
Home » Posts tagged 'Antineoplastics'
Tag Archives: Antineoplastics
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Linrodostat BMS 986205, ONO 7701
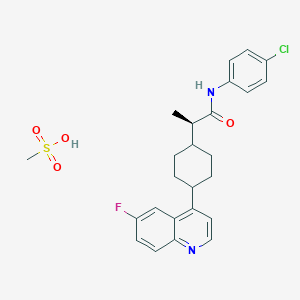
cas 2221034-29-1
- Linrodostat
- (2R)-N-(4-chlorophenyl)-2-(cis-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
- Linrodostat mesylate
- Linrodostat [USAN]
- UNII-OS7OBU191R
- OS7OBU191R
- Linrodostat mesylate [USAN]
- BMS-986205-04
- 2221034-29-1
- Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-alpha- methyl-, (alphaR,1alpha,4alpha)-, methanesulfonate (1:1)
Linrodostat; (2R)-N-(4-chlorophenyl)-2-(cis-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide; Linrodostat mesylate; Linrodostat [USAN]; UNII-OS7OBU191R; OS7OBU191R


BMS 986205
CAS: 1923833-60-6
Phase III Head and neck cancer; Malignant melanoma
BMS-986205, ONO-7701, F- 001287
- Molecular Formula C24H24ClFN2O
- Average mass 410.912 Da
BMS986205, BMS 986205, ONO-7701
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-(αR)-
A potent and selective IDO1 (indoleamine 2,3-dioxygenase 1) inhibitor.
| Alternate Name | (R)-N-(4-chlorophenyl)-2-((1s,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propenamide |
|---|---|
| Appearance | Crystalline solid |
| CAS # | 1923833-60-6 |
| Molecular Formula | C₂₄H₂₄ClFN₂O |
| Molecular Weight | 410.92
|
- Originator Bristol-Myers Squibb
- Developer Bristol-Myers Squibb; Ono Pharmaceutical
- Class Antineoplastics; Cyclohexanes; Quinolines; Small molecules
- Mechanism of Action Indoleamine-pyrrole 2,3-dioxygenase inhibitors
Highest Development Phases
- Phase II IHead and neck cancer; Malignant melanoma
- Phase I/II Cancer
- Phase I Solid tumours
Most Recent Events
- 01 Jun 2018Efficacy and adverse events data from a phase I/IIa trial in Bladder cancer (Combination therapy, Late-stage disease) presented at the 54th Annual Meeting of the American Society of Clinical Oncology (ASCO- 2018)
- 08 May 2018Bristol-Myers Squibb plans the CheckMate 9UT phase II trial for Bladder Cancer in USA, Canada, Italy, Mexico, Netherlands, Spain and United Kingdom , (NCT03519256)
- 30 Apr 2018Bristol-Myers Squibb withdraws a phase III trial for Non-small cell lung cancer (First-line therapy, Combination therapy, Late-stage disease) in USA, Austria, Australia, Brazil, Canada, Czech Republic, France, Germany, Greece, Italy, Japan, South Korea, Mexico, Spain, Switzerland, Taiwan and Turkey prior to enrolment (NCT03417037)
BMS , following its acquisition of Flexus Biosciences , and licensee Ono Pharmaceutical are developing linrodostat, a once-daily, indoleamine 2,3-dioxygenase 1 inhibitor for the potential oral treatment of cancer including renal cell carcinoma, muscle-invasive bladder cancer and melanoma. In October 2018, the trial was initiated in the US, Europe, Israel and Brazil.
WO2015031295 product pat
WO2016073770 first disclosed
WO2018209049
- WO 2016073770


Bristol-Myers Squibb, following its acquisition of Flexus Biosciences, is developing BMS-986205 (previously F- 001287), the lead from an immunotherapy program of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors for the potential treatment of cancer. In February 2016, a phase I/IIa trial was initiated .
BMS-986205 (ONO-7701) is being evaluated at Bristol-Myers Squibb in phase I/II clinical trials for the oral treatment of adult patients with advanced cancers in combination with nivolumab. Early clinical development is also ongoing at Ono in Japan for the treatment of hematologic cancer and for the treatment of solid tumors.
In April 2017, data from the trial were presented at the 108th AACR Annual Meeting in Washington DC. As of February 2017, the MTD had not been reached, but BMS-986205 plus nivolumab treatment was well tolerated, with only two patients discontinuing treatment due to DLTs. The most commonly reported treatment-related adverse events (TRAEs) were decreased appetite, fatigue, nausea, diarrhea, and vomiting. Grade 3 TRAEs were reported in three patients during the combination therapy; however, no grade 3 events were reported during BMS-986205 monotherapy lead-in. No grade 4 or 5 TRAEs were reported with BMS-986205 alone or in combination with nivolumab
Indoleamine 2,3-dioxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO is an oxidoreductase and one of two enzymes that catalyze the first and rate-limiting step in the conversion of tryptophan to N-formyl-kynurenine. It exists as a 41kD monomer that is found in several cell populations, including immune cells, endothelial cells, and fibroblasts. IDO is relatively well-conserved between species, with mouse and human sharing 63% sequence identity at the amino acid level. Data derived from its crystal structure and site-directed mutagenesis show that both substrate binding and the relationship between the substrate and iron-bound dioxygenase are necessary for activity. A homolog to IDO (ID02) has been identified that shares 44% amino acid sequence homology with IDO, but its function is largely distinct from that of IDO. (See, e.g., Serafini P, et al, Semin. Cancer Biol, 16(l):53-65 (Feb. 2006) and Ball, H.J. et al, Gene, 396(1):203-213 (Jul. 2007)).
IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. Importantly, IDO regulates immunity at the T cell level, and a nexus exists between IDO and cytokine production. In addition, tumors frequently manipulate immune function by upregulation of IDO. Thus, modulation of IDO can have a therapeutic impact on a number of diseases, disorders and conditions.
A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res., 12(4): 1144-1151 (Feb. 15, 2006)).
Treatment of cancer commonly entails surgical resection followed by chemotherapy and radiotherapy. The standard treatment regimens show highly variable degrees of long-term success because of the ability of tumor cells to essentially escape by regenerating primary tumor growth and, often more importantly, seeding distant metastasis. Recent advances in the treatment of cancer and cancer-related diseases, disorders and conditions comprise the use of combination therapy incorporating immunotherapy with more traditional chemotherapy and radiotherapy. Under most scenarios, immunotherapy is associated with less toxicity than traditional chemotherapy because it utilizes the patient’s own immune system to identify and eliminate tumor cells.
In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g. , rheumatoid arthritis). Thus, suppression of tryptophan degradation by inhibition of IDO activity has tremendous therapeutic value. Moreover, inhibitors of IDO can be used to enhance T cell activation when the T cells are suppressed by pregnancy, malignancy, or a virus (e.g., HIV). Although their roles are not as well defined, IDO inhibitors may also find use in the treatment of patients with neurological or neuropsychiatric diseases or disorders (e.g., depression).
Small molecule inhibitors of IDO have been developed to treat or prevent IDO-related diseases. For example, the IDO inhibitors 1-methyl-DL-tryptophan; p-(3-benzofuranyl)-DL-alanine; p-[3-benzo(b)thienyl]-DL-alanine; and 6-nitro-L-tryptophan have been used to modulate T cell-mediated immunity by altering local extracellular concentrations of tryptophan and tryptophan metabolites (WO 99/29310). Compounds having IDO inhibitory activity are further reported in WO 2004/094409.
In view of the role played by indoleamine 2,3-dioxygenase in a diverse array of diseases, disorders and conditions, and the limitations (e.g., efficacy) of current IDO inhibitors, new IDO modulators, and compositions and methods associated therewith, are needed.
In April 2017, preclinical data were presented at the 108th AACR Annual Meeting in Washington DC. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively
In April 2017, preclinical data were presented at the 253rd ACS National Meeting and Exhibition in San Francisco, CA. BMS-986205 showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. A good pharmacokinetic profile was seen at oral and iv doses in rats, dogs and monkeys. The compound showed good oral exposure and efficacy in in vivo assays
Preclinical studies were performed to evaluate the activity of BMS-986205, a potent and selective optimized indoleamine 2, 3-dioxygenase (IDO)- 1inhibitor, for the treatment of cancer. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. BMS-986205 was also found to be potent when compared with IDO-1from other species (human < dog equivalent monkey equivalent mouse > rat). In cell-free systems, incubation of inhibitor lead to loss of heme absorbance of IDO-1 which was observed in the presence of BMS-986205 (10 microM), while did not observed with epacadostat (10 microM). The check inhibitory activity and check reversibility (24 h after compound removal) of BMS-986205 was found to be < 1 and 18% in M109 (mouse) and < 1 and 12% SKOV3 (human) cells, respectively. In human whole blood IDO-1, human DC mixed lymphocyte reaction and human T cells cocultured with SKOV3 cells- cell based assays, BMS-986205 showed potent cellular effects (inhibition of kynurenine and T-cell proliferation 3H-thymidine) with IC50 values of 2 to 42 (median 9.4 months), 1 to 7 and 15 nM, respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively. In vivo human-SKOV3 and hWB-xenografts, IC50 values of BMS-986205 were 3.4 and 9.4 NM, respectively. The ADME of BMS-986205 at parameters iv/po dose was 0.5/2, 0.5/1.5 and 0.5/1.2 mg/kg, respectively; iv/clearance was 27, 25 and 19 ml, min/kg, respectively; iv Vss was 3.8, 5.7 and 4.1 l/kg, respectively; t1/2 (iv) was 3.9, 4.7 and 6.6 h, respectively; fraction (po) was 64, 39 and 10%, respectively. At the time of presentation, BMS-986205 was being evaluated in combination with nivolumab.
The chemical structure and preclinical profile was presented for BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.
BMS-986158: a BET inhibitor for cancerAshvinikumar Gavai of Bristol Myers Squibb (BMS) gave an overview of his company’s research into Bromodomian and extra-terminal domain (BET) as oncology target for transcriptional suppression of key oncogenes, such as MYC and BCL2. BET inhibition has been defined as strong rational strategy for the treatment of hematologic malignancies and solid tumors. From crystal-structure guided SAR studies, BMS-986158, 2-{3-(1,4-Dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol, was chosen as a potent BET inhibitor, showing IC50 values for BRD2, BRD3 and BRD4 activity of 1 nM; it also inhibited Myc oncogene (IC50 = 0.5 nM) and induced chlorogenic cancer cell death. In vitro the compound also displayed significant cytotoxicity against cancer cells. When administered at 0.25, 0.5 and 1 mg/kg po, qd to mice bearing human lung H187 SCLC cancer xenograft, BMS-986158 was robust and showed efficacy as a anticancer agent at low doses. In metabolic studies, it showed t1/2 of 36, 40 and 24 min in human, rat and mice, respectively, and it gave an efflux ratio of 3 in Caco-2 permeability assay. In phase 1/II studies, BMS-986158 was well tolerated at efficacious doses and regimens, and drug tolerable toxicity at efficacy doses and regimens. Selective Itk inhibitors for inflammatory disordersThe development of highly selective Itk inhibitors for the treatment of diseases related to T-cell function, such as inflammatory disorders, was described by Shigeyuki Takai (Ono Pharmaceutical). Inhibitory properties of a hit compound, ONO-8810443, were modified via X-ray structure and Molecular Dynamics stimulation to get ONO-212049 with significant kinase selectivity (140-fold) against Lck, a tyrosine kinase operating upstream of Itk in the TCR cascade. Further modifications identified final lead compound ONO-7790500 (N-[6-[3-amino-6-[2-(3-methoxyazetidin-1-yl)pyridin-4-yl]pyrazin-2-yl]pyridin-3-yl]-1-(3-methoxyphenyl)-2,3-dimethyl-5-oxopyrazole-4-carboxamide), which selectively inhibited Itk (IC50 = < 0.004 microM) over Lck (IC50 = 9.1 microM; SI 2000-fold) and suppressed Jurkat T-cell proliferation (IC50 = 0.014 microM). This compound suppressed alphaCD3/CDP28 CD4+T-cell stimulation (IC50 = 0.074 microM) with selectivity over PMA/Ionomycin (IC50 = > 10 microM). ONO-7790500 also exhibited in vivo IL-2 inhibitory properties (62% inhibition at 30 mg/kg po) in mice. In pharmacokinetic studies in balb/c mice, the compound administered orally (10 mg/kg) showed a Cmax of 1420 ng/ml, AUClast of 11,700 ng*h/ml, t1/2 of 5.3 h and oral bioavailability of 68%. Administration iv at 0.3 mg/kg gave an AUC last of 610 ng*h/ml, t1/2 of 3.8 h, Vss of 1260 ml/kg and Cl of 5.1 ml/min/kg. ADMET data showed ONO-7790500 did not have relevant activity in cytochromes and hERG channels (IC50 > 10 microM) in toxicological studies, and gave a PAMPA value of 5.0 x 10(-6) cm/s. Fused imidazole and pyrazole derivatives as TGF-beta inhibitorsDual growth and differentiation factor-8 (GDF-8; also known as myostatin) and TGF-beta inhibitors were described. Both targets belong to TGF-beta superfamily consisting of a large group of structurally related cell regulatory proteins involved in fundamental biological and pathological processes, such as cell proliferation or immunomodulation. Myostatin (GDF8) is a negative regulator negative regulator of skeletal muscle growth and has also been related to bone metabolism. Investigators at Rigel Pharmaceuticals found that compounds designed to be GDF-8 inhibitors were able to inhibit TGF-beta as well, this could be an advantage for the treatment of diseases associated with muscle and adipose tissue disorders, as well as potentially immunosuppressive disorders. Jiaxin Yu from the company described new fused imidazole derivatives, of which the best compound was 6-[2-(2,4,5-Trifluorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl]quinoxaline. This compound was very potent at TGF-beta Receptor Type-1 (ALK5) inhibition with an IC50 value of 1nM. In an in vivo mouse assay this compound showed good activity at 59.7 mg/kg, po, and good plasma exposure; inhibition of GDF-8 and TGFbeta growth factors was 90 and 81.6 %, respectively.Rigel’s Ihab Darwish described a series of fused pyrazole derivatives, with the best compound being 6-[2-(2,4-Difluorophenyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridine. This compound showed an IC50 of 0.06 and 0.23 microM for GDF-8 and TGFbeta, respectively, in the pSMAD (MPC-11) signaling inhibition test. The compound had a good pharmacokinetic profile, with 40% of bioavailability in mice after a 5-mg/kg po dose. An iv dose of 1 mg/kg showed t1/2 of 0.7 h and Vss of 1.0 l/h/kgDiscovery of selective inhibitor of IDO BMS-986205 for cancerIndoleamine-2,3-dioxygenase (IDO)-1 enzyme initiates and regulates the first step of the kynurenine pathway (KP) of tryptophan metabolism, and evidence has shown that overexpression of IDO-1 in cancer tumors is a crucial mechanism facilitating tumor immune evasion and persistence. The chemical structure and preclinical profile of BMS-986205 was presented by Aaron Balog from BMS. BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), is a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.Three further reports have been published from this meeting .The website for this meeting can be found at https://www.acs.org/content/acs/en/meetings/spring-2017.html.
SYNTHESIS
1 Wittig NaH
2 REDUCTION H2, Pd, AcOEt, 4 h, rt, 50 psi
3 Hydrolysis HCl, H2O, Me2CO, 2 h, reflux
4 4-Me-2,6-(t-Bu)2-Py, CH2Cl2, overnight, rt
5 SUZUKI AcOK, 72287-26-4, Dioxane, 16 h, 80°C
6 Heck Reaction, Suzuki Coupling, Hydrogenolysis of Carboxylic Esters, Reduction of Bonds, HYDROGEN
7 Et3N, THF, rt – -78°C , Pivaloyl chloride, 15 min, -78°C; 1 h, 0°C ,THF, 0°C – -78°C, BuLi, Me(CH2)4Me, 15 min, -78°C, R:(Me3Si)2NH •Na, THF, 10 min, -50°C , HYDROLYSIS, (PrP(=O)O)3, C5H5N, AcOEt, 5 min, rt

Product Patent
WO2016073770
Example 19
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
Example 19 : (i?)-N-(4-chlorophenyl)-2-(cz5-4-(6-fluoroquinolin-4- yl)cyclohexyl)propanamide
[0277] Prepared using General Procedures K, B, E, L, M, N, and O. General Procedure L employed 2-(4-(6-fluoroquinolin-4-yl)-cyclohexyl)acetic acid (mixture of
diastereomers), and ( ?)-2-phenyl-oxazolidinone. General Procedure M employed the cis product and iodomethane. The auxiliary was removed following General Procedure N and the desired product formed employing General Procedure O with 4-chloroaniline.
Purified using silica gel chromatography (0% to 100% ethyl acetate in hexanes) to afford Example 19. 1H NMR of czs-isomer (400 MHz; CDC13): δ 9.14 (s, 1H), 8.70 (d, J= 4.6 Hz, 1H), 8.06 (dd, J= 9.2 Hz, J= 5.6 Hz, 1H), 7.58-7.64 (m, 3H), 7.45 (ddd, J= 9.3 Hz, J= 7.8 Hz, J= 2.7 Hz, 1H), 7.19-7.24 (m, 2H), 7.15 (d, J= 4.6Hz, 1H), 3.16-3.26 (m, 1H), 2.59-2.69 (m, 1H), 2.08-2.16 (m, 1H), 1.66-1.86 (m, 7H), 1.31-1.42 (m, 1H), 1.21 (d, J= 6.8Hz, 3H) ppm. m/z 411.2 (M+H)+.
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(3), 319-329.
https://www.sciencedirect.com/science/article/pii/S0960894X17312180

