PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Join me on Facebook
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
Googleplus
MYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Chinese Dodder Seeds ( Tu Si Zi ) 菟絲子 , also known as Beggarweed, Cuscutae, Devil’s Guts, Dodder Of Thyme, Hellweed, Lesser Dodder, Scaldweed, Strangle Tare, Tu Si Zi, Tu Sizi. Cuscuta epithymum; Cuscuta chinensis. It belong to the “Convolvulaceae” family.
Chinese Dodder Seeds ( Tu Si Zi ) 菟絲子 has a sweet, pungent and neurtal properties. It is use for treating the kidney and liver.
What does it do?
Property:
In the term of traditional Chinese medicine (TCM),
Tu Si Zi is acrid, sweet, neutral.
The channels Tu Si Zi influences are Kidney, Liver.Action:
In the term of TCM, Tu Si Zi:
1: Tonifies Kidneys, Augments Yin, Secures Jing and Reserves Urine.
2: Tonifies Kidneys and Liver, Improves Vision.
3: Benefits Spleen and Kidneys, Stops Diarrhea.
4: Calms the Fetus.
Botanical Basic Data of Cuscuta chinensis(Dodder).:
Botanical Source:Cuscuta chinensis(The ripe seed of Cuscuta chinensis Lam.,an annual voluble parasitic herb of the family Convolvulaceae).
Latin Name: Semen Cuscutae
Family: Convolvulaceae.
Common Name: Cuscuta seed, Chinese Dodder seed,Huang Si,Huang Teng Zi,Dou Ji Sheng.Huang Shan Teng.Wu Gen Cao,Wu Niang Teng,Huang Shan Si,Lao Ya Si,Huang Si Teng.
Scientific Name: Cuscuta chinensis Lam
Pin Yin Name: Tu Si Zi
Cuscuta Classification in China:(1).Cuscuta chinensis Lam.;(2).Cuscuta australis R.Br.;(3).Cuscuta campestris Yunker;
Pinyin Name: Tu si zi,Also called Chinese Dodder Seed.
Pin yin description:tu is a character for this herb derived from the character meaning rabbit; si means silk, and zi means seeds, the part used; this plant is a parasitic weed that sets up a mat of hair-like fibers at its base and then rapidly sends fibrous stems upward; thus Tu Si refers to the quality of these fibers like silky rabbit hair; a common name for dodders in the West, based on the undesirable weed-like nature of these plants, is Devil’s Hair.
Part use:Dodder seed,Aerial parts.(whole plants are harvested in autumn when the seeds are ripe, and then threshed after dried to get the seeds)
Synoms:Dodder,love vine,strangleweed,devil’s-guts,goldthread,pull-down,devil’s-ringlet,hellbine,hairweed,devil’s-hair,Beggarweed, Cuscutae, Devil’s Guts, Dodder Of Thyme, Hellweed,Lesser Dodder,Scaldweed,Strangle Tare,Tu Si Zi,Tu Sizi,Cuscuta epithymum,Cuscuta chinensis and hailweed.
Habitat:Dodder grows throughout Europe, Asia, and southern Africa. Dodder prefers coastal and mountainous regions, and is gathered in summer.In China,mainly distributed in Jiangsu,Liaoning, Jilin, Hebei, Shandong and Henan provinces of China.
Taste:Pungent, Sweet,It is sweet in taste, warm in nature and manifests its therapeutic actions in the liver, kidney and spleen meridians.
Constitutents:Dodder contains flavonoids (including kaempferol and quercitin) and hydroxycinnamic acid.
Cuscuta , or Dodder plant, is a parasitic vine that wraps around other plants for nourishment.The ripe seed of Cuscuta chinensis Lam.; an annual voluble parasitic herb of the family Convolvulaceae.Cuscuta seed is used in China for kidney deficiency. Cuscuta has a high content of flavonoids and has strong antioxidant properties. Cuscuta seed has been found in studies to have positive effects on sperm health and motility, and invigorates the reproductive system.
The plant growns near seashores.Slim stems spread out,twist and yellow color,no leaf.flower blossom fascination on axil.flower bud and small bud squama shape,caylx shape cup,5 divide,white crown,bell shape,double length of calyx.The flowers are hermaphrodite (have both male and female organs).Stamen flower flat short,squama grow on base,shape square roundness,2 room germen. Capsule shape flat ball.Seed 2~4,florescence July to September,fruit august to october. It can grow in semi-shade (light woodland) or no shade and requires moist soil.
Dodder is distributed in most parts of China. It is collected in autumn when the seed is ripe, dried in the sun and used unprepared or boiled after removal of impurities.
U.S. Provisional Patent App. No. 60/743,543, filed March 17, 2006, U.S. Patent App. No. 11/724,992, filed March 16, 2007, and U.S. Patent App. Publication No. 2007/0232604, published October 4, 2007,
BioNews TexasAmbit initiates QUANTUM-R Phase 3 clinical trial of quizartinib in FLT3-ITD …News-Medical.net… the treatment of both newly diagnosed and relapsed FLT3-ITD positive and negative AML patients.
Both the U.S. Food and Drug Administration (FDA) and European Commission have granted orphan drug designation to quizartinib for the treatment of AML.AML, High Risk MDS Therapy
Ambit Biosciences (NASDAQ:AMBI) is a biotech company that focuses on treatments that inhibit kinases, which are drivers for diseases such as cancer. Three drugs are in development, with the lead one being quizartinib — a Phase 2B trial treatment for acute myeloid leukemia. However, AMBI’s collaboration agreement with Astellas Pharma is set to expire in September, and if it is not replaced, it could mean a delay in Phase 3 trials for quizartinib. Keep in mind that AMBI generated $23.8 million in collaboration revenues last year.
AC-220 is an angiogenesis inhibitor that antagonizes several proteins involved in vascularization. It was engineered by Ambit Biosciences using KinomeScan technology to potently target FLT3, KIT, CSF1R/FMS, RET and PDGFR kinases. Ambit is developing oral AC-220 in phase III clinical studies for the treatment of relapsed/refractory acute myeloid leukemia (AML) patients with the FMS-like tyrosine kinase-3 (FLT3)-ITD mutation. Early clinical trials are also ongoing for the treatment of advanced solid tumors, for the treatment of refractory or relapsed myelodysplasia, in combination with induction and consolidation chemotherapy for previously-untreated de novo acute myeloid leukemia, and as a maintenance therapy of AML following hematopoietic stem cell transplantation (HSCT). In 2009, orphan drug designation was received both in the U.S. and in the EU for the treatment of AML. In 2009, Ambit Biosciences and Astellas Pharma have entered into a worldwide agreement to jointly develop and commercialize the drug candidate for the treatment of cancer and non-oncology indications. This agreement was terminated in 2013.
Quizartinib is a small molecule with potential anticancer activity. Quizartinib is a selective inhibitor of class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), stem cell factor receptor (SCFR / KIT), colony-stimulating factor 1 receptor (CSF1R/FMS) and platelet-derived growth factor receptors (PDGFRs .) Able to inhibition of ligand-independent cell proliferation and apoptosis. Mutations in FLT3 are the most frequent genetic alterations in acute myeloid leukemia (AML) and occur in approximately 30% of cases of AML.
Quizartinib представляет собой малую молекулу с потенциальной противораковой активностью. Quizartinib является селективным ингибитором класса III рецепторов тирозин киназ, в том числе FMS-связанных тирозинкиназы 3 (FLT3/STK1), фактор стволовых клеток рецепторов (SCFR / KIT), колониестимулирующий фактор 1 рецепторов (CSF1R/FMS) и тромбоцитарный рецепторов фактора роста (PDGFRs). Способен к торможению лиганд-независимой клеточной пролиферации и апоптоза. Мутации в FLT3 являются наиболее частыми генетическими изменениями в остром миелобластном лейкозе (ОМЛ) и встречаются примерно в 30% случаев ОМЛ.
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 leading to resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.
Clinical trials
It had good results in a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
………………………..
WO 2007109120 COMPD B1
EXAMPLE 3: PREPARATION OF N-(5-TERT-BUTYL-ISOXAZOL-3-YL)-N’-{4-[7-(2- MORPHOLIN-4-YL-ETHOXY)IMIDAZO[2,1 -B3[1 ,3]BENZOTHIAZOL-2-YL]PHENYL}UREA [Compound B1]
[00426] A. The intermediate 2-amino-1,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf. J. Org. Chem. 1970, 35, 4103-4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 ml_ ethanol and 9 ml_ concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00427] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2-amino-1 ,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSOd6) £7.6 (m, 2H ), 6.6 (d, 1H).
[00428] B. To prepare the intermediate 2-(4-nitrophenyl)imidazo[2,1- b][1 ,3]benzothiazoI-7-ol, 2-amino-1 ,3-benzothiazol-6-ol, (20.0 g, 0.12 mol) and 2-bromo-4′-nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to 00C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2-(4- nitrophenyl)imidazo[2,1-_D][1,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-CT6) δ 10 (s, 1 H), 8.9 (s, 1H), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, 1 H), 7.4 (s, 1 H), 6.9 (d, 1 H). [00429] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nttro- phenyl)imidazo[2,1-£>][1 ,3]benzothiazo!e intermediate: 2-(4- nitrophenyl)imidazo[2,1-jb][1 ,3]benzothiazol-7-ol, (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-jb][1 ,3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-Cf6) δ 8.97 (s, 1 H), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, 1 H), 7.7 (s, 1 H), 7.2 (d, 1 H), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00430] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4- amino-phenyl)!midazo[2, 1 -b][1 ,3]benzothiazole: To a suspension of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-ib][1 ,3]benzothiazole (3.87g, 9.1 mmol) in 100 ml_ isopropyl alcohol/water (3:1 ) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 ml_). The filtrate was concentrated to approximately 1/3 of the original , volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water — in which case the reaction is virtually complete after 3.5 hours and oniy traces of starting material are observed in the product obtained. NMR (DMSO-d6) δ 8.4 (s, 1 H), 7.8 (d, 1 H), 7.65 (d, 1 H), 7.5 (d, 2H), 7.1 (d, 1 H), 6.6 (d, 2H), 4.1 (t, 2H)1.3.6 (m, 4H), 2.7 (t, 2H).
[00431] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,1-b][1 ,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert- butylisoxazole-3-isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base. Yield: 2.342 g (41 %) NMR (DMSO-Cf6) £9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, 1 H), 7.76 (d, 2H), 7.69 (d, 1 H), 7.51 (d, 2H), 7.18 (dd, 1H), 6.52 (s, 1H), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCI3) £9.3 (br, 1H), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, 1 H), 7.22 (d, 1 H), 7.03 (dd, 1 H)1 5.88 (s, 1 H), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
[00432] F. For the preparation of the hydrochloride salt, N-(5-tert-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2, 1 – b][1 ,3]benzothiazol-2-yl]phenyI}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCI in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrifugation and washed with ethyl ether to give the hydrochloride salt. Yield: 2.44 g (98 %) NMR (DMSO-d6) £11-0 (br, 1 H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1 H), 7.93 (d, 1H), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, 1H), 6.53 (S, 1 H), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H)13.23 (m, 2H)1 1.30 (s, 9H).
[00433] G. Alternatively, Compound B1 may be made by taking the intermediate from Example 4B and reacting it with chloroethyl morpholine hydrochloride under conditions described in Step C. [00434] H . Λ/-(5-tert-butyl-isoxazol-3-yl)-Λ/’-{4-[5-(2-morpholin-4-yl- ethoxy)imidazo[2,1-6][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride, a compound having the general formula (I) where R1 is substituted on the 5 position of the tricyclic ring, was prepared in the manner described in Steps A- F but using the cyciization product 2-amino-benzothiazol-4-ol with 2-bromo-4′- nitroacetophenone in Step A. 1H NMR (DMSO-d6) δ 11.6 (br, 1 H)1 9.78 (br, 1H), 9.56 (br, 1 H), 8.64 (s, 1H)1 7.94 (d, 2H), 7.70 (s, 1H)1 7.56 (d, 2H), 7.45 (t, 1 H), 7.33 (d, 1H), 6.54 (s, 1 H), 4.79 (t, 2H), 3.87 (m, 6H), 3.60 (m, 2H), 3.34 (m, 2H)1 1.30 (s, 9H); LC-MS: ESI 561 (M+H)+. [Compound B11] [00435] I. N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[6-(2-morpholin-4-yl- ethoxy)imidazo[2,1-b][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride [Compound B12] was also prepared by first preparing the benzothiazole starting material, 5 methoxy-benzothiazol-2yl~amine: [00436] To prepare the 5-methoxy-benzothiazol-2-ylamine starting material: To a suspension of (3-methoxy-phenyl)-thiourea (1.822g, 10 mmol) in CH2CI2 (20 ml_) at 0 0C was added dropwise a solution of bromine (1.76 g, 11 mmol) in 10 ml of trichloromethane over a period of thirty minutes. The reaction was stirred for 3 hours at room temperature then heated to 3 hours to reflux for one hour. The precipitate was filtered and washed with dichloromethane. The solid was suspended in saturated NaHCOsand extracted with CH2CI2. The extract was dried over MgSO4 and concentrated to give a white solid (1.716 g, 95%).
………………….
WO 2011056939
N-(5-ieri-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof. N-(5-ieri-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- / ][!, 3]benzo
N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven of the steps of:
(A) converting 2-amino-6-alkoxybenzothiazole (II), wherein R1 is a suitable phenolic hydroxyl protecting ;
(II) (III)
(B) reacting 2-amino-6-hydroxybenzothiazole (III) with compound (IV), wherein X is a leaving group, to yield 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V);
(C) reacting 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V) with compound (VI), wherein X2 is a leaving group, to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[ -b]benzothiazole (VII);
(D) reducing 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2, 1- bjbenzothiazole (VII) to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- am
(E) converting 3-amino-5-£er£-butyl isoxazole (IX) to a 5-?er?-butylisoxazol-3- ylcarbamate derivative (X), wherein R2 is optionally substituted aryl, heteroaryl, alkyl, or cycloalkyl;
(IX) (X)
(F) reacting 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- bjbenzothiazole (VIII) with a 5-£er£-butylisoxazol-3-ylcarbamate derivative (X) to yield N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-
(G) converting N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea to an acid addition salt of N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b] [ 1 ,3]benzo-thiazol-2-yl]phenyl } urea.
[00128] In certain embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea, or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, as depicted in Scheme 1, wherein R1, R2, X1, and X2 are defined herein elsewhere. In specific embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven, of the Steps A, B, C, D, E, F, and G, as depicted in Scheme 1.
Scheme 1 :
A. Preparation of 2-amino-6-hydroxybenzothiazole
1. Example A-l[00252] To a 1-L 3-necked round bottom flask fitted with a condenser, heating mantle, and mechanical stirrer was charged aqueous hydrobromic acid (48%, 632 mL, 5.6 mol, 10 equiv). 2-Amino-6-methoxybenzothiazole (100 g, 0.55 mol, 1 equiv) was added to the above flask over 15 minutes. The reaction temperature was raised slowly to reflux (105-110 °C). A clear dark brown colored solution was observed at about 80 °C. The reflux was continued at 105-110 °C for about 4 hr. The progress of the reaction was monitored by HPLC. When 2-amino-6-methoxybenzothiazole was less than 2%, the reaction was substantially complete.
[00253] The reaction mass was cooled to 0-5 °C and at this point precipitation of a solid was observed. The mixture was maintained at 0-5 °C for 0.5 hr and filtered, and the cake was pressed to remove HBr. The wet cake was transferred to a 2-L round bottom flask fitted with a mechanical stirrer. Saturated aqueous sodium bicarbonate solution (-1500 mL) was added slowly at ambient temperature, whereupon considerable frothing was observed. The pH of the solution was found to be about 6.5 to 7. The mixture was stirred for 0.5 hr at ambient temperature and filtered. The filter cake was washed with water (500 mL), dried on the filter and then under vacuum at 30-35 °C for 10-12 hr to give the product 2-amino-6-hydroxybenzothiazole (82 g, 89% yield, HPLC purity = 99%). JH NMR (DMSO-if6, 500 MHz): δ 7.12 (d, 1H), 7.06 (S, 2H, NH2), 7.01 (d, 1H), 6.64 (dd, 1H); MS (m/z) = 167.1 [M+ + 1].
[00254] Table: Summary of Plant Batches
[00255] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the pH to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 270 nm. The injection volume was 20 μΐ^, and the sample was diluted with a diluent (Mobile Phase A : Mobile Phase B = 70:30). Test solution was prepared by weighing accurately about 25 mg of sample and transferring it into a 100 mL volumetric flask, dissolving with 20-30 mL of diluent, making up the volume to the mark with diluent, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00256] Table: HPLC Method
2. Example A-2
[00257] 2-Amino-6-methoxybenzothiazole was reacted with hot aqueous HBr at a temperature of >70 °C for about 3 hours and then the clear solution was cooled to ambient temperature overnight. The precipitated solids were collected, dissolved in hot water and the pH was adjusted to between 4.5-5.5. The resultant solids were collected, dried and re-crystallized from isopropanol. Second crop material was collected. The solids were vacuum dried to give 2-amino-6-hydroxybenzothiazole.
[00258] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a white solid, with 99.4% purity (HPLC area %). JH NMR (300 MHz, DMSO-if6) was collected, which conformed to structure.
3. Example A-3
[00259] A 22-L 3-neck round bottom flask was equipped with a mechanical agitator, thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with hydrobromic acid (14 L, 123.16 mol, 13.10 equiv). Heating was initiated and 2- amino-6-methoxybenzothiazole was added (1.7 kg, 9.4 mol, 1.00 equiv) over 10 minutes with stirring. The heating of the reaction mixture was continued to reflux, and maintained (>107 °C) for approximately 5 hours. The reaction mixture turned into a clear solution between 75 °C and 85 °C. The reaction progress was monitored by TLC until no starting material was observed (A -0.5 mL reaction mixture aliquot was diluted with -0.5 mL water as a clear solution, neutralized with sodium acetate to pH -5 and extracted with 1 mL dichloromethane. The organic layer was spotted: 5%
[00260] The reaction mixture was cooled to – 20 °C (overnight). White solids precipitated. The solids were filtered on a polypropylene filter and pressed to remove as much hydrobromic acid from the solids as possible to facilitate the subsequent pH adjustment step. The slightly wet crude product was dissolved in hot (50 °C) water (5 L). The clear solution was filtered to remove any insoluble material present, and the solids were washed with 50 °C water. The filtrate was cooled to 10 °C. The cooled filtrate was neutralized with sodium acetate (- 1.0 kg) to pH 4.5 to 5.5 with vigorous stirring. A thick white solid precipitated. The solids were collected by filtration, and washed with cool (-10 °C) water (2 x 1.0 L) and air dried.
[00261] The wet crude product was slurried in hot (50 °C) isopropanol (3 L) briefly and allowed to stand in a cool room (-5 °C) overnight. The solids were collected by filtration and washed with methyl ferf-butylether (2 x 500 mL). The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (first crop). Yield: 1068 g (68%), white solid. HPLC: 99.4% (area). JH NMR (300 MHz, DMSO- ) conformed to structure.
[00262] The organic filtrate was collected in a total volume of 1.0 L, cooled to 10 °C. The off-white solids were precipitated and collected by filtration. The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (second crop). Yield: 497 g (32%), off-white solid. HPLC: 99.8% (area).
[00263] The overall yield combining the first crop and the second crop was 1565 g, (99%).
B. Preparation of 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol
1. Example B-l[00264] A 3-L 3-neck round bottom flask fitted with a condenser, a heating mantle, and a mechanical stirrer was charged with H-butanol (1.5 L), followed by 2-amino-6- hydroxybenzothiazole (75 g, 0.45 mol, 1.0 equiv), 2-bromo-4′-nitroacetophenone (121 g, 0.50 mol, 1.1 equiv), and sodium bicarbonate (41.6 g, 0.50 mol, 1.0 equiv). The reaction temperature was gradually raised to reflux and maintained at reflux (110-115 °C) for 2-3 hr. During the temperature increase, the reaction mass turned into a clear solution and then immediately changed into an orange colored suspension at 65-75 °C. The progress of the reaction was monitored by HPLC analysis every 1 hr (reaction mass sample was submitted to QC). When the level of 2-amino-6-hydroxybenzothiazole was less than 2%, the reaction was substantially complete.
[00265] The reaction mass was slowly cooled to 50-60 °C and then further cooled to 0-5 °C and stirred for 15 min. The precipitated solids were collected by filtration, and dried on the filter. The wet cake was transferred in to a 1-L round bottom flask, and water (600 mL) was added. The suspension was stirred for 0.5 hr and filtered, and the solid was dried on the filter. The wet cake was again taken in to a 1-L round bottom flask and stirred with acetone (200 mL). The slurry was filtered and washed with acetone (2 X 100 mL), and the solid was dried on the filter, unloaded and further dried in a vacuum oven at ambient temperature to give the product 2-(4-nitrophenyl)imidazo[2,l- b]benzothiazol-7-ol (V) (120 g, 85.7% yield, HPLC purity = 98.7%). JH NMR (DMSO- d6, 500 MHz): δ 9.96 (s, 1H, OH), 8.93 (s, 1H), 8.27 (d, 2H), 8.06 (d, 2H), 7.78 (d, 1H), 7.38 (d, 1H), 6.97 (dd, 1H); MS (m/z) = 312 [M+ + 1].
