
LINEZOLID
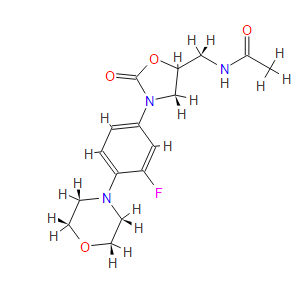
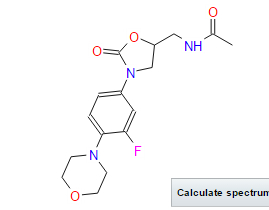
N- [[(5S) – 3 – [3 -Fluoro-4- (4-morpholinyl)phenyl] -2-oxo- 5 -oxazolidinyl] methyl] acetamide and marketed by Pfizer in US under brand name Zyvox. Linezolid is a synthetic antibacterial agent of the oxazolidinone class. It is used for the treatment of infections caused by multi-resistant bacteria including streptococci and methicillin-resistant Staphylococcus aureus.

(S)-N-[[3-(3-fluoro-4-morpholinylphenyl)-2-oxo-5-oxazolidinyl]methyl] acetamide.
N-[[(5s)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide
PRODUCT PATENTUS5688792 (1997 to Pharmacia & Upjohn) |
| CAS No.: |
165800-03-3 |
| Synonyms: |
|
| Formula: |
C16H20FN3O4 |
| Exact Mass: |
337.14400 |
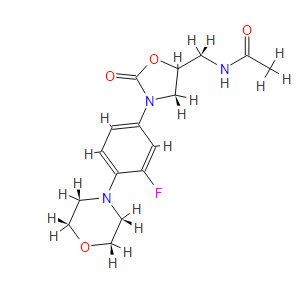

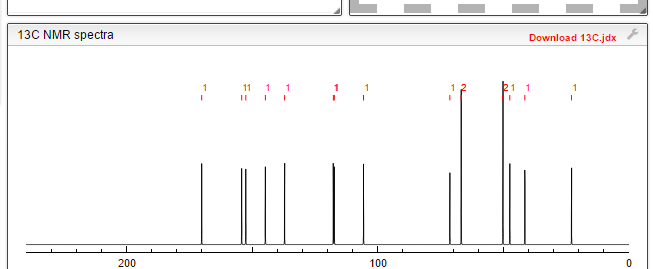
13C
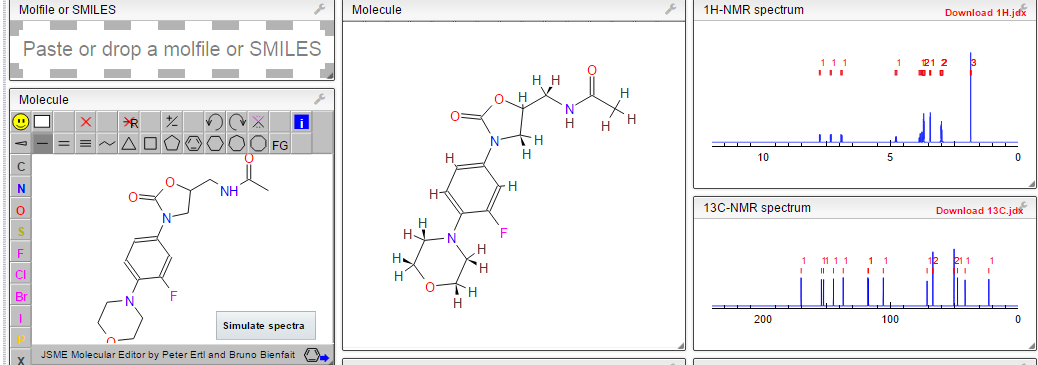
1H NMR AND 13C PREDICT
1H NMR PREDICT
![N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide NMR spectra analysis, Chemical CAS NO. 165800-03-3 NMR spectral analysis, N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-19/000/030/236/165800-03-3-1h.png)
13C NMR PREDICT
![N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide NMR spectra analysis, Chemical CAS NO. 165800-03-3 NMR spectral analysis, N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-19/000/030/236/165800-03-3-13c.png)
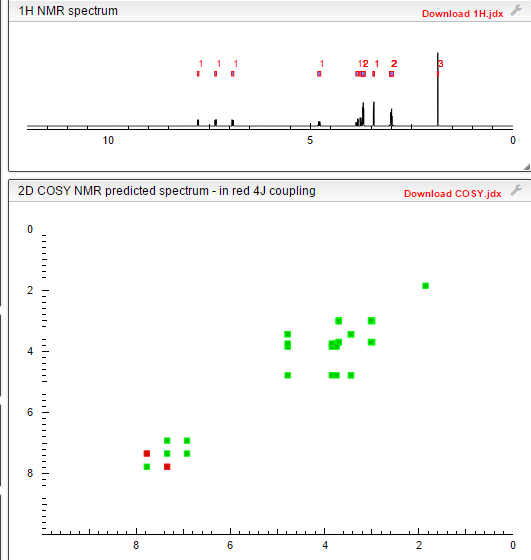
COSY
PREDICT
HMBC
ORIGINAL 1H NMR…………...http://www.selleckchem.com/products/Linezolid(Zyvox).html
INTERMEDIATES USED
Arkivoc, , vol. 2012, # 6 p. 45 – 56
WO2011/137222 A1, ;

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 11 ;

THE REGENTS OF THE UNIVERSITY OF CALIFORNIA; GARG, Neil K.; RAMGREN, Stephen D.; SILBERSTEIN, Amanda L.; QUASDORF, Kyle W. Patent: WO2012/94622 A2, 2012 ; Location in patent: Page/Page column 31-32 ;

Lianhe Chemical Technology Co., Ltd. Patent: EP2388251 A1, 2011 ; Location in patent: Page/Page column 11 ;
Tammana, Rajesh; Vemula, Kiran Kumar; Guruvindapalli, Ramadasu; Yanamandr, Ramesh; Gutta, Madhusudhan Arkivoc, 2012 , vol. 2012, # 6 p. 45 – 56

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 10 ;

Song, Lirong; Chen, Xiaobei; Zhang, Shilei; Zhang, Haoyi; Li, Ping; Luo, Guangshun; Liu, Wenjing; Duan, Wenhu; Wang, Wei Organic Letters, 2008 , vol. 10, # 23 p. 5489 – 5492

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 10 ;

JUBILANT LIFE SCIENCES LIMITED; BISWAS, Sujay; PANDA, Atulya, Kumar; GUPTA, Ashish, Kumar; SINGH, Shishupal; TIWARI, Praveen; VIR, Dharam; THOMAS, Saji Patent: WO2013/111048 A1, 2013 ; Location in patent: Page/Page column 24; 25 ;

Perrault, William R.; Pearlman, Bruce A.; Godrej, Delara B.; Jeganathan, Azhwarsamy; Yamagata, Koji; Chen, Jiong J.; Lu, Cuong V.; Herrinton, Paul M.; Gadwood, Robert C.; Chan, Lai; Lyster, Mark A.; Maloney, Mark T.; Moeslein, Jeffery A.; Greene, Meredith L.; Barbachyn, Michael R. Organic Process Research and Development, 2003 , vol. 7, # 4 p. 533 – 546

US6362334 B1, ; Example 13 ;

