
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

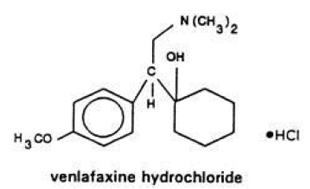
VENLAFAXINE PART 1/3


 SEE
SEEpart 1………http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine.html / https://newdrugapprovals.org/2015/04/09/venlafaxine-part-12/
part 2……..https://newdrugapprovals.org/2015/04/09/venlafaxine-22/
PART 3…..http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine-part-33.html
SEE ALSO
http://www.google.com/patents/WO2008059525A2?cl=en
WILL BE UPDATED………..
Chemistry

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (RS)-1-[2-dimethylamino-1-(4-methoxyphenyl)-ethyl]cyclohexanol | |
| Clinical data | |
| Trade names | Effexor XR, Effexor, Trevilor |
| AHFS/Drugs.com | monograph |
| Licence data | US Daily Med:link |
|
|
|
|
| Oral | |
| Pharmacokinetic data | |
| Bioavailability | 42±15%[1] |
| Protein binding | 27±2% (parent compound), 30±12% (active metabolite,desvenlafaxine)[2] |
| Metabolism | Hepatic (~50% of the parent compound is metabolised on first pass through the liver)[1][2] |
| Half-life | 5±2 h (parent compound for immediate release preparations), 15±6 h (parent compound for extended release preparations), 11±2 h (active metabolite)[1][2] |
| Excretion | Renal (87%; 5% as unchanged drug; 29% asdesvenlafaxine and 53% as other metabolites)[1][2] |
| Identifiers | |
| 93413-69-5 |
|
| N06AX16 | |
| PubChem | CID 5656 |
| DrugBank | DB00285 |
| ChemSpider | 5454 |
| UNII | GRZ5RCB1QG |
| ChEBI | CHEBI:9943 |
| ChEMBL | CHEMBL637 |
| Chemical data | |
| Formula | C17H27NO2 |
| 277.402 g/mol | |

HSQC

1H NMR PREDICT OF HCL

13C NMR PREDICT OF HCL


BASE


External links[Drug information
- U.S. Food and Drug Administration information on Effexor
- Efexor patient information leaflet (UK)
- Effexor XR prescribing information for healthcare professionals (pdf) (USA only) Archived from the original on 17 September 2006.
- Detailed Patient/Parent Information on Effexor
- List of international brand names for Venlafaxine
- U.S. National Library of Medicine: Drug Information Portal -Venlafaxine
Diagnostic tools
Patient experiences
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
amcrasto@gmail.com
VENLAFAXINE PART 2/3

part 1………http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine.html / https://newdrugapprovals.org/2015/04/09/venlafaxine-part-12/
part 2……..https://newdrugapprovals.org/2015/04/09/venlafaxine-22/
PART 3…..http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine-part-33.html
http://www.google.com/patents/WO2008059525A2?cl=en
WILL BE UPDATED………..
………………….

……………………….
PAPER
DOI: 10.1039/C4RA00840E
http://pubs.rsc.org/en/content/articlelanding/2014/ra/c4ra00840e#!divAbstract
A protecting group free asymmetric total synthesis of (−)-venlafaxine is reported. The strategy employs Sharpless epoxidation and regio-selective epoxide ring opening by an in situgenerated Gilman reagent as key steps. This paper reports a 53% overall yield in 6 steps for total synthesis of (−)-venlafaxine.


…………………
http://www.google.com/patents/EP2181982B1?cl=en
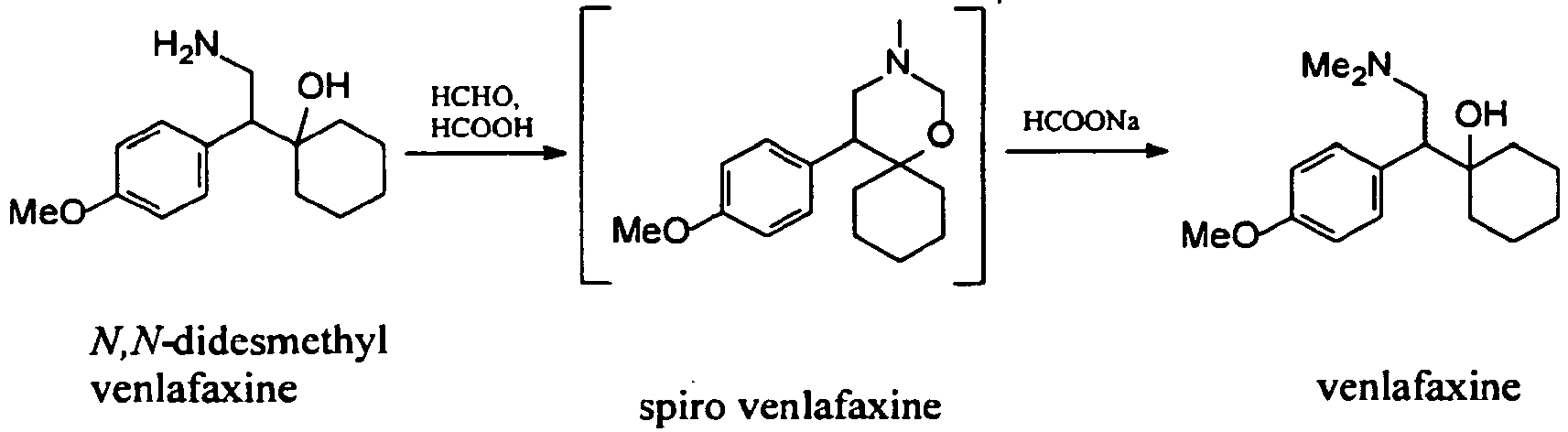
Examples:Example 1 – Preparation of venlafaxine from N,N-didesmethyl venlafaxine hydrochloride
-
A 50 % aqueous NaOH solution (4 ml, 74 mmol) was added to a stirred solution of N,N-didesmethyl venlafaxine hydrochloride (5.72 g, 20 mmol) in water (16 ml) at room temperature. Formic acid (98 %, 11.5 ml, 305 mmol) and 37 % aqueous solution of formaldehyde (8.4 ml, 113 mmol) were added to this mixture. The mixture was stirred under reflux temperature and the conversion was completed in 5 h (HPLC: 98.67 area %). Then the solution was cooled to room temperature and adjusted with 50 % aequous NaOH to pH 12. The mixture was extracted twice with 66 ml of isopropyl acetate. The collected organic phases were washed three times with water (66 ml). The isolated solution of venlafaxine base was very pure (HPLC: 98.9 area%).
Example 2 – Preparation of venlafaxine hydrochloride form I from the solution of venlafaxine base in isopropyl acetate
-
To the solution of venlafaxine base in isopropyl acetate from example 1 (66 ml, 10 mmol) 5 ml of 2 M aqueous HCl were added. The mixture was heated and water was removed by azeotropic distillation using a Dean-Starck trap. When all water was removed from the mixture, the product began slowly to crystallize. The obtained suspension was heated under reflux temperature for 1.5 h, then cooled and filtered. 2.75 g (88 % from N,N-didesmethyl venlafaxine hydrochloride) of pure venlafaxine hydrochloride form I (HPLC: 99.65 area %) were obtained.
Example 3 (exemplary) – Preparation of venlafaxine hydrochloride form I from the solution of venlafaxine base in isopropyl acetate
-
The solution of venlafaxine in isopropyl acetate from example 1 (66 ml, 10 mmol) was concentrated to ½ of the volume. Then 10 to 50 mg of venlafaxine hydrochloride form I was added to the solution. Subsequently, a 2.5 M solution of HCl in ethanol (4.0 ml) was slowly added within 30 min. After the whole amount of acid was added, the obtained suspension was stirred for another 2 h. Then the mixture was filtered and the product was washed with isopropyl acetate and dried. We obtained 2.69 g (86 % from N,N-didesmethyl venlafaxine hydrochloride) of pure venlafaxine hydrochloride form I (HPLC: 99.65 area %).
…………………..
PATENT
http://www.google.com/patents/WO2006035457A1?cl=en
Venlafaxine is known by the chemical name 1-[2-dimethylamino-1-(4 methoxyphenyl ethyl Cyclohexanol hydrochloride and structure of formula (V).

(V)
Venlafaxine is a useful pharmaceutical agent as an antidepressant. Venlafaxine, the intermediates in the manufacture of Venlafaxine, the process of preparing said Venlafaxine and their intermediates are well known from US Patents 4,535,186, US Patent No. 6,350,912, and CN 1225356.
Further International Publication No. WO03/050074 discloses the manufacture of Venlafaxine Hydrochloride and crystalline polymorphs Form I, Form II, Form III and optically pure (R) and (S) enantiomers exhibiting different crystalline structures of Venlafaxine hydrochloride. The preparation of all the forms of Venlafaxine and their inter-conversion are also described in said WO03/0500074 publication. U.S. Patents 4535186, 4761501 disclose a process for manufacture of 1-[2- amino-i-(p-methoxyphenyl) ethyljcyclohexanol (free base of formula IV), an intermediate produced during the preparation of Venlafaxine in two stages by reacting p-methoxyphenyl acetonitrile with cyclohexanone in the presence of n- butyl lithium (Stage 1) to form 1-[cyano(p-methoxyphenyl) methyl] Cyclohexanol of formula III

(111)
This process is commonly used for the preparation of formula III. The US Patent 4,535,186 produces a yield of about 30% based on p-methoxyphenyl acetonitrile.
WO/03/050074 suggests an alternate way of preparing compound of formula III without using butyl lithium i.e. by reacting p-methoxyphenyl acetonitrile with cyclohexanone in the presence of alkali metal hydroxide in a mixture of toluene and hexane. The publication WO/03/050074 also suggests a material yield of 74% based upon p-methoxyphenyl acetonitrile and purity.
The drop wise addition of butyl lithium to p-methoxyphenyl acetonitrile is hazardous and hence it requires skill and safety measures to be taken by the person skilled in the art for handling butyl lithium over the addition period to avoid any accidents during the preparation process.
The second stage i.e. conversion of compound of formula III to formula IV described in US patent US 4,535,186 is by hydrogenating compound of formula III using Rhodium on alumina. The catalyst Rhodium is recycled by filtering and washing the catalyst with ethanol and the combined filtrate evaporated and dried under vacuum yielding free base as an oil. However, the cost of Rhodium catalyst is very high and hence the catalyst has to be recovered.
WO/02/500017 suggests the use of a Nickel or cobalt catalyst for the hydrogenation, which is highly economical when compared with the Rhodium catalyst as suggested by US Patent No. 4,535,186. The International Publication WO/02/500017 teaches that the hydrogenation reaction of Stage Il may be carried out in the presence of an organic solvent preferably an alcohol. The international publication also suggests the pretreatment of the catalyst with ethanol.
The US Patent 4,535,186 describes the third stage in the process of preparing Venlafaxine i.e. conversion of compound of formula IV (free base) to compound V i.e. Venlafaxine by methylating the compound of formula IV (free base) with a mixture of formaldehyde and formic acid in water.

(IV)
US Patent Publication No. 2005/0033088 describes a process for preparing phenylethylamine derivative, an intermediate of Venlafaxine hydrochloride; said process comprising steps of reduction of compound of formula III with palladium on charcoal in an organic acid selected from formic acid, acetic acid or propionic acid, preferably acetic acid in an autoclave at a pressure of 5 to 25 kg/cm2 preferably 10 to 15 kg/cm2 at a temperature in the range of 30 to 75°C, preferably at 50 to 55°C till the hydrogenation substantially complete, filtering the palladium catalyst and evaporating the filtrate. Extracting the filtrate with halogenated hydrocarbon solvent and purifying the same. The process also describes the preparation of Venlafaxine hydrochloride without isolation of freebase.
The route of synthesis for Venlafaxine (formula V) and intermediate of Venlafaxine (formula IV) is depicted in the following scheme:
Step -I

(I) (II) (III) Step-ll

…………..(III) ……………………(IV)
Step-ll I

(IV) (V)
Accordingly, the present invention relates to an improved process for the preparation of compound of formula IV

(IV)
comprising the step of hydrogenating a compound of formula

(Hi)
in the presence of toluene, water, and a catalyst wherein the said process yields 66% formula (IV) with 99% HPLC purity.
The compound of formula IV is further methylated using formaldehyde and formic acid mixture to form Venlafaxine (formula V) followed by the treatment with HCL gas dissolved in Isopropanol.

(IV) (V)
According to the preferred embodiment of the present invention, nickel catalyst, preferably Raney nickel catalyst is used. The catalyst is washed in water to remove the alkali. No pretreatment of the nickel catalyst is required.

(HI) (IV)
According to another embodiment of the present invention, compound of formula IV is prepared by hydrogenating compound of formula III in the presence of Raney Nickel and water. According to another embodiment of the present invention, compound of formula III is prepared by charging p-methoxyphenyl acetonitrile into butyl lithium at -70 to -75°C and tetrahydrofuran; cooling the reaction mixture to about -50°C to -750C; adding cyclohexanone at a temperature below -500C quenching with ice and saturated ammonium chloride solution below 0°C; and stirring and filtering the product of formula (III) wherein the said process yields 89% of compound of formula III with 99.8% purity. The reaction scheme is depicted as follows:

…………(I) …………(H)………………………………. (III)
Example 1 :
Preparation of 1-[cyano-(4-methoxyphenyl) methyl Icvclohexanol.
In a 2 ltr 4 necked round bottom flask equipped with a overhead stirrer, thermometer and dropping funnel, 100 ml dry THF followed by 210 ml Butylithium (1.6 M solution in Hexane) was charged. The reaction mixture was cooled to – 700C. Added gradually a solution of 50 gm p-methoxyphenyl acetonitrile dissolved in 50 ml dry THF at -70 to -75°C. After 30 min, added solution of 33.1 gm Cyclohexanone in 50 ml THF. After the addition, maintained at -65 to -700C and monitored by TLC. After 4 hrs, reaction mixture was gradually added over mixture of ice and 150 ml saturated ammonium chloride solution below 0°C and adjusted pH to 7 with dilute Hydrochloric acid. Stirred for 1 hr and filtered the product. Washed the product with 200 ml hexane and dried to obtain 74.3 gm. (The yield based on p-methoxyphenyl acetonitrile 89%, Melting range 123- 125°C, HPLC purity of 99.8%).
Example 2:
Preparation of 1-[2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate
In an autoclave are charged 100 gm 1-[cyano(4- methoxyphenyl)ethyl]cyclohexanol, 100 ml toluene and 400 ml water at RT. Stirred and cooled to 10°C. Charged 20 gm Raney Nickel (which was prewashed with water to make it free of Alkali) and 100 ml liquor ammonia (20%). Then pressurized the autoclave with hydrogen to 4 – 5 kg pressure and maintained for 120 minutes below 120C. Then the reaction temperature slowly raised to bout 500C along with the increase in the hydrogen pressure to 7 to 8 kg. Maintained between 45 – 50°C for 8 hr. After the completion of the reaction, cooled the reaction to RT, released the hydrogen pressure and charged 400 ml toluene. Filtered the catalyst and washed bed with 100 ml toluene. Separated the organic layer from the filtrate. The organic layer was washed with 10% Sodium chloride solution. To the organic layer was added 40 ml methanol followed by 10 ml acetic acid. Stirred for 15 minutes and then again charged 10 ml acetic acid. Then heated to 75-8O0C and maintained for 15 minutes. Cooled to 0 – 50C. Filtered the product. Washed the product with 100 ml ethyl acetate and dried: 83.5 gm (Yield 66%, Melting range 164-166°C, HPLC purity 99%).
Example 3:
Preparation of H2-dimethylamino-1-(4-methoxyphenyl) ethvH Cyclohexanol Hydrochloride
To a stirred solution of 100 gm of 1-[2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate in 300 ml water was added 117 gm of formic acid (88%) and 91 gm of formaldehyde (40% solution). The solution was heated to 98°C and maintained for 20 hrs. Reaction mixture was cooled to about 100C and added 500 ml ethyl acetate. The pH was adjusted to about 7 with sodium hydroxide solution and further to 10 – 10.5 with ammonium hydroxide solution. Layers were separated. Aqueous layer was extracted with ethyl acetate. Combined organic layers were washed with water. Combined organic extract was stirred with activated carbon (5 gm) and filtered. Filtrate was concentrated in vacuum to completely remove ethyl acetate. Residue was dissolved in isopropanol (300 ml) and acidified at 300C (pH 1-1.5) with the solution of HCI in isopropanol. Temperature was then raised to 600C and maintained for 60 to 90 min. The reaction mass was cooled under agitation to 10°C and maintained under agitation at 10°C for 60 min. Product was isolated by filtration. Finally it was washed with isopropanol and dried at 60°C.
Dry wt. : 85 gm (84% yield, HPLC 99.9% purity with all individual impurities below 0.1% concentration). This material exhibited following characteristic x-ray powder diffraction pattern with characteristic peaks expressed in d-values (A) at.
(The abbreviations in brackets mean : (vs) = Relative intensity above 80%; (s) = 30% – 80%; (m) = 15% – 30%; ![]() = 8% to 15% and (vw) = below 8%.) 2.23 (VW), 2.29(VW), 2.32 (VW)1 2.35(VW), 2.38(VW), 2.43(VW), 2.46(VW), -2.48(VW)12.55(M), 2.64(W), 2.69(W), 2.73(VW), 2.8(W), 2.83(W), 2.88(W), 2.93(VW), 3.09(VW), 3.12(M), 3.26(VM), 3.31(VM), 3.38(W), 3.45(VW), 3.5(VW), 3.55(M), 3.69(VW), 3.87(VW), 3.99(VW), 4.07(M), 4.18(S), 4.35(VS), 4.48(VW), 4.68(M), 5.1 (VW), 5.27(W), 5.42(VW), 5.55(VW), 5.63(M), 5.68(M), 5.76(VW), 6.5(S), 6.95(VS), 8.65(VW), 10.56(M), 13.06(M).
= 8% to 15% and (vw) = below 8%.) 2.23 (VW), 2.29(VW), 2.32 (VW)1 2.35(VW), 2.38(VW), 2.43(VW), 2.46(VW), -2.48(VW)12.55(M), 2.64(W), 2.69(W), 2.73(VW), 2.8(W), 2.83(W), 2.88(W), 2.93(VW), 3.09(VW), 3.12(M), 3.26(VM), 3.31(VM), 3.38(W), 3.45(VW), 3.5(VW), 3.55(M), 3.69(VW), 3.87(VW), 3.99(VW), 4.07(M), 4.18(S), 4.35(VS), 4.48(VW), 4.68(M), 5.1 (VW), 5.27(W), 5.42(VW), 5.55(VW), 5.63(M), 5.68(M), 5.76(VW), 6.5(S), 6.95(VS), 8.65(VW), 10.56(M), 13.06(M).
Example 4 :
Preparation of 1-f2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate (IV)
In an autoclave charged 150 gm 1-[cyano(4-methoxyphenyl)ethyl]cyclohexanol, and 675 ml water at RT. Stirred and cooled to 1O0C. Charged 30 gm Raney Nickel (prewashed with water to make it free of Alkali) and 150 ml liquor Ammonia (20%). Then pressurized the autoclave with hydrogen to 4 – 5 kg pressure and maintaind for 120 minutes below 12°C. After completion of 120 minutes slowly raised the temperature to about 500C along with the increase in the hydrogen pressure to 7 to 8 kg. Maintained between 45 – 5O0C for about 20 hrs. Monitored reaction by TLC to ensure disappearance of starting material. After the completion of the reaction cooled the reaction to RT, released the hydrogen pressure and filtered through celite bed. Washed bed with 300 ml toluene. To the filtrate added 300 ml toluene. Shaken well and separated the organic layer. The organic layer was washed with 5% Sodium chloride solution. To the organic layer was added 45 ml methanol and 15 ml acetic acid. Stirred for 15 minutes and then again charged 15 ml acetic acid. Then heated to 75-8O0C and maintained for 15 mins. Cooled to 0 – 5°C. Filtered the product. Washed the product with 100 ml ethyl acetate and dried: 104 gm (Yield 53%, Melting range 152-153°C, HPLC purity 90%).
……………….
PATENT
http://www.google.com/patents/WO2008059525A2?cl=en
Venlafaxine acts by inhibiting re-uptake of norepinephrine and serotonin. It has been reported that its (-) enantiomer is a more potent inhibitor of norepinephrine synaptosomal uptake while its (+) enantiomer is more selective in inhibiting serotonin uptake (J. Med. Chem. 1990, 33(10), 2899-2905) In humans, venlafaxine is transformed by a metabolic pathway into two minor metabolites, N-desmethylvenlafaxine of formula II, N,O-di- desmethylvenlafaxine of formula IV and one major metabolite, O-desmethylvenlafaxine of formula III.

formula I formula II

formula III formula IV
In the literature there are several processes reported for the synthesis of venlafaxine of formula I and venlafaxine hydrochloride of formula Ia.

formula Ia
The synthesis of venlafaxine from 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile (hereinafter called as cyano-intermediate and represented by formula V) involving two step synthesis is known in the prior art.

formula V
US 4,535,186 discloses the preparation of venlafaxine of formula I by the reaction of p- methoxyphenylacetonitrile with cyclohexanone at -78 0C in the presence of n-butyllithium as a base which yields 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile of formula V. Reduction of the cyano-intermediate under hydrogen pressure with rhodium on alumina catalyst gives l-[2-amino-l-(4-methoxyphenyl)ethyl]cyclohexanol. N-Methylation of the amino compound is accomplished employing formaldehyde and formic acid (Eschweiler- Clarke reaction) to give venlafaxine of formula I.
The reaction is as shown in the Scheme- 1.

Reduction


Scheme-1
Another prior art reference, Zhou Jinpei et.al, J. China Pharm. University, 1999, 30(4), 249- 50) discloses the preparation of venlafaxine starting from anisole. Anisole is acylated to the chloroacetyl derivative, which is then animated using N,N-dirnethylamine. The carbonyl group of this compound is reduced to the alcohol using KBH4 and is converted to the bromo- derivative using PBr3 which in turn when reacted with Mg and cyclohexanone undergoes a Grignard reaction to provide venlafaxine of formula I.
The reaction is as shown in the Scheme-2.

Scheme-2
US 2005033088 discloses a two step process for venlafaxine starting from the cyano- intermediate. The cyano-intermediate is reduced in the presence of palladium on charcoal in acetic acid at a hydrogen pressure of 5-25 kg/cm2 at a temperature in the range of 30-75 0C. The product of step 1 is N-methylated using formic acid, formaldehyde solution at a temperature of 90-98 0C for 19 hrs to yield venlafaxine, which is then converted to its hydrochloride salt.
WO2006035457 also discloses a process of making venlafaxine and its intermediates. The process comprises the step of hydrogenating the cyano-intermediate in the presence of toluene, water, and Raney nickel where in the said process yields 66% of an intermediate with 99% HPLC purity. This reaction is carried out at 10-12 0C and at 4-5 kg/cm2 of hydrogen pressure for 2 hrs and further at 50 0C at 7-8 kg/cm2 for 7-8 hrs. This intermediate is N-methylated using formaldehyde and formic acid mixture to form venlafaxine, which is treated with IPA/HC1 to get venlafaxine hydrochloride.
US 6,350,912 discloses a one pot process for the preparation of venlafaxine in 15-28 % yield from the cyano-intermediate. In the said patent venlafaxine has been prepared by the reduction of cyano-intermediate in the presence of Raney nickel and without isolation of intermediate l-[2-amino-l-(4-methoxyphenyl) ethyl] cyclohexanol
CN 1850781 discloses a process for the preparation of venlafaxine by following steps: (1) carrying out condensation of 4-methoxyphenylacetonitrile and cyclohexanone in presence of base to obtain 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile, (2) reacting 2-(l- hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile with cuprous chloride and dimethylamine to obtain 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)-N,N- dimethylacetimidamide, and (3) reacting 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)- N,N-dimethylacetimidamide with KBH4 to obtain 1 – [2-dimethylamino)- 1 -(4-methoxyphenyl) ethyl)cyclohexanol (venlafaxine).
The reaction is as shown in the Scheme-3.

Scheme-3
The processes disclosed in the prior art have many disadvantages. Most of the prior art processes employ formaldehyde as a reactant for N-methylation step which is known to be a carcinogen. Acute exposure of the same is highly irritating to the eyes, nose and throat. Ingestion of formaldehyde is fatal and long term exposure causes respiratory problems and skin irritation.
Another disadvantage is the formation of an impurity (represented by formula VI), which is formed during N-methylation step using formaldehyde as a reagent.

formula VI
As may be appreciated, all the above well-known processes share the same strategy of synthesis, consisting of two steps for the synthesis of venlafaxine from cyano-intermediate or are prepared in one pot with poor yield.
Yet another drawback of the processes disclosed in the prior art is the use of expensive catalysts like rhodium on alumina and use OfBF3 etherate which is highly corrosive.
The prior art references disclose the synthesis of alkoxyphenylethyldimethylamine from alkoxyphenylacetonitrile using excess dimethylamine and palladium catalyst in methanol solution which is firstly reported by Kindler and Hensse. (1. Kindler and Hesse; Arch. Pharm., 1933, 271, 439. 2. Johannes S. Buck, Richard Baltzly and Walter S. Ide; J. Am. Chem. Soc. 1938, 60(8), 1789-1792; 3. Albert J. Schuster and Eugene R. Wagner; J. Labelled Compounds and Radiopharmaceuticals 1992, XXXIII(3), 213-217).
The reaction is as shown in the Scheme-5.

Scheme-5
According to an aspect of the invention there is provided a novel single step process for the synthesis of venlafaxine of formula I and N-desmethylvenlafaxine of formula II from 2-(l- hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile of formula V comprising reaction of 2- (l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile with an alkylamine and/or its salt in a solvent in the presence of a transition metal catalyst, under hydrogen atmosphere.

formula I formula II formula V DETAILED DESCRIPTION OF THE INVENTION
The present invention describes a single step process for venlafaxine and its analog starting from the cyano-intermediate. The present invention circumvents the difficulties encountered in the prior art and is an economically viable process.
The present invention particularly relates to a single step synthesis of venlafaxine of formula I, and N-desmethylvenlafaxine of formula II from the cyano-intermediate of formula V.
The reaction of the present invention is as shown in Scheme-4:

formula V formula L R = CH3 (Venlafaxine) formula II. R = H (N-desmethylvenlafaxine)

formula Ia. R = CH3, X = Cl formula Ha. R = H, X = Cl
Example-1:
General procedure for synthesis of venlafaxine and N-desmethylvenlafaxine To the, stirred solution of 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile (1.0 equiv.) in methanol (10-20 volumes), alkylamine (3-5 equiv.) was added and the mixture was stirred for 5-10 minutes to get a clear solution. Palladium catalyst (10-50 wt %) was added under nitrogen atmosphere to the above reaction mixture. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 5 to 40 hrs. The progress of the reaction was monitored by TLC and HPLC. After completion of the reaction, the catalyst was filtered through celite and washed with methanol. The combined filtrate was concentrated to dryness under reduced pressure and the residue was poured in water. The aqueous layer was basifϊed with 10% aq. NaOH to pH 8-10 and extracted with ethyl acetate (3 times). The combined ethyl acetate layers were washed with brine and dried over sodium sulphate. Ethyl acetate was evaporated under vacuum to obtain the title compound.
Example-2: Venlafaxine
To the stirred solution of 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile (20.0 g, 1 equiv.) in methanol (460.0 ml), dimethylamine hydrochloride (26.6 g, 4 equiv.) was added and the mixture was stirred for 5-10 minutes at room temperature to obtain a clear solution. 5% Palladium on alumina (4.0 g, 20 wt %) was added under nitrogen to the above clear solution. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 20 hrs. After completion of the reaction product was isolated by the procedure as described in Example- 1 above to obtain light yellow viscous liquid (18.Og) which was directly converted to its hydrochloride salt using IPA/HC1. HPLC purity of the crude reaction mixture (18.Og) = 81.64%.
Venlafaxine-free base:
1H NMR in CDCl3 (300 MHz): δ 7.05 (d, 2H), 6.81 (d, 2H), 3.79 (s, 3H), 3.27 (t, IH), 2.94 , (dd, IH), 2.31 (s, 6H), 2.28 (dd, IH), 1.71-0.94 (m, 10H). 13C NMR in CDCl3: δ 158.16,’ 132.65, 130.01, 113.20, 74.14, 61.14, 55.05, 51.52, 45.37, 37.99, 31.07, 25.91, 21.52, 21.23.
IR (KBr): 3152, 2980, 2941, 2895, 1728, 1607, 1512, 1462, 1439, 1358, 1279, 1204, 1186, 1177, 1146, 1103, 1040, 1011, 968, 851 cm“1. HPLC Purity: 99.37 % (area %). GC-MS: 178 (M+H+).
Venlafaxine hydrochloride salt:
1H NMR in D2O (300 MHz): δ 7.23 (d, 2H), 6.91 (d, 2H), 3.71 (s, 3H), 3.54 (t, IH), 3.48 (dd, IH), 2.97 (dd, IH), 2.69 (s, 6H), 1.41-1.0 (m, 10H).
13C NMR in D2O: δ 158.52, 130.86, 128.16, 114.25, 73.23, 58.26, 55.25, 50.38, 44.87, 41.41, 35.07, 33.34, 24.76, 21.08, 20.86.
IR (KBr): 3321, 2941, 2928, 2675, 2644, 2623, 2611, 2587, 2521, 2482, 2359, 2330, 1512, 1441, 1242, 1179, 1038, 829 cm“1. HPLC Purity: 99.64 % (area %). GC-MS: 178 (M+H+).
Example-3: N-Desmethyl venlafaxine To the stirred solution of 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile (20.0 g, 1 equiv.) in methanol (350.0 ml), monomethylamine hydrochloride (27.7 g, 4 equiv.) was added and the mixture was stirred for 5-10 minutes at room temperature to obtain a clear solution. 5% Palladium on alumina (4.0 g, 20 wt %) was added under nitrogen to the above clear solution. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 24 hrs. After completion of the reaction product was isolated by the procedure as described in Example- 1 above to obtain light yellow viscous liquid (18.5g) which was directly converted to its hydrochloride salt using IPA/HCl.
HPLC conversion to N-desmethylvenlafaxine (in the crude reaction mixture = 18.5g): 35.46%. N-Desmethyl venlafaxine-free base:
1H NMR in CDCl3 (300 MHz): δ 7.23 (d, 2H), £91 (d, 2H), 3.71 (s, 3H), 3.54 (t, IH), 3.48 (dd, IH), 2.97 (dd, IH), 2.69 (s, 3H), 1.41-1.0 (m, 10H)
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003050074A1 * | Mar 19, 2002 | Jun 19, 2003 | Cadila Healthcare Ltd | Manufacture of venlafaxine hydrochloride and crystalline polymorphs thereof |
| US4535186 * | Oct 26, 1983 | Aug 13, 1985 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008059525A2 * | Oct 1, 2007 | May 22, 2008 | Calyx Chemicals And Pharmaceut | An improved process for the preparation of venlafaxine and its analogs |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006035457A1 * | Sep 16, 2005 | Apr 6, 2006 | Amoli Organics Ltd | A process for the manufacture of venlafaxine and intermediates thereof |
| US4535186 * | Oct 26, 1983 | Aug 13, 1985 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| US6350912 * | Feb 28, 2001 | Feb 26, 2002 | Council Of Scientific And Industrial Research | One pot process for the preparation of 1-[2-dimethylamino-(4-methoxyphenyl)-ethyl]cyclohexanol |
| US20050033088 * | Jun 7, 2004 | Feb 10, 2005 | Dr. Reddy’s Laboratories Limited | Catalytic hydrogenation of phenylacetonitrile using palladium on carbon supports |
| Reference | ||
|---|---|---|
| 1 | * | CHAVAN S P ET AL: “An efficient and green protocol for the preparation of cycloalkanols: a practical synthesis of venlafaxine” TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 45, no. 39, 20 September 2004 (2004-09-20), pages 7291-7295, XP004558985 ISSN: 0040-4039 |
| 2 | * | J.S. BUCK ET AL: “beta-Phenylethylamine Derivatives. Tertiary and quaternary salts” J.AM.CHEM.SOC., 1938, pages 1789-1792, XP002478651 cited in the application |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010100520A1 * | Mar 4, 2009 | Sep 10, 2010 | Hikal Limited | A process for preparation of phenethylamine derivative |
| WO2011124190A2 | Apr 6, 2011 | Oct 13, 2011 | Zentiva, K.S. | Method of producing 4-(2-(substituted)-1-(1-hydroxycyclohexyl)ethyl)phenols by o- demethylation of their methylethers by means of inodorous aromatic thiols |



Chernivtsi central square

KIEV

Independence Square (Maidan Nezalezhnosty)

DONETSK
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMRDRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
OLANZEPINE VISITED PART 3/3

PART 1…..https://newdrugapprovals.org/2015/04/08/olanzepine/
PART 2….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-22/
PART 3…….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-33/
review……http://cosmos.ucdavis.edu/archives/2011/cluster8/JAIN_VINEET.pdf
WATCHOUT………..
…………….
DOI: 10.1039/C3OB27424A
http://pubs.rsc.org/en/Content/ArticleLanding/2013/OB/c3ob27424a#!divAbstract
A new strategy for converting antipsychotic drug olanzapine into PDE4 inhibitors is describedvia the design and Pd/C mediated synthesis of novel N-indolylmethyl olanzapine derivatives. One compound showed good inhibition (IC50 1.1 μM) and >10 fold selectivity towards PDE4B over D that was supported by docking studies. This compound also showed significant inhibition of TNF-α and no major toxicities in cell lines and a zebrafish embryo model except the teratogenic effects to be re-assessed in rodents.

