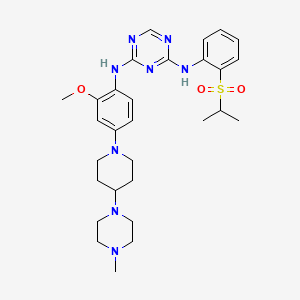
ASP3026
ASP3026;
CAS 1097917-15-1; ASP-3026; ASP 3026; UNII-HP4L6MXF10;
N2-[2-Methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl]phenyl]-N4-[2-[(1-methylethyl)sulfonyl]phenyl]-1,3,5-triazine-2,4-diamine;
2-N-[2-methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl]-4-N-(2-propan-2-ylsulfonylphenyl)-1,3,5-triazine-2,4-diamine
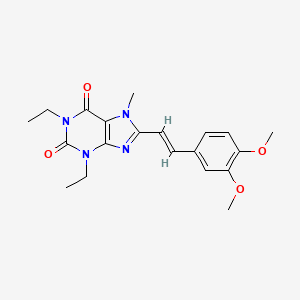
(N-{2-methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl}-N′-[2-(propane-2-sulfonyl)phenyl]-1,3,5-triazine-2,4-diamine) was developed as a novel selective inhibitor of the fusion protein EML4-ALK.
1H NMR (CDCl3, 400 MHz) (ppm) = 1.31 (d, 6H, J = 6.8 Hz), 1.58–1.80 (m, 4H), 1.90–2.04 (m, 2H), 2.16–2.84 (m, 12H), 3.18–3.32 (m, 1H), 3.66–3.76 (m, 2H), 3.88 (s, 3H), 6.48–6.60 (m, 2H), 7.18–7.26 (m, 1H), 7.50–7.72 (m, 2H), 7.86–7.92 (dd, 1H, J = 1.2 Hz, J = 7.6 Hz), 8.06–8.16 (m, 1H), 8.28–8.48 (m, 1H), 8.48–8.62 (m, 1H), 9.28 (s, 1H).
| Molecular Formula: |
C29H40N8O3S |
| Molecular Weight: |
580.7447 g/mol |
ASP3026 is a novel and selective inhibitor for the ALK kinase. ASP3026 potently inhibited ALK kinase activity and was more selective than crizotinib in a Tyr-kinase panel. In an anchorage independent in vitro cell growth assay, ASP3026 inhibited the growth of NCI-H2228, a human NSCLC tumor cell line endogenously expressing EML4-ALK variant 3 and that of 3T3 cells expressing EML4-ALK variant 1, 2 and 3. The plasma and tumor concentrations of ASP3026 in mice xenografted with NCI-H2228 tumor were determined using high-performance liquid chromatography-tandem mass spectrometry. Significant tumor penetration was observed. The antitumor activities were evaluated using mice bearing subcutaneous NCI-H2228 tumor xenografts.
ASP-3026 was studied in P1 clinical trials at Astellas Pharma for the oral treatment of advanced solid tumors and advanced B-cell lymphoma. In 2014 the product was discontinued by Astellas due to strategic reasons

JP 2012153674
WO 2012102393
WO 2011145548
WO 2009008371

PATENT
WO2012102393
The compound of the formula (1) has an excellent EML4-ALK fusion protein and inhibitory activity of the kinase of the mutant EGFR protein, we are already reported to be useful as an active ingredient of a pharmaceutical composition for cancer treatment (Patent Document 1). Further, it is the compound of formula (1) there are five polymorphs shown as A01 ~ A05 type, among others A04 type crystal is in finding reported that the most stable type crystals (Japanese Patent Document 2).
[Formula 1] a compound of formula (1) described in Patent Document 1 production method of (Patent Document 1 of Example 23), referring to Production Examples and Examples described in this document, the reaction formula (I) It is shown in. That is, 2,4-dichloro-1,3,5-triazine (hereinafter, may be referred to as “compound of formula (15)”.), 2- (isopropylsulfonyl) aniline (hereinafter, “the formula (8) sometimes referred to compound “.) using, by reacting according to the method described in production example 7 of this document, to give compounds of formula (14) to (production example 22 of Patent Document 1), then , the resulting compound of formula (14), a known method (e.g., International Publication No. 2005/016894 pamphlet reference) was prepared by 2-methoxy-4- [4- (4-methylpiperazin-1- yl) piperidin-1-yl] aniline (hereinafter, may be referred to as “formula (13) compounds of.”) is used to react according to the method described in example 1 of the document, and the target it is a method for producing a compound of formula (1) to.