NMR OF INTERMEDTIATES
………….
…….
………
……………
……………

-
Linezolid is a pharmaceutically active compound useful as an antibacterial agent, e.g. for the treatment of diabetic food infections caused by Gram-positive bacteria. It is represented by the formula (I),
-
[0003]
The marketed pharmaceutical compositions are a sterile isotonic solution for an i.v. infusion, a tablet for oral administration and an aqueous suspension for oral administration. They are marketed, i.e., under brand name ZYVOX by Pfizer.
-
[0004]
The molecule of linezolid has one asymmetric carbon in the molecule allowing for 2 enantiomers; the marketed compound is the (S)-enantiomer. In the above-marketed compositions, linezolid is present as a free base.
-
[0005]
Hereinunder, the name linezolid will be used as the generic name for N-(3-(3-fluoro-4-(morpholin-4-yl)phenyl)-2-oxooxazolidin-5(S)-ylmethyl)acetamide, unless indicated to the contrary.
-
[0006]
-
[0007]
Various processes for making linezolid are known in the art. In particular, the important ones are these, the final step of which comprises acetylation of an amine precursor of the formula (II) with an acetylhalide or acetic anhydride (see, e.g.,
WO 2005 099353 ),
-
[0008]
This amine precursor (II) may be made from various starting materials, e.g.:
- a) By a reduction of an azide compound of formula (III) by a suitable reductant ( WO2006/091731 , WO 95/07271 , US 5837870 , WO2009/063505 , US 7291614 ),
The starting compound (III) may be made from the corresponding tosylate or chloride of general formula (VII) below ( WO 2005/099353 ).
- b) By a decomposition of a phthalimide compound of formula (IV), e.g. by methylamine ( WO95/07271 ) or by hydrazine ( US 5837870 ),
The starting compound (IV) may be made from the same tosylate or chloride as sub a) ( WO2005/099353 ) or by a cyclization of the oxazolidine ring ( WO 99/24393 , WO2006/008754 ).
- c) From a sulfonate compound of formula (V),
by treatment with ammonium hydroxide in isopropanol or THF ( WO 95/07271 ) or by treatment with ammonia under enhanced pressure ( WO 97/37980 ).
- d) By a reduction of an imine (VI),
wherein R2 is a chlorophenyl, bromophenyl or 2,4,-dichlorophenyl moiety ( WO 2007/116284 ).
-
[0009]
Except of the imine (VI), each of the preceded synthetic approaches is based on a step of converting a starting material of the general formula (VII),

wherein L is a suitable leaving group, for instance a halogen or an alkyl-or aryl sulfonyloxy group,
by a reaction with a nitrogen nucleophile (an azide salt, phthalimide salt, ammonia or ammonium hydroxide), followed, if necessary, by a next step of conversion of the formed reaction intermediate (e.g., compound (III) or compound (IV)) into the amino/compound (II). Apparently, making the starting amine-compound (II) in a good yield and purity is the key aspect of commercial success of any of the above synthetic routes yielding linezolid. However, the known approaches have various drawbacks, for instance serious toxicity and explosion hazard of the azide salts, long reaction times and hazardous agents (hydrazine, methyl amine) in using the phthalimide intermediate, low yields and many side products at the ammonium hydroxide approach, or harsh reaction conditions in reaction with ammonia.
Linezolid [(S)-N-[[3-(3-Fluoro-4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide] is an antimicrobial agent. Linezolid is an oxazolidinone, having the empirical formula C16H20FN3O4 and the following structure (1):

Linezolid (1) is described in The Merck Index (13th edition, Monograph number: 05526, CAS Registry Number: 165800-03-3) as white crystals, with a melting point of 181.5-182.5°. Linezolid (1), as well as a process for its preparation, is disclosed in U.S. Pat. No. 5,688,792 (Example 5), European Patent No. 717738, Israeli Patent No. 110,802, Canadian Patent No. 2,168,560, and International Patent Publication WO 95/07271.
U.S. Pat. No. 5,688,792 discloses the antibacterial agent linezolid as well as a process for its preparation. EXAMPLE 5 reports the linezolid produced had a mp of 181.5-182.5°.
There are many other disclosures of processes to prepare linezolid. J. Med. Chem., 39(3), 673-9 (1996) reports the linezolid was, “recrystallized from ethyl acetate and hexanes . . . white crystals, m.p. 181.5-182.5C.” It also sets forth the IR spectrum as “3284, 3092, 1753, 1728, 1649, 1565, 1519, 1447, 1435”.
Tetrahedron Lett., 40(26), 4855 (1999) discloses linezolid and a process to prepare linezolid. However, this document does not set forth the melting point or IR spectrum of the linezolid prepared.
U.S. Pat. No. 5,837,870 (International Publication WO97/37980 of PCT/US97/03458) discloses a process to prepare linezolid. Linezolid is described in EXAMPLE 18, which does not set forth the melting point or IR spectrum of the linezolid prepared.
International Publication WO99/24393 of PCT/US98/20934 discloses a process to prepare linezolid. Linezolid is described in EXAMPLES 8, 9 and 12 which do no set forth the melting point or IR spectrum of the linezolid prepared.
The form of linezolid being used in the clinical trials to support the filing of the NDA is Form II.
Linezolid (1) is marketed in the United States by Pfizer, Inc. as an injection, tablets, and oral suspension under the name ZYVOX®. Its main indications are nosocomial pneumonia, skin and skin-structure infections, and vancomycin-resistant Enterococcus faecium infections.
U.S. Pat. No. 5,688,792 claims linezolid (1) and its use for the treatment of microbial infections. This patent also discloses, but does not claim, the following method of preparation:
This method of preparation was also disclosed in Bricker, et al., J. Med. Chem., 39 673 -679 (1996), where it was stated that the above route avoids the use of phosgene to make the carbamate precursor of the oxazolidinone ring. The authors also disclose that the use of NaN3 can be avoided by using potassium phthalimide, followed by deblocking of the phthalimide with aqueous methyl amine.
In the above-described synthesis, the intermediate amine, S-N-(4-morpholinyl-3-fluorophenyl)-2-oxo-5-oxazolidinyl-methyl amine (2)
is reacted without isolation with acetic anhydride as an oily product, or in solution, to produce the acetamide, linezolid (1). This is followed by procedures for isolating the linezolid (1) such as those described in U.S. Pat. No. 5,688,792, at col. 15, 11. 22-28 (chromatography and separation of the desired fraction, followed by evaporation and trituration of the product to obtain pure linezolid (1)).
In the above-described syntheses, the intermediate azide R-N-(4-morpholinyl-3-fluorophenyl)-2-oxo-5-oxazolidinyl-methyl azide (3)
is reduced to its corresponding amine, S-N-(4-morpholinyl-3-fluorophenyl)-2-oxo-5-oxazolidinyl-methyl amine (2) in the solvent ethyl acetate by hydrogenation using hydrogen gas and a palladium/carbon catalyst. These reaction conditions lead to the production of an undesirable level of reaction by-products, and, following the acetylation of the intermediate amine (2) to linezolid (1), to undesirably high levels of bis-linezolid (4)
http://www.google.com/patents/US20060252932
FIG. 1 shows the 1H-NMR spectrum of bis-linezolid (4)
FIG. 2 shows the 13C-NMR spectrum of bis-linezolid (4)
FIG. 3 shows the IR spectrum of bis-linezolid (4)

A Novel Synthesis of Oxazolidinone Derivatives (A Key Intermediate of Linezolid)
Pingili Krishna Reddy1,2, K. Mukkanti2 and Dodda Mohan Rao1*
1Symed Research Centre, Plot No. 89/A, Phase-I, Shapoornagar, IDA Jeedimetla, Hyderabad, Andhra Pradesh, India
2Center for Pharmaceutical sciences, Jawaharlal Nehru Technological University, Kukatpally, Hyderabad, India
http://www.orientjchem.org/vol29no3/a-novel-synthesis-of-oxazolidinone-derivatives-a-key-intermediate-of-linezolid/