…………..
http://www.google.com/patents/US7329747
Olanzapine or 2-methyl-4-[4-methyl-1-piperazinyl]-10H-thieno[2,3b][1,5]-benzodiazepine is a pharmaceutically active compound that can be represented by the formula (1).

It was disclosed in EP 454436 and corresponding U.S. Pat. No. 5,229,382 as a useful antipsychotic agent. Olanzapine acts as a serotonin (5-HT2) and dopamine (D1/D2) receptor antagonist with anticholinergic activity. In commercially available final forms, the active substance is marketed as a free base, which is a white to yellow crystalline solid that is insoluble in water.
One synthetic route for making olanzapine starts from “des-methylpiperazine olanzapine precursor” of formula (3), which reacts with piperazine to form a “des-methyl olanzapine precursor” of formula (2) (see Jun-Da Cen, Chinese Journal of Pharmaceuticals 2001, 32(9),391-393). The compound (2) can be methylated to form olanzapine (see U.S. Pat. No. 4,115,568 for such suggestion). The methylation reaction can be carried out using formaldehyde under conditions of Eschweiler-Clarke reaction (see Jun-Da Cen) or by classical methylation agents such as methyl iodide (see WO 04-000847).

This synthetic pathway has the disadvantage that the reaction with piperazine may lead to formation of dimeric impurities and that the methylation with formaldehyde or other methylation agent may lead to side products, e.g. products of multiple methylation. All these contaminants are difficult to remove from the product. Also, methylation agents are, in general, toxic and mutagenic compounds.
An alternative of the above process was suggested in WO 04/000847 and comprises converting the compound (2) into a “formyl-olanzapine precursor” of formula (4) by a reaction with a methyl formate, and converting the compound (4) into olanzapine by a reduction with a metal borohydride.

In comparison with the preceding procedure, the alternate procedure is one step longer and suffers from the same problems in the step of making compound (2). Furthermore, the reported purity of the actually obtained olanzapine product is only 88%, which is not sufficient for pharmaceutical applications.
The present invention relates to the formation, purification and/or use of an N-formyl olanzapine. Accordingly, a first aspect of the present invention relates to a process, which comprises reacting a des-piperazine olanzapine of formula (3) or a salt thereof

with an N-formyl piperazine of formula (5)

to form an N-formyl olanzapine of formula (4) or a salt thereof

The reaction can be carried out in an inert solvent, generally a dipolar aprotic solvent, and is typically accomplished by heating. The N-formyl olanzapine can be converted to olanzapine.
Another aspect of the invention relates to a process for making an olanzapine salt, which comprises: reducing an N-formyl olanzapine of formula (4) or a salt thereof

with a reducing agent in a solvent to form olanzapine or a salt thereof dissolved in said solvent; reacting said dissolved olanzapine or a salt thereof with an acid to form an acid addition salt of olanzapine; and precipitating said olanzapine acid addition salt from said solution. Precipitating the salt of olanzapine can avoid the formation of technical grade olanzapine. That is, the olanzapine salt can be obtained in a purified state and then converted to olanzapine base, if desired, in high purity.
A further aspect of the present invention relates to purifying the N-formyl olanzapine, which process which comprises:
(1) dissolving and/or slurrying an N-formyl olanzapine of formula (4)

or a salt thereof in a solvent selected from the group consisting of an aliphatic alcohol, an aromatic hydrocarbon, and mixtures thereof, at a temperature of at least 35° C. to form a crystallization treatment medium;
(2) cooling said crystallization treatment medium; and
(3) isolating solid N-formyl olanzapine of formula (4) having improved purity. The steps (1)-(3) can be repeated if necessary until the desired purity is reached. Generally, such a process can achieve purity of greater than 95% and preferably greater than 98%.
An overall synthetic scheme for making olanzapine, which combines various aspects of the present invention, is set forth below:

Example 1
N-Formyl Olanzapine (4)
In a 1000 ml flask, a mixture of 12.0 g of “des-methylpiperazine olanzapine precursor” (compound of formula (2)) hydrochloride and 40 ml of N-formyl piperazine in a mixture of 60 ml of dimethylsulfoxide and 60 ml of toluene was heated at reflux under a nitrogen atmosphere overnight. Progress was monitored by HPLC. After cooling to 40° C., 160 ml of water was added. The resulting mixture was cooled and stirred at 0° C. The solid material was isolated by filtration and washed with 2×40 ml of water. Wet solid was dried overnight at ambient conditions and subsequently at 40° C. under vacuum.
Isolated yield: 12.19 gram, Purity (HPLC): 91.6%
Example 2
Crystallization of the Compound (4)
8.0 g of crude N-formyl olanzapine precursor (compound (4)) of a purity of about 89% (HPLC) was suspended in 50 ml of methanol and heated at 60° C. for 3 hours. The hot suspension was allowed to cool to room temperature and was subsequently cooled to 5° C. under stirring. The solid material was isolated by filtration, washed with 5 ml of cold methanol and 10 ml of cold diethyl ether and dried overnight at 40° C. under vacuum.
Yield: 3.97 g, purity 96.7% (HPLC)
……………….
PATENT
http://www.google.com/patents/WO2004056833A1?cl=en
…………………………….
http://www.google.com/patents/WO2002018390A1?cl=en
Olanzapine is represented by the following structure.

Olanzapine
Olanzapine is useful for treating psychotic patients and mild anxiety states. Preparation of Olanzapine and its acid salts, having pharmaceutical properties particularly in the treatment of disorders ofthe central nervous system has been discussed in U.S. Patent No. 5,229,382.
U.S. Patent No. 5,229,382 does not refer to any specific polymorphic crystalline form of Olanzapine. European patent specification No. 733635A1 claims Form-2 of Olanzapine. The process under this patent describes preparation of Form-2 from ethyl acetate. This patent also designated the product obtained according to the process described in U.S. Patent No. 5,229,382 as Form-1. Furthermore, EP 733635 Al discloses the d values for Form-1 and Form-2 from their X-ray Diffractograms. The values are: d value d value
Form-1 Form-2 9.94 10.26
8.55 8.57 8.24 7.47 6.88 7.12 6.37 6.14 6.24 6.07 5.58 5.48 5.30 5.21 4.98 5.12 4.83 4.98 4.72 4.76 4.62 4.71 4.53 4.47 4.46 4.33 4.29 4.22 4.23 4.14 4.08 3.98 3.82 3.72 3.74 3.56 3.69 3.53 3.58 3.38 3.50 3.25 3.33 3.12 3.28 3.08
3.21 3.06
3.11 3.01
3.05 2.87 2.94 2.81
2.81 ‘ 2.72
2.75 2.64
2.65 2.60
2.63 2.59
It is noteworthy to mention that EP 0 831 098 A2 discloses the preparation of a series of dihydrates of olanzapine namely Dihydrate B, Dihydrate D and Dihydrate E.
The d values from the X-ray Diffractograms for these forms are listed in EP 0 831
098 A2. We conducted experiments to obtain Olanzapine Form I by recrystallization of olanzapine from acetonitrile using the process described in Example 1, sub example 4 of U.S. Patent No. 5,229,382. The process is described herein for reference: A mixture of 4-amino-2-methyl-10H-thieno-[ 2,3-b] [l,5]benzodiazepine HCl (100 g),
N-methyl piperizine (350ml), DMSO (465 ml) and toluene (465 ml) was heated to reflux. The reaction mass was maintained at reflux for 19 hours and then cooled to
50°C and water was added. The reaction mass was cooled to 0-10°C and stirred at the same temperature for 6 hours. The crude Olanzapine separated was filtered and dried in oven to a constant weight (76.5 g). The crude compound was added to acetonitrile (750 ml) at boiling temperature. The mixture was boiled for further 5 minutes. The mixture was filtered to remove the undissolved solid. The filtrate was treated with carbon and filtered. The filtrate was distilled to a minimum volume, cooled to 0-5 °C and maintained at the same temperature for 1.0 hour and filtered.
The compound was dried to a constant weight in an oven (51.6g).
The polymorphic form obtained from these experiments was characterized for its X-ray Powder Diffraction on Rigaku D / Max 2200. As clearly observed, the d values for this product (Fig. 1) matched with those of Olanzapine Form-2 claimed in EP 733635A1. It is therefore inferred that the recrystallization of Olanzapine in acetonitrile produces Form-2 and not Form-1.
Accordingly, the present invention provides a novel method for preparation of hydrates of olanzapine, which are different from those reported in the literature. These hydrates are named Olanzapine monohydrate-I and Olanzapine dihydrate-I for convenience.
Accordingly, the present invention also provides a novel method for preparation of Olanzapine Form-1 by recrystallization of olanzapine or its hydrates in dichloromethane . The present invention also provides a novel method for converting Olanzapine
Form-2 to Olanzapine Form-1
PREPARATION OF OLANZAPINE MONOHYDRATE-1
EXAMPLE 1 A mixture of 4-amino-2-methyl-10H-thieno-[2,3-b][l,5]benzodiazepine hydrochloride (20 Kg), N-methyl piperazine (42 lit), dimethyl sulfoxide (40 lit) and toluene (95 lit) was heated to reflux. The reaction mass was maintained at reflux for
17 hours and 15 minutes and then cooled to 40-50°C. Water (95 lit) was added slowly at40-50°C. The reaction mass was cooled to -0.6 to 1.2°C and stirred at the same temperature for six hours. The Olanzapine crude that separated was filtered and washed with water (10 lit). The product was dried at 30.5 to 31.8°C for 10 hrs and 50 minutes. Yield: 20 Kg. A 20 gm sample from the above material after prolonged heating for an additional 72 hours gave the product with a moisture content of 5.22%.
PREPARATION OF OLANZAPINE DIHYDRATE-I EXAMPLE 2
A mixture of 4-amino-2-methyl-l OH-thieno- [2,3-b] [ 1 ,5]benzodiazepine hydrochloride (200 g), N-methyl piperazine (420 ml), dimethyl sulfoxide (200 ml) and toluene (940 ml) was heated to reflux. The reaction mass was maintained at reflux for 12 hours and then cooled to 40°C. Water (940 ml) was added slowly at 40-44°C. The reaction mass was cooled to 0-5°C and stirred at the same temperature for five hours. The Olanzapine crude that separated was filtered and washed with water (100 ml). The solid obtained was dried atmospherically (25-35°C) for 24 hours (Yield :
241 g).
PREPARATION OF FORM-1
EXAMPLE 3 Crude 2-methyl-4-(4-methyl- 1 -piperazinyl)- 1 OH-thieno- [2,3 -b] [ 1 ,5] benzodiazepine (35.0 g) was suspended in dichloromethane (160.0 ml). The suspension was heated to reflux to obtain a clear solution. The resultant solution was then treated with carbon (3.5 g) followed by filtration. Upon completion of this step the filtrate was cooled to 0 to 5°C and stirred at the same temperature for one hour. The separated solid was filtered and washed with chilled dichloromethane (10.0ml). The product obtained on drying in oven at 65 to 70°C to a constant weight gave Form-1 of Olanzapine (Yield 22.0 g).
CONVERSION OF FORM-2 TO FORM-1 EXAMPLE 4 The stirred suspension of pure form-2 of 2-methyl-4-(4-methyl-l-piperazinyl)-
10H-thieno-[2,3-b][l,5]benzodiazepine (20.0 g) in dichloromethane (90.0 ml) was heated to reflux to obtain a clear solution. The clear solution was filtered and the filtrate was then cooled to 3 to 5°C and stirred at same temperature for one hour. The crystalline solid separated was filtered and washed with dichloromethane (4.0 ml). Subsequent drying at 60 to 70°C to a constant weight yielded Olanzapine Form-1. (Yield: 12.7 g).
PREPARATION OF FORM-1 FROM MONOHYDRATE-I OF OLANZAPINE
EXAMPLE 5 Monohydrate-I of 2-methyl-4-(4-methyl-l-piperazinyl)-10H-thieno-[2,3- b][l,5] benzo- diazepine (25.0 g) prepared as per Example- 1 was suspended in dichloromethane (325.0 ml). The suspension was heated to reflux to obtain a clear solution. The resultant solution was then treated with carbon (2.5 g) followed by filtration. Upon completion of this step the filtrate was distilled to a minimum volume and then cooled to 2 to 4°C and stirred at the same temperature for 90 minutes. The product separated was filtered arid washed with chilled dichloromethane (10 ml). The product obtained on drying in oven at 60 to 70°C to a constant weight gave Form-1 of Olanzapine (Yield 16.5 g)
PREPARATION OF FORM-1 FROM DIHYDRATE-I OF OLANZAPINE
EXAMPLE 6 Dihydrate-I of 2-methyl-4-(4-methyl-l-piperazinyl)-10H-thieno-[2,3-b][l,5] benzodiazepine (40.0 g) prepared as per Example-2 was suspended in dichloromethane (520.0 ml). The suspension was heated to reflux to obtain a clear solution. The resultant solution was then treated with carbon (4.0 g) followed by filtration. Upon completion of this step the filtrate was distilled to a minimum volume and the left over reaction mass was cooled to 0 to 2°C and stirred at the same temperature for one hour. The separated solid was filtered and washed with dichloromethane (10.0ml). The product obtained on drying in oven at 65 to 70°C to a constant weight renders Form-1 of Olanzapine (Yield 26.0 g).
The aforementioned crystalline forms in examples 1 to 6 have been examined for their structural and analytical data viz., Powder X-Ray Diffraction, Differential Scanning Calorimetry, and Infrared Absorption Spectroscopy. The results obtained are discussed and the respective drawings attached (Fig. 2 -19).
………………………………..
http://www.google.com/patents/EP2292624A1?cl=en
-
Olanzapine is a pharmaceutical active substance from the group of antipsychotics, applicable for the treatment of different mental diseases and conditions, such as, for example, disorders of the central nervous system, schizophrenia, hallucination, acute mania, depression, and the like.
-
Chemically, it belongs to the group of the benzodiazepines and is 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine (formula 1).

-
Olanzapine and analogues thereof are encompassed for the first time within a general formula in the patent GB 1,533,235 and specifically described in EP 454 436 B1 . The patents disclose two different one-step processes for olanzapine preparation. The first described process is a reaction of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride with N-methylpiperazine in an organic solvent, such as anisole, toluene, dimethyl formamide or dimethyl sulfoxide, preferably at a temperature from 100 to 150°C to yield olanzapine (Scheme 1).

-
The second process disclosed in EP 454 436 B1 is the reaction of N-methylpiperazine with methyl-2-(2-aminoanilino)-5-methylthiophene-3-carboxylate in the presence of titanium tetrachloride (Scheme 2).

-
The same patent also mentions the formation of acid addition salts of olanzapine and their potential use as intermediates in olanzapine purification process and for a pharmaceutical use.
-
As disclosed in EP 454 436 B1 and US equivalent US 5,229,382 , olanzapine obtained according to the first synthesis (Scheme 1) is purified by recrystallization from acetonitrile, whereas olanzapine prepared according to the second route (Scheme 2) is further purified by column chromatography on Florisil® and recrystallization from acetonitrile. This purification procedure lacks on industrial applicability.
-
Some other synthetic approaches for the preparation of olanzapine describe two steps for creating 4-methylpiperazinyl side chain (Scheme 3). Firstly, 4-amino-2-methyl-10H-thieno [2,3-b][1,5] benzodiazepine hydrochloride reacts with piperazine to yield 2-methyl-4-(1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine (i.e. N-desmethylolanzapine; Bioorganic & Medicinal Che-mistry Letters, Vol. 7, No. 1, pp. 25-30, 1997), then the methyl group is introduced either by reductive N-methylation (using formaldehyde and metal boron hydride -WO 04/000847) or by nucleophylic substitution reaction with methyl iodide (WO 05/090359). The two-step approach reduces the dark colour appearance but it does not solve the problem of purification from similar by-products.

-
It is well known to a skilled person that most chemical reactions are not completely finished, may be reversible or are driven simultaneously with some other parallel reactions. Starting materials or side reaction products are usually found as impurities in the isolated main product which should therefore be further purified. The simplest way of purification includes various recrystallization and precipitation procedures which are usually less effective if the impurities have physico-chemical properties very similar to the main product.
-
In the case where olanzapine is prepared according to the one step processes disclosed in EP 454 436 B1 , the starting material, 4-amino-2-methyl-10H-thieno[2,3-b][1,5]-benzodiazepine, is found as an impurity in the final product olanzapine.
-
In the case of preparation of olanzapine via the two-step process, as disclosed also in the patent application WO 04/000847 , the presence of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]-benzodiazepine hydrochloride is not critical but various other similar compounds could be found as impurities, such as 4-(4-formylpiperazinyl)-2-methyl-10H-thieno[2,3-b][1,5]-benzo diazepine and N-desmethylolanzapine. In the case of preparation of olanzapine by a two-step process with methyl iodide, an overmethylated N,N-dimethylpiperazinium analogue can be formed.
-
A further undesired impurity which accompanies olanzapine is obtained when olanzapine compound is dissolved in methylene chloride. It is so called olanzapine-CM, being (E)-1-(chloromethyl)-1-methyl-4-(2-methyl-10H-benzo[b]thieno[2,3-e][1,4]diazepin-4-yl)piperazin-1-ium chloride). Olanzapine-CM is formed by alkylation of olanzapine with methylene chloride in methylene chloride solution, for example during evaporation of methylene chloride before the crystallization (Scheme 4).

-
According to the Regulatory Toxicology and Pharmacology 44, pp. 198-211, 2006, classification of potential genotoxic impurities, olanzapine-CM is a potential genotoxic substance due to its R-CH2-Cl structural element, which is known to be involved in reactions with DNA.
-
For all impurities that have a thienobenzodiazepine ring system as a part of the molecule skeleton and because it represents a great part of the molecule, said ring system is crucial for the similarity of physico-chemical properties of said impurities compared to olanzapine.
-
Different salts and crystal forms of an active pharmaceutical ingredient are an important tool for modulating pharmacokinetic properties but can be also a tool for purification.
-
WO 04/089313 discloses olanzapine acid salts, solvates and co-crystals and their use as active pharmaceutical ingredients in formulations. The preparation of fumaric, maleic and malonic acid addition salts of olanzapine is disclosed in WO 04/089313 . Olanzapine acid addition salts disclosed in this application exhibit specific aqueous solubility from 50 µg/ml to 100 mg/ml.
-
WO 05/090359 discloses a method for the purification of olanzapine which has on one hand been prepared by the one-step-process according to Scheme 1, and on the other hand by the process according to Scheme 3, by preparing an addition salt of at least one carboxylic salt, purification of said salt and transfer into purified olanzapine.
-
Neutral olanzapine can be isolated in various crystal forms, hydrates and other solvates. However crystal forms I and II are the most often mentioned as an active pharmaceutical ingredient. Both forms were first disclosed in EP 733 635 alleging that form II is thermodynamically more stable than form I which had been prepared already by the basic patent procedures ( EP 454 436 B1 ).
-
Crystal Growth & Design, Vol. 3, No. 6, pp. 897-907, 2003 discloses anhydrates and hydrates of olanzapine.
-
Olanzapine form I is thermodynamically less stable but it can possess specific kinetic properties which can be applied in designing a final dosage form. Many procedures are known how to prepare it but a person skilled in the art can soon find that the use of methylene chloride is unavoidable to develop a repeatable process. Because this solvent is used in the final step of preparation of olanzapine form I, previous purification methods cannot prevent the presence of the impurity olanzapine-CM in the final product.
-
WO 03/101997 A1 discloses a process for the preparation of a pure olanzapine form I by an addition of methyl-piperazine to the hydrochloride salt of the corresponding benzodiazepine derivative. The purification of olanzapine is conducted by recrystallization. Olanzapine-CM is not disclosed as a disturbing side product which can be removed by recrystallization.
-
WO 02/18390 discloses a process for the preparation of hydrates of olanzapine and the conversion thereof into crystalline forms of olanzapine by recrystallization from methylene dichloride. It is not disclosed that essentially pure olanzapine can be obtained by the removal of olanzapine-CM.
-
WO 04/056833 A1 discloses a process for the preparation of essentially pure olanzapine by removing olanzapine-CM, which is obtained from a solution of olanzapine in methylene chloride, by treating this solution with SiO2, followed by the removal of SiO2. According to the examples, olanzapine is obtained having a purity of 99.92 %, whereas olanzapine-CM is present in an amount of 0.05 %, corresponding to 500 ppm. But SiO2 is acidic, so it adsorbs well also the basic olanzapine what leads to considerable losses of the mother compound.
-
Olanzapin-CM has to be removed in order to obtain essentially pure olanzapine with a high purity which can be further used in pharmaceutical applications. It has been found that olanzapine cannot be efficiently separated from its highly related impurities, in particular from olanzapine-CM, using repeated crystallizations of crude olanzapine. It would therefore be desirable to develop a purification process, in order to provide pharmaceutically acceptable pure and discoloured olanzapine, in particular to provide pharmaceutically acceptable pure olanzapine, being essentially free of the olanzapine-CM impurity. A further object of the present invention is to provide a process for the purification of olanzapine which can also and preferably be conducted in a large scale synthesis.





