
Home » Posts tagged 'Atopic dermatitis'
Tag Archives: Atopic dermatitis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tezepelumab-ekko

(Heavy chain)
QMQLVESGGG VVQPGRSLRL SCAASGFTFR TYGMHWVRQA PGKGLEWVAV IWYDGSNKHY
ADSVKGRFTI TRDNSKNTLN LQMNSLRAED TAVYYCARAP QWELVHEAFD IWGQGTMVTV
SSASTKGPSV FPLAPCSRST SESTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSNFGT QTYTCNVDHK PSNTKVDKTV ERKCCVECPP CPAPPVAGPS
VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVQFNWYV DGVEVHNAKT KPREEQFNST
FRVVSVLTVV HQDWLNGKEY KCKVSNKGLP APIEKTISKT KGQPREPQVY TLPPSREEMT
KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPMLD SDGSFFLYSK LTVDKSRWQQ
GNVFSCSVMH EALHNHYTQK SLSLSPGK
(Light chain)
SYVLTQPPSV SVAPGQTARI TCGGNNLGSK SVHWYQQKPG QAPVLVVYDD SDRPSWIPER
FSGSNSGNTA TLTISRGEAG DEADYYCQVW DSSSDHVVFG GGTKLTVLGQ PKAAPSVTLF
PPSSEELQAN KATLVCLISD FYPGAVTVAW KADSSPVKAG VETTTPSKQS NNKYAASSYL
SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS
(Disulfide bridge: H22-H96, H136-L213, H149-H205, H224-H’224, H225-H’225, H228-H’228, H231-H’231, H262-H322, H368-H426, H’22-H’96, H’136-L’213, H’149-H’205, H’262-H’322, H’368-H’426, L22-L87, L136-L195, L’22-L’87, L’136-L’195)
Tezepelumab-ekko
テゼペルマブ (遺伝子組換え)
| Formula | C6400H9844N1732O1992S52 |
|---|---|
| CAS | 1572943-04-4 |
| Mol weight | 144588.4306 |
PEPTIDE
UD FDA APPROVED, 12/17/2021, To treat severe asthma as an add-on maintenance therapy , Tezspire
Monoclonal antibody
Treatment of asthma and atopic dermatitis
Tezepelumab, sold under the brand name Tezspire, is a human monoclonal antibody used for the treatment of asthma.[4][5]
It blocks thymic stromal lymphopoietin (TSLP),[2] an epithelial cytokine that has been suggested to be critical in the initiation and persistence of airway inflammation.[6]
It was approved for medical use in the United States in December 2021.[2][3]
Medical uses
Tezepelumab is indicated for the add-on maintenance treatment of people aged twelve years and older with severe asthma.[2]
Research
In Phase III trials, tezepelumab demonstrated efficacy compared to placebo for patients with severe, uncontrolled asthma.[7][8]
Structural studies by X-ray crystallography showed that Tezepelumab competes against a critical part of the TSLPR binding site on TSLP.[1]
It is being studied for the treatment of chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE).[3]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b Verstraete K, Peelman F, Braun H, Lopez J, Van Rompaey D, Dansercoer A, et al. (April 2017). “Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma”. Nature Communications. 8 (1): 14937. Bibcode:2017NatCo…814937V. doi:10.1038/ncomms14937. PMC 5382266. PMID 28368013.
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761224s000lbl.pdf
- ^ Jump up to:a b c “Tezspire (tezepelumab) approved in the US for severe asthma”. AstraZeneca (Press release). 17 December 2021. Retrieved 17 December 2021.
- ^ Marone G, Spadaro G, Braile M, Poto R, Criscuolo G, Pahima H, et al. (November 2019). “Tezepelumab: a novel biological therapy for the treatment of severe uncontrolled asthma”. Expert Opinion on Investigational Drugs. 28 (11): 931–940. doi:10.1080/13543784.2019.1672657. PMID 31549891. S2CID 202746054.
- ^ Matera MG, Rogliani P, Calzetta L, Cazzola M (February 2020). “TSLP Inhibitors for Asthma: Current Status and Future Prospects”. Drugs. 80 (5): 449–458. doi:10.1007/s40265-020-01273-4. PMID 32078149. S2CID 211194472.
- ^ “Tezepelumab granted Breakthrough Therapy Designation by US FDA”. AstraZeneca (Press release). 7 September 2018.
- ^ “Studies found for: Tezepelumab”. ClinicalTrials.Gov. National Library of Medicine, National Institutes of Health, U.S. Department of Health and Human Services.
- ^ Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. (May 2021). “Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma”. New England Journal of Medicine. 384 (19): 1800–09. doi:10.1056/NEJMoa2034975. PMID 33979488. S2CID 234484931.
External links
- “Tezepelumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02054130 for “Study to Evaluate the Efficacy and Safety of MEDI9929 (AMG 157) in Adult Subjects With Inadequately Controlled, Severe Asthma” at ClinicalTrials.gov
- Clinical trial number NCT03347279 for “Study to Evaluate Tezepelumab in Adults & Adolescents With Severe Uncontrolled Asthma (NAVIGATOR)” at ClinicalTrials.gov
| Structural basis for inhibition of TSLP-signaling by Tezepelumab (PDB 5J13)[1] | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | thymic stromal lymphopoietin (TSLP) |
| Clinical data | |
| Trade names | Tezspire |
| Other names | MEDI9929, AMG 157, tezepelumab-ekko |
| License data | US DailyMed: Tezepelumab |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2][3] |
| Identifiers | |
| CAS Number | 1572943-04-4 |
| DrugBank | DB15090 |
| ChemSpider | None |
| UNII | RJ1IW3B4QX |
| KEGG | D11771 |
| Chemical and physical data | |
| Formula | C6400H9844N1732O1992S52 |
| Molar mass | 144590.40 g·mol−1 |
////////////Tezepelumab-ekko, Tezspire, PEPTIDE, APPROVALS 2021, FDA 2021, Monoclonal antibody
, asthma, atopic dermatitis, ANTI INFLAMATORY, テゼペルマブ (遺伝子組換え)

NEW DRUG APPROVALS
ONE TIME
$10.00
Delgocitinib

Delgocitinib
デルゴシチニブ
3-[(3S,4R)-3-methyl-7-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,7-diazaspiro[3.4]octan-1-yl]-3-oxopropanenitrile
1,6-Diazaspiro(3.4)octane-1-propanenitrile, 3-methyl-beta-oxo-6-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-, (3S,4R)-
3-((3S,4R)-3-methyl-6-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-1,6-diazaspiro(3.4)octan-1-yl)-3-oxopropanenitrile
| Formula |
C16H18N6O
|
|---|---|
| CAS |
1263774-59-9
|
| Mol weight |
310.3537
|
Approved, Japan 2020, Corectim, 2020/1/23, atopic dermatitis, Japan Tobacco (JT)
Torii
7/23/2025 fda approved, Anzupgo
| To treat moderate-to-severe chronic hand eczema when topical corticosteroids are not advisable or produce an inadequate response |
UNII-9L0Q8KK220, JTE-052, LP-0133, ROH-201, 9L0Q8KK220, LEO 124249A, LEO 124249, HY-109053
CS-0031558, D11046, GTPL9619, JTE-052A, JTE052
Delgocitinib, also known as LEO-124249 and JTE052, is a potent and selective JAK inhibitor. JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation.
Delgocitinib is a JAK inhibitor first approved in Japan for the treatment of atopic dermatitis in patients 16 years of age or older. Japan Tobacco is conducting phase III clinical trials for the treatment of atopic dermatitis in pediatric patients. Leo is developing the drug in phase II clinical trials for the treatment of inflammatory skin diseases, such as atopic dermatitis, and chronic hand eczema and for the treatment of discoid lupus erythematosus. Rohto is evaluating the product in early clinical development for ophthalmologic indications.
In 2014, the drug was licensed to Leo by Japan Tobacco for the development, registration and marketing worldwide excluding Japan for treatment of inflammatory skin conditions. In 2016, Japan Tobacco licensed the rights of co-development and commercialization in Japan to Torii. In 2018, Japan Tobacco licensed the Japanese rights of development and commercialization to Rohto for the treatment of ophthalmologic diseases.
Delgocitinib, sold under the brand name Corectim among others, is a medication used for the treatment of autoimmune disorders and hypersensitivity, including inflammatory skin conditions.[3] Delgocitinib was developed by Japan Tobacco and approved in Japan for the treatment of atopic dermatitis.[3] In the United States, delgocitinib is in Phase III clinical trials and the Food and Drug Administration has granted delgocitinib fast track designation for topical treatment of adults with moderate to severe chronic hand eczema.[4]
Delgocitinib works by blocking activation of the JAK-STAT signaling pathway which contributes to the pathogenesis of chronic inflammatory skin diseases.[5]
PATENTS
WO 2018117151
IN 201917029002
IN 201917029003
IN 201917029000
PATENTS
WO 2011013785
https://patents.google.com/patent/WO2011013785A1/en
[Production Example 6]: Synthesis of Compound 6

(1) Optically active substance of 2-benzylaminopropan-1-ol

To a solution of (S)-(+)-2-aminopropan-1-ol (50.0 g) and benzaldehyde (74 ml) in ethanol (500 ml) was added 5% palladium carbon (5.0 g) at room temperature and normal pressure. Hydrogenated for 8 hours. The reaction mixture was filtered through celite and concentrated under reduced pressure to give the title compound (111.2 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.23-7.18 (1H, m), 4.53-4.47 (1H, m), 3.76 (1H, d, J = 13.5 Hz) , 3.66 (1H, d, J = 13.5 Hz), 3.29-3.24 (2H, m), 2.65-2.55 (1H, m), 1.99 (1H, br s), 0.93 (3H, d, J = 6.4 Hz) .
(2) Optically active substance of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester

To a mixture of optically active 2-benzylaminopropan-1-ol (111.2 g), potassium carbonate (111.6 g) and N, N-dimethylformamide (556 ml) cooled to 0 ° C., tert-butyl bromoacetate was added. Ester (109 ml) was added dropwise over 20 minutes and stirred at room temperature for 19.5 hours. The mixture was acidified to pH 2 by adding 2M aqueous hydrochloric acid and 6M aqueous hydrochloric acid, and washed with toluene (1000 ml). The separated organic layer was extracted with 0.1 M aqueous hydrochloric acid (300 ml). The combined aqueous layer was adjusted to pH 10 with 4M aqueous sodium hydroxide solution and extracted with ethyl acetate (700 ml). The organic layer was washed successively with water (900 ml) and saturated aqueous sodium chloride solution (500 ml). The separated aqueous layer was extracted again with ethyl acetate (400 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the title compound (160.0 g).
1 H-NMR (DMSO-D 6 ) δ: 7.37-7.26 (4H, m), 7.24-7.19 (1H, m), 4.26 (1H, dd, J = 6.9, 3.9 Hz), 3.76 (1H, d, J = 14.1 Hz), 3.68 (1H, d, J = 13.9 Hz), 3.45-3.39 (1H, m), 3.29-3.20 (1H, m), 3.24 (1H, d, J = 17.2 Hz), 3.13 ( 1H, d, J = 17.0 Hz), 2.84-2.74 (1H, m), 1.37 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
(3) Optically active substance of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester

(3)-(1) Optically active form of [benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester

To a solution of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (160.0 g) cooled to 0 ° C. in chloroform (640 ml) was added thionyl chloride (50.0 ml). Was added dropwise and stirred at 60 ° C. for 2 hours. The reaction mixture was cooled to 0 ° C., saturated aqueous sodium hydrogen carbonate solution (1000 ml) and chloroform (100 ml) were added and stirred. The separated organic layer was washed with a saturated aqueous sodium chloride solution (500 ml), and the aqueous layer was extracted again with chloroform (450 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain the title compound (172.9 g).
1 H-NMR (CDCl 3 ) δ: 7.40-7.22 (5H, m), 4.05-3.97 (0.4H, m), 3.93-3.81 (2H, m), 3.70-3.65 (0.6H, m), 3.44- 3.38 (0.6H, m), 3.29 (0.8H, s), 3.27 (1.2H, d, J = 2.4 Hz), 3.24-3.15 (0.6H, m), 3.05-2.99 (0.4H, m), 2.94 -2.88 (0.4H, m), 1.50 (1.2H, d, J = 6.4 Hz), 1.48 (3.6H, s), 1.45 (5.4H, s), 1.23 (1.8H, d, J = 6.8 Hz) .
(3)-(2) Optically active form of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester

[Benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (172.9 g) was dissolved in N, N-dimethylformamide (520 ml) and stirred at 80 ° C. for 140 minutes. did. The reaction mixture was cooled to 0 ° C., water (1200 ml) was added, and the mixture was extracted with n-hexane / ethyl acetate (2/1, 1000 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (400 ml), and the separated aqueous layer was extracted again with n-hexane / ethyl acetate (2/1, 600 ml). The combined organic layers were concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 50/1 to 40/1) to give the title compound (127.0 g )
1 H-NMR (CDCl 3 ) δ: 7.37-7.29 (4H, m), 7.28-7.23 (1H, m), 4.05-3.97 (1H, m), 3.91 (1H, d, J = 13.5 Hz), 3.86 (1H, d, J = 13.7 Hz), 3.29 (2H, s), 3.03 (1H, dd, J = 13.9, 6.6 Hz), 2.91 (1H, dd, J = 13.9, 6.8 Hz), 1.50 (3H, d, J = 6.4 Hz), 1.48 (9H, s).
(4) Optically active substance of 1-benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester

To a solution of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester optically active substance (60.0 g) cooled to −72 ° C. and hexamethylphosphoramide (36.0 ml) in tetrahydrofuran (360 ml), Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 242 ml) was added dropwise over 18 minutes, and the temperature was raised to 0 ° C. over 80 minutes. A saturated aqueous ammonium chloride solution (300 ml) and water (400 ml) were sequentially added to the reaction mixture, and the mixture was extracted with ethyl acetate (500 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (500 ml), and the separated aqueous layer was extracted again with ethyl acetate (300 ml). The combined organic layers were dried over anhydrous sodium sulfate, concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: n-hexane / ethyl acetate = 50/1 to 4/1). To give the title compound (50.9 g).
1 H-NMR (CDCl 3 ) δ: 7.34-7.21 (5H, m), 3.75 (1H, d, J = 12.6 Hz), 3.70-3.67 (1H, m), 3.58 (1H, d, J = 12.6 Hz ), 3.05-3.01 (1H, m), 2.99-2.95 (1H, m), 2.70-2.59 (1H, m), 1.41 (9H, s), 1.24 (3H, d, J = 7.1 Hz).
(5) Optically active substance of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester

1-Benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester optically active substance (43.5 g) and di-tert-butyl dicarbonate (38.2 g) in tetrahydrofuran / methanol (130 ml / 130 ml) solution 20% Palladium hydroxide carbon (3.5 g) was added thereto, and hydrogenated at 4 atm for 2 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (48.0 g).
1 H-NMR (DMSO-D 6 ) δ: 4.44 (1H, d, J = 8.8 Hz), 3.99-3.77 (1H, m), 3.45-3.37 (1H, m), 3.00-2.88 (1H, m) , 1.45 (9H, s), 1.40-1.30 (9H, m), 1.02 (3H, d, J = 7.2 Hz).
(6) Optically active substance of 3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester

Optically active substance (48.0 g) of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester cooled to -69 ° C. and 1-bromo-3-methyl-2-butene (25.4 ml) Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 200 ml) was added to a tetrahydrofuran solution (380 ml). The reaction mixture was warmed to −20 ° C. in 40 minutes and further stirred at the same temperature for 20 minutes. A saturated aqueous ammonium chloride solution (200 ml) and water (300 ml) were successively added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1 / 1,500 ml). The separated organic layer was washed successively with water (200 ml) and saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 15/1 to 8/1) to give the titled compound (44.5 g).
1 H-NMR (CDCl 3 ) δ: 5.29-5.21 (1H, m), 3.77-3.72 (1H, m), 3.49-3.44 (1H, m), 2.73-2.52 (3H, m), 1.76-1.74 ( 3H, m), 1.66-1.65 (3H, m), 1.51 (9H, s), 1.43 (9H, s), 1.05 (3H, d, J = 7.3 Hz).
(7) Optically active substance of 3-methyl-2- (2-oxoethyl) azetidine-1,2-dicarboxylic acid di-tert-butyl ester

3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (44.5 g) in chloroform / cooled to −70 ° C. An ozone stream was passed through the methanol solution (310 ml / 310 ml) for 1 hour. To this reaction mixture, a solution of triphenylphosphine (44.7 g) in chloroform (45 ml) was added little by little, and then the mixture was warmed to room temperature. To this mixture were added saturated aqueous sodium thiosulfate solution (200 ml) and water (300 ml), and the mixture was extracted with chloroform (500 ml). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to obtain the title compound (95.0 g). This product was subjected to the next step without further purification.
1 H-NMR (DMSO-D 6 ) δ: 9.65 (1H, t, J = 2.6 Hz), 3.79-3.74 (1H, m), 3.45-3.40 (1H, m), 2.99-2.80 (3H, m) , 1.46 (9H, s), 1.34 (9H, s), 1.06 (3H, d, J = 7.2 Hz).
(8) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester

To a solution of the residue (95.0 g) obtained in (7) in tetrahydrofuran (300 ml) was added benzylamine (34 ml) at room temperature, and the mixture was stirred for 2 hours. The mixture was cooled to 0 ° C., sodium triacetoxyborohydride (83.3 g) was added, and the mixture was stirred at room temperature for 1.5 hours. Water (300 ml) was added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1/3, 600 ml). The separated organic layer was washed with water (300 ml) and saturated aqueous sodium chloride solution (200 ml), and then extracted twice with 5% aqueous citric acid solution (300 ml, 200 ml) and three times with 10% aqueous citric acid solution (250 ml × 3). . The combined aqueous layers were basified to pH 10 with 4M aqueous sodium hydroxide solution and extracted with chloroform (300 ml). The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (46.9 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.26 (4H, m), 7.22-7.17 (1H, m), 3.74-3.65 (2H, m), 3.61 (1H, t, J = 7.8 Hz) , 3.28 (1H, t, J = 7.5 Hz), 2.76-2.66 (2H, m), 2.57-2.45 (1H, m), 2.15 (1H, br s), 2.05-1.89 (2H, m), 1.42 ( 9H, s), 1.27 (9H, s), 0.96 (3H, d, J = 7.1 Hz).
(9) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride

2- (2-Benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (46.5 g), 4M hydrochloric acid 1,4-dioxane (230 ml) and water (4.1 ml) was mixed and stirred at 80 ° C. for 2 hours. The mixture was concentrated under reduced pressure, azeotroped with toluene, and then slurry washed with n-hexane / ethyl acetate (1/1, 440 ml) to give the title compound (30.1 g).
1 H-NMR (DMSO-D 6 ) δ: 10.24 (1H, br s), 9.64 (2H, br s), 8.90 (1H, br s), 7.58-7.53 (2H, m), 7.47-7.41 (3H , m), 4.21-4.10 (2H, m), 4.02-3.94 (1H, m), 3.46-3.37 (1H, m), 3.20-3.10 (1H, m), 2.99-2.85 (2H, m), 2.69 -2.54 (2H, m), 1.10 (3H, d, J = 7.2 Hz).
(10) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one

To a solution of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride optically active substance (29.1 g) and N, N-diisopropylethylamine (65 ml) in chloroform (290 ml), At room temperature, O- (7-azabenzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (41.3 g) was added and stirred for 4 hours. To this reaction mixture were added saturated aqueous sodium hydrogen carbonate solution (200 ml) and water (100 ml), and the mixture was extracted with chloroform (200 ml). The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 20/1 to 10/1) to give the titled compound (21.3 g).
1 H-NMR (DMSO-D 6 ) δ: 7.38-7.31 (2H, m), 7.30-7.22 (3H, m), 4.52 (1H, d, J = 14.8 Hz), 4.29 (1H, d, J = 14.8 Hz), 3.35-3.27 (2H, m), 3.22-3.17 (1H, m), 3.05 (2H, dd, J = 9.5, 4.0 Hz), 2.77-2.66 (1H, m), 2.16-2.10 (1H , m), 1.96-1.87 (1H, m), 0.94 (3H, d, J = 7.1 Hz).
(11) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester

Concentrated sulfuric acid (4.8 ml) was slowly added dropwise to a suspension of lithium aluminum hydride (6.8 g) in tetrahydrofuran (300 ml) under ice cooling, and the mixture was stirred for 30 minutes. To this mixture was added dropwise a solution of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one optically active substance (21.3 g) in tetrahydrofuran (100 ml) at the same temperature. Stir for 45 minutes. Water (7.0 ml), 4M aqueous sodium hydroxide solution (7.0 ml) and water (14.0 ml) were sequentially added to the reaction mixture, and the mixture was stirred as it was for 30 minutes. To this mixture was added anhydrous magnesium sulfate and ethyl acetate (100 ml), and the mixture was stirred and filtered through celite. Di-tert-butyl dicarbonate (23.4 g) was added to the filtrate at room temperature and stirred for 3 hours. The mixture was concentrated under reduced pressure to a half volume and washed twice with a saturated aqueous ammonium chloride solution (200 ml × 2). N-Hexane (200 ml) was added to the separated organic layer, and the mixture was extracted 5 times with a 10% aqueous citric acid solution. The separated aqueous layer was basified with 4M aqueous sodium hydroxide solution and extracted with chloroform. The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 40/1 to 20/1) to give the titled compound (15.6 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.26-7.21 (1H, m), 3.84-3.69 (1H, m), 3.62-3.47 (2H, m), 3.19- 3.05 (1H, m), 3.02-2.92 (1H, m), 2.76-2.69 (1H, m), 2.47-2.24 (4H, m), 1.95-1.77 (1H, m), 1.36 (9H, s), 1.03 (3H, d, J = 7.0 Hz).
(12) Optically active substance of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester

20% of optically active form of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (10.0 g) in tetrahydrofuran / methanol (50 ml / 50 ml) solution Palladium hydroxide on carbon (2.0 g) was added and hydrogenated at 4 atm for 24 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (7.3 g).
1 H-NMR (DMSO-D 6 ) δ: 3.88-3.71 (1H, m), 3.44-3.06 (2H, m), 3.02-2.64 (4H, m), 2.55-2.38 (1H, m), 2.31- 2.15 (1H, m), 1.81-1.72 (1H, m), 1.37 (9H, s), 1.07 (3H, d, J = 7.0 Hz).
(13) Optical activity of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester body

The optically active substance (6.9 g) of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester was converted into 4-chloro-7H-pyrrolo [2,3-d] pyrimidine ( 4.3 g), potassium carbonate (7.7 g) and water (65 ml) and stirred for 4 hours at reflux. The mixture was cooled to room temperature, water (60 ml) was added, and the mixture was extracted with chloroform / methanol (10/1, 120 ml). The organic layer was washed successively with water, saturated aqueous ammonium chloride solution and saturated aqueous sodium chloride solution, and dried over anhydrous sodium sulfate. To this mixture, silica gel (4 g) was added, stirred for 10 minutes, filtered through celite, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / ethyl acetate = 1/1, then chloroform / methanol = 50/1 to 20/1) to give the title compound (10.0 g). Obtained.
1 H-NMR (DMSO-D 6 ) δ: 11.59 (1H, br s), 8.09 (1H, s), 7.12-7.09 (1H, m), 6.64-6.59 (1H, m), 4.09-3.66 (5H , m), 3.39-3.21 (1H, m), 2.64-2.44 (2H, m), 2.27-2.06 (1H, m), 1.36 (3H, s), 1.21 (6H, s), 1.11 (3H, d , J = 6.5 Hz).
(14) Optically active form of 4- (3-methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride

Optically active form of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (9 0.5 g), 4M hydrochloric acid 1,4-dioxane (50 ml), chloroform (50 ml) and methanol (100 ml) were mixed and stirred at 60 ° C. for 30 minutes. The mixture was concentrated under reduced pressure and azeotroped with toluene to give the title compound (9.3 g).
1 H-NMR (DMSO-D 6 ) δ: 12.91 (1H, br s), 9.97-9.64 (2H, m), 8.45-8.35 (1H, m), 7.58-7.47 (1H, m), 7.04-6.92 (1H, m), 4.99-4.65 (1H, m), 4.32-3.21 (7H, m), 3.04-2.90 (1H, m), 2.46-2.31 (1H, m), 1.27 (3H, d, J = 6.0 Hz).
(15) 3- [3-Methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] oct-1-yl] -3-oxo Optically active form of propionitrile

