
Home » Uncategorized (Page 86)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA Says Chinese Pfizer Plant Hid Failures, Used Old Ingredients
DRUG REGULATORY AFFAIRS INTERNATIONAL


October 31, 2015 — 2:22 AM IST,
The Pfizer Ltd. research and development plant in Dalian, China.
Bernardo De Niz/Bloomberg
A Pfizer Inc. plant in China that was being inspected by Food and Drug Administration regulators in order to ship drugs to the U.S. kept a second set of quality and manufacturing records that didn’t match official ones, according to an FDA review of the facility.
read
![]()
/////
крисаборол , كريسابورول , Crisaborole, AN 2728
Crisaborole
Treatment for Inflammatory Skin Diseases, including Atopic Dermatitis and Psoriasis
C14H10BNO3, Average mass251.045 Da
4-[(1-Hydroxy-1,3-dihydro-2,1-benzoxaborol-5-yl)oxy]benzonitrile ,
4-((1-Hydroxy-1,3-dihydrobenzo(c)(1,2)oxaborol-6-yl)oxy)benzonitrile
CAS 906673-24-3, AN-2728
Benzonitrile, 4-[(1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy]-
1,3-Dihydro-1-hydroxy-5-(4-cyanophenoxy)-2,1-benzoxaborole
5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole
crisaborol, crisaborole, Crisaborole, crisaborolum
UNII-Q2R47HGR7P
крисаборол
كريسابورول
In phase 3 for treatment of mild to moderate atopic dermatitis……Anacor Pharmaceuticals, Inc.
Psoriasis is a chronic skin disorder caused by inflammatory cell infiltration into the dermis and epidermis, and is accompanied by keratinocyte hyperproliferation. Once triggered, a strong T-cell response is mounted, and a cascade of cytokine and chemokine production is induced.
Down-regulation of certain cytokines and chemokines is considered to be a good approach to treatment, and indeed, the biologics targeting TNF-α demonstrate the effectiveness of this approach.However, biologics have intrinsic challenges, such as limited administration route, side effects, quality control and production cost.
Small molecule approaches to treat psoriasis include systemic or topical steroids, cyclosporine, psoralen plus UVA (PUVA), retinoids, methotrexete, and vitamin D3 analogs.Atopic dermatitis is an allergic skin disorder, which is typically treated with topical steroids, antihistamines, and calcineurin inhibitors.
However, there is still a need for new treatment with improved safety profile. Recently phosphodiesterase 4 (PDE4) inhibitors have been in development for such skin diseases. CC-10004 is in development as an oral treatment for psoriasis and atopic dermatitis. AWD-12-281 was, until recently, in development for the topical treatment of atopic dermatitis. In addition, roflumilast is under Phase 1 development for both diseases.

Figure 1.
PDE4 inhibitors aiming at skin inflammatory diseases.

Anacor’s lead product candidate is crisaborole, an investigational non-steroidal topical PDE-4 inhibitor in development for the potential treatment of mild-to-moderate atopic dermatitis and psoriasis
crisaborole is an investigational topical antiinflammatory drug in phase III clinical development by Anacor Pharmaceuticals for the treatment of mild to moderate atopic dermatitis and in phase II clinical trials in mild to moderate psoriasis
A novel boron-containing small molecule, Crisaborole inhibits the release of pro-inflammatory cytokines including TNF-alpha, IL-12, and IL-23, known mediators of the inflammation associated with psoriasis.
Synthesis
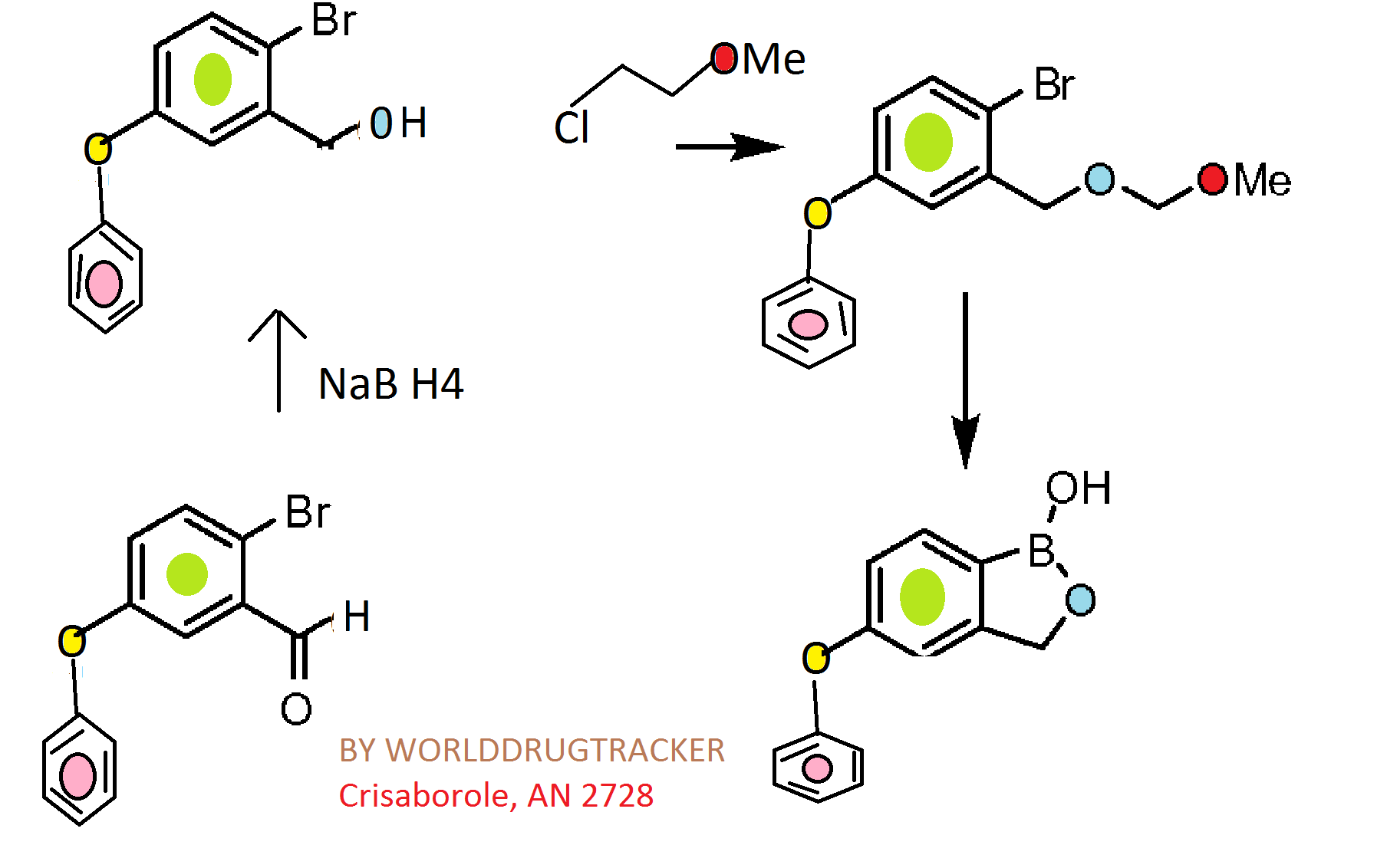
CKICK ON IMAGE FOR CLEAR VIEW
| Originator | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Therapeutic Claim | |||||||||||||||||||||
| Class | |||||||||||||||||||||
| Mechanism of action | |||||||||||||||||||||
| WHO ATC code(s) | |||||||||||||||||||||
| EPhMRA code(s) | |||||||||||||||||||||
| Clinical trial(s) |
|
PAPER
Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis
Bioorg Med Chem Lett 2009, 19(8): 2129
http://www.sciencedirect.com/science/article/pii/S0960894X09002996
- Anacor Pharmaceuticals, Inc., 1020 E. Meadow Circle, Palo Alto, CA 94303, USA
A series of phenoxy benzoxaboroles were synthesized and screened for their inhibitory activity against PDE4 and cytokine release. 5-(4-Cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2728) showed potent activity both in vitro and in vivo. This compound is now in clinical development for the topical treatment of psoriasis and being pursued for the topical treatment of atopic dermatitis


Scheme 1.
Reagents and conditions: (a) ethylene glycol, p-TsOH, toluene, reflux, 6 h (quant.); (b) K2CO3, DMF, 100 °C, overnight (82–96%); (c) 3 M HCl, THF, reflux, 2 h (80–100%); (d) NaBH4, MeOH, rt, 1 h (quant.); (e) 3,4-dihydro-2H-pyran, camphorsulfonic acid, CH2Cl2, rt, 2 h (quant.); (f) (i-PrO)3B, n-BuLi, THF, −78 °C to rt, 3 h; (g) 6 M HCl, THF, rt, 3 h (37–44%); (h) 6 M NaOH, MeOH, 1,4-dioxane, reflux, 6 days (79%); (i) diethylamine (for 5f) or morpholine (for 5g), EDCI, HOBt, DMAP, DMF, rt, overnight (41–70%).
PATENT
http://www.google.co.in/patents/WO2006089067A2?cl=en
4.2. q 5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole (C17) [0264] 1H-NMR (300 MHz,
δ ppm 4.95 (s, 2H), 7.08 (dd, J= 7.9, 2.1 Hz, IH), 7.14 (d, J= 8.8 Hz, IH), 7.15 (d, J= 2.1 Hz, IH), 7.78 (d, J= 7.9 Hz, IH), 7.85 (d, J= 9.1 Hz, 2H), 9.22 (s, IH).
PATENT
EXAMPLE 15
http://www.google.com/patents/WO2007095638A2?cl=en
4-(4-Cvanophenoxy)phenylboronic acid (C97)

(a) (4-cyanophenyl) (4-bromophenyl) ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 1000C for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with IN NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4- bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-de): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p.l94-198°C. MS: m/z = 239 (M+), 240 (M+ 1) (ESI+) and m/z = 238 (M-I) (ESI-). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6 + D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.
FURTHER METHOD

2-Bromo-5-(4-cvanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J= 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
PATENT
http://www.google.im/patents/EP1976536A2?cl=en
2.2.a 2-Bromo-5-(4-cyanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J- 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
2.2.b 2-Bromo-4-(4-cyanophenoxγ)benzyl Alcohol
1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.58 (d, IH), 7.39 (d, IH), 7.18 (dd, IH), 7.11- (d, 2H), 5.48 (t, IH) and 4.50 (d, 2H) ppm.
2.2.c 5- (4-Cyanophenoxy) -1 -Indanol
M.p.50-53°C. MS (ESI+): m/z = 252 (M+l). HPLC: 99.7% purity at 254 nm and 99.0% at 220 nm. 1H NMR (300 MHz, DMSOd6): δ 7.80 (d, 2H), 7.37 (d, IH), 7.04 (d, 2H), 6.98-6.93 (m, 2H), 5.27 (d, IH)5 5.03 (q, IH), 2.95-2.85 (m, IH), 2.75-2.64 (m, IH), 2.39-2.29 (m, IH) and 1.85-1.74 (m, IH) ppm.
2.2. d 2-Bromo-5-(tert-butyldimethylsiloxy)benzyl Alcohol [0429] 1H-NMR (300 MHz, CDCl3) δ (ppm) 0.20 (s, 6H), 0.98 (s, 9H), 4.67 (br s,lH), 6.65 (dd, J= 8.2, 2.6 Hz, IH), 6.98 (d, J= 2.9 Hz, IH), 7.36 (d, J= 8.8 Hz, IH).
3.2.k 2-Bromo-5-(2-cyanophenoχy)-l-(methoxymethoxymethyl)benzene [0443] 1H-NMR (300 MHz, CDCl3) δ (ppm) 3.41 (s, 3H), 4.64 (s, 2H), 4.76 (s, 2H), 6.8-6.9 (m, 2H), 7.16 (td, J= 7.6, 0.9 Hz, IH), 7.28 (d, J= 2.9 Hz, IH), 7.49 (ddd, J= 8.8, 7.6, 1.8 Hz, IH)5 7.56 (d, J= 8.5 Hz, IH), 7.67 (dd, J= 7.9, 1.8 Hz, IH).
EXAMPLE 32
Alternative Preparation of C17 -Intermediate

The procedure described in Example II I was followed for 1H NMR characterization of the current alcohol-borate intermediate. 1H NMR determination indicated there were 72.7 mol% of the desired alcohol-borate intermediate [2-bromo- 5-(4-cyanophenoxy)benzyl] diisopropyl borate, 20.7 mol% of an unknown intermediate and 6.5 mol% of unreacted alcohol. 1H NMR (CDCl3, 300 MHz) of [2- bromo-5-(4-cyanophenoxy)benzyl] diisopropyl borate: δ= 7.61 (d, J= 9.0 Hz, 2H), 7.52 (d, J= 8.4 Hz, IH), 7.15 (d, J= 3.0 Hz, IH), 7.03 (d, J= 8.7 Hz, 2H), 6.84 (dd, J= 8.7 Hz, J= 3.0 Hz, IH), 4.85 (s, 2H), 4.35 (septet, J= 6.1 Hz, 2H), 1.11 (d, J= 6.1 Hz, 12H) ppm.
PATENT
http://www.google.com/patents/US20090291917
- Example 154-(4-Cyanophenoxy)phenylboronic acid (C97)
-

-
(a) (4-cyanophenyl)(4-bromophenyl)ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 100° C. for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with 1N NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4-bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
-
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p. 194-198° C. MS: m/z=239 (M+), 240 (M+1) (ESI+) and m/z=238 (M−1) (ESI−). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6+D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.

see
http://www.google.co.in/patents/WO2006089067A2?cl=en
see
http://www.google.com/patents/US20090291917
| US5688928 * | Jun 7, 1995 | Nov 18, 1997 | Prolinx, Inc. | Phenylboronic acid complexing reagents derived from aminosalicylic acid |
| US5880188 * | May 26, 1995 | Mar 9, 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US5962498 * | Dec 2, 1994 | Oct 5, 1999 | Procyon Pharmaceuticals, Inc. | Protein kinase C modulators. C. indolactam structural-types with anti-inflammatory activity |
| US6369098 * | Oct 4, 2000 | Apr 9, 2002 | Bethesda Pharmaceuticals, Inc. | Dithiolane derivatives |
| US20030032673 * | Jul 19, 2002 | Feb 13, 2003 | Isis Innovation Limited | Therapeutic strategies for prevention and treatment of alzheimer’s disease |
| US20050239170 * | Jul 16, 2001 | Oct 27, 2005 | Hedley Mary L | Alpha-MSH related compounds and methods of use |
| US20060009386 * | May 12, 2005 | Jan 12, 2006 | The Brigham And Women’s Hospital, Inc. | Use of gelsolin to treat infections |
|
Methods of treating anti-inflammatory conditions through the use of boron- containing small molecules are disclosed.
|
|
… Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … Francisco, CA Mar. 6-10, 2009. 7 , “AN2728 … Kyoto, Japan, May 14-18, 2008. 10, “AN2728 …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3- dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
… Dermatology Annual Meeting, San Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … 7, “AN2728 … Francisco, CA May 6-10, 2009. 10, “AN2728 …
|
|
… from the group consisting of AN-2728, AN-2898, CBS- 3595, apremilast, ELB- 353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, …
|
|
“AN2728” is the compound 4-(l-hydroxy-l,3-dihydro-2 … GSK256066, oglemilast, tetomilast, apremilast, AN2728, Compound A, Compound B, …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
85.用于治疗疼痛的UK-500,001。 85. for the treatment of pain UK-500,001. 86.用 于治疗疼痛的AN2728。 86. for the treatment of pain AN2728.
|
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
///////////crisaborole, AN 2728, PHASE 3, Anti-inflammatory, Phosphodiesterase, Oxaborole, Psoriasis, Atopic dermatitis, borole
GSK 2251052, Epetraborole, AN3365

(S)-3-(Aminomethyl)-7-(3-hydroxypropoxy)-1-hydroxy-1,3-dihydro- 2,1-benzoxaborole (GSK2251052) is a novel boron-containing antibiotic that inhibits bacterial leucyl tRNA synthetase, and that has been in development for the treatment of serious Gramnegative infections
(S)-3-aminomethylbenzoxaborole; ABX; AN-3365; GSK ‘052; GSK-052; GSK-2251052, GSK2251052, Epetraborole
[(S)-3-(aminomethyl)-7-(3-hydroxypropoxy)-1-hydroxy- 1,3-dihydro-2,1-benzoxaborole hydrochloride],
(S)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride
1-Propanol, 3-(((3S)-3-(aminomethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaborol-7-yl)oxy)-
AN3365,
MW 237.0614,
cas 1093643-37-8
UNII: 6MC93Z2DF9
Anacor Pharmaceuticals, Inc., INNOVATOR
Glaxosmithkline Llc DEVELOPER
Biomedical Advanced Research and Development Authority (BARDA)
GSK 2251052 • $38.5M over the 1st two years; up to $94M……..http://www.idsociety.org/uploadedFiles/IDSA/Policy_and_Advocacy/Current_Topics_and_Issues/Advancing_Product_Research_and_Development/Bad_Bugs_No_Drugs/Press_Releases/FIS%20Slides.pdf

Originally came from Anacor about ten years ago, then was picked up by GlaxoSmithKline, and it’s an oxaborole heterocycle that inhibits leucyl tRNA synthetase
GlaxoSmithKline recently announced a contract with the Biomedical Advanced Research and Development
Authority (BARDA), a US government preparedness organization , The award guarantees GSK $38.5 million over 2 years towards development of GSK2251052, a molecule co-developed with Anacor Pharma a few years back, as a counter-bioterrorism agent. The full funding amount may later increase to $94 million, pending BARDA’s future option.
The goal here is to develop “GSK ‘052”, as it’s nicknamed among med-chemists, into a new antibiotic against especially vicious and virulent Gram negative bacteria, such as the classic foes plague (Yersinia pestis) or anthrax (Bacillus anthracis).
Look closely at GSK’052 (shown above): that’s a boron heterocycle there! Anacor, a company specializing in boron based lead compounds, first partnered with GSK in 2007 to develop novel benzoxaborole scaffolds. This isn’t the first company to try the boron approach to target proteins; Myogenics (which, after several acquisitions, became Millennium Pharma) first synthesizedbortezomib, a boronic acid peptide, in 1995.


Stephen Benkovic (a former Anacor scientific board member) and coworkers at Penn State first discovered Anacor’s early boron lead molecules in 2001, with a screening assay. The molecules bust bacteria by inhibiting leucyl-tRNA synthetase, an enzyme that helps bacterial cells to correctly tag tRNA with the amino acid leucine. Compounds with cyclic boronic acids “stick” to one end of the tRNA, rendering the tRNA unable to cycle through the enzyme’s editing domain. As a result, mislabeled tRNAs pile up, eventually killing the bacterial cell.
Inhibition of synthetase function turns out to be a useful mechanism to conquer all sorts of diseases. Similar benzoxaborozoles to GSK ‘052 show activity against sleeping sickness (see Trypanosoma post by fellow Haystack contributor Aaron Rowe), malaria, and various fungi.
Boron-containing molecules such as benzoxaboroles that are useful as antimicrobials have been described previously, see e.g. “Benzoxaboroles – Old compounds with new applications” Adamczyk-Wozniak, A. et al., Journal of Organometallic Chemistry Volume 694, Issue 22, 15 October 2009, Pages 3533-3541 , and U.S. Pat. Pubs. US20060234981 and US20070155699. Generally speaking, a benzoxaborole has the following structure and substituent numbering system:

Certain benzoxaboroles which are monosubstituted at the 3-, 6-, or 7-position, or disubstituted at the 3-/6- or 3-/7- positions are surprisingly effective antibacterials, and they have been found to bind to the editing domain of LeuRS in association with tRNALeu Such compounds have been described in US7, 816,344. Using combinations of certain substituted benzoxaboroles with norvaline and/or other amino acid analogs and their salts to: (a) reduce the rate of resistance that develops; and/or (b) decrease the frequency of resistance that develops; and/or (c) suppress the emergence of resistance, in bacteria exposed to compounds
Epetraborole R-mandelate
1234563-15-5
Epetraborole hydrochloride
1234563-16-6