PATENT
WO 2018022992
PATENT
WO 2018071500
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018071500&redirectedID=true
WO-2019006292
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006292&tab=PCTDESCRIPTION&maxRec=1000
Improved methods for the preparation of substituted quinolinycyclohexylpropanamide compounds, such as linrodostat claiming substituted pyridine compounds as IDO1 inhibitors, useful for treating cancers.
Indoleamine 2,3 -di oxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res. , 12(4): 1144-1151 (Feb. 15, 2006)). In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g., rheumatoid arthritis).
Substituted quinolinylcyclohexylpropanamide pharmaceutical compounds that inhibit IDO and are useful for the treatment of cancer have been previously described. See, e.g., WO2016/073770. Improved methods of making such compounds, which reduce production costs and improve production safety, are, therefore, needed.
Scheme 4
[0076] The disclosure is also directed to methods of preparing intermediate compounds of formula IV. Methods to produce compounds of formula IV are depicted in Schemes 5 and 6.
Scheme 5
IX-A
Scheme 6
IX-B IV
Compounds of the disclosure that include one or more radioisotopes can be used in imaging. See, e.g., WO2018017529. For example, radiolabeled compounds of the disclosure can be used in Positron Emission Tomography (PET). Such methods are useful in the imaging of cancer in a subject. A preferred radiolabeled compound is
1
Pharmaceutically acceptable salts of [18F]-Compound 1 are also within the scope of the disclosure. An exemplary method for the preparation of [18F]-Compound 1 is depicted in Scheme below.
1 . reaction
[18F]-Compound 1
Example 9
(R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
[00258] To a 10 L glass-lined reactor under a blanket of nitrogen was charged 349 g Ν,Ν,Ν’,Ν’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and 2 L acetonitrile. 245 g N-methylimidazole was added followed by 0.3 L acetonitrile. 300 g (R)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanoic acid was added followed by 0.3 L acetonitrile. The mixture was held for 0.5 h then 139 g 4-chloroaniline charged followed by 0.4 L acetonitrile. The mixture was maintained at 20 °C until the reaction was deemed complete by HPLC analysis. The solution was then heated to 60°C, and 1.2 L water was charged. The solution was then cooled to 40 °C, seeds (3 g) were charged, and the resulting slurry was maintained for 1 h. The slurry was then cooled to 20 °C and 2.7 L water was charged. The slurry was filtered and the cake was washed three times with 3 L of 2: 1 water: acetonitrile. The cake was dissolved with 5.1 L ethyl acetate and the solution was distilled to a volume of 4.2 L at 41 °C under vacuum. The slurry was cooled to 20 °C, 4.14 g seeds were charged, and a solution of 95.7 g methanesulfonic acid in 2.9 L ethyl acetate was added. The slurry was then filtered and washed two times with 1.65 L ethyl acetate and dried under vacuum at 50°C to yield 445 g of (R)-N-(4-chlorophenyl)-2-((l s,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide methanesulfonate as a white solid in 88% yield.
[00259] ¾ NMR (600 MHz, DMSO-de) δ 10.19 (s, IH), 9.24 (d, J=5.7 Hz, IH), 8.40 (dd, J=10.3, 2.6 Hz, IH), 8.33 (dd, J=9A, 5.3 Hz, IH), 8.09 (d, J=5.7 Hz, IH), 8.04 (t, J=8.6 Hz, IH), 7.71 – 7.64 (m, 2H), 7.37 – 7.30 (m, 2H), 3.64 (ddt, J=10.8, 7.3, 3.8 Hz, IH), 2.98 – 2.89 (m, IH), 2.43 (s, 3H), 2.05 – 1.60 (m, 9H), 1.14 (d, J=6.7 Hz, 3H); 13C NMR (126 MHz, DMSO-de) δ 175.0, 162.7, 161.1 , 145.4, 138.2, 136.8, 128.6, 128.1 , 126.7, 126.4, 123.3, 120.8, 119.8, 109.0, 39.8, 39.7, 38.6, 35.5, 28.3, 27.6, 27.2, 26.1 , 16.2 MS (ESI): calcd for C24H24CIFN2O
([M + H]+), 410.16; found, 410.15.
[00260] HPLC analysis: Column: Sigma-Aldrich Supelco Ascentis Express CI 8 2.7um, 150 x 4.6 mm ID; Solvent A: 0.05% TFA with MeCN:water (5/95 v/v); Solvent B: 0.05% TFA with MeCN: water (95/5 v/v); Gradient: %B: 0 Min. 15%; 1 Min. 15%; 13 Min. 55%; 19 Min. 65%; 24 Min. 100%; 24.1 15%; 28 Min. 15%; Stop Time: 24 Min; Flow Rate: 1.0 ml/min;
Column temperature: 30 °C; wavelength: 218 nm. The retention time (R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide peak was 12.6 min.
Example 7
(R)-N-(4-chlorophenyl)-2-((ls, -4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
[00252] To a 50 L glass-lined reactor under a blanket of nitrogen was charged 13.75 kg acetonitrile, then 2.68 Kg Ν,Ν,Ν’,Ν’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and rinsed with 2.0 Kg acetonitrile. 2.03 Kg N-methylimidazole was added followed by 1.95 Kg acetonitrile. 2.48 Kg (R)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanoic acid was added followed by 1.05 Kg acetonitrile. The mixture was held for 0.5 h then 1.21 Kg 4-chloroaniline charged followed by 1.0 Kg acetonitrile. The mixture was maintained at 20 °C until the reaction was deemed complete by HPLC analysis. The solution was then heated to 60°C, and 9.25 Kg water was charged. The solution was then cooled to 40 °C, the mixture was aged
for 1 h, seeds (32 g) were charged and rinsed with 1.15 Kg 2: 1 water: acetonitrile, and the resulting slurry was maintained for 1 h. The slurry was then cooled to 20 °C and 25.75 Kg water was charged. The slurry was filtered and the cake was washed three times with 6.9 Kg of 2: 1 water: acetonitrile. The cake was dried under vacuum at 50°C to yield 3.33 Kg of (R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide hydrate as a white solid in 94.1% yield.
[00253] ¾ NMR (600 MHz, DMSO-de) δ 10.09 (s, 1H), 8.86 (d, J=4.5 Hz, 1H), 8.08 (dd, J=9.0, 5.6 Hz, 1H), 7.95 (dd, J=10.9, 2.6 Hz, 1H), 7.70 – 7.60 (m, 3H), 7.54 (d, J=4.5 Hz, 1H), 7.33 (d, J=9.0 Hz, 2H), 3.43 – 3.31 (m, 3H), 2.90 – 2.80 (m, 1H), 1.99 – 1.55 (m, 9H), 1.13 (d, J=6.8 Hz, 3H); 13C NMR (151 MHz, DMSO-de) δ 175.0, 159.9, 152.4, 149.7, 145.2, 138.1, 132.7, 128.5, 127.2, 126.7, 120.8, 119.0, 118.6, 107.2, 40.2, 37.4, 35.6, 28.5, 27.6, 27.4, 26.3, 16.1 ; HRMS (ESI); calcd for C24H24CIFN2O ([M + H]+), 411.1619; found 411.1649.
WO-2019006283
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006283&redirectedID=true
Novel crystalline forms of linrodostat , its salts and hydrates, designated as Forms 1, 2 and 4 (first disclosed in WO2016073770 ), processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating prostate cancer, liver cancer, brain cancer, bladder cancer, ovary cancer and breast cancer.
(R)-N-(4-chlorophenyl)-2-((l S,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanami the below structure:
[0003] Compound 1 is a potent inhibitor of indoleamine 2,3-dioxygenase (IDO; also known as IDOl), which is an IFN-γ target gene that plays a role in immunomodulation.
Compound 1 is being investigated as a treatment for cancer and other diseases. Compound 1 has been previously described in WO2016/073770.
[0004] A compound, as a free base, hydrate, solvate, or salt, can exist in amorphous form and/or one or more crystalline forms, each having different physical properties, for example, different X-ray diffraction patterns (XRPD or PXRD) and different thermal behavior. The free base, hydrate, solvate, and salt forms of a compound can also differ with respect to their individual stabilities, processing, formulation, dissolution profile, bioavailability, and the like. [0005] New forms of (R)-N-(4-chlorophenyl)-2-((l S,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide, having desirable and beneficial chemical and physical properties are needed. There is also a need for reliable and reproducible methods for the manufacture, purification, and formulation of Compound 1 (and its hydrates, solvates, salt,, and hydrated salt forms) to facilitate commercialization. The present disclosure is directed to these, as well as other important aspects.
REFERENCES
23-Feb-2015
Bristol-Myers Squibb To Expand Its Immuno-Oncology Pipeline with Agreement to Acquire Flexus Biosciences, Inc
Bristol-Myers Squibb Co; Flexus Biosciences Inc
17-Dec-2014
Flexus Biosciences, a Cancer Immunotherapy Company Focused on Agents for the Reversal of Tumor Immunosuppression (ARTIS), Announces $38M Financing
Flexus Biosciences Inc
2015106thApril 21Abs 4290
Potent and selective next generation inhibitors of indoleamine-2,3-dioxygenase (IDO1) for the treatment of cancer
American Association for Cancer Research Annual Meeting
Jay P. Powers, Matthew J. Walters, Rajkumar Noubade, Stephen W. Young, Lisa Marshall, Jan Melom, Adam Park, Nick Shah, Pia Bjork, Jordan S. Fridman, Hilary P. Beck, David Chian, Jenny V. McKinnell, Maksim Osipov, Maureen K. Reilly, Hunter P. Shunatona, James R. Walker, Mikhail Zibinsky, Juan C. Jaen
2017108thApril 04Abs 4964
Structure, in vitro biology and in vivo pharmacodynamic characterization of a novel clinical IDO1 inhibitor
American Association for Cancer Research Annual Meeting
John T Hunt, Aaron Balog, Christine Huang, Tai-An Lin, Tai-An Lin, Derrick Maley, Johnni Gullo-Brown, Jesse Swanson, Jennifer Brown
2017253rdApril 05Abs MEDI 368
Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer
American Chemical Society National Meeting and Exposition
Aaron Balog
April 2-62017
American Chemical Society – 253rd National Meeting and Exhibition (Part IV) – OVERNIGHT REPORT, San Francisco, CA, USA
Casellas J, Carceller V
////////////////PHASE 1, BMS 986205, 1923833-60-6, BMS-986205, ONO-7701,Bristol-Myers Squibb, Antineoplastics, F- 001287
C[C@H]([C@H]1CC[C@@H](C2=CC=NC3=CC=C(F)C=C23)CC1)C(NC4=CC=C(Cl)C=C4)=O
Wrapping up #MEDI‘s 1st time disclosures is Aaron Balog of @bmsnews talking about an IOD-1 inhibitor to treat cancer #ACSSanFran
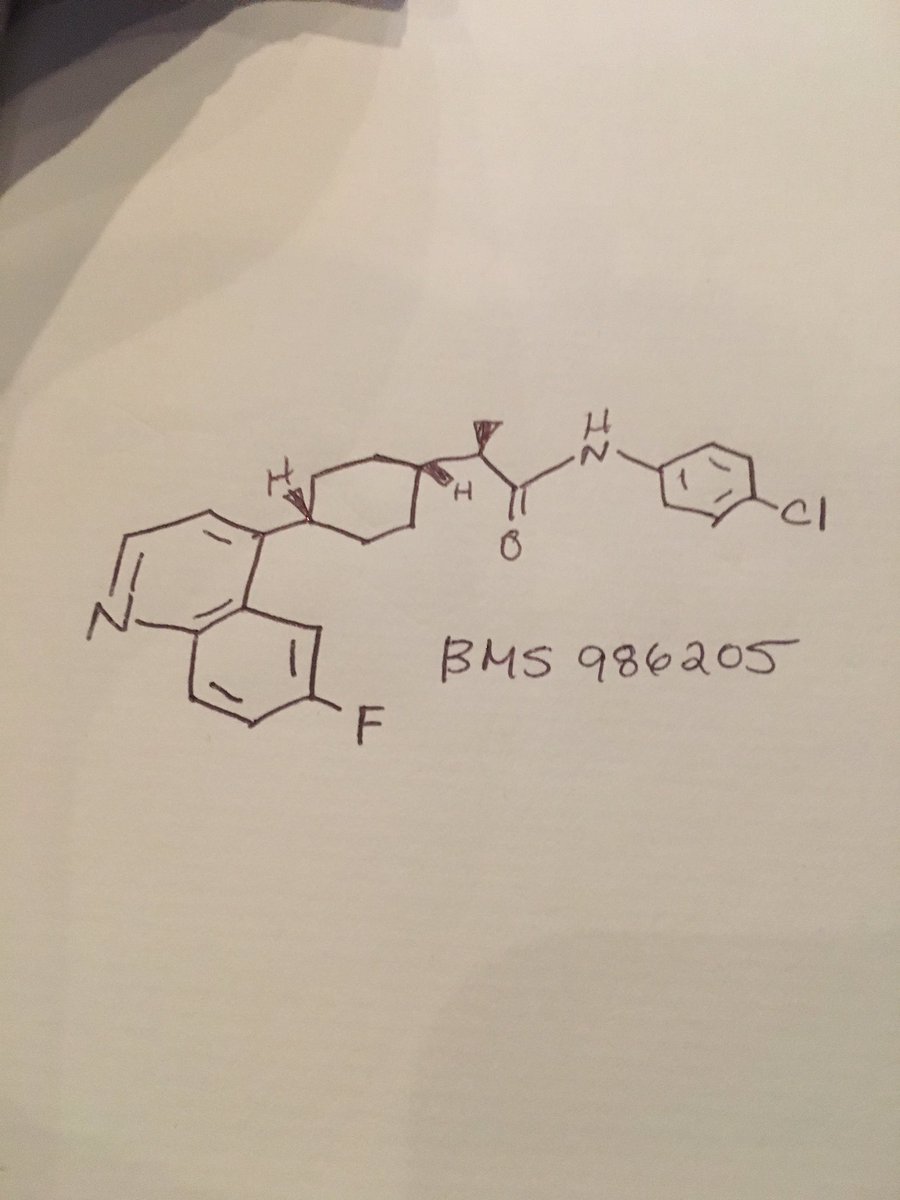
////////////////BMS986205, BMS 986205, BM-986205, ONO-7701, Phase III, Head and neck cancer, Malignant melanoma, 1923833-60-6, Linrodostat
CC(C1CCC(CC1)C2=C3C=C(C=CC3=NC=C2)F)C(=O)NC4=CC=C(C=C4)Cl
BMS 986205


BMS 986205
CAS: 1923833-60-6
Phase 1 cancer
BMS-986205, ONO-7701, F- 001287
- Molecular Formula C24H24ClFN2O
- Average mass 410.912 Da
- Originator Bristol-Myers Squibb
- Class Antineoplastics
- 01 Feb 2016 Phase-I/II clinical trials in Cancer (Combination therapy, Late-stage disease, Second-line therapy or greater) in Canada (PO) (NCT02658890)
- 31 Jan 2016 Preclinical trials in Cancer in USA (PO) before January 2016
- 01 Jan 2016 Bristol-Myers Squibb plans a phase I/IIa trial for Cancer (Late-stage disease, Combination therapy, Second-line therapy or greater) in USA, Australia and Canada (PO) (NCT02658890)

Hilary Beck
 EX Principal Investigator, Company NameFLX Bio, Inc.,
EX Principal Investigator, Company NameFLX Bio, Inc.,
CURRENTLY Director, Medicinal Chemistry at IDEAYA Biosciences, IDEAYA Biosciences, The University of Texas at Austin
![]()

