
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

BENAZEPRIL HYDROCHLORIDE SYNTHESIS AND REVIEW

BENAZEPRIL HYDROCHLORIDE, CAS NO 86541-74-4
Benazepril, brand name Lotensin (Novartis), is a medication used to treat high blood pressure (hypertension), congestive heart failure, and chronic renal failure. Upon cleavage of its ester group by the liver, benazepril is converted into its active form benazeprilat, a non-sulfhydryl angiotensin-converting enzyme (ACE) inhibitor.
Dosage forms
Benazepril is available as oral tablets, in 5-, 10-, 20-, and 40-mg doses.
Benazepril is also available in combination with hydrochlorothiazide, under the trade name Lotensin HCT, and with amlodipine(trade name Lotrel).

Side effects
Most commonly, headaches and cough can occur with its use. Anaphylaxis, angioedema and hyperkalemia, the elevation of potassium levels, can also occur.
Benazepril may cause harm to the fetus during pregnancy.
According to coverage of the study on WebMD:
| “ | ACE inhibitors can pose a potential threat to kidneys as well. The key question was whether damaged kidneys would worsen if patients took ACE inhibitors. In a nutshell, concerns centered on blood levels of potassium andcreatinine, waste products that are excreted by the kidneys. Testing creatinine levels in the blood is used as a way to monitor kidney function (…) kidney problems worsened more slowly in those taking Lotensin. Overall, there were no major differences in side effects between patients taking Lotensin or the placebo.[2] | ” |
This study marks the first indication that benazepril, and perhaps other ACE inhibitors, may actually be beneficial in the treatment of hypertension in patients with kidney disease.
The Benazepril hydrochloride, with the CAS registry number 86541-74-4, is also known as (3S)-3-(((1S)-1-Carboxy-3-phenylpropyl)amino)-2,3,4,5-tetrahydro-2-oxo-1H-1-benzazepine-1-acetic acid, 3-ethyl ester, monohydrochloride; Benazepril HCl; Cibacen; Cibacen CHF; Labopol. It belongs to the product categories of Intermediates & Fine Chemicals; Pharmaceuticals; Amines; Aromatics; Heterocycles. This chemical’s molecular formula is C24H29ClN2O5 and molecular weight is 460.96. What’s more, its IUPAC name 2-[(3S)-3-[[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino]-2-oxo-4,5-dihydro-3H-1-benzazepin-1-yl]acetic acid hydrochloride. In addition, Benazepril hydrochloride (CAS 86541-74-4) is crystalline solid which is soluble in DMSO. It is used in high blood pressure and congestive heart failure. When you are using this chemical, you should not breathe dust and avoid contact with skin and eyes.
Veterinary use
Under the brand names Fortekor (Novartis) and VetACE (Jurox Animal Health), benazepril hydrochloride is used to treat congestive heart failure in dogs and chronic renal failure in dogs and cats.
- ^ Hou F, Zhang X, Zhang G, Xie D, Chen P, Zhang W, Jiang J, Liang M, Wang G, Liu Z, Geng R (2006). “Efficacy and safety of benazepril for advanced chronic renal insufficiency”. N Engl J Med 354 (2): 131–40. doi:10.1056/NEJMoa053107. PMID 16407508.
- ^ a b Hitti, Miranda; Chang, Louise (January 11, 2006). “Drug May Treat Advanced Kidney Disease”. WebMD. Retrieved 2006-09-07.

| Benazepril hydrochloride, TWT-8154, CGS-14824A, Cibacene, Briem, Cibacen, Lotensin | |
| 1-Carboxymethyl-3(S)-[1(S)-ethoxycarbonyl-3-phenylpropylamino]-2,3,4,5-tetrahydro-1H-1-benzazepin-2-one monohydrochloride; 3(S)-[1(S)-Ethoxycarbonyl-3-phenylpropylamino]-2-oxo-2,3,4,5-tetrahydro-1-benzazepine-1-acetic acid monohydrochloride | |
| 【CAS】 | 86541-74-4, 86541-75-5 (free base) |
| MF | C24-H28-N2-O5.Cl-H |
| MW | 460.9551rot–[Alpha] 20 D -141.0 °. (C = 0.9, ethanol) |
| Cardiovascular Drugs, Hypertension, Treatment of, Angiotensin-I Converting Enzyme (ACE) Inhibitors | |
| Launched-1990 | |
| Novartis (Originator), Pierre Fabre (Licensee), Andrx (Generic), Eon Labs (Generic), KV Pharmaceutical (Generic), Mylan (Generic) |

Above Preparation of Benazepril hydrochloride (CAS 86541-74-4): The reaction of 2(R)-hydroxy-4-phenyl butyric acid ethyl ester (I) with trifluoromethanesulfonic anhydride in dichloromethane gives the corresponding triflate (II), which is then condensed with the amino benzazepinone (III) by means of NMM in the same solvent to provide the target benazepril.

ABOVE SCHEME-EP 1891014 B1
BACKGROUND
-
Benazepril (CAS REGISTRY No. 86541-75-5) first disclosed inUS 4,410,520 is one of the well-known ACE inhibitors and is used for the treatment of hypertension.
-
Chemically, Benazepril, is (3S)-1-(carboxymethyl-[[(1(S)-1-(ethoxycarbonyl)-3-phenylpropyl]amino]-2,3,4,5-tetrahydro-1H-[1]benzazepine-2-one.
-
Benazepril is administered orally in the form of hydrochloride salt (CAS REGISTRY No. 86541-74-4) represented by formula (I).

-
The preparation of benazepril disclosed in US 4,410,520 , J. Med. Chem. 1985, 28, 1511-1516, and Helvetica Chimica Acta (1988) 71, 337-342, as given in scheme 1, involves reductive amination of ethyl 2-oxo-4-phenyl butyrate (IV) with sodium salt of (3S)-3-amino-1-carboxymethyl-2,3,4,5-tetrahydro-1 H-benzazepin-2-one (III).

