pathway respectively will be discussed.
PATENT
2448/CHE/2015 15.05.2015 IN
PATENT
PATENT
This application claims the benefit of Indian provisional application number 5641/CHE/2013 filed on 06th December 2013 which hereby incorporated by reference.
PATENT
- KOTRABASAIAH UJJINAMATADA, Ravi
- PANDIT, Chetan
2049005-13-0
2-Quinolinecarboxamide, 6-(2,6-dimethyl-4-pyrimidinyl)-N-[[4-(ethylsulfonyl)phenyl]methyl]-5,6,7,8-tetrahydro-6-methyl-5-oxo-, (6S)-
Molecular Weight492.59, C26 H28 N4 O4 S

EXAMPLE
PATENT
CLIP
https://www.sciencedirect.com/science/article/abs/pii/S0223523419301011
2013239366 CA 170

NEW DRUG APPROVALS
ONE TIME
$10.00
///////////////////////AUR 101, AURIGENE, ROR, IL-17, PHASE 2, CDSCO, Ravi Ujjinamatada, KOTRABASAIAH UJJINAMATADA Ravi, PANDIT Chetan, AUR101-201, plaque-type psoriasis


Ravi Ujjinamatada
XL 114, AUR 104 and XL 102, AUR 102 (NO CONCLUSIONS, ONLY PREDICTIONS)

XL 114
FOR BOTH, JUST PREDICTION
PREDICTIONS
or


N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
![(2S)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117943691&t=l)

(2S)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
CAS 2305027-62-5
C12 H20 N6 O7, 360.32Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-, (2S,3ξ)-N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
ALSO SEE


![(2S,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://drugapprovalsint.com/wp-content/uploads/2021/10/str1-10.jpg)
1673534-76-3C12 H20 N6 O7, 360.32
L-Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]
(2S,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acidN-[[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-L-threonine
CAS 1673534-76-3
PD-1-IN-1 free base, EX-A1918, CS-6240, NSC-799645, CA-170 (AUPM-170)|PDL1 inhibitor, HY-101093, PD-1-IN-1
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)[C@@H](C)O)CC(N)=O
XL 114, AUR 104
A novel covalent inhibitor of FABP5 for cancer therapy
XL 102, AUR 102
A potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7)
NO CONCLUSIONS, ONLY PREDICTIONS
PREDICTIONS MORE
![(2R,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155529077&t=l)

(2R,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
C12H20N6O7, 360.32
![(2S,3S)-2-[[(1S)-3-Amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155339943&t=l)

(2S,3S)-2-[[(1S)-3-amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
XL102, AUR 102
XL102 is a potent, selective and orally bioavailable covalent inhibitor of CDK7, which is an important regulator of the cellular transcriptional and cell cycle machinery. CDK7 helps regulate cell cycle progression, with overexpression observed in multiple cancers, such as breast, prostate and ovarian cancers. In preclinical studies, XL102 revealed potent anti-proliferative activity, induced cell death in a large panel of cancer cell lines and caused tumor growth inhibition and regression in xenograft models, demonstrating its potential as a targeted antitumor agent.
In late 2020, Exelixis exercised its option to in-license XL102 (formerly AUR102) from Aurigene per the companies’ July 2019 collaboration, option and license agreement. Exelixis has assumed responsibility for the future clinical development, manufacturing and commercialization of XL102. Aurigene retains limited development and commercial rights for India and Russia.
SYN

ABOUT Fatty acid-binding proteins (FABPs)
Fatty acid-binding proteins (FABPs) are involved in binding and storing hydrophobic ligands such as long-chain fatty acids, as well as transporting them to the appropriate compartments in the cell. Epidermal fatty acid-binding protein (FABP5) is an intracellular lipid-binding protein that is abundantly expressed in adipocytes and macrophages. Previous studies have revealed that the FABP5 expression level is closely related to malignancy in various types of cancer. However, its precise functions in the metabolisms of cancer cells remain unclear. Here, we revealed that FABP5 knockdown significantly induced downregulation of the genes expression, such as hormone-sensitive lipase (HSL), monoacylglycerol lipase (MAGL), elongation of long-chain fatty acid member 6 (Elovl6), and acyl-CoA synthetase long-chain family member 1 (ACSL1), which are involved in altered lipid metabolism, lipolysis, and de novo FA synthesis in highly aggressive prostate and breast cancer cells. Moreover, we demonstrated that FABP5 induced inflammation and cytokine production through the nuclear factor-kappa B signaling pathway activated by reactive oxygen species and protein kinase C in PC-3 and MDA-MB-231 cells. Thus, FABP5 might regulate lipid quality and/or quantity to promote aggressiveness such as cell growth, invasiveness, survival, and inflammation in prostate and breast cancer cells. In the present study, we have revealed for the first time that high expression of FABP5 plays a critical role in alterations of lipid metabolism, leading to cancer development and metastasis in highly aggressive prostate and breast cancer cells.
Fatty acid-binding protein, epidermal is a protein that in humans is encoded by the FABP5 gene
Function
This gene encodes the fatty acid binding protein found in epidermal cells, and was first identified as being upregulated in psoriasis tissue. Fatty acid binding proteins are a family of small, highly conserved, cytoplasmic proteins that bind long-chain fatty acids and other hydrophobic ligands. It is thought that FABPs roles include fatty acid uptake, transport, and metabolism.[6]
The phytocannabinoids (THC and CBD) inhibit endocannabinoid anandamide (AEA) uptake by targeting FABP5, and competition for FABPs may in part or wholly explain the increased circulating levels of endocannabinoids reported after consumption of cannabinoids.[7] Results show that cannabinoids inhibit keratinocyte proliferation, and therefore support a potential role for cannabinoids in the treatment of psoriasis.[8]
Interactions
FABP5 has been shown to interact with S100A7.[
ABOUT CD47/SIRPa axis
CD47/SIRPa axis is established as a critical regulator of myeloid cell activation and serves as an immune checkpoint for macrophage mediated phagocytosis. Because of its frequent upregulation in several cancers, CD47 contributes to immune evasion and cancer progression. CD47 regulates phagocytosis primarily through interactions with SIRPla expressed on macrophages. Blockade of SIRPla/CD47 has been shown to dramatically enhance tumor cell phagocytosis and dendritic cells maturation for better antigen presentation leading to substantially improved antitumor responses in preclinical models of cancer (M. P. Chao et al. Curr Opin Immunol. 2012 (2): 225-232). Disruption of CD47-SIRPa interaction is now being evaluated as a therapeutic strategy for cancer with the use of monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys.
CD47 is expressed on virtually all non-malignant cells, and blocking the CD47 or the loss of CD47 expression or changes in membrane distribution can serve as markers of aged or damaged cells, particularly on red blood cells (RBC). Alternatively, blocking SIRPa also allows engulfment of targets that are not normally phagocytosed, for those cells where pre-phagocytic signals are also present. CD47 is a broadly expressed transmembrane glycoprotein with a single Ig-like domain and five membrane- spanning regions, which functions as a cellular ligand for SIRPa with binding mediated through the NH2-terminal V-like domain of SIRPa. SIRPa is expressed primarily on myeloid cells, including macrophages, granulocytes, myeloid dendritic cells (DCs), mast cells, and their precursors, including hematopoietic stem cells.
CD47 is also constitutively upregulated on a number of cancers such as Non-Hodgkin Lymphoma (NHL), Acute myeloid leukemia (AML), breast, colon, glioblastoma, glioma, ovarian, bladder and prostate cancers, etc. Overexpression of CD47 by tumor cells, which efficiently helps them to escape immune surveillance and killing by innate immune cells. However, in most of the tumor types, blockade of the CD47-SIRPa interaction as a single agent may not be capable of inducing significant phagocytosis and antitumor immunity, necessitating the need to combine with other therapeutic agents. The concomitant engagement of activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors (collectively known as “eat-me” signals) may be necessary for exploiting the maximum potential of the CD-47-SIPRa pathway blockade.
The role of engagement of prophagocytic receptors is proved by inefficiency to trigger phagocytosis either by anti-CD47 F(ab) fragments, single chain variable fragments of CD-47 or non-Fc portion- containing SIRPa proteins in blocking of the CD47-SIRPa interaction. When activating prophagocytic receptors are engaged, as evident in the case of using Fc portion-containing blocking anti-CD47 antibodies, CD47- SIRPa blockade is able to trigger more efficient phagocytosis. Combining CD47-SIRPa blocking agents with therapeutic antibodies (Fc-containing) targeting tumor antigens stimulate activating Fc receptors (FcRs) leading to efficient phagocytosis. The Fc portion of therapeutic antibody targeting tumor antigen also induces antibody-dependent cellular cytotoxicity (ADCC), which also adds to the therapeutic efficacy. Hence antibodies selected from the group consisting of rituximab, herceptin, trastuzumab, alemtuzumab, bevacizumab, cetuximab and panitumumab, daratumumab due to its tumor targeting nature and ADCC, can trigger more efficient phagocytosis.
Earlier approaches to disrupt CD47- SIRPa interaction utilized monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys fused to Fc fragment. However, a concern with this approach is that CD47 is highly expressed on both hematopoietic and non-hematopoietic normal cells. Hence along with tumor cells CD47-SIRPa blocking agents containing Fc-portion may also target many normal cells potentially leading to their elimination by macrophages. The interaction of blocking antibodies with normal cells is considered as a major safety issue resulting in anemia, thrombocytopenia, and leukopenia. These agents may also affect solid tissues rich in macrophages such as liver, lung, and brain. Hence it may be ideal to block the CD47- SIRPa interaction by agents devoid of Fc portion, such as small
molecules, peptides, Fab fragments etc. while activating prophagocytic receptors in tumor cells by appropriate combinations to induce efficient phagocytosis of tumor cells.
Apart from Fc Receptors, a number of other prophagocytic receptors are also reported to promote engulfment of tumor cells in response to CD47-SIRPa blockade by triggering the phagocytosis. These include receptors for SLAMF7, Mac-l, calreticulin and possibly yet to identified receptors. B cell tumor lines such as Raji and other diffuse large B cell lymphoma express SLAMF7 and are implicated in triggering prophagocytic signals during CD47-SIRPa blockade.
Therapeutic agents known to activate prophagocytic receptors are also therefore ideal partners for use in combination with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These agents include proteasome inhibitors (bortezomib, ixazomib and carfilzomib), Anthracyclines (Doxorubicin, Epirubicin, Daunorubicin, Idarubicin, Mitoxantrone) Oxaliplatin, Cyclophosphamide, Bleomycin, Vorinostat, Paclitaxel, 5-Fluorouracil, Cytarabine, BRAF inhibitory drugs (Dabrafenib, Vemurafenib), PI3K inhibitor, Docetaxel, Mitomycin C, Sorafenib, Tamoxifen and oncolytic viruses.
Apart from the specific agents known to have effect on‘eat me’ signals other agents including Abiraterone acetate, Afatinib, Aldesleukin, Aldesleukin, Alemtuzumab, Anastrozole, Axitinib, Belinostat, Bendamustine, Bicalutamide, Blinatumomab, Bosutinib, Brentuximab, Busulfan, Cabazitaxel, Capecitabine, Carboplatin, Carfilzomib, Carmustine, Ceritinib, Clofarabine, Crizotinib, Dacarbazine, Dactinomycin, Dasatinib, Degarelix, Denileukin, Denosumab, Enzalutamide, Eribulin, Erlotinib, Everolimus, Exemestane, Exemestane, Fludarabine, Fulvestrant, Gefitinib, Goserelin, Ibritumomab, Imatinib, Ipilimumab, Irinotecan, Ixabepilone, Lapatinib, Lenalidomide, Letrozole, Leucovorin, Leuprolide, Lomustine, Mechlorethamine, Megestrol, Nelarabine, Nilotinib, Nivolumab, Olaparib, Omacetaxine, Palbociclib, Pamidronate, Panitumumab, Panobinostat, Pazopanib, Pegaspargase, Pembrolizumab, Pemetrexed Disodium, Pertuzumab, Plerixafor, Pomalidomide, Ponatinib, Pralatrexate, Procarbazine, Radium 223, Ramucirumab, Regorafenib, rIFNa-2b, Romidepsin, Sunitinib, Temozolomide, Temsirolimus, Thiotepa, Tositumomab, Trametinib, Vinorelbine, Methotrexate, Ibrutinib, Aflibercept, Toremifene, Vinblastine, Vincristine, Idelalisib, Mercaptopurine and Thalidomide could potentially have effect on‘eat me’ signal pathway on combining with CD-47-SIRPa blocking agents.
In addition to the therapeutic agents mentioned above, other treatment modalities that are in use in cancer therapy also activate prophagocytic receptors, and thus can be combined with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These include Hypericin-based photodynamic therapy (Hyp-PDT), radiotherapy, High-hydrostatic pressure, Photofrin-based PDT and Rose Bengal acetate -based PDT.
However, there is an unmet need for combining small molecule CD-47-SIRPa pathway inhibitors with agents capable of stimulating activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors, or combining with other treatment modalities that are in use in cancer therapy to activate prophagocytic receptors for exploiting the maximum potential of the CD-47- SIRPa pathway blockade.
CLIP
Exelixis In-Licenses Second Anti-Cancer Compound from Aurigene Following FDA Acceptance of Investigational New Drug Application for Phase 1 Clinical Trial in Non-Hodgkin’s Lymphoma
– Robust preclinical data support Exelixis’ clinical development of XL114, with phase 1 trial in Non-Hodgkin’s lymphoma expected to begin in the coming months –
– Exelixis will make an option exercise payment of $10 million to Aurigene –
https://www.businesswire.com/news/home/20211014005549/en/Exelixis-In-Licenses-Second-Anti-Cancer-Compound-from-Aurigene-Following-FDA-Acceptance-of-Investigational-New-Drug-Application-for-Phase-1-Clinical-Trial-in-Non-Hodgkin%E2%80%99s-LymphomaOctober 14, 2021 08:00 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today announced that Exelixis has exercised its exclusive option under the companies’ July 2019 agreement to in-license XL114 (formerly AUR104), a novel anti-cancer compound that inhibits the CARD11-BCL10-MALT1 (CBM) signaling pathway, which promotes lymphocyte survival and proliferation. Exelixis has now assumed responsibility for the future clinical development, commercialization and global manufacturing of XL114. Following the U.S. Food and Drug Administration’s (FDA) recent acceptance of its Investigational New Drug (IND) application, Exelixis will soon initiate a phase 1 clinical trial evaluating XL114 monotherapy in patients with Non-Hodgkin’s lymphoma (NHL). At the American Association of Cancer Research Annual Meeting in April of this year, Aurigene presented preclinical data (Abstract 1266) demonstrating that XL114 exhibited potent anti-proliferative activity in a large panel of cancer cell lines ranging from hematological cancers to solid tumors with excellent selectivity over normal cells. In addition, oral dosing of XL114 resulted in significant dose-dependent tumor growth inhibition in diffuse large B-cell lymphoma (DLBCL) and colon carcinoma models.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline”
XL114 is the second molecule that Exelixis in-licensed from Aurigene under the companies’ July 2019 collaboration, option and license agreement. Exelixis previously exercised its option to in-license XL102, a potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7), from Aurigene in December 2020 and initiated a phase 1 trial of XL102 as a single agent and in combination with other anti-cancer agents in patients with advanced or metastatic solid tumors in January 2021.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline,” said Peter Lamb, Ph.D., Executive Vice President, Scientific Strategy and Chief Scientific Officer, Exelixis. “XL114 has shown potent anti-proliferative activity in lymphoma cell lines that have aberrant activation of the CBM signaling pathway and may have a differentiated profile and potential as a best-in-class molecule that could improve outcomes for patients with Non-Hodgkin’s lymphoma and other hematologic cancers.”
XL114 was identified to have anti-proliferative activity in cell lines with constitutive activation of CBM signaling, including activated B-cell-like DLBCL (ABC-DLBCL), mantle cell lymphoma and follicular lymphoma cell lines. Further characterization of XL114 in cell-based assays demonstrated a functional role in B-cell (BCR) signaling pathways. Additionally, XL114 showed dose-dependent tumor growth inhibition in an ABC-DLBCL mouse xenograft tumor model. In preclinical development, XL114 also demonstrated a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. While the precise molecular mechanism underlying XL114’s function in repressing BCR signaling and MALT1 activation has yet to be characterized, the fatty acid-binding protein 5 (FABP5) has been identified as a prominent XL114-binding target.
“XL114 is the second molecule that Exelixis has opted to in-license under our July 2019 agreement, underscoring the significant potential of our approach to the discovery and preclinical development of innovative cancer therapies that target novel mechanisms of action,” said Murali Ramachandra, Ph.D., Chief Executive Officer, Aurigene. “Exelixis has a track record of success in the clinical development and commercialization of anti-cancer therapies that provide patients with important new treatment options, and we are pleased that the continued advancement of XL114 will be supported by the company’s extensive clinical, regulatory and commercialization infrastructure.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to obtain an exclusive license from Aurigene to three preexisting programs, including the compounds now known as XL102 and XL114. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for an additional upfront payment of $2.5 million per program. The collaboration was expanded in 2021 to include three additional early discovery programs. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all nine programs. Exelixis may exercise its option for a program at any time up until the first IND for the program becomes effective. Having exercised options on two programs thus far (XL102 and XL114), if and when Exelixis exercises a future option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. To exercise its option for XL114, Exelixis will make an option exercise payment to Aurigene of $10 million. Once Exelixis exercises its option for a program, Aurigene will be eligible for clinical development, regulatory and sales milestones, as well as royalties on future potential sales of the compound. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene Discovery Technologies Limited is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY, NSEIFSC: DRREDDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the U.S. and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of the Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. In November 2020, the company was named to Fortune’s 100 Fastest-Growing Companies list for the first time, ranking 17th overall and the third-highest biopharmaceutical company. For more information about Exelixis, please visit www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.