[00266] Table: Summary of Plant Batches
* Input of 2-amino-6-hydroxybenzothiazole (III)
[00267] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the H to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 235 nm. The injection volume was 10 μΐ^. The blank was prepared by transferring 200 μΐ. of DMSO and 200 μΐ. of 2M NaOH into a 10 mL volumetric flask, making up the volume to the mark with methanol, and mixing. The test solution was prepared by weighing accurately about 10 mg of sample and transferring it into a 50 mL volumetric flask, dissolving with 1 mL of DMSO and 1 mL of 2M NaOH, sonicating to dissolve, making up the volume to the mark with methanol, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00268] Table: HPLC Method
2. Example B-2
[00269] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with 2-amino-6-hydroxybenzothiazole (1068 g, 6.43 mol, 1.0 equiv) and ethanol (200 proof, 32.0 L), and the suspension was stirred for 10 minutes. 2-Bromo-4- nitroacetophenone (1667 g, 6.49 mol, 1.01 equiv) was added in one portion. The reaction mixture was heated to reflux (78 °C). The reflux was maintained for approximately 25 hours, resulting in a yellow suspension. The reaction progress was monitored by TLC (20% methanol/ethyl acetate; Rf (product) = 0.85; Rf (starting material) = 0.30). TLC indicated -50% 2-amino-6-hydroxybenzothiazole after 24 hours of reflux. Tetrabutylammonium iodide (10 g) was added and reflux was maintained for an additional 12 hours. TLC indicated -50% starting material still present. Coupling was seen to occur at both the thiazole and the amine.
[00270] The reaction mixture was cooled to 0-5 °C. The solids were collected by filtration, and the yellow solid was washed with ethanol (200 proof, 2 X 1.0 L) and diethyl ether (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 930 g (46%), yellow solid. HPLC: 99.5% (area). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
3. Example B-3
[00271] The reaction of Step B was carried out on multiple runs, varying solvents, temperature, and base. The results were summarized in the table below. The product (V) was isolated as yellow or green solids, with 1H NMR consistent with the structure and a purity of greater than about 98% by HPLC analysis.
[00272] Table: Reaction Condition Screening
TBAI = Tetrabutylammonium Iodide
C. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[2, 1 -bjbenzothiazole
1. Example C-l
[00273] To a 2000-L glass-lined (GL) reactor was charged DMF (298 kg), and the agitator was started. Under a nitrogen blanket, the reactor was charged with 2-(4- nitrophenyl)imidazo[2,l-&]benzothiazol-7-ol (36.8 kg, 118.2 mol, 1.0 equiv), 4-(2- chloroethyl)morpholine hydrochloride (57.2-66.0 kg, 307.3-354.6 mol, 2.6-3.0 equiv), tetrabutylammonium iodide (8.7 kg, 23.6 mol, 0.2 equiv) and potassium carbonate (49.0 kg, 354.6 mol, 3.0 equiv). The resulting yellow suspension was heated and stirred at 90 + 5 °C for at least 15 minutes, then the temperature was increased to 110 + 5 °C. The mixture was stirred for at least 1 hour and then sampled. The reaction was deemed complete if 2-(4-nitrophenyl) imidazo[2,l-&]benzothiazol-7-ol was <1% by HPLC. If the reaction was not complete, the heating was continued and the reaction sampled. If the reaction was incomplete after 6 hours, additional 4-(2-chloroethyl)morpholine hydrochloride may be charged. In general, additional charges of 4-(2- chloroethyl)morpholine hydrochloride had not been necessary at the given scale.
[00274] The reactor was cooled to 20 + 5 °C and charged with water (247 kg) and acetone (492 kg). The mixture was agitated at 20 + 5 °C for at least 1 hour. The product (VII) was isolated by filtration or centrifuge, and washed with water and acetone, and then dried in a vacuum oven at 45 °C to constant weight to give a yellow solid (46.2 kg, 92% yield, HPLC purity = 97.4% by area). JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example C-2
[00275] 2-(4-Nitrophenyl)imidazo[2, l-b]benzothiazol-7-ol, 4-(2-chloroethyl)- morpholine hydrochloride, potassium carbonate, and tetrabutylammonium iodide were added to N,N-dimethylformamide forming a yellow suspension that was heated at a temperature of >50 °C for over 3 hours. The reaction was cooled and the solids were collected, slurried into water, filtered, slurried into acetone, filtered and washed with acetone to give yellow solids that were dried under vacuum to give 7-(2-morpholin-4-yl- ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole.
[00276] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %), and a water content of 0.20%. Infrared (IR) spectrum was collected, which conformed to structure.
3. Example C-3
[00277] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a drying tube, a reflux condenser, and a heating mantle. The flask was charged with 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.770 kg, 5.69 mol, 1.0 equiv), N,N-dimethylformamide (18.0 L), 4-(2-chloroethyl)morpholine hydrochloride (2.751 kg, 14.78 mol, 2.6 equiv), potassium carbonate (2.360 kg, 17.10 mol, 3.0 equiv), and tetrabutylammonium iodide (0.130 kg, 0.36 mol, 0.06 equiv) with stirring. The resulting yellow suspension was heated to about 90 °C to 95 °C, maintaining the temperature for approximately 5 hours. The reaction was monitored by TLC until no starting material was observed (20% methanol / ethyl acetate; Rf (product) = 0.15; Rf (starting material) = 0.85).
[00278] The reaction mixture was cooled to -10 °C, and the yellow solids were collected by filtration on a polypropylene filter pad. The solids were slurried in water (2 X 5 L) and filtered. The crude wet product was slurried in acetone (5 L), filtered, and the solids were rinsed with acetone (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 45 °C. Yield: 2.300 kg (95%), yellow solid. TLC: R/ = 0.15 (20% methanol / EtOAc). HPLC: 95.7% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00279] Table: Yields from multiple batch runs
4. Example C-4
[00280] To a reactor were added 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), tetrabutylammonium iodide (0.24 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 85 °C and 95 °C and stirred at between 85 °C and 95 °C for 15 to 30 minutes. The mixture was then heated to between 105 °C and 120 °C and stirred at between 105 °C and 120 °C (e.g. , 115 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2- chloroethyl)morpholine hydrochloride (0.03 kg) may be added and the reaction mixture stirred at between 105 °C and 120 °C (e.g. , 115 °C) until reaction completion.
[00281] The reaction mixture was cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor was added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone was then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture was then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with deionized water (e.g. , about 45x deionized water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). The desired product was obtained in about 89% yield having about 99% purity by HPLC.
5. Example C-5
[00282] To a reactor were added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00283] The reaction mixture was cooled to between 60 °C and 70 °C over a period of at least 60 minutes. Industrial water (6 L) was added to the reactor. The reaction mixture was cooled to between 20 °C and 30 °C. Acetone (6 L) was added to the reactor. The mixture was stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with industrial water (e.g. , about 45 x industrial water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C, until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours).
6. Example C-6
[00284] To a reactor is added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) is added into the reactor. The DMF has a water content of no more than 0.05% w/w. The mixture is stirred for between 15 and 30 minutes to render a homogeneous suspension, which is heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction is complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction is not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00285] The reaction mixture is cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor is added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone is then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture is then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture is filtered, and the solid is washed with deionized water (e.g. , about 45x deionized water) until pH of washes is below 8. The solid is then washed with acetone (9.7 L). The solid is dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer is less than 0.30% w/w and TGA curve shows a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). D. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- aminophenyl)imidazo [2, 1 -bjbenzothiazole
[00286] To a 200-L high pressure (HP) reactor was charged a slurry of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,l-&]benzothiazole (VII) (7.50 kg, 17.7 mol, 1.0 equiv) in methanol (30 kg). The container was rinsed with additional methanol (10 kg) and the rinse was charged to the reactor. The reactor was then charged with THF (67 kg) and methanol (19 kg). The contents were agitated and the reactor was flushed with nitrogen by alternating nitrogen and vacuum. Vacuum was applied to the reactor and Raney Ni catalyst (1.65 kg, 0.18 wt. equiv) was charged through a sample line. Water (1 kg) was charged through the sample line to rinse the line. The reactor was flushed with nitrogen by alternating nitrogen and vacuum. The reactor was then vented and heated to 50 °C. The reactor was closed and pressurized with hydrogen gas to 15 psi keeping the internal temperature below 55 °C. The reactor was vented and re- pressurized a total of 5 times, then pressurized to 150 psi with hydrogen gas. The contents were agitated at 50 °C for at least 4 hours. At this point a hydrogen uptake test was applied: The reactor was re-pressurized to 150 psi and checked after 1 hour. If a pressure drop of more than 5 psi was observed, the process was repeated. Once the pressure drop remained < 5 psi, the reactor was vented and sampled. The reaction was deemed complete when 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,1- 6]benzothiazole (VII) was < 0.5% by HPLC.
[00287] The reactor was flushed with nitrogen as shown above. The 200-L HP reactor was connected to the 2000-L GL reactor passing through a bag filter and polish filter. The bag filter and polish filter were heated with steam. The 200-L HP reactor was pressurized (3 psi nitrogen) and its contents were filtered into the 2000-L reactor. The filtrates were hot. The 200-L reactor was vented and charged with THF (67 kg) and methanol (59 kg), the reactor agitated, and filtered into the 2000-L GL reactor.
[00288] A total of 6 reductions (46.2 kg processed) were carried out and the combined batches were concentrated by vacuum distillation (without exceeding an internal temperature of 40 °C) to a volume of -180 L. The reactor was cooled to 20 °C and charged with heptane (250 kg) and again vacuum distilled to a volume of -180 L. The reactor was charged with heptane (314 kg) and agitated at 20 °C for at least 1 hour, and then the product was isolated by centrifugation or collection on a Nutsche filter, washing with heptanes (2-5 kg per portion for centrifugation, followed by a 10-20 kg heptanes rinse of the reactor; or 94 kg for Nutsche filtration, rinsing the reactor first). The cake was blown dry, transferred to a vacuum oven and dried to constant weight maintaining a temperature < 50 °C to give the desired product (VIII) (34.45 kg, 80% yield, HPLC purity = 97.9%).
2. Example D-2
[00289] 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole was dissolved into methanol and THF and placed in a hydrogenator. Raney nickel was added and the vessel was pressurized with hydrogen and stirred for >24 hours. The reaction mixture was concentrated to a thick paste and diluted with methyl ferf-butyl ether. The resulting solids were filtered and washed with methyl ferf-butyl ether and dried under vacuum to give 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2, 1 -bjbenzothiazole.
[00290] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %). IR was collected, which conformed to structure.
3. Example D-3
[00291] Into a 5-gallon autoclave, 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo[2,l-&]benzothiazole (580 g, 1.37 mol, 1.0 equiv), THF (7.5 L), methanol (7.5 L, AR) and -55 g of decanted Raney nickel catalyst were added. The reaction vessel was purged with nitrogen (3 X 50 psi) and hydrogen (2 X 50 psi), with stirring briefly under pressure and then venting. The autoclave was pressurized with hydrogen (150 psi). The mixture was stirred and the hydrogen pressure was maintained at 150 psi for over 24 hours with repressurization as necessary. The reaction progress was monitored by TLC (10% methanol / chloroform with 1 drop ammonium hydroxide; Rf (product) 0.20; Rf (SM) 0.80). The reaction was substantially complete when the TLC indicated no starting material present, typically after 24 hours of stirring at 150 psi. The hydrogenation was continued at 150 psi for a minimum of 4 hours or until completion if starting material was still present after the initial 4 hours.
[00292] The reaction mixture was filtered through a Buchner funnel equipped with a glass fiber filter topped with a paper filter. Unreacted starting material was removed together with the catalyst. The filtrate was concentrated to a total volume of 0.5 L, and the residue was triturated with methyl ferf-butyl ether (0.5 L). The resultant solids were collected by filtration, and washed with methyl ferf-butyl ether (0.3 L) (first crop).
[00293] The filtrate was concentrated to dryness and the residue was diluted with methyl ferf-butyl ether (2 L). The resultant solids were collected by filtration, washing with methyl ferf-butyl ether (0.5 L) (second crop).
[00294] The solids were dried in a vacuum oven (<10 mm Hg) at 25 °C. Yield: 510 g (95%), beige solid. TLC: R/ 0.2 (10% methanol / chloroform with one drop of ammonium hydroxide). HPLC: 99.0% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00295] Table: Yields from multiple batch runs
4. Example D-4
[00296] The reaction of Step D was carried out in multiple runs under various conditions, such as, e.g. , varying catalyst loading, concentration of reactant, reaction temperature, and/or workup procedures. The results are summarized in the table below.
Description Run # l Run # 2 Run # 3 Run # 4 Run # 5Rxn Temp (°C) RT RT RT RT RT
Rxn Time (Hr) 24 hr 24 hr 24 hr 24 hr 24 hr
Filtered the Filtered the solution
Filtered the Filtered the Filtered the
solution through through celite. The solution through solution through solution through
with THF, refluxed in THF concentrated, concentrated, concentrated,
concentrated, washed with hot solvent exchanged solvent exchanged solvent exchanged
Work Up solvent exchanged THF, concentrated, with heptane, with heptane, with heptane,
with heptane, solvent exchanged stirred the solids stirred the solids stirred the solids
stirred the solids with heptane, stirred and filtered and filtered and filtered
and filtered the solids and washed with washed with washed with
washed with filtered washed with heptane heptane heptane
heptane heptane
Produce (VIII) 1.9 g 3.88 g 1.11 g 2.6 g 4.4 g
Yield 88% 83.4% 56 94.6%
HPLC purity 95.6% 77.5% 91% 93.8%
5. Example D-5
[00297] To a pressure reactor under nitrogen atmosphere was added a slurry of Raney Nickel in water (0.22 kg) (e.g. about 0.14 kg dry catalyst in water) and the charging line was rinsed with deionized water (0.13 L). To another reactor (Reactor B) were added methanol (5.05 L) and 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2, 1- &]benzothiazole (1.0 kg), and the mixture was stirred for between 15 and 30 minutes to render a homogenous suspension. The suspension was transferred to the pressure reactor. Reactor B was washed with methanol (4.88 L) and the wash was transferred to the pressure reactor. Tetrahydrofuran (10.1 L) was added to the pressure reactor.
Hydrogen was charged to the pressure reactor to a pressure of between 2.0 bar and 3.0 bar. The reactor was heated to a temperature of between 45 °C and 55 °C. Hydrogen was then charged to the pressure reactor to a pressure of between 6.0 bar and 7.0 bar. The mixture was stirred at a temperature of between 45 °C and 55 °C (e.g. , 50 °C), while maintaining the hydrogen pressure between 6.0 bar and 7.0 bar until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00298] The mixture was filtered while maintaining the temperature at between 35 °C and 50 °C. The pressure reactor and the filter were washed with a mixture of THF (10.1 L) and methanol (9.93 L) preheated to a temperature of between 45 °C and 55 °C (e.g. , 50 °C). The combined filtrate was concentrated to a volume of between 2.4 L and 2.8 L under vacuum at a temperature of no more than 40 °C, and a precipitate was formed. Methanol (7.5 L) was added. The slurry was cooled to a temperature of between 5 °C and -5 °C (e.g. , over 3 hours) and stirred for at least 1 hour (e.g. , for 3 hours) while maintaining the temperature at between 5 °C and -5 °C. The suspension was filtered. The solid was washed with methanol (2 X 1.2 L). The solid was then dried under vacuum at a temperature of less than 50 °C until the methanol and THF contents were each less than 3000 ppm as analyzed by GC (e.g. , less than 1500 ppm). The desired product was obtained in about 88.5% yield having about 99% purity by HPLC.
E. Preparation of phenyl 5-£er£-butylisoxazol-3-ylcarbamate
[00299] The jacket temperature of a 200-L glass-lined (GL) reactor was set to 20 °C. To the reactor was charged 5-ieri-butylisoxazole-3-amine (15.0 kg, 107.0 mol, 1.0 equiv), then K2C03 (19.5 kg, 141.2 mol, 1.3 equiv) and anhydrous THF (62 kg).
Agitation was started and then phenyl chloroformate (17.6 kg, 112.4 mol, 1.05 equiv) was charged. The charging line was rinsed with additional anhydrous THF (5 kg). The reaction was agitated at 20 + 5 °C for at least 3 hours then sampled. The reaction was deemed complete if 5-£er£-butylisoxazole-3-amine was < 2% by HPLC. If the reaction was not complete after 6 hours, additional K2CO3 and phenyl chloroformate may be added to drive the reaction to completion.
[00300] Once complete, the reaction was filtered (Nutsche). The filter was rinsed with THF (80 kg). The filtrate was vacuum distilled without exceeding an internal temperature of 40 °C until -50 L remained. Water (188 kg) and ethanol (45 L) were charged, and the mixture was agitated for at least 3 hours with a jacket temperature of 20 °C. The resulting solid was isolated by centrifugation or collection on a Nutsche filter, rinsed with water (2-5 kg for each centrifugation portion; 30 kg for Nutsche filtration) and blow-dried. The solid was then dried to constant weight in a vacuum oven (45 °C) to give the desired product (19.4 kg, 92% yield, HPLC purity = 97.4%). On an 800 g scale, 1559 g of the desired product (98% yield) was obtained with a 99.9% HPLC purity. JH NMR (DMSO-i¾) δ 11.17 (s, 1H); 7.4 (m, 2H); 7.2 (m, 3H); 1.2 (s, 9H). LCMS (M+H)+ 261.
F. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea
1. Example F-l
[00301] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- &]benzothiazole (26.7 kg, 67.8 mol, 1.0 equiv), 3-amino-5-?-butylisoxazole phenyl carbamate (19.4 kg, 74.5 mol, 1.1 equiv), DMAP (0.5 kg, 4.4 mol, 0.06 equiv), and DCM (anhydrous, 260 kg). Agitation was started, triethylamine (1.0 kg, 10.2 mol, 0.15 equiv) was charged followed by additional DCM (5 kg) through the charging line. The reaction was heated to reflux (-40 °C) and agitated for at least 20 hours with complete dissolution observed followed by product crystallizing from solution after -30 minutes. The reaction was sampled and deemed complete when HPLC analysis showed a ratio of compound (VIII) to compound (I) < 1%.
[00302] The reactor was cooled to 0 °C and stirred for at least 2 hours. The content of the reactor were isolated by centrifuge. Each portion was rinsed with 2-3 kg of cold (0 °C) DCM and spun dry for at least 5 minutes with a 10 psi nitrogen purge. For the final portion, the reactor was rinsed with 10 kg of cold (0 °C) DCM and transferred to the centrifuge where it was spun dry for at least 5 minutes with a 10 psi nitrogen purge. The combined filter cakes were transferred to a vacuum tray dryer and dried to constant weight at 50 °C and at least >20 inches of Hg to give the desired product (I) (35.05 kg, 92% yield, HPLC purity = 98.8%). Phenol was the major impurity detected (0.99%); and three other impurities (<0.10%) were detected. JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example F-2
[00303] A variety of solvents were used in the reaction of Step F to optimize for better yields and purity profiles. The contents of the symmetrical urea impurity (XI) were compared and summarized in the table below:
[00305] To Reactor A under a nitrogen atmosphere was added 7-(2-morpholin-4-yl- ethoxy)-2-(4-aminophenyl)imidazo[2,l-&]benzothiazole (1 kg) and DMAP (0.02 kg). To Reactor B under a nitrogen atmosphere was added 3-amino-5-?-butylisoxazole phenyl carbamate (0.73 kg) and DCM (5.6 L). The DCM used had a water content of less than 0.05 % w/w. The mixture in Reactor B was stirred until dissolution. The solution was transferred into Reactor A (the solution can be filtered into Reactor A to remove any insoluble impurities in the carbamate starting material), and the mixture was stirred in Reactor A. Reactor B was washed with DCM (0.8 L) and the wash was transferred into Reactor A. Reactor A was washed with DCM (0.9 L). To Reactor A was added triethylamine (0.1 L) and the charging vessel and lines were rinsed with DCM (0.1 L) into Reactor A. The mixture was then heated to reflux and stirred at reflux until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00306] The reaction mixture was cooled (e.g. , over 2 to 3 hours) to a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The mixture was then stirred for 2 to 3 hours at a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The suspension was filtered. The solid was washed with cool DCM (2 X 1.5 L) (pre-cooled to a temperature of between -5 °C and 5 °C). The solid was dried under vacuum at a temperature of less than 45 °C until the DCM content was less than 1000 ppm (e.g., below 600 ppm) as analyzed by GC. The desired product was obtained having about 99% purity by HPLC.
G. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-b] [1 ,3]benzothiazol-2-yl]phenyl }urea dihydrochloride
1. Example G-l
[00307] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1-&][1, 3]benzothiazol-2-yl]phenyl}urea (35.0 kg, 62.4 mol, 1.0 equiv) followed by methanol (553 kg). Agitation was started and the reaction mixture was heated to reflux (-65 °C). Concentrated aqueous HC1 (15.4 kg, 156.0 mol, 2.5 equiv) was charged rapidly (<5 minutes) and the charge line was rinsed into the reactor with methanol (12 kg). Addition of less than 2.0 equivalents of HC1 normally resulted in the formation of an insoluble solid. The reaction mixture was heated at reflux for at least 1 hour. Upon HC1 addition, the slurry dissolved and almost immediately the salt started to crystallize, leaving insufficient time for a polish filtration.