-
In example 12 of US 4,410,520 , the crude benazepril (II) obtained in a diastereomeric ratio of SS: SR=70:30 was dissolved in dichloromethane and treated with HCl gas to obtain benazepril hydrochloride. The benazepril hydrochloride of formula (I) obtained as a foam was crystallized from methyl ethyl ketone to obtain in a SS: SR=95:5 diastereomeric ratio. Benazepril hydrochloride was further purified by recrystallization from a mixture of 3-pentanone/methanol (10:1), melting point: 188-190 °C.
-
Alternatively, in example 27 of US 4,410,520 , benazepril hydrochloride was purified by refluxing in chloroform, filtering, and washing first with chloroform and then with diethyl ether. The melting point of benazepril hydrochloride obtained as per this example is 184-186 °C.
-
An alternative process disclosed in US 4,785,089 involves nucleophilic substitution of (3S)-3-amino-1-t-butoxycarbonylmethyl-2,3,4,5-tetrahydro-1H-benzazepine-2-one (V), using the chiral substrate ethyl (2R)-2-(4-nitrobenzenesulfonyl)-4-phenyl butyrate (VI) in presence of N-methylmorpholine (scheme 2). The benazepril t-butyl ester (IIa) obtained in a diastereomeric ratio of SS: SR=96:4 was hydrolyzed to benazepril (II) and converted to hydrochloride salt by treating with HCl gas in ethyl acetate. The crystalline suspension of benazepril hydrochloride in ethyl acetate was diluted with acetone and filtered to obtain in a diastereomeric ratio of SS: SR=99.1:0.9. Further purification by refluxing in ethyl acetate afforded benazepril hydrochloride in a diastereomeric ratio of SS: SR=99.7:0.3, melting point of 181 °C.
-
The above documents do not disclose the crystalline form of benazepril hydrochloride obtained by following the purification processes disclosed in the examples.
-
The Merck Index., 12th edition reports benazepril hydrochloride crystals obtained from 3-pentanone+methanol (10:1), melting point 188-190 °C
-
The crystallization methods taught in the prior art does not consistently produce a constant diastereomeric composition of SS:SR diastereomer. This is evident from the variation in the melting points of the benazepril hydrochloride reported in three different working examples, which varies between 181 to 190°C.
-
The variation in diastereomeric composition of a pharmaceutical substance is not desirable as it would affect its efficacy. Hence there is a need for a crystallization process that consistently produce a constant diastereomeric composition of SS diastereomer in greater than 99.8%.
-
Coming to the crystalline form, it is well known in the art that the solid form of a pharmaceutical substance affect the dissolution rate, solubility and bioavailability. The solid form may be controlled by process employed for the manufacture of the pharmaceutical substance. In particular the process of purification of the solid substance by crystallization is used to control the solid form (Organic Process Research & Development, 2003, 7, 958-1027).
-
It has been found that the crystalline form of benazepril hydrochloride obtained from processes of prior art documents is designated as crystalline Form A as evident from the following documents.
-
In a monograph published by Al-badar et al in Profiles of Drug Substances, Excipients, and Related Methodology, Vol. 31, 2004, p117-161; benazepril hydrochloride prepared by the process disclosed in US 4,410,520 , and J. Med. Chem. 1985, 28, 1511-1516, has been characterized by powder X-ray diffraction pattern having 2θ peaks at 6.6, 9.9, 11.9, 13.7, 14.0, 14.9, 15.3, 16.4, 17.3, 18.9, 19.6, 20.2, 20.9, 21.5, 22.2, 25.2, 25.5, 26.4, 26.6, 27.1, 27.9, 29.8, 30.4, 31.0, 32.6, 33.3, 33.8, 34.4, 35.5, 38.2, 39.9, 43.9, 48.9.
-
The major peaks are at 6.6, 9.9, 11.9, 13.7, 14.9, 16.4, 17.3, 18.9, 19.6, 20.2, 20.9, 21.5, 25.2, 25.5, 26.4, 26.6, 27.9, 31.0, and 32.6.
-
WO 2004/013105 A1 also discloses that by following the processes of the prior art mentioned above, crystalline benazepril hydrochloride is isolated in a form designated as Form A having a powder X-ray diffraction pattern with 2θ values at 6.7, 10.1, 12.0, 13.8, 15.1, 16.4, 17.4, 19.0, 19.6, 20.2, 20.9, 21.0, 25.3, 25.5, 26.4, 26.6, 27.6, 28.0, 31.0, 32.7.
-
WO 2004/013105 A1 discloses that benazepril hydrochloride Form A may be prepared from a concentrated solution of the benazepril hydrochloride in a solvent selected from C1-C10 alcohol, N,N-dimethylformamide, N-methylpyrrolidone by adding an anti-solvent selected from C4-C12 alkane or C1-C10 acetate, preferably, hexane or ethyl acetate.
-
WO 2004/013105 A1 in Example 5 describes a process of making crystalline form A of benazepril hydrochloride by passing HCl gas into a solution of benazepril free base in diethyl ether and filtering the resulting suspension.
-
Similarly, in Example 6, the benazepril hydrochloride was dissolved in water free ethanol and the resulting solution was added to heptane at 20° C to obtain the crystalline Form A.
-
Further, WO 2004/013105 A1 , mentions a list of solvents and anti-solvents that can be used to make benazepril hydrochloride crystalline Form A. However, there is no enabling disclosure and the document is silent on the diastereomeric purity of the crystalline form A obtainable by the process disclosed.
-
The processes of crystallization and/or recrystallization disclosed in the prior art do not consistently produce benazepril hydrochloride with constant diasteromeric content as evident from the variation in the melting point of the crystalline benazepril hydrochloride obtained from crystallization from various solvents.