Example 1 – Synthesis of olanzapine oxalic salt
-
[0093]A solution of 12.0 g of N-desmethylolanzapine in 90 mL of THF and 36 mL of dimethylacetamide (DMAc) is cooled to approx. -20 °C. At -20 °C, 8.19 g of diisopropylamine is added to the solution and afterwards, 8.14 g of methyl iodide is added over 30 min. After stirring the reaction mixture for 65 minutes at -20 °C, 6.4 mL of concentrated hydrochloric acid in 50 mL of water and a solution of 6.36 g of thiourea in 50 mL of water is added and the reaction mixture is stirred for 15 minutes at 20 °C. Then THF is evaporated off and 120 mL of methylene chloride is added and the pH is adjusted to 8.6 with a 40 % water solution of NaOH. After the separation of the phases, the water phase is washed twice with 60 and 30 mL of methylene chloride. The organic phases are combined and 380 mg of acetic anhydride is added and the mixture is stirred for 5 minutes. Then 6.20 g of oxalic acid in 24 mL of methanol is added within 15 minutes. The resulting suspension is stirred for about 1 hour at approx. 20 °C and afterwards 1 hour at approx. 0 °C. The product is isolated by filtration, washed with 100 mL of methylene chloride and dried for 15 hours at 25 °C in vacuo. Yield: 15 g (93 %).
Example 2 – Formation of olanzapine form I
-
[0094]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. After the solution is concentrated in vacuo to the volume of 15 mL, the solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of -20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 380 ppm IR Form I XRD Form I
Example 3 – Formation of olanzapine form I (scale up)
-
[0095]24 kg of olanzapine oxalate is dissolved in 240 L of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 2.4 kg of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 L of water. The filtrate and wash water are combined and after the addition of 300 L of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 15 minutes, the layers are separated and the water phase is extracted twice with 50 L of methylene chloride. The organic layers are combined and washed twice with 100 L of water. After the solution is concentrated in vacuo to the volume of 50 L, the solution is immediately cooled to -15 °C. The resulting suspension is stirred for 30 minutes, then olanzapine is isolated by filtration. The wet cake is washed with 10 L of methylene chloride of the temperature of -20 °C. The product is dried for 10 hours at 100 °C in vacuo.
HPLC-Purity: 99.7 % olanzapine-CM 1000 ppm IR Form I XRD Form I
Example 4 – Formation of olanzapine form I
- (
Al2O3 fluidized bed adsorption
-
[0096]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. After 0.2 g of Al2O3 is added and the methylene chloride suspension is stirred for 5 minutes, Al2O3 is filtered off and the methylene chloride solution is concentrated to the volume of 15 mL. The solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of -20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 214 ppm IR Form I XRD Form I
- )
Example 5 – Formation of olanzapine form I (Al2O3 fluidized bed adsorption
-
[0097]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. Afterwards, the methylene chloride solution is concentrated to 30 mL and 0.2 g of Al2O3is added. After stirring for 5 minutes, Al2O3 is filtered off and the methylene chloride solution is concentrated to the volume of 15 mL. The solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of -20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % Olanzapine-CM 321 ppm IR Form I XRD Form I
- )
Example 6 – Formation of olanzapine form I (Al2O3 fluidized bed adsorption
-
[0098]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. Then the methylene chloride solution is concentrated to 30 mL and 1 g of Al2O3 is added. After stirring for 5 minutes, Al2O3 is filtered off and the methylene chloride solution is concentrated to the volume of 15 mL. The solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of a temperature of -20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 138 ppm IR Form I XRD Form I
- )
Example 7 – Formation of olanzapine form I (Al2O3 short column adsorption)
-
[0099]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. Then the methylene chloride solution is concentrated to 30 mL and filtered through 20 g of Al2O3 (h = 2 cm). After the methylene chloride solution is concentrated to the volume of 15 mL, the solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of-20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 162 ppm IR Form I XRD Form I
Example 8 – Formation of olanzapine form I (Al2O3 short column adsorption)
-
[0100]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. Afterwards, the methylene chloride solution is concentrated to 30 mL and filtered through 20 g of Al2O3 (h = 5 cm). After the methylene chloride solution is concentrated to the volume of 15 mL, the solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of-20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 73 ppm IR Form I XRD Form I Table 1. Comparison and overview of beneficial effect of Al2O3 treatment
Ex. Scale Treatment Mass (Al2O3) [g] Al2O3 pad height [cm] CM assay [ppm] 2 5 g none / / 380 3 24 kg none / / 1000 4 5 g fluidized bed adsorption 0.2 / 214 5 5 g fluidized bed adsorption (concentrated to 30 mL) 0.2 / 321 6 5 g fluidized bed adsorption (concentrated to 30 mL) 1.0 / 138 7 5 g short column adsorption (concentrated to 30 mL) 20 2 162 8 5 g short column adsorption (concentrated to 30 mL) 20 5 73 Table 2. As comparative data, olanzapine obtained by processes according to the prior art comprises the following amounts of olanzapine-CM:
No. prior art amount of olanzapine-CM 1 WO 2005/090359 , lab. scale 300 – 400 ppm 2 WO 2005/090359 , industrial scale 800 – 2000 ppm 3 WO 2004/056833 <0.15%
………………………………
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0454436B1 | Apr 24, 1991 | Sep 13, 1995 | Lilly Industries Limited | Pharmaceutical compounds |
| EP0733635A1 | Mar 22, 1996 | Sep 25, 1996 | Eli Lilly And Company | Crystal forms of a thieno(2,3-B)(1,5) benzodiazepine derivative and process for their preparation |
| GB1533235A | Title not available | |||
| US5229382 | May 22, 1992 | Jul 20, 1993 | Lilly Industries Limited | 2-methyl-thieno-benzodiazepine |
| WO2002018390A1 | Mar 7, 2001 | Mar 7, 2002 | Ramesh Chakka | Process for preparation of hydrates of olanzapine and their conversion into crystalline forms of olanzapine |
| WO2003055438A2 * | Dec 23, 2002 | Jul 10, 2003 | Vijay Chhangamal Chhabada | Crystalline form i of 2-methyl-4-(4-methyl-1-piperazinyl) 10h thieno [2,3-b][1,5]benzodiazepine |
| WO2003101997A1 | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004000847A1 | Jun 10, 2003 | Dec 31, 2003 | Adamed Sp Zoo | A process for the preparation of olanzapine and an intermediate therefor |
| WO2004056833A1 | Dec 15, 2003 | Jul 8, 2004 | Adamed Sp Zoo | A process for the preparation of a pharmaceutically pure polymorphic form i of olanzapine |
| WO2004089313A2 | Mar 31, 2004 | Oct 21, 2004 | Magali Bourghol Hickey | Novel olanzapine forms and related methods of treatment |
| WO2005090359A2 | Mar 17, 2005 | Sep 29, 2005 | Lek Pharmaceuticals | Synthesis of 2-methyl-4-(4-methyl-1-piperazinyl)-10h-thieno[2, 3-b][1,5]benzodiazepine and salts thereof |
| WO2008139228A2 * | May 14, 2008 | Nov 20, 2008 | Generics Uk Ltd | Process for the purification of olanzapine |
| Reference | ||
|---|---|---|
| 1 | BIOORGANIC & MEDICINAL CHE-MISTRY LETTERS vol. 7, no. 1, 1997, pages 25 – 30 | |
| 2 | BIOORGANIC & MEDICINAL CHEMISTRY LETTERS vol. 7, no. 1, 1997, pages 25 – 30 | |
| 3 | CRYSTAL GROWTH & DESIGN vol. 3, no. 6, 2003, pages 897 – 907 | |
| 4 | REGULATORY TOXICOLOGY AND PHARMACOLOGY vol. 44, 2006, pages 198 – 211 | |
| WO1998011893A1 * | Sep 18, 1997 | Mar 26, 1998 | Lilly Co Eli | Olanzapine dihydrate d |
| EP0733635A1 * | Mar 22, 1996 | Sep 25, 1996 | Eli Lilly And Company | Crystal forms of a thieno(2,3-B)(1,5) benzodiazepine derivative and process for their preparation |
| EP0831098A2 * | Sep 22, 1997 | Mar 25, 1998 | Eli Lilly And Company | Intermediates and process for preparing olanzapine |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003037903A1 * | Oct 29, 2002 | May 8, 2003 | Cord Janet I | Olanzapine dihydrate-ii a process for its preparation and use thereof |
| WO2003090730A1 * | Apr 25, 2002 | Nov 6, 2003 | Generics Uk Ltd | Novel crystalline forms of celecoxib and other compounds |
| WO2003091260A1 * | Apr 22, 2003 | Nov 6, 2003 | Ramesh Chakka | Novel crystalline polymorph form-vi of olanzapine and a process for preparation thereof |
| WO2003097650A1 * | May 16, 2003 | Nov 27, 2003 | Urszula Fraczek | Methods for preparation of olanzapine polymorphic form i |
| WO2003101997A1 | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004006933A2 | Jul 14, 2003 | Jan 22, 2004 | Krka D D Novo Mesto | Crystal forms of olanzapine and processes for their preparation |
| WO2004056833A1 * | Dec 15, 2003 | Jul 8, 2004 | Adamed Sp Zoo | A process for the preparation of a pharmaceutically pure polymorphic form i of olanzapine |
| WO2004058773A1 * | Dec 24, 2003 | Jul 15, 2004 | Judith Aronhime | Novel crystal forms of olanzapine, methods for their preparation and method for the preparation of known olanzapine crystal forms |
| WO2005070937A1 * | Jan 26, 2005 | Aug 4, 2005 | Rolf Keltjens | A process for making olanzapine in a polymorph form i |
| WO2005080401A1 * | Jul 16, 2004 | Sep 1, 2005 | Shriprakash Dhar Dwivedi | Process for the preparation of olanzapine form 1 useful as antipsychotic drug |
| WO2005107375A2 * | Apr 4, 2005 | Nov 17, 2005 | Jyothi Basu Abbineni | Process for the preparation of olanzapine form-i |
| WO2006006185A1 * | Jul 13, 2005 | Jan 19, 2006 | Saravanan Govindaraju | Improved process for making form i of olanzapine. |
| WO2006025065A1 * | Aug 31, 2004 | Mar 9, 2006 | Lee Pharma Private Ltd | A process for the preparation of anhydrous olanzopine hydrochloride of form-1 |
| WO2008091169A2 | Jan 22, 2008 | Jul 31, 2008 | Tomasz Kozluk Nobilus Ent | Process for preparation of substantially pure polymorphic form i of olanzapine |
| WO2008139228A2 * | May 14, 2008 | Nov 20, 2008 | Generics Uk Ltd | Process for the purification of olanzapine |
| WO2011009831A1 | Jul 19, 2010 | Jan 27, 2011 | Lek Pharmaceuticals D.D. | Process for the purification of olanzapine |
| EP1863775A2 * | Mar 20, 2006 | Dec 12, 2007 | Dr. Reddy’s Laboratories Ltd. | Process for preparing crystalline form i of olanzapine |
| EP1997822A1 * | Nov 10, 2006 | Dec 3, 2008 | EGIS Gyógyszergyár Nyilvánosan Müködõ Részvénytársaság | Process for the preparation of 2-methyl-4-(4-methylpiperazin-1-yl)-10h-thieno-[2,3-b] [1,5] benzodiazepine dihydrochloride trihydrate, 2-methyl-4-(4-methyl-piperazin-1-yl) -10h-thieno[2,3-b] [1,5] benzodiazepine dihydrochloride trihydrate, pharmaceutical compositions comprising it and its use |
| EP2264016A2 | Jul 14, 2004 | Dec 22, 2010 | Jubilant Organosys Limited | A process for producing pure form form of 2-Methyl-4-(4-Methyl-1-Piperazinyl)-10h-thieno[2,3-B][1,5] benzodiazepine |
| EP2292624A1 | Jul 20, 2009 | Mar 9, 2011 | LEK Pharmaceuticals d.d. | Process for the purification of olanzapine |
| US7297789 | May 30, 2003 | Nov 20, 2007 | Sandoz, Inc. | From 4-amino-2-methyl-10H-thieno(2,3-b)(1,5) benzodiazpine HCl and 1-methylpiperazine in an aprotic high boiling solvent; purifying in an acid medium; basifying to pH of 7.5-9; and extracting with a low boiling organic solvent |
| US7323459 | Dec 24, 2003 | Jan 29, 2008 | Teva Pharmaceutical Industries Ltd. | Dissolving olanzapine in a solution of acetic acid and water; filtering the solution; stirring the solution; precipitating the crystalline form by adding a base; and isolating the crystals |
| US7329747 | Jan 27, 2005 | Feb 12, 2008 | Synthon Ip Inc. | Synthesis of olanzapine and intermediates thereof |
| US7425627 | Dec 22, 2004 | Sep 16, 2008 | Teva Pharmaceutical Industries Ltd. | Methods of synthesizing olanzapine |
| US7459449 | Jan 27, 2005 | Dec 2, 2008 | Rolf Keltjens | Olanzapine malonate, olanzapine glycolate, and olanzapine benzoate; antipsychotic agents; water soluble; can form tablets by direct compression; may be mixed with dibasic calcium phosphate and/or hydroxypropyl cellulose |
| US7538213 | May 16, 2003 | May 26, 2009 | Institut Farmaceutyczny | Condensation of molar excess of N-methylpiperazine with 4-amine-2-methyl-10H-thieno [2,3-b][1,5]benzodiazepine hydrochloride in dimethylsulfoxide; recovery and purification with methylene chloride |
| US7745429 | Jul 14, 2003 | Jun 29, 2010 | Krka, D.D. Novo Mesto | Crystal forms of olanzapine and processes for their preparation |
| US7759484 | Jul 7, 2005 | Jul 20, 2010 | Inke, S.A. | Mixed solvate of olanzapine, method for preparing it and method for preparing form I of olanzapine therefrom |
| US7829700 | Sep 5, 2005 | Nov 9, 2010 | Shasun Chemicals And Drugs Limited | Process for preparation of a pharmaceutically pure polymorphic form I of Olanzapine |
| US7834176 | Jan 26, 2006 | Nov 16, 2010 | Sandoz Ag | Polymorph E of Olanzapine and preparation of anhydrous non-solvated crystalline polymorphic Form I of 2-methyl-4(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5] benzodiazepine (Olanzapine Form I) from the polymorphic Olanzapine Form E |
| WO2002018390A1 * | Mar 7, 2001 | Mar 7, 2002 | Ramesh Chakka | Process for preparation of hydrates of olanzapine and their conversion into crystalline forms of olanzapine |
| WO2003097650A1 * | May 16, 2003 | Nov 27, 2003 | Urszula Fraczek | Methods for preparation of olanzapine polymorphic form i |
| WO2003101997A1 * | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004006933A2 * | Jul 14, 2003 | Jan 22, 2004 | Krka D D Novo Mesto | Crystal forms of olanzapine and processes for their preparation |
| EP0733367A1 * | Mar 22, 1996 | Sep 25, 1996 | Eli Lilly And Company | Oral olanzapine formulation |
| US5229382 * | May 22, 1992 | Jul 20, 1993 | Lilly Industries Limited | 2-methyl-thieno-benzodiazepine |
| US5703232 * | Jan 16, 1996 | Dec 30, 1997 | Eli Lilly And Company | Process and solvate of 2-methyl-thieno-benzodiazepine |
| US5736541 * | Jul 25, 1996 | Apr 7, 1998 | Eli Lilly And Company | Slurrying impure material in anhydrous ethyl acetate, then crystallization yields pure stable drug for treatment of anxiety, psychological disorders and gastrointestinal disorders |
| US6348458 * | Mar 31, 2000 | Feb 19, 2002 | U & I Pharmaceuticals Ltd. | Polymorphic forms of olanzapine |
| WO2006025065A1 * | Aug 31, 2004 | Mar 9, 2006 | Lee Pharma Private Ltd | A process for the preparation of anhydrous olanzopine hydrochloride of form-1 |
| WO2007052167A2 | Oct 27, 2006 | May 10, 2007 | Actavis Group Ptc Ehf | Stable composition for a pharmaceutical formulation containing olanzapine |
| WO2007144901A1 * | Jun 12, 2007 | Dec 21, 2007 | Jubilant Organosys Ltd | Process for stabilization of olanzapine polymorphic form i |
| WO2008091169A2 | Jan 22, 2008 | Jul 31, 2008 | Tomasz Kozluk Nobilus Ent | Process for preparation of substantially pure polymorphic form i of olanzapine |
| WO2011009831A1 | Jul 19, 2010 | Jan 27, 2011 | Lek Pharmaceuticals D.D. | Process for the purification of olanzapine |
| EP2292624A1 | Jul 20, 2009 | Mar 9, 2011 | LEK Pharmaceuticals d.d. | Process for the purification of olanzapine |
| US7323459 | Dec 24, 2003 | Jan 29, 2008 | Teva Pharmaceutical Industries Ltd. | Dissolving olanzapine in a solution of acetic acid and water; filtering the solution; stirring the solution; precipitating the crystalline form by adding a base; and isolating the crystals |
| US7829700 | Sep 5, 2005 | Nov 9, 2010 | Shasun Chemicals And Drugs Limited | Process for preparation of a pharmaceutically pure polymorphic form I of Olanzapine |
COCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1996038151A1 | May 30, 1995 | Dec 5, 1996 | Lilly Co Eli | Method for treating cognitive dysfunction |
| WO1999016312A1 * | Sep 25, 1998 | Apr 8, 1999 | Lilly Co Eli | Method for treating sexual dysfunction |
| WO2001047933A1 | Dec 22, 2000 | Jul 5, 2001 | Cipla Ltd | New polymorphic forms of olanzapine |
| WO2002018390A1 | Mar 7, 2001 | Mar 7, 2002 | Ramesh Chakka | Process for preparation of hydrates of olanzapine and their conversion into crystalline forms of olanzapine |
| WO2003097650A1 | May 16, 2003 | Nov 27, 2003 | Urszula Fraczek | Methods for preparation of olanzapine polymorphic form i |
| WO2003101997A1 * | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004056833A1 | Dec 15, 2003 | Jul 8, 2004 | Adamed Sp Zoo | A process for the preparation of a pharmaceutically pure polymorphic form i of olanzapine |
| WO2005107375A2 * | Apr 4, 2005 | Nov 17, 2005 | Jyothi Basu Abbineni | Process for the preparation of olanzapine form-i |
| WO2006006180A1 * | Jul 14, 2004 | Jan 19, 2006 | Akshat Bhatnagar | A PROCESS FOR PRODUCING PURE FORM OF 2-METHYL-4-(4-METHYL-1-PIPERAZINYL)-10H-THIENO[2,3-b][1,5]BENZODIAZEPINE |
| WO2006010620A2 | Jul 28, 2005 | Feb 2, 2006 | Krka Tovarna Zdravil D D Novo | Olanzapine salts and their conversion to olanzapine free base |
| EP0454436A1 | Apr 24, 1991 | Oct 30, 1991 | Lilly Industries Limited | Pharmaceutical compounds |
| EP0454436B1 | Apr 24, 1991 | Sep 13, 1995 | Lilly Industries Limited | Pharmaceutical compounds |
| EP0733635B1 | Mar 22, 1996 | Aug 16, 2001 | Eli Lilly And Company | Crystal forms of a thieno(2,3-B)(1,5) benzodiazepine derivative and process for their preparation |
| US5637584 | Mar 24, 1995 | Jun 10, 1997 | Eli Lilly And Company | Solvate of olanzapine |
Data Integrity – Again Import Alert issued for Indian company IPCA
DRUG REGULATORY AFFAIRS INTERNATIONAL

Data Integrity – Again Import Alert issued for Indian company IPCA…http://www.gmp-compliance.org/enews_04789_Data-Integrity—Again-Import-Alert-issued-for-Indian-company-IPCA_9193,S-QSB_n.html
![]()
Data Integrity has become one of the most important GMP compliance issues in past two years. This has enormous consequences for the concerned companies but also for companies and authorities in EU and US. It was the US FDA that has first experienced huge data integrity problems in companies worldwide. Many sites in India have been found to violate GMP requirements by Data Integrity issues. Tests have been repeated and original data have been deleted. This is called “testing into compliance”. At the Webpage of the US FDA IPCA products are listed which are impacted by the Import Alert. Two facilities from IPCA have been found to be out of GMP compliance: One in Pithampur (Madhya Pradesh) and one in Piparia (Silvassa) (see also report by FiercePharma).
Products manufactured at those facilities might cause high risks to patients. The quality of the products…
View original post 207 more words
OLANZEPINE VISITED PART 2/3
PART 1…..https://newdrugapprovals.org/2015/04/08/olanzepine/
PART 2….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-22/
PART 3…….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-33/
WATCHOUT………..


…………………….
PATENT
http://www.google.com/patents/US7459449
Olanzapine is a pharmaceutically active compound that can be represented by formula (1).

It was disclosed in EP 454436 and corresponding U.S. Pat. No. 5,229,382 as a useful antipsychotic agent. Olanzapine acts as a serotonin (5-HT2) and dopamine (D1/D2) receptor antagonist with anticholinergic activity. In commercially available final forms, the active substance is marketed as a free base, which is a white to yellow crystalline solid that is insoluble in water; i.e., solubility at pH 6.8=0.02 mg/ml.
The olanzapine base is known to exist in various crystalline modifications and in various hydrated forms that are generally stable at ambient conditions; see for example EP 733635 and corresponding U.S. Pat. No. 5,736,541; WO 98-11893; and EP 831098. Having so many different forms is considered to be a disadvantage as repeated production of olanzapine substance may give rise to unpredictable amounts of the respective modifications in the product, which in turn can influence the properties of the product such as in tabletting and/or releasing of the active from the tablets after ingestion.
WO 99-16313 discloses olanzapine pamoate as a pharmaceutical agent. It is a compound that is also insoluble in water and is useful particularly in intramuscular depot forms. However, like the free base, the pamoate salt exists in several forms including hydrates, solvates, and in different counter ion ratios.
WO 03-007912 discloses an amorphous lyophilized olanzapine in a reconstitutable parenteral formulation. The olanzapine is “intimately mixed” with a stabilizer and a solubilizer. The stabilizer is preferably lactose and the solubilizer includes organic acids and most preferably tartaric acid. The composition is formed by lyophilizing, i.e. a type of freeze drying, a solution of olanzapine, the stabilizer and the solubilizer to form the intimate mixture. The resulting lyophilized amorphous product can be reconstituted with parenteral diluents to make an injectable composition. Whether the tartaric acid salt of olanzapine is present in the lyophilized product is unclear.
For instance, the “des-methyl olanzapine” (2) may be methylated by formic acid/formaldehyde (Chinese Journal of Pharmaceuticals 2001, 32, 391-393) to form an olanzapine reaction mixture.

Similarly, the “des-piperazine olanzapine” (3) can be treated with N-methylpiperazine in DMSO under conditions of olanzapine formation to produce a reaction mixture containing olanzapine.

In a third process, the “formyl-olanzapine” (4) is reduced by a reducing agent, for instance by a borohydride agent (WO 2004/000847) or by hydrogen under the presence of a hydrogenation catalyst.

The starting compounds (2), (3), and (4) are known compounds and may be obtained by procedures known in the art.
EXAMPLE 16A
Olanzapine Benzoate by Methylation of Desmethyl Olanzapine

In a 100 ml flask, equipped with a magnetic stirrer, 0.5 g desmethyl olanzapine was dissolved in 5 ml DMSO. Then, 0.142 g formic acid (37% in water) and 0.082 g formic acid (98%) were added and the resulting mixture was heated at 80° C. for 2 hours. After cooling to room temperature, 20 ml ethyl acetate and 20 ml water were added. The organic layer was washed with 2*20 ml water and 20 ml saturated aqueous NaCl, dried (Na2SO4) and concentrated at reduced pressure to a volume of about 10 ml. Then, 0.200 g benzoic acid dissolved in 2 ml ethyl acetate was added dropwise to the crude olanzapine solution. An off-white/yellow solid formed which was isolated by filtration and dried over weekend at 40° C. in vacuo. Isolated yield: 0.58 gram (80%). 1H-NMR: expected compound; trace of ethyl acetate present.
EXAMPLE 16B
Synthesis of Olanzapine and Isolation of Olanzapine as the Benzoate Salt

In a 250 ml flask, equipped with a magnetic stirrer, 4.3 g of N-desmethylpiperazine-olanzapine was refluxed in a mixture of 15 ml N-methylpiperazine, 20 ml DMSO, and 20 ml toluene under a nitrogen atmosphere for 20 hours. The mixture was cooled and 50 ml water was added. The aqueous layer was extracted three times with 150 ml ethyl acetate and the combined organic layers were washed 3 times with 100 ml water and once with 100 ml aqueous saturated sodium chloride. After drying over Na2SO4, the organic layer was concentrated to about 100 ml and 1.6 g benzoic acid was added. After a few minutes, a yellow solid was formed. Stirring was continued at 4° C. for 1 hour. The solid material was isolated by filtration, washed with 5 ml ethyl acetate and 10 ml diethyl ether, and dried overnight at 40° C. in vacuum. Isolated yield: 4.61 g (80%; based on benzoic acid).
EXAMPLE 16C
Synthesis of Olanzapine and Isolation of Olanzapine as the Benzoate Salt

In a 2000 ml flask, 50 g of desmethyl olanzapine was dissolved in 450 ml of DMSO. Then, 13.04 g of formaldehyde (37% in water) and 7.59 g of formic acid (98%) were added and the resulting mixture was heated at 80° C. for 2 hours. The crude reaction mixture was poured into a mixture of 1000 ml of ethyl acetate and 1000 ml of ice-cooled water. The aqueous layer was separated and extracted with 2×500 ml of ethyl acetate. The combined organic layers were washed with 3×500 ml of water and 500 ml of saturated aqueous NaCl, dried (Na2SO4) and concentrated at reduced pressure to a volume of about 1000 ml. To the warm solution, 20.4 g of benzoic acid was added in one portion under stirring. An off-white/yellow solid was formed. Stirring was continued overnight at room temperature and subsequently for 2 hours at 4 C. The yellow solid was isolated by filtration, washed with 30 ml of cold ethyl acetate and 100 ml of diethyl ether and dried overnight at 60° C. in vacuo. Isolated yield: 60.25 g. Assay (HPLC): 99.1%.
EXAMPLE 16D
Synthesis of Olanzapine and Isolation of Olanzapine as the Benzoate Salt

In a 3000 ml flask, 86 g of desmethylpiperazine-olanzapine hydrochloride was refluxed in a mixture of 300 ml of N-methylpiperazine, 400 ml of DMSO, and 400 ml of toluene under a nitrogen atmosphere for 5 hours. The mixture was cooled to 50° C. and poured into a mixture of 2000 ml of ethyl acetate and 2000 ml of ice-cooled water. The aqueous layer was extracted with 2×500 ml of ethyl acetate and the combined organic layers were washed with 3×500 ml of water and with 500 ml of aqueous saturated sodium chloride. After drying over Na2SO4, the organic layer was concentrated to about 1500 ml and 39.6 g of benzoic acid was added in one portion. After a few minutes, a yellowish solid was formed. Stirring was continued overnight at room temperature. The solid material was isolated by filtration, washed with 50 ml of ethyl acetate and 200 ml of diethyl ether, and dried overnight at 60° C. in vacuum. Yield: 86.35 gram.
EXAMPLE 16E
Olanzapine Benzoate from Formyl Olanzapine

In a 250 ml flask, 3.0 g of N-formyl olanzapine precursor (compound (4)) was suspended in 45 ml of dry toluene and cooled to 0C. Under nitrogen atmosphere, 5.4 ml of Red-Al™ solution (70 wt % solution of sodium dihydrido-bis(2-methoxyethoxy) aluminate in toluene) was added dropwise under stirring. The resulting mixture was allowed to warm up to room temperature. Then 5.0 ml of Red-Al solution was added dropwise at this temperature. After stirring for 5 hours at room temperature, the reaction mixture was poured into 100 ml of water and immediately 100 ml of ethyl acetate was added. The mixture was filtered over a P3-filter to remove insoluble material. The biphasic filtrate was allowed to stand for separating the layers and the aqueous layer was removed and washed with 2×50 ml of ethyl acetate. The combined organic layers were washed with 2×50 ml of water, dried over anhydrous sodium sulfate and concentrated at reduced pressure to a volume of about 50-60 ml. Then, 1.12 g of benzoic acid was added in one portion and the resulting mixture was stirred at 4° C. for 4 hours. The formed solid was isolated by filtration, washed with 5 ml of cold ethyl acetate and 10 ml of cold diethyl ether, and dried overnight at 40 C under vacuum. Yield: 2.75 gram, purity (HPLC): 94.8%.
………………
PATENT
http://www.google.com/patents/WO2003101997A1?cl=en
EXAMPLE 1 ULTRA-PURE OLANZAPINE FORM I

A three necked flask, fitted with a nitrogen gas inlet and a water condenser with calcium chloride guard tube, is charged with 4-amino-2-methyl-10H-theino[2,3-b][1 ,5] benzodiazepine HCI (5.0 g, 0.0188 mol), 1-methylpiperazine (13.0 mL, 0.11 mol, 99.0%, Aldrich Chemicals, USA) and anhydrous dimethyl sulfoxide (30.0 mL, Aldrich Chemicals, USA, water< 0.1%). The reaction mixture is stirred at 112-115°C (oil bath temperature 115°C) for 16 hours under continuous flow of nitrogen to drive away the ammonia gas generated during the reaction. The reaction is monitored by HPLC and it is found that within 16 hours 97% product is formed. The reaction mixture is cooled to room temperature (24- 25°C) and added dropwise to a mixture of dichloromethane.water-methanol (190:190:15, 395 mL). After addition, the reaction mixture is stirred for 30 minutes at room temperature. The resulting mixture is yellowish hazy with a dark brown organic layer settled at the bottom of the flask. The dark brown colored dichloromethane layer is separated from the aqueous hazy phase.
After separating the organic layer, the aqueous hazy phase is again extracted with dichloromethane (1×100.0 mL). The combined dichloromethane phases (total volume 290.0 mL) are extracted twice with 50 % aqueous acetic acid solution (1×100 mL, 1×75.0 mL). A dark orange color acetic acid layer is separated. The pH of the acetic acid solution is found to be around 3.0-3.5 when tested by litmus paper. Combined aqueous acetic acid solution is basified, to pH 7.5-8.5, using 40% aqueous sodium hydroxide solution under cold conditions (0-10°C). After attaining the desired pH of the solution, 200 mL dichloromethane is added and stirred. The content is transferred to a separating funnel and is vigorously shaken. The dichloromethane layer is separated and the aqueous phase is again extracted with dichloromethane (1×75.0 mL). The combined dichloromethane extracts are washed with cold saturated sodium chloride solution (1×30.0 mL) and dried over anhydrous sodium sulfate. Removal of solvent on a rotary evaporator with a water bath temperature of 45°C, gives a dark orange brown viscous liquid. To this viscous liquid, 80-85.0 mL dry toluene is added.
The toluene containing crude olanzapine is transferred into a dry 250 mL single necked round bottom flask. Methanolic sodium hydroxide solution (0.32 g sodium hydroxide dissolved in 3.0-4.0 mL methanol by sonication) is added and the mixture is heated in an oil bath at 60°C for 2 hours. After the stipulated time, 20-25 % of the total volume of solvent is evaporated on a rotary evaporator, with a 55-60°C water bath, to ensure the complete removal of dichloromethane and trace amounts of water, resulting in a final volume of between 55-60 mL. The hot solution is removed from the water bath and cooled in an ice bath with stirring. Within 2-3 minutes, the solution is quickly seeded with previously prepared ultra pure olanzapine Form I, as determined by X-Ray and IR, with stirring. Stirring is continued for 40-45 minutes. The yellowish solid obtained in the solution is filtered off, washed with 1.5-2.0 mL dichloromethane and dried on a vacuum pump for 50-60 minutes to give 4.85 g ( 82.4 % yield) of olanzapine Form I. The solid obtained is crushed to a fine powder and air dried to remove traces of dichloromethane. Karl Fisher analysis indicates 5000-8000 ppm water content. The material is dried in an oven at 65°C for 1.5-2.0 hours and analyzed for water (670-860 ppm water). The weight of the title product is 4.80 g (82 % yield), HPLC purity = 99.83%, polymorphic purity is 100% as no detectable polymorph II is observed by X-ray and IR, as shown in Example 3.
The HPLC conditions are as follows: Column: SymmetryC18 , 4.6 x 250 mm λmax 254 nm
Flowrate : 1.0 mL/min. Run Time: 70 minutes
The buffer comprises 5.4 g potassium phosphate; 0.5 g heptanesulfonic acid sodium salt and 0.5 g 1-octanesulfonic acid sodium salt dissolved in 500 mL Dl water and adjusted the pH to 2.6 using cone, phosphoric acid. The mobile phase was 500 mL buffer/300 mL acetonitrile/200 mL methanol. The final pH of the mobile phase is about 3.6. The concentration of the standard is 100μg/mL; the injection volume is 15 μl; and RT = 4.6—4.7 min.
EXAMPLE 2 RECRYSTALLIZATION
From the dried yellowish solid prepared according to Example 1 , 2.0 g (0.0064 mol) is transferred into a single necked round bottom flask provided with a magnetic stirrer. To the solid, 40.0 mL dry toluene and methanolic sodium hydroxide solution (0.052 g sodium hydroxide pellets dissolved in 2.0 L methanol by sonication) are added. To this mixture 3.5-4.0 mL dichloromethane is added.
The mixture is heated for 5-10 minutes in an oil-bath at 60-65°C until a clear solution is obtained. After heating, the solution is transferred into an ice bath and seeded with previously prepared ultra-pure olanzapine Form I. The solution is stirred for 30-35 minutes at 0-10°C. The yellowish solid obtained is filtered on vacuum pump and washed with 2.0-2.5 mL dichloromethane. The solid is dried on a vacuum pump for 40-45 minutes. The solid obtained is crushed into a fine powder and air dried to remove traces of dichloromethane. The air dried material is further dried in the oven at 65°C for 1.5-2.0 hours and analyzed for water content. Karl Fisher study shows 670-860 ppm water content. The weight of olanzapine Forml is 1.93 g (95.0 % crystallization yield) of 99.96 % HPLC purity. EXAMPLE 3 X-RAY POWDER DIFFRACTOMETRY STUDY
Olanzapine Form I prepared according to Example 1 is analyzed by X-ray, IR, and DSC and found to conform to a commercially available reference standard olanzapine Form I. DSC of the olanzapine Form I prepared according to the present invention shows an endotherm peak at 195°C.
…………………
http://www.google.com/patents/CN102268010A?cl=en
Currently, the preparation of olanzapine, mainly in the following three ways:
1.4-amino-2-methyl–10H- thieno [2,3-b] [1,5] – benzodiazepine hydrochloride and N- methylpiperazine, in a nitrogen atmosphere Toluene and DMSO as solvent at reflux for 20h, after-treatment to give the product, recrystallized from acetonitrile to give crystals of olanzapine, a yield of 33% (US5229382).