Anacor Pharmaceuticals is out to change that. The Palo Alto, Calif.-based biotechnology company is developing a family of boron-containing small-molecule drugs. And with the assistance of Naeja Pharmaceutical, a Canadian contract research organization, Anacor has licensed one of those molecules to GlaxoSmithKline and taken another one into Phase III clinical trials.
Anacor was founded in 2002 to develop technology created by Lucy Shapiro, a Stanford University bacterial geneticist, and Stephen J. Benkovic, a Pennsylvania State University organic chemist. Through a long-standing scientific collaboration, the two researchers had discovered boron-containing compounds that inhibited specific bacterial targets.
 Lucy Shapiro is a Professor in the Department of Developmental Biology at Stanford University School of Medicine where she holds the Virginia and D. K. Ludwig Chair in Cancer Research and is the Director of the Beckman Center for Molecular and Genetic Medicine. She is a member of the Scientific Advisory Board of Ludwig Institute for Cancer Research and is a member of the Board of Directors of Pacific Biosciences, Inc. She founded the anti-infectives discovery company, Anacor Pharmaceuticals, and is a member of the Anacor Board of Directors. Professor Shapiro has been the recipient of multiple honors, including: election to the American Academy of Arts and Sciences, the US National Academy of Sciences, the US Institute of Medicine, the American Academy of Microbiology, and the American Philosophical Society. She was awarded the FASEB Excellence in Science Award, the 2005 Selman Waksman Award from the National Academy of Sciences, the Canadian International 2009 Gairdner Award, the 2009 John Scott Award, the 2010 Abbott Lifetime Achievement Award, the 2012 Horwitz Prize and President Obama awarded her the National Medal of Science in 2012. Her studies of the control of the bacterial cell cycle and the establishment of cell fate has yielded valuable paradigms for understanding the bacterial cell as an integrated system in which the transcriptional circuitry is interwoven with the three-dimensional deployment of key regulatory and morphological proteins, adding a spatial dimension to the systems biology of regulatory networks.
Lucy Shapiro is a Professor in the Department of Developmental Biology at Stanford University School of Medicine where she holds the Virginia and D. K. Ludwig Chair in Cancer Research and is the Director of the Beckman Center for Molecular and Genetic Medicine. She is a member of the Scientific Advisory Board of Ludwig Institute for Cancer Research and is a member of the Board of Directors of Pacific Biosciences, Inc. She founded the anti-infectives discovery company, Anacor Pharmaceuticals, and is a member of the Anacor Board of Directors. Professor Shapiro has been the recipient of multiple honors, including: election to the American Academy of Arts and Sciences, the US National Academy of Sciences, the US Institute of Medicine, the American Academy of Microbiology, and the American Philosophical Society. She was awarded the FASEB Excellence in Science Award, the 2005 Selman Waksman Award from the National Academy of Sciences, the Canadian International 2009 Gairdner Award, the 2009 John Scott Award, the 2010 Abbott Lifetime Achievement Award, the 2012 Horwitz Prize and President Obama awarded her the National Medal of Science in 2012. Her studies of the control of the bacterial cell cycle and the establishment of cell fate has yielded valuable paradigms for understanding the bacterial cell as an integrated system in which the transcriptional circuitry is interwoven with the three-dimensional deployment of key regulatory and morphological proteins, adding a spatial dimension to the systems biology of regulatory networks.
Stephen J. Benkovic
- Evan Pugh University Professor and Eberly Chair in Chemistry
Office:
414 Wartik Laboratory
University Park, PA 16802
Email: sjb1@psu.edu
(814) 865-2882
http://chem.psu.edu/directory/sjb1
Websites,Benkovic Research Group
Naeja was a three-year-old contract research firm run by Ronald Micetich and his son Christopher Micetich. Based in Edmonton, Alberta, the firm is staffed by chemists and biologists from a variety of nations who have found Canada welcoming to highly educated immigrants.
GSK. Last July, the British firm paid Anacor $15 million and exercised its option to take over development of AN3365. David J. Payne, vice president of GSK’s antibacterial drug discovery unit, lauded the compound, now renamed GSK2251052, as having “the potential to be the first new-class antibacterial to treat serious hospital gram-negative infections in 30 years.” GSK chemists have since developed a stereospecific synthesis for commercial-scale production
david.j.payne@gsk.com
David J. Payne, vice president of GSK’s antibacterial drug discovery unit
David J Payne Dr Payne holds a BSc in Biochemistry from Brunel University, UK, and a PhD and DSc from The Medical School, University of Edinburgh, UK. Dr Payne has 20 years of experience in antibacterial drug discovery and is currently Vice President and Head of the Antibacterial Discovery Performance Unit (DPU) within the Infectious Diseases Centre of Excellence in Drug Discovery (ID CEDD) where he is responsible for GSK’s antibacterial research effort from discovery to clinical proof of concept (up to Phase II clinical trials). At GSK, Dr Payne has played a leading role in redesigning the strategy for antibacterial research and has helped create long-term alliances with innovative biotechnology companies which has expanded GSK’s discovery pipeline. Furthermore, he has created industry-leading partnerships with the Wellcome Trust and the Defense Threat Reduction Agency (US Department of Defense) to accelerate GSK’s antibacterial programmes. To date, Dr Payne has been involved with the progression of a broad diversity of novel mechanism antibacterial agents into development. Dr Payne has authored more than 190 papers and conference presentations.
PATENT
http://www.google.com/patents/US20090227541
- General procedure for Chiral Synthesis of 3-aminomethylbenzoxaboroles
-




- 4-(3-Aminomethyl-1-hydroxy-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyramide acetate salt (A5)

4-[2-(5,5-Dimethyl-[1,3,2]dioxaborinan-2-yl)-3-formyl-phenoxy]-butyric acid ethyl ester
-

-
A mixture of 4-(2-bromo-3-formyl-phenoxy)-butyric acid ethyl ester (5.50 g, 17.5 mmol), bis(neopentyl glucolato)diboron (6.80 g, 30.1 mmol), PdCl2(dppf).CH2Cl2 (1.30 g, 1.79 mmol), and KOAc (5.30 g, 54.1 mmol) in anhydrous THF (600 mL) was heated with stirring at 80° C. (bath temp) O/N under an atmosphere of N2. The mixture was then filtered through Celite and concentrated in vacuo to approximately one quarter of the original volume. The resulting precipitate was isolated by filtration. The precipitate was washed with THF and EtOAc and the combined filtrate was concentrated in vacuo to give an oily residue which was used directly in the next reaction without further purification.
-
1H NMR (400 MHz, CDCl3) δ (ppm): 9.95 (s, 1H), 7.47-7.39 (m, 2H), 7.09-7.07 (m, 1H), 4.14 (q, J=7.2 Hz, 2H), 4.09-4.01 (m, 2H), 3.83 (s, 3H), 3.66 (s, 3H), 2.53 (t, J=8.0 Hz, 2H), 2.19-2.07 (m, 2H), 1.32-1.22 (m, 3H), 0.98 (s, 6H).

4-(1-Hydroxy-3-nitromethyl-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyric acid ethyl ester
-

-
MeNO2 (1.3 mL, 25 mmol) was added dropwise to a stirred solution of crude 4-[2-(5,5-dimethyl-[1,3,2]dioxaborinan-2-yl)-3-formyl-phenoxy]-butyric acid methyl ester (9.4 g), NaOH (1.0 g, 25 mmol) and H2O (35 mL) in MeCN (90 mL) at rt. The mixture was stirred at rt O/N and then acidified (pH 2) using 4 M HCl. The THF was removed in vacuo and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography (10% to 30% EtOAc in hexane) to give the title compound as a yellow oil: yield 2.52 g (45% over 2 steps).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.04 (s, 1H), 7.46-7.42 (m, 1H), 7.07-7.05 (m, 1H), 6.88-6.86 (m, 1H), 5.87 (d, J=8.2 Hz, 1H), 5.69 (dd, J=9.2, 2.5 Hz, 1H), 5.29 (dd, J=13.3, 2.7 Hz, 1H), 4.14-3.94 (m, 5H), 2.55-2.44 (m, 2H), 2.02-1.88 (m, 2H), 1.16 (t, J=7.2 Hz, 3H); MS (ESI) m/z=322 (M−1, negative).

4-(1-Hydroxy-3-nitromethyl-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyric acid
-

-
A mixture of 4-(1-hydroxy-3-nitromethyl-1,3-dihydro-benzo[c][1,2]oxaborol-7-yloxy)-butyric acid ethyl ester (2.51 g, 7.78 mmol), 10% NaOH (17 mL), and 1:1 MeOH/H2O (70 mL) was stirred at rt for 5 h. The MeOH was removed in vacuo and the remaining aqueous layer was acidified to pH 1 using 2 M HCl. The aqueous layer was then extracted with EtOAc. The organic fractions were washed with brine, dried (MgSO4), and concentrated in vacuo to give the title compound as a pale yellow foam: yield 1.85 g (81%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.08 (bs, 1H), 9.01 (bs, 1H), 7.46-7.41 (m, 1H), 7.06-7.04 (m, 1H) 6.89-6.87 (m, 1H), 5.70 (dd, J=7.0, 2.3 Hz, 1H), 5.30 (dd, J=13.3, 2.3 Hz, 1H), 4.55 (dd, J=13.6, 4.2 Hz, 1H), 4.03 (t, J=6.6 Hz, 2H), 2.40 (t, J=7.5 Hz, 2H), 1.95-1.89 (m, 2H); MS (ESI) m/z=296 (M+1, positive).

- 3-Aminomethyl-6-(2-hydroxy-propoxy)-3H-Benzo[c][1,2]oxaborol-1-ol acetate salt (A31)

4-(2-Benzyloxy propoxy-2-bromobenzaldehyde
-

-
A mixture of 2-bromo-4-fluoro-benzaldehyde (30.0 g, 148 mmol), Na2CO3 (78.31 g, 738.8 mmol) and 2-benzyloxy propanol (24.56 g, 147.8 mmol) in anhydrous DMSO (300 mL) was heated with stirring at 130° C. (bath temp) for 72 h under N2. The reaction mixture was cooled to rt and diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O then brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography (hexane to 30% EtOAc in hexane) to give the title compound: yield 3.84 g (7%).
-
1H NMR (400 MHz, CDCl3) δ (ppm): 10.22 (s, 1H), 7.88 (d, J=8.6 Hz, 1H), 7.42-7.20 (m, 5H), 7.12 (d, J=2.3 Hz, 1H), 6.92 (dd, J=8.8, 2.2 Hz, 1H), 4.52 (s, 2H), 4.16 (t, J=6.2 Hz, 2H), 3.65 (t, J=6.1 Hz, 2H), 2.10 (q, J=6.2 Hz, 2H).

4-(2-Benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[/, 3, 2]dioxaborolan-2-yl)-benzaldehyde
-

-
General procedure 5: 4-(2-benzyloxy propoxy-2-bromobenzaldehyde (4.84 g, 13.9 mmol), B2pin2 (5.27 g, 20.8 mmol), KOAc (4.08 g, 41.6 mmol), PdCl2(dppf).CH2Cl2 (811 mg, 8 mol %), and 1,4-dioxane (50 mL). Purification: Biotage (gradient from 2% EtOAc/hexane to 20% EtOAc/hexane): yield 4.0 g (70%).
-
1H NMR (400 MHz, CDCl3) δ (ppm): 10.36 (s, 1H), 7.93 (d, J=8.6 Hz, 1H), 7.43-7.14 (m, 6H), 7.01 (dd, J=8.6, 2.7 Hz, 1H), 4.53 (s, 2H), 4.18 (t, J=6.2 Hz, 2H), 3.66 (t, J=6.1 Hz, 2H), 2.11 (q, J=6.1 Hz, 2H), 1.40 (s, 12H).

6-(2-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol
-

-
General procedure 8: 4-(2-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (3.0 g, 7.6 mmol), MeNO2 (924 mg, 15.1 mmol), NaOH (605 mg, 15.1 mmol), and H2O (10 mL). Purification: flash chromatography (10% EtOAc/hexane to 40% EtOAc): yield 820 mg (30%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.46 (bs, 1H), 7.45 (d, J=8.2 Hz, 1H), 7.41-7.18 (m, 6H), 7.09 (dd, J=8.6, 2.3 Hz, 1H), 5.71 (dd, J=9.2, 2.5 Hz, 1H), 5.31 (dd, J=13.3, 2.7 Hz, 1H), 4.58-4.40 (m, 3H), 4.08 (t, J=6.2 Hz, 2H), 3.60 (t, J=6.2 Hz, 2H), 2.08-1.94 (m, 2H).

3-Aminomethyl-6-(2-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol acetate salt (A31)
-

-
General procedure 13: 6-(2-benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (820 mg, 2.29 mmol), 20% Pd(OH)2 (850 mg, 1 equiv w/w), and AcOH (40 mL). Purification: preparative HPLC: yield 120 mg (22%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.32 (d, J=8.2 Hz, 1H), 7.22 (s, 1H), 7.02 (d, J=7.8 Hz, 1H), 4.98 (bs, 1H), 4.04 (t, J=6.2 Hz, 2H), 3.56 (t, J=6.2 Hz, 2H), 3.03-2.85 (m, 1H), 2.61 (dd, J=12.9, 7.0 Hz, 1H), 1.89 (s, 3H), 1.97-1.67 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 97.44% (MaxPlot 200-400 nm), 97.77% (220 nm).
-
- 7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (A47)
3-(3-Benzyloxy-propoxy)-2-hydroxy-benzaldehyde
-

-
NaH (2.95 g, 72.4 mmol) was added to an ice-cold solution of 2,3-dihydroxybenzaldehyde (5.0 g, 36 mmol) in anhydrous DMSO (45 mL). Benzyl-3-bromopropyl ether (6.45 mL, 36.2 mmol) was then added and the mixture was stirred at rt for 12 h. The mixture was neutralized using 1 N HCl and then extracted with EtOAc. The organic fraction was washed with H2O and concentrated in vacuo. The residue was purified by flash chromatography (8:2 hexane/EtOAc) to give the title compound as a brown oil: yield 8.40 g (81%).
-
[0891]1H NMR (400 MHz, CDCl3) δ (ppm): 9.93 (s, 1H), 7.36-7.23 (m, 6H), 7.20-7.16 (m, 2H), 6.98-6.91 (m, 1H), 4.53 (s, 2H), 4.19 (t, J=6.2 Hz, 2H), 3.70 (t, J=6.1 Hz, 2H), 2.19-2.16 (m, 2H).
3-(3-Benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[/, 3, 2]dioxaborolan-2-yl)-benzaldehyde
-

-
[0893]General procedure 6: 3-(3-benzyloxy-propoxy)-2-hydroxy-benzaldehyde (7.6 g, 26 mmol), pyridine (3.42 mL, 42.5 mmol), Tf2O (4.60 mL, 27.9 mmol), and CH2Cl2 (200 mL): yield 8.60 g (77%).
-
[0894]1H NMR (400 MHz, CDCl3) δ (ppm): 10.23 (s, 1H), 7.54-7.47 (m, 1H), 7.43 (t, J=8.0 Hz, 1H), 7.36-7.22 (m, 6H), 4.52 (s, 2H), 4.23 (t, J=6.3 Hz, 2H), 3.71 (t, J=6.1 Hz, 2H), 2.21-2.17 (m, 2H).
-
[0895]General procedure 5: trifluoro-methanesulfonic acid 2-(3-benzyloxy-propoxy)-6-formyl-phenyl ester (8.0 g, 19 mmol), B2pin2 (9.71 g, 38.2 mmol), KOAc (5.71 g, 57.4 mmol), PdCl2(dppf).CH2Cl2 (1.39 g, 1.89 mmol), and anhydrous dioxane (160 mL). Purification: flash chromatography (9:1 hexane/EtOAc): yield 4.80 g (43%)-some pinacol contamination, used without further purification.
-
[0896]1H NMR (400 MHz, CDCl3) δ (ppm): 9.93 (s, 1H), 7.46 (t, J=7.8 Hz, 1H), 7.41-7.36 (m, 1H), 7.35-7.24 (m, 5H), 7.08 (d, J=7.8 Hz, 1H), 4.50 (s, 2H), 4.10 (t, J=6.3 Hz, 2H), 3.67 (t, J=6.3 Hz, 2H), 2.11 (quin, J=6.2 Hz, 2H), 1.43 (s, 12H).
7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (A47)
-

-
General procedure 8: 3-(3-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (36 g, 91 mmol), MeNO2 (16.6 g, 273 mmol), NaOH (3.64 g, 83 mmol), H2O (180 mL), and THF (50 mL). Purification: flash chromatography (1:1 hexane/EtOAc). A47 was isolated as a light yellow oil: yield 15.9 g (50%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.05 (s, 1H), 7.44 (t, J=7.8 Hz, 1H), 7.35-7.20 (m, 5H), 7.06 (d, J=7.4 Hz, 1H), 6.88 (d, J=8.2 Hz, 1H), 5.70 (dd, J=9.4, 2.3 Hz, 1H), 5.29 (dd, J=13.7, 2.7 Hz, 1H), 4.53 (dd, J=13.3, 9.4 Hz, 1H), 4.45 (s, 2H), 4.11 (t, J=6.1 Hz, 2H), 3.60 (t, J=6.3 Hz, 2H), 2.04-1.91 (m, 2H); MS (ESI): m/z=356 (M−1, negative); HPLC purity: 99.35% (MaxPlot 200-400 nm), 97.32% (220 nm).
Alternative synthesis of 3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride (A46)
-

-
General procedure 13: A47 (0.50 g, 1.4 mmol), 20% Pd(OH)2/C (0.5 g, 1:1 w/w), AcOH (20 mL), and H2O (0.24 mL). The filtrate was concentrated and treated with 4 N HCl to give the title compound as a colorless solid: yield 0.22 g (47%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.42 (t, J=7.8 Hz, 1H), 6.97-6.90 (m, 1H), 6.86 (d, J=8.2 Hz, 1H), 5.20 (dd, J=9.2, 2.5 Hz, 1H), 4.02 (t, J=6.2 Hz, 2H), 3.54 (t, J=6.2 Hz, 2H), 3.40 (dd, J=13.3, 2.7 Hz, 1H), 2.68 (dd, J=13.1, 9.2 Hz, 1H), 1.88-1.78 (m, 2H); MS (ESI): m/z=238 (M+1, positive).
-






- 3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride (A46)

Synthesis of 3-(3-Benzyloxy-propoxy)-2-bromo-benzaldehyde (C)
-

-
To a 5° C. solution of compound A (15.0 g, 0.075 mol), B (12.0 ml, 0.075 mol) and triphenylphosphine (19.6 g, 0.075 mol) in 200 ml of anhydrous THF was added DIAD (14.8 ml, 0.075 mol) drop by drop over a period of 15 minutes. The resulting solution was warmed to room temperature over a period of 5 h and the solvent was evaporated in vacuo. The residue was dissolved in 150 ml of EtOAc and the organic layer washed with water, brine and dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of hexane to 5% EtOAc/hexane) generating 13.0 g (50% yield) of C [3-(3-benzyloxy-propoxy)-2-bromo-benzaldehyde].
-
1H NMR (400 MHz, DMSO-d6) δ (ppm) 10.41 (s, 1H), 7.49 (d, J=7.2 Hz, 1H), 7.32-7.25 (m, 6H), 7.08 (d, J=8.0 Hz, 1H), 4.54 (s, 2H), 4.16 (t, J=6.0 Hz, 2H), 3.74 (t, J=5.8 Hz, 2H), 2.19-2.14 (m, 2H).

3-(3-Benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (D)
-

-
Compound C (8.9 g, 0.025 mol), KOAc (7.5 g, 0.076 mol), and bis(pinacolato)diboron (12.9 g, 0.051 mol) were dissolved in 50 ml of dry DMF and degassed for 30 minutes. To this was added PdCl2(dppf).DCM (0.56 g, 0.76 mmol) and the contents were again degassed for 10 minutes and then heated to 90° C. for 4 h. An additional quantity of PdCl2(dppf).DCM (0.2 g, 0.27 mmol) was added and heating was continued for an additional 2 h. The reaction was cooled to RT, filtered through celite and the solvent evaporated in vacuo. The residue was dissolved in DCM, washed with brine and the organic layer dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of hexane to 5% EtOAc/hexane) provided 5.4 g (53% yield) of D [3-(3-benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde].
-
1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.91 (s, 1H), 7.43 (t, J=7.8 Hz, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.32-7.27 (m, 5H), 7.06 (d, J=8.4 Hz, 1H), 4.49 (s, 2H), 4.08 (t, J=6.0 Hz, 2H), 3.67 (t, J=6.2 Hz, 2H), 2.11-2.08 (m, 2H), 1.44 (s, 12H). ESI+MS m/z, 397 (M+H)+.

7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (E)
-

-
To an ice cold solution of NaOH (0.68 g, 0.017 mol) in 10 ml of water was added a solution of compound D (6.8 g, 0.017 mol) dissolved in 5 ml of THF. After 15 minutes, nitromethane (0.93 ml, 0.017 mol) was added drop by drop and the content stirred at RT overnight. The THF was evaporated under reduced pressure and the contents acidified to pH-3 with 2N HCl. The aqueous layer was extracted with EtOAc several times, and the combined ethyl acetate layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The product was purified by silica gel column chromatography (gradient of 10% EtOAc/hexane to 30% EtOAc/hexane) provided 3.7 g (55% yield) of E [7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol] 3.7 g.
-
1H NMR (400 MHz, DMSO-d6+D2O (0.01 ml)) δ (ppm) 7.49 (t, J=7.8 Hz, 1H), 7.34-7.25 (m, 5H), 7.08 (d, J=7.6 Hz, 1H), 6.92 (d, J=8.0 Hz, 1H), 5.71 (d, J=6.4 Hz, 1H), 5.23 (dd, J=13.2, 2.4 Hz, 1H), 4.58-4.53 (m, 1H), 4.47 (s, 2H), 4.12 (t, J=6.2 Hz, 2H), 3.63 (t, J=6.0 Hz, 2H), 2.04-2.00 (m, 2H). ESI-MS m/z, 356 [M−H]−. HPLC purity: 97.12% (MaxPlot 200-400 nm).

- 3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol (A46)
-

-
Compound E (6.0 g, 0.016 mol) was dissolved in 50 ml of glacial acetic acid and to it was added Pd(OH)2 on Carbon (20% metal content, 50% weight-wet) (5.2 g) and the content set for hydrogenation in a Parr shaker at 45 psi for 2 h. The reaction was checked for completion and the contents were filtered through Celite. The solvent was evaporated under reduced pressure at ambient temperature to yield a gummy material. To this three times was added 15 ml of dry toluene and evaporated yielding a fluffy solid. Purification was accomplished by preparative HPLC (C18 column, using acetonitrile and 0.1% AcOH/water solution) provided 1.5 g (45% yield) of compound A46 [3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol] with 0.33 mol % acetic acid (by HNMR).
-
1H NMR (400 MHz, DMSO-d6+D2O (0.01 ml)) δ (ppm) 7.52 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.2 Hz, 1H), 6.95 (d, J=8.4 Hz, 1H), 5.29 (dd, J=9.2, 2.4, 1H), 4.12 (t, J=6.2 Hz, 2H), 3.62 (t, J=6.2 Hz, 2H), 3.48 (dd, J=13.2, 2.8 Hz, 1H), 2.80-2.74 (m, 1H), 1.92 (t, J=6.2 Hz, 2H). ESI+MS m/z, 238 [M+H]+. HPLC purity: 95.67% (MaxPlot 200-400 nm) and 96.22% (220 single wavelength).