SYNTHETIC SCHEMES
| Benzazepin-2-ones, process for their preparation, pharmaceutical preparations containing these compounds and the compounds for therapeutical use | |
| Watthey, J.W.H. (Novartis AG) | |
| EP 0072352; GB 2103614; JP 8338260 | |
 |
|
| The reaction of 2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (I) with PCl5 in hot xylene gives 3,3-dichloro-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (II), which is treated with sodium acetate and reduced with H2 over Pd/C in acetic acid yielding 3-chloro-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (III). The reaction of (III) with sodium azide in DMSO affords 3-azido-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (IV), which is condensed with benzyl bromoacetate (V) by means of NaH in DMF giving 3-azido-1-(benzyloxycarbonylmethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VI). The treatment of (VI) with Raney-Ni in ethanol-water yields 3-amino-1-(benzyloxycarbonylmethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VII), which is debenzylated by hydrogenation with H2 over Pd/C in ethanol affording 3-amino-1-(carboxymethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VIII). Finally, this compound is condensed with ethyl 3-benzylpyruvate (IX) by means of sodium cyanoborohydride in methanol acetic acid. | |
| Process for the preparation of benazepril | |
| Kumar, Y.; De, S.; Thaper, R.K.; Kumar, D.S.M. (Ranbaxy Laboratories Ltd.) | |
| WO 0276375 | |
 |
|
| The reaction of 2(R)-hydroxy-4-phenyl butyric acid ethyl ester (I) with trifluoromethanesulfonic anhydride in dichloromethane gives the corresponding triflate (II), which is then condensed with the amino benzazepinone (III) by means of NMM in the same solvent to provide the target benazepril. | |
| CGS-14824 A | |
| Casta馿r, J.; Serradell, M.N. | |
| Drugs Fut 1984,9(5),317 | |
|
|
| The reaction of 2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (I) with PCl5 in hot xylene gives 3,3-dichloro-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (II), which is treated with sodium acetate and reduced with H2 over Pd/C in acetic acid yielding 3-chloro-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (III). The reaction of (III) with sodium azide in DMSO affords 3-azido-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (IV), which is condensed with benzyl bromoacetate (V) by means of NaH in DMF giving 3-azido-1-(benzyloxycarbonylmethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VI). The treatment of (VI) with Raney-Ni in ethanol-water yields 3-amino-1-(benzyloxycarbonylmethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VII), which is debenzylated by hydrogenation with H2 over Pd/C in ethanol affording 3-amino-1-(carboxymethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VIII). Finally, this compound is condensed with ethyl 3-benzylpyruvate (IX) by means of sodium cyanoborohydride in methanol acetic acid. | |
| Synthesis of 14C-labeled 3-([1-ethoxycarbonyl-3-phenyl-(1S)-propyl]amino)-2,3,4,5-tetrahydro-2-oxo-1H-1-(3S)-benzazepine-1-acetic acid hydrochloride ([14C]CGS 14824A) | |
| Chaudhuri, N.K.; Patera, R.; Markus, B.; Sung, M.-S. | |
| J Label Compd Radiopharm 1987,24(10),1177-84 | |
 |
|
| A new synthesis of CGS-14824A is given: The reaction of 3-bromo-1-phenylpropane (I) with KCN gives 4-phenylbutyronitrile (II), which is hydrolyzed to the corresponding butyric acid (III). The cyclization of (III) with polyphosphoric acid affords 1-tetralone (IV), which is brominated to 2-bromo-1-tetralone (V) and treated with hydroxylamine to give the oxime (VI). The Beckman rearrangement of (VI) yields 3-bromo-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VII), which is treated with sodium azide to afford the azide derivative (VIII). The N-alkylation of (VIII) with ethyl bromoacetate (IX) by means of KOH and tetrabutylammonium bromide in THF gives the N-alkylated azide (X), which is reduced by catalytic hydrogenation to the corresponding amine (XI). The hydrolysis of the ester group of (XI) with NaOH yields the free acetic acid derivative (XII), which is finally reductocondensed with ethyl 2-oxo-4-phenylbutyrate (XIII) by means of sodium cyanoborohydride. | |

US 6548665 B2– above

see translated vesrsion————-First, 2,3,4,5 – tetrahydro-1H-[1] azepin-2 phenyl – one (2) Preparation of
the dry reaction flask, add α- tetralone 20g (0.137mol), stacked acid 7.36g (0.171mol) and chloroform 140ml, was stirred at 40 ℃ in 1h concentrated sulfuric acid was slowly added dropwise 36ml, acid layer was separated and poured into 900ml water to give a creamy solid. Recrystallization with hot water to give white crystals (2) 15.5g (70%), mp141 ℃. (Acidic filtrate and after a small amount of product can be obtained.)
Second, 3,3 – dichloro-2, 3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one (3) of the prepared
in a dry reaction flask, (2) 48.3g (0.3mol) and xylene solution of 1300ml, phosphorus pentachloride 188g (0.9mol), stirred and gradually heated to at 0.5h 90 ℃, (Caution! When phosphorus pentachloride dissolved hydrogen chloride gas had severe.) 90 ℃ the reaction was continued for 0.5h, filtered to remove a small amount of suspended solids, solvent recovery under reduced pressure, to the residue was added saturated sodium bicarbonate solution, 100ml, stirred until a solid precipitate complete, filtered and the cake washed with ethanol (100ml × 2), diethyl ether (50ml) and dried to give (3) 69.0g (90%), mp185 ~ 187 ℃.
III.3 – chloro-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one (4) Preparation of
the reaction flask (3) 10g (0.087mol), Sodium acetate 77g (0.11mol), acetic acid 460ml and 5% Pd-C 0.86g, under atmospheric pressure at room temperature to a hydrogen-absorbing up total of 950ml (about 0.5h). Filtration, recycling the catalyst recovered solvent, the residue was dried under reduced pressure, and then added 900ml of 10% sodium bicarbonate solution and dichloromethane 300ml, stirring, standing, the organic layer was separated and the aqueous layer extracted with dichloromethane (300ml × 3) extracted organic layers were combined, dried over anhydrous sodium sulfate, the solvent recovered under reduced pressure. Diethyl ether was added to the cured 350ml, and mashed, filtered and dried to give (4) 8.19g (95%), mp163 ~ 167 ℃.
4 (3) – azido-2, 3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one (5) Preparation of
the dry reaction flask (4) 15.9g ( 0.08mol), sodium azide 6.4g (0.10mol) and 320ml solution of dimethyl sulfate, the reaction was stirred at 80 ℃ 3h, cooled to room temperature, poured into ice-water (1L) to precipitate a pale yellow solid , filtered and dried under reduced pressure at 75 ℃ to give (5) 14.7g (90%), mp142 ~ 145 ℃.
V.3 – azido-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one-1 – acetate (6) Preparation of
the dry reaction flask, (5) 3.0g (0.015mol), tetrabutylammonium bromide, 0.5g (0.0015 mol), powdered potassium hydroxide 1.1g (0.016mol) and 30ml of tetrahydrofuran solution of ethyl bromoacetate was added 1.9ml ( 0.016mol), stirred rapidly at room temperature for 1.5h (nitrogen). Water was added: dichloromethane (50:100 ml), stirred, allowed to stand, the organic layer separated. Washed with water, dried over anhydrous sodium sulfate, the solvent recovered under reduced pressure to give a pale yellow oil (6) 4.1g (96%) (can be used directly in the next step).
VI.3 – amino-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one-1 – acetate (7) Preparation of
the dry reaction flask, (6 ) 20.0g (0.070mol), ethanol 100ml, 10% Pd-C 1.0g stirring, at room temperature, 303.9kPa hydrogenated under a hydrogen pressure 1.5h, intermittent deflated to remove the generated nitrogen gas, after the reaction was collected by filtration Pd / C, recovery of solvents under reduced pressure to give a yellow oil, add ether l00ml, mashed, filtered and dried to give a white solid (7) 17.0g (93%) mp101 ~ 102 ℃.
Seven, (3S) -3 – amino-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one-1 – acetate (8) Preparation of
the reaction flask, adding (7) 25.1g (0.096mol), L – tartaric acid 14.4g (0.096mol) and hot ethanol 200ml, stirring to dissolve, cooled at room temperature overnight, filtered and dried under reduced pressure to give a white powder 30.7g, with ethanol Recrystallization twice (each 200ml), to give (8) tartaric acid salt of 13.6g (34%), mp168 ~ 169 ℃, with 10% ammonium hydroxide, to give a white solid (8) 8.0g (95%) mp104 ~ 106 ℃.
Eight, (3S) -3 – amino-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one-1 – acetate (9) Preparation of
the reaction flask, (8) 4.0g (0.056mol) and 150ml of methanol solution of sodium hydroxide 2.1g (0.056moI) and a solution of 5ml of water, stirred at room temperature for 2h, the solvent recovered under reduced pressure, the residue was dried and diethyl ether was added 100ml, trace broken, filtered, and dried to give (9) 12.9g (89%) (used directly in the next step).
IX benazepril (1) Synthesis of
the reaction flask (9) 12.9g (0.050mol), 2 – oxo-4 – phenylbutyrate 31.0g (0.15mol), acetic acid and 100ml methanol 75ml, the reaction was stirred at room temperature for 1h (nitrogen). Of sodium borohydride cyanide was slowly added dropwise 3.8g (0.062mol) and 30ml of methanol solution of (4h was completed within), stirred overnight, heat. Concentrated hydrochloric acid 10ml, 1h stirring at room temperature, the solvent was recovered under reduced pressure, water was added to the residue and diethyl ether 400ml l00ml, dissolved with concentrated ammonium hydroxide and the pH adjusted to 9.3, the organic layer was separated and the aqueous layer acidified with concentrated hydrochloric to pH 4.3, extracted with ethyl acetate (100ml × 3) extracted organic layers were combined, dried over anhydrous magnesium sulfate, the solvent recovered under reduced pressure, to the residue was added methylene chloride (150ml) to dissolve. And pass into dry hydrogen chloride after 5min recovered solvent under reduced pressure, to the residue was added hot ethyl ketone 100ml, stirring to dissolve, cooled and precipitated solid was filtered to give crude product (1). A 3 – amyl ketone / methanol (volume ratio 10:1) (110ml) was recrystallized (1) 5.8 g, mp 188 ~ 190 ℃, [alpha] D 20 -141.0 (C = 0.9, C 2 H 5 OH )
[Spectral Data] (free base) [2]
MS: m / Z (%) 424 (M + , 2), 351 (100), 190 (22), 91 (65)
] [other synthetic routes
described in the reference literature.
[References]
[1] Briggs LH et al. J Chem Soc, 1937, 456
[2] Watthey WH et al. J Med Clmm, 1985, 28:1511
[3] EP 1986, 206933 (CA, 1987, 107: 77434e)
[4] EP 1983, 72352 (CA, 1983, 99:53621 d)
[5] package insert: Lotensin
[6] property protection case I: Lotensin
[7] property protection case II: Lotensin
[8] Drug Monograph information: BENAZEPRIL
more info
Partition Coefficient.
Gas Chromatography.
High Performance Liquid Chromatography.
Ultraviolet Spectrum.
Clarke’s Analysis of Drugs and Poisons
Watthey, J.W.H. et al.: J. Med. Chem. (JMCMAR) 28, 1511 (1985).
US 4 410 520 (Ciba-Geigy; 18.10.1983; prior. 11.8.1981, 9.11.1981, 19.7.1982).
EP 72 352 (Ciba-Geigy; appl. 5.8.1982; USA-prior. 11.8.1981, 9.11.1981).
A significant number of new specialty medications are on track to be approved in 2013, and some will provide increased competition in certain therapy classes.