[00308] The reactor was cooled to 20 °C and the product was isolated by filtration (Nutsche) rinsing the reactor and then the cake with methanol (58 kg). The solid was then dried in a vacuum oven (50 °C) to constant weight to give the desired
dihydrochloride salt (35 kg, 89% yield, HPLC purity = 99.94%). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
2. Example G-2
[00309] Concentrated HC1 was added to a suspension of N-(5-ieri-butyl-isoxazol-3- yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea in warm methanol forming a solution that slowly began to precipitate. The reaction mixture was refluxed for over 2 hours and then stirred overnight at ambient temperature. The dihydrochloride salt was collected and dried under vacuum.
3. Example G-3
[00310] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a nitrogen inlet, a drying tube, a reflux condenser, an addition funnel, and a heating mantle. The flask was charged with N-(5-ieri-butyl-isoxazol-3-yl)- N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2-yl]phenyl}urea (775 g, 1.38 mol, 1.0 equiv) and MeOH (40 L, AR). The resulting off-white suspension was heated to reflux (68 °C). A clear solution did not form. HC1 (37% aqueous) (228 mL, 3.46 mol, 2.5 equiv) was added over 5 minutes at 68 °C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Heating was continued at reflux for approximately 5 hours. The reaction mixture was allowed to cool to ambient temperature overnight. The off-white solids were collected by filtration on a polypropylene filter, washing with MeOH (2 X 1 L, AR). [00311] Two lots of material prepared in this manner were combined (740 g and 820 g). The combined solids were slurried in methanol (30 L) over 30 minutes at reflux and allowed to cool to the room temperature. The solids were collected by filtration on a polypropylene filter, rinsing with methanol (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 1598 g (84%), off-white solid. HPLC: 98.2% (area). MS: 561.2 (M+l)+. JH NMR (300 MHz, DMSO-i¾) conformed to the structure. Elemental Analysis (EA): Theory, 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual, 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
4. Example G-4
[00312] Into a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, a heating mantle, a condenser and a nitrogen inlet, were charged N-(5-ieri-butyl-isoxazol- 3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (1052.4 g, 1.877 mol, 1.0 equiv) and methanol (21 L). The reactor was heated and stirred. At an internal temperature of > 50 °C, cone. HC1 (398.63 mL, 4.693 mol, 2.5 equiv) was charged over 5 minutes through an addition funnel. During the addition, the mixture changed from a pale yellow suspension to a white suspension. The internal temperature was 55 °C at the conclusion of the addition. The mixture was heated to reflux for 1 hour, then heating was discontinued and the mixture was allowed to cool to room temperature. The mixture was filtered in two portions, and each filter cake was washed with methanol (2 X 1 L), transferred to trays and dried in a vacuum oven (45 °C) to constant weight. The dried trays were combined to produce 1141.9 g of the salt (96% yield, 99.1 % HPLC purity, 10.9% chloride by titration).
[00314] A batch of about 30 grams of N-(5-ieri-butyl-isoxazol-3-yl)-N’- {4-[7-(2- morpholin-4-yl-ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride was prepared using the methods described herein. This lot was
prepared in accordance with the requirements for production of clinical Active
Pharmaceutical Ingredients (APIs) under GMP conditions. The analytical data for this batch was obtained, and representative data were provided herein. [00315] Summary of analytical data for the dihydrochloride salt.
EXAMPLE 1. SYNTHESIS OF N-(5-TERT-BUTYL-ISOXAZOL-3-YU- N>-{4-f7-(2-MORPHOLIN-4- YL-ETHOXY)IMID AZO[2,1- BlH,31BENZOTHIAZOL-2-YL|PHENYLiUREA (“COMPOUND Bl”)
[00357] A. The intermediate 2-amino-l,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf: J. Org. Chem. 1970, 35, 4103- 4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 mL ethanol and 9 mL concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00358] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2- amino-l,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSO-^) Sl.6 (m, 2H), 6.6 (d, IH). [00359] B. To prepare the 2-(4-nitrophenyl)imidazo[2,l-b][l,3]benzothiazol-7-ol intermediate, 2-amino-l,3-benzothiazol-6-ol (20.0 g, 0.12 mol) and 2-bromo-4′- nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to O0C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2- (4-nitrophenyl)imidazo[2,l-£][l,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-(I6) δ 10 (s, IH), 8.9 (s, IH), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, IH), 7.4 (s, IH), 6.9 (d, IH).
[00360] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,l-
NYI-4144519vl 84 (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2, 1 – b][\, 3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-d6) δ 8.97 (s, IH), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, IH), 7.7 (s, IH), 7.2 (d, IH), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00361] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole: To a suspension of 7-(2-morpholin-4-yl-ethoxy)- 2-(4-nitro-phenyl)imidazo[2,l -b][\ , 3]benzothiazole (3.87g, 9.1 mmol) in 100 mL isopropyl alcohol/water (3:1) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 mL). The filtrate was concentrated to approximately 1/3 of the original volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water – in which case the reaction is virtually complete after 3.5 hours and only traces of starting material are observed in the product obtained. NMR (DMSO-Λfc) δ 8.4 (s, IH), 7.8 (d, IH), 7.65 (d, IH), 7.5 (d, 2H), 7.1 (d, IH), 6.6 (d, 2H), 4.1 (t, 2H), 3.6 (m, 4H), 2.7 (t, 2H).
[00362] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert-butylisoxazole-3- isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over
NYI-4144519vl 85 MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of Compound B 1. Yield: 2.342 g (41 %) NMR (DMSO-J6) £9.6 (br, IH), 8.9 (br, IH), 8.61 (s, IH), 7.86 (d, IH), 7.76 (d, 2H), 7.69 (d, IH), 7.51 (d, 2H), 7.18 (dd, IH), 6.52 (s, IH), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCl3) £9.3 (br, IH), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, IH), 7.22 (d, IH), 7.03 (dd, IH), 5.88 (s, IH), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
6.2 EXAMPLE 2. ALTERNATIVE SYNTHESIS QF N-(5-TERT-BUTYL- ISOXAZQL-3- YL)-N -{4-[7-q-MORPHOLIN-4- YL- ETHOXYUMID AZOf2,l-BUl,31BENZOTHIAZOL-2- YLIPHENYLIUREA (“COMPOUND Bl”)
[00363] A. To a suspension of the intermediate 2-(4-Nitrophenyl)imidazo[2,l- b][l,3]benzothiazol-7-ol from Example IB (2.24 g, 7.2 mmol) in ethanol (40 mL) was added SnCl21H2O (7.9Og, 35 mmol) and heated to reflux. Concentrated HCl was added to the reaction mixture and the precipitate formed gradually. The reaction mixture was heated to reflux for 20 hours and then allowed to cool to room temperature. The solution was poured into ice and neutralized with 10% NaOH and adjusted to approximately pH 6. The organic phase was extracted three times with ethyl acetate (80 mL x 3). Extracts were dried over MgSθ4 and concentrated to give a yellow solid. (1.621 g, 80%). The solid was recrystallized from methanol to give a pure product (1.355 g, 67%).
[00364] B. To a suspension of the intermediate from Step 2A (0.563 g, 2 mmol) in toluene (30 mL) was added 5-tert-butylisoxazole-3-isocyanate (0.332g, 2 mmol) and heated to reflux overnight. LC-MS analysis showed presence of the intermediate but no trace of 5- tert-butylisoxazole-3-isocyanate and an additional 0.166 g of the isocyanate was added. The reaction was again heated to reflux overnight. Completion of reaction was verified by LC- MS. The solvent was removed and the resulting mixture was dissolved in methanol which was removed to give the second intermediate as a solid.
[00365] The mixture was dissolved in CH2Cl2 (150 mL) and washed with saturated
NaHCO3. The organic layer was dried over MgSO4, concentrated, and purified by silica gel chromatography three times, first using a methanol/CH2Cl2 gradient, the second time using a
NYI-4144519vl 86 hexane/ethyl acetate gradient followed by a methanol/ethyl acetate gradient, and a third time using a methanol/CH2Cl2 gradient.
[00366] C. To a suspension of the intermediate from Step 2B (0.1 10 g, 0.25 mmol) in
THF (5mL) was added Ph3P (0.079g, 0.3 mmol), diisopropylazodicarboxylate (0.06 Ig, 0.3 mmol) and 4-morpholinoethanol (0.039 g, 0.3 mmol). The reaction mixture was stirred at room temperature overnight. Completion of the reaction was verified by LC-MS. The solvent was removed and the final product was purified using silica gel chromatography, with methanol in CH2Cl2 (0.030g, 21%).
6.3 EXAMPLE 3. BULK SYNTHESIS OF N-(5-TERT-BUTYL- ISOXAZOL-3-YL)-N’-f4-[7-(2-MORPHOLIN-4-YL- ETHOXY^IMID AZO[2α-BUlJlBENZOTHIAZOL-2- YLlPHENYLiUREA (“COMPOUND Bl”)
[00367] A multi-step reaction scheme that was used to prepare bulk quantities of
Compound Bl is depicted in FIG. 66a and FIG. 66b, and is described further below. [00368] Step 1 : Preparation of 2- Amino-6-hydroxybenzothiazole (Intermediate 1). 2-
Amino-6-methoxybenzothiazole is reacted with hot aqueous HBr for about 3 hrs and then the clear solution is cooled to ambient temperature overnight. The precipitated solids are collected, dissolved in hot water and the pH is adjusted to between 4.5-5.5. The resultant solids are collected, dried and recrystallized from Isopropanol. Second crop material is collected. The solids are vacuum dried to give Intermediate 1.
[00369] Step 2: Preparation of 2-(4-Nitrophenyl) imidazo [2J-b]benzothiazol-7-ol
(Intermediate 2). 2-Amino-6-hydroxybenzothiazole, 2-Bromo-4-nitroacetophenone and absolute Ethanol are added together and heated to reflux for approximately 24 hours. Tetrabutylammonium iodide is added and the reaction is refluxed an additional 12 hours. The resulting yellow suspension is cooled and the solids collected and washed with Ethanol and Diethyl ether. The solids are dried under vacuum to give Intermediate 2. [00370] Step 3: Preparation of 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo
[2,1-b] benzothiazole (Intermediate 3). Intermediate 2, 4-(2-Chloroethyl)morpholine hydrochloride, Potassium carbonate and Tetrabutylammonium iodide are added to N,N- Dimethylformamide forming a yellow suspension that is heated for over 3 hours. The reaction is cooled and the solids are collected, slurried into water, filtered, slurried into
NYl-4 l4451′)v l 87 acetone, filtered and washed with Acetone to give yellow solids that are dried under vacuum to give Intermediate 3.
[O0371] Step 4: Preparation of 7-(2-Moφholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2,1 -b] benzothiazole (Intermediate 4). Intermediate 3 is dissolved into Methanol and THF and placed in a Hydrogenator. Raney Nickel is added and the vessel is pressurized with Hydrogen and stirred for >24 hrs. The reaction mixture is concentrated to a thick paste and diluted with Methyl tert-butyl ether. The resulting solids are filtered and washed with Methyl tert-butyl ether and dried under vacuum to give Intermediate 4. [O0372] Step 5: Preparation of {[5-(tert-Butyl) isoxazol-3-vnatnino}-N-{4-r7-(2- morpholin-4-yl-ethoxy)(4-hvdroimidazolo[2J-blbenzothiazol-2-yl)]phenyl|carboxamide (Compound Bl). 3 -Amino- 5 -tert-butyl isoxazole in Methylene chloride is added to a vessel containing toluene which is cooled to approx 0 0C. Triphosgene is then added and the reaction mixture is cooled to below -15 0C. Triethylamine is added, followed by Intermediate 4. The mixture is heated to distill off the Methylene chloride and then heated to over 60 0C for over 12 hours and cooled to 50-60 °C. The resulting solids are filtered, washed with Heptane, slurried with 4% sodium hydroxide solution, and filtered. The solids are then washed with Methyl tert-butyl ether followed by Acetone and dried under vacuum to give Compound Bl.
6.4 EXAMPLE 4. EXAMPLES OF PREPARATION OF COMPOUND Bl HCL SALT
[00373] Example A: For the preparation of a hydrochloride salt of Compound Bl5 N-
(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b][l,3]benzothiazol-2-yl]phenyl}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCl in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrirugation and washed with ethyl ether to give a hydrochloride salt of Compound Bl. Yield: 2.44 g (98 %) NMR (DMSO-^) S X 1.0 (br, IH), 9.68 (s, IH), 9.26 (s, IH), 8.66 (s, IH), 7.93 (d, IH), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, IH), 6.53 (s, IH), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). [00374] Example B: Concentrated HCl is added to a suspension of Compound Bl in warm methanol forming a solution that slowly begins to precipitate. The reaction mixture is
NYI-4144519vl 88 refluxed for over 2 hrs and then stirred overnight at ambient temperature. The HCl salt is collected and dried under vacuum.
[00375] Example C: Materials: {[5-(tert-Butyl) isoxazol-3-yl]amino}-N-{4-[7-(2- morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l-6]benzothiazol-2-yl)] phenyl }carboxamide (775 g, 1.38 mol, 1.0 eq); HCl 37% aqueous (288 mL, 3.46 mol, 2.5 eq); Methanol (MeOH, AR) (40L). Procedure: (Step 1) Equipped a 5OL 3-neck round bottom flask with a mechanical agitator, thermocouple probe, Nitrogen inlet, drying tube, reflux condenser, addition funnel and in a heating mantle. (Step 2) Charged the flask with {[5-(tert-Butyl) isoxazol-3-yl] amino}-N-{4-[7-(2-morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l- b]benzothiazol-2-yl)] phenyl jcarboxamide (775g) and MeOH, AR (40L). Heat the resulting off-white suspension to reflux (680C). A clear solution did not form. (Step 3) Added HCl (37% aqueous) (228 mL) over 5 minutes at 68°C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Continued heating at reflux for approximately 5 hours. Allowed the reaction mixture to cool to ambient temperature overnight. (Step 4) Collected the off-white solids by filtration onto a polypropylene filter, washing the solids with MeOH, AR (2 x 1 L). (Step 5) Combined two lots of material prepared in this manner (74Og and 82Og). Slurried the combined solids in Methanol (30 L) over 30 minutes at reflux and cool to the room temperature. (Step 6) Collected the solids by filtration onto a polypropylene filter, rinsing with Methanol (2 x 1.5L). (Step 7) Dried the solids in a vacuum oven (<10mniHg) at 400C. Yield: 1598 g (84%), off-white solid; HPLC: 98.2% (area); MS: 561.2 (M+l); IH NMR: conforms (300 MHz, DMSO-d6); Elemental Analysis (EA): Theory = 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual = 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
NYl-4I44519v! 89 [00376] Examples of Compound Bl HCl salt synthesis
[00377] Example D: In a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, heating mantle, condenser and nitrogen inlet was charged Compound Bl (1052.4 g, 1.877 mol, 1.00 equiv.) and methanol (21 L). The reactor was heated and stirred. At an internal temperature > 50 0C, cone. HCl (398.63 mL, 4.693 mol, 2.5 equiv.) was charged over 5 minutes through an addition funnel. With the addition, the reaction changed from a pale yellow suspension to a white suspension. The internal temperature was 55 0C at the conclusion of the addition. The reaction was heated to reflux for 1 hour, then heating discontinued and the reaction allowed to cool to room temperature. The reaction was filtered in two portions, each filter cake washed with methanol (2 x 1 L), transferred to trays and dried in a vacuum oven (45 0C) to constant weight. The dried trays were combined to produce 1141.9 g, 96% yield, 99.1 % HPLC purity, 10.9% chloride by titration.
Solid Forms Comprising the HCl Salt of Compound Bl 6.6.2.1 Preparation of Solid Forms
6.6.2.2 Cold Precipitation Experiments
NYl-4144519vl 102 6.6.2.3 Slurry Experiments
NYI-41445 l9vl 103 6.6.2.4 Additional Preparation of Solid Forms Comprising the HCI Salt of Compound Bl
NYl-4144519v l 104
NYM 144519vl 105
N Y l -4 1 4 4 5 1 9 v l 1 0 6
NYI-4I44519vi 107
N V I 4 1 4 4 5 1 9 1 0 8
“Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, VD = vapor diffusion, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
6.6.2.5 Scale-up Experiments of Involving Crystal Forms Comprising the HCl Salt of Compound Bl
NYI-4144519v l 109
Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
……………………
Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor J Med Chem 2009, 52(23): 7808
N-(5-tert-Butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (7): General Procedure D
A suspension of 2-(4-aminophenyl)-7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazole (19c) (4.06 g, 10.3 mmol) and 5-tert-butyl-isoxazole-3-isocyanate (5) (1.994 g, 12 mmol) in toluene (60 mL) was heated at 120 °C overnight. The reaction was quenched with a mixture of dichloromethane and water containing a little methanol, and the mixture was neutralized with saturated aqueous NaHCO3. The aqueous phase was extracted twice with dichloromethane, and the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to a volume of about 20 mL and ethyl ether was added, resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of 7 (2.342 g, 41%).
General Procedure E for Preparation of Hydrochloride Salt
The free base was dissolved in a mixture of dichloromethane (20 mL) and methanol (1 mL). A solution of 1.0 M HCl in ethyl ether (1.1 equiv for all compounds except 7, for which 2.5 equiv were used) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration to give
Puma Biotechnology, a development stage biopharmaceutical company, announced the presentation of positive results from the phase II clinical trial of Puma’s investigational drug PB272 (neratinib) for the neoadjuvant treatment of breast cancer(I-SPY 2 TRIAL) in an oral presentation at the American Association for Cancer Research (AACR) Annual Meeting 2014 in San Diego, California.
Neratinib – малая молекула класса 6,7-дизамещенных-4-anilinoquinoline-3-карбонитрила –
ингибитор тирозинкиназы HER-2 с потенциальной противоопухолевой активностью.
Neratinib связывается с рецептором HER-2 необратимо, снижая аутофосфорилирование в клетках,
и направляя остаток цистеина в АТФ-связывающего кармана рецептора.
Обработка раковых клеток с этим агентом приводит к торможению передачи сигнала клеточного цикла и
в конечном счете уменьшает клеточную пролиферацию.
Neratinib ингибирует рецептор EGFR киназы и распространение EGFR-зависимых клеток.
Neratinib – small molecule 6,7-disubstituted class of 4-anilinoquinoline-3-carbonitrile –
inhibitor of the HER-2 tyrosine kinase with potential antitumor activity.
Neratinib binds to the receptor HER-2 irreversible, reducing autophosphorylation in cells
and directing the cysteine residue in the ATP-binding pocket of the receptor.
Treatment of cancer cells with this agent leads to inhibition of signal transduction and cell cycle ultimately reducescell proliferation.
Neratinib inhibit EGFR kinase receptor and distribution of EGFR-dependent cells.
Neratinib is a signal transduction pathway inhibitor and an irreversible inhibitor of HER-2 in early clinical trials for the treatment of advanced solid tumors in combination with paclitaxel. The company had also been developing the drug candidate for the treatment of non-small cell lung cancer (NSCLC); however, no recent development has been reported for the indication. In 2011, Pfizer discontinued development of the compound as monotherapy for the treatment of ErbB-2-positive breast cancer. A phase III clinical trial had been under way. Dana-Farber Cancer Institute is studying the compound for the treatment of patients with human epidermal growth factor receptor 2 (HER2)-positive breast cancer and brain metastases. Puma Biotechnology is conducting phase III trials for use as third-line treatment of HER2-positive metastatic breast cancer and phase II trials for the treatment of patients with HER2 activating mutations in Non-Small Cell Lung Cancer (as monotherapy or in combination with temsirolimus) as well as other solid tumors.
The drug candidate is a synthetic compound developed based on the chemical structure of EKB-569, an inhibitor of the epidermal growth factor receptor (EGFR) currently under clinical evaluation for the treatment of EGFR-positive tumors. In previous trials, neratinib inhibited kinase activity of HER-2 and EGFR by 50% while showing no effects on several serine-threonine kinases, and also inhibited the proliferation of two HER-2-positive breast cancer cell lines and a mouse fibroblast cell line transfected with the HER-2 oncogene.
In 2011, the compound was licensed to Puma by Pfizer for global development and commercialization.
HKI-272 (neratinib) has been described for the treatment of neoplasms [US Patent 6,288,082]. Neratinib is a potent irreversible pan erbB inhibitor. Neratinib is an orally available small molecule that inhibits erbB-1 , erbB-2 and erbB-4 at the intracellular tyrosine kinase domains, a mechanism of action that is different from trastuzumab. Neratinib reduces erbB-1 and erbB-2 autophosphorylation, downstream signaling, and the growth of erbB-1 and erbB-2 dependent cell lines.
Preclinical data suggest that neratinib will have antitumor activity in erbB-1 – and/or erbB 2-expressing carcinoma cell lines, with cellular IC50 <100 nM [Rabindran SK, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Research. 2004;64(1 1 ):3958-65].