4- (3-Methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride optically active substance (8.8 g) was converted to 1- The mixture was mixed with cyanoacetyl-3,5-dimethylpyrazole (6.8 g), N, N-diisopropylethylamine (20 ml) and 1,4-dioxane (100 ml) and stirred at 100 ° C. for 1 hour. The mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform / methanol (10/1). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 30/1 to 9/1). The residue obtained by concentration under reduced pressure was slurry washed with n-heptane / ethanol (2/1, 90 ml) to obtain a solid (7.3 g). The solid was slurried again with n-heptane / ethanol (5/1, 90 ml) to give the title compound as crystals 1 (6.1 g).
1 H-NMR (DMSO-D 6 ) δ: 11.60 (1H, br s), 8.08 (1H, s), 7.11 (1H, dd, J = 3.5, 2.4 Hz), 6.58 (1H, dd, J = 3.4 , 1.9 Hz), 4.18-4.14 (1H, m), 4.09-3.93 (3H, m), 3.84-3.73 (1H, m), 3.71 (1H, d, J = 19.0 Hz), 3.66 (1H, d, J = 18.7 Hz), 3.58 (1H, dd, J = 8.2, 6.0 Hz), 2.70-2.58 (2H, m), 2.24-2.12 (1H, m), 1.12 (3H, d, J = 7.1 Hz).
[Α] D = + 47.09 ° (25 ° C., c = 0.55, methanol)
1-Butanol (39 ml) was added to the obtained crystal 1 (2.6 g), and the mixture was heated and stirred at 100 ° C. After complete dissolution, the solution was cooled to room temperature by 10 ° C. every 30 minutes and further stirred at room temperature overnight. The produced crystals were collected by filtration, washed with 1-butanol (6.2 ml), and dried under reduced pressure to give crystals 2 (2.1 g) of the title compound.
PATENTS
WO 2017006968
WO 2018117152
WO 2018117151
PATENT
WO 2018117153
https://patentscope.wipo.int/search/zh/detail.jsf?docId=WO2018117153&tab=FULLTEXT
Janus kinase (JAK) inhibitors are of current interest for the treatment of various diseases including autoimmune diseases, inflammatory diseases, and cancer. To date, two JAK inhibitors have been approved by the U.S. Food & Drug Administration (FDA). Ruxolitinib has been approved for the treatment of primary myelofibrosis and polycythemia vera (PV), and tofacitinib has been approved for the treatment of rheumatoid arthritis. Other JAK inhibitors are in the literature. The compound 3-((3S,4R)-3-methyl-6-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,6-diazaspiro[3.4]octan-1-yl)-3-oxopropanenitrile (Compound A) (see structure below) is an example of a spirocyclic JAK inhibitor reported in U.S. Pat. Pub. Nos. 2011/0136778 and International Pat. Pub. No. PCT/JP2016/070046.
[Chem. 1]
[Chem. 2]
Step 1
[Chem. 3]
A crude product of S-BBMO which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.36-7.13 (5H, m), 4.26 (1H, dd, J = 6.8, 3.9 Hz), 3.72 (2H, dd, J = 14.2, 6.8 Hz), 3.47-3.38 (1H, m), 3.30-3.08 (3H, m), 2.79 (1H, sext, J = 6.8 Hz), 1.35 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
MS: m/z = 280 [M+H] +
[Chem. 4]
A crude product of R-BCAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.11 (5H, m), 4.24-4.11 (1H, m), 3.80 (2H, d, J = 3.6 Hz), 3.24 (2H, d, J = 3.6 Hz), 2.98-2.78 (2H, m), 1.46-1.37 (12H, m).
MS: m/z = 298 [M+H] +
[Chem. 5]
A crude product of S-MABB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.25 (10H, m), 3.75 (1H, d, J = 12.7 Hz), 3.68 (1H, d, J = 1.4 Hz), 3.66 (1H, d, J = 6.7 Hz), 3.46 (2H, d, J = 12.7 Hz), 3.30-3.17 (2H, m), 2.95 (1H, dd, J = 6.2, 1.2 Hz), 2.77 (1H, dd, J = 6.1, 2.2 Hz), 2.65-2.55 (1H, m), 2.48-2.40 (2H, m), 1.35 (9H, s), 1.35 (9H, s), 1.12 (3H, d, J = 7.2 Hz), 1.09 (3H, d, J = 6.2 Hz).
MS: m/z = 262 [M+H] +
[Chem. 6]
S-MABB-HC which was prepared by the same process was measured about NMR, MS, and Cl-content.
1H-NMR (DMSO-d 6) δ: 11.08 (1H, br s), 10.94 (1H, br s), 7.52-7.42 (10H, m), 5.34 (1H, t, J = 8.4 Hz), 4.90 (1H, br s), 4.45-4.10 (5H, m), 3.92-3.49 (3H, br m), 3.10-2.73 (2H, br m), 1.35 (9H, s), 1.29 (9H, s), 1.24 (3H, d, J = 6.7 Hz), 1.17 (3H, d, J = 7.4 Hz).
MS: m/z = 262 [M+H-HCl] +
Cl content (ion chromatography): 11.9 % (in theory: 11.9 %).
[Chem. 7]
A crude product of S-MACB-HC which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 9.60 (br s, 1H), 4.97 (d, 1H, J = 9.2 Hz), 4.61 (d, 1H, J = 8.4 Hz), 4.01 (dd, 1H, J = 10.0, 8.4 Hz), 3.78-3.74 (m, 1H), 3.54 (dd, 1H, J = 9.6, 8.4 Hz), 3.35 (dd, 1H, J = 10.0, 6.0 Hz), 3.15-3.03 (m, 1H), 3.00-2.88 (m, 1H), 1.49 (s, 9H), 1.47 (s, 9H), 1.22 (d, 3H, J = 6.8 Hz), 1.14 (d, 3H, J = 7.2 Hz).
MS: m/z = 172 [M+H] + (free form)
[Chem. 8]
A crude product of S-ZMAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.38-7.28 (m, 10H), 5.16-5.04 (m, 4H), 4.60 (d, 1H, J = 9.2 Hz), 4.18-4.12 (m, 2H), 4.04 (t, 1H, J = 8.6 Hz), 3.66 (dd, 1H, J = 7.6, 7.2 Hz), 3.50 (dd, 1H, J = 8.0, 5.2 Hz), 3.05-2.94 (m, 1H), 2.60-2.50 (m, 1H), 1.43 (br s, 18H), 1.33 (d, 3H, J = 6.5 Hz), 1.15 (d, 3H, J = 7.2 Hz).
MS: m/z = 328 [M+Na] +.
[Chem. 9]
A crude product of RS-ZMBB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.38-7.29 (m, 5H), 5.09-4.96 (m, 2H), 3.91 (t, 0.4H, J = 8.0 Hz), 3.79 (t, 0.6H, J = 8.0 Hz), 3.55 (t, 0.4H, J = 7.2 Hz), 3.46 (t, 0.6H, J = 7.5 Hz), 3.14-3.04 (m, 1H), 2.83-2.72 (m, 2H), 1.38 (br s, 9H), 1.37 (br s, 3.6H), 1.34 (br s, 5.4H), 1.12-1.09 (m, 3H).
MS: m/z = 420 [M+H] +.
[Chem. 10]
RS-ZMAA-DN .2H 2O which was prepared by the same process was measured about NMR, MS, Na-content, and water-content.
1H-NMR (DMSO-d 6) δ: 7.32-7.22 (m, 5H), 4.97 (d, 1H, J = 12.7 Hz), 4.84 (d, 1H, J = 12.7 Hz), 3.79 (t, 1H, J = 8.0 Hz), 3.29 (d, 1H, J = 14.8 Hz), 3.16-3.12 (m, 1H), 2.17-2.09 (m, 2H), 1.07 (d, 3H, J = 6.9 Hz).
MS: m/z = 352 [M+H] + (anhydrate)
Na content (ion chromatography): 13.3 % (after correction of water content)(13.1 % in theory)
Water content (Karl Fischer’s method): 9.8 % (9.3 % in theory)
[Chem. 11]
RS-ZMAA which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.35-7.28 (m, 5H), 5.06-4.94 (m, 2H), 3.86 (dt, 1H, J = 48.4, 7.9 Hz), 3.50 (dt, 1H, J = 37.9, 7.4 Hz), 3.16-3.02 (br m, 1H), 2.91-2.77 (br m, 2H), 1.08 (d, 3H, J = 6.9 Hz)
MS: m/z = 308 [M+H] +.
[Chem. 12]
RS-ZMOO which was prepared by the same process was measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.39-7.30 (m, 5H), 5.10 (s, 2H), 4.15-4.01 (br m, 2H), 3.83-3.73 (br m, 3H), 3.48 (dd, 1H, J = 8.3, 6.4 Hz), 2.59-2.50 (br m, 1H), 2.46-2.40 (br m, 1H), 2.07-1.99 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 280 [M+H]+.
[Chem. 13]
RS-ZMSS which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.37-7.27 (br m, 5H), 5.10-4.98 (m, 2H), 4.58-4.22 (br m, 4H), 3.84 (dt, 1H, J = 45.6, 8.1 Hz), 3.48-3.33 (br m, 1H), 3.17-3.10 (m, 6H), 2.81-2.74 (br m, 1H), 2.22-2.12 (m, 2H)
MS: m/z = 436 [M+H] +.
[Chem. 14]
1H-NMR (CDCl 3) δ: 7.35-7.20 (m, 10H), 5.08 (d, 2H, J = 23.6 Hz), 3.94 (q, 1H, J = 7.9 Hz), 3.73-3.42 (br m, 2H), 3.30-3.23 (m, 1H), 3.05 (dd, 1H, J = 19.7, 9.5 Hz), 2.79 (dt, 1H, J = 69.6, 6.1 Hz), 2.57-2.32 (br m, 4H), 1.96-1.89 (m, 1H), 1.09 (d, 3H, J = 6.9 Hz)
MS: m/z = 351 [M+H] +.
[Chem. 15]
SR-MDOZ which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.37-7.28 (m, 5H), 5.08 (dd, 2H, J = 16.8, 12.8 Hz), 4.00 (dd, 1H, J = 17.1, 8.3 Hz), 3.40-3.31 (m, 1H), 3.24 (d, 1H, J = 12.7 Hz), 3.00 (dd, 1H, J = 54.9, 12.4 Hz), 2.87-2.57 (m, 3H), 2.47-2.27 (m, 1H), 1.91-1.80 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 261 [M+H] +.
[Chem. 16]
SR-MDOZ-OX which was prepared by the same process was measured about NMR, MS, and elementary analysis.
1H-NMR (DMSO-D 6) δ: 7.37-7.30 (m, 5H), 5.15-5.01 (m, 2H), 3.92 (dt, 1H, J = 43.5, 8.4 Hz), 3.48-3.12 (br m, 5H), 2.67-2.56 (m, 1H), 2.46-2.35 (m, 1H), 2.12-2.05 (m, 1H), 1.13 (d, 3H, J = 6.9 Hz)
MS: m/z = 261 [M+H] +
elementary analysis: C 58.4wt % , H 6.4wt % , N 7.9 % wt % (theoretically, C 58.3wt % , H 6.3wt % , N 8.0wt % )
[Chem. 17]
SR-MDPZ which was prepared by the same process was isolated as a solid from a mixture of ethyl acetate and n-heptane, and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.41-7.26 (br m, 3H), 7.22-7.08 (br m, 3H), 6.64-6.51 (br m, 1H), 5.07-4.91 (br m, 2H), 4.09-3.67 (br m, 5H), 3.47-3.32 (br m, 1H), 2.67-2.55 (br m, 2H), 2.21-2.15 (br m, 1H), 1.11 (d, 3H, J = 6.9 Hz).
MS: m/z = 378 [M+H] +
[Chem. 18]
SR-MDOP which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.57 (br s, 1H), 8.07 (s, 1H), 7.10 (d, 1H, J = 3.2 Hz), 6.58 (d, 1H, J = 3.2 Hz), 3.92-3.59 (br m, 4H), 3.49 (dd, 1H, J = 8.3, 7.2 Hz), 2.93 (dd, 1H, J = 7.2, 6.1 Hz), 2.61-2.53 (m, 2H), 2.12-2.01 (br m, 2H), 1.10 (d, 3H, J = 6.9 Hz).
MS: m/z = 244 [M+H] +.
[Chem. 19]
Compound A mono-ethanolate which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.34 (t, 1H, J = 5.1 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.92 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 3.44 (dq, 2H, J = 6.7, 5.1 Hz), 2.69-2.60 (m, 2H), 2.23-2.13 (br m, 1H), 1.12 (d, 3H, J = 7.1 Hz), 1.06 (t, 3H, J = 6.7 Hz).
MS: m/z = 311 [M+H] +
[Chem. 20]
Compound A which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.5 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.69-2.59 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H] +
(1) Preparation of Single crystal
To 10 mg of Compound A in a LaPha ROBO Vial(R) 2.0 mL wide-mouthed vial was added 0.5 mL of chloroform. The vial was covered with a cap, in which Compound A was completely dissolved. In order to evaporate the solvent slowly, a hole was made on the septum attached in the cap with a needle of a TERUMO(R) syringe, and the vial was still stood at room temperature. The resulting single crystal was used in the structural analysis.
(2) Measuring instrument
Beam line: SPring-8 BL32B2
Detector: Rigaku R-AXIS V diffractometer
(3) Measuring method
The radiant light of 0.71068Å was irradiated to the single crystal to measure X-ray diffraction data.
(4) Assay method
Using the X-ray anomalous scattering effect of the chlorine atom in the resulting Compound A chloroform-solvate, the absolute configuration of Compound A was identified as (3S,4R). Based on the obtained absolute configuration of Compound A, the absolute configurations of each process intermediate were identified.
REFERENCES
1: Nakagawa H, Nemoto O, Yamada H, Nagata T, Ninomiya N. Phase 1 studies to assess the safety, tolerability and pharmacokinetics of JTE-052 (a novel Janus kinase inhibitor) ointment in Japanese healthy volunteers and patients with atopic dermatitis. J Dermatol. 2018 Jun;45(6):701-709. doi: 10.1111/1346-8138.14322. Epub 2018 Apr 17. PubMed PMID: 29665062; PubMed Central PMCID: PMC6001687.
2: Nakagawa H, Nemoto O, Igarashi A, Nagata T. Efficacy and safety of topical JTE-052, a Janus kinase inhibitor, in Japanese adult patients with moderate-to-severe atopic dermatitis: a phase II, multicentre, randomized, vehicle-controlled clinical study. Br J Dermatol. 2018 Feb;178(2):424-432. doi: 10.1111/bjd.16014. Epub 2018 Jan 15. PubMed PMID: 28960254.
3: Tanimoto A, Shinozaki Y, Yamamoto Y, Katsuda Y, Taniai-Riya E, Toyoda K, Kakimoto K, Kimoto Y, Amano W, Konishi N, Hayashi M. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. Exp Dermatol. 2018 Jan;27(1):22-29. doi: 10.1111/exd.13370. Epub 2017 Jul 3. PubMed PMID: 28423239.
4: Nomura T, Kabashima K. Advances in atopic dermatitis in 2015. J Allergy Clin Immunol. 2016 Dec;138(6):1548-1555. doi: 10.1016/j.jaci.2016.10.004. Review. PubMed PMID: 27931536.
5: Amano W, Nakajima S, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. JAK inhibitor JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation. J Dermatol Sci. 2016 Dec;84(3):258-265. doi: 10.1016/j.jdermsci.2016.09.007. Epub 2016 Sep 13. PubMed PMID: 27665390.
6: Tanimoto A, Shinozaki Y, Nozawa K, Kimoto Y, Amano W, Matsuo A, Yamaguchi T, Matsushita M. Improvement of spontaneous locomotor activity with JAK inhibition by JTE-052 in rat adjuvant-induced arthritis. BMC Musculoskelet Disord. 2015 Nov 6;16:339. doi: 10.1186/s12891-015-0802-0. PubMed PMID: 26546348; PubMed Central PMCID: PMC4636776.
7: Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, Dainichi T, Honda T, Otsuka A, Kimoto Y, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. 2015 Sep;136(3):667-677.e7. doi: 10.1016/j.jaci.2015.03.051. Epub 2015 Jun 24. PubMed PMID: 26115905.
8: Tanimoto A, Ogawa Y, Oki C, Kimoto Y, Nozawa K, Amano W, Noji S, Shiozaki M, Matsuo A, Shinozaki Y, Matsushita M. Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflamm Res. 2015 Jan;64(1):41-51. doi: 10.1007/s00011-014-0782-9. Epub 2014 Nov 12. PubMed PMID: 25387665; PubMed Central PMCID: PMC4286029.
References
- “Anzupgo EPAR”. European Medicines Agency. 25 July 2024. Retrieved 25 July 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Anzupgo PI”. Union Register of medicinal products. 23 September 2024. Retrieved 27 September 2024.
- Dhillon S (April 2020). “Delgocitinib: First Approval”. Drugs. 80 (6): 609–615. doi:10.1007/s40265-020-01291-2. PMID 32166597. S2CID 212681247.
- Park B (5 August 2020). “Delgocitinib Cream Gets Fast Track Status for Chronic Hand Eczema”. empr.com.
- Szalus K, Trzeciak M, Nowicki RJ (November 2020). “JAK-STAT Inhibitors in Atopic Dermatitis from Pathogenesis to Clinical Trials Results”. Microorganisms. 8 (11): 1743. doi:10.3390/microorganisms8111743. PMC 7694787. PMID 33172122.
- “Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 22-25 July 2024”. European Medicines Agency (Press release). 25 July 2024. Retrieved 29 July 2024.
/////////Delgocitinib, デルゴシチニブ , JAPAN 2020, 2020 APPROVALS, Corectim, UNII-9L0Q8KK220, JTE-052, 9L0Q8KK220, LEO 124249A, LEO 124249, HY-109053, CS-0031558, D11046, GTPL9619, JTE-052A, JTE052, LP-0133 , ROH-201, atopic dermatitis
CC1CN(C12CCN(C2)C3=NC=NC4=C3C=CN4)C(=O)CC#N

 |
|
| Clinical data | |
|---|---|
| Trade names | Corectim, others |
| Other names | JTE-052; JTE-052A |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C16H18N6O |
| Molar mass | 310.361 g·mol−1 |
| 3D model (JSmol) | |
https://pubs.acs.org/doi/10.1021/acs.oprd.1c00031
https://www.chemicalbook.com/article/synthesis-of-delgocitinib.htm
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