read all at
………

Aimee Tharaldson, PharmD, is a senior clinical consultant in the emerging therapeutics department at Express Scripts. She is responsible for monitoring and analyzing the specialty pharmaceutical pipeline. The emerging therapeutics department produces several proprietary reports on the pipeline for use by Express Scripts’ employees and clients. It is also responsible for the safety program that alerts patients, physicians, and clients to important information regarding serious drug safety alerts and market withdrawals. She contributes to Express Scripts’ Drug Trend Report and plays a key role in developing and maintaining Express Scripts’ specialty drug list. She received her doctor of pharmacy degree from the University of Minnesota, College of Pharmacy, and completed a pharmacy practice residency at the Minneapolis VA Medical Center. –
Sanofi to withdraw the lixisenatide New Drug Application (NDA) in the U.S., The company plans to resubmit the NDA in 2015, after completion of the ELIXA CV study.
lixisenatide
Sanofi Provides Update on Lixisenatide New Drug Application in U.S.
Paris, France – September 12, 2013 – Sanofi (EURONEXT: SAN and NYSE: SNY) announced today its decision to withdraw the lixisenatide New Drug Application (NDA) in the U.S., which included early interim results from the ongoing ELIXA cardiovascular (CV) outcomes study. The company plans to resubmit the NDA in 2015, after completion of the ELIXA CV study.
The decision to withdraw the lixisenatide application follows discussions with the U.S. Food and Drug Administration (FDA) regarding its proposed process for the review of interim data. Sanofi believes that potential public disclosure of early interim data, even with safeguards, could potentially compromise the integrity of the ongoing ELIXA study. Sanofi’s decision is not related to safety issues or deficiencies in the NDA………………………read all at
http://www.pharmalive.com/sanofi-pulls-diabetes-drug-nda
EU

FDA Study: Some Imported Spices Contaminated With Salmonella
FDA Study: Some Imported Spices Contaminated With Salmonella
Donna Pierce was a 69-year-old grandmother who loved to laugh and thought that laughter would add years to her life.
Unfortunately, Pierce was one of 87 people who contracted Salmonella Rissen between 2008 and 2009, and she subsequently died from the infection after spending the last month of her life in the hospital.
An investigation into the source of the outbreak pinpointed Salmonella-contaminated white pepper that had been processed by U.F. Union facility in Union City, CA, and originally imported from Vietnam.
This outbreak, along with two other large-scale outbreaks related to Salmonella-contaminated spices between 2007 and 2010, prompted FDA to begin a major investigation into spice safety.
Since imported spices account for more than 80 percent of the U.S. supply, they were an important part of FDA’s investigation and, in a study released in June, the agency found that
View original post 114 more words
FDA Approves Botox Cosmetic to Improve the Appearance of Crow’s Feet Lines
WEDNESDAY, September 11, 2013 — The U.S. Food and Drug Administration today approved a new use for Botox Cosmetic (onabotulinumtoxinA) for the temporary improvement in the appearance of moderate to severe lateral canthal lines, known as crow’s feet, in adults. Botox Cosmetic is the only FDA approved drug treatment option for lateral canthal lines.
The FDA approved Botox Cosmetic in 2002 for the temporary improvement of glabellar lines (wrinkles between the eyebrows, known as frown lines), in adults. Botox Cosmetic works by keeping muscles from tightening so wrinkles are less prominent
READ ALL AT

