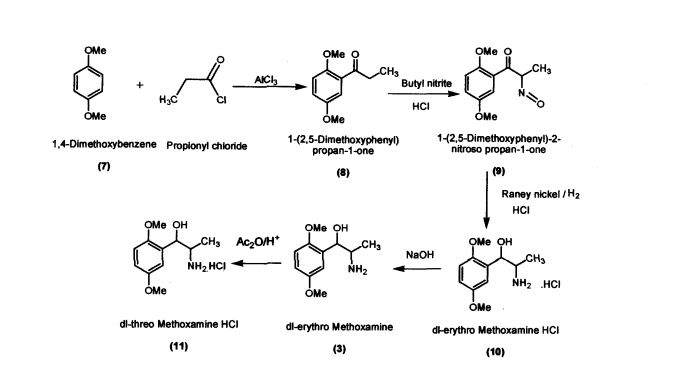
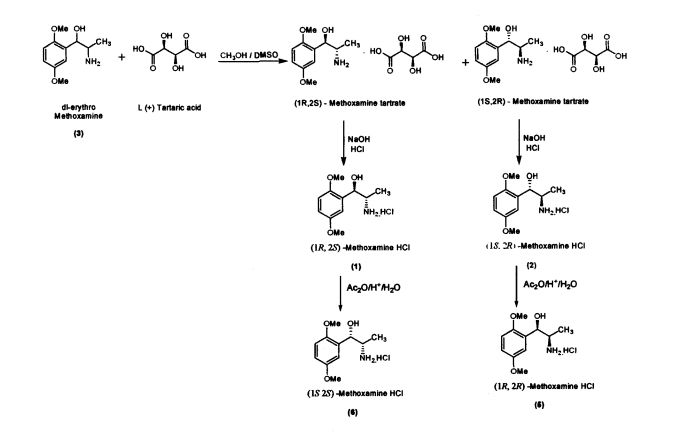
 .
.
PIC CREDIT.BETHANY HALFORD




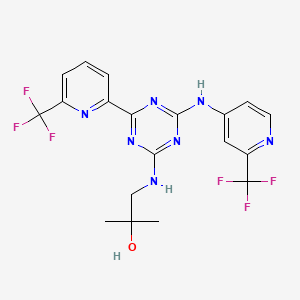
Name: AMG-337(AMG337; AMG 337)
Cas 1173699-31-4
Formula: C23H22FN7O3
M.Wt: 463.46
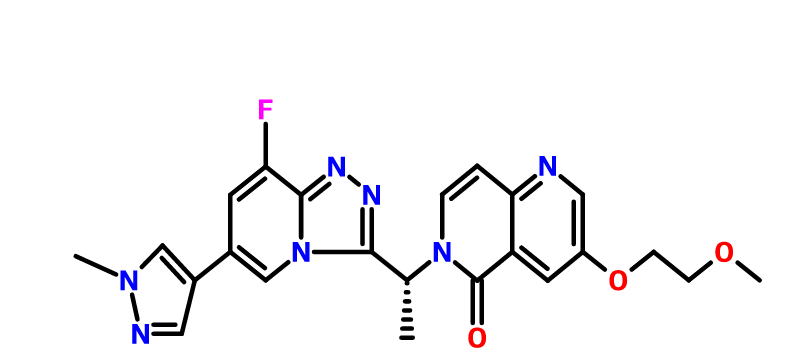
Chemical Name: 6-[(1R)-1-[8-fluoro-6-(1-methylpyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl]ethyl]-3-(2-methoxyethoxy)-5-methylidene-1,6-naphthyridine
(R)-6-(1-(8-fluoro-6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-1,6-naphthyridin-5(6H)-one
(R)-6-(1-(8-Fluoro-6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-1,6-naphthyridin-5(6H)-one
6-{ (lR)-l-[8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)[l,2,4]triazolo[4,3-a]pyridin-3-yl]ethyl}-3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one (“Compound M”),
PHASE 2 CANCER OF ESOPHAGUS
AMG-337 is a potent and highly selective small molecule ATP-competitive MET kinase inhibitor. AMG 337 inhibits MET kinase activity with an IC50 of < 5nM in enzymatic assays.
IC50 value: < 5nM [1]
Target: MET
in vitro: AMG-337 demonstrates exquisite selectivity for MET when profiled against a diverse panel of over 400 protein and lipid kinases in a competitive binding assay. In cellular assays, AMG 337 inhibits HGF-dependent MET phosphorylation with an IC50 of < 10 nM. [1] AMG 337 is a selective inhibitor of Met, which inhibits multiple mechanisms of Met activation. [2]
in vivo: AMG-337 demonstrates robust activity in MET-dependent cancer models. Oral administration of AMG 337 results in robust dose-dependent anti-tumor efficacy in MET amplified gastric cancer xenograft models, with inhibition of tumor growth consistent with the pharmacodynamic modulation of MET signaling
AMG 337 is a potent and highly selective small molecule ATP-competitive MET kinase inhibitor that demonstrates robust activity in MET-dependent cancer models. In enzymatic assays, AMG 337 inhibited MET kinase activity with an IC50 less than 5 nM. AMG 337 demonstrated exquisite selectivity for MET when profiled against a diverse panel of over 400 protein and lipid kinases in a competitive binding assay. In cellular assays, AMG 337 inhibited HGF-dependent MET phosphorylation with an IC50 of less than 10 nM [1].
AMG 337 was profiled in cell viability assays using a diverse panel of over 200 cancer cell lines where on treatment with AMG 337 affected the viability of only two gastric cancer cell lines (SNU-5 and Hs746T), both of which harbor amplification of the MET gene. The AMG 337 IC50 in the two sensitive cell lines was less than 50 nM, and greater than 10 µM in all other tested cell lines.
The receptor tyrosine kinase c-Met and its natural ligand, hepatocyte growth factor (HGF), are involved in cell proliferation, migration, and invasion and are essential for normal embryonic development. Deregulation of c-Met/HGF signaling can lead to tumorigenesis and metastasis and has been implicated in a variety of cancers. Several mechanisms lead to deregulation, including overexpression of c-Met and/or HGF, amplification of the MET gene, or activating mutations of c-Met, all of which have been found in human cancers.
AMG 337 is a potent and highly selective inhibitor of wild-type and some mutant forms of MET. In a competitive binding assay conducted on 402 human kinases, AMG 337 bound only to MET. In a cell viability study, the only cell lines that responded to an AMG 337 analog were gastric cancer cells harboring MET gene amplification. None of the other cell lines were sensitive to the AMG 337 analog and none harbored MET gene amplification. In secondary pharmacology assays with transporters, enzymes, ion channels, and receptors, binding to the adenosine transporter was the only activity inhibited.
In vivo, oral administration of AMG 337 resulted in robust dose-dependent anti-tumor efficacy in MET amplified gastric cancer xenograft models, with inhibition of tumor growth consistent with the pharmacodynamic modulation of MET signaling. Further studies in an expanded panel of additional cancer cell lines derived from gastric, NSCLC, and esophageal cancer confirmed that the in-vitro anti-proliferative activity of AMG 337 correlated with amplification of MET. In those cell lines, treatment with AMG 337 inhibited downstream PI3K and MAPK signaling pathways, which translated into growth arrest as evidenced by an accumulation of cells in the G1 phase of the cell cycle, a concomitant reduction in DNA synthesis, and the induction of apoptosis [1].
In a small subset of patients with MET-amplified gastrointestinal (GI) tumors, monotherapy with the investigational agent AMG 337 produced a “dramatic” response. Of the 13 patients with MET-amplified gastric and esophageal cancers, eight experienced a response. The overall response rate in this group of patients was 62%. Response was rapid, with time to response being 4 weeks in most cases. Patients achieved tumor shrinkage and symptomatic improvement. One patient achieved a complete response and is still on treatment at 155 weeks; the others achieved partial responses or stable disease. This has led to further trials, including Phase II trials MET amplified gastric/esophageal adenocarcinoma or other solid tumors.
PAPER
Discovery of (R)-6-(1-(8-Fluoro-6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-1,6-naphthyridin-5(6H)-one (AMG 337), a Potent and Selective Inhibitor of MET with High Unbound Target Coverage and Robust In Vivo Antitumor Activity.
Boezio, A.A., Copeland, K.W., Rex, K., K Albrecht, B., Bauer, D., Bellon, S.F., Boezio, C., Broome, M.A., Choquette, D., Coxon, A., Dussault, I., Hirai, S., Lewis, R., Lin, M.H., Lohman, J., Liu, J., Peterson, E.A., Potashman, M., Shimanovich, R., Teffera, Y., Whittington, D.A., Vaida, K.R., Harmange, J.C.
(2016) J.Med.Chem. 59: 2328-2342
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01716
Deregulation of the receptor tyrosine kinase mesenchymal epithelial transition factor (MET) has been implicated in several human cancers and is an attractive target for small molecule drug discovery. Herein, we report the discovery of compound 23 (AMG 337), which demonstrates nanomolar inhibition of MET kinase activity, desirable preclinical pharmacokinetics, significant inhibition of MET phosphorylation in mice, and robust tumor growth inhibition in a MET-dependent mouse efficacy model.
(R)-6-(1-(8-Fluoro-6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-1,6-naphthyridin-5(6H)-one (23)
Step 1: Coupling
9c and
13c in MeCN for 30 min at room temperature resulted in 86% yield. LRMS (ESI):
m/
z (M + H) 482.2. Step 2: THF for 50 min at room temperature resulted in 48% yield. The racemate was purified by supercritical fluid chromatography (SFC) by repeating 0.75 mL injections of a 30 mg/mL solution onto a Chiralpak AS-H, 2 cm × 15 cm (i.d. × length) column, eluting with 20%
i-PrOH and 80% CO
2 at a flow rate of 50 mL/min to provide 120 mg peak 1 (
23) with >99% ee and 150 mg of peak 2 (
ent-23) with >99% ee.
(29) 1H NMR (400 MHz, Chloroform-
d): δ 8.72 (d,
J = 2.93 Hz, 1H), 8.31 (d,
J = 0.78 Hz, 1H), 8.15 (d,
J = 2.84 Hz, 1H), 7.72 (s, 1H), 7.61 (s, 1H), 7.42 (d,
J = 7.82 Hz, 1H), 7.09 (dd,
J = 0.73, 10.61 Hz, 1H), 7.05 (q,
J= 7.00 Hz, 1H), 6.82 (d,
J = 7.82 Hz, 1H), 4.26–4.37 (m, 2H), 3.97 (s, 3H), 3.80–3.88 (m,
J = 3.80, 5.10 Hz, 2H), 3.49 (s, 3H), 2.15 (d,
J = 7.14 Hz, 3H). HRMS (ESI):
m/
z (M + H) calcd, 464.1859; found, 464.1841. The solid was recrystallized in EtOH followed by the addition of H
2O to form crystalline free base monohydrate form I with a dehydration event at 40–55 °C followed by a melt at 151–153 °C. The solid could also be recrystallized in EtOH under anhydrous conditions to form crystalline anhydrous free base form I with a melting point of 151–153 °C.
PATENT
WO 2009091374
http://www.google.com/patents/WO2009091374A2?cl=en
Example 515
(SV6-(l-f8-fluoro-6-(3-methvIisoxazol-5-vn-|l,2,41triazoIo[4,3-a1pyridin-3-vncthvn-3-(f2- methoxyethoxy)methv.)-l,6-naphthyridin-5(6HVone Synthesized in the same general manner as that previously described for example 509 using General Method N. Chiral separation by preparative SFC (Chiralpak® AD-H (20 x 150 mm, 5Dm), 25% MeOH, 75% CO2, 0.2% DEA; 100 bar system pressure; 75 mL/min; tr 4.75min). On the basis of previous crystallographic data and potency recorded for related compound in the same program, the absolute stereochemistry has been assigned to be the S enantiomer. M/Z – 465.2 [M+H], calc 464.16 for C23H2iFN6O4