- (S)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol hydrochloride (A49)
-

-
A solution of 1 (160 g, 962.58 mmol) and triphenylphosphine (277.72 g, 1.1 eq, 1058.83 mmol) was dissolved in dichloromethane (800 mL) and cooled to 0° C. (ice/water). A solution of carbon tetrabromide (351.16 g, 1.1 eq, 1058.83 mmol) in dichloromethane (200 mL) was added dropwise and the mixture was left to stir at rt for 18 h. The dichloromethane solvent was evaporated to obtain a white solid. The solid was treated with an excess of hexanes, stirred for 1 h, filtered off and the solvent was evaporated to yield a crude product. The crude product was purified by silica gel column chromatography using 5-10% ethyl acetate and hexane to obtain 2 (199 g, 91%) as a colorless liquid.
- (3-Benzyloxy)-1-bromo-propane (2)

3-(3-Benzyloxy-propoxy)-2-hydroxy-benzaldehyde (4)
-

-
To a solution of aldehyde 3 (27.47 g, 1 eq, 198.88 mmol) in 0.5 L of anhydrous DMSO was added sodium tertiary-butoxide (42.3 g, 2.2 eq, 440.31 mmol) portionwise. The reaction mixture was stirred at rt for 30 minutes. A brown color solution was formed. The reaction mixture was cooled to 0° C. and added bromide (56 g, 1.2 eq, 244.41 mmol) dropwise. The mixture was stirred at rt O/N. 90% of aldehyde 3 was converted to product. The reaction mixture was acidified to pH-3 and then extracted into EtOAc and washed with water. The organic layer was concentrated, the product was purified on silica gel column (EtOAc:hexane 80:20), to yield as compound 4 (48 g, 84.31% yield) (viscous oil).

Trifluoro-methanesulfonic acid 2-(3-benzyloxy-propoxy)-6-formyl-phenyl ester (5)
-

-
To an ice cold solution of 4 (48 g, 1.0 eq, 167.72 mmol) in 200 mL of dry DCM was added pyridine (22 mL, 1.62 eq, 272.11 mmol). To the reaction mixture trifluoromethanesulfonic anhydride (33 mL, 1.16 eq, 196.14 mol) was added drop by drop. The mixture was stirred for 3 h at 0° C. The mixture was quenched with 500 mL of 1N HCl. The compound was then extracted into DCM (300 mL) and passed through a small silica gel column and concentrated to give compound 5 (57 g, 82% yield) as a pale yellow thick oil.

3-(3-Benzyloxy-propoxy)-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzaldehyde (6)
-

-
Compound 5 (65 g, 1.0 eq, 155.5 mmol), bis(pinacolato)diboron (86.9 g, 2.2 eq, 342.11 mmol), KOAc (45.7 g, 3.0 eq, 466.5 mmol) were mixed together and 600 mL of dioxane was added. The mixture was degassed with N2 for 30 minutes and PdCl2(dppf).DCM (5.7 g, 0.05 eq, 7.77 mmol) was added. The resulting slurry was heated to 90° C. overnight. The solvent was evaporated, EtOAc was added and then filtered through a pad of Celite. The organic layer was then washed with water (2×150 mL) and the solvent was evaporated. Column chromatography using 15% EtOAc/hexanes gave compound 6 (37.1 g, 61% yield).

7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (A47)
-

-
A solution of compound 6 (36 g, 1.0 eq, 90.91 mmol) in 50 mL of THF was cooled to 0° C. Nitromethane (16.6 g, 3.0 eq, 272.72 mmol) was added, followed by an aqueous solution of NaOH (3.64 g in 180 mL of H2O). The reaction mixture was stirred at room temperature overnight. The starting material disappeared. The cyclization was afforded by adding 1N HCl until the solution was acidified and then extracted into EtOAc. The EtOAc was evaporated and the mixture was triturated with water and decanted. Column chromatography using 50% EtOAc/hexanes gave compound A47 (15.9 g, 50% yield).

(R) and (S) 7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol
-

-
4.82 g of (A47) was resolved via chiral HPLC using CHIRALPAK ADH column and CO2:methanol (86:14) as eluent (25° C. UV detection was monitored at 230 nm. Two peaks, (S)-7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol and (R)-7-(3-Benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol were collected and evaporated to yellow oils. Analysis of the pooled fractions using a CHIRALPAK ADH 4.6 mm ID×250 mm analytical column and the same mobile phase provided the (S) enantiomer [0.7 g (29% yield)] with a retention time of 6.11 min and a 98.2% ee. The (R) enantiomer [1.0 g (41% yield)] had a retention time of 8.86 min and a 99.6% ee.

(S)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol (A49)
-

-
(A47) (550 mg, 1.57 mmol) was dissolved in 15 mL of glacial acetic acid. 280 mg of 20 wt % palladium hydroxide on carbon (Pearlman’s catalyst) was added and the reaction mixture was flushed with hydrogen 3× and hydrogenated at 55 psi for 3.5 hours. The mixture was filtered through Celite to remove catalyst and rinsed with methanol. Acetic acid was evaporated to obtain the crude product. HPLC purification gave 128 mg of the acetate salt of (A49). The acetate salt was treated with 10 mL of 2N HCl and stirred for 3 hours. The material was lyophilized overnight to obtain 93 mg of the hydrochloride salt of (A49) (Yield 22%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.48 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.4 Hz, 1H), 6.92 (d, J=8.2 Hz, 1H), 5.27 (d, J=9.4 Hz, 1H), 4.11 (t, J=6.3 Hz, 2H), 3.58 (t, J=5.9 Hz, 2H), 2.82 (dd, J=13.3, 9.0 Hz, 1H), 1.95-1.83 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 98.74% (MaxPlot 200-400 nm), 98.38% (220 nm); Chiral HPLC=95.14% ee.

OTHER ISOMER
- (R)-3-Aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[c][1,2]oxaborol-1-ol (A50)
-

-
(R)-7-(3-benzyloxy-propoxy)-3-nitromethyl-3H-benzo[c][1,2]oxaborol-1-ol (0.70 g, 2.0 mmol) was dissolved in 20 mL of glacial acetic acid. 350 mg of 20 wt % palladium hydroxide on carbon (Pearlman’s catalyst) was added and the reaction mixture was flushed with hydrogen 3× and hydrogenated at 55 psi for 3.5 hours. The mixture was filtered through Celite to remove catalyst and rinsed with methanol. Acetic acid was evaporated to obtain the crude product. HPLC purification gave 65 mg of pure compound. After purification, this acetate salt was combined with material from another reaction. This product was treated with 2N HCl (10 mL) and stirred for 3 h at rt. The material was lyophilized overnight to obtain 74 mg of the hydrochloride salt of (A50) (Yield 14%).
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.48 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.4 Hz, 1H), 6.92 (d, J=8.2 Hz, 1H), 5.27 (d, J=9.4 Hz, 1H), 4.11 (t, J=6.3 Hz, 2H), 3.58 (t, J=5.9 Hz, 2H), 2.83 (dd, J=13.3, 8.6 Hz, 1H), 1.94-1.82 (m, 2H); MS (ESI): m/z=238 (M+1, positive); HPLC purity: 99.12% (MaxPlot 200-400 nm), 98.74% (220 nm); Chiral HPLC=98.82% ee.

REFERENCES
https://pubs.acs.org/cen/coverstory/89/8912cover3.html
| US20040203094 * | Sep 20, 2002 | Oct 14, 2004 | Martinis Susan A. | Eucyl-tRNA synthetases and derivatives thereof that activate and aminoacylate non-leucine amino acids to tRNA adaptor molecules |
| US20070155699 * | Aug 16, 2006 | Jul 5, 2007 | Anacor Pharmaceuticals | Boron-containing small molecules |
| US20090227541 * | Jun 19, 2008 | Sep 10, 2009 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
 Anacor
Anacor

![]()
Dr. R. G. Micetich’s research career began in 1963 as a Research Scientist with R & L Molecular Research Ltd. (established by Dr. R. U. Lemieux). This company later became Raylo Chemicals Ltd. Dr. Micetich served as the Research Director (Pharmaceutical Research) of Raylo. During the period from 1963 to 1980 Dr. Micetich’s group was involved in pharmaceutical research and process development work in antibiotics and in NSAI’s (non-steroidal anti-inflammatory agents). This work produced a drug “Mofezolac” – a NSAI which is now marketed in Japan by the Japanese company “Yoshitomi”. Market ~ U.S. $60 million.
In 1980, Dr. R. G. Micetich joined the Faculty of Pharmacy, University of Alberta as an Adjunct Professor working on projects for international big Pharma companies. The work with Taiho Pharmaceutical Company in Japan has produced another drug – a beta-lactamase inhibitor – “TAZOBACTAM” which is now marketed worldwide. This drug now produces annual sales of over US$ 1 billion.
In 1987, Dr. Micetich established a joint venture research company with Taiho, Japan called SynPhar. SynPhar had numerous patents worldwide in various therapeutic areas and many compounds and classes of compounds at various stages of development up to late preclinical.
In view of the significant growth opportunities for SynPhar and in response to the changing international market place for pharmaceuticals, Dr. Micetich acquired and transferred all the assets including intellectual property, equipment and fixtures from SynPhar to NAEJA Pharmaceutical Inc. in 1999. NAEJA is a private Albertan company, founded by the Micetich family which from an initial staff in August 1999 of 40, has grown to 130 and is still growing. NAEJA is a completely self-supporting private company with no venture capital, nor private, nor government funding. The majority of NAEJA employees hold Ph.D.’s. NAEJA has collaborative agreements with pharmaceutical companies around the world. Based on its own intellectual property, NAEJA also has a number of co-development agreements with biotech companies worldwide. Dr. Micetich laid the seeds of foundation for NAEJA and the company continues after his passing, building his legacy.
Dr. R. G. Micetich boasted over 100 publications in well know scientific journals and composed over 100 patents taken out in many countries…………..http://www.bioalberta.com/ron-micetich
more……….
RONALD G. MICETICH (1931-2006): A Scientific Career Ronald Micetich was born in Podanur, Coimbatore (South India). Following receipt of B.Sc. Honors (Chemistry, Loyola College, Madras) and M.A. (Chemistry, Madras University) degrees in India, Ron obtained a Ph.D. (Organic Chemistry, University of Saskatchewan, Canada) in 1962. Ron initiated his interest in microbiology while he was a postdoctoral fellow at the National Research Council of Canada. During the period 1963-1980, Ron held a number of industrial appointments where he rapidly advanced his industrial scientific career (research scientist → associate research director → acting research director → director pharmaceutical / agricultural research) at Raylo Chemicals in Edmonton, Alberta. In 1981 Ron joined the Faculty of Pharmacy & Pharmaceutical Sciences at the University of Alberta as an Adjunct Professor at which time a highly successful drug development program was established with Taiho Pharmaceuticals. This joint industrial collaboration led to the birth of SynPhar with Dr. Micetich as Chairman of the Board, President, CEO and Research Director (1987-1999). Ron, again as Chairman of the Board, CEO and Research Director, established NAEJA (North America, Europe, Japan, Asia) Pharmaceuticals in 1999 with a rollover of assets, including staff, equipment and intellectual property, from SynPhar Laboratories. What began as a full fledged pharmaceutical company with an extensive intellectual property portfolio and a proven track record evolved into an internationally respected pharmaceutical outsource service provider. NAEJA has carved a unique niche in the outsource industry offering extensive discovery experience and expertise. Today, NAEJA has over 120 staff that consists of over 90% scientists holding PhD degrees.,………….see link below
[PDF]RONALD G. MICETICH – University of Alberta – Journal …



Founder, President & CEO, Board Chairman
Fedora Pharmaceuticals Inc.
January 2012 – Present (3 years 10 months)Edmonton, Alberta, Canada
See us at: http://www.fedorapharma.com
Fedora Pharmaceuticals has developed a family of beta-lactamase inhibitors designed to have activity against pathogens containing all four classes of beta-lactamases. These promising novel beta-lactamase inhibitors, used in combination with various beta-lactam antibiotics to treat those antibiotic infections currently resistant to therapy, have recently been licensed to Swiss-based pharmaceutical giant, Hoffman La Roche Ltd. in what is being touted as one of the largest biotech licensing deals ever signed in Canadian history!
Fedora Pharmaceuticals in Canada and Meiji Seika in Japan, with shared world-wide rights, have teamed and jointly entered into this significant tripartite agreement with Roche. Under the terms of the agreement, Roche will obtain exclusive rights from both companies to develop and commercialize the agent worldwide excluding Japan. Fedora and Meiji will receive from Roche; an upfront payment, development, regulatory and sales event milestone payments in addition to royalties on sales of products. While the details of the amounts have not been disclosed a total deal value of US$750 Million in addition to royalties has been announced.
Fedora was founded in 2012 and is headquartered in Edmonton, Alberta, Canada.

President & CEO, Founder
Naeja
August 1999 – Present (16 years 3 months)
NAEJA Pharmaceuticals Inc. is a privately controlled pharmaceutical CRO specializing in early stage drug discovery research with particular expertise in the area of Medicinal Chemistry. NAEJA employs highly skilled PhD scientists recruited from around the globe.
The company boasts a very long and successful track record of rapidly advancing drugs through discovery into the clinic. Several drugs in latter stages of clinical development and on the market today have originated from NAEJA. Most recently is Fedora Pharmaceuticals US $750M licensing deal to Hoffman La Roche Ltd that originated from the laboratories of NAEJA.
As a privately controlled company, NAEJA are very responsive to client’s needs offering many years of drug discovery experience to successfully find ways to advance research programs in a timely, cost effective and efficient manner. NAEJA’s longstanding track record is a testimony in itself!
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
////////////GSK 2251052, Epetraborole, Christopher Micetich, Ronald Micetich, Naeja Pharmaceuticals
B1(c2c(cccc2OCCCO)[C@H](O1)CN)O
TRAMADOL
 |
|||
|
|||


2-[(Dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol
Tramadol (marketed as Ultram, and as generics) is an opioid pain medication used to treat moderate to moderately severepain.[1] When taken as an immediate-release oral formulation, the onset of pain relief usually occurs within about an hour.[5]It has two different mechanisms. First, it binds to the μ-opioid receptor. Second, it inhibits the reuptake of serotonin andnorepinephrine.[6][7]
Serious side effects may include seizures, increased risk of serotonin syndrome, decreased alertness, and drug addiction. Common side effects include: constipation, itchiness and nausea, among others. A change in dosage may be recommended in those with kidney or liver problems. Its use is not recommended in women who are breastfeeding or those who are at risk of suicide.[1]
Tramadol is marketed as a racemic mixture of both R– and S–stereoisomers.[2] This is because the two isomers complement each other’s analgesic activity.[2] It is often combined with paracetamol (acetaminophen) as this is known to improve the efficacy of tramadol in relieving pain.[2] Tramadol is metabolised to O-desmethyltramadol, which is a more potent opioid.[8] It is of the benzenoid class.
Tramadol was launched and marketed as Tramal by the German pharmaceutical company Grünenthal GmbH in 1977 inWest Germany, and 20 years later it was launched in countries such as the UK, U.S., and Australia.[7]
Developed (from 1962) by the German company Grünenthal, and is marketed through much of the world under various trade names, including Acugersic (Malaysia), Mabron (some Eastern European countries as well as parts of the Middle and Far East), Ultram (USA), Zaldiar (France and much of Europe, as well as Russia) and Zydol (UK and Ireland).
CONFUSION ON CIS TRANS
There is some confusion within the literature as to what should be called cis and what should be called trans. For purposes of this disclosure, what is referred to herein as the trans form of Tramadol includes the R,R and S,S isomers as shown by the following two structures:
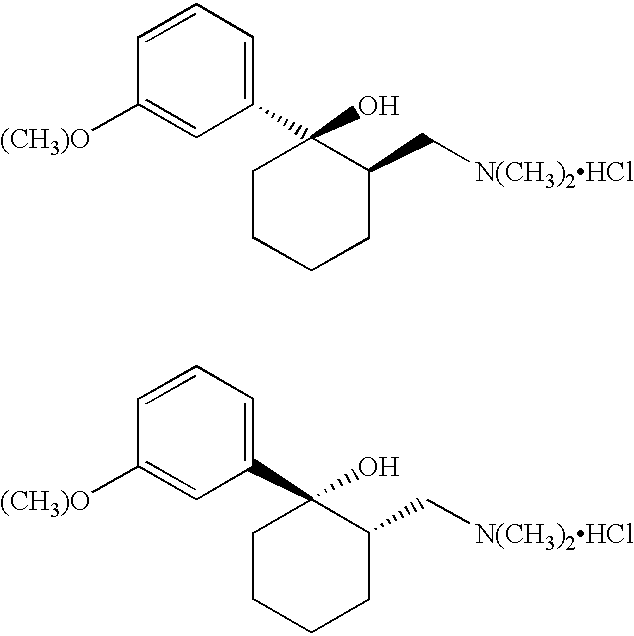
The cis form of Tramadol, as that phrase is used herein, includes the S,R and the R,S isomers which are shown by the following two structures:

Tramadol is marketed as a racemic mixture of both R and S stereoisomers. It is a μ-opioid receptor agonist, like morphine, but much less active. It inhibits reuptake of the neurotransmitters serotonin and norepinephrine, suggesting that it lifts mood and thereby may dull the brain’s perception of pain.
 |
 |
|
1R,2R-Tramadol |
1S,2S-Tramadol |
In the body, tramadol undergoes demethylation to several metabolites by a Cytochrome P450 enzyme (CYP2D6) in the liver, the most important of these products being O-desmethyltramadol. O-desmethyltramadol has a much stronger (200x) affinity for the μ-opioid receptor than tramadol, so in effect tramadol is a prodrug.
 |
 |
|
1R,2R–O-desmethyltramadol |
1S,2S–O-desmethyltramadol |
Not everyone’s liver works identically. Around 6% of the Caucasian population has a reduced CYP2D6 activity (hence reducing metabolism), so there is a reduced analgesic effect with Tramadol. These people require a dose increase of 30% to get the same pain relief as the norm. A case has been reported of a patient where, following an overdose, their ultrarapid tramadol metabolism led to excessive norepinephrine levels, with near-fatal consequences.
However, it has recently been discovered at relatively high concentrations in the roots of the African peach or pin cushion tree (Nauclea latifolia), which has a long tradition as a folk remedy. As usual, Nature got there first.

The African pin-cushion tree (Nauclea latifolia)
In the area of “legal highs”, a disturbing development is a drug blend known as “Krypton”. This isn’t the noble gas, but a mixture of O-desmethyltramadol withKratom (Mitragyna speciosa, a medicinal plant that originates in SE Asia, seemingly the local equivalent of khat), which contains an alkaloid mitragynine which is also a μ-receptor agonist. Several fatalities have been linked with its use, notably in Sweden.
SYNTHESIS
-Tramadol.svg)
-Tramadol_gespiegelt.svg)
(1R,2R)-Tramadol (1S,2S)-Tramadol
-Tramadol.svg)
-Tramadol_gespiegelt.svg)
(1R,2S)-Tramadol (1S,2R)-Tramadol
The chemical synthesis of tramadol is described in the literature.[35] Tramadol [2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol] has two stereogenic centers at thecyclohexane ring. Thus, 2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol may exist in four different configurational forms:
- (1R,2R)-isomer
- (1S,2S)-isomer
- (1R,2S)-isomer
- (1S,2R)-isomer
The synthetic pathway leads to the racemate (1:1 mixture) of (1R,2R)-isomer and the (1S,2S)-isomer as the main products. Minor amounts of the racemic mixture of the (1R,2S)-isomer and the (1S,2R)-isomer are formed as well. The isolation of the (1R,2R)-isomer and the (1S,2S)-isomer from the diastereomeric minor racemate [(1R,2S)-isomer and (1S,2R)-isomer] is realized by the recrystallization of the hydrochlorides. The drug tramadol is a racemate of the hydrochlorides of the (1R,2R)-(+)- and the (1S,2S)-(–)-enantiomers. The resolution of the racemate [(1R,2R)-(+)-isomer / (1S,2S)-(–)-isomer] was described[36] employing (R)-(–)- or (S)-(+)-mandelic acid. This process does not find industrial application, since tramadol is used as a racemate, despite known different physiological effects[37] of the (1R,2R)- and (1S,2S)-isomers, because the racemate showed higher analgesic activity than either enantiomer in animals[38] and in humans.[39]
Synthesised by chemists at the German company Grünenthal and brought to the market in 1977. It can readily be made by nucleophilic attack of a Grignard or RLi species upon a carbonyl group.