BOTOX Cosmetic (onabotulinum toxin A) For Injection, is a sterile, vacuum-dried purifiedbotulinum toxin type A, produced from fermentation of Hall strain Clostridium botulinumtype A grown in a medium containing casein hydrolysate, glucose, and yeast extract, intended for intramuscular use. It is purified from the culture solution by dialysis and a series of acid precipitations to a complex consisting of the neurotoxin, and several accessory proteins. The complex is dissolved in sterile sodium chloride solution containing Albumin Human and is sterile filtered (0.2 microns) prior to filling and vacuum-drying.
The primary release procedure for BOTOX Cosmetic uses a cell-based potency assay to determine the potency relative to a reference standard. The assay is specific to Allergan’s products BOTOX and BOTOX Cosmetic. One Unit of BOTOX Cosmetic corresponds to the calculated median intraperitoneal lethal dose (LD50) in mice. Due to specific details of this assay such as the vehicle, dilution scheme and laboratory protocols, Units of biological activity of BOTOX Cosmetic cannot be compared to nor converted into Units of any other botulinum toxin or any toxin assessed with any other specific assay method. The specific activity of BOTOX Cosmetic is approximately 20 Units/nanogram of neurotoxin protein complex.
Each vial of BOTOX Cosmetic contains either 50 Units of Clostridium botulinum type A neurotoxin complex, 0.25 mg of Albumin Human, and 0.45 mg of sodium chloride; or 100 Units of Clostridium botulinum type A neurotoxin complex, 0.5 mg of Albumin Human, and 0.9 mg of sodium chloride in a sterile, vacuum-dried form without a preservative.
Since the approval of BOTOX® Cosmetic by the U.S. Food and Drug Administration in 2002, Allergan has virtually changed the face of medical aesthetics. Men and women between the ages of 18 to 65 now have the ability to choose science-based, non-invasive medical aesthetic solutions, including BOTOX® Cosmetic and the JUVÉDERM® family of dermal fillers, to achieve their own results. Over the last seven years, there have been nearly 11.8 million BOTOX® Cosmetic treatments recorded in the United States.1 More importantly, its 97 percent satisfaction rating (survey of 117 patients)2,3 is just one indication of the trust consumers have placed in Allergan.
BOTOX® Cosmetic is a simple, non-surgical procedure for temporarily reducing the appearance of moderate to severe glabellar lines – the vertical frown lines between the eyebrows that look like an “11” – in adult women and men aged 18 to 65. BOTOX® Cosmetic reduces the activity of the muscles that cause the “11s” to form by blocking nerve impulses that trigger wrinkle-causing muscle contractions, creating an improved appearance between the brows. Results can last up to four months and may vary with each patient. Ask your doctor if BOTOX® Cosmetic is right for you.
Gilead Submits New Drug Application to U.S. FDA for Idelalisib for the Treatment of Indolent Non-Hodgkin’s Lymphoma
CAL 101, IDELALISIB
SEPT 2013
Gilead Submits New Drug Application to U.S. FDA for Idelalisib for the Treatment of Indolent Non-Hodgkin’s Lymphoma
FOSTER CITY, Calif.–(BUSINESS WIRE)–Sep. 11, 2013– Gilead Sciences, Inc. today announced that the company has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of idelalisib, an investigational, targeted, oral inhibitor of PI3K delta, for the treatment of indolent non-Hodgkin’s lymphoma (iNHL). The data submitted in this NDA support the use of idelalisib for patients with iNHL that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy.
read all at
Idelalisib ….US FDA Accepts NDA for Gilead’s Idelalisib for the Treatment of Refractory Indolent Non-Hodgkin’s Lymphoma
JANUARY 14, 2014 8:35 AM / LEAVE A COMMENT