[0013] The process route fewer steps, simple process, raw materials, but a long reaction time, the use of toxic solvents, pollution, low yield, industrial production adversely.
[0014] CN1906201A discloses the use of no solvent or low boiling organic solvent method for preparing olanzapine, pointed N- methylpiperazine with 4-amino-2-methyl -10H- thieno [2,3-b] [l, 5] molar ratio _ benzodiazepine hydrochloride of 3: 1~8: 1,110~145 ° C after the reaction at least polonium, water was added, at least two organic solvents, or water and at least one organic solvent precipitation olanzapine. This improved process reaction time is shortened, reducing energy consumption and cost, but an excess of starting material N- methylpiperazine unrecovered, resource waste problems still exist.
[0015] 2. 4-amino-2-methyl -10H- thieno [2,3_b] [1,5] – benzodiazepine hydrochloride, to generate demethylolanzapine piperazine is reacted with level, and then obtained by methylation of olanzapine, recrystallized from ethanol to obtain refined product yield of 68% (CN1420117A).
[0016]

[0017] The method for preparing olanzapine via a two-step process is relatively complicated and the reaction time is still long; Further, more by-products: The first step is easy to form dimeric product, a second step Iddo methylation.
[0018] 3. 4-amino-2-methyl -10H- thieno [2,3-b] [1,5] – benzodiazepine and N, N- two – (2_ haloethyl ) _ methylamine in alkaline catalyst, solvent reflux 3~IOh obtained crude olanzapine, yield 75% to 92%. Wherein, X = Br or Cl, the catalyst is sodium alkoxide, sodium hydroxide, sodium amide, sodium hydroxide, inorganic bases (CN101168544A).
[0019]

[0020] This method is simple, shorten the reaction time and therefore reduce energy consumption, but the raw material N, N- two – (2-halo-ethyl) _ methylamine not easy, if more raw material preparation is bound to increase the cost of production.
The present invention is olanzapine preparation method:
[0024]

Example 1 olanzapine [0030] Implementation
[0031] To a 250ml three-necked flask of 4-amino-2-methyl -10H- thieno [2,3_b] [1,5] – benzodiazepine hydrochloride 20. OOg (0 075mol.) , N_-methylpiperazine 75. 30g (0. 75mol), nitrogen and stirred and heated to reflux the reaction cell. Under stirring, the reaction mixture was poured into 200ml of water to precipitate a pale yellow solid powder, stirring was continued for lh, filtered and dried to give olanzapine product 23. 30g, yield 99.4%, purity 99. 0% (HPLC).
2 olanzapine refined example [0032] Implementation
[0033] Example 1 was 23. 30g olanzapine product was transferred into 250ml single neck flask was added MOml ethanol, stirred and heated to reflux to make the product the whole solution. 0. 20g of activated carbon was added to the system, reflow bleaching treatment 30min, filtered, and the filtrate cooled to room temperature and crystallization, filtration and dried to give a yellow crystalline powder 16. 32g, yield 70.0%, the purity of 99. 8% (HPLC), high performance liquid phase chromatogram, see photos.
Olanzapine Preparation Example 3 [0034] Implementation
[0035] To a three-necked flask IOOOml 4-amino-2-methyl -10H- thieno [2,3_b] [1,5] – benzodiazepine hydrochloride 150. 03g (. 0 56mol) , N- methylpiperazine 339. 29g (3. 39mol), nitrogen and stirred and heated to reflux the reaction cell. Cooling, vacuum distillation recovery more than 70% excess N- methylpiperazine to give Olanzapine crude solid.
[0036] 400ml of ethanol was added to the three-necked flask and heated to reflux for solid all dissolved. Then dissolved under stirring in ethanol olanzapine solution was poured IOOOml water to precipitate a pale yellow solid powder was filtered and dried to give olanzapine product 173. 87g, yield 99.4%, purity 98. 9% (HPLC).
Preparation 4 olanzapine [0037] Implementation
[0038] To a 250ml three-necked flask of 4-amino-2-methyl -10H- thieno [2,3_b] [1,5] – benzodiazepine hydrochloride 10. OOg (0 038mol.) , N_-methylpiperazine 39. 63g (0. 40mol) and diethylene glycol dimethyl ether 30ml, nitrogen and stirred and heated to reflux the reaction cell. Cooling, vacuum distillation recovery more than 80% excess of N- methylpiperazine and diethylene glycol dimethyl ether mixture to give solid crude olanzapine.
[0039] 40ml of ethanol was added to the three-necked flask and heated to reflux all dissolved solids. Then under stirring to dissolve olanzapine solution was poured into 200ml of water in ethanol, a yellow powder precipitated solid was filtered and dried to give olanzapine product 11.79g, yield 99.3%, purity 98. 7% (HPLC).
Olanzapine Preparation Example 5 [0040] Implementation
[0041] To a 250ml bottle of three 4-amino-2-methyl–10H- thieno [2,3_b] [1,5] – benzodiazepine hydrochloride 20. OOg (0. 075mol) , N_-methylpiperazine 62. 34g (0. 62mol), nitrogen and stirred and heated to reflux the reaction cell. Cooling, vacuum distillation recovery more than 75% excess N- methylpiperazine to give Olanzapine crude solid.
[0042] 60ml of ethanol was added to the three-necked flask and heated to reflux all dissolved solids. Then under stirring to dissolve olanzapine solution was poured into 600ml of ethanol in water to precipitate a pale yellow solid powder was filtered and dried to give olanzapine product 23. 37g, yield 99.7%, purity 99. 0% (HPLC). [0043] Example 6 olanzapine refined
[0044] Example 5 was 23. 37g olanzapine product was transferred into 250ml single neck flask was added ^ Oml ethanol, stirred and heated to reflux to make the product the whole solution. 0. 03g of activated carbon is added to the system and 0. 03g diatomite, reflow bleaching treatment 15min, filtered, and the filtrate cooled to room temperature and crystallization, filtration and dried to give a yellow crystalline powder 16. 76g, yield 71.7%, purity 99.7% ( HPLC).
………………….
http://www.google.com/patents/EP0733635B1?cl=en
Example 1
-

-
In a suitable three neck flask the following was added:
- Dimethylsulfoxide (analytical):
- 6 volumes
- Intermediate 1 :
- 75 g
- N-Methylpiperazine (reagent) :
- 6 equivalents
Intermediate 1 can be prepared using methods known to the skilled artisan. For example, the preparation of the Intermediate 1 is taught in the ‘382 patent.
-
A sub-surface nitrogen sparge line was added to remove the ammonia formed during the reaction. The reaction was heated to 120°C and maintained at that temperature throughout the duration of the reaction. The reactions were followed by HPLC until ≤ 5% of the intermediate 1 was left unreacted.
-
After the reaction was complete, the mixture was allowed to cool slowly to 20°C (about 2 hours). The reaction mixture was then transferred to an appropriate three neck round bottom flask and water bath. To this solution with agitation was added 10 volumes reagent grade methanol and the reaction was stirred at 20°C for 30 minutes. Three volumes of water was added slowly over about 30 minutes. The reaction slurry was cooled to zero to 5°C and stirred for 30 minutes. The product was filtered and the wet cake was washed with chilled methanol. The wet cake was dried in vacuo at 45°C overnight. The product was identified as technical olanzapine.
Yield: 76.7%; Potency: 98.1%
- Technical Grade olanzapine

Example 2
- Form II
-
A 270 g sample of technical grade 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine was suspended in anhydrous ethyl acetate (2.7 L) . The mixture was heated to 76°C and maintained at 76°C for 30 minutes. The mixture was allowed to cool to 25°C. The resulting product was isolated using vacuum filtration. The product was identified as Form II using x-ray powder analysis. Yield: 197 g.
-
The process described above for preparing Form II provides a pharmaceutically elegant product having potency > 97%, total related substances < 0.5% and an isolated yield of > 73%.
………………….
Impurities

Olanzapine N-oxide (Olanzapine Impurity D)
…………..
Synthesis and characterization of impurities of an anti-psychotic drug substance, Olanzapine (08-3022UP)
Poornachander Thatipalli, Ramesh Kumar, Chandrasekhar Bulusu, Ramesh Chakka, Pratap R. Padi, Anjaneyulu Yerra and Satyanarayana Bollikonda
Full Text: PDF (226K)
pp. 195 – 201
| EP1965773A1 † | Dec 19, 2006 | Sep 10, 2008 | Laboratorios Lesvi, S. L. | Oral formulation of anhydrous olanzapine form i |
| US7297789 | May 30, 2003 | Nov 20, 2007 | Sandoz, Inc. | From 4-amino-2-methyl-10H-thieno(2,3-b)(1,5) benzodiazpine HCl and 1-methylpiperazine in an aprotic high boiling solvent; purifying in an acid medium; basifying to pH of 7.5-9; and extracting with a low boiling organic solvent |
| US7329747 | Jan 27, 2005 | Feb 12, 2008 | Synthon Ip Inc. | Synthesis of olanzapine and intermediates thereof |
| US7459449 | Jan 27, 2005 | Dec 2, 2008 | Rolf Keltjens | Olanzapine malonate, olanzapine glycolate, and olanzapine benzoate; antipsychotic agents; water soluble; can form tablets by direct compression; may be mixed with dibasic calcium phosphate and/or hydroxypropyl cellulose |
| US7538213 | May 16, 2003 | May 26, 2009 | Institut Farmaceutyczny | Condensation of molar excess of N-methylpiperazine with 4-amine-2-methyl-10H-thieno [2,3-b][1,5]benzodiazepine hydrochloride in dimethylsulfoxide; recovery and purification with methylene chloride |
| US7829700 | Sep 5, 2005 | Nov 9, 2010 | Shasun Chemicals And Drugs Limited | Process for preparation of a pharmaceutically pure polymorphic form I of Olanzapine |
| WO2004006933A2 | Jul 14, 2003 | Jan 22, 2004 | Krka D D Novo Mesto | Crystal forms of olanzapine and processes for their preparation |
| WO2006027800A1 * | Sep 5, 2005 | Mar 16, 2006 | T Chanidran | A novel process for preparation of a pharmaceutically pure polymorphic form 1 of olanzapine |
| WO2008091169A2 | Jan 22, 2008 | Jul 31, 2008 | Tomasz Kozluk Nobilus Ent | Process for preparation of substantially pure polymorphic form i of olanzapine |
| CN1420117A * | Nov 16, 2001 | May 28, 2003 | 连云港豪森制药有限公司 | Method for preparing olanzapine |
| CN1906201A * | Dec 22, 2004 | Jan 31, 2007 | 特瓦制药工业有限公司 | Methods of preparing olanzapine |
| CN101735239A * | Nov 6, 2008 | Jun 16, 2010 | 齐鲁制药有限公司 | Preparation method of anhydrous olanzapine crystal form II |
| PL203992B1 * | Title not available | |||
| WO2003101997A1 * | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2002018390A1 | Mar 7, 2001 | Mar 7, 2002 | Ramesh Chakka | Process for preparation of hydrates of olanzapine and their conversion into crystalline forms of olanzapine |
| WO2003097650A1 | May 16, 2003 | Nov 27, 2003 | Urszula Fraczek | Methods for preparation of olanzapine polymorphic form i |
| EP0454436A1 * | Apr 24, 1991 | Oct 30, 1991 | Lilly Industries Limited | Pharmaceutical compounds |
| EP0733635A1 | Mar 22, 1996 | Sep 25, 1996 | Eli Lilly And Company | Crystal forms of a thieno(2,3-B)(1,5) benzodiazepine derivative and process for their preparation |
| EP0911028A2 | Sep 30, 1998 | Apr 28, 1999 | Eli Lilly And Company | Method for treating sexual dysfunction |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2004056833A1 * | Dec 15, 2003 | Jul 8, 2004 | Adamed Sp Zoo | A process for the preparation of a pharmaceutically pure polymorphic form i of olanzapine |
| WO2005070937A1 * | Jan 26, 2005 | Aug 4, 2005 | Rolf Keltjens | A process for making olanzapine in a polymorph form i |
| WO2005090359A2 * | Mar 17, 2005 | Sep 29, 2005 | Lek Pharmaceuticals | Synthesis of 2-methyl-4-(4-methyl-1-piperazinyl)-10h-thieno[2, 3-b][1,5]benzodiazepine and salts thereof |
| WO2006010620A2 * | Jul 28, 2005 | Feb 2, 2006 | Krka Tovarna Zdravil D D Novo | Olanzapine salts and their conversion to olanzapine free base |
| WO2007020080A1 * | Aug 16, 2006 | Feb 22, 2007 | Synthon Bv | A process for making olanzapine form i |
| WO2007054750A2 | Nov 10, 2006 | May 18, 2007 | Egis Gyogyszergyar Nyrt | Process for the preparation of olanzapine |
| WO2007144901A1 * | Jun 12, 2007 | Dec 21, 2007 | Jubilant Organosys Ltd | Process for stabilization of olanzapine polymorphic form i |
| WO2008091169A2 * | Jan 22, 2008 | Jul 31, 2008 | Tomasz Kozluk Nobilus Ent | Process for preparation of substantially pure polymorphic form i of olanzapine |
| WO2011009831A1 | Jul 19, 2010 | Jan 27, 2011 | Lek Pharmaceuticals D.D. | Process for the purification of olanzapine |
| CN102268010A * | Jun 17, 2011 | Dec 7, 2011 | 大连理工大学 | 一种奥氮平的制备方法和精制方法 |
| EP1781665A2 * | Jul 28, 2005 | May 9, 2007 | KRKA, tovarna zdravil, d.d., Novo mesto | Olanzapine salts and their conversion to olanzapine free base |
| EP1965773A1 † | Dec 19, 2006 | Sep 10, 2008 | Laboratorios Lesvi, S. L. | Oral formulation of anhydrous olanzapine form i |
| EP2264016A2 * | Jul 14, 2004 | Dec 22, 2010 | Jubilant Organosys Limited | A process for producing pure form form of 2-Methyl-4-(4-Methyl-1-Piperazinyl)-10h-thieno[2,3-B][1,5] benzodiazepine |
| EP2292624A1 | Jul 20, 2009 | Mar 9, 2011 | LEK Pharmaceuticals d.d. | Process for the purification of olanzapine |
| US7329747 | Jan 27, 2005 | Feb 12, 2008 | Synthon Ip Inc. | Synthesis of olanzapine and intermediates thereof |
| US7459449 | Jan 27, 2005 | Dec 2, 2008 | Rolf Keltjens | Olanzapine malonate, olanzapine glycolate, and olanzapine benzoate; antipsychotic agents; water soluble; can form tablets by direct compression; may be mixed with dibasic calcium phosphate and/or hydroxypropyl cellulose |
| US7759484 | Jul 7, 2005 | Jul 20, 2010 | Inke, S.A. | Mixed solvate of olanzapine, method for preparing it and method for preparing form I of olanzapine therefrom |
| US7829700 | Sep 5, 2005 | Nov 9, 2010 | Shasun Chemicals And Drugs Limited | Process for preparation of a pharmaceutically pure polymorphic form I of Olanzapine |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4115568 | Apr 13, 1977 | Sep 19, 1978 | Lilly Industries Limited | Thieno[3,2-b]-[1,5]benzodiazepines |
| US5229382 | May 22, 1992 | Jul 20, 1993 | Lilly Industries Limited | 2-methyl-thieno-benzodiazepine |
| US5457101 | Jun 3, 1994 | Oct 10, 1995 | Eli Lilly And Company | Thieno[1,5]benzoidiazepine use |
| US5605897 | Feb 13, 1995 | Feb 25, 1997 | Eli Lilly And Company | Nervous system disorders, psychological disorders |
| US5736541 | Jul 25, 1996 | Apr 7, 1998 | Eli Lilly And Company | Slurrying impure material in anhydrous ethyl acetate, then crystallization yields pure stable drug for treatment of anxiety, psychological disorders and gastrointestinal disorders |
| US6063802 | Jan 28, 1999 | May 16, 2000 | Glaxco Wellcome Inc. | Ondansetron freeze-dried dosage form compositions for oral administration |
| US6348458 | Mar 31, 2000 | Feb 19, 2002 | U & I Pharmaceuticals Ltd. | Polymorphic forms of olanzapine |
| US20020086993 | Dec 19, 2001 | Jul 4, 2002 | Julian Davies | Crystal modification |
| US20040265375 | Apr 15, 2004 | Dec 30, 2004 | Platteeuw Johannes J. | Matrix of silicified microcrystalline cellulose; antiinflammatory, antirheumatic, antiemetic, analgesic, antiepileptic, antipsychotic, antidepressant, hypnotic, antiulceric, prokinetic, antiasthmatic , vasodilator, antidiabetic, and/or hypolipidemic agents; Parkinson*s disease; cardiovascular disorders |
| US20050267099 | Jan 27, 2005 | Dec 1, 2005 | Rolf Keltjens | Reacting a des-piperazine olanzapine with an N-formyl piperazine to form an N-formyl olanzapine |
| US20050272720 | Jan 27, 2005 | Dec 8, 2005 | Rolf Keltjens | Process for making olanzapine Form I |
| EP0454436B1 | Apr 24, 1991 | Sep 13, 1995 | Lilly Industries Limited | Pharmaceutical compounds |
| EP0733635B1 | Mar 22, 1996 | Aug 16, 2001 | Eli Lilly And Company | Crystal forms of a thieno(2,3-B)(1,5) benzodiazepine derivative and process for their preparation |
| EP0828494A1 | May 30, 1995 | Mar 18, 1998 | Eli Lilly And Company | Method for treating cognitive dysfunction |
| EP0831098B1 | Sep 22, 1997 | Nov 21, 2001 | Eli Lilly And Company | Intermediates and process for preparing olanzapine |
| WO1998011893A1 | Sep 18, 1997 | Mar 26, 1998 | Lilly Co Eli | Olanzapine dihydrate d |
| WO1999016313A1 | Sep 30, 1998 | Apr 8, 1999 | Douglas James Allen | 2-methyl-thieno-benzodiazepine formulation |
| WO2001047933A1 | Dec 22, 2000 | Jul 5, 2001 | Cipla Ltd | New polymorphic forms of olanzapine |
| WO2002018390A1 | Mar 7, 2001 | Mar 7, 2002 | Ramesh Chakka | Process for preparation of hydrates of olanzapine and their conversion into crystalline forms of olanzapine |
| WO2003007912A2 | Jul 5, 2002 | Jan 30, 2003 | Dekemper Kurt Douglas | Lyophilized formulation comprising olanzapine |
| WO2003091260A1 | Apr 22, 2003 | Nov 6, 2003 | Ramesh Chakka | Novel crystalline polymorph form-vi of olanzapine and a process for preparation thereof |
| WO2003097650A1 | May 16, 2003 | Nov 27, 2003 | Urszula Fraczek | Methods for preparation of olanzapine polymorphic form i |
| WO2003101997A1 | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004000847A1 | Jun 10, 2003 | Dec 31, 2003 | Adamed Sp Zoo | A process for the preparation of olanzapine and an intermediate therefor |
| WO2004006933A2 | Jul 14, 2003 | Jan 22, 2004 | Krka D D Novo Mesto | Crystal forms of olanzapine and processes for their preparation |
| Reference | ||
|---|---|---|
| 1 | “Anhydrates and Hydrates of Olanzapine: Crystallization, Solid-State Characterization, and Structural Relationships“, Crystal Growth & Design, 2003, vol. 3, No. 6, pp. 897-907. | |
| 2 | “Catalytic Transfer Hydrogenation of Aromatic Nitro-compounds“, Chinese Journal of Pharmaceuticals, 2001, 32(9), pp. 391-393. | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7951798 * | Apr 22, 2004 | May 31, 2011 | Egis Hyogyszergyar Nyrt. | Polymorphs of olanzapine hydrochloride |
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
Cadila reports Stable amorphous form of vortioxetine hydrobromide…WO 2015044963

Vortioxetine
O N Sept. 30, 2013 — The U.S. Food and Drug Administration today approved Brintellix (vortioxetine) to treat adults with major depressive disorder.
Major depressive disorder (MDD),

Commonly referred to as depression, is a mental disorder characterized by mood changes and other symptoms that interfere with a person’s ability to work, sleep, study, eat and enjoy once-pleasurable activities. Episodes of depression often recur throughout a person’s lifetime, although some may experience a single occurrence.
READ ALL AT
http://www.drugs.com/newdrugs/fda-approves-brintellix-major-depressive-disorder-3918.html
SYNTHESIS……..https://newdrugapprovals.org/2013/10/01/vortioxetine-fda-approves-brintellix-to-treat-major-depressive-disorder/

An amorphous vortioxetine and salts thereof
Cadila Healthcare Ltd
Singh, Kumar Kamlesh; Gajera, Jitendra Maganbhai; Raikwar, Dinesh Kumar; Khera, Brij; Dwivedi, Shri Prakash Dhar
| The present invention relates to an amorphous vortioxetine and salts thereof. In particular, the invention relates to a process for the preparation of an amorphous vortioxetine hydrobromide. Further, the invention also relates to a process for preparation of amorphous vortioxetine free base. The invention also relates topharmaceutical compositions comprising an amorphous vortioxetine or hydrobromide salt thereof for oral administration for treatment of major depressive disorder (MDD) and generalized anxiety disorder (GAD). |
Stable amorphous form of vortioxetine hydrobromide, useful for treating depression, major depressive disorder (MDD) and generalized anxiety disorder. Also claims a process for preparing the amorphous form and solid dispersions comprising the same.
This API, which was originally developed and launched by Lundbeck and Takeda for treating MDD.
A phase IV trial (NCT02357797) for schizophrenia was scheduled to begin in March 2015. Family members of the product case, WO03029232, hold SPC protection in the EP until 2027 and one of its Orange Book listed filings, US7144884, expire in the US in 2023 with US154 extension.
The US FDA Orange Book also lists patents describing crystalline forms of vortioxetine/Brintellix, US8722684 and US8969355, that are due to expire in 2030 and 2027 respectively. The drug also has NCE exclusivity expiring in September 2018.
Cadila is potentially interested in vortioxetine hydrobromide.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

 amcrasto@gmail.com
amcrasto@gmail.com
OLANZEPINE VISITED PART 1/3


PART 1…..https://newdrugapprovals.org/2015/04/08/olanzepine/
PART 2….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-22/
PART 3…….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-33/

Olanzapine (sold under the brand names Zyprexa, Zypadhera and Lanzek or in combination with fluoxetine, Symbyax) is anatypical antipsychotic. It is approved by the U.S. Food and Drug Administration (FDA) for the treatment of schizophrenia and bipolar disorder.[4]
Olanzapine is structurally similar to clozapine and quetiapine, but is classified as a thienobenzodiazepine. The olanzapine formulations are manufactured and marketed by the pharmaceutical company Eli Lilly and Company; the drug went generic in 2011. Sales of Zyprexa in 2008 were $2.2B in the US, and $4.7B worldwide.[5]

Zyprexa (olanzapine) 10 mg tablets (AU)
Olanzapine has a higher affinity for 5-HT2A serotonin receptors than D2 dopamine receptors, which is a common property of all atypical antipsychotics, aside from the benzamide antipsychotics such as amisulpride. Olanzapine also had the highest affinity of any second-generation antipsychotic towards the P-glycoprotein in one in vitro study.[60] P-glycoprotein transports a number of drugs across a number of different biological membranes including the blood-brain barrier, which could mean that less brain exposure to olanzapine results from this interaction with the P-glycoprotein.[61]
Olanzapine is a potent antagonist of the muscarinic M3 receptor,[65] which may underlie its diabetogenic side effects.[64][66] Additionally, olanzapine also exhibits a relatively low affinity for serotonin 5-HT1, GABAA, beta-adrenergic receptors, and benzodiazepine binding sites.[67] [27]
Dosage forms
Olanzapine is marketed in a number of countries, with tablets ranging from 2.5 to 20 milligrams. Zyprexa (and generic olanzapine) is available as an orally-disintegrating “wafer” which rapidly dissolves in saliva. It is also available in 10 milligram vials for intramuscular injection.[4]
Research
Olanzapine has been investigated for use as an antiemetic, particularly for the control of chemotherapy-induced nausea and vomiting (CINV). A 2007 study demonstrated its successful potential for this use, achieving a complete response in the acute prevention of nausea and vomiting in 100% of patients treated with moderately and highly-emetogenic chemotherapy, when used in combination with palonosetron and dexamethasone.[85]
Olanzapine has been considered as part of an early psychosis approach for schizophrenia. The Prevention through Risk Identification, Management, and Education (PRIME) study, funded by the National Institute of Mental Health and Eli Lilly, tested the hypothesis that olanzapine might prevent the onset of psychosis in people at very high risk forschizophrenia. The study examined 60 patients with prodromalschizophrenia, who were at an estimated risk of 36–54% of developing schizophrenia within a year, and treated half with olanzapine and half with placebo.[86] In this study, patients receiving olanzapine did not have a significantly lower risk of progressing to psychosis. Olanzapine was effective for treating the prodromal symptoms, but was associated with significant weight gain.[87]
1H NMR PREDICT

13C NMR PREDICT


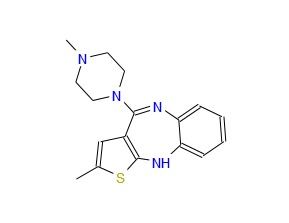
COSY

HMBC
…………………………………..WILL BE UPDATED
UV
IR
1H NMR
13C NMR
MASS
…………………………………….
INTERMEDIATE/S USED IN SYNTHESIS AND REFERENCE


LEK PHARMACEUTICALS D.D. Patent: WO2005/90359 A2, 2005 ; Location in patent: Page/Page column 21 ;


Apotex Pharmachem Inc. Patent: US2008/319189 A1, 2008 ; Location in patent: Page/Page column 2 ;

LEK PHARMACEUTICALS D.D. Patent: WO2005/90359 A2, 2005 ; Location in patent: Page/Page column 21 ;

Leyva-Perez, Antonio; Cabrero-Antonino, Jose R.; Corma, Avelino Tetrahedron, 2010 , vol. 66, # 41 p. 8203 – 8209
SEE
WATSON PHARMACEUTICALS, INC. Patent: WO2004/94390 A1, 2004 ; Location in patent: Page 15 ;

WO2006/6180 A1, ; Page/Page column 11 ;
 Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
WO2006/6180 A1, ; Page/Page column 11 ;
 US5605897 A1, ;
US5605897 A1, ;
………………………………………
PATENT
http://www.google.com.na/patents/EP1730153B1?cl=en


Figure 2 shows the NMR spectrum of the solvate according to the invention. The peaks were assigned as follows (1H NMR; CDCl3, 300 MHz) :

| Chemical shift δ | Assignement |
| 1.20 (3H, d) | CH3 – isopropanol |
| 2.30 (3H, s) | 4′-CH3 |
| 2.34 (3H, s) | 2- CH3 |
| 2.20-2.40 (2H, br s) | H – water |
| 2.49 (4H, m) | 3′-CH2 |
| 3.52 (4H, m) | 2′-CH2 |
| 4.03 (0.5H, dq) | CH – isopropanol |
| 5.02 (H, broad s) | 10-NH |
| 6.29 (H, broad s) | 3-CH |
| 6.29-7.05 (4H, m) | 6, 7, 8, 9-H |
-
Olanzapine has shown to have high activity with regard to the central nervous system and is also useful for the treatment of schizophrenia, schizophreniform disorders, acute mania, mild anxiety states and psychosis.
-
Various polymorphic and pseudopolymorphic forms, such as solvates, of olanzapine have become known. Some of them are useful for conversion to other desirable forms.
-
The British patent GB 1 533 235 discloses antipsychotically effective thienobenzodiazepines by a generic formula which also covers olanzapine.
-
US patent 5,229,382 discloses olanzapine explicitly. The described process for its synthesis involves a crystallization from acetonitrile.
-
EP-B-733 635 claims crystalline form II olanzapine, and this polymorphic form is said to be more stable than the material obtained according to US 5,229,382 which is designated “form I olanzapine”. Both the form I and the form II of olanzapine are characterized by e. g. X-ray data. The preparation of the more stable form II of olanzapine is effected by dissolving technical grade olanzapine in ethyl acetate and crystallization from the resulting solution by any conventional process such as seeding, cooling, scratching the glass of the reaction vessel or other common techniques.
-
WO 02/18390 discloses the monohydrate form I and the dihydrate form I of olanzapine, a process for production thereof and a process for production of form I of olanzapine which comprises the steps of stirring olanzapine monohydrate form I or crude olanzapine or form II of olanzapine in methylene chloride at reflux, cooling, filtering and drying. It is also described that a repeating of the process described in US 5,229,382 Example 1, subexample 4 did not lead to formation of form I of olanzapine.
-
WO 03/101997 relates to processes for preparation of form I of olanzapine by regulation of the pH-value of the solution.
-
WO 03/055438 discloses the preparation of form I olanzapine by crystallization from ethanol and subsequent conversion of the obtained ethanol solvate.
-
US patent 5,637,584 discloses the (mono)methylene chloride solvate form of olanzapine and a method for its conversion to the polymorphic form I of olanzapine.
-
EP-B-733 634 discloses three specific solvates of olanzapine, namely the methanol, ethanol and 1-propanol solvates and a process for production of form II olanzapine by drying such a solvate.
-
WO 03/097650 describes two new, mixed solvate forms, the water/methylene chloride solvate and the water/DMSO solvate, methods for preparing them, and their transformation to polymorphic form I.
-
WO 2004/006933 discloses a process for the preparation of form I olanzapine, as well as various pseudopolymorphic forms, namely the isopropanol solvate, and the acetonitrile/methylene chloride/water and acetonitrile/water mixed solvates of olanzapine, and the polymorphic form A.

Preparation of the water-isopropanol mixed solvate of olanzapine Example 1
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (120 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 200 ml were distilled off under vacuum. The residue was cooled to room temperature, isopropanol (180 ml) was added, and the solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After the crystallization was completed, the precipitate was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 25°C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C and after completion of the crystallization the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 22.84 g. Loss on drying (140°C): 13.6%. Water content (Karl Fischer): 5.12%.
Example 2
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (36 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 80 ml were distilled off under vacuum. The residue was cooled to room temperature, and isopropanol (180 ml) was added. The solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After the crystallization was completed, the precipitate was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 35 °C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C, upon finalization of the crystallization, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 21.98 g. Loss on drying (140°C): 13.2 %. Water content (Karl Fischer): 5.09%. Assay of isopropanol (GC): 8.55 %.
Example 3
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (36 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 120 ml were distilled off under vacuum. The residue was cooled to room temperature, and isopropanol (180 ml) was added. The solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After completion of the crystallization, the precipitate.was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 35°C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C, upon completion of the crystallization, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 24.35 g. Loss on drying (140°C): 13.5%. Water content (Karl Fischer): 5.05%.
Example 4
-
Anhydrous olanzapine (10 g) was suspended in isopropanol (108 ml) and heated to reflux to obtain a clear solution. The solution was slowly cooled. Water (6 ml) was added at 57°C to start crystallization. The suspension was cooled to 0°C, upon finalization of the crystallization, the product was filtered off and washed with isopropanol (5 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 10.97 g. Loss on drying (140°C): 13.3%. Water content (Karl Fischer): 5.13%.
Example 5
-
60 g of olanzapine obtained from mother liquors was suspended in isopropanol (650 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (7.9 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 25°C and water (16 ml) was added to start crystallization. The suspension was cooled to 0°C and, upon completion of the crystallization, the product was filtered off and washed with isopropanol (50 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 57.64 g. Loss on drying (140°C): 13.5%. Water content (Karl Fischer): 5.26%.
Example 6
-
The solution of 2,4-bis(4-methyl-1-piperazinyl)-3-propylidene-3H-[1,5]benzodiazepine (41.86 g, 0.11 mmol) (prepared according to WO 2004/065390 ), pyridinium p-toluenesulfonate (55.29 g, 0.22 mmol) and sulfur (11.99 g, 0.374 mmol) in benzonitrile (1100 mL) was stirred at 140°C for 11 h, cooled to 90°C and concentrated to an oily residue. The residue was diluted with dichloromethane and isopropanol (250 mL, 1 : 1). The precipitate was filtered off and washed with dichloromethane and isopropanol (20 ml, 1 : 1). The filtrate was extracted with HCl (250 ml, 2 M). The organic phase was further extracted with HCl (2 X 100 ml, 1 M). The combined aqueous phases were cooled in an ice bath and made alkaline by using 5 M NaOH. The obtained turbid solution was left in a refrigerator over night resulting in a suspension. This was separated by filtration and washed with isopropanol (2 X 25 ml). The wet material was suspended in isopropanol (215 ml) and heated to reflux to obtain a clear solution. The solution was hot filtered. Water (6.5 ml) was added to induce crystallization. The obtained’suspension was cooled to 0°C, and upon completion of crystallisation, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 18.61 g. Loss on drying (140°C): 12.8 %. Water content (Karl Fischer): 5.29 %.
Example 7
-
The solution of 2,4-bis(4-methyl-1-piperazinyl)-3-propylidene-3H-[1,5]benzodiazepine (3.805 g, 10 mmol) (prepared according to WO 2004/065390 ), pyridinium p-toluenesulfonate (5.026 g, 20 mmol) and sulfur (1.122 g, 35 mmol) in benzonitrile (100 ml.) was stirred at 140°C for 8.5 h, cooled to 90°C and concentrated to an oily residue. The residue was diluted with isopropanol (50 ml) and dimethyl sulfoxide (5 ml). The precipitate was filtered off and washed with isopropanol (5 ml). Water (10 ml) and sodium hydroxide (1.00 g, 25 mmol) were added to the filtrate. The mixture was stirred at room temperature until the sodium hydroxide had dissolved. The turbid solution was left in a refrigerator over night resulting in a suspension. This was filtered off and washed with isopropanol (5 mL). The wet material was suspended in isopropanol (25 mL) and the suspension was heated to reflux. Then solids were hot filtered. Water (0.75 mL) was added to the filtrate to induce crystallization. The resulting suspension was cooled to 0°C, and upon completion of crystallisation, the product was filtered off and washed with isopropanol (1 mL). The product was then dried at room temperature under vacuum to a constant weight. Yield: 0.738 g.
………………………………..
PAPER
http://www.biomedcentral.com/1471-2210/12/8