Dinesh Chikkanna
Director, Medicinal Chemistry Aurigene Discovery Technologies

Murali Ramachandra
CEO at Aurigene Discovery Technologies

CLIP
https://cancerres.aacrjournals.org/content/81/13_Supplement/1266
Abstract 1266: Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapyDinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar and Murali Ramachandra
DOI: 10.1158/1538-7445.AM2021-1266 Published July 2021
Proceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Dysregulated fatty acid metabolism is thought to be a hallmark of cancer, wherein fatty acids function both as an energy source and as signals for enzymatic and transcriptional networks contributing to malignancy. Fatty acid-binding protein 5 (FABP5) is an intracellular protein that facilitates transport of fatty acids and plays a role in regulating the expression of genes associated with cancer progression such as cell growth, survival, and metastasis. Overexpression of FABP5 has been reported to contribute to an aggressive phenotype and a poor survival correlation in several cancers. Therefore, inhibition of FABP5 is considered as a therapeutic approach for cancers. Phenotypic screening of a library of covalent compounds for selective sensitivity of cancer cells followed by medicinal chemistry optimization resulted in the identification of AUR104 with desirable properties. Chemoproteomic-based target deconvolution revealed FABP5 as the cellular target of AUR104. Covalent adduct formation with Cys43 of FABP5 by AUR104 was confirmed by mass spectrometry. Target occupancy studies using a biotin-tagged AUR104 demonstrated potent covalent binding to FABP5 in both cell-free and cellular conditions. Ligand displacement assay with a fluorescent fatty acid probe confirmed the competitive binding mode of AUR104 with fatty acids. Binding at the fatty acid site and covalent bond formation with Cys43 were also demonstrated by crystallography. Furthermore, AUR104 showed a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. AUR104 exhibited potent anti-proliferative activity in a large panel of cell lines derived from both hematological and solid cancers with a high degree of selectivity over normal cells. Anti-proliferative activity in lymphoma cell lines correlated with inhibition of MALT1 pathway activity, cleavage of RelB/Bcl10 and secretion of cytokines, IL-10 and IL-6. AUR104 displayed desirable drug-like properties and dose-dependent oral exposure in pharmacokinetic studies. Oral dosing with AUR104 resulted in dose-dependent anti-tumor activity in DLBCL (OCI-LY10) and NSCLC (NCI-H1975) xenograft models. In a repeated dose MTD studies in rodents and non-rodents, AUR104 showed good tolerability with an exposure multiple of >500 over cellular EC50 for up to 8 hours. In summary, we have identified a novel covalent FABP5 inhibitor with optimized properties that showed anti-tumor activity in in vitro and in vivo models with acceptable safety profile. The data presented here strongly support clinical development of AUR104.
Citation Format: Dinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar, Murali Ramachandra. Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapy [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1266.
Patent
US20200147054 – COMBINATION OF SMALL MOLECULE CD-47 INHIBITORS WITH OTHER ANTI-CANCER AGENTS
Muralidhara Ramachandra
Pottayil Govindan Nair Sasikumar
Girish Chandrappa Daginakatte
Kiran Aithal Balkudru
PATENT
WO 2020095256
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020095256
Example- 1: The synthetic procedures for the preparation of compounds described in the present invention were described in co-pending Indian provisional patent application 201841001438 dated 12* Jan 2018, which is converted as PCT application
PCT/IB2019/050219, the contents of which are hereby incorporated by reference in their entirety.
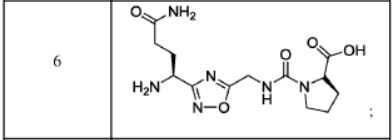
PATENT
WO 2018178947https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018178947&tab=PCTDESCRIPTION
PATENT
WO 2019138367
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019138367
PATENT
WO 2019073399
https://patents.google.com/patent/WO2019073399A1/en
Priority to IN201741036169
Example 4 of WO 2015/033299
PATENT
https://patents.google.com/patent/BR112020014202A2/en
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
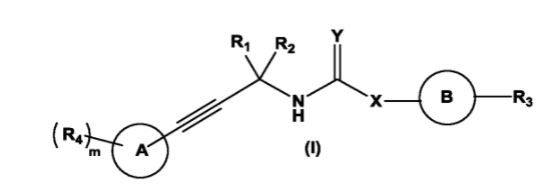
Patent
Example 1
(((S)-4-amino-1-(3-((S)-1,5-diaminopentyl)-1,2,4-oxadiazol-5-yl)-4-oxobutyl)carbamoyl)-L-proline (Compound 1)
Synthesis of Compound 1 b
Synthesis of Compound 1C
Synthesis of Compound 1d
Synthesis of Compound 1f
Synthesis of Compound 1g
Synthesis of Compound 1h
Synthesis of Compound 1i
Synthesis of compound 1j
Synthesis of Compound 1
Example 2
(S)-4-(3-((S)-1-amino-4-guanidinobutyl)-1,2,4-oxadiazol-5-yl)-4-(3-((S)-1-carboxy-2-phenylethyl) ureido)butanoic acid (Compound 7)
Synthesis of Compound 2b
Synthesis of Compound 2c
Synthesis of Compound 2d
Synthesis of Compound 2f
Synthesis of Compound 2g
Synthesis of Compound 2h
Synthesis of Compound 2i
Synthesis of Compound 2j
Synthesis of Compound 7
PATENT
WO 2015/033299
https://patents.google.com/patent/WO2015033299A1/en?oq=WO+2015%2f033299
Pottayil Govindan Nair SasikumarMuralidhara RamachandraSeetharamaiah Setty Sudarshan Naremaddepalli
Example 1: Synthesis of Compound 1

Step la:

Ethylchloroformate (1.5 g, 13.78 mniol) and N-Methylmorpholine ( 1.4 g, 13.78 mmol) were added to a solution of compound la (3 g, 11.48 mmol) in THE (30 mL) arid stirred at -20 °C. After 20 min. Liquid ammonia (0.77 g, 45.92 mmol) was added to the active mixed anhydride formed in- situ and stirred at 0-5 °C for 20 min. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOs, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to get 2.9 g of compound lb (Yield: 96.3%). LCMS: 261.0 ( Vi+H ; .
Step lb:

1 b 1cTrifluroacetic anhydride (9.7 g, 46.0 mmol) was added to a solution of compound lb (8 g, 30.7 mmol) in pyridine (24.3 g, 307.0 mmol) and stirred at room temperature for 3 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCO?,, citric acid, brine solution, dried over Na2-S04 and evaporated under reduced pressure to afford 7 g of compound lc (Yield: 94.0%). LCMS: 187.2 (M-¾u )+.
Step lc:

1 c 1dHydroxylamine hydrochloride (3 g, 43.37 mmol) and potassium carbonate (6 g, 43.37 mmol) were added to a solution of compound lc (7 g, 28.91 mmol) in EtOH (70 m L) and stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with brine solution, dried over Na2S04 and evaporated under reduced pressure. The crude compound was purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to get 4.2 g of compound Id (Yield: 52.8%). LCMS: 276.4 (M+H)+.Step Id:

Deoxo-Fluor® (1.83 g, 8.3 mmol) was added to a solution of Fmoc-Asn(Trt)-OH (4.5 g, 7.5 mmol) in CH2Q2 (50 mL) and stirred at 0 °C for 3 h. Then CH2CI2 was evaporated and triturated with hexane, decanted and evaporated under vacuum to get the corresponding acid fluoride. NMM (1.17 g, 1 1.6 mmol) and compound Id (1.6 g, 5.8 mmol) in THF were added to the acid fluoride and stirred at room temperature for 12 h. Then THF was evaporated and sodium acetate (0.72 g, 8.7 mmol) was added followed by EtOH (50 mL). The reaction mixture was stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOa, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure, which was further purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to afford 2.8 g of compound le (Yield: 44.4%). LCMS: 836.4 (M+Hf .Step le:
Ph3

To compound le (2.3 g, 2.7 mmol) in CH2CI2 (10 mL) diethyiarnine (10 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting solution was concentrated in vacuum to get gummy residue. The crude compound was purified by neutral alumina column chromatography (Eluent: 0-50% ethyl acetate in hexane then 0-5% methanol in chloroform) to get 1.4 g of If (Yield: 90 %). LCMS: 636.5 (M+Na)+.

1f 1To a solution of compound If (0.45 g) in CH2CI2 (5 mL), trifluoroacetic acid (5 mL) and catalytic amount of triisopropylsilane were added and stirred for 3 h at room temperature to remove the acid sensitive protecting groups. The resulting solution was concentrated in vacuum to afford 0.29 g of crude compound 1 which was purified using prep-HPLC method described under experimental conditions. \H NMR (DMSQ-d6, 400 MHz): δ 2.58 (m, 2H), 3.53 (m, 3H), 3.91 (t, 1H), 4.36 (t, 1H), 6.91 (s, 1H), 7.45 (s, 1H); 1 C NMR (DMSO-de, 400 MHz): δ 20.85, 45.71 , 50.23, 65.55, 171.03, 171 .41, 181.66. LCMS: 216.2 (Μ+ΗΓ; HPLC: tR = 13.1 min.Example 2: Synthesis of Co

Step 2a:

1f2a
The urea linkage was carried out by the coupling compound If (2.7 g, 4.39 mmoi) in THF (30 mL) at room temperature with compound 2b (1.67 g, 4.39 mmoi). The coupling was initiated by the addition of TEA (0.9 g, 8.78 mmoi) in THF (10 m L) and the resultant mixture was stirred at room temperature. After completion of 20 h, THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get compound 2a, which was further purified by silica gel column chromatography (Fluent: 0-50% ethyl acetate in hexane) to afford 3.46 g of compound 2a (Yield: 92.10%). LCMS 857.4 (M+H)+.