Brian Wong
Chief Executive Officer at FLX Bio, Inc.
Bristol-Myers Squibb, following its acquisition of Flexus Biosciences, is developing BMS-986205 (previously F- 001287), the lead from an immunotherapy program of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors for the potential treatment of cancer. In February 2016, a phase I/IIa trial was initiated .
BMS-986205 (ONO-7701) is being evaluated at Bristol-Myers Squibb in phase I/II clinical trials for the oral treatment of adult patients with advanced cancers in combination with nivolumab. Early clinical development is also ongoing at Ono in Japan for the treatment of hematologic cancer and for the treatment of solid tumors.
In April 2017, data from the trial were presented at the 108th AACR Annual Meeting in Washington DC. As of February 2017, the MTD had not been reached, but BMS-986205 plus nivolumab treatment was well tolerated, with only two patients discontinuing treatment due to DLTs. The most commonly reported treatment-related adverse events (TRAEs) were decreased appetite, fatigue, nausea, diarrhea, and vomiting. Grade 3 TRAEs were reported in three patients during the combination therapy; however, no grade 3 events were reported during BMS-986205 monotherapy lead-in. No grade 4 or 5 TRAEs were reported with BMS-986205 alone or in combination with nivolumab
Indoleamine 2,3-dioxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO is an oxidoreductase and one of two enzymes that catalyze the first and rate-limiting step in the conversion of tryptophan to N-formyl-kynurenine. It exists as a 41kD monomer that is found in several cell populations, including immune cells, endothelial cells, and fibroblasts. IDO is relatively well-conserved between species, with mouse and human sharing 63% sequence identity at the amino acid level. Data derived from its crystal structure and site-directed mutagenesis show that both substrate binding and the relationship between the substrate and iron-bound dioxygenase are necessary for activity. A homolog to IDO (ID02) has been identified that shares 44% amino acid sequence homology with IDO, but its function is largely distinct from that of IDO. (See, e.g., Serafini P, et al, Semin. Cancer Biol, 16(l):53-65 (Feb. 2006) and Ball, H.J. et al, Gene, 396(1):203-213 (Jul. 2007)).
IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. Importantly, IDO regulates immunity at the T cell level, and a nexus exists between IDO and cytokine production. In addition, tumors frequently manipulate immune function by upregulation of IDO. Thus, modulation of IDO can have a therapeutic impact on a number of diseases, disorders and conditions.
A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res., 12(4): 1144-1151 (Feb. 15, 2006)).
Treatment of cancer commonly entails surgical resection followed by chemotherapy and radiotherapy. The standard treatment regimens show highly variable degrees of long-term success because of the ability of tumor cells to essentially escape by regenerating primary tumor growth and, often more importantly, seeding distant metastasis. Recent advances in the treatment of cancer and cancer-related diseases, disorders and conditions comprise the use of combination therapy incorporating immunotherapy with more traditional chemotherapy and radiotherapy. Under most scenarios, immunotherapy is associated with less toxicity than traditional chemotherapy because it utilizes the patient’s own immune system to identify and eliminate tumor cells.
In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g. , rheumatoid arthritis). Thus, suppression of tryptophan degradation by inhibition of IDO activity has tremendous therapeutic value. Moreover, inhibitors of IDO can be used to enhance T cell activation when the T cells are suppressed by pregnancy, malignancy, or a virus (e.g., HIV). Although their roles are not as well defined, IDO inhibitors may also find use in the treatment of patients with neurological or neuropsychiatric diseases or disorders (e.g., depression).
Small molecule inhibitors of IDO have been developed to treat or prevent IDO-related diseases. For example, the IDO inhibitors 1-methyl-DL-tryptophan; p-(3-benzofuranyl)-DL-alanine; p-[3-benzo(b)thienyl]-DL-alanine; and 6-nitro-L-tryptophan have been used to modulate T cell-mediated immunity by altering local extracellular concentrations of tryptophan and tryptophan metabolites (WO 99/29310). Compounds having IDO inhibitory activity are further reported in WO 2004/094409.
In view of the role played by indoleamine 2,3-dioxygenase in a diverse array of diseases, disorders and conditions, and the limitations (e.g., efficacy) of current IDO inhibitors, new IDO modulators, and compositions and methods associated therewith, are needed.
In April 2017, preclinical data were presented at the 108th AACR Annual Meeting in Washington DC. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively
In April 2017, preclinical data were presented at the 253rd ACS National Meeting and Exhibition in San Francisco, CA. BMS-986205 showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. A good pharmacokinetic profile was seen at oral and iv doses in rats, dogs and monkeys. The compound showed good oral exposure and efficacy in in vivo assays
Preclinical studies were performed to evaluate the activity of BMS-986205, a potent and selective optimized indoleamine 2, 3-dioxygenase (IDO)- 1inhibitor, for the treatment of cancer. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. BMS-986205 was also found to be potent when compared with IDO-1from other species (human < dog equivalent monkey equivalent mouse > rat). In cell-free systems, incubation of inhibitor lead to loss of heme absorbance of IDO-1 which was observed in the presence of BMS-986205 (10 microM), while did not observed with epacadostat (10 microM). The check inhibitory activity and check reversibility (24 h after compound removal) of BMS-986205 was found to be < 1 and 18% in M109 (mouse) and < 1 and 12% SKOV3 (human) cells, respectively. In human whole blood IDO-1, human DC mixed lymphocyte reaction and human T cells cocultured with SKOV3 cells- cell based assays, BMS-986205 showed potent cellular effects (inhibition of kynurenine and T-cell proliferation 3H-thymidine) with IC50 values of 2 to 42 (median 9.4 months), 1 to 7 and 15 nM, respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively. In vivo human-SKOV3 and hWB-xenografts, IC50 values of BMS-986205 were 3.4 and 9.4 NM, respectively. The ADME of BMS-986205 at parameters iv/po dose was 0.5/2, 0.5/1.5 and 0.5/1.2 mg/kg, respectively; iv/clearance was 27, 25 and 19 ml, min/kg, respectively; iv Vss was 3.8, 5.7 and 4.1 l/kg, respectively; t1/2 (iv) was 3.9, 4.7 and 6.6 h, respectively; fraction (po) was 64, 39 and 10%, respectively. At the time of presentation, BMS-986205 was being evaluated in combination with nivolumab.
The chemical structure and preclinical profile was presented for BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.
BMS-986158: a BET inhibitor for cancerAshvinikumar Gavai of Bristol Myers Squibb (BMS) gave an overview of his company’s research into Bromodomian and extra-terminal domain (BET) as oncology target for transcriptional suppression of key oncogenes, such as MYC and BCL2. BET inhibition has been defined as strong rational strategy for the treatment of hematologic malignancies and solid tumors. From crystal-structure guided SAR studies, BMS-986158, 2-{3-(1,4-Dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol, was chosen as a potent BET inhibitor, showing IC50 values for BRD2, BRD3 and BRD4 activity of 1 nM; it also inhibited Myc oncogene (IC50 = 0.5 nM) and induced chlorogenic cancer cell death. In vitro the compound also displayed significant cytotoxicity against cancer cells. When administered at 0.25, 0.5 and 1 mg/kg po, qd to mice bearing human lung H187 SCLC cancer xenograft, BMS-986158 was robust and showed efficacy as a anticancer agent at low doses. In metabolic studies, it showed t1/2 of 36, 40 and 24 min in human, rat and mice, respectively, and it gave an efflux ratio of 3 in Caco-2 permeability assay. In phase 1/II studies, BMS-986158 was well tolerated at efficacious doses and regimens, and drug tolerable toxicity at efficacy doses and regimens. Selective Itk inhibitors for inflammatory disordersThe development of highly selective Itk inhibitors for the treatment of diseases related to T-cell function, such as inflammatory disorders, was described by Shigeyuki Takai (Ono Pharmaceutical). Inhibitory properties of a hit compound, ONO-8810443, were modified via X-ray structure and Molecular Dynamics stimulation to get ONO-212049 with significant kinase selectivity (140-fold) against Lck, a tyrosine kinase operating upstream of Itk in the TCR cascade. Further modifications identified final lead compound ONO-7790500 (N-[6-[3-amino-6-[2-(3-methoxyazetidin-1-yl)pyridin-4-yl]pyrazin-2-yl]pyridin-3-yl]-1-(3-methoxyphenyl)-2,3-dimethyl-5-oxopyrazole-4-carboxamide), which selectively inhibited Itk (IC50 = < 0.004 microM) over Lck (IC50 = 9.1 microM; SI 2000-fold) and suppressed Jurkat T-cell proliferation (IC50 = 0.014 microM). This compound suppressed alphaCD3/CDP28 CD4+T-cell stimulation (IC50 = 0.074 microM) with selectivity over PMA/Ionomycin (IC50 = > 10 microM). ONO-7790500 also exhibited in vivo IL-2 inhibitory properties (62% inhibition at 30 mg/kg po) in mice. In pharmacokinetic studies in balb/c mice, the compound administered orally (10 mg/kg) showed a Cmax of 1420 ng/ml, AUClast of 11,700 ng*h/ml, t1/2 of 5.3 h and oral bioavailability of 68%. Administration iv at 0.3 mg/kg gave an AUC last of 610 ng*h/ml, t1/2 of 3.8 h, Vss of 1260 ml/kg and Cl of 5.1 ml/min/kg. ADMET data showed ONO-7790500 did not have relevant activity in cytochromes and hERG channels (IC50 > 10 microM) in toxicological studies, and gave a PAMPA value of 5.0 x 10(-6) cm/s. Fused imidazole and pyrazole derivatives as TGF-beta inhibitorsDual growth and differentiation factor-8 (GDF-8; also known as myostatin) and TGF-beta inhibitors were described. Both targets belong to TGF-beta superfamily consisting of a large group of structurally related cell regulatory proteins involved in fundamental biological and pathological processes, such as cell proliferation or immunomodulation. Myostatin (GDF8) is a negative regulator negative regulator of skeletal muscle growth and has also been related to bone metabolism. Investigators at Rigel Pharmaceuticals found that compounds designed to be GDF-8 inhibitors were able to inhibit TGF-beta as well, this could be an advantage for the treatment of diseases associated with muscle and adipose tissue disorders, as well as potentially immunosuppressive disorders. Jiaxin Yu from the company described new fused imidazole derivatives, of which the best compound was 6-[2-(2,4,5-Trifluorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl]quinoxaline. This compound was very potent at TGF-beta Receptor Type-1 (ALK5) inhibition with an IC50 value of 1nM. In an in vivo mouse assay this compound showed good activity at 59.7 mg/kg, po, and good plasma exposure; inhibition of GDF-8 and TGFbeta growth factors was 90 and 81.6 %, respectively.Rigel’s Ihab Darwish described a series of fused pyrazole derivatives, with the best compound being 6-[2-(2,4-Difluorophenyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridine. This compound showed an IC50 of 0.06 and 0.23 microM for GDF-8 and TGFbeta, respectively, in the pSMAD (MPC-11) signaling inhibition test. The compound had a good pharmacokinetic profile, with 40% of bioavailability in mice after a 5-mg/kg po dose. An iv dose of 1 mg/kg showed t1/2 of 0.7 h and Vss of 1.0 l/h/kgDiscovery of selective inhibitor of IDO BMS-986205 for cancerIndoleamine-2,3-dioxygenase (IDO)-1 enzyme initiates and regulates the first step of the kynurenine pathway (KP) of tryptophan metabolism, and evidence has shown that overexpression of IDO-1 in cancer tumors is a crucial mechanism facilitating tumor immune evasion and persistence. The chemical structure and preclinical profile of BMS-986205 was presented by Aaron Balog from BMS. BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), is a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.Three further reports have been published from this meeting .The website for this meeting can be found at https://www.acs.org/content/acs/en/meetings/spring-2017.html.
SYNTHESIS
1 Wittig NaH
2 REDUCTION H2, Pd, AcOEt, 4 h, rt, 50 psi
3 Hydrolysis HCl, H2O, Me2CO, 2 h, reflux
4 4-Me-2,6-(t-Bu)2-Py, CH2Cl2, overnight, rt
5 SUZUKI AcOK, 72287-26-4, Dioxane, 16 h, 80°C
6 Heck Reaction, Suzuki Coupling, Hydrogenolysis of Carboxylic Esters, Reduction of Bonds, HYDROGEN
7 Et3N, THF, rt – -78°C , Pivaloyl chloride, 15 min, -78°C; 1 h, 0°C ,THF, 0°C – -78°C, BuLi, Me(CH2)4Me, 15 min, -78°C, R:(Me3Si)2NH •Na, THF, 10 min, -50°C , HYDROLYSIS, (PrP(=O)O)3, C5H5N, AcOEt, 5 min, rt
Patent
WO2016073770
Example 19
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
Example 19 : (i?)-N-(4-chlorophenyl)-2-(cz5-4-(6-fluoroquinolin-4- yl)cyclohexyl)propanamide
[0277] Prepared using General Procedures K, B, E, L, M, N, and O. General Procedure L employed 2-(4-(6-fluoroquinolin-4-yl)-cyclohexyl)acetic acid (mixture of
diastereomers), and ( ?)-2-phenyl-oxazolidinone. General Procedure M employed the cis product and iodomethane. The auxiliary was removed following General Procedure N and the desired product formed employing General Procedure O with 4-chloroaniline.
Purified using silica gel chromatography (0% to 100% ethyl acetate in hexanes) to afford Example 19. 1H NMR of czs-isomer (400 MHz; CDC13): δ 9.14 (s, 1H), 8.70 (d, J= 4.6 Hz, 1H), 8.06 (dd, J= 9.2 Hz, J= 5.6 Hz, 1H), 7.58-7.64 (m, 3H), 7.45 (ddd, J= 9.3 Hz, J= 7.8 Hz, J= 2.7 Hz, 1H), 7.19-7.24 (m, 2H), 7.15 (d, J= 4.6Hz, 1H), 3.16-3.26 (m, 1H), 2.59-2.69 (m, 1H), 2.08-2.16 (m, 1H), 1.66-1.86 (m, 7H), 1.31-1.42 (m, 1H), 1.21 (d, J= 6.8Hz, 3H) ppm. m/z 411.2 (M+H)+.
REFERENCES
23-Feb-2015
Bristol-Myers Squibb To Expand Its Immuno-Oncology Pipeline with Agreement to Acquire Flexus Biosciences, Inc
Bristol-Myers Squibb Co; Flexus Biosciences Inc
17-Dec-2014
Flexus Biosciences, a Cancer Immunotherapy Company Focused on Agents for the Reversal of Tumor Immunosuppression (ARTIS), Announces $38M Financing
Flexus Biosciences Inc
2015106thApril 21Abs 4290
Potent and selective next generation inhibitors of indoleamine-2,3-dioxygenase (IDO1) for the treatment of cancer
American Association for Cancer Research Annual Meeting
Jay P. Powers, Matthew J. Walters, Rajkumar Noubade, Stephen W. Young, Lisa Marshall, Jan Melom, Adam Park, Nick Shah, Pia Bjork, Jordan S. Fridman, Hilary P. Beck, David Chian, Jenny V. McKinnell, Maksim Osipov, Maureen K. Reilly, Hunter P. Shunatona, James R. Walker, Mikhail Zibinsky, Juan C. Jaen
2017108thApril 04Abs 4964
Structure, in vitro biology and in vivo pharmacodynamic characterization of a novel clinical IDO1 inhibitor
American Association for Cancer Research Annual Meeting
John T Hunt, Aaron Balog, Christine Huang, Tai-An Lin, Tai-An Lin, Derrick Maley, Johnni Gullo-Brown, Jesse Swanson, Jennifer Brown
2017253rdApril 05Abs MEDI 368
Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer
American Chemical Society National Meeting and Exposition
Aaron Balog
April 2-62017
American Chemical Society – 253rd National Meeting and Exhibition (Part IV) – OVERNIGHT REPORT, San Francisco, CA, USA
Casellas J, Carceller V

Juan Jaen

Jordan Fridman
Chief Scientific Officer at FLX Bio, Inc.