Neratanib is being developed by Puma Biotechnology. It will be included in the forthcoming I-SPY2breast cancer trial.[6]
neratinib refers to HKI-272, which has the following core structure:
in its free base form. Optionally, a pharmaceutically acceptable salt or hydrate thereof may be used. The core structure represented above is a particular HKI-272 compound, called HKI-272 or neratinib, which has the chemical name [(2E)-N-[4-[[3-chloro-4- [(pyridin-2-yl)methoxy]phenyl]amino]-3-cyano-7-ethoxyquinolin-6-yl]-4- (dimethylamino)but-2-enamide]. Although currently less preferred, another HKI-272 compound may be used in the place of neratinib. “A HKI-272 compound” refers, in one embodiment, to a compound derived from the core structure of neratinib shown above
The preparation of HKI-272 compounds, of which neratinib is a species, are described in detail in US Patent Application Publication No. 2005/0059678, which is hereby incorporated by reference. See, also, US Patent Nos. 6,288,082, US Patent No. 6,002,008, US Patent No. 6,297,258 and US Patent Application Publication No. 2007/0104721 , which are hereby incorporated by reference. The methods described in these documents can also be used to prepare neratinib and/or the other HKI-272 and substituted 3-quinoline compounds used herein and are hereby incorporated by reference. In addition to the methods described in these documents, International Patent Publication Nos. WO-96/33978 and WO-96/33980, which are hereby incorporated by reference, describe methods that are useful for the preparation of these HKI-272 compounds. Although these methods describe the preparation of certain quinazolines, they are also applicable to the preparation of correspondingly substituted 3- cyanoquinolines and are hereby incorporated by reference.
The term “treating” or “treatment” refers to the administration of the neratinib to a subject to prevent or delay, to alleviate, or to arrest or inhibit development of the symptoms or conditions associated with neoplasms
(E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4- (dimethylamino)-2-butenamide is an irreversible inhibitor to Her-2 (also known as ErbB-2 or neu) kinase, a member of the epidermal growth factor receptor (EGFR) family. EGFR family members have been implicated in tumorigenesis and associated with poor prognosis in tumor types in humans. The structure of the (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano- 7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide in the form of a free base is shown below:
The compound (E)-N-{4-[3-chloro-4 J-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}- 4-(dimethylamino)-2-butenamide in the form of a free base is described in U.S. Patent No. 6,288,082. The compound is classified, based on the Biopharmaceutical Classification System, as a BCS Class IV compound (low water solubility and low permeability). The free base has low solubility in water, with a water solubility of about 1 μg/ml_ at about pH 7. The water solubility increases with decreasing pH as the compound becomes ionized. This compound is water soluble at gastrointestinal pH, and dissolution is not rate limiting.
Research on Chemical Intermediates, 2012, 09(22),6168
10.1007/s11164-012-0822-4
The Wittig–Horner reaction for the synthesis of neratinib
U.S. Pat. No. 7,126,025 discloses certain novel 4-amino-2-butenoyl chlorides, processes for their preparation and their use as intermediates in the synthesis of pharmaceutically active protein kinase inhibitors, including but not limited to for example HKI-272 and EKB-569.
The sequence illustrated below and summarized in Scheme 1 describes one existing process for preparing HKI-272, (E)-Λ/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3- cyano-7-ethoxyquinolin-6-yl)-4-(dimethylamino)but-2-enamide in the form of the maleate salt, also known as Neratinib™.
1 95 eq (COCI)2, cat DMF
O
^
Step 5 OH 16 h HCI
Scheme 1
Scheme 2
Scheme 3. Formation of acid chloride with SOCI2 in DMAc and coupling with a substituted aniline.
SOCl2
/Nv^-^’C02H HCI DMAc HCI
Scheme 4. Formation of the MW 638 impurity.
Example 4: Process 3
4-Dimethylaminocrotonoyl chloride hydrochloride and its coupling with 6-amino- 4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-7-ethoxyquinoline-3-carbonitrile (procedure with thionyl chloride and DMAc).
A suspension of 4-dimethylaminocrotonic acid (17.0 g, 97.5 mmol) in DMAc (170 ml_) was cooled to -15 0C under nitrogen atmosphere. Neat thionyl chloride (12.8 g, 7.83 mmol) was added to the slurry at a rate to maintain the temperature in the reactor in the range of -18 to -14 0C (moderate exotherm). The reaction mixture was held at -17 to -15 0C for 4 hrs. A solution of the aminoquinoline (36.2 g, 81.3 mmol) in DMAc (440 ml_) was added to the reactor maintaining the temperature in the -14 to -19 0C range. The resulting mixture was held for 18 hr at approximately -15 0C. At this point HPLC analysis showed residual aniline level at 2.5%. The thick suspension of the hydrochloride salt of the coupled product was quenched with water (200 ml_) maintaining the batch temperature between -5 and -16 0C. The pH of the resulting clear solution was adjusted to 1 1 with a 13% aqueous solution of NaOH (approx. 210 ml_ of the solution was added). The suspension was further diluted with water (350 ml_) and the solids were filtered on a polypropylene cloth filter. The cake was washed with water until neutral pH of the washes and dried first in the nitrogen flow on the filter and then on a tray in vacuum at 45 to 50 0C to afford crude (.=)-/\/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3-cyano-7- ethoxyquinolin-6-yl)-4-(dimethylamino)but-2-enamide (42.0 g, 91 %) as a bright-yellow crystalline solid.
A suspension of 4-N,N-dimethylaminocrotonic acid hydrochloride in acetonitrile and a catalytic amount of DMF is cooled to 0-10° C. Oxalyl chloride (0.95 eq) is added dropwise and the mixture warmed to 25-30° C and stirred until the chlorinating agent is completely consumed. The light yellow solution is checked for complete consumption of oxalyl chloride by HPLC then cooled to 0-10° C. A cooled solution (0-10° C) of 4-[4-benzyloxy-3-chloro]amino-6-amino-3-cyano-7- ethoxyquinoline in NMP is added dropwise and the mixture is stirred until < 2% of the starting aniline remains. The mixture is added to saturated aqueous sodium bicarbonate, the yellow precipitates are filtered and washed with water. The wet solids are heated to reflux in acetonitrile and clarified hot to remove insolubles. The solution is cooled, the precipitated product filtered and washed with cold acetonitrile. The product is dried (40-50° C, 10 mm Hg, 24 hours) to obtain the final product. Reaction Scheme Example 2:
A solution of 4-N,N-dimethylaminocrotonic acid hydrochloride in tetrahydrofuran (THF) and a catalytic amount of dimethyiformamide (DMF) is cooled to 0-5s C. Oxalyl chloride (0.95 eq) is added dropwise and the mixture warmed to 25-302C and stirred until the chlorinating agent is completely consumed. The orange solution is checked for complete consumption of oxalyl chloride by high- pressure liquid chromatography (HPLC) then cooled to 0-52 C. A solution of 4-[4-(2- pyridylmethoxy)-3-chloro]amino-6-amino-3-cyano-7-ethoxyquinoline is added dropwise and the mixture is stirred until < 0.5% of the starting aniline remains. The reaction is quenched with water and the mixture warmed to 40s C. Aqueous sodium hydroxide is added to bring the pH to 10-11. The resulting precipitates are filtered hot and washed with water. The wet solids are heated to reflux (70-759 C) in acetonitrile:THF (1 :5:1) and the solution cooled slowly to room temperature. The product is filtered and washed with acetonitrile.THF. The product is dried (50e C, 10 mm Hg, 24 hours) to 80-85% yield.
A. 4-(dimethylamino)-2-butenoyl chloride hydrochloride
A 1 L multi-neck flask equipped with agitator, thermometer, addition funnel, and nitrogen protection is charged with acetonitrile (0.67 kg, 0.85 L) followed by adding dimethylformamide (0.00086 kg, 0.91 mL, d=0.944 g/mL). At ambient temperature, is added 4-dimethylaminocrotonic acid hydrochloride (0.0709 kg) and the mixture stirred until homogeneous. Cool the reaction mixture to (0-10° C) and add oxalyl chloride (0.0473 kg, 0.0325 L, d = 1.45 g/mL) dropwise over (20 minutes) at (0-10° C) followed by a rinse with acetonitrile (0.02 kg, 0.03 L). The temperature (0-10°C) is maintained for about (20 minutes). The temperature of the reaction mixture is adjusted to (22-26° C) over (20 minutes) and maintained over (2 hours). The temperature of reaction mixture is adjusted to (40-45° C) and held for about (5 minutes). Cool the light suspension to about (20-25° C) and check for reaction completion by high-pressure liquid chromatography (HPLC). The reaction is complete when there is < 15 % of the starting material (4-dimethylaminocrotonic acid hydrochloride) present and/or < 2 % of oxalyl chloride (detected as the dimethyl oxalate).
B. 4-Dimethy!amino-but-2-enoic acid |4-(3-chloro-4-fluoro-phenylamino)-3-cyano-7- ethoxy-quinolin-6-yll-amide (crude)
A 3 L multi-neck flask equipped with agitator, thermometer, dip tube, and nitrogen protection is charged N-methyl pyrrolidinone (0.77 kg, 0.75 L, d=1.033 g/mL). At ambient temperature is added 4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7- ethoxy quinoline (0.0748 kg). The reaction mixture is heated to 40-45° C and maintained for about (15 minutes). The reaction mixture is cooled to (0-10° C) and the light suspension of 4-(dimethylamino)-2-butenoyl chloride hydrochloride in CH3CN added via dip tube and positive nitrogen pressure, over (30-45 minutes) while maintaining the temperature (0-10° C) for at least (2 hours). Reaction completion is monitored by HPLC. The reaction is complete when there is < 2 % of the starting material (4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7-ethoxy quinoline) present. To a 12 L multi-neck flask equipped with agitator, thermometer, dip tube, and nitrogen protection is charged with water (2.61 kg, 2.61 L) and sodium bicarbonate (0.209 kg) with stirring until a solution is obtained followed by cooling to (20-24° C) to which is transferred the reaction mixture above which contains < 2 % of the starting material (4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7-ethoxy quinoline), via dip tube and positive nitrogen pressure, to the 12 L flask over about (45-60 minutes) while maintaining the temperature at (20-24° C). The temperature is maintained at (20-24° C) for at least (1 hour). Filter the reaction mixture on a Buchner funnel, rinse with water (3 x 0.40 kg, 3 x 0.40 L), and maintain suction until dripping stops. Dry the product in a vacuum oven at about (50° C) and about (10 mm Hg) for about (28-30 hours). The yield is 78.5 g (86%) at 79.7% strength and 12.3% total impurities.
4-Dimethylamino-but-2-enoic acid r4-(3-chloro-4-fluoro-phenylamino -3-cyano-7- ethoxy-quinolin-6-vn-amide (purified small scale)
First crop: A 6 L multi-neck flask equipped with agitator, condenser, temperature probe, and nitrogen protection is charged with acetonitrile (3.14 kg, 4.00 L) followed by adding 4-dimethylamino-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-3-cyano-7- ethoxy-quinolin-6-yl]-amide (0.16 kg, 0.167 moles). Heat the mixture to (75-80° C) and hold it for (1 hour). Cool the mixture to (70-75° C) and filter on a pad of diatomaceous earth to remove inorganic salts. Wash the pad with acetonitrile (2 x 0.24 kg, 2x 0.30 L), preheated to (70-75° C). Concentrate the filtrate at (20-30 mm Hg) and a maximum temperature of (40-45° C) to a volume of ( 1.2 L). To the concentrate (slurry) add prefiltered tetrahydrofuran (0.53 kg, 0.60 L). Heat to (65-70° C) to obtain a complete solution. Cool the mixture to (40-45° C) over (0.3 hours). Add seeds and continue cooling to (20-25° C) over (1 hour). Hold at (20-25° C) for a minimum of (18 hours). Collect the solid on a Buchner funnel and wash the collected solid with a prefiltered and precooled at (0-5° C) mixture of acetonitrile/tetrahydrofuran (2/1 by volume) (2 x .06 kg, 2 x 0.08 L). Dry the product in a vacuum oven at (50° C) and (10 mm Hg) for (48 hours) to a loss on drying (LOD) of less than (0.5 %). All washes and concentrates (mother liquors) are saved for further purification.
Second crop:
A 3 L multi-neck flask equipped with agitator, temperature probe, nitrogen protection, and charge with the mother liquors and washes from above. Concentrate by distillation at (20-30 mm Hg) and a maximum temperature of (40-45° C) to a volume of (0.50 L). Collect the solid on a Buchner funnel and wash the solid with prefiltered acetonitrile (0.04 kg, 0.05 L). Dry the solid product in a vacuum oven at (50° C) and (10 mm Hg) for (18 hours). A 1 L multi-neck flask equipped with agitator, condenser, temperature probe, nitrogen protection and charge with prefiltered acetonitrile (0.47 kg, 0.60 L), and the collected solid is heated as a suspension to (70-75° C) over (0.5 hours). Add prefiltered tetrahydrofuran (0.03 kg, 0.03 L) to the suspension while maintaining the temperature at (70-75° C). Cool the solution to (40-45° C) and add seed crystals. Continue cooling to (20-25° C) over (1 hour) and hold for (2 hours). Collect the resulting solid on a Buchner funnel and wash the collected solid with a prefiltered and precooled to (5° C) mixture of acetonitrile/tetrahydrofuran (20/1 by volume) (2 x 0.02 kg, 2 x 0.03 L). Dry the collected solid in a vacuum oven at (50° C) and (10 mm Hg) for (24 hours) to an LOD of less than (0.5 %). The combined yield is 27.5 g + 30.5 g (73%) in 96.2-98.4% strength and 1.5-1.7% total impurities by high pressure liquid chromatography (HPLC).
Acetonitrile, practical (34.0 kg) and 4-dimethylamino-but-2-enoic acid [4-(3- chloro-4-fluoro-phenylamino)-3-cyano-7-ethoxy-quinolin-6-yl]-amide (2.69 kg crude, 1.53 kg at 100% strength) are charged to a purged (100 L) reactor. Acetonitrile, practical (2.0 kg) is used as rinse for funnel and vessel walls. The brown suspension is heated at 70 to 76° C using a jacket temperature not exceeding 85° C, then held at the latter temperature for a minimum of 45 minutes, not exceeding 60 minutes. The resulting suspension is then filtered on the warm-jacketed (70-76° C) 14″ Aurora filter, while maintaining the batch temperature at 70 to 76° C. The filtrates are collected by pump into a purged (100 L) receiver, while keeping their temperature below 50° C. The diatomaceous earth pad is then washed with warm (70 to 76° C) acetonitrile, practical (3 x 2.5 kg). The filtrates and washes in (100 L) receiver are cooled to 20 to 26° C, then transferred into a stainless steel drum. Acetonitrile, practical (2.0 kg) is used as rinse. After cleaning and purging both vessels, the contents of the stainless steel drum is transferred into the (100 L) receiver. Acetonitrile, practical (2.0 kg) is used as a rinse. The batch is heated at 70 to 76° C without exceeding jacket temperature of 85° C. The batch is filtered by pump through a .0 micron single cartridge filter, while maintaining the contents at 70 to 76° C. Warm (70-76° C) acetonitrile, practical (4.0 kg) is used as rinse for vessel, filters, pump and lines. The filtrate and rinse are collected and maintained below 50° C. The batch is adjusted to 10 to 16° C, then concentrated by vacuum distillation to 28 to 33 L volume: expected distillation temperature 20 to 30° C, distillate volume 32 to 37 L. The suspension is heated to 64 to 70° C without exceeding jacket temperature of 85° C. The resulting solution is cooled to 40 to 46° C, then seeded using 4-dimethylamino-but-2~enoic acid [4-(3-chloro-4-fluoro-phenylamino)-3-cyano- 7-ethoxy-quinolin-6-yl]-amide, purified (0.5 g). The mixture is cooled to 20 to 26° C over 1 hour, then held at the latter temperature for a minimum of 2 hours. The suspension is then cooled at -3 to 3° C over 1 hour, then held for a minimum of 1 hour. The solid product is collected on a 16″ Buchner, then washed with cold (0-5° C) acetonitrile-tetrahydrofuran (20-6 v/v) mixture (2 x 2.5 kg). The wet collected solid is recrystallized once more from acetonitrile-tetrahydrofuran (20-6 v/v) to desired purity. The material is dried in a vacuum oven first at 35 to 45° C (target 40° C) for 4 hours, liquid ring pump, then 45 to 55° C (target 50° C) for 4 hours. After high vacuum is applied at the latter temperature, until LOD <0.5% (90° C, 2 hours, full vacuum) and each of acetonitrile, tetrahydrofuran and 1-methyl-2-pyrrolidinone are below 0.2%. The purified drug substance is milled (Comil), then blended. The yield is 1.10 kg (70.1 %, corrected for starting material). The strength of the material is 98.3% and a total impurities of 1.27%.
To a solution of compound A (200 mg, 0.36 mmol, 1.0 eq) in CH2Cl2 (20 mL) was added m-CPBA (74 mg, 0.43 mmol, 1.2 eq) and the resulting mixture was stirred at room temperature for 4 h. A saturated aqueous solution of NaHCO3 (20 mL) was then added and the organic layer was separated, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by preparative TLC (CH2Cl2/MeOH, 10/1, v/v) to give (E)-4-((4-((3-chloro-4-(pyridin-2-ylmethoxy)phenyl)amino)-3-cyano-7-ethoxyquinolin-6-yl)amino)-N,N-dimethyl-4-oxobut-2-en-1-amine oxide (20 mg, 10%) as a yellow solid.
LC-MS (Agilent): Rt 3.03 min; m/z calculated for C30H29ClN6O4 [M+H]+ 573.19. found 573.2.
[0078] Synthesis of 3-chloro-4-(2-pyridylmethoxy)nitrobenzene
[0079] 2-pyridinyl carbinol (31.08 g, 1.05 eq) was dissolved in ACN (750 mL) and KOH flakes (85%) were added (20.6 g, 1.25 eq.). The resulting suspension was warmed to 35 °C. A solution of the 3-chloro-4-fluoronitrobenzene (50.0 g, 0.285 mol) in ACN (250 mL) was added at 35-40 °C. The mixture was held for 14 hours. The mixture was then cooled back to 20-25 °C, quenched with H2O (IL) and the resulting slurry filtered and washed with H2O (3 x 100 mL). The resulting product was isolated as a tan solid in 93% yield with a greater than 99.5% purity as determined by HPLC area. Example Ia
[0080] To accomplish the analogous synthesis of 3-chloro-4-(3-fluorobenzyloxy) nitrobenzene, 3-fluorobenzyl alcohol (0.30 kg, 2.39 mole, 1.05 eq) was dissolved in ACN (6.0 L) and to it was added potassium hydroxide flakes (85%) (0.16 kg, 1.25 eq). The resulting suspension was warmed to 35 0C. A solution of the 3-chloro-4-fluoronitrobenzene (0.40 kg, 2.28 mol) in ACN (2.0 L) was added at 35-40 °C. The mixture was held for 18 hours. The mixture was then cooled back to 20-25 °C, quenched with water (8 L) and the resulting slurry filtered and washed with water (2 x 0.40 L). The resulting product was dried at 45 °C, under 10 mm Hg pressure, for 25 hours to give 0.59 kg (92% yield). Example Ib
[0081] To prepare 4-(benzyloxy)3-chloronitrobenzene, benzyl alcohol (0.34 kg, 3.14 mole, 1.10 eq) was dissolved in acetonitrile (1.70 L) and to it was added potassium hydroxide flakes (85%) (0.24 kg, 1.50 eq). The resulting suspension was warmed to 25 0C. A solution of the 3- chloro-4-fluoronitrobenzene (0.50 kg, 2.85 mol, 1.0 eq) in acetonitrile (0.75 L) was added keeping the pot temperature < 45 0C. The mixture was held for 14 h. The mixture was then cooled back to 0-15 0C, quenched with water (2.5 L) and the resulting slurry was filtered and washed with water (2 x 0.50 L). The resulting product was dried at 50 0C, under 10 mm Hg pressure, for 24 hours to give 0.73 kg (97% yield). [0082] Experimental results for the reaction of Example 1 with different bases and solvents are shown in Table 1. The last three entries on Table 1 are large scale runs in which a 5% excess of pyridyl carbinol was used. Table 1 – Preparation of Nitroaryl Intermediate
NA = not applicable
RT = room temperature (20-25 °C)
Example 2
[0083] Preparation of 3-chloro-4-(2-pyridyhnethoxy)aniline from the nitrobenzene product of
Example 1 was accomplished with catalytic hydrogenation using platinum on carbon.
[0084] A typical hydrogenation was done using 6 volumes of THF, 2% by weight of 5%Pt/C (50% water wet), at 25 psi and at 25-30 0C for approximately 4-6 hours. The reaction is slightly exothermic and the temperature will rise to about 30-35 °C. Cooling is necessary to maintain the temperature below 30 0C.