ALSO

Paper
http://www.jmcs.org.mx/PDFS/V49/N4/04-Alvarado.pdf
Tramadol hydrochloride (1). To a solution of 3-bromoanisol 13 (0.823 g, 4.4 mmol) in dry THF (10 mL), 1.75 M n-BuLi (2.5 mL, 4.4 mmol) was added dropwise at -78°C under argon atmosphere. The mixture was stirred at the same temperature during 45 minutes and a solution of 2-dimethylaminomethylcyclohexanone 6a (0.62 g, 4mmol) in dry THF was added dropwise. The resulting mixture was stirred at -78°C for 2 h. and the solvent was removed in vacuo. Water (30 mL) was added and the product was extracted with ethyl ether (3X30 mL). The extracts were dried over sodium sulfate, filtered and evaporated in vacuum. The residue was treated with 5mL of ethyl ether saturated with hydrogen chloride; the ethyl ether was evaporated in vacuo and the resulting solid was purified by crystallization from acetone. Tramadol hydrochloride 1 was obtained as white crystals (0.94 g, 78.6%),
MP 168- 175°C.
IR (KBr): 3410, 3185, 2935, 2826, 2782, 1601, 1249, 702 cm-1;
1H NMR (CDCl3, 300 MHz) δ 1.2-1.9 (10H, m), 2.15 (6H, s), 2.45 (1H, dd, J = 15.1, 4.4 Hz), 3.82 (3H, s), 6.76 (1H, dd, J = 8, 2.4 Hz), 7.04 (1H, d, 7.6 Hz), 7.14 (1H, s), 7.26 (1H, t, 3.9 Hz), 11.4 (1H, bs);
MS (EI): m/z 263 M+ (28), 58 (100).
PATENT
http://www.google.com.ar/patents/WO1999003820A1?cl=en
Tramadol is the compound cis(+/-)-2-[(dimethylamino)-methyl]-l-(3- methoxyphenyl) cyclohexanol which, in the form of the hydrochloride salt is widely used as an analgesic
Tramadol means the racemic mixture of cis-Tramadol as shown by the following chemical structures:

(1 R, 2R) (1S. 2S) cis-Tramadol
Step l Formation of Dimethylaminomethyl Cyclohexanone Hydrochloride

Dimethylaminomethyl Cyclohexanone Hydrochloride
Step 2 Formation of Tramadol Mannich Base
NaOH, Water
Toluene, TBME


Tramadol Mannich Base
Step 3 Formation of Tramadol Base Hydrate

Tramadol Base Hydrate (crude) Step 4 Purification of Tramadol Base Hydrate

Tramadol Base Hydrate (pure)
Step 5 Formation of Tramadol Hydrochloride

Tramadol Hydrochloride
Example 1
To produce the Tramadol base hydrate, a reaction vessel is charged successively with 69 Kg of Magnesium, 400 1 of dry Tetrahydrofuran (THF) and 15 1 of 3- bromoanisole.
With careful heating, the reactor temperature is brought up to ca. 30°C. The Grignard initiates at this point and exotherms to approximately 50°C. A further 5 1 of bromoanisole are added which maintains reflux. 400 1 of THF are then added before the remainder of the bromoanisole. This addition of the remainder of the bromoanisole is carried out slowly so as to sustain a gentle reflux. The reaction is refluxed after complete addition of 3-bromoanisole. The vessel is cooled and Mannich base is added. When addition is complete, the vessel is reheated to reflux for 30 minutes to ensure complete reaction. After cooling to ca. 10°C, 2,300 1 of water are added to quench the reaction. When complete, part of the solvents are distilled under vacuum. Approximately 260 1 of concentrated HC1 is added at a low temperature until a pH of 0 – 1 is reached. This aqueous phase is extracted with toluene. The toluene phases are discarded and ethyl acetate is added to the aqueous phase. 30% Ammonia solution is then charged to reach pH 9 – 10 and the phases are separated. The aqueous phases are extracted again with ethyl acetate and finally all ethyl acetate layers are combined and washed twice with water. Ethyl acetate is then distilled from the reaction solution at atmospheric pressure. Process water is added and the solution cooled to 20°C and seeded. After crystallisation, the vessel is cooled to -5 to 0°C and stirred for one hour.
The product is centrifuged at this temperature and washed with cold ethyl acetate 5 x 50 1. Approximately 310 – 360 Kg of moist cis-Tramadol base hydrate are obtained.
Purification
A reactor vessel is charged successively with cis-Tramadol base hydrate (crude) 200 Kg and ethyl acetate 300 1 and the contents of the vessel heated to 50°C until all solids are in solution. The vessel is then cooled to -5 to 0°C and the product crystallises. Stirring is continued for two hours and the product is then centrifuged and washed with cold ethyl acetate, 2 x 25 1. Approximately 165 – 175 Kg (moist) of cis(+/-) Tramadol base hydrate are obtained from this procedure.
The overall process produced high yields of cis-Tramadol with a trans isomer content of less than 0.03%. Analytical data of the base hydrate of cis-Tramadol
Melting point: 79 – 80°C (in comparison cis-Tramadol base anhydrous is an oil). Water content (KF) : 6.52% (= monohydrate) IR-spectrum of the base hydrate of cis-Tramadol (see Fig. 1).
IR-spectrum (=cis-Tramadol base anhydrous, see Fig. 2).
The invention provides a unique process in which a base hydrate of cis-Tramadol is selectively crystallised without impurities. The base hydrate is processed to readily form cis-Tramadol hydrochloride. The process is substantially simpler than known processes and does not require the use of potentially toxic solvents. Thus the process is environmentally friendly.
The base hydrate of cis-Tramadol prepared may also be used in various formulations.
The base hydrate of cis-Tramadol may be formulated in the form of a solid with a slow release profile. For example, slow release pellets may be prepared by coating a suitable core material with a coating, for example, of ethylcellulose/schellack solution (4:1) and suitable pharmaceutical excipients. The pellets have typical average diameter of 0.6 to 1.6 mm. The pellets may be readily converted into gelatine capsules or pressed into tablet form using well-known techniques.
Alternatively the base hydrate of cis-Tramadol may be formulated into effervescent tablets by forming granules of the base hydrate with acidity/taste modifiers and a suitable effervescent base such as sodium hydrogen carbonate /anhydrous sodium carbonate (12:1). The ingredients are typically blended in a mixer /granulator and heated until granulation occurs. The resulting granules may be pressed into tablet form, on cooling. Of particular interest is the use of the base hydrate of cis-Tramadol in a form for parenteral use/injectables. The base hydrate is typically dissolved in water together with suitable excipients (as necessary). The solution is filtered through a membrane to remove solid fibres or particles. The filtered solution may then be filled into ampoules, typically containing 10.0 mg of the active compound. Usually the formulation is prepared for intramuscular injection.
PATENT
http://www.google.com/patents/EP1346978A1?cl=en
-
Various processes for the synthesis of tramadol hydrochloride have been described in the prior art. For example, US 3 652 589 and British patent specification no. 992 399 describe the preparation of tramadol hydrochloride. In this method, Grignard reaction of 2-dimethylaminomethyl cyclohexanone (Mannich base) with metabromo-anisole gives an oily mixture of tramadol and the corresponding cis isomer, along with Grignard impurities. This oily reaction mixture is subjected to high vacuum distillation at high temperature to give both the geometric isomers of the product base as an oil. This oil, on acidification with hydrogen chloride gas, furnishes insufficiently pure tramadol hydrochloride as a solid. This must then be purified, by using a halogenated solvent and 1,4-dioxane, to give sufficiently pure tramadol hydrochloride. The main drawback of this process is the use of large quantities of 1,4-dioxane and the need for multiple crystallizations to get sufficiently pure trans isomer hydrochloride (Scheme – 1).
-
[0004]
The use of dioxane for the separation of tramadol hydrochloride from the corresponding cis isomer has many disadvantages, such as safety hazards by potentially forming explosive peroxides, and it is also a category 1 carcinogen (Kirk and Othmer, 3rd edition, 17, 48). Toxicological studies of dioxane show side effects such as CNS depression, and necrosis of the liver and kidneys. Furthermore, the content of dioxane in the final tramadol hydrochloride has been strictly limited; for example, the German Drug Codex (Deutscher Arzneimittel Codex, DAC (1991)) restricts the level of dioxane in tramadol hydrochloride to 0.5 parts per million (ppm).

-
In another process, disclosed in US patent specification no. 5 414 129, the purification and separation of tramadol hydrochloride is undertaken from a reaction mixture containing the trans and cis isomers, and Grignard reaction side products, in which the reaction mixture is diluted in isopropyl alcohol and acidified with gaseous hydrogen chloride to yield (trans) tramadol hydrochloride (97.8%) and its cis isomer (2.2%), which is itself crystallized twice with isopropyl alcohol to give pure (trans) tramadol hydrochloride (Scheme – 2). This process relies on the use of multiple solvents to separate the isomers (ie butylacetate, 1-butanol, 1-pentanol, primary amyl alcohol mixture, 1-hexanol, cyclohexanol, 1-octanol, 2-ethylhexanol and anisole). The main drawback of this process is therefore in using high boiling solvents; furthermore, the yields of tramadol hydrochloride are still relatively low and the yield of the corresponding cis hydrochloride is relatively high in most cases.

-
PCT patent specification no. WO 99/03820 describes a method of preparation of tramadol (base) monohydrate, which involves the reaction of Mannich base with metabromo-anisole (Grignard reaction) to furnish a mixture of tramadol base with its corresponding cis isomer and Grignard impurities. This, on treatment with an equimolar quantity of water and cooling to 0 to -5°C, gives a mixture of tramadol (base) monohydrate with the corresponding cis isomer (crude). It is further purified with ethyl acetate to furnish pure (trans) tramadol (base) monohydrate, which is again treated with hydrochloric acid in the presence of a suitable solvent to give its hydrochloride salt (Scheme – 2). The drawback of this method is that, to get pure (trans) tramadol hydrochloride, first is prepared pure (trans) tramadol (base) monohydrate, involving a two-step process, and this is then converted to its hydrochloride salt. The overall yield is low because of the multiple steps and tedious process involved.

-
More recently, a process for the separation of tramadol hydrochloride from a mixture with its cis isomer, using an electrophilic reagent, has been described in US patent specification no. 5 874 620. The mixture of tramadol hydrochloride with the corresponding cis isomer is reacted with an electrophilic reagent, such as acetic anhydride, thionyl chloride or sodium azide, using an appropriate solvent (dimethylformamide or chlorobenzene) to furnish a mixture of tramadol hydrochloride (93.3 to 98.6%) with the corresponding cis isomer (1.4 to 6.66%), (Scheme – 3). The product thus obtained is further purified in isopropyl alcohol to give pure (trans) tramadol hydrochloride. However, the drawback of this process is that a mixture of tramadol base with its cis isomer is first converted into the hydrochloride salts, and this is further reacted with toxic, hazardous and expensive electrophilic reagents to get semi-pure (trans) tramadol hydrochloride. The content of the cis isomer is sufficiently high to require further purification, and this therefore results in a lower overall yield.
-
Therefore, all the known methods require potentially toxic solvents and/or reagents, and multiple steps to produce the desired quality and quantity of tramadol hydrochloride. By contrast, the present invention requires a single step process (or only two steps when tramadol hydrochloride is made via the tramadol (base) monohydrate route) using a natural solvent (ie water) in the absence of carcinogenic solvents (such as the category 1 carcinogen, 1,4-dioxane) to produce pure tramadol hydrochloride, so it is ‘ecofriendly’ and easily commercialized to plant scale without any difficulties.


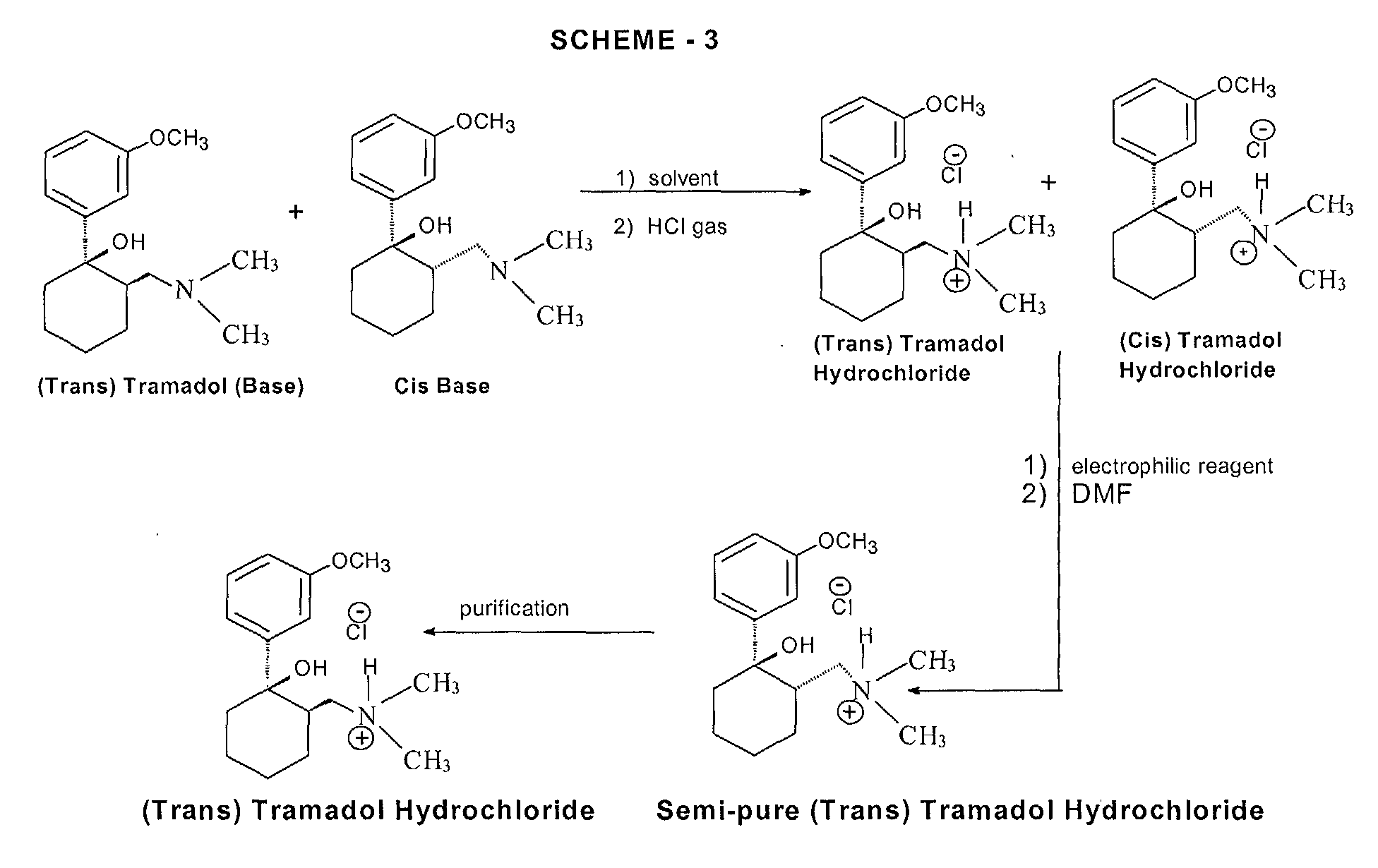
PATENT
http://www.google.co.in/patents/US6399829

EXAMPLE 8 Hydrochloride Formed without Improvement of the Trans:Cis Ratio
Whether a recrystallization step improves the trans:cis ratio of Tramadol depends upon the solvent composition from which the recrystallization is performed. When the hydrochloride form of Tramadol is produced and then crystallized in the presence of a solvent with a high toluene concentration, the ratio of trans:cis remains essentially unchanged. This is in contrast to the recrystallization from a solvent which has a high acetonitrile concentration as was the case in Examples 5-7.
A 21 mL solution of 1.8 g of HCl gas (bubbled at 5° C.) in acetonitrile (yielding a 2.0 M solution), was added to 10.2 g of Grignard product C (90/10 of trans/cis) in 30 mL of toluene and stirred mechanically for 3 hours. The mixture was filtered and washed with toluene. Drying in vacuo yielded 11.2 g (96% recovery). The resulting hydrochloride had a trans/cis ratio of 92:8, essentially the same trans:cis ratio as did the 10.2 g of Grignard product C.
Recrystallization from 90 mL of acetonitrile yielded 8.83 g, which was 96.6/3.4 of trans/cis by HPLC. Of this, 8.6 g was recrystallized from 75 mL of acetonitrile to give 7.44 g, trans/cis ratio of 99.6/0.4.
This example shows that the formation of the hydrochloride in the presence of a relatively large amount of toluene (here about 60%) and crystallization from toluene-acetonitrile does not improve the trans:cis ratio. As the percentage of toluene present in the mixture of toluene and acetonitrile in a crystallization step is decreased, the trans:cis ratio of the recovered product will increase. Steps in which the hydrochloride is recrystallized from acetonitrile do yield an improved trans:cis ratio.
PATENT
https://www.google.com/patents/EP0831082A1
The synthesis of Tramadol is described in U.S. Patent No. 3,652,589 and in British Patent No. 992,399. The synthesis of Tramadol consists of a Grignard reaction between 2-dimethylaminomethylcyclohexanone and 3-methoxyphenyl magnesium bromide (Equation 1). From the reaction scheme, it is clear that both isomers (RR,SS) (Structure 1) and (RS, SR) (Structure 2) are obtained in variable ratios, depending on the reaction conditions.

The original patents assigned to Gruenenthal GmbH describe the isolation of the (RR,SS) isomer, as follows:
The complex mixture of products containing both isomers of Tramadol obtained from the Grignard reaction is distilled under reduced pressure. The isomers are distilled together at 138-140°C (0.6 mm Hg). The distillate is dissolved in ether and is reacted with gaseous HCl. The resulting mixture of both isomers of Tramadol is precipitated as hydrochlorides and filtered. The resulting mixture contains about 20% of the (RS,SR) isomer. The isomer mixture is then refluxed twice with five volumes of moist dioxane, and filtered. The cake obtained consists of pure (RR,SS) isomer. The residual solution consists of “a mixture of about 20-30% of the cis (i.e. RS,SR), which cannot be further separated by boiling dioxane” [U.S. Patent 3,652,589, Example 2].
Dioxane, used in large quantities in this process, possesses many undesirable properties. It has recently been listed as a Category I carcinogen by OSHA [Kirk & Othmer, 3rd Ed., Vol. 9, p. 386], and it is known to cause CNS depression and liver necrosis [ibid., Vol. 13, p. 267]; in addition, it tends to form hazardous peroxides [ibid., Vol 17, p. 48]. As a result, the concentration of dioxane in the final product has been strictly limited to several ppb’s, and the DAC (1991) restricted the level of dioxane in Tramadol to 0.5 ppm.
A different separation method, described in Israeli Specification No. 103096, takes advantage of the fact that the precipitation of the (RR,SS) isomer of Tramadol from its solution in medium chained alcohols (C4-C8) occurs faster than the precipitation of the (RS,SR) isomer, which tends to separate later. The main disadvantage of this method is, that the time interval between the end of separation of the (RR,SS) isomer and the beginning of the (RS,SR) isomer separation is variable, and seems to depend sharply on the composition of the crude mixture. Therefore, variations in the yield and the quality of the product often occur. Furthermore, about 40% of the (RR,SS) isomer does not separate and remains in solution, along with the (RS,SR) isomer. This remaining mixture cannot be further purified by this method.
Another method, described in Israeli Specification No. 116281, relies on the fact that the (RS,SR) isomer of Tramadol undergoes dehydration much faster then the (RR,SS) isomer, when treated with 4-toluenesulfonic acid, or sulfuric acid; furthermore, when the reaction is carried out in an aqueous medium, a certain amount (up to 50%) of the (RS,SR) isomer is converted to the (RR,SS) isomer. This may, of course increase the efficiency of the process.
The unreacted (RR, SS) isomer is then separated from the dehydrated products and from other impurities by simple crystallization.
While further examining the results of the latter process, it was surprisingly found that the hydroxyl group of the (RS,SR) isomer of Tramadol reacts faster than the same group of the (RR,SS) with various reagents. A plausible explanation for this observation can be supplied by comparing the structures of both isomers, and their ability to form hydrogen bonds.
Looking closely at Fig. 1 [(RR,SS) Tramadol hydrochloride] and at Fig. 2 [(RS,SR) Tramadol hydrochloride], one can provide a plausible explanation for the difference in the OH group’s activity, as follows: The proton attached to the nitrogen atom of the protonated (RR,SS) isomer of Tramadol is capable of forming a stable hydrogen bonding with the oxygen atom of the hydroxyl group (see Fig. 1). Thus, any reaction involving protonation of the hydroxyl group (such as dehydration), or any reaction in which the hydroxyl group reacts as a nucleophile (such as a nucleophilic substitution or esterification process) is less favored to occur.
In the (RS,SR) isomer, on the other hand, there is no possible way of forming a stable intramolecular hydrogen bond, and therefore, any of the above-mentioned types of reactions can easily occur, considering the fact that this particular hydroxyl group is tertiary and benzyllic.