Rekha Hemrajani
Chief Operating Officer at FLX Bio, Inc

Max Osipov
////////////////PHASE 1, BMS 986205, 1923833-60-6, BMS-986205, ONO-7701,Bristol-Myers Squibb, Antineoplastics, F- 001287
C[C@H]([C@H]1CC[C@@H](C2=CC=NC3=CC=C(F)C=C23)CC1)C(NC4=CC=C(Cl)C=C4)=O
Wrapping up #MEDI‘s 1st time disclosures is Aaron Balog of @bmsnews talking about an IOD-1 inhibitor to treat cancer #ACSSanFran
FGF 401


FGF 401
NVP-FGF-401
CAS 1708971-55-4
N-[5-Cyano-4-[(2-methoxyethyl)amino]-2-pyridinyl]-7-formyl-3,4-dihydro-6-[(4-methyl-2-oxo-1-piperazinyl)methyl]-1,8-naphthyridine-1(2H)-carboxamide
/V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide
Phase I/II Hepatocellular carcinoma; Solid tumours
- Originator Novartis
- Developer Novartis Oncology
- Class Antineoplastics
- Mechanism of Action Type 4 fibroblast growth factor receptor antagonists
- 26 Jan 2016 Phase-I/II clinical trials in Solid tumours and Hepatocellular carcinoma in USA, Hong Kong, Japan, Taiwan, France, Germany and Spain (PO)
- 26 Dec 2014 Phase-I/II clinical trials in Hepatocellular carcinoma in Singapore (PO)
- 26 Dec 2014 Phase-I/II clinical trials in Solid tumours in Singapore (PO)
Activation of FGFRs (fibroblast growth factor receptors) has an essential role in regulating cell survival, proliferation, migration and differentiation.1 Dysregulation of the FGFR signaling pathway has been associated with human cancer.1 FGFRs represent an important target for cancer therapeutics because a growing body of evidence indicates that they can act in an oncogenic fashion to promote multiple steps of cancer progression, including induction of mitogenic and survival signals
FGF-401 is a FGFR4 inhibitor in phase I/II clinical studies at Novartis for the treatment of positive FGFR4 and KLB expresion solid tumors and hepatocellular carcinoma
Normal growth, as well as tissue repair and remodeling, require specific and delicate control of activating growth factors and their receptors. Fibroblast Growth Factors (FGFs) constitute a family of over twenty structurally related polypeptides that are developmental^ regulated and expressed in a wide variety of tissues. FGFs stimulate proliferation, cell migration and differentiation and play a major role in skeletal and limb development, wound healing, tissue repair, hematopoiesis, angiogenesis, and tumorigenesis (reviewed in Ornitz, Novartis Found Symp 232: 63-76; discussion 76-80, 272-82 (2001)).
The biological action of FGFs is mediated by specific cell surface receptors belonging to the Receptor Protein Tyrosine Kinase (RPTK) family of protein kinases. These proteins consist of an extracellular ligand binding domain, a single transmembrane domain and an intracellular tyrosine kinase domain which undergoes phosphorylation upon binding of FGF. Four FGFRs have been identified to date: FGFR1 (also called Fig, fms-like gene, fit- 2, bFGFR, N-bFGFR or Cek1 ), FGFR2 (also called Bek-Bacterial Expressed Kinase-, KGFR, Ksam, Ksaml and Cek3), FGFR3 (also called Cek2) and FGFR4. All mature FGFRs share a common structure consisting of an amino terminal signal peptide, three extracellular immunoglobulin-like domains (Ig domain I, Ig domain II, Ig domain III), with an acidic region between Ig domains (the “acidic box” domain), a transmembrane domain, and intracellular kinase domains (Ullrich and Schlessinger, Cell 61 : 203,1990 ; Johnson and Williams (1992) Adv. Cancer Res. 60: 1 -41). The distinct FGFR isoforms have different binding affinities for the different FGF ligands.
Alterations in FGFRs have been associated with a number of human cancers including myeloma, breast, stomach, colon, bladder, pancreatic and hepatocellular carcinomas. Recently, it was reported that FGFR4 may play an important role in liver cancer in particular (PLoS One, 2012, volume 7, 36713). Other studies have also implicated FGFR4 or its ligand FGF19 in other cancer types including breast, glioblastoma, prostate, rhabdomyosarcoma, gastric, ovarian, lung, colon (Int. J. Cancer 1993; 54:378-382; Oncogene 2010; 29:1543-1552; Cancer Res 2010; 70:802-812; Cancer Res 201 1 ; 71 :4550-4561 ; Clin Cancer Res 2004; 10:6169-6178; Cancer Res 2013;
73:2551 -2562; Clin Cancer Res 2012; 18:3780-3790; J. Clin. Invest. 2009; 1 19:3395-3407; Ann Surg Oncol 2010; 17:3354-61 ; Cancer 201 1 ; 1 17:5304-13; Clin Cancer Res 2013; 19:809-820; PNAS 2013; 1 10:12426-12431 ; Oncogene 2008; 27:85-97).
Therapies involving FGFR4 blocking antibodies have been described for instance in
WO2009/009173, WO2007/136893, WO2012/138975, WO2010/026291 , WO2008/052798 and WO2010/004204. WO2014/144737 and WO2014/01 1900 also describe low molecular weight FGFR4 inhibitors.
in spite of numerous treatment options for patients with cancer, there remains a need for effective and safe therapeutic agents and a need for new combination therapies that can be administered for the effective long-term treatment of cancer.
Liver cancer or hepatic cancer is classified as primary liver cancer (i.e. cancer that forms in the tissues of the liver) and secondary liver cancer (i.e. cancer that spreads to the liver from another part of the body). According to the National Cancer Institute at the National Institutes of Health, the number of estimated new cases and deaths from liver and intrahepatic bile duct cancer in the United States in 2014 was 33,190 and 23,000, respectively. Importantly, the percent surviving five years or more after being diagnosed with liver and intrahepatic bile duct cancer is only about 16%.
It has now been found that a combination of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in free form or in pharmaceutically acceptable salt form and at least one further active ingredient, as defined herein, shows synergistic combination activity in an in vitro cell proliferation assay as shown in the experimental section and may therefore be effective for the delay of progression or treatment of a proliferative disease, such as cancer, in particular liver cancer.
| Inventors | Nicole Buschmann, Robin Alec Fairhurst, Pascal Furet, Thomas Knöpfel, Catherine Leblanc, Robert Mah, Pierre NIMSGERN, Sebastien RIPOCHE, Lv LIAO, Jing XIONG, Xianglin ZHAO, Bo Han, Can Wang, |
| Applicant | Novartis Ag |

Nicole Buschmann

Drawn by worlddrugtracker, helping millions………………..
PATENT
WO 2015059668
https://www.google.com/patents/WO2015059668A1?cl=en
PATENT
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1-yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid salt form has the following structure:

Example 1 – A/-(5-cvano-4 (2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1-yl)methyl)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid salt form (1 :1).