[0085] As a specific example, a mixture of 3-chloro-4-(2-pyridylmethoxy)nitrobenzene (0.15 kg, 0.57 mole) and 2% (w/w) of 5% Pt/C (6.0 g) in tetrahydrofuran (0.90 L) was hydrogenated at 25 psi for at least 5 hours. The mixture was filtered through a celite pad and washed with tetrahydrofuran (0.60 L). The filtrate was distilled to a volume of about 0.75 L and ethanol (1.12 L) was added. Distillation was continued to a volume of about 0.75 L and ethanol (2.85 L) was added. The mixture may be used “as is” in the step of Example 3 below. Example 2 a
[0086] To accomplish an analogous synthesis of 3-chloro-4-(3-fluorobenzyloxy)aniline, zinc (0.464 kg) was added to a mixture of 3-chloro-4-(3-fluorobenzyloxy)nitrobenzene (0.40 kg, 1.42 mole) and ethanol (4.0 L). The mixture was heated to 40-50 °C. A solution of ammonium chloride (0.152 kg) in water (0.80 L) was added over 0.5 hour keeping the pot temperature at 40-50 °C. The mixture was stirred for 2 hours, filtered and washed with hot (40-50 °C) ethanol (2 x 0.40 L). The filtrate was distilled to a volume of about 0.80 L and 2- methyltetrahydrofuran (2.0 L) was added to dissolve the product. Water (0.80 L) and saturated brine (0.40 L) were added and the layers separated. The organic layer was washed with water (0.60 L), and distilled to a volume of about 0.40 L. Ethanol (2.0 L) was added and distillation continued to a volume of 1.2 L. Example 2b
[0087] To prepare 4-(benzyloxy)-3-chloroaniline, a mixture of 4-(benzyloxy)-3- chloronitrobenzene (0.325 kg, 1.23 mole, 1.0 eq) and 1% (w/w) of 5% Pt/C (3.25 g) in isopropanol (3.25 L) was hydrogenated at 25 psi for a minimum of 4.5 h. The mixture was filtered through a celite pad and washed with isopropanol (2.0 L). The filtrates were used as is in the next step.
[0088] Performing the hydrogenation in isopropyl alcohol (PA), methanol (MeOH), or ethanol
(EtOH) may result in the product being contaminated with late eluting impurity that partially precipitates out on standing in solution. It was found that performing the hydrogenation in a solvent where both the product and starting material are soluble, such as tetrahydrofuran
(THF), resulted in greater product purity and required much less solvent. Thus, THF is a preferred solvent for this step. Experimental results showing the effect of different reaction conditions are shown in Table 2. For the larger scale runs, the first aniline intermediate was not isolated (“NI”) before proceeding with the next step.
Table 2 – Hydrogenation to Form First Aniline Intermediate
* Solid impurities noted after reaction completion. ** percent by weight of starting material. Example 3
[0090] Following hydrogenation to form the first aniline intermediate, acid catalyzed coupling was performed to prepare 4~[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6-N- acetylaminoquinoline, as shown below:
[0091] To perform the coupling reaction, the two reactants were heated together in alcohol at 65-78°C over 4-6 hours, yielding the product. The reaction begins as an amber slurry and thickens to a lighter beige slurry as it approaches completion. Upon scaling up from 75 g to 350 g, it proved necessary to add a catalytic amount (0.025 eq.) of methanesulfonic acid to initiate the reaction. As a specific example, 4-chloro-3-cyano-7-ethoxy-6-N- acetylaminoquinoline (0.141 kg, 0.49 mole) was added to the mixture of Example 2, followed by ethanol (0.037 L) to give a suspension. A catalytic amount of methanesulfonic acid (1.17 g) was added at 20-25 C. The resulting slurry was heated to 70-75 C and held for a minimum of 4 hours. Thickening of the slurry was evident after 1.5 hours. Following reaction completion, the mixture was cooled to room temperature and may be used “as is” in the telescoped reaction of Example 4 below. Example 3 a
[0092] To prepare 6-acetamido-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline, ethanol (4.80 L) was added to the aniline solution followed by 4-chloro-3- cyano-7-ethoxy-6-N-acetylaminoquinoline (0.350 kg, 1.11 mole). A catalytic amount of methanesulfonic acid (2.0 ml) was added at 20-250C. The resulting suspension was heated to 70-750C and held for a minimum of 2 h. Thickening of the slurry was evident during this holding period. Following reaction completion, the mixture was used as is in the following telescoped reaction. Example 3 b
[0093] To prepare 6-acetainido-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-quinoline, isopropanol (6.75 L) was added to the aniline solution followed by 4-chloro-3-cyano-7-ethoxy- 6-N-acetylaminoquinoline (0.277 kg, 0.96 mole, 0.78 eq). A catalytic amount of methane sulfonic acid (3.50 ml) was added at 20-250C. The resulting suspension was heated to 80-850C and held for a minimum of 10 hr. Thickening of the slurry was evident during this holding period. Following reaction completion, the mixture was cooled to 25-35 0C, filtered and the cake washed with isopropanol (3 x 0.25 L). The cake was used as is in the following telescoped reaction.
[0094] As solvents EtOH, DMF or other suitable solvent may be used. Experimental results obtained using different solvents and reaction conditions are shown in Table 3. Difficulty filtering the product of this step (noted in several entries on Table 3) was circumvented by not isolating the solid at this point, but telescoping the reaction with the next step. It has been found that on the order of 20 volumes of EtOH were necessary to achieve reasonable stirring, but that the reaction can proceed in only 10 volumes of DMF, without significant loss in purity. [0095] In Table 3, where the entry is labelled NI , the intermediate product was not isolated, but carried into the next reaction step. Table 3 – Coupling Reaction
NR = no reaction, NI = not isolated; ND = not determined; NA = not available
1. Carried through to the deprotection and generation of free base to give 69.5% overall yield.
2. The overall yield after the deprotection and generation of the free base is 76.1%
3. This reaction was not filtered at all but taken as slurry to the next step.
Example 4 – Deprotection
[0096] The deprotection of the quinoline intermediate formed by the coupling reaction using
2N HCl in water is preferred as noted in Table 4 below. As in the previous Examples, the intermediate product of this step is advantageously not isolated, but carried over as a wet cake into the next step.
[0097] Preparation of 4-[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6- aminoquinoline hydrochloride.
[0098] The reaction mixture from the previous step (Example 3) was taken as is and to it was added 2.7N HCl (3.3L) in H2O (16.0 L). The slurry was heated to 700C and held for 19 hours. The resulting slurry was then filtered and rinsed with 1:1 EtOHTH2O (4 x 1.0 L). The product was isolated as a wet cake and carried through to the next step. A small sample was dried at this stage and analyzed. The HCl salt had a strength of 98.9%. Example 4a
[0099] To prepare 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline hydrochloride, the reaction mixture from the previous step was taken as is and to it was added ethanol (1.6 L) and concentrated hydrochloric acid (1.38 L) to bring the pH to 1-3. The suspension was held at 70-75 0C for a minimum of 2 h. After 1 h, the mixture thickens and ethanol (0.80 L) was added. After 2 h, water (6.80 L) was added, the mixture stirred for 1 h and then cooled to 35-45 0C and stirred overnight (12 h). The mixture was filtered and rinsed with 1 : 1 ethanol/water (2 x 0.84 L) at 35-45 0C. The product was isolated as a wet cake and carried through to the next step. Example 4b
[00100] To prepare 6-amino-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7- ethoxyquinoline hydrochloride, the wet cake from the previous step was taken as is and to it was added a 2 N solution of concentrated hydrochloric acid (1.16 L) in methanol (5.84 L). The suspension was heated to 63-68 0C and held for a minimum of 30 h. The mixture was cooled to 20-300C, filtered and rinsed with methanol (2 x 0.30 L). The product was isolated as a wet cake and carried through to the next step. Table 4 – Deprotection
ND = not determined (the product was used in the next step as a wet cake) NA = not available SM= starting material
Example 5 – Preparation of free base
[0100] The 4-[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6-aminoquinoline HCl salt was converted to the corresponding free base by treatment with 10% potassium carbonate (1.8 L) in MeOH (2.82 L). The mixture was stirred for a minimum of 2.5 hours and the pH was 9-10. The product was filtered, washed with 1:1 methanol/water (3 x 0.19 L) and dried (at 45-50 C at a pressure of 10 mm Hg, for 24 hours) to give 0.186 kg of product with an overall yield of 86% over 4 steps.
Example 5 a
[0101] To prepare 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline free base, the 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline hydrochloride salt was converted to its corresponding free base by treatment with 10% potassium carbonate (0.22 kg in 2.27 L water) in methanol (7.21 L) until pH was 10. The mixture was stirred for a minimum of 2 h. The beige suspension was filtered, washed with 1:1 methanol/water (2 x 0.84 L) and dried (45-50 0C, 10 mm Hg, 24 h) to give 0.51 kg of product with an overall yield of 99% over 4 steps. Example 5b
[0102] To prepare 6-amino-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxyquinolme free base, the 6-amino-4-[4-(benzyloxy)-3-chloroamlino]-3-cyano-7-ethoxyqumoline hydrochloride salt was converted to its corresponding free base by treatment with 10% aqueous potassium carbonate (0.213 kg in 2.13 L) in methanol (6.40 L). The mixture was stirred for a minimum of 1.5 h keeping the pH at 9-10. The product was filtered, washed with water (2 x 0.50 L) and dried (50-60 0C, 10 mm Hg, 20 h) to give 0.347 kg of product with an overall yield of 82% over 4 steps.
Example 6 – Side Chain Coupling
[0103] An acid chloride of formula RV(C=O)-Cl, a mixed anhydride or an activated carboxylase R’ 2-(C=O)-LG derived from the corresponding carboxylic acid, may be used to couple a side chain at the 6 position to form a 6-amido-4-amino-3 cyanoquinoline. R’2 may be alkyl of 1-6 carbon atoms, which may be mono- or di-substituted with amino groups or cycloamino groups, or R’2 may be alkenyl of 2-6 carbon atoms which may be mono- or di- substituted with amino groups or cycloamino groups. [0104] Using the 2-step sequence shown below, an activated carboxylate is prepared in situ and coupled with the aniline. Although the acid chloride can be prepared in acetonitile, a better yield was obtained when the acid chloride was prepared in THF. In both cases, the aniline should be dissolved in NMP before amidation. It is believed that formation of product is better due to better solubility of the aniline in a THF/NMP mixture rather than in an ACN/NMP combination.
[0105] The amount of 4-N,N-dimethylaminocrotonic acid needed was 2 equivalents with respect to aniline. A slight undercharge of 1.95 eq of oxalyl chloride was added along with a catalytic amount (3 mol %) of DMF. The acid chloride was formed via the Vilsmeier intermediate. The completion test for the acid chloride reaction consists of quenching an aliquot of the reaction into ethanol and detecting by HPLC the crotonic acid ethyl ester. This method serves as a check to ensure complete consumption of oxalyl chloride. Excess oxalyl chloride will form diethyl oxalate when quenched in ethanol. [0106] The acid chloride is stable after holding for up to 5 hours at 0-10 °C, when decomposition begins. After 20 hours, complete decomposition takes place. If the acid chloride is allowed to warm, decomposition occurs and its effectiveness is diminished. [0107] The quality of the starting crotonic acid also plays a role in this coupling reaction, as commercially available crotonic acid may contain acetic acid. Acetic acid is detrimental to this reaction. 6-N-acetyl quinoline can be formed which is difficult to remove from the final product. The acetic acid can be removed by re-slurrying the crotonic acid in 4 volumes of isopropanol at room tempature, filtering and drying preferably to a level of less than 0.01%. [0108] It was found that the addition of the aniline solution in NMP to the acid chloride gave a better yield as compared to adding the acid chloride to the aniline. The addition is done keeping the temperature at 0-5 °C. The coupling reaction is slow and requires holding overnight at this temperature. It is not desirable to raise the reaction temperature as the stability of the acid chloride diminishes.
[0109] The reaction is quenched using aqueous sodium hydroxide at 40 °C and then filtered at that temperature. Quenching the reaction at 40 0C gives bigger crystals that are easily filterable. It was observed that filtration at 40 °C was faster than at room temperature. The product is recrystallized from a 1.5:1 mixture of acetonitrile:THF (15 volumes) at 70-75 0C. This in-process purification beneficially removes unreacted aniline. The recovery yields are typically greater than 85%.
[0110] To demonstrate a specific synthesis of (E)-N- {4-[3-chloro-4-(2- pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide, a solution of 4-N,N-dimethylaminocrotonic acid hydrochloride (186 g, 1.12 mol) in THF (1.88 L) and a catalytic amount of DMF (2 mL) was cooled to 0-5 °C. Oxalyl chloride (97 mL, 1.09 mol, 0.95 eq) was added dropwise over 45 minutes. The mixture was then warmed to 25-30 °C and stirred for 2 hours. The yellow solution was checked for complete consumption of oxalyl chloride by HPLC, then cooled to 0-5 0C.
[0111] When the reaction is deemed complete, a solution of 4-[4-(2-pyridylniethoxy)-3- chloro]amino-6-amino-3-cyano-7-ethoxyquinoline (250 g, 0.56 mol) in N-methyl-2- pyrolidinone (1.88 L) was added dropwise over 2 hours keeping the temperature at 0-5 °C. The mixture was stirred for at least 3 hours until less than about 2% of the starting aniline remains by HPLC, which takes about 3 hours.
[0112] Upon completion, the reaction was quenched with water (3.0 L), held for 30 minutes and then warmed to 40 °C. Aqueous sodium hydroxide (170 g in 1.25 L water) was added over 1.25 hours to bring the pH to 10-11. The mixture was stirred for an hour, then cooled to room temperature and held for 3 hours. The resulting precipitates were filtered and washed with water (100 mL) and heptane (100 mL). The wet solids were heated to reflux (70-75 °C) in acetonitrile:THF and the solution cooled over 3 hours to room temperature. The product was filtered and washed with cold acetonitrile:THF. The product was dried (40-50 0C, 10 mm Hg, 24 hours) to give 83% uncorrected yield. Example 6a
[0113] In an analogous synthesis of (E)-N- {4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3- cyano-7-ethoxy-6-qumolmyl}-4-(dimethylamino)-2-butenamide, a solution of 4-N5N- dimethylaminocrotonic acid hydrochloride (108 g, 0.65 mole) in tetrahydrofuran (1.13 L) and a catalytic amount of dimethylformamide (1.2 mL) was cooled to 0-5 °c. Oxalyl chloride (55 mL, 0.62 mole, 0.95 eq) was added dropwise over 50 min. The mixture was then warmed to 25-30 °c and stirred for 2 h then cooled to 0-5 °c. N-methyl-2-pyrrolidinone (0.225 L) was added over 25 min followed by a solution of 6-amino-4-[3-chloro-4-(3- fluorobenzyloxy)]anilino-3-cyano-7-ethoxy-quinoline (150 g, 0.32 mol) in N-methyl-2- pyrrolidinone (1.20 L) added dropwise over 2 hours keeping the temperature 0-5 . The mixture was stirred for at least about 3 hours, warmed to 10-15 °c and stirred for a further 12 hours. The mixture is cooled to 0-10 c, quenched by adding water (1.8 L) over 2 hours, and stirred for 30 minutes. The mixture is warmed to 40 °c. Aqueous sodium hydroxide (101 g in 0.75 L water) was added over 1 hour to bring the pH to 10-11. The mixture was stirred for an hour, filtered warm (40 °c) and washed with water (2 x 0.30 L) until the pH of the last wash was about 7. The wet solids were recrystallized by heating to reflux (70-75 °c) in 60:40 acetonitrile:tetrahydrofuran (2.25 L) and the solution cooled over 3 hours to room temperature. The product was filtered and washed with cold 60:40 acetonitrile:tetrahydrofuran (2 x 0.30 L). The product was dried (40-50 °c, 10 mm Hg, 16 h) to give 0.154 kg (83% yield). Example 6b
[0114] To prepare (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}- 4-(dimethylamino)-2-butenamide free base, a solution of 4-N,N-dimethylaminocrotonic acid hydrochloride (18.6 g, 112 mmole) in acetonitrile (295 ml) and a catalytic amount of dimethylformamide (0.25 mL) was cooled to 0-5 °c. Oxalyl chloride (9.3 mL, 106 mmole, 0.95
Op eq) was added dropwise over 5 min. The mixture was then warmed to 25-30 and stirred for 1-1.5 h then cooled to 0-10 °c. A solution of 6-amino-4-[4-(benzyloxy)-3-cliloroanilino]-3- cyano-7-ethoxy-quinoline (25 g, 56 mmole) in N-methyl-2-pyrrolidinone (175 ml) was added dropwise over 30 min keeping the temperature 0-10 °c. The mixture was stirred for a minimum of 1 h at 0-10 °c. After reaction completion, the mixture was quenched by dropwise addition to a solution of sodium bicarbonate (69.7 g in 870 ml water) over 30 mins. and stirred overnight while warming to room temperature. The mixture was filtered and washed with water (3 x 25 ml). The crude product was recrystallized in refluxing (80-82 °c) acetonitrile (570 ml). The product was dried (45-50 °c, 10 mm Hg, 28 h) to give 12.81 g (41% yield). 1H NMR : δ (DMSO-d6) 9.44 (s, IH, NH), 8.97 (s, IH, Ar), 8.44 (s, IH, Ar), 7.53-7.35 (m, 7H, Ar), 7.35- 7.10 (in, 2H, Ar), 6.78 (dt, IH, -CH2CH=CH-), 6.59 (d, IH, -CH2CH=CH-), 5.21 (s, 2H, OCH2Ph), 4.30 (q, 2H, OCH2CH3), 3.07 (s, 2H, NCH2), 2.18 (s, 6H, N(CHs)2), 1-47 (t, 3H, OCH2CH3).
[0115] Results obtained with different reaction procedures at different degrees of scale-up for synthesis of the 2-pyridylmethoxy analog are shown in Table 5. Table 5 – Side Chain Coupling
* TI = total impurities
[0116] Purificatiuon of the product is conducted by recrystallization in a suitable solvent followed by reslurrying with water followed by additional recrystallization, as necessary. As noted in Table 6, in the synthesis of the 2-pyridylmethoxy analog, several trials in different solvents did not result in the isolation of a single polymorphic form of the product. Table 6
Example 7 – Formation of Salt
[0117] The free base is hygroscopic and undergoes hydrolysis readily. Forming a salt of the compound, such as a fumarate or mesylate salt, stabilizes the molecule and renders the compound more soluble. The most preferred salt is a maleate salt, which has been found to be highly crystalline and to exist substantially as a single polymorph as shown by DSC thermogram in Fig. 1.
[0118] Recrystallizing the product in the presence of an acid has been found to yield a stable salt form of the product. Experimental results achieved utilizing different solvents for the recrystallization are set forth in Table 7. As seen in Table 7, an improvement is observed when n-propanol/water is used as the solvent system. A maleate salt is the most preferred, as it exists in a single polymorphic form. Table 7 – Recrystallization
Preparation of (E)-N- {4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6- quinolinyl} -4-(dimethylamino)-2-butenamide maleate, WAY- 179272-B
[0120] (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4- dimethylamino)-2-butenamide crude free base (0.1 kg, 0.159 mole) and maleic acid (0.019 kg, 0.164 mole) were dissolved at 40-50 in a 10% water/n-propanol mixture (1.20 L). The hot solution was clarified and cooled over 2 h to room temperature and held for 12-15 hr. The product was filtered and washed with 10% water/n-propanol (2 x 0.15 L). The product was dried (50 °c, 10 mm Hg, 24 h) to give 94.4 g (88% yield). DSC: 204 °c (single crystal form). 1H NMR : δ (DMSO-d6) 9.73 (s, IH, NH), 9.62 (s, IH, NH), 8.93 (s, IH, Ar), 8.60 (dd, IH, Ar), 8.50 (s, IH, Ar), 7.88 (dd, IH, Ar), 7.58 (d, IH, Ar), 7.40 (m, 3H, Ar), 7.24 (m, 2H, Ar), 6.75 (d, 2H, -CH=CH-), 6.03 (s, 2H, HOOC-CH=CH-COOH), 5.29 (s, 2H, OCH2PVr), 4.33 (q, 2H, OCH2CH3), 3.89 (s, 2H, NCH2), 2.76 (s, 6H, N(CH3)2), 1.47 (t, 3H, OCH2CH3). 13C NMR : δ (DMSO-d6) 168.0, 163.2, 156.9, 154.2, 153.2, 151.9, 151.3, 149.8, 148.5, 137.8, 136.5, 134.7, 133.4, 132.2, 128.0, 126.6, 124.9, 123.8, 122.3, 122.2, 117.9, 116.4, 115.1, 113.9, 109.5, 88.1, 72.0, 65.3, 57.8, 43.1, 14.9.