Example 516 ri?)-6-ri-(8-fluoro-6-(l-methyl-lH-pyrazol-4-vn-H.2.41triazolo[4,3-alpyridin-3-yl)ethyl)- 3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one The title compound was synthesized using General Method N. Chiral separation by preparative SFC (Chiralpak® AS-H (20 x 150 mm, 5 Dm), 20% iPrOH, 80% CO2; 100 bar system pressure, 50 mL/min; tr 1.67 min). On the basis of previous crystallographic data and potency recorded for related compound in the same program, the absolute stereochemistry has been assigned to be the R enantiomer. M/Z = 464.2 [M+H], calc 463.18 for C23H22FN7O3. 1H NMR (400 MHz, CHLOROFORM-^ D ppm 2.15 (d, J=7.14 Hz, 3 H) 3.49 (s, 3 H) 3.80 – 3.90 (m, 2 H) 3.97 (s, 3 H) 4.27 – 4.39 (m, 2 H) 6.83 (d, J=7.73 Hz, 1 H) 7.00 – 7.13 (m, 2 H) 7.42 (d, J=7.82 Hz, 1 H) 7.61 (s, 1 H) 7.72 (s, 1 H) 8.15 (d, J=2.84 Hz, 1 H) 8.31 (s, 1 H) 8.72 (d, J=3.03 Hz, 1 H).
PATENT
WO 2015161152
6-{ (lR)-l-[8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)[l,2,4]triazolo[4,3-a]pyridin-3-yl]ethyl}-3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one (“Compound M”), which is a selective inhibitor of the c-Met receptor, and useful in the treatment, prevention, or amelioration of cancer:
PATENT
https://www.google.com/patents/WO2014210042A2?cl=en
The overall scheme for the preparation of Compound A is shown below. The optical purity of Compound A is controlled during the synthetic process by both the quality of the incoming starting materials and the specific reagents used for the transformations. Chiral purity is preserved during both the coupling reaction (the second step) and the dehydration reaction (the third step).

NAPH (S)-halopropionic NAPA
acid/ester

PREPARATION OF COMPOUND A
In one aspect, provided herein is a method for preparing Compound A, salts of Compound A, and the monohydrate form of Compound A. Compound A can be prepared from the NAPH, PYRH, and S-propionic acid/ester starting materials in three steps. First, NAPH and ^-propionic acid/ester undergo an S 2 alkylation reaction to result in (R)-2-(3-(2-methoxyethoxy)-5-oxo-l,6-naphthyridin-6(5H)-yl)propanoic acid/ester. The ^-propionic acid starting material produces (R)-2-(3-(2-methoxyethoxy)-5-oxo-l,6-naphthyridin-6(5H)-yl)propanoic acid (“NAPA”) in one step. The ^-propionic ester starting material first produces the ester analog of NAPA, and is subsequently hydrolyzed to form NAPA. During workup, the acid can optionally form a salt (e.g., HC1 or 2-naphthalenesulfonic acid).
Step 1:

NAPH (S)-2-halopropionic
acid/ester
1 2
wherein R is Br, CI, I, or OTf; and R is COOH or Ci-salkyl ester, and
when R is Ci^alkyl ester, the method of forming the NAPA or salt thereof further comprises hydrolyzing the Ci-salkyl ester to form an acid.
Second, NAPA and PYRH are coupled together to form (R)-N’-(3-fluoro-5-(lmethyl-lH-pyrazol-4-yl)pyridin-2-yl)-2-(3-(2-methoxyethoxy)-5-oxo- l,6-naphthyridin- 6(5H)yl)propanehydrazide (“HYDZ”).
Step 2:

Third, HYDZ is dehydrated to form Compound A.

The free base form of Compound A can be crystallized as a salt or a monohydrate.
Step 1: Alkylation of NAPH to form NAPA
The first step in the preparation of Compound A is the alkylation of NAPH to form NAPA. The NAPA product of the alkylation reaction is produced as a free base and is advantageously stable.
Thus, one aspect of the disclosure provides a method for preparing NAPA comprising admixing 3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one (“NAPH”):

Me
‘1 R2 , and a base, under conditions sufficient to form NAPA:

wherein R1 is Br, CI, I, or OTf; and
R2 is COOH or C^alkyl ester;
and when R2 is Ci_3alkyl ester, the method of forming the NAPA or salt thereof further comprises hydrolyzing the Ci-3alkyl ester to form an acid.
Me
The compound, R1 R2 , represents an (^-propionic acid and/or (S)- propionic ester
Me
(“(S)-propionic acid/ester”). When R1 R2 is an acid (i.e., R2 is COOH), NAPA is formed in one step:

-prop on c ac
Me
When R1 R2 is an ester (i.e., R2 is C1-3 alkyl ester), then the NAPA ester analog is formed, which can be hydrolyzed to form NAPA.


The SN2 alkylation of NAPH to form NAPA occurs with an inversion of
EXAMPLE 1
SYNTHESIS OF (R)-2-(3-(2-METHOXYETHOXY)-5-OXO-l,6-NAPHTHYRIDIN-6(5H)- YL)PROPANOIC ACID NAPHTHALENE-2-SULFONATE (NAPA)

Scheme 1: Synthesis of naphthyridinone acid 2-napsylate (NAPA)
NAPA was synthesized according to Scheme 1 by the following procedure. A jacket reactor (60 L) was charged with 3000 g (1.0 equivalent) of 3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one and 4646 g (2.0 equivalents) of magnesium ie/t-butoxide. 12 L (4.0 Vol) tetrahydrofuran was added to the reactor and an N2sweep and stirring were initiated. 2213 g (1.5 equivalents) of S-2-bromopropionic acid was added over at least 30 min, controlling the addition such that the batch temperature did not rise above 30 °C. The charge port was rinsed with tetrahydrofuran (0.5 Vol) after addition. The batch was then aged for at least 5 min at 25 °C. 1600 g (1.05 equivalents) of potassium iert-butoxide was added to the reactor in four portions (approximately equal) such that the batch temperature did not rise above 30 °C. The charge port was again rinsed with tetrahydrofuran (1.5 L, 0.5 Vol). The batch temperature was adjusted to 35+5 °C and the batch was aged for at least 12 h.
A separate 100 L reactor was charged with 6 L of 2-Metetrahydrofuran (2-MeTHF) (2.0 Vol), 8.4 L of water (1.5 Vol) and 9.08 L (4.0 equivalents) of 6 N HC1. The mixture from the 60 L reactor was pumped into the 100 L reactor, while maintaining the batch temperature at less than 45 °C.
The batch temperature was then adjusted to 20+5°C. The pH of the batch was adjusted with 6N HC1 (or 2N NaOH) solution until the pH was 1.4 to 1.9. The aqueous layer was separated from the product-containing organic layer. The aqueous layer was extracted with 2-MeTHF (2 Vol), and the 2-MeTHF was combined with the product stream in the reactor. The combined organic stream was washed with 20% brine (1 Vol). The organic layer was polish-filtered through a < ΙΟμιη filter into a clean vessel.
In a separate vessel, 1.1 equivalents of 2-Naphthalenesulfonic acid hydrate was dissolved in THF (2 Vol). The solution was polish-filtered prior to use. The 2-naphthalenesulfonic acid hydrate THF solution was added into the product organic solution in the vessel over at least 2 h at 25+5 °C. The batch temperature was adjusted to 60+5 °C and the batch was aged for 1+0.5 h. The batch temperature was adjusted to 20+5 °C over at least 2 h. The batch was filtered to collect the product. The collected filter cake was washed with THF (5.0 Vol) by displacement. The product cake was dried on a frit under vacuum/nitrogen stream until the water content was < lwt% by LOD.
The yield of the product (R)-2-(3-(2-methoxyethoxy)-5-oxo- l,6-naphthyridin-6(5H)-yl)propanoic acid naphthalene-2-sulfonate, was 87%. The chiral purity was determined using chiral HPLC and was found to be 98-99% ee. The purity was determined using HPLC, and was found to be > 98%.
Thus, Example 1 shows the synthesis of NAPA according to the disclosure.
EXAMPLE 2
SYNTHESIS OF (R)-N’-(3-FLUORO-5-(l-METHYL-lH-PYRAZOL-4-YL)PYRIDIN-2- YL)-2-(3-(2-METHOXYETHOXY)-5-OXO-l,6-NAPHTHYRIDIN-6(5H)- YL)PROPANEHYDRAZIDE (HYDZ)