Synthesis of Delgocitinib
Dec 26,2023
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

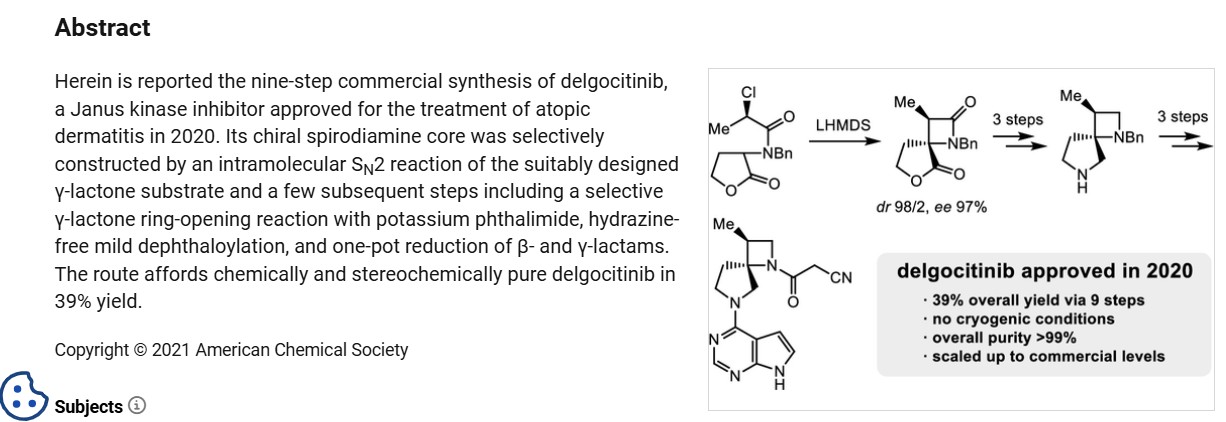
A stereocontrolled kilogram scale synthesis of delgocitinib has been disclosed, beginning with an SN2 reaction involving bromolactone 128 and benzyl amine to provide α-amino lactone 129, which was isolated as the HCl salt after precipitation from hydrochloric acid in ethyl acetate. Amine 129 was then acylated with enantiomerically pure acid chloride 131 (prepared by thionyl chloride treatment of commercial acid 130) to furnish lactone 132. In the crucial spirocyclic ring ringforming sequence of the synthesis, lactone 132 was treated with LHMDS to form an enolate that underwent SN2 displacement of the chloride, forming the spirolactone 133 and establishing both stereocenters with 98:2 dr and 96% ee.
The lactone ring of 133 was then opened by an attack of potassium phthalimide on the γ- carbon, and the resulting carboxylic acid was converted to the ethyl ester by treatment with ethyl iodide. Finally, treatment with diethylenetriamine released phthalimide, providing a free amine for subsequent cyclization to spirolactam 134 via the corresponding ethyl ester intermediate. This sequence took place in 80% yield over four steps and provided the spirolactam in >99% de after recrystallization.
The carbonyl groups within spirolactam 134 were then reduced with lithium aluminum hydride and aluminum chloride in THF, and the resulting diamine 135 was crystallized as a succinic acid salt in 86% yield. The SNAr reaction of 135 with chloropyrrolopyrimidine 136 followed by hydrogenative removal of the benzyl protecting group provided amine 137 in 92% yield over 2 steps. Finally, amine 137 was acylated with cyanoacetyl pyrazole 138 and recrystallized from n-butanol with 3 wt % BHT to provide delgocitinib in 86% yield, >99% ee, and >99% de.
PF 04965842, Abrocitinib
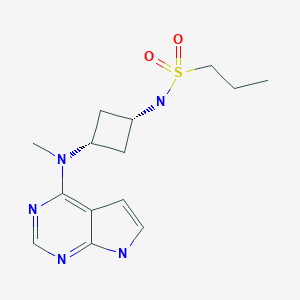

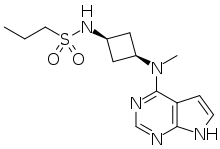
PF-04965842
PF 04965842, Abrocitinib
UNII: 73SM5SF3OR
CAS Number 1622902-68-4, Empirical Formula C14H21N5O2S, Molecular Weight 323.41
N-[cis-3-(Methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)cyclobutyl]-1-propanesulfonamide,
N-((1s,3s)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)cyclobutyl)propane-1-sulfonamide
1-Propanesulfonamide, N-(cis-3-(methyl-7H-pyrrolo(2,3-d)pyrimidin-4-ylamino)cyclobutyl)-
N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide
PHASE 3, for the potential oral treatment of moderate-to-severe atopic dermatitis (AD)
Jak1 tyrosine kinase inhibitor
UPDATE…… JAPAN APPROVED, 2021, 2021/9/27, CIBINQO
ALSO
fda 2022, APPROVALS 2022, 1/14/2022
THE US
In February 2018, the FDA granted Breakthrough Therapy designation for the treatment of patients with moderate-to-severe AD
PHASEIII
In December 2017, a randomized, double-blind, placebo-controlled, parallel-group, phase III trial (NCT03349060; JADE Mono-1; JADE; B7451012; 2017-003651-29) of PF-04965842 began in patients aged 12 years and older (expected n = 375) with moderate-to-severe AD
PRODUCT PATENT
| Pub. No.: | WO/2014/128591 | International Application No.: | PCT/IB2014/058889 | |||
| Publication Date: | 28.08.2014 | International Filing Date: | 11.02.2014 |
EXPIRY Roughly 2034
| form | powder |
| color | white to beige |
| solubility | DMSO: 10 mg/mL, clear |
| storage temp. | room temp |
- Biochem/physiol Actions
-
- PF-04965842 is a Janus Kinase (JAK) inhibitor selective for JAK1 with an IC50value of 29 nM for JAK1 compared to 803 nM for JAK2, >10000 nM for JAK3 and 1250 nM for Tyk2. JAKs mediate cytokine signaling, and are involved in cell proliferation and differentiation. PF-04965842 has been investigated as a possible treatment for psoriasis.
- Originator Pfizer
- Class Skin disorder therapies; Small molecules
- Mechanism of Action Janus kinase 1 inhibitors
Highest Development Phases
- Phase IIIAtopic dermatitis
- DiscontinuedLupus vulgaris; Plaque psoriasis
Most Recent Events
- 08 Mar 2018Phase-III clinical trials in Atopic dermatitis (In children, In adults, In adolescents) in USA (PO) (NCT03422822)
- 14 Feb 2018PF 4965842 receives Breakthrough Therapy status for Atopic dermatitis in USA
- 06 Feb 2018Pfizer plans the phase III JADE EXTEND trial for Atopic Dermatitis (In children, In adults, In adolescents) in March 2018 (PO) (NCT03422822)
This compound was developed by Pfizer for Kinase Phosphatase Biology research. To learn more about Sigma′s partnership with Pfizer and view other authentic, high-quality Pfizer compounds,
PF-04965842 is an oral Janus Kinase 1 inhibitor being investigated for treatment of plaque psoriasis.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified into tyrosine and serine/threonine kinases. Inappropriate kinase activity, arising from mutation, over-expression, or inappropriate regulation, dys-regulation or de-regulation, as well as over- or under-production of growth factors or cytokines has been i mplicated in many diseases, including but not limited to cancer, cardiovascular diseases, allergies, asthma and other respiratory diseases, autoimmune d iseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative disorders such as Alzheimer’s disease. Inappropriate kinase activity triggers a variety of biological cellular responses relating to cell growth, cell differentiation , survival, apoptosis, mitogenesis, cell cycle control, and cel l mobility implicated in the aforementioned and related diseases.
Thus, protein kinases have emerged as an important class of enzymes as targets for therapeutic intervention. In particular, the JAK family of cellular protein tyrosine kinases (JAK1, JAK2, JAK3, and Tyk2) play a central role in cytoki ne signaling (Kisseleva et al., Gene, 2002, 285 , 1; Yamaoka et al. Genome Biology 2004, 5, 253)). Upon binding to their receptors, cytokines activate JAK which then phosphorylate the cytokine receptor, thereby creating docking sites for signaling molecules, notably, members of the signal transducer and activator of transcription (STAT) family that ultimately lead to gene expression. Numerous cytokines are known to activate the JAK family. These cytokines include, the IFN family (IFN-alpha, IFN-beta, IFN-omega, Limitin, IFN-gamma, IL- 10, IL- 19, IL-20, IL-22), the gp 130 family (IL-6, IL- 11, OSM, LIF, CNTF, NNT- 1//SF-3, G-CSF, CT- 1, Leptin, IL- 12 , I L-23), gamma C family (IL-2 , I L-7, TSLP, IL-9, IL- 15 , IL-21, IL-4, I L- 13), IL-3 family (IL-3 , IL-5 , GM-CSF), single chain family (EPO, GH, PRL, TPO), receptor tyrosine kinases (EGF, PDGF, CSF- 1, HGF), and G-protein coupled receptors (ATI).
Abrocitinib, sold under the brand name Cibinqo, is a Janus kinase inhibitor medication used for the treatment of atopic dermatitis (eczema).[2] It was developed by Pfizer.[2]
Medical uses
Abrocitinib is indicated for the treatment of moderate-to-severe atopic dermatitis in adults who are candidates for systemic therapy.[2]
Side effects
The most common adverse effects in studies were upper respiratory tract infection, headache, nausea, and diarrhea.[3]
Pharmacology
Mechanism of action
It is a selective inhibitor of the enzyme janus kinase 1 (JAK1).[3]
Pharmacokinetics
Abrocitinib is quickly absorbed from the gut and generally reaches highest blood plasma concentrations within one hour. Only 1.0 to 4.4% of the dose are found unmetabolized in the urine.[4]
History
- April 2016: initiation of Phase 2b trial
- December 2017: initiation of JADE Mono-1 Phase 3 trial[5]
- May 2018: Results of Phase 2b trial posted
- October 2019: Results of Phase 3 trial presented[6]
- June 2020: Results of second Phase 3 trial published[7]
Society and culture
Legal status
In October 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Cibinqo, intended for the treatment of atopic dermatitis.[8] The applicant for this medicinal product is Pfizer Europe MA EEIG.[8] In December 2021, the European Commission approved abrocitinib for the treatment of atopic dermatitis.[2][9]
In January 2022, the United States Food and Drug Administration (FDA) approved abrocitinib for adults with moderate-to-severe atopic dermatitis.[10]
////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
EU
Click to access cibinqo-epar-public-assessment-report_en.pdf
Introduction
The finished product is presented as immediate release film-coated tablets containing 50 mg, 100 mg
or 200 mg of abrocitinib as active substance.
Other ingredients are:
Tablet core: microcrystalline cellulose (E460i), anhydrous dibasic calcium phosphate (E341ii), sodium
starch glycolate and magnesium stearate (E470b).
Film-coat: hypromellose (E464), titanium dioxide (E171), lactose monohydrate, macrogol (E1521),
triacetin (E1518) and red iron oxide (E172).
The product is available in high-density polyethylene (HDPE) bottles with polypropylene closure or
polyvinylidene chloride (PVDC) blisters with aluminium foil lidding film, as described in section 6.5 of
the SmPC.
The chemical name of abrocitinib is N-((1S,3S)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-
yl)amino)cyclobutyl)propane-1-sulfonamide corresponding to the molecular formula C14H21N5O2S. It
has a relative molecular mass of 323.42 Daltons and the following structure depicted in Figure 1:

The chemical structure of abrocitinib was elucidated by a combination of UV/VIS and IR spectroscopy,
mass spectrometry, NMR spectroscopy and X-ray diffraction.
The active substance is a white to pale-purple or pale pink crystalline powder. It is non-hygroscopic
and its solubility is pH dependent. Abrocitinib is classified as BCS Class II. The impact of particle size
on finished product uniformity of dosage units and dissolution has been studied (see finished product
section). Based on the abrocitinib finished product biopharmaceutics performance, stability, and
manufacturing experience, the active substance particle size specification was established.
Abrocitinib is an achiral molecule, but with 2 stereocentres.
Only one crystalline anhydrous form (Form 1) of abrocitinib has been identified. This form has been the
only form used in all toxicology and clinical studies. Extensive polymorph and hydrate screening have
been conducted to investigate if additional solid forms of abrocitinib could be discovered. Abrocitinib,
Form 1 was the only anhydrous crystalline form identified from these studies. No new anhydrous
polymorphs, hydrates or amorphous solids of abrocitinib were isolated from these screens.
Experiments with 1,4 dioxane and dimethyl sulfoxide yielded solvated forms of abrocitinib. When these
solvated structures were subjected to high temperature, these materials desolvated and converted to
Form 1, free base anhydrous form of abrocitinib. However, these are not relevant since the commercial
crystallisation step does not utilise either of these solvent systems.
It has been confirmed that the manufacturing process consistently yields polymorphic form I. This form
is physically and chemically stable under normal manufacturing and storage conditions as well as
under accelerated conditions. Hence the absence of control of form I is justified.
FDA
U.S. FDA Approves Pfizer’s CIBINQO® (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis
CIBINQO is a once-daily oral treatment with proven efficacy to manage symptoms for adults who have not yet found relief with current options
NEW YORK–(BUSINESS WIRE)– Pfizer Inc. (NYSE: PFE) announced today that the United States (U.S.) Food and Drug Administration (FDA) approved CIBINQO® (abrocitinib), an oral, once-daily, Janus kinase 1 (JAK1) inhibitor, for the treatment of adults living with refractory, moderate-to-severe atopic dermatitis (AD) whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
CIBINQO is approved at the recommended doses of 100 mg and 200 mg, with the 200 mg dose being recommended for patients who are not responding to the 100 mg dose. Additionally, a 50 mg dose was approved to treat moderate-to-severe AD specifically in patients with moderate renal impairment (kidney failure), certain patients receiving treatment with inhibitors of cytochrome P450 (CYP) 2C19, or patients who are known or suspected to be poor metabolizers of CYP2C19. For patients with moderate renal impairment who are not responding to 50 mg once daily, 100 mg once daily may also be prescribed.
“The reality for patients living with chronic inflammatory skin disease such as moderate-to-severe atopic dermatitis is that many experience debilitating symptoms that are not managed by current treatment options. Today’s approval of CIBINQO will provide an important new oral option that could help those who have yet to find relief,” said Jonathan Silverberg, MD, PhD, MPH, Department of Dermatology, The George Washington University School of Medicine and Health Sciences. “In multiple large-scale clinical trials, CIBINQO demonstrated strong efficacy at clearing skin, improving itch, and managing the extent and severity of eczema, offering a benefit-risk profile that supports the use of this treatment in the FDA-approved patient population.”
The FDA approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The safety and efficacy of CIBINQO was evaluated in three randomized, placebo-controlled, Phase 3 trials. Additionally, safety was evaluated through a randomized, placebo-controlled, dose-ranging trial and an ongoing long-term open-label extension trial. Across the trials, CIBINQO demonstrated a consistent safety profile and profound improvements in skin clearance, extent of disease, and severity, as well as rapid improvement in itch after two weeks, for some people living with AD versus placebo. In addition, a higher proportion of subjects treated with CIBINQO in two monotherapy trials achieved improvement in itching at week 12 compared to placebo.
“The FDA’s approval offers hope to the millions of patients across the U.S. who are suffering daily with an immuno-inflammatory condition that can cause intense and persistent itching, pain, discomfort, and distress if left uncontrolled,” said Mike Gladstone, Global President of Pfizer Inflammation & Immunology. “CIBINQO, an efficacious once-daily pill, is a medical breakthrough made possible by Pfizer researchers and the people living with moderate-to-severe atopic dermatitis who participated in our clinical trials.”
“Atopic dermatitis is so much more than just a rash, and it goes beyond the surface of the skin. It’s a chronic condition that can both significantly disrupt patients’ daily lives and negatively impact their emotional well-being,” said Julie Block, President and CEO, National Eczema Association. “We appreciate Pfizer’s commitment to this resilient patient community and eagerly await the positive impact CIBINQO could have on the treatment landscape for moderate-to-severe atopic dermatitis.”
The most common adverse events reported in ≥5% of patients with CIBINQO included nasopharyngitis (12.4% with CIBINQO 100 mg, 8.7% with CIBINQO 200 mg, and 7.9%, with placebo), nausea (6%, 14.5%, and 2.1%, respectively), and headache (6%, 7.8%, and 3.5%, respectively).
The full prescribing information for CIBINQO can be found here. CIBINQO will be made available in the coming weeks.
Additional Details on the CIBINQO Clinical Trial Program
Five clinical trials in the CIBINQO JAK1 Atopic Dermatitis Efficacy and Safety (JADE) global development program were included in the New Drug Application (NDA) to support the FDA approval.
The safety and efficacy of CIBINQO was evaluated in three Phase 3, randomized, placebo-controlled clinical trials. The trials evaluated measures of improvements in skin clearance, itch, disease extent, and severity, including the Investigator Global Assessment (IGA), Eczema Area and Severity Index (EASI), and Peak Pruritus Numerical Ratings Scale (PP-NRS). In each of the trials, over 40% of patients had prior exposure to a systemic therapy:
- JADE MONO-1 and JADE MONO-2: A pair of randomized, double-blind, placebo-controlled trials designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO monotherapy in 778 patients 12 years of age and older with moderate-to-severe AD. The trials assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
- JADE COMPARE: A randomized, double-blind, placebo-controlled trial designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO in 837 adult patients with moderate-to-severe AD on background topical medicated therapy. The trial also included an active control arm with dupilumab, a biologic treatment administered by subcutaneous injection, compared with placebo. The trial assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
Select findings for CIBINQO 100 mg, 200 mg, and placebo follow (*p<0.01 or **p<0.001):
- JADE MONO-1:
- IGA Response Rate (Week 12): 24%*, 44%**, and 8%, respectively
- EASI-75 Response Rate (Week 12): 40%**, 62%**, and 12%, respectively
- JADE MONO-2
- IGA Response Rate (Week 12): 28%**, 38%**, and 9%, respectively
- EASI-75 Response Rate (Week 12): 44%**, 61%**, and 10%, respectively
- JADE COMPARE
- IGA Response Rate (Week 12): 36%**, 47%**, and 14%, respectively
- EASI-75 Response Rate (Week 12): 58%**, 68%**, and 27%, respectively
Safety was additionally evaluated through a randomized dose-ranging trial and a long-term, open-label, extension trial (JADE EXTEND).
U.S. IMPORTANT SAFETY INFORMATION
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, AND THROMBOSIS
Serious Infections
Patients treated with CIBINQO may be at increased risk for developing serious infections that may lead to hospitalization or death. The most frequent serious infections reported with CIBINQO were herpes simplex, herpes zoster, and pneumonia.
If a serious or opportunistic infection develops, discontinue CIBINQO and control the infection.
Reported infections from Janus kinase (JAK) inhibitors used to treat inflammatory conditions:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Test for latent TB before and during therapy; treat latent TB prior to use. Monitor all patients for active TB during treatment, even patients with initial negative, latent TB test.
- Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral (including herpes zoster), and other infections due to opportunistic pathogens.
Avoid use of CIBINQO in patients with an active, serious infection, including localized infections. The risks and benefits of treatment with CIBINQO should be carefully considered prior to initiating therapy in patients with chronic or recurrent infections or those who have resided or traveled in areas of endemic tuberculosis or endemic mycoses.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with CIBINQO, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
Consider yearly screening for patients in highly endemic areas for TB. CIBINQO is not recommended for use in patients with active TB. For patients with a new diagnosis of latent TB or prior untreated latent TB, or for patients with a negative test for latent TB but who are at high risk for TB infection, start preventive therapy for latent TB prior to initiation of CIBINQO.
Viral reactivation, including herpes virus reactivation (eg, herpes zoster, herpes simplex), was reported in clinical studies with CIBINQO. If a patient develops herpes zoster, consider interrupting CIBINQO until the episode resolves. Hepatitis B virus reactivation has been reported in patients receiving JAK inhibitors. Perform viral hepatitis screening and monitoring for reactivation in accordance with clinical guidelines before starting therapy and during therapy with CIBINQO. CIBINQO is not recommended for use in patients with active hepatitis B or hepatitis C.
Mortality
In a large, randomized postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another JAK inhibitor to TNF blocker treatment, a higher rate of all-cause mortality (including sudden cardiovascular death) was observed with the JAK inhibitor. CIBINQO is not approved for use in RA patients.
Malignancies
Malignancies, including non-melanoma skin cancer (NMSC), were reported in patients treated with CIBINQO. Lymphoma and other malignancies have been observed in patients receiving JAK inhibitors used to treat inflammatory conditions. Perform periodic skin examination for patients who are at increased risk for skin cancer. Exposure to sunlight and UV light should be limited by wearing protective clothing and using broad-spectrum sunscreen.
In a large, randomized postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. CIBINQO is not approved for use in RA patients. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Major Adverse Cardiovascular Events
Major adverse cardiovascular events were reported in patients treated with CIBINQO. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients. Patients who are current or past smokers are at additional increased risk. Discontinue CIBINQO in patients that have experienced a myocardial infarction or stroke.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur.
Thrombosis
Deep vein thrombosis (DVT) and pulmonary embolism (PE) have been reported in patients treated with CIBINQO. Thrombosis, including PE, DVT, and arterial thrombosis have been reported in patients receiving JAK inhibitors used to treat inflammatory conditions. Many of these adverse reactions were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of overall thrombosis, DVT, and PE were observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients.
Avoid CIBINQO in patients that may be at increased risk of thrombosis. If symptoms of thrombosis occur, discontinue CIBINQO and treat patients appropriately.
Contraindication
CIBINQO is contraindicated in patients taking antiplatelet therapies, except for low-dose aspirin (≤81 mg daily), during the first 3 months of treatment.
Laboratory Abnormalities
Hematologic Abnormalities: Treatment with CIBINQO was associated with an increased incidence of thrombocytopenia and lymphopenia. Prior to CIBINQO initiation, perform a complete blood count (CBC). CBC evaluations are recommended at 4 weeks after initiation and 4 weeks after dose increase of CIBINQO. Discontinuation of CIBINQO therapy is required for certain laboratory abnormalities.
Lipid Elevations: Dose-dependent increase in blood lipid parameters were reported in patients treated with CIBINQO. Lipid parameters should be assessed approximately 4 weeks following initiation of CIBINQO therapy, and thereafter patients should be managed according to clinical guidelines for hyperlipidemia. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Immunizations
Prior to initiating CIBINQO, complete all age-appropriate vaccinations as recommended by current immunization guidelines, including prophylactic herpes zoster vaccinations. Avoid vaccination with live vaccines immediately prior to, during, and immediately after CIBINQO therapy.
Renal Impairment
Avoid use in patients with severe renal impairment or end stage renal disease, including those on renal replacement therapy.
Hepatic Impairment
Avoid use in patients with severe hepatic impairment.
Adverse Reactions
Most common adverse reactions (≥1%) in subjects receiving 100 mg and 200 mg include: nasopharyngitis, nausea, headache, herpes simplex, increased blood creatinine phosphokinase, dizziness, urinary tract infection, fatigue, acne, vomiting, oropharyngeal pain, influenza, gastroenteritis.
Most common adverse reactions (≥1%) in subjects receiving either 100 mg or 200 mg also include: impetigo, hypertension, contact dermatitis, upper abdominal pain, abdominal discomfort, herpes zoster, and thrombocytopenia.
Use in Pregnancy
Available data from pregnancies reported in clinical trials with CIBINQO are not sufficient to establish a drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Advise females of reproductive potential that CIBINQO may impair fertility.
There will be a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to CIBINQO during pregnancy. Pregnant women exposed to CIBINQO and health care providers are encouraged to call 1-877-311-3770.
Lactation
Advise women not to breastfeed during treatment with CIBINQO and for one day after the last dose.
Indication
CIBINQO is indicated for the treatment of adults with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
Limitations of Use: CIBINQO is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
About CIBINQO® (abrocitinib)
CIBINQO is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD, including interleukin IL-4, IL-13, IL-31, IL-22, and thymic stromal lymphopoietin (TSLP).
In addition to receiving regulatory approval in the U.S., CIBINQO has received marketing authorization in the European Union, Great Britain, Japan, Korea, the United Arab Emirates, Norway, Iceland, and Singapore.
About Atopic Dermatitis
AD is a chronic skin disease characterized by inflammation of the skin and skin barrier defects.i,ii Most people know AD is a skin condition. But many don’t realize it can be caused in part by an abnormal immune response beneath the skin. This dysregulated immune response is thought to contribute to inflammation within the skin and the signs of AD on the surface. Lesions of AD are characterized by erythema (red/pink or discolored skin patches, depending on normal skin color), itching, lichenification (thick/leathery skin), induration (hardening)/papulation (formulation of papules), and oozing/crusting.i,ii
AD is one of the most common inflammatory skin diseases, affecting approximately 5-10% of adults in the U.S.iii,iv Approximately 1 in 3 adults with AD have moderate-to-severe disease.v,vi
About Pfizer Inflammation & Immunology
At Pfizer Inflammation & Immunology, we strive to deliver breakthroughs that enable freedom from day-to-day suffering for people living with autoimmune and chronic inflammatory diseases, which can be debilitating, disfiguring and distressing, dramatically affecting what they can do. With a focus on immuno-inflammatory conditions in Rheumatology, Gastroenterology and Medical Dermatology, our current portfolio of approved medicines and investigational molecules spans multiple action and delivery mechanisms, from topicals to small molecules, biologics and biosimilars. The root cause of many immunological diseases is immuno-inflammation, which requires specifically designed agents. Our differentiated R&D approach resulted in one of the broadest pipelines in the industry, where we purposefully match molecules to diseases where we believe they can make the biggest difference. Building on our decades-long commitment and pioneering science, we continue to advance the standard of care for patients living with immuno-inflammatory diseases and are working hand-in-hand with patients, caregivers and the broader healthcare community on healthcare solutions for the many challenges of managing chronic inflammatory diseases, allowing patients to live their best lives.
Pfizer Inc.: Breakthroughs that Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety, and value in the discovery, development, and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments, and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments, and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
There remains a need for new compounds that effectively and selectively inhibit specific JAK enzymes, and JAK1 in particular, vs. JAK2. JAK1 is a member of the Janus family of protein kinases composed of JAK1, JAK2, JAK3 and TYK2. JAK1 is expressed to various levels in all tissues. Many cytokine receptors signal through pairs of JAK kinases in the following combinations: JAK1/JAK2, JAK1/JAK3, JAK1/TYK2 , JAK2/TYK2 or JAK2/JAK2. JAK1 is the most broadly
paired JAK kinase in this context and is required for signaling by γ-common (IL-2Rγ) cytokine receptors, IL—6 receptor family, Type I, II and III receptor families and IL- 10 receptor family. Animal studies have shown that JAK1 is required for the development, function and homeostasis of the immune system. Modulation of immune activity through inhibition of JAK1 kinase activity can prove useful in the treatment of various immune disorders (Murray, P.J.
J. Immunol., 178, 2623-2629 (2007); Kisseleva, T., et al., Gene, 285 , 1-24 (2002); O’Shea, J . J., et al., Ceil , 109, (suppl .) S121-S131 (2002)) while avoiding JAK2 dependent erythropoietin (EPO) and thrombopoietin (TPO) signaling (Neubauer H., et al., Cell, 93(3), 397-409 (1998);
Parganas E., et al., Cell, 93(3), 385-95 (1998)).