[Formula 2]

Patent Document 1: International Publication No. 2009/008371 pamphlet
Patent Document 2: WO 2011/145548 pamphlet

Example 1
The first step 4,4-dimethoxy-1- (3-methoxy-4-nitrophenyl) piperidine (R 1 and R 2 Synthesis of methyl Any compound of formula (10))
4,4 – N and dimethoxy piperidine monohydrochloride (35.9 g), N-dimethylformamide and (75 mL) were mixed, and the mixed solution of 1,8-diazabicyclo [5.4.0] undec-7-ene (57.5 mL) was added It was. It was separately prepared here 5-fluoro-2-nitroanisole (30.0 g) and N, N-dimethylformamide (30 mL) was stirred for 5 hours at room temperature. Water (120 mL) was added at room temperature to the reaction mixture, after stirring for 4 hours, the precipitated crystals were collected by filtration. The resulting crystals N, N-dimethylformamide and a mixed solution of water (1: 1) (60mL) , water (60 mL), was further washed sequentially with water (60 mL), and dried under reduced pressure at 40 ° C. to give 4,4-dimethoxy-1- (3-methoxy-4-nitrophenyl) piperidine (49.9 g, 96.1% yield) as crystals.
D2: 1.72-1.80 (4H, m) , 3.14 (6H, s), 3.44-3.50 (4H, m), 3.91 (3H, m), 6.52 (1H, d, J = 2.4Hz), 6.60 (1H, dd, J = 2.4,9.2Hz), 7.88 (1H, D, J = 9.2Hz)
ESI Tasu: 297
The second step 4- (R (4,4-dimethoxy-1-yl) -2-methoxyaniline 1 and R 2 is methyl none has the formula (Compound 6)) Synthesis of
4,4-dimethoxy – 1- (3-methoxy-4-nitrophenyl) piperidine and (45.0 g) in tetrahydrofuran and a (225 mL) were mixed, 5% palladium carbon (about 50% wet product, 4.5 g) to this mixed solution was added at room temperature, under a hydrogen atmosphere (2.4821×10 5 Pa), and the mixture was stirred for 5 hours and a half at room temperature. Then filtered off and palladium-carbon, washed with tetrahydrofuran (90mL), was concentrated under reduced pressure filtrate until total volume of about 90mL obtain a slurry. After the slurry was stirred for 1 hour at 40 ° C., n- heptane (135 mL) was added and after stirring for 1 hour at 40 ° C., cooled to 0 ° C., was added n- heptane (405 mL), precipitated crystals It was collected by filtration.The obtained crystals were washed with a mixed solution of tetrahydrofuran (9 mL) and n- heptane (54 mL), and dried in vacuo at 40 ℃, 4- (4,4- dimethoxy-1-yl) -2-methoxy to give aniline (37.9g, 93.7% yield) as crystals.
D2: 1.72-1.80 (4H, m) , 2.90-2.97 (4H, m), 3.11 (6H, s), 3.73 (3H, m), 4.21 (1H, br), 6.30 (1H, d, J = 2.4 , 8.4Hz), 6.46_6.56 (2H, M)
ESI Tasu: 267
The third step 4,6-dichloro-N- [2-(propane-2-sulfonyl) phenyl] -1,3,5-triazin-2-amine (Lv is Cl any, compounds of formula (7) synthesis of)
cyanuric chloride (25.0 g), sodium bicarbonate (13.7 g), were mixed 2- (isopropylsulfonyl) aniline (29.7 g) and acetone (200 mL), and stirred at room temperature for 25 hours. After adding water (200 mL) at room temperature the reaction mixture was stirred for 19 hours, the precipitated crystals were collected by filtration. The resulting crystals acetone and a mixed solution of water (1: 1) was washed with (100 mL), and dried in vacuo at 40 ° C., 4,6-dichloro-N- [2-(propane-2-sulfonyl) to give phenyl] -1,3,5-triazin-2-amine (45.1g, 95.8% yield) as crystals.