Scheme 2: Synthesis of (R)-N’-(3-fluoro-5-(l-methyl- lH-pyrazol-4-yl)pyridin-2-yl)-2-(3-(2-methoxyethoxy)-5-oxo-l,6-naphthyridin-6(5H)-yl)propanehydrazide
HYDZ was synthesized according to Scheme 2 by the following procedure. A 60 L jacket reactor was charged with 2805.0 g (1.0 equivalent) of (R)-2-(3-(2-methoxyethoxy)-5-oxo-l,6-naphthyridin-6(5H)-yl)propanoic acid 2-napsylate (NAPA) and N,N-dimethylacetamide (DMAC) (4.6 mL DMAC per gram of NAPA). Stirring and an N2 sweep were initiated. 1.05 equivalents of N,N-diisopropylethylamine (DIPEA) was added while maintaining the batch temperature at less than 35°C. Initially the NAPA dissolves. A white precipitate formed while aging, but the precipitate had no impact on the reaction performance. 2197 g (1.10 equivalents) of 3-fluoro-2-hydrazinyl-5-(l-methyl- lH-pyrazol-4-yl)pyridine (PYRH) was added to the batch. The batch temperature was adjusted to 10+5 °C. 2208 g (1.2 equivalents) of N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC) was added in four portions (approximately equal) over at least 1 h (about 20 min interval per portion) at 10+5 °C.
The batch was aged until the amide conversion target was met. If the amide conversion target was not reached within 2 h, additional EDC was added until the conversion target was met. Once the target was met, the batch was heated to 55 °C until the solution was homogeneous. The batch was filtered through a <20 μ in-line filter into a reactor. The vessel and filter were rinsed with DMAC (0.2 mL DMAC/g of NAPA). The batch temperature was adjusted to 45+5 °C.
The reactor was charged with a seed slurry of (R)-N’-(3-fluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridin-2-yl)-2-(3-(2-methoxyethoxy)-5-oxo-l,6-naphthyridin-6(5H)-yl)propanehydrazide (HYDZ) (0.01 equivalents) in water (0.3 mL/g).
The batch was aged at 50+5 °C for at least 30 min. The batch temperature was adjusted to 20+5°C over at least 2 h. The batch was aged at 20+5°C for at least 30 min. 2.90 mL water per g was added at 25+5 °C over at least 2 h. The batch was aged at 20+5 °C for at least 1 h. The batch slurry was filtered to collect the product. The product was washed with 30% DMAC/H20 (0.5 Vol) by displacement. The product cake was washed with water (3 Vol) by displacement. The product cake was dried on the frit under vacuum/nitrogen stream until the water content was < 0.2 wt% as determined by Karl Fischer titration (KF). The product was a white, crystalline solid. The yield was about 83-84%. The ee was measured by HPLC and was found to be > 99.8%ee. The purity was determined by HPLC and was found to be >99.8 LCAP (purity by LC area percentage).
Thus, Example 2 demonstrates the synthesis of HYDZ according to the disclosure.
EXAMPLE 3
SYNTHESIS OF (R)-6-(l-(8-FLUORO-6-(l-METHYL-lH-PYRAZOL-4-YL)- [l,2,4]TRIAZOLO[4,3-A]PYRIDIN-3-YL)ETHYL)-3-(2-METHOXYETHOXY)-l,6- NAPHTHYRIDIN-5(6H)-ONE HYDROCHLORIDE SALT (COMPOUND A-HCL) – ROUTE 1

Scheme 3 Route 1 – Synthesis of (R)-6-(l-(8-fluoro-6-(l-methyl- lH-pyrazol-4-yl)- [l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)- l,6-naphthyridin-5(6H)-one hydrochloride
(R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)- l,6-naphthyridin-5(6H)-one hydrochloride salt (Compound A-
HC1) was synthesized according to Scheme 3, Route 1 by the following procedure. A 15 L reactor, Reactor 1, was charged with 750 g HYDZ and the reactor jacket temperature was adjusted to 20+5 °C. A nitrogen sweep was initiated in Reactor 1 and the condenser coolant (at 5+5 °C) was started. Acetonitrile (3.4 L, 4.5 Vol) was added to Reactor 1 and stirring was initiated. 420 g (2.5 equivalents) of 2,6-lutidine was added to the reactor.
A solution of diphenylphosphinyl chloride Ph2P(0)(Cl) was prepared by combining 850 g (2.3 equivalents) of Ph2P(0)(Cl) and 300 g acetonitrile in an appropriate container. The contents of the PH2P(0)(C1) solution were added to Reactor 1. The jacket temperature was adjusted over 60+30 min until the reflux temperature of the batch (approximately 85 °C) was reached. The reaction was stirred for 14+6 h. The batch temperature was reduced to 75+5 °C and the batch was sampled for IPT analysis. The expected result was < 2% HYDZ remaining. If the target was not met, the heating at reflux temperature was continued for 9+6 h. Sampling, analysis, and heating was repeated until a satisfactory conversion assay result was obtained (< 10% HYDZ was considered satisfactory, < 1% was actually achieved). The final sample was assayed for optical purity by HPLC, and was found to be > 99.5% ee.
A K2CO3/KCI quench solution (5.0 Vol) was prepared in advance by combining 555 g (3.1 equivalents) of potassium carbonate with 335 g (2.9 equivalents) of potassium chloride and 3450 g of water in an appropriate container. The quench solution was added to Reactor 1 over at least 15 min, maintaining the batch temperature at 60+5 °C. As the aqueous base reacted with excess acid some bubbling (C02) occurred. 3.0 L (4.0 Vol) of toluene was added to Reactor 1 at 65+5 °C. A sample of the batch was taken for IPT analysis. The lower (aqueous) phase of the sample was assayed by pH probe (glass electrode). The pH was acceptable if in the range of pH 8-11. The upper (organic) phase of the sample was assayed by HPLC.
The batch was agitated for 20+10 min at 65+5 °C. Stirring was stopped and the suspension was allowed to settle for at least 20 min. The aqueous phase was drained from Reactor 1 via a closed transfer into an appropriate inerted container. The remaining organic phase was drained from Reactor 1 via a closed transfer to an appropriate inerted container. The aqueous phase was transferred back into Reactor 1.
An aqueous cut wash was prepared in advance by combining 2.3 L (3.0 Vol) acetonitrile and 2.3 L(3.0 Vol) toluene in an appropriate container. The aqueous cut wash was added to Reactor 1. The batch was agitated for 20+10 min at 65+5 °C. The stirring was stopped and the suspension was allowed to settle for at least 20 min. The lower (aqueous) phase was drained from Reactor 1 via a closed transfer into an appropriate inerted container. The organic phase was drained from Reactor 1 via a closed transfer to the inerted container containing the first organic cut. The combined mass of the two organic cuts was measured and the organic cuts were transferred back to Reactor 1. Agitation was initiated and the batch temperature was adjusted to 60+10 °C. A sample of the batch was taken and tested for Compound A content by HPLC. The contents of Reactor 1 were distilled under vacuum (about 300-450 mmHg) to approximately 8 volumes while maintaining a batch temperature of 60+10 °C and a jacket temperature of less than 85 °C. The final volume was between 8 and 12 volumes.
The nitrogen sweep in Reactor 1 was resumed and the batch temperature adjusted to 70+5 °C. A sample of the batch was taken to determine the toluene content by GC. If the result was not within 0-10% area, the distillation was continued and concomitantly an equal volume of 2-propanol, up to 5 volumes, was added to maintain constant batch volume. Sampling, analysis, and distillation was repeated until the toluene content was within the 0-10% area window. After the distillation was complete, 540 g (450 mL, 3.5 equivalents) of hydrochloric acid was added to Reactor 1 over 45+15 min while maintaining a batch temperature at 75+5 °C.
A Compound A-HC1 seed suspension was prepared in advance by combining 7.5 g of Compound A-HC1 and 380 mL (0.5 Vol) of 3 propanol in an appropriate container. The seed suspension was added to Reactor 1 at 75+5 °C. The batch was agitated for 60+30 min at 75+5 °C. The batch was cooled to 20+5 °C over 3+1 h. The batch was agitated for 30+15 min at 20+5 °C. 2.6 L (3.5 Vol) of heptane was added to the batch over 2+1 h. The batch was then agitated for 60+30 min at 20+5 °C. A sample of the batch was taken and filtered for IPT analysis. The filtrate was assayed for Compound A-HC1. If the amount of Compound A-HC1 in the filtrate was greater than 5.0 mg/mL the batch was held at 20 °C for at least 4 h prior to filtration. If the amount of Compound A-HC1 in the filtrate was in the range of 2-5 mg.ML, the contents of Reactor 1 were filtered through a < 25 μιη PTFE or PP filter cloth, sending the filtrate to an appropriate container.
A first cake wash was prepared in advance by combining 1.5L (2.0 Vol) of 2-propanol and 1.5L (2.0 Vol) of heptane in an appropriate container. The first cake wash was added to Reactor 1 and the contents were agitated for approximately 5 min at 20+5 °C. The contents of Reactor 1 were transferred to the cake and filter. A second cake wash of 3.0L (4.0 Vol) of heptane was added to Reactor 1 and the contents were agitated for approximately 5 min at 20+5 °C. The contents of Reactor 1 were transferred to the cake and filter. The wet cake was dried under a flow of nitrogen and vacuum until the heptane content was less than 0.5 wt% as determined by GC. The dried yield was 701g, 85% as a yellow powder. The dried material was assayed for chemical purity and potency by HPLC and for residual solvent content by GC. The isolated product was 88.8% Compound A-HC1, having 99.8% ee and 0.6% water.
Thus, Example 3 shows the synthesis of Compound A-HCL according to the disclosure.
EXAMPLE 4
SYNTHESIS OF (R)-6-(l-(8-FLUORO-6-(l-METHYL-lH-PYRAZOL-4-YL)- [l,2,4]TRIAZOLO[4,3-A]PYRIDIN-3-YL)ETHYL)-3-(2-METHOXYETHOXY)-l,6- NAPHTHYRIDIN-5(6H)-ONE HYDROCHLORIDE SALT (COMPOUND A-HCL) – ROUTE 2