Step 1 : 2-(dimethoxymethyl)-1 ,8-naphthyridine.
The procedure described in J. Org. Chem., 2004, 69 (6), pp 1959-1966 was used. Into a 20 L 4-necked round-bottom flask was placed 2-aminopyridine-3-carbaldehyde (1000 g, 8.19 mol), 1 , 1-dimethoxypropan-2-one (1257 g, 10.64 mol), ethanol (10 L), and water (2 L). This was followed by the addition of a solution of sodium hydroxide (409.8 g, 10.24 mol) in water (1000 mL) drop wise with stirring at 0-15 °C. The solution was stirred for 3 h at 0-20 °C and then concentrated under vacuum. The resulting solution was extracted with 3×1200 mL of ethyl acetate and the organic layers were combined. The mixture was dried over sodium sulfate and concentrated under vacuum. The residue was washed with 3×300 mL of hexane and the solid was collected by filtration. This resulted in the title compound as a yellow solid. 1 H-NMR (400 MHz, DMSO-cf6) δ 9.1 1 (dd, 1 H), 8.53 (d, 1 H), 8.50 (dd, 1 H), 7.73 (d, 1 H), 7.67 (dd, 1 H), 5.44 (s, 1 H), 3.41 (s, 6H).
Step 2: 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 5-L pressure tank reactor (5 atm) was placed 2-(dimethoxymethyl)-1 ,8-naphthyridine (200 g, 979 mmol), ethanol (3 L), Pt02 (12 g). The reactor was evacuated and flushed three times with nitrogen, followed by flushing with hydrogen. The mixture was stirred overnight at 23 °C under an
atmosphere of hydrogen. This reaction was repeated four times. The solids were filtered out and the resulting mixture was concentrated under vacuum to give the title compound as a yellow solid. 1 H-NMR (400 MHz, DMSO-d6) δ 7.14 (d, 1 H), 6.51 (d, 1 H), 6.47 – 6.41 (m, 1 H), 4.98 (s, 1 H), 3.28 -3.19 (m, 2H), 3.23 (s, 6H), 2.64 (t, 2H), 1 .73 – 1.79 (m, 2H).
Step 3: 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
Into a 3 L 4-necked round-bottom flask was placed 7-(dimethoxymethyl)-1 ,2,3, 4-tetrahydro-1 ,8-naphthyridine (1 14.6 g, 550.3mmol) in acetonitrile (2 L). This was followed by the addition of NBS (103 g, 578 mol) in portions with stirring at 25 °C. The resulting solution was stirred for 30 min at 25 °C. The resulting mixture was concentrated under vacuum and the residue was diluted with 1000 mL of diethylether. The mixture was washed with 3×100 mL of ice/water. The aqueous phase was extracted with 2×100 mL of diethylether and the organic layers were combined. The resulting mixture was washed with 1×100 mL of brine, dried over sodium sulfate and concentrated under vacuum to give the title compound as a light yellow solid. LC-MS: (ES, m/z): 286.03 [M+H]+. 1 H-NMR: (300MHz, CDCI3) δ 1 .86 – 1 .94 (2H, m), 2.70 – 2.74 (2H, m), 3.9 – 3.43 (2H, m), 3.47 (6H, s), 5.23 (1 H, s), 5.58 (1 H, s), 7.29 (1 H, s).
Step 4: 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde.
To a solution of 6-bromo-7-(dimethoxymethyl)-1 ,2,3, 4-tetrahydro-1 ,8-naphthyridine (15.0 g, 52.2 mmol) in THF (400 mL) at -78 °C under argon, was added MeLi (1 .6 M in Et20, 32.6 mL, 52.2 mmol), the solution was stirred for 5 min, then n-BuLi (1 .6 M in hexane, 35.9 mL, 57.5 mmol) was added slowly and the solution was stirred for 20 min. THF (100 mL) was added to the reaction at -78 °C. Subsequently, n-BuLi (1 .6 M in hexane, 49.0 mL, 78 mmol) was added and the reaction mixture was stirred for 20 min, then again n-BuLi (1 .6 M in hexane, 6.53 mL, 10.45 mmol) was added and the mixture was stirred for 10 min at – 78 °C. DMF (2.10 mL, 27.2 mmol) was added and the reaction mixture was stirred at -78 °C for 45 min, then it was allowed to warm to room temperature, poured into sat. aq. NH4CI and extracted twice with DCM. The combined organic phases were dried over Na2S04, filtered and evaporated to give the title compound as an orange oil. (UPLC-MS 3) tR 0.63 min; ESI-MS 237.2 [M+H]+.
Step 5: ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate.
Ethyl bromoacetate (1.27 mL, 1 1 .48 mmol) was added to a mixture of tert-butyl (2-(methylamino)ethyl)carbamate (2.0 g, 1 1 .48 mmol), triethylamine (4.81 mL) and THF (24 mL) at 0 °C. After stirring 24 h at room temperature the reaction mixture was partitioned between saturated aqueous NaHC03 and DCM, extracted 2x with DCM, the organic layers dried over Na2S04 and
evaporated to give the title compound as a clear pale-yellow oil. 1H NMR (400 MHz, CDCI3) δ 5.20 (s, br, 1 H), 4.18 (q, 2H), 3.24 (s, 2H), 3.22 – 3.16 (m, 2H), 2.65 – 2.61 (m, 2H), 2.38 (s, 3H), 1 .42 (s, 9H), 1 .24 (t, 3H).
Step 6: ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride.
Concentrated hydrochloric acid (10 mL) was added to a solution of ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate (3.05 g, 1 1 .13 mmol) in THF (20 mL) and EtOH (100 mL) at room temperature. After stirring 1 h at room temperature the reaction mixture was evaporated, ethanol (20 mL) added, evaporated, further ethanol (50 mL) added and then stirred at 60 °C for 70 min. The cooled reaction mixture was then evaporated to give the title compound as a pale-yellow glass. 1 H NMR (400 MHz, DMSO-d6) δ 8.58 (s, br, 3H), 4.19 (q, 2H), 4.26 – 4.15 (m, 2H), 3.44 (s, br, 2H), 3.21 (s, br, 2H), 2.88 (s, 3H), 1 .21 (t, 3H).
Step 7: 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one.
Sodium triacetoxyborohydride (3.10 g, 14.61 mmol) was added to a mixture of 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde (obtained in step 4, 2.30 g, 9.74 mmol), ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride (obtained in step 6, 2.6 g, 14.61 mmol) and triethylamine (6.75 mL, 48.7 mmol) in 1 ,2-dichloroethane (20 mL) at room temperature. The reaction mixture was stirred for 21 h at room temperature and additional sodium triacetoxyborohydride (2.6 g, 9.74 mmol) was added. After a further 4 h stirring at room temperature, again additional sodium triacetoxyborohydride (1 .3 g, 4.87 mmol) was added and the reaction maintained at 4 °C for 2.5 days. The reaction mixture was then warmed to room temperature, saturated aqueous NaHC03 solution added, the mixture extracted with DCM (3x), the combined organic layers dried over Na2S04 and evaporated. The residue was applied to a 120 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 10% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1 H NMR (400 MHz, CDCI3) δ 7.08 (s, 1 H), 5.30 (s, br, 1 H), 5.20 (s, 1 H), 4.69 (s, 2H), 3.44 – 3.34 (m, 2H), 3.40 (s, 6H), 3.22 – 3.15 (m, 2H), 3.24 (s, 2H), 2.71 – 2.64 (m, 2H), 2.58 – 2.50 (m, 2H), 2.31 (s, 3H), 1 .98 – 1.82 (m, 2H). (UPLC-MS 6) tR 0.33; ESI-MS 335.3 [M+H]+.
Step 8: 4-fluoro-5-iodopyridin-2-amine.
A suspension of 4-fluoropyridin-2-amine (336 g, 2.5 mol) and NIS (745 g, 2.75 mol) in MeCN (9 L) was treated with TFA (1 14 g, 1 mol). The reaction mixture was then stirred at room temperature for 8 h. The reaction mixture was diluted with EtOAc (10 L), washed with sat. aq. Na2S203 (2 x 5 L), brine (4 x 5 L). The combined organic layers were dried over Na2S04, filtered and concentrated to get the crude product. The crude product was purified by recrystallization from EtOAc/pentane (1/10) to afford the title compound as a white solid. 1H NMR (400 MHz, DMSO-cf6) δ 8.14 (d, 1 H), 6.45 (s, 2H), 6.33 (d, 1 H).
Step 9: 6-amino-4-fluoronicotinonitrile.
4-fluoro-5-iodopyridin-2-amine (obtained in step 8, 240 g, 1 mol), zinc cyanide (125 g, 1.05 mol), zinc (13 g, 0.2 mol), Pd2(dba)3 (25 g, 25 mmol) and dppf (55 g, 0.1 mol) in DMA (800 mL) were degassed and charged into the round bottom flask under nitrogen. The mixture was stirred at 100 °C for 3 h. The reaction mixture was diluted with 5% NaHC03 (2 L), extracted with EtOAc (4 x 600 mL). The combined organic layers were washed with 5% NaOH (1 L), dried over Na2S04, concentrated to 700 mL. The resulting organic phase was eluted through silica gel column with EtOAc (1.7 L). The combined organic filtrate was washed with 2 M HCI (3 x 800 mL). The pH of the aqueous phase was adjusted to 10 with saturated NaHC03. The aqueous phase was extracted whit DCM (3 x 500 mL). The combined DCM was dried over Na2S04 and concentrated. The residue was further purified by column chromatography (eluted with pentane: EtOAc 10: 1 to 3:2) followed by recrystallization from pentane/EtOAc 3/1 to give the title compound as white solid. 1 H NMR (400 MHz, DMSO-d6) δ 8.40 (d, 1 H), 7.40 (s, 2H), 6.34 (d, 1 H).
Step 10: tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate.
A mixture of 2,4-dichloro-5-cyanopyridine (1 Og, 57.8 mmol), fe/f-butyl carbamate (8.2 g, 70.5 mmol), Pd(OAc)2 (0.26 g, 1 .1 mmol), Xantphos (1 .34 g, 2.3mmol) and K2C03 (12 g, 87 mmol) in THF (150 mL) was degassed 3x with nitrogen. The mixture was then heated at 70 °C for 4-5 h and monitored by chromatography until complete conversion. Following completion of the reaction, additional THF (100 mL) was added and heated the mixture at 70 °C for additional 1 h and then cooled to room temperature. The suspension was then filtered through a pad of celite to remove the solid. The filtrate was then concentrated and azotropically distilled with ethyl acetete before filtering to give the title compound. 1 H NMR (DMSO-d6, 400 MHz): δ 10.82 (s, 1 H), 8.79 (s, 1 H), 8.09 (s, 1 H), 1 .49 (s, 9H).
Step 1 1 : fe/f-butyl N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)carbamate.
A mixture of tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate (obtained in step 10, 9.8 g, 38.6 mmol), 2-methoxyethylamine (5.8 g, 77.3 mmol) and DIPEA (6 g, 46.4 mmol) in DMSO (80 mL) was heated at 65-70 °C for 24 h and monitored by chromatography until complete conversion. The
solution was then cooled to room temperature and a white solid precipitated gradually. Water (20 mL) was then added slowly within 1 h. The suspension was stirred for a further 1 h, filtered and dried to give the title compound as a white solid. 1 H NMR (DMSO-d6, 400 MHz): δ 9.87 (s, 1 H), 8.18 (s, 1 H), 7.20 (s, 1 H), 6.86 (s, 9H), 3.51 (t, 2H), 3.36 (t, 2H), 3.28 (s, 3H), 1.47 (s, 9H).
Step 12: 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile.
A solution of 6-amino-4-fluoronicotinonitrile (obtained in step 9, 1 .10 g, 8.02 mmol) in DMA (20 mL) was treated with 2-methoxyethylamine (2.07 mL, 24.1 mmol) and DIPEA (4.20 mL, 24.1 mmol), heated to 50 °C and stirred for 15 h. The reaction mixture was cooled to room temperature and concentrated. The crude material was purified by normal phase chromatography (24 g silica gel cartridge, heptanes/EtOAc 100:0 to 0:100). The product containing fractions were concentrated and dried under vacuum to give the title compound as an off-white solid.
An alternative synthesis of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile is outlined below:
To tert-butyl N-{5-cyano-4-[(2-methoxyethyl)amino]pyridin-2-yl}carbamate (obtained in step 1 1 , 7g) was added 30-36% aqueous HCI (40 mL), the mixture stirred at room temperature for 30 minutes and monitored by chromatography until complete conversion. The solution was then basified with 20-30% NaOH solution to pH=9-10 and filtered to give a white solid. The solid was added to ethyl acetate (15 mL) and heated to 50-55 °C to form a clear solution. The solution was then cooled to 3-6 °C, stirred for 2-3 h and filtered. The wet cake was then dried to give the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.92 (s, 1 H), 6.39 (s, 2H), 6.15 (t, 1 H), 5.61 (s, 1 H), 3.46 (t, 2H), 3.27 (s, 3H), 3.24 (q, 2H). (UPLC-MS 3) tR 0.62; ESI-MS 193.1 [M+H]+.
Step 13: N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
A solution of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile (obtained in step 12, 481 mg, 2.50 mmol) in anhydrous DMF (1.5 mL) was added drop wise over 10 minutes to a mixture of di(1 H-1 ,2,4-triazol-1 -yl)methanone (410 mg, 2.50 mmol) and DMF (1 .5 mL) cooled at 0 °C. After stirring for 45 minutes at 0 °C the reaction mixture was allowed to warm to room temperature and after a further 90 minutes at room temperature a solution of 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one (obtained in step 7, 418 mg, 1.00 mmol) in DMF (2 mL) was added. The reaction mixture was stirred for 17.5 h at room temperature, quenched by the addition of MeOH and evaporated. The residue was applied to a 80 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 2% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, DMSO-d6) δ 13.50 (s, 1 H), 8.27 (s,
1 H), 7.52 (s, 1 H), 7.39 (s, 1 H), 6.93 (t, 1 H), 5.45 (s, 1 H), 4.65 (s, 2H), 3.94 – 3.89 (m, 2H), 3.54 -3.50 (m, 2H), 3.40 – 3.35 (m, 2H), 3.38 (s, 6H), 3.29 (s, 3H), 3.20 – 3.16 (m, 2H), 3.05 (s, 2H), 2.86 – 2.80 (m, 2H), 2.61 – 2.55 (m, 2H), 2.22 (s, 3H), 1 .94 – 1 .88 (m, 2H). (UPLC-MS 6) tR 0.72; ESI-MS 553.3 [M+H]+.
Step 14: /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-form
yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide
Concentrated hydrochloric acid (0.40 mL) was added to a solution of A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 13, 470 mg, 0.808 mmol) in THF (3 mL) and water (1 mL) at room temperature. After stirring for 3 h at room temperature saturated aqueous NaHC03 was added, the mixture extracted with DCM (3x), the organic layers dried over Na2S04 and evaporated. The residue was sonicated with EtOAc (6 mL) and pentane (6 mL) and then filtered. The white solid obtained was then dissolved in DCM (6 mL), EtOAc added (3 mL), the solution warmed, sealed and allowed to stand at room temperature for 2 h. Filtration and drying gave A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide as a white solid.
1 H NMR (400 MHz, DMSO-d6) δ 13.43 (s, 1 H), 10.06 (s, 1 H), 8.24 (s, 1 H), 7.49 (s, 1 H), 7.47 (s, 1 H), 6.96 (t, br, 1 H), 4.86 (s, 2H), 3.96 – 3.90 (m, 2H), 3.52 – 3.46 (m, 2H), 3.39 – 3.33 (m, 2H), 3.30 – 3.21 (m, 2H), 3.37 (s, 3H), 3.02 (s, 2H), 2.93 – 2.86 (m, 2H), 2.61 – 2.56 (m, 2H), 2.21 (s, 3H), 1 .95 – 1.85 (m, 2H). (UPLC-MS 6) tR0.70, ESI-MS 507.2, [M+H]+.
Step 15: A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 :1 ).
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 14, 4g, 7.896 mmol) was stirred in propionic acid (29.3 g, 29.60mL) at 70 °C until dissolution was complete (20 minutes). The solution was cooled to 55 °C and a solution of citric acid in acetone (23% w/w) was added to it. Separately, a seed suspension was prepared by adding acetone (0.2 g, 0.252mL) to A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (0.0185 g, 0.026 mmol). The seed suspension was added to the solution at 50 °C and the resulting suspension was left to stir at 50 °C for 40 minutes. A further solution of citric acid in acetone (26.6g, 2.51 % w/w, 33.63 mL) was added to the reaction over 380 minutes. The resulting suspension was stirred for a further 120 minutes and cooled to 20 °C with stirring over 4 hours. The suspension was stirred for another 12 hours
before filtering the suspension under vacuum and washing the resulting solid with a propionic acid: acetone solution (1 : 1 , 7g, 7.96ml_) at room temperature. The solid was further washed with acetone (7g, 8.85ml_) at room temperature. The resulting solid was dried in an oven at 40 °C and 5mbar to give the title compound as a light orange solid (5.2g, 7.443 mmol). (mw 698.70), mp (DSC) 168.8 °C (onset).
XRPD analysis showed the same pattern as with particles obtained by a process described in PCT/I B2014/065585 (reference example 1 ) – see Figure 5.
Example 1a
Steps 1 to 14 were carried out as described in example 1 .
Step 15a: A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 : 1 )
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 14, 5g, 9.930 mmol) was stirred in propionic acid (33.5 g, 33.84ml_) at 60 °C. Once A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide had dissolved, anhydrous citric acid powder (0.19g, 0.9889 mmol) was added. The resulting suspension was heated to 70 °C and sonicated for 5 minutes to ensure full dissolution. The resulting solution was cooled to 50 °C and a solution of citric acid in ethyl acetate (3.7 g, 1 .3% citric acid in ethyl acetate) was added over 20 minutes. Seeds of N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (0.02 g) were added to the solution and the suspension was aged for 15 minutes. Another aliquot of citric acid in ethyl acetate (128g, 1 .3% citric acid in ethyl acetate) was added to the suspension over 1 1 .85hours. The suspension was left to stir for over 4 hours. The suspension was then filtered under vacuum (500mbar) and the resulting solid was washed firstly with a propionic acid: ethyl acetate solution (1 : 1 , 7g, 7.44ml_) at room temperature and then with ethyl acetate (12g, 13.38ml_) at room temperature. The resulting solid was dried in an oven at 40 °C and 5mbar to give the title compound as a light orange solid (6.3 g, 9.074 mmol).
XRPD analysis showed the same pattern as with particles obtained by a process described in PCT/I B2014/065585 (reference example 1 ) – see Figure 5.
Reference example 1 (described in PCT/IB2014/065585) – V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 :1 )
Steps 1 to 14 were carried out as described in example 1.
Reference Step 15 – /V-(5-cvano-4-((2-methoxyethyl‘)amino‘)pyridin-2-yl‘)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl‘)methyl‘)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H‘)-carboxamide in citric acid form (1 :1 )
A solution of citric acid (96.9 mg) in acetone (5 mL) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in acetone solution (2 mL) was then added to a suspension of Λ/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in acetone (4 mL) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h before slowly cooling to room temperature. The white solid was then collected by filtration, washing 2x with acetone (2 mL), and dried for 18 h at 40 °C under vacuum to give the title salt.
Alternatively, N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (6.5 g, 12.83 mmol) was placed in a 500ml 4-flask reactor. 49 mL of glacial acetic acid was added and the resulting suspension was stirred at 23 °C until a clear mixture was obtained. In a separate flask, anhydrous 2-hydroxypropane-1 ,2,3-tricarboxylic acid (2.59 g, 13.47 mmol, 1 .05 equiv.) was dissolved in 49 mL of glacial acetic acid at 50 °C until a clear solution was obtained. This solution was then added at 23°C to the N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide solution previously prepared. This mixture was stirred for 30 min at 23 °C and then added dropwise over 1 h to 192 mL of ethyl acetate warmed to 75 °C. The temperature remained constant over the addition. At the end of the addition, the temperature of the mixture was cooled slowly to 23 °C and let 16h at this temperature under gentle stirring. The suspension was cooled to 5-10 °C and filtered. The cake was washed with 15 mL of ethyl acetate and 15 mL of acetone. The wet cake (ca 8.5g) was transferred in a 500 mL flask containing 192 mL of dry acetone. The resulting suspension was refluxed for 24h. The suspension was filtered and the cake was washed with 2 times 15 mL of dry acetone then dried at 50 °C under vacuum for several hours to give the title salt.
PATENT
The synthesis of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (abbreviated herein as CPi and also named as Example 83) and salts thereof is disclosed in PCT/IB2014/065585, the content of which are incorporated by reference, as described herein below:
Example 83: /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
Concentrated hydrochloric acid (0.40 ml) was added to a solution of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (intermediate 80, 470 mg, 0.808 mmol) in THF (3 ml) and water (1 ml) at room temperature. After stirring for 3 h at room temperature saturated aqueous NaHC03 was added, the mixture extracted with DCM (3x), the organic layers dried over Na2S04 and evaporated. The residue was sonicated with EtOAc (6 ml) and pentane (6 ml) and then filtered. The white solid obtained was then dissolved in DCM (6 ml), EtOAc added (3 ml), the solution warmed, sealed and allowed to stand at room temperature for 2 h. Filtration and drying gave the title compound as a white solid.
1H NMR (400 MHz, DMSO-c/6) δ 13.43 (s, 1 H), 10.06 (s, 1 H), 8.24 (s, 1 H), 7.49 (s, 1 H), 7.47 (s, 1 H), 6.96 (t, br, 1 H), 4.86 (s, 2H), 3.96 – 3.90 (m, 2H), 3.52 – 3.46 (m, 2H), 3.39 – 3.33 (m, 2H), 3.30 – 3.21 (m, 2H), 3.37 (s, 3H), 3.02 (s, 2H), 2.93 – 2.86 (m, 2H), 2.61
– 2.56 (m, 2H), 2.21 (s, 3H), 1 .95 – 1 .85 (m, 2H).
(UPLC-MS 6) tR 0.70, ESI-MS 507.2, [M+H]+.
The following salts were prepared from the above free form form of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide by precipitation with the appropriate counterions.
Malate with 1 :1 stoichiometry (mw 640.66), mp (DSC) 181 .1 °C (onset): Acetone (2 ml) was added to a mixture of malic acid (26.4 mg, 0.197 mmol) and /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg, 0.197 mmol) and the mixture heated on a mini-block with heating-cooling cycles from 55 to 5 °C for 7 repeat cycles (heating rate: 1 .5 °C/min, cooling rate: 0.25 °C/min). The white solid was collected by centrifugation and dried for 18 h at 40 °C to give the title salt.
Tartrate with 1 :0.5 stoichiometry (mw 581 .72), mp (DSC) 176.7 °C (onset). A solution of tartaric acid (75.7 mg) in methanol (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M tartaric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h. The white solid was then collected by filtration, washing 2x with methanol (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Tartrate with 1 :1 stoichiometry (mw 656.66), mp (DSC) 169.9 °C (onset): A solution of tartaric acid (75.7 mg) in acetone (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M tartaric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Citrate with 1 :0.5 stoichiometry (mw 602.73), mp (DSC) 168.4 °C (onset): A solution of citric acid (96.9 mg) in methanol (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in methanol solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with
stirring for 2 h. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Citrate with 1 :1 stoichiometry (mw 698.70), mp (DSC) 168.8 °C (onset): A solution of citric acid (96.9 mg) in acetone (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in acetone (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h before slowly cooling to room temperature. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Alternatively, N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (6.5 g, 12.83 mmol) was placed in a 500ml 4-flask reactor. 49 ml of glacial acetic acid was added and the resulting suspension was stirred at 23 °C until a clear mixture was obtained. In a separate flask, anhydrous 2-hydroxypropane-1 ,2,3-tricarboxylic acid (2.59 g, 13.47 mmol, 1 .05 equiv.) was dissolved in 49 ml of glacial acetic acid at 50 °C until a clear solution was obtained. This solution was then added at 23°C to the N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide solution previously prepared. This mixture was stirred for 30 min at 23 °C and then added dropwise over 1 h to 192 ml of ethyl acetate warmed to 75 °C. The temperature remained constant over the addition. At the end of the addition, the temperature of the mixture was cooled slowly to 23 °C and let 16h at this temperature under gentle stirring. The suspension was cooled to 5-10 °C and filtered. The cake was washed with 15 ml of ethyl acetate and 15 ml of acetone. The wet cake (ca 8.5g) was transferred in a 500 ml flask containing 192 ml of dry acetone. The resulting suspension was refluxed for 24h. The suspension was filtered and the cake was washed with 2 times 15 ml of dry acetone then dried at 50 °C under vacuum for several hours to give the title salt.
Intermediate 80: N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7- (dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
A solution of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile (intermediate 75, 481 mg, 2.50 mmol) in anhydrous DMF (1 .5 ml) was added drop wise over 10 minutes to a mixture of di(1 H-1 ,2,4-triazol-1 -yl)methanone (410 mg, 2.50 mmol) and DMF (1 .5 ml) cooled at 0 °C. After stirring for 45 minutes at 0 °C the reaction mixture was allowed to warm to room temperature and after a further 90 minutes at room temperature a solution of 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one (intermediate 81 , 418 mg, 1 .00 mmol) in DMF (2 ml) was added. The reaction mixture was stirred for 17.5 h at room temperature, quenched by the addition of MeOH and evaporated. The residue was applied to a 80 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 2% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, DMSO-c/6) δ 13.50 (s, 1 H), 8.27 (s, 1 H), 7.52 (s, 1 H), 7.39 (s, 1 H), 6.93 (t, 1 H), 5.45 (s, 1 H), 4.65 (s, 2H), 3.94 – 3.89 (m, 2H), 3.54 – 3.50 (m, 2H), 3.40 – 3.35 (m, 2H), 3.38 (s, 6H), 3.29 (s, 3H), 3.20 – 3.16 (m, 2H), 3.05 (s, 2H), 2.86 – 2.80 (m, 2H), 2.61 – 2.55 (m, 2H), 2.22 (s, 3H), 1 .94 – 1 .88 (m, 2H). (UPLC-MS 6) tR 0.72; ESI-MS 553.3 [M+H]+.
Intermediate 81 : 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one.
Sodium triacetoxyborohydride (3.10 g, 14.61 mmol) was added to a mixture of 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde (intermediate 41 , 2.30 g, 9.74 mmol), ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride (intermediate 82, 2.6 g, 14.61 mmol) and triethylamine (6.75 ml, 48.7 mmol) in 1 ,2-dichloroethane (20 ml) at room temperature. The reaction mixture was stirred for 21 h at room temperature and additional sodium triacetoxyborohydride (2.6 g, 9.74 mmol) was added. After a further 4 h stirring at room temperature, again additional sodium triacetoxyborohydride (1 .3 g, 4.87 mmol) was added and the reaction maintained at 4 °C for 2.5 days. The reaction mixture was then warmed to room temperature, saturated aqueous NaHC03 solution added, the mixture extracted with DCM (3x), the combined organic layers dried over Na2S04 and evaporated. The residue was applied to a 120 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 10% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, CDCI3) δ 7.08 (s, 1 H), 5.30 (s, br, 1 H), 5.20 (s, 1 H), 4.69 (s, 2H), 3.44 – 3.34 (m, 2H), 3.40 (s, 6H), 3.22 – 3.15 (m, 2H), 3.24 (s, 2H), 2.71 -2.64 (m, 2H), 2.58 – 2.50 (m, 2H), 2.31 (s, 3H), 1 .98 – 1 .82 (m, 2H). (UPLC-MS 6) tR 0.33; ESI-MS 335.3 [M+H]+.
Intermediate 82: ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride.
Concentrated hydrochloric acid (10 ml) was added to a solution of ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate (intermediate 83, 3.05 g, 1 1 .13 mmol) in THF (20 ml) and EtOH (100 ml) at room temperature. After stirring 1 h at room temperature the reaction mixture was evaporated, ethanol (20 ml) added, evaporated, further ethanol (50 ml) added and then stirred at 60 °C for 70 min. The cooled reaction
mixture was then evaporated to give the title compound as a pale-yellow glass. 1H NMR (400 MHz, DMSO-c/6) δ 8.58 (s, br, 3H), 4.19 (q, 2H), 4.26 – 4.15 (m, 2H), 3.44 (s, br, 2H), 3.21 (s, br, 2H), 2.88 (s, 3H), 1 .21 (t, 3H).
Intermediate 83: ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate.
Ethyl bromoacetate (1 .27 ml, 1 1 .48 mmol) was added to a mixture of tert-butyl (2-(methylamino)ethyl)carbamate (2.0 g, 1 1 .48 mmol), triethylamine (4.81 ml) and THF (24 ml) at 0 °C. After stirring 24 h at room temperature the reaction mixture was partitioned between saturated aqueous NaHC03 and DCM, extracted 2x with DCM, the organic layers dried over Na2S04 and evaporated to give the title compound as a clear pale-yellow oil. 1 H NMR (400 MHz, CDCI3) δ 5.20 (s, br, 1 H), 4.18 (q, 2H), 3.24 (s, 2H), 3.22 -3.16 (m, 2H), 2.65 – 2.61 (m, 2H), 2.38 (s, 3H), 1 .42 (s, 9H), 1 .24 (t, 3H).
Intermediate 41 : 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde.
To a solution of 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine
(intermediate 12, 15.0 g, 52.2 mmol) in THF (400 ml) at -78 °C under argon, was added MeLi (1 .6 M in Et20, 32.6 ml, 52.2 mmol), the solution was stirred for 5 min, then n-BuLi (1 .6 M in hexane, 35.9 ml, 57.5 mmol) was added slowly and the solution was stirred for 20 min. THF (100 ml) was added to the reaction at – 78 °C. Subsequently, n-BuLi (1 .6 M in hexane, 49.0 ml, 78 mmol) was added and the reaction mixture was stirred for 20 min, then again n-BuLi (1 .6 M in hexane, 6.53 ml, 10.45 mmol) was added and the mixture was stirred for 10 min at – 78 °C. DMF (2.10 ml, 27.2 mmol) was added and the reaction mixture was stirred at -78 °C for 45 min, then it was allowed to warm to room
temperature, poured into sat. aq. NH4CI and extracted twice with DCM. The combined organic phases were dried over Na2S04, filtered and evaporated to give the title compound as an orange oil. (UPLC-MS 3) tR 0.63 min; ESI-MS 237.2 [M+H]+.
Intermediate 12: 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
Into a 3 I 4-necked round-bottom flask was placed 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine (intermediate 4, 1 14.6 g, 550.3mmol) in acetonitrile (2 I). This was followed by the addition of NBS (103 g, 578 mol) in portions with stirring at 25 °C. The resulting solution was stirred for 30 min at 25 °C. The resulting mixture was concentrated under vacuum and the residue was diluted with 1000 ml of diethylether. The mixture was washed with 3×100 ml of ice/water. The aqueous phase was extracted with 2×100 ml of diethylether and the organic layers were combined. The resulting mixture was washed with 1 x100 ml of brine, dried over sodium sulfate and concentrated under vacuum to give the title compound as a light yellow solid. LC-MS: (ES, m/z):
286.03 [M+H]+. 1H-NMR: (300MHz, CDCI3) δ 1 .86 – 1 .94 (2H, m), 2.70 – 2.74 (2H, m), 3.9 – 3.43 (2H, m), 3.47 (6H, s), 5.23 (1 H, s), 5.58 (1 H, s), 7.29 (1 H, s).
Intermediate 4: 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 5-I pressure tank reactor (5 atm) was placed 2-(dimethoxymethyl)-1 ,8-naphthyridine (intermediate 5, 200 g, 979 mmol), ethanol (3 I), Pt02 (12 g). The reactor was evacuated and flushed three times with nitrogen, followed by flushing with hydrogen. The mixture was stirred overnight at 23 °C under an atmosphere of hydrogen. This reaction was repeated four times. The solids were filtered out and the resulting mixture was concentrated under vacuum to give the title compound as a yellow solid.
Intermediate 5: 2-(dimethoxymethyl)-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 20 I 4-necked round-bottom flask was placed 2-aminopyridine-3-carbaldehyde (1000 g, 8.19 mol), 1 ,1 -dimethoxypropan-2-one (1257 g, 10.64 mol), ethanol (10 I), and water (2 I). This was followed by the addition of a solution of sodium hydroxide (409.8 g, 10.24 mol) in water (1000 ml) drop wise with stirring at 0-15 °C. The solution was stirred for 3 h at 0-20 °C and then concentrated under vacuum. The resulting solution was extracted with 3×1200 ml of ethyl acetate and the organic layers were combined. The mixture was dried over sodium sulfate and concentrated under vacuum. The residue was washed with 3×300 ml of hexane and the solid was collected by filtration. This resulted in the title compound as a yellow solid. 1H-NMR (400 MHz, DMSO-c/6) δ 9.1 1 (dd, 1 H), 8.53 (d, 1 H), 8.50 (dd, 1 H), 7.73 (d, 1 H), 7.67 (dd, 1 H), 5.44 (s, 1 H), 3.41 (s, 6H).
Intermediate 75: 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile.
A solution of 6-amino-4-fluoronicotinonitrile (intermediate 21 , 1 .10 g, 8.02 mmol) in DMA (20 ml) was treated with 2-methoxyethylamine (2.07 ml, 24.1 mmol) and DIPEA (4.20 ml_, 24.1 mmol), heated to 50 °C and stirred for 15 h. The reaction mixture was cooled to room temperature and concentrated. The crude material was purified by normal phase chromatography (24 g silica gel cartridge, heptanes/EtOAc 100:0 to 0:100). The product containing fractions were concentrated and dried under vacuum to give the title compound as an off-white solid.
An alternative synthesis of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile is outlined below:
To fe/ -butyl N-{5-cyano-4-[(2-methoxyethyl)amino]pyridin-2-yl}carbamate (intermediate 287, 7g) was added 30-36% aqueous HCI (40 ml), the mixture stirred at room temperature for 30 minutes and monitored by chromatography until complete conversion. The solution was then basified with 20-30% NaOH solution to pH=9-10 and filtered to give a white solid. The solid was added to ethyl acetate (15 ml) and heated to 50-55 °C to form a clear solution. The solution was then cooled to 3-6 °C, stirred for 2-3 h and filtered. The wet cake was then dried to give the title compound as a white solid. 1H NMR (400 MHz, DMSO-c/6) δ 7.92 (s, 1 H), 6.39 (s, 2H), 6.15 (t, 1 H), 5.61 (s, 1 H), 3.46 (t, 2H), 3.27 (s, 3H), 3.24 (q, 2H). (UPLC-MS 3) tR 0.62; ESI-MS 193.1 [M+H]+.
1H-NMR (400 MHz, DMSO-c/6) δ 7.14 (d, 1 H), 6.51 (d, 1 H), 6.47 – 6.41 (m, 1 H), 4.98 (s, 1 H), 3.28 – 3.19 (m, 2H), 3.23 (s, 6H), 2.64 (t, 2H), 1 .73 – 1 .79 (m, 2H).
Intermediate 21 : 6-amino-4-fluoronicotinonitrile.
4-fluoro-5-iodopyridin-2-amine (intermediate 22, 240 g, 1 mol), zinc cyanide (125 g, 1 .05 mol), zinc (13 g, 0.2 mol), Pd2(dba)3 (25 g, 25 mmol) and dppf (55 g, 0.1 mol) in DMA (800 ml) were degassed and charged into the round bottom flask under nitrogen. The mixture was stirred at 100 °C for 3 h. The reaction mixture was diluted with 5% NaHC03 (2 I), extracted with EtOAc (4 x 600 ml). The combined organic layers were washed with 5% NaOH (1 I), dried over Na2S04, concentrated to 700 ml. The resulting organic phase was eluted through silica gel column with EtOAc (1 .7 I). The combined organic filtrate was washed with 2 M HCI (3 x 800 ml). The pH of the aqueous phase was adjusted to 10 with saturated NaHC03. The aqueous phase was extracted whit DCM (3 x 500 ml). The combined DCM was dried over Na2S04 and concentrated. The residue was further purified by column chromatography (eluted with pentane: EtOAc 10:1 to 3:2) followed by recrystallization from pentane/EtOAc 3/1 to give the title compound as white solid. 1H NMR (400 MHz, DMSO-c/6) δ 8.40 (d, 1 H), 7.40 (s, 2H), 6.34 (d, 1 H).
Intermediate 22: 4-fluoro-5-iodopyridin-2-amine.
A suspension of 4-fluoropyridin-2-amine (336 g, 2.5 mol) and NIS (745 g, 2.75 mol) in MeCN (9 I) was treated with TFA (1 14 g, 1 mol). The reaction mixture was then stirred at room temperature for 8 h. The reaction mixture was diluted with EtOAc (10 I), washed with sat. aq. Na2S203 (2 x 5 I), brine (4 x 5 I). The combined organic layers were dried over Na2S04, filtered and concentrated to get the crude product. The crude product was purified by recrystallization from EtOAc/pentane (1/10) to afford the title compound as a white solid. 1H NMR (400 MHz, DMSO-c/6) δ 8.14 (d, 1 H), 6.45 (s, 2H), 6.33 (d, 1 H).
Intermediate 287: fe/ -butyl (5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)carbamate.
A mixture of tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate (intermediate 288, 9.8 g, 38.6 mmol), 2-methoxyethylamine (5.8 g, 77.3 mmol) and DIPEA (6 g, 46.4 mmol) in DMSO (80 ml) was heated at 65-70 °C for 24 h and monitored by chromatography until complete conversion. The solution was then cooled to room temperature and a white solid precipitated gradually. Water (20 ml) was then added slowly within 1 h. The suspension was stirred for a further 1 h, filtered and dried to give the title compound as a white solid. 1H NMR (DMSO-d6, 400 MHz): δ 9.87 (s, 1 H), 8.18 (s, 1 H), 7.20 (s, 1 H), 6.86 (s, 9H), 3.51 (t, 2H), 3.36 (t, 2H), 3.28 (s, 3H), 1 .47 (s, 9H).
Intermediate 288: tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate.
A mixture of 2,4-dichloro-5-cyanopyridine (10g, 57.8 mmol), fe/ -butyl carbamate (8.2 g, 70.5 mmol), Pd(OAc)2 (0.26 g, 1 .1 mmol), Xantphos (1 .34 g, 2.3mmol) and K2C03 (12 g, 87 mmol) in THF (150 ml) was degassed 3x with nitrogen. The mixture was then heated at 70 °C for 4-5 h and monitored by chromatography until complete conversion. Following completion of the reaction, additional THF (100 ml) was added and heated the mixture at 70 °C for additional 1 h and then cooled to room temperature. The suspension was then filtered through a pad of celite to remove the solid. The filtrate was then concentrated and azotropically distilled with ethyl acetete before filtering to give the title compound. 1H NMR (DMSO-d6, 400 MHz): δ 10.82 (s, 1 H), 8.79 (s, 1 H), 8.09 (s, 1 H), 1 .49 (s, 9H).
/////////////FGF 401, 1708971-55-4, PHASE 1, Hepatocellular carcinoma, Solid tumours, Novartis, Novartis Oncology, Antineoplastics, Type 4 fibroblast growth factor receptor antagonists, NVP-FGF-401, Nicole Buschmann, Robin Alec Fairhurst, Pascal Furet, Thomas Knöpfel, Catherine Leblanc, Robert Mah, Pierre NIMSGERN, Sebastien RIPOCHE, Lv LIAO, Jing XIONG, Xianglin ZHAO, Bo Han, Can Wang,
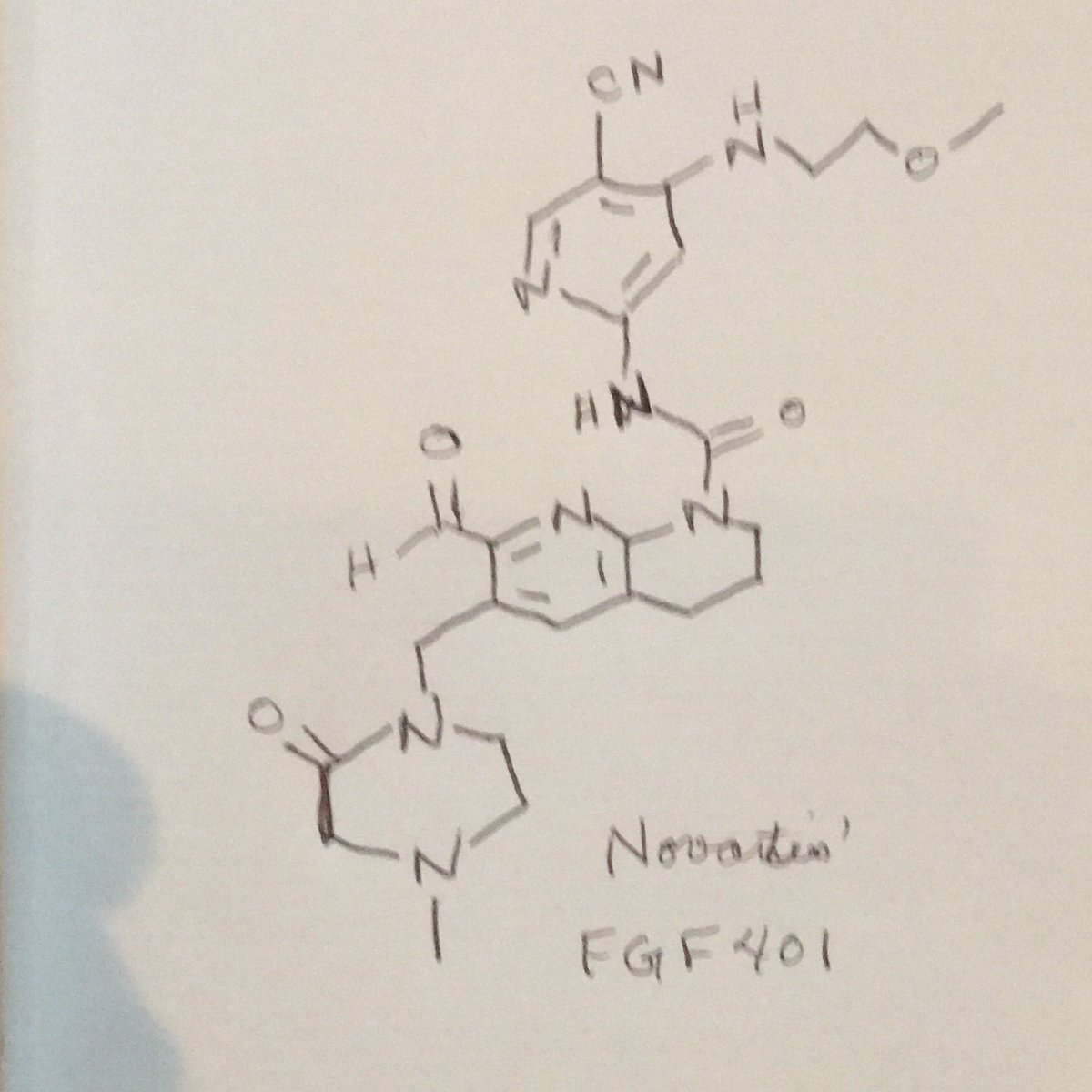
Now in #MEDI 1st time disclosures Robin Fairhurst of @Novartis will also talk about an FGFR inhibitor. They are popular! #ACSSanFran
CN4CC(=O)N(Cc1cc(C=O)nc2N(CCCc12)C(=O)Nc3cc(NCCOC)c(C#N)cn3)CC4
DAROLUTAMIDE даролутамид , دارولوتاميد , 达罗他胺 , ダロルタミド
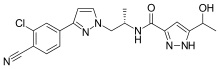