D1: 1.32 (6H, d, J = 6.8Hz), 3.22 (1H, sept, J = 6.8Hz), 7.37 (1H, m), 7.74 (1H, m), 7.93 (1H, m), 8.44 (1H , M), 10.02 (1H, Br)
ESI-: 345, 347
Fourth step 6-chloro -N- [4- (4,4- dimethoxy-1-yl) -2-methoxy-phenyl] -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3 , (a Lv is Cl, R 5- triazine-2,4-diamine 1and R 2 none is methyl, the formula (compound 5)) synthesis of
4,6-dichloro-N- [2-( propane-2-sulfonyl) phenyl] -1,3,5-triazin-2-amine (40.0 g) was mixed with tetrahydrofuran (400 mL), to this mixed solution 4- (4,4-dimethoxy-piperidin-1 yl) -2-methoxyaniline (32.2 g) and N, N- diisopropylethylamine (16.38g) was stirred for 4 hours at room temperature.Thereafter, isopropyl acetate (40 mL), then extracted by adding a mixed solution of potassium carbonate (2.0 g) and water (40 mL). The obtained organic layer was concentrated under reduced pressure until the total volume of about 200 mL, as a seed crystal, 6-chloro -N- [4- (4,4- dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- inoculated with [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-crystalline diamine (4 mg), to give a slurry and stirred for about 15 minutes. The slurry n- heptane (200 mL) was added and filtered off cooled to 18 hours with stirring to precipitate crystals to 0 ° C.. The resulting crystals were washed with a mixed solution of tetrahydrofuran (40 mL) and n- heptane (40 mL), and dried in vacuo at 40 ° C., 6- Chloro -N- [4- (4,4- dimethoxy-piperidine – 1-yl) -2-methoxyphenyl] -N ‘- [2- (the propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (61.4 g, 92.4% yield) It was obtained as a crystal.
D1: 1.30 (6H, d, J = 6.8Hz), 1.88-1.92 (4H, m), 3.18-3.26 (1H, m), 3.23 (3H, s), 3.87 (1H, br), 6.53 (2H, br), 7.21-7.23 (1H, m ), 7.62 (1H, br), 7.88 (1H, d, J = 7.9Hz), 8.05 (1H, br), 8.48 (1H, br), 9.41 (1H, br )
ESI-: 575,577
The fourth alternative process (e.g. without using a seed crystal) 6-Chloro-N- [4- (4,4-dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- [2- (propane 2-sulfonyl) phenyl] (a Lv is Cl, R-1,3,5-triazine-2,4-diamine 1 and R 2 none is methyl, the formula (5) synthesis of compound of)
4 , and mixed 6-dichloro -N- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazin-2-amine (23.0 g) in tetrahydrofuran (230 mL), to this mixed solution 4- (4,4-dimethoxy-1-yl) -2-methoxyaniline (18.5 g) and N, N- diisopropylethylamine (12.7 mL) was stirred for 2 hours at room temperature. Thereafter, isopropyl acetate (57.5 mL), then extracted by adding potassium carbonate (5.75 g) and a mixed solution of water (115 mL). The resulting organic layer was concentrated under reduced pressure. The resulting residue is added and stirred in tetrahydrofuran (50mL) to obtain a slurry. After stirring for 1 hour at the slurry was added tetrahydrofuran (75 mL) and n- heptane (75mL) 40 ℃, cooled to 0 ° C., and stirred for a further 18 hours.Thereafter, n- heptane (50 mL) was added, and the precipitated crystals were collected by filtration. The resulting crystals tetrahydrofuran and n- heptane mixed solution (5: 7) After washing with (24 mL), and dried in vacuo at 40 ° C., 6- chloro-N- [4- (4,4-dimethoxy piperidin-1-yl) -2-methoxyphenyl] -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (30.6g, 80.0% yield ) was obtained as a crystal.