Reddy P. K, Mukkanti K, Rao D. M. A Novel Synthesis of Oxazolidinone Derivatives (A Key Intermediate of Linezolid). Orient J Chem 2013;29(3). doi :
http://dx.doi.org/10.13005/ojc/290322N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide (7a):
IR (KBr, cm-1): 3338 (N-H stretching), 3117, 3066 (aromatic C-H stretching), 2971, 2863, 2818 (aliphatic C-H stretching), 1738, 1662 (C=O stretching), 1545, 1516,1453 (aromatic C=C stretching), 1425 (C-N stretching), 1381 (aliphatic C-H bending), 1334 (C-F stretching), 1274 (C-O stretching), 1198, 1177 (C-N bending), 1117, 1081 (aromatic C-H bending).
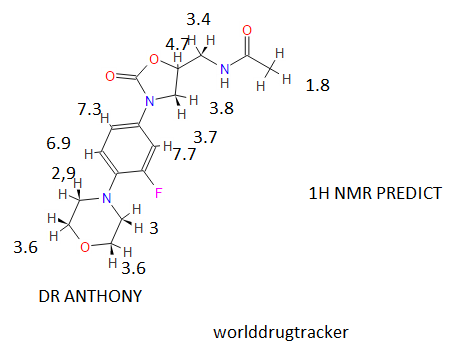
1H NMR (CDCl3) δ ppm: 7.44 (m, 1H), 7.26 (m, 1H), 6.99 (m, 1H), 6.01 (t,1H), 4.76 (m, 1H), 4.02 (m, 2H), 3.80 (m, 4H), 3.61(m, 2H), 3.05 (m, 4H), 2.02 (t, 3H):
C13NMR(CDCl3) δppm: 171.33, 156.87, 154.44, 136.40, 132.84, 118.67, 113.81, 107.52, 71.96, 66.76, 50.79, 47.46, 41.68, 22.81. MS: 338 (M++H);
……………………………………………………………………….
ARKIVOC 2012 (vi) 45-56 Page 45 ©ARKAT-USA, Inc.
An expeditious construction of 3-aryl-5-(substituted methyl)-2- oxazolidinones: a short and efficient synthesis of Linezolid
Rajesh Tammana,a,b Kiran Kumar Vemula,a Ramadasu Guruvindapalli,a Ramesh Yanamandra,c and Madhusudhan Gutta* a
aDepartment of Research & Development, Inogent Laboratories Pvt. Ltd.,
A GVK BIO Company, 28A, IDA, Nacharam, Hyderabad 500 076, Andhra Pradesh, India
bCentre for Pharmaceutical Sciences, Institute of Science and Technology, Jawaharlal Nehru Technological University, Hyderabad 500 072, Andhra Pradesh, India
cDepartment of Analytical Research & Development, GVK Biosciences Pvt. Ltd., 28A, IDA, Nacharam, Hyderabad 500 076, Andhra Pradesh, India
E-mail: madhusudhan.gutta@inogent.com
http://www.arkat-usa.org/get-file/42622/
N-(((S)-3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)acetamide 1 (Linezolid) 1 was prepared according to the method described in literature.12,15
Mp 182-183 °C, (lit.12a 181.5- 182.5 °C); enantiomeric purity 99.9% (by chiral HPLC);
IR (KBr): ν 3343 (NH), 3075 (Ar-H), 2967 (CH), 1741 (C═O), 1660 (C═O) cm-1 ;
1H NMR (CDCl3): δ 2.03 (s, 3H), 3.04-3.07 (t, 4H), 3.56-3.77 (m, 3H), 3.86-3.89 (t, 4H), 4.00-4.06 (t, 1H), 4.74-4.79 (m, 1H), 5.96 (s, 1H), 6.90- 6.96 (t, 1H), 7.06-7.10 (d, 1H), 7.43-7.48 (d, 1H).
13C NMR (DMSO-d6): δ 22.4, 41.4, 47.3, 50.6, 66.1, 71.5, 106.4, 114.0, 119.1, 133.3, 135.5, 154.0, 156.2, 170.0;
ESI-MS (C16H20FN3O4): m/z (%) 338.18 (100, M+ +1).
12. (a) Brickner, S. J.; Hutchinson, D. K.; Barbachyn, M. R.; Manninen, P. R.; Ulanowicz, D. A.;
Garmon, S. A.; Grega, K. C.; Hendges, S. K.; Toops, D. S.; Ford, C. W.; Zurenko, G. E. J.
Med. Chem. 1996, 39, 673. (b) Barbachyn, M. R.; Brickner, S. J.; Hutchinson, D. K. U.S.
patent 5688792; 1997; Chem. Abstr. 1995, 123, 256742. (c) Dhananjay, G. S.; Nandu, B. B.;
Avinash, V. N.; Kamlesh, D. S.; Anindya, S. B.; Tushar, A. N. PCT Int. Appl. 063505, 2009;
Chem. Abstr. 2009, 150, 515152.
13. (a) Imbordino, R. J.; Perrault, W. R.; Reeder, M. R. PCT Int. Appl. 116284, 2007; Chem.
Abstr. 2007, 147, 469356. (b) Pearlman, B. A.; Perrault, W. R.; Barbachyn, M. R.;
Manninen, P. R.; Toops, D. S.; Houser, D. J.; Fleck, T. J. U.S. Patent 5837870, 1998; Chem.
Abstr. 1998, 130, 25061. (c) Perrault, W. R.; Pearlman, B. A.; Godrej, D. B.; Jeganathan, A.;
Yamagata, K.; Chen, J. J.; Lu, C. V.; Herrinton, P. M.; Gadwood, R. C.; Chan, L.; Lyster, M.
A.; Maloney, M. T.; Moeslein, J. A.; Greene, M. L.; Barbachyn, M. R. Org. Proc. Res. Dev.
2003, 7, 533.
14. (a) Yu, D. S.; Huang, L.; Liang, H.; Gong, P. Chin. Chem. Lett. 2005, 16, 875. (b) Pearlman,
B. A. PCT Int. Appl. 9924393, 1999; Chem. Abstr. 1999, 130, 338099. (c) Weigert, F. J. J.
Org. Chem. 1973, 38, 1316.
15. (a) Wang, M.; Tong, H. CN patent 101220001, 2008. (b) Mohan Rao, D.; Krishna Reddy, P.
PCT Int. Appl. 099353, 2005; Chem. Abstr. 2005, 143, 440395. (c) Mohan Rao, D.; Krishna
Reddy, P. PCT Int. Appl. 008754, 2006; Chem. Abstr. 2006, 144, 170978.
……………………………………
Org. Proc. Res. Dev., 2003, 7 (4), pp 533–546
DOI: 10.1021/op034028h
Organic Process Research and Development, 2003 , vol. 7, # 4 p. 533 – 546
http://pubs.acs.org/doi/abs/10.1021/op034028h

Since 1993, a significant process research and development effort directed towards the large-scale synthesis of oxazolidinone antibacterial agents has been ongoing in both Early Chemical Process Research and Development, and Chemical Process Research and Development at Pharmacia. This work has led to the successful development of the current commercial process to produce Zyvox (linezolid), recently approved by the FDA as an antibacterial. While this synthesis is appropriate for the preparation of linezolid in particular, a more convergent and versatile synthesis was developed for the rapid preparation of numerous other oxazolidinone analogues. Toward this end, economical methods for the large-scale preparation of N-[(2S)-2-(acetyloxy)-3-chloropropyl]acetamide 3 and tert-butyl [(2S)-3-chloro-2-hydroxypropyl]carbamate 27 from commercially available (S)-epichlorohydrin via the common intermediate (2S)-1-amino-3-chloro-2-propanol hydrochloride 2a were developed. Also, general methods for coupling these reagents with N-aryl carbamates to giveN-aryl-5(S)-aminomethyl-2-oxazolidinone derivatives in one step were developed. These reagents and procedures have proven widely applicable in the preparation of a diverse array of oxazolidinone analogues such as 23 and 28 in both process and medicinal chemistry research.

