



| Patent | Submitted | Granted |
|---|---|---|
| COMPOUNDS AND METHODS FOR TREATING INFLAMMATORY AND FIBROTIC DISORDERS [US2009318455] | 2009-12-24 | |
| COMPOSITION AND METHOD FOR TREATING PROTEINURIA [US2010099719] | 2010-04-22 | |
| COMPOSITION AND METHOD FOR TREATING FIBROTIC DISEASES [US2009258911] | 2009-10-15 | |
| Composition and Method for Treating Fibrotic Diseases [US2008319027] | 2008-12-25 | |
| METHODS FOR TREATING ACUTE MYOCARDIAL INFARCTIONS AND ASSOCIATED DISORDERS [US2010190731] | 2010-07-29 | |
| Methods for Treating Acute Myocardial Infarctions and Associated Disorders [US2011218515] | 2011-09-08 | |
| METHODS OF TREATING HIV PATIENTS WITH ANTI-FIBROTICS [US2012014917] | 2012-01-19 | |
| Composition and Method for Treating Fibrotic Diseases Composition and Method for Treating Fibrotic Diseases [US2009005424] | 2007-08-30 | |
| Crystalline 1-(3-fluorophenyl)-5-methyl-2-(1H)pyridone, the preparation methods, compositions and applications thereof [US8232408] | 2009-03-10 | 2012-07-31 |
Home » Uncategorized (Page 78)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New patent, WO 2016001885, Dr Reddy’s Laboratories Ltd, Eliglustat hemitartarate
![]()
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills, Telangana, India Hyderabad 500034 (IN)
VELAGA, Dharma Jagannadha Rao; (IN).
PEDDY, Vishweshwar; (IN).
VYALA, Sunitha; (IN)
(WO2016001885) AMORPHOUS FORM OF ELIGLUSTAT HEMITARTARATE
Chemically Eliglustat is named N-[(1 R,2R)-2-(2,3-dihydro-1 ,4-benzodioxin-6-yl)-2-hydroxy-1 -(1 -pyrrolidinylmethyl)ethyl]-Octanamide(2R!3R)-2,3-dihydroxybutanedioate and the hemitartarate salt of eliglustat has the structural formula as shown in Formula I.

Formula I
Eliglustat hemitartrate (Genz-1 12638), currently under development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy. Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase, and is currently under development by Genzyme.
U.S. patent No. 7,196,205 discloses a process for the preparation of Eliglustat or a pharmaceutically acceptable salt thereof.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 discloses process for preparation of Eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of Eliglustat, (ii) a hemitartrate salt of Eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
It has been disclosed earlier that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailablity patterns compared to crystalline forms [Konne T., Chem pharm Bull., 38, 2003(1990)]. For some therapeutic indications one bioavailabihty pattern may be favoured over another. An amorphous form of Cefuroxime axetil is a good example for exhibiting higher bioavailability than the crystalline form.
Solid amorphous dispersions of drugs are known generally to improve the stability and solubility of drug products. However, such dispersions are generally unstable over time. Amorphous dispersions of drugs tend to convert to crystalline forms over time, which can lead to improper dosing due to differences of the solubility of crystalline drug material compared to amorphous drug material. The present invention, however, provides stable amorphous dispersions of eliglustat hemitartrate. Moreover, the present invention provides solid dispersions of eliglustat hemitartrate which may be reproduced easily and is amenable for processing into a dosage form.
There remains a need to provide solid state forms of eliglustat hemitartarate which are advantageous in a cost effective and environment friendly manner.

EXAMPLES
Example 1 : Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 14 mL of dichloromethane at 26°C and stirred for 15 min. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 45°C. After distillation the solid was dried under vacuum at 45°C.
Example 2: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 70 mL of ethanol and stirred for 15 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid was dried under vacuum at 48°C.
Example 3: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 20 mL of methanol and stirred for 15 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid was dried under vacuum at 48°C.
Example 4: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and PVP-K30.
500mg of eliglustat hemitartarate and 500mg of PVP-K30 was dissolved in 20 mL of methanol and stirred for 10 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid is dried under vacuum at 48°C.
Example 5: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and hydroxy propyl cellulose.
500mg of eliglustat hemitartarate and 500 mg of hydroxy propyl cellulose was dissolved in 30 ml of methanol and stirred for 10 min at 25° – 30°C. The solution is distilled under reduced pressure at 49°C. After distillation the solid is dried under vacuum at 49°C.
Example 6: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and hydroxy propyl methyl cellulose.
500mg of eliglustat hemitartarate and 500 mg of hydroxy propyl methyl cellulose was dissolved in 30 mL of methanol and stirred for 10 min at 25° – 30°C. The solution is distilled under reduced pressure at 48°C. After distillation the solid is dried under vacuum at 48°C.
Example 7 Preparation of amorphous form of eliglustat hemitartarate.
3g of eliglustat hemitartarate was dissolved in 75 mL of methanol and stirred at 25°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected for spray drying at inlet temperature of 70°C and outlet temperature of 42°C to afford the title compound.
Example 8: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 30 mL of isopropanol and stirred at 56°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected to complete distillation under reduced pressure and drying at about 56°C to afford the title compound.
Example 9: Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 40 mL of ethyl acetate and stirred at about 63°C. Then methanol (5 mL) is added at the same temperature to obtain clear solution which was filtered to remove the undissolved particles. Then additional quantity of methanol (5mL) is added to the filtrate and the filtrate was again filtered to remove particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 10: Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 40 mL of acetone and stirred at about 55°C followed by addition of methanol (15 mL). The mixture is stirred at 55°C for clear solution and filtered to remove the undissolved particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 11 : Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 25 mL of isopropyl alcohol and 25 mL of ethanol. The mixture was stirred at about 58°C for dissolution and filtered to remove the undissolved particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 12 Preparation of amorphous form of eliglustat hemitartarate.
5g of eliglustat hemitartarate was provided in 300 mL of isopropyl alcohol and stirred at about 59°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected for spray drying at inlet temperature of 65°C and outlet temperature of 37°C to afford the title compound according to Fig. 6
Example 13: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Copovidone
500mg of eliglustat hemitartarate and 500mg of Copovidone were dissolved in 30 mL of methanol and stirred for clear solution, then filtered to make it particle free. The solvent from the filtrate was evaporated under reduced pressure at 45°C and obtained solid was subjected to drying at 45°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction.
Example 14: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Copovidone
2g of eliglustat hemitartarate and 2g of Copovidone were dissolved in 100 mL of methanol and stirred for clear solution, then filtered to make it particle free. The solvent from the filtrate was subjected to spray drying at inlet temperature of 70 at 45°C and outlet temperature of 42°C to afford the title compound. The resulting dispersion was found to be amorphous by X-ray powder diffraction.
Example 15: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate
2g of eliglustat hemitartarate was charged in 40 mL of methanol followed by addition of 2g of PVP K-30. The mixture was stirred for clear solution and filtered to make it particle free, the bed was washed with 20 mL of methanol. Then 2g of Syloid is added to the filtrate and filtrate is subjected to distillation under reduced pressure at about 57°C and obtained solid was subjected to drying at about 57°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 7a. The said dispersion is kept at 25°C under 40% relative humidity for 24 hours and PXRD was recorded and found to be amorphous according to Fig 7b.
Example 16: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate
2g of eliglustat hemitartarate was charged in 40 mL of methanol followed by addition of 2g of Copovidone. The mixture was stirred for clear solution and filtered to make it particle free, the bed was washed with 20 mL of methanol. Then 2g of Syloid is added to the filtrate and filtrate is subjected to distillation under reduced pressure at about 57°C and obtained solid was subjected to drying at about 57°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 8a. The said dispersion is kept at 25°C under 40% relative humidity for 24 hours and PXRD was recorded and found to be amorphous according to Fig. 8b and D90 of the resultant solid is about 437 microns.
Example 17: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Syloid
1 g of eliglustat hemitartarate was dissolved in 25 ml_ of methanol and filtered to make it particle free. Then 1 g of Syloid 244 FPNF was added to the filtrate and solvent from the filtrate was evaporated under reduced pressure at 56°C and obtained solid was subjected to drying at 56°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 9 and D90 of the resultant solid is about 4 microns.
Example 18: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Syloid
1 g of eliglustat hemitartarate was dissolved in 25 ml_ of methanol and filtered to make it particle free. Then 500mg of Syloid 244 FPNF was added to the filtrate and solvent from the filtrate was evaporated under reduced pressure at 56°C and obtained solid was subjected to drying at 56°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction.

PATENT
(WO2015059679) IMPROVED PROCESS FOR THE PREPARATION OF ELIGLUSTAT
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills Hyderabad 500034 (IN)
JAVED, Iqbal; (IN).
DAHANUKAR, Vilas Hareshwar; (IN).
ORUGANTI, Srinivas; (IN).
KANDAGATLA, Bhaskar; (IN)
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.

Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy. Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence: (i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate, (ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone, (iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine, (iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
It is also an objective of the present application to provide an improved process for the preparation of eliglustat and a pharmaceutically acceptable salt thereof which is high yielding, simple, cost effective, environment friendly and commercially viable by avoiding repeated cumbersome and lengthy purification steps. It is a further objective of the present application to provide crystalline forms of eliglustat free base and its salts.
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %

G.V. Prasad, chairman, Dr Reddy’s Laboratories
//////////////New patent, WO 2016001885, Dr Reddy’s Laboratories Ltd, Eliglustat hemitartarate, WO 2015059679
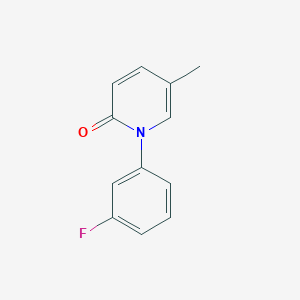
AZD 2716
AZD2716
CAS 1845753-81-2
MF C24 H23 N O3, MW 373.44
[1,1′-Biphenyl]-3-propanoic acid, 2′-(aminocarbonyl)-α-methyl-5′-(phenylmethyl)-, (αR)-
- Antiplaque candidate drug
AstraZeneca INNOVATOR
(R)-7(AZD2716) a novel, potent secreted phospholipase A2 (sPLA2) inhibitor with excellent preclinical pharmacokinetic properties across species, clear in vivo efficacy, and minimized safety risk. Based on accumulated profiling data, (R)-7 was selected as a clinical candidate for the treatment of coronary artery disease.
Chiral HPLC using a Chiralcel OJ 5 μm 20×250 mm
column with heptane/EtOH/formic acid ((10:90:0.1; 15 ml/min, 40 °C, 260 nm) as mobile
phase to yield (S)-7 and (R)-7
(R)-7:tR=5.8 min [α]D20 15.4 (c 0.5, ACN), 99.7 %ee. desired
(S)-7: tR=9.2 min. 99.0 % ee. undesired
LINK
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.6b00188
![]()
SYNTHESIS
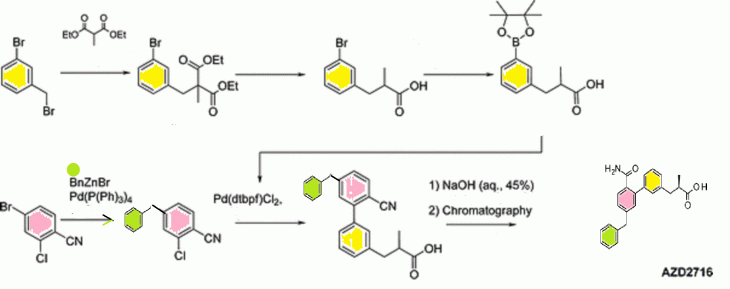
1H NMR (400 MHz, DMSO-d6): δ 1.04 (d, J = 6.6 Hz, 3H), 2.55–2.68 (m, 2H), 2.95 (dd, J = 6.1, 12.8 Hz, 1H), 4.00 (s, 2H), 7.13–7.37 (m, 13H), 7.49–7.54 (m, 1H), 12.2 (s, br, 1H).
13C NMR (151 MHz, DMSO): δ 16.7, 39.1, 40.7, 41.0, 126.3, 126.4, 127.3, 127.8, 128.0, 128.2, 128.7, 128.9, 129.2, 130.3, 135.3, 139.2, 139.5, 140.5, 141.2, 142.7, 171.3, 177.1.
HRMS (ESI): [M + H]+ m/z calcd for C24H24NO3 374.1751, found 374.1748.
1H NMR