Tofacitinib (1), baricitinib (2), and ruxolitinib (3)
SYNTHESIS 5+1 =6 steps
Main synthesis
Journal of Medicinal Chemistry, 61(3), 1130-1152; 2018

INTERMEDIATE
CN 105732637
ONE STEP

CAS 479633-63-1, 7H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-7-[(4- methylphenyl)sulfonyl]-
![]()
Pfizer Receives Breakthrough Therapy Designation from FDA for PF-04965842, an oral JAK1 Inhibitor, for the Treatment of Patients with Moderate-to-Severe Atopic Dermatitis
Dateline:
Public Company Information:
NEW YORK–(BUSINESS WIRE)–Pfizer Inc. (NYSE:PFE) today announced its once-daily oral Janus kinase 1 (JAK1) inhibitor PF-04965842 received Breakthrough Therapy designation from the U.S. Food and Drug Administration (FDA) for the treatment of patients with moderate-to-severe atopic dermatitis (AD). The Phase 3 program for PF-04965842 initiated in December and is the first trial in the J AK1 A topic D ermatitis E fficacy and Safety (JADE) global development program.
“Achieving Breakthrough Therapy Designation is an important milestone not only for Pfizer but also for patients living with the often devastating impact of moderate-to-severe atopic dermatitis, their providers and caregivers,” said Michael Corbo, Chief Development Officer, Inflammation & Immunology, Pfizer Global Product Development. “We look forward to working closely with the FDA throughout our ongoing Phase 3 development program with the hope of ultimately bringing this important new treatment option to these patients.”
Breakthrough Therapy Designation was initiated as part of the Food and Drug Administration Safety and Innovation Act (FDASIA) signed in 2012. As defined by the FDA, a breakthrough therapy is a drug intended to be used alone or in combination with one or more other drugs to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If a drug is designated as a breakthrough therapy, the FDA will expedite the development and review of such drug.1
About PF-04965842 and Pfizer’s Kinase Inhibitor Leadership
PF-04965842 is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD including interleukin (IL)-4, IL-13, IL-31 and interferon gamma.
Pfizer has established a leading kinase research capability with multiple unique kinase inhibitor therapies in development. As a pioneer in JAK science, the Company is advancing several investigational programs with novel selectivity profiles, which, if successful, could potentially deliver transformative therapies for patients. Pfizer has three additional kinase inhibitors in Phase 2 development across multiple indications:
- PF-06651600: A JAK3 inhibitor under investigation for the treatment of rheumatoid arthritis, ulcerative colitis and alopecia areata
- PF-06700841: A tyrosine kinase 2 (TYK2)/JAK1 inhibitor under investigation for the treatment of psoriasis, ulcerative colitis and alopecia areata
- PF-06650833: An interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor under investigation for the treatment of rheumatoid arthritis
Working together for a healthier world®
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products. Our global portfolio includes medicines and vaccines as well as many of the world’s best-known consumer health care products. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 150 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
DISCLOSURE NOTICE: The information contained in this release is as of February 14, 2018. Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments.
This release contains forward-looking information about PF-04965842 and Pfizer’s ongoing investigational programs in kinase inhibitor therapies, including their potential benefits, that involves substantial risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Risks and uncertainties include, among other things, the uncertainties inherent in research and development, including the ability to meet anticipated clinical trial commencement and completion dates and regulatory submission dates, as well as the possibility of unfavorable clinical trial results, including unfavorable new clinical data and additional analyses of existing data; risks associated with preliminary data; the risk that clinical trial data are subject to differing interpretations, and, even when we view data as sufficient to support the safety and/or effectiveness of a product candidate, regulatory authorities may not share our views and may require additional data or may deny approval altogether; whether regulatory authorities will be satisfied with the design of and results from our clinical studies; whether and when drug applications may be filed in any jurisdictions for any potential indication for PF-04965842 or any other investigational kinase inhibitor therapies; whether and when any such applications may be approved by regulatory authorities, which will depend on the assessment by such regulatory authorities of the benefit-risk profile suggested by the totality of the efficacy and safety information submitted, and, if approved, whether PF-04965842 or any such other investigational kinase inhibitor therapies will be commercially successful; decisions by regulatory authorities regarding labeling, safety and other matters that could affect the availability or commercial potential of PF-04965842 or any other investigational kinase inhibitor therapies; and competitive developments.
A further description of risks and uncertainties can be found in Pfizer’s Annual Report on Form 10-K for the fiscal year ended December 31, 2016 and in its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results”, as well as in its subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov and www.pfizer.com .

# # # # #
1 Food and Drug Administration Fact Sheet Breakthrough Therapies at https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDASIA/ucm329491.htmaccessed on January 25, 2018
PATENT
CA 2899888
PATENT
WO 2014128591
PFIZER INC. [US/US]; 235 East 42nd Street New York, New York 10017 (US)
BROWN, Matthew Frank; (US).
FENWICK, Ashley Edward; (US).
FLANAGAN, Mark Edward; (US).
GONZALES, Andrea; (US).
JOHNSON, Timothy Allan; (US).
KAILA, Neelu; (US).
MITTON-FRY, Mark J.; (US).
STROHBACH, Joseph Walter; (US).
TENBRINK, Ruth E.; (US).
TRZUPEK, John David; (US).
UNWALLA, Rayomand Jal; (US).
VAZQUEZ, Michael L.; (US).
PARIKH, Mihir, D.; (US)
COMPD 2

Example 2 : N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane- l -sulƒonamide
This compound was prepared using 1-propanesulfonyl chloride. The crude compound was purified by chromatography on silica gel eluting with a mixture of dichloromethane and methanol (93 : 7) to afford the title compound as a tan sol id (78% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.60 (br s, 1 H), 8.08 (s, 1 H), 7.46 (d, 1 H), 7.12 (d, 1 H), 6.61 (d, 1 H), 4.81-4.94 (m, 1 H), 3.47-3.62 (m, 1 H), 3.23 (s, 3 H), 2.87-2.96 (m, 2 H), 2.52-2.63 (m, 2 H), 2.14-2.27 (m, 2 H) 1.60- 1.73 (m, 2 H) 0.96 (t, 3 H). LC/MS (exact mass) calculated for C14H21N5O2S;
323.142, found (M + H+); 324.1.
PAPER
Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.

https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.7b01598
N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (25)
Schmieder, G.; Draelos, Z.; Pariser, D.; Banfield, C.; Cox, L.; Hodge, M.; Kieras, E.; Parsons-Rich, D.; Menon, S.; Salganik, M.; Page, K.; Peeva, E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study Br. J. Dermatol. 2017, DOI: 10.1111/bjd.16004
Compound 25, N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide is available through MilliporeSigma (cat. no. PZ0304).
CLIP
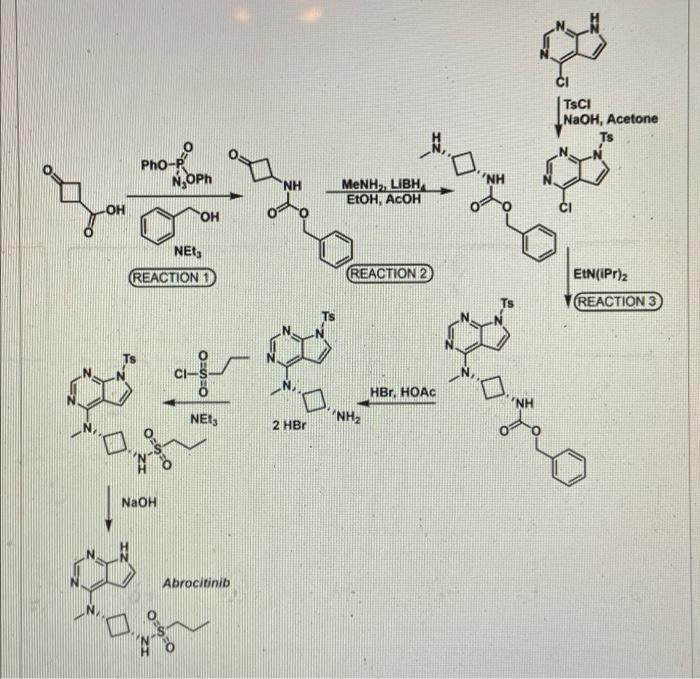
REFERENCES
1: Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, Kieras E, Parsons-Rich D, Menon S, Salganik M, Page K, Peeva E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study. Br J Dermatol. 2017 Sep 26. doi: 10.1111/bjd.16004. [Epub ahead of print] PubMed PMID: 28949012
2 Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.
- Originator Pfizer
- Class Anti-inflammatories; Antipsoriatics; Pyrimidines; Pyrroles; Skin disorder therapies; Small molecules; Sulfonamides
- Mechanism of Action Janus kinase 1 inhibitors
- Phase III Atopic dermatitis
- Discontinued Lupus vulgaris; Plaque psoriasis
- 21 May 2019Pfizer initiates enrolment in a phase I trial in Healthy volunteers in USA (PO) (NCT03937258)
- 09 May 2019 Pfizer plans a phase I pharmacokinetic and drug-drug interaction trial in healthy volunteers in May 2019 (NCT03937258)
- 30 Apr 2019 Pfizer completes a phase I trial (In volunteers) in USA (PO) (NCT03626415)
References[
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/213871s000lbl.pdf
- ^ Jump up to:a b c d e “Cibinqo EPAR”. European Medicines Agency (EMA). 11 October 2021. Retrieved 17 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. (October 2019). “Efficacy and Safety of Oral Janus Kinase 1 Inhibitor Abrocitinib for Patients With Atopic Dermatitis: A Phase 2 Randomized Clinical Trial”. JAMA Dermatology. 155 (12): 1371–1379. doi:10.1001/jamadermatol.2019.2855. PMC 6777226. PMID 31577341.
- ^ Peeva E, Hodge MR, Kieras E, Vazquez ML, Goteti K, Tarabar SG, et al. (August 2018). “Evaluation of a Janus kinase 1 inhibitor, PF-04965842, in healthy subjects: A phase 1, randomized, placebo-controlled, dose-escalation study”. British Journal of Clinical Pharmacology. 84 (8): 1776–1788. doi:10.1111/bcp.13612. PMC 6046510. PMID 29672897.
- ^ Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
- ^ “Pfizer Presents Positive Phase 3 Data at the 28th Congress of the European Academy of Dermatology and Venereology for Abrocitinib in Moderate to Severe Atopic Dermatitis”. Drugs.com. 12 October 2019.
- ^ Silverberg, J. I.; Simpson, E. L.; Thyssen, J. P.; Gooderham, M.; Chan, G.; Feeney, C.; Biswas, P.; Valdez, H.; Dibonaventura, M.; Nduaka, C.; Rojo, R. (3 June 2020). “Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial”. JAMA Dermatology. 156 (8): 863–873. doi:10.1001/jamadermatol.2020.1406. PMC 7271424. PMID 32492087.
- ^ Jump up to:a b “Cibinqo: Pending EC decision”. European Medicines Agency. 15 October 2021. Retrieved 15 October 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “European Commission Approves Pfizer’s Cibinqo (abrocitinib) for the Treatment of Adults with Moderate-to-Severe Atopic Dermatitis”. Pfizer Inc. (Press release). 10 December 2021. Retrieved 17 December 2021.
- ^ “U.S. FDA Approves Pfizer’s Cibinqo (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis”. Pfizer Inc. (Press release). 14 January 2022. Retrieved 16 January 2022.
External links
- “Abrocitinib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
- Clinical trial number NCT03575871 for “Study Evaluating Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-2)” at ClinicalTrials.gov
- {{ClinicalTrialsGov|NCT03720470|Study Evaluating Efficacy and Safety of PF-04965842 and Dupilumab in Adult Subjects With Moderate to Severe Atopic Dermatitis on Background Topical Therapy (JADE Compare)}
 |
|
| Clinical data | |
|---|---|
| Trade names | Cibinqo |
| Other names | PF-04965842 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Elimination half-life | 2.8–5.2 h |
| Excretion | 1.0–4.4% unchanged in urine |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.251.498 |
| Chemical and physical data | |
| Formula | C14H21N5O2S |
| Molar mass | 323.42 g·mol−1 |
| 3D model (JSmol) | |
/////////PF 04965842, Abrocitinib, Phase III, Atopic dermatitis, pfizer, fda 2022, APPROVALS 2022
CCCS(=O)(N[C@H]1C[C@@H](N(C)C2=C3C(NC=C3)=NC=N2)C1)=O
CCCS(=O)(=O)N[C@@H]1C[C@@H](C1)N(C)c2ncnc3[nH]ccc23
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
PF 06650833


PF-06650833
1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide
CAS 1817626-54-2
Chemical Formula: C18H20FN3O4
Molecular Weight: 361.3734
- Originator Pfizer
- Class Anti-inflammatories; Antirheumatics
- Mechanism of Action Interleukin-1 receptor-associated kinase inhibitors
- Phase II Rheumatoid arthritis
- Phase I Lupus vulgaris
- 01 Aug 2018 Pfizer completes a phase II trial in Rheumatoid arthritis (Treatment-experienced) in USA, Ukraine, Taiwan, Serbia, Russia, Romania, Poland, Mexico, South Korea, Georgia, Bosnia-Herzegovina, Australia, Croatia, Spain, Slovakia, Czech Republic, Hungary, Germany, Bulgaria (PO) (NCT02996500)
- 28 Jul 2018 No recent reports of development identified for phase-I development in Lupus(In volunteers) in USA (PO, Controlled release)
- 28 Jul 2018 No recent reports of development identified for phase-I development in Lupus(In volunteers) in USA (PO, Immediate release)
- PF-06650833 is an inhibitor of Interleukin-1 receptor associated kinase 4 (IRAK4). RAK4 is located proximal to TLR/IL-1 receptors, and in preclinical studies, inhibits downstream signaling from these receptors. The development of novel small molecule inhibitors of this kinase has the potential to lead to new therapeutics to treat diseases such as rheumatoid arthritis, lupus, and lymphomas.
Interleukin-1 receptor associated kinase 4 (IRAK-4) is a serine threonine kinases that plays a key role in innate immune signaling. IRAK-4 is activated by the interleukin (IL-1) family receptors (IL-1R, IL-18R, and IL-33R), as well as the Toll-like receptors (TLRs). Inhibition of IRAK-4 blocks the production of inflammatory cytokines such as type I interferons, tumor necrosis factor (TNF), IL-1, IL-6, and IL-12 that are key drivers of autoimmune and inflammatory diseases. IRAK-4 is an attractive therapeutic target for diseases associated with dysregulated inflammation, such as systemic lupus erythematosus and rheumatoid arthritis.