D1: 1.30 (6H, d, J = 6.8Hz), 1.88-1.92 (4H, m), 3.18-3.26 (1H, m), 3.23 (3H, s), 3.87 (1H, br), 6.53 (2H, br), 7.21-7.23 (1H, m ), 7.62 (1H, br), 7.88 (1H, d, J = 7.9Hz), 8.05 (1H, br), 8.48 (1H, br), 9.41 (1H, br )
ESI-: 575,577
The fifth step and the sixth step (continuous process) 1- [3-methoxy-4 – ({4- [2- (propane-2-sulfonyl) anilino] -1,3,5-triazin-2-yl} amino ) phenyl] piperidin-4-one synthesis of compound) (formula (3)
6-chloro-N- [4- (4,4-dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- [ 2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (60.0 g), tetrahydrofuran (540 mL) and 10% palladium carbon (about 50% wet product, 10.7 g) and mixed, N to the mixture, added to N- diisopropylethylamine (16.11g) and 2-propanol (60 mL), under a hydrogen atmosphere (2.4131X10 5 of 5 Pa), and stirred for 7 hours at 40 ° C.. Filtration of the palladium-carbon, and washed with tetrahydrofuran (120 mL), the resulting filtrate activated carbon (12.0 g) was added to, and stirred at room temperature overnight. Then filtered off and the activated carbon, and washed with tetrahydrofuran (120mL), N- [4- ( 4,4- dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- [2- (propane – to obtain a solution containing 2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine. To this solution was added a mixed solution of 35% hydrochloric acid (21.7 g) and water (120 mL), and stirred for 21 hours at room temperature. To the reaction mixture, it was added a mixed solution of potassium carbonate (35.9 g) and water (120 mL), and extracted. Activated carbon (12.0 g) was added to the obtained organic layer was stirred for 16 h, filtered, washed with activated carbon in tetrahydrofuran (120 mL). The filtrate obtained total amount was concentrated under reduced pressure to approximately 120 mL. After addition of acetone (180 mL) to the resulting mixture, as a seed crystal, 1- [3-methoxy-4 – ({4- [2- (propane-2-sulfonyl) anilino] -1,3,5 after stirring for 1 hour and inoculated triazin-2-yl} amino) phenyl] piperidin-4-one crystals (60 mg), water (480 mL) was stirred for 20 hours was added, and the precipitated crystals were collected by filtration . The obtained crystals were washed with a mixed solution of acetone (36 mL) and water (96 mL), and dried in vacuo at 40 ℃, 1- [3- methoxy-4 – ({4- [2- (propane -2 – was obtained sulfonyl) anilino] -1,3,5-triazine-2-yl} amino) phenyl] piperidine-4-one (45.8g, 88.7% yield (yield in a continuous two steps)) as crystals .
D2,343K: 1.17 (6H, d, J = 6.8Hz), 2.46-2.50 (4H, m), 3.40 (1H, sept, J = 6.8Hz), 3.61 (4H, dd, J = 6.1,6.2Hz) , 3.79 (3H, s), 6.57 (1H, dd, J = 2.6,8.7Hz), 6.70 (1H, d, J = 2.6Hz), 7.25-7.29 (1H, m), 7.38 (1H, d, J 8.7 Hz =), 7.61 (1H, br), 7.77-7.80 (1H, yd), 8.28 (1H, s), 8.50 (1H, br), 8.66 (1H, br), 9.25 (1H, br)
ESI +: 497
Fifth Step N- [4- (4,4- dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine 2,4-diamine (R 1 and R 2 is methyl any formula (4) of compound) synthesis of
6-chloro-N- [4- (4,4-dimethoxy-1-yl) – 2-methoxyphenyl] -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (5.0 g), tetrahydrofuran (45 mL), 2-propanol (5mL ), 10% palladium-carbon (about 50% wet product, 1.0 g) were mixed, added N, N- diisopropylethylamine (1.81 mL) to this mixed solution, under a hydrogen atmosphere (2.4821X10 5 of 5 Pa), 40 ° C. in and the mixture was stirred for 5 hours and a half. Filtration of the palladium-carbon was washed with tetrahydrofuran (10 mL), and extraction was performed with 10% brine (20 mL). The resulting organic layer was concentrated under reduced pressure. Acetone to the concentrated residue (10 mL), was added diisopropyl ether (40 mL), it was collected by filtration stirred precipitated crystals 30 minutes. The obtained crystals were washed with diisopropyl ether (20 mL), and dried in vacuo at 40 ℃, N- [4- (4,4- dimethoxy-1-yl) -2-methoxyphenyl]-N’- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (4.31 g, 91.6% yield) as crystals.