13C NMR
An Enantioselective Hydrogenation of an Alkenoic Acid as a Key Step in the Synthesis of AZD2716
† CVMD iMed, Medicinal Chemistry, AstraZeneca R&D Mölndal, SE-431 83 Mölndal, Sweden
‡SP Process Development, Box 36, SE-151 21 Södertälje, Sweden
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00382………..http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00382
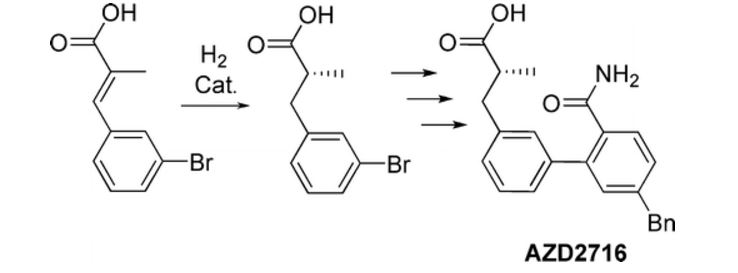
A classical resolution of a racemic carboxylic acid through salt formation and an asymmetric hydrogenation of an α,β-unsaturated carboxylic acid were investigated in parallel to prepare an enantiomerically pure alkanoic acid used as a key intermediate in the synthesis of an antiplaque candidate drug. After an extensive screening of rhodium- and ruthenium-based catalysts, we developed a rhodium-catalyzed hydrogenation that gave the alkanoic acid with 90% ee, and after a subsequent crystallization with (R)-1-phenylethanamine, the ee was enriched to 97%. The chiral acid was then used in sequential Negishi and Suzuki couplings followed by basic hydrolysis of a nitrile to an amide to give the active pharmaceutical ingredient in 22% overall yield.
Paper

Expedited structure-based optimization of the initial fragment hit 1 led to the design of (R)-7(AZD2716) a novel, potent secreted phospholipase A2 (sPLA2) inhibitor with excellent preclinical pharmacokinetic properties across species, clear in vivo efficacy, and minimized safety risk. Based on accumulated profiling data, (R)-7 was selected as a clinical candidate for the treatment of coronary artery disease.
Discovery of AZD2716: A Novel Secreted Phospholipase A2 (sPLA2) Inhibitor for the Treatment of Coronary Artery Disease
†Cardiovascular and Metabolic Diseases, Innovative Medicines and Early Development Biotech Unit Departments of Medicinal Chemistry, ‡Bioscience, §DMPK, ∥Discovery Sciences Departments of Structure & Biophysics, ⊥Reagents and Assay Development, and #Screening Sciences and Sample Management, Astrazeneca, Mölndal, Pepparedsleden 1, SE-431 83 Mölndal, Sweden
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.6b00188
*(F.G.) Phone: +1-212-4780-822. E-mail: fabrizio.giordanetto@deshawresearch.com., *(D.P.) Phone: +46 31 7065 663. E-mail:daniel.pettersen@astrazeneca.com.
http://pubs.acs.org/doi/full/10.1021/acsmedchemlett.6b00188

akenoic acid as a key step in the sysnthesis of AZD2716. Org. Proc. Res. Dev. 2016, 20(2),
262-269).
/////////atherosclerosis, coronary artery disease, fragment screening, fragment-based drug discovery, Secreted phospholipase A2, sPLA2, AZD2716, AZD-2716, AZD 2716, PRECLINICAL
c1c(cc(c(c1)C(=O)N)c2cccc(c2)CC(C(=O)O)C)Cc3ccccc3
FK-3311
FK-3311
FK 3311; 116686-15-8; FK-3311; N-[4-acetyl-2-(2,4-difluorophenoxy)phenyl]methanesulfonamide; COX-2 Inhibitor V, FK3311; FK3311;
A prostaglandin receptor antagonist potentially for the treatment of rheumatoid arthritis.

cas 116686-15-8
| Molecular Formula: | C15H13F2NO4S |
|---|---|
| Molecular Weight: | 341.329826 g/mol |

This compound has been obtained by two different ways: 1) The oxidation of 4′-amino-3′-chloroacetophenone (I) with NaNO2 and HCl in water gives 3′-chloro-4-nitroacetophenone (II), which is condensed with 2,4-difluorophenol (III) by means of K2CO3 in xylene yielding 3′-(2,4-difluorophenyl)-4′-nitroacetophenone (IV). The reduction of (IV) with Fe and NH4Cl in ethanol affords the corresponding 4′-amino compound (V), which is finally treated with methanesulfonyl chloride and pyridine. 2) The reaction of 4′-aminoacetophenone (VI) with methanesulfonyl chloride as before gives the corresponding sulfonamide (VII), which is brominated with Br2 in acetic acid yielding N-(4-acetyl-3-bromophenyl)methanesulfonamide (VIII). Finally, this compound is condensed with 2,4-difluorophenol (III) by means of K2CO3 and CuCl as before.
Chem Pharm Bull 1992,40(9),2399
|
1 to 4 of 4
|
|---|
| Patent | Submitted | Granted |
|---|---|---|
| METHODS TO TREAT INFECTIONS [US2014329777] | 2014-04-22 | 2014-11-06 |
| NOVEL NIMESULIDE COMPOSITIONS [US2012063996] | 2011-07-28 | 2012-03-15 |
| NANOPARTICULATE MELOXICAM FORMULATIONS [US2014141083] | 2013-07-12 | 2014-05-22 |
| Alkanesulfonanilide derivatives, processes for preparation thereof and pharmaceutical composition comprising the same [US4866091] | 1989-09-12 | |
\\\\\\\\\\\\\CC(=O)C1=CC(=C(C=C1)NS(=O)(=O)C)OC2=C(C=C(C=C2)F)F
SEE………..http://apisynthesisint.blogspot.in/2016/01/fk-3311.html
Fluorofenidone
Fluorofenidone
1- (3-fluorophenyl) -5-methyl – 2 (1H) pyridone
2(1H)-Pyridinone, 1-(3-fluorophenyl)-5-methyl-
1- (3_ fluorophenyl) -5_ methylpyridine _2 (IH) – one
C12 H10 F N O, 203.2123
PRECLINICAL, IND Filing
An anti-inflammatory agent potentially for the treatment of organ fibrosis.
![]()
CAS No. 848353-85-5
Synthesis
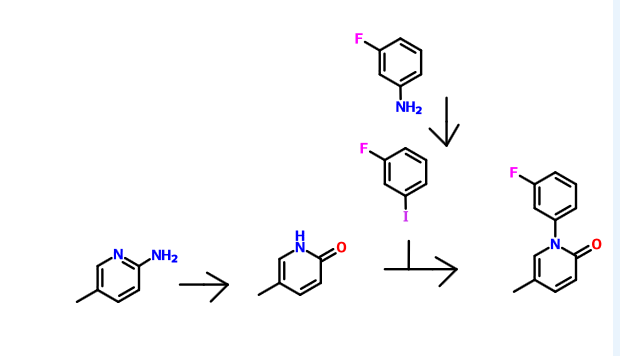
PATENT
WO 2006108354
http://www.google.co.in/patents/WO2006108354A1?cl=en
PATENT
http://www.google.com/patents/CN102241625A?cl=zh
(Compound 1)
A. (3_ fluorophenyl) methyl pyridine _2 (IH) 1- -5_ – -one
9. 6gDMF, 45 0g (0 2mol.) Inter-fluoro-iodobenzene, 21 8g (0. 2mol) 5_ methylpyridine _2_ (IH) -.. -one, 28g of anhydrous potassium carbonate and 1. Og copper powder, 160 ° -170 °, the reaction was stirred at reflux for 20 hours, the natural cooling to 110~120 ° C, was slowly added to about 330ml 80~90 ° C hot water, cooled to 20 ° C. Suction filtered, the filter cake was washed with about 20ml of water, remove the cake, with about 300ml of ethyl acetate ultrasound 30min, suction filtered, the filter residue was washed with 20ml of ethyl acetate. The combined ethyl acetate, washed with water three times (50ml * 3), and the filtrate layers were separated and allowed to stand for 15min, ethyl acetate fraction was concentrated to a non-steamed, hot added under stirring for about 85ml of petroleum ether, cooling to 15~20 ° C insulation ~ 1.5 hours. Filtration, the filter cake was washed twice with petroleum ether (about 20ml * 2) used to give 34. 9g crude. Recrystallized from 20% ethanol to give the product 1- (3_ fluorophenyl) -5_ methylpyridine _2 (IH) – one as a white solid # 30. Ig0 Μ P.: 132 · 1 ~133 7 °.. C.
PATENT
http://www.google.co.in/patents/WO2009149188A1?cl=zh-CN
PATENT
CN 102241625
http://www.google.com/patents/CN102241625A?cl=zh
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009111947
PAPER
| CN1386737A * | Jun 11, 2002 | Dec 25, 2002 | 中南大学湘雅医学院 | Antifibrosis pyridinone medicine and its prepaing process |
| CN1846699A | Apr 13, 2005 | Oct 18, 2006 | 中南大学湘雅医院 | Application of 1-(substituted phenyl)-5-methyl-2-(1H)-pyridone compound in preparing medicine for anti-other organifibrosis and tissue fibrosis except renal interstitial fibrosis |
| CN101235013A* | Mar 10, 2008 | Aug 6, 2008 | 广东东阳光药业有限公司;张中能 | Crystallized 1-(3-fluorophenyl)-5-methyl-2-(1H)pyridine and its preparation method composition and application |
| US20070203203 | May 1, 2007 | Aug 30, 2007 | Tao Li J | Composition and Method for Treating Fibrotic Diseases |
/////////
CC1=CN(C(=O)C=C1)C2=CC(=CC=C2)F
PNQ 201 from Advinus for for potential treatment of IBD.
formula I

PNQ 201
STRUCTURE COMING……
Adenosine A2b receptor antagonist
Advinus Therapeutics Ltd
KEEP WATCHING THIS POST……………
PNQ-201 is a proprietary orally active A2B Adenosine receptor (A2BAdoR) antagonist, currently in pre-clinical development for potential treatment of IBD. Advinus is looking for partnering/co-development opportunities.
A2BAdenosine Receptor (A2BAdoR) Antagonist PNQ-201 for IBD
Inflammatory Bowel Disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), is a multifactorial disease of an etiology not fully understood. It includes chronic inflammation of the gut, characterized by dysfunction of mucosal immunity. Current oral therapies are ineffective, non-specific, and have significant adverse effects. As such, there is a large unmet medical need for the development of new and specific therapies for IBD.
Adenosine is a stimulator of pro-inflammatory effects in the gastro-intestinal tract. Adenosine regulates tissue function by activating its receptors: A1AdoR and A2AAdoR are high affinity receptors and A2BAdoR and A3AdoR are low affinity receptors. A2BAdoR is highly expressed in cecum and colon, with expression increased even further in epithelial cells in human and murine colitis. A2BAdoR, agonized by adenosine induces cytokine secretion at the mucosal surface, inflammatory cell infiltration into intestinal wall, focal crypt damage and ulceration. Therefore, A2BAdoR antagonists are expected to be beneficial in IBD patients.
PNQ-201 is a proprietary orally active A2BAdoR antagonist, currently in pre-clinical development for the potential treatment of IBD. PNQ-201 is a potent and selective A2B antagonist. It is selected for development on the basis of poor systemic bioavailability and high exposure in colon/cecum. Negligible systemic bioavailability and maximum exposure at the sites of action in the lower gastrointestinal tract is expected to offer maximum therapeutic benefits while minimizing potential side effects. PNQ-201 has shown a robust efficacy profile in standard models of IBD, namely, the mouse DSS-induced colitis model and the rat TNBS-induced colitis model. PNQ-201 was found to be safe in exploratory safety studies including a Drug Matrix Screen, mini-AMES test, and a 14- day repeat dose toxicology study in rats.
PATENT
Example 1 : 8-(l-Benzyl~lH-pyrazol-4-yl)-l-propyl-l,4,5,7-tetrahydro-purin-6-one

Step 1: l-Benzyl-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide
A mixture of 5,6-diamino-3-propyl-lH-pyrimidine-2,4-dione (1.6g, 8.55mmol), 1-benzyl-lH-pyrazole-4-carboxylic acid (1.75g, 8.65mmol) in methanol (10ml) were cooled to 0 0C and added EDCLHCl (2.32g, 12.11mmol). The reaction mixture was stirred at 25 0C for 20 hours and the solvents were removed under reduced pressure. To this residue water (10ml) was added and the precipitate was filtered off, and was washed sequentially with cold water (20ml) and DCM (25ml) to obtain l-Benzyl-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3 -propyl- 1 ,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (1.5 g, 47 %) as a pale yellow solid.
1HNMR^OOMHZ5 DMSO d6): δ 0.82 (t, J=7.6Hzs 3H); 1.46-1.51 (m, 2H); 3.64 (t, J=7.2Hz, 2H);^5.36 (s, 2H); 6.01 (s, 2H); 7.26-7.38 (m, 5H); 7.96 (s, IH); 8.31 (s, IH); 8.54 (s, IH); 10.43 (s, IH).
Step 2 : 8-(l-Benzyl-lH-pyrazol-4-yl)-2-chloro-l-propyH,7-dihydro-purin-6-one A mixture of l-benzyl-lH-pyrazole-4-carboxylicacid(6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-ρyrimidin-5-yl)-amide (0.5g, 13.5mmol)s POCl3 (10ml) and DMF (0.1ml) were heated at 125-130 0C for 20 hours. Reaction mixture was cooled to 20-25 0C. It was then concentrated under vacuum. The residue was triturated with diethyl ether, dried. The crude product was purified by column chromatography using silica gel (100-200 mesh) and 2 to 4 % methanol in DCM as an eluent to obtain 8-( 1 -Benzyl- 1 H-pyrazol-4-yl)-2-chloro-l -propyl- l,7-dihydro-purin-6-one (0.04g, 8%) as a pale brown solid.
1HNMR^OOMHZ5 DMSO d6): δ 0.93 (t, J=7.6Hz, 3H); 1.67-1.73 (m, 2H); 4.15 (t, J=7.6Hz, 2H); 5.42 (s, 2H); 7.29-7.39 (m, 5H); 8.14 (s, IH); 8.49 (s, IH); 13.68 (bs, IH). Step 3: 8-(l-Benzyl-lH-pyrazol-4-yl)-l-propyl-l,7-dihydro-purin-6-one
A mixture of 8-(l -benzyl- lH-pyrazol-4-yl)-2-chloro-l -propyl- l,7-dihydro-purin-6-one (0.035 g, 0.094 mmol), Pd\C (10%) (0.025g), in ethanol (20ml) were stirred under hydrogen atmosphere for 20 hours. Reaction mixture was filtered through celite bed washed with methanol (20ml), and the solvents were removed under vacuum. The crude product was purified by column chromatography using silica gel (100-200 mesh) and 2 to 4 % methanol in DCM as an eluent to obtain 8-(l-Benzyl-lH-pyrazol-4-yl)-l-propyl-l,7-dihydro-purin-6-one (0.012g, 39%) as off white solid.
1HNMR^OOMHZ, DMSO d6): δ 0.89 (t, J=7.2Hz, 3H); 1.66-1.72 (m, 2H); 3.94 (t, J=7.6Hz, 2H); 5.41 (s, 2H); 7.302-7.38 (m, 5H); 8.03 (s, IH); 8.16 (s, IH); 8.34 (s, IH).
PATENT
WO 2011055391
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011055391
Preparation 1: 2-chloro-8-cyclopentyl-l-propyI-l, 7-dihydro-purin-6-one:

Step 1: Cyclopentane carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetra hydro-pyriniidin-S-y -amide
To a solution of 5, 6-diamino-3-propyI-lH-pyrimidine-2, 4-dione (0.6 g, 2.72 mmol) in methanol (50 ml) was added cyclopentane carboxylic acid (0.310 g, 2.72 mmol). The reaction mixture was cooled to 0°C and then l-ethyl-3(3′-dimethylaminopropyl) carbodiimide hydrochloride (EDCI.HC1) (0.78 g, 4.1 mmol) was added. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in water. The solid was filtered and washed thoroughly with water followed by diethyl ether. The product obtained was dried under high vacuum. The crude product (0.40 g) was used for the next step without further purification.
Step 2: Preparation of 8-cyclopentyI-2-chloro-l-propyl-l, 7-dihydro-purin-6-one
To a suspension of cyclopentanecarboxylic acid (6-amino-2,4-dioxo-3-propyl- 1,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (0.40 g, crude) obtained from step 1 in phosphorus oxychloride (25 ml) was added phosphorus pentachloride (0.10 g) and the resulting reaction mixture was refluxed overnight. Phosphorus oxychloride was evaporated under reduced pressure. The residue was slowly quenched with water. Ethyl acetate was added and the organic layer was separated and washed thoroughly with water followed by brine. The ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under vacuum. The crude product was purified by preparative TLC using dichloromethane, methanol (9:1) as the solvent system to give 0.075 g (19% over two steps) of the product as a white solid.
•H MR (400 MHz, DMSO d6): δ 0.9 (t, J = 8 Hz, 3H), 1.59-1.82 (m, 8H), 1.99 (m, 2H), 3.15 (t, J = 8 Hz, 1H), 4.12 (t, J = 8 Hz, 2H).
Preparations 2 to 7 were prepared following the experimental procedure as given for Preparation 1.
Preparation 2: 2-Chloro-8-cyclohexyl- 1 -propyl- 1 ,7-dihydro-purin-6-one,
Preparation 3: 2-Chloro-8-cyclopropyl-l -propyl- 1 ,7-dihydro-purin-6-one,
Preparation^ 2-Chloro-8-(hexahydro-2,5-methano-pentalen-3a-yl)-l -propyl- 1,7- dihydro-purin-6-one,
Preparation 5: 8-Bicyclo-[2.2.1]-hept-2-yl-2-chloro-l -propyl- 1, 7-dihydro-purin-6- one,
Preparation 6: 8-Adamantan-2-yl-2-chloro-l -propyl- 1, 7-dihydro-purin-6-one, Preparation7:3-[4-(2-Chloro-6-ox0-l-propyl-6,7-dihydro-lH-purin-8-yl)- bicyclo[2.2.2]oct-l-yl]-propionic acid.
Example 1: 8-Cyclopentyl-2-(3, 4-difluoro-phenoxy)-l-propyl-l, 7-dihydro-purin- 6-one:

To a solution of 8-cyclopentyl-2-chloro- 1 -propyl- l,7-dihydro-purin-6-one (0.06 g, 0.21 mmol) in N-methyl-2-pyrrolidone (0.2 ml) was added K2CO3 (0.044g, 0.32 mmol) followed by 3, 4-difluoro phenol and the reaction mixture was heated at 130 °C overnight. The reaction mixture was diluted with ethyl acetate and water. The layers were separated and ethyl acetate layer was washed with water. The ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under vacuum. The crude product was purified by preparative TLC using 3% methanol in DCM to give the product (0.015 g, 19 %) as a white solid.
‘HNMR (400 MHz, DMSO d6): δ 0.94 (t, J = 8 Hz, 3H), 1.59-1.74 (m, 6H), 1.94 (br.s, 2H), 3.12 (m, 2H), 4.09 (br. s, 2H), 7.21 (d, J = 8 Hz, 1H), 7.53-7.65 (m, 2H), 12.74 (br.s, 1H).
AN 2898

AN2898
(5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole)
1,2-Benzenedicarbonitrile, 4-((1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy)-,
AN-2898
cas: 906673-33-4
UNII: 6O60L94RMB,
MW 276.0581, MF C15 H9 B N2 O3
A PDE4 inhibitor potentially for the treatment of fungal infection.
AN-2898, a novel topical anti-inflammatory compound that inhibits phosphodiesterase 4 and 7 enzyme activit
PHASE 2 FUNGAL INFECTION, Anacor Pharmaceuticals for the treatment of atopic dermatitis
![]()
| Anacor Pharmaceuticals Inc. | |
| Description | Boron-containing small molecule phosphodiesterase-4 (PDE-4) inhibitor that reduces the production of tumor necrosis factor (TNF) alpha, IL-12 and IL-23 |
| Molecular Target | Phosphodiesterase-4 (PDE-4) |
| Mechanism of Action | Phosphodiesterase-4 (PDE-4) inhibitor |
| Therapeutic Modality | Small molecule |
AN2898 (5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole) is a broad spectrum anti-inflammatory compound currently in development for the topical treatment of plaque and atopic psoriasis.
AN2898 inhibited phosphodiesterase 4 (PDE4) enzyme activity (IC50 0.060 μM) and the release of multiple cytokines including TNF-α (IC50 0.16 μM) from peripheral blood mononuclear cells (hPBMCs) stimulated by lipopolysaccharide (LPS) or phytohemag- glutinin.
Further, AN2898 was also found to inhibit IL-23 release (IC50 1.0 μM) from THP-1 cells stimulated by LPS and IFN-γ. Investigation of the structure-activity relation-ship around this compound was reported to identify a more potent dual TNF-α/IL-23 inhibitor
( ref………. Akama T, Antunes J, Freund Y, Kimura R, Dong C, Sanders V, et al. Structure-activity studies of novel oxaborole dual inhibitors of PDE4 and IL-23 release. 69th Annu Meet Soc Invest Dermatol (May 6-9, Montreal) 2009 Abst 282. ).
PATENT
WO 2007095638
https://www.google.co.in/patents/WO2007095638A2?cl=en
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en
US 7582621
http://www.google.co.in/patents/US7582621
WO 2009111676
http://www.google.im/patents/WO2009111676A2?cl=en
WO 2007078340
http://www.google.im/patents/WO2007078340A2?cl=en
US 20070286822
http://www.google.com/patents/US20070286822
REFERENCES
1 Structure-activity studies led to the discovery of AN2898 in development for topical treatment of psoriasis and atopic dermatitis, J Am Acad Dermatol 2009, 60(3, Suppl. 1): Abst P1317
2 FEBS Letters (2012), 586(19), 3410-3414
See all Bboroles at………http://apisynthesisint.blogspot.in/p/borole-compds.html
/////////AN2898, AN 2898, ANACOR, BOROLE
B1(c2ccc(cc2CO1)Oc3ccc(c(c3)C#N)C#N)O
RO-28-1675 for Type 2 Diabetes

RO-28-1675
- (2R)-3-Cyclopentyl-2-[4-(methanesulfonyl)phenyl]-N-(thiazol-2-yl)propionamide
- Ro 028-1675
- Ro 0281675
- Ro 28-1675
3-Cyclopentyl-2(R)-[4-(methylsulfonyl)phenyl]-N-(2-thiazolyl)propionamide
| MW | 378.51 | .-70.4 °
Conc 0.027 g/100mL; chloroform, 589 nm; 23 °C
|
|
|---|---|---|---|
| Formula | C18H22N2O3S2 | ||
| CAS No | 300353-13-3 |
Glucokinase Activators
Ro 28-1675 (Ro 0281675) is a potent allosteric GK activator with a SC1.5 value of 0.24± 0.0019 uM.
Roche (Innovator)
PHASE 1 Type 2 DIABETES,
IC50 value: 0.24± 0.0019 uM (SC1.5) [1]
Target: Glucokinase activator
The R stereoisomer Ro 28-1675 activated GK with a SC1.5 of 0.24 uM, while the S isomer did not activated GK up to 10 uM. Oral administration of Ro 28-1675 (50 mg/Kg) to male C57B1/6J mice caused a statistically significant reduction in fasting glucose levels and improvement in glucose tolerance relative to the vehicle treated animals [1].
Comparison of rat PK parameters indicated that Ro 28-1675 displayed lower clearance and higher oral bioavailability compared to 9a.
Following a single oral dose, Ro 28-1675 reduced fasting and postprandial glucose levels following an OGTT, was well tolerated, and displayed no adverse effects related to drug administration other than hypoglycemia at the maximum dose (400 mg).
 .
.
RO-28-1675 as glucokinase activator.
Joseph Grimsby et al., of Roche have recently discovered activators of glucokinase that increase kcat and decrease the S0.5 for glucose, and these may offer a treatment for type II diabetes. Glucokinase (GK) plays a key role in whole-body glucose homeostasis by catalyzing the phosphorylation of glucose in cells that express this enzyme, such as pancreatic β cells and hepatocytes.
By screening of a library of 120,000 structurally diverse synthetic compounds, they found one small molecule that increased the enzymatic activity of GK. Chemical optimization of this initial molecule led to the synthesis of RO-28-0450 as a lead GK activator which is a class of antidiabetic agents that act as nonessential, mixed-type GK activators (GKAs) that increase the glucose affinity and maximum velocity (Vmax) of GK. RO-28-0450 is a racemic compound.
Activation of GK was exquisitely sensitive to the chirality of the molecule: The R enantiomer, RO-28-1675, was found to be a potent GKA, whereas the S enantiomer, RO-28-1674, was inactive. RO-28-1675 also reversed the inhibitory action of the human glucokinase regulatory protein (GKRP). The activators binding in a glucokinase regulatory site originally was discovered in patients with persistent hyperinsulinemic hypoglycemi.
The result of RO-28-1675 as a potent small molecule GKA may shed light to the chemical biologists to devise strategy for developing activators. Thus for a success to this end we must focus on highly regulated enzymes, or cooperative enzymes such as glucokinase, where nature has provided binding sites that are designed to modulate catalysis.
.SYNTHESIS


Paper
J. Med. Chem., 2010, 53 (9), pp 3618–3625
DOI: 10.1021/jm100039a

Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl-propionamide (2.10 g, 74%) as a white foam.
[α] 23 589 = –70.4° (c=0.027, chloroform).
EI-HRMS m/e calcd for C18H22N2O3S2 (M+ ) 378.1072, found 378.1081.
1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 10.48 (br. s., 1 H), 7.88 (d, J=8.6 Hz, 2 H), 7.53 (d, J=8.6 Hz, 2 H), 7.50 (d, J=3.5 Hz, 1 H), 7.06 (d, J=3.5 Hz, 1 H), 3.76 (t, J=7.7 Hz, 1 H), 3.03 (s, 3 H), 2.28 (dt, J=13.6, 7.7 Hz, 1 H), 1.88 – 1.98 (m, 1 H), 1.42 – 1.84 (m, 7 H), 1.07 – 1.19 (m, 2 H).
Anal. Calcd for C18H22N2O3S2: C, 56.94; H, 5.59; N, 7.28. Found: C, 57.12; H, 5.86; N, 7.40.
PATENT
WO 2000058293
http://www.google.com/patents/WO2000058293A2?cl=en
Example 3 (A) 3-CyclopentyI-2-(4-methanesulfonyl-phenyI)-N-thiazol-2-yI-propionamide