The general purification procedure of the present invention consists of reacting a mixture of both geometrical isomers of Tramadol hydrochloride with a potential electrophile under such conditions that the (RS,SR) isomer reacts almost exclusively, while the (RR,SS) isomer remains practically intact. The resulting mixture is evaporated and the resulting solid substance is then recrystallized from isopropanol or any other suitable solvent.
Example 1
11.1 g of a mixture consisting of 77% (RR,SS) Tramadol hydrochloride and 23% of the corresponding (RS,SR) isomer were dissolved in 30 ml DMF. 1.3 g acetic anhydride were added and the reaction mixture was stirred at room temperature for 12 hours. The solvent was partly evaporated under reduced pressure and 15 ml toluene were added. The suspension obtained was filtered and washed with 5 ml toluene. 5.8 g of crystals were obtained, in which the (RR,SS):(RS,SR) isomer ratio was 70:1. The product obtained was crystallized from 12 ml isopropanol and 4 g of pure (RR,SS) Tramadol hydrochloride were obtained.
Example 2
19.5 g of a mixture consisting of 60.5% (RR,SS) Tramadol hydrochloride and 40.5% of the corresponding (RS,SR) isomer were suspended in 55 ml chlorobenzene. A solution of 4 ml thionyl chloride in 15 ml chlorobenzene was added dropwise for two hours. The suspension was partly evaporated, the residue was filtered and rinsed with toluene, and 8.1 g of crystals were obtained, in which the (RR,SS):(RS, SR) isomer ratio was 14:1. The product obtained was recrystallized from isopropanol, and 6.7 g of pure (RR,SS) Tramadol hydrochloride were obtained.
Example 3
33.4 g of a mixture consisting of 45% (RR,SS) Tramadol hydrochloride and 55% of the corresponding (RS,SR) isomer was immersed in 50 ml trifluoroacetic acid, 5.2 g of sodium azide was added, and the reaction mixture was stirred for 24 hours. The reaction mixture was then evaporated under reduced pressure, 50 ml water was added, and the solution was brought to pH 12 with solid potassium carbonate. The suspension was extracted with 50 ml toluene, the solvent was evaporated and 25 ml hydrogen chloride solution in isopropanol were added. The solution was cooled and filtered. 9.5 g of crude (RR,SS) Tramadol were obtained, and the crude product was purified by recrystallization from isopropanol.
The hitherto unknown (RS,SR)-2-(dimethylaminomethyl)-1-azido-1-(3-methoxyphenyl)-cyclohexane hydrochloride was isolated from the reaction mixture, recrystallized from isopropanol and characterized as follows:
(RS,SR)-2-(dimethylaminomethyl)-1-azido-1-(3-methoxyphenyl)-cyclohexane hydrochloride
- ms : 288 m+
- IR : 2050 cm-1 (N3)
- 1H-NMR (DMSO): 10.42 ppm: (acidic proton); 1H; 7.40-6.90 ppm: (aromatic protons) 4H; 3.79 ppm; (OCH3 ), 3H; 2.78, 2.42 ppm: NCH2 ; 2H; 2.58, 2.37 ppm: [N(CH3 )2], 6H; 2.30-1.40 ppm: cyclohexane ring protons, 9H.
- 13C-NMR (DMSO): 159.74 ppm: C1; 144.12 ppm: C5; 130.08 ppm: C3; 117.36 ppm: C4; 112.97, 111.35 ppm: C2, C6; 69.61 ppm: C8; 59.29 ppm: C14; 55.21 ppm: C7; 44.6 ppm: C15; 40.12 ppm: C13; 35.94, 27.02, 23.56, 21.63 ppm: cyclohexane ring carbon nucleii.

AZIDE COMPD
Bibliography
Synthesis of Tramadol
http://www.nioch.nsc.ru/icnpas98/pdf/posters1/156.pdf
- http://www.opioids.com/tramadol/synthesis/index.html
- Patents to Grünenthal G.m.b.H.:- K. Flick and E. Frankus, U.S. Patent 3,652,589, March 28, 1972; Chem. Abs., 1972, 76, 153321; K. Flick and E. Frankus, U.S. Patent. 3,830,934, August 20, 1974; Chem. Abs., (1974), 82, 21817 (synth)
- C. Alvarado, Á. Guzmán, E. Díaz and R. Patiño, J. Mex. Chem. Soc., 49, (2005) 324-327 (synth)
- G. Venkanna, G. Madhusudhan, K. Mukkanti, A. Sankar, V. M. Kumar and S. A. Ali, J. Chem. Pharm. Res., 4 (2012) 4506-4513 (synth)
- A. Boumendjel, G. S.Taïwe, E. N. Bum, T. Chabrol, C. Beney, V. Sinniger, R. Haudecoeur, L. Marcourt, S. Challal, E. F. Queiroz, F. Souard, M. Le Borgne, T. Lomberget, A. Depaulis, C. Lavaud, R. Robins, J.-L. Wolfender, B. Bonaz and M. De Waard, Angew. Chem. Int. Ed., 52 (2013) 11780 –11784 (Tramadol in nature)
Action
- R. B. Raffa, E. Friderichs, W. Reimann, R. P. Shank, E. E. Codd and J. L. Vaught, J. Pharmacol. Exp. Ther., 260 (1992) 275-285 (means of action)
- P. Dayer, L. Collart and J. Desmeules, Drugs (Suppl. 1), (1994) 3-7.
- T. A. Bamigbade, C. Davidson, R. M. Langford and J. A. Stamford, Br. J. Anaesthes., 79 (1997) 352-356. (action of enantiomers)
- P. W. Keeley, G. Foster and L. Whitelaw, BMJ, 321 (2000) 1608 (auditory hallucinations with tramadol)
- U. M. Stamer, F. Musshoff, M. Kobilay, B. Madea, A. Hoeft and F. Stuber, Clin. Pharmacol. Ther., 82 (2007) 41-47 (different CYP2D6 Genotypes and metabolism)
- W. Leppert, Pharmacology, 87 (2011)274–285 (metabolism of tramadol to O-desmethyltramadol by CYP2D6)
- A. Elkalioubie, D. Allorge, L. Robriquet, J.-F. Wiart, A. Garat, F. Broly and F. Fourrier, Eur. J. Clin. Pharmacol., 67 (2011) 855–858 (ultrarapid metabolism of tramadol)
- C. F. Samer, K. I. Lorenzini, V. Rollason, Y. Daali and J. A. Desmeules, Molecular Diagnosis & Therapy, 17 (2013), 165–84 (variations in tramadol response)
Tramadol abuse
- M. K. Wedge, Can. Pharm. J., 142 (2009) 71-73. (tramadol and antidepressants)
- ACMD advice on O-desmethyltramadol
- Y. Progler, J. Res. Med. Sci., 15 (2010) 185-188 (Tramadol abuse and smuggling into Gaza)
- M. M. Fawzi, Egypt. J. Forensic Sci., 1 (2011) 99–102 (Tramadol abuse in Egypt)
- I. Giraudon, K. Lowitz, P. I. Dargan, D. M. Wood and R. C. Dart, Br. J. Clin. Pharmacol., 76, (2013) 823–824 (prescription opioid abuse in the UK)
- C. Stannard, BMJ, 347 (2013) f5108 (tramadol problem)
- N. Hawkes, BMJ, 347 (2013) f5336 (deaths from tramadol and legal highs)
- A. Winstock, J. Bell and R. Borschmann, BMJ 347 (2013) f5599 (monitoring Tramadol abuse)
- S. H. Park, R. C. Wackernah and G. L. Stimmel, J. Pharm. Pract., 27 (2014) 71-78.
- Unemployment in Gaza and Tramadol addiction.
Tramadol in “highs”
- T. Arndt, U. Claussen, B. Güssregen, S. Schröfel, B. Stürzer, A. Werle and G. Wolf, For. Sci. Int., 208 (2011) 47-52. (Kratom alkaloids and O-desmethyltramadol in urine of a “Krypton” herbal mixture consumer)
- R. Kronstrand, M. Roman, G. Thelander and A. Eriksson, J. Anal. Toxicol., 35 (2011) 242–247. (fatalities from mitragynine and O-desmethyltramadol combinations in Krypton herbal mixture)
- C. D. Rosenbaum, S. P. Carreiro and K. M. Babu, J. Med. Toxicol., 8 (2012) 15-32 (review of herbal marijuana alternatives, including Kratom)
Tramadol in cycling
- Michael Barry, “Shadows on the Road: Life at the Heart of the Peloton, from US Postal to Team Sky”, Faber 2014.
- Chris Froome, “The Climb: The Autobiography”, Viking, 2014.
- http://cyclingtips.com.au/2014/05/wada-proposes-tramadol-remains-a-monitored-rather-than-a-banned-substance-in-2015/
- Tramadol blamed for crashes
- Tramadol use for pain in cycling
| EP0778262A2 * | 19 Nov 1996 | 11 Jun 1997 | Chemagis Ltd. | Process for the purification of (RR-SS)-2-dimethyl-aminomethyl-1-(3-methoxyphenyl)cyclohexanol and its salts |
| EP0787715A1 * | 21 Dec 1996 | 6 Aug 1997 | Grünenthal GmbH | Process for the optical resolution of tramadol |
| EP0831082A1 * | 19 Aug 1997 | 25 Mar 1998 | Chemagis Ltd. | Process for the purification of (RR-SS)-2-dimethylaminomethyl-1-(3-methoxyphenyl)cyclohexanol hydrochloride |
| US5414129 * | 8 Sep 1993 | 9 May 1995 | Chemagis, Ltd. | Process for the purification of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol and its salts |
| Reference | ||
|---|---|---|
| 1 | * | CHEMICAL ABSTRACTS, vol. 126, no. 26, 30 June 1997 Columbus, Ohio, US; abstract no. 343383v, page 597; column 1; XP002079824 & IL 103 096 A (CHEMAGIS LTD) 5 December 1996 |
| 2 | * | CHEMICAL ABSTRACTS, vol. 127, no. 20, 17 November 1997 Columbus, Ohio, US; abstract no. 278028n, QIAO, BEN-ZHI ET AL.: “Synthesis and structure of 1-(m-methoxyphenyl)-2-(dimethylaminomethyl )cyclohexanol.” page 683; column 2; XP002079825 & GAODENG XUEXIAO HUAXUE XUEBAO , vol. 18, no. 6, 1997, pages 902-905, |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999036389A1 * | 14 Jan 1999 | 22 Jul 1999 | Nicholas Archer | Purification of tramadol |
| WO2000078705A1 * | 22 Jun 1999 | 28 Dec 2000 | Bernhard Akteries | Method for separating the diastereomer bases of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)-cyclohexanol |
| WO2003078380A2 * | 20 Mar 2003 | 25 Sep 2003 | Shahid Akhtar Ansari | Process for preparing tramadol hydrochloride and/or tramadol momohydrate |
| WO2004020390A1 * | 7 Aug 2003 | 11 Mar 2004 | Bernhard Akteries | Method for the production of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol |
| EP1346978A1 * | 21 Mar 2002 | 24 Sep 2003 | Jubilant Organosys Limited | Process for preparing tramadol hydrochloride and/or tramadol monohydrate |
| US6521792 * | 21 Dec 2001 | 18 Feb 2003 | Gruenenthal Gmbh | Process for separating the diastereomeric bases of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)-cylohexanol |
| US6649783 | 3 Oct 2001 | 18 Nov 2003 | Euro-Celtique, S.A. | Synthesis of (+/-)-2-((dimethylamino)methyl)-1-(aryl)cyclohexanols |
| US6784319 | 15 Sep 2003 | 31 Aug 2004 | Euro-Celtique, S.A. | Synthesis of (±)-2-((dimethylamino)methyl)-1-(aryl)cyclohexanols |
| US7030276 | 9 Feb 2005 | 18 Apr 2006 | Gruenenthal Gmbh | Process for preparing 2-[(dimethylamino)-methyl]-1-(3-methoxyphenyl)cyclohexanol |
| US8221792 | 7 Jul 2006 | 17 Jul 2012 | Farnam Companies, Inc. | Sustained release pharmaceutical compositions for highly water soluble drugs |
| EP0940385A1 * | Mar 3, 1999 | Sep 8, 1999 | Dinamite Dipharma S.p.A. | Process for the separation of the (RR,SS)-2-(dimethylamino)methyl-1-(3-methoxyphenyl)-cyclohexanol isomer from the (RS,SR) isomer by selective precipitation |
| WO1999003820A1 * | Jun 26, 1998 | Jan 28, 1999 | Nikolopoulos Angelo | Tramadol, salts thereof and process for their preparation |
| WO1999036390A1 * | Jan 14, 1999 | Jul 22, 1999 | Nicholas Archer | Purification of tramadol |
| WO2000078705A1 * | Jun 22, 1999 | Dec 28, 2000 | Bernhard Akteries | Method for separating the diastereomer bases of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)-cyclohexanol |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7470816 | Nov 13, 2006 | Dec 30, 2008 | Ipac Laboratories Limited | Tramadol recovery process |
| US3652589 * | 27 Jul 1967 | 28 Mar 1972 | Gruenenthal Chemie | 1-(m-substituted phenyl)-2-aminomethyl cyclohexanols |
| US5414129 * | 8 Sep 1993 | 9 May 1995 | Chemagis, Ltd. | Process for the purification of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol and its salts |
| EP0778262A2 * | 19 Nov 1996 | 11 Jun 1997 | Chemagis Ltd. | Process for the purification of (RR-SS)-2-dimethyl-aminomethyl-1-(3-methoxyphenyl)cyclohexanol and its salts |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6828345 * | 31 Mar 2003 | 7 Dec 2004 | Gruenenthal Gmbh | O-substituted 6-methyltramadol derivatives |
| US7030276 | 9 Feb 2005 | 18 Apr 2006 | Gruenenthal Gmbh | Process for preparing 2-[(dimethylamino)-methyl]-1-(3-methoxyphenyl)cyclohexanol |
| US7470816 | 13 Nov 2006 | 30 Dec 2008 | Ipac Laboratories Limited | Tramadol recovery process |
| US20050215821 * | 9 Feb 2005 | 29 Sep 2005 | Gruenenthal Gmbh | Process for preparing 2-[(dimethylamino)-methyl]-1-(3-methoxyphenyl)cyclohexanol |
| US20070112074 * | 13 Nov 2006 | 17 May 2007 | Ashok Kumar | Tramadol recovery process |
| EP0778262A2 * | Nov 19, 1996 | Jun 11, 1997 | Chemagis Ltd. | Process for the purification of (RR-SS)-2-dimethyl-aminomethyl-1-(3-methoxyphenyl)cyclohexanol and its salts |
| US3652589 * | Jul 27, 1967 | Mar 28, 1972 | Gruenenthal Chemie | 1-(m-substituted phenyl)-2-aminomethyl cyclohexanols |
| US5414129 * | Sep 8, 1993 | May 9, 1995 | Chemagis, Ltd. | Process for the purification of 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol and its salts |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0940385A1 * | Mar 3, 1999 | Sep 8, 1999 | Dinamite Dipharma S.p.A. | Process for the separation of the (RR,SS)-2-(dimethylamino)methyl-1-(3-methoxyphenyl)-cyclohexanol isomer from the (RS,SR) isomer by selective precipitation |
| DE10218862A1 * | Apr 26, 2002 | Nov 6, 2003 | Gruenenthal Gmbh | Verfahren zur Chlorierung tertiärer Alkohole |
| US6169205 | Mar 4, 1999 | Jan 2, 2001 | Dipharma S.P.A. | Process for the purification of (RR,SS)-2-(dimethylamino) methyl-1-(3-methoxyphenyl)-cyclohexanol from (RS,SR)-2-(dimethylamino)methyl-1-(3-methoxyphenyl) cyclohexanol |
| US6469213 | Jan 14, 2000 | Oct 22, 2002 | Russinsky Limited | Tramadol, salts thereof and process for their preparation |
| US6649783 | Oct 3, 2001 | Nov 18, 2003 | Euro-Celtique, S.A. | Synthesis of (+/-)-2-((dimethylamino)methyl)-1-(aryl)cyclohexanols |
| US6784319 | Sep 15, 2003 | Aug 31, 2004 | Euro-Celtique, S.A. | Synthesis of (±)-2-((dimethylamino)methyl)-1-(aryl)cyclohexanols |
| US7235693 | Oct 26, 2004 | Jun 26, 2007 | Gruenenthal Gmbh | Process for chlorinating tertiary alcohols |
| US7470816 | Nov 13, 2006 | Dec 30, 2008 | Ipac Laboratories Limited | Tramadol recovery process |
| WO1999003820A1 * | Jun 26, 1998 | Jan 28, 1999 | Nikolopoulos Angelo | Tramadol, salts thereof and process for their preparation |
| Systematic (IUPAC) name | |
|---|---|
|
2-[(Dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol
|
|
| Clinical data | |
| Trade names | Ryzolt, Tramal, Ultram |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a695011 |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Dependence liability |
Present[1] |
| Routes of administration |
Oral, IV, IM, rectal |
| Pharmacokinetic data | |
| Bioavailability | 70–75% (oral), 77% (rectal), 100% (IM)[2] |
| Protein binding | 20%[3] |
| Metabolism | Liver-mediated demethylation andglucuronidation via CYP2D6 &CYP3A4[2][3] |
| Biological half-life | 6.3 ± 1.4 hr[3] |
| Excretion | Urine (95%)[4] |
| Identifiers | |
| CAS Registry Number | 27203-92-5 |
| ATC code | N02AX02 |
| PubChem | CID: 33741 |
| DrugBank | DB00193 |
| ChemSpider | 31105 |
| UNII | 39J1LGJ30J |
| KEGG | D08623 |
| ChEBI | CHEBI:9648 |
| ChEMBL | CHEMBL1066 |
| Chemical data | |
| Formula | C16H25NO2 |
| Molecular mass | 263.4 g/mol |
DMF
Tramadol hydrochloride..CEP
| Product Name | Country | Manufacture | Chemical formula | CAS # | CEP | DMF |
| Tramadol hydrochloride | India | IPCA Laboratories Ltd | C16H26ClNO2 | 22204-88-2 | R0-CEP 2008-189-Rev 00 | – – – |
| Tramadol hydrochloride | India | Dishman Pharmaceuticals and Chemicals Ltd. | C16H26ClNO2 | 22204-88-2 | R0-CEP 2003-148-Rev 01 | – – – |
| Tramadol hydrochloride | India | Sun Pharmaceutical Industries Ltd. | C16H26ClNO2 | 22204-88-2 | R1-CEP 2002-232-Rev 02 | – – – |
| Tramadol hydrochloride | China | CSPC OUYI PHARMACEUTICAL CO., LTD. | C16H26ClNO2 | 22204-88-2 | R0-CEP 2005-227-Rev 02 | – – – |
| Tramadol hydrochloride | India | JUBILANT LIFE SCIENCES LIMITED | C16H26ClNO2 | 22204-88-2 | R1-CEP 2002-199-Rev 03 | – – – |
| Tramadol hydrochloride | India | Cadila Pharmaceuticals Ltd. | C16H26ClNO2 | 22204-88-2 | R1-CEP 2004-098-Rev 01 | – – – |
| Tramadol hydrochloride | Germany | AREVIPHARMA GMBH | C16H26ClNO2 | 22204-88-2 | R0-CEP 2005-020-Rev 02 | – – – |
| Tramadol hydrochloride New process | India | Inogent Laboratories Private Limited | C16H26ClNO2 | 22204-88-2 | R0-CEP 2007-129-Rev 00 | – – – |
| Tramadol hydrochloride | India | SPIC Limited, Pharmaceuticals Division | C16H26ClNO2 | 22204-88-2 | R0-CEP 2004-245-Rev 00 | – – – |
| Tramadol hydrochloride | Switzerland | Cilag AG CH | C16H26ClNO2 | 22204-88-2 | R0-CEP 2006-262-Rev 00 | – – – |
| Tramadol hydrochloride | Italy | Dipharma Francis S.r.l. | C16H26ClNO2 | 22204-88-2 | R0-CEP 2002-105-Rev 01 | – – – |
| Tramadol hydrochloride | Israel | Chemagis Ltd | C16H26ClNO2 | 22204-88-2 | R1-CEP 2003-146-Rev 00 | – – – |
| Tramadol hydrochloride | India | Wanbury LTD | C16H26ClNO2 | 22204-88-2 | R0-CEP 2005-151-Rev 01 | – – – |
| Tramadol hydrochloride | Germany | Excella GmbH | C16H26ClNO2 | 22204-88-2 | R0-CEP 2003-137-Rev 02 | – – – |
| Tramadol hydrochloride | Switzerland | Proto Chemicals AG | C16H26ClNO2 | 22204-88-2 | R1-CEP 2002-204-Rev 01 | – – – |
| Tramadol hydrochloride | Czech Republic | ZENTIVA K.S. | C16H26ClNO2 | 22204-88-2 | R0-CEP 2009-214-Rev 00 | – – – |
| Tramadol hydrochloride | United States | Noramco, Inc. | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | Germany | GRUNENTHAL GMBH | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | Ireland | IROTEC LABORATORIES | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | Switzerland | HELSINN CHEMICALS SA | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | Israel | Chemagis Ltd | US – – | |||
| Tramadol hydrochloride | Slovakia | “ZENTIVA, A.S.” | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | United States | WYCKOFF CHEMICAL CO INC | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | Germany | Excella GmbH | US – – | |||
| Tramadol hydrochloride USP (BULK) | China | SHIJIAZHUANG PHARMACEUTICAL GROUP CO LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | Germany | EVONIK DEGUSSA GMBH | US – – | |||
| Tramadol hydrochloride | Italy | RECORDATI S.p.A. | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | India | Dishman Pharmaceuticals and Chemicals Ltd. | US – – | |||
| TRAMADOL HYDROCHLORIDE | India | Sun Pharmaceutical Industries Ltd. | US – – | |||
| TRAMADOL HYDROCHLORIDE | India | Cadila Pharmaceuticals Ltd. | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | Switzerland | PROTO CHEMICALS AG | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride EP | India | DAI ICHI KARKARIA LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | PEARL ORGANICS LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride EP | China | SHIJIAZHUANG PHARMACEUTICAL GROUP HUASHENG PHARMA CO LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | India | JUBILANT LIFE SCIENCES LIMITED | US – – | |||
| TRAMADOL HYDROCHLORIDE | Germany | AREVIPHARMA GMBH | US – – | |||
| Tramadol hydrochloride | India | TONIRA PHARMA LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | INOGENT LABORATORIES PRIVATE LIMITED | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | China | ZHEJIANG HISOAR PHARMACEUTICAL CO LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| TRAMADOL HYDROCHLORIDE | India | IPCA Laboratories Ltd | US – – | |||
| Tramadol hydrochloride | China | HEBEI ZHONGSHENG PHARMACEUTICAL CO LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride BP | India | KAMUD DRUGS PVT LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | Raks Pharma Pvt Ltd. | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride USP n/sterile | India | Aurobindo Pharma Ltd. | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride (YT3 PROCESS) | India | IPCA Laboratories Ltd | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | PIRAMAL HEALTHCARE LTD | C16H26ClNO2 | 22204-88-2 | US – – | |
| Tramadol hydrochloride | India | Asence Pharma Private Ltd | C16H26ClNO2 | 22204-88-2 | – – – |
SYNOPSIS FOR M. PHARM DISSERTATION
Formulated and Optimized Hydrodynamically balanced Oral Controlled release Bioadhesive
Tablets of Tramadol Hydrochloride using different polymers like Carbopol 971P (CP) and …
////////TRAMADOL,
Study Demonstrates Efficacy of New Tumor Treatment