HYDZ A HCI
Scheme 4: Route 2 – Synthesis of (R)-6-(l-(8-fluoro-6-(l-methyl- lH-pyrazol-4-yl)- [l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)- l,6-naphthyridin-5(6H)-one hydrochloride
(R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)- l,6-naphthyridin-5(6H)-one hydrochloride salt was synthesized according to Scheme 4, Route 2, by the following procedure. A clean and dry 60 L reactor was fitted with a reflux condenser, nitrogen inlet, and vented to a scrubber (Reactor 1). The jacket temperature of Reactor 1 was set to 20 °C. A scrubber was set up to the vent of Reactor 1, and aqueous bleach solution was charged to the scrubber. The circulating pump (commercial 5.25% NaOCl) was initiated. The scrubber pump was turned on and N2 sweep on Reactor 1 was started. Reactor 1 was charged with 2597 g (0.52 equivalents) of Lawesson’s reagent. Reactor 1 was then charged with 6000 g (1.0 equivalent) of HYDZ and 30 L (5.0 vol) acetonitrile (MeCN). Agitation of Reactor 1 was initiated. The reactor was heated to 50+5 °C and aged until an LC assay showed consumption of HYDZ (> 99% conversion).
The jacket temperature of a second clean and dry reactor, Reactor 2, was set to 50 °C. The contents of Reactor 1 were transferred to Reactor 2 through a 5 micron inline filter. Reactor 1 was rinsed with MeCN, and the rinse was transferred through the inline filter to Reactor 2. Reactor 2 was charged with toluene. (31.7 Kg)
In a separate container a solution of 16.7% K2C03 was prepared by adding 7200 g K2C03 and 36 L water to the container and shaking the container well until all the solid was dissolved. Half of the contents of the K2C03 solution was added to Reactor 2 over at least 10 min. The batch temperature of Reactor 2 was adjusted to 50+5 °C. The batch in Reactor 2 was agitated at 50+5 °C for at least 1 h. The agitation was stopped and the batch in Reactor 2 was allowed to phase separate. The aqueous phase was removed. The remaining contents of the K2C03 solution was added to Reactor 2 over at least 10 min. The batch temperature in Reactor 2 was adjusted to 50+5 °C. The batch in Reactor 2 was agitated at 50+5 °C for at least 1 h. The agitation was stopped and the batch in Reactor 2 was allowed to phase separate. The aqueous phase was removed.
The jacket temperature of a clean and dry reactor, Reactor 3, was set to 50 °C. The contents of Reactor 2 were transferred to Reactor 3 through a 5 micron in-line filter. The contents of Reactor 3 were distilled at reduced pressure. Isopropyl alcohol (IP A, 23.9 kg) was charged to Reactor 3 and then the batch was distilled down. IPA (23.2 kg) was again added to Reactor 3. The charge/distillation/charge cycle was repeated. The batch temperature in Reactor 3 was adjusted to 70+15 °C. Reactor 3 was then charged with DI water (1.8 L). Concentrated HC1 (1015 mL) was added to Reactor 3 over at least 15 min at 70+15 °C.
A seed of the Compound A-HCl was prepared by combining a seed and IPA in a separate container. The Compound A-HCl seed was added to Reactor 3 as a slurry. The batch in Reactor 3 was aged at 70+15 °C for at least 15 min to ensure that the seed held. The batch in Reactor 3 was cooled to 20+5 °C over at least 1 h. Heptane (24.5 kg) was added to Reactor 3 at 20+5 °C over at least 1 h. The batch was aged at 20+5 °C for at least 15 min. The contents of Reactor 3 were filtered through an Aurora filter fitted with a <25 μιη PTFE or PP filter cloth. The mother liquor was used to rinse Reactor 3.
A 50% v/v IP A/heptane solution was prepared, in advance, in a separate container by adding the IPA and heptane to the container and shaking. The filter cake from Reactor 3 was washed with the 50% IP A/heptane solution. If needed, the IP A/heptane mixture, or heptane alone, can be added to Reactor 3 prior to filtering the contents through the Aurora filter. The cake was washed with heptane. The cake was dried under nitrogen and vacuum until there was about < 0.5 wt% heptane by GC analysis. The product was analyzed for purity and wt% assay by achiral HPLC, for wt% by QNMR, for water content by KF, for form by XRD, for chiral purity by chiral HPLC, and for K and P content by ICP elemental analysis.
Compound A-HCl had a purity of 99.56 area% and 88.3 wt% assay by achiral HPLC, and 89.9 wt% by QNMR. The water content was 0.99 wt% as determined by KF. The chiral purity was 99.9%ee as determined by chiral HPLC. The P and K content was found to be 171 ppm and 1356 ppm, respectively, as determined by ICP elemental analysis.
Thus, Example 4 shows the synthesis of Compound A-HCl according to the disclosure.
EXAMPLE 5
SYNTHESIS OF (R)-6-(l-(8-FLUORO-6-(l-METHYL-lH-PYRAZOL-4-YL)- [l,2,4]TRIAZOLO[4,3-A]PYRIDIN-3-YL)ETHYL)-3-(2-METHOXYETHOXY)-l,6- NAPHTHYRIDIN-5(6H)-ONE (COMPOUND A) – ROUTE 3

Scheme 5: Route 3 – Synthesis of (R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)- l,6-naphthyridin-5(6H)-one (compound A)
(R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)- l,6-naphthyridin-5(6H)-one was synthesized according to Scheme 5, Route 3, by the following procedure. 0.760 g (1.6 mmol) N’-iS-fluoro-S-il-methyl-lH-pyrazol-4-yl)pyridin-2-yl)-2-(3-(2-methoxyethoxy)-5-oxo- l,6-naphthyridin-6(5H)-yl)propanehydrazide (HYDZ) and 0.62 g (2.4 mmol) triphenylphosphine were taken up in 16 mL THF. 0.31 mL (2.4 mmol) trimethylsilyl (TMS)-azide was added, followed by addition of 0.37 mL (2.4 mmol) DEAD, maintaining the reaction temperature below 33 °C. The reaction was stirred at room temperature for 50 minutes. The reaction mixture was concentrated in vacuo.
The crude material was taken up in dichloromethane and loaded onto silica gel. The crude material was purified via medium pressure liquid chromatography using a 90: 10: 1 DCM : MeOH : NH4OH solvent system. 350 mg, (48% yield) of (R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one was collected as a tan solid. The (S) isomer was also collected. The product had a purity of 97% by HPLC.
Thus, Example 5 shows the synthesis of enantiomerically pure Compound A according to the disclosure.
EXAMPLE 6
SYNTHESIS OF (R)-6-(l-(8-FLUORO-6-(l-METHYL-lH-PYRAZOL-4-YL)- [l,2,4]TRIAZOLO[4,3-A]PYRIDIN-3-YL)ETHYL)-3-(2-METHOXYETHOXY)-l,6-NAPHTHYRIDIN-5(6H)-ONE (COMPOUND A) AND THE HYDROCHLORIDE SALT- ROUTE 3

Scheme 6: Route 3 – Synthesis of (R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one (compound A) and the hydrochloride salt
(R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one was synthesized according to Scheme 6, Route 3, by the following procedure. Benzothiazyl disulfide (3.31 g, 9.97 mmol), HYDZ (4.0 g, 8.31 mmol), and a stir bar were added to a 50 mL 3-neck flask fitted with a reflux condenser topped with a nitrogen inlet, a thermocouple and a septum. The flask headspace was purged with nitrogen, and the solids were suspended in MeCN (20.00 mL, 5 mL/g) at ambient conditions. The flask contents were heated to 50 °C on a heating mantle. Finally,
trimethylphosphine, solution in THF (9.97 ml, 9.97 mmol) was added dropwise by syringe pump with stirring over 1 h. An ice pack was affixed to the side of the flask in lieu of a reflux condenser. After about 0.5 h from addition, the resulting suspension was sampled and analyzed by, showing about 99% conversion of penultimate, and about 94% Compound A vs.
benzothiazole-2-thiol (“BtSH”) adduct selectivity.
After about 0.75 h from addition, the yellow reaction mixture was cooled to 0 °C in an ice bath, and 30% hydrogen peroxide in water (2.037 mL, 19.94 mmol) was added dropwise over 2 hours. The reaction solution was allowed to warm to room temperature overnight.
The suspension was heated to 30 °C, held at that temperature for 3 h and then cooled to room temperature. After cooling was complete, an aliquot was filtered and the filtrate was analyzed by liquid chromatography, showing 99% Compound A vs. BtSH adduct (91% purity for Compound A overall).
A Celite filtration pad about 0.5″ thick was set up on a 50 mL disposable filter frit and wetted with toluene (32.0 mL, 8 mL/g). The reaction suspension was transferred to the Celite pad and filtered to remove BtSH-related byproducts, washing with MeCN (2.000 mL, 0.5 mL/g). The filtrate was transferred to a 100 mL round bottom flask, and treated with 30 mL (7.5 Vol) of an aqueous quench solution consisting of sodium bicarbonate (7.5 ml, 8.93 mmol) and sodium thiosulfate (3.75 ml, 4.74 mmol) at overall about 5 wt% salt. The suspension was stirred for about 15 min and then the layers were allowed to separate. Once the layers were cut, the aqueous waste stream was analyzed by LC, showing 8% loss. The organic stream was similarly analyzed, showing 71% assay yield, implying about 20% loss to waste cake.
The organic cut was transferred to a 3-neck 50 mL round bottom flask with magnetic stir bar, thermocouple, and a shortpath distillation head with an ice-cooled receiving flask. The boiling flask contents were distilled at 55 °C and 300 torr pressure. The volume was reduced to 17 mL. The distillation was continued at constant volume with concomitant infusion of IPA (about 75 mL). The resulting thin suspension was filtered into a warm flask and water (0.8 mL) was added. The solution was heated to 80 °C. After this temperature had been reached, hydrochloric acid, 37% concentrated (0.512 ml, 6.23 mmol) was added, and the solution was seeded with about 30 mg (about 1 wt%) Compound A-HC1 salt. The seed held for 15 min. Next the suspension was cooled to 20 °C over 2 h. Finally heptane (17 mL, 6 Vol) was added over 2 h by syringe pump. The suspension was allowed to stir under ambient conditions overnight.
The yellow-green solid was filtered on an M-porosity glass filter frit. The wet cake was washed with 1: 1 heptane/IPA (2 Vol, 5.5 mL) and then with 2 Vol additional heptane (5.5 mL). The cake was dried by passage of air. The dried cake (3.06 g , 78.5 wt%, 94 LC area% Compound A, 62% yield) was analyzed by chiral LC showing optical purity of 99.6% ee.
Thus, Example 6 shows the synthesis of enantiomerically pure Compound A and the hydrochloric salt thereof, according to the disclosure.
EXAMPLE 7
RE-CRYSTALLIZATION OF COMPOUND A

