
186497-07-4, ZD4054, ZD-4054, Zd 4054, ZD4054, Zibotentan
Molecular Weight:424.43314 g/mol
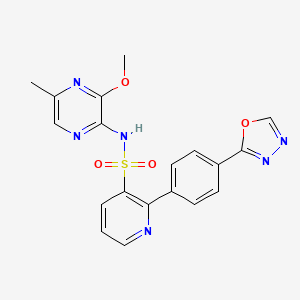
N-(3-methoxy-5-methylpyrazin-2-yl)-2-[4-(1,3,4-oxadiazol-2-yl)phenyl]pyridine-3-sulfonamide
Oncolytic Drugs, Prostate Cancer Therapy, Solid Tumors Therapy, Antimitotic Drugs, Endothelin ETA Receptor Antagonists
Zibotentan (INN) (earlier code name ZD4054) is an anti-cancer candidate.[1] It is an endothelin receptor antagonist.[2]
It failed a phase III clinical trial for prostate cancer[3] but other trials are planned.[4] Tolerability of zibotentan plus docetaxel has been evaluated.[5]
SYN

https://www.google.com/patents/WO1996040681A1?cl=en
Bromination of 2-amino-5-methylpyrazine (I) with Br2 in CHCl3 affords the bromopyrazine (II). Subsequent bromide displacement in (II) by means of sodium methoxide gives rise to the methoxypyrazine (III). The amino group of (III) is then protected by acylation with isobutyl chloroformate, to produce carbamate (IV). Diazotization of 3-amino-2-chloropyridine (V), followed by treatment with sulfur dioxide in the presence of CuCl furnishes sulfonyl chloride (VI). Carbamate (IV) is then acylated by means of NaH and sulfonyl chloride (VI) in DMF to furnish the N-sulfonyl carbamate (VII). Esterification of 4-carboxyphenylboronic acid (VIII) with H2SO4 in MeOH gives 4-(methoxycarbonyl)phenylboronic acid (IX). Mitsunobu coupling between boronic acid (IX) and chloropyridine (VII) furnishes adduct (X). Methyl ester (X) is converted into hydrazide (XI) by treatment with hydrazine hydrate in refluxing methanol. Then, cyclization of the acyl hydrazide (XI) with boiling triethyl orthoformate gives rise to the target oxadiazole derivative.
https://www.google.com/patents/WO1996040681A1?cl=en
Example 36
Hydrazine hydrate (1.2 ml) was added to a solution of N-(isobutoxycarbonyl)-2- (4-memoxycarbonylphenyl)-N-(3-metJ oxy-5-methylpyrazin-2-yl)pyridine-3-sulphonamide (1.54 g) in methanol (15 ml) and the mixture was heated and stiπed under reflux for 24 hours then cooled. The solid was collected and dried under reduced pressure to give the free sulphonamido-acylhydrazide (0.857 g); 1H NMR (cVDMSO): 2.2 (s, 3H), 3.7 (s, 3H), 6.7 (br s, 2H), 7.3 (s, IH), 7.5 (m, 3H), 7.8 (d, 2H), 8.4 (d, IH), 8.75 (dd, IH), 9.8 (br s, IH). A solution of this acylhydrazide (207 mg) in triethylorthoformate (5 ml) was heated under reflux for 17 hours then cooled. The resultant solid was collected and purified by chromatography on a silica gel Mega Bond Elut column, eluting with 0-10% methanol/dichloromethane to give N-(3-methoxy-5-mef ylpyrazin-2-yl)-2-(4-[l,3,4-oxadiazol-2-yl]phenyl)pyridine-3- sulphonamide (39 mg) as a solid; 1H NMR (DMSO-do): 2.2 (br s, 3H), 3.8 (s, 3H), 7.4 (br s, IH), 7.6-7.8 (m, 3H), 8.0 (m, 2H), 8.5 (dd, IH), 8.9 (dd, IH), 9.4 (s, IH); mass spectrum (+ve ESP): 425 (M+H)+.
………………………….
http://www.google.im/patents/EP1904490A1?cl=en
N-(3-methoxy-5-methylpyrazin-2-yl)-2- (4-[l,3,4-oxadiazol-2-yl]phenyl)pyridine-3-sulphonamide (hereafter “Compound (I)). More specifically the invention relates to the ethanolamine salt of Compound (I) (hereafter “Compound (I) ethanolamine salt), and to pharmaceutical compositions containing it. The invention further relates to the use of Compound (I) ethanolamine salt in the manufacture of medicament for use in treating cancer and to methods of treating cancer in a warm blooded animal such as man using this salt. The invention further relates to the use of Compound (I) ethanolamine salt in producing Compound (I) during manufacture.
Compound (I) is an endothelin antagonist. The endothelins are a family of endogenous 21 amino acid peptides comprising three isoforms, endothelin-1 (ET-I), endothelin-2 and endothelin-3. The endothelins are formed by cleavage of the Trp2I-Val22 bond of their corresponding proendothelins by an endothelin converting enzyme. The endothelins are among the most potent vasoconstrictors known and have a characteristic long duration of action. They exhibit a wide range of other activities including cell proliferation and mitogenesis, extravasation and chemotaxis, and also interact with a number of other vasoactive agents.