(S)-N-[[3-[3-Fluoro-4-(4-morpholinyl)phenyl]-2-oxo- 5-oxazolidinyl]methyl]acetamide: Linezolid: Zyvox
HPLC analyses showed the first and second crops to be 98.9 and 94.6 wt % linezolid, respectively, with <0.2% enantiomer in each; also, an additional 9.7% yield of linezolid was detected in the filtrate by external standard HPLC (total ) 80.6%). Analysis data for 1st crop material: mp ) 73-76 °C;
1 H NMR (CDCl3, 400 MHz)
δ 7.43 (dd, J ) 14.4, 2.4 Hz, 1H), 7.07 (dd, J ) 8.8, 2.0 Hz, 1H), 6.91 (t, J ) 8.8 Hz, 1H), 6.43 (br t, 1H), 4.77 (m, 1H), 4.02 (t, J ) 9.2 Hz, 1H), 3.86 (t, J ) 4.4 Hz, 4H), 3.76 (dd, J ) 8.8, 6.8 Hz, 1H), 3.66 (m, 2H), 3.05 (t, J ) 4.8 Hz, 4H), 2.02 (s, 3H);
13C NMR (CDCl3, 100 MHz)
δ 23.07 (q), 41.93 (t), 47.66 (t), 51.00 (t), 66.95 (t), 71.99 (d), 107.56 (dd, JC-F ) 26.16 Hz), 113.97 (dd, JC-F ) 3.02 Hz), 118.85 (dd, JC-F ) 4.03 Hz), 132.90 (sd, JC-F ) 4.03 Hz), 136.58 (sd, JC-F ) 9.06 Hz), 154.42 (s), 155.50(sd, JC-F ) 246.53 Hz), 171.19 (s)
MS (EI) m/z (relative intensity) 337 (90), 293 (81), 209 (100);
[R]25D ) -16 (c ) 1.05, ethanol).
Anal. Calcd for C16H20FN3O4: C, 56.97; H, 5.97; N, 12.46; found: C, 56.86; H, 6.05; N, 12.44
HPLC (99.0 wt %, 98.9 area % linezolid, tR 1.60 min) conditions: InertsilODS-2 5.0 µm 150 mm × 4.6 mm, flow rate ) 2.0 mL/ min, gradient elution from 40:60 A:B to 80:20 A:B over 10 min; A ) acetonitrile; B ) water. External standard HPLC analysis of the filtrate showed
d 12.9% and 7.6% yield of linezolid and 8, respectively.
SEE HPLC AT http://file.selleckchem.com/downloads/hplc/S140801-Linezolid-Zyvox-HPLC-Selleck.pdf
………………………….
http://www.google.com/patents/WO2007064818A1?cl=en
Linezolid [(S)-N-[[3-(3-Fluoro-4-morpholinyl)phenyl]-2-oxo-5- oxazolidinyljmethyl] acetamide} is an antimicrobial agent. Linezolid is an oxazolidinone, having the empirical formula C16H20FN3O4 and the following structure:
Linezolid
Linezolid is described in The Merck Index (13th edition, Monograph number: 05526, CAS Registry Number: 165800-03-3) as white crystals, with a melting point of 181.5-182.50C. Linezolid, as well as a process for its preparation, is described in U.S. Patent No. 5,688,792 (Example 5), European Patent No. 717738, Israeli Patent No. 110,802, Canadian Patent No. 2,168,560, and International Patent Publication WO 95/07271. Linezolid is marketed in the United States by Pfizer, Inc. as an injection, as tablets, and as an oral suspension under the name ZYVOX®. Its main indications are nosocomial pneumonia, skin and skin-structure infections, and vancomycin-resistant Enterococcus faecium infections.
U.S. Patent No. 5,688,792 describes linezolid and its use for the treatment of microbial infections. This patent also describes the following method for the preparation of linezolid:
This method of preparation was also described in Bricker, et al., J. Med. Chem., 39, 673 — 679 (1996), where it was stated that the above route avoids the use of phosgene to make the carbamate precursor of the oxazolidinone ring. The authors also disclose that the use OfNaN3 can be avoided by using potassium phthalimide, followed by deblocking of the phthalimide with aqueous methyl amine.
An analysis of the commercial tablet ZYVOX® shows the presence of desfluoro linezolid as an impurity of linezolid. An HPLC chromatogram of ZYVOX® is depicted in Figure 1. The desfluoro linezolid haviong a relative retention time (RRT) of 0.69 compared to the retention time of linezolid.
desfluoro linezolid of the following structure:
Desfluoro linezolid
As illustrated in Figure 1, this impurity is ideal for use as a reference standard since it is detectable by HPLC, and yet it is present in much less amounts than linezolid, having a RRT of 0.69 compared to the retention time of linezolid.
The isolated desfluoro linezolid is pure. Preferably it has about 95% purity by weight with respect to other compounds, including linezolid. Preferably, the desfluoro linezolid is isolated in about 99.3% purity by weight. Thus, the isolated desfluoro linezolid contains less than about 5%, preferably less than about 2%, and even more preferably less than about 1%, by weight, linezolid.
The isolated desfluoro linezolid of the present invention can be characterized by data selected from: 1H NMR (400MHz, DMSO-d6) δ (ppm): 1.8a (s), 3.04 (brt), 3.40 (t), 3.68 (m), 3.72 (brt), 4.04 (t), 4.67 (m), 6.95 (d), 6.95 (d), 737 (d), 7.37 (d) and 8.21 (t); 13C NMR (lOOMHz, DMSO-d6) δ (ppm): 22.8, 41.9, 48.0, 49.2, 66.5, 71.7, 115.9, 115.9, 119.9, 119.9, 130.9, 148.0, 154.7, 170.0; EI+m/z (MH+): 319; and IR spectra on KBr at 1523, 1555, 1656, 1731, 2830, 2926, 2968 and 3311 cm‘1.
The isolated desfluoro linezolid of the present invention may be characterized by a 1H NMR, substantially as depicted in figure 2. The isolated desfluoro linezolid of the present invention may be characterized by 13C NMR, substantially as depicted in figure 3. The isolated desfluoro linezolid of the present invention may be characterized by an IR spectrum substantially as depicted in figure 4. The isolated, desfluoro linezolid of the present invention may be characterized by an Mass spectrum substantially as depicted in figure 5. The isolated desfluoro linezolid of the present invention may be prepared by performing the process described in U.S. Patent No. 5,688,792, with l-fluoro-4- nitrobenzene instead of 3,4-difluoronitrobenzene, according to the following scheme:
Desfluoro Linezolid
The desfluoro linezolid of the present invention is isolated by a process comprising the following steps; a) combining (5R)-[[3-[4-(4-morpholinyl)phenyl]-2- oxo-5-oxazolidinyl]methyl]azide with an organic solvent, preferably a C1-C4 alkyl ester or a C6 to C12 aromatic hydrocarbon, more preferably toluene or ethylacetate, most preferably toluene, and hydrogen gas in the presence of a catalyst to obtain a reaction mixture containing (5S)-[[3-[4-(4-morpholinyl)phenyl]-2-oxo-5- oxazolidinyl]methyl] amine; b) filtering the reaction mixture to obtain a solution containing (5S)-[[3-[4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl}methyl]amine; c) adding acetic anhydride to the solution to obtain a precipitate; and d) recovering and drying the precipitate to obtain isolated desfluoro linezolid. Preferably, recovering of the precipitate in step d) is carried out by filtering or decanting. Preferably, the catalyst in step a) is selected from the group consisting of Pd/C, Raney Nickel, and noble metal catalysts, more preferably the catalyst is Pd/C. The isolated desfluoro linezolid of the present invention is useful as a reference marker for linezolid. As such, it may be used in order to detect the desfluoro linezolid impurity in a linezolid sample.
Step 7. Preparation of N-rr(5S)-3-r4-(4-moφholinyl)phenyl1-2-oxo-5- oxazolidinyl]methyl1acetamide (des-fluoro-linezolid). In a IL reactor, 6 g (5R)-[[ 3-(4-morpholinyl)phenyl]-2-oxo-5- oxazolidinyl]methyl]azide were charged with 0.7L toluene followed by 0.6 g Pd/C (10% Pd/C containing 52% water). The system was bubbled with ammonia (gas) during 2 h, and then flushed three times with nitrogen and 3 times with hydrogen. The pressure of hydrogen was set to 1.5 arm. The reaction mixture was stirred at RT and the reaction followed up until completion. The reaction mixture was filtered and the solution was treated with 60 ml acetic anhydride at RT. The precipitate was filtered and dried to obtain 3.3 g of desfluoro linezolid (purity: 99.3%). Desfluorolϊnezolid 1H-NMR and 13C-NMR identification
……………………………………….
http://www.google.com/patents/EP2690100A1?cl=en
Example 3
-
[0034]
To a 25 ml, round-bottomed flask equipped with a magnetic stirring bar was charged “amine” (0.49 g) followed by water (8.30 ml). A heterogeneous mixture was stirred and hydrochloric acid (0.12 mL, 35 %) was added. A homogenous solution was obtained. The solution was cooled down in an ice-water bath to 0°C. Acetic anhydride (0.31 mL) was added followed by sodium bicarbonate (0.45 g). Carbon dioxide was immediately released and a formation of white precipitate was observed. The precipitate was filtered off and the filter cake was washed with water (10 ml). The filter cake was collected and dried (100 mbar) at 70°C overnight. An off-white solid linezolid (0.26 g) was isolated.
…………………………..
PATENT
http://www.google.com/patents/WO2007116284A1?cl=en
Example 3 Preparation of (S)~N-[3-(3-fiuoro~4~morpholin-4-yI-ρhenyI)~2-oxo- oxazolidin-5~ylmethyl]-acetarnide (Linezo!id)
Method A
To (S)-5-{[(4-chloro-benzylidene)-amino]-methyl}-3-(3-fluoro-4-morpholin-4-yl- phenyl)-oxazolidin-2-one (129.5g, 31 mmol, 1.0 eq.) is added ethyl acetate (935 mL) and water (935 mL). To the heterogeneous mixture is added 12M aq. HCl (51.58 mL, 620 mmol, 2.0 eq.). Within minutes, the solid went into solution and the reaction mixture is biphasic. After stirring the emulsion at ambient temperature for 2 hours, HPLC assay showed the hydrolysis reaction to be complete (HPLC conditions: YMC 5μ ODS-AM 150 nm X 4.6 nm column, eluting with CH3CN /water + 0.1% TFA from 20% CH3CN to 80% CH3CN in 8 min at 0.5 mL/min, detecting at 254nm, Retention time of (S)-N-[3-(3-fluoro-4-morpholin-4-yl- phenyl)-2-oxo-oxazolidin-5-ylmethyl]-amine is 3.2 min). The phases are separated, the organic layer is discarded, and the aqueous layer is washed with ethyl acetate (500 mL). CH2Cl2 (900 mL) is added and the pH is adjusted to 6.7 with ~ 25 mL aq. 50% aq. NaOH. With constant stirring, Ac2O (58.49 mL, 620 mmol, 2.0 eq.) is added in one portion and the pH dropped to 2. The pH is then readjusted to 6 using 50% aq. NaOH. The pH is adjusted to ca. 7.1 with 50% aq. NaOH and the phases separated. The aqueous phase is extracted with CHiCl2 (800 mL) and the organics are combined and concentrated to ~1L in volume. Ethyl acetate (IL) is added and the volume is reduced to 1.5 L under vacuum. Another IL of ethyl acetate is added and volume is reduced again to IL under vacuum. The resultant slurry is cooled to 00C and the precipitate collected by vacuum filtration. The resulting solid is washed with ethyl acetate (250 mL). The crude product is dried under vacuum at 500C for 2 hours to give the title compound as Hnezolid crystalline Form I.