A-HCI A monohydrate
Scheme 7: Re-crystallization of Compound A
Compound A-HCI was recrystallized to Compound A. A (60 L) jacketed reactor, Reactor 1, with a jacket temperature of 20 °C was charged with 5291 g, 1.0 equivalent of Compound A-HCI. 2 Vol (10.6 L) of IPA and 1 Vol (5.3 L) of water were added to Reactor 1 and agitation of Reactor 1 was initiated.
An aqueous NaHC03 solution was prepared in advance by charging NaHC03 (1112 g) and water (15.87 L, 3 Vol) into an appropriate container and shaking well until all solids were dissolved. The prepared NaHC03 solution was added to Reactor 1 over at least 30 min, maintaining the batch temperature below 30 °C. The batch temperature was then adjusted to about 60 °C. The reaction solution was filtered by transferring the contents of Reactor 1 through an in-line filter to a second reactor, Reactor 2, having a jacket temperature of 60+5 °C. Reactor 2 was charged with water (21.16 L) over at least 30 min through an in-line filter, maintaining the batch temperature at approximately 60 °C. After the addition, the batch temperature was adjusted to approximately 60 °C.
A seed was prepared by combining Compound A seed (0.01 equivalents) and IP A/water (20:80) in an appropriate container, in an amount sufficient to obtain a suspension. The seed preparation step was performed in advance. Reactor 2 was charged with the seed slurry. The batch was aged at 55-60 °C for at least 15 min. The batch was cooled to 20+5 °C over at least 1 h. The batch from Reactor 2 was recirculated through a wet mill for at least 1 h, for example, using 1 fine rotor stator at 60 Hz, having a flow rate of 4 L/min, for about 150 min.
The reaction mixture was sampled for particle size distribution during the milling operation. The solids were analyzed by Malvern particle size distribution (PSD) and
microscopic imaging. At the end of the milling operation a sample of the reaction mixture was again analyzed. The supernatant concentration was analyzed by HPLC, and the solids were analyzed by Malvern PSD and microscopic imaging to visualize the resulting crystals.
The batch temperature was adjusted to 35+5 °C and the batch was aged for at least 1 h. The batch was cooled to 20+5 °C over at least 2 h. The reaction mixture was sampled to determine the amount of product remaining in the supernatant. The supernatant concentration was analyzed by HPLC for target of <5 mg/mL Compound A in the supernatant. The contents of Reactor 2 were filtered through an Aurora filter fitted with a <25 μιη PTFE or PP filter cloth.
A 20% v/v IP A/water solution was prepared and the filter cake from Reactor 2 was washed with the 20% IP A/water solution. The cake was then washed with water. If needed, the IP A/water solution, or water alone, can be added to Reactor 2 prior to filtering to rinse the contents of the reactor. The cake was dried under moist nitrogen and vacuum until target residual water and IPA levels were reached. The product had 3.2-4.2% water by KF analysis. The product was analyzed by GC for residual IPA (an acceptable about less than or equal to about 5000 ppm). The yield and purity were determined to be 100% and 99.69% (by HPLC), respectively.
Thus, Example 6 shows the recrystallization of Compound A from the HC1 salt, Compound A-HC1, according to the disclosure.
EXAMPLE 8
SYNTHESIS OF (R)-6-(l-(8-FLUORO-6-(l-METHYL-lH-PYRAZOL-4-YL)- [l,2,4]TRIAZOLO[4,3-A]PYRIDIN-3-YL)ETHYL)-3-(2-METHOXYETHOXY)-l,6- NAPHTHYRIDIN-5(6H)-ONE (COMPOUND A)

HYDZ A
Scheme 8 Synthesis of (R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one
(R)-6-(l-(8-fluoro-6-(l-methyl-lH-pyrazol-4-yl)-[l,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-l,6-naphthyridin-5(6H)-one was synthesized according to Scheme 8 by the following procedure. A clean and dry 60 L reactor was fitted with a reflux condenser, nitrogen inlet, and vented to a scrubber (Reactor 1). The jacket temperature of Reactor 1 was set to 20 °C. A scrubber was set up to the vent of Reactor 1, and aqueous bleach solution was charged to the scrubber. The circulating pump (commercial 5.25% NaOCl) was initiated. The scrubber pump was turned on and N2 sweep on Reactor 1 was started. Reactor 1 was charged with 1599.5 g (0.52 equivalents) of Lawesson’s reagent. Reactor 1 was then charged with 24.4 L acetonitrile (MeCN). Agitation of Reactor 1 was initiated. 3664.7 g (1.0 equivalent) of HYDZ was added to the reactor in portions over 1+0.5 h, using acetonitrile (5 L) as rinse. The reactor was heated to 50+5 °C and aged until an LC assay shows consumption of HYDZ (> 99% conversion).
The reactor was cooled to 20 °C and the reaction was assayed by HPLC for
Compound A. The assay showed a 99% crude yield of Compound A.
The contents of Reactor 1 were transferred to second reactor, Reactor 2, through a 1 micron inline filter. Reactor 2 was charged with 2 L of water. Reactor 2 was connected to a batch concentrator and vacuum distilled until a final volume of about 10 L. The jacket temperature was 50 °C during distillation and the pot temperature was maintained below 50 °C. The batch was then cooled to 20 °C.
In a separate container a solution of 10% K2CO3 was prepared by adding 1160 g K2CO3 and 10450 mL water to the container and shaking the container well until all the solid was dissolved. The K2CO3 solution was added to Reactor 2 through an in-line filter (5 μηι). 13 kg of purified water was added to the reactor through the in-line filter (5 μηι).
A Compound A seed was added to the reactor through an addition port. The resulting slurry was aged for one hour during which crystallization was observed. The reactor was placed under vacuum and charged with 16 L of water. The resulting slurry was aged at 20 °C overnight. The product slurry was filtered through a 25 μιη filter cloth and washed with 10 L of a 10% MeCN in water solution, followed by 12 L of water. The product was dried on a frit under a stream of ambient humidity filtered air.
Compound A was isolated as a monohydrate crystalline solid which reversibly dehydrates at < 11% RH. After drying, there was 3.9 wt.% water present in constant weight solid as determined by KF. 3.317 kg, 89% yield, of Compound A was isolated as a pale yellow solid. The product had a purity of 99.4 wt.% as determined by LCAP.
EXAMPLE 9
SYNTHESIS OF NAPH – ROUTE 1
CuBr (5-10%)

ethyl 5-bromo-2- Bromonaphthyridinone Naphthyridinone ether methylnicotinate
Scheme 9: Synthesis of NAPH – Route 1
The NAPH starting material for the synthesis of Compound A was synthesized according to Scheme 9, Route 1 by the following procedure. The jacket temperature of a 6 L jacketed reactor, Reactor 1, was set to 22 °C. 2409 g (1.0 equiv) of ethyl 5-bromo-2-methylnicotinate, 824 g (1.0 equivalent) of triazine, and 3.6 L dimethyl sulfoxide (DMSO) were added to the reactor. The jacket temperature was adjusted to 45 °C. The reactor was agitated until a homogenous solution resulted. Once complete dissolution has occurred (visually) the jacket of Reactor 1 was cooled to 22 °C.
A second, 60 mL reactor, Reactor 2, was prepared. 8.0 L of water was charged to a scrubber. 4.0 L of 10 N sodium hydroxide was added to the scrubber and the scrubber was connected to Reactor 2. The cooling condenser was started. 6411.2 g of cesium carbonate and 12.0 L of DMSO were added to Reactor 2. Agitation of Reactor 2 was initiated. The batch temperature of Reactor 2 was adjusted to 80 °C. The solution from Reactor 1 was added slowly over 1 h at 80 °C, while monitoring the internal temperature. 1.2 L of DMSO was added to Reactor 1 as a rinse. The DMSO rinse was transferred from Reactor 1 to Reactor 2 over 6 min. Reactor 2 was agitated for more than 1 h and the conversion to 3-bromo-l,6-naphthyridin-5(6H)-one was monitored by HPLC until there was < 1.0% ethyl 5-bromo-2-methylnicotinate remaining. When the reaction was complete the batch temperature was adjusted to 60 °C. 24.0 L (10V) of water was added to Reactor 2 over 2 h, maintaining a reaction temperature of 60+5 °C, using a peristaltic pump at 192 mL/min. Reactor 2 was cooled to 22 °C over 1 h 10 min. Stirring was continued at 22+5 °C until the supernatant assays for less than 3mg/mL of 3-bromo-l,6-naphthyridin-5(6H)-one (analyzed by HPLC). The crystallized product was filtered through an Aurora filter fitted with 25 μιη polypropylene filter cloth. The reactor and filter cake were washed with a 75 wt% H20-DMSO solution (3 Vol made from 1.6 L DMSO and 5.6 L water), followed by water (7.2 L, 3 Vol), and finally toluene (7.2 L, 3 Vol). The product cake was dried on the aurora filter under vacuum with a nitrogen stream at ambient temperature. The product was determined to be dry when the KF was < 2.0 wt% water. 2194 g of 3-bromo-l,6-naphthyridin-5(6H)-one was isolated as a beige solid. The chemical purity was 99.73%. The adjusted yield was 2031.6 g (91.9%).
The jacket temperature of a 100 L reactor, Reactor 3, was set to 15+5 °C. 6.45 L of 2-methoxyethanol was added to the reactor and agitation was initiated. (8107 g) lithium tert-butoxide was added portion- wise to the reactor, maintaining the reactor temperature in a range of 15 °C to 24 °C. 3795 g of 3-bromo-l,6-naphthyridin-5(6H)-one was added to the reactor. 4 mL of 2-methoxyethanol was added to rinse the solids on the wall of the reactor. The reactor contents were stirred for at least 5 min. The reaction mixture was heated to distillation to remove i-BuOH and water, under 1 atm of nitrogen (jacket temperature 145 °C). Distillation continued until the pot temperature reached 122+3 °C. The reactor contents were sampled and analyzed for water content by KF. The reaction mixture was cooled to less than 35 °C. 243 g CuBr was added to the reactor. The reaction mixture was de-gassed by applying vacuum to 50 torr and backfilling with nitrogen three times. The batch was heated to 120+5 °C while maintaining the jacket temperature below 150 °C. The batch was agitated (174 RPM) for 15.5 h. A sample of the reaction was taken and the reaction progress was monitored by HPLC. When the remaining 3-bromo-l,6-maphthyridin-5(6H)-one was less than 1%, the jacket temperature was cooled down to 25 °C.
An Aurora filter was equipped with a 25 μιη PTFE cloth and charged with Celite®. The reactor content was transferred onto the filter cloth and the filtrate was collected in the reactor. 800 mL of 2-methoxyethanol was added to the reactor and agitated. The reactor contents were transferred onto the filter and the filtrate was collected in the reactor. 5.6 L of acetic acid was added to the reactor to adjust the pH to 6.5, while maintaining the temperature at less than 32 °C. The batch was then heated to 80 °C. The reaction mixture was concentrated to 3.0+5 Vol (about 12 L) at 80+5 °C via distillation under vacuum.
In a separate container labeled as HEDTA Solution, 589.9 g of N-(2-hydroxyethyl)ethylenediaminetriacetic acid trisodium salt hydrate and 7660 mL water were mixed to prepare a clear solution. The HEDTA solution was slowly added to the reactor while maintaining the temperature of the batch at about 80-82 °C. The batch was then cooled to 72 °C.
An aqueous seed slurry of NAPH (31.3g) in 200 mL of water was added to the reactor. The slurry was aged for 30+10 min. 20 L of water was slowly added to the reactor to maintain the temperature at 65+5 °C. The batch was aged at 65+5 °C for 30 min. The batch was cooled to 20 °C over 1 h. The reactor contents were purged with compressed air for 1 h, and then the batch was further cooled to – 15 °C and aged for 12.5 h. The batch was filtered through a centrifuge fitted with 25 μιη PTFE filter cloth. 5.31 Kg of wet cake was collected (60-62 wt ). The wet cake was reslurried in 6V HEDTA solution and filtered through the centrifuge. The collected wet cake was dried in the centrifuge, and transferred to an Aurora filter for continued drying.
2.82 kg (76% isolated yield) of NAPH was collected having a 2.7% water content by KF.
Thus, Example 8 shows the synthesis of NAPH according to the examples.
EXAMPLE 10
SYNTHESIS OF NAPH – ROUTE 2