The endothelins are released from a range of tissue and cell sources including vascular endothelium, vascular smooth muscle, kidney, liver, uterus, airways, intestine and leukocytes. Release can be stimulated by hypoxia, shear stress, physical injury and a wide range of hormones and cytokines. Elevated endothelin levels have been found in a number of disease states in man including cancers.
Recently, endothelin A receptor antagonists have been identified as potentially of value in the treatment of cancer (Cancer Research, 56, 663-668, February 15th, 1996 and Nature Medicine, Volume 1, Number 9, September 1999, 944-949).
Cancer affects an estimated 10 million people worldwide. This figure includes incidence, prevalence and mortality. More than 4.4 million cancer cases are reported from Asia, including 2.5 million cases from Eastern Asia, which has the highest rate of incidence in the world. By comparison, Europe has 2.8 million cases, North America 1.4 million cases, and Africa 627,000 cases. In the UK and US, for example, more than one in three people will develop cancer at some point in their life, Cancer mortality in the U.S. is estimated to account for about 600,000 a year, about one in every four deaths, second only to heart disease in percent of all deaths, and second to accidents as a cause of death of children 1-14 years of age. The estimated cancer incidence in the U.S. is now about 1,380,000 new cases annually, exclusive of about 900,000 cases of non-melanotic (basal and squamous cell) skin cancer.
Cancer is also a major cause of morbidity in the UK with nearly 260,000 new cases (excluding non-melanoma skin cancer) registered in 1997. Cancer is a disease that affects mainly older people, with 65% of cases occurring in those over 65. Since the average life expectancy in the UK has almost doubled since the mid nineteenth century, the population at risk of cancer has grown. Death rates from other causes of death, such as heart disease, have fallen in recent years while deaths from cancer have remained relatively stable. The result is that 1 in 3 people will be diagnosed with cancer during their lifetime and 1 in 4 people will die from cancer. In people under the age of 75, deaths from cancer outnumber deaths from diseases of the circulatory system, including ischaemic heart disease and stroke. In 2000, there were 151,200 deaths from cancer. Over one fifth (22 per cent) of these were from lung cancer, and a quarter (26 per cent) from cancers of the large bowel, breast and prostate.
Worldwide, the incidence and mortality rates of certain types of cancer (of stomach, breast, prostate, skin, and so on) have wide geographical differences which are attributed to racial, cultural, and especially environmental influences. There are over 200 different types of cancer but the four major types, lung, breast, prostate and colorectal, account for over half of all cases diagnosed in the UK and US. Prostate cancer is the fourth most common malignancy among men worldwide, with an estimated 400,000 new cases diagnosed annually, accounting for 3.9 percent of all new cancer cases. Current options for treating cancers include surgical resection, external beam radiation therapy and / or systemic chemotherapy. These are partially successful in some forms of cancer, but are not successful in others. There is a clear need for new therapeutic treatments. Compound (I) is exemplified and described in WO96/40681 as Example 36. WO96/40681 claims the endothelin receptors described therein for the treatment of cardiovascular diseases. The use of Compound (I) in the treatment of cancers and pain is described in WO04/018044. Compound (I) has the following structure:

Compound (I)
In WO04/018044 an endothelin human receptor binding assay is described. The pICjo (negative log of the concentration of compound required to displace 50% of the ligand) for Compound (I) at the ETA receptor was 8.27 [8.23 – 8.32] (n=4). Compound (I) is thus an excellent endothelin antagonist.
WO96/40681 and WO04/018044 disclose, in general terms, certain pharmaceutically acceptable salts of the compounds disclosed therein. Specifically it is stated that suitable pharmaceutically-acceptable salts include, for example, salts with alkali metal (such as sodium, potassium or lithium), alkaline earth metals (such as calcium or magnesium), ammonium salts, and salts with organic bases affording physiologically acceptable cations, such as salts with methylamine, dimethylamine, trimethylamine, piperidine and morpholine. In addition, it was stated that suitable pharmaceutically-acceptable salts include, pharmaceutically-acceptable acid- addition salts with hydrogen halides, sulphuric acid, phosphoric acid and with organic acids such as citric acid, maleic acid, methanesulphonic acid and p-toluenesulphonic acid.
Example 2 Formation of Compound (I) using ethanolamine
The above organic layer from Example 1 was adjusted to 42°C and isopropyl alcohol (114 ml), water (170ml) and ethanolamine (28.2 ml) were added and stirred at 42°C for 90 mins. The reaction mixture was allowed to cool to 2O0C and the lower aqueous phase separated and filtered through a 1 μm filter. The aqueous phase was then charged over 40min to a stirred solution of acetic acid (141 g) and water (33.5 g) at 500C and then cooled to 2O0C over 60 mins. The product was isolated by filtration and washed with a mixture of isopropyl alcohol (48.5 ml) and water (48.5 ml) and then isopropyl alcohol (48.5 ml). The product was dried overnight in a vacuum oven at 55°C. Weight 43.08g, Strength = 100%, 86.7%yield. 1H NMR (400 MHz5 DMSOd6) 9.87 (IH, s), 9.14 (IH, s), 8.81 (lH,d), 8.52 (IH, d), 7.98 (2H, d), 7.65 (2H, d), 7.62 (IH, dd), 7.41 (IH, bs), 3.80 (3H, s), 2.23 (3H, s). Mass Spectra MH+ 425.1036 (Ci9Hi7N6O4S calculated 425.1032).
| Patent |
Submitted |
Granted |
| Substituted pyrazin-2-yl-sulphonamide-(3-pyridyl) compounds and uses thereof [US6060475] |
2000-05-09 |
|
| COMPOSITION 064 [US8168221] |
2009-04-16 |
2012-05-01 |
| THERAPEUTIC TREATMENT-014 [US2009062246] |
2009-03-05 |
|
| Ethanolamine Salt of N- (3-Methoxy-5-Methylpyrazin-2Yl) -2- (4-[1, 3, 4-Oxadiazole-2-Yl] Phenyl) Pyridine-3-Sulphonamide [US2008221124] |
2008-09-11 |
|
| N-HETEROARYL-PYRIDINESULFONAMIDE DERIVATIVES AND THEIR USE AS ENDOTHELIN ANTAGONISTS [WO9640681] |
1996-12-19 |
References
External links

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
















![(1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol NMR spectra analysis, Chemical CAS NO. 1194508-25-2 NMR spectral analysis, (1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/570/465/1194508-25-2-1h.png)
![(1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol NMR spectra analysis, Chemical CAS NO. 1194508-25-2 NMR spectral analysis, (1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/570/465/1194508-25-2-13c.png)












 .
.

.png)


