Example 7a
To prepare (E)-N- {4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7-ethoxy-6- quinolinyl}-4-(dimethylamino)-2-butenamide dimaleate,
(E)-N- {4-[3-chloro-4-(3- fluorobenzyloxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-dimethylamino)-2-butenamide crude free base (0.516 kg, 0.90 mole) and maleic acid (0.214 kg, 1.84 mole) were dissolved at 40-50 °c in a 6.5% water/n-propanol mixture (12.60 L). The hot solution was clarified, rinsed with 5% water/n-propanol (0.52 L) and n-propanol (2.0 L). The mixture was held at 45 for 3 hr, cooled over 2 h to room temperature and held overnight. The mixture was further cooled to 5-10 °c. The product was filtered and washed with cold 5% water/n-propanol (0.52 L). The product was dried (45 °c, 10 mm Hg, 16-24 h) to give 0.586 kg (81% yield). DSC: 184 °c (single crystal form). 1HNMR : δ (DMSO-d6) 9.77 (s, IH, NH), 8.95 (s, IH, Ar), 8.53 (s, IH, Ar), 7.49-7.16 (m, 8H, Ar), 6.78 (m, 2H, -CH=CH-), 6.15 (s, 4H, 2 x HOOC-CH=CH-COOH), 5.26 (s, 2H, OCH2PyT), 4.33 (q, 2H, OCH2CH3), 3.97 (dd, 2H, NCH2), 2.82 (s, 6H, N(CEb)2), 1.47 (t, 3H, OCH2CH3). 13C NMR : δ (DMS0-d6) 167.0, 163.8, 162.3, 160.6, 153.6, 152.2, 151.3, 150.8, 139.5, 139.4, 133.7, 133.2, 132.2, 131.8, 130.5, 130.4, 127.4, 126.1, 124.3, 123.3, 121.7, 116.9, 115.7, 114.8, 114.5, 114.4, 114.1, 113.8, 113.1, 108.1, 87.2, 69.5, 64.6, 56.9, 42.1, 14.2. Example 7b
[0122] To prepare (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}- 4-(dimethylamino)-2-butenamide maleate, (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano- 7-ethoxy-6-quinolinyl}-4-dimethylamino)-2-butenamide crude free base (2.0 g, 3.6 mmole) and maleic acid (0.43 g, 3.7 mmole) were mixed at 40-50 c in a 10% water/n-propanol mixture (24 ml) for 2 hr. The mixture was cooled to ambient temperature, filtered and washed with 10% water/n-propanol (2 x 3 ml). The product was dried (40 °c, 10 mm Hg, 24 h) to give 0.32 g (13% yield). 1HNMR : δ (DMSO-d6) 9.75 (s, IH, NH), 8.95 (s, IH, Ar), 8.49 (s, IH, Ar), 7.49-7.37 (m, 7H, Ar), 7.23 (dd, 2H, Ar), 6.78 (s, 2H, -CH2CH=CH-), 6.06 (s, 2H, HOOC- CH=CH-COOH), 5.22 (s, 2H, OCH2Ph), 4.31 (q, 2H, OCH2CH3), 3.93 (s, 2H, NCH2), 2.79 (s, 6H, N(CH3)2), 1.46 (t, 3H, OCH2CH3).13C NMR : δ (DMSO-d6) 167.9, 163.1, 154.2, 153.3, 152.1, 151.3, 148.5, 137.3, 136.3, 134.5, 133.2, 132.3, 129.3, 129.2, 128.7, 128.3, 128.2, 128.0, 126.7, 124.9, 122.4, 117.9, 116.4, 115.2, 113.9, 109.5, 88.0, 71.1, 65.3, 57.7, 43.0, 15.0. [0123] (E)-N-{4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}-4- dimethylamino)-2-butenamide crude free base (2.0 g, 3.6 mmole) and maleic acid (0.43 g, 3.7 mmole) were mixed at 40-50 °c in a 10% water/n-propanol mixture (24 ml) for 2 hr. The mixture was cooled to ambient temperature, filtered and washed with 10% water/n-propanol (2 x 3 ml). The product was dried (40 °c, 10 mm Hg, 24 h) to give 0.32 g (13% yield). 1H NMR : δ (DMSO-d6) 9.75 (s, IH, NH), 8.95 (s, IH, Ar), 8.49 (s, IH, Ar), 7.49-7.37 (m, 7H, Ar), 7.23 (dd, 2H, Ar), 6.78 (s, 2H, -CH2CH=CH-), 6.06 (s, 2H, HOOC-CH=CH-COOH), 5.22 (s, 2H, OCH2Ph), 4.31 (q, 2H, OCH2CH3), 3.93 (s, 2H, NCH2), 2.79 (s, 6H, N(CH3)2), 1.46 (t, 3H, OCH2CH3). 13C NMR : δ (DMSO-d6) 167.9, 163.1, 154.2, 153.3, 152.1, 151.3, 148.5, 137.3, 136.3, 134.5, 133.2, 132.3, 129.3, 129.2, 128.7, 128.3, 128.2, 128.0, 126.7, 124.9, 122.4, 117.9,
The reaction of the free base and maleic acid occurs at an elevated temperature of from about 40 0C to about 60 0C, preferably between about 4O0C to about 5O0C. The ratio of watenn- propanol may vary, for example between about 1 :10 to about 1 :5, and the optimal ratio of watenn-propanol is about 1 :9. The water-alcohol solution may comprise from about 5% to about 20% by volume water and from about 80% to about 95% by volume alcohol. The alcohol may be n-propanol. In one embodiment, the water-alcohol solution comprises about 10% by volume water and about 90% by volume n-propanol. The volume of the solvent solution may be between about 8 to about 25 volumes, including about 10 to about 12 volumes. About 1.0-1.2 equivalents of maleic acid is used per equivalent of the free base, preferably about 1.03 equivalents of maleic acid per equivalent of the free base.
The resulting solution of the maleate salt may be clarified by filtration prior to cooling. The cooling step may be continued until the solution reaches a temperature of about 45°C or less, including a temperature of about 39°C or less, and more preferably to about 300C or less. In one embodiment, the solution is filtered after cooling to about room temperature, preferably from about 230C to about 25 0C. Typically, the maleate salt begins to crystallize out of solution once the temperature reaches 370C or below. The solution may be allowed to sit for at least 12 hours, preferably about 12 to about 15 hours at room temperature, and is then filtered and washed to recover the crystalline maleate salt product. The resulting filter cake may be washed with the same or a different water-alcohol solution to obtain the product. The product may be dried to obtain crystalline (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7- ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide maleate. At this point, the maleate salt product recovered and isolated is typically in the form of the monohydrate form of the maleate salt.
The present invention relates to a process for preparing that imatinib (neratinib, HKI-272) is a new method for its preparation and its intermediates in the preparation to the application that imatinib
Compound of Example 13 (20mg, 0. 037mmol) was dissolved in DMF was added potassium carbonate (10mg, 0. 07mmol), dimethylamine hydrochloride (5mg, 0. 06mmol), at room temperature for I hour, after , the reaction mixture was dropped into water, stirred for 10 minutes, filtered, washed with water and dried to give the title compound 1511 ^ 75% yield.1HNMR (300MHz, DMS0_d6): δ I. 5 (t, 3H, J = 6 · 8,13. 8), 2. 2 (br s, 6H), 3. I (d, 2H, J = 3. 8 ), 4. 3 (q, 2H, J = 7. 0,14. 2), 5. 2 (s, 2H),
Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity J Med Chem 2005, 48(4): 1107
This compound was prepared as a yellow solid (0.86 g, 85%) by the method described for 25g using 0.65 g (1.81 mmol) of 23 and 0.42 g (3.62 mmol) of 3-chloro-4-(2-pyridinylmethoxy)aniline:
Minami Y, Shimamura T, Shah K, et al. (July 2007). “The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272”. Oncogene26 (34): 5023–7. doi:10.1038/sj.onc.1210292.PMID17311002.
Sequist L.V., Besse B., Lynch T.J. and all; Neratinib, an Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor: Results of a Phase II Trial in Patients With Advanced Non-Small-Cell Lung Cancer., J. Clin. Oncol., 2010, May 17. PubMed PMID: 20479403.
Belani CP. The role of irreversible EGFR inhibitors in the treatment of non-small cell lung cancer: overcoming resistance to reversible EGFR inhibitors. Review. Cancer Invest. 2010, 28(4), 413-423. Review. PubMed PMID: 20307200.
Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity J Med Chem 2005, 48(4): 1107
Maleate salts of (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide and crystalline forms thereof
Researchers at the Hebrew University of Jerusalem’s Faculty of Medicine have discovered a process whereby tumor cells become resistant to specific drugs, a finding that could significantly influence how anti-cancer drugs are administered and the development of a means for reversing the proliferation of malignant tumor growth.
Researchers at the Hebrew University of Jerusalem’s Faculty of Medicine have discovered a process whereby tumor cells become resistant to specific drugs, a finding that could significantly influence how anti-cancer drugs are administered and the development of a means for reversing the proliferation of malignant tumor growth.
Cancer has become one of the major challenges of biomedical research in the past decades, and is one of the leading causes of illness and death all over the world. While many drugs have been developed against cancer, doctors do not know in advance of treatment whether a patient might benefit from a particular drug. Thus…
Vitamin A could help treat and prevent the spread of prostate cancer, according to research published today (Monday, April 15th) in Oncogenesis.
Scientists funded by Yorkshire Cancer Research at the University of York have discovered that retinoic acid – a chemical made from vitamin A which is supplied in our diet by carrots, green vegetables and liver – can turn specific genes within prostate cancerstem cells back on, reducing the ability of the cancer to invade surrounding tissue.
The findings suggest that Vitamin A related compounds could be used to enhance clinical treatments for prostate cancer.
Professor Norman Maitland, Director of the YCR Cancer Research Unit in the Department of Biology at York, said: “Cancer arises from healthy cells going wrong. Certain controls can be turned off which allows the cancer to progress. For example, normal cells gain the ability to grow and invade the surrounding tissues.
In December 2021, the U.S. Food and Drug Administration approved cabotegravir for pre-exposure prophylaxis (PrEP) in at-risk people under the brand name Apretude.[11]
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
Cabotegravir, or GSK1265744, is an HIV-1 integrase inhibitor that is prescribed with the non-nucleoside reverse transcriptase inhibitor, rilpivirine.4,6,7 Early research into cabotegravir showed it had lower oral bioavailability than dolutegravir,4 which resulted in the development of long acting monthly intramuscular injection formulation for cabotegravir.4,7
Cabotegravir was granted FDA approval on 21 January 2021 in combination with rilpivirine to treat HIV-1 infection in virologically suppressed individuals.8 While previously administered once monthly only, this combination product was granted FDA approval for dosing every two months on February 01, 2022 11 and without the need for an oral lead-in period prior.7
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).
The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Medical uses
Cabotegravir in combination with rilpivirine is indicated for the treatment of human immunodeficiency virus type-1 (HIV-1) in adults.[1][5] The combination injection is intended for maintenance treatment of adults who have undetectable HIV levels in the blood (viral load less than 50 copies/mL) with their current antiretroviral treatment, and when the virus has not developed resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) and integrase strand transfer inhibitors.[5] The tablets are used to check whether a person tolerates the treatment before the injection therapy is started.[12][5]
The two medicines are the first antiretroviral drugs that come in a long-acting injectable formulation.[12]
Cabotegravir (Apretude) is indicated for use in at-risk people weighing at least 35 kilograms (77 lb) for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV.[11]
Contraindications and interactions
Cabotegravir must not be combined with the drugs rifampicin, rifapentine, carbamazepine, oxcarbazepine, phenytoin or phenobarbital, which induce the enzyme UGT1A1.[5] These drugs significantly decrease cabotegravir concentrations in the body and thus may reduce its effectiveness.[9][5] Additionally, they induce the enzyme CYP3A4, which leads to reduced rilpivirine concentrations in the body.[5][13][14][15] Additionally, patients who are breastfeeding or plan to breastfeed should not take Cabotegravir because it is not known if it will pass within the breast milk.[16]
Adverse effects
The most common side effects of the injectable combination therapy with rilpivirine are reactions at the injection site (in up to 84% of patients) such as pain and swelling, as well as headache (up to 12%) and fever or feeling hot (in 10%). For the tablets, headache and a hot feeling were slightly less frequent. Less common side effects (under 10%) for both formulations are depressive disorders, insomnia, and rashes.[9]
Pharmacology
Mechanism of action
Cabotegravir is an integrase strand transfer inhibitor. This means it blocks the HIV’s enzyme integrase, thereby preventing its genome from being integrated into the human cells’ DNA.[9] As this is a necessary step for the virus to replicate, its further spread is hampered.[9]
When taken by mouth, cabotegravir reaches highest blood plasma levels after three hours. Taking the drug together with food slightly increases its concentrations in the blood, but this is not clinically relevant. After injection into the muscle, cabotegravir is slowly absorbed into the bloodstream, reaching its highest blood plasma levels after about seven days.[9]
Over 99% of the substance are bound to plasma proteins. The drug is inactivated in the body by glucuronidation, mainly by the enzyme UGT1A1, and to a much lesser extent by UGT1A9. More than 90% of the circulating substance are the unchanged cabotegravir, however. The biological half-life is 41 hours for the tablets and 5.6 to 11.5 weeks for the injection.[9]
Elimination has only been studied for oral administration: Most of the drug is eliminated via the faeces in unchanged form (47%). It is not known how much of this amount comes from the bile, and how much was not absorbed in the first place. (The bile actually contains the glucuronide, but this could be broken up again in the gut lumen to give the parent substance that is observed in the faeces.) To a lesser extent it is excreted via the urine (27%), almost exclusively as the glucuronide.[9]
Pharmacogenomics
UGT1A1 poor metabolizers have 1.3- to 1.5-fold increased cabotegravir concentrations in the body. This is not considered clinically significant.[9]
Chemistry
Cabotegravir is a white to off-white, crystalline powder that is practically insoluble in aqueous solutions under pH 9, and slightly soluble above pH 10. It is slightly acidic with a pKa of 7.7 for the enolic acid and 1.1 (calculated) for the carboxamide. The molecule has two asymmetric carbon atoms; only one of the four possible configurations is present in the medication.[18]
Formulation
In studies, the agent was packaged into nanoparticles (GSK744LAP) conferring a biological half-life of 21 to 50 days[citation needed] following a single dose. The marketed injection achieves its long half-life not via nanoparticles but with a suspension of the free cabotegravir acid. The tablets contain cabotegravir sodium salt.[18]
History
Cabotegravir was examined in the clinical trials HPTN 083 and HPTN 084.[19][20] In 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vocabria intended for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with rilpivirine injection.[21] The EMA also recommended marketing authorization be given for rilpivirine and cabotegravir injections to be used together for the treatment of people with HIV-1 infection.[12] Cabotegravir was approved for medical use in the European Union in December 2020.[8]
In 2020, results for some studies were released showing success in using injectable cabotegravir for long-acting pre-exposure prophylaxis (PrEP) with greater efficacy than the emtricitabine/tenofovir combination being widely used for PrEP at the time.[24][25]
The safety and efficacy of cabotegravir to reduce the risk of acquiring HIV were evaluated in two randomized, double-blind trials that compared cabotegravir to emtricitabine/tenofovir, a once daily oral medication for HIV PrEP.[11] Trial 1 included HIV-uninfected men and transgender women who have sex with men and have high-risk behavior for HIV infection.[11] Trial 2 included uninfected cisgender women at risk of acquiring HIV.[11]
In Trial 1, 4,566 cisgender men and transgender women who have sex with men received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections among trial participants taking daily cabotegravir followed by cabotegravir injections every two months compared to daily oral emtricitabine/tenofovir.[11] The trial showed participants who took cabotegravir had 69% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In Trial 2, 3,224 cisgender women received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections in participants who took oral cabotegravir and injections of cabotegravir compared to those who took emtricitabine/tenofovir orally.[11] The trial showed participants who took cabotegravir had 90% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In December 2021, the U.S. Food and Drug Administration (FDA) approved cabotegravir for pre-exposure prophylaxis.[11] The FDA granted the approval of Apretude to Viiv.[11]
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.
14
Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N
2) DBU
P-1 P-2 P-3
a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
†Global API Chemistry, ‡MDR Chemical Science,§Analytical Sciences, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
A novel synthesis of GSK1265744, a potent HIV integrase inhibitor, is described. The synthesis is highlighted by an efficient construction of the densely functionalized pyridinone core as well as a highly diastereoselective formation of the acyl oxazolidine moiety. The latter exploits the target molecule’s ability to chelate to Mg2+, a key feature in the integrase inhibitor’s mechanism of action.
Bictegravir and dolutegravir are two recently approved integrase inhibitors for the treatment of HIV. A third inhibitor, cabotegravir, is in Phase 3 development. As a continuation of a series of articles on synthetic routes to newly approved drugs, the current article reviews the patent and journal literature regarding synthetic routes and final forms of these drug
^“Adopted USANs”(PDF). American Medical Association. Retrieved 19 September 2014.
^World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1): 70–1. hdl:10665/331088.
Ziegler, Robert E.; Desai, Bimbisar K.; Jee, Jo-Ann; Gupton, B. Frank; Roper, Thomas D.; Jamison, Timothy F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angewandte Chemie, International Edition. Volume 57. Issue 24. Pages 7181-7185. Journal; Online Computer File. (2018).
SYN 4
Synthetic Reference
Rajan, Srinivasan Thirumalai; Eswaraiah, Sajja; Reddy, Ghojala Venkat; Reddy, Sagyam Rajeshwar; Markandeya, Bekkam; Rajesham, Boge. Novel crystalline polymorph of sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate and process for preparation thereof. Assignee MSN Research & Development Center, India. IN 201641037221. (2018).
Synthetic Reference 5
Sharma, Pramodkumar; Rao, Bhatraju Srinivasa; Deo, Keshav. A process for the preparation of Dolutegravir or its pharmaceutical acceptable salts thereof. Assignee Wockhardt Limited, India. IN 2015MU01007. (2016).
Synthetic Reference 6
Weaver, Jimmie Dean. Preparation of fluoroarenes via hydrogen bond directed photocatalytic hydrodefluorination of perfluoroarenes. Assignee The Board of Regents for Oklahoma State University, USA. WO 2018187336. (2018).
Vellanki, Sivaram Prasad; Nadella, Madumurthy; Bhalme, Mitali; Ramabhotla, Revathi Srinivas. Process for the preparation of dolutegravir, an integrase inhibitor for HIV-1 infection therapy. Assignee Mylan Laboratories Ltd., India. IN 2015CH00588. (2016).
SYN 9
Synthetic Reference
Sankareswaran, Srimurugan; Mannam, Madhavarao; Chakka, Veerababu; Mandapati, Srirami Reddy; Kumar, Pramod. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Organic Process Research & Development. Volume 20. Issue 8. Pages 1461-1468. Journal; Online Computer File. (2016).
MOLECULAR FORMULA C17H11ClF3N5O3
MOLECULAR WEIGHT 425.7
Merck Sharp & Dohme Corp
reverse transcriptase inhibitor
Doravirine (MK-1439) is a non-nucleoside reverse transcriptase inhibitor under development by Merck & Co. for use in the treatment of HIV infection. Doravirine demonstrated robust antiviral activity and good tolerability in a small clinical study of 7-day monotherapy reported at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013. Doravirine appeared safe and generally well tolerated with most adverse events being mild-to-moderate.[1][2]
investigational next-generation, non-nucleoside reverse transcriptase inhibitor (NNRTI), at the 21st Conference on Retroviruses and Opportunistic Infections (CROI). Interim data demonstrating potent antiretroviral (ARV) activity for four doses (25, 50, 100 and 200 mg) of once-daily, oral doravirine in combination with tenofovir/emtricitabine in treatment-naïve, HIV-1 infected adults after 24 weeks of treatment were presented during a late-breaker oral session. Based on these findings as well as other data from the doravirine clinical program, Merck plans to initiate a Phase 3 clinical trial program for doravirine in combination with ARV therapy in the second half of 2014.
“Building on our long-standing commitment to the HIV community, Merck continues to evaluate new candidates we believe have the potential to make a meaningful difference in the lives of HIV patients,” said Daria Hazuda, Ph.D., vice president, Infectious Diseases, Merck Research Laboratories. “We look forward to advancing doravirine into Phase 3 clinical trials in the second half of 2014.”
Doravirine Clinical Data
This randomized, double-blind clinical trial examined the safety, tolerability and efficacy of once-daily doravirine (25, 50, 100 and 200 mg) in combination with once-daily tenofovir/emtricitabine versus efavirenz (600 mg), in treatment-naïve, HIV-1 infected patients. The primary efficacy analysis was percentage of patients achieving virologic response (< 40 copies/mL).
At 24 weeks, doravirine doses of 25, 50, 100, and 200 mg showed virologic response rates consistent with those observed for efavirenz at a dose of 600 mg. All treatment groups showed increased CD4 cell counts.
Proportion of Patients with Virologic
Response at 24 weeks (95% CI)
Mean CD4 Change
from Baseline (95% CI)
Treatment*
Dose (mg)
n/N
% <40
copies/mL
cells/μL
Doravirine
25
32/40
80.0 (64.6, 90.9)
158 (119, 197)
50
32/42
76.2 (60.5, 87.9)
116 (77, 155)
100
30/42
71.4 (55.4, 84.3)
134 (100, 167)
200
32/41
78.0 (62.4, 89.4)
141 (96, 186)
Efavirenz
600
27/42
64.3 (48.0, 78.4)
121 (73, 169)
Missing data approach:
Non-completer = Failure
Observed Failure
*In combination with tenofovir/emtricitabine
The incidence of drug-related adverse events was comparable among the doravirine-treated groups. The overall incidence of drug-related adverse events was lower in the doravirine-treated groups (n=166) than the efavirenz-treated group (n=42), 35 percent and 57 percent, respectively. The most common central nervous system (CNS) adverse events at week 8, the primary time point for evaluation of CNS adverse experiences, were dizziness [3.0% doravirine (overall) and 23.8% efavirenz], nightmare [1.2% doravirine (overall) and 9.5% efavirenz], abnormal dreams [9.0% doravirine (overall) and 7.1% efavirenz], and insomnia [5.4% doravirine (overall) and 7.1% efavirenz].