An antineoplastic agent and p110delta inhibitor
(S)-2-(1-(9H-purin-6-ylamino)propyl)-5-fluoro-3-phenylquinazolin-4(3H)-one
Icos (Originator)
- CAL-101
- GS-1101
- Idelalisib
- UNII-YG57I8T5M0
M.Wt: 415.43
Formula: C22H18FN7O
CAS No.: 870281-82-6
CAL-101 Solubility: DMSO ≥80mg/mL Water <1.2mg/mL Ethanol ≥33mg/mL
5-Fluoro-3-phenyl-2-[(1S)-1-(7H-purin-6-ylamino)propyl]-4(3H)-quinazolinone
idelalisib
Idelalisib (codenamed GS-1101 or CAL-101) is a drug under investigation for the treatment of chronic lymphocytic leukaemia. It is in Phase III clinical trials testing drug combinations with rituximab and/or bendamustine as of 2013. The substance acts as aphosphoinositide 3-kinase inhibitor; more specifically, it blocks P110δ, the delta isoform of the enzyme phosphoinositide 3-kinase.[1][2]
GDC-0032 is a potent, next-generation beta isoform-sparing PI3K inhibitor targeting PI3Kα/δ/γ with IC 50 of 0.29 nM/0.12 nM/0.97nM,> 10 fold over Selective PI3K [beta].
GS-1101 is a novel, orally available small molecule inhibitor of phosphatidylinositol 3-kinase delta (PI3Kdelta) develop by Gilead and is waiting for registration in U.S. for the treatment of patients with indolent non-Hodgkin’s lymphoma that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy and for the treatment of chronic lymphocytic leukemia. The compound is also in phase III clinical evaluation for the treatment of elderly patients with previously untreated small lymphocytic lymphoma (SLL) and acute myeloid leukemia. Clinical trials had been under way for the treatment of inflammation and allergic rhinitis; however, no recent development has been reported. Preclinical studies have shown that GS-1101 has desirable pharmaceutical properties. The compound was originally developed by Calistoga Pharmaceuticals, acquired by Gilead on April 1, 2011.
clinical trials, click link
http://clinicaltrials.gov/search/intervention=CAL-101%20OR%20GS-1101%20OR%20Idelalisib
FOSTER CITY, Calif.–(BUSINESS WIRE)–Jan. 13, 2014– Gilead Sciences, Inc. (Nasdaq: GILD) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the company’s New Drug Application (NDA) for idelalisib, a targeted, oral inhibitor of PI3K delta, for the treatment of refractory indolent non-Hodgkin’s lymphoma (iNHL). FDA has granted a standard review for the iNHL NDA and has set a target review date under the Prescription Drug User Fee Act (PDUFA) of September 11, 2014.
The NDA for iNHL, submitted on September 11, 2013, was supported by a single arm Phase 2 study (Study 101-09) evaluating idelalisib in patients with iNHL that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy. Following Gilead’s NDA submission for iNHL, FDA granted idelalisib a Breakthrough Therapy designation for relapsed chronic lymphocytic leukemia (CLL). The FDA grants Breakthrough Therapy designation to drug candidates that may offer major advances in treatment over existing options. Gilead submitted an NDA for idelalisib for the treatment of CLL on December 6, 2013.
About Idelalisib
Idelalisib is an investigational, highly selective oral inhibitor of phosphoinositide 3-kinase (PI3K) delta. PI3K delta signaling is critical for the activation, proliferation, survival and trafficking of B lymphocytes and is hyperactive in many B-cell malignancies. Idelalisib is being developed both as a single agent and in combination with approved and investigational therapies.
Gilead’s clinical development program for idelalisib in iNHL includes Study 101-09 in highly refractory patients and two Phase 3 studies of idelalisib in previously treated patients. The development program in CLL includes three Phase 3 studies of idelalisib in previously treated patients. Combination therapy with idelalisib and GS-9973, Gilead’s novel spleen tyrosine kinase (Syk) inhibitor, also is being evaluated in a Phase 2 trial of patients with relapsed or refractory CLL, iNHL and other lymphoid malignancies.
Additional information about clinical studies of idelalisib and Gilead’s other investigational cancer agents can be found at http://www.clinicaltrials.gov. Idelalisib and GS-9973 are investigational products and their safety and efficacy have not been established.
About Indolent Non-Hodgkin’s Lymphoma
Indolent non-Hodgkin’s lymphoma refers to a group of largely incurable slow-growing lymphomas that run a relapsing course after therapy and can lead ultimately to life-threatening complications such as serious infections and marrow failure. Most iNHL patients are diagnosed at an advanced stage of disease, and median survival from time of initial diagnosis for patients with the most common form of iNHL, follicular lymphoma, is 8 to 10 years. The outlook for refractory iNHL patients is significantly poorer.
About Gilead Sciences
Gilead Sciences is a biopharmaceutical company that discovers, develops and commercializes innovative therapeutics in areas of unmet medical need. The company’s mission is to advance the care of patients suffering from life-threatening diseases worldwide. Headquartered in Foster City, California, Gilead has operations in North and South America, Europe and Asia Pacific.
The delta form of PI3K is expressed primarily in blood-cell lineages, including cells that cause or mediate hematologic malignancies, inflammation, autoimmune diseases and allergies. By specifically inhibiting only PI3K delta, a therapeutic effect is exerted without inhibiting PI3K signalling that is critical to the normal function of healthy cells. Extensive studies have shown that inhibition of other PI3K forms can cause significant toxicities, particularly with respect to glucose metabolism, which is essential for normal cell activity.
In 2011, orphan drug designation was assigned to GS-1101 in the U.S. for the treatment of CLL. In 2013, several orphan drug designations were assigned to the compound in the E.U. and U.S.: for the treatment of follicular lymphoma, for the treatment of mucosa-associated lymphoid tissue lymphoma (MALT), for the treatment of nodal marginal zone lymphoma, for the treatment of splenic marginal zone lymphoma, and for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma. Orphan drug designation was also assigned in the U.S. for the treatment of lymphoplasmacytic lymphoma with or without Walenstom’s macroglobulinemia and, in the E.U., for the treatment of Waldenstrom’s macroglobulinemia (lymphoplasmacytic lymphoma).
Later in 2013, some of these orphan drug designations were withdrawn in the E.U.; for the treatment of chronic lymphocytic leukemia / small lymphocytic lymphoma, for the treatment of extranodal marginal-zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma), for the treatment of of nodal marginal-zone lymphoma and for the treatment of splenic marginal-zone lymphoma. In 2013, the FDA granted a breakthrough therapy designation for the treatment of chronic lymphocytic leukemia.
- H. Spreitzer (13 May 2013). “Neue Wirkstoffe – Ibrutinib und Idelalisib”. Österreichische Apothekerzeitung (in German) (10/2013): 34.
- Wu, M.; Akinleye, A.; Zhu, X. (2013). “Novel agents for chronic lymphocytic leukemia”.Journal of Hematology & Oncology 6: 36. doi:10.1186/1756-8722-6-36.PMC 3659027. PMID 23680477.
CAL-101 is an Oral Delta Isoform-Selective PI3 Kinase Inhibitor.
| US8207153 | 6-27-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2012015964 | 1-20-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2011306622 | 12-16-2011 | METHODS OF TREATING HEMATOLOGICAL DISORDERS WITH QUINAZOLINONE COMPOUNDS IN SELECTED SUBJECTS |
| US7932260 | 4-27-2011 | Quinazolinones as Inhibitors of Human Phosphatidylinositol 3-Kinase Delta |
| US2011044942 | 2-25-2011 | METHODS OF TREATMENT FOR SOLID TUMORS |
| US2010256167 | 10-8-2010 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2010202963 | 8-13-2010 | THERAPIES FOR HEMATOLOGIC MALIGNANCIES |
| WO2005113556A1 * | 12 May 2005 | 1 Dec 2005 | Icos Corp | Quinazolinones as inhibitors of human phosphatidylinositol 3-kinase delta |
| WO2005117889A1 * | 12 Nov 2004 | 15 Dec 2005 | Didier Bouscary | Methods for treating and/or preventing aberrant proliferation of hematopoietic |
| WO2005120511A1 * | 4 Jun 2005 | 22 Dec 2005 | Joel S Hayflick | Methods for treating mast cell disorders |
| WO2006089106A2 * | 16 Feb 2006 | 24 Aug 2006 | Icos Corp | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US20060106038 * | 25 May 2005 | 18 May 2006 | Icos Corporation | Methods for treating and/or preventing aberrant proliferation of hematopoietic cells |
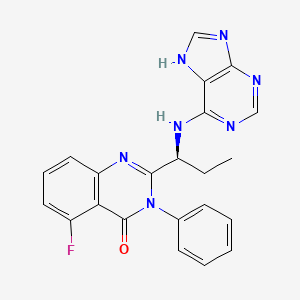
The synthesis of a compound in accordance with formula I is first exemplified using steps A-E below, which provide a synthetic procedure for compound 107, the structure of which is shown below.

(107) is idelalisib
……………….
Synthesis of 2-fluoro-6-nitro-N-phenyl-benzamide (108)
Step A: A solution of 2-fluoro-6- nitrobenzoic acid (100 g, 0.54 mol) and dimethylformamide (5 mL) in dichloromethane (600 mL) was treated dropwise with oxalyl chloride (2 M in dichloromethane, 410 mL, 0.8 mol, 1.5 eq) over 30 min. After stirring 2 h at room temperature, the reaction was concentrated to an orange syrup with some solids present. The syrup was dissolved in dry dioxane (80 mL) and slowly added to a suspension of aniline (49 mL, 0.54 mol, 1 eq) and sodium bicarbonate (90 g, 1.08 mol, 2 eq) in a mixture of dioxane (250 mL) and water (250 mL) at 6 0C. The temperature reached 27°C at the end of the addition. After 30 min, the reaction mixture was treated with water (1.2 L). The precipitate was collected by vacuum filtration, washed with water (300 mL) , air dried in the funnel, and dried in vacuo at 50°C for 24 h to afford an off-white solid product (139 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 10.82 (s, IH), 8.12 (d, J = 7.7 Hz, IH), 7.91-7.77 (m, 2H), 7.64 (d, J = 7.7 Hz, 2H), 7.38 (t, J = 7.9 Hz, 2H), 7.15 > (t, J = 7.4 Hz, IH), ESI-MS m/z 261 (MH+). The reaction described above and compound 108 are shown below.