2aTo a solution of compound 2a (0.22 g, 0.25 mmol) in 0¾ί¾ (5 m L), trifluoroaeetic acid (5 mL) and catalytic amount of triisopropyisilane were added and stirred for 3h at room, temperature. The resulting solution was concentrated under reduced pressure to obtain 0.35 g of crude compound. The crude solid material was purified using preparative- HPLC method described under experimental conditions. LCMS: 347.1 (M+H)+; HPLC: tR = 12.9 min.
Synthesis of

2bTo the compound H-Ser(tBu)-OiBu (2 g, 9.2 mmol) in C I I■■(.‘{■ (20 mL), triethylamine (1.39 g, 13.8 mmol) was added and the solution was stirred at room temperature for 5-10 min. To this mixture, solution of 4-Nitrophenyl chioro formate (2.22 g, 11.04 mmol) in CH2CI2 was added and the resultant mixture was stirred at room temperature for 30 min. The completion of the reaction was confirmed by TLC analysis. After completion of reaction, reaction mixture was diluted with CH2CI2 and washed with water and 5.0 M citric acid solution, dried over Na2SC>4 and evaporated under reduced pressure to get crude compound 2b, which was further purified by silica gel column chromatography (Eiuent: 0-20% ethyl acetate in hexane) to yield 2.1 g (58.9%) of 2b.Example 3: Synthesis of Compound 3

The compound was synthesised using similar procedure as depicted in Example 1 (compound 1) and D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH (compound la, Example 1) and Fmoc-D- Asn(trt)-OH in place of Fmoc-Asn(trt)-OH to yield 0.15 g crude material of the title compound 3. LCMS: 230.1 (M+H)+.Example 4: Synthesis of Co
The compound was synthesised using similar procedure as depicted in Example 2 for synthesising compound 2 using

instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.35 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.2 (M+H)+, HPLC: tR = 12.19 min.Example 5: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 4 (compound 4) using D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH, Fmoc-D-Asn(trt)-OH in place of Fmoc-Asn(trt)- OH and H-D-Ser(‘Bu)-0’Bu was used in place of H-Thr^Bu^O’Bu to yield 0.3 g crude material of the title compound. The cmde solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.3 (M+H)+. HPLC: tR = 13.58 min.Example 6: Synthesis of Compound 6

The compound was synthesised using similar procedure as depicted in Example 2 by using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.2 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 375.1 (M+H)+, HPLC: tR = 1.84 min.Example 7: Synthesis of Compound 7

Step 7a:

1f7aThe compound 7a was synthesised using similar procedure as for compound 2a (Example 2, step 2a) using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-OtBu to get crude material which was further purified by silica gel column chromatography (Eluent: 0-50% ethyl acetate in he ane) to get 2.0 g of compound 7a (Yield: 74 %). LCMS: 829.2 (M+H)+.Step 7b:

7a 7bTo a solution of compound 7a (0.35 g, 4.0 mmol) in THF (5 mL) was added lithium hydroxide (0.026 g, 0.63 mmol) at 0 °C and the mixture was stirred for 2 h at room temperature. The completion of the reaction was confirmed by TLC analysis. THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to afford 7b, which was further purified by silica gel column chromatography (Eluent: 0-5% methanol in DCM) to get 0.3 g of product 7b (Yield: 86.7%). LCMS 815.2 (M+H)+.
Step 7c:

7b 7Compound 7b (0.295 g, 0.39 mmol) was anchored to Rink amide resin (0.7 g, 0.55 mmol/g) using HOBT (0.072 g, 0.54 mmol) and DIC (0.068 g, 0.54 mmol) method in DMF (10 mL). The resin was stirred for 12 h at room temperature. The resin was washed with DCM, DMF and DCM and dried. The target compound was cleaved from the rink amide resin using TFA (5 mL) and catalytic amount of TIPS. The resin was allowed to remain at room temperature for 2 h with occasional stirring. After 2 h, TFA and TIPS were evaporated under nitrogen atmosphere and the resulting residue was washed with diethyl ether to yield 0.1 g crude material of the title compound 7. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 360.0 (M+H)+, HPLC: tR = 13.88 min.Example 8: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 2 (compound 2) using Fmoc-Glu(0’Bu)-OH instead of Fmoc-Asn(Trt)-OH to get 0.4 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 362.1 (M+H)+. HPLC: tR = 13.27 min.
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324&tab=FULLTEXT
Patenthttps://patents.google.com/patent/WO2019067678A1/enPATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324
PATENThttps://patents.google.com/patent/WO2018073754A1/en
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
PAPERSScientific Reports (2019), 9(1), 1-19. https://www.nature.com/articles/s41598-019-48826-6
Chemical structures of PD-L1 inhibitors developed by Aurigene (Aurigene-1) and Bristol-Meyers Squibb (BMSpep-57, BMS-103, and BMS-142). Chemical structures were generated using ChemDraw Professional 15. PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
L-threonine’ mentioned in compound of formula (I) thereof can be represented by any one of the following formulae:
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020289477-A1 | Conjoint therapies for immunomodulation | 2017-11-06 | |
| WO-2019073399-A1 | CRYSTALLINE FORMS OF 1,2,4-OXADIAZOLE SUBSTITUTED IN POSITION 3 | 2017-10-11 | |
| AU-2018341583-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| WO-2019061324-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 | |
| WO-2019067678-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020247766-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| US-2020061030-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| WO-2018073754-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| US-2020361880-A1 | 1,2,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators | 2015-03-10 | |
| EP-3041827-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2018-04-18 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3363790-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-02-19 |
| US-10173989-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2019-01-08 |
| US-10590093-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-03-17 |
| US-2015073024-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2017101386-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2018072689-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2019144402-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2020199086-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-9771338-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2017-09-26 |
| WO-2015033299-A1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 |
////////////Investigational New Drug Application, Phase 1, Clinical Trial, Non-Hodgkin’s Lymphoma, XL 114, AUR 104, aurigene, Exelixis
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
https://patentscope.wipo.int/search/en/result.jsf?inchikey=HFOBENSCBRZVSP-WHFCDURNSA-N

NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT


The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
XL 102
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
Exelixis Forward-Looking Statements
This press release contains forward-looking statements, including, without limitation, statements related to: Exelixis’ and Aurigene’s plans to present preclinical data in support of the continued development of AUR102 in a poster as part of the 32nd ENA Symposium; the potential for AUR102 to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma; the potential for AUR102 to be the subject of an Investigational New Drug filing later in 2020; Exelixis’ potential future financial and other obligations under the exclusive collaboration, option and license agreement with Aurigene; and Exelixis’ plans to reinvest in its business to maximize the potential of the company’s pipeline, including through targeted business development activities and internal drug discovery. Any statements that refer to expectations, projections or other characterizations of future events or circumstances are forward-looking statements and are based upon Exelixis’ current plans, assumptions, beliefs, expectations, estimates and projections. Forward-looking statements involve risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in the forward-looking statements as a result of these risks and uncertainties, which include, without limitation: the availability of data at the referenced times; the level of costs associated with Exelixis’ commercialization, research and development, in-licensing or acquisition of product candidates, and other activities; uncertainties inherent in the drug discovery and product development process; Exelixis’ dependence on its relationship with Aurigene, including Aurigene’s adherence to its obligations under the exclusive collaboration, option and license agreement and the level of Aurigene’s assistance to Exelixis in completing clinical trials, pursuing regulatory approvals or successfully commercializing partnered compounds in the territories where they may be approved; the continuing COVID-19 pandemic and its impact on Exelixis’ research and development operations; complexities and the unpredictability of the regulatory review and approval processes in the U.S. and elsewhere; Exelixis’ and Aurigene’s continuing compliance with applicable legal and regulatory requirements; Exelixis’ and Aurigene’s ability to protect their respective intellectual property rights; market competition; changes in economic and business conditions; and other factors affecting Exelixis and its product pipeline discussed under the caption “Risk Factors” in Exelixis’ Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission (SEC) on August 6, 2020, and in Exelixis’ future filings with the SEC. All forward-looking statements in this press release are based on information available to Exelixis as of the date of this press release, and Exelixis undertakes no obligation to update or revise any forward-looking statements contained herein, except as required by law.
Exelixis, the Exelixis logo, CABOMETYX, COMETRIQ and COTELLIC are registered U.S. trademarks. MINNEBRO is a registered Japanese trademark.
Curis and Aurigene’s CA 4948, AU 4948
![]()

Example 13 WO2015104688
6-(6-aminopyridin-3-yl)-N-(2-morpholin-4-yl-1,3-benzothiazol-6-yl)pyridine-2-carboxamide
| Molecular Formula: | C22H20N6O2S |
|---|---|
| Molecular Weight: | 432.4982 g/mol |
PROBABLE STRUCTURE

Example 1 ……..6′-amino-N-(2-morpholinooxazolo[4,5-b]pyridin-6-yl)-[2,3′-bipyridine]-6-carboxamideWO2015104688