Following the general procedure of method A and making non-critical variations, but substituting (S)-5- { [2,4-dichloro-benzylidene)-amino]-methyl } -3-(3-fluoro-4-morphoIin-4-yl- phenyl)-oxazolidin-2-one (example 11) for (S)-5-{[(4-chloro-benzylidene)-amino]- methyl}-3-(3-fluoro-4-morρholin-4-yl-phenyl)-oxazolidin-2-one, the title compound is obtained.
Following the general procedure of method B and making non-critical variations, but substituting (S)-5-{ [4-bromo-benzylidene)-amino] -methyl }-3-(3-fluoro-4-morpholin-4-yl~ phenyl)-oxazolidin-2-one (example 9) for (S)-5-{[(4-chloro-benzylidene)-amino]- methyl}-3-(3-fluoro-4-morph.olin-4-yl-phenyl)-oxazoIidin-2-one, the title compound is obtained.
Example 4 Trituration (convert linezolid crystalline Form I to linezolid crystalline Form E) The product from Example (89.18 g) is transferred to a 3L round bottom flask equipped with a mechanical stirrer, thermocouple and heating mantel. Ethyl acetate (2.23 L, 15 mL/g) is added and seeded with Linezolid form II crystals and the slurry is heated to ca. 500C. A slight exotherm of 30C is observed. After 30 minutes of heating the form change is observable as the solid is changing to long needles. Stirring is continued for 2 hours at 500C, at which time the contents are cooled to ambient temperature and stirred for an additional 30 minutes. The contents are then cooled to 30C for 1.5 hours, filtered and washed with cold ethyl acetate (300 mL total). The resultant solids are dried under vacuum at 50°C for 18 hours to give Linezolid (78.12 g) Form II by XRD, 99.8 wt%, 99.9% ee. HPLC conditions: YMC 5μ ODS-AM 150 nm X 4.6 nm column, etuting with CH3CN /water + 0.1% TFA from 20% CH3CN to 80% CH3CN in 8 min at 0.5 mL/min, detecting at 254nm. TR (Linezolid) = 4.4 min; HPLC conditions: Chiralcel OJ-H 250 nm X 4.6 nm column, eluting with 90% CO2/ 10%MeOH at 3.0 mL/min, detecting at 255 nm. TR [title compound] = 3.6 min; TR (enantiomer of title compound) = 4.1 rain
……………………………………..
http://www.google.com/patents/EP2516408A1?cl=en
The polymorphic form obtained by following process disclosed in U.S. Pat. No. 5,688,792 is designated as Form I. Figure- 1 depicts the PXRD graph of Form I obtained by following prior art process. [15] Disadvantage of the process disclosed in U.S. Pat. No. 5,688,792 is that it involves use of n-butyl lithium. Due to its explosive nature it is difficult to handle at plant scale. Also, the said reaction is carried out at temperature of -78°C, which is difficult to attain during commercial production. Further the intermediate obtained requires purification by column chromatography. Column chromatography is a cumbersome technique and difficult to practice during commercial scale production.
The process for the preparation of Linezolid is also disclosed in Journal of Medicinal Chemistry (1996), 39(3), 673-9, U.S. Pat. Nos. 6,492,555, 5,837,870, 6,887,995, 7,307,163, 7,429,661, etc.
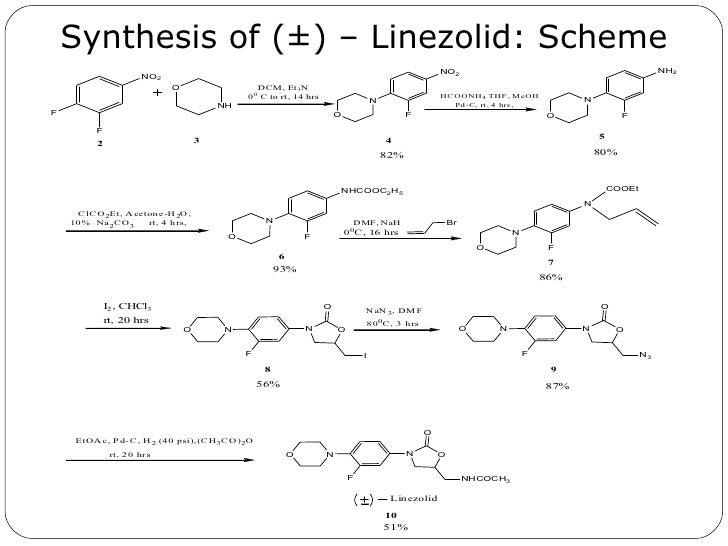
Linezolid was first disclosed in U.S. Pat. No. 5,688,792. The process for synthesis is as disclosed in Scheme-I