Scheme 1
Synthesis of compounds 8a, 8b, and 8c. Reagents and conditions: (i) 1-fluoro-2-nitrobenzene, NaH, THF, rt, 20 h, 60%; (ii) SnCl2, EtOH, 80°C, 1 h, 80%; (iiia) N-methylhomopiperazine (5 equiv), no solvent, microwave heating, 80°C, 4 h,65%; (iiib) N-methylhomopiperazine (5 equiv), no solvent, microwave heating, 120°C, 3 h, 55%; (iiic) N-methylpiperazine (10 equiv), N-methylpiperazine hydrochloride (10 equiv), DMSO, 110–120°C, 20 h, 56%.
References
- Burton, Michael E.; Shaw, Leslie M.; Schentag, Jerome J.; Evans, William E. (May 1, 2005). Applied Pharmacokinetics & Pharmacodynamics: Principles of Therapeutic Drug Monitoring (4th ed.). Lippincott Williams & Wilkins. p. 815. ISBN 978-0-7817-4431-7.
- “PRODUCT INFORMATION OLANZAPINE SANDOZ® 2.5mg/5mg/7.5mg/10mg/15mg/20mg FILM-COATED TABLETS” (PDF). TGA eBusiness Services. Sandoz Pty Ltd. 8 June 2012. Retrieved 26 November 2013.
- “Zyprexa, Zyprexa Relprevv (olanzapine) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 26 November 2013.
- ^ Jump up to:a b c “Olanzapine Prescribing Information” (PDF). Eli Lilly and Company. 2009-03-19. Retrieved 2009-09-06.
- “Lilly 2008 Annual Report” (PDF). Lilly. 2009. Retrieved 2009-08-06.
- National Collaborating Centre for Mental Health (25 March 2009). “Schizophrenia: Full national clinical guideline on core interventions in primary and secondary care”. Retrieved 25 November 2009.
- Duggan L, Fenton M, Dardennes RM, El-Dosoky A, Indran S (2005). Duggan, Lorna, ed. “Olanzapine for schizophrenia”. Cochrane Database of Systematic Reviews (2): CD001359.doi:10.1002/14651858.CD001359.pub2. PMID 10796640.
- “Psychosis and schizophrenia in adults: treatment and management | Guidance and guidelines | NICE”. National Institute for Health and Care Excellence.
- Barnes TR (2011). “Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology”. J. Psychopharmacol. (Oxford) 25 (5): 567–620. doi:10.1177/0269881110391123.PMID 21292923.
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Thibaut F, Möller HJ (2013). “World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects”. World J. Biol. Psychiatry 14 (1): 2–44. doi:10.3109/15622975.2012.739708. PMID 23216388.
- Abou-Setta, AM; Mousavi, SS; Spooner, C; Schouten, JR; Pasichnyk, D; Armijo-Olivo, S; Beaith, A; Seida, JC; Dursun, S; Newton, AS; Hartling, L (August 2012).PMID 23035275. Missing or empty
|title=(help) - Zhang JP, Gallego JA, Robinson DG, Malhotra AK, Kane JM, Correll CU (July 2013).“Efficacy and safety of individual second-generation vs. first-generation antipsychotics in first-episode psychosis: a systematic review and meta-analysis”. Int. J. Neuropsychopharmacol. 16 (6): 1205–18. doi:10.1017/S1461145712001277.PMC 3594563. PMID 23199972.
- Citrome L (August 2012). “A systematic review of meta-analyses of the efficacy of oral atypical antipsychotics for the treatment of adult patients with schizophrenia”. Expert Opin Pharmacother 13 (11): 1545–73. doi:10.1517/14656566.2011.626769.PMID 21999805.
- Lepping P, Sambhi RS, Whittington R, Lane S, Poole R (May 2011). “Clinical relevance of findings in trials of antipsychotics: systematic review”. Br J Psychiatry 198 (5): 341–5.doi:10.1192/bjp.bp.109.075366. PMID 21525517.
- Désaméricq G, Schurhoff F, Meary A et al. (February 2014). “Long-term neurocognitive effects of antipsychotics in schizophrenia: a network meta-analysis”. Eur. J. Clin. Pharmacol. 70 (2): 127–34. doi:10.1007/s00228-013-1600-y. PMID 24145817.
- Leucht S, Cipriani A, Spineli L et al. (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”.Lancet 382 (9896): 951–62. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019.
- Osser DN, Roudsari MJ, Manschreck T (2013). “The psychopharmacology algorithm project at the Harvard South Shore Program: an update on schizophrenia”. Harv Rev Psychiatry 21 (1): 18–40. doi:10.1097/HRP.0b013e31827fd915. PMID 23656760.
- [+http://www.nice.org.uk/guidance/cg185/chapter/1-recommendations “Bipolar disorder: the assessment and management of bipolar disorder in adults, children and young people in primary and secondary care | 1-recommendations | Guidance and guidelines | NICE”].
- Yatham LN, Kennedy SH, O’Donovan C et al. (December 2006). “Canadian Network for Mood and Anxiety Treatments (CANMAT) guidelines for the management of patients with bipolar disorder: update 2007”. Bipolar Disord 8 (6): 721–39. doi:10.1111/j.1399-5618.2006.00432.x. PMID 17156158.
- Selle V, Schalkwijk S, Vázquez GH, Baldessarini RJ (March 2014). “Treatments for acute bipolar depression: meta-analyses of placebo-controlled, monotherapy trials of anticonvulsants, lithium and antipsychotics”. Pharmacopsychiatry 47 (2): 43–52.doi:10.1055/s-0033-1363258. PMID 24549862.
- Maglione M, Maher AR, Hu J et al. (2011). “Off-Label Use of Atypical Antipsychotics: An Update”. PMID 22132426.
- Review of olanzapine in the management of bipolar disorders Neuropsychiatr Dis Treat. 2007 October; 3(5): 579–587.
- Scott, Lisa (Winter 2006). “Genetic and Neurological Factors in Stuttering”. Stuttering Foundation of America.
- “Important Safety Information for Olanzapine”. Zyprexa package insert. Eli Lilly & Company. 2007. Archived from the original on 2007-11-23. Retrieved 2007-12-03.
Elderly patients with dementia-related psychosis treated with atypical antipsychotic drugs are at an increased risk of death compared to placebo. […] ZYPREXA (olanzapine) is not approved for the treatment of elderly patients with dementia-related psychosis.
- “Doctors ‘ignoring drugs warning'”. BBC News. 17 June 2008. Retrieved 2008-06-22.
- L-Z\In re Zyprexa\Documents Leak\Injunction Memo & Order\FINAL INJUNCTION MEMO 2.13.07.wpd
- EFF.org
- Press Releases: January, 2007 | Electronic Frontier Foundation
- Eli Lilly was Concerned by Zyprexa Side-Effects from 1998,[dead link] The Times (London), January 23, 2007
- Navari RM, Einhorn LH, Loehrer PJ, Passik SD, Vinson J, McClean J, Chowhan N, Hanna NH, Johnson CS (2007). “A phase II trial of olanzapine, dexamethasone, and palonosetron for the prevention of chemotherapy-induced nausea and vomiting: A Hoosier oncology group study”. Supportive Care in Cancer 15 (11): 1285–91. doi:10.1007/s00520-007-0248-5. PMID 17375339.
- McGlashan TH, Zipursky RB, Perkins D, Addington J, Miller TJ, Woods SW, Hawkins KA, Hoffman R, Lindborg S, Tohen M, Breier A (2003). “The PRIME North America randomized double-blind clinical trial of olanzapine versus placebo in patients at risk of being prodromally symptomatic for psychosis”. Schizophrenia Research 61 (1): 7–18.doi:10.1016/S0920-9964(02)00439-5. PMID 12648731.
- McGlashan TH, Zipursky RB, Perkins D, Addington J, Miller T, Woods SW, Hawkins KA, Hoffman RE, Preda A, Epstein I, Addington D, Lindborg S, Trzaskoma Q, Tohen M, Breier A (2006). “Randomized, Double-Blind Trial of Olanzapine Versus Placebo in Patients Prodromally Symptomatic for Psychosis”. American Journal of Psychiatry 163 (5): 790–9.
Literature References:
Serotonin (5-HT2) and dopamine (D1/D2) receptor antagonist with anticholinergic activity. Prepn: J. K. Chakrabarti et al., EP 454436; eidem, US 5229382 (1991, 1993 both to Lilly). PRODUCT PATENT
Comparative pharmacology: N. A. Moore et al., Curr. Opin. Invest. Drugs 2, 281 (1993). HPLC determn in human plasma: J. T. Catlow et al., J. Chromatogr. B 668, 85 (1995).
Clinical evaluation in schizophrenia: D. S. Baldwin, S. A. Montgomery, Int. Clin. Psychopharmacol. 10, 239 (1995); in mania of bipolar disorder: M. Tohen et al., Am. J. Psychiatry 156, 702 (1999).
Review of pharmacology and clinical experience: B. C. Lund, P. J. Perry, Expert Opin. Pharmacother. 1, 305-323 (2000).
External links
- Zyprexa.com – official Zyprexa brand website from Eli Lilly and Company
- Zyprexa package insert
- Berenson, Alex (December 17, 2006). “Lilly Settles With 18,000 Over Zyprexa”. New York Times.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
TELMISARTAN PART 3/3

A direct and efficient total synthesis has been developed for telmisartan, a widely prescribed treatment for hypertension. This approach brings together two functionalized benzimidazoles using a high-yielding Suzuki reaction that can be catalyzed by either a homogeneous palladium source or graphene-supported palladium nanoparticles. The ability to perform the cross-coupling reaction was facilitated by the regio-controlled preparation of the 2-bromo-1-methylbenzimidazole precursor. This convergent approach provides telmisartan in an overall yield of 72% while circumventing many issues associated with previously reported processes.
……………………
PAPER
International Journal of Research in Pharmaceutical and Biomedical Sciences (2013), 4(1), 293-295
telmisartan1. [Yield 87%, Purity 99.97% by HPLC.M.P. 260 – 262°C, Sulphated ash < 0.01%].
1H NMR (DMSO-d6): δ 0.98-1.03 (t,3H), 1.73- 1.86 (m, 2H), 2.5 – 2.63 (s, 3H), 2.90-2.95 (s, 2H),3.82 (s, 3H), 5.62 (s, 2H), 7.16-7.34 (m,7H), 7.40-7.59 (m,4H), 7.68-7.70 (m, 3H), 12.86 (s, 1H).
M/Z: 515.50 [M + H]+
………………………..
PATENT
WO 2014027280
http://www.google.com/patents/WO2014027280A1?cl=en
Scheme 1 given below:
Formula .
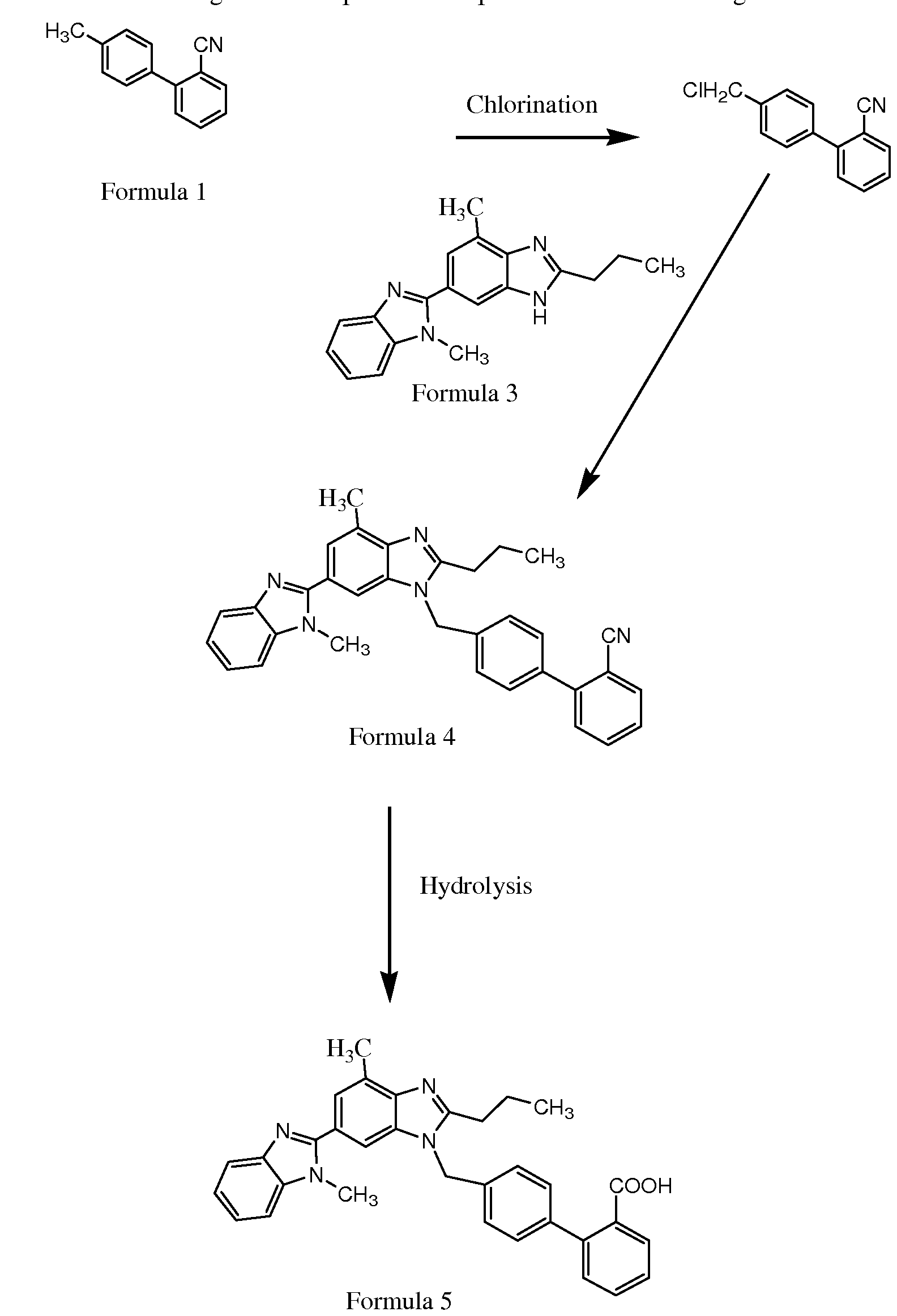
Example 1:
4′-[2-n^ropyl-4-methyl-6-(l-methylbenzimidazol-2-yl)benzimidazol-l-ylmeth^
carboxylic acid
In a 2 litre reaction flask was added 400 ml methylene chloride, followed by 100 gm of 2- cyano-4′ -methyl biphenyl. The reaction mass was stirred to get a clear solution and cooled to 20 °C. Chlorine gas was sparged into the reaction mass for a period of 15 hours till completion of the reaction. The reaction was monitored by TLC using mobile phase n-hexane: ethyl acetate (8:2). The excess chlorine from the reaction mass was removed by flushing with nitrogen. The solvent was distilled out completely by distillation at atmospheric pressure and removal of the final traces under vacuum. To the residual mass, 500 ml of methyl isobutyl ketone was added. The reaction mass was stirred and washed with a solution of 300 ml of 5% sodium bicarbonate solution. The lower aqueous layer was separated and the upper organic layer was washed with 300 ml water. The lower aqueous layer was separated. To the organic layer containing 4-chloromethyl-2′-cyanobiphenyl, the compound 2-n-propyl-4-methyl-6-(l’- methylbenzimidazol-2′-yl)benzimidazole was added, followed by a solution of 40 gm sodium hydroxide in 300 ml water. The reaction mass was stirred for 10 minutes and 10 gm of tetrabutyl ammonium hydrogen sulphate was added. The reaction mass was heated to 80 UC and maintained at 80 to 85 °C for 4 hours.
The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1). After completion of reaction, the lower aqueous layer was separated. The solvent was distilled out till mass temperature 120 °C and final traces were removed completely under vacuum. To the residual mass, 50 ml of n-butanol was added and the solvent distilled out under vacuum below 100 °C to remove all traces of methyl isobutyl ketone. The residue was dissolved in 750 ml of n-butanol and 83 gm sodium hydroxide added. The reaction mass was heated to reflux and maintained for 24 hours at 123 to 126 °C. The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1). The solvent was distilled out at atmospheric pressure till the mass temperature reached 140 C. The residual mass was cooled to 100 °C and 300 ml water was added. The solvent was distilled out azeotropically till the mass temperature reached 120 °C. To the reaction mass 750 ml of water was added, the solution warmed to 80 °C. The pH of the reaction mass was adjusted to 8.0 with hydrochloric acid. Finally the pH was adjusted to 6.0 with acetic acid, and the reaction mass maintained at 80 to 85 °C for one hour. The product obtained was filtered, washed with water and dried to yield 120 gm of 4′-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid, which can be purified as per the procedure described mentioned in Example 5. Example 2:
4-chloromethyl-2 ‘-cyanobiphenyl
In a 1 litre reaction flask 400 ml of methylene chloride was added followed by 100 gm of 2- cyano-4′ -methyl biphenyl. The reaction mass was stirred to get a clear solution and cooled to 20 °C. Chlorine gas was sparged into the reaction mass for a period of 15 hours at 20 to 25 °C till completion of the reaction. The reaction was monitored by TLC using mobile phase n- hexane: ethyl acetate (8:2). The excess chlorine from the reaction mass was removed by flushing with nitrogen. The solvent was distilled out completely by distillation at atmospheric pressure and removal of the final traces under vacuum. To the residual mass, 400 ml of n- heptane was added. The reaction mass was stirred and warmed to 60 °C. The clear solution obtained was cooled to 10 °C and the product precipitated was filtered, washed with n-heptane and dried. Further crystallization with n-heptane yielded 80 gm of pure 4-chloromethyl-2’- cyanobiphenyl.
C 73.87%, H 4.41%, N 6.19%; m/z 192.25; 1H NMR DMSO d6400 Mhz : 5ppm 4.84 (s, 2H) 7.32 – 7.66 (aromatic 8H). Example 3:
2-cyano-4,-(2,,-n-propyl-4,,-methyl-6,,-{V”-methylbenzim
ylmethyl) biphenyl
In a 2 litre reaction flask 500 ml of methyl isobutyl ketone was added followed by 100 gm of 2-n-propyl-4-methyl-6-( -methylbenzimidazol-2′-yl)benzimidazole. The reaction mass was stirred and a solution of 40 gm sodium hydroxide in 300 ml water was added. To this solution, 10 gm tetra butyl ammonium hydrogen sulphate and 80 gm of 4-chloromethyl-2′- cyanobiphenyl was added. The reaction mass was warmed to 80 °C and maintained for 4 hours at 80 to 85 °C.
The completion of the reaction was monitored by TLC using mobile phase chloroform : methanol (9:1). After completion of the reaction, the mass was cooled to 20 °C, maintained 3 hours at 15 to 20 °C. The product which precipitated out was filtered, washed with methyl isobutyl ketone, followed by water to yield 126 gm of 2-cyano-4′-(2″-n-propyl-4″-methyl- 6″-(r”-methylbenzimidazol-2″‘-yl)benzimidazol-l”- ylmethyl) biphenyl, melting at 196 – 198 °C. C 80.53%, H 5.70%, N 14.20%; m/z = 496.64 lH NMR DMSO d6 400 Mhz : 5ppm 0.96 – 0.99 (t, 3H) 1.75 – 1.84 (m, 2H) 2.62 (s, 3H) 2.89 – 2.93 (t, 2H) 3.80 (s, 3H) 5.67 (s, 2H) 7.18 – 7.92 (m, 14H)
Example 4:
4′-[2-n^ropyl-4-methyl-6-(l-methylbenzi idazol-2-yl)benzi idazol-ylmethyl]bipheny carboxylic acid
126 gm of 2-cyano-4′-(2″-n-propyl-4″-methyl-6″-(l “‘-methylbenzimidazol-2″‘-yl) benzimidazol-1”- ylmethyl) biphenyl was dissolved in 750 ml of n-butanol and 83 gm sodium hydroxide added. The reaction mass was heated to reflux and maintained for 15 hours at 123 to 126 °C. The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1).
The solvent was distilled out at atmospheric pressure till the mass temperature reached 140 °C. The residual mass was cooled to 100 °C and 300 ml water was added. The solvent was distilled out azeotropically till the mass temperature reached 120 °C. To the reaction mass 750 ml of water was added, the solution warmed to 80 °C. The pH of the reaction mass was adjusted to 8.0 with hydrochloric acid. Finally the pH was adjusted to 6.0 with acetic acid, and the reaction mass maintained at 80 to 85 °C for one hour. The product obtained was filtered, washed with water and dried to yield 120 gm of 4’-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid. Example 5:
Purification of 4′-[2-n^ropyl-4-methyl-6-(l-methylbenzimidazol-2-yl)benzimidazol-l- ylmethyl]biphenyl-2-carboxytic acid
In a 3 litre reaction flask, 1000 ml of methanol was added followed by the addition of 120 gm of 4′-[2-n-propyl-4-methyl-6-(l-methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl- 2-carboxylic acid obtained by procedure described in Example 4. The solution was warmed to 50 °C and pH adjusted to 10.0 to 10.5 with 100 ml of a 10% methanolic potassium hydroxide solution. The reaction mass became a clear solution, and 6 gm activated carbon was added. The mass was maintained at 50 to 55 °C for one hour and filtered through hyflo supercel to remove the activated carbon. The clear filtrate obtained was collected and its pH adjusted to 6.0 to 6.5 with 130 ml of acetic acid, maintaining the temperature between 50 to 55 °C. The mass was cooled to 15 °C and maintained one hour at 10 to 15 °C. The product which precipitated out was filtered, washed with 50 ml of methanol followed by 500 ml of water. The wet product was dried to yield 107 gm of 4′-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid.
C 76.49%; H 5.74%, N 11.02%; m/z 515.45.; 1H NMR DMSO d6 400 Mhz : 5ppm 0.97 – 1.01 (t, 3H) 1.76 – 1.85 (m, 2H) 2.62 (s, 3H) 2.90 – 2.94 (t, 3H) 3.81 (s, 3H) 5.61 (s, 2H) 7.15 – 7.71 (14H aromatic);
Melting point of purified telmisartan: 269 °C.
…………………
PAPER
Journal of Organic Chemistry (2014), 79(21), 10568-10580
http://pubs.acs.org/doi/abs/10.1021/jo501665e

On the basis of our recently reported aniline aqueous borylation, molecular diversity was achieved in a one-pot process by combining other reactions such as esterification, Suzuki–Miyaura coupling, hydrogenolysis, or Petasis borono-Mannich.
TELMISARTAN IS COMPD 9
……………….
PATENT
US 20150031768
(EN)
Methods of halogenating a carbon containing compound having an sp3 C—H bond are provided. Methods of fluorinating a carbon containing compound comprising halogenation with Cl or Br followed by nucleophilic substitution with F are provided. Methods of direct oxidative C—H fluorination of a carbon containing compound having an sp3 C—H bond are provided. The halogenated products of the methods are provided.
…………………..
PATENT
WO 2014067237
http://www.google.com/patents/WO2014067237A1?cl=en
Telmisartan Preparation: 12 Examples
The title compound (III, R = COOCH 3) (52.8g, O. lmol) of Example 11 with glacial acetic acid
(200ml) and concentrated hydrochloric acid (250ml) mixing, 100 ° C to react for 5 to 6 hours. Evaporated to most mixed acid, residue slowly poured into crushed ice, under ice cooling with saturated K 2 CO ^ solution to adjust the pH to neutral, solid precipitation, filtration, filtrate was washed with water, was for Mischa Tan crude, recrystallization telmisartan (40.1g), liquid purity greater than 99%.
Example 13: Preparation of telmisartan of formula I compound (0.62g, leq) was added to acetonitrile (10ml). After stirring evenly, the KOH (0.14g, 1. leq) was slowly added, after stirring for 10 plus minutes, the title compound of Example 10 of the embodiment (11, R = COOCH 3) (0.5g, leq) was slowly added, stirred for 3-4 hours, TLC the reaction was complete, the direct addition of 50% ethanol (30mL), reflux The reaction for 6 hours. After completion of the reaction by TLC, recovering the organic solvent under reduced pressure, the remaining solution was added dropwise hydrochloric acid (1: 1) to neutral pH. The precipitated solid was filtered, washed with water to give crude telmisartan, telmisartan recrystallized (yield 75.1%), the liquid phase is greater than 98% purity.
Chloromethyl biphenyl -2- (II, R = CN) Preparation of 4′-nitrile:
…………………
Journal of Pharmaceutical and Biomedical Analysis (2015), 108, 86-96.
………………..
IN 262831/EP 1912975
……………………….
JP 2014201585
……………………..
IN 2013KO00463/WO 2014174397
…………………………….
PATENT
http://www.google.com/patents/CN1768044A?cl=en
Example 7: Telmisartan make 5.51 g telmisartan × HCl was dissolved in 50 ml of 40% acetic acid while refluxing. The brown solution was then filtered hot through 1.1 g of carbon, 2.5 ml of 40% acetic acid and washed, and at 80-90 ℃ 2.5 ml of 4N NaOH was added dropwise with stirring to light brown filtrate. Telmisartan crystallization, the suspension was diluted with 30 ml of water, and slowly cooled to ambient temperature. Telmisartan suction filtration, and washed with 50 ml of water. And dried in vacuo at 80 ℃ drying cabinet telmisartan.
Yield: 4.80 g (93.3% of the theoretical yield).
……………………………….
PATENT
http://www.google.com/patents/CN102731407A?cl=en


Example 4 Preparation of telmisartan
[0031] 2-n-propyl group as shown in Formula I-4-methyl-6- (benzimidazol-2-yl-methyl 1’_) benzimidazole (30. 4g, O. 10mol), 4_ bromomethyl-biphenyl-2-carboxylic acid (43. 6g, O. 15mol), three ko amine (12. Ig, O. 15mol) and ko ni ni ether 500ml alcohol were mixed and reacted at 100 ° C for 6 inches The reaction solution was poured into ice water, acidified with dilute hydrochloric acid and slowly adjusted PH2-3, to precipitate a solid. Filtration, 70 ° C drying crude, the resulting crude product ko ko acid ester 300ml heating beating again. Filtered, 70 ° C dry. Recrystallization from DMF telmisartan of formula III as shown in 25. Ig, yield: 50%.
…………………….
PATENT
http://www.google.com/patents/WO2010146187A2?cl=en
For example, WO 2004/087676 describes the hydrolysis of a compound with the chemical name 4 ‘-((1,7’- dimethyl-2 ‘ -propyl-lH, 3 ‘H-2, 5 ‘ -bibenzo [d] imidazol-3 ‘ -yl) – methyl) biphenyl-2-carbonitrile and having formula 2

which is hereinafter referred to as cyanotelmisartan . In par- ticular, the hydrolysis of cyanotelmisartan is carried out at elevated temperatures using strong alkaline conditions. Also, CN 1412183 discloses the hydrolysis of cyanotelmisartan.
US 2006/0264491 Al discloses the hydrolysis of 4′-((l,7′- dimethyl-2 ‘ -propyl-lH, 3 ‘H-2, 5 ‘ -bibenzo [d] imidazol-3 ‘ – yl) methyl) biphenyl-2-carboxamide having formula 3