A solution of dπsopropylamine (3.3 mL, 23.5 mmol) in dry tetrahydrofuran (50 mL) and 1.3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdιnone (10 mL) was cooled to -78°C under nitrogen and then treated with a 10M solution of n-butyllithium m hexanes (2.35 mL, 23 5 mmol) The yellow reaction mixture was stiπed at -78°C for 30 mm and then treated dropwise with a solution of 4-methylsulfonylphenylacetιc acid (2.40 g, 11.2 mmol) in a small amount of dry tetrahydrofuran. After approximately one-half of the 4- methylsulfonylphenylacetic acid m dry tetrahydrofuran was added, a precipitate formed Upon further addition of the remaining 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture became thick in nature After complete addition of the 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture was very thick and became difficult to stir An additional amount of dry tetrahydrofuran (20 mL) was added to the thick reaction mixture, and the reaction mixture was stirred at –
78 C for 45 mm, at which time, a solution of lodomethylcyclopentane (2.35 g, 11.2 mmol) in a small amount of dry tetrahydrofuran was added dropwise The reaction mixture was allowed to warm to 25°C where it was stiπed for 15 h. The reaction mixture was quenched with water (100 mL), and the resulting yellow reaction mixture was concentrated in vacuo to remove tetrahydrofuran. The aqueous residue was acidified to pH = 2 using concentrated hydrochloπc acid The aqueous layer was extracted with ethyl acetate The organic phase was dπed over magnesium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 230-400 mesh, 1/3 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (1.80 g, 52%) as a white solid: mp 152-154°C; EI-HRMS m/e calcd for C15H20O4S (Nf) 296.1082, found 296.1080
A solution of 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (4.91 g, 16.56 mmol) and tnphenylphosphine (6.52 g, 24.85 mmol) m methylene chloπde (41 mL) was cooled to 0°C and then treated with N-bromosuccinimide (5.01 g, 28.16 mmol) m small portions The reaction mixture color changed from light yellow to a darker yellow then to brown After the complete addition of N-bromosuccinimide, the reaction mixture was allowed to warm to 25°C over 30 min. The brown reaction mixture was then treated with 2-aminothiazole (4.98 g, 49.69 mmol). The resulting reaction mixture was stiπed at 25°C for 19 h. The reaction mixture was then concentrated in vacuo to remove methylene chloride. The remaining black residue was diluted with a 10% aqueous hydrochloric acid solution (400 mL) and then extracted with ethyl acetate (3 x 200 mL). The combined organic layers were washed with a saturated aqueous sodium chloride solution (1 x 200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate then 1/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)-N-thiazol-2- yl-propionamide (4.49 g, 72%) as a white solid: mp 216-217°C; EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1071.
Example 13
(2R)-3-Cyclopentyl-2-(4-methanesuIfonylphenyl)-N-thiazol-2-yl-propionamide

A solution of ^-( ethanesulfonyl)phenyl acetic acid (43 63 g, 0.204 mol) in methanol (509 mL) was treated slowly with concentrated sulfunc acid (2 mL) The resulting reaction mixture was heated under reflux for 19 h The reaction mixture was allowed to cool to 25°C and then concentrated in vacuo to remove methanol The residue was diluted with ethyl acetate (800 mL) The organic phase was washed with a saturated aqueous sodium bicarbonate solution (1 x 200 mL), washed with a saturated aqueous sodium chlonde solution (1 x 200 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 1/1 hexanes/ethyl acetate) afforded 4-(methanesulfonyl)phenyl acetic acid methyl ester (45.42 g, 98%) as a yellow oil which solidified to a cream colored solid upon sitting over time at 25°C mp 78-80°C, EI-HRMS m/e calcd for Cι0H12O4S (M+) 228 0456, found 228 0451.
A mechanical stiπer was used for this reaction A solution of dnsopropylamme (29.2 mL, 0.21 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro- 2(lH)-pyπmιdιnone (62 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (83 4 mL, 0.21 mol) The yellow-orange reaction mixture was stiπed at -78°C for 35 min and then slowly treated with a solution of 4- (methanesulfonyl)phenyl acetic acid methyl ester (45.35 g, 0.20 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdmone (62 mL) The reaction mixture turned dark in color. The reaction mixture was then stiπed at -78°C for 50 mm, at which time, a solution of lodomethylcyclopentane (50.08 g, 0.24 mol) in a small amount of dry tetrahydrofuran was added slowly. The reaction mixture was then stiπed at -78°C for 50 mm, and then allowed to warm to 25°C, where it was stirred for 36 h. The reaction mixture was quenched with water (100 mL), and the resulting reaction mixture was concentrated in vacuo to remove tetrahydrofuran The remaining residue was diluted with ethyl acetate (1.5 L). The organic phase was washed with a saturated aqueous sodium chloπde solution (1 x 500 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid methyl ester (41.79 g, 68%) as a yellow viscous oil EI-HRMS m/e calcd for Cι6H22O4S (M+) 310.1239. found 310.1230.
A solution of 3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid methyl ester (50 96 g, 0.16 mol) in methanol (410 mL) was treated with a IN aqueous sodium hydroxide solution (345 mL, 0.35 mol). The reaction mixture was stirred at 25°C for 24 h. The reaction mixture was concentrated in vacuo to remove methanol. The resulting aqueous residue was acidified to pH = 2 with concentrated hydrochlonc acid and then extracted with ethyl acetate (5 x 200 mL) The combined organic layers were dned over sodium sulfate, filtered, and concentrated in vacuo to afford pure 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (43 61 g, 90%) as a white solid which was used without further puπfication. mp 152-154°C, EI-HRMS m e calcd for C15H20O4S (M+) 296.1082, found 296.1080.
Two separate reactions were setup in parallel: (1) A solution of (R)-(+)-4-benzyl-2- oxazohdmone (3.67 g, 20.73 mmol) m dry tetrahydrofuran (35 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (7.9 mL, 19.86 mmol). The resulting reaction mixture was stiπed at -78°C for 30 mm and then allowed to warm to 25°C, where it was stirred for 1.5 h (2) A solution of racemic 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (5.12 g, 17.27 mmol) in dry tetrahydrofuran (35 mL) was cooled to 0°C and then treated with tnethylamme (2.8 mL, 19.86 mmol). The reaction mixture was stiπed at 0°C for 10 nun and then treated dropwise with tπmethylacetyl chlonde (2.6 mL, 20.73 mmol). The resulting reaction mixture was stiπed at 0°C for 2 h and then cooled to -78°C for the addition of the freshly prepared chiral oxazolidmone. The reaction mixture containing the oxazolidmone was then added to the cooled (-78°C) mixed anhydπde solution The resulting reaction mixture was stiπed as -78°C for 1 h and allowed to gradually warm to 25°C. The reaction mixture was then stiπed at 25°C for 3 d. The resulting reaction mixture was quenched with water (100 mL) and then concentrated in vacuo to remove tetrahydrofuran. The resulting aqueous residue was diluted with ethyl acetate (600 mL). The organic layer was washed with a saturated aqueous sodium chloπde solution (1 x 300 mL), dπed over sodium sulfate, filtered, and concentrated in vacuo Thin layer chromatography using 13/7 hexanes/ethyl acetate as the developing solvent indicated the presence of two products The higher moving product had a Rf =0.32 and the lower moving product had a Rf = 0.19. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 9/1 then 13/7 hexanes/ethyl acetate) afforded two products: (1) The higher Rf product (4R, 2’S)-4-benzyl-3-[3- cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazohdm-2-one (2.12 g, 54%) as a white foam- mp 62-64°C; [c.]23 589 = +6.3° (c=0.24, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M+) 455.1766, found 455.1757. (2) The lower Rf product (4R, 2R)-4- benzyl-3-[3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.88 g, 99%) as a white foam: mp 59-61°C; [α]23 589 = -98.3° (c=0.35, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M +) 455.1766, found 455.1753. The combined mass recovery from the two products was 6.00 g, providing a 76% conversion yield for the reaction
An aqueous solution of lithium hydroperoxide was freshly prepared from mixing a solution of anhydrous lithium hydroxide powder (707.3 mg, 16.86 mmol) m 5.27 mL of water with a 30% aqueous hydrogen peroxide solution (3.44 mL, 33.71 mmol). This freshly prepared aqueous lithium hydroperoxide solution was cooled to 0°C and then slowly added to a cooled (0°C) solution of (4R, 2’R)-4-benzyl-3-[3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.84 g, 8.43 mmol) in tetrahydrofuran (33 mL) and water (11 mL). The reaction mixture was stiπed 0°C for 1.5 h The reaction mixture was then quenched with a 1.5N aqueous sodium sulfite solution (25 mL) The reaction mixture was further diluted with water (300 mL) The resulting aqueous layer was continuously extracted with diethyl ether until thm layer chromatography indicated the absence of the recovered chiral oxazolidmone in the aqueous layer The aqueous layer was then acidified to pH = 2 with a 10% aqueous hydrochlonc acid solution and extracted with ethyl acetate (300 mL) The organic extract was dned over sodium sulfate, filtered, and concentrated in vacuo to afford (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white solid (2.23 g, 89%) which was used without further puπfication Flash chromatography (Merck Silica gel 60, 70-230 mesh, 30/1 methylene chlonde/methanol then 10/1 methylene chlonde/methanol) was used to obtain a punfied sample for analytical data and afforded pure (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white foam- mp 62-64°C (foam to gel), [α]23 589 = -50.0° (c=0.02, chloroform), EI-HRMS m/e calcd for C15H20O4S (M+) 296 1082, found 296 1080
A solution of tnphenylphosphme (3.35 g, 12.79 mmol) m methylene chloπde (19 mL) was cooled to 0°C and then slowly treated with N-bromosuccmimide (2.28 g, 12.79 mmol) in small portions. The reaction mixture was stiπed at 0°C for 30 mm, and dunng this time penod, the color of the reaction mixture changed from light yellow to a darker yellow then to a purple color. The cooled purple reaction mixture was then treated with the (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid (2.23 g, 7.52 mmol) The resulting reaction mixture was then allowed to warm to 25°C over 45 mm, at which time, the reaction mixture was then treated with 2-amιnothιazole (1.88 g, 18.81 mmol) The resulting reaction mixture was stiπed at 25°C for 12 h. The reaction mixture was then concentrated in vacuo to remove methylene chloπde The remaining black residue was diluted with ethyl acetate (300 mL) and then washed well with a 10% aqueous hydrochlonc acid solution (2 x 100 mL), a 5% aqueous sodium bicarbonate solution (3 x 100 mL), and a saturated aqueous sodium chloride solution (1 x 200 mL). The organic layer was then dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl- propionamide (2.10 g, 74%) as a white foam: mp 78-80°C (foam to gel); [α]23 589 = -70.4° (c=0.027, chloroform); EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1081.
REFERENCES
Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
http://www.nature.com/nrd/journal/v8/n5/fig_tab/nrd2850_T2.html
NMR…..http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-NMR-HY-10595-13569-2014.pdf
http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-Lcms_Ms-HY-10595-13569-2014.pdf
///////////RO-28-1675, Ro 0281675
O=C(Nc1nccs1)[C@H](CC2CCCC2)c3ccc(cc3)S(C)(=O)=O

Chemical structures of Roche’s glucokinase activators (GKAs) RO-28-1675 and piragliatin, as well as the related GKA 1.
PF-04191834 for Patients With Osteoarthritis Of The Knee


PF 4191834
CAS 1029317-21-2
UNII-YX55DXP4T1; PF-4191834; DVNQWYLVSNPCJZ-UHFFFAOYSA-N;
4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio]phenyl) tetrahydro-2H-pyran-4-carboxamide;
4-[3-[4-(2-methylpyrazol-3-yl)phenyl]sulfanylphenyl]oxane-4-carboxamide
| Molecular Formula: | C22H23N3O2S |
|---|---|
| Molecular Weight: | 393.50192 g/mol |
PF-04191834 works in animal models by inhibiting one of the enzymes, 5-lipoxygenasein which is involved in the pathway that causes inflammation and pain. The purpose of this study is to test how effective, safe and tolerated PF-04191834 is in patients with osteoarthritis of the knee by itself or with naproxen, particularly to test if patients have less pain.
Mechanism:
5-Lipoxygenase (5-LO) inhibitor
Original Development Indication:
AsthmaChronic osteoarthritis pain

PATENT
US 20080125474
formula (Ib):

A compound of formula (Ib) may be prepared according to the following process:

- Example 1

4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio}phenyl)tetrahydro-2H-pyran-4 carboxamideStep 1: Preparation of 4-(3-bromophenyl)-tetrahydro-2H-pyran-4-carboxamide
-
4-(3-bromophenyl)tetrahydro-2H-pyran-4-carbonitrile made by the procedures described in EP 108114 (1.05 kg, 3.95 mole) was stirred in 98% H2SO4 (3.00 L) at room temperature for about 40 h. The mixture was then poured onto ice and the very fine suspension was filtered and washed with H2O thoroughly until pH of wash is neutral. The white solid was washed with hexanes and was then dried in vacuo at 35-40° C. to give 1119 g (99.8% yield) of product in 99.9% purity. LC/MS: 5%-100% CH3CN:H20-0.01% TFA gradient over 10 minutes: 4.68 min. (M+H)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.50-7.49 (m, 1H), 7.43-7.40 (m, 1H), 7.36-7.30 (m, 1H), 7.27 (d, J=7.92 Hz, 1H) 7.06 (s, 1H), 5.00 (brs, 1H) 3.71 (dt, J=11.7, 3.7 Hz, 2H), 3.42 (t, J=10.7 Hz, 2H), 2.38 (d, J=13.6 Hz, 2H), 1.75 (td, J=12.2, 4.3 Hz, 2H).
Step 2: Preparation of 4-(3-(triisopropylsilylthio)phenyl)-tetrahydro-2H-pyran-4-carboxamide
-
Alternative 1
-
4-(3-Bromophenyl)-tetrahydro-2H-pyran-4-carboxamide prepared in step 1 (300 g (1.06 mole), sodium tert-butoxide (122 g, 1.27 mole), Pd(OAc)2 (4.74 g 0.0211 mole) and DIPPF (1,1-bis(diisopropylphosphino)ferrocene) (10.6 g 0.0253 mole) were placed in a flask which was evacuated and filled with N2 3 times. Anhydrous dioxane (2.3 L) was added and the mixture was stirred at room temperature for 1 h. To the mixture was added triisopropylsilane thiol (221 g 1.16 mole) and the resulting mixture was heated to reflux. Reflux was stopped after 1 h and the mixture was allowed to cool to room temperature. The mixture was then poured into ethyl acetate (7 L) which was then washed with H2O (2×4 L) and brine (2 L). The combined aqueous washes were back extracted with ethyl acetate (3 L) which was then washed with H2O (2×2 L) and brine (1 L). The combined organic layers were dried over MgSO4, filtered and concentrated to dryness. Ethyl acetate (0.5 L) was added to the solid and the mixture was stirred on the rotary evaporator to give a fine suspension. Hexanes (1.5 L) was then added and the suspension was allowed to stand for 1 hour. The solid was filtered, washed with 1:1 ethyl acetate-hexanes (1 L) and then hexanes. The resulting brown solid was dried in vacuo to give 334 g (80% yield) of the product in 99% purity. A second crop was obtained from the filtrate which was washed as before and dried to give an additional 15 g product for a total yield of 84%. LC/MS: 5%-100% CH3CN:H20-0.01% TFA gradient over 10 minutes: 9.35 min. 394.1 (M+H)+. 1H NMR (400 MHz, CDCl3) δ ppm 7.52-7.51 (m, 1H) 7.42-7.39 (m, 1H), 7.22-7.21 (m, 2H), 5.35 (brs, 1H), 5.13 (brs, 1H) 3.78-3.75 (m, 4H) 2.36-2.32 (m, 2H), 2.06-2.00 (m, 2H), 1.27-1.16 (m, 3H), 1.05 (d, J=7.25 Hz, 18H).
Step 2: Preparation of 4-(3-(triisopropylsilylthio)phenyl)-tetrahydro-2H-pyran-4-carboxamide
-
Alternative 2
-
Purge a 3-neck flask (overhead stirrer, nitrogen inlet, serum cap) with nitrogen. Add 4-(3-Bromophenyl)-tetrahydro-2H-pyran-4-carboxamide prepared in step 1 (10 g, 0.03519 mole). Add sodium t-butoxide (4.1 g, 0.04223 moles). Add anhydrous toluene. Toluene should be as dry as possible, <0.01% water by KF is sufficient. Initiate stirring. Purge the reaction mixture with 4 vacuum/nitrogen purge cycles, maintaining 60 torr vacuum for 30 seconds with each cycle. Add the thiol (9.1 g, 0.04223 moles) assuring that oxygen is not introduced into the vessel. Heat to 75° C. Add PdCl2(diphenyl-phosphino ferrocene) (0.258 g, 0.00035 moles). Continue heating to reflux (reaction temperature about 107° C.) for a minimum of 1 hour. The mixture should reach reflux within 30 minutes.
-
Cool the reaction mixture to 25° C. Add ethyl acetate (300 mL, 30 mL/g) and stir the resulting suspension for 30 min. Filter the suspension through celite (30 g). Rinse the celite with ethyl acetate for rinse (100 mL, of product to be rinsed), combining filtrates. Concentrate the filtrate via vacuum distillation at 70 torr at 30° C. until 80% of the filtrate volume has been removed. Add hexane (200 mL, 20 mL/g of product to be crystallized) for crystallization to the slurry over 5 minutes. Stir and cool the mixture to 5° C. Maintain the mixture at 5° C. for a minimum of 1 hour. Isolate product by filtration. Rinse the cake with hexane (100 mL, of product to be rinsed). Dry the cake on the filter to LOD of no more than 5%. Dry the solid at 45-50° C. under vacuum to an LOD of no more than 1.5%. Yield 12 grams (85% yield).
-
Any mL/g amount indicated above is referred to grams of bromo carboxamide.
Step 3: Preparation of 5-(4-bromophenyl)-1-methyl-1H-pyrazole
-
Alternative 1
-
A N,N′-dimethylformamide (15 mL) solution of 4-bromoacetophenone (10.60 g, 53.25 mmols) and N,N′-dimethylformamide dimethyl acetal (2.5 equivalents) was heated at 125 degrees Celcius for 3 hours. The dark red solution was cooled to room temperature. The volatiles were removed by rotary evaporation providing a red viscous oil. To this substance was added anhydrous N,N′-dimethylformamide (15 mL) and methyl hydrazine (7.6 g, 160 mmols, 3 equivalents). The mixture was stirred at room temperature for 1 hour and then heated at 75 degrees Celcius for 4 hours. The volatiles were removed by rotary evaporation and the crude residue was taken up in a small volume of methylene chloride. This red solution was applied to a cartridge of silica gel. The cartridge was eluted with a 20:80 mixture of ethyl acetate and hexanes, respectively. The appropriate fractions were combined and concentrated to produce 12.5 g of a white solid.
-
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 3.87-3.95 (m, J=2.22 Hz, 3H) 6.29-6.36 (m, 1H) 7.31 (dd, J=8.36 Hz, 2H) 7.52-7.56 (m, 1H) 7.62 (dd, J=2.05 Hz, 2H).
Step 3: Preparation of 5-(4-bromophenyl)-1-methyl-1H-pyrazole
-
Alternative 2
-
4-bromoacetophenone (20.0 g; 0.10 mole) and N,N-dimethylformamide dimethylacetal (28.5 mL; 0.20 mole) were mixed together in DMF (12 mL) and heated to 110° C. for 4 hours. The methanol and water that were generated during the reaction were distilled (6.2 mL). The mixture was cooled to 25° C. Methyl t-butyl ether (100 mL) and methylhydrazine (21.2 mL; 0.40 moles) were added and the mixture was stirred over night. The reaction mixture was washed with 1 M aqueous ammonium chloride (3×40 mL) and water (40 mL). The organic phase was dried by azeotropic distillation using a Dean-Stark apparatus. As an alternative to distillation, the solution was dried through an anhydrous magnesium sulfate cartridge. The solution was filtered through a silica gel cartridge (60 g). The product was flushed from the cartridge with methyl t-butyl ether. The fraction(s) containing product were combined and concentrated to about 70 mL by distillation. Heptane (120 mL) was added and distillation was continued until the pot temperature reached 98.4° C. About 100 mL of distillate was collected. The mixture was cooled to 40° C. The mixture was seeded and the temperature was maintained at 40° C. for 30 minutes while crystallization was initiated. The mixture was slowly chilled to 0° C. over 90 minutes. The mixture was held at 0° C. for 30 minutes. The mixture was filtered and the solid was washed (3×) with chilled (0° C.) heptane. The solid was dried on the filter. A cream-colored, crystalline solid (16.3 g; 68% yield) was obtained. The NMR data of the title compound are as per alternative 1.
Step 4: Preparation of 4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio]phenyl) tetrahydro-2H-pyran-4-carboxamide
-
A mixture of 5-(4-bromophenyl)-1-methyl-1H-pyrazole (0.50 g, 2.10 mmols,), 4-{3-[(tri-isopropylsilyl)thio]phenyl}tetrahydro-2H-pyran-4-carboxamide (0.83 g, 2.10 mmols), Tetrakis(triphenylphosphine)palladium(0) (243 mg, 0.10 equivalents), bis[(2-diphenyl-phosphino)]phenyl ether (113 mg, 0.10 equivalents), and 1.0 M potassium tert-butoxide in THF (6.3 mmols, 3 equivalents) in iPrOH (15 mL) that contained 5% water was heated for 4 hours at 90 degrees Celcius in an atmosphere of nitrogen. The reaction mixture was cooled to room temperature and 7 mL of 1N HCl was added. The product was precipitated by the addition of water (30 mL). The precipitate was collected by suction filtration and washed with water (2×20 mL) and cold ethyl ether (4×20 mL). The tan brown solid was dissolved in a small volume of methylene chloride containing 1% methanol and applied to a 140 g cartridge of silica gel. The cartridge was eluted with an acetone:hexane gradient. The appropriate fractions were concentrated and triturated with methanol to produce a white solid (710 mg) as product. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.75-1.84 (m, 3H) 2.40 (d, J=13.54 Hz, 3H) 3.43-3.51 (m, 1H) 3.72 (d, J=11.34 Hz, 3H) 3.84 (s, 3H) 6.40 (d, J=1.46 Hz, 1H) 7.02 (s, 1H) 7.22-7.30 (m, 2H) 7.34 (d, J=8.05 Hz, 1H) 7.38-7.43 (m, 2H) 7.45-7.52 (m, 3H). HRMS calc M+H, 394.1589, found 394.1630.
Step 4: Preparation of 4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio]phenyl) tetrahydro-2H-pyran-4-carboxamideScale-Up Alternative
-
4-{3-[(tri-isopropylsilyl)thio]phenyl}tetrahydro-2H-pyran-4-carboxamide (200 g, 0.51 moles), 5-(4-bromophenyl)-1-methyl-1H-pyrazole (126 g, 0.53 moles), and 2-methyltetrahydrofuran (2,000 mL, 10 mL/g of tips carboxamide) were put into the reactor and sparged with nitrogen while heating to 60° C. The sodium methoxide (244.0 mL, 1.07 moles, added as sodium methoxide in methanol solution 25% w/w) was added to the reactor and sparging was continued for another 30 minutes. PdCl2DPPF (3.7 g, 0.005 moles) was added to the reactor and the mixture was heated to 70° C. Once the amount of tips carboxamide was less than 1% of starting amount, the mixture was cooled to 0° C. The mixture was held at 0° C. for one hour. The mixture was filtered and the solid was washed with 2-methyltetrahydrofuran (3×2.5 mL/g). The solid was dried on the filter. The solid was returned to a clean reactor and triturated with water (2,000 mL, 10 mL/g) for two hours at 20° C. The mixture was filtered and the solid was washed with water (2,000 mL, 2×5 mL/g). The solid was dried on the filter. The solid was returned to a clean reactor with the Si-thiol (90.0 g, 0.5 g/g) and THF (about 12.8 L, 70 mL/g). The mixture was heated to 60-65° C. and held for two hours. The mixture was cooled to 25° C. and filtered. The Si-thiol was washed with THF (about 0.9 L, 5 mL/g). The solution was distilled to a concentration of 10 mL/g. The mixture was cooled to 25° C. and hexanes (422.5 mL, 5 mL/g) was added. The mixture was filtered and the solid was washed with hexanes (422.5 mL, 5 mL/g). The solid was dried in a vacuum oven at 70° C.
-
For 2-methyltetrahydrofuran and water, mL/g are referred to grams of tips carboxamide. For Si-thiol, tetrahydrofuran and hexanes, mL/g are referred to grams of title compound.
Step 5: Purification of 4-(3-{[4-(1-methyl-1H-pyrazol-5-yl)phenyl]thio]phenyl) tetrahydro-2H-pyran-4-carboxamide
Crude title compound (181.0 g, 1.0 eq.) obtained from step 4, scale-up version, was returned to a clean reactor with Si-thiol (0.5 g/g of title compound) and THF (75 mL/g of title compound). The mixture was heated to 60-65° C. and held overnight. The mixture was cooled to 25° C. and filtered. The Si-thiol was washed with THF (5 mL/g of title compound). The solution was distilled to a concentration of 10 mL/g. Product may cake on reactor wall during the distillation. The mixture was cooled to 25° C. Hexanes (5 mL/g of title compound) was added and the mixture was held for 30 minutes. The mixture was filtered and the solid was dried on the filter. The reactor was rinsed with methanol to remove residual THF. The solid was returned to the reactor with methanol (20 mL/g of title compound). The mixture was heated to reflux and held over night. The mixture was cooled to 20° C. and held for 2 hours. The mixture was filtered. The solid was dried in a vacuum oven at 70° C. 162 g of purified title compound was obtained (85% yield). The NMR data of the title compound are as per Step 4.
Any mL/g amount indicated above is referred to grams of crude title compound.
PAPER
Transition Metal-Catalyzed Couplings in Process Chemistry (2013), 253-266
Book Title