The synthetic reaction scheme of the present invention is as shown below.
Scheme-ll
Example 6: Synthesis of Linezolid Crude.
[140] Ethyl acetate (3500ml) and 10% palladium on carbon catalyst (6.0g) are added in autoclave having (R)- [N- 3 – (3 -Fluoro-4-morpholinylphenyl) -2-oxo- 5 -oxazolidinyl] methyl azide (lOOg) at 20-30°C. Cool the reaction mass & maintain 2-3kg hydrogen pressure at 15-20°C for 6-7 hrs. Filter it & wash the hyflo bed by Ethyl acetate
(100mlx2). Then add the Triethyl amine (35. lg) & Acetic anhydride (29.9g) slowly at 25-30°C under stirring. Cool the mix, filter it and wash the solid with chilled (0-5°C) Ethyl acetate (100 ml) followed by water (100mlx2). Finally product is dried at 55-60° C. Yield: 0.85.: Percentage 81%w/w.
[141]
[142] Example 7: Synthesis of Linezolid Pure
[143] Reflux the Acetone (1020ml) and Linezolid crude (lOOg) at 55-60°C for the 30
minutes. Filter the hot turbid solution & wash it with hot (55-60°C) acetone (50ml). Cool the reaction mixture at -5 to 0°C for 1 hour, wash the solid with chilled (-5 to 0°C) acetone (50ml). After drying the Linezolid semi pure (77g) add n-Propanol (308ml) reflux it at 95-100°C for 30 min & filter it by hot solution through hyflo bed. Cool the mix to 0-5°C for 1 hour and wash the solid with chilled (0-5°C) n-Propanol (77ml). Dry the material at 55-60°C. Yield: 0.73.: Percentage 73%w/w.
[144]
[145] Example 8: Synthesis of Linezolid
[146] Ethyl acetate (3500ml) and 10% palladium on carbon catalyst (6.0g) are added in autoclave having (R)- [N- 3 – (3 -Fluoro-4-morpholinylphenyl) -2-oxo- 5 -oxazolidinyl] methyl azide (lOOg) at 20-30°C. Cool the reaction mass & maintain 2-3kg hydrogen pressure at 15-20°C for 6-7 hrs. Filter it & wash the hyflo bed by Ethyl acetate. Distill out ethyl acetate at 75-90°C and then cool the reaction mass to 0-5°C. Add acetone (1000ml) & acetic anhydride (29.9g) at 0-5°C. Further, add Triethyl amine (37.8g) slowly at 0-5°C under stirring. Maintain the reaction mass at 0-5°C for 1-2 hrs. Heat the reaction mass to reflux at 65-75°C for 1 hr. Again cool the reaction mass to 0-5°C fori hr. Filter the solid wash it with acetone and water and dry it at 55-60C. Yield: 0.80.: Percentage 80 w/w.
Example 9: Synthesis of Linezolid Form I
[149] Reflux n-propanol (400ml) and Linezolid (lOOg) at 95-100°C till all solid gets
dissolved. Add activated charcoal (2.0g) and heat for 30 mins. Filter thro hyflo bed. Heat the filtrate and concentrate the solution by partially removing n-propanol. Cool to 0-5°C and filter the solid and dry it at 55-60°C under vacuum. Yield: 0.9. : Percentage 90 w/w.
………………………………………..
https://acs.confex.com/acs/green08/techprogram/P52019.HTM
William R. Perrault, James B. Keeler, William C. Snyder, Christian L. Clark, Michael R. Reeder, Richard J. Imbordino, Rebecca M. Anderson, Nabil Ghazal, Stephen L. Seacrest, and Bruce A. Pearlman. Pfizer, Kalamazoo, MI
Pfizer has developed a novel, convergent, green, second generation synthesis of Linezolid (the active ingredient in ZyvoxTM). The second generation process will replace the launch process after approval by appropriate regulatory agencies and has numerous green chemistry benefits: overall yield is increased by 8%; total waste is reduced by 56%; non-recycled w is eliminated. At current volumes, total waste will be reduced 1.9 million kilograms per year and 1.7 million kg per year non-recyclable waste will be eliminated. The improved process utilizes a highly efficient low dilution convergent synthesis to replace the more dilute linear synthesis utilized in the launch process. The key chlorohydrin imine reagent 1 contains both the chiral center and the key 5-S-aminomethyl moiety of linezolid. In the launch process, S-1-chloro-2,3-propanediol was utilized to install the oxazolidinone functionality. However, this yielded a 5-S-hydroxymethyl group which required activation as the 3-nitrobenzenesulfonate and displacement with excess ammonia to generate the corresponding aminomethyl group of linezolid. The second generation process affords the oxazolidinone imine 3 in the convergent step. The penultimate 5-S-aminomethyl oxazolidinone 4 is then easily formed via hydrolysis with stoichiometric hydrochloric acid. Acylation of this amine with acetic anhydride, utilizing an improved Schotten Baumann reaction, affords high purity linezolid.
………………………………….

……………………………………..

……………………………………….
http://www.google.com/patents/EP2072505A2?cl=en
-
WO 95/07271 , which specifically describes the synthesis of linezolid, namely [(S)-N-[[3-(3-fluoro-4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide], according to the following scheme:
-
[0003]
Other synthetic routes for the preparation of linezolid are reported for example in
US 6107519 and in
Tetrahedron Letters, Vol 37, N° 44, pages 7937-7940, wherein the chiral compound shown below is used instead of glycidyl butyrate as a synthon containing the molecule stereocenter.
-
[0004]
It should be appreciated that all of the known approaches to the preparation of linezolid make use of chiral synthons for the construction of the stereocenter. These are small molecules characterized by a high cost, therefore they are not suitable for the production of the compound on an industrial scale.
-
[0005]
There is therefore the need for an alternative synthesis which provides oxazolidinone derivatives, linezolid included, from inexpensive starting materials, and which does not require a chiral synthon for the construction of the molecule, so that it can be used for the industrial preparation of such derivatives.
………………………………….
http://pubs.rsc.org/en/content/articlelanding/2010/md/c0md00015a/unauth

………………………………….

…………………………………………

…………………………………
RSC Adv., 2013,3, 24946-24951
DOI: 10.1039/C3RA45186K
http://pubs.rsc.org/en/content/articlelanding/2013/ra/c3ra45186k#!divAbstract


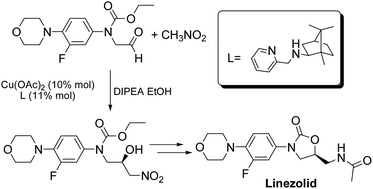
A new asymmetric synthesis of the antibiotic Linezolid was performed through a copper-catalyzed Henry reaction as the key step. The use of camphor-derived aminopyridine ligands helped to improve the yields of the chiral precursor and to obtain Linezolid in good overall yield and enantiomeric excess.
Linezolid 1. Mp: 181–182 C [lit. 181.5–182.5 C];
1 H-NMR (300 MHz; CDCl3) d 2.02 (s, 3H), 3.06 (t, J ¼ 4.7 Hz, 4H), 3.61– 3.78 (m, 3H), 3.87 (t, J ¼ 4.7 Hz, 4H), 4.03 (t, J ¼ 9.0 Hz, 1H), 4.72–4.82 (m, 1H), 6.17 (bt, 1H, exch. with D2O), 6.93 (t, J ¼ 9.0 Hz, 1H), 7.08 (dd, J1 ¼ 9.0 Hz, J2 ¼ 2.5 Hz, 1H), 7.44 (dd, J1 ¼ 14.4 Hz, J2 ¼ 2.5 Hz, 1H); ee ¼ 71%;
HPLC (Daicel CHIRALPAK-IA, hexane/i-PrOH ¼ 70 : 30, ow rate 0.8 mL min 1 , l ¼ 254 nm); tR (major) ¼ 14.1 min; tR (minor) ¼ 16.4 min. A true sample of (S)-Linezolid (ee > 98%) under the same HPLC conditions gave a tR ¼ 14.1 min.
………………………………..
http://www.slideshare.net/vishwajeeta/introduction-new-ppt
………………………………

http://www.slideshare.net/pushechnikov/linezolid-case-study
…………………………………
http://pubs.rsc.org/en/content/articlelanding/2011/cc/c1cc15503b#!divAbstract