Compound-6: 6′-amino-N-(5-(cyclopropyIamino)-2-morpholinobenzo [d]oxazoI-6-yl)-[2,3′-bipyridine]-6-carboxamide.WO2013042137
PROBABLE CA 4948, AU 4948,AU-4948, CA-4948
STRUCTURE AND SYNTHESIS COMING……..
| Latest Stage of Development | Preclinical |
| Standard Indication | B cell lymphoma |
| Indication Details | Treat diffuse large B cell lymphoma (DLBCL) |
| Regulatory Designation | |
| Partner | Curis Inc. |
Interleukin-1 Receptor Associated Kinase-4 (IRAK-4) is a serine/threonine protein kinase belonging to tyrosine like kinase (TLK) family. IRAK-4 is one of the important signalling components downstream of IL-1/Toll family of receptors (IL-1R, IL-18R, IL-33R, Toll-like receptors). Recent studies have reported occurrence of oncogenic mutations in MYD88 in 30% of ABC diffuse large B cell lymphomas (ABC DLBCL) and 90% of Waldenstrom’s macroglobulinemia (WM). Most of ABC DLBCLs have a single amino acid substitution of proline for the leucine at position 265 (L265P) in the TIR domain of MYD88 protein resulting in constitutive activation of IRAK-4. Thus, IRAK4 is an attractive therapeutic target for the treatment of B-cell lymphomas with activating MYD88 L265P mutation. We have designed, synthesized and tested small molecule IRAK-4 inhibitors based on hits originating from Aurigene’ s compound library. These novel compounds were profiled for IRAK4 kinase inhibition, anti-proliferative activity, kinase selectivity, and drug-like properties. Furthermore, selected compounds were tested in a proliferation assay and pIRAK1 mechanistic assay using ABC-DLBCL cell lines with activating MYD88 L265P mutation, OCI-lLy10 and OCI-lLy3. We have identified a series of novel bicyclic heterocycles as potent inhibitors of IRAK-4. Aurigene Lead compound exhibited potent inhibitory activity for IRAK-4 with an IC50 of 3nM in biochemical assay. Aurigene Lead compound inhibited pIRAK1 levels, and proliferation of OCI-Ly3 and OCI-Ly10 cells with an IC501of 132nM and 52nM respectively. To the best of our knowledge, Aurigene Lead compound represents the most potent IRAK4 inhibitor reported for target modulation and anti-proliferative activity in DLBCL cell lines with activating MYD88 L265P mutation. Aurigene Lead compound has good oral pharmacokinetic profile in mice and has demonstrated excellent pharmacodynamic effect in an in vivo LPS induced TNF-α model with an ED50 of 3.8 mg/Kg in mice. Preliminary in vitro tox studies indicated clean safety profile. Demonstration of efficacy in OCI-lLy10 mouse tumor model is ongoing. In summary, a series of potent IRAK-4 inhibitors belonging to 3 different chemical series have been discovered and are being evaluated for treatment of B-cell lymphomas.
Curis with the option to exclusively license Aurigene’s orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers.
![]()
Curis and Aurigene Announce Collaboration, License and Option Agreement to Discover, Develop and Commercialize Small Molecule Antagonists for Immuno-Oncology and Precision Oncology Targets
— Agreement Provides Curis with Option to Exclusively License Aurigene’s Antagonists for Immuno-Oncology, Including an Antagonist of PD-L1 and Selected Precision Oncology Targets, Including an IRAK4 Kinase Inhibitor —
— Investigational New Drug (IND) Application Filings for Both Initial Collaboration Programs Expected this Year —
— Curis to issue 17.1M shares of its Common Stock as Up-front Consideration —
— Management to Host Conference Call Today at 8:00 a.m. EST —
LEXINGTON, Mass. and BANGALORE, India, Jan. 21, 2015 (GLOBE NEWSWIRE) — Curis, Inc. (Nasdaq:CRIS), a biotechnology company focused on the development and commercialization of innovative drug candidates for the treatment of human cancers, and Aurigene Discovery Technologies Limited, a specialized, discovery stage biotechnology company developing novel therapies to treat cancer and inflammatory diseases, today announced that they have entered into an exclusive collaboration agreement focused on immuno-oncology and selected precision oncology targets. The collaboration provides for inclusion of multiple programs, with Curis having the option to exclusively license compounds once a development candidate is nominated within each respective program. The partnership draws from each company’s respective areas of expertise, with Aurigene having the responsibility for conducting all discovery and preclinical activities, including IND-enabling studies and providing Phase 1 clinical trial supply, and Curis having responsibility for all clinical development, regulatory and commercialization efforts worldwide, excluding India and Russia, for each program for which it exercises an option to obtain a license.
The first two programs under the collaboration are an orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field and an orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
“We are thrilled to partner with Aurigene in seeking to discover, develop and commercialize small molecule drug candidates generated from Aurigene’s novel technology and we believe that this collaboration represents a true transformation for Curis that positions the company for continued growth in the development and eventual commercialization of cancer drugs,” said Ali Fattaey, Ph.D., President and Chief Executive Officer of Curis. “The multi-year nature of our collaboration means that the parties have the potential to generate a steady pipeline of novel drug candidates in the coming years. Addressing immune checkpoint pathways is now a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients. Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers. We look forward to advancing these programs into clinical development later this year.”
Dr. Fattaey continued, “Aurigene has a long and well-established track record of generating targeted small molecule drug candidates with bio-pharmaceutical collaborators and we have significantly expanded our drug development capabilities as we advance our proprietary drug candidates in currently ongoing clinical studies. We believe that we are well-positioned to advance compounds from this collaboration into clinical development.”
CSN Murthy, Chief Executive Officer of Aurigene, said, “We are excited to enter into this exclusive collaboration with Curis under which we intend to discover and develop a number of drug candidates from our chemistry innovations in the most exciting fields of cancer therapy. This unique collaboration is an opportunity for Aurigene to participate in advancing our discoveries into clinical development and beyond, and mutually align interests as provided for in our agreement. Our scientists at Aurigene have established a novel strategy to address immune checkpoint targets using small molecule chemical approaches, and have discovered a number of candidates that modulate these checkpoint pathways, including PD-1/PD-L1. We have established a large panel of preclinical tumor models in immunocompetent mice and can show significant in vivo anti-tumor activity using our small molecule PD-L1 antagonists. We are also in the late stages of selecting a candidate that is a potent and selective inhibitor of the IRAK4 kinase, demonstrating excellent in vivo activity in preclinical tumor models.”
In connection with the transaction, Curis has issued to Aurigene approximately 17.1 million shares of its common stock, or 19.9% of its outstanding common stock immediately prior to the transaction, in partial consideration for the rights granted to Curis under the collaboration agreement. The shares issued to Aurigene are subject to a lock-up agreement until January 18, 2017, with a portion of the shares being released from the lock-up in four equal bi-annual installments between now and that date.
The agreement provides that the parties will collaborate exclusively in immuno-oncology for an initial period of approximately two years, with the option for Curis to extend the broad immuno-oncology exclusivity.
In addition Curis has agreed to make payments to Aurigene as follows:
- for the first two programs: up to $52.5 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any;
- for the third and fourth programs: up to $50 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any; and
- for any program thereafter: up to $140.5 million per program, including $87.5 million per program in approval and commercial milestones, plus specified approval milestone payments for additional indications, if any.
Curis has agreed to pay Aurigene royalties on any net sales ranging from high single digits to 10% in territories where it successfully commercializes products and will also share in amounts that it receives from sublicensees depending upon the stage of development of the respective molecule.
About IRAK4:
Interleukin-1 receptor-associated kinase 4, or IRAK4 is a signaling kinase that becomes inappropriately activated in certain cancers including activated B cell-diffuse large B cell lymphoma (ABC-DLBCL), an aggressive form of lymphoma with poor prognosis. There appears to be a mechanistic link with IRAK4 in ABC-DLBCL where these tumors from approximately 35% of patients harbor oncogenic mutations in the MYD88 gene, which encodes an adaptor protein that interacts directly with IRAK4. MYD88 mutations appear to constitutively activate the IRAK4 kinase complex, driving pro-survival pathways in ABC-DLBCL disease. Oncogenic MYD88 mutations have also been identified in other cancers, including in over 90% of patients with Waldenström’s Macroglobulinemia as well as in a subset of patients with chronic lymphocytic leukemia (CLL).
About Curis, Inc.
Curis is a biotechnology company focused on the development and commercialization of novel drug candidates for the treatment of human cancers. Curis’ pipeline of drug candidates includes CUDC-907, a dual HDAC and PI3K inhibitor, CUDC-427, a small molecule antagonist of IAP proteins, and Debio 0932, an oral HSP90 inhibitor. Curis is also engaged in a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are developing and commercializing Erivedge®, the first and only FDA-approved medicine for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at www.curis.com.
About Aurigene
Aurigene is a specialized, discovery stage biotechnology company, developing novel and best-in-class therapies to treat cancer and inflammatory diseases. Aurigene’s Programmed Death pathway program is the first of several immune checkpoint programs that are at different stages of discovery and preclinical development. Aurigene has partnered with several large- and mid-pharma companies in the United States and Europe and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies. Aurigene is an independent, wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (NYSE:RDY). For more information, please visit Aurigene’s website at http://aurigene.com/.
Small Molecule IRAK4 Kinase Inhibitor)
Innate immune responses mediated through Toll-like receptors or certain interleukin receptors are important mediators of the body’s initial defense against foreign antigens, while their dysregulation is associated with certain inflammatory conditions. Toll-like receptor and interleukin receptor signaling through the adaptor protein MYD88, results in the assembly and activation of IRAK4, initiating a signaling cascade that induces cytokine and survival factor expression mediated by the transcription factor NFκB. More recently, components of this pathway are recognized to be genetically altered and have important roles in specific human cancers. Toll-like receptor and interleukin receptor signaling through the adaptor protein MYD88, results in the assembly and activation of IRAK4, initiating a signaling cascade that induces cytokine and survival factor expression mediated by the transcription factor NFκB. MYD88 gene mutations are shown to occur in approximately 30% of Activated B-Cell (ABC) subtype of diffuse large B-cell lymphomas (DLBCL)1,2 and in over 90% of the B-cell malignancy Waldenstrom’s macroglobulinemia.3 Due to IRAK4’s central role in these signaling pathways, it is considered an attractive target for generation of therapeutics to treat these B-cell malignancies as well as certain inflammatory diseases.
As part of the collaboration with Aurigene, in October 2015 we exercised our option to exclusively license a program of orally-available, small molecule inhibitors of IRAK4 kinase, including the development candidate, CA-4948. Curis expects to file an IND and initiate clinical testing of CA-4948 in patients with advanced hematologic cancers during the second half of 2016.
1Nature. 2011; 470(7332):115–1192Immunology and Cell Biology. 2011; 89(6):659–6603N Engl J Med. 30, 2012; 367(9):826–833
CLIP
In November 2015, preclinical data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
Aurigene Collaboration (IRAK4 Inhibitor):
In October 2015, Curis exercised its option to exclusively license a program of orally available small molecule inhibitors of IRAK4 kinase, a serine/threonine kinase involved in innate immune responses as well as in certain hematologic cancers. The Company has since designated the development candidate as CA-4948 and expects to file an IND application for this molecule during 2016.
In November 2015, Curis’ collaborator Aurigene presented preclinical data from the IRAK4 program at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA. This presentation included data from chemically distinct series of small molecule compounds with potent IRAK4 inhibitory activity in biochemical assays as well as in in vivo preclinical models, including MYD88 mutant DLBCL xenograft tumor models as well as a model of inflammatory disease.
CLIP
In April 2014, preclinical data presented at the CHI’s Ninth Drug Discovery Chemistry Conference in San Diego, CA, showed the compounds in vivo to have activity down to 10 mg/kg .
CLIP

 Susanta Samajdar, Ph.D., Research Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Susanta Samajdar, Ph.D., Research Director, Medicinal Chemistry, Aurigene Discovery Technologies LimitedApril 24-25 2014
Drug Discovery Chemistry – CHI’s Ninth Annual Conference: Fifth Annual Kinase inhibitor Chemistry, San Diego, CA, USA
Novel IRAK4 inhibitors
Susanta Samajdar from Aurigene Discovery Technologies presented the discovery of new IRAK4 (IL-1 receptor-associated kinase 4) inhibitors. Research began with a HTS campaign using two types of libraries: rationally designed novel scaffolds by hopping and morphing of known IRAK4 inhibitors and novel scaffolds identified by virtual screening of drug-like commercial library. A benzoxazol series was identified and crystallography was used to help their design. Lead optimization culminated in the identification of very potent compounds (AU-2807 and AU-2202) in cell assay (inflammation pathway and oncology pathway, respectively). The compounds were also active against Flt3 and KDR. Some PD in vivo data using LPS and TNFalpha release were presented in which the compound showed activity down to 10 mg/kg: no other in vivo model data were disclosed, but it was mentioned that studies in the CIA (collagen induced arthritis) model was ongoing. Dr Samajdar answered to three questions, one related to IRAK1 selectivity (the answer was that the compound is fully selective against IRAK1 and IRAK2). It was also mentioned that the compounds have a PBB higher than 98%. And the last question was related to the synergetic effect with BTK inhibitor in activated B-cell like diffuse large B-cell lymphoma, and this effect was observed with these compounds.

Research Director at Aurigene Discovery Technologies
PATENT
http://www.google.com/patents/WO2013042137A1?cl=en
Compound-6: Synthesis of 6′-amino-N-(5-(cyclopropyIamino)-2-morpholinobenzo [d]oxazoI-6-yl)-[2,3′-bipyridine]-6-carboxamide.
Step_l^N-cyclopropyl-2-morpholino-6-nitrobenzo[d]oxazol-5-amine.

N-cyclopropyl-2-moφholino-6-nitrobenzo[d]oxazol-5-amine(0.7g,70%) was prepared from 5-fluoro-2-mo holino-6-nitrobenzo[d]oxazole(lg,Intermediate-2) by treating with cyciopropanamine in sealed tube at 100°C for 8-14h. The progress of the reaction was monitored by TLC. After the reaction was completed, it was extracted with water (15ml) and dichioromethane (2x 15ml). The organic layer was collected, washed with brine, dried over sodium sulfate and concentrated under reduced pressure to get the crude. MS (ES) m/e 305(M+1, 50%).
Steg2:6-bromo-N-(5-(cyclopropylamino)-2-morpholinobenzo[d]oxazol-6-yl)
picolinamide.

Step Π and ii):The process of these steps are adopted from step 2 and step 3 of compound- 1.
Step3:6′-amino-N-(5-(cvclopropvlamino)-2-morpholinobenzord]oxazol-6-yl)-r2,3′- bipyridine]-6-carboxamide.

(i) N-(4-methoxybenzyl)-5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin
Na2C03, Pd(dppf)Cl2, ACN, H20, 80-100°C, 8-14h; TFA, 60-70°C, 8-14h.
6′-amino-N-(5-(cyclopropylamino)-2-mo holinobenzo[d]oxazol-6-yl)-[2,3′-bipyridine]-6- carboxamide (0.03g,61%) was prepared from 6-bromo-N-(5-(cyclopropyIamino)-2- moφholinobenzo[d]o azoI-6-yl)picolinamide(0.07g, step-3) by following the same process used in step-1 and 2 of compound-3.
Ή NMR (400 MHz, DMSO-< ):6 1 1.63 (s, IH), 8.90 (s, IH), 8.61 (s, IH), 8.55 (s, IH), 8.37- 8.03 (m, 2H), 7.39 (s, IH), 6.80-6.62 (s, IH), 3.80-3.59 (m, 15H), 2.88-2.64 (m, 2H). MS (ESI): 472 (M+l , 60%).
PATENT
Example 13
6′-amino-N-(2-morphol ne]-6-carboxamide

Step-1: Synthesis of 6-chloro thiazolo[4,5-c]pyridine-2(3H)-thione
Using the same reaction conditions as described in step 1 of example 1, 4,6-dichloropyridin-3-amine (1.3 g, 7 mmol) was cyclised using potassium ethyl xanthate (2.55 g, 15 mmol) in DMF (25mL) at 150°C for 8h to afford the title compound (1.3 g, 86.6 %) as a light brown solid.
1HNMR (400 MHz, DMSO-d6): δ 14.2-14.0 (b, 1H), 8.274 (s, 1H), 7.931 (s, 1H); LCMS: 100%, m/z = 201.3 (M+l)+.
Step-2: Synthesis of 4-(6-chloro thiazolo[4,5-c]pyridin-2-yl) morpholine
To a suspension of 6-chlorothiazolo[4,5-c]pyridine-2(3H)-thione (0.3 g, 1.16 mmol) in
DCM (4 mL), oxalyl chloride (0.2 mL, 2.38 mmol) and DMF (1.5 mL) were added at 0°C. The resulting mixture was slowly allowed to warm to room temperature and stirred there for 1 h. The reaction mixture was again cooled to 0°C and triethyl amine (0.66 mL, 4.76 mmol) and morpholine (0.13 mL, 1.75 mmol) were added. The reaction mixture was stirred at RT for 1 h and quenched with water and extracted with ethyl acetate. The combined organic layers were washed with water, brine, dried over sodium sulphate and concentrated under reduced pressure. The crude material was purified by column chromatography (EtOAc/n-hexanes 3:7) to afford the title compound (0.14 g, 39.6 %) as a light brown solid.
1H NMR (400 MHz, DMSO-d6): δ 8.47 (s, 1H), 8.04 (s, 1H), 3.74-3.72 (m, 4H), 3.61-3.59 (m, 4H); LCMS: m/z = 256.1 (M+l)+.
Step-3: Synthesis of 6′-amino-/V-(2-morpholino thiazolo [4,5-c]pyridin-6-yl)-[2,3′-bipyridine]-6-carboxamide
Using the same reaction conditions as described in step 4 of example 12, 4-(6-chlorothiazolo[4,5-c] pyridin-2-yl) morpholine (0.081 g, 0.32 mmol), was coupled with tert-butyl (6-carbamoyl-[2,3′-bipyridin]-6′-yl)carbamate (intermediate 2) (0.1 g, 0.32 mmol) using cesium carbonate (0.21 g, 0.64 mmol), XantPhos (0.028g, 0.047mmol) and Pd2(dba)3 (0.015 mg, 0.015 mmol) in toluene : dioxane (2:2mL) to get the crude product. The resultant crude was purified by 60-120 silica gel column chromatography using 2% methanol in DCM as eluent. Further the resultant crude was purified by prep HPLC to afford title compound (0.01 g, 6 %) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 10.65 (s, 1H), 8.88 (d, 1H), 8.85 (dd, 1H), 8.71 (s, 1H), 8.55 (s, 1H), 8.22-8.13 (m, 4 H), 7.09 (d, 1H), 3.73 (t, 4H), 3.58 (t, 4H). LCMS: 100%, m/z = 434.2 (M+l)+.
Example 11
(S)-2-(2-methylpyridin-4-yl)-N-(2-morpholino-5-(pyrrolidin-3-ylamino)oxazolo[4,5-b]pyridin-6-yl)oxazole-4-carboxamide