Conditions: (a) LDA (1.2 equiv), TMSCl (1.3 equiv), THF, −60 °C, 30 min; (b) allyl methyl carbonate (1.1 equiv), Pd(OAc)2 (0.05 equiv), THF, 65 °C, 2 h, 73% (2 steps); (c) LiThCN (1.5 equiv), EtMgCl (1.5 equiv), TMSCl (2.0 equiv), THF, −78 °C, 6 h, 90%; (d) LDA (1.8 equiv), NFSI (1.25 equiv), THF, −78 °C, 1 h, 23% (8), 45% (9); (e) pTsOH (0.05 equiv), MeCN, H2O, 90 °C, 2 h, 97%; (f) 3 (0.9 equiv), KHMDS (2.0 equiv), DMF, THF, −10 °C, 30 min, 84%; (g) H2O2 (10 equiv), K2CO3 (4.0 equiv), DMSO, 20 °C, 2 h, 97%.
CLIP
Target: Interleukin-1 receptor associated kinase 4 (IRAK4): This kinase is important in innate immunity, and its inhibition is predicted to be beneficial in treating inflammatory diseases.
Disease: Rheumatoid arthritis, inflammatory bowel disorder
Notes: PF06650833 came from a screening assay that used nuclear magnetic resonance spectroscopy to determine binding between molecular fragments and IRAK4. The initial hit, which bound weakly to IRAK4, was optimized with structure- and property-based medicinal chemistry to generate a series of potent inhibitors, said Katherine Lee, an associate research fellow at Pfizer.
Paper
Improvements to Enable the Large Scale Synthesis of 1-{[(2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide (PF-06650833)
 , Bryan Li‡ , Zhihui Peng‡, Lulin Wei‡, Emma McInturff‡, David Place‡, David B. Damon‡, and Robert A. Singer‡
, Bryan Li‡ , Zhihui Peng‡, Lulin Wei‡, Emma McInturff‡, David Place‡, David B. Damon‡, and Robert A. Singer‡ 

https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00386/suppl_file/op8b00386_si_001.pdf

An improved process for the large scale synthesis of 1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide (1), a candidate currently in clinical development, was developed. Key objectives were to eliminate chromatographic purifications, to maximize the reproducibility of each step, and to improve the yield and efficiency of each step relative to the previous discovery syntheses of 1. This work was focused on improvements to the synthesis of the stereochemically complex lactam 2. Steps of particular concern were the preparation of the unsaturated lactam 6, the cuprate conjugate addition reaction to produce 7, and the conversion of 7 to 8 with a high degree of diastereoselection. The solutions to these challenges have permitted the synthesis of 2 in excess of 100 kg, which in turn has permitted 1 to be prepared in sufficient amounts to support further development.
1 (31.3 kg, 91%, 82% overall) as a white, free-flowing powder.
1H NMR (500 MHz, DMSO): δ 8.86 (s, 1H), 8.16 (s, 1H), 7.90 (d, J = 5.9 Hz, 1H), 7.84 (br. s., 1H), 7.74 (s, 1H), 7.70 (br. s., 1H), 7.42 (d, J = 5.9 Hz, 1H), 4.90 (dd, J = 5.9, 53.8 Hz, 1H), 4.54 (dd, J = 3.5, 11.1 Hz, 1H), 4.26 (dd, J = 6.4, 11.0 Hz, 1H), 4.13–4.05 (m, 1H), 3.97 (s, 3H), 2.69–2.54 (m, 1H), 1.68–1.53 (m, 2H), 1.02 (t, J = 7.3 Hz, 3H).
13C NMR{1H} (126 MHz, DMSO): δ 171.0 (d, J = 19.4 Hz), 166.4, 158.4, 155.1, 137.7, 131.8, 130.3, 128.4, 120.3, 115.2, 103.2 (d, J = 4.2 Hz), 90.0 (d, J = 179.2 Hz), 66.3, 56.0, 54.1, 42.2 (d, J = 19.4 Hz), 16.4 (d, J = 8.4 Hz), 12.1.
19F NMR (H decoupled, 376 MHz, DMSO-d6): δ −199.26.
LCMS: 362 (MH+).


//////////////PF-06650833, PF 06650833, PF06650833, PF-6650833, PF 6650833, PF6650833.
O=C(C1=CC2=C(C(OC[C@H]([C@H](CC)[C@@H]3F)NC3=O)=NC=C2)C=C1OC)N
/////////////////PF-06650833, PF 06650833, Phase 3, Atopic dermatitis, PFIZER, Breakthrough Therapy Designation
крисаборол , كريسابورول , Crisaborole, AN 2728
Crisaborole
Treatment for Inflammatory Skin Diseases, including Atopic Dermatitis and Psoriasis
C14H10BNO3, Average mass251.045 Da
4-[(1-Hydroxy-1,3-dihydro-2,1-benzoxaborol-5-yl)oxy]benzonitrile ,
4-((1-Hydroxy-1,3-dihydrobenzo(c)(1,2)oxaborol-6-yl)oxy)benzonitrile
CAS 906673-24-3, AN-2728
Benzonitrile, 4-[(1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy]-
1,3-Dihydro-1-hydroxy-5-(4-cyanophenoxy)-2,1-benzoxaborole
5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole
crisaborol, crisaborole, Crisaborole, crisaborolum
UNII-Q2R47HGR7P
крисаборол
كريسابورول
In phase 3 for treatment of mild to moderate atopic dermatitis……Anacor Pharmaceuticals, Inc.
Psoriasis is a chronic skin disorder caused by inflammatory cell infiltration into the dermis and epidermis, and is accompanied by keratinocyte hyperproliferation. Once triggered, a strong T-cell response is mounted, and a cascade of cytokine and chemokine production is induced.
Down-regulation of certain cytokines and chemokines is considered to be a good approach to treatment, and indeed, the biologics targeting TNF-α demonstrate the effectiveness of this approach.However, biologics have intrinsic challenges, such as limited administration route, side effects, quality control and production cost.
Small molecule approaches to treat psoriasis include systemic or topical steroids, cyclosporine, psoralen plus UVA (PUVA), retinoids, methotrexete, and vitamin D3 analogs.Atopic dermatitis is an allergic skin disorder, which is typically treated with topical steroids, antihistamines, and calcineurin inhibitors.
However, there is still a need for new treatment with improved safety profile. Recently phosphodiesterase 4 (PDE4) inhibitors have been in development for such skin diseases. CC-10004 is in development as an oral treatment for psoriasis and atopic dermatitis. AWD-12-281 was, until recently, in development for the topical treatment of atopic dermatitis. In addition, roflumilast is under Phase 1 development for both diseases.

Figure 1.
PDE4 inhibitors aiming at skin inflammatory diseases.

Anacor’s lead product candidate is crisaborole, an investigational non-steroidal topical PDE-4 inhibitor in development for the potential treatment of mild-to-moderate atopic dermatitis and psoriasis
crisaborole is an investigational topical antiinflammatory drug in phase III clinical development by Anacor Pharmaceuticals for the treatment of mild to moderate atopic dermatitis and in phase II clinical trials in mild to moderate psoriasis
A novel boron-containing small molecule, Crisaborole inhibits the release of pro-inflammatory cytokines including TNF-alpha, IL-12, and IL-23, known mediators of the inflammation associated with psoriasis.
Synthesis
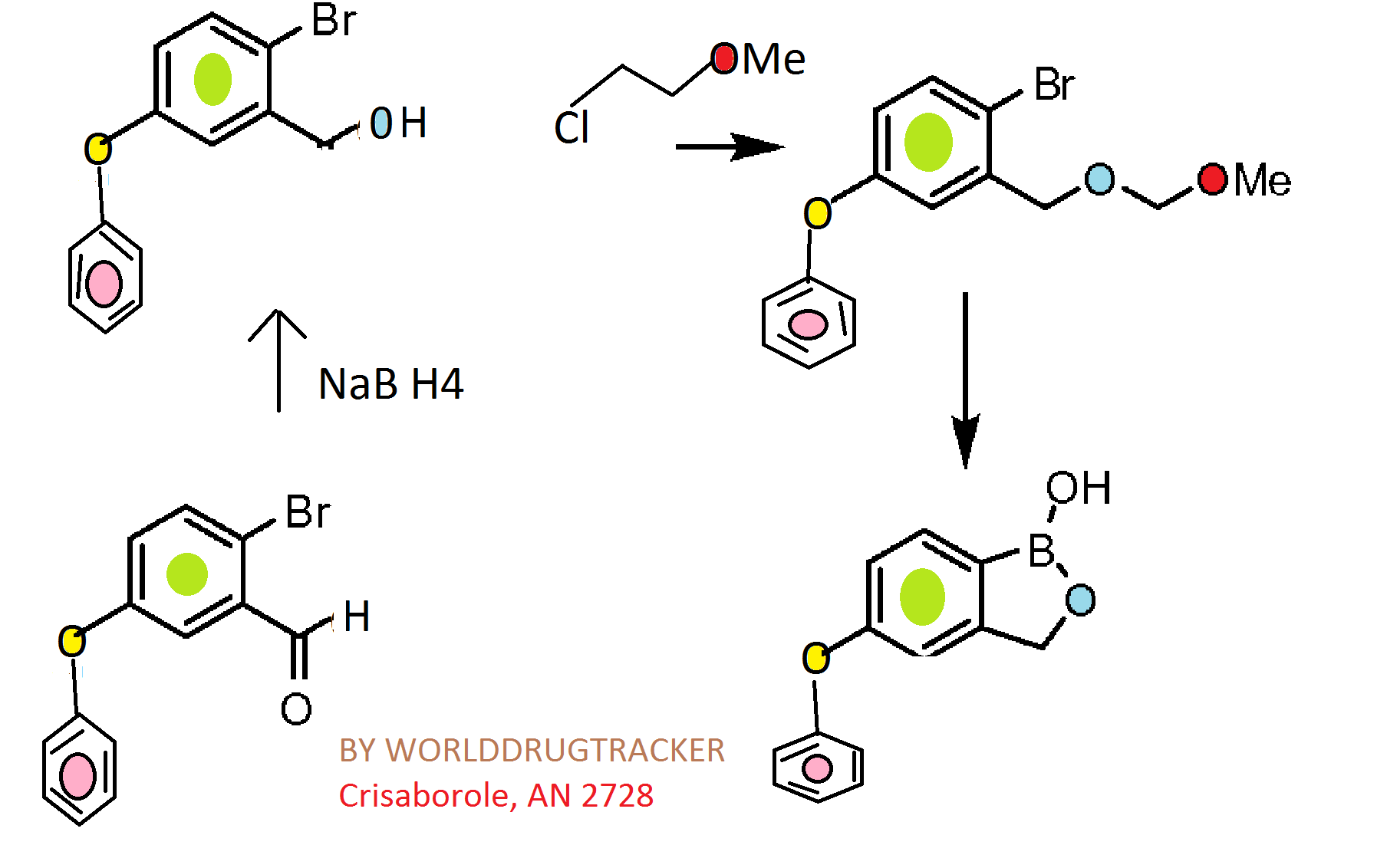
CKICK ON IMAGE FOR CLEAR VIEW
| Originator | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Therapeutic Claim | |||||||||||||||||||||
| Class | |||||||||||||||||||||
| Mechanism of action | |||||||||||||||||||||
| WHO ATC code(s) | |||||||||||||||||||||
| EPhMRA code(s) | |||||||||||||||||||||
| Clinical trial(s) |
|
PAPER
Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis
Bioorg Med Chem Lett 2009, 19(8): 2129
http://www.sciencedirect.com/science/article/pii/S0960894X09002996
- Anacor Pharmaceuticals, Inc., 1020 E. Meadow Circle, Palo Alto, CA 94303, USA
A series of phenoxy benzoxaboroles were synthesized and screened for their inhibitory activity against PDE4 and cytokine release. 5-(4-Cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2728) showed potent activity both in vitro and in vivo. This compound is now in clinical development for the topical treatment of psoriasis and being pursued for the topical treatment of atopic dermatitis


Scheme 1.
Reagents and conditions: (a) ethylene glycol, p-TsOH, toluene, reflux, 6 h (quant.); (b) K2CO3, DMF, 100 °C, overnight (82–96%); (c) 3 M HCl, THF, reflux, 2 h (80–100%); (d) NaBH4, MeOH, rt, 1 h (quant.); (e) 3,4-dihydro-2H-pyran, camphorsulfonic acid, CH2Cl2, rt, 2 h (quant.); (f) (i-PrO)3B, n-BuLi, THF, −78 °C to rt, 3 h; (g) 6 M HCl, THF, rt, 3 h (37–44%); (h) 6 M NaOH, MeOH, 1,4-dioxane, reflux, 6 days (79%); (i) diethylamine (for 5f) or morpholine (for 5g), EDCI, HOBt, DMAP, DMF, rt, overnight (41–70%).
PATENT
http://www.google.co.in/patents/WO2006089067A2?cl=en
4.2. q 5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole (C17) [0264] 1H-NMR (300 MHz,
δ ppm 4.95 (s, 2H), 7.08 (dd, J= 7.9, 2.1 Hz, IH), 7.14 (d, J= 8.8 Hz, IH), 7.15 (d, J= 2.1 Hz, IH), 7.78 (d, J= 7.9 Hz, IH), 7.85 (d, J= 9.1 Hz, 2H), 9.22 (s, IH).
PATENT
EXAMPLE 15
http://www.google.com/patents/WO2007095638A2?cl=en
4-(4-Cvanophenoxy)phenylboronic acid (C97)