Scheme 10: Synthesis of NAPH via Route 2
The NAPH starting material for the synthesis of Compound A was synthesized according to Scheme 10, Route 2, by the following procedure.
Preparation of protected 2-methoxy-pyridin-4ylamine. A 1600 L reactor was flushed with nitrogen and charged with 120 L of N,N-dimethylacetamide, 100.0 kg 2-methoxy-pyridin-4-ylamine, and 89.6 kg triethylamine, maintaining the temperature of the reactor at less than 20 °C. In a separate container, 103.0 kg pivaloyl chloride was dissolved in 15.0 L of N,N-dimethylacetamide and cooled to less than 10 °C. The pivaloyl chloride solution was added to the reactor using an addition funnel over 3.2 hours while maintaining the reactor temperature between 5 °C and 25 °C. The addition funnel was washed with 15.0 L of N,N-dimethylacetamide, which was added to the reactor. The reaction was stirred for 2.3 hours at 20-25 °C. A sample of the reaction was taken and analyzed for 2-methoxy-pyridin-4ylamine by TLC. No 2-methoxy-pyridin-4ylamine remained in the solution and the reaction was aged at 20-25 °C under nitrogen over night. 1200 L of deionized water was added to the reaction over 2
hours at while the reaction was maintained at 5-15 °C. The resulting mixture was stirred at 15 °C for 2 hours and then cooled to 5 °C. The reaction was centrifugated at 700-900 rpm in 3 batches. Each batch was washed 3 times with deionized water (3x 167 L) at 800 rpm. The wet solids obtained were dried under vacuum at 55 °C for 18 hours in 2 batches, sieved and dried again under vacuum at 55 °C for 21 hours until the water content was < 0.2% as determined by KF. 80.4 kg (89.7% yield) of the protected 2-methoxy-pyridin-4ylamine was collected as a white solid.
Preparation of protected 3-formyl-4-amino-2-methoxypyridine. A 1600 L reactor was flushed with nitrogen and charged with 1000 L of THF and 70.5 kg of the protected 2-methoxy-pyridin-4ylamine. The reaction was stirred for 10 min at 15-25°C. The reaction was cooled to -5 °C and 236.5 kg of w-hexyllithium (solution in hexane) was added over 11.5 hours while maintaining the temperature of the reaction at <-4°C. The reaction was maintained at <-4°C for 2 hours. A sample of the reaction was quenched with D20 and the extent of the ortho-lithiation was determined by 1H NMR (98.2% conversion). 61.9 kg dimethylforaiamide (DMF) was added at <-4°C over 3.2 h. After stirring 7.5 hours at <-4°C, a sample of the reaction was assayed for conversion by HPLC (98.5% conversion).
A 1600 L reactor, Reactor 2, was flushed with nitrogen and charged with 145 L THF and 203.4 kg of acetic acid. The resulting solution was cooled to -5 °C. The content of the first reactor was transferred to Reactor 2 over 2.5 hours at 0 °C. The first reactor was washed with 50 L THF and the washing was transferred into Reactor 2. 353 L deionized water was added to Reactor 2 while maintaining the temperature at less than 5 °C. After 15 min of decantation, the aqueous layer was removed and the organic layer was concentrated at atmospheric pressure over 5 hours until the volume was 337 L. Isopropanol (350 L + 355 L) was added and the reaction was again concentrated at atmospheric pressure until the volume was 337 L. Distillation was stopped and 90 L of isopropanol was added to the reactor at 75-94 °C. 350 L of deionized water was added to the reactor at 60-80 °C over 1 h (the temperature was about 60-65 °C at the end of the addition). The reaction was cooled to 0-5 °C. After 1 hour, the resulting suspension was filtered. Reactor 2 was washed twice with deionized water (2x 140L). The washings were used to rinse the solid on the filter. The wet solid was dried under vacuum at 50 °C for 15 h. 71.0 kg (80% yield) of the protected 3-formyl-4-amino-2-methoxypyridine was produced. The purity of the formyl substituted pyridine was found to be 92.7% by LCAP.
A 1600 L reactor, Reactor 3, was flushed with nitrogen and successively charged with 190 L ethanol, 128.7 kg of protected 3-formyl-4-amino-2-methoxypyridine, 144 L of deionized water and 278.2 kg of sodium hydroxide. The batch was heated to 60-65°C and 329.8 kg of the bisulfite adduct was added over 1 h. After lh of stirring, a sample was taken for HPLC analysis which showed 100% conversion. The batch was aged 2 hours at 60-65 °C, then was allowed to slowly cool down to 20-25 °C. The batch was aged 12 h at 20-25 °C. The batch was filtered and the reactor was washed with water (2x 125 L). The washings were used to rinse the solid on the filter. The wet solid was transferred to the reactor with 500 L deionized water and heated to 45-50 °C for 1 h. The batch was allowed to return to 20-25 °C (24 h). The solid was filtered and the reactor was washed with deionized water (2x 250 L). The washings were used to rinse the solid on the filter. 112.5 kg of wet white solid was obtained (containing 85.1 Kg (dry) of the naphthyridine, 72.3% yield, greater than 97% purity as determined by HPLC). The wet product was used directly in the next step, without drying.
A 1600 L reactor was flushed with nitrogen and charged with 417 L of deionized water and 112.5 kg of the wet napthyridine. The scrubber was filled with 700 L of water and 92.2 kg monoethanolamine. A solution of hydrochloric acid (46.6 kg diluted in 34 L of deionized water) was added to the reactor at 15-20 °C over 10 minutes. The batch was heated to 60-65 °C for 3 h. A sample of the batch was taken and contained no remaining starting material as determined by TLC. A solution of concentrated sodium hydroxide (58.2 kg in 31 L of deionized water) was added to the reactor at 60-65 °C. 65% of the solution was added over 15 min and then the batch was seeded with crystallized NAPH. Crystallization was observed after 2.5 h and then the remaining35% of the sodium hydroxide solution was added (pH – 11.1). The batch was cooled to 25-30 °C and a solution of sodium phosphate monobasic (1.8 kg in 2.9 L of deionized water) was added over 25 min at 25-30 °C) (pH = 6.75). The batch was stirred at 15-20 °C for 12 hours and filtered. The reactor was washed twice with deionized water (2x 176 L). The washings were used to rinse the solid on the filter. The wet solid was dried under vacuum at 50 °C until the water content was < 5% (by KF), to give 78.1 kg (73.8% yield, > 95%)) of NAPH as a beige powder.
Thus, Example 9 shows the synthesis of NAPH according to the disclosure.
EXAMPLE 11
SYNTHESIS OF (R)-2-(3-(2-METHOXYETHOXY)-5-OXO-l,6-NAPHTHYRIDIN-6(5H)- YL)PROPANOIC ACID NAPHTHALENE-2-SULFONATE (NAPA)

6N HCI/ THF 80C

Scheme 11: Synthesis of NAPA, Route 3
NAPA was synthesized according to Scheme 11, Route 3 by the following procedure. 4.75 g of 3-(2-Methoxyethoxy)-l,6-naphthyridin-5(6H)-one was suspended in 45 mL of DMF. 2.58 mL (s)-methyl lactate and 9.05 g triphenylphosphine were added to the suspension. The reaction mixture was cooled to 0 °C. 5.12 mL diethyl azodicarboxylate (DEAD) was added dropwise via syringe. The mixture was stirred at 0 °C for 1 h. A sample of the reaction was taken and the reaction was determined to be complete by LCMS. The reaction mixture was concentrated under vacuum to give crude material as a yellow oil.
1 g of the crude material was loaded in dichloromethane onto a silia gel pre-column. The sample was purified using the Isco Combi-Flash System; column 40 g, solvent system hexane/ethyl acetate, gradient 0-100% ethyl acetate over 15 minutes. Product eluted at 100% ethyl acetate. The product fractions were combined and concentrated under vacuum. 256 mg of (R)-methyl 2-(3-(2-methoxyethoxy)-5-oxo-l,6-naphthyridin-6(5H)-yl)propanoate was collected as a pale yellow oil.
The remaining residue was partitioned between benzene and 6N aq hydrochloric acid (35.9 mL). The acidic layer was extracted with benzene (3x), diethyl ether (2x), ethyl acetate (2x) and dichloromethane (lx). The dichloromethane layer was back extracted with 6N aq. Hydrochloric acid (2x). The aqueous layer was diluted with THF (80 mL). The mixture was heated at 80 °C for 3 h. The reaction mixture was concentrated to remove the THF. The remaining acidic water layer was extracted with ethyl acetate and dichloromethane. The aqueous layer was concentrated under vacuum. The remaining solid was triturated with methanol. The mixture was filtered to remove the solid (naphthyridone). The methanol layer was concentrated under vacuum. The remaining solid was dried overnight on a freeze drier. 10.2 g of material was collected as a yellow solid. NAPA made up 72% of the material as determined by HPLC.
1.0 g of the crude material was dissolved in minimal hot iPrOH then filtered and cooled to RT. Crystallization didn’t occur; therefore the solution was cooled in the freezer overnight. A yellow precipitate formed. The solid was collected on a glass frit and was washed with minimal iPrOH. 171 mg of yellow solid was collected, which was NAPA with a small amount of naphthyridone by LC-MS and 1H NMR.
Acid-base extraction. About 1 g of the crude material was dissolved in saturated aqueous sodium bicarbonate. The crude material was extracted with dichloromethane. The pH of the aqueous layer was adjusted to 6-7 with acetic acid then extracted with dichloromethane. 11 mg of the product was isolated; the majority of the product remained in the aqueous layer. The pH was reduced to approximately 4-5 with additional acetic acid. The aqueous layer was extracted with dichloromethane, ethyl acetate, and 15% methanol/dichloromethane. The organic layers were concentrated under vacuum to yield 260 mg of NAPA as the free base, as determined by LC-MS.
Thus, Example 10 shows the synthesis of NAPA according to the disclosure.
EXAMPLE 12
SYNTHESIS OF BISULFITE ADDUCT
DMSO
(COCI)2
MeCX ,ΟΗ Et3N
O