D2,343K: 1.17 (6H, d, J = 6.8Hz), 1.80 (4H, dd, J = 5.5,5.7Hz), 3.15 (6H, s), 3.21 (4H, dd, J = 5.5,5.7Hz) , 3.77 (3H, s), 6.50 (1H, dd, J = 2.5,8.7Hz), 6.62 (1H, d, J = 2.5Hz), 7.25-7.28 (1H, m), 7.34 (1H, d, J 8.7 Hz =), 7.58 (1H, br), 7.77-7.79 (1H, yd), 8.28 (1H, s), 8.49 (1H, br), 8.63 (1H, br), 9.25 (1H, br)
ESI +: 543
Sixth Step 1- [3-methoxy-4 – ({4- [2- (propane-2-sulfonyl) anilino] -1,3,5-triazin-2-yl} amino) phenyl] piperidin-4-one (equation (3) a compound of) synthesis of
N- [4- (4,4- dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- [2- (propane-2-sulfonyl) phenyl] – 1,3,5-triazine-2,4-diamine (4.0 g), and tetrahydrofuran (36 mL) and 2-propanol (4 mL) solution of 35% hydrochloric acid containing (1.44 g) a mixture of water (4 mL) was added on, and the mixture was stirred for 17 hours at room temperature. To the reaction mixture, it was added a mixed solution of potassium carbonate (2.4 g) and water (4 mL), and extracted.The resulting organic layer was concentrated under reduced pressure. After stirring for 30 minutes by addition of acetone (12 mL) and water (4 mL) to the concentrated residue, add water (28 mL) was stirred for 1 hour, the precipitated crystals were collected by filtration. The obtained crystals were washed with a mixed solution of acetone (8 mL) and tetrahydrofuran (3 mL), and dried in vacuo at 40 ℃, 1- [3- methoxy-4 – ({4- [2- (propane -2 – give sulfonyl) anilino] -1,3,5-triazin-2-yl} amino) phenyl] piperidin-4-one (3.42g, 99.2% yield) as crystals.
D2,343K: 1.17 (6H, d, J = 6.8Hz), 2.46-2.50 (4H, m), 3.40 (1H, sept, J = 6.8Hz), 3.61 (4H, dd, J = 6.1,6.2Hz) , 3.79 (3H, s), 6.57 (1H, dd, J = 2.6,8.7Hz), 6.70 (1H, d, J = 2.6Hz), 7.25-7.29 (1H, m), 7.38 (1H, d, J 8.7 Hz =), 7.61 (1H, br), 7.77-7.80 (1H, yd), 8.28 (1H, s), 8.50 (1H, br), 8.66 (1H, br), 9.25 (1H, br)
ESI +: 497
Seventh Step N- {2- methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] – 1,3,5-triazine-2,4-diamine (formula (1) compounds) synthesis
of 1- [3-methoxy-4 – ({4- [2- (propane-2-sulfonyl) anilino] -1 , 3,5-triazin-2-yl} amino) phenyl] piperidin-4-one (20.0 g), methyl piperazine (8.07 g), were mixed in toluene (200 mL) and acetic acid (9.0 mL), 1 hour at room temperature It stirred. To this mixture solution was added sodium triacetoxyborohydride (17.06 g), and stirred at room temperature for 20 hours. To the reaction mixture, water (60 mL) and methanol (20 mL) was added, extraction to give an organic layer and an aqueous layer 1. This organic layer, water (20 mL) and re-extracted to give a water layer 2. After mixing the aqueous layer 1 and aqueous layer 2 was extracted by adding isopropyl acetate (200 mL). Methanol (220 mL) to the resulting aqueous layer, a mixed solution of sodium hydroxide (9.68 g) and water (48 mL) was added, as a seed crystal, N-{2-methoxy-4- [4- (4-methylpiperazin- 1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-crystal of diamine (2.0mg) inoculated, after stirring at room temperature for 1.5 hours, add water (220 mL), further stirred for 2 hours at room temperature, the precipitated crystals were collected by filtration. The resulting crystals were washed with a mixed solution of methanol (40mL) and water (40mL), and then dried under reduced pressure at 50 ℃, N- {2- methoxy-4- [4- (4-methyl-piperazine -1 – yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (20.15g, 86.1% yield) It was obtained as A06-form crystals.
D1: 1.31 (6H, d, J = 6.8Hz), 1.59-1.78 (2H, m), 1.90-2.01 (2H, m), 2.24-2.80 (11H, m), 2.30 (3H, s), 3.19- 3.32 (1H, m), 3.65-3.75 (2H, m), 3.88 (3H, s), 6.50-6.59 (2H, m), 7.18-7.30 (1H, m), 7.53-7.70 (2H, m), 7.88 (1H, dd, J = 1.5,8.3Hz), 8.10 (1H, br), 8.37 (1H, br), 8.53 (1H, br), 9.29 (1H, s)
ESI +: 581
Alternatively 1 (Example not using seed crystals) N-{2-methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} -N seventh step ‘- [ 2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (compound of formula (1))
1- [3-methoxy-4 – ({4- [2 – (propane-2-sulfonyl) anilino] -1,3,5-triazin-2-yl} amino) phenyl] piperidin-4-one (5.0 g), methyl piperazine (2.02 g), toluene (50 mL) and acetic acid (1.5 mL) were mixed and stirred at room temperature for 1 hour. To this mixture solution was added sodium triacetoxyborohydride (4.72 g), and stirred at room temperature for 18 hours. To the reaction mixture, water (15 mL) and methanol (5 mL) was added, extraction to give an organic layer and an aqueous layer 1. This organic layer, water (5 mL) and re-extracted to give a water layer 2. After mixing the aqueous layer 1 and aqueous layer 2 was extracted by adding isopropyl acetate (50 mL). The resulting aqueous layer methanol (55 mL), a mixed solution was added sodium hydroxide (2.0 g) and water (10 mL), was stirred for 62 hours at room temperature, add water (55 mL), at room temperature for a further 2 hours stirring, the formed crystals were separated by filtration. The obtained crystals were washed with a mixed solution of methanol (5 mL) and water (5 mL), and dried in vacuo at 40 ℃, N- {2- methoxy-4- [4- (4-methylpiperazin–1 – yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (4.56g, 78.0% yield) It was obtained as A06-form crystals.