Step l:Preparation of (S)-tert-butyl 3-((2-morpholino-6-nitrooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine- 1 -carboxylate
A solution of 5-chloro-2-morpholino-6-nitrooxazolo[4,5-b]pyridine (300mg, 1.0563 mmol) (S)-tert-butyl 3 -aminopyrrolidine- 1 -carboxylate (237mg, 1.267 mmol) and potassium carbonate (292mg, 2.112 mmol) in DMF (2mL) was heated at 100°C for 2h. Reaction was quenched with ice water and filtered the solid. The resultant crude was purified by 60-120 silica gel column chromatography using 1 % methanol in DCM as eluent to obtain the title compound (350mg, 76.25%). LCMS: m/z: 435.4 (M+l)+.
Step 2:Preparation of (S)-tert-butyl 3-((6-amino-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine- 1 -carboxylate
Using the same reaction conditions as described in step 5 of example 1, (S)-tert-butyl 3- ((2-morpholino-6-nitrooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (350mg, 0.806 mmol) was reduced with zinc dust (422mg, 6.451 mmol) and ammonium chloride (691mg, 12.903 mmol) in THF/methanol/H20 (10mL/2mL/lmL) to get the title compound (240mg, 71.8%). LCMS: m/z: 405.2 (M+l)+.
Step 3:Preparation of (S)-tert-butyl 3-((6-(2-(2-methylpyridin-4-yl)oxazole-4-carboxamido)-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l-carboxylate
Using the same reaction conditions as described in step 6 of example 1, (S)-tert-butyl 3-((6-amino-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (115mg, 0.284 mmol), was coupled with 2-(2-methylpyridin-4-yl)oxazole-4-carboxylic acid (70mg, 0.341 mmol) using EDCI.HCl (82mg, 0.426 mmol), HOBt (58mg, 0.426 mmol), DIPEA (0.199mL, 1.138 mmol) in DMF (2mL) to afford the title compound (lOOmg, 59.52%). LCMS: m/z: 591.4 (M+l)+.
Step 4: Preparation of (S)-2-(2-methylpyridin-4-yl)-N-(2-morpholino-5-(pyrrolidin-3-ylamino)oxazolo[4,5-b]pyridin-6-yl)oxazole-4-carboxamide
Using the same reaction conditions as described in step 8 of example 1, (S)-tert-butyl 3- ((6-(2-(2-methylpyridin-4-yl)oxazole-4-carboxamido)-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (lOOmg, 0.169 mmol) was deprotected using methanolic HC1 (5mL) to get the crude product. This was then purified by prep HPLC to get the title compound (9mg, 10.84%).
1HNMR (CDCI3, 400MHz): δ 9.91 (s, 1H), 8.78 (s, 1H), 8.74-8.73 (d, 1H), 8.45 (s, 1H), 7.82 (s, 1H), 7.76-7.74 (d, 1H), 4.50 (s, 1H), 4.04-4.03 (d, 4H), 3.30-3.00 (m, 7H), 2.70 (s, 3H), 2.40-1.80 (m, 4H), 1.00-0.08 (m, 1H). LCMS: 100%, m/z = 491.3 (M+l)+.
REFERENCES
http://www.curis.com/images/stories/pdfs/posters/Aurigene_IRAK4_AACR-NCI-EORTC_2015.pdf
http://www.curis.com/images/stories/pdfs/posters/Aurigene_IRAK4_AACR_20150421.pdf
1Nature. 2011; 470(7332):115–119
2Immunology and Cell Biology. 2011; 89(6):659–660
3N Engl J Med. 30, 2012; 367(9):826–833
April 2014, preclinical data presented at the CHI’s Ninth Drug Discovery Chemistry Conference in San Diego, CA
November 2015, preclinical data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
http://pubs.acs.org/doi/abs/10.1021/jm5016044
http://cancerres.aacrjournals.org/content/75/15_Supplement/3646
2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 3646. doi:10.1158/1538-7445.AM2015-3646
////////IRAK4 Kinase Inhibitor, Curis, Aurigene, CA 4948, AU 4948, CA-4948, AU-4948, 1428335-77-6
c21ccc(cc1sc(n2)N3CCOCC3)NC(c4nc(ccc4)c5ccc(nc5)N)=O
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on Facebook 


 LIONEL MY SON
LIONEL MY SON
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
Curis and Aurigene’s AUPM 170, CA 170
1,2,4-oxadiazole and 1 ,2,4-thiadiazole compounds of formula (I):
ONE EXAMPLE
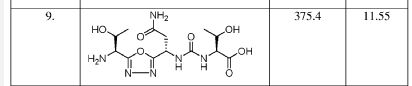
EXAMPLES
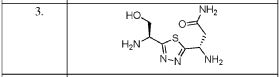
PREDICTED AUPM 170, CA 170, AUPM-170, CA-170
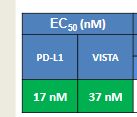
Synthesis coming………….
WATCH THIS SPACE
Aurigene Discovery Technologies Limited INNOVATOR
Curis with the option to exclusively license Aurigene’s orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field
Addressing immune checkpoint pathways is a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients.
Through its collaboration with Aurigene, Curis is now engaged in the discovery and development of the first ever orally bioavailable, small molecule antagonists that target immune checkpoint receptor-ligand interactions, including PD-1/PD-L1 interactions. In the first half of 2016, Curis expects to file an IND application with the U.S. FDA to initiate clinical testing of CA-170, the first small molecule immune checkpoint antagonist targeting PD-L1 and VISTA. The multi-year collaboration with Aurigene is focused on generation of small molecule antagonists targeting additional checkpoint receptor-ligand interactions and Curis expects to advance additional drug candidates for clinical testing in the coming years. The next immuno-oncology program in the collaboration is currently targeting the immune checkpoints PD-L1 and TIM3.
In November 2015, preclinical data were reported. Data demonstrated tha the drug rescued and sustained activation of T cells functions in culture. CA-170 resulted in anti-tumor activity in multiple syngeneic tumor models including melanoma and colon cancer. Similar data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
By August 2015, preclinical data had been reported. Preliminary data demonstrated that in in vitro studies, small molecule PD-L1 antagonists induced effective T cell proliferation and IFN-gamma production by T cells that were specifically suppressed by PD-L1 in culture. The compounds were found to have effects similar to anti-PD1 antibodies in in vivo tumor models
(Oral Small Molecule PD-L1/VISTAAntagonist)
Certain human cancers express a ligand on their cell surface referred to as Programmed-death Ligand 1, or PD-L1, which binds to its cognate receptor, Programmed-death 1, or PD-1, present on the surface of the immune system’s T cells. Cell surface interactions between tumor cells and T cells through PD-L1/PD-1 molecules result in T cell inactivation and hence the inability of the body to mount an effective immune response against the tumor. It has been previously shown that modulation of the PD-1 mediated inhibition of T cells by either anti-PD1 antibodies or anti-PD-L1 antibodies can lead to activation of T cells that result in the observed anti-tumor effects in the tumor tissues. Therapeutic monoclonal antibodies targeting the PD-1/PD-L1 interactions have now been approved by the U.S. FDA for the treatment of certain cancers, and multiple therapeutic monoclonal antibodies targeting PD-1 or PD-L1 are currently in development.
In addition to PD-1/PD-L1 immune regulators, there are several other checkpoint molecules that are involved in the modulation of immune responses to tumor cells1. One such regulator is V-domain Ig suppressor of T-cell activation or VISTA that shares structural homology with PD-L1 and is also a potent suppressor of T cell functions. However, the expression of VISTA is different from that of PD-L1, and appears to be limited to the hematopoietic compartment in tissues such as spleen, lymph nodes and blood as well as in myeloid hematopoietic cells within the tumor microenvironment. Recent animal studies have demonstrated that combined targeting/ blockade of PD-1/PD-L1 interactions and VISTA result in improved anti-tumor responses in certain tumor models, highlighting their distinct and non-redundant functions in regulating the immune response to tumors2.
As part of the collaboration with Aurigene, in October 2015 Curis licensed a first-in-class oral, small molecule antagonist designated as CA-170 that selectively targets PD-L1 and VISTA, both of which function as negative checkpoint regulators of immune activation. CA-170 was selected from the broad PD-1 pathway antagonist program that the companies have been engaged in since the collaboration was established in January 2015. Preclinical data demonstrate that CA-170 can induce effective proliferation and IFN-γ (Interferon-gamma) production (a cytokine that is produced by activated T cells and is a marker of T cell activation) by T cells that are specifically suppressed by PD-L1 or VISTA in culture. In addition, CA-170 also appears to have anti-tumor effects similar to anti-PD-1 or anti-VISTA antibodies in multiple in vivo tumor models and appears to have a good in vivo safety profile. Curis expects to file an IND and initiate clinical testing of CA-170 in patients with advanced tumors during the first half of 2016.
![]()
Curis and Aurigene Announce Collaboration, License and Option Agreement to Discover, Develop and Commercialize Small Molecule Antagonists for Immuno-Oncology and Precision Oncology Targets
— Agreement Provides Curis with Option to Exclusively License Aurigene’s Antagonists for Immuno-Oncology, Including an Antagonist of PD-L1 and Selected Precision Oncology Targets, Including an IRAK4 Kinase Inhibitor —
— Investigational New Drug (IND) Application Filings for Both Initial Collaboration Programs Expected this Year —
— Curis to issue 17.1M shares of its Common Stock as Up-front Consideration —
— Management to Host Conference Call Today at 8:00 a.m. EST —
LEXINGTON, Mass. and BANGALORE, India, Jan. 21, 2015 (GLOBE NEWSWIRE) — Curis, Inc. (Nasdaq:CRIS), a biotechnology company focused on the development and commercialization of innovative drug candidates for the treatment of human cancers, and Aurigene Discovery Technologies Limited, a specialized, discovery stage biotechnology company developing novel therapies to treat cancer and inflammatory diseases, today announced that they have entered into an exclusive collaboration agreement focused on immuno-oncology and selected precision oncology targets. The collaboration provides for inclusion of multiple programs, with Curis having the option to exclusively license compounds once a development candidate is nominated within each respective program. The partnership draws from each company’s respective areas of expertise, with Aurigene having the responsibility for conducting all discovery and preclinical activities, including IND-enabling studies and providing Phase 1 clinical trial supply, and Curis having responsibility for all clinical development, regulatory and commercialization efforts worldwide, excluding India and Russia, for each program for which it exercises an option to obtain a license.
The first two programs under the collaboration are an orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field and an orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
“We are thrilled to partner with Aurigene in seeking to discover, develop and commercialize small molecule drug candidates generated from Aurigene’s novel technology and we believe that this collaboration represents a true transformation for Curis that positions the company for continued growth in the development and eventual commercialization of cancer drugs,” said Ali Fattaey, Ph.D., President and Chief Executive Officer of Curis. “The multi-year nature of our collaboration means that the parties have the potential to generate a steady pipeline of novel drug candidates in the coming years. Addressing immune checkpoint pathways is now a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients. Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers. We look forward to advancing these programs into clinical development later this year.”
Dr. Fattaey continued, “Aurigene has a long and well-established track record of generating targeted small molecule drug candidates with bio-pharmaceutical collaborators and we have significantly expanded our drug development capabilities as we advance our proprietary drug candidates in currently ongoing clinical studies. We believe that we are well-positioned to advance compounds from this collaboration into clinical development.”
CSN Murthy, Chief Executive Officer of Aurigene, said, “We are excited to enter into this exclusive collaboration with Curis under which we intend to discover and develop a number of drug candidates from our chemistry innovations in the most exciting fields of cancer therapy. This unique collaboration is an opportunity for Aurigene to participate in advancing our discoveries into clinical development and beyond, and mutually align interests as provided for in our agreement. Our scientists at Aurigene have established a novel strategy to address immune checkpoint targets using small molecule chemical approaches, and have discovered a number of candidates that modulate these checkpoint pathways, including PD-1/PD-L1. We have established a large panel of preclinical tumor models in immunocompetent mice and can show significant in vivo anti-tumor activity using our small molecule PD-L1 antagonists. We are also in the late stages of selecting a candidate that is a potent and selective inhibitor of the IRAK4 kinase, demonstrating excellent in vivo activity in preclinical tumor models.”
In connection with the transaction, Curis has issued to Aurigene approximately 17.1 million shares of its common stock, or 19.9% of its outstanding common stock immediately prior to the transaction, in partial consideration for the rights granted to Curis under the collaboration agreement. The shares issued to Aurigene are subject to a lock-up agreement until January 18, 2017, with a portion of the shares being released from the lock-up in four equal bi-annual installments between now and that date.
The agreement provides that the parties will collaborate exclusively in immuno-oncology for an initial period of approximately two years, with the option for Curis to extend the broad immuno-oncology exclusivity.
In addition Curis has agreed to make payments to Aurigene as follows:
- for the first two programs: up to $52.5 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any;
- for the third and fourth programs: up to $50 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any; and
- for any program thereafter: up to $140.5 million per program, including $87.5 million per program in approval and commercial milestones, plus specified approval milestone payments for additional indications, if any.
Curis has agreed to pay Aurigene royalties on any net sales ranging from high single digits to 10% in territories where it successfully commercializes products and will also share in amounts that it receives from sublicensees depending upon the stage of development of the respective molecule.
About Immune Checkpoint Modulation and Programmed Death 1 Pathway
Modulation of immune checkpoint pathways has emerged as a highly promising therapeutic approach in a wide range of human cancers. Immune checkpoints are critical for the maintenance of self-tolerance as well as for the protection of tissues from excessive immune response generated during infections. However, cancer cells have the ability to modulate certain immune checkpoint pathways as a mechanism to evade the immune system. Certain immune checkpoint receptors or ligands are expressed by various cancer cells, targeting of which may be an effective strategy for generating anti-tumor activity. Some immune-checkpoint modulators, such as programmed death 1 (PD-1) protein, specifically regulate immune cell effector functions within tissues. One of the mechanisms by which tumor cells block anti-tumor immune responses in the tumor microenvironment is by upregulating ligands for PD-1, such as PD-L1. Hence, targeting of PD-1 and/or PD-L1 has been shown to lead to the generation of effective anti-tumor responses.
About Curis, Inc.
Curis is a biotechnology company focused on the development and commercialization of novel drug candidates for the treatment of human cancers. Curis’ pipeline of drug candidates includes CUDC-907, a dual HDAC and PI3K inhibitor, CUDC-427, a small molecule antagonist of IAP proteins, and Debio 0932, an oral HSP90 inhibitor. Curis is also engaged in a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are developing and commercializing Erivedge®, the first and only FDA-approved medicine for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at www.curis.com.
About Aurigene
Aurigene is a specialized, discovery stage biotechnology company, developing novel and best-in-class therapies to treat cancer and inflammatory diseases. Aurigene’s Programmed Death pathway program is the first of several immune checkpoint programs that are at different stages of discovery and preclinical development. Aurigene has partnered with several large- and mid-pharma companies in the United States and Europe and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies. Aurigene is an independent, wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (NYSE:RDY). For more information, please visit Aurigene’s website at http://aurigene.com/.
POSTER
WO2011161699, WO2012/168944, WO2013144704 and WO2013132317 report peptides or peptidomimetic compounds which are capable of suppressing and/or inhibiting the programmed cell death 1 (PD1) signaling pathway.
PATENT
Example 5: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 4 (compound 4) using D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH, Fmoc-D-Asn(trt)-OH in place of Fmoc-Asn(trt)-OH and H-D-Ser(‘Bu)-0’Bu was used in place of H-Thr^Bu^O’Bu to yield 0.3 g crude material of the title compound. The cmde solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.3 (M+H)+. HPLC: tR = 13.58 min.
Example 8: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 2 (compound 2) using Fmoc-Glu(0’Bu)-OH instead of Fmoc-Asn(Trt)-OH to get 0.4 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 362.1 (M+H)+. HPLC: tR = 13.27 min.
PATENT
Example 3: Synthesis of compound 3
Step 3a:

3a
Lawesson’s reagent (2.85 g, 7.03 mmol) was added to a solution of compound 2e (4 g, 4.68 mmol) in THF (40 mL) and stirred at 75°C for 4 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced
pressure and the obtained residue was partitioned between ice water and ethyl acetate. The organic layer was washed with NaHCC>3 solution followed brine solution. The organic layer was dried over Na2S04, filtered and evaporated under reduced pressure to get residue which was further purified by silica gel column chromatography (eluent: 0-5% ethyl acetate in hexane) to afford 2.7 g of compound 3a (Yield: 67.66%). LCMS: 852.3 (M+H)+,
Step 3

3a 3b
Fmoc group on compound 3a was deprotected by adding diethylamine (3.8 mL) to the solution of compound 3a (1 g, 1.17 mmol) in CH2CI2 (3.8 mL). The reaction mixture was stirred at room temperature for 30 min. The resulting solution was concentrated in vacuum to get a thick gummy residue. The crude compound was purified by neutral alumina column chromatography (eluent: 0-50% ethyl acetate in hexane then 0-5% methanol in chloroform) to attain 0.62 g of compound 3b. LCMS: 630.5 (M+H)+.
Step 3c

To a solution of compound 3b (0.6 g) in CH2CI2 (7.5 mL), trifluoroacetic acid (2.5 mL) and catalytic amount of triisopropylsilane were added and stirred at room temperature for 3 h. The resulting solution was concentrated in vacuum to get 0.13 g of compound 3 which was purified by preparative HPLC method described under experimental conditions. LCMS: 232.3 (M+H)+.
Example 1: Synthesis of compound 1
Step la:

Potassium carbonate (7.9 g, 57.39 mmol) and Methyl iodide (1.3 mL, 21.04 mmol) were added to a solution of compound la (5.0 g, 19.13 mmol) in DMF (35 mL) and stirred at room temperature for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get 5.0 g of compound lb (Yield: 96.1%). LCMS: 176.1 (M-Boc)+.
Step lb:

Hydrazine hydrate (7.2 mL) was added to a solution of compound lb (5.0 g, 18.16 mmol) in methanol (30 mL) and stirred at room temperature for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure, the residue obtained was partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get 4.0 g of compound lc (Yield: 80.0%). LCMS: 276.3 (M+H)+. Step lc:

NMM (0.67 ml, 6.52 mmol) was slowly added to a stirred solution of lc (1.2 g, 4.35 mmol), Id (1.43 g, 4.35 mmol), HOBt (0.7 g, 5.22 mmol) and EDC.HC1 (0.99 g, 5.22 mmol) in DMF (15 mL) at 0°C. The reaction mixture was stirred at room temperature for 12 h. The completeness of the reaction was confirmed by TLC analysis. The reaction was quenched with ice and the solid precipitated was filtered and dried under vacuum to obtain 2.0 g of pure product le (Yield: 83.3%). LCMS: 591.5 (M+Na)+.
St

1 e
1f
To a stirred solution of le (1.5 g, 2.63 mmol) in dry THF (15.0 mL) and DMF (5.0 mL) triphenylphosphine (1.38 g, 5.27 mmol) and iodine (1.33 g, 5.27 mmol) were added at 0°C. After the iodine was completely dissolved, Et3N (1.52 mL, 10.54 mmol) was added to this reaction mixture at ice cold temperature. Reaction mixture was allowed to attain room temperature and stirred for 4 h. The completeness of the reaction was confirmed by TLC analysis. The reaction was quenched with ice water and extracted with ethyl acetate. Organic layer was washed with saturated sodium thiosulphate and brine solution.
The separated Organic layer was dried over Na2SC>4 and evaporated under reduced pressure to get residue, which was further purified by silica gel column chromatography (eluent: 30% ethyl acetate in hexane) to afford 0.8 g of compound If (Yield: 55%). LCMS: 551.3 (M+H)+.
Step le:

1f i g
Fmoc group was deprotected by the addition of diethylamine (20.0 mL) to a solution of compound If (0.8 g, 1.45 mmol) in CH2CI2 (20.0 mL) at 0°C. The reaction was stirred at room temperature for 2 h. The resulting solution was concentrated in vacuum to get a thick gummy residue. The crude compound was purified by neutral alumina column chromatography (eluent: 2% methanol in chloroform) to afford 0.38 g of compound lg (Yield: 80.0%): LCMS: 329.4 (M+H)+.
Step If:

ig 1 i
Compound lg (0.38 g, 1.16 mmol), TEA (0.33 mL, 2.32 mmol) dissolved in DMF (10 mL) were added drop wise to a solution of lh (0.55 g, 1.39 mmol) at 0°C for urea bond formation and the mixture was stirred at room temperature for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction was quenched with ice water, the solid precipitated was filtered and dried under vacuum to get crude compound, which was further purified by silica gel column chromatography (eluent: 0-35% ethyl acetate in hexane) to get 0.4 g of product li (Yield: 59.7%). LCMS: 586.4 (M+H)+.
Step lg:
BocHN’ IJ, H LT Y~™
![]()
1
To a solution of compound li (0.4 g, 0.68 mmol) in CH2CI2 (5 m L), trifluoro acetic acid (5 mL) and catalytic amount of triisopropylsilane were added and stirred at room temperature for 3 h to remove the acid sensitive protecting groups. The resulting solution was concentrated under nitrogen and the solid material was purified by preparative HPLC method as described under experimental conditions (Yield: 0.05 g). LCMS: 318.0 (M+H)+; HPLC: tR= 10.96 min.
Synthesis of compound lh (N02-C6H4-OCO-Thr(tBu)- 0¾u):

To a solution of 4-nitrophenylchloroformate (4.79 g, 23.77 mmol) in DCM (25.0 mL) was added a solution of H-Thr(tBu)-OtBu (5.0 g, 21.61 mmol) TEA (6.2 mL, 43.22 mmol) in CH2CI2 (25 mL) slowly at 0°C and allowed to stir for 30 min. The completion of the reaction was confirmed by TLC analysis. After completion of reaction it was diluted with DCM and washed with 1.0 M of citric acid followed by 1.0 M sodium carbonate solution. The organic layer was dried over Na2S04 and evaporated under reduced pressure to afford crude compound 1 h, which was further purified by silica gel column chromatography (eluent: 0-5% ethyl acetate in hexane) to get 3.0 g of product lh. jH NMR (CDCI3, 400 MHz): £1.17 (s, 9H), 1 .28 (d, 3H), .50 (s, 9H), 4.11 (m, 1 H), 4.28 (m, 1H , 5.89 (d, 1H), 7.37 (d, 2H), 8.26 (d, 2H).
Brahma Reddy V, Thomas Antony, Murali Ramachandra, Venkateshwar Rao G, Wesley Roy Balasubramanian, Kishore Narayanan, Samiulla DS, Aravind AB, and Shekar Chelur.
REFERENCES
US20150073024
| WO2011161699A2 | 27 Jun 2011 | 29 Dec 2011 | Aurigene Discovery Technologies Limited | Immunosuppression modulating compounds |
| WO2012168944A1 | 21 Dec 2011 | 13 Dec 2012 | Aurigene Discovery Technologies Limited | Therapeutic compounds for immunomodulation |
| WO2013132317A1 | 4 Mar 2013 | 12 Sep 2013 | Aurigene Discovery Technologies Limited | Peptidomimetic compounds as immunomodulators |
| WO2013144704A1 | 28 Mar 2013 | 3 Oct 2013 | Aurigene Discovery Technologies Limited | Immunomodulating cyclic compounds from the bc loop of human pd1 |
http://www.curis.com/images/stories/pdfs/posters/Aurigene_PD-L1_VISTA_AACR-NCI-EORTC_2015.pdf
////////Curis and Aurigene, AUPM 170, CA 170, AUPM-170, CA-170, PD-L1, VISTA antagonist
CFG 920, Novartis Scientists team up with Researchers at Aurigene, Bangalore, India,
CFG920,
Inhibitor Of Prostate Cancer With Fewer Cardiac Side Effects
Cas 1260006-20-9
Novartis
Target: CYP17/CYP11B2
Disease: Castration-resistant prostate cancer
MF C14H13ClN4O
MW: 288.0778
Elemental Analysis: C, 58.24; H, 4.54; Cl, 12.28; N, 19.40; O, 5.54
Steroid 17-alpha-hydroxylase inhibitors
CFG920 is a CYP17 inhibitor, is also an orally available inhibitor of the steroid 17-alpha-hydroxylase/C17,20 lyase (CYP17A1 or CYP17), with potential antiandrogen and antineoplastic activities. Upon oral administration, CYP17 inhibitor CFG920 inhibits the enzymatic activity of CYP17A1 in both the testes and adrenal glands, thereby inhibiting androgen production. This may decrease androgen-dependent growth signaling and may inhibit cell proliferation of androgen-dependent tumor cells.
https://clinicaltrials.gov/ct2/show/NCT01647789
NCT01647789: A Study of Oral CFG920 in Patients With Castration Resistant Prostate Cancer2012
- 09 Nov 2015Adverse events, efficacy and pharmacokinetics data from the phase I part of a phase I/II trial in Prostate cancer (Metastatic disease) presented at the 27th AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics (AACR-NCI-EORTC-2015)
- 29 Jan 2013Phase-I clinical trials in Prostate cancer in Spain (PO)
- 10 Dec 2012Phase-I clinical trials in Prostate cancer in Canada (PO)
In August 2015, preclinical data were presented at the 250th ACS meeting in Boston, MA. In monkeys, treatment with CFG-920 (3 mg/kg, po) showed good bioavailability with F value of 93%, Tmax of 0.5 h, Cmax of 1382 nM.dn and AUC of 2364 nM.h, while CFG-920 (10 mg/kg, po) showed F value of 183%, Cmax of 1179 nM.dn and Tmax of 1.04 h
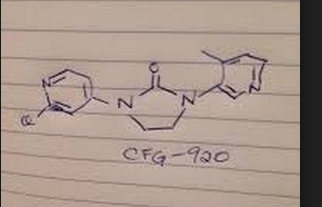
Bethany Halford on Twitter: “CFG920 – @Novartis CMOS for …