aqueous solution
Scheme 12: Synthesis of bisulfite adduct
Method 1
The bisulfite adduct was synthesize according to Method 1 of Scheme 12 by the following procedure. A 2L round-bottom flask (RBF) was purged with nitrogen and charged with 73.1 mL of reagent grade oxalyl chloride and 693 mL methylene chloride. The batch was cooled to less than -40 °C. 88 mL of dimethyl sulfoxide was added to the flask via an addition funnel at less than -40 °C. After the addition, the batch was stirred for 10 in at -60 °C. 97 mL diethylene glycol monomethyl ether was added to the flask at less than -50 °C over 10 min. The resulting white slurry was stirred at -60 °C for 30 min. 229 mL triethylamine was added to the flask via an addition funnel at less than -30 °C over 1 h. The batch was warmed to RT. 300 mL MTBE was added to the flask and the batch was stirred for 15 min. The slurry was filtered through a fritted funnel and the cake was washed with 300 mL MTBE. The filtrate was concentrated to 350-400g and then filtered again to remove triethylamine-HCl salt, and the solid was rinsed with MTBE, resulting in 357.7 g of a slightly yellow filtrate solution. The solution was assayed by QNMR and comprised 19 wt (68 g) of the desired aldehyde (70% crude yield). The solution was concentrated to 150.2 g.
A 500 mL RBF was charged with 60.0 g sodium bisulfite and 150 mL of water to give a clear solution. The concentrated aldehyde solution was added to the aqueous bisulfite solution over 5 min. An exothermic temperature rising was observed up to 60 °C from 18 °C. The solution was rinsed with 15 mL water. The resulting yellow solution was cooled to RT and was stirred under a sweep of nitrogen overnight.. A QNMR of the solution was taken. The solution contained 43 wt.% of the bisulfite adduct (300 g, 70% yield).
Method 2
The bisulfite adduct was synthesized according to Method 2 of Scheme 12 by the following procedure. A 2500 L reactor was flushed with nitrogen and charged with 657.5 L of 2-methoxyethanol. 62.6 kg of lithium hydroxide monohydrate was added to the reactor while maintaining the temperature at less than 30 °C. The reactor was heated to 113+7 °C. 270 L of solvent were distilled over 1 h and then the reactor temperature was adjusted to 110 °C. 269.4 kg of bromoacetaldehyde diethyl acetal was added over 16 minutes, maintaining the temperature between 110 and 120 °C. The reaction was heated to reflux (115-127°C) for 13 hours. A sample of the reaction was assayed and conversion to 2-(2-methoxyethoxy)acetaldehyde was found to be 98.3%. The reaction was cooled to 15-20°C and 1305 L of methyl ie/t-butyl ether (MTBE) and 132 L of deionized water was added to the reactor. The reaction was stirred for 20 min and then was decanted. The aqueous layer was transferred into a 1600 L reactor and the organic layer was kept in the first reactor. The aqueous layer was extracted with 260 L of MTBE for 10 min. After 10 min decantation, the aqueous layer was removed and the organic layer was transferred to the first reactor. The mixed organic layers were washed twice, 15 min each, with a mixture of concentrated sodium hydroxide solution (2x 17.3 kg) diluted in deionized water (2x 120 L). The aqueous layers were removed, and the organic layer was concentrated at atmospheric pressure at 60-65 °C until the volume was 540 L. The organic layer was cooled down to 15-20 °C to give 2-(2-methoxyethoxy)acetaldehyde as an orange liquid solution (417.4 kg) containing 215.2 kg of pure product (87.3% yield) as determined by 1H NMR and HPLC assay.
A 1600 L reactor, Reactor 3, was flushed with nitrogen and charged with 595 L deionized water followed by 37.8 kg sulfuric acid over 25 minutes via addition funnel, while maintaining the temperature below 25 °C. The addition funnel was washed with 124 L of deionized water and the washing was added to Reactor 3.
A 2500 L reactor, Reactor 4, was flushed with nitrogen and charged with 417.4 kg of the solution of the 2-(2-methoxyethoxy)acetaldehyde. The content of Reactor 3 was transferred into Reactor 4 over 25 min while maintaining the temperature of Reactor 4 below 35 °C. The batch was aged at 30-35 °C for 3 hours. A sample of the batch was taken and assayed for 2-(2- methoxyethoxy)acetaldehyde. No 2-(2-methoxyethoxy)acetaldehyde remained. The batch was aged 5 h then cooled to 15-20 °C.
A solution of sodium carbonate (39.2 kg) in deionized water (196 L) was prepared in Reactor 3. The sodium carbonate solution was transferred to Reactor 4 over 25 min while maintaining the temperature of Reactor 4 below 30 °C. The pH of the resulting mixture was pH 5-6. 1.0 kg sodium carbonate was added by portion until the pH was about 7-8. A solution of sodium bisulfite (116.5 kg) in deionized water (218 L) was prepared in Reactor 3. The sodium bisulfite solution was transferred to Reactor 4 over 20 min while maintaining the temperature of Reactor 4 below 30 °C. Reactor 3 was washed with deionized water (15 L) and the washing was added to Reactor 4. The batch was stirred for 1.2 hours. 23.3 kg sodium bisulfite was added to Reactor 4 and the batch was aged overnight. The batch was concentrated under vacuum at 30-50 °C over 6.5 hours until precipitation was observed. The batch was cooled to 0-10°C at atmospheric pressure. After 30 min at 0-10 °C, the suspension was filtered on 2 filters. Reactor 4 was washed with deionized water (2x 23 L). The first washing was used to rinse the solid on the first filter and the second washing was used to rinse the solid on the second filter. Filtrates were joined to give 473.9 kg of an aqueous solution of the bisulfite adduct (202.5 kg of pure product, 76.3% yield) as a yellow liquid.
Thus, Example 11 shows the synthesis of the bisulfite adduct according to the invention.
EXAMPLE 13
SYNTHESIS OF 2,3-DIFLUORO-5-(l-METHYL-lH-PYRAZOL-4-YL)PYRIDINE

Scheme 13: Synthesis of 2,3-difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine, precursor to PYRH
2,3-Difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine was synthesized according to Scheme 13 by the following procedure. A boronic-ate complex slurry was prepared in a first 3-neck-2-L round-bottom flask (RBF #1). RBF #1 was charged with 141 g (66.4 wt%, 0.9 equivalents based on boronic ester) of lithium 2-hydroxy-4,4,5,5-tetramethyl-2-(l-methyl-lH-pyrazol-4-yl)-l,3,2-dioxaborolan-2-uide. 120 mL (1.6 Vol relative to 5-chloro-2,3-difluoropyridine) of nitrogen- sparged (2 h) 2-BuOH and 120 mL (1.6 Vol) nitrogen-sparged (2 h) water were added to RBF #1. Agitation and N2 sweep were initiated. The reaction was aged at 20 °C for at least 30 min (reactions aged to 24 h were also successful).
] A second 3-neck-2-L round-bottom flask (RBF #2) was charged with 1.48 g (0.004 equivalents) of Xphos-palladacycle and 450 mL (6 Vol relative to 5-chloro-2,3-difluoropyridine) of nitrogen- sparged (2 h) 2-BuOH. Vacuum/N2 flush was cycled through RBF #2 three times to inert the RBF with N2. The batch in RBF #2 was heated to 80 °C. 75 g (1.0 equivalents) of 5-chloro-2,3-difluoropyridine was added to RBF #2.
The slurry of boronic-ate complex was transferred from RBF #1 to a 500 mL dropping funnel. RBF #1 was rinsed with 30 mL (0.4 Vol) 2-BuOH. Using the dropping funnel, the slurry of boronic-ate complex was added over 1 h to the hot solution mixture in RBF #2. After 1 h, 95% conversion was observed. If greater than 90% conversion was not observed, additional boronic-ate complex slurry was added (0.1 equivalents at a time with 1.6 Vol of 1: 1 2-BuOH/water relative to boronic-ate complex). After the conversion was complete, the batch was cooled to 50 °C. While cooling, 600 mL (8 Vol) of toluene was added to RBF #2. 300 mL (4 Vol) of 20% w/v NaHS03 in water was added to RBF #2 and the batch was stirred at 50 °C for at least 1 h. The batch was polish filtered using a 5 micron Whatman filter at 50 °C, into a 2-L Atlas reactor. RBF #2 was rinsed with 30 mL (4.0 Vol) of a 1: 1 2-BuOH:toluene solution. The temperature of the batch was adjusted to 50 °C in the Atlas reactor while stirring. The stirring was stopped and the phases were allowed to settle for at least 15 min while maintaining the batch at 50 °C. The bottom, aqueous layer was separated from the batch. The Atlas reactor was charged with 300 mL (4 Vol) of a 20% w/v NaHS03 solution and the batch was stirred at 50°C for 1 h. The agitation was stopped and the phases were allowed to settle for at least 15 min at 50 °C. The bottom, aqueous layer was removed. Agitation was initiated and the Atlas reactor was charged with 200 mL (4 Vol) of 0.5 M KF while keeping the batch at 50 °C for at least 30 min. The agitation was stopped and the phases were allowed to settle for at least 15 min at 50 °C. The bottom, aqueous layer was removed. Agitation was initiated and the reactor was charged with 300 mL (4 Vol) of water. The batch was aged at 50 °C for at least 30 min. Agitation was stopped and the phases were allowed to settle for at least 15 min at 50 °C. The bottom, aqueous later was removed.
The organic phase was concentrated by distillation under reduced pressure (180 torr, jacket temp 70°C, internal temp about 50 °C) to a minimal stir volume (about 225 mL). 525 mL (7 Vol) of 2-BuOH was added to the Atlas reactor. The organic batch was again concentrated using reduced pressure (85-95 torr, jacket temp 75 °C, internal temp about 55 °C) to a minimal stir volume (about 125 mL). The total volume of the batch was adjusted to 250 mL with 2-BuOH.
525 mL (7 Vol) heptane was added to the slurry mixture in the Atlas reactor. The jacket temperature was adjusted to 100 °C and the batch was aged for more than 15 min, until the batch became homogeneous. The batch was cooled to 20 °C over at least 3 h. A sample of the mixture was taken and the supernatant assayed for 2,3-difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine. If the concentration was greater than 10 mg/mL, the aging was continued for at least 1 h until the supernatant concentration was less than 10 mg/mL. The batch was filtered using a medium frit. The filter cake was washed with 150 mL (2 Vol) 30% 2-BuOH/heptane solution followed by 150 mL (2 Vol) heptane. The filter cake was dried under N2/vacuum. 76.64 g of 2,3-difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine was isolated as a white solid (87% yield).
A 60 L jacketed reactor was fitted with a reflux condenser. The condenser cooling was initiated at 0+5 °C. The reactor was charged with 2612 g (1 equivalent) of 2,3-difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine and placed under an atmosphere of nitrogen. 31.7 L (12.2 Vol) water was added to the reactor and the resulting slurry was nitrogen sparged for 1 h with agitation. 7221 mL (6 equivalents) of hydrazine (35 wt% in water) was added to the reactor under a nitrogen atmosphere. The reactor was heated to 100 °C for 2+2 h until reaction was complete by HPLC analysis. The reactor was cooled to 20 °C over 2+1 h at a rate of 40°C/h. The reactor contents were stirred for 10+9 hours until the desired supernatant assay (< 2mg/mL PYRH in mother liquor). The reactor contents were filtered through an Aurora filter fitted with 25 μιη polypropylene filter cloth. The collected filter cake was washed with 12.0 L (4.6 V) of water in three portions. The filter cake was dried on the Aurora filter for 4-24 h at 22+5 °C, or until the product contained less than 0.5% water as determined by KF. The dry product was collected. 2.69 kg (97% yield) 2,3-Difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine was collected as a white crystalline solid. The solid had a water content of 12 ppm as determined by KF.
Thus, Example 12 shows the synthesis of 2,3-Difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine, a precursor to PYRH, according to the disclosure.
EXAMPLE 14
SYNTHESIS OF PYRH – ROUTE 2