(a) (4-cyanophenyl) (4-bromophenyl) ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 1000C for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with IN NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4- bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-de): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p.l94-198°C. MS: m/z = 239 (M+), 240 (M+ 1) (ESI+) and m/z = 238 (M-I) (ESI-). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6 + D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.
FURTHER METHOD

2-Bromo-5-(4-cvanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J= 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
PATENT
http://www.google.im/patents/EP1976536A2?cl=en
2.2.a 2-Bromo-5-(4-cyanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J- 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
2.2.b 2-Bromo-4-(4-cyanophenoxγ)benzyl Alcohol
1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.58 (d, IH), 7.39 (d, IH), 7.18 (dd, IH), 7.11- (d, 2H), 5.48 (t, IH) and 4.50 (d, 2H) ppm.
2.2.c 5- (4-Cyanophenoxy) -1 -Indanol
M.p.50-53°C. MS (ESI+): m/z = 252 (M+l). HPLC: 99.7% purity at 254 nm and 99.0% at 220 nm. 1H NMR (300 MHz, DMSOd6): δ 7.80 (d, 2H), 7.37 (d, IH), 7.04 (d, 2H), 6.98-6.93 (m, 2H), 5.27 (d, IH)5 5.03 (q, IH), 2.95-2.85 (m, IH), 2.75-2.64 (m, IH), 2.39-2.29 (m, IH) and 1.85-1.74 (m, IH) ppm.
2.2. d 2-Bromo-5-(tert-butyldimethylsiloxy)benzyl Alcohol [0429] 1H-NMR (300 MHz, CDCl3) δ (ppm) 0.20 (s, 6H), 0.98 (s, 9H), 4.67 (br s,lH), 6.65 (dd, J= 8.2, 2.6 Hz, IH), 6.98 (d, J= 2.9 Hz, IH), 7.36 (d, J= 8.8 Hz, IH).
3.2.k 2-Bromo-5-(2-cyanophenoχy)-l-(methoxymethoxymethyl)benzene [0443] 1H-NMR (300 MHz, CDCl3) δ (ppm) 3.41 (s, 3H), 4.64 (s, 2H), 4.76 (s, 2H), 6.8-6.9 (m, 2H), 7.16 (td, J= 7.6, 0.9 Hz, IH), 7.28 (d, J= 2.9 Hz, IH), 7.49 (ddd, J= 8.8, 7.6, 1.8 Hz, IH)5 7.56 (d, J= 8.5 Hz, IH), 7.67 (dd, J= 7.9, 1.8 Hz, IH).
EXAMPLE 32
Alternative Preparation of C17 -Intermediate

The procedure described in Example II I was followed for 1H NMR characterization of the current alcohol-borate intermediate. 1H NMR determination indicated there were 72.7 mol% of the desired alcohol-borate intermediate [2-bromo- 5-(4-cyanophenoxy)benzyl] diisopropyl borate, 20.7 mol% of an unknown intermediate and 6.5 mol% of unreacted alcohol. 1H NMR (CDCl3, 300 MHz) of [2- bromo-5-(4-cyanophenoxy)benzyl] diisopropyl borate: δ= 7.61 (d, J= 9.0 Hz, 2H), 7.52 (d, J= 8.4 Hz, IH), 7.15 (d, J= 3.0 Hz, IH), 7.03 (d, J= 8.7 Hz, 2H), 6.84 (dd, J= 8.7 Hz, J= 3.0 Hz, IH), 4.85 (s, 2H), 4.35 (septet, J= 6.1 Hz, 2H), 1.11 (d, J= 6.1 Hz, 12H) ppm.
PATENT
http://www.google.com/patents/US20090291917
- Example 154-(4-Cyanophenoxy)phenylboronic acid (C97)
-

-
(a) (4-cyanophenyl)(4-bromophenyl)ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 100° C. for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with 1N NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4-bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
-
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p. 194-198° C. MS: m/z=239 (M+), 240 (M+1) (ESI+) and m/z=238 (M−1) (ESI−). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6+D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.

see
http://www.google.co.in/patents/WO2006089067A2?cl=en
see
http://www.google.com/patents/US20090291917
| US5688928 * | Jun 7, 1995 | Nov 18, 1997 | Prolinx, Inc. | Phenylboronic acid complexing reagents derived from aminosalicylic acid |
| US5880188 * | May 26, 1995 | Mar 9, 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US5962498 * | Dec 2, 1994 | Oct 5, 1999 | Procyon Pharmaceuticals, Inc. | Protein kinase C modulators. C. indolactam structural-types with anti-inflammatory activity |
| US6369098 * | Oct 4, 2000 | Apr 9, 2002 | Bethesda Pharmaceuticals, Inc. | Dithiolane derivatives |
| US20030032673 * | Jul 19, 2002 | Feb 13, 2003 | Isis Innovation Limited | Therapeutic strategies for prevention and treatment of alzheimer’s disease |
| US20050239170 * | Jul 16, 2001 | Oct 27, 2005 | Hedley Mary L | Alpha-MSH related compounds and methods of use |
| US20060009386 * | May 12, 2005 | Jan 12, 2006 | The Brigham And Women’s Hospital, Inc. | Use of gelsolin to treat infections |
|
Methods of treating anti-inflammatory conditions through the use of boron- containing small molecules are disclosed.
|
|
… Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … Francisco, CA Mar. 6-10, 2009. 7 , “AN2728 … Kyoto, Japan, May 14-18, 2008. 10, “AN2728 …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3- dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
… Dermatology Annual Meeting, San Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … 7, “AN2728 … Francisco, CA May 6-10, 2009. 10, “AN2728 …
|
|
… from the group consisting of AN-2728, AN-2898, CBS- 3595, apremilast, ELB- 353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, …
|
|
“AN2728” is the compound 4-(l-hydroxy-l,3-dihydro-2 … GSK256066, oglemilast, tetomilast, apremilast, AN2728, Compound A, Compound B, …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
85.用于治疗疼痛的UK-500,001。 85. for the treatment of pain UK-500,001. 86.用 于治疗疼痛的AN2728。 86. for the treatment of pain AN2728.
|
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
///////////crisaborole, AN 2728, PHASE 3, Anti-inflammatory, Phosphodiesterase, Oxaborole, Psoriasis, Atopic dermatitis, borole
Regeneron and Sanofi’s dupilumab gets FDA breakthrough therapy status for atopic dermatitis
// // //

Regeneron Pharmaceuticals and Sanofi’s dupilumab has received breakthrough therapy designation from US Food and Drug Administration (FDA) to treat adults with moderate-to-severe atopic dermatitis (AD).
Sanofi And Regeneron Report Positive Proof-of-Concept Data For Dupilumab, An IL-4R Alpha Antibody, In Atopic Dermatitis
| Monoclonal antibody | |
|---|---|
| Source | Human |
| Target | IL4 receptor alpha |
Treatment of atopic diseases
Immunoglobulin, anti-(human interleukin 4 receptor α) (human REGN668 heavy chain),
disulfide with human REGN668 κ-chain, dimer
Immunoglobulin G4, anti-(human interleukin-4 receptor subunit alpha (IL-4R-alpha,
CD124)); human monoclonal REGN668 des-452-lysine{CH3107K>-}-[233-
proline{H10S>P}]γ4 heavy chain (139-219′)-disulfide with human monoclonal REGN668
κ light chain, dimer (231-231”:234-234”)-bisdisulfide
1190264-60-8 cas no
REGN668, SAR231893
MOLECULAR FORMULA- C6512H10066N1730O2052S46
Dupilumab is a monoclonal antibody designed for the treatment of atopic diseases.[1]
This drug was developed by Regeneron Pharmaceuticals.
- Statement On A Nonproprietary Name Adopted By The USAN Council – Dupilumab,American Medical Association.
Phase 1b Data Presented at Late Breaking Session of 71st Annual Meeting of the American Academy of Dermatology
PARIS and TARRYTOWN, N.Y., March 2, 2013 – Sanofi and Regeneron Pharmaceuticals, Inc. today announced that pooled data from two Phase 1b trials with dupilumab (REGN668/SAR231893), an investigational, high-affinity, subcutaneously administered, fully-human antibody targeting the alpha subunit of the interleukin 4 receptor (IL-4R alpha), were presented at the 71st Annual Meeting of the American Academy of Dermatology (AAD) in Miami.
The primary objective of the Phase 1b studies was to assess the safety profile of dupilumab. Other exploratory endpoints included pharmacokinetic, biomarker, and efficacy parameters. The efficacy data showed that treatment with four weekly subcutaneous injections of dupilumab at either 150 milligrams (mg) or 300mg per week, significantly improved the signs and symptoms of patients with moderate-to-severe atopic dermatitis (AD) whose disease was not adequately controlled with topical medications. Specifically, patients treated with dupilumab had significant improvements in body surface area (BSA) score, Investigator Global Assessment (IGA) score, and Eczema Area Severity Index (EASI) from baseline to week 4 compared to placebo (p<0.05 vs. placebo for all measures and doses). The significant improvements in BSA, IGA, and EASI scores were maintained at week 8 in the 300mg dose group (p<0.05 vs. placebo). A responder analysis demonstrated that at week 4, 54.5% of patients treated with the 150mg dose and 71.4% of patients treated with the 300mg dose achieved a reduction in EASI score of 50% or greater compared to 18.8% with placebo (p<0.05). The most common adverse events (AEs) were nasopharyngitis (19.6% vs 12.5% for placebo) and headache (11.8% vs 6.3% for placebo).
“Despite existing therapies, a significant proportion of patients with moderate-to-severe atopic dermatitis continue to suffer from inflamed skin and intractable itch, which significantly impacts their quality of life,” said Dr. Eric Simpson, Associate Professor, Director of Clinical Studies, Oregon Health and Science University, Portland, Oregon, USA, and Principal Investigator of the study. ”The early phase results with this biologic therapy, which has a novel mechanism of action, are encouraging to those of us who treat these patients and warrant further clinical investigation.”
“Through blockade of IL-4R alpha, dupilumab modulates signaling of both the IL-4 and IL-13 pathway, which have been implicated in the pathophysiology of allergic disease,” said George D. Yancopoulos, M.D., Ph.D., Chief Scientific Officer of Regeneron and President of Regeneron Laboratories. ”We look forward to presenting additional data from a 12-week, Phase 2a trial in atopic dermatitis, as well as starting a larger Phase 2b trial with dupilumab in patients with atopic dermatitis, later this year.”
Presented today in a late-breaking clinical trials session at the AAD meeting, the Phase 1b trials included 67 patients randomized to three different doses of dupilumab (75mg, n=8; 150mg, n=22; 300mg, n=21) and placebo (n=16). The primary objective of the Phase 1b studies was to assess the safety profile of dupilumab. Other endpoints included pharmacokinetic, biomarker, and efficacy parameters. Following the 4-week treatment period, patients in the studies were followed for an additional 4 weeks for a total of 8 weeks.
About IL-4R and the IL-4/IL-13 Pathway
Atopic dermatitis and some types of asthma are characterized by the induction of a specific type of an immune response that is driven by a subset of immune cells called Type 2 helper T cells, or Th2 cells. IL-4 and IL-13 are key cytokines that are required for the initiation and maintenance of this Th2 immune response. Both IL-4 and IL-13 signaling occurs through two different IL-4 receptors (Type I and II), which both contain a common IL-4R alpha subunit.
About Dupilumab (SAR231893/REGN668)
Dupilumab is a fully human monoclonal antibody directed against IL-4R alpha and is administered via subcutaneous injection. By blocking IL-4R alpha dupilumab modulates signaling of both IL-4 and IL-13, drivers of a Th2 immune response. Dupilumab was created using Regeneron’s pioneering VelocImmune® technology and is being co-developed with Sanofi. Dupilumab is currently being studied in both atopic dermatitis and asthma.
About Atopic Dermatitis
Atopic dermatitis (AD) is a chronic, immune-mediated, inflammation of the skin that is characterized by poorly defined erythema (redness) with edema (swelling), weeping in the acute stage, and skin thickening (lichenification) in the chronic stage. Chronic and/or relapsing lesions, along with pruritus (itching) and scratching are the hallmarks of the disease. The prevalence of AD is estimated to be between 1% and 3% of adults. For many patients, topical therapies are not effective for keeping the disease under control and the only approved systemic therapies to treat AD are prednisone and cyclosporine (in Europe). Moderate-to-severe atopic dermatitis can negatively impact patients’ lives and is associated with a high burden to society both in terms of direct costs of medical care and prescription drugs, as well as loss of productivity.
About Sanofi
Sanofi, a global and diversified healthcare leader, discovers, develops and distributes therapeutic solutions focused on patients’ needs. Sanofi has core strengths in the field of healthcare with seven growth platforms: diabetes solutions, human vaccines, innovative drugs, consumer healthcare, emerging markets, animal health and the new Genzyme. Sanofi is listed in Paris (EURONEXT: SAN) and in New York (NYSE:SNY).
About Regeneron Pharmaceuticals, Inc.
Regeneron is a leading science-based biopharmaceutical company based in Tarrytown, New York that discovers, invents, develops, manufactures, and commercializes medicines for the treatment of serious medical conditions. Regeneron markets medicines for eye diseases, colorectal cancer, and a rare inflammatory condition and has product candidates in development in other areas of high unmet medical need, including hypercholesterolemia, rheumatoid arthritis, asthma, and atopic dermatitis. For additional information about the company, please visitwww.regeneron.com.

{kind=link}
{kind=link}