Darolutamide
N-((S)-1-(3-(3-Chloro-4-cyanophenyl)-1H-pyrazol-1-yl)-propan-2-yl)-5-(1-hydroxyethyl)-1H-pyrazole-3-carboxamide
N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)-propan-2-yl)-5-(l-hydroxyethyl)-lH-pyrazole-3-carboxamide
- MF C19H19ClN6O2
- MW 398.846
BAY 1841788; ODM-201
| ダロルタミド Darolutamide  C19H19ClN6O2 : 398.85 [1297538-32-9] |

Orion and licensee Bayer are codeveloping darolutamide (ODM-201, BAY-1841788), an androgen receptor antagonist, for the potential treatment of castration-resistant prostate cancer (CRPC) and metastatic hormone-sensitive prostate cancer (HSPC) .
In September 2014, a phase III trial (ARAMIS) was initiated for non-metastatic CRPC; in April 2018, the trial was ongoing . In November 2016, a phase III trial in metatstic HSPC (ARASENS) was initiated .
PRODUCT PATENT
US-09657003 provides patent protection until May 2032.
Priority date 2009-10-27
InventorGerd WohlfahrtOlli TörmäkangasHarri SaloIisa HöglundArja KarjalainenPia KoivikkoPatrik HolmSirpa RaskuAnniina Vesalainen Current Assignee Orion Corp Original AssigneeOrion Corp
05-May-2011 WO-2011051540-A1, Priority date 2009-10-27
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8921378 | Androgen receptor modulating carboxamides |
2012-04-20
|
2014-12-30
|
| US8975254 | ANDROGEN RECEPTOR MODULATING COMPOUNDS |
2010-10-27
|
2012-09-06
|
| US2017260206 | ANDROGEN RECEPTOR MODULATING COMPOUNDS |
2017-04-13
|
|
| US9657003 | ANDROGEN RECEPTOR MODULATING COMPOUNDS |
2015-01-16
|
2015-07-23
|
PHASE III
In September 2014, the double-blind, randomized, placebo-controlled, phase III trial (NCT02200614; ; ARAMIS) began to evaluate the safety and efficacy of darolutamide in patients (expected n = 1500, Taiwanese n = 20) in the US, Argentina, Australia, Brazil, Canada, Europe, Israel, Japan, Peru, South Korea, Russian Federation, South Africa, Taiwan and Turkey with non-metastatic CRPC. The primary endpoint was metastasis-free survival (MFS), defined as time between randomization and evidence of metastasis or death from any cause . In April 2018, the trial was expected to complete in September 2018