Transition Metal-Catalyzed Couplings in Process Chemistry: Case Studies from the Pharmaceutical Industry
18. Development of Migita Couplings for the Manufacture of a 5-Lipoxygenase Inhibitor
Published Online: 19 JUL 2013
DOI: 10.1002/9783527658909.ch18
- 5-lipoxygenase inhibitor;
- isooctyl 3-mercaptopropionate;
- Migita couplings;
- one-pot process;
- triisopropylsilanethiol (TIPS-SH)
Summary
The biggest shortcoming of the medicinal chemistry route is the introduction of the sulfur source for the first of two Migita couplings. The authors felt that the initial Migita coupling was a better candidate for a kinetic study on the formation of impurity, as it was harder to maintain a constant concentration of active Pd for the second coupling with two sources of Pd/ligand in this step. As depicted in the mechanism of the Migita coupling, the catalytic cycle is composed of three steps: oxidative addition, transmetalation, and reductive elimination. This chapter develops a three-step, one-pot process for the synthesis of 5-lipoxygenase inhibitor via a sequence of two Migita couplings. This strategy employed cheap, odorless, and readily available isooctyl 3-mercaptopropionate as the sulfur source for the initial Migita coupling as a general alternative to the popular triisopropylsilanethiol (TIPS-SH) for the formation of diaryl thioethers.
PAPER

Brian Chekal, David Damon, Danny LaFrance, Kyle Leeman, Carlos Mojica, Andrew Palm, Michael St. Pierre, Janice Sieser, Karen Sutherland, Rajappa Vaidyanathan, John Van Alsten, Brian Vanderplas, Carrie Wager, Gerald Weisenburger, Gregory Withbroe, and Shu Yu
Publication Date (Web): July 17, 2015 (Article)
DOI: 10.1021/op500412a
A de novo three-step-one-pot process for the formation of PF-04191834 was developed. This methodology employed inexpensive, odorless, and readily available commodity chemical iso-octyl-3-mercaptopropionate as a sulfur source, which could be a general alternative to the popular TIPS-SH in the formation of diarylthioethers via Migita coupling. A kinetic study revealed that, at high temperature, reductive elimination could be the rate-limiting step in the catalytic cycle, which opens pathways for the generation of undesired impurities. By proper control of the reaction conditions, the desired API was synthesized in >70% crude yield and in 55% isolated yield after vigorous purifications. This process was successfully demonstrated on a 20 kg scale.
Pure API after drying under vacuum. Mp 173 °C.
1H NMR (400 MHz, DMSO-d6) 7.52 (2H, m), 7.48 (2H, m), 7.42 (2H, m), 7.35 (2H, m), 7.29 (2H, m), 7.07 (1H, br. s), 6.42 (1H, d, J = 1.8 Hz), 3.85 (3H, s), 3.74 (2H, dt, J = 11.7, 3.7 Hz), 3.47 (2H, br. t, J = 11.7 Hz), 2.41 (2H, br. d, J = 13.3 Hz), 1.80 (2H, m).
13C NMR (100.6 MHz, DMSO-d6) 174.6, 146.0, 141.9, 137.9, 136.0, 133.2, 130.1, 129.7, 129.4, 129.3, 128.6, 125.6, 105.9, 64.6, 47.8, 37.6, 33.9.
LCMS: found m/z 394.17 [M + H]+. Anal. Calcd for C22H23N3O2S: C, 67.15; H, 5.89; N, 10.68; S, 8.15. Found: C, 67.09; H, 5.93; N, 10.69; S, 8.16.
After pd removal
Mp 173 °C.
1H NMR (400 MHz, DMSO-d6) 7.52 (2H, m), 7.48 (2H, m), 7.42 (2H, m), 7.35 (2H, m), 7.29 (2H, m), 7.07 (1H, br. s), 6.42 (1H, d, J = 1.8 Hz), 3.85 (3H, s), 3.74 (2H, dt, J = 11.7, 3.7 Hz), 3.47 (2H, br. t, J = 11.7 Hz), 2.41 (2H, br. d, J = 13.3 Hz), 1.80 (2H, m).
13C NMR (100.6 MHz, DMSO-d6) 174.6, 146.0, 141.9, 137.9, 136.0, 133.2, 130.1, 129.7, 129.4, 129.3, 128.6, 125.6, 105.9, 64.6, 47.8, 37.6, 33.9.
LCMS: found m/z 394.17 [M + H]+. Anal. Calcd for C22H23N3O2S: C, 67.15; H, 5.89; N, 10.68; S, 8.15. Found: C, 67.09; H, 5.93; N, 10.69; S, 8.16.
| Patent | Submitted | Granted |
|---|---|---|
| Pyrazole Analogs [US7772269] | 2008-05-29 | 2010-08-10 |
| Pyrazole Derivatives as 5-LO-Inhibitors [US8097733] | 2009-09-10 | 2012-01-17 |
| NOVEL TREATMENT FOR AGE RELATED MACULAR DEGENERATION AND OCULAR ISCHEMIC DISEASE ASSOCIATED WITH COMPLEMENT ACTIVATION BY TARGETING 5-LIPOXYGENASE [US2011269807] | 2011-11-03 | |
| TREATMENT AND PREVENTION OF DISEASES MEDIATED BY MICROORGANISMS VIA DRUG-MEDIATED MANIPULATION OF THE EICOSANOID BALANCE [US2014171445] | 2012-08-02 | 2014-06-19 |
////////
c1c(cc(cc1)C2(C(=O)N)CCOCC2)Sc3ccc(cc3)c4ccnn4C or
CN1C(=CC=N1)C2=CC=C(C=C2)SC3=CC=CC(=C3)C4(CCOCC4)C(=O)N
Lixivaptan


Lixivaptan
CRTX-080; VPA-985; WAY-VPA-985
N-[3-chloro-4-(6,11-dihydropyrrolo[2,1-c][1,4]benzodiazepine-5-carbonyl)phenyl]-5-fluoro-2-methylbenzamide
- 5-fluoro-2-methyl-N-(4-(5H-pyrrolo(2,1-c)-(1,4)benzodiazepin-10-(11H)-ylcarbonyl)-3-chlorophenyl)benzamide
- N-4-(3-chloro-4-(5H-pyrrolo(2,1-C)(1,4)benzodiazepin-10(11H)-ylcarbonyl)phenyl)-5-fluoro-2-methylbenzamide
- N-[3-chloro-4-(5H-pyrrolo[2,1-c][1,4]benzodiazepin-10(11H)-ylcarbonyl)phenyl]-5-fluoro-2-methylBenzamide
- N-(3-CHLORO-4-{3,9-DIAZATRICYCLO[8.4.0.0(3),?]TETRADECA-1(10),4,6,11,13-PENTAENE
CAS 168079-32-1
MW 473.9,
| MF C27H21ClFN3O2 |
NDA Filing
A vasopressin (AVP) V2 antagonist potentially for treatment of heart failure and hyponatremia.
![]()
Lixivaptan (VPA-985) is a phase III pharmaceutical being developed by Cardiokine, Inc., a specialty pharmaceutical company based in Philadelphia, PA, focused on the development of pharmaceuticals for the treatment and prevention of cardiovascular diseases. Lixivaptan is, as of May 2010, in Phase III clinical trials involving patients with hyponatremia, including those with concomitant heart failure.[1] Hyponatremia is an electrolyte disturbance in which the sodium concentration in the serum is lower than normal. Lixivaptan may help some patients eliminate excess fluids while retaining electrolytes.
ChemistryLixivaptan is synthesized as follows:[2]


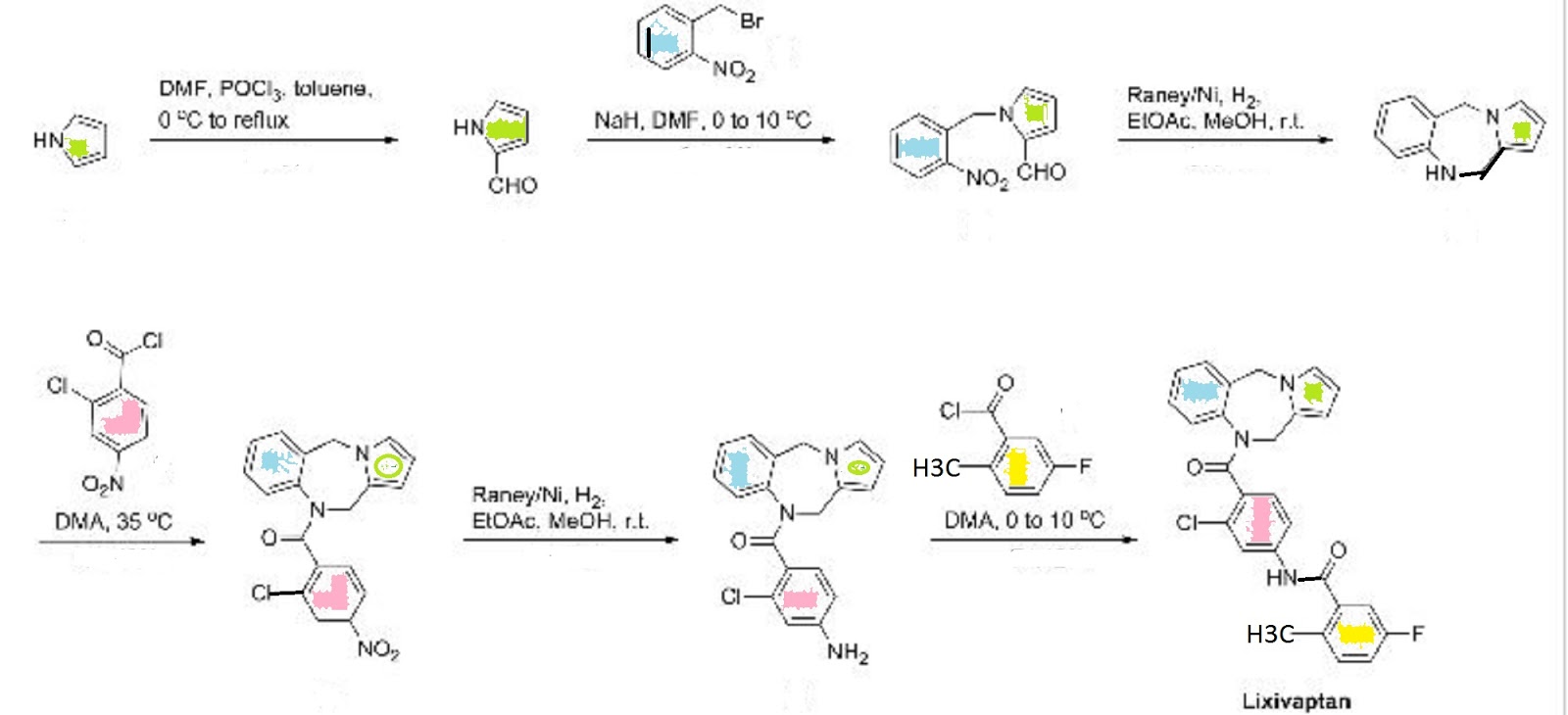
Mechanism of action
Lixivaptan is a potent, non-peptide, selective vasopressin 2 receptor antagonist. The oral capsule works by reducing the action of the hormone vasopressin that blocks fluid excretion. Lixivaptan acts by blocking vasopressin, an anti-diuretic hormone that causes the kidneys to retain water. When the body needs to remain hydrated under certain conditions, vasopressin can have protective effects. But an excess of vasopressin is counterproductive in a body retaining too much fluid. The drug shows promise in treating heart failure in patients with hyponatremia.
THE BALANCE study
In February 2008, Cardiokine and its worldwide partner, Biogen Idec, initiated THE BALANCE (Treatment of HyponatrEmia BAsed on LixivAptan in N Yha class III/IV Cardiac patient Evaluation) study. THE BALANCE study is a 650-patient Phase III, global, multi-center, randomized, placebo-controlled, double-blind, study of lixivaptan for hyponatremia in patients with heart failure. The primary objective is to evaluate the safety and effectiveness of lixivaptan, when compared to the placebo, in increasing serum sodium from baseline in heart failure patients with hyponatremia.[3][4]
Previous studies
In previous studies, lixivaptan improved blood sodium levels, lowered body weight and increased urine volume. Those studies suggest that lixivaptan may play an important role in treating hyponatremia and the signs and symptoms of water retention associated with heart failure, Syndrome of Inappropriate Anti-Diuretic Hormone(SIADH), and Liver Cirrhosis with Ascites (LCWA). In clinical trials involving patients with water volume overload, lixivaptan resulted in correction of hyponatremia together with marked aquaresis.
Vaptans
The vasopressin receptor antagonists, dubbed vaptans, target the vasopressin hormonal feedback system. Vasopressin, also called the anti-diuretic hormone or ADH, is an important part of regulation in the circulatory system and is integral to the balance of water in the body. As a fundamental part of hormonal control in the body, it is implicated in many different conditions. Vaptans can be administered orally or intravenously. They work by competing for the active sites on cells meant for vasopressin binding—in this way, the vasopressin is blocked from acting, which earns the title of vasopressing antagonists.
SYNTHESIS COMING………………..
JMC 1998, 41, 2442
US 5516774
CN103694240
Lixiputan (Lixivaptan, I) is pressurized by a Wyeth (wyeth) research and development of non-peptide hormone arginine oral selective V2 receptor antagonist, chemical name N- [3- chloro-4- (10, 11- dihydro -5H- pyrrolo [2,1-c] [1,4] benzodiazepine-10-yl carbonyl) phenyl] -5-fluoro-2- methylbenzamide. Clinical studies have shown that, compared with traditional diuretic, Lixiputan for the treatment of congestive heart failure (CHF), cirrhosis of hyponatremia and syndrome of inappropriate antidiuretic hormone secretion disorders (SIADH) patients, its in increase free water clearance without affecting renal sodium discharge, it will not activate the neuroendocrine system, and has a high safety and tolerability. Lixiputan V2 receptor selectivity higher than in May 2009 the FDA approved tolvaptan, Phase III clinical studies for the treatment of hyponatremia have been completed in the United States, in the pre-registration stage.
Document (Journalof medicinal chemistry, 1998,41 (14):. 2442-2444) reported Lixiputan there are two synthetic routes, one route to 10,11-dihydro -5H- pyrrolo [2, ι-c] [1,4] benzodiazepine (2) as raw materials, in turn with 2-chloro-4-nitrobenzoyl and 5-fluoro-2-methylbenzoyl docking, to obtain I; the second is the first line of 2-chloro-4-amino benzoic acid methyl ester (5) and 5-fluoro-2-methylbenzoyl chloride (7) butt, by hydrolysis, acylation reaction of 2-chloro-like -4 – [(5-fluoro-2-methylbenzoyl) amino] benzoyl chloride (10), and then with 2 reaction of I. 2 As the raw material is expensive, Route One to two as the starting material, the multi-step reaction, its low efficiency, high cost of production. Therefore, this study reference line two, 2-chloro-4-nitro-benzoic acid (3) as the starting material, by esterification, hydrogenation, acylation, hydrolysis, chloride, and so the reaction of 10; 10 and then with 2 After acylation reaction of N- I. I synthetic route follows.