Example 2: Preparation and isolation of telmisartan
Into a reaction vessel 20.5g (40 mmol) 4 ‘ – ( (1, 7 ‘ -dimethyl-2 ‘ – propyl-IH, 3 ‘ H-2 , 5 ‘ -bibenzo [d] imidazol-3 ‘ -yl) methyl) biphenyl-2- carboxamide and 20 ml (lδOmmol) H2SO4 (1:1) were added. The re- action mixture was heated to about 125°C and stirred at this temperature for 28 h. A sample of the reaction mixture was analyzed by Area% HPLC (starting compound below 0.1%, telmis- artan over 97%) . The reaction mixture was cooled below 800C and 250 ml of water were added. Then, 200 ml of dichloro- methane were added and pH value of mixture was adjusted to 5.4 by addition of 6M NaOH. The mixture was stirred for approximately 5 min and then the phases were separated. The water phase was reextracted by 136 ml of dichloromethane . Collected organic phases were washed with water (2χl36ml) and then treated with activated charcoal (5.3 g) . Subsequently, the organic phase was evaporated an oily residue (26g) . 264 ml of acetone were added. The mixture was stirred at room temperature for at least 6 hours. The precipitated product was sepa- rated and washed with fresh acetone and dried at 65°C under reduced pressure for 3 hours. Yield: 18.3g (89%) Area % HPLC: Telmisartan 99.80%
Example 3: Isolation of telmisartan
Into a reaction vessel 7.5g (15 mmol) of cyanotelmisartan, 30 ml of propylene glycol, 0.8 ml of water and 3g (45 mmol) of 85% KOH were added. The reaction mixture was heated to around 1600C to 170 0C and stirred at this temperature for 24 h. The reaction mixture was cooled below 800C and 75 ml of water were added. Then, pH value of the mixture was adjusted to 4.8 (by addition of 6M HCl) and then 150 ml of dichloromethane were added. The mixture was stirred for approximately 5 min and then the phases were separated. The water phase is reextracted by 50 ml of dichloromethane. Collected organic phases were washed with water (2χ50ml) and then treated with activated charcoal (2 g) . After that the organic phase was evaporated to an oily residue (9.8g) . 100 ml of acetone were added. The mix- ture was stirred at room temperature for at least 6 hours. The precipitated product was separated and washed with fresh acetone and dried at 65°C under reduced pressure for 3 hours. Yield: 6.8g (88%) Area % HPLC: Telmisartan 99.60%
…………………….
PATENT
http://www.google.com/patents/CN1548421A?cl=en
Specific embodiments
14 ‘Example – [(1,4′-dimethyl-2′-propyl [2,6′- two-1H – benzoimidazol] 1′-yl) methyl] – [1, 1’-biphenyl] -2-carboxylic acid sodium salt in 250ml reaction flask, telmisartan 10g (0.0195mol), NaOH0.75g (0.0189mol) and water 100ml, stirred for 1 hour (30 ℃), filtered insoluble materials are removed and concentrated to a small volume, plus ethanol 30ml, concentrated, washed with 30ml of n-hexane, decanted, plus ethanol 30ml, concentrated, and then repeat again, and concentrated to dryness to obtain telmisartan sodium salt 9.9g yield 95.2%. Melting point: 223-225 ℃.
Elemental analysis: C33H29N4O2Na · H2O Calcd: C71.48 H5.10 N10.11 Found: C71.42 H5.08 N10.22 Example 24 ‘- [(1,4′-dimethyl-2′-n propyl [2,6′- two-1H – benzoimidazol] 1′-yl) methyl] – [1,1’-biphenyl] -2-carboxylic acid potassium salt in 250ml reaction flask, Telmisartan 10g (0.0195mol), KOH1.06g (0.0188mol) and water 100ml, stirred for 1 hour (30 ℃), filtered to remove insolubles, and concentrated to a small volume, ethanol 30ml, concentrated, hexane 30ml washed, decanted, plus ethanol 30ml, concentrated, and then repeat again, and concentrated to dryness to obtain telmisartan potassium 10.6g, yield 95.6%. Melting point: 203-205 ℃.
Elemental analysis: C33H29N4O2K · H2O Calcd: C69.04 H5.40 N9.76 Found: C69.01 H5.28 N9.88 Example 3 starting material and the mixed powder was sieved excipients, 5% polyethylene pyrrolidone was granulated and dried. After dried particles were sieved magnesium stearate was added mixed tabletted.
mg / tablet of telmisartan sodium salt 20 Lactose 170 Sodium carboxymethyl starch 10 mg Magnesium stearate 8 meglumine 25% polyvinyl pyrrolidone solution q.s. Example 4 A mixed powder of raw materials and auxiliary materials sieved, added 5 % solution of polyvinylpyrrolidone is granulated and dried. After dried particles were sieved magnesium stearate was added mixed tabletted.
mg / tablet telmisartan sodium Lactose 200 40 140 DCP sodium carboxymethyl starch 16 mg Magnesium stearate 45% povidone solution appropriate amount of Example 5 of this product, according to the dissolution assay (Chinese Pharmacopoeia 2000 edition Appendix II XC second method), phosphate buffer 900ml solvent, the speed of 75 revolutions per minute, operate according to the law, after 30 minutes, take the solution as spectrophotometry (Chinese Pharmacopoeia 2000 edition of the test solution, according to the spectrophotometric two Appendix IVA), absorbance was measured at 295nm wavelength. Another reference standard stock solution 10ml precise amount of determination under set 100ml flask, diluted with phosphate buffer to the mark, then the precise amount of 5ml, set 10ml volumetric flask, dilute to the mark with phosphate buffer , shake, the same method absorption, calculated for each piece of the dissolution of the limits of 80% scalar, should be specified. Dissolution test results in Table.
Table dissolution test results Dissolution (%) telmisartan sodium 97.29 99.65 102.55 95.83 101.10 98.92 99.20 ± 2.45
…………………..
PATENT
http://www.google.com/patents/CN1412183A?cl=en
Example 5 4 ‘- [(1,4′-dimethyl-2′-propyl [2,6′- two -1H- benzimidazol] -1′-yl) methyl] – [1,1’ – biphenyl] -2-carboxylic acid (III) IV (24.8g, 0.05mol) was added ethylene glycol (100ml) and water (150ml) (or other previously described a mixed solvent), sodium ethoxide (or as previously said other alcohols sodium) (13.6g, 0.2mol), was refluxed for 10 hours. After no starting material by TLC was cooled to room temperature, hydrochloric acid was added dropwise (1/1) to pH 5-6, the precipitated solid was filtered, washed with water to give III.
……………………
PATENT
http://www.google.com/patents/CN101550107B?cl=en
Example 3
[0047] 1) Preparation of telmisartan crude methyl ester
Compound II into 50g in 500mL reaction flask, 200mL of methyl isobutyl ketone (MIBK), 25 ° C _30 ° C with stirring until dissolved, was added dropwise 35mL of triethylamine was added 55. Og After the completion of the compound III, 5 (T60 ° C or so for about 4_5 hours, TLC monitoring completion of the reaction, filtered and the filter cake washed with a small amount of MIBK, and then washed with water, dried to give 70. 3g of crude product. 81% yield, purity of about 98%.
(TLC test conditions: ethyl acetate: methanol = 8: 1)
2) preparation of high purity methyl telmisartan
IOOOmL reaction flask, the input step to give the crude methyl ester telmisartan, add 500mL of isopropanol was heated to dissolve, 2gX 2 activated bleaching filtrate was heated to about 90 ° C, added dropwise with stirring 150mL 7jC insulation 0. 5~Ih, cooled slowly to room temperature with stirring. Filtered, and the filter cake washed sequentially with MIBK and water washing, and drying, the yield of about 82%, HPLC purity 99.5%, the single impurities less than 0.1%.
3) Preparation of telmisartan with high purity
[0053] A reaction flask was put in a 500mL high purity 15g telmisartan ester, 3. Og sodium, 200mL of isopropanol, water, 80ml, was heated to reflux for 5 ~ 7 h, TLC monitoring of the reaction was complete, the distillation Isopropanol was removed, and water was added to completely dissolve the solid 40ml, 0. 5g of activated carbon bleaching, the filtrate was added 50ml of water, heated to 80 ° C, lmol / L of acetic acid to adjust the pH to 5. (Γ5. 5, filtered, and the filter cake dried to give 13. 14g of solid, yield 90%, HPLC purity 99.7%, the single impurities less than 0.1%.
(TLC test conditions: ethyl acetate: methanol = 8: 1)
………………………..
http://www.google.com/patents/CN101172968B?cl=en
Example 1
[0023] 1, 100gPPA, 21. 8g (0. Lmol) 2_ n-propyl _4_ _6_ carboxyl methyl benzimidazole and 21. 5gN- methyl-o-phenylenediamine added to the reaction flask in under N2 protection feeding, heated to IO (TC _1601 :, reaction 8-20 hours, down 70-80.C 200ml water was added and the reaction with hydrochloric acid to adjust ffl = 1~2, put charcoal 5_8%,, 8 (TC about 5 to 10 minutes, filtered, and the reaction repeated, the adjustment ra 12-14 with NaOH, for several hours, and filtered to give the crude intermediate 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl ) benzimidazole sodium salt. [0024] 2, the product of the previous step, 2-n-propyl -4-methyl–6_ (methyl benzimidazol-_2_ yl) benzimidazole sodium salt crude product was dissolved into 200 ml of ethanol , and dissolved by heating, cooling to room temperature, 400 ml 1N NaOH, to precipitate the compound 2-n-propyl -4-methyl-6- (methyl benzimidazol-2-yl) benzimidazole .50-8 ( TC dried in vacuo. [0025] 3, product of the previous step -4-methyl-2-n-propyl -6_ (methyl benzimidazol-_2_ yl) benzimidazole into 200 ml of dimethyl sulfoxide was stirred was added at room temperature and 4-bromomethyl – biphenyl-2-carboxylic acid methyl ester 33.55 g, was stirred for 14 hours, extracted with dichloromethane (200, 100, 100), and evaporated to dryness under reduced pressure, 300 ml of methanol and 10% potassium hydroxide (240 ml, 0. 6mo1) mixture was refluxed for 6 hours, cooled, washed with 80 ml of methylene chloride, adjusted with glacial acetic acid ffl = 6, a lot of white floc precipitated precipitate was filtered and dried to give a white Tilmicosin 49.6 g of crude product, the crude product was added 100 ml of chloroform was heated to reflux, activated carbon decolorization, crystallization, filtration, 8 (TC dried in vacuo to give a white pure telmisartan (HLPC> 99. 0%) 41 克, purification yield 82%. mp 261~263.C, H-NMR (d6-DMS0) S 1. 05t, 3H), 1. 83 (m, 2H), 2. 71 (s, 3H), 2. 94 (t, 2H), 3. 81 (s, 3H), 5. 57 (s, 2H), 7. 16-7. 83 (m, 14H) • C33H33N402 [0026] Example 2 Preparation of telmisartan
1, 100gPPA, 21. 8g (0. 1) 2_ [4-methyl-n-propyl-benzimidazole and _6_ 21. 5gN- carboxy-o-phenylenediamine added to the reaction flask in N2 Under the protection of feeding, heated to 100 ° C _160 ° C, the reaction for 8-20 hours, down 70-80. C, the reaction was added 200ml of water, adjusted with hydrochloric acid ffl = 1~2, into charcoal 5_8%, about 8 (TC 5_10 minutes filtered again reacted with K0H ra adjusted to 12-14 for several hours and filtered to give Intermediate crude 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt.
2, the product of the previous step, 2-n-propyl -4-methyl–6_ (methyl benzimidazol-_2_ yl) benzimidazole potassium salt of the crude product into 200 ml of ethanol, and dissolved by heating, cooling to room temperature was added 400 ml 1N K0H, a precipitated compound is 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt. 50-8 (TC dried in vacuo. [0029] 3, 2-n-propyl prepared in the previous step -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt and 27.2 g of 4-bromomethyl-2-cyanobiphenyl, 10.4 g of triethylamine and DMF (DMA, dichloromethane, dichloroethane) were mixed and reacted for 5-10 hours at 35-40 °, TLC detection After no starting material the reaction mixture was poured into 600 g of ice water, extracted with ethyl acetate (300ml * 3), the combined organic phases were washed with water (300ml * 2), dried and desolvation, and then petroleum ether was added and stirred until a solid precipitated was The crude product was 45.6 g.
4, the upper step of the solid 45.6 grams, was added 200ml of ethylene glycol, 150ml water, 12 g of sodium hydroxide, the reaction was refluxed for 10 hours, TLC detected no starting material and then cooled to room temperature, acidified with hydrochloric ra is 5 to 6, there is solid precipitation, filtration, washing, telmisartan was crude, DMF and recrystallized to give 44.5 g of telmisartan pure product (HLPC> 99. 0%) mp261~263 ° C. Force -NMR (de-DMS0) S 1. 05t, 3H), 1. 83 (m, 2H), 2. 71 (s, 3H), 2. 94 (t, 2H), 3. 81 (s, 3H ), 5. 57 (s, 2H), 7. 16-7. 83 (m, 14H) • C33H33N402 [0031]
……………………….
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN100460396C | Mar 8, 2007 | Feb 11, 2009 | 杭州盛美医药科技开发有限公司 | Intermediate of telmisartan, preparation and use thereof |
| CN100506799C | Apr 19, 2007 | Jul 1, 2009 | 北京理工大学 | [(2-n-propyl-4-methyl-1H-Benzimidazole)6-radical] carboxamide-1-radical] methylbiphenyl compound, synthesizing method and usage |
| CN100548293C | Aug 8, 2003 | Oct 14, 2009 | 贝塞斯达药物股份有限公司 | Novel PPAR ligands that do not cause fluid retention, edema or congestive heart failure |
| CN101550107B | Apr 2, 2009 | Jan 12, 2011 | 宁波九胜创新医药科技有限公司 | Method for preparing telmisartan |
| CN101743228B | Jul 3, 2008 | Jan 29, 2014 | 新梅斯托克尔卡托瓦纳兹德拉韦尔公司 | Process for preparing telmisartan |
| CN101891735B | Nov 25, 2009 | Jul 18, 2012 | 北京理工大学 | Biphenyl sulfafurazole compound, synthesis method and application thereof |
| CN102093297B | Jan 28, 2011 | Aug 1, 2012 | 海南美兰史克制药有限公司 | Telmisartan compound and new preparation method thereof |
| EP1805146A2 * | Oct 18, 2005 | Jul 11, 2007 | Dr. Reddy’s Laboratories Ltd. | Process for preparing telmisartan |
| EP2123648A1 | May 20, 2008 | Nov 25, 2009 | Chemo Ibérica, S.A. | A process for the preparation of Telmisartan. |
| US7501448 | Oct 13, 2005 | Mar 10, 2009 | Teva Pharmaceutical Industries, Ltd. | high yields, low cost process; easy solvent extraction; nontoxic, safe, environmentally friendly, low boiling point organic solvents; 1,7′-dimethyl-2′-propyl-1H,3’H-[2,5′]bibenzoimidazolyl is reacted with 4’bromomethyl-biphenyl-2-carboxylic acid alkyl ester; industrial scale; hydrolysis |
| US8691999 * | May 10, 2005 | Apr 8, 2014 | Cipla Limited | Process for the preparation of telmisartan |
| WO2006044648A1 * | Oct 13, 2005 | Apr 27, 2006 | Teva Pharma | Process for preparing telmisartan |
| WO2007010558A1 * | Jul 19, 2006 | Jan 25, 2007 | Satyanarayana Chava | A process for the preparation of telmisartan |
| WO2009123483A1 | Mar 30, 2009 | Oct 8, 2009 | Zaklady Farmaceutyczne Polpharma Sa | Process for preparation of telmisartan |
| WO2012028925A2 | Aug 29, 2011 | Mar 8, 2012 | Ogene Systems (I) Pvt Ltd | An improved process for the preparation of telmisartan |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan |
| CN1620437A | Jan 15, 2003 | May 25, 2005 | 贝林格尔英格海姆法玛两合公司 | Method for the production and purification of 1, 7′-dimethyl-2′-propyl-2, 5′-bi-1h-benzimidazole |
| CN1768044A | Mar 26, 2004 | May 3, 2006 | 贝林格尔·英格海姆国际有限公司 | Process for manufacture of telmisartan |
| WO2005108375A1 | May 10, 2005 | Nov 17, 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| CN1344712A | Jul 30, 2001 | Apr 17, 2002 | 中国科学院上海药物研究所 | Synthesis path of Timisatem | |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan | |
| US2006/0094883 | Title not available | ||||
| WO03/059890A1 | Title not available | ||||
| WO2005/108375A1 | Title not available |
| US7501448 | Oct 13, 2005 | Mar 10, 2009 | Teva Pharmaceutical Industries, Ltd. | high yields, low cost process; easy solvent extraction; nontoxic, safe, environmentally friendly, low boiling point organic solvents; 1,7′-dimethyl-2′-propyl-1H,3’H-[2,5′]bibenzoimidazolyl is reacted with 4’bromomethyl-biphenyl-2-carboxylic acid alkyl ester; industrial scale; hydrolysis |
| WO2007147889A2 * | Jun 22, 2007 | Dec 27, 2007 | Krka Tovarna Zdravil D D Novo | Preparation of telmisartan salts |
| WO2010146187A2 | Jun 21, 2010 | Dec 23, 2010 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Process for the preparation of telmisartan |
| WO2012055941A1 | Oct 26, 2011 | May 3, 2012 | Krka,Tovarna Zdravil, D. D., Novo Mesto | Multilayer pharmaceutical composition comprising telmisartan and amlodipine |
| WO2003007876A2 | Jun 25, 2002 | Jan 30, 2003 | Sumner H Burstein | N-fatty acid-amino acid conjugates and therapeutic uses |
| WO2003059327A1 | Jan 16, 2002 | Jul 24, 2003 | Boehringer Ingelheim Pharma | Bilayer pharmaceutical tablet comprising telmisartan and a diuretic and preparation thereof |
| WO2004028505A1 | Sep 18, 2003 | Apr 8, 2004 | Boehringer Ingelheim Int | Solid pharmaceutical formulations comprising telmisartan |
| WO2004087676A1 | Mar 26, 2004 | Oct 14, 2004 | Boehringer Ingelheim Int | Method for the production of telmisartan |
| WO2004096215A1 | Apr 27, 2004 | Nov 11, 2004 | Boehringer Ingelheim Int | Pharmaceutical formulation of the sodium salt of telmisartan |
| WO2005108375A1 * | May 10, 2005 | Nov 17, 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| WO2006044754A2 | Oct 18, 2005 | Apr 27, 2006 | Muthulingam Arunagiri | Process for preparing telmisartan |
| WO2006050509A2 | Nov 3, 2005 | May 11, 2006 | Teva Pharma | Amorphous and polymorphic forms of telmisartan sodium |
| WO2006050921A2 | Nov 9, 2005 | May 18, 2006 | Lek Pharmaceuticals | Preparation of telmisartan salts with improved solubility |
| WO2006063737A1 | Dec 9, 2005 | Jun 22, 2006 | Boehringer Ingelheim Int | Combination therapy comprising telmisartan and hydrochlorothiazide |
| WO2006136916A2 | Jun 20, 2006 | Dec 28, 2006 | Glenmark Pharmaceuticals Ltd | Substantially pure micronized particles of telmisartan and pharmaceutical compositions containing same |
| WO2007010559A2 | Jul 19, 2006 | Jan 25, 2007 | Panacea Biotec Ltd | Novel pharmaceutical modified release dosage form cyclooxygenase enzyme inhibitor |
| WO2007060170A2 | Nov 22, 2006 | May 31, 2007 | Boehringer Ingelheim Int | Bilayer tablet comprising telmisartan and diuretic |
| WO2007144175A2 | Jun 14, 2007 | Dec 21, 2007 | Lek Pharmaceuticals | Pharmaceutical composition comprising hydrochlorothiazide and telmisartan |
| WO2007147889A2 | Jun 22, 2007 | Dec 27, 2007 | Krka Tovarna Zdravil D D Novo | Preparation of telmisartan salts |
| WO2009004064A1 | Jul 3, 2008 | Jan 8, 2009 | Krka Tovarna Zdravil D D Novo | Process for preparing telmisartan |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan |
| CN1548421A | May 22, 2003 | Nov 24, 2004 | 上海医药工业研究院 | Tilmisartan salt and its prepn |
| EP0502314A1 | Jan 31, 1992 | Sep 9, 1992 | Dr. Karl Thomae GmbH | Benzimidazol, medicaments containing them and process for their preparation |
| EP1144386A1 | Jan 7, 2000 | Oct 17, 2001 | Boehringer Ingelheim Pharma KG | Telmisartan polymorphs, methods for producing same and their use in the preparation of a medicament |
| EP1719766A2 | Apr 18, 2006 | Nov 8, 2006 | Dipharma S.p.A. | A process for the preparation of telmisartan |
| US20060264491 | Jun 8, 2006 | Nov 23, 2006 | Chemagis Ltd. | Telmisartan production process |
| CN101172968B | Nov 1, 2006 | May 12, 2010 | 浙江天宇药业有限公司 | 2-propyl-4 methyl-6-(tolimidazole-2group) benzoglioxaline salt and method for producing the same |
| CN101743228B | Jul 3, 2008 | Jan 29, 2014 | 新梅斯托克尔卡托瓦纳兹德拉韦尔公司 | Process for preparing telmisartan |
| CN102015690B | Mar 19, 2009 | Apr 30, 2014 | 力奇制药公司 | Catalyzed carbonylation in the synthesis of angiotensin II antagonists |
| CN102036937B | Mar 19, 2009 | Jun 4, 2014 | 力奇制药公司 | 2′-halobiphenyl-4-yl intermediates in the synthesis of angiotensin ii antagonists |
| WO2014067237A1 * | Oct 31, 2013 | May 8, 2014 | Topharman Shanghai Co., Ltd. | Telmisartan preparation method and intermediate thereof |
| WO2010018441A2 * | Aug 10, 2009 | Feb 18, 2010 | Cadila Pharmaceuticals Ltd. | An improved process for the preparation of substantially pure telmisartan |
| WO2010146187A2 * | Jun 21, 2010 | Dec 23, 2010 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Process for the preparation of telmisartan |
| WO2011077444A1 * | May 28, 2010 | Jun 30, 2011 | Inogent Laboratories Private Limited | A new process for the preparation of pure telmisartan |
| WO2012028925A2 * | Aug 29, 2011 | Mar 8, 2012 | Ogene Systems (I) Pvt Ltd | An improved process for the preparation of telmisartan |
| CN1768044A * | Mar 26, 2004 | May 3, 2006 | 贝林格尔·英格海姆国际有限公司 | Process for manufacture of telmisartan |
| CN102731407A * | Jul 4, 2012 | Oct 17, 2012 | 宁波九胜创新医药科技有限公司 | Method for preparing telmisartan |
| EP0627433A1 * | Dec 7, 1993 | Dec 7, 1994 | Eisai Co., Ltd. | Process for producing imidazopyridine derivative and intermediate |
| EP2123648A1 * | May 20, 2008 | Nov 25, 2009 | Chemo Ibérica, S.A. | A process for the preparation of Telmisartan. |
| EP2305650A1 * | Sep 21, 2009 | Apr 6, 2011 | Chemo Ibérica, S.A. | Novel process for the preparation of telmisartan |
| KR20090000113A * | Title not available | |||
| US20040236113 | Mar 17, 2004 | Nov 25, 2004 | Boehringer Ingelheim International Gmbh | Process for manufacture of telmisartan |
| US20130137878 | Jan 25, 2013 | May 30, 2013 | Boehringer Ingelheim International Gmbh | Process for manufacture of telmisartan |
……..
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
COCK WILL TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
amcrasto@gmail.com
Omeprazole spectral visit

Omeprazole
CAS NO. 119141-89-8
(RS)-5-methoxy-2-((4-methoxy-3,5-dimethylpyridin-2-yl) methylsulfinyl)-1H-benzo[d]imidazole
| Omeprazole | |
| CAS No.: | 119141-89-8 |
|---|---|
| Synonyms: |
|
| Formula: | C17H19N3O3S |
| Exact Mass: | 345.11500 |
Ome is a chemical substance (C17H19N3O3S), its molecular weight is 345.42g/mol, the color is white, has weak alkaline properties, melts at 156oC

NMR……………http://file.selleckchem.com/downloads/nmr/S138902-Omeprazole-Prilosec-HNMR-Selleck.pdf
NMR………..file:///C:/Users/anthonyc/Downloads/233-434-1-SM.pdf

1H NMR PREDICT

13C NMR PREDICT

COSY

HMBC


………………………………..
ARKIVOC 2006 (v) 5-11
The structure of Omeprazole in the solid state: a 13C and 15N NMR/CPMAS study
Rosa M. Claramunt,a Concepción López,a and José Elguerob *
a Departamento de Química Orgánica y Bio-Orgánica, Facultad de Ciencias, UNED, Senda del Rey 9, E-28040 Madrid, Spain
b Instituto de Química Médica, CSIC, Juan de la Cierva, 3. E-28006 Madrid, Spain
E-mail: iqmbe17@iqm.csic.es
http://www.arkat-usa.org/get-file/22955/
Abstract
The 13C and 15N CPMAS spectra of a solid sample of Omeprazole have been recorded and all the signals assigned. The sample consists uniquely of the 6-methoxy tautomer. For analytical purposes, the signals of the other tautomer, the 5-methoxy one, were estimated from the data in solution (Magn. Reson. Chem. 2004, 42, 712).
Keywords: Omeprazole, NMR, 13C, 15N, CPMAS, tautomerism, benzimidazole
Omeprazole, 5(6)-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl}-1H-benz – imidazole [1(2)], is an important ulcer drug,1 that has been classified amongst the blockbuster drugs.2 This compound presents two sources of structural differentiation. First, Omeprazole is chiral (a vs. b) 3 since it has a stereogenic center on the sulfur atom but the commercial form has been sold, until recently, as a racemate. In 2001, Esomeprazole magnesium, the S enantiomer was approved.4 The second source of diversity is that these compounds present tautomerism (1 vs. 2). We have already devoted a paper to the tautomerism of Omeprazole in solution using 1 H and 13C NMR spectroscopy.5 In this paper a complete assignment of the signals was carried out and the tautomeric equilibrium constant, KT = [2]/[1], was determined in THF at 195 K, to be 0.59 in favor of the 6-methoxy tautomer 2.
References
1. Carlsson, E.; Lindberg, P.; von Unge, S. Chem. Brit. 2002, 38, 42 and references therein.
2. Berkowitz, B. A.; Sachs, G. Mol. Interventions 2002, 2, 6.
3. von Unge, S.; Langer, V.; Sjölin, L. Tetrahedron: Asymmetry 1997, 8, 1967.
4. Olbe, L.; Carlsson, E.; Lindberg, P. Nature Reviews Drug Discovery 2003, 2, 132.
5. Claramunt, R. M.; López, C.; Alkorta, I.; Elguero, J.; Yang, R.; Schulman, S. Magn. Reson. Chem. 2004, 42, 712.
6. Elguero, J.; Katritzky, A. R.; Denisko, O. Adv. Heterocycl. Chem. 2000, 76. 1.
7. Allen, F. H. Acta Crystallogr. Sect. B 2002, 58, 380.
8. Braga, S. S.; Ribeiro-Claro, P.; Pillinger, M.; Gonçalves, I. S.; Fernandes, A. C.; Pereira, F.; Romåo, C. C.; Correia, P. B.; Teixeira-Dias, J. J. C. J. Incl. Phenom. Macro. Chem. 2003, 47, 47.
9. Berger, S.; Braun, S. 200 and More NMR Experiments. Wiley-VCH, Weinheim, 2004.
DSC OF OMEPRAZOLE
UV
UV Study: The Ultraviolet spectrum was recorded from 200 nm to 400 nm, with API concentration of 0.0015% in methanol. The spectrum showed two λmax at 207 and 301 nm. As seen below.
FTIR Study The FTIR of spectrum of Omeprazole was recorded by preparation of pellet with KBr.
NMR
13 C NMR
mass
The mass spectrum of Omeprazole was recorded on 4000-Q trap LCMSMS system. The sample is introduced into the system through HPLC by bypassing the column. The ESI +ve ionization spectrum of Omeprazole displayed a protonated molecular ion at m/z= 346 which corresponds to the molecular formula C17H17N3O3S. The fragmentation pattern was observed with product ion scan.
Raman
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
TELMISARTAN PART 2/3

4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan)
PART 1……..http://orgspectroscopyint.blogspot.in/2015/04/telmisartan-part-13.html
PART 2……..http://orgspectroscopyint.blogspot.in/2015/04/telmisartan-part-23.html
OR https://newdrugapprovals.org/2015/04/06/telmisartan-part-23/
PART3…… http://orgspectroscopyint.blogspot.in/2015/04/telmisartan-part-33.html
GENERAL DESCRIPTION
Telmisartan is currently available as oral tablets in 20, 40, and 80 mg strengths for use in the treatment of hypertension. It is also marketed as Micardis® HCT which is a fixed dose combination with Hydrochlorothiazide (HCTZ) in 40/12.5, 80/12.5, 80/25 mg/mg strengths, and Twynsta® its fixed dose combination with Amlodipine in 40/5, 80/5, 40/10, 80/10 mg/mg strengths.
In 2009, Boehringer Ingelheim (Boehringer) gained approval to extend the market authorised indication of the Telmisartan 80 mg strength to include reducing the risk of myocardial infarction, stroke or death from cardiovascular disorders.
The Telmisartan molecule was discovered and developed by Boehringer, and was launched in Europe and the US in 1998. Boehringer has co-marketing agreements with Bayer Schering Pharma and GlaxoSmithKline in certain countries.
Telmisartan (1) is an angiotensin II receptor antagonist useful in the treatment of hypertension, heart diseases, heart strokes, and bladder diseases.1 Telmisartan (1) is currently available in the market as an antihypertensive drug2 under the brand name of MICARDIS. The first reported synthetic method3 for this molecule consists of 8 steps (Scheme 1) involving condensation of 4-amino-3-methyl benzoic acid methyl ester (2) with butyryl chloride (3) in chlorobenzene to yield 4. Nitration of 4 followed by reduction of the resulting 5-substituted nitro compound 5 over Pd-C in methanol yielded amine 6. Cyclisation of 6 in acetic acid reflux affords the monobenzimidazole derivative 7, which upon further hydrolysis yielded an acid intermediate 8 by a saponification process. Condensation of compound 8 with diamine derivative 9 in polyphosphoric acid yielded the dibenzimidazole compound 10, which was further alkylated with 4′-bromomethyl-biphenyl-2-carboxylic acid tert-butyl ester (11)4 to afford product 12. Finally, hydrolysis of ester12 in trifluoracetic acid yielded telmisartan (1) in an overall yield of around 21% with several impurities. This process suffers from disadvantages such as (a) a multistep synthesis for compound 8 (3 steps from compound 5); (b) the solvents dimethyl formamide (DMF) or dimethylsulfoxide (DMSO) used in the penultimate stage are unrecoverable, while the use of potassium tert-butoxide resulted in high organic volatile impurities (OVI) in telmisartan; (c) deprotection of the tert-butyl group using trifluoroacetic acid in DMF lead to the formation of several byproducts; (d) residue on ignition (ROI) in API obtained from this process is always >1.0% (ICH limit <0.1%), and there is no specified process mentioned in the literature to control the ash content. This is mainly due to very poor solubility of the telmisartan in most of the solvents including water; and (e) the overall yield (21%) of this process is discouraging, which makes the process less viable for commercial production.
(1) (a) Battershill, A. J.; Scott, L. J. Drugs 2006, 66 (1), 51-83. (b) Norbert, H.; Berthold, N.; Uwe, R.; Jacobus, C. A.; Van, M.; Wolfgang, W.; Michael, E. U.S. Patent 5,591,762, 1997. (c) Ruth, R. W.; William, J. C.; John, D. I.; Michael, R. C.; Kristine, P.; Ronald, D. S.; Pieter, B. M. W. M. T. J. Med. Chem. 1996, 39 (3), 625-656.
(2) http://www.rxlist.com/cgi/generic2/telmisartan.htm.
(3) (a) Uwe, J. R.; Gerhard, B. N.; Kai, M. H.; Helmut, W.; Michael, E.; Jacobus, C. A.; Van, M.; Wolfgang, W.; Norbert, H. H. J. Med. Chem. 1993, 36, 4040-4051. (b) Merlo
(4) Carini, D. J.; Dunicia, J. V. Eu. Patent 2,53,310, 1988. (5) Venkataraman, S.; Mathad, V. T.; Kikkuru, S. R.; Neti, S.; Chinta, R. R.; Arunagiri, M.; Routhu, L. K PCT WO 06/044754A2, 2006.
(6) The intermediate 9 is prepared via monomethylation of o-nitroaniline (15) using dimethylsulfate followed by hydrogenation over Pd-C catalyst in methanol with 75% of overall yield. Of the several methylating agents such as CH3I, DMS, HCOOH, and H2CO explored
(7) Structures of these impurities were tentatively proposed based on MS-MS data and a probable reaction mechanism and then synthesized as shown in Scheme 3. These impurities were characterized by NMR, mass, and IR techniques and further confirmed to be present in the sample by HPLC coinjection and spiking methods (0.1%). (8) Shen, J.; Li, J.; Yan, T.; Li, H.; Ji, R. CN 1,344,712, 2002.
(9) Several brominating agents such as molecular bromine, N-bromosuccinimide (NBS), and 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) resulted in 13 along with the dibromo impurity 26. The formation of the dibromo impurity 26 is varying from 20-45% by HPLC. The content of 26 is nearly 45% in the case of NBS bromination, whereas the same is in the range of 15%- 20% in the case of DBDMH. Hence, DMDBH has been utilized as the brominating agent in the process. However impurity 26 did not participated in the next step and was easily washed out to a nondetected level during the isolation of 14 in the condensation step.
(10) Robert, E. D.; Peter, S.; Herbert, N.; Kenneth, S.; William, I. F. D. J. Pharm. Sci. 2000, 89 (11), 1465-1479.
Whilst patent protection for Telmisartan molecule, DE4103492A, has expired in Canada, it is still in force in the US until January 2014, receiving the longer term based on 17 years from the issue date for patents filed prior to June 8 1995. The equivalent European patent, EP0502314 (‘314), has been extended by SPC in France, Germany, Spain and the UK until December 2013 (see Figure 3).
Boehringer, seeking to protect its Telmisartan franchise, has also filed SPC applications for its Telmisartan-HCTZ and Telmisartan-Amlodipine products, for the basic patent ‘314, in France, Germany, Spain and the UK, potentially extending protection until January 2017 (see Figure 3). GenericsWeb’s proprietary SPC analyser has identified the basic patent as a ‘C3’ category, suggesting the claims of the basic patent do not protect the combinations and therefore the SPC may be invalid. The response by the national IPOs in respect to the invalidity of SPCs for the Telmisartan combinations has varied. The French SPC application (FR02C0028) for Telmisartan-HCTZ was initially rejected by the Institut National de la Propriété Industrielle (INPI) in December 2010, finding the claims of the basic patent did not protect a medicine comprising Telmisartan in association with HCTZ. The Paris Court of Appeal upheld INPI’s decision in June 2012, denying Boehringer’s request for appeal. Similarly, on June 2012, the Juzgado de lo Mercantil de Pamplona (the Court) held the Spanish SPC (C20020018) for Telmisartan-HCTZ invalid following a revocation suit filed by Cinfa and Actavis against Boehringer in April 2010. The Court’s decision relied on the ECJ’s findings in the ‘Medeva’ decision relating to SPCs for combination products, which concluded that to satisfy article 3(a) of SPC regulation 469/2009 the wording of the claims of the basic patent had to specify all active ingredients. Therefore, the Court found the SPC to be invalid on the grounds of article 15.1(a) in regard to 3(a), finding ‘314 did not specify a composition of Telmisartan in association with HCTZ. In February 2013, revocation proceedings were filed in the Bundespatentgericht for the German SPC (DE10299029) for Telmisartan-HCTZ. This raises the question of whether the SPC will prevent a generic Telmisartan-HCTZ product in Germany until conclusion of the revocation proceedings or will generic companies launch their products ‘at risk’ upon expiry of the SPC for Telmisartan, therefore assuming invalidity based on the ‘Medeva’ decision and similar findings by other PTOs and Courts in the matter.
The French (FR11C0008), German (DE122011000013) and Spanish (C201100010) SPCs for the Telmisartan-Amlodipine combination have been withdrawn. However, the UKIPO has granted the SPCs for both combination products (see Figure 3). No litigation proceedings have been detected in the UK. This may be due to amendments, under section 27 of the Patents Act 1977, of the specification for the UK designation of ‘314, in 2004 and 2011. The amendments were in the form of amended claim pages which included a pharmaceutical composition comprising HCTZ or a calcium channel blocker. Patents in the family with priority GB9722026A protect authorised indicated uses of the 80 mg dosage form of Telmisartan for reducing cardiac tissue damage associated with myocardial infarction and prevention or treatment of stroke, so are considered to be a constraint only for those indicated uses.
The family with the priority DE19901921A protects the crystalline form used in the commercially available product but are not considered to be a constraint to generic competition because the protected technology is likely to be circumvented. Families AU2002242676A, DE10301371A and EP04026234A protect Telmisartan combination products (see Figure 2). AU2002242676A and EP04026234A claim bilayer tablets comprising Telmisartan and HCTZ or Amlodipine, respectively. They are not considered to be a constraint to generic competition because the protected technologies are likely to be circumvented by generic reformulation. However, patents in the family DE19901921A expiring in July 2024, claiming composition of Telmisartan and several other drugs, including Amlodipine, are considered to be a constraint to generic competition for the Telmisartan-Amlodipine product. The family was deemed key due to its Canadian member 2534006 being listed on Health Canada’s patent register. Equivalent patents in the US have not been granted yet, but claims listed in the image wrapper in USPTO appear to limit the claims to a currently unauthorised use of Telmisartan and Amlodipine, therefore may not be a constraint for generic entry in the US.
Amongst the US approvals, Watson is the only company to have obtained tentative market approvals for all dosage strengths for the Telmisartan tablets and the fixed dose combination of Telmisartan and HCTZ. Lupin has gained a tentative approval for the Telmisartan and Amlodipine fixed dose combination. No generics are currently on the market in the UK due to unexpired patent protection, however several companies including Egis, Sandoz and Glenmark have obtained market authorisation for Telmisartan tablets in all dosage strengths. Actavis and Teva have obtained market authorisations via the centralised procedure. Dr Reddy’s Lab and Krka hold generic authorisations for both Telmisartan and Telmisartan-HCTZ fixed dose combination tablets. These generic approvals are suggestive of the competition Micardis® will face across Europe upon molecule patent expiry. Currently no generic Telmisartan-Amlodipine approvals have been identified in Europe. This is due to data exclusivity previsions in Europe, preventing the filing of generic market authorisation until October 2018, and a further 2 year market exclusivity period could prevent the launch of a generic equivalent until October 2020. In Canada, Mylan was one of the generic competitors to launch Telmisartan and Telmisartan-HCTZ following molecule patent expiry. This is likely to be mirrored in other territories upon expiry of the molecule patent.
Figure 4: Marketing Authorisations for products containing Telmisartan in Key Countries