…………………………………
http://www.mdpi.com/1424-8247/3/7/1988/htm

………………………………

Numbered structure of linezolid, showing the pharmacophore required for good activity (in blue) and desirable structural features (in orange).
Title: Linezolid
CAS Registry Number: 165800-03-3
CAS Name:N-[[(5S)-3-[3-Fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide
Manufacturers’ Codes: PNU-100766; U-100766
Trademarks: Zyvox (Pharmacia & Upjohn); Zyvoxid (Pharmacia & Upjohn)
Molecular Formula: C16H20FN3O4
Molecular Weight: 337.35
Percent Composition: C 56.96%, H 5.98%, F 5.63%, N 12.46%, O 18.97%
Literature References: Prototype of the oxazolidinone antimicrobials; inhibits bacterial mRNA translation. Prepn: M. R. Barbachyn et al.,WO9507271 (1995 to Upjohn); eidem,US5688792 (1997 to Pharmacia & Upjohn); S. J. Brickner et al.,J. Med. Chem.39, 673 (1996).
Antibacterial spectrum: C. W. Ford et al.,Antimicrob. Agents Chemother.40, 1508 (1996). Mechanism of action study: D. L. Shinabarger et al.,ibid.41, 2132 (1997).
HPLC determn in plasma: C. Buerger et al.,J. Chromatogr. B796, 155 (2003). Clinical comparison with vancomycin, q.v., for MRSA infections: D. L. Stevens et al., Clin. Infect. Dis.34, 1481 (2002).
Review of pharmacology: L. D. Dresser, M. J. Rybak, Pharmacotherapy18, 456-462 (1998); and clinical experience: R. Norrby, Expert Opin. Pharmacother.2, 293-302 (2001).
Properties: White crystals from ethyl acetate and hexanes, mp 181.5-182.5°. [a]D20 -9° (c = 0.919 in chloroform).
Melting point: mp 181.5-182.5°
Optical Rotation: [a]D20 -9° (c = 0.919 in chloroform)
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic); Oxazolidinones.
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO1995007271A1 * |
Aug 16, 1994 |
Mar 16, 1995 |
Michael R Barbachyn |
Substituted oxazine and thiazine oxazolidinone antimicrobials |
| AU2001100437A4 * |
Title not available |
| EP0963980A2 * |
Mar 10, 1999 |
Dec 15, 1999 |
The Wellcome Foundation Limited |
1,2,4-Triazine derivative, its preparation and its use as reference marker for testing purity and stability of “lamotrigine” |
| Reference |
| 1 |
* |
[Online] August 2002 (2002-08), XP002388488 Retrieved from the Internet: URL:www.emea.eu.int/pdfs/human/ich/273799e n.pdf> [retrieved on 2006-07-03] |
| 2 |
* |
[Online] June 1995 (1995-06), XP002388489 Retrieved from the Internet: URL:www.emea.eu.int/pdfs/human/ich/38195en .pdf> [retrieved on 2006-07-03] |
| 3 |
* |
DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, JUN ET AL: “Preparation of oxazolidone derivatives as antibacterial agents” XP002429969 retrieved from STN Database accession no. 2003:576097 -& CN 1 355 165 A (INSTITUTE OF MEDICAL AND BIOLOGICAL TECHNOLOGY, CHINESE ACADEMY OF MED) 26 June 2002 (2002-06-26) |
| 4 |
* |
GLEAVE D M ET AL: “Synthesis and antibacterial activity of [6,5,5] and [6,6,5] tricyclic fused oxazolidinones” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 8, no. 10, 19 May 1998 (1998-05-19), pages 1231-1236, XP004137053 ISSN: 0960-894X |
| 5 |
* |
REDDY K V S R K ET AL: “Isolation and characterization of process related impurities in linezolid” JOURNAL OF PHARMACEUTICAL AN BIOMEDICAL ANALYSIS, vol. 30, no. 3, 15 October 2003 (2003-10-15), pages 635-642, XP002388486 |
| WO2001057035A1 * |
Jan 29, 2001 |
Aug 9, 2001 |
Upjohn Co |
Linezolid-crystal form ii |
| WO2002032857A1 * |
Oct 17, 2001 |
Apr 25, 2002 |
Robert C Gadwood |
Methods of producing oxazolidinone compounds |
| WO2002085849A2 * |
Apr 15, 2002 |
Oct 31, 2002 |
Delara B Godrej |
Process to prepare oxazolidinones |
| WO2005099353A2 * |
Apr 19, 2004 |
Oct 27, 2005 |
Reddy Pingili Krishna |
A novel process for the preparation of linezolid and related compounds |
| WO2006008754A1 |
Jul 20, 2004 |
Jan 26, 2006 |
Reddy Pingili Krishna |
Novel intermediates for linezolid and related compounds |
| WO2006031179A1 * |
Sep 12, 2005 |
Mar 23, 2006 |
Astrazeneca Ab |
Process for preparation of phtalimide |
| WO2007116284A1 * |
Mar 26, 2007 |
Oct 18, 2007 |
Pfizer Prod Inc |
Process for preparing linezolid |
| WO2010081404A1 |
Jan 8, 2010 |
Jul 22, 2010 |
Lianhe Chemical Technology Co., Ltd. |
Method for preparing linezolid and intermediates thereof |
| WO2012019632A1 |
Aug 11, 2010 |
Feb 16, 2012 |
Synthon B.V. |
Process for making linezolid |
| WO2012019862A1 |
Jul 14, 2011 |
Feb 16, 2012 |
Synthon B.V. |
Process for making linezolid |
| WO2012114354A1 |
Feb 21, 2012 |
Aug 30, 2012 |
Lee Pharma Limited |
Anhydrous linezolid crystalline form-ii |
| WO2013072923A1 |
Sep 18, 2012 |
May 23, 2013 |
Cadila Healthcare Limited |
Process for the preparation of crystalline linezolid |
| WO2013111048A1 |
Jan 22, 2013 |
Aug 1, 2013 |
Jubilant Life Sciences Limited |
Improved process for the preparation of stable crystalline form-i of linezolid, substantially free of residual solvent |
| WO2014071990A1 |
Nov 9, 2012 |
May 15, 2014 |
Synthon Bv |
Process for making linezolid |
| EP1403267A1 * |
Sep 25, 2003 |
Mar 31, 2004 |
Daiso Co., Ltd. |
Process for preparing glycidylphthalimide |
| EP1564215A1 * |
Sep 25, 2003 |
Aug 17, 2005 |
Daiso Co., Ltd. |
Process for preparing glycidylphthalimide |
| EP2100884A1 |
Oct 16, 2003 |
Sep 16, 2009 |
Symed Labs Limited |
Crystalline form of linezolid |
| EP2690100A1 |
Jul 14, 2011 |
Jan 29, 2014 |
Synhton B.V. |
Process for making linezolid |
| US6444813 |
Jan 29, 2001 |
Sep 3, 2002 |
Pharmacia & Upjohn Company |
Mixing linezolid of an >98% enantomeric purity in a solvent at >80 degrees; separating a crystal (ii) of >99% purity; analysis by the powder x-ray diffraction spectrum/infrared spectrum as a mineral oil mull; bactericides; stability |
| US6514529 |
Mar 15, 2001 |
Feb 4, 2003 |
Pharmacia & Upjohn Company |
A compressed tablet of antibacterial oxazolidinone selected from the group consisting of linezolid, eperezolid and (S)-N-((3-(3-fluoro-4-(tetrahydro-2H-thiopyran-4-yl)phenyl-2-o xo-5-oxazolidinylmethyl)acetamide S,S-dioxide |
| US6544991 |
Jun 21, 2001 |
Apr 8, 2003 |
Pharmacia & Upjohn Company |
Compositions and methods for treating bacterial infections |
| US6559305 |
May 23, 2002 |
May 6, 2003 |
Pharmacia & Upjohn Company |
Linezolid—crystal form II |
| US6617339 |
Jun 3, 1999 |
Sep 9, 2003 |
Syngenta Limited |
Oxazolidinone derivatives, process for their preparation and pharmaceutical compositions containing them |
| US6796975 |
Mar 15, 2001 |
Sep 28, 2004 |
Pharmacia & Upjohn Company |
Container for linezolid intravenous solution |
| US6833453 |
Oct 17, 2001 |
Dec 21, 2004 |
Pharmacia & Upjohn Company |
As an example, manufacturing a 5-(tert-butylcarbamoyl)-amino-methyl-oxazolidinone by condensing a carbamate with a tert-butylcarbamoyl protected derivative of glycidylamine or a 3-amino-1-halopropanol |
| US6875875 |
Sep 25, 2003 |
Apr 5, 2005 |
Daiso Co., Ltd. |
Process for preparing glycidylphthalimide |
| US6887995 |
Apr 15, 2002 |
May 3, 2005 |
Pharmacia & Upjohn Company |
Reacting N-aryl-O-alkylcarbamate with an amide derivative in the presence of a lithium cation, a base, and a nucleophile |
| US6989381 |
Aug 20, 2001 |
Jan 24, 2006 |
Pharmacia Corporation |
Containing s cyclodextrin compound in a concentration sufficient to maintain the drug in solution at such a drug concentration. |
| US7087784 |
Mar 25, 2004 |
Aug 8, 2006 |
Pharmacia & Upjohn |
Process to prepare oxazolidinones |
| US7128928 |
Feb 20, 2003 |
Oct 31, 2006 |
Pharmacia Corporation |
Ophthalmic formulation with novel gum composition |
| US7135576 |
Jan 7, 2005 |
Nov 14, 2006 |
Daiso Co., Ltd. |
Process for preparing glycidylphthalimide |
| US7307163 |
Apr 19, 2004 |
Dec 11, 2007 |
Symed Labs Limited |
Process for the preparation of linezolid and related compounds |
| US7351824 |
Oct 8, 2007 |
Apr 1, 2008 |
Symed Labs Limited |
Intermediates for oxazolidinone antibacterials; N-[3-Chloro-2-(R)-hydroxypropyl]-3-fluoro-4-morpholinyl aniline |
| US7429661 |
Jul 20, 2004 |
Sep 30, 2008 |
Symed Labs Limited |
Intermediates for linezolid and related compounds |
| US7524954 |
Oct 8, 2007 |
Apr 28, 2009 |
Symed Labs Limited |
Reacting 3-fluoro-4-morpholinyl aniline derivative with epichlorohydrin; converting chloromethyl oxazolidinone to aminomethyl oxazolidinone; carbonylation ; reacting with potassium phthalimide, hydrazine hydrate, and acetic anhydride; cyclization, carbamylation |
| US7714128 |
Oct 16, 2003 |
May 11, 2010 |
Symed Labs Limited |
crystalline linezolid form III (N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide) an antibacterial agent; thermal stability |
| US7718799 |
Sep 26, 2007 |
May 18, 2010 |
Symed Labs Limited |
Crystalline form of linezolid |
| US7718800 |
Sep 26, 2007 |
May 18, 2010 |
Symed Labs Limited |
Prepared by mixing linezolid with solvent or mixture of solvents, cooling contents to below 15 degrees C., optionally seeding contents with linezolid form III, stirring, and collecting linezolid form III crystals by filtration or centrifugation; antibacterial agent; thermally stable |
| US7732597 |
Sep 26, 2007 |
Jun 8, 2010 |
Symed Labs Limited |
Prepared by acetylating (S)-N-[[3-[3-fluoro-4-[4-morpholinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]amine in a solvent, optionally in presence of an organic base to form linezolid, seeding reaction mixture, and isolating linezolid form III; antibacterial agent; thermally stable |
| US7741480 |
Oct 8, 2007 |
Jun 22, 2010 |
Symed Labs Limited |
Process for the preparation of linezolid and related compounds |
| US8658789 |
Jan 8, 2010 |
Feb 25, 2014 |
Lianhe Chemical Technology Co., Ltd. |
Method for preparing linezolid and intermediates thereof |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2009032294A2 * |
Sep 5, 2008 |
Mar 12, 2009 |
Teva Pharma |
Processes for the preparation of a linezolid intermediate, linezolid hydroxide |
| WO2011076678A1 * |
Dec 17, 2010 |
Jun 30, 2011 |
F. Hoffmann-La Roche Ag |
Substituted benzamide derivatives |
……………