The chemical structure:

formula = C27H21ClFN3O2
Molecular Weight: 473.93
The method for producing foreign products have been reported, such as the literature Journal of medicinalchemistry, 1998,41 (14):. 2442-2444 and US, 5516774 [P], 1996-5-14. Currently, Lixiputan (Iixivaptan) abroad in Phase III clinical studies, there are good prospects for development, given the value of the pharmaceutical compounds, high purity, with a very determined and reproducible crystalline compounds are important .
The present inventors have repeated the document US, 5,516,774 Lixiputan method of purity, obtained was 97.5%, mpl91-195 ° C, by the study of a plurality of batches, the melting point of the same, by a powder X- ray diffraction pattern See
preparation of Lixiputan solvate Lixiputan, by two synthetic methods. As literature Journalof medicinal chemistry, 1998, 41 (14):. 2442-2444 and US, 5516774 [P],
The method reported in [0026] 1996-5-14. Preclude the use of the route of the present invention is represented by the following reaction:

synthetic Lixiputan by proton nuclear magnetic resonance spectroscopy (1H-NMRX mass spectrometry (MS), infrared spectroscopy (IR) and other confirmed its chemical structure (see Figure 3 MS). Test equipment for nuclear magnetic resonance Bruker AV400 meter, gas generation agent for CambridgeIsotope Laboratories Company DMS0_d6.
ES1-HRMS (m / z): 474.17 [M + H] + NMR (400MHz, DMS0_d6) δ: 10.49 (s, 1H), 7.84 (s, 1H), 7.40 (d, J = 6.8Hz, 2H), 7.33 (d, J = 8.4Hz, 3H), 7.23 (t, J = 8.4Hz, 1H), 7.13 (t, J = 5.6Hz, 2H), 7.05 (d, J = 6.8Hz, 1H) , 6.82 (s, 1H), 5.94 (d, J = 32Hz, 2H), 5.23 (br, 4H), 2.30 (s, 3H).
The product obtained, with a purity of 97.5%, mp 191-195 ° C.

Lixiputan solvates H NMR spectrum, δ: 1.147-1.182 “3” methyl hydrogens; δ: 1.971-1.977 for the “I” position methyl hydrogen; δ: 3.994-4.047 “2” position methylene hydrogen.
CN104059070
CN104140429
IN 2012 MUM 03309


References
- 1 Lixivaptan: a novel vasopressin receptor antagonist
- 2 Albright, J. D.; Reich, M. F.; Delos Santos, E. G.; Dusza, J. P.; Sum, F. W.; Venkatesan, A. M.; Coupet, J.; Chan, P. S.; Ru, X.; Mazandarani, H.; Bailey, T. (1998). “5-Fluoro-2-methyl-N-[4-(5H-pyrrolo[2,1-c]- [1,4]benzodiazepin-10(11H)-ylcarbonyl)-3- chlorophenyl]benzamide (VPA-985): An Orally Active Arginine Vasopressin Antagonist with Selectivity for V2Receptors”. Journal of Medicinal Chemistry 41 (14): 2442–2444. doi:10.1021/jm980179c. PMID 9651149.
- 3 http://www.clinicaltrials.gov/ct2/show/NCT00578695?term=Lixivaptan+Heart+Failure&rank=3
- 4 http://onlinelibrary.wiley.com/doi/10.1111/j.1752-8062.2010.00217.x/pdf
| Patent | Submitted | Granted |
|---|---|---|
| AURIS FORMULATIONS FOR TREATING OTIC DISEASES AND CONDITIONS [US2009306225] | 2009-12-10 | |
| Vasopressin antagonist and diuretic combination [US6656931] | 2003-04-10 | 2003-12-02 |
| Pharmaceutical carrier formulation [US6437006] | 2002-08-20 | |
| Vasopressin antagonist formulation and process [US6352718] | 2002-03-05 | |
| Nonpeptide agonists and antagonists of vasopressin receptors [US2002128208] | 2002-09-12 | |
| Tricyclic diazepine vasopressin antagonists and oxytocin antagonists [US5968930] | 1999-10-19 | |
| Tricyclic diazepine vasopressin antagonists and oxytocin antagonists [US5968937] | 1999-10-19 | |
| Tricyclic diazepine vasopressin antagonists and oxytocin antagonists [US5516774] | 1996-05-14 | |
| Tricyclic diazepine vasopressin antagonists and oxytocin antagonists [US5733905] | 1998-03-31 | |
| Tricyclic diazepine vasopressin antagonists and oxytocin antagonists [US5736540] | 1998-04-07 |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[3-chloro-4-(6,11-dihydropyrrolo[2,1-c][1,4]benzodiazepine-5-carbonyl)phenyl]-5-fluoro-2-methylbenzamide
|
|
| Identifiers | |
| CAS Number | 168079-32-1 |
| ATC code | None |
| PubChem | CID: 172997 |
| IUPHAR/BPS | 2238 |
| ChemSpider | 151067 |
| UNII | 8F5X4B082E |
| ChEMBL | CHEMBL49429 |
| Chemical data | |
| Formula | C27H21ClFN3O2 |
| Molecular mass | 473.926 g/mol |
| CN102020609A * | Sep 17, 2009 | Apr 20, 2011 | 北京本草天源药物研究院 | Tolvapta crystal or amorphous substance and preparation method thereof |
| CN102918038A * | Mar 31, 2011 | Feb 6, 2013 | 万梯雅有限公司 | New polymorph |
| US5516774 * | Jun 13, 1994 | May 14, 1996 | American Cyanamid Company | Tricyclic diazepine vasopressin antagonists and oxytocin antagonists |
| 1 | * | 吕扬 等: “《晶型药物》”, 31 October 2009, article “”第七章 晶型药物的研究方法”“, pages: 136-139 |
//////////Lixivaptan, CRTX-080, VPA-985, WAY-VPA-985
CC1=C(C=C(C=C1)F)C(=O)NC2=CC(=C(C=C2)C(=O)N3CC4=CC=CN4CC5=CC=CC=C53)Cl
CC1=C(C=C(C=C1)F)C(=O)NC2=CC(=C(C=C2)C(=O)N3CC4=CC=CN4CC5=CC=CC=C53)Cl
Uridine triacetate, ウリジントリアセタート FDA approves first emergency treatment for overdose of certain types of chemotherapy

12/11/2015 12:05 PM EST
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
UPDATED IN SEPT 2016…………..
Uridine triacetate
Uridine, 5-hydroxy-, 2′,3′,5′-triacetate
Orphan drug
FAST TRACK
MF C15H18N2O9, MW 370.314
|
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
|
Vistogard [Trade name]
Xuriden [Trade name]
(2R,3R,4R,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl diacetate
Wellstat (Originator)
PN-401; RG-2133; TAU
MOA:Pyrimidine analog
Indication:Hereditary orotic aciduria; Chemotherapy induced poisoning
To treat patients with hereditary orotic aciduria
| Drug Name(s) | XURIDEN |
| FDA Application No. | (NDA) 208169 |
| Active Ingredient(s) | URIDINE TRIACETATE |
| Company | WELLSTAT THERAP |
| Original Approval or Tentative Approval Date | September 4, 2015 |
FDA APPROVAL SUMMARY
Chemotherapy induced poisoning, VISTOGARD, FDA 2015-12-11
Hereditary orotic aciduria, Xuriden, FIRST APPROVAL, 2015-09-04
|
|||||||
| External Identifiers |
|
|---|
Uridine triacetate is a drug used in the treatment of hereditary orotic aciduria[1] and to treat patients following an overdose ofchemotherapy drugs 5-fluorouracil or capecitabine, or in patients exhibiting early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of 5-fluorouracil or capecitabine administration.[2][3]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics and it is marketed in USA by BTG. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine triacetate is a prodrug of uridine.[4]
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTP incorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [3]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidine metabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
Marketed as the product Xuriden (FDA), uridine triacetate is indicated for the treatment of hereditary orotic aciduria. Marketed as the product Vistogard (FDA), uridine triacetate is indicated for the emergency treatment of adult and pediatric patients in the following situations: following a fluorouracil or capecitabine overdose regardless of the presence of symptoms; or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration.

Uridine Triacetate was approved by the U.S. Food and Drug Administration (FDA) on Sep 4, 2015. It was developed by Wellstat Therapeutics, then marketed as Xuriden® by Wellstat Therapeutics in US. Then it was also approved by FDA for overdose of certain types of chemotherapy on Dec 11, 2015 and marketed as Vistogard®.
Uridine Triacetate is a prodrug of the nucleoside uridine used to treat hereditary orotic aciduria. Hereditary orotic aciduria is inherited from a recessive gene. The disease is due to a defective or deficient enzyme, which results in the body being unable to normally synthesize uridine, a necessary component of ribonucleic acid (RNA). Signs and symptoms of the disease include blood abnormalities (anemia, decreased white blood cell count, decreased neutrophil count), urinary tract obstruction due to the formation of orotic acid crystals in the urinary tract, failure to thrive, and developmental delays.
Xuriden® is approved as oral granules that can be mixed with food or in milk or infant formula, and is administered once daily. The starting dosage is 60 mg/kg once daily; the dose may be increased to 120 mg/kg (not to exceed 8 grams) once daily for insufficient efficacy.
Mechanism Of Action
Uridine triacetate is an acetylated form of uridine. Following oral administration, uridine triacetate is deacetylated by nonspecific esterases present throughout the body, yielding uridine in the circulation (Figure 1).
Figure 1: Uridine Triacetate Conversion to Uridine

URIDEN provides uridine in the systemic circulation of patients with hereditary orotic aciduria who cannot synthesize adequate quantities of uridine due to a genetic defect in uridine nucleotide synthesis.
Uridine triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578] such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Uridine triacetate is also used for replacement therapy in the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase (UMPS) deficiency. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria.
Route 1


Reference:1. J. Am. Chem. Soc. 1953, 75, 2017-2019.
2. Angew. Chem. internat. Edit. 1971, 10, 75.
3. US3116282.
PATENT
Production Example 1

5.6 g of uracil and 0.1 g of ammonium sulfate were dissolved in 22.4 ml of 1,1,1,3,3,3-hexamethyldisilazane and reacted at 120° C. for 2.5 hours. After the completion of the reaction, the reaction mixture was distilled to give 11.8 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine. 1H-NMR (400 MHz, in C2D6CO): δ=0.29 (s, 9H), 0.31 (s, 9H), 6.35 (d, J=5.6 Hz, 1H), 8.19 (d, J=5.5Hz, 1H)
Referential Example 11.21 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 1.15 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.8 ml of acetonitrile and cooled to 5° C. Next, 0.94 g of SnCl4 was added dropwise thereinto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted for 3 hours. The reaction mixture was analyzed by HPLC. Thus, β-uridine triacetate was obtained with a reaction yield of 83%.
Example 1

0.93 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 0.92 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.7 ml of acetonitrile and cooled to 4° C. Then 0.49 g of FeCl3 was added thereto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted. The reaction was monitored by HPLC. After the completion of the reaction, the reaction mixture was added dropwise at 4° C. into a cold aqueous solution of sodium hydrogencarbonate which had been preliminarily prepared. After filtering off the catalyst residue, the filtrate was separated and the aqueous layer was extracted with 20 ml portions of ethyl acetate thrice. The organic layers were combined, washed with a saturated aqueous solution of sodium chloride and dried over sodium sulfate. After distilling off the solvent, 1.2 g (purity 80%) of the target compound was obtained as a viscous white solid.
Namely, the target compound could be obtained at a yield comparable to REFERNTIAL EXAMPLE 1 wherein SnCl4 was employed as the catalyst. 1H-NMR (400 MHz, in CDCl3): δ=2.11 (s, 3H), 2.14 (s, 3H), 2.15 (s, 3H), 4.35 (m, 3H), 5.33 (m, 2H), 5.79 (d, J=8.2 Hz, 1H), 6.04 (d, J=4.9 Hz, 1H), 7.39 (d, J=8.2 Hz, 1H)

CLIP
12/11/2015 12:05 PM EST
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
CLIP
With support from Almac, Wellstat delivers for a rare disease.
Proximity of API and finished drug development helps uridine triacetate to market for two indications
By Rick Mullin
“The initial contact was a cold call by Almac in 2010 or 2011,” recalls Mike Bamat, senior vice president of R&D at Wellstat Therapeutics, a small drug company in Gaithersburg, Md. “There were probably a couple of calls. It was one of those things where timing is everything.”
Almac, a Craigavon, Northern Ireland-based pharmaceutical services company, was looking to get in on Wellstat’s development of uridine triacetate, a synthetic pyrimidine analog, as an antidote for fluorouracil and capecitabine toxicity and overdose in cancer patients receiving those chemotherapies. And the calls, which Almac records indicate followed some communication between the companies, happened to come just when Wellstat was looking to change service partners as it moved toward commercial development of the drug.