Novartis is developing CFG-920 (structure shown), an oral CYP17 inhibitor, for the potential treatment of metastatic castration-resistant prostate cancer. In March 2013, a phase I/II trial was initiated and at that time, the study was expected to complete in January 2015; in August 2015, clinical data were presented
2015 250th (August 19) Abs MEDI 341
Discovery of CFG920, a dual CYP17/CYP11B2 inhibitor, for the treatment of castration resistant prostate cancer
American Chemical Society National Meeting and Exposition
Christoph Gaul, Prakash Mistry, Henrik Moebitz, Mark Perrone, Bjoern Gruenenfelder, Nelson Guerreiro, Wolfgang Hackl, Peter Wessels, Estelle Berger, Mark Bock, Saumitra Sengupta, Venkateshwar Rao, Murali Ramachandra, Thomas Antony, Kishore Narayanan, Samiulla Dodheri, Aravind Basavaraju, Shekar Chelur


| Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, CSN Murthy is chief executive officer |

WO 2010149755

Prostate cancer is the most commonly occurring cancer in men. Doctors often treat the metastatic stage of the disease by depriving the patient of sex hormones via chemical or surgical castration. But if it progresses far enough, the cancer can survive this therapy, transforming into the castration-resistant form. “Once the cancer becomes castration-resistant, the prognosis is poor,” said Novartis’s Christoph Gaul.
In recent years, CYP17, a bifunctional 17α-hydroxylase/17,20-lyase cytochrome P450 enzyme, has emerged as a target for treating castration-resistant prostate cancer. The enzyme catalyzes the biosynthesis of sex hormones, including testosterone, and blocking it can starve prostate cancer of the androgens it needs to thrive.
Johnson & Johnson’s CYP17 inhibitor, abiraterone acetate (Zytiga), a steroid that binds irreversibly to CYP17, was approved by the Food & Drug Administration in 2011. But Novartis scientists thought they could make a better CYP17 inhibitor, Gaul told C&EN. They teamed up with researchers at Aurigene, in Bangalore, India, and came up with their clinical candidate, CFG920.
Unlike abiraterone, CFG920 isn’t a steroid, and it inhibits CYP17 reversibly. It also reversibly inhibits another cytochrome P450 enzyme, CYP11B2, which is involved in the synthesis of the mineralocorticoids, hormones that regulate cardiac function.
Treating prostate cancer patients by lowering their androgen levels turns out to have negative cardiac side effects: Patients’ lipid metabolism is thrown off and their mineralocorticoid levels jump, leading to increases in blood pressure. Those changes can be stressful for the heart. “If prostate cancer patients don’t die because of the cancer, a lot of times they die because of cardiac disease,” Gaul said.
Because CFG920 also keeps mineralocorticoid levels in check, Novartis is hoping the drug candidate will ameliorate some of the cardiac side effects of inhibiting CYP17. The compound is currently in Phase I clinical trials.
PATENT
WO 2010149755
Example 58
Prύpιn”ation ofI'(2’ChIoroψ}ri(ibi-^’\l)’3’f4’metMψ}τUin’3’yl)-imiJazoliJin’2’θne (5HA)-

Using the same reaction conditions as in Example 14. 1-(4-methyl-pyridin-3-yl)- itnida/olidin-2-onc ().-.!.4b: 600 mg. 3.3898 mmol) uas reacted with 2-chloro-4-iodo- py.idine (974 mg.4.067 mmol). 1 , 4-dioxane (60 mL). copper iodide (65 mg, 0.3398 mmol), /r<w.v-1.2-diamino cycK)hexane (0.12 ml,, 1.0169 mmol) and potassium phosphate (2.15 g, 10.1694 mmol) to afford 810 mg of the product (83% yield).
1H NMR (C1DCI3. 300 Mi l/): 6 8.5-8.4 (m. 211). 8.3 (d. IH), 7.6-7.5 (m, 2H). 7.2 (S. 111). 4.1-3.9 (ni. 4H), 2.35 <s. 3H)
LCVIS puιϊt>: 90.8%. nι-7 – 289.1 (M M)
HPl C: 97.14%
REFERENCES
1: Gomez L, Kovac JR, Lamb DJ. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids. 2015 Mar;95:80-7. doi: 10.1016/j.steroids.2014.12.021. Epub 2015 Jan 3. Review. PubMed PMID: 25560485; PubMed Central PMCID: PMC4323677.
2: Yin L, Hu Q, Hartmann RW. Recent progress in pharmaceutical therapies for castration-resistant prostate cancer. Int J Mol Sci. 2013 Jul 4;14(7):13958-78. doi: 10.3390/ijms140713958. Review. PubMed PMID: 23880851; PubMed Central PMCID: PMC3742227.
///////CFG-920, CYP17 inhibitor (prostate cancer), Novartis, CFG 920, Novartis scientists, team up , researchers , Aurigene, Bangalore, India,
AUNP-12 from Aurigene Discovery Technologies Limited
AUNP-12
AUR-012; Aurigene-012; NP-12, Aurigene; PD-1 inhibitor peptide (cancer), Aurigene; PD-1 inhibitor peptide (cancer), Aurigene/ Pierre Fabre; W-014A
| Latest Stage of Development | Preclinical |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat cancer |
| Regulatory Designation | |
| Partner | Laboratoires Pierre Fabre S.A. |
Aurigene Discovery Technologies Limited

-
Programmed Cell Death 1 or PD-1 (also referred to as PDCD1) is a 50 to 55 kD type I membrane glycoprotein (Shinohara T et al, Genomics, 1994, Vol. 23, No. 3, pp. 704-706). PD-1 is a receptor of the CD28 superfamily that negatively regulates T cell antigen receptor signalling by interacting with the specific ligands and is suggested to play a role in the maintenance of self tolerance.
-
PD-1 peptide relates to almost every aspect of immune responses including autoimmunity, tumour immunity, infectious immunity, transplantation immunity, allergy and immunological privilege.
-
The PD-1 protein’s structure comprise of—
-
- an extracellular IgV domain followed by
- a transmembrane region and
- an intracellular tail
-
-
The intracellular tail contains two phosphorylation sites located in an immunoreceptor tyrosine-based inhibitory motif and an immunoreceptor tyrosine-based switch motif, which suggests that PD-1 negatively regulates TCR signals. Also, PD-1 is expressed on the surface of activated T cells, B cells, and macrophages, (Y. Agata et al., Int Immunol 8, 765, May 1996) suggesting that compared to CTLA-4 ((Cytotoxic T-Lymphocyte Antigen 4, also known as CD152 (Cluster of differentiation 152) is a protein that also plays an important regulatory role in the immune system), PD-1 more broadly negatively regulates immune responses.
-
PD-1 has two ligands, PD-L1 (Programmed Death Ligand for PDCD1L1 or B7-H1) (Freeman G J et al, Journal of Experimental Medicine, 2000, Vol. 19, No. 7, pp. 1027-1034) and PD-L2 (Programmed Death Ligand 2 or PDCD1L2 or B7-DC) (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267), which are members of the B7 family. PD-L1 is known to be expressed not only in immune cells, but also in certain kinds of tumour cell lines (such as monocytic leukaemia-derived cell lines, mast cell tumour-derived cell lines, hematoma-derived cell lines, neuroblastoma-derived cell lines, and various mammary tumour-derived cell lines) and in cancer cells derived from diverse human cancer tissues (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267) and on almost all murine tumour cell lines, including PA1 myeloma, P815 mastocytoma, and B16 melanoma upon treatment with IFN-γ (Y. Iwai et al., Proc Natl Acad Sci USA 99, 12293, Sep. 17, 2002 and C. Blank et al., Cancer Res 64, 1140, February, 2004). Similarly PD-L2 expression is more restricted and is expressed mainly by dendritic cells and a few tumour cell lines. PD-L2 expression has been verified in Hodgkin’s lymphoma cell lines and others. There is a hypothesis that some of the cancer or tumour cells take advantage from interaction between PD-1 and PD-L1 or PD-L2, for suppressing or intercepting T-cell immune responses to their own (Iwai Y et al, Proceedings of the National Academy of Science of the United States of America, 2002, Vol. 99, No. 19, pp. 12293-12297).
-
Tumour cells and virus (including HCV and HIV) infected cells are known to express the ligand for PD-1 (to create Immunosuppression) in order to escape immune surveillance by host T cells. It has been reported that the PD-1 gene is one of genes responsible for autoimmune diseases like systemic lupus erythematosis (Prokunina et al, Nature Genetics, 2002, Vol. 32, No. 4, 666-669). It has also been indicated that PD-1 serves as a regulatory factor for the onset of autoimmune diseases, particularly for peripheral self-tolerance, on the ground that PD-1-deficient mice develop lupus autoimmune diseases, such as glomerulonephritis and arthritis (Nishimura H et al, International Immunology, 1998, Vol. 10, No. 10, pp. 1563-1572; Nishimura H et al, Immunity, 1999, Vol. 11, No. 2, pp. 141-151), and dilated cardiomyopathy-like disease (Nishimura H et al, Science, 2001, Vol. 291, No. 5502, pp. 319-332).
-
Hence, in one approach, blocking the interaction of PD-1 with its ligand (PD-L1, PD-L2 or both) may provide an effective way for specific tumour and viral immunotherapy.
-
Wood et al in U.S. Pat. No. 6,808,710 discloses method for down modulating an immune response comprising contacting an immune cell expressing PD-1 with an antibody that binds to PD-1, in multivalent form, such that a negative signal is transduced via PD-1 to thereby down modulate the immune response. Such an antibody may be a cross-linked antibody to PD-1 or an immobilized antibody to PD-1.
-
Freeman et al in U.S. Pat. No. 6,936,704 and its divisional patent U.S. Pat. No. 7,038,013 discloses isolated nucleic acids molecules, designated B7-4 nucleic acid molecules, which encode novel B7-4 polypeptides, isolated B7-4 proteins, fusion proteins, antigenic peptides and anti-B7-4 antibodies, which co-stimulates T cell proliferation in vitro when the polypeptide is present on a first surface and an antigen or a polyclonal activator that transmits an activating signal via the T-cell receptor is present on a second, different surface.
-
There are some reports regarding substances inhibiting immunosuppressive activity of PD-1, or interaction between PD-1 and PD-L1 or PD-L2, as well as the uses thereof. A PD-1 inhibitory antibody or the concept of a PD-1 inhibitory peptide is reported in WO 01/14557, WO 2004/004771, and WO 2004/056875. On the other hand, a PD-L1 inhibitory antibody or a PD-L1 inhibitory peptide is reported in WO 02/079499, WO 03/042402, WO 2002/086083, and WO 2001/039722. A PD-L2 inhibitory antibody or a PD-L2 inhibitory peptide is reported in WO 03/042402 and WO 02/00730.
-
WO2007005874 describes isolated human monoclonal antibodies that specifically bind to PD-L1 with high affinity. The disclosure provides methods for treating various diseases including cancer using anti-PD-L1 antibodies.
-
US2009/0305950 describes multimers, particularly tetramers of an extracellular domain of PD-1 or PD-L1. The application describes therapeutic peptides.
-
Further, the specification mentions that peptides can be used therapeutically to treat disease, e.g., by altering co-stimulation in a patient. An isolated B7-4 or PD-1 protein, or a portion or fragment thereof (or a nucleic acid molecule encoding such a polypeptide), can be used as an immunogen to generate antibodies that bind B7-4 or PD-1 using standard techniques for polyclonal and monoclonal antibody preparation. A full-length B7-4 or PD-1 protein can be used, or alternatively, the invention provides antigenic peptide fragments of B7-4 or PD-1 for use as immunogens. The antigenic peptide of B7-4 or PD-1 comprises at least 8 amino acid residues and encompasses an epitope of B7-4 or PD-1 such that an antibody raised against the peptide forms a specific immune complex with B7-4 or PD-1. Preferably, the antigenic peptide comprises at least 10 amino acid residues, more preferably at least 15 amino acid residues, even more preferably at least amino acid residues, and most preferably at least 30 amino acid residues.
-
Freeman et al in U.S. Pat. No. 7,432,059 appears to disclose and claim methods of identifying compounds that up modulate T cell activation in the presence of a PD-1-mediated signal. Diagnostic and treatment methods utilizing compositions of the invention are also provided in the patent.
-
Further, Freeman et al in U.S. Pat. No. 7,709,214 appears to cover methods for up regulating an immune response with agents that inhibit the interactions between PD-L2 and PD-1.
-
Despite existence of many disclosures as discussed above, however, a significant unmet medical need still exists due to the lack of effective peptides or modified peptides as therapeutic agents as alternatives in the therapeutic area. It is known that synthetic peptides offer certain advantages over antibodies such as ease of production with newer technologies, better purity and lack of contamination by cellular materials, low immunogenicity, improved potency and specificity. Peptides may be more stable and offer better storage properties than antibodies. Moreover, often peptides possess better tissue penetration in comparison with antibodies, which could result in better efficacy. Peptides can also offer definite advantages over small molecule therapeutics counterparts such as lesser degree of toxicity and lower probability of drug-drug interaction.
-
The present invention therefore may provide the solution for this unmet medical need by offering novel synthetic peptide and its derivatives which are based on the PD1 ectodomain.