D1: 1.31 (6H, d, J = 6.8Hz), 1.59-1.78 (2H, m), 1.90-2.01 (2H, m), 2.24-2.80 (11H, m), 2.30 (3H, s), 3.19- 3.32 (1H, m), 3.65-3.75 (2H, m), 3.88 (3H, s), 6.50-6.59 (2H, m), 7.18-7.30 (1H, m), 7.53-7.70 (2H, m), 7.88 (1H, dd, J = 1.5,8.3Hz), 8.10 (1H, br), 8.37 (1H, br), 8.53 (1H, br), 9.29 (1H, s)
ESI +: 581
alternative seventh step 2 (example using reducing catalyst) N-{2-methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane -2 – sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine synthesis of compounds of formula (1)
1- [3-methoxy-4 – ({4- [2- (propan-2 sulfonyl) anilino] -1,3,5-triazin-2-yl} amino) phenyl] piperidin-4-one (5.0 g), tetrahydrofuran (30 mL), methylpiperazine (1.81 g) and 10% palladium carbon (about 50 % wet product, were mixed 0.8 g), under a hydrogen atmosphere (2.4821X10 5 of 5Pa), and stirred for 7 hours at 40 ° C.. Filtration of the palladium-carbon, and washed with tetrahydrofuran (10 mL), the resulting filtrate was concentrated under reduced pressure. To the concentrated residue 2-butanone (9 mL) was added, followed by stirring at 60 ° C. 30 minutes, cooled slowly, at 30 ° C. n-heptane (9 mL) was added, and stirred for 19 hours at room temperature, the precipitated crystals were collected by filtration did.The resulting crystals of 2-butanone and (1 mL) was washed with a mixture of n- heptane (1 mL), and dried in vacuo at 40 ℃, N- {2- methoxy-4- [4- (4-methyl piperazin-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (3.09 g, yield: 88.0%) was obtained.
D1: 1.31 (6H, d, J = 6.8Hz), 1.59-1.78 (2H, m), 1.90-2.01 (2H, m), 2.24-2.80 (11H, m), 2.30 (3H, s), 3.19- 3.32 (1H, m), 3.65-3.75 (2H, m), 3.88 (3H, s), 6.50-6.59 (2H, m), 7.18-7.30 (1H, m), 7.53-7.70 (2H, m), 7.88 (1H, dd, J = 1.5,8.3Hz), 8.10 (1H, br), 8.37 (1H, br), 8.53 (1H, br), 9.29 (1H, s)
ESI +: 581
N- {2- methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3 , 5-triazine-2,4-diamine by recrystallization purification steps (formula (1 compound of))
(the a method) N-{2-methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (8.80 g), 2-butanone (211 mL) after mixing and confirmation of dissolution and stirring at 65 ° C. 30 minutes for clarifying filtration. After filtrate was total volume concentrated normal pressure to approximately 70 mL, and cooled to 70 ° C., as a seed crystal N- {2- methoxy-4- [4- (4-methylpiperazin-1-yl) piperidine-1 yl] phenyl} -N ‘- [2- inoculated with (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-crystalline diamine (0.9 mg), and stirred for about 10 minutes to obtain a slurry. After stirring for 3 hours at 70 ° C., cooled to 5 ℃ at a rate of 20 ° C. / h and stirred for 17 hours, the precipitated crystals were collected by filtration. The resulting crystals were washed with 2-butanone were cooled with ice water (35.2 mL), and dried in vacuo at 50 ℃, N- {2- methoxy-4- [4- (4-methylpiperazin-1- yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (7.88 g, 89.5% yield, purity 99.4%) was obtained as a A04 type crystal (A04 type ratio 98.9%).