Scheme 14: Synthesis of 3-fluoro-2-hydrazinyl-5-(l-methyl-lH-pyrazol-4-yl)-pyridine (PYRH)
3-fluoro-2-hydrazinyl-5-(l-methyl-lH-pyrazol-4-yl)-pyridine was synthesized according to Scheme 14 by the following procedure. A 60 L jacketed reactor was fitted with a 5 L addition funnel and the jacket temperature was set to 20+5 °C. 36.0 L (15 Vol) of 2-methyltetrahydrofuran was added to the reactor via a 20 μιη inline filter with vacuum using polypropylene transfer lines. The solution was sparged by bubbling nitrogen through a dipstick in the solution for 1+0.5 h with agitation. After 1 h the dipstick was removed but the nitrogen sweep continued. 1.55 kg of sparged 2-MeTHF was removed to be used as rinse volumes. 36.7 g of Pd2dba3, 75.6 g X-Phos, 259 g of tetrabutylammonium bromide, and 7397 g of potassium phosphate tribasic were added to the reactor. The manhole was rinsed with 0.125 kg of sparged 2-MeTHF. The reactor was agitated and the nitrogen sweep continued for 1+0.5 h. Then the nitrogen sweep was stopped and the reaction left under a positive pressure of nitrogen.
3.6 L (1.5 Vol) of sparged water was prepared in advance by bubbling nitrogen through a 4 L bottle of water for 1+0.5 h. The nitrogen sparged water was transferred to the 5 L addition funnel via a 20 μηι inline filter with vacuum using polypropylene transfer lines, then slowly added to the reaction while maintaining the internal temperature at 20+5 °C. The 5 L addition funnel was replaced with a 2 L addition funnel. 2412 g of 5-chloro-2,3-difluoropyridine was added to the 2 L addition funnel. The 5-chloro-2,3-difluoropyridine was then added to the reaction through the 2 L addition funnel. The 2L addition funnel was rinsed with 0.060 kg of sparged 2-MeTHF. 83.8 g (1.15 equivalents) of l-methylpyrazole-4-boronic acid, pinacol ester was added to reactor, the reactor was swept with nitrogen for 1+0.5 h, then left under a positive pressure of nitrogen. The internal temperature of the reactor was adjusted to 70+5 °C. The batch was agitated at 70+5 °C for at least 4 hours after the final reagent was added. A sample was taken from the reaction and the reaction progress assayed for conversion. The progress of the reaction was checked every 2 hours until the reaction was completed (e.g., greater than 99% conversion). The batch was cooled to 20+5 °C.
A 20% w/v sodium bisulfite solution (12.0 L, 5 Vol) was prepared by charging 12.0 L of water then 2411 g sodium bisulfite to an appropriate container and agitating until
homogeneous. The 20% sodium bisulfite solution was transferred into the reactor and agitated for 30 minutes. The agitation was stopped, the phases allowed to settle, and the aqueous phase was removed. A 0.5 M potassium fluoride solution (12.0 L, 5 Vol) was prepared by charging 12.0 L of water and 348 g of potassium fluoride to an appropriate container and agitating until homogenous. The 0.5 M potassium fluoride solution was transferred into the reactor and agitated for 30 min. The agitation was stopped, the phases were allowed to settle, and the aqueous phase was removed. A 25% w/v sodium chloride solution (12.0 L, 5 Vol) was prepared by charging an appropriate container with 12.0 L of water and 2999 g of sodium chloride and agitating until homogeneous. The 25% sodium chloride solution was transferred into the reactor and agitated for 30 min. The agitation was stopped, the phases were allowed to settle, and the aqueous phase was removed from the reactor.
The organic phase was distilled at constant volume (36 L, 15 Vol) while maintaining the internal temperature of the reactor at 50+5 °C by adjusting the vacuum pressure until no more than 0.3% of water remained. 2-Methyltetrahydrofuran was added to the reactor as needed to
maintain constant volume. The batch was cooled to 20 °C and transferred into drums. The batch was transferred using a polish filter (using a 5 μιη inline filter) into a 60 L jacketed reactor with a batched concentrator attached. 1.2 L of 2-MeTHF was used to rinse the drums. The batch was concentrated to about 9 Vol while maintaining the internal temperature of the vessel at 50+5 °C by adjusting the vacuum pressure. The batch was then distilled at constant volume (22.0 L, 9Vol) while maintaining the internal temperature of the vessel at 50+5 °C by adjusting the vacuum pressure. Heptane was added with residual vacuum until a 15% 2-MeTHF:heptane supernatant mixture was obtained. The pressure was brought to atmospheric pressure under nitrogen. The reactor was cooled to 20+5 °C over 2+2 h. The batch was agitated at 20+5 °C until an assay of the supernatant indicated that the amount of product was 7 mg/mL 2,3-difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine.
A 10% 2-MeTHF:heptane (7.2 L, 3 Vol) wash solution was prepared by mixing 720 mL of 2-MeTHF and 6.5 L of heptane. The batch slurry was filtered through an Aurora filter fitted with a 25 μιη polypropylene filter cloth, resulting in heavy crystals that required pumping with a diaphragm pump using polypropylene transfer lines through the top of the reactor while stirring. The mother liquor was recycled to complete the transfer. The reactor and filter cake were washed with two portions of the 10% 2-MeTHF:heptane wash solution (3.6 L each). The product cake was dried on a frit under a nitrogen stream at ambient temperature. The 2,3-difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine was determined to be dry when the 1H NMR assay was < 0.05+0.05. 2.635 kg was isolated as an off white crystalline solid (85% yield).
A 60 L jacketed reactor was fitted with a reflux condenser. The condenser cooling was initiated at 0+5 °C. The reactor was charged with 2612 g (1 equivalent) of 2,3-difluoro-5-(l-methyl-lH-pyrazol-4-yl)pyridine and placed under an atmosphere of nitrogen. 31.7 L (12.2 Vol) water was added to the reactor and the resulting slurry was nitrogen sparged for 1 h with agitation. 7221 mL (6 equivalents) of hydrazine (35 wt% in water) was added to the reactor under a nitrogen atmosphere. The reactor was heated to 100 °C for 2+2 h until reaction was complete by HPLC analysis. The reactor was cooled to 20 °C over 2+1 h at a rate of 40°C/h. The reactor contents were stirred for 10+9 hours until the desired supernatant assay was reached (< 2mg/mL PYRH in mother liquor). The reactor contents were filtered through an Aurora filter fitted with 25 μιη polypropylene filter cloth. The collected filter cake was washed with 12.0 L
(4.6 V) of water in three portions. The filter cake was dried on the Aurora filter for 4-24 h at 22+5 °C, or until the product contained less than 0.5% water as determined by KF. The dry product was collected. 2.69 kg was isolated as a white crystalline solid (97% yield). The water content was determined to be 12 ppm by KF.
| WO2007075567A1 * |
Dec 18, 2006 |
Jul 5, 2007 |
Janssen Pharmaceutica, N.V. |
Triazolopyridazines as tyrosine kinase modulators |
| WO2007138472A2 * |
May 18, 2007 |
Dec 6, 2007 |
Pfizer Products Inc. |
Triazolopyridazine derivatives |
| WO2008008539A2 * |
Jul 13, 2007 |
Jan 17, 2008 |
Amgen Inc. |
Fused heterocyclic derivatives useful as inhibitors of the hepatocyte growth factor receptor |
| WO2008051805A2 * |
Oct 18, 2007 |
May 2, 2008 |
Sgx Pharmaceuticals, Inc. |
Triazolo-pyridazine protein kinase modulators |
| WO2008155378A1 * |
Jun 19, 2008 |
Dec 24, 2008 |
Janssen Pharmaceutica Nv |
Polymorphic and hydrate forms, salts and process for preparing 6-{difluoro[6-(1-methyl-1h-pyrazol-4-yl)[1,2,4]triazolo[4,3-b]pyridazin-3-yl]methyl}quinoline |
References:
1. Hughes, P. E.; et. al. Abstract 728: AMG 337, a novel, potent and selective MET kinase inhibitor, has robust growth inhibitory activity in MET-dependent cancer models. Cancer Res 2014, 74, 728.
2. Boezio, A. A.; et. al. Discovery and optimization of potent and selective triazolopyridazine series of c-Met inhibitors. Bioorg Med Chem Lett 2009, 19(22), 6307-6312.
3. ClinicalTrials.gov Phase 2 Study of AMG 337 in MET Amplified Gastric/Esophageal Adenocarcinoma or Other Solid Tumors. NCT02016534 (retrieved 10-06-2015)
4. ClinicalTrials.gov A Study of AMG 337 in Subjects With Advanced Solid Tumors. NCT01253707 (retrieved 10-06-2015)
/////////// AMG-337, AMG337, AMG 337, 1173699-31-4, AMGEN, ESOPHAGUS
O=C1C2=C(N=CC(OCCOC)=C2)C=CN1[C@@H](C3=NN=C4C(F)=CC(C5=CN(C)N=C5)=CN43)C

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

























 Drug Authorization Procedures in the EU
Drug Authorization Procedures in the EU 


















































 L. Nathan Tumey, Ph.D., Principal Research Scientist, Pfizer Global R&D
L. Nathan Tumey, Ph.D., Principal Research Scientist, Pfizer Global R&D





 Susanta Samajdar, Ph.D., Research Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Susanta Samajdar, Ph.D., Research Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited









 LIONEL MY SON
LIONEL MY SON