Patent
http://www.google.com/patents/US20110318373
| 8. | SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2 (SEQ ID NO: 49) |
|

Example 2 Synthesis of

Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
-
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows
-
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 m L). The resin was washed with DMF (6×15 m L), DCM (6×15 m L) and DMF (6×15 m L). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 m mol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 m L; 5 equiv, 1 m mol) and HOBT (0.08 g; 5 equiv, 0.6 m mol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 m mol, 8 equivalent excess of HOBt (0.22 g, 1.6 m mol) and 10 equivalent excess of DIC (0.32 m L, 2 m mol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 m mol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 m mol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B: Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT-12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+); 816.2[M/4+H]+;
STRUCTURE , READER DISCRETION IS NEEDED
N2,N6-Bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-alpha-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-L-alpha-glutamine
C142 H226 N40 O48, 3261.553
SEE ALSO
US 2015087581
Compound 8 (SEQ ID NO: 49) SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2
Example 2Synthesis of Sequence Shown in SEQ ID NO: 49
Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows.
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 mmol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 mL; 5 equiv, 1 mmol) and HOBT (0.08 g; 5 equiv, 0.6 mmol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 mmol, 8 equivalent excess of HOBt (0.22 g, 1.6 mmol) and 10 equivalent excess of DIC (0.32 mL, 2 mmol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 mmol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 mmol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B:Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT—12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+;); 816.2[M/4+H]+.
SMILES
O=C(N[C@@H](CCCCNC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)N)[C@@H](C)O)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(=O)O)C(N)=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)N)[C@@H](C)O
NEXT………..
- Molecular Weight, 3261.55
NEXT……….

Clips
Aurigene and Pierre Fabre Pharmaceuticals Announce a Licensing Agreement for a New Cancer Therapeutic in Immuno-oncology: AUNP12, an Immune Checkpoint Modulator Targeting the PD-1 Pathway
Pierre Fabre are thus reinforcing their oncology portfolio which already enjoys a combination of chemotherapies, monoclonal antibodies and immuno-conjugates assets at various development phases
Feb 13, 2014, 03:14 ET from Aurigene and Pierre Fabre Pharmaceuticals
CASTRES, France and BANGALORE, India, February 13, 2014 /PRNewswire/ —
Pierre Fabre, the third largest French pharmaceutical company, and Aurigene, a leading biotech company based in India, today announced that the two companies have entered into a collaborative license, development and commercialization agreement granting Pierre Fabre global Worldwide rights (excluding India) to a new immune checkpoint modulator, AUNP-12.
AUNP-12 offers a breakthrough mechanism of action in the PD-1 pathway compared to other molecules currently in development in the highly promising immune therapy cancer space. AUNP-12 is the only peptide therapeutic in this pathway and could offer more effective and safer combination opportunities with emerging and established treatment regimens. AUNP-12 will be in development for numerous cancer indications.
Under the terms of this agreement, Aurigene will receive an upfront payment from Pierre Fabre. Aurigene will also receive additional milestone payments based upon the continued development, regulatory progresses and commercialization of AUNP-12.
“We are pleased that Pierre Fabre see the PD-1 program as a strategic asset in their portfolio. Overall, the deal structure, in line with the financial terms that have been seen in this space, demonstrate the importance that Pierre Fabre attach to the program,” said CSN Murthy, CEO, Aurigene.
“The plans that Pierre Fabre have detailed for the development of this differentiated asset highlight the long-term opportunities for this novel cancer therapeutic,” added Murali Ramachandra, Sr VP, Research, Aurigene.
“This agreement, in the field of oncology, is fully consistent with our vision to build Pierre Fabre’s future in prescription drugs, from a combination of cutting-edge internal R&D capabilities and license partnerships with innovative biotech companies like Aurigene,” stated Bertrand Parmentier, CEO, Pierre Fabre.
“With this deal, Pierre-Fabre Pharmaceuticals are reinforcing their portfolio of oncology assets and capitalizing on their proven capabilities in developing biological compounds such as monoclonal antibodies and immuno-conjugates. We have been impressed by the science at Aurigene and encouraged by the differentiated profile reported for AUNP-12,” added Frédéric Duchesne, President, Pierre Fabre Pharmaceuticals.
About immuno-oncology
Immuno-oncology is an emerging field in cancer therapy, where the body’s own immune system is harnessed to fight against cancer. This approach of targeting cancer through immune response has had a breakthrough when robust and sustained responses were obtained only upon blocking the immune checkpoint targets (such as PD-1 and CTLA4). Recent successes in clinical trials performed with such therapies suggest that immunotherapy should be considered alongside surgery, chemotherapy, radiotherapy and targeted therapy as the fifth cornerstone of cancer treatment.
PD-1 (Programmed cell Death 1) is a receptor that negatively regulates T-cell activation by interacting with specific ligands PD-L1 and PD-L2. Tumor cells express these ligands and thereby escape from the action of T-cells.
About AUNP-12
AUNP-12 is a branched 29-amino acid peptide sequence engineered from the PD-L1/ L2 binding domain of PD-1 It blocks the PD-1/PD-L1, PD-1/PD-L2 and PD-L1/CD80 pathways. AUNP-12 is highly effective in antagonizing PD-1 signaling, with desirable in vivo exposure upon subcutaneous dosing. It inhibits tumor growth and metastasis in preclinical models of cancer and is well tolerated with no overt toxicity at any of the tested doses.
About Aurigene
Aurigene is a biotech focused on development of innovative small molecule and peptide therapeutics for Oncology and Inflammation; key focus areas for Aurigene are Immuno-oncology, Epigenetics and the Th17 pathway. Aurigene’s PD-1 program is the first of several peptide-based immune checkpoint programs that are at different stages of Discovery.
Aurigene has partnered with several big pharma and mid-pharma companies in the US and Europe, and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies.
Aurigene’s pre-clinical pipeline includes (1) Selective and pan-BET Bromodomain inhibitors (2) RoR gamma reverse agonists (3) EZH2 inhibitors (4) NAMPT inhibitors and (5) Several immune check point peptide inhibitor programs.
For more information: http://aurigene.com/
About Pierre Fabre:
Pierre Fabre is a privately-owned health care company created in 1961 by Mr Pierre Fabre. It is the second largest French independent pharmaceutical group with 2013 sales amounting to about €2 billion (yet to be audited) across 140 countries. The company is structured around two divisions: Pharmaceuticals (Prescription drugs, OTC, Oral care) and Dermo-cosmetics. Prescription drugs are organized around four main franchises: oncology, dermatology, women’s health and neuropsychiatry. Pierre Fabre employs some 10 000 people worldwide, including 1 300 in R&D. The company allocates about 20% of its pharmaceuticals sales to R&D and relies on more than 25 years of experience in the discovery, development and global commercialization of innovative drugs in oncology. Pierre Fabre has a long commitment to oncology and immunology with major R&D centers in France: the Pierre Fabre immunology Centre (CIPF) in Saint Julien en Genevois and the Pierre Fabre Research Institute (IRPF) located on the Toulouse-Oncopole campus which has been officially recognized as a National Center of Excellence for cancer research since 2012.
REFERENCES
http://slideplayer.com/slide/5760496/
P. Sasikumar, R. Shrimali, S. Adurthi, R. Ramachandra, L. Satyam, A. Dhudashiya, D. Samiulla, K. B. Sunilkumar and M. Ramachandra, “A novel peptide therapeutic targeting PD1 immune checkpoint with equipotent antagonism of both ligands and a potential for better management of immune-related adverse events,” Journal for ImmunoTherapy of Cancer, vol. 1, no. Suppl 1, O24, 2013.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Vemula, L. K. Satyam, K. Subbarao, K. R. Shrimali and S. Kandepudu (Aurigene Discovery Technologies Ltd, Bangalore, India), “Immunosuppression modulating compounds”, US Patent application US 2011/0318373, 29 Dec 2011.
P. G. Sasikumar, L. K. Satyam, R. K. Shrimali, K. Subbarao, R. Ramachandra, S. Vadlamani, A. Reddy, A. Kumar, A. Srinivas, S. Reddy, S. Gopinath, D. S. Samiulla and M. Ramachandra, “Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway,” Cancer Research, vol. 72, no. 8 Suppl. 1, Abstract 2850, 2012.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Shrimali and K. Subbarao, “Therapeutic compounds for immunomodulation” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO 2012/168944, 13 Dec 2012.
P. G. N. Sasikumar and M. Ramachandra, “Immunomodulating cyclic compounds from the BC loop of human PD1” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO/2013/144704, 3 Oct 2013.
P. G. N. Sasikumar, M. Ramachandra and S. S. S. Naremaddepalli, “Peptidomimetic compounds as immunomodulators” (Aurigene Discovery Technologies Ltd, Bangalore, India), US Patent Application US 2013/0237580, 12 Sep 2013.
A. H. Sharpe, M. J. Butte and S. Oyama (Harvard College), “Modulators of immunoinhibitory receptor PD-1, and methods of use thereof”, PCT Patent Application WO/2011/082400, 7 Jul 2011.
M. Cordingley, “Battle of PD-1 blockade is on”, February 7, 2014 : http://discoveryview.ca/battle-of-pd-1-blockade-is-on/ [Accessed 25 February 2014].
Mr. CSN Murthy
Chief Executive Officer, Aurigene Discovery Technologies Ltd.

Mr. CSN Murthy began his career with ICICI Ventures, India’s first Venture Capital fund. He was subsequently a management consultant to the Pharma and Chemical sectors. Later, he worked in the Business Development and General Management functions in Pharmaceutical companies, including as the Chief Operating Officer of Gland Pharma Ltd. CSN holds a Bachelors degree in Chemical Engineering from the Indian Institute of Technology (IIT), Madras and an MBA from the Indian Institute of Management (IIM), Bangalore.
Dr.Thomas Antony
Associate Research Director, Aurigene Discovery Technologies Ltd.

Dr.Thomas Antony did his Ph.D in Biophysical Chemistry from University of Delhi and had his postdoctoral training at Jawaharlal Nehru University- Delhi, The University of Medicine and Dentistry of New Jersey- USA, and Max Planck Institute for Biophysical Chemistry- Germany. He is the recipient of many research fellowships, including Max Planck Fellowship and Humboldt Research Fellowship. He has more than 20 years of research experience. Dr.Thomas has published 24 research papers and he is the co-author of three international patents. His core area of expertise is in assay development and screening. At Aurigene, Dr.Thomas leads the Biochemistry and Structural Biology Divisions. He was the coordinator of Aurigene-University of Malaya collaboration programs.
Dr. Kavitha Nellore
Associate Research Director, Aurigene Discovery Technologies Ltd.

Dr. Kavitha Nellore obtained her PhD in Bioengineering from Pennsylvania State University, USA. During this time, she was a fellow of the Huck’s Institute of Life Sciences specializing in Biomolecular Transport Dynamics. She has been at Aurigene for more than a decade, and is currently leading a group of cell biologists at both Bangalore and Kuala Lumpur. At Aurigene, she leads multiple drug discovery programs in the therapeutic areas of inflammation, oncology and immuno-oncology. She plays a key role in target selection as well as validation efforts to add to Aurigene’s pipeline. Kavitha also played a key role in coordinating the Aurigene-University of Malaya collaboration.
To print: Click here or Select File and then Print from your browser's menu “Strategic partnerships will boost drug discovery activities in India”Wednesday, May 16, 2007 08:00 IST
What are the factors led to the scores of partnerships in research and drug discovery space? Biology demands expertise in areas such as DMPK, cell and assay biology and vivo expertise, which is not as common as chemistry expertise in India. There is also a paradigm shift in assignments for CROs, as they are gearing up to gain higher value from existing projects, including Intellectual Property (IP) generation. Probably, this is where Aurigene has a head start over other CROs. Although we are in research services, our success is in IP generated work. We work with world-class companies like Novo Nordisk, Rheoscience, Debiopharm, Forest Labs and Merck Serono on integrated discovery projects. What do you think is the uniqueness of Aurigene that sets it apart from other companies? It is understood that a major chunk of the business for CROs is from global customers. What is the reason for this? The whole concept of drug discovery outsourcing business has caught on in the last 18-month. Could you give us an overview of the market? In India, CROs operate differently. The innovation driven companies are taking on ‘collaborative drug discovery’ projects, leveraging the mutual strengths each has to offer the other (cost and expertise respectively). Therefore, the complexion of business is different from that of the Western countries. We would see large outsourcing companies positioning themselves as offshore partners for pharma-biotech majors. They could also go in for a BOT (Build Operate Transfer) model. Under this model, companies will set-up the facility and manage it to make it a ‘centre of excellence’, focusing on certain disease segments. Another option is that some companies can take up their own programmes to develop compounds and enter into licensing partnerships. This is similar to the US and Europe biotech style of working. Both of these practices are likely to happen in India. How much do you acknowledge science and strategy in Aurigene’s growth? What according to you is the future of the drug discovery sector? |
/////////AUNP-12, Aurigene, Pierre Fabre Pharmaceuticals, Licensing Agreement, New Cancer Therapeutic, Immuno-oncology, AUNP 12, Immune Checkpoint Modulator Targeting the PD-1 Pathway, PEPTIDES
FEW MORE COMPDS FROM PATENT
C142 H225 N39 O49
L-Glutamic acid, N2,N6-bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3262.54, Sequence Length: 29, 22, 7
multichain; modified (modifications unspecified)
SNTSESFK FRVTQ LAPKAQIKE, 1353564-66-5
SNTSESF
C142 H225 N39 O49
L-Glutamic acid, N2,N6-bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3262.54

NEXT……………………
SNTSESFK FRVTQ LAPKAQI KE
SNTSESF
CAS 1353564-64-3
C142 H226 N40 O48
L-α-Glutamine, L-seryl-D-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6-(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
MW 3261.55, Sequence Length: 29, 22, 7
multichain; modified

smiles
O=C(N[C@@H](CCCCNC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@@H](C)O)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(=O)O)C(N)=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@@H](C)O
NEXT……………..
CAS 1353564-60-9
C142 H226 N40 O48
L-α-Glutamine, D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6-(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3261.55
Sequence Length: 29, 22, 7multichain; modified
SNTSESFKFR VTQLAPKAQI KE
NRXT…………………….
. CAS 1353564-61-0
C142 H226 N40 O48
L-α-Glutamine, N2,N6-bis(D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3261.55
Sequence Length: 29, 22, 7multichain; modified
SNTSESFK FRVTQ LAPKAQI KE
SNTSESF

/////////////