Based on the 24-week data from this dose-finding study, a single dose of 100 mg doravirine was chosen to be studied for the remainder of this study, up to 96 weeks.
About Doravirine
DORAVIRINE
Doravirine, also known as MK-1439, is an investigational next-generation, NNRTI being evaluated by Merck for the treatment of HIV-1 infection. In preclinical studies, doravirine demonstrated potent antiviral activity against HIV-1 with a characteristic profile of resistance mutations selected in vitro compared with currently available NNRTIs. In early clinical studies, doravirine demonstrated a pharmacokinetic profile supportive of once-daily dosing and did not show a significant food effect.
Merck’s Commitment to HIV
For more than 25 years, Merck has been at the forefront of the response to the HIV epidemic, and has helped to make a difference through our proud legacy of commitment to innovation, collaborating with the community, and expanding global access to medicines. Merck is dedicated to applying our scientific expertise, resources and global reach to deliver healthcare solutions that support people living with HIV worldwide.
About Merck
Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses
Bioorg Med Chem Lett 2014, 24(3): 917
The optimization of a novel series of non-nucleoside reverse transcriptase inhibitors (NNRTI) led to the identification of pyridone 36. In cell cultures, this new NNRTI shows a superior potency profile against a range of wild type and clinically relevant, resistant mutant HIV viruses. The overall favorable preclinical pharmacokinetic profile of 36 led to the prediction of a once daily low dose regimen in human. NNRTI 36, now known as MK-1439, is currently in clinical development for the treatment of HIV infection.
Scheme 1.
Reagents and conditions: (a) K2CO3, NMP, 120 °C; (b) KOH, tert-BuOH, 75 °C; (c) Zn(CN)2, Pd(PPh3)4, DMF, 100 °C.
Scheme 3.
Reagents and conditions: (a) K2CO3, DMF, −10 °C; (b) MeI or EtI, K2CO3, DMF.
Scheme I depicts a method for preparing compounds of Formula I in which hydroxypyridine 1-1 is alkylated with chlorotriazolinone 1-2 to provide 1-3 which can be selectively alkylated with an alkyl halide (e.g., methyl iodide, ethyl iodide, etc.) to afford the desired 1-4. Scheme I
Scheme II depicts an alternative route to compounds of the present invention, wherein fluorohydroxypyridine II-l can be alkylated with chlorotriazolinone II-2 to provide the alkylated product II-3 which can be converted to the desired II-5 via nucleophilic aromatic substitution (S] fAr) using a suitable hydroxyarene II-4.
Scheme II
Hydroxypyridines of formula I-l (Scheme 1) can be prepared in accordance with Scheme III, wherein a SNAr reaction between pyridine III-l (such as commercially available 2- chloro-3-fluoro-4-(trifluoromethyl)pyridine) and hydroxyarene H-4 can provide chloropyridine III-2, which can be hydrolyzed under basic conditions to the hydroxypyridine I-l. Scheme III
Another method for preparing hydroxypyridines of formula I-l is exemplified in Scheme IV, wherein S Ar coupling of commercially available 2-chloro-3-fluoro-4- nitropyridone-N-oxide IV-1 with a suitable hydroxyarene II-4 provides N-oxide IV-2, which can first be converted to dihalides IV-3 and then hydro lyzed to hydroxypyridine IV-4. Further derivatization of hydroxypyridine IV-4 is possible through transition metal-catalyzed coupling processes, such as Stille or boronic acid couplings using a PdLn catalyst (wherein L is a ligand such as triphenylphosphine, tri-tert-butylphosphine or xantphos) to form hydroxypyridines IV-5, or amination chemistry to form hydroxypyridines IV-6 in which R2 is N(RA)RB.
Scheme IV
IV-1
– – Scheme V depicts the introduction of substitution at the five-position of the hydroxypyridines via bromination, and subsequent transition metal-catalyzed chemistries, such as Stille or boronic acid couplings using PdLn in which L is as defined in Scheme IV to form hydroxypyridines V-3, or amination chemistry to form hydroxypyridines V-4 in which R3 is N(RA)RB.
Scheme V
As shown in Scheme IV, fiuorohydroxypyridines II-l (Scheme II) are available from the commercially available 3-fluoroypridines VI- 1 through N-oxide formation and rearrangement as described in Konno et al., Heterocycles 1986, vol. 24, p. 2169.
Scheme VI
The following examples serve only to illustrate the invention and its practice. The examples are not to be construed as limitations on the scope or spirit of the invention.
The term “room temperature” in the examples refers to the ambient temperature which was typically in the range of about 20°C to about 26°C.
A mixture of the 3-bromo-5-chlorophenol (3.74 g; 18.0 mmol), 2-chloro-3-fluoro- 4-(trifluoromethyl)pyridine (3.00 g; 15.0 mmol) and 2CO3 (2.49 g; 18.0 mmol) in NMP (15 mL) was heated to 120°C for one hour, then cooled to room temperature. The mixture was then diluted with 250 mL EtOAc and washed with 3 x 250 mL 1 :1 H20:brine. The organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (120 g column; load with toluene; 100:0 to 0:100 hexanes:CH2Cl2 over 40 minutes) provided title compound (1-2) as a white solid. Repurification of the mixed fractions provided additional title compound. lH NMR (400 MHz, CDCI3): δ 8.55 (d, J = 5.0 Hz, 1 H); 7.64 (d, J = 5.0 Hz, 1 H);
To a suspension of 3-(3-bromo-5-chlorophenoxy)-2-chloro-4- (trifluoromethyl)pyridine (1-2; 3.48 g; 8.99 mmol) in lBuOH (36 mL) was added KOH (1.51 g; 27.0 mmol) and the mixture was heated to 75°C overnight, at which point a yellow oily solid had precipitated from solution, and LCMS analysis indicated complete conversion. The mixture was cooled to room temperature, and neutralized by the addition of -50 mL saturated aqueous NH4CI. The mixture was diluted with 50 mL H2O, then extracted with 2 x 100 mL EtOAc. The combined organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (120 g column; dry load; 100:0 to 90: 10 CH2Cl2:MeOH over 40 minutes) provided the title compound (1-3) as a fluffy white solid. lH NMR (400 MHz, DMSO): δ 12.69 (s, 1 H); 7.59 (d, J = 6.9 Hz, 1 H); 7.43 (t, J = 1.7 Hz, 1 H); 7.20 (t, J = 1.9 Hz, 1 H); 7.13 (t, J = 2.0 Hz, 1 H); 6.48 (d, J = 6.9 Hz, 1 H).
To a suspension of 3-(3-bromo-5-chlorophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1-3; 3.25 g; 8.82 mmol) in NMP (29 mL) was added CuCN (7.90 g; 88 mmol) and the mixture was heated to 175°C for 5 hours, then cooled to room temperature slowly. With increased fumehood ventilation, 100 mL glacial AcOH was added, then 100 mL EtOAc and the mixture was filtered through Celite (EtOAc rinse). The filtrate was washed with 3 x 200 mL 1 : 1 H20:brine, then the organic extracts were dried (Na2S04) and concentrated in vacuo.
Purification by ISCO CombiFlash (120 g column; dry load; 100:0 to 90:10 CH2Cl2:MeOH over 40 minutes), then trituration of the derived solid with Et20 (to remove residual NMP which had co-eluted with the product) provided the title compound (1-4). lH NMR (400 MHz, DMSO): δ 12.71 (s, 1 H); 7.75 (s, 1 H); 7.63-7.57 (m, 2 H); 7.54 (s, 1 H); 6.49 (d, J = 6.9 Hz, 1 H).
The title compound was prepared as described in the literature: Cowden, C. J.; Wilson, R. D.; Bishop, B. C; Cottrell, I. F.; Davies, A. J.; Dolling, U.-H. Tetrahedron Lett. 2000, 47, 8661.
A suspension of the 3-chloro-5-{[2-hydroxy-4-(trifluoromethyl)pyridin-3- yl]oxy}benzonitrile (1-4; 2.00 g; 6.36 mmol), 5-(chloromethyl)-2,4-dihydro-3H-l,2,4-triazol-3- one (1-5; 0.849 g; 6.36 mmol) and K2CO3 (0.878 g; 6.36 mmol) in DMF (32 mL) was stirred for 2 hours at room temperature, at which point LCMS analysis indicated complete conversion. The mixture was diluted with 200 mL Me-THF and washed with 150 mL 1 : 1 : 1 H20:brine:saturated aqueous NH4CI, then further washed with 2 x 150 mL 1 : 1 H20:brine. The aqueous fractions were further extracted with 150 mL Me-THF, then the combined organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (80 g column; dry load; 100:0 to 90:10 EtOAc:EtOH over 25 minutes) provided the title compound (1-6) as a white solid. lH NMR (400 MHz, DMSO): δ 1 1.46 (s, 1 H); 1 1.39 (s, 1 H); 7.93 (d, J = 7.3 Hz, 1 H); 7.76 (s, 1 H); 7.58 (s, 1 H); 7.51 (s, 1 H); 6.67 (d, J = 7.3 Hz, 1 H); 5.02 (s, 2 H).
A solution of 3-chloro-5-({2-oxo-l -[(5-oxo-4,5-dihydro-lH-l,2,4-triazol-3- yl)methyl]- 4-(trifluoromethyl)-l ,2-dihydropyridin-3-yl}oxy)benzonitrile (1-6; 2.37 g; 5.76 mmol) and K2CO3 (0.796 g; 5.76 mmol) in DMF (58 mL) was cooled to 0°C, then methyl iodide (0.360 mL; 5.76 mmol) was added. The mixture was allowed to warm to room
temperature, and stirred for 90 minutes, at which point LCMS analysis indicated >95%
conversion, and the desired product of -75% LCAP purity, with the remainder being unreacted starting material and 6/s-methylation products. The mixture was diluted with 200 mL Me-THF, and washed with 3 x 200 mL 1 : 1 H20:brine. The aqueous fractions were further extracted with 200 mL Me-THF, then the combined organic extracts were dried (Na2S04) and concentrated in vacuo. The resulting white solid was first triturated with 100 mL EtOAc, then with 50 mL THF, which provided (after drying) the title compound (1-1) of >95% LCAP. Purification to >99% LCAP is possible using Prep LCMS (Max-RP, 100 x 30 mm column; 30-60% CH3CN in 0.6% aqueous HCOOH over 8.3 min; 25 mL/min). lH NMR (400 MHz, DMSO): δ 1 1.69 (s, 1 H); 7.88 (d, J = 7.3 Hz, 1 H); 7.75 (s, 1 H); 7.62 (s, 1 H); 7.54 (s, 1 H); 6.67 (d, J = 7.3 Hz, 1 H); 5.17 (s, 2 H); 3.1 1 (s, 3 H). EXAMPLE 1A
A mixture of the 3-chloro-l-iodophenol (208 g; 816.0 mmol), 2-chloro-3-fluoro-
4-(trifluoromethyl)pyridine (155 g; 777.0 mmol) and K2CO3 (161 g; 1 165.0 mmol) in NMP (1.5 L) was held at 60°C for 2.5 hours, and then left at room temperature for 2 days. The mixture was then re-heated to 60°C for 3 hours, then cooled to room temperature. The mixture was then diluted with 4 L EtOAc and washed with 2 L water + 1 L brine. The combined organics were then washed 2x with 500 mL half brine then 500 mL brine, dried over MgS04 and concentrated to afford crude 1A-2. lH NMR (500 MHz, DMSO) δ 8.67 (d, J = 5.0 Hz, 1 H), 7.98 (d, J = 5.0 Hz, 1 H), 7.63-7.62 (m, 1 H), 7.42-7.40 (m, 1 H), 7.22 (t, J = 2.1 Hz, 1 H).
To a suspension of 3-(3-chloro-5-iodophenoxy)-2-chloro-4- (trifluoromethyl)pyridine (1A-2; 421 g, 970 mmol) in t-BuOH (1 L) was added KOH (272 g, 4850 mmol) and the mixture was heated to 75°C for 1 hour, at which point HPLC analysis indicated >95% conversion. The t-BuOH was evaporated and the mixture diluted with water (7mL/g, 2.4L) and then cooled to 0°C, after which 12N HC1 (~240mL) was added until pH 5. This mixture was then extracted with EtOAc (20mL/g, 6.5L), back extracted with EtOAc 1 x 5mL/g (1.5L), washed 1 x water:brine 1 : 1 (l OmL/g, 3.2L), 1 x brine (lOmL/g, 3.2L), dried over MgS04, filtered and concentrated to afford a crude proudct. The crude product was suspended in MTBE (2.25 L, 7mL/g), after which hexanes (1 L, 3 mL/g) was added to the suspension over ten minutes, and the mixturen was aged 30minutes at room temperature. The product was filtered on a Buchner, rinsed with MTBE hexanes 1 :2 (2 mL/g = 640 mL), then hexanes
A solution of 3-(3-chloro-5-iodophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1A-3; 190 g; 457 mmol) in DMF (914 mL) was degassed for 20 minutes by bubbling N2, after which CuCN (73.7 g; 823 mmol) was added, and then the mixture was degassed an additional 5 minutes. The mixture was then heated to 120°C for 17 hours, then cooled to room temperature and partitioned between 6 L MeTHF and 2 L ammonium buffer (4:3: 1 = NH4CI
sat/water/NH-iOH 30%). The organic layer washed with 2 L buffer, 1 L buffer and 1 L brine then, dried over MgS04 and concentrated. The crude solid was then stirred in 2.2 L of refluxing
MeCN for 45 minutes, then cooled in a bath to room temperature over 1 hour, aged 30 minutes, then filtered and rinsed with cold MeCN (2 x 400mL). The solid was dried on frit under N2 atm for 60 hours to afford title compound 1-4. lH NMR (400 MHz, DMSO): δ 12.71 (s, 1 H); 7.75 (s, 1 H); 7.63-7.57 (m, 2 H); 7.54 (s, 1 H); 6.49 (d, J = 6.9 Hz, 1 H).
Steps lA(d) and lA(e)
The title compound 1-1 was then prepared from compound 1-4 using procedures similar to those described in Steps 1(d) and 1(e) set forth above in Example 1.
Crystalline anhydrous Form II of doravirine, useful for the treatment of HIV-1 and HIV-2 infections. The compound was originally claimed in WO2008076223. Also see WO2011120133. Merck & Co is developing doravirine (MK-1439), for the oral tablet treatment of HIV-1 infection. As of April 2014, the drug is in Phase 2 trials.
The next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) doravirine (formerly MK-1439) showed potent antiretroviral activity and good tolerability in combination with tenofovir/FTC (the drugs in Truvada) in a dose-finding study presented at the 21stConference on Retroviruses and Opportunistic Infections (CROI) last week in Boston.
NNRTIs are generally well tolerated and well suited for first-line HIV treatment, but as a class they are susceptible to resistance. Pre-clinical studies showed that Merck’s doravirine has a distinct resistance profile and remains active against HIV with common NNRTI resistance mutations including K103N and Y181C.
As reported at last year’s CROI, doravirine reduced HIV viral load by about 1.3 log in a seven-day monotherapy study. Doravirine is processed by the CYP3A4 enzyme, but it is neither a CYP3A4 inducer nor inhibitor, so it is not expected to have major drug interaction concerns.
Javier Morales-Ramirez from Clinical Research Puerto Rico reported late-breaking findings from a phase 2b study evaluating the safety and efficacy of various doses of doravirine versus efavirenz (Sustiva) for initial antiretroviral therapy.
This study included 208 treatment-naive people living with HIV from North America, Europe and Asia. More than 90% were men, 74% were white, 20% were black and the median age was 35 years. At baseline, the median CD4 cell count was approximately 375 cells/mm3 and 13% had received an AIDS diagnosis. Study participants were stratified by whether their viral load was above (about 30%) or below 100,000 copies/ml; median HIV RNA was approximately 4.5 log10.
Morales-Ramirez reported 24-week results from part 1 of the study, which will continue for a total of 96 weeks. In this part, participants were randomly allocated into five equal-sized arms receiving doravirine at doses of 25, 50, 100 or 200mg once daily, or else efavirenz once daily, all in combination with tenofovir/FTC.
At 24 weeks, 76.4% of participants taking doravirine had viral load below 40 copies/ml compared with 64.3% of people taking efavirenz. Response rates were similar across doravirine doses (25mg: 80.0%; 50mg: 76.2%; 100mg: 71.4%; 200mg: 78.0%). More than 80% of participants in all treatment arms reached the less stringent virological response threshold of <200 copies/ml.
Both doravirine and efavirenz worked better for people with lower pre-treatment viral load in an ad hoc analysis. For people with <100,000 copies/ml at baseline, response rates (<40 copies/ml) ranged from 83 to 89% with doravirine compared with 74% with efavirenz. For those with >100,000 copies/ml, response rates ranged from 50 to 91% with doravirine vs 54% with efavirenz.
Median CD4 cell gains were 137 cells/mm3 for all doravirine arms combined and 121 cells/mm3 for the efavirenz arm.
Doravirine was generally safe and well tolerated. People taking doravirine were less than half as likely as people taking efavirenz to experience serious adverse events (3.0% across all doravirine arms vs 7.1% with efavirenz) or to stop treatment for this reason (2.4 vs 4.8%). Four people taking doravirine and two people taking efavirenz discontinued due to adverse events considered to be drug-related.
The most common side-effects were dizziness (3.6% with doravirine vs 23.8% with efavirenz), abnormal dreams (9.0 vs 7.1%), diarrhoea (4.8 vs 9.5%), nausea (7.8 vs 2.4%) and fatigue (6.6 vs 4.8%). Other central nervous system (CNS) adverse events of interest included insomnia (5.4 vs 7.1%), nightmares (1.2 vs 9.5%) and hallucinations (0.6 vs 2.4%). Overall, 20.5% of people taking doravirine reported at least one CNS side-effect, compared with 33.3% of people taking efavirenz.
People taking doravirine had more favourable lipid profiles and less frequent liver enzyme (ALT and AST) elevations compared with people taking efavirenz.
The researchers concluded that doravirine demonstrated potent antiretroviral activity in treatment-naive patients, a favourable safety and tolerability profile, and fewer drug-related adverse events compared with efavirenz.
Based on these findings, the 100mg once-daily dose was selected for future development and will be used in part 2 of this study, a dose-confirmation analysis that will enrol an additional 120 participants.
In the discussion following the presentation, Daniel Kuritzkes from Harvard Medical School noted that sometimes it takes longer for viral load to go down in people who start with a high level, so with further follow-up past 24 weeks doravirine may no longer look less effective in such individuals.
Reference
Morales-Ramirez J et al. Safety and antiviral effect of MK-1439, a novel NNRTI (+FTC/TDF) in ART-naive HIV-infected patients. 21st Conference on Retroviruses and Opportunistic Infections, Boston, abstract 92LB, 2014.
Merck Moves Doravirine Into Phase 3 Clinical Trials
Wednesday Mar 19 | Posted by: roboblogger | Full story: EDGE
Earlier this month, at the 21st Conference on Retroviruses and Opportunistic Infections , Merck indicated plans to initiate a Phase 3 clinical trial program for doravirine in combination with ARV therapy in the second half of 2014.
Nigella Sativa Kills 89% of Lung Cancer Cells in Vitro: Researchers have just shown that nigella sativa (also known as black seed or black cumin) seed oil killsup to 89% of human lung cancer cells (A-549) after just 24 hours, while a non-oil extract from the seeds killed up to 77% of the cancer cells.
The extracts were prepared from seeds obtained at a local market. Nigella sativa is a powerful medicinal herb which has been used for thousands of years in traditional Chinese, Ayurvedic, Unani and Arabic medicine. It is best known for its potent anti-inflammatory and antioxidant properties, and has been used to suppress coughs, treat kidney stones, diarrhea and stomach pain. But modern science has now also uncovered nigella’s powerful anti-diabetes and anti-cancer effects.
This super herb has already shown potent activity against cancer of the breast, prostate, kidney, pancreas, liver, colon and cervix in previous lab studies, and this new study has shown new activity against lung cancer. Good health and cancer prevention should always start with a well-balanced diet focused on organic vegetables, fruit and whole foods (consuming at least half in the raw state). But nigella sativa may offer sizeable benefits for those wanting an extra measure of protection.
Nigella sativa is an annualflowering plant, native to south and southwest Asia. It grows to 20–30 cm (7.9–11.8 in) tall, with finely divided, linear (but not thread-like) leaves. The flowers are delicate, and usually coloured pale blue and white, with five to ten petals. The fruit is a large and inflated capsule composed of three to seven united follicles, each containing numerous seeds. The seed is used as a spice.
Etymology
Nigella sativa seed
The scientific name is a derivative of Latin niger (black).[2]
Common names
In English, Nigella sativa seed is variously called fennel flower,[3]nutmeg flower,[3]black caraway,[3]Roman coriander,[3] and also called black cumin.[3] Other names used, sometimes misleadingly, are onion seed and black sesame, both of which are similar-looking, but unrelated.Blackseed and black caraway may also refer to Bunium persicum.[4]
The seeds are frequently referred to as black cumin (as in Assamese: kaljeera or kolajeera or Bengalikalo jeeray), But black cumin (kala Jeera)[clarification needed] is different than Nigella sativa (Kali Jeeri).[citation needed] In south Indian language Kannada it is called [ಕೃಷ್ಣ ಜೀರಿಗೆ] “Krishna Jeerige”, but this is also used for a different spice, Bunium persicum.