………………………..
Synthesis of(S) – [1- (2-fluoro-6-nitro-benzoyl) -phenyl-aminocarbonyl] – propyl-carbamic acid tert-butyl ester (109)
Step B: A suspension of compound 108 (0.5 mol) and dimethylformamide (5 mL) in thionyl chloride (256 mL, 2.5 mol, 5 eq) was stirred at 85°C for 5 hours. The reaction mixture was concentrated in vacuo to a brown syrup. The syrup was dissolved in dichloromethane (200 mL) and was slowly added to a solution of N-BOC-L-2-aminobutyric acid (112 g, 0.55 mol, 1.1 eq) and triethylamine (77 mL, 0.55 mol, 1.1 eq) in dichloromethane (600 mL) at 10 0C. After stirring at room temperature for 3 h, salts were removed by filtration, and the solution was washed with 100 mL of water, saturated sodium bicarbonate, water, 5% citric acid, and saturated sodium chloride. The organic phase was dried with magnesium sulfate and concentrated to a red syrup. The syrup was dissolved in dichloromethane (450 mL) and purified by flash chromatography on a silica gel plug (15 x 22 cm, 4 L dry silica) eluted with hexanes/ethyl acetate (10%, 8 L; 15%, 8 L; 20%, 8 L; 25%, 4 L) to yield the compound 109 as an off-white solid (147 g, 66%). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (d, J = 8.0 Hz, IH), 7.84 (t, J = 8.6 Hz, IH), 7.78- 7.67 (m, IH), 7.65-7.49 (m, 3H), 7.40-7.28 ( m, 2H), 7.19 (d, J = 7.5 Hz, IH), 4.05 (broad s, IH), 1.75- 1.30 (m, 2H), 1.34 (s, 9H), 0.93 (broad s, 3H). ESI- MS m/z 446.3 (MH+) . The reaction described above and compound 109 are shown below.

Synthesis of(S) – [1- (5-fluoro-4-oxo-3-phenyl-3 , 4-dihydro-quinazolin-2- yl) -propyl] -carbamic acid tert-butyl ester (110)
Step C: A solution of compound 109 (125 mmol, 1 eq) in acetic acid (500 mL) was treated with zinc dust (48.4 g, 740 mmol, 6 eq) added in 3 portions, and the reaction mixture was allowed to cool to below 35°C between additions. After stirring for 2 h at ambient temperature, solids were filtered off by vacuum filtration and washed with acetic acid (50 mL) . The filtrate was concentrated in vacuo, dissolved in EtOAc (400 mL) , washed with water (300 mL) , and the water layer was extracted with EtOAc (300 mL) . The combined organic layers were washed with water (200 mL) , sat’d sodium bicarbonate (2 x 200 mL) , sat’d NaCl (100 mL) , dried with MgSO4, and concentrated to a syrup. The syrup was dissolved in toluene (200 mL) and purified by flash chromatography on a silica gel plug (13 x 15 cm, 2 L dry silica) eluted with hexanes/ethyl acetate (10%, 4 L; 15%, 4 L; 17.5%, 8 L; 25%, 4 L) to yield compound 110 as an off-white foamy solid (33.6 g, 69%). 1H NMR (300 MHz, DMSO-d6) δ 7.83 (td, J = 8.2, 5.7 Hz, IH), 7.64-7.48 (m, 5H), 7.39 (broad d, J = 7.6 Hz, IH), 7.30 (dd, J = 8.3 Hz, IH), 7.23 (d, J = 7.6 Hz, IH), 4.02-3.90 (m, IH), 1.76-1.66 (m, IH), 1.62-1.46 (m, IH), 1.33 (s, 9H), 0.63 (t, J= 7.3 Hz, 3H). ESI-MS m/z 398.3 (MH+). The reaction described above and compound 110 are shown below.

…………..
Syn of (S) -2- (1-amino-propyl) -5-fluoro-3-phenyl-3H-quinazolin-4- one (111)
Step D: A solution of compound 110 (85 mmol) in dichloromethane (60 mL) was treated with trifluoroacetic acid (60 mL) . The reaction mixture was stirred for 1 h, concentrated in vacuo, and partitioned between dichloromethane (150 mL) and 10% K2CO3 (sufficient amount to keep the pH greated than 10) . The aqueous layer was extracted with additional dichloromethane (100 raL) , and the combined organic layers were washed with water (50 mli) and brine (50 mL) . After drying with Mg SO4, the solution was concentrated to provide compound 111 as an off-white solid (22 g, 88%) . 1H NMR (300 MHz,
CDCl3) δ 7.73-7.65 (m, IH), 7.62-7.49 (m, 4H), 7.32- 7.22 (m, 2H), 7.13-7.06 (m, IH), 3.42 (dd, J= 7.5, 5.2 Hz, IH), 1.87-1.70 (m, IH), 1.58-1.43 (m, IH), 0.80 (t, J = 7.4 Hz, 3H) . ESI-MS m/z 298.2 (MH+) . The reaction described above and compound 111 are shown below.

………………
syn of (S) -5-fluoro-3-phenyl-2- [1- (9H-purin-6-ylamino) -propyl] – 3H-quinazolin-4-one (107)
Step E: A suspension of compound 111(65.6 mmol, 1 eq) , 6-bromopurine (14.6 g, 73.4 mmol, 1.1 eq) , and DIEA (24.3 mL, 140 mmol, 2 eq) in tert- butanol (40 mL) was stirred for 24 h at 800C. The reaction mixture was concentrated in vacuo and treated with water to yield a solid crude product that was collected by vacuum filtration, washed with water, and air dried. Half of the obtained solid crude product was dissolved in MeOH (600 mL) , concentrated onto silica gel (300 mL dry) , and purified by flash chromatography (7.5 x 36 cm, eluted with 10 L of 4% MeOH/CH2Cl2) to yield a solid product. The solid product was then dissolved in EtOH (250 mL) and concentrated in vacuo to compound 107 idelalisib as a light yellow solid (7.2 g, 50%).
1H NMR (300 MHz, 80 0C, DMSO-d5) δ 12.66 (broad s, IH), 8.11 (s, IH), 8.02 (broad s, IH), 7.81-7.73 (m, IH),7.60-7.42 (m, 6H), 7.25-7.15 (m, 2H), 4.97 (broad s, IH), 2.02-1.73 (m, 2H), 0.79 (t, J= 7.3 Hz, 3H).
ESI-MS m/z 416.2 (MH+).
C, H, N elemental analysis (C22Hi8N7OF-EtOH- 0.4 H2O).
Chiral purity 99.8:0.2 (S:R) using chiral HPLC (4.6 x 250 mm Chiralpak ODH column, 20 °C, 85:15 hexanes : EtOH, 1 rnL/min, sample loaded at a concentration of 1 mg/mL in EtOH) . The reaction described above and compound 107 idelalisib are shown below.