(B method): N- {2- methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl ] -1,3,5-triazine-2,4-diamine (8.80g), was mixed activated carbon (0.88 g) and 2-butanone (211 mL), after stirring for 1 hour at 75 ° C., was subjected to activated carbon filtration .The filtrate activated carbon (0.88g) in addition to, and the mixture was stirred for 1 hour at 75 ℃, was activated carbon filtration. The filtrate activated carbon (0.88g) in addition to, and the mixture was stirred for 1 hour at 75 ℃, was activated carbon filtration. After filtrate was total volume concentrated normal pressure to approximately 70 mL, and cooled to 70 ° C., as a seed crystal N- {2- methoxy-4- [4- (4-methylpiperazin-1-yl) piperidine-1 yl] phenyl} -N ‘- [2- inoculated with (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-crystalline diamine (0.9 mg), and stirred for about 10 minutes to obtain a slurry. After stirring for 3 hours at 70 ° C., cooled to 5 ℃ at a rate of 20 ° C. / h and stirred for 16 hours, the precipitated crystals were collected by filtration. The resulting crystals were washed with 2-butanone were cooled with ice water (35.2 mL), and dried in vacuo at 50 ℃, N- {2- methoxy-4- [4- (4-methylpiperazin-1- yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (6.60 g, 75.0% yield, purity 99.3%) was obtained as A04 type crystal (A04 type ratio 100%).
Example 2
The first step 4,4-dimethoxy-1- (3-methoxy-4-nitrophenyl) piperidine (R 1 and R 2 is methyl Any formula (Compound 10)) Synthesis of
4,4 – dimethoxy piperidine monohydrochloride (69.9kg) and N, N-dimethylformamide (125.7kg) was mixed, to this mixed solution 1,8-diazabicyclo [5.4.0] undec-7-ene and (117.3kg) N It was added N- dimethylformamide (17.0kg). N of separately prepared here 5-fluoro-2-nitroanisole (60.0kg), the N- dimethylformamide (57.0kg) was added at room temperature, N, N- dimethylformamide (29.0 kg) solution was added 5 hours It stirred. At room temperature with a seed crystal of 4,4-dimethoxy-1- (3-methoxy-4-nitrophenyl) piperidine (about 6 g) was added to the reaction mixture was stirred at room temperature for 14 hours. Water (240 kg) was added at room temperature to the reaction mixture, after stirring for 22 hours, the precipitated crystals were collected by filtration. The obtained crystals N, washed with a mixed solution of N- dimethylformamide (56.9kg) and water (60kg), washed twice with water (120 kg), and dried in vacuo at 50 ° C., 4, 4 – to give dimethoxy-1- (3-methoxy-4-nitrophenyl) piperidine (99.7kg, 96.0% yield) as crystals.
D2: 1.72-1.80 (4H, m) , 3.14 (6H, s), 3.44-3.50 (4H, m), 3.91 (3H, m), 6.52 (1H, d, J = 2.4Hz), 6.60 (1H, dd, J = 2.4,9.2Hz), 7.88 (1H, D, J = 9.2Hz)
ESI Tasu: 297
The second step 4- (R (4,4-dimethoxy-1-yl) -2-methoxyaniline 1 and R 2 is methyl none has the formula (Compound 6)) Synthesis of
4,4-dimethoxy – 1- (3-methoxy-4-nitrophenyl) piperidine (99.0kg), 5% palladium carbon (about 50% wet product, 10.5 kg), were mixed at room temperature in tetrahydrofuran (440 kg), under a hydrogen atmosphere (200 ~ 300 kPa ), and stirred at room temperature for 3 hours. Then filtered off and palladium-carbon, tetrahydrofuran and washed with (180.5Kg), the filtrate was concentrated under reduced pressure until the total volume of about 220L, as a seed crystal 4- (4,4-dimethoxy-1-yl) – crystals of 2-methoxyaniline was inoculated (approximately 10g). To the resulting slurry n- heptane (205.4kg) was added at 40 ° C., after stirring for 1 h, was stirred and cooled to 0 ° C. 16 hours. To this slurry was added n- heptane (613.5kg), After stirring for 2 hours, the crystals were collected by filtration. The obtained crystals were washed with a mixed solution of tetrahydrofuran (17.8 kg) and n- heptane (81.5kg), and dried in vacuo at 50 ℃, 4- (4,4- dimethoxy-1-yl) -2 – give methoxyaniline (84.1kg, 94.5% yield) as crystals.