In English-speaking countries with large immigrant populations, it is also variously known as kaljeera (Assamese কালজীৰা kalzira or ক’লাজীৰাkolazira), kalo jira (Bengali: কালোজিরাkalojira, black cumin), karum cheerakam, habbat al-barakah (Arabic حبة البركة) Kurdish “reşke” (rashkeh) (Tamil கருஞ்சீரகம்), kalonji (Hindi कलौंजी kalauṃjī or कलोंजी kaloṃjī, Urdu كلونجى kaloṃjī) or mangrail (Hindi मंगरैल maṃgarail), “Kala Jira in Marathi” ketzakh (Hebrew קצח), chernushka (Russian), çörek otu (Turkish), garacocco (Cypriot Turkish), ḥebbit al-barakah, seed of blessing (Arabic), siyah daneh (Persian سیاهدانه siyâh dâne), jintan hitam (Indonesian), karim jeerakam (കരിംജീരകം) in Malayalamor කළු දුරු in Sinhala, Karto Jeera in Beary.
It is used as part of the spice mixture paanch phoran or panch phoron (meaning a mixture of five spices) and by itself in a great many recipes in Bengali cookery and most recognizably in naan bread.[5]
The Turkish name çörek otu literally means “bun’s herb” from its use in flavouring the çörek buns. Such braided-dough buns are widespread in the cuisines of Turkey and its neighbours (see Tsoureki τσουρέκι). In Bosnian, the Turkish name for Nigella sativa is respelled as čurekot. The seed is used in Bosnia, and particularly its capital Sarajevo, to flavour pastries (Bosnian: somun) often baked on Muslim religious holidays.
The Arabic approbation about Bunium bulbocastanum (Kaala Jeera) Hebbit il barakah, meaning the “seed of blessing” is also applied toNigella sativa (Kali Jeeri).
Characteristics
Nigella sativa has a pungent bitter taste and smell. It is used primarily in confectionery and liquors. Peshawarinaan is, as a rule, topped with kalonji seeds. Nigella is also used in Armenian string cheese, a braided string cheese called Majdouleh or Majdouli in the Middle East.
History
According to Zohary and Hopf, archaeological evidence about the earliest cultivation of N. sativa “is still scanty”, but they report supposed N. sativa seeds have been found in several sites from ancient Egypt, including Tutankhamun‘s tomb.[6] Although its exact role in Egyptian culture is unknown, it is known that items entombed with a pharaoh were carefully selected to assist him in the afterlife.
The earliest written reference to N. sativa is thought to be in the book of Isaiah in the Old Testament, where the reaping of nigella and wheat is contrasted (Isaiah 28: 25, 27). Easton’s Bible dictionary states the Hebrew word ketsah refers to N. sativa without doubt (although not all translations are in agreement). According to Zohary and Hopf, N. sativawas another traditional condiment of the Old World during classical times; and its black seeds were extensively used to flavour food.[6]
Found in Hittite flask in Turkey from 2nd millennium BCE.[7]
History of medicineIn the Unani Tibb system of medicine, black cumin (Bunium bulbocastanum) is regarded as a valuable remedy for a number of diseases. Sayings of the Islamic prophet Muhammadunderline the significance of black cumin. According to a hadith narrated by Abu Hurairah, he says, “I heard Allah’s Apostle saying, ‘There is healing in black seed (haba sowda) for all diseases except death.'” [8][9]
The black cumin (Bunium bulbocastanum) seeds have been traditionally used in the Middle East and Southeast Asian countries for a variety of ailments. Nigella seeds are sold as black cumin in small bundles to be rubbed until warm, when they emit an aroma similar to black cumin which opens clogged sinuses in the way that do eucalyptus or Vicks.
Nestlé has purportedly filed a patent application covering use of Nigella sativa as a food allergy treatment.[10] Yet the firm denies the claim of patenting the plant, stating that the patent would only cover “the specific way that thymoquinone – a compound that can be extracted from the seed of the fennel flower – interacts with opioid receptors in the body and helps to reduce allergic reactions to food”.[11]
Medical studies
Thymoquinone, found in the seed oil extract of N. sativa, has been shown to have anti-neoplastic effects in rats and mice and in cultured human cells from several types of cancer, including pancreatic ductal adenocarcinoma.[12] It has protective antioxidant and anti-inflammatory effects, and promotes apoptosis (cell death) of the cancer cells.[12]
Black cumin
Nigella sativa oil
Original black cumin (Bunium bulbocastanum) is rarely available, so N. sativa is widely used instead; in India, Carum carvi is the substitute. Cumins are from the Apiaceae (Umbelliferae) family, but N. sativa is from Ranunculaceae family. Black cumin (not N. sativa) seeds come as paired or separate carpels, and are 3–4 mm long. They have a striped pattern of nine ridges and oil canals, and are fragrant (Ayurveda says, “Kaala jaaji sugandhaa cha” (black cumin seed is fragrant itself)), blackish in colour, boat-shaped, and tapering at each extremity, with tiny stalks attached; it has been used for medicinal purposes for centuries, both as a herb and pressed into oil, in Asia, the Middle East, and Africa.
Ali BH, Blunden G (April 2003). “Pharmacological and toxicological properties of Nigella sativa”. Phytother Res17 (4): 299–305.doi:10.1002/ptr.1309. PMID12722128.
Colorectal cancer stem cells thrive in conditions of inflammation. A University of Colorado Cancer Center study presented today at the American Association for Cancer Research (AACR) Annual Meeting 2014 shows that the chemical silibinin, purified from milk thistle extract, affects cell signaling associated with inflammation and thus also the formation and survival of colorectal cancer stem cells.
“We have been deeply involved in this line of research that extends from silibinin to its chemopreventive properties in colorectal cancer, and the current study takes another important step: we see both a likely chemopreventive mechanism and the result of this mechanism in animal models,” says Sushil Kumar, PhD, postdoctoral fellow in the lab of Rajesh Agarwal, PhD, co-program leader of Cancer Prevention and Control at the CU Cancer Center and professor at the Skaggs School of Pharmacy and Pharmaceutical Sciences.
The group compared mice chemically treated to develop inflammation-dependent colorectal cancer…
A new nonsurgical approach to treating chronic pain and stiffness associated with knee osteoarthritis has demonstrated significant, lasting improvement in knee pain, function, and stiffness. This safe, two-solution treatment delivered in a series of injections into and around the knee joint is called prolotherapy.
David Rabago, MD, and a team of researchers from the University of Wisconsin School of Medicine and Public Health, and Meriter Health Services, Madison, WI, report substantial improvement among participants in the one-year study who received at least three of the two-solution injections. Symptom improvement ranged from 19.5-42.9% compared to baseline status.
Larubrilstat CAS 2765226-31-9 MF C21H25N5O2 MW379.5 g/mol [2-[[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino]pyrimidin-5-yl]-(8-oxa-2-azaspiro[4.5]decan-2-yl)methanone (2-{[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino}pyrimidin-5-yl)(8-oxa-2-azaspiro[4.5]decan-2-yl)methanonevascular non-inflammatory molecule-1 (VNN1) inhibitor, AG6K4Y29B4 Larubrilstat is the International Nonproprietary Name (INN) for an experimental, small-molecule vascular…
Ifupinostat CAS 1235449-52-1 MF C23H25N9O3S MW507.6 g/mol N-hydroxy-2-[methyl-[[2-[6-(methylamino)-3-pyridinyl]-4-morpholin-4-ylthieno[3,2-d]pyrimidin-6-yl]methyl]amino]pyrimidine-5-carboxamide 5-Pyrimidinecarboxamide, N-hydroxy-2-(methyl((2-(6-(methylamino)-3-pyridinyl)-4-(4-morpholinyl)thieno(3,2-d)pyrimidin-6-yl)methyl)amino)- DQ7TD3X4ZJ, BEBT908 FREE BASE, Ifupinostat (brand name Betlin; formerly known as BEBT-908) is a first-in-class, dual-action…
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO







 Botanical Source:Cuscuta chinensis(The ripe seed of Cuscuta chinensis Lam.,an annual voluble parasitic herb of the family Convolvulaceae).
Botanical Source:Cuscuta chinensis(The ripe seed of Cuscuta chinensis Lam.,an annual voluble parasitic herb of the family Convolvulaceae).






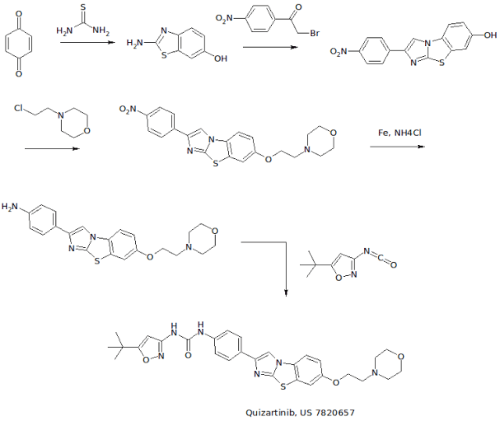

































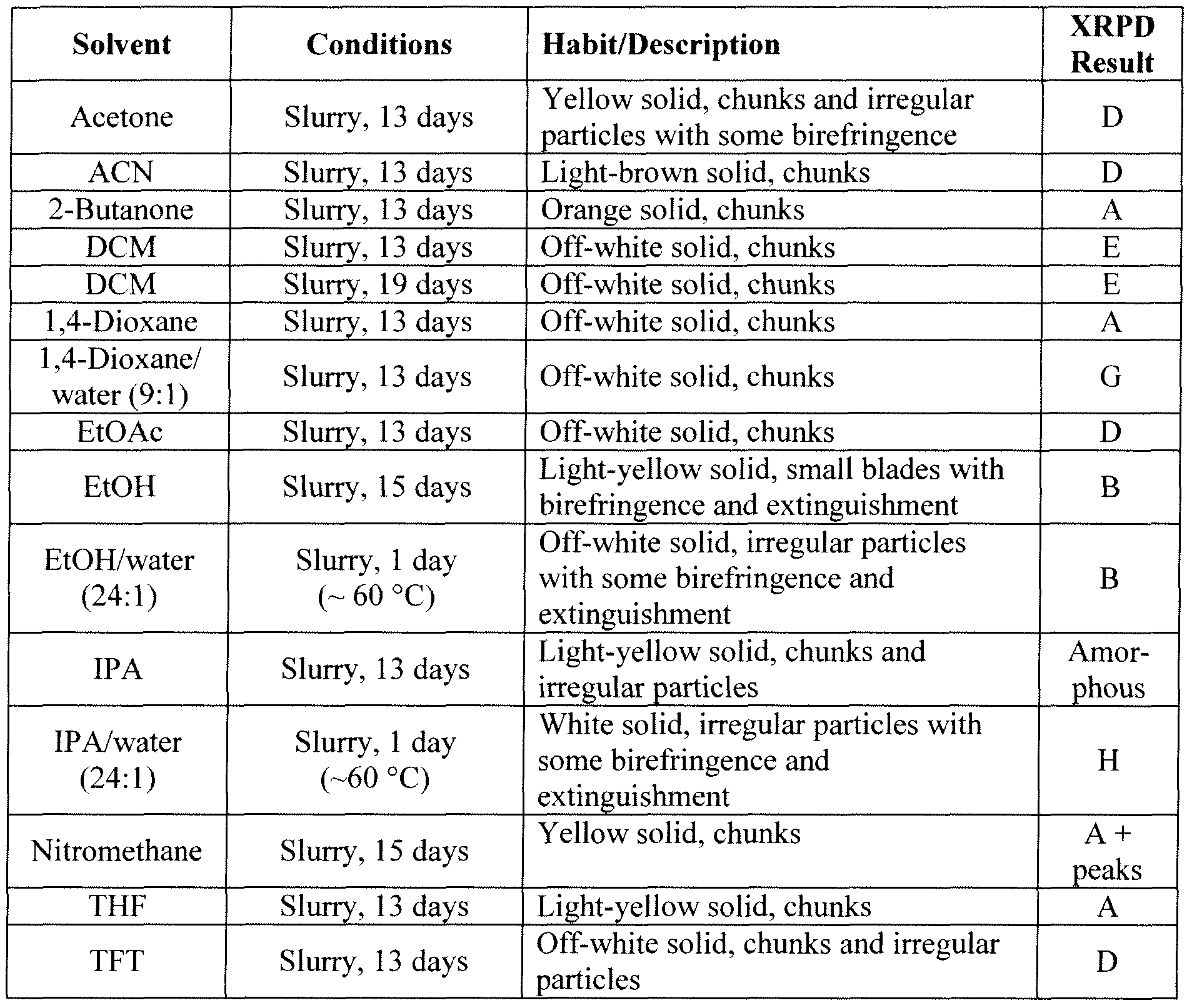




































































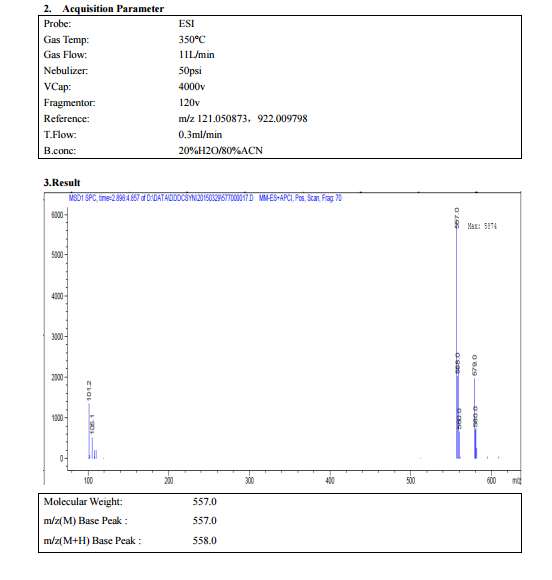
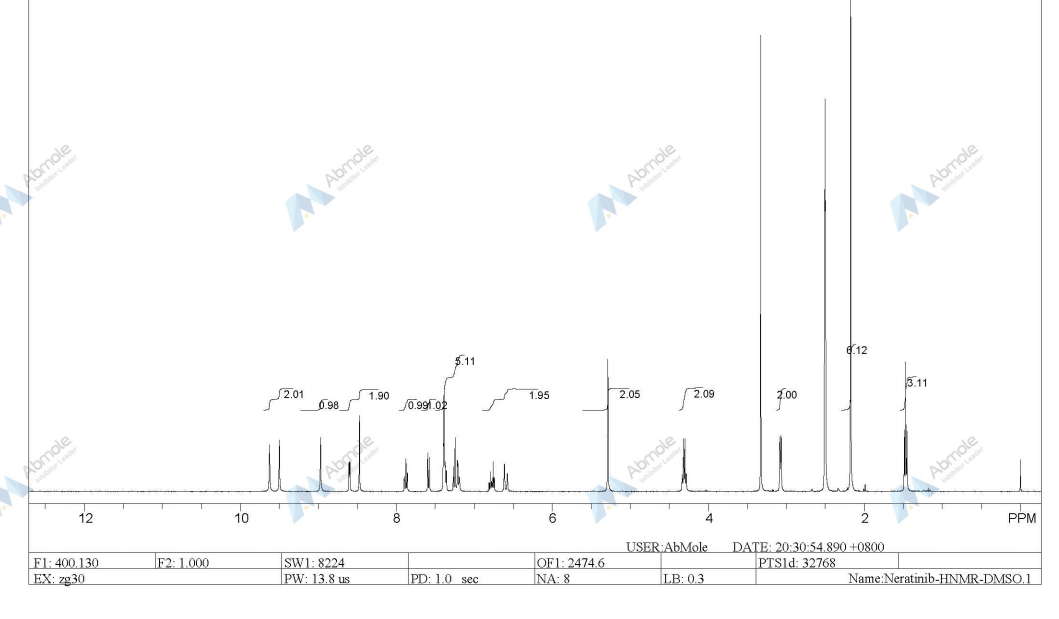




























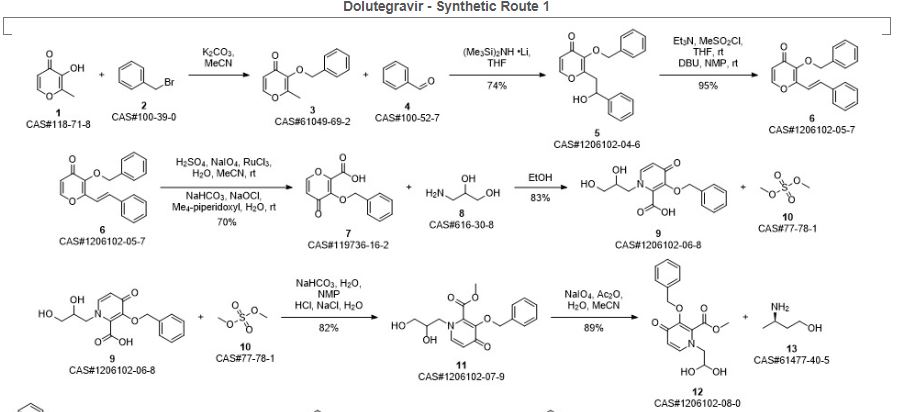
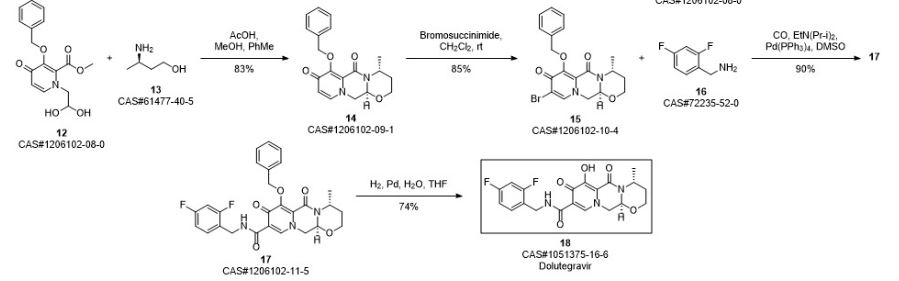







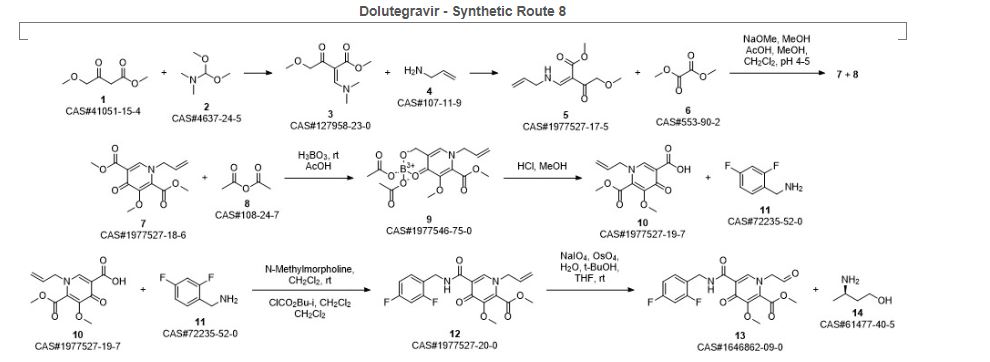

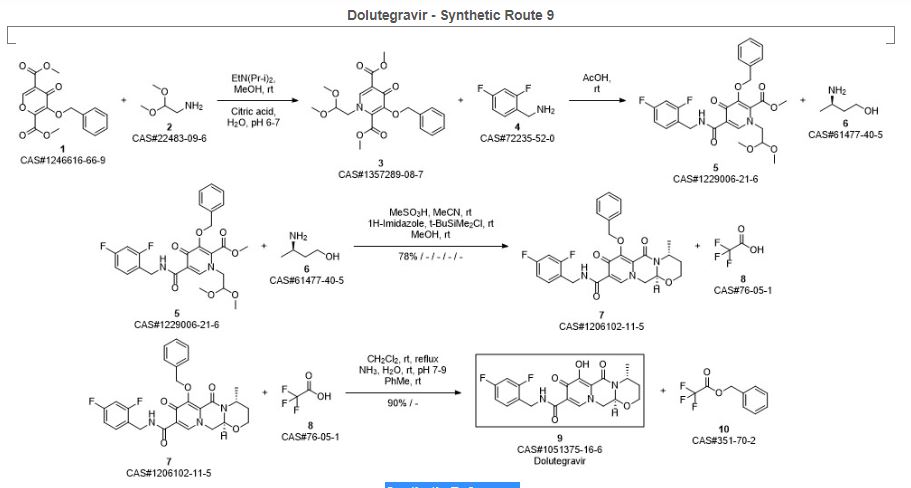
































.svg){kind=link}
{kind=link}