Uridine triacetate
Discovery: Wellstat Therapeutic’s research on the therapeutic potential of exogenous uridine leads to a determination that uridine triacetate is a safe means of delivering the agent
Applications: Treatment of hereditary orotic aciduria (HOA), an extremely rare disease in which the body does not produce uridine, causing overproduction of orotic acid; emergency treatment of toxic reaction to or overdose of the cancer treatments fluorouracil and capecitabine
Methods of action: Treating HOA, uridine triacetate restores intracellular nucleotide concentrations, normalizing orotic acid production; as a chemotherapy antidote, it increases intracellular levels of uridine to dilute fluorouracil and capecitabine
Years in development: Since 2008 for chemotherapy antidote, and 2013 for HOA
Approved: Xuriden for HOA, Sept. 4, 2015; Vistogard for chemotherapy antidote, Dec. 11, 2015
The job went to Almac, as did work that sprang up as the result of another phone call to Wellstat—this one from the U.S. Food & Drug Administration.
As Bamat explains, uridine triacetate caught FDA’s attention regarding another potential indication—an extremely rare and life-threatening disease called hereditary orotic aciduria, or HOA. A consequence of the body’s inability to produce uridine, a necessary component of ribonucleic acid, HOA can manifest in a range of symptoms including blood abnormalities, developmental delays, and urinary tract obstruction caused by overproduction of orotic acid. There have been 20 reported cases of HOA since the 1950s. Only four cases are currently known in the U.S., Bamat says, and likely fewer than 20 in the world.
Wellstat landed approvals for Xuriden, the HOA treatment, in September of last year and Vistogard, the chemotherapy antidote, in December.
The story of Xuriden centers on a raft of FDA incentives for super-rare diseases that enabled Wellstat to move forward on an expedited application for a drug that will never be made in any great volume. But bringing Xuriden and Vistogard to market may also be viewed as the story of a drug discovery firm becoming a commercial enterprise thanks to its partnership with a service provider.
As Wellstat began late-stage development of the chemotherapy antidote, its research partner at the time, QS Pharma, was acquired by the service firm WIL Research. The look and feel of the partnership changed, according to Bamat.
“We kind of lost the small, easy-to-work-with relationship we had with them,” he says. Wellstat also needed support on development and manufacturing of a finished drug product composed of granules delivered in packets or sachets. The drug is administered orally, usually sprinkled on food such as applesauce or yogurt.
Almac was deemed a good fit because of its experience with developing drugs in granule form for “sachet presentation,” a packaging method more common in Europe than in the U.S. The Northern Ireland firm’s ability to develop and manufacture the active pharmaceutical ingredient (API) and the drug product in one location—at its headquarters—would also prove to be a significant advantage.
The distance between Gaithersburg and Craigavon, however, was a concern, according to Bamat. “We debated it. Especially those of us who knew we would be going there,” he says. “We couldn’t just jump in a car and go. But we looked at a variety of things, including cost and value, and it was all very positive at Almac.”
According to David Downey, vice president of commercial operations at Almac, bringing Wellstat’s work on uridine triacetate to commercial production posed several challenges, the first being to secure supply of uridine starting material, which is extracted from sugar beets by Euticals, an Italian firm. Next was developing a method to control particle size in both the API and the finished product. Almac also had to validate process equipment as it scaled up production.
“Uridine triacetate is Wellstat’s first commercial product,” Downey says. “So we were provided with a process more fit for development than for commercial production.”
The basic formulation of a granule drug product is simple, according to Downey: The API and excipient are mixed in a dry blender. The challenge is developing an analytical regimen to assure the granules are blended uniformly. Meeting the challenge required a high level of coordination between API and drug product process development.
“Wellstat needed a partner that could support them from the API to the drug product,” Downey says. The physical proximity between the Almac facilities in Craigavon conducting API and drug product work was a key advantage, he claims.

Uridine triacetate is formulated into granules presented in packets and sprinkled on food.
Credit: Wellstat Therapeutics
“If you listen to our business development people, you’ll hear them use the term, ‘crossing car parks as opposed to crossing oceans,’ ” Downey says, explaining that many competitors who offer API and finished drug services run these operations thousands of kilometers apart from each other, sometimes on different continents.
Before it signed on with Almac, Wellstat had been working with uridine triacetate for about 10 years. Its focus on developing the antidote drug started in 2008. Branching into the HOA treatment, however, upped the stakes.
Clinical study development for an HOA therapy was expedited via a full house of regulatory incentives from FDA, according to Bamat. “We had orphan drug designation, rare pediatric designation, breakthrough therapy designation, and priority review,” he says. “So they really went all out in helping us develop this.”
Although Wellstat was interested in developing a life saving drug for children, it was concerned about paying for it, given the tiny market. “At that time, the rare pediatric disease priority review voucher program was just on the radar,” Bamat says. “FDA said, ‘Consider this new program. Maybe it’s a way that at some risk you could recoup some of your costs.’ We looked at it and were willing to take the risk.”
It paid off. Wellstat was able to sell its priority review voucher—which entitles a company that brings a rare pediatric drug to market to receive expedited review of a subsequent drug—to AstraZeneca last year for an undisclosed amount. Other vouchers sold in 2015 brought high sums, including $350 million for one that AbbVie bought from United Therapeutics in August.
Bamat says Wellstat is not likely to change focus after its success with uridine triacetate. It continues to investigate new indications for the compound and will likely work with Almac on anything going into commercial development.
He emphasizes the importance of maintaining an effective working relationship with an outsourcing partner. “My main consideration is that these are people we can really work with on a day-to-day, week-to-week basis,” Bamat says. “Will the communication be good? Will they be honest and transparent with us, and will we be the same for them? That was a key factor, and we felt it was a plus with Almac.”
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2-3 hours |
| Biological half-life | 2-2.5 hours |
| Excretion | Renal |
| Identifiers | |
| DrugBank | DB09144 |
| Chemical data | |
| Formula | C15H18Cl0N2O9S0 |
| Molar mass | 370.31 g·mol−1 |
References
- HIGHLIGHTS OF PRESCRIBING INFORMATION OF XURIDEN
- Jump up^ BTG Announces FDA Approval of VISTOGARD® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine
- Jump up^ “FDA Approved Drugs:Uridine Triacetate”. FDA. 2015-12-11. Retrieved 2016-04-29.
- “Uridine triacetate”. DrugBank.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7807654 | 2010-10-05 | Compositions and methods for treatment of mitochondrial diseases |
| US2010222296 | 2010-09-02 | PYRIMIDINES, SUCH AS URIDINE, IN TREATMENTS FOR PATIENTS WITH BIPOLAR DISORDER |
| US7737128 | 2010-06-15 | Pyrimidines, such as uridine, in treatments for patients with bipolar disorder |
| US2010098678 | 2010-04-22 | Methods of Treatment of Mitochondrial Disorders |
| US2010041620 | 2010-02-18 | METHODS FOR IMPROVING FRONTAL BRAIN BIOENERGETIC METABOLISM |
| US2010041621 | 2010-02-18 | METHODS AND COMPOSITIONS FOR IMPROVING COGNITIVE PERFORMANCE |
| US7582619 | 2009-09-01 | Compositions and methods for treatment of mitochondrial diseases |
| US2008226684 | 2008-09-18 | METHOD AND PROCESS FOR THE PRODUCTION OF MULTI-COATED RECOGNITIVE AND RELEASING SYSTEMS |
| US7105498 | 2006-09-12 | Acylated uridine and cytidine and uses thereof |
| US6956028 | 2005-10-18 | Compositions and methods for treatment of mitochondrial diseases |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015307542 | 2015-10-29 | MODIFIED NUCLEIC ACID MOLECULES AND USES THEREOF |
| US2015167017 | 2015-06-18 | ALTERNATIVE NUCLEIC ACID MOLECULES AND USES THEREOF |
| US8821899 | 2014-09-02 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8771713 | 2014-07-08 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8741316 | 2014-06-03 | Highly porous, recognitive polymer systems |
| US2012294869 | 2012-11-22 | Methods for Treating Fatty Liver Disease |
| US2012078529 | 2012-03-29 | DETERMINING THE SEVERITY OF 5-FLUOROURACIL OVERDOSE |
| US8067392 | 2011-11-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7915233 | 2011-03-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7884202 | 2011-02-08 | Nucleobase Having Perfluoroalkyl Group and Process for Producing the Same |
Uridine triacetate
- Molecular FormulaC15H18N2O9
- Average mass370.311 Da
ウリジントリアセタート
[(2R,3R,4R,5R)-3,4-diacetyloxy-5-(2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl acetate
Uridine, 2′,3′,5′-triacetate
uridini triacetas
Vistogard [Trade name]
Xuriden [Trade name]
(2R,3R,4R,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl diacetate
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
FDA APPROVED2015/9/4 . AS Xuriden
Uridine triacetate (INN),[1] formerly known as vistonuridine, is an orally active tri-acetylated prodrug of uridine[2] used:
- in the treatment of hereditary orotic aciduria (brand name Xuriden /ˈzʊərədɛn/ ZOOR-ə-den);[3]
- to treat patients following an overdose of chemotherapy drugs 5-fluorouracil (5-FU) or capecitabine regardless of the presence of symptoms, or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration (brand name Vistogard).[4][5][6]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine Triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. Uridine, a pyrimidine nucleotide, has been used in a variety of diseases including depressive disorders and inherited myopathies. (NCI04)
Pharmacology from NCIt
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTPincorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidinemetabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
from DrugBank
Uridine triacetate is an acetate ester that is uracil in which the three hydroxy hydrogens are replaced by acetate group. A prodrug for uridine, it is used for the treatment of hereditary orotic aciduria and for management of fluorouracil toxicity. It has a role as a prodrug, a neuroprotective agent and an orphan drug. It is a member of uridines and an acetate ester.
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 65” (PDF). World Health Organization. p. 92. Retrieved 12 March 2017.
- ^ “Uridine triacetate — DrugBank Page”. 12 March 2017.
- ^ “Xuriden (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “Vistogard (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “BTG Announces FDA Approval of Vistogard® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine”. BTG International Ltd. 11 December 2015. Retrieved 12 March 2017.
- ^ “Approved Drugs — Uridine Triacetate”. U.S. Food and Drug Administration. Retrieved 12 March 2017.
External links
Patents
- US7776838
- US5968914
- US6258795
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7776838 |
| Expiration | Aug 17, 2027 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
from FDA Orange Book
| FDA Orange Book Patents: 2 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6258795 |
| Expiration | Jul 10, 2019 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
from FDA Orange Book
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2–3 hours |
| Elimination half-life | 2–2.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.021.710 |
| Chemical and physical data | |
| Formula | C15H18N2O9 |
| Molar mass | 370.31 g·mol−1 |
| 3D model (JSmol) | |
////////////Uridine triacetate, ウリジントリアセタート , FDA 2015, breakthrough therapy designation ,
//////////174105-38-8, Priority review drug , Orphan drug, FDA 2015, Vistogard, uridine triacetate, fast track designations, PN-401, RG-2133, TAU, XURIDEN
|
CC(=O)OC[C@H]1O[C@H]([C@H](OC(C)=O)[C@@H]1OC(C)=O)N1C=CC(=O)NC1=O
|