- Originator Orion
- Developer Bayer HealthCare; Orion
- Class Antineoplastics
- Mechanism of Action Androgen receptor antagonists
- Phase III Prostate cancer
-
Most Recent Events
- 03 Jun 2016 Bayer and Orion plan the phase III ARASENS trial for Prostate cancer
- 03 Jun 2016 Bayer and Orion expand the licensing agreement to include joint development of ODM 201 for Metastatic hormone-sensitive prostate cancer (mHSPC)
- 06 May 2016 Long-term combined adverse events data from the the ARADES (phase I/II) and the ARAFOR (phase I) trials in Prostate cancer presented at the 111th Annual Meeting of the American Urological Association (AUA -2016)
Darolutamide (INN) (developmental code names ODM-201, BAY-1841788) is a non-steroidal antiandrogen, specifically, a full and high-affinity antagonist of the androgen receptor (AR), that is under development by Orion and Bayer HealthCare[1] for the treatment of advanced, castration-resistant prostate cancer (CRPC).[2][3]
Orion and licensee Bayer are co-developing darolutamide, an androgen receptor antagonist, for treating castration-resistant prostate cancer and metastatic hormone-sensitive prostate cancer. In August 2016, darolutamide was reported to be in phase 3 clinical development. The drug appears to be first disclosed in WO2011051540, claiming novel heterocyclic derivatives as tissue-selective androgen receptor modulators, useful for the treatment of prostate cancer.
![]()
Mode of action
Relative to enzalutamide (MDV3100 or Xtandi) and apalutamide (ARN-509), two other recent non-steroidal antiandrogens, darolutamide shows some advantages.[3] Darolutamide appears to negligibly cross the blood-brain-barrier.[3] This is beneficial due to the reduced risk of seizures and other central side effects from off-target GABAA receptor inhibition that tends to occur in non-steroidal antiandrogens that are structurally similar to enzalutamide.[3] Moreover, in accordance with its lack of central penetration, darolutamide does not seem to increase testosterone levels in mice or humans, unlike other non-steroidal antiandrogens.[3] Another advantage is that darolutamide has been found to block the activity of all tested/well-known mutant ARs in prostate cancer, including the recently-identified clinically-relevant F876L mutation that produces resistance to enzalutamide and apalutamide.[3] Finally, darolutamide shows higher affinity and inhibitory efficacy at the AR (Ki = 11 nM relative to 86 nM for enzalutamide and 93 nM for apalutamide; IC50 = 26 nM relative to 219 nM for enzalutamide and 200 nM for apalutamide) and greater potency/efficaciousness in non-clinical models of prostate cancer.[3]
ORM-15341 is the main active metabolite of darolutamide.[3] It, similarly, is a full antagonist of the AR, with an affinity (Ki) of 8 nM and an IC50 of 38 nM.[3]

Clinical trials
Darolutamide has been studied in phase I and phase II clinical trials and has thus far been found to be effective and well-tolerated,[4] with the most commonly reported side effects including fatigue, nausea, and diarrhea.[5][6] No seizures have been observed.[6][7] As of July 2015, darolutamide is in phase III trials for CRPC.[3]
Representative binding affinities of ODM-201, ORM-15341, enzalutamide, and ARN-509 measured in competition with [3H]mibolerone using wtAR isolated from rat ventral prostates (C). All data points are means of quadruplicates ±SEM. Ki values are presented in parentheses. D. Antagonism to wtAR was determined using AR-HEK293 cells treated with ODM-201, ORM-15341, enzalutamide, or ARN-509 together with 0.45 nM testosterone in steroid-depleted medium for 24 hours before luciferase activity measurements. All data points are means of triplicates ±SEM. IC50 values are presented in parentheses.
WHIPPANY, N.J., Sept. 16, 2014 /PRNewswire/ — Bayer HealthCare and Orion Corporation, a pharmaceutical company based in Espoo, Finland, have begun to enroll patients in a Phase III trial with ODM-201, an investigational oral androgen receptor inhibitor in clinical development. The study, called ARAMIS, evaluates ODM-201 in men with castration-resistant prostate cancer who have rising Prostate Specific Antigen (PSA) levels and no detectable metastases. The trial is designed to determine the effects of the treatment on metastasis-free survival (MFS).
“The field of treatment options for prostate cancer patients is evolving rapidly. However, once prostate cancer becomes resistant to conventional anti-hormonal therapy, many patients will eventually develop metastatic disease,” said Dr. Joerg Moeller, Member of the Bayer HealthCare Executive Committee and Head of Global Development. “The initiation of a Phase III clinical trial for ODM-201 marks the starting point for a potential new treatment option for patients whose cancer has not yet spread. This is an important milestone for Bayer in our ongoing effort to meet the unmet needs of men affected by prostate cancer.”
Earlier this year, Bayer and Orion entered into a global agreement under which the companies will jointly develop ODM-201, with Bayer contributing a major share of the costs of future development. Bayer will commercialize ODM-201 globally, and Orion has the option to co-promote ODM-201 in Europe. Orion will be responsible for the manufacturing of the product.
About the ARAMIS Study
The ARAMIS trial is a randomized, Phase III, multicenter, double-blind, placebo-controlled trial evaluating the safety and efficacy of oral ODM-201 in patients with non-metastatic CRPC who are at high risk for developing metastatic disease. About 1,500 patients are planned to be randomized in a 2:1 ratio to receive 600 mg of ODM-201 twice a day or matching placebo. Randomisation will be stratified by PSA doubling time (PSADT less than or equal to 6 months vs. > 6 months) and use of osteoclast-targeted therapy (yes vs. no).
The primary endpoint of this study is metastasis-free survival (MFS), defined as time between randomization and evidence of metastasis or death from any cause. The secondary objectives of this study are overall survival (OS), time to first symptomatic skeletal event (SSE), time to initiation of first cytotoxic chemotherapy, time to pain progression, and characterization of the safety and tolerability of ODM-201.
About ODM-201
ODM-201 is an investigational androgen receptor (AR) inhibitor that is thought to block the growth of prostate cancer cells. ODM-201 binds to the AR and inhibits receptor function by blocking its cellular function.
About Oncology at Bayer
Bayer is committed to science for a better life by advancing a portfolio of innovative treatments. The oncology franchise at Bayer now includes three oncology products and several other compounds in various stages of clinical development. Together, these products reflect the company’s approach to research, which prioritizes targets and pathways with the potential to impact the way that cancer is treated.
About Bayer HealthCare Pharmaceuticals Inc.
Bayer HealthCare Pharmaceuticals Inc. is the U.S.-based pharmaceuticals business of Bayer HealthCare LLC, a subsidiary of Bayer AG. Bayer HealthCare is one of the world’s leading, innovative companies in the healthcare and medical products industry, and combines the activities of the Animal Health, Consumer Care, Medical Care, and Pharmaceuticals divisions. As a specialty pharmaceutical company, Bayer HealthCare provides products for General Medicine, Hematology, Neurology, Oncology and Women’s Healthcare. The company’s aim is to discover and manufacture products that will improve human health worldwide by diagnosing, preventing and treating diseases.
Bayer® and the Bayer Cross® are registered trademarks of Bayer.
SYNTHESIS
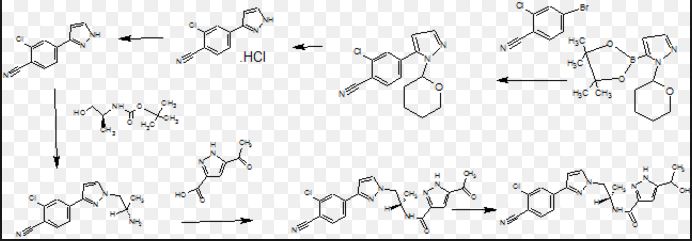
cas 1297538-32-9
Synthesis
WO 2016162604

POLYMORPH
CRYSTALLINE FORM I, I’, I” IN WO-2016120530


PATENTS
WO2011051540
https://www.google.com/patents/WO2011051540A1?cl=en
PATENT
US 2015203479
http://www.google.com/patents/WO2011051540A1?cl=en
PATENT
WO 2012143599
http://www.google.com/patents/US20140094474?cl=de
PATENT
IN 2011KO00570
PATENT
Compound of (I) (5 g) was dissolved in an acetonitrile and distilled water. The reaction mixture was heated at 75 °C and then slowly cooled down at RT and stirred at RT for 3 days. The solid obtained was filtered, washed twice with the acetonitrile: water and dried under vacuum at 40 °C and 60 °C to yield crystalline form of (I) (4.42 g) with 88% of yield (example 1, page 10).
Compound (I) can be synthetized using the procedures described in WO
201 1/051540.

Pure diastereomers (la) and (lb) can be suitably synthetized, for example, using ketoreductase enzymes (KREDs) for both S- and R-selective reduction of compound 1 to compound 2 as shown in Scheme 1, wherein R is H or Ci_6 alkyl.


Scheme 1.
For example, Codexis KRED-130 and KRED -NADH-110 enzymes are useful for obtaining excellent stereoselectivity, even stereospecificity. In Scheme 1 the starting material 1 is preferably an ester (R= Ci_6 alkyl), for example ethyl ester (R=ethyl), such as to facilitate extraction of the product into the organic phase as the compound where R=H has a tendency to remain in the water phase. Intermediate 2 can be protected, preferably with silyl derivatives such as tert-butyldiphenylsilyl, in order to avoid esterification in amidation step. In the case of R=Ci_6 alkyl, ester hydrolysis is typically performed before amidation step, preferably in the presence of LiOH, NaOH or KOH. Amidation from compound 3 to compound 5_is suitably carried out using EDCI HBTU, DIPEA system but using other typical amidation methods is also possible. Deprotection of 5 give pure diastereomers (la) and (lb).
Pyrazole ring without NH substitution is known tautomerizable functionality and is described here only as single tautomer but every intermediate and end product here can exist in both tautomeric forms at the same time.
The stereochemistry of the compounds can be confirmed by using optically pure starting materials with known absolute configuration as demonstrated in Scheme 2, wherein R=H or Ci_6 alkyl, preferably alkyl, for example ethyl. The end products of Scheme 2 are typically obtained as a mixture of tautomers at +300K 1H-NMR analyses in DMSO.


Scheme 2. Synthesis pathway to stereoisomers by using starting materials with known absolute configuration
The crystalline forms I, Γ and Γ ‘ of compounds (I), (la) and (lb), respectively, can be prepared, for example, by dissolving the compound in question in an
acetonitrile: water mixture having volume ratio from about 85: 15 to about 99: 1, such as from about 90: 10 to about 98:2, for example about 95:5, under heating and slowly cooling the solution until the crystalline form precipitates from the solution. The concentration of the compound in the acetonitrile: water solvent mixture is suitably about 1 kg of the compound in 5-25 liters of acetonitrile: water solvent mixture, for example 1 kg of the compound in 10-20 liters of acetonitrile: water solvent mixture. The compound is suitably dissolved in the acetonitrile: water solvent mixture by heating the solution, for example near to the reflux temperature, for example to about 60-80 °C, for example to about 75 °C, under stirring and filtering if necessary. The solution is suitably then cooled to about 0-50 °C, for example to about 5-35 °C, for example to about RT, over about 5 to about 24 hours, for example over about 6 to 12 hours, and stirred at this temperature for about 3 to 72 hours, for example for about 5 to 12 hours. The obtained crystalline product can then be filtered, washed, and dried. The drying is suitably carried out in vacuum at about 40 to 60 °C, for example at 55 °C, for about 1 to 24 hours, such as for about 2 to 12 hours, for example 2 to 6 hours.
The crystalline forms I, Γ and I” of compounds (I), (la) and (lb), respectively, are useful as medicaments and can be formulated into pharmaceutical dosage forms, such as tablets and capsules for oral administration, by mixing with pharmaceutical excipients known in the art.
The disclosure is further illustrated by the following examples.
Example 1. Crystallization of N-((S)- 1 -(3 -(3 -chloro-4-cyanophenyl)- 1 H-pyrazol- 1 -yl)-propan-2-yl)-5 -( 1 -hydroxyethyl)- 1 H-pyrazole-3 -carboxamide (I)
N-((iS)- 1 -(3 -(3 -chloro-4-cyanophenyl)- 1 H-pyrazol- 1 -yl)-propan-2-yl)-5 -( 1 -hydroxyethyl)-! H-pyrazole-3 -carboxamide (I) (5 g), 71.25 ml of acetonitrile, and 3.75 ml of distilled water were charged to a flask, and the mixture was heated up to 75 °C. The mixture was slowly cooled down to RT and stirred at RT for 3 days. The solid obtained was filtered and washed twice with acetonitrile: water (9.5 ml:0.5 ml). The product was dried under vacuum at 40 °C and finally at 60°C to obtain 4.42 g of crystalline title compound (yield of 88 %) which was used in X-ray diffraction study.
Example 3. Synthesis of N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)-propan-2-yl)-5-((S)- 1 -hy droxy ethyl)- lH-pyrazole-3-carboxamide (la)
a) Ethyl-5 -((S) 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxylate


HO
MgS04 x7H20 (341 mg), NADP monosodium salt (596 mg), D(+)-glucose (9.26 g) and optimized enzyme CDX-901 lyophilized powder (142 mg) were added to 0.2 mM of KH2P04 buffer (pH 7.0, 709 ml) to prepare solution I. To this solution I was added solution II which contained ethyl-5 -acetyl- 1 H-pyrazole-3 -carboxylate (8.509 g; 46.70 mmol), EtOH (28 ml) and K ED-130 (NADPH ketoreductase, 474 mg). The mixture was agitated at 30-32°C for 5.5 h (monitoring by HPLC) and allowed to cool to RT. The mixture was evaporated to smaller volume and the residue was agitated with diatomaceous earth and filtered. The mother liquid was extracted with 3×210 ml of EtOAc and dried. The solution was filtered through silica (83 g) and evaporated to dryness to give 7.40 g of the title compound. The optical purity was 100 % ee.
b) Ethyl 5-((S)-l -((tert-butyldiphenylsilyl)oxy)ethyl)- 1 H-pyrazole-3 -carboxylate


Diphenyl-tert-butyl chlorosilane (7.48 g, 27.21 mmol) was added in 26 ml of DMF to a mixture of compound of Example 3(a) (5.00 g, 27.15 mmol) and imidazole (2.81 g, 41.27 mmol) in DMF (50 ml) at RT. The mixture was stirred at RT for 24 h.
Saturated aqueous NaHC03 (56 ml) and water (56 ml) were added and the mixture was stirred at RT for 20 min. The mixture was extracted with 2×100 ml of EtOAc. Combined organic phases were washed with water (1×100 ml, 1×50 ml), dried (Na2S04), filtered and concentrated to give 10.92 g of crude title compound.
c) 5-((S)-l -((tert-Butyldiphenylsilyl)oxy)ethyl)- 1 H-pyrazole-3 -carboxylic acid


2 M NaOH (aq) (38.8 ml; 77.5 mmol) was added to a solution of the compound of Example 3(b) (10.9 g, 25.8 mmol) in 66 ml of THF. The mixture was heated up to reflux temperature. Heating was continued for 2.5 h and THF was removed in vacuum. Water (40 ml) and EtOAc (110 ml) were added. Clear solution was obtained after addition of more water (10 ml). Layers were separated and aqueous phase was extracted with 100 ml of EtOAc. Combined organic phases were dried (Na2S04), filtered and concentrated to give 9.8 g of the title compound.
d) 5-((S)- 1 -((tert-Butyldiphenylsilyl)oxy)ethyl)-N-((S)- 1 -(3-(3-chloro-4-cyano-phenyl)- 1 H-pyrazol- 1 -yl)propan-2-yl)- 1 H-pyrazole-3 -carboxamide


Under nitrogen atmosphere HBTU (0.84 g; 2.22 mmol), EDCIxHCl (3.26 g; 17.02 mmol) and (S)-4-(l-(2-aminopropyl)-lH-pyrazol-3-yl)-2-chlorobenzonitrile (3.86 g; 14.80 mmol) were added to a mixture of crude compound of Example 3(c) (8.68g; purity 77.4 area-%) and DIPEA (2.20 g; 17.02 mmol) in DCM (50 ml). The mixture was stirred at RT for 46 h (6 ml of DCM was added after 20 h). The mixture was washed with 3×20 ml of water, dried (Na2S04), filtered and concentrated to give 13.7 g of crude title compound.
e) N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((S)- 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxamide (la)
TBAF hydrate (Bu4NF x 3H20; 2.34 g; 7.40 mmol) in 10 ml of THF was added to the solution of the compound of Example 3(d) (9.43 g; 14.79 mmol) in THF (94 ml) at 0 °C under nitrogen atmosphere. Stirring was continued at RT for 21.5 h and the mixture was concentrated. DCM (94 ml) was added to the residue and the solution was washed with 3×50 ml of water, dried (Na2S04), filtered and concentrated. Crude product was purified by flash chromatography (EtOAc/n-heptane) to give 2.1 g of the title compound. 1H-NMR (400MHz; d6-DMSO; 300K): Major tautomer (-85 %): δ 1.11 (d, 3H), 1.39 (d, 3H), 4.24-4.40 (m, 2H), 4.40-4.50 (m, 1H), 6.41(s, 1H), 6.93 (d, 1H), 7.77-7.82 (m, 1H), 7.88-8.01 (m, 2H), 8.08 (s, 1H), 8.19 (d, 1H), 13.02 (broad s, 1H). Minor tautomer (-15 %) δ 1.07-1.19 (m, 3H), 1.32-1.41 (m, 3H), 4.24-4.40 (m, 2H), 4.40-4.50 (m, 1H), 6.80 (broad s, 1H), 6.91-6-94 (m, 1H), 7.77-7.82 (m, 1H), 7.88-8.01 (m, 2H), 8.05-8.09 (m, 1H), 8.31 (d, 1H), 13.10 (broad s, 1H).
Example 4. Crystallization of N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((S)- 1 -hy droxy ethyl)- lH-pyrazole-3-carboxamide (la)
N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((S)- 1 -hydroxyethyl)-lH-pyrazole-3-carboxamide (la) (5.00 g; 12.54 mmol) was mixed with 47.5 ml of ACN and 2.5 ml of water. The mixture was heated until compound (la) was fully dissolved. The solution was allowed to cool slowly to RT to form a precipitate. The mixture was then further cooled to 0 °C and kept in this temperature for 30 min. The mixture was filtered and the precipitate was dried under vacuum to obtain 4.50 g of crystalline title compound which was used in the X-ray diffraction study.
Example 6. Synthesis of N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)-propan-2-yl)-5-((R)- 1 -hy droxy ethyl)- lH-pyrazole-3-carboxamide (lb)
a) Ethyl-5 -((R)- 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxylate


Potassium dihydrogen phosphate buffer (Solution I) was prepared by dissolving potassium dihydrogen phosphate (11.350 g, 54.89 mmol) to water (333 ml) and adjusting pH of the solution to 7.0 by addition of 5 M solution of NaOH. MgS04 x 7 H20 (1.650 g), NAD monosodium salt (0.500 g), D(+)-glucose (10.880 g) and optimised enzyme CDX-901 lyophilised powder (0.200 g) were added to Solution I. To this solution (Solution II) were added KRED-NADH- 110 (0.467 g), ethyl-5-acetyl-1 H-pyrazole-3 -carboxylate (10.00 g; 54.89 mmol) and 2-methyltetrahydro-furan (16 ml). The mixture was agitated at 30° C for 11 h and allowed to cool to RT overnight. The pH of the mixture was kept at 7 by addition of 5 M solution of NaOH. The mixture was evaporated to a smaller volume. The evaporation residue was agitated for 10 min with diatomaceous earth (40 g) and activated charcoal (0.54 g), and filtered. Material on the filter was washed with water (40 ml) and the washings were combined with the filtrate. Layers were separated and aqueous phase was extracted with EtOAc (450 ml and 2×270 ml). Combined organic phases were dried over Na2S04, filtered and evaporated to dryness to give 9.85 g of the title compound (100 % ee).
b) Ethyl-5 -((R)- 1 -((tert-butyldiphenylsilyl)oxy)ethyl)- 1 H-pyrazole-3 -carboxylate


Imidazole (5.32 g; 78.08 mmol) was added to a DCM (67 ml) solution of the compound of Example 6(a) (9.85 g; 53.48). The mixture was stirred until all reagent was dissolved and tert-butyldiphenyl chlorosilane (13.21 ml; 50.80 mmol) was added to the mixture. The mixture was stirred for 1.5 h, 70 ml of water was added and stirring was continued for 15 min. Layers were separated and organic phase was washed with 2×70 ml of water and dried over Na2S04, filtered and concentrated to give 22.07 g of crude title compound.
c) 5 -((R)- 1 -((tert-Butyldiphenylsilyl)oxy)ethyl)- 1 H-pyrazole-3 -carboxylic acid


Compound of Example 6(b) (11.3 g; 26.74 mmol; theoretical yield from the previous step) was dissolved in 34 ml of THF and 50 ml of 2 M NaOH (aq.) was added. The mixture was heated under reflux temperature for 70 min. The mixture was extracted with 2×55 ml of EtOAc and combined organic phases were washed with brine, dried over Na2S04, filtered and concentrated. Evaporation residue was triturated in 250 ml of n-heptane, filtered and dried to give 17.58 g of crude title compound.
d) 5-((R)- 1 -((tert-Butyldiphenylsilyl)oxy)ethyl)-N-((S)- 1 -(3-(3-chloro-4-cyano-phenyl)- 1 H-pyrazol- 1 -yl)propan-2-yl)- 1 H-pyrazole-3 -carboxamide


A mixture of the compound of Example 6(c) (11.14 g; 26.75 mmol; theoretical yield from the previous step), 91 ml of DCM, HBTU (1.52 g; 4.01 mmol), EDCIxHCl
(5.90 g; 30.76 mmol), (S)-4-(l-(2-aminopropyl)-lH-pyrazol-3-yl)-2-chlorobenzo-nitrile (6.97 g; 26.75 mmol) and DIPEA (3.98 g; 30.76 mmol) was stirred at RT for 3 h and at 30° C for 22 h. The mixture was washed with 2×90 ml of 0.5 M HC1 and 4×90 ml of water, dried over Na2S04, filtered and concentrated. Crude product was purified by flash column chromatography (n-heptane-EtOAc) to give 16.97 g of title compound.
e) N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((R)- 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxamide (lb)
A mixture of the compound of Example 6(d) (6.09 g; 9.56 mmol), 61 ml of THF and TBAF was stirred at 40 °C for 6.5 h. The mixture was concentrated and 61 ml of EtOAc was added to the evaporation residue. Solution was washed with 2×50 ml of 0.5 M HC1 and 4×50 ml of water, dried over Na2S04, filtered and concentrated. Crude product was purified by flash column chromatography (n-heptane-EtOAc) to give 1.71 g of the title compound. 1H-NMR (400MHz; d6-DMSO; 300K): Major tautomer (~85%): 5 1.10 (d, 3H), 1.38 (d, 3H), 4.14-4.57 (m, 2H), 5.42 (d, 1H),
6.39(s, 1H), 6.86-6.98 (m, 1H), 7.74-7.84 (m, 1H), 7.86-8.02 (m, 2H), 8.08 (s, 1H), 8.21 (d, 1H), 13.04 (broad s, 1H). Minor tautomer (-15%) δ 0.95-1.24 (m, 3H), 1.25-1.50 (m, 3H), 4.14-4.57 (m, 2H), 4.60-4.90 (m, 1H), 5.08 (d, 1H), 6.78 (broad s, 1H), 6.86-6.98 (m, 1H), 7.77-7.84 (m, 1H), 7.86-8.02 (m, 2H), 8.02-8.12 (m, 1H), 8.32 (d, 1H), 13.1 1 (broad s, 1H).
Example 7. Crystallization of N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((R)- 1 -hy droxy ethyl)- 1 H-pyrazole-3 -carboxamide (lb)
N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((R)- 1 -hydroxyethyl)-l H-pyrazole-3 -carboxamide (lb) (3.7 g; 9.28 mmol) was mixed with 70 ml of ACN and 3.5 ml of water. The mixture was heated to reflux temperature until compound (lb) was fully dissolved. The solution was allowed to cool slowly. The mixture was filtered at 50 °C to obtain 6.3 mg of the precipitate. Mother liquid was cooled to 41 °C and filtered again to obtain 20.7 mg of the precipitate. Obtained mother liquid was then cooled to 36 °C and filtered to obtain 173 mg of the precipitate. The final mother liquid was cooled to RT, stirred overnight, cooled to 0 °C, filtered, washed with cold ACN: water (1 : 1) and dried to obtain 2.71 g of the precipitate. The precipitates were checked for optical purity and the last precipitate of crystalline title compound (optical purity 100 %) was used in the X-ray diffraction study.
Example 9. Synthesis of Ethyl-5 -((S) 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxylate


HO
Zinc trifluoromethanesulfonate (0.259 g; 0.713 mmol) and (S)-(-)-3-butyn-2-ol (0.25 g; 3.57 mmol) were added to 0.75 ml (5.35 mmol) of Et3N under nitrogen
atmosphere. Ethyldiazoacetate (0.45 ml; 4.28 mmol) was added slowly and the
mixture was heated at 100 °C for 2 h. The mixture was cooled to RT and 5 ml of water was added. The mixture was washed with 15 ml of DCM, 5 ml of water was added and phases were separated. Water phase was washed twice with DCM, all organic layers were combined, dried with phase separator filtration and evaporated to dryness to give 0.523 g of crude material. The product was purified by normal phase column chromatography (0-5 % MeOH:DCM) to give 0.165 mg of the title compound. 1H-NMR (400MHz; d6-DMSO; temp +300 K): Tautomer 1 (major 77%): δ 1.28 (t, 3H), 1.39 (d, 3H), 4.20-4.28 (m, 2H), (d, 1H), 4.75-4.85 (m, 1H) 5.43 (broad d, 1H), 6.54 (broad s, 1H), 13.28 (broad s, 1H). Tautomer 2 (minor 23%): δ 1.28 (t, 3H), 1.39 (d, 3H), 4.20-4.28 (m, 2H), 4.66-4.85 (m, 1H), 5.04-5.15 (broad s, 1H), 6.71 (broad s, 1H), 13.60 (broad s, 1H).
Exam le 10. Ethyl-5 -((R)- 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxylate

Zinc trifluoromethanesulfonate (1.037 g; 2.85 mmol) and (R)-(+)-3-butyn-2-ol (1.00 g; 14.27 mmol) were added to 2.98 ml (21.40 mmol) of Et3N under nitrogen atmosphere. Ethyldiazoacetate (1.80 ml; 21.40 mmol) was added slowly and then refluxed for 3 h. The mixture was cooled to RT and 45 ml of water was added. The mixture was extracted with 3×50 ml of DCM, organic layers were combined, dried with phase separator filtration and evaporated to dryness to give 2.503 g of crude material which was purified by normal phase column chromatography (0-10 % MeOH:DCM) to give 0.67 lmg of the title compound. 1H-NMR (400MHz; d6-DMSO; temp +300 K): Tautomer 1 (major 78%): δ 1.28 (t, 3H), 1.39 (d, 3H), 4.18-4.35 (m, 2H), (d, 1H), 4.75-4.85 (m, 1H) 5.42 (broad d, 1H), 6.54 (s, 1H), 13.29 (broad s, 1H). Tautomer 2 (minor 22%): δ 1.28 (t, 3H), 1.39 (d, 3H), 4.18-4.35 (m, 2H), 4.66-4.85 (m, 1H), 5.09 (broad s, 1H), 6.71 (broad s, 1H), 13.61 (broad s, 1H).
References
- “Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies.”. Sci Rep. 5: 12007. 2015. doi:10.1038/srep12007. PMC 4490394
 . PMID 26137992.
. PMID 26137992. - Fizazi K, Albiges L, Loriot Y, Massard C (2015). “ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer”. Expert Rev Anticancer Ther. 15(9): 1007–17. doi:10.1586/14737140.2015.1081566. PMID 26313416.
- Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, Nykänen PS, Törmäkangas OP, Palvimo JJ, Kallio PJ (2015). “Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies”. Sci Rep. 5: 12007.doi:10.1038/srep12007. PMC 4490394. PMID 26137992.
- “ODM-201 is safe and active in metastatic castration-resistant prostate cancer”. Cancer Discov. 4 (9): OF10. 2014. doi:10.1158/2159-8290.CD-RW2014-150. PMID 25185192.
- Pinto Á (2014). “Beyond abiraterone: new hormonal therapies for metastatic castration-resistant prostate cancer”. Cancer Biol. Ther. 15 (2): 149–55. doi:10.4161/cbt.26724.PMC 3928129. PMID 24100689.
- Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, Garcia JA, Protheroe A, Tammela TL, Elliott T, Mattila L, Aspegren J, Vuorela A, Langmuir P, Mustonen M (2014). “Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial”. Lancet Oncol. 15 (9): 975–85. doi:10.1016/S1470-2045(14)70240-2. PMID 24974051.
- Agarwal N, Di Lorenzo G, Sonpavde G, Bellmunt J (2014). “New agents for prostate cancer”. Ann. Oncol. 25 (9): 1700–9. doi:10.1093/annonc/mdu038. PMID 24658665.
External links
Fenner A. Prostate cancer: ODM-201 tablets complete phase I. Nat Rev Urol. 2015 Dec;12(12):654. doi: 10.1038/nrurol.2015.268. Epub 2015 Nov 3. PubMed PMID: 26526759.
2: Massard C, Penttinen HM, Vjaters E, Bono P, Lietuvietis V, Tammela TL, Vuorela A, Nykänen P, Pohjanjousi P, Snapir A, Fizazi K. Pharmacokinetics, Antitumor Activity, and Safety of ODM-201 in Patients with Chemotherapy-naive Metastatic Castration-resistant Prostate Cancer: An Open-label Phase 1 Study. Eur Urol. 2015 Oct 10. pii: S0302-2838(15)00964-1. doi: 10.1016/j.eururo.2015.09.046. [Epub ahead of print] PubMed PMID: 26463318.
3: Fizazi K, Albiges L, Loriot Y, Massard C. ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev Anticancer Ther. 2015;15(9):1007-17. doi: 10.1586/14737140.2015.1081566. PubMed PMID: 26313416; PubMed Central PMCID: PMC4673554.
4: Bambury RM, Rathkopf DE. Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide. Urol Oncol. 2015 Jul 7. pii: S1078-1439(15)00269-0. doi: 10.1016/j.urolonc.2015.05.025. [Epub ahead of print] Review. PubMed PMID: 26162486.
5: Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, Nykänen PS, Törmäkangas OP, Palvimo JJ, Kallio PJ. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. 2015 Jul 3;5:12007. doi: 10.1038/srep12007. PubMed PMID: 26137992; PubMed Central PMCID: PMC4490394.
6: Thibault C, Massard C. [New therapies in metastatic castration resistant prostate cancer]. Bull Cancer. 2015 Jun;102(6):501-8. doi: 10.1016/j.bulcan.2015.04.016. Epub 2015 May 26. Review. French. PubMed PMID: 26022286.
7: Bjartell A. Re: activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Eur Urol. 2015 Feb;67(2):348-9. doi: 10.1016/j.eururo.2014.11.019. PubMed PMID: 25760250.
8: De Maeseneer DJ, Van Praet C, Lumen N, Rottey S. Battling resistance mechanisms in antihormonal prostate cancer treatment: Novel agents and combinations. Urol Oncol. 2015 Jul;33(7):310-21. doi: 10.1016/j.urolonc.2015.01.008. Epub 2015 Feb 21. Review. PubMed PMID: 25708954.
9: Boegemann M, Schrader AJ, Krabbe LM, Herrmann E. Present, Emerging and Possible Future Biomarkers in Castration Resistant Prostate Cancer (CRPC). Curr Cancer Drug Targets. 2015;15(3):243-55. PubMed PMID: 25654638.
10: ODM-201 is safe and active in metastatic castration-resistant prostate cancer. Cancer Discov. 2014 Sep;4(9):OF10. doi: 10.1158/2159-8290.CD-RW2014-150. Epub 2014 Jul 9. PubMed PMID: 25185192.
11: Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, Garcia JA, Protheroe A, Tammela TL, Elliott T, Mattila L, Aspegren J, Vuorela A, Langmuir P, Mustonen M; ARADES study group. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol. 2014 Aug;15(9):975-85. doi: 10.1016/S1470-2045(14)70240-2. Epub 2014 Jun 25. PubMed PMID: 24974051.
12: Agarwal N, Di Lorenzo G, Sonpavde G, Bellmunt J. New agents for prostate cancer. Ann Oncol. 2014 Sep;25(9):1700-9. doi: 10.1093/annonc/mdu038. Epub 2014 Mar 20. Review. PubMed PMID: 24658665.
13: Pinto Á. Beyond abiraterone: new hormonal therapies for metastatic castration-resistant prostate cancer. Cancer Biol Ther. 2014 Feb;15(2):149-55. doi: 10.4161/cbt.26724. Epub 2013 Nov 1. Review. PubMed PMID: 24100689; PubMed Central PMCID: PMC3928129.
14: Yin L, Hu Q, Hartmann RW. Recent progress in pharmaceutical therapies for castration-resistant prostate cancer. Int J Mol Sci. 2013 Jul 4;14(7):13958-78. doi: 10.3390/ijms140713958. Review. PubMed PMID: 23880851; PubMed Central PMCID: PMC3742227.
15: Leibowitz-Amit R, Joshua AM. Targeting the androgen receptor in the management of castration-resistant prostate cancer: rationale, progress, and future directions. Curr Oncol. 2012 Dec;19(Suppl 3):S22-31. doi: 10.3747/co.19.1281. PubMed PMID: 23355790; PubMed Central PMCID: PMC3553559.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-((S)-1-(3-(3-chloro-4-cyanophenyl)-1H-pyrazol-1-yl)propan-2-yl)-5-(1-hydroxyethyl)-1H-pyrazole-3-carboxamide[1]
|
|
| Identifiers | |
| ChemSpider | 38772320 |
| UNII | X05U0N2RCO |
| Chemical data | |
| Formula | C19H19ClN6O2 |
| Molar mass | 398.85 g·mol−1 |
//////////// Bayer HealthCare, Orion, Antineoplastics, Androgen receptor antagonists, Phase III, Prostate cancer, BAY 1841788, ODM-201, даролутамид , دارولوتاميد , 达罗他胺 , دارولوتاميد , ダロルタミド
O=C(N[C@@H](C)Cn1ccc(n1)c2ccc(C#N)c(Cl)c2)c3cc(nn3)C(O)C