| WO2001030768A1 * | 26 Oct 2000 | 3 May 2001 | Gustave Bergnes | Methods and compositions utilizing quinazolinones |
| WO2001081346A2 * | 24 Apr 2001 | 1 Nov 2001 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2003035075A1 * | 27 Aug 2002 | 1 May 2003 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2005016348A1 * | 13 Aug 2004 | 24 Feb 2005 | Jason Douangpanya | Method of inhibiting immune responses stimulated by an endogenous factor |
| WO2005016349A1 * | 13 Aug 2004 | 24 Feb 2005 | Thomas G Diacovo | Methods of inhibiting leukocyte accumulation |
| WO2005067901A2 * | 7 Jan 2005 | 28 Jul 2005 | Carrie A Northcott | Methods for treating and preventing hypertension and hypertension-related disorders |
3 Herbs you Need to Know for Healing Depression: Interview with Sarah Josey
Depression is a condition that reportedly affects one in ten Americans. It is a conversation often avoided, but I was lucky enough to interview Sarah Josey Herbalist, Nutritionist and Founder of Golden Poppy Herbal Apothecary. She shared three herbs that everyone should know about when it comes to depression and one to avoid, while on medication. Watch video.
Find out more about Sarah Josey and Golden Poppy Herbal Apothecary at http://www.goldenpoppyherbs.com/
Join the conversation below. Have you tried any of these herbs? Let me know in the comments.
SAMIDIRECT -Healthy, Wealthy & Wise, FREE OF COST CONSULTATION on Diabetes, Cancer, Arthritis, Osteoporosis, Heart- Liver -Lung & Kidney problems, Low Immunity, Alzheimer, Weight Management, Weak Memory, Neutritional Deficiency, UTI problems
![]()

Healthy, Wealthy & Wise
For over 25 years the Sami Group has been unlocking the mystery of herbs, extracting the goodness, and gifting the world with good health.
Now the Sami Group provides YOU an opportunity to unlock the mystery of Success, Wealth & Better living.
FREE OF COST CONSULTATION on Diabetes, Cancer, Arthritis, Osteoporosis, Heart- Liver -Lung & Kidney problems, Low Immunity, Alzheimer, Weight Management, Weak Memory, Neutritional Deficiency, UTI problems etc.
(REVOLUTIONARY AYURVEDIC SOLUTIONS with ISO 22000 Certified Indian MNC after 25 years of R & D by 120 Scientists.
Numberless Testimonials.)
http://www.samidirect.com/products/
Do visit the website www.samidirect.com & have the study in detail. Have a look at the Sami Direct Corporate Video on you tube. If you get the wonderful potential of the brightest future…do call me for ‘How to get started?’




…..









DISTRIBUTOR ENQUIRY WELCOME
CONTACT MR JAY DESAI
REGARDS
+91 9699952526
Mumbai, INDIA
email-jaydesai1502@gmail.com
 |
Bussines Sami Direct Seminar ppt.ppt1.pdf 9204K View Download |
samidirect corporate video

DR MAJID, FOUNDER , SAMIDIRECT
Dr. Muhammed Majeed
Dear Friend, Congratulations on your decision!
A little over three decades ago I went from a small town in South India to the United States Of America seeking fulfillment of my dreams. Today with a business conglomerate spread across the globe, I can confidently say that the future belongs to those who believe in the beauty of their dreams.
The aspiration to dream and the conviction to follow their dreams is what sets apart the extraordinary from the ordinary. Congratulations for choosing to be among the extraordinary. Now we are in it together. You have chosen the right place and the right means. The awesome combination of extensively researched products and a revolutionary business plan is a definite formula for success. We are with you at every step to help you fulfill your dreams and reach greater heights.
Dr. Muhammed Majeed
Welcome home again!
– See more at: http://www.samidirect.com/about/founder-desk/#sthash.rrOCRiJ1.dpuf
Sami Direct, as a part of the Sami Group, is the culmination of relentless Research and Development for more than two decades. We at Sami Direct are committed to offer you an unrivalled range of nutraceuticals, soon to be followed by cosmeceutical products, which have been acknowledged by the world over for its highest quality and safety standards.
Sami Direct is supported by its very own R&D facility- SAMI LABS LTD., located in Bangalore. This state-of-the-art, multi-disciplinary division pursues diverse fields of research with over 120 scientists focusing all efforts towards creating effective and safe products. With six highly advanced cutting-edge manufacturing units adhering to the strictest quality and safety standards, Sami Direct ensures that the highest quality of products are being produced.
Today the Sami Group holds a strong intellectual property portfolio with over 70 US and International Patents to its credit including awards and recognitions worldwide.
With the perfect blend of world class products and a revolutionary business plan, it is a lifetime opportunity not just to enhance your health, but also a fruitful and lasting career heightening your income.
DISTRIBUTOR ENQUIRY WELCOME
CONTACT MR JAY DESAI
REGARDS
+91 9699952526
Mumbai, INDIA
email; jaydesai1502@gmail.com
“5TH PHARMACOVIGILANCE CONGREGATION 2013–20th November 2013, Kohinoor Continental Hotel, Mumbai, India.
DRUG REGULATORY AFFAIRS INTERNATIONAL

“5TH PHARMACOVIGILANCE CONGREGATION 2013”
“Ensuring safer drugs to market by analyzing latest developments in pharmacovigilance, drug safety and risk management”
20th November 2013, Kohinoor Continental Hotel, Mumbai, India.
Greetings from Virtue Insight,
I am happy to invite you and your colleagues to be a sponsor/ delegate for our upcoming “5th Pharmacovigilance Congregation 2013” The conference will be held on 20th November 2013, Kohinoor Continental Hotel, Mumbai, India.
KEY SPEAKERS:-
- Deepa Arora, Global Head, Drug Safety & Risk Management, Lupin
- Arun Bhatt, President, Clininvent Research
- Moin Don, Executive Director, PVCON Pharmacovigilance Auditing & Consulting Services
- Bhaswat Chakraborty, Senior Vice President, Research & Development, Cadila Pharmaceuticals
- Parminder Kaur, Owner & Regulatory Affairs & PhV Consultant (QPPV), RegPak BioPharma Consulting (Netherlands, UK)
- Babita Kirodian, Head – Pharmacovigilance, Bristol-Myers Squibb
- Rajani Rokade, Head – Pharmacovigilance, Sanofi Aventis
- Veena Rajan, Head – Patient Safety, AstraZeneca
- Sofi Joseph, Head –…
View original post 465 more words
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