In summary, patent protection remains a significant barrier to generic entry for the Telmisartan products in most major markets due to the molecule patent being in force. Boehringer’s lifecycle management attempts to maintain a monopoly for their blockbuster drug, including combination products and extensions of indications. Patent protection for its products, apart from the molecule patent, include a ‘use’ patent and combination patents which may pose a barrier to generic competition and may see Boehringer retain some of their market share. SPCs for the Telmisartan-HCTZ combination have been the subject of litigation in France and Spain, resulting in their invalidation, a revocation proceeding is on-going in Germany. Data exclusivity provisions in Europe will prevent the launch of a generic Telmisartan-Amlodipine fixed dose combination. In Canada, generic competition for Telmisartan and Telmisartan-HCTZ entered the market shortly after the expiry of the molecule patent. This is likely to be mirrored in other territories with generic companies already holding market authorisations for both products.
……………………………………..
PATENT
WO 2010018441
http://www.google.im/patents/WO2010018441A2?cl=en
Telmisartan is chemically named as 4′-[(1,4′-Dimethyl-2I-propyl[2l6′-bi-1H- benzimidazol]-1′-yl)methyl][1 ,1′-biphenyl]-2-carboxylic acid; or 4′-[[4-methyl-6-(1-methyl-2- benzimidazolyO^-propyl-i-benzimidazolyllmethyll^-biphenylcarboxylic acid.

The key raw material used to prepare Telmisartan is Bltyl, chemically named as 1,7′- dimethyl-2′-propyl-2,5′-bi-1H-benzimidazole, also known by other names, i.e 2-Propyl-4- methyl-6-(1 -methylbenzimidazol-2-yl)benzimidazole; 4-Methyl-6-(1 -methyl benzimidazol-2- yl)-2-propylbenzimidazole, and the structure shown as below:

BIM WO2006136916 describes substantially pure micronized particles of Telmisartan or a pharmaceutically acceptable salt, ester or derivative. The “substantially pure” is further defined as “Telmisartan or pharmaceutically acceptable salt, ester or derivative thereof having a purity of greater than or equal to about 98%, preferably a purity of greater than or equal to about 99% and more preferably a purity of greater than or equal to about 99.5%.° The substantially pure Telmisartan or a pharmaceutically acceptable salt, ester or derivative has an effective average particle size of less than about 300 microns.
A Journal of http://www.IP.com (2005), 5(7B), 4 – describes a process for purification of 4′-(2-propyl-4-methyl-6-(1-methylbenzimidazol-2-yl)benzimidazol-1-ylmethyl)biphenyl-2- carboxylic acid (Telmisartan). The pure compound was isolated by filtration under reduced pressure.
US20060276525 claims Telmisartan form A having HPLC purity > 99.5 %. It further provides a process for preparing Telmisartan form A by crystallization from a polar organic solvent selected from the group consisting of dimethyl sulfoxide, DMF, N.N-dimethyl acetamide, N-methyl 2-pyrrolidone, water and mixtures thereof. The process provides Telmisartan with a limit of DMSO at a level of < 1000 ppm. The process uses high boiling solvent in the last step for getting required purity, and which is also an extra purification step, which limits its commercial application.
US5591762 ( column-37,38 ) described the general process for the preparation of compound of formula-V

wherein bromine in structure IV is leaving group. There are several other leaving groups such as chlorine, iodine, a substituted sulphonyloxy group, e.g. a methane sulphonyloxy, phenylsulphonyloxy or p-toluenesulphonyloxy group are reported.
US5591762 describes preparation of Telmisartan from Telmisartan tert. butyl ester using trifluoroacetic acid in DMF as a solvent in 63.9 % yield. (Example-9) The resulting product had a melting point of 261-2630C.

The process for the preparation of tert. Butyl ester of Telmisartan is not commercially viable and deprotection involving the use of trifluoro acetic acid is not eco-friendly.
US 6385986 describes polymorphs of 4′-[2-n-propyl-4-methyl-6-(1-methylbenzimid- azol-2-yl) benzimidazol-1-ylmethyl] biphenyl-2-carboxylic acid (Telmisartan) i.e. polymorphic form B, mixtures of the polymorphs. The processes for preparing Telmisartan containing form B and the use for preparing a pharmaceutical composition. US ‘986 further describes that Telmisartan obtained process of as described in EP502314B1 to give a solid in the form of long needles which is difficult to filter, wash and isolate. It is further characterized that it requires a long time for drying due to the presence of solvent which forms large and hard fragments during the drying process. The fragments on grinding produce a dry powder which exhibits strong tendency to electrostatic charging and is virtually impossible to pour. The polymorphic form B of Telmisartan shows virtually no tendency to electrostatic charging and easy for suction filtration, centrifuge, washing, drying and is free-flowing even without being ground up.
Therefore, as a consequence of the alleged unsuitability of Telmisartan form A for pharmaceutical use, only a mixture of crystalline Telmisartan form A and form B is claimed in the ‘986 patent, wherein Telmisartan form A is characterized by having an endothermic maximum at 269±2°C, and Telmisartan form B is characterized by having an endothermic maximum at 183±2°C.
Apparently Telmisartan form A is similar to the original form characterized by its’ melting point in the ‘762 patent. The differences between the DSC value and the measured melting point may be attributed to the different methodologies used-the DSC maxima can be slightly different than the visually observed melting point.
Hence, the prior art teaches a lengthy, complicated and industrially disadvantageous process for obtaining crystalline Telmisartan form A. The need to further reprocess the re- crystallized Telmisartan, as taught in the examples of the ‘986 patent, shows that the product was not highly-pure and/or that it contained residual solvents, because the solvents used therein have high boiling point. JMC-1993, vol-36, No25 pg-4040-4051 describes preparation of Telmisartan tert. butyl ester using BIM and 2-(4’bromomethyl phenyl) tert. butyl benzoate using pot. Tert butoxide as a base in DMSO as solvent.

Formula 6
The preparative details for compound of formula-VII on page-4049, coloumn-3, compound 33, paragraph-4; line1-4 reads as follows.
Potassium tert-butoxide was added to the solution of BIM in DMSO at room temperature followed by the addition of the compound of formula Vl. Upon stirring for 14 hrs, the mixture was poured into water and extracted with ethyl acetate, the combined extract was dried on MgSO4 and evaporated. Residue was purified by silica gel column chromatography to give compound of formula-VII. The above mentioned process uses chromatographic purification, which is generally cumbersome and time consuming process and also requires solvents in high volume.
US20060094883 describes a process for the preparing Telmisartan, wherein Telmisartan alkyl ester – a

compound of formula-ll is prepared , comprising the steps of :
(a) combining i.y-dimethyl^’-propyl-IH.S’H-p.S1 ] bibenzimidazole (referred to as BIM) of formula III,

Formula 3 with 4′-bromomethyl-biphenyl-2-carboxylic acid alkyl ester (referred to as BMBP alkyl ester) of formula IV1

Formula 4 an inorganic base and a low boiling point organic solvent, to obtain a mixture;
(b) heating the mixture obtained in step (a) to a temperature of about 55°C. to about 1200C;
(c) maintaining the mixture obtained in step (b) for about 1 hour to about 8 hours, to obtain Telmisartan alkyl ester of formula II; and

(d) recovering Telmisartan alkyl ester of formula II, wherein, R is a straight or branched chain C1-C4 alkyl.
WO2005108375 describes process for the preparation of Telmisartan, characterized in that 1H-Benzimidazole-2-n-propyl-4-methyl-6-(1 ‘-methyl benzimidazole- 2’yl) of formula (II) and methyl-4- (bromo methyl)biphenyl 2-carboxylate of formula (III) are subjected to

WO 2007/010558 describes a method for the preparation of Telmisartan involving
Telmisartan dihydrochloride which comprises, i) condensing 4-Methyl-2-n-propyl-IH- benzimidazole-6-carboxylic acid with N-Methyl- O-phenylene diamine dihydrochloride to yields 4-methyl-6 (1 -methyl benzimidazol-2- yl)-2-n-propyl IH- benzimidazole, ii) treating 4- methyl-6-(l -methyl benzimidazol-2-yl)-2-n-propyl-IH-benzimidazole with
4*– (bromomethyl)-2-biphenyl-2-carboxylate in presence of a base in an organic solvent and isolating the ester as acid addition salt, iii) converting ester acid addition salt to Telmisartan dihydrochloride and iv) converting Telmisartan dihydrochloride to Telmisartan. CN1344712 describes method comprising reaction of 4-methyl-6-(1-methyl-2(1H)- benzimidazolyl)-1H-benzimidazole with 4′-bromomethyl-biphenyl-2-carboxylic acid alkyl ester [wherein alkyl is methyl or ethyl] in solvent i.e. DMF, DMSO, THF, dioxane, chloroform, dichloroethane, etc. in the presence of base [such as Na alcoholate, triethylamine, tributylamine, tripropylamine, KOH, NaOH, CsOH, Ba(OH)2 etc.] as acid capturer at 20- 1000C for 8-10 hrs, and then hydrolyzing with acid (such as H2SO4, HCI, HBr, HOAc, etc) at room temp, to reflux temp, or with base in Ci-5 alc.-water at 20-1600C for 1-10 hour. WO 2006/125592 describes a new process for the preparation of saltans 2-butyl-3- [[2″-[1 -(triphenylmethyl)-i H- tetrazol-5-yl][1 , 1 ‘-biphenyl]-4-yl]methyl]-1 ,3-diazaspiro[4.4] non- 1-en-4-one is disclosed, which proceeds via novel intermediate, 4-[(2-butyl-4-oxo-1 ,3- diazaspiro[4.4]non-1-en-3-yl)methyl]phenylboronic acid (Formula (H)) or its analogs. Compound (II) reacts with 5-(2-bromophenyl)-1-(triphenylmethyl)-1H-tetrazole (III) in the presence of catalyst, using conditions of Suzuki reaction, to give trityl irbesartan (I), whereas analogs to compound (II) may give candesartan, valsartan, Telmisartan, losartan and olmesartan.
WO 2006/050509 describes the amorphous form of Telmisartan sodium and the preparation thereof. Also provided are the Telmisartan sodium polymorph crystal Forms 0 to
XIII and XV to XX and preparations thereof. Also provided are pharmaceutical composition of amorphous and polymorphic forms of Telmisartan sodium or mixtures thereof, and methods of treatment of a mammal in need thereof.
WO 2006/044754 describes a process for preparing Telmisartan and intermediates formed in the process.
WO 2004/087676 describes a novel method for the production of Telmisartan by reacting 2-n-propyl-4-methyl-6-(1′-methylbenzimidazol-2’-yl)-benzimidazol with a compound of general formula (IV)1 in which Z is a leaving group, wherein the compound 2-cyano-4′-[2″- n-propyl-4″-methyl-6″-(1 ‘”-methylbenzimidazol-2l“-yl)benzimidazol-1 “-ylmethyl]biphenyl is obtained, and subsequently conducting hydrolysis of the nitrile to acid function.
WO2000/043370 describes polymorphs of 4′-[2-n-propyl-4-methyl-6(1 -methyl benzimidazol -2-yl) benzimidazol -1-ylmethyl] biphenyl-2-carboxylic acid (INN: Telmisartan), and in particular the polymorphous form B of formula (I), characterized by an endothermic peak at 183 ± 2°C during thermal analysis by differential scanning calorimetry. The invention also relates to mixtures of said polymorphs, methods for producing Telmisartan containing form B and to the use thereof in the preparation of a medicament.
Example-5 : Preparation of 4′-[[2-n-propyl-4methyl-6-(1-methylbenzimidazol-2-yl)- benzimidazol-1-yl]-methyl] biphenyl carboxylic acid [Telmisartan]
90 gm of ethyl-4′-[[2-n-propyl-4-methyl-6-(1-methylbenzimidazol-2-yl)-benzimidazol- 1-yl]-methyl] biphenyl carboxylate was stirred with 810 ml aq. HCI [32-35 % wt/ vol] at 95±2°C for about 8-10 hours. The reaction mixture was cooled to 25-300C. 180 ml of Dichloromethane and 1350 ml of water were added, pH of the reaction mixture was adjusted to -9.0 to 10.0 using 20 % aq. NaOH. The reaction mixture was stirred at 30-350C for about 30 minutes and the layer was allowed to separate. 1800 ml of MDC was added to aqueous phase at 25-300C. pH of the solution was adjusted to ~3 to 3.5 with acetic acid. The mixture was stirred for about 20 minutes and the layer was allow to separate. The aqueous layer was extracted with 900 ml DCM and organic layer was separated and washed with 2 X 900 ml water. The organic phase was dried over anhy. Sodium sulfate and charcoalized followed by distillation to remove about 80-85 % of DCM at 40-420C. The reaction mixture was slowly cooled to 80C and stirred at 8-120C for about 1Hr. 2700 ml of acetone (100C was slowly added and temperature is maintained at 8-12°C.The reaction mixture was stirred for 2 hours with slow RPM. The mixture was filtered at 8-120C and washed with 2×180 ml of acetone. The product was obtained through suction drying for 30-45 minutes, and under vacuum at 85-900C. 70.0 gm of Telmisartan is obtained having purity of 99.84%.
…………………………
PATENT
WO2009006860A2
http://www.google.im/patents/WO2009006860A2?cl=en
Telmisartan (I) is produced in accordance with the original patent of Boehringer Ingelheim (US 5 591 762) from telmisartan tert-butyl ester (II). The hydrolysis is carried out using of trifluoroacetic acid in the toxic solvent N,N-dimethylformamide.

According to another patent applied by the same company (US 2004 236113) the manufacture was problematic and this is why this procedure was replaced with hydrolysis of the corresponding nitrile (III). However, during the hydrolysis, which is carried out with potassium hydroxide in ethylene glycol, a high temperature (160 0C) is used, which causes browning of the product, which must be subsequently purified by means of activated carbon. Also, the energy demands of several-ton production would be considerably high.

In a newer application of Cipla (WO 2005/10837) the last two synthetic steps (iii+iv) are combined and telmisartan is isolated after alkaline hydrolysis by acidifying of the reaction mixture in water or extraction with dichloromethane and precipitation with acetone. Both the ways of isolation are unsuitable for industrial production. In the case of telmisartan of crystalline form A its isolation from water or aqueous solutions of organic solvents is very difficult since a hardly filterable product is formed. Extraction of the product with dichloromethane and precipitation with acetone brings a well-filterable product, but the use of dichloromethane is virtually impossible from the point of view of environment protection.

Another method has been described by Dr. Reddy (WO 2006/044754), which starts from telmisartan methylester hydrochloride, which is hydrolyzed to produce the potassium salt of termisartan, which is further acidified in aqueous acetonitrile; after isolation it crystallizes from a dichloromethane/methanol mixture and finally from methanol alone, and wherein a pressure apparatus is used for the dissolution in methanol at a temperature above its boiling point (80 °C). The result of this complex procedure, which manifests the already above mentioned shortcomings, is a low yield of the product.
The method of Teva (WO 2006/044648) is in many aspects similar to the above mentioned procedure of Cipla, wherein the last two steps of the synthesis are also combined. The method comprises phase separations, which lead to low yields (69 % – 80 %) besides increased tediousness. Matrix starts from telmisartan tert-butyl ester (II), which is first converted to telmisartan dihydrochloride, which in turn, by action of aqueous ammonia in methanol, provides telmisartan with a low total yield of 73%.
http://www.google.im/patents/WO2009006860A2?cl=en
Example 1
4′-[[4-methyl-6-(l-methyl-lH“-benzimidazol-2-yl)-2-proρyl-lH-benzimidazol- lyl]methyl]biphenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (VI) (40 g) was refluxed in methanol (440 ml) with potassium hydroxide (14.9 g) for 24 hours. To the boiling solution, methanol (240 ml) and then acetic acid (45.5 g) were added. While boiling, the mixture was stirred for another 1 hour, after cooling to 4°C the product was aspirated within 1 hour and washed with methanol (2 x 80 ml). After drying at the laboratory temperature (24 h) 35.18 g (90 %) of the product were obtained.
Analytic assessment: HPLC purity: 99.90 %,
Content of residual solvents: methanol (below the detection limit) acetic acid (360 ppm) Titration content: 100.9 % Sulfate ash content: 0.04 % DSC: form A
Example 2
4′-[[4-Methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-lH-benzimidazol- lyl]methyl]biphenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (VI) (20 g) was refluxed in methanol (300 ml) with potassium hydroxide (7 g ) for 24 h. After addition of formic acid (17 g) and after cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 ml). After drying at the laboratory temperature (24 h) 18.7 g (96 %) of the product were obtained.
Example 3
4′-[[4-methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-l/J-benzimidazol- lyl]methyl]biρhenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (VT) (20 kg) was refluxed in methanol (400 1) with potassium hydroxide (7 kg) for 24 h. After addition of acetic acid (20 kg) and cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 1). After drying at the laboratory temperature (24 h) 18.5 kg (95 %) of the product were obtained. Example 4
4′-[[4-methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-lH‘-benzimidazol- lyl]methyl]biphenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (40 g) was refluxed in methanol (240 ml) with potassium hydroxide (14.9 g) for 24 h. To the boiling solution methanol (240 ml) and then acetic acid (45.5 g) were added. After cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 ml). After drying at the laboratory temperature (24 h) 36 g (92%) of the product were obtained.
…………………………..
PAPER

Telmisartan (1), a substituted dibenzimidazole derivative, is an antihypertensive drug, essentially used to control blood pressure. An improved, cost-effective, and impurity-free process for telmisartan (1) suitable for large-scale production is described here by addressing various process development issues. The overall yield obtained from this newly developed process is around 50% (over five steps) compared to the literature reported process (21%, over eight steps).
4′-[(1,4′-Dimethyl-2′-propyl-[2,6′-bi-1H-benzimidazol]- 1′-yl)methyl]-[1,1′-biphenyl]-2-carboxylic Acid (1).
Telmisartan (1) as a white crystalline powder. Yield 7 g (77%); purity by HPLC 99.9%; mp 260- 262 °C; Pd content not detected; Heavy metals <10 ppm; MS m/z 515 (M+ + H);
1 H NMR (CDCl3) δ 12.8 (s, 1H), 7.05-7.5 (m, 14H), 5.60 (s, 2H), 3.82 (s, 3H), 2.97 (t, J ) 7.5, 2H), 2.63 (s, 3H), 1.88 (q, J ) 7.3, 2H), 1.04 (t, J ) 7.3, 3H);
13C NMR (DMSO-d6) δ 13.5, 16.7, 20.6, 27.6, 32.7, 47.1, 51.7, 112.0, 112.7, 114.7, 118.6, 125.3, 125.7, 125.8, 127.0, 127.4, 128.6, 129.3, 130.4, 130.6, 131.5, 132.3, 133.1, 133.7, 134.5, 140.2, 140.5, 150.2, 157.3, 168.1.
Anal. Calcd for C33H30N4O2: C, 77.02; H, 5.88; N, 10.89; O, 6.22. Found: C, 77.0; H, 5.82; N, 10.89; O, 6.20.
………………….
PATENT
EP1719766A2
http://www.google.im/patents/EP1719766A2?cl=en
he present invention provides a process for the preparation of a compound of formula (I) or a salt thereof

comprising the reaction of a compound of formula (II) or a salt thereof

with a synthon of formula (III) or a salt thereof

prepared by reaction of a compound of formula (IV)

with a compound of formula (V)

-
Telmisartan, 4′-[(1,7′ -dimethyl-2′ -propyl[2,5′ -bis-l H-benzimidazol]-3′-yl)methyl][1,1′-biphenyl]-2-carboxylic acid is a known ACE inhibitor useful in therapy as antihypertensive agent. Its preparation is disclosed inEP 502314 and comprises the alkylation of 4-methyl-6-(1-methyl-benzimidazol-2-yl)-2-propylbenzimidazole (A) with t-butyl 4′-(bromomethyl)biphenyl-2-carboxylate (B)

-
However, compound (B) is not commercially available and its synthesis requires a number of steps, among them the protection of the carboxylic function which is finally removed by hydrolysis. There is therefore the need for an alternative synthesis for the industrial preparation of telmisartan, which makes use of commercially available or easy to prepare intermediates and which, if possible, avoids the additional steps of protection and deprotection of the carboxylic function.

Example 4. 4′-[[4-Methyl-6-(1-methyl-2-benzimidazolyl)-2-propyl-1-benzimidazolyl]methyl]-2-biphenylcarboxylic acid (telmisartan)
-
(4′-Methyl-2′-propyl-1H-benzimidazol-6′-yl)-1-methyl benzimidazole (3.0 g, 9.8 mmol), 4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)benzyl methanesulfonate (3.12 g, 10 mmol), tetrahydrofuran (15 ml) and potassium carbonate (1.38 g, 10 mmol) are loaded into a round-bottom flask equipped with magnetic stirrer, condenser and under nitrogen atmosphere. The mixture is stirred at room temperature for 8 hours, then 10% hydrochloric acid is added to pH=2.
-
THF is evaporated off, which causes precipitation of boronic acid. After recrystallization from ethyl acetate, 4.2 g of product are obtained.
-
The boronic acid (3.5 g, 8.0 mmol), ethyl 2-bromoacetate (1.83 g, 8.0 mmol), sodium hydroxide (1.28 g, 32 mmol), water (5 ml), tetrahydrofuran (20 ml), triphenylphosphine (315 mg, 1.2 mmol) and palladium acetate (90 mg, 0.4 mmol) are loaded into a round-bottom flask equipped with magnetic stirrer and condenser. All the residual air is removed with nitrogen and then the mixture is heated at 60°C for 18 hours, thereafter is cooled, added with water (30 ml) and tetrahydrofuran is evaporated off. Ethyl acetate is added (30 ml) and the mixture is acidified with acetic acid to pH=5. The product is filtered and washed with water, to obtain 2.8 g of crude telmisartan, which is purified by dissolution in concentrated ammonia (2 ml), addition of acetone and reprecipitation with acetic acid.
……………………
PATENT

http://www.google.im/patents/EP2305650A1?cl=en
-
Telmisartan and its physiologically acceptable salts have valuable pharmacological properties. Telmisartan is an angiotensin-II-antagonist, which may be used to treat hypertension and cardiac insufficiency, ischaemic peripheral circulatory disorders, myocardial ischaemia (angina). Furthermore, Telmisartan may be used to prevent the progression of cardiac insufficiency after myocardial infarct, to treat diabetic neuropathy, glaucoma, gastrointestinal diseases and bladder diseases. Telmisartan is also suitable for treating pulmonary diseases, e. g. lung oedema and chronic bronchitis, for preventing arterial restenosis after angioplasty, for preventing thickening of blood vessel walls after vascular operations, and for preventing arteriosclerosis and diabetic angiopathy. In view of the effects of angiotensin on the release of acetyl-choline and dopamine in the brain, Telmisartan is also suitable for alleviating central nervous system disorders, e. g. depression, Alzheimer’s disease, Parkinson syndrome, bulimia and disorders of cognitive function.
-
Telmisartan is a compound of formula (I)

chemically known as 4′-((1,7′-dimethyl-2′-propyl-1H,3′H-2,5′,-bibenzo[d]imidazol-3′-yl)methyl)biphenyl-2-carboxylic acid, which is disclosed in EP 502314 B1 and marketed under the trade name Micardis®.
-
Several methods have been used to prepare Telmisartan.
-
The process described inEP 502314 B1 comprises the alkylation of 4-methyl-6-(1-methyl-benzimidazol-2-yl)-2-propylbenzimidazole (III)

with t-butyl 4′-(bromomethyl)biphenyl-2-carboxylate and subsequently hydrolysis to Telmisartan. t-Butyl 4′-(bromomethyl)biphenyl-2-carboxylate is not commercially available and its synthesis requires a number of steps, among them the protection of the carboxylic function which is finally removed by hydrolysis.
-
The patent application WO 2006044648 relates to a method for the production of Telmisartan by reacting 4-methyl-6-(1-methyl-benzimidazol-2-yl)-2-propylbenzimidazole (III) with 4′-(bromomethyl)biphenyl-2-carboxylic acid alkyl ester and subsequently hydrolysis.
-
The patent application WO 2004087676 relates to a method for the production of Telmisartan by reacting 4-methyl-6-(1-methyl-benzimidazol-2-yl)-2-propylbenzimidazole (III) with 4-bromomethyl-2′-cyanobiphenyl and subsequently hydrolysis of the nitrile to the acid function.
-
The patent application EP 1719766 relates to a method for the production of Telmisartan, by coupling with a Suzuki reaction the N-4-bromobenzyl derivative of the compound of formula (III) with 2-carboxylphenyl boronic acid. As described in EP 1878735 , 2-carboxyphenyl boronic acid requires a very laborious process to separate it, since it is extremely soluble in water, making the process unattractive for an industrial application. Thus, the active substance prepared by the process known up till now can only be obtained in a satisfactory quality after running through a number of process steps, wherein additional steps of protection and deprotection of the carboxylic function or additional steps to obtain the carboxylic function are often present.


Example 2 4′-((1,7′-dimethyl-2′-propyl-1H,3′H-2,5′-bibenzo[d]imidazol-3′-yl)methyl)biphenyl-2-carboxylic acid (I)
-

-
A 2L four-necked glass reactor, fitted with mechanical stirrer, thermometer, dropping funnel, under nitrogen atmosphere, was charged with sodium hydride (60% in mineral oil) (12.5 g, 312 mmol) and toluene (450 mL). The suspension was stirred and trimethylsilanol (31 g, 343 mmol) was added dropwise. After stirring for 15 minutes, methyl 4′-((1,7′-dimethyl-2′-propyl-1H, 3′H-2,5′-bibenzo[d]imidazol-3′-yl)methyl)biphenyl-2-carboxylate (V) (145 g, 274 mmol) was added, the mixture was stirred for 5 hours at about 100°C and monitored by quantitative TLC (elution with 5% MeOH in EtOAc) until complete conversion. The mixture was then cooled at room temperature, water (130 mL) was added, and the mixture was brought at 50°C. The phases were separated and the aqueous phase was stripped under vacuum to remove residual toluene.
350 g of an aqueous solution were obtained and used as such in the next step. -
A 1L four-necked glass reactor, fitted with mechanical stirrer, thermometer, dropping funnel, under nitrogen atmosphere, was charged with the aqueous solution in MeOH (600 mL). The mixture was heated under stirring at 40°C until dissolution and charcoal (7 g) was added. The suspension was stirred at 40°C for 30 minutes, filtered through a pad of Celite and the resulting solid was washed with a mixture of MeOH/water 4/1 (100 mL). The filtrate and the washings were combined, the resulting solution was heated to reflux temperature and acetic acid (17.7 g, 295 mmol) was added dropwise over 1 hour. The suspension was then cooled, filtered and the solid was washed with a mixture MeOH/water 4/1 (3 x 50 mL). The collected solid was then dried at 55°C under reduced pressure affording the title compound (130 g) as a white solid.

………………………
PATENT
WO2014067237A1
http://www.google.im/patents/WO2014067237A1?cl=en
Telmisartan is a novel non-peptide angiotensin Π (ΑΤ Π) receptor antagonist, for the clinical treatment of hypertension, its chemical name is 4 ‘- [(1,4′-dimethyl – 2′-propyl [2,6’- two -1Η- benzoimidazol] -Γ–yl) methyl] biphenyl] -2-carboxylic acid, knot

Telmisartan
Telmisartan synthetic route has mainly 3-methyl-4-amino-benzoic acid methyl ester as the starting material by N- acylation, nitration, reduction, cyclization, ester hydrolysis and condensation reaction intermediates 2-n-propyl-4-methyl – 6 – (1 – methyl-benzimidazol-2-yl) benzimidazole-α), Ϊ with 4′-bromomethyl-biphenyl-2-carboxylate (V) via nucleophilic substitution, hydrolysis reaction to give the final product two Bu telmisartan (reaction formula 1) (J Med Chem, 1993, 36: 4040-4051).
Reaction Scheme 1

After has been reported by 4′-bromomethyl-biphenyl-2-carboxylic acid methyl ester (or ethyl ester) (VI) or 4′-bromomethyl-biphenyl-2-carbonitrile (VII) Preparation of telmisartan (CN01126367 .9, CN01131915.1).