Study Demonstrates Efficacy of New Tumor Treatment |
||||
|
The results of a study demonstrate the efficacy of this drug in treating nonfunctional neuroendocrine tumors of lung or gastrointestinal origin.
|
What are “complex manufacturing processes”? A recent reply from the EMA
DRUG REGULATORY AFFAIRS INTERNATIONAL
Sometimes a clear definition of terms is crucial in the communication between authorities and pharmaceutical companies. Find out what the European Medicines Agency EMA defines as “complex manufacturing steps” and what authorisation holders providing a variation application need to consider.
The Variations Regulation (EC) no. 1234/2008 of the European Commission defines the procedure for variations of existing marketing authorisations. The “detailed guidelines for the various categories of variations“, which were published in the consolidated version in August 2013 in the European Official Journal, explain the interpretation and application of this Variations Regulation.
Although the “detailed guidelines” describe a number of scenarios of possible variations in some detail, there are formulations in the Guideline text which require clarification due to their blur. The EMA adopted such a case in a recent update of itsquestions and answers collection “Quality of Medicines Questions and Answers: Part 1”…
View original post 258 more words
New “mTOR” inhibitor from Exelixis, Inc., XL 388

XL 388
A Novel Class of Highly Potent, Selective, ATP-Competitive, and Orally Bioavailable Inhibitors of the Mammalian Target of Rapamycin (mTOR)
Benzoxazepine-Containing Kinase Inhibitor

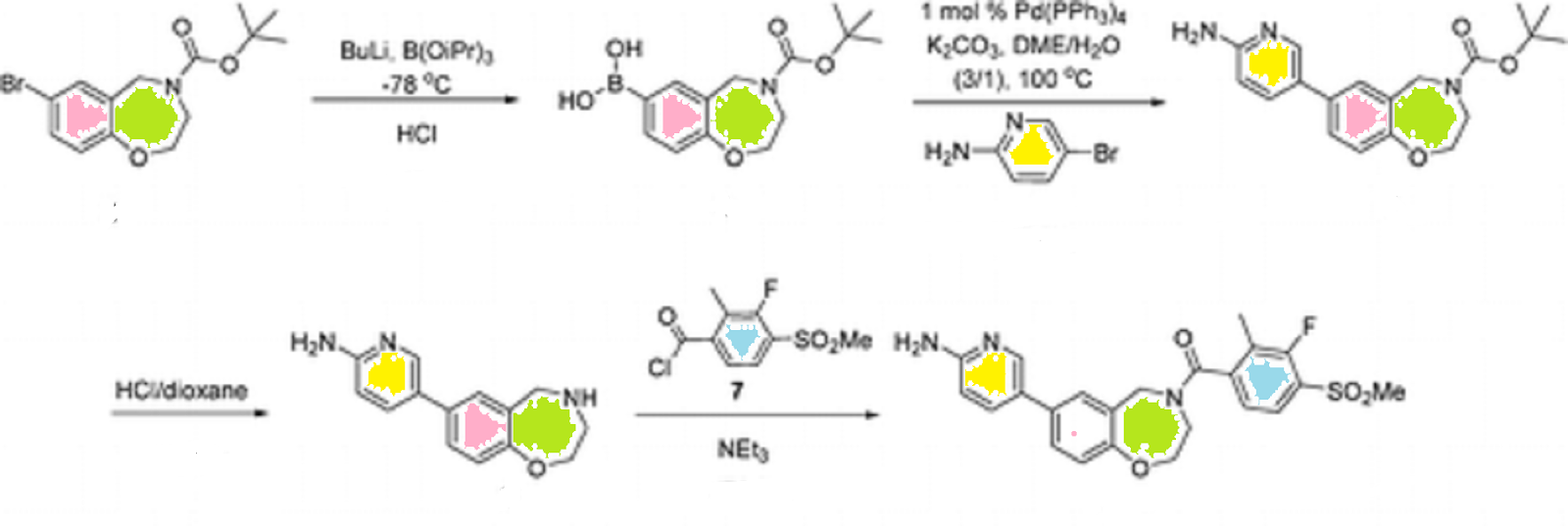


PATENT
Example 2:
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-l,4-benzoxazepin-4(5H)-yl] [3-fluoro- 2-methyl-4-(methylsulfonyl)phenyl]methanone

tørt-Butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[/] [l,4]oxazepine-4(5H)- carboxylate. To a mixture of 4-(te/t-butoxycarbonyl)-2,3,4,5- tetrahydrobenzo[/][l,4]oxazepin-7-ylboronic acid (1.52 g, 5.2 mmol), prepared as described in Reference Example 5, 2-amino-5-bromopyridine (900 mg, 5.2 mmol), and potassium carbonate (1.73 g, 12.5 mmol) in 1 ,2-dimethoxyethane/water (30 mL/10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 1.5 mol%) and the reaction mixture was purged with nitrogen and stirred at reflux for 3 h. The reaction was cooled to rt, diluted with water/ethyl acetate (50 mL/50 mL), and the separated aqueous layer was extracted with ethyl acetate. The resulting emulsion was removed by filtration. The combined organic layer was washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure, and the residue was triturated with toluene for 1 h. The resulting off-white solid was isolated by filtration to give the desired product (1.37 g, 77 %) as an off-white solid. MS (EI) for Ci9H23N3O3: 342 (MH+).
5-(2,3,4,5-Tetrahydrobenzo[/] [l,4]oxazepin-7-yl)pyridine-2-amine. To a stirred solution of tert-butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[/][l,4]oxazepine- 4(5H)-carboxylate (1.36 g, 3.98 mmol) in 1,4-dioxane (5 mL) was added 4 N hydrogen chloride in 1 ,4-dioxane (5 mL) and the reaction mixture was stirred at rt overnight. The reaction was concentrated on a rotary evaporator and the residue was triturated with ether. The solid was isolated by filtration. This solid was dissolved in water (5 mL) and made basic with 5 N sodium hydroxide to pH 11-12. The brownish sticky oil that aggregated at the bottom was isolated and the aqueous layer was extracted with 5 % methanol in ethyl acetate. The extracts were dried with sodium sulfate and concentrated on a rotary evaporator. The brownish sticky oil was dissolved with a mixture of methanol/ethyl acetate, combined with the isolated organic residue and concentrated under reduced pressure to give a yellow solid. This solid was triturated with dichloromethane (10 mL) for 1 h and a yellow solid was isolated by filtration and dried under high vacuum to give amine the desired product (920 mg, 96 %). MS (EI) for Ci4Hi5N3O: 242 (MH+).
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-l,4-benzoxazepin-4(5H)-yl][3-fluoro-2- methyl-4-(methylsulfonyl)phenyl]methanone.
To a stirred suspension of 5-(2, 3,4,5- tetrahydrobenzo[/][l,4]oxazepin-7-yl)pyridine-2-amine (85 mg, 352 μmol) and triethylamine (54 μL, 387 μmol) in dichloromethane (10 mL) was added 3-fluoro-2-methyl-4- (methylsulfonyl)benzoyl chloride (91 mg, in 3 mL of dichloromethane), prepared as described in Reference Example 1, at 0 0C for 2 h. After stirring for an additional 1 h at rt, the reaction mixture was diluted with water (5 mL) and the separated aqueous layer was extracted with dichloromethane. The combined extracts were dried with sodium sulfate, filtered and concentrated under reduced pressure to give a light-yellow solid that was purified via silica gel chromatography to give the desired product (113 mg, 70%) as a white solid.
1H NMR (400 MHz, DMSO-d6): δ 8.24-8.03 (dd, IH), 7.79-7.71 (m, IH), 7.71-7.69 (dd, 0.5H), 7.57-7.57 (d, 0.5H), 7.44-7.40 (m, 1.5H), 7.29-7.19 (dd, IH), 7.05-7.01 (dd, IH), 6.64-6.63 (d, 0.5H), 6.54-6.45 (dd, IH), 6.06 (s, 2H), 4.93-4.31 (m, 2H), 4.31-3.54 (m, 4H), 3.37-3.36(d, 3H), 2.12-1.77 (d, 3H).
MS (EI) C23H22FN3O4S: 456 (MH+).
PAPER

A series of novel, highly potent, selective, and ATP-competitive mammalian target of rapamycin (mTOR) inhibitors based on a benzoxazepine scaffold have been identified. Lead optimization resulted in the discovery of inhibitors with low nanomolar activity and greater than 1000-fold selectivity over the closely related PI3K kinases. Compound 28 (XL388) inhibited cellular phosphorylation of mTOR complex 1 (p-p70S6K, pS6, and p-4E-BP1) and mTOR complex 2 (pAKT (S473)) substrates. Furthermore, this compound displayed good pharmacokinetics and oral exposure in multiple species with moderate bioavailability. Oral administration of compound 28 to athymic nude mice implanted with human tumor xenografts afforded significant and dose-dependent antitumor activity.
(7-(6-Aminopyridin-3-yl)-2,3-dihydrobenz[f][1,4]oxazepin-4(5H)-yl)(3-fluoro-2-methyl-4-(methylsulfonyl)phenyl)methanone (28)
PATENT
- SYNTHETIC EXAMPLES
-

-
1-Bromo-3,4-difluoro-2-methylbenzene. To a stirred mixture of 2,3-difluorotoluene (1.9 g, 14.8 mmol) and iron (82.7 mg, 1.48 mmol) in chloroform (10 mL) at rt was added bromine (76 μL, 14.8 mmol) over 2 h. The resulting mixture was stirred at rt overnight. Excess water (10 mL) was added and the reaction mixture was diluted with ether (20 mL). The separated organic layer was washed with aqueous sodium thiosulfate, brine, dried over sodium sulfate and concentrated on a rotary evaporator. The residue was distilled to give the desired product (2.49 g, 81%) as a colorless oil.
-
3,4-Difluoro-2-methylbenzoic acid. To a stirred solution of 1-bromo-3,4-difluoro-2-methylbenzene (940 mg, 4.54 mmol) in tetrahydrofuran (5 mL) was added isopropylmagnesium bromide (3.0 mL, 6.0 mmol) over 1 h at 0° C. The resulting mixture was stirred at rt for 24 h. Carbon dioxide (CO2), generated from dry ice, was introduced to the reaction mixture over 2 h and the resulting mixture was stirred for an additional 30 min. The reaction mixture was quenched with addition of an excess amount of water (5 mL) and the tetrahydrofuran was removed on a rotary evaporator. The resulting aqueous layer was diluted with water (5 mL) and acidified with concentrated hydrochloric acid to pH 1-2. The white precipitate was filtered and washed with water and cold hexanes and dried under high vacuum to give the desired product (630 mg, 81%) as a white powder. MS (EI) for C8H6F2O2: 171 (MH−).
-
3-Fluoro-2-methyl-4-(thiomethyl)benzoic acid. To a stirred solution of acid 3,4-difluoro-2-methylbenzoic acid (700 mg, 4.1 mmol) in dimethylsulfoxide (5 mL) was added powdered potassium hydroxide (274 mg, 4.9 mmol) and the mixture was stirred at rt for 30 min. Sodium thiomethoxide (342 mg, 4.9 mmol) was added to the mixture and the resulting mixture was stirred at 55-60° C. for 4 h. Additional powdered potassium hydroxide (70 mg, 1.2 mmol), sodium thiomethoxide (60 mg, 0.8 mmol), and dimethylsulfoxide (2 mL) were added to the reaction mixture. After stirring for 4 h, the mixture was cooled to 0° C. and quenched with excess water (10 mL). The resulting suspension was acidified at 0° C. with concentrated hydrochloric acid to pH 1-2. The white precipitate was collected by suction filtration, washed with water and dried under vacuum overnight to give the desired product (870 mg, 100%). The intermediate sulfide was used in the next step without further purification. MS (EI) for C9H9FO2S: 199.1 (MH−).
-
3-Fluoro-2-methyl-4-(methylsulfonyl)benzoic acid. To a stirred suspension of 3-fluoro-2-methyl-4-(thiomethyl)benzoic acid in an acetone/water (1 mL/10 mL) mixture was added sodium hydroxide (330 mg, 8.25 mmol) and sodium bicarbonate (680 mg, 8.1 mmol). Oxone (˜4 g) was added portionwise to the reaction mixture at 0° C. over 2 h. The reaction was monitored by LC/MS. Concentrated hydrochloric acid was added to adjust the pH 2-3 and the white precipitate was collected by suction filtration, washed with water, and dried under vacuum. Dried precipitate was suspended in water (10 mL), stirred vigorously at rt for 1 h, filtered, washed with water, and hexanes and dried under vacuum to give the desired product (886 mg, 94%) as a white powder. MS (EI) for C9H9FO4S: 231 (MH−).
-
3-Fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride. A mixture of 3-fluoro-2-methyl-4-(methylsulfonyl)benzoic acid (860 mg, 3.7 mmol) in thionyl chloride (10 mL) was heated to reflux for 3 h. (the reaction mixture became homogenous). The reaction mixture was concentrated on a rotary evaporator to give the crude acid chloride. This acid chloride was triturated with dichloromethane (2 mL) and concentrated under reduced pressure. The trituration process was repeated 3 times until the product (900 mg, 98%) was obtained as a white powder.
- Reference Example 13-Fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride

Reference Example 2Ethyl 4-(2,3,4,5-tetrahydro-1,4-benzoxazepin-7-yl)benzoate hydrochloride salt
-

-
4-(ethoxycarbonyl)phenylboronic acid (22.16 g, 114 mmol), tert-butyl 7-bromo-2,3-dihydrobenzo[f][1,4]oxazepin-4(5H)-carboxylate (34.08 g, 104 mmol), prepared as described in Reference Example 4, Pd(dppf)Cl2 and TEA (21 g, 208 mmol) were combined in a mixture of dioxane (200 mL) and water (20 mL). The reaction mixture was heated to 90° C. for 2 h, then cooled and the solvent removed. Purification of the residue by silica chromatography gave the desired product ester (31.3 g, 69% yield).
-
To the solution of tert-butyl 7-(4-(ethoxycarbonyl)phenyl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (10.3 g, 25.93 mmol) in MeOH (120 mL) was added a solution of 4 N HCl in dioxane (50 mL). The reaction mixture was heated to 50° C. for 3 h (monitored by LC/MS). The reaction mixture was allowed to cool to rt. Ethyl 4-(2,3,4,5-tetrahydro-1,4-benzoxazepin-7-yl)benzoate as the hydrochloride salt (8.8 g, 99% yield) was collected by suction filtration.

-

-
tert-Butyl-5-bromo-2-hydroxybenzyl(2-hydroxyethyl)carbamate. Commercially-available 5-bromo-2-hydroxybenzaldehyde (4.0 g, 10 mmol) and 2-aminoethanol were combined in THF/MeOH (100 mL, 10:1) and sodium borohydride (0.76 g, 2.0 mmol) was added with stirring. The resulting reaction mixture was stirred at 40° C. for 4 h, concentrated on a rotary evaporator then diluted with EtOAc (50 mL) and saturated NaHCO3 (30 mL). To this suspension was added di-tert-butyl dicarbonate (2.83 g, 13 mmol). The mixture was stirred at rt overnight. The organic layer was washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated on a rotary evaporator. Hexane was subsequently added to the crude reaction product which resulted in the formation of a white solid. This slurry was filtered to obtain the desired product (6.8 g, 98%) as a white solid. MS (EI) for C14H20BrNO4, found 346 (MH+).
-
tert-Butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate. tert-Butyl-5-bromo-2-hydroxybenzyl(2-hydroxyethyl)carbamate (3.46 g, 10 mmol) and triphenylphosphine (3.96 g, 15 mmol) were combined in DCM (100 mL) and diisopropyl azodicarboxylate (3.03 g, 15 mmol) was added. The resulting reaction mixture was stirred at rt for 12 h. The reaction mixture was washed with water, dried, filtered, and concentrated on a rotary evaporator. The resulting crude product was purified via silica gel chromatography eluting with 8:2 hexane/ethyl acetate to give the desired product (1.74 g, 53%) as a white solid. MS (EI) for C14H18BrNO3, found 328 (MH+).
- Reference Example 4tert-Butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate

Reference Example 54-(tert-Butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid
-

-
To a stirred solution of tert-butyl-7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (10 g, 30.5 mmol), prepared as described in Reference Example 4, and triisopropylborate (9.1 mL, 40 mmol) in dry tetrahydrofuran (100 mL) was added dropwise n-butyllithium in tetrahydrofuran (1.6 M, 25 mL, 40 mmol) while maintaining the temperature below −60° C. Upon completion of addition, the reaction mixture was stirred for 30 min, then quenched with 1 N aqueous hydrochloric acid (35 mL) and allowed to warm to rt. The reaction mixture was extracted with ethyl acetate, dried over anhydrous magnesium sulfate, filtered and concentrated on a rotary evaporator. Hexane was subsequently added to the crude reaction product which resulted in the formation of a white solid. This slurry was stirred for 1 h and filtered to obtain 4-(tert-butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid (8.6 g, 95%) as a white solid. MS (EI) for C14H20BNO5: 194 (M-Boc).

- Example 2[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone
-

-
tert-Butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate. To a mixture of 4-(tert-butoxycarbonyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-ylboronic acid (1.52 g, 5.2 mmol), prepared as described in Reference Example 5, 2-amino-5-bromopyridine (900 mg, 5.2 mmol), and potassium carbonate (1.73 g, 12.5 mmol) in 1,2-dimethoxyethane/water (30 mL/10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 1.5 mol %) and the reaction mixture was purged with nitrogen and stirred at reflux for 3 h. The reaction was cooled to rt, diluted with water/ethyl acetate (50 mL/50 mL), and the separated aqueous layer was extracted with ethyl acetate. The resulting emulsion was removed by filtration. The combined organic layer was washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure, and the residue was triturated with toluene for 1 h. The resulting off-white solid was isolated by filtration to give the desired product (1.37 g, 77%) as an off-white solid. MS (EI) for C19H23N3O3: 342 (MH+).
-
5-(2,3,4,5-Tetrahydrobenzo[f][1,4]oxazepin-7-yl)pyridine-2-amine. To a stirred solution of tert-butyl 7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (1.36 g, 3.98 mmol) in 1,4-dioxane (5 mL) was added 4 N hydrogen chloride in 1,4-dioxane (5 mL) and the reaction mixture was stirred at rt overnight. The reaction was concentrated on a rotary evaporator and the residue was triturated with ether. The solid was isolated by filtration. This solid was dissolved in water (5 mL) and made basic with 5 N sodium hydroxide to pH 11-12. The brownish sticky oil that aggregated at the bottom was isolated and the aqueous layer was extracted with 5% methanol in ethyl acetate. The extracts were dried with sodium sulfate and concentrated on a rotary evaporator. The brownish sticky oil was dissolved with a mixture of methanol/ethyl acetate, combined with the isolated organic residue and concentrated under reduced pressure to give a yellow solid. This solid was triturated with dichloromethane (10 mL) for 1 h and a yellow solid was isolated by filtration and dried under high vacuum to give amine the desired product (920 mg, 96%). MS (EI) for C14H15N3O: 242 (MH+).
-
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone. To a stirred suspension of 5-(2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-7-yl)pyridine-2-amine (85 mg, 352 μmol) and triethylamine (54 μL, 387 μmol) in dichloromethane (10 mL) was added 3-fluoro-2-methyl-4-(methylsulfonyl)benzoyl chloride (91 mg, in 3 mL of dichloromethane), prepared as described in Reference Example 1, at 0° C. for 2 h. After stirring for an additional 1 h at rt, the reaction mixture was diluted with water (5 mL) and the separated aqueous layer was extracted with dichloromethane. The combined extracts were dried with sodium sulfate, filtered and concentrated under reduced pressure to give a light-yellow solid that was purified via silica gel chromatography to give the desired product (113 mg, 70%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.24-8.03 (dd, 1H), 7.79-7.71 (m, 1H), 7.71-7.69 (dd, 0.5H), 7.57-7.57 (d, 0.5H), 7.44-7.40 (m, 1.5H), 7.29-7.19 (dd, 1H), 7.05-7.01 (dd, 1H), 6.64-6.63 (d, 0.5H), 6.54-6.45 (dd, 1H), 6.06 (s, 2H), 4.93-4.31 (m, 2H), 4.31-3.54 (m, 4H), 3.37-3.36 (d, 3H), 2.12-1.77 (d, 3H). MS (EI) C23H22FN3O4S: 456 (MH+).

PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00037

The benzoxazepine core is present in several kinase inhibitors, including the mTOR inhibitor 1. The process development for a scalable synthesis of 7-bromobenzoxazepine and the telescoped synthesis of 1 are reported. Compound 1 consists of three chemically rich, distinct fragments: the tetrahydrobenzo[f][1,4]oxazepine core, the aminopyridyl fragment, and the substituted (methylsulfonyl)benzoyl fragment. Routes were developed for the preparation of 3-fluoro-2-methyl-4-(methylsulfonyl)benzoic acid (17) and tert-butyl 7-bromo-2,3-dihydrobenzo[f][1,4]oxazepine-4(5H)-carboxylate (2). The processes for the two compounds were scaled up, and over 15 kg of each starting material was prepared in overall yields of 42% and 58%, respectively.
A telescoped sequence beginning with compound 2 afforded 7.5 kg of the elaborated intermediate 5-(2,3,4,5-tetrahydrobenzo[f][1,4]oxazepin-2-amine dihydrochloride (6) in 63% yield. Subsequent coupling with benzoic acid 17 gave 7.6 kg of the target compound 1 in 84% yield. The preferred hydrochloride salt was eventually prepared. The overall yield for the synthesis of inhibitor 1 was 21% over eight isolated synthetic steps, and the final salt was obtained with 99.7% HPLC purity.
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone (1)
[7-(6-Aminopyridin-3-yl)-2,3-dihydro-1,4-benzoxazepin-4(5H)-yl][3-fluoro-2-methyl-4-(methylsulfonyl)phenyl]methanone Hydrochloride (1·HCl)
REFERENCES
Anand, N.; Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of their Use and Manufacture. U.S. Patent 8,648,066, Feb 11, 2014.
| US8637499 * | May 25, 2010 | Jan 28, 2014 | Exelixis, Inc. | Benzoxazepines as inhibitors of PI3K/mTOR and methods of their use and manufacture |
| US20120258953 * | May 25, 2010 | Oct 11, 2012 | Exelixis, Inc. | Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of Their Use and Manufacture |
PROFILE

Senior Director
Chemical Development at Dermira, Inc.
Lives San jose caifornia

https://www.linkedin.com/pub/sriram-naganathan/3/50a/5b6
https://www.facebook.com/sriram.naganathan.5
snaganat@exelixis.com, sriramrevathi@yahoo.com
Summary
Chemical process-development and CMC professional offering 20 years of experience from preclinical development through commercialization of small molecules and peptides.
Hands-on experience in multi-step synthesis, route-scouting, process development, scale-up, tech transfer to CRO/CMO, including manufacture under cGMP and process validation.
Extensive knowledge of CMC regulatory landscape (FDA, EMEA) including preparation of CMC sections of IND, IMPD, NDA and MAA
Experience
Senior Director, Chemical Development
Dermira, Inc.
January 2015 – Present (10 months)Menlo Park, CA

Consultant
Intarcia Therapeutics
December 2014 – January 2015 (2 months)

Senior Director
Exelixis
March 2013 – November 2014 (1 year 9 months)South San Francisco, CA
210 E. Grand Ave
United States
Senior Scientist II
Exelixis
August 2004 – January 2008 (3 years 6 months)
Associate Director
CellGate, Inc.
2000 – 2004 (4 years)
Research Scientist
Roche Bioscience
1997 – 2000 (3 years)
Research Scientist
Cultor
1995 – 1997 (2 years)

Research Scientist
Pfizer
1994 – 1997 (3 years)

Research Assistant Professor
University of Pittsburgh
April 1992 – October 1994 (2 years 7 months)
Worked on Vitamin K mechanism in the labs of (Late) Prof Paul Dowd
Education


Vivekananda College (University of Madras), India
Bachelor of Science (B.Sc.), Chemistry
1980 – 1983
![]()

Sriram Naganathan, Ph.D. 1992, snaganat@exelixis.com, sriramrevathi@yahoo.com

As many things change, many things remain constant. One such constant is the frequent reminder that “You can take the boy out of sulfur chemistry but you cannot take sulfur chemistry out of the boy”. At every stage of my professional career organic chemistry of sulfur and sulfur-containing compounds have followed me (or is it the other way around?). Not many can point to the cover of an Angewandte Chemie issue as a synopsis of his/her thesis work – I will be forever grateful for that opportunity received in the Block Group.
As a post-doc in the late Prof. Paul Dowd’s lab at the University of Pittsburgh we used sulfur-containing analogs of vitamin K to probe the mechanism of action. I was then hired at Pfizer Central Research in Groton, CT in the Specialty Chemicals Division to investigate possible decomposition pathways of sulfur-containing high-intensity artificial sweeteners.
At Roche Bioscience (Palo Alto, CA) and Exelixis (South San Francisco, CA – my current job………CHANGED……Dermira) I was involved in process development for the preparation of therapeutic agents, several of them sulfur-containing molecules. Between those two positions I was a Senior Scientist at CellGate (Sunnyvale, CA).
We attempted to exploit the chemistry of sulfur-containing linkers to target the delivery active pharmaceutical agents, using the transport properties of polyarginines. Although I thought I was only training to become a synthetic organic chemist, I did not realize that my passion was really organic reaction mechanisms until I arrived in the Block lab – the two arms of the science are truly inseparable.
I realize after many years that the seed was really sown and nurtured during the many friendly and sometimes-fiery discussions in the lab, and further solidified in my post-doc years. I learned that every “blip-in-the-baseline” cannot to be ignored, and is part of the whole story.
As a process chemist in the pharma industry, I can attribute much of my success to lessons about careful and critical evaluation of primary data and thorough knowledge of reaction mechanisms. I am currently Director, Chemical Development, at Exelixis.NOW DERMIRA.
My primary responsibility involves the manufacture and potential commercialization of our primary product, cabozantinib. It was only natural that I developed a strong interest in the science of cooking and food. I have been pursuing this avenue since moving to Northern California.
I am also an avid gardener, experimenting with growing interesting varieties of chilies, tomatoes and then combining those with all sorts of alliums. It does help that I live close enough to Gilroy, CA, that I can often smell what they are famous for as I walk out of the front door!! I have shared my knowledge in several lectures at the Tech Museum (San Jose, CA) where I was a volunteer exhibit explainer.
My family (my wife Revathi and our two high-school-age daughters Swetha and Sandhya) like to travel and also enjoy the outdoor recreation so abundant in Northern California. We try to take in a new country each year and accomplish personal challenges. After many interesting years in the tech-industry, Revathi is a full-time mom. She is also a fitness instructor at the Y. Swetha and Sandhya are part of the water polo and swim teams at their school.
Swetha is very active in a leadership role for the robotics team, and Sandhya belongs to the quiz team. Revathi and I climbed Half Dome (Yosemite) a few years ago and I just completed a 100-mile bicycle ride around Lake Tahoe.
I remain a highly-opinionated baseball and college basketball fan (favorite teams: in order, Kansas, North Carolina and whoever happens to be playing Missouri and Duke). I am still an avid photographer, although I spend no money on film (I thought I was going to be the last guy on the planet still shooting film!!). I greatly value the many friendships developed during my stay in Albany and keep in touch with many.
In fact, one of my roommates from the SUNY days was instrumental in me getting my present position. Of course, this also means that I have lost touch with several friends during the past decades. If you are reading this and haven’t contacted me in a few years, please do, via e-mail.
We enjoy entertaining guests who drop by – so now you have no excuse not to contact us, especially when you visit the SF Bay Area.

OLD PROFLE……Dr Sriram Naganathan received his Ph.D. from SUNY-Albany where he studied organosulfur chemistry. He is currently an Associate Director at CellGate, Inc. located in Sunnyvale, California. CellGate is involved in the commercialization of novel medicines by utilizing proprietary transporter technology, based on oligomers of arginine, to enhance the therapeutic potential of existing drugs. His responsibilities include process development, scale-up and GMP production of clinical candidates, as well some basic research. He previously held positions at Pfizer Central Research and Roche Bioscience.
Dermira


Thomas G. Wiggans | Founder & Chief Executive Officer……..http://dermira.com/about-us/management-team/

CEO TOM WIGGANS, LEFT AND CMO GENE GAUER, RIGHT


Exelixis, Inc.
![]()

210 East Grand Avenue
So. San Francisco, CA 94080
(650) 837-7000 phone
(650) 837-8300 fax
Directions to Exelixis, Inc.
101 Northbound from San Francisco Airport:
- Take 101 North toward San Francisco.
- Take the Grand Avenue exit, exit 425A, toward So San Francisco.
- Turn right onto East Grand Ave.
- 210 East Grand Ave is on your right-hand side.
101 Southbound from San Francisco:
- Take 101 South.
- Take the Grand Avenue exit. Turn left at the first light.
- Immediately turn left at the first light onto Grand Avenue (which will become East Grand Avenue)
- 210 East Grand Ave is on your right-hand side.

////////////mTOR inhibitor, Exelixis, Inc., PI3K, phosphatidylinositol-3-kinase, XL 388, XL388, IND Filed
IPI 504, Retaspamycin, Retaspimycin

IPI 504, Retaspamycin, Retaspimycin
CAS 857402-63-2
Cas 857402-23-4 ( Retaspimycin); 857402-63-2 ( Retaspimycin HCl).
MF C31H45N3O8 BASE
MW: 587.32067 BASE
Infinity Pharmaceuticals Inc, INNOVATOR
[(3R,5S,6R,7S,8E,10S,11S,12Z,14E)-6,20,22-trihydroxy-5,11-dimethoxy-3,7,9,15-tetramethyl-16-oxo-21-(prop-2-enylamino)-17-azabicyclo[16.3.1]docosa-1(22),8,12,14,18,20-hexaen-10-yl] carbamate;hydrochloride
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride
| Retaspimycin hydrochloride; 8,21-didehydro-17-demethoxy-18,21-dideoxo-18,21-dihydroxy-17-(2-propenylamino)-geldanamycin monohydrochloride | |
| Application: | A novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90) |
| Molecular Weight: | 624.17 ……….HCl salt |
| Molecular Formula: | C31H46ClN3O8……….HCl salt |
Introduction
IPI-504 is a novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90).
Orphan drug designation was assigned to the compound by the FDA for the treatment of gastrointestinal stromal cancer (GIST).

Retaspimycin Hydrochloride is the hydrochloride salt of a small-molecule inhibitor of heat shock protein 90 (HSP90) with antiproliferative and antineoplastic activities. Retaspimycinbinds to and inhibits the cytosolic chaperone functions of HSP90, which maintains the stability and functional shape of many oncogenic signaling proteins and may be overexpressed or overactive in tumor cells. Retaspimycin-mediated inhibition of HSP90 promotes the proteasomal degradation of oncogenic signaling proteins in susceptible tumor cell populations, which may result in the induction of apoptosis.
Phase I study of Retaspimycin: A phase 1 study of IPI-504 (retaspimycin hydrochloride) administered intravenously twice weekly for 2 weeks at 22.5, 45, 90, 150, 225, 300 or 400 mg/m(2) followed by 10 days off-treatment was conducted to determine the safety and maximum tolerated dose (MTD) of IPI-504 in patients with relapsed or relapsed/refractory multiple myeloma (MM). Anti-tumor activity and pharmacokinetics were also evaluated. Eighteen patients (mean age 60.5 years; median 9 prior therapies) were enrolled. No dose-limiting toxicities (DLTs) were reported for IPI-504 doses up to 400 mg/m(2).
The most common treatment-related adverse event was grade 1 infusion site pain (four patients). All other treatment-related events were assessed as grade 1 or 2 in severity. The area under the curve (AUC) increased with increasing dose, and the mean half-life was approximately 2-4 h for IPI-504 and its metabolites. Four patients had stable disease, demonstrating modest single-agent activity in relapsed or relapsed/refractory MM. (source: Leuk Lymphoma. 2011 Dec;52(12):2308-15.)

Figure Hsp90 protein partners and clients destabilized by Hsp90 inhibition (Jackson et al., 2004).
In a different approach, Infinity Pharmaceuticals has developed IPI504 (retaspimycin or 17-AAG hydroquinone, Figure 4) (Adams et al., 2005; Sydor et al., 2006), a new GA analogue, in which the quinone moiety was replaced by a dihydroquinone one. Indeed, the preclinical data suggested that the hepatotoxicity of 17-AAG was attributable to the ansamycin benzoquinone moiety, prone to nucleophilic attack.
Furthermore, it was recently reported that the hydroquinone form binds Hsp90 with more efficiency than the corresponding quinone form (Maroney et al., 2006). In biological conditions, the hydroquinone form can interconvert with GA, depending on redox equilibrium existing in cell. It has been recently proposed, that NQ01 (NAD(P)H: quinone oxidoreductase) can produce the active hydroquinone from the quinone form of IPI504 (Chiosis, 2006).
However, Infinity Pharmaceuticals showed that if the overexpression of NQ01 increased the level of hydroquinone and cell sensitivity to IPI504, it has no significant effect on its growth inhibitory activity. These results suggest that NQ01 is not a determinant of IPI504 activity in vivo (Douglas et al., 2009).

Figure 4: GA, 17-AAG, 17-DMAG and IPI504.
PATENT
http://www.google.com/patents/EP2321645A1?cl=en
Geldanamycin (IUPAC name ([18S-(4E,5Z,8R*.9R*.10E,12R*.13S*,14R*,l6S*)]- 9- [(aminocarbonyl)oxy]- 13- hydroxy- 8,14,19- trimetoxy- 4,10,12,16- tetramethyl- 2- azabicyclo[16.3.1.]docosa- 4,6.10,18,21- pentan- 3.20,22trion) is a benzoquinone ansamycin antibiotic which may be produced by the bacterium Streptorayces hygroscopicus. Geldanamycin binds specifically to HSP90 (Heat Shock Protein 90) and alters its function.
While Hsp90 generally stabilizes folding of proteins and, in particular in tumor cells, folding of overexpressed/mutant proteins such as v-Src. Bcr-Abl and p53. the Hsp90 inhibitor Geldanamycin induces degradation of such proteins.
The respectiv e formula of geldanamycin is given herein below:

E\en though geldanamycin is a potent antitumor agent, the use of geldanamycin also shows some negathe side-effects (e.g. hepatotoxicity) which led to the dev elopment of geldanamycin analogues/derivatives, in particular analogues/deriv atives containing a derivatisation at the 17 position. Without being bound by theory , modification at the 17 position of geldanamycin may lead to decreases hepatotoxicity.
Accordingly geldanamycin analogues/derivatives which are modified at the 17 position, such as 17-AAG (17-N-Allylamino-17-demethoxygeldanamycin), are preferred in context of the present invention. Also preferred herein are geldanamycin derivatives to be used in accordance with the present invention which are water-soluble or which can be dissoh ed in water completely (at least 90 %. more preferably 95 %. 96 %. 97 %, 98 % and most preferably 99 %). 17-AAG ([QS.5S,6RJS$EΛ0R,l \SΛ2E,14E)-2\- (allylamino)-6-hydroxy-5.11-diraethoxy-
3.7.9,15-tetramethyl-16.20.22-trioxo-17-azabicyclo[16.3.1]docosa-8,12.14,18,21-pentaen-10- yl] carbamate) is. as mentioned above a preferred derivative of geldanamycin. 17- AAG is commercially available under the trade name “Tanespimycin“ (also known as KOS-953) for example from Kosan Biosciences Incorporated (Acquired by Bristol-Myers Squibb Company). Tanespimycin is presently studied in phase II clinical trials for multiple myeloma and breast cancer and is usually administered intravenously.
The respective formula of 17- AAG is given herein below:

Preferred geldanamycin-derh ative (HSP90 inhibitor) to be used in context of the present invention are IPI-504 (also known as retaspiimcin or Mcdi-561 : lnfinin Pharmaceuticals (Medlmmunc/ Astra Zeneca)). Clinical trials on the use of IPI-504 (which is usually administered intravenously) in the treatment of non-small cell lung cancer (NSCLC) and breast cancer are performed. Also alvespimycin hy drochloride (Kosan Biosciences Incorporated Acquired By : Bristol-Myers Squibb Company) is a highly potent, water-soluble and orally acti\e derivative of geldanamycin preferably used in context of the present invention.

IPI-504
PATENT
WO 2005063714
http://www.google.co.ug/patents/WO2005063714A1?cl=en
Example 24
Preparation of Air-stable Hydroquinone Derivatives of the Geldanamycin Family of Molecules
,

17-Allylamino-17-Demethoxygeldanamycin (10.0 g, 17.1 mmol) in ethyl acetate
(200 mL) was stirred vigorously with a freshly prepared solution of 10% aqueous sodium hydrosulfite (200 mL) for 2 h at ambient temperature. The color changed from dark purple to bright yellow, indicating a complete reaction. The layers were separated and the organic phase was dried with magnesium sulfate (15 g). The drying agent was rinsed with ethyl acetate (50 mL). The combined filtrate was acidified with 1.5 M hydrogen chloride in ethyl acetate (12 mL) to pH 2 over 20 min. The resulting slurry was stirred for 1.5 h at ambient temperature. The solids were isolated by filtration, rinsed with ethyl acetate (50 mL) and dried at 40 °C, 1 mm Hg, for 16 h to afford 9.9 g (91%) of off-white solid. Crude hydroquinone hydrochloride (2.5 g) was added to a stirred solution of 5% 0.01 N aq. hydrochloric acid in methanol (5 mL). The resulting solution was clarified by filtration then diluted with acetone (70 mL). Solids appeared after 2-3 min. The resulting slurry was stirred for 3 h at ambient temperature, then for 1 h at 0-5 °C. The solids were isolated by filtration, rinsed with acetone (15 mL) and dried
PAPER

17-Allylamino-17-demethoxygeldanamycin (17-AAG)1 is a semisynthetic inhibitor of the 90 kDa heat shock protein (Hsp90) currently in clinical trials for the treatment of cancer. However, 17-AAG faces challenging formulation issues due to its poor solubility. Here we report the synthesis and evaluation of a highly soluble hydroquinone hydrochloride derivative of 17-AAG, 1a (IPI-504), and several of the physiological metabolites. These compounds show comparable binding affinity to human Hsp90 and its endoplasmic reticulum (ER) homologue, the 94 kDa glucose regulated protein (Grp94). Furthermore, the compounds inhibit the growth of the human cancer cell lines SKBR3 and SKOV3, which overexpress Hsp90 client protein Her2, and cause down-regulation of Her2 as well as induction of Hsp70 consistent with Hsp90 inhibition. There is a clear correlation between the measured binding affinity of the compounds and their cellular activities. Upon the basis of its potent activity against Hsp90 and a significant improvement in solubility, 1a is currently under evaluation in Phase I clinical trials for cancer.
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride Ia
17-AAG hydroquinone hydrochloride (1a) as an off-white solid (11 g, 18 mmol, 80% yield). HPLC purity: 99.6%;
IR (neat): 3175, 2972, 1728, 1651, 1581, 1546, 1456, 1392, 1316, 1224, 1099, 1036 cm-1;
1H NMR (CDCl3:d6-DMSO, 6:1, 400 MHz):
δ 10.20 (1H, br), 9.62 (2H, br), 8.53 (1H, s), 8.47 (1H, s), 7.74 (1H, s), 6.72 (1H, d, J= 11.6 Hz), 6.28 (1H, dd, J = 11.6, 11.2 Hz), 5.73 (1H, dddd, J = 17.2, 10.0, 3.2, 2.4 Hz), 5.53 (1H, d, J = 10.4 Hz), 5.49 (1H, dd, J = 10.8, 10.0 Hz), 5.32 (2H, s), 5.04 (1H, d, J = 4.8 Hz), 5.02 (1H, d, J = 16.0 Hz), 4.81 (1H, s), 4.07 (1H, d, J = 9.6 Hz), 3.67 (2H, d, J = 6.4 Hz), 3.31 (1H, d,J = 8.8 Hz), 3.07 (3H, s), 3.07−3.04 (1H, m), 2.99 (3H, s), 2.64 (1H, m), 2.52−2.49 (1H, m), 1.76 (3H, s), 1.61−1.39 (3H, m), 0.78 (3H, d, J = 6.4 Hz), 0.64 (3H, d, J = 7.2 Hz);
13C NMR (CDCl3:d6-DMSO, 6:1, 100 MHz): δ 167.3, 155.8, 143.3, 136.3, 135.0, 134.2, 132.9, 132.1, 128.8, 127.6, 125.9, 125.3, 123.7, 123.0, 115.1, 104.5, 80.9, 80.7, 80.1, 72.5, 56.2, 56.2, 52.4, 34.6, 33.2, 31.1, 27.2, 21.6, 12.1, 12.1, 11.7;
HRMS calculated for C31H45N3O8 (M+ + H): 588.3285, Found 588.3273.
POSTER
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
MEDI 210 |
| IPI-504 is the hydroquinone hydrochloride salt of 17-allylamino-17-demethoxy-geldanamycin (17-AAG), an Hsp90 inhibitor that is currently in clinical trials for the treatment of cancer.
IPI-504 demonstrates high aqueous solubility (>200 mg/mL). Interestingly, in vitro and in vivo IPI-504 interconverts with 17-AAG and exists in a pH and enzyme-mediated redox equilibrium. This occurs due to oxidation of the hydroquinone (IPI-504) to the quinone (17-AAG) at physiological pH and the reduction of 17-AAG by quinone reductases such as NQO1 to IPI-504. Here we report the design and synthesis of the stabilized hydroquinone IPI-504 and its inhibitory effect against Hsp90 and Grp94. Although IPI-504 was originally designed to be a soluble prodrug of 17-AAG, the hydroquinone is more potent than the quinone in the biochemical Hsp90 binding assay. Various hydroquinone analogs have been prepared to investigate the structure activity relationship of hydroquinone binding to Hsp90. Hydroquinone and quinone forms of 17-AAG metabolites show comparable binding affinities for Hsp90 and in cancer cell lines, hydroquinone analogs elicit specific responses consistent with Hsp90 inhibition. The desirable pharmacological properties as well as in vitro and in vivo activity of our lead compound, IPI-504, has led to the initiation of Phase I clinical trials in multiple myeloma. |
| http://oasys2.confex.com/acs/231nm/techprogram/P945016.HTM |
|
Geldanamycin and Beyond: Progress Toward the Development of HSP90 Inhibitors
8:30 AM-12:05 PM, Wednesday, 29 March 2006 Georgia World Congress Center — Georgia Ballroom 1, OralDivision of Medicinal ChemistryThe 231st ACS National Meeting, Atlanta, GA, March 26-30, 2006 |
References
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
231st Am Chem Soc (ACS) Natl Meet (March 26-30, Atlanta) 2006, Abst MEDI 210
Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90
J Med Chem 2006, 49(15): 4606
http://www.biotechduediligence.com/retaspamycin-hcl-ipi-504.html
///////////////////Hsp90, IPI-504, infinity pharma, Retaspamycin, Retaspimycin
Regulation of Herbal (Traditional) Medicinal Products in the European Union