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO

































































 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR












 .
.


 MANUDEVI
MANUDEVI







 .
. .
.













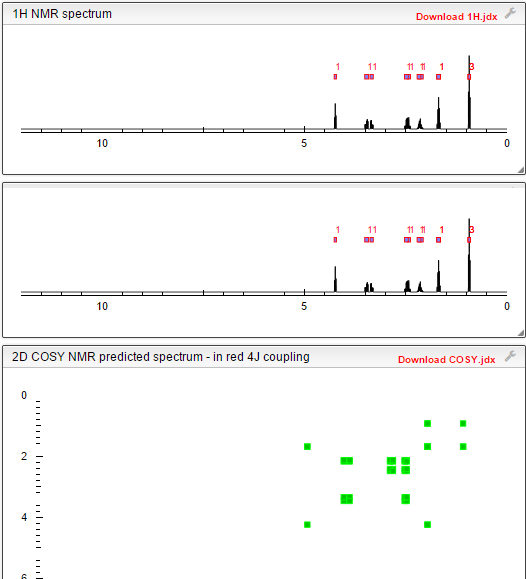
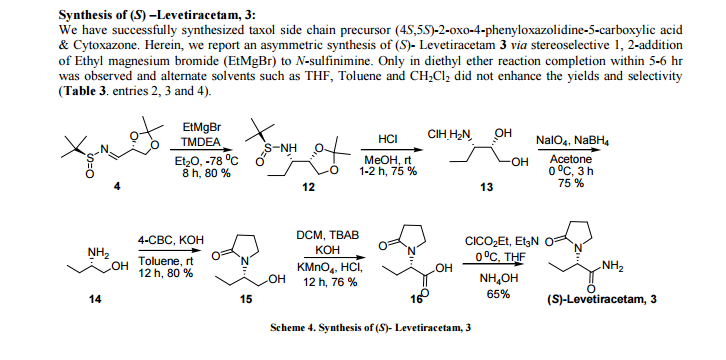








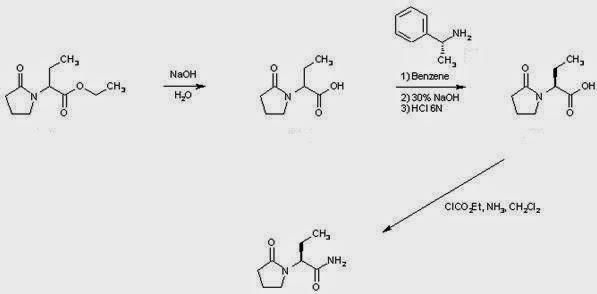






 US 7902380, Levetiracetam
US 7902380, Levetiracetam




 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..


 dalat city
dalat city hanoi
hanoi