Example 16: Preparation of telmisartan
The title compound of Example 15 (III, R = CN) (24.8g, 0.05mol) was added propylene glycol (100ml) and water (100ml) (or other previously described embodiments will be an aqueous mixed solvent :), potassium hydroxide (or e.g. prior to said other inorganic bases) (0.2mol), was refluxed for 10 hours. After no starting material by TLC was cooled to room temperature, concentrated under reduced pressure to a small volume, was added dropwise hydrochloric acid adjusted to pH 5 to 6, the precipitated solid was filtered, washed with water to obtain telmisartan.
Telmisartan Preparation: 17 Examples
The title compound I (30.4g, 0.10moi>, embodiments of Example 14 4′-chloro-methyl-biphenyl-2-carbonitrile (0.12mol), sodium ethoxide (or other organic bases as previously described) (0.3mol) and DMF (or other solvent as previously described) (200ml) mixed, 65 ° C for about 5 hours. TLC detected no starting material, was added ethylene glycol (100ml and water (50ml) (or other aqueous solvent), and heated to 160 ° C. TLC detected no starting material, concentrated hydrochloric acid under ice cooling to adjust pH to 5-6, to precipitate a solid, the resulting solid was filtered, washed with water to give crude telmisartan, by recrystallization in telmisartan.
Examples 18 to 24: Preparation of telmisartan reference method of Example 8, the title compound of Example 6 to the compound of formula I (10g, leq) and implemented as a reactant, with NaH as a base, the reaction temperature under different conditions the reaction, the reaction solution was subjected to phase detection by conventional post-treatment to give telmisartan (crude), yield was calculated, and the purity of the liquid phase detection telmisartan. The test results are shown in Table 2.
Table 2 compares the reaction conditions


1 Telmisartan ……………………………….2 Impurity B
Table 1 : Preparation of Telmisartan and 2 with reported synthetic schemes

process for the preparation of telmisartan, comprising: condensation of -n-propyl-4-methyl-6-(l’-methylbenzimidazol-2′-yl)benzimidazole (I)

with a compound of general formula II)

wherein Z denotes a leaving group such as a halogen atom, for example, a chlorine, bromine, or iodine atom to obtain the compound 2-cyano-4′-[2″-n-propyl-4″-methyl-6″-( 1 “‘-methylbenzimidazol-2″‘-yl)benzimidazol- 1 “-ylmethyl]biphenyl (III), and subsequent

hydrolysis of nitrile in the presence of excess base and solvent followed by acid/base purification to obtain pure telmisartan.

Scheme-I


EXAMPLES:
Experiment-1: Preparation of 2-cyano-4-[2-n-propyl-4-methyI-6-(l- methylbenzimidazol-2-yl) bnzimidazol-l-ylmethyl] biphenyl.
Add 2-n-propyl-4-methyl-6-(l ‘-methylbenzimidazol-2′-yl) benzimidazole 100 g in 1000ml of acetone and of potassium hydroxide 22.0 g with stirring at 20-25°C. Then of 4-bromomethyl-2′-cyanobiphenyl 92g is added at 20-25°C. Monitor the reaction on thin layer chromatography, after compilation reaction, the crystals are suction filtered, washed with chilled acetone, water, and then dried in a air drying cupboard at 80° C. Yield: 135.0 g (82.82% of theory); melting point: 196° C.-197° C; HPLC: 99.30%. N-3 isomer: 0.08%.
Experiment-2: Preparation of 2-cyano-4-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl) benzimidazol-l-ylmethyl] biphenyl.
Add 2-n-propyl-4-methyl-6-( -methylbenzimidazol-2′-yl) benzimidazole 100 g in 1000ml of acetone and of potassium hydroxide 22.0 g with stirring at 20-25°C. Then of 4-bromomethyl-2′-cyanobiphenyl 92g is added at 20-25 °C. Monitor the reaction on thin layer chromatography, after the reaction is completed, cooled to 0 to 5.0° C. and stirred for another hour at this temperature. The material is filtered, washed with chilled acetone, then wash with water, and then dried in a air drying cupboard at 80° C. Yield: 141.50 g (87.73% of theory); melting point: 196° C.-197° C; HPLC: 99.50%. N-3 isomer: 0.16%
Experiment-3: Preparation of Telmisartan.
Add potassium hydroxide 80g in 500ml of ethylene glycol then add 2-cyano-4′- [2-n-propyl-4-methyl-6-( 1 -methyl benzimidazol-2-yl) benzimidazol- 1 -ylmethyl] biphenyl lOOgm at room temperature. Stir the reaction mixture and raise temperature to 150- 155° C. The mixture is stirred for 15 to 18 hours at this temperature and monitor reaction mass by HPLC. After compilation of reaction cool to 30 to 35°C then diluted with 800 ml methanol then telmisartan precipitates by adding of acetic acid at 25 to 30°C and further diluted with water. Then stirred for further 90min at 25 to 30°C. After the crystals have been suction filtered. The wet material dissolve in 500ml methanol with 12gm potassium hydroxide then after treatment of charcoal crystallize the telmisartan to adjusting of pH 6.0 to 6.4 by acetic acid then dilute with 400ml water. Filtered and then dried in a vacuum tray drier at 85°C. Yield: 90g (87.37% of theory); HPLC: 99.91%.
Experiment-4: Preparation of Telmisartan.
Add potassium hydroxide lOOg in 500ml of ethylene glycol then add 2-cyano- 4’-[2-n-propyl-4-methyl-6-(l -methyl benzimidazol-2-yl) benzimidazol- 1 -ylmethyl] biphenyl 1 OOgm at room temperature. Stir the reaction mixture and raise temperature to 150-155° C. The mixture is stirred for 15 to 18 hours at this temperature and monitor reaction mass by HPLC. After compilation of reaction cool to 30 to 35°C then diluted with 800ml methanol then telmisartan crystallize by adding of acetic acid at 25 to 30°C then dilute with 300ml water. Stir for further 90min at 25 to 30°C. Filter and then dried in a vacuum drying cupboard at 85°C. Yield: 101 g (1.03% of theory); HPLC: 99.90%.
Experiment-5: Preparation of pure Telmisartan.
Crude telmisartan 101 g (from example 4) & activated carbon lOg is added in methanol 100ml , dichloromethane 500ml at 25 to 30°C. Stir the reaction mixture then the brown solution is filtered through hyflow bed, Completely distilled out filtrate below 50°C then add 800ml water at that temperature and stir for lhr. The telmisartan is hot filtered and washed with water. The telmisartan is dried at 80° C. in a vacuum drying cupboard. Yield: 90 g (87.37% of theory); HPLC: >99.95%.
Experiment-6: Preparation of Telmisartan.
2-cyano-4′- [2-n-propyl-4-methyl-6-( 1 -methyl benzimidazol-2-yl) benzimidazol- 1 – ylmethyl] biphenyl lOOgm is added in 500ml of ethylene glycol with lOOg of potassium hydroxide powder at 20-25°C. Stir and raise temperature to 160° C. to 165° C. The mixture is stirred for 15 to 18 hours at this temperature and monitor reaction mass by HPLC. After compilation of reaction cool to 70 to 75°C then diluted with methanol and water then telmisartan crystallize by adding of acetic acid to adjust the pH 5.5 to 6.0 at 25 to 30°C. Stir for further 60min at 25 to 30°C. After the crystals have been suction filtered. The wet material dissolve in methanol with potassium hydroxide 12gm then after treatment of charcoal crystallize the telmisartan by adding of acetic acid by adjusting of pH 6.0 to 6.4 then stir for further 60min. The material is filtered and dried in a vacuum drying cupboard at 85°C. Yield: 86.56g (84.03% of theory); HPLC: >99.96%.
Experiment-7: Preparation of Telmisartan.
of 2-n-propyl-4-methyl-6-(r-methylbenzimidazol-2′-yl) benzimidazole 100 g is add in 1000 ml of acetone, and of potassium hydroxide 22 gm with stirring at 20-25° C and then 90.0 g of 4-bromomethyl-2′-cyanobiphenyl is added at 20-25°C. The temperature of the reaction mixture is maintained at 20 to 25° C. Stir for a further 6.0 to 8.0 hours at 20 to 25° C. Monitor the reaction on thin layer chromatography, after compilation reaction distil out acetone. Add ethylene glycol 500ml and potassium hydroxide lOOgm to residue Stir the reaction mixture and raise temperature to 150° C. to 155° C. The mixture is stirred for 15 to 18 hours at this temperature and monitor reaction mass by HPLC. After compilation of reaction cool to 30 to 35°C. Reaction mass diluted with methanol and stir for 30min then telmisartan precipitated by adding of acetic acid to adjust the pH 6.0 to 6.5 at 25 to 30°C. Then dilute with water and filter, wash with of methanol. Wet telmisartan is dissolved in methanolic potassium hydroxide, filtered to remove un dissolved material. Acetic acid is added to adjust the pH 6.0 to 6.4 , water added for complete precipitation of material. Finally telmisartan is suction filter and wash with water at 40 to 45 °C. The telmisartan is dried at 80° C. in a vacuum drying cupboard. Yield: 130g
HPLC: 99.4%.
1H NMR (DMSO-d6) δ 1.0 (t,3H), 1.9 (q, 2H), 2.95 (t, 2H), 2.4 (s, 3H), 3.95 (s, 3H), 5.8 (s, 2H), 7.28 (s,lH),7.80 (s,lH), 7.75 (d, 2H), 7.25 (t, 2H), 7.10 (d, 2H), 7.30 (d, 2H), 7.40 (d, 1H), 7.40 (t, 1H), 7.30 (t, 1H), 7.45 (d, 1H). 12.9 (s, 1H).
m/z 514.7 [ M + H]+.

Telmisartan (I) is produced in accordance with the original patent of Boehringer Ingelheim (US 5 591 762) from telmisartan tert-butyl ester (II). The hydrolysis is carried out using of trifluoroacetic acid in the toxic solvent N,N-dimethylformamide.
According to another patent applied by the same company (US 2004 236113) the manufacture was problematic and this is why this procedure was replaced with hydrolysis of the corresponding nitrile (III). However, during the hydrolysis, which is carried out with potassium hydroxide in ethylene glycol, a high temperature (160 0C) is used, which causes browning of the product, which must be subsequently purified by means of activated carbon. Also, the energy demands of several-ton production would be considerably high.
In a newer application of Cipla (WO 2005/10837) the last two synthetic steps (iii+iv) are combined and telmisartan is isolated after alkaline hydrolysis by acidifying of the reaction mixture in water or extraction with dichloromethane and precipitation with acetone. Both the ways of isolation are unsuitable for industrial production. In the case of telmisartan of crystalline form A its isolation from water or aqueous solutions of organic solvents is very difficult since a hardly filterable product is formed. Extraction of the product with dichloromethane and precipitation with acetone brings a well-filterable product, but the use of dichloromethane is virtually impossible from the point of view of environment protection.
Another method has been described by Dr. Reddy (WO 2006/044754), which starts from telmisartan methylester hydrochloride, which is hydrolyzed to produce the potassium salt of termisartan, which is further acidified in aqueous acetonitrile; after isolation it crystallizes from a dichloromethane/methanol mixture and finally from methanol alone, and wherein a pressure apparatus is used for the dissolution in methanol at a temperature above its boiling point (80 °C). The result of this complex procedure, which manifests the already above mentioned shortcomings, is a low yield of the product.
The method of Teva (WO 2006/044648) is in many aspects similar to the above mentioned procedure of Cipla, wherein the last two steps of the synthesis are also combined. The method comprises phase separations, which lead to low yields (69 % – 80 %) besides increased tediousness. Matrix starts from telmisartan tert-butyl ester (II), which is first converted to telmisartan dihydrochloride, which in turn, by action of aqueous ammonia in methanol, provides telmisartan with a low total yield of 73%.
Example 3
4′-[[4-methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-l/J-benzimidazol- lyl]methyl]biρhenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (VT) (20 kg) was refluxed in methanol (400 1) with potassium hydroxide (7 kg) for 24 h. After addition of acetic acid (20 kg) and cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 1). After drying at the laboratory temperature (24 h) 18.5 kg (95 %) of the product were obtained.
Example 4
4′-[[4-methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-lH‘-benzimidazol- lyl]methyl]biphenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (40 g) was refluxed in methanol (240 ml) with potassium hydroxide (14.9 g) for 24 h. To the boiling solution methanol (240 ml) and then acetic acid (45.5 g) were added. After cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 ml). After drying at the laboratory temperature (24 h) 36 g (92%) of the product were obtained.
Telmisartan was first disclosed in US 5,591,762. US 5,591,762 also discloses a process for the preparation of Telmisartan by reacting l,4′-dimethyl-2′-propyl[2,6′-bi-lH- benzimidazole (II) with 4′-(bromomethyl)[l,r-biphenyl]-2-carboxylic acid 1,1- dimethylethyl ester (III) in a solvent optionally in the presence of an acid binding agent to produce the intermediate 4′-[(l,4′-dimethyl-2′-propyl[2,6l-bi-lH-benzimidazol]-l- yl)methyl]-[l,l’-biphenyl]-2-carboxylic acid 1,1-dimethylethyl ester (IV), which is further hydrolysed to produce crude Telmisartan. The crude product obtained is purified over a silica gel column and finally crystallized from acetone. The process is shown in Scheme 1 :

Hydrolysis

(I)
The disadvantage with the above process is the use of column chromatography in the purification of Telmisartan. Employing column chromatography technique is tedious and laborious and also involves use of large quantities of solvents, and hence is not suitable for industrial scale operations.
US 6,358,986 describes two crystalline forms of Telmisartan donated as Form A, Form B. In US 6,358,986, the process for preparing crystalline Telmisartan Form A comprises mixing the Telmisartan with ethanol, adding activated charcoal and aqueous ammonia and mixing for one hour, then filtering to another stirring apparatus and washing with ethanol. Resulting solution is heated to 70~80°C, adding glacial acetic acid and stirring for further 1.5-2 hours at the same temperature, cooling to 0-10°C, stirring for further 2 hours, isolating the product by centrifugation, washing with ethanol then with water and drying at 70-90°C. According to the detailed description given in the US ‘986 patent, in addition to the disadvantageously prolonged drying process of the Telmisartan Form A, very hard particles are obtained. The grinding process of these particles produces a dry powder, which has strong tendency to electrostatic charging and which is virtually impossible to pour and manipulate for pharmaceutical preparations. On the other hand, Telmisartan Form B is free from the above-mentioned limitations. However, the inventors of the US ‘986 patent could not obtain pure, dry Form B because upon drying, some of Form B transformed into Form A. According to the teachings of the US ‘986 patent, mixtures of Telmisartan Form A and Form B ranging from 90:10 to 60:40 are suitable for industrial scaling-up, and even a content of 10% of Form B is sufficient to ensure that the product will have the positive qualities required for large-scale production.
US 2006/0276525 Al describes a process for the preparation of crystalline solid of Telmisartan Form A by dissolving Telmisartan in a polar solvent such as dimethylsulfoxide (DMSO), N,N-dimethylformamide (DMF), ΛζiV-dimethylacetamide (DMA)5 iV-methyl-2-pyrrolidone (ΝMP) and cooling the solution for sufficient time to produce Telmisartan Form A crystals, which are filtered and dried.
EXAMPLE-8
PREPARATION OF 4′-[[4-METHYL-O-(I-METHYL-Z-BENZIMIDAZOLYL) ^- PROPYL-1-BENZIMIDAZOLYL] METHYL]-Z-BIPHENYLCARBOXYLIC
ACID [TELMISARTAN]
Powdered sodium hydroxide (6.83 g) was added in N,N~dimethylformamide (175 ml) at 4°C followed by 4-methyl-6-(l-methyl-2-benzimidazolyl)-2 -propyl- 1- benzimidazole monohydrate (50 g) and stirred for 5 min. Thereafter, methyl-2-[4′- (bromomethylphenyl)]benzoate (54.76 g) was added at 0°C and stirred to the reaction mass till completion of the reaction. Methylene chloride (250 ml) was added, followed by water (500 ml) at 20C and stirred for 10 min. The aqueous layer was separated and extracted with methylene chloride (50 ml). The combined organic extract was washed with water (250 ml) to obtain 380 ml of the organic solution containing Telmisartan methyl ester. 320 ml of this organic layer was concentrated at ambient pressure to collect 210 ml of the distillate. Methanol (120 ml) was added to the concentrated mass and distilled to collect 96 ml of the distillate. The concentrated mass was diluted with 160 ml of methanol at 5O0C. Thereafter, aqueous sodium hydroxide solution (17.4 g of NaOH in 40 ml of water) was added at 5O0C and heated to reflux at 69-7O0C and stirred at reflux temperature till completion of hydrolysis reaction. Thereafter, the reaction mass was concentrated under reduced pressure at 60-650C till no more solvent distils. Water (600 ml) and methylene chloride (200 ml) was added to this solution. pH was adjusted to 4 with hydrochloric acid (22 ml, 35% w/w) at 27-28°C. The aqueous layer was separated and extracted with methylene chloride (40 ml). The combined organic layer was washed with water (80 ml) to obtain 280 ml of the organic solution. This is divided in to four parts and taken for isolation of Telmisartan as given below.
Part-1 The organic layer (70 ml) as obtained above was diluted with N,N-dimethylformamide (500 ml) at 27°C and seeded with Telmisartan form-A. The solution was left on standing without stirring for 30 min. The resulting suspension was stirred at 27-28°C for 30 min at this temperature. Solid was filtered, washed with precooled N5N- dimethylformamide (15 ml, -5°C) followed by precooled ethanol (10 ml, -2°C) and dried at 85-900C under reduced pressure to afford 10.1 g of Telmisartan.
Part-2
The organic layer (70 ml) as obtained above was diluted with N,N-dimethylformamide (50 ml) at 27°C and seeded with Telmisartan form-A. The solution was left on standing without stirring for 30 min. The resulting suspension was concentrated under reduced pressure at 65-700C to collect 30 ml of the distillate. Thereafter, the concentrated mass was cooled to -5°C and stirred for 30 min at this temperature. Product was filtered, washed with precooled N,N-dimethylformamide (15 ml, -3°C) followed by precooled ethanol (10 ml, -2°C) and dried at 85-900C under reduced pressure to afford 11.4 g of Telmisartan.
Part-3
The organic layer (70 ml) as obtained above was diluted with N,N-dimethylformamide (60 ml) at 27°C and seeded with Telmisartan form-A. The solution was left on standing without stirring for 30 min. The resulting suspension was concentrated under reduced pressure at 65-70°C to collect 50 ml of the distillate. Thereafter, stirred at 30°C for 15 min, cooled to -5°C and stirred for 30 min at this temperature. Product was filtered, washed with precooled N,N-dimethylformamide (15 ml, -5°C) followed by precooled ethanol (10 ml, -20C) and dried at 85-900C under reduced pressure to afford 11.7 g of Telmisartan.
Part-4
The organic layer (70 ml) as obtained above was diluted with N,N-dimethylformamide (40 ml) at 27°C arid seeded with Telmisartan form-A. The solution was left on standing without stirring for 30 min. The resulting suspension was concentrated under reduced pressure at 65-700C to collect 45 ml of the distillate. Thereafter, stirred at 300C for 15 min, cooled to -5°C and stirred for 30 min at this temperature. Product was filtered, washed with precooled N,N-dimethylformamide (15 ml, -5°C) followed by precooled ethanol (10 ml, -20C) and dried at 85-900C under reduced pressure to afford 12.3 g of Telmisartan.
| DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, LI-YAN: “Study on the optimization synthesis of telmisartan” XP002548425 retrieved from STN Database accession no. 2007:587025 & HUAXUE GONGCHENGSHI , 21(3), 60-61 CODEN: HGUOAP; ISSN: 1002-1124, 2007, |
| WO2006044754A2 * | 18 Oct 2005 | 27 Apr 2006 | Muthulingam Arunagiri | Process for preparing telmisartan |
| WO2009006860A2 * | 8 Jul 2008 | 15 Jan 2009 | Zentiva As | A method of manufacturing 4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan) |
| EP0502314A1 * | 31 Jan 1992 | 9 Sep 1992 | Dr. Karl Thomae GmbH | Benzimidazol, medicaments containing them and process for their preparation |
| US20060211866 * | 21 Mar 2006 | 21 Sep 2006 | Glenmark Pharmaceuticals Limited | Process for the preparation of angiotensin receptor blockers and intermediates thereof |
| US6737432 | 30 Oct 2002 | 18 May 2004 | Boehringer Ingelheim Pharma Kg | Crystalline form of telmisartan sodium |
| US7193089 | 17 Mar 2004 | 20 Mar 2007 | Boehringer Ingelheim International Gmbh | Process for manufacture of telmisartan |
| US7501448 | 13 Oct 2005 | 10 Mar 2009 | Teva Pharmaceutical Industries, Ltd. | high yields, low cost process; easy solvent extraction; nontoxic, safe, environmentally friendly, low boiling point organic solvents; 1,7′-dimethyl-2′-propyl-1H,3’H-[2,5′]bibenzoimidazolyl is reacted with 4’bromomethyl-biphenyl-2-carboxylic acid alkyl ester; industrial scale; hydrolysis |
| US7511145 | 30 Jul 2004 | 31 Mar 2009 | Genelabs Technologies, Inc. | Bicyclic heteroaryl derivatives |
| US7943781 | 18 Oct 2005 | 17 May 2011 | Dr. Reddy’s Laboratories Limited | Process for preparing telmisartan |
| US8541593 * | 20 Jun 2008 | 24 Sep 2013 | Amgen Inc. | Process for making substituted 2-amino-thiazolones |
| US8691999 | 10 May 2005 | 8 Apr 2014 | Cipla Limited | Process for the preparation of telmisartan |
| US20100222402 * | 8 Jul 2008 | 2 Sep 2010 | Jan Stach | Method of manufacturing 4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan) |
| US20100280255 * | 20 Jun 2008 | 4 Nov 2010 | Amgen Inc. | Process for making substituted 2-amino-thiazolones |
| US20120196896 * | 17 Sep 2010 | 2 Aug 2012 | Georgetown University | Treatment for Oxidative Stress and/or Hypertension |
| EP2420231A2 | 4 Oct 2007 | 22 Feb 2012 | Boehringer Ingelheim Vetmedica GmbH | Angiotensin II receptor antagonist for the prevention or treatment of cardiovascular disease in cats |
| EP2420232A2 | 4 Oct 2007 | 22 Feb 2012 | Boehringer Ingelheim Vetmedica GmbH | Angiotensin II receptor antagonist for the prevention or treatment of cardiovascular diseases in cats |
| EP2420233A2 | 4 Oct 2007 | 22 Feb 2012 | Boehringer Ingelheim Vetmedica GmbH | Angiotensin II receptor antagonist for the prevention or treatment of systemic diseases in cats |
| WO2005108375A1 * | 10 May 2005 | 17 Nov 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| WO2009006860A2 * | 8 Jul 2008 | 15 Jan 2009 | Zentiva As | A method of manufacturing 4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan) |
| WO2010075347A2 | 22 Dec 2009 | 1 Jul 2010 | Takeda Pharmaceutical Company Limited | Methods of treating hypertension with at least one angiotensin ii receptor blocker and chlorthalidone |
| EP0502314B1 | 31 Jan 1992 | 20 May 1998 | Dr. Karl Thomae GmbH | Benzimidazol derivatives, medicaments containing them and process for their preparation |
| WO2010018441A2 * | 10 Aug 2009 | 18 Feb 2010 | Cadila Pharmaceuticals Ltd. | An improved process for the preparation of substantially pure telmisartan |
| WO2010146187A2 * | 21 Jun 2010 | 23 Dec 2010 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Process for the preparation of telmisartan |
| WO2011077444A1 * | 28 May 2010 | 30 Jun 2011 | Inogent Laboratories Private Limited | A new process for the preparation of pure telmisartan |
| WO2012028925A2 * | 29 Aug 2011 | 8 Mar 2012 | Ogene Systems (I) Pvt Ltd | An improved process for the preparation of telmisartan |
| CN1768044A * | 26 Mar 2004 | 3 May 2006 | 贝林格尔·英格海姆国际有限公司 | Process for manufacture of telmisartan |
| CN102731407A * | 4 Jul 2012 | 17 Oct 2012 | 宁波九胜创新医药科技有限公司 | Method for preparing telmisartan |
| EP0627433A1 * | 7 Dec 1993 | 7 Dec 1994 | Eisai Co., Ltd. | Process for producing imidazopyridine derivative and intermediate |
| EP2123648A1 * | 20 May 2008 | 25 Nov 2009 | Chemo Ibérica, S.A. | A process for the preparation of Telmisartan. |
| EP2305650A1 * | 21 Sep 2009 | 6 Apr 2011 | Chemo Ibérica, S.A. | Novel process for the preparation of telmisartan |
| EP0502314B1 | 31 Jan 1992 | 20 May 1998 | Dr. Karl Thomae GmbH | Benzimidazol derivatives, medicaments containing them and process for their preparation |
| EP1719766A2 | 18 Apr 2006 | 8 Nov 2006 | Dipharma S.p.A. | A process for the preparation of telmisartan |
| EP1878735A1 | 28 Jun 2007 | 16 Jan 2008 | Dipharma Francis S.r.l. | Process for the preparation of boronic acids and derivatives thereof |
| WO2004087676A1 | 26 Mar 2004 | 14 Oct 2004 | Boehringer Ingelheim Int | Method for the production of telmisartan |
| WO2006044648A1 | 13 Oct 2005 | 27 Apr 2006 | Teva Pharma | Process for preparing telmisartan |
| WO2006136916A2 * | 20 Jun 2006 | 28 Dec 2006 | Glenmark Pharmaceuticals Ltd | Substantially pure micronized particles of telmisartan and pharmaceutical compositions containing same |
| EP1878735A1 * | 28 Jun 2007 | 16 Jan 2008 | Dipharma Francis S.r.l. | Process for the preparation of boronic acids and derivatives thereof |
| EP2103609A1 * | 20 Mar 2008 | 23 Sep 2009 | Lek Pharmaceuticals D.D. | Catalyzed carbonylation in the synthesis of angiotensin ii antagonists |
| EP2123648A1 | 20 May 2008 | 25 Nov 2009 | Chemo Ibérica, S.A. | A process for the preparation of Telmisartan. |
| EP2149566A1 | 15 Jul 2008 | 3 Feb 2010 | Chemo Ibérica, S.A. | A process for the preparation of telmisartan |
| EP2305650A1 | 21 Sep 2009 | 6 Apr 2011 | Chemo Ibérica, S.A. | Novel process for the preparation of telmisartan |
| CN102015690B | 19 Mar 2009 | 30 Apr 2014 | 力奇制药公司 | Catalyzed carbonylation in the synthesis of angiotensin II antagonists |
| US8410285 | 19 Mar 2009 | 2 Apr 2013 | Lek Pharmaceuticals D.D. | 2′-halobiphenyl-4-yl intermediates in the synthesis of angiotensin II antagonists |
| US8445693 | 19 Mar 2009 | 21 May 2013 | Lek Pharmaceuticals D.D. | Catalyzed carbonylation in the synthesis of angiotensin II antagonists |
| WO2009115585A1 * | 19 Mar 2009 | 24 Sep 2009 | Lek Pharmaceuticals D.D. | Catalyzed carbonylation in the synthesis of angiotensin ii antagonists |
| WO2009123483A1 | 30 Mar 2009 | 8 Oct 2009 | Zaklady Farmaceutyczne Polpharma Sa | Process for preparation of telmisartan |
| WO2010018441A2 * | 10 Aug 2009 | 18 Feb 2010 | Cadila Pharmaceuticals Ltd. | An improved process for the preparation of substantially pure telmisartan |
| WO2010146187A2 | 21 Jun 2010 | 23 Dec 2010 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Process for the preparation of telmisartan |
| EP0502314A1 * | 31 Jan 1992 | 9 Sep 1992 | Dr. Karl Thomae GmbH | Benzimidazol, medicaments containing them and process for their preparation |
| DE4142366A1 * | 20 Dec 1991 | 24 Jun 1993 | Thomae Gmbh Dr K | New phenylalkyl derivs. – are angiotensin II antagonists used to treat hypertension, coronary insufficiency, angina, cns disorders etc. |
| US20040162327 * | 12 Feb 2004 | 19 Aug 2004 | Boehringer Ingelheim Pharma Kg | Treatment of hypertension and cardiac insufficiency, ischaemic peripheral circulatory disorders, diabetic neuropathy, glaucoma, gastrointestinal diseases and bladder diseases |
| WO2004087676A1 * | 26 Mar 2004 | 14 Oct 2004 | Boehringer Ingelheim Int | Method for the production of telmisartan |
| WO2005108375A1 | 10 May 2005 | 17 Nov 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| WO2006044648A1 * | 13 Oct 2005 | 27 Apr 2006 | Teva Pharma | Process for preparing telmisartan |
| WO2006044754A2 * | 18 Oct 2005 | 27 Apr 2006 | Muthulingam Arunagiri | Process for preparing telmisartan |
| US6358986 * | 10 Jan 2000 | 19 Mar 2002 | Boehringer Ingelheim Pharma Kg | Polymorphs of telmisartan |
| US20040236113 | 17 Mar 2004 | 25 Nov 2004 | Boehringer Ingelheim International Gmbh | Process for manufacture of telmisartan |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010004385A1 * | 15 Jun 2009 | 14 Jan 2010 | Aurobindo Pharma Limited | Process for the preparation of pure 4′-[4-methyl-6-(1-methyl-2-benzimidazolyl)-2-propyl-1-benzimidazolyl]methyl]-2-biphenylcarboxylic acid |
| WO2010018441A2 * | 10 Aug 2009 | 18 Feb 2010 | Cadila Pharmaceuticals Ltd. | An improved process for the preparation of substantially pure telmisartan |
| WO2012055941A1 | 26 Oct 2011 | 3 May 2012 | Krka,Tovarna Zdravil, D. D., Novo Mesto | Multilayer pharmaceutical composition comprising telmisartan and amlodipine |
| WO2005108375A1 * | 10 May 2005 | 17 Nov 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| WO2006044754A2 * | 18 Oct 2005 | 27 Apr 2006 | Muthulingam Arunagiri | Process for preparing telmisartan |
| WO2009006860A2 * | 8 Jul 2008 | 15 Jan 2009 | Zentiva As | A method of manufacturing 4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan) |
| EP1719766A2 * | 18 Apr 2006 | 8 Nov 2006 | Dipharma S.p.A. | A process for the preparation of telmisartan |
| US20060211866 * | 21 Mar 2006 | 21 Sep 2006 | Glenmark Pharmaceuticals Limited | Process for the preparation of angiotensin receptor blockers and intermediates thereof |
| US20060276525 * | 17 May 2006 | 7 Dec 2006 | Itai Adin | Processes of preparing highly pure telmisartan form A, suitable for pharmaceutical compositions |
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