Regulation of Herbal (Traditional) Medicinal Products in the European Union
| Introduction | |
| The European Union (EU) regulatory framework for medicinal products is complex and is based on the need of a marketing authorization before placing medicines in the market. The main objective is to protect public health by assuring quality, efficacy and safety. The requirements and procedures to obtain a marketing authorization are laid down in regulations, directives and scientific guidelines which are contained in the “Rules Governing Medicinal Products in the European Union”. Several volumes are included which are supported by other publications with complementary information such as scientific or Good Manufacturing Practice (GMP) guidelines, between others [1]. | |
 |
|
| Medicinal plants have been used since Ancient times in all parts of the world. Nonetheless, regulation of herbal medicines in a legal environment was introduced in the 20th century. The EU regulatory framework includes specific requirements for herbal medicinal products (HMP) which are independent from their legal status: traditional herbal medicinal product (THMP) or products based on clinical evidence – well established use (WEU). | |
| Before a HMP is placed in the market, it must be approved by a MS or by the European Commission by one of the existing types of application: full marketing authorization application, well-established use marketing authorization application or Traditional use marketing registration (Table 1).
|
|
| The applicant has to submit adequate quality, non-clinical and clinical documentation of the product, irrespectively of the procedure used. Quality requirements of the pharmaceutical product are the same, regardless of the type of application, while efficacy documentation differs between them. The full marketing application is chosen for new medicinal products (new chemical entity) and it has to be completed with the results of pharmaceutical tests (quality documentation), nonclinical (toxicological and pharmacological) studies and clinical trials. Safety data have to be of sufficient size according the existing guidelines; efficacy is demonstrated by results from the clinical trials which have to be in conformity with the guidelines of the corresponding therapeutic area. This type of application is open for HMP, but only a few examples of herbal products have obtained a marketing authorization in the EU in this way. | |
| EU Pharmaceutical Legislation for Herbal Medicinal Products for Human Use | |
 |
|
| Quality requirements | |
| The principles to assure quality of medicinal products are defined mainly in two Directives of volume 1: Directive 2001/83/EC (which was emended by Directive 2004/24/EC) and Directive 2003/63/EC. | |
| The basic legislation lay down in Directive 2001/83/EC describes the general requirements and provides legal definitions of herbal substances, herbal preparations and herbal medicinal products (Table 2). These concepts are essential for setting quality standards for HMP, as they are by definition complex in nature and so quality requirements set for purified compounds are not suitable for herbal products. | |
| According to the Directive 2001/83/EC, monographs in the European Pharmacopoeia (Eur. Ph.) are legally binding and applicable to all substances which are included in it. For substances which do not have a Eur. Ph. monograph, each Member State (MS) may apply its own national pharmacopoeia. Constituents which are not given in any pharmacopoeia shall be described in the form of a monograph under the same headings included in any monograph in the Eur. Ph., i.e., the name of the substance supplemented by any trade or scientific synonyms; the definition of the substance, set down in a form similar to that used in the European Pharmacopoeia; methods of identification and purity tests. | |
| Moreover, all medicinal products have to be manufactured according to the principles and guidelines of GMP for medicinal products. GMP are applicable to both finished HMP and active substances and, according to Article 46 (f) of Directive 2001/83/EC as amended, marketing authorization holders are required to use as starting materials only active substances which have been manufactured in accordance with the guidelines on the GMP for starting materials as adopted by the Community and distributed in accordance with good distribution practices for active substances. | |
| |
|
| Additional requirements are found in the Directive 2003/63/EC, as Herbal medicinal products differ substantially from conventional medicinal products in so far as they are intrinsically associated with the very particular notion of herbal substances and herbal preparations. It is therefore appropriate to determine specific requirements in respect of these products with regards to the standardized marketing authorization requirements. Then, detailed information on the herbal medicinal product, herbal substances and herbal preparations has to be included, such as the name, address and responsibility of each herbal substance supplier or description of the plant production process, geographical source or drying and storage conditions. The application dossier of a HMP should include specifications and details of all the analytical methods used for testing herbal substances and herbal preparations, results of batch analyses and analytical validation, together with the justification for the specifications. | |
| Most of the quality requirements for HMP are laid down in soft laws (considered as EU measures such as scientific guidelines) which do not have legal force but provide practical harmonization between the MS and the European Medicines Agency (EMA). | |
| The guideline on quality of HMP/THMP covers the general quality aspects of HMP for human and veterinary use, including THMP for human use (EMA, 2014) and indicates which information has to be included in the application dossier. It provides definitions to be taken in account such as genuine (native) herbal preparations, markers, drug to extract ratio (DER) and specifications. Which is more important, it states that the herbal substance or herbal preparation is considered as the whole active substance. In consequence, the quality control of these products has to include appropriate fingerprint analysis to cover not only the content of markers or constituents with known therapeutic activity but also a wider range of chemical constituents. | |
| |
|
| Efficacy requirements | |
| Before a HMP is placed in the market, it must be approved by a MS or by the European Commission by one of the existing types of application: full authorization application, well-established use authorization application or Traditional use registration (Table 3). | |
| The applicant has to submit adequate quality, non-clinical and clinical documentation of the product, irrespectively of the procedure used. Quality requirements of the pharmaceutical product are the same, regardless of the type of application, while efficacy documentation differs between them. The full marketing application is chosen for new medicinal products (new chemical entity) and it has to be completed with the results of pharmaceutical tests (quality documentation), nonclinical (toxicological and pharmacological) studies and clinical trials. Safety data have to be of sufficient size according the existing guidelines; efficacy is demonstrated by results from the clinical trials which have to be in conformity with the guidelines of the corresponding therapeutic area. This type of application is open for HMP, but only a few examples of herbal products have obtained a marketing authorization in the EU in this way. | |
| The well-established medicinal use (WEU) in the EU can be applied to medicinal products for which there exists a wide clinical experience within the EU (not only HMP). The assessment may be based in published controlled clinical trials, non-clinical studies and epidemiological studies. In this type of application, there are no limitations to the therapeutic indication, as this will be derived from the available documentation. | |
| |
|
| (Traditional) Herbal Medicinal Products | |
| Under the Traditional use registration for herbal medicinal product (article 16e), there exist some herbal products that not fulfill the efficacy requirements for a marketing authorization but are endorsed with a long tradition of use. In this case, no clinical trials on these products have been conducted and the efficacy is based on the long-standing use and experience. This simplified registration procedure is limited to products which are intended for use without medical supervision, with a specified strength and posology, to be used by oral, external or inhalation ways, and which can demonstrate a period of use equal or superior to 30 years, including at least 15 years within the EU. In this case, therapeutic indications are limited to those which can be considered safe for use without the supervision of a physician such as minor disorders or symptoms that are benign or self- limiting. In case the applicant should consider another kind of indication, the product must be documented with results of clinical and non-clinical studies, so a full application would be necessary. | |
| Simplified registration of THMP is described in Chapter 2a of Directive 2004/24/EC with three main objectives: a) to protect public health by allowing access to safe and high-quality HMP; b) to allow European citizens the access to medicines of their choice, even those HMP with a long tradition of use and which efficacy hasn’t been proved by clinical trials performed according the modern standards; c) to facilitate movement of medicinal products on the European market. | |
| Directive 2004/24/EC has two different dimensions: the evaluation by National Competent Authorities (NCA) of applications submitted by companies at any MS in the EU and at the EMA, and the establishment of advisory scientific opinions on the medicinal use of herbal substances or preparations. The directive on THMP also established a new scientific committee, the Herbal Medicinal Products Committee (HMPC) at the EMA in London, in 2004, to replace the previous Working Party on Herbal Medicinal Products (CPMP) with the following aims: to elaborate Community monographs and List entries for herbal substances/preparations; to publish scientific guidelines useful for the application of European legal framework; to publish its scientific opinion on questions related to herbal medicinal products and coordinate its work with the European Quality group. The HMPC is made up by 33 members, one member (and one alternate) nominated by each MS of the EU and by Iceland and Norway (the EFAEFTA states). Among them, also five experts are included, representing specific fields of expertise as clinical and non-clinical pharmacology, toxicology or pediatrician medicine. | |
| The guidelines and the monographs developed and approved by the HMPC are accepted by both companies and NCAs and are used for TUR and WEU marketing authorizations. This committee plays a key role in the harmonization of the regulation of HMP whereby Community herbal monographs have a fundamental role. | |
| Usage of Community herbal monographs in the EU regulation of traditional HMP | |
| These documents are established for HMP with regards to bibliographic applications (art. 10 a Directive 2001/83/EC) as well as THPMs. Community monographs reflect the scientific opinion of the HMPC on safety and efficacy data concerning a herbal substance. Any single plant or herbal preparation is assessed individually, according to the available information and includes qualitative and quantitative composition, pharmaceutical form(s), therapeutic indication(s), posology and method of administration, contraindications, special warnings and precautions of use, interactions, use in special population (pregnancy, lactation), effects on ability to drive and use machines, undesirable effects, overdose, pharmacological,pharmacodynamics, pharmacokinetic properties and preclinical safety data. | |
| Community list entry | |
| In the EU, a community list of herbal substances, preparations and combinations thereof for use in THMPs has been established. This list is based in the proposals form HMPC and is gradually developed. Substances or preparations which are included in the list have the main advantage that applicants do not need to provide evidence on the safe or traditional use for its registration at the NCA in the intended use and indication. | |
| A Public statement for one herbal substance/preparation is published because of safety reasons or lack of data to comply with the conditions in the Directive 2004/24/EC (the assessment work didn’t allow a monograph to be published) [2]. | |
| Community monographs are published by the EMA while list entries are approved and published by the European Commission because they are endorsed with a wider legal status: list entries are legally binding and NCAs should not request additional data on safety and traditional use. | |
| The establishment of monographs and list entries is based on the assessment of the published scientific data, together with the existing products in the market. Most of the assessment work is developed by the Monograph and List Working Party (MLWP) at the HMPC, which was established in 2006. In this working group, a member is designed as rapporteur and is responsible of drafting a monograph and/or list entry which will be later on considered and approved by the HPC and then, by the EMA. The documents are published on the EMA website: Community monographs have to be taken in account by the MS when assessing the application of any company. Monographs are note legally binding and MS are not obliged to follow the monographs. | |
| More than 100 species are included in the priority list with the following data: a) scientific data being assessed (R- Rapporteur assigned); b) evaluation report in progress and discussion in the MLWP (D- Draft under discussion); c) scientific opinion under public consultation (PDraft Published); d) comments after public consultation period being evaluated (PF- Assessment close to finalization – pre-final); e) final opinion adopted (F- Final opinion adopted). | |
| MLWP is also responsible of developing guidelines related to legal requirements for TU and WEU, as well as evaluating hazards and problems related to HMP. For the latter, coordination is established with the Safety Working Party (SWP) from the Committee for Medicinal Products for Human Use (CHMP). | |
| Community herbal monographs to support HMPC authorization | |
| A community monograph reflects the scientific opinion from the HMPC in relation to safety and efficacy of one herbal substance/ preparation for medicinal use. AS stated before, a community monograph may be used by a company for a TU or WEU application. That’s the reason why monographs are divided in to two columns: Well Established Use and Traditional Use (simplified application) (Figure 1). WEU is based in the existence of safety data of sufficient size and efficacy data derived from good-quality clinical trials. Traditional use is accepted for those applications which fulfill the criteria shown in the Directive 2004/24/EC. | |
| Each herbal substance/preparation is assessed individually, as the available information may be different for each one. As a result, some substances/preparations may be included in the WEU side, while others will be included in the TU side. If no enough data are available for the substance/preparation, it won’t be included in the monograph. | |
| The approved draft art he HMPC is published for public consultation for 3 month at the EMA website. Comments received are discussed and taken in account when necessary to achieve the final version of the monograph which will be finally published at the MA website. | |
| By the end of 2014, 126 monographs have been adopted and published by the EMA: 104 of them for TU only; 9 of the monographs refer only to WEU (Aloe vera, Cimicifuga racemosa, Rhamnus frangula, Plantago ovata – seed and tegumentum-, Plantago afra, Rheum palmatum, Cassia senna – leaves and fruits-.among them 13 monograph include both TU and WEU. | |
| The main application of a community monograph is to serve as a reference material for the marketing application, both for TU or WEU. Simplified registration is carried on at a national level, so the company gives the dossier to the NCA. With the aim of improving harmonization, the other MSs should recognize the first authorization granted in the first MS, considering that this is based in the European list. | |
| Directive 2004/24/CE established an adaptation period for those herbal products which were on the European market at the moment the Directive was approved. This seven-year period finalized last April 30th, 2011 and implies that nowadays those herbal preparations that not fulfill the actual legislation will not be marketed any more. | |
| In the public report form the EMA last June 2014, the status of updating the medicines registration in the EU was shown. The number Traditional use registrations (TUR) and Well-established use marketing authorizations (WEU) grouped for mono component and combination products has increased in the last years (Figure 2). | |
| The European market for HMP is increasing during the last years and even exceeds prescription medicines. The indications approved cover a wide range of therapeutic areas, most of them characteristic of self-medication diseases: the main therapeutic areas are respiratory tract disorders (cough and cold), mental stress and mood disorders, urinary tract and gynecology disorders, sleep disorders and temporary insomnia (Figure 3). Most of the approved THMP until now were updates of existing authorizations and were based on Community monographs. The Summary of Product Characteristics (SoPC) reflects the items in the corresponding monograph [3]. | |
| A good correlation between the HMPC work and the evaluation of the dossiers from the companies was detected. The relevance of these documents (as shown by the accepted dossiers) is reflected in the HMPC working plan; as an example, last December 2012, 54 among the 56 species with more than 3 marketing authorizations were listed in the priority list. | |
| Conclusión | |
| European legal framework for medicinal products does also include herbal medicinal products to assure their quality, efficacy and safety. The specific characteristics of these products led to the development of a simplified procedure to assure pharmaceutical quality, while keeping safety and efficacy criteria according to marketing authorization granted. | |
| Although the starting point was quite different for the MS, nowadays there exist Community monographs for most of the herbal substances/ preparations that are used in the European market and which form the basis for a harmonization scenario. Moreover, HMPC acts as an International Regulatory Body for herbal medicinal products in order to achieve global standards for this type of medicines, according to other International organization such as the International Conference on Harmonization (ICH). The main tasks the HMPC has to face are those related to herbal medicinal products which have been previously marketed abroad the EU and the increasing existence of combination products within the MS. | |
| References | |
|
|
|
|
||||||||||||
| Table 1: Types of applications for marketing authorization for a HMP in the EU according the Directive 2001/83/EC. |
|
||||||
| Table 2: Definitions applicable to herbal medicinal products (Directive 2001/83/EC). |
|
||||||
| Table 3: Definitions applicable to herbal medicinal products (Directive 2001/83/EC). |
 |
| Figure 1: European Community Monograph for Valeriana officinalis L., radix, for WEU and TU |
Ruiz-Poveda OMP*
Department of Pharmacology, Faculty of Pharmacy, Universidad Complutense de Madrid, 28040 Madrid, Spain
Ruiz-Poveda OMP
Department of Pharmacology, Faculty of Pharmacy
Universidad Complutense de Madrid, Madrid
Ciudad Universitaria s/n. 28040 Madrid, Spain
Tel: 913 941 767
Fax: 913 941 726
E-mail: olgapalomino@farm.ucm.es
Citation: Ruiz-Poveda OMP (2015) Regulation of Herbal (Traditional) Medicinal Products in the European Union. Pharmaceut Reg Affairs 4:142. doi: 10.4172/2167-7689.1000142
/////////Herbal medicines, Good manufacturing practices, Traditional uses
EMA: European Medicines Agency; DER: Drug to Extract Ratio; HMP: Herbal Medicinal Products; THMP: Traditional Herbal Medicinal Products
Dr. Ashok Kumar, President – Research and Development (Chemical) at IPCA LABORATORIES LTD

Dr. Ashok Kumar
The 25-year process patent regime allowed a large number of generic companies in India to reap rich dividends, but there were few who believed in the need to go beyond the horizon of process development to tap into unexplored terrains.
In the year 2000, when Dr Ashok Kumar joined the board of Mumbai-based IPCA Labs, he was determined to implement a different strategy to accelerate IPCA’s R&D initiatives. Having seen IPCA grow from a 350-crore company to one clocking an annual turnover of over 2,500 crore, Dr Ashok Kumar, president- Center for Research and Development, IPCA Labs, is now leaving no stone unturned to exploit the biotech and drug discovery space.

Dr Kumar completed his M Sc in Chemistry from Kumaun University, now in Uttarakhand. He then decided to pursue his PhD in organic chemistry and joined Banaras Hindu University (BHU), but opted out three months later to do PhD from the Central Drug Research Institute (CDRI), Lucknow, under the guidance of the then director of the CDRI, Dr Nityanand.
Dr Kumar did his post doctoral studies from the University of Sussex, UK. “During the 1980s, jobs in scientific research were not available in India. It was always good to go for higher studies abroad,” he says about the reason for going abroad. “Dr Nityanand taught me to be explorative and think of new ways to approach a subject. I still follow that process,” he says. In 1984, Dr Kumar decided to return to India and took up a job at the Imperial Chemical Laboratories (ICI), Mumbai. In 1994, he joined Lupin Labs where he was once again involved in process development of small molecules.

In 2000, he joined IPCA Labs where he immediately focused on bringing about two changes – introducing a library and bringing in systems like a nuclear magnetic resonance (NMR), which at that time was the costliest instrument. “I understood the importance of high-end technologies since my PhD years at the CDRI (which housed a couple of NMRs) and then in the UK. You do not enjoy organic chemistry without an NMR. My main objective at IPCA was cost reduction along with process development.”
In the last few years, IPCA’s R&D team has brought out over 100 products. Under Dr Kumar, IPCA has an R&D center in Mumbai and another parallel R&D center in Ratlam, Madhya Pradesh. “In Mumbai, we have around 60 people. The Mumbai team takes care of basic chemistry and small-scale development. Scaling up is done in Ratlam,” he explains. IPCA is also coming up with a facility at Vadodara, Gujarat, that will look into large-scale manufacture of both organic and biotech drugs. The facility will have a strategic importance for the company. “We are growing at a rate of 20 per cent year-on-year and, next year, we intend to add 500 crore to our revenue. For that, we need more products to come to the market and more volume,” he adds.
Apart from organic chemistry, Dr Kumar is currently aligning his attention to two promising but high risk segments – fermentation-based products and biosimilars. “We are working on five-to-six molecules, mainly active metabolites that are intermediates or biotech drugs,” he adds. IPCA has also collaborated with two companies in India for the development of biosimilars. Currently, there are three biosimilar products in the pipeline.
The R&D team at IPCA ventured into drug discovery three years ago. It has two products in the pipeline; one anti-malarial and the other anti-thrombotic. “We will be filing the investigational new drug application for one molecule this year and for the other next year. The success rate here is 99 per cent,” adds Dr Kumar. IPCA has also joined hands with the CDRI and licensed two molecules in the anti-malarial space. One of these molecules is currently in phase-I stage.
Innogen summit India 2016, 18-19 Aug, Mumbai, India, HOTEL HOLIDAY INN, Mumbai International Airport,Organised by Inventicon Business Intelligence Pvt. Ltd………topic is Supergenerics, Innovation in Generics, commercialization, regulatory, other insights,


Dr. Ashok Kumar, President – Centre for Research & Development, Ipca Laboratories Ltd, at Innogen summit India 2016, 18-19 Aug, Mumbai, India,, HOTEL HOLIDAY INN, Mumbai International Airport,Organised by Inventicon Business Intelligence Pvt. Ltd — with DR ASHOK KUMAR OF IPCA at Holiday Inn-Mumbai Intl Airport.

PANEL DISCUSSION, Dr. Ashok Kumar, President – Centre for Research & Development, Ipca Laboratories Ltd , Dr. Nilima A. Kshirsagar, National Chair Clinical Pharmacology, ICMR Government of India, Yugal Sikri, Chairman – Pharmaceutical Management, School of Business Management, SVKM’s Narsee Monjee Institute of Management Studies — with Yugal Sikri,, Nilima A. Kshirsagarand ASHOK KUMAR OF IPCA at Holiday Inn-Mumbai Intl Airport.
JOURNEY




Experience
PRESIDENT – R&D
IPCA LABORATORIES LTD
2005 – Present (10 years)
Patent 1
chlorthalidone is 3-hydroxy-3-(3′-sulfamyI-4′- chlorophenyl)phtalimidine and is represented by the structural formula shown below.


Chlorthalidone Formula 1
(Scheme 2). The starting material, 2-(4′-chlorobenzoyl) benzoic acid, of Formula (2) and its preparation was reported earlier for example in patents US 4500636, US 30555904, US4379092, US 3764664.


Formula 9 CIS03H


Chlorthalidone Formula 10 Formula 1

PATENT 2
https://www.google.co.in/patents/US7847094Quetiapine and its process for preparation is first disclosed in the patent specification EP0240228 and various other processes for the preparation are disclosed in EP0282236, WO0155125, WO9906381, WO2004076431.

PATENT 3
http://www.google.com/patents/US20080009635
. The chemical name of Ondansetron is 1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl)-1H-imidazole-1-yl)methyl]-4H-carbazol-4-one and is represented by the structural formula given below:

PATENT 4
-
scheme 2.
-






Chemical Research & Development Centre
123-AB, Kandivli Industrial Estate, Kandivli (West)
Mumbai 400 067, Maharashtra