D2: 1.72-1.80 (4H, m) , 2.90-2.97 (4H, m), 3.11 (6H, s), 3.73 (3H, m), 4.21 (1H, br), 6.30 (1H, d, J = 2.4 , 8.4Hz), 6.46_6.56 (2H, M)
ESI Tasu: 267
The third step 4,6-dichloro-N- [2-(propane-2-sulfonyl) phenyl] -1,3,5-triazin-2-amine (Lv is Cl any, compounds of formula (7) synthesis of)
cyanuric acid chloride (40.0kg) and acetone (249.2kg) was mixed at a 17 ℃. Sodium hydrogen carbonate in the mixed solution (21.9 kg), 2-a (isopropylsulfonyl) aniline (47.5Kg) was added, and stirred at room temperature for 23 hours. After adding to the reaction mixture water (320 kg) at room temperature, and stirred for 3.5 hours, the precipitated crystals were collected by filtration. After washing the obtained crystals with a mixed solution of acetone (63.0kg) and water (80 kg), and dried in vacuo at 50 ° C., 4,6-dichloro -N- [2- (propane-2-sulfonyl) phenyl ] -1,3,5-triazin-2-amine (71.6kg, 95.1% yield) was obtained as crystals.
D1: 1.32 (6H, d, J = 6.8Hz), 3.22 (1H, sept, J = 6.8Hz), 7.37 (1H, m), 7.74 (1H, m), 7.93 (1H, m), 8.44 (1H , M), 10.02 (1H, Br)
ESI-: 345, 347
Fourth step 6-chloro -N- [4- (4,4- dimethoxy-1-yl) -2-methoxy-phenyl] -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3 , (a Lv is Cl, R 5- triazine-2,4-diamine 1and R 2 none is methyl, the formula (compound 5)) synthesis of
4,6-dichloro-N- [2-( propane-2-sulfonyl) phenyl] -1,3,5-triazin-2-amine (70.9 kg) in tetrahydrofuran (611.1kg) was mixed at room temperature, to this mixed solution 4- (4,4-dimethoxy-piperidine 1-yl) -2-methoxyaniline (57.1kg), N, N- diisopropylethylamine (29.1 kg) was stirred for 4 hours at room temperature. Thereafter, isopropyl acetate (61.0kg), then extracted by adding potassium carbonate (3.6 kg) and a mixed solution of water (71 kg).The resulting organic layer total amount was concentrated under reduced pressure at an external temperature of about 40 ° C. to approximately 360 L, as a seed crystal, 6-chloro -N- [4- (4,4- dimethoxy-1-yl) -2 – methoxyphenyl] -N ‘- [2- was inoculated with (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-crystalline diamine (approximately 7 g) to give a slurry. To this slurry of 2-propanol (111.0kg), n- heptane (243.1kg) was added and after cooling for 2 hours at room temperature, was collected by filtration stirred precipitated crystals were cooled to 0 ℃ 18 hours. The resulting crystals tetrahydrofuran (74.9kg), 2- propanol (44.6kg), was washed with a mixed solution of n- heptane (97.6kg), and then dried under reduced pressure at 50 ℃, 6- chloro -N- [ 4- (4,4-dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine It was obtained (108.9kg, 92.4% yield) as crystals.
D1: 1.30 (6H, d, J = 6.8Hz), 1.88-1.92 (4H, m), 3.18-3.26 (1H, m), 3.23 (3H, s), 3.87 (1H, br), 6.53 (2H, br), 7.21-7.23 (1H, m ), 7.62 (1H, br), 7.88 (1H, d, J = 7.9Hz), 8.05 (1H, br), 8.48 (1H, br), 9.41 (1H, br )
ESI -: 575,577
fifth step and the sixth step (continuous process) 1- [3-methoxy-4 – ({4- [2- (propane-2-sulfonyl) anilino] -1,3,5-triazine – 2-yl} amino) phenyl] piperidin-4-one synthesis of compound) (formula (3)
6-chloro-N- [4- (4,4-dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (108.2kg), tetrahydrofuran (866.0kg), 10% palladium carbon (about 50% wet goods, 23.3 kg) were mixed, N to this mixed solution was added to N- diisopropylethylamine (28.9 kg) and 2-propanol (85.5kg), under a hydrogen atmosphere (100 ~ 300kPa), 4 hours at 40 ° C. did. Filtration of the palladium-carbon was washed with tetrahydrofuran (193.3kg), N- [4- ( 4,4- dimethoxy-1-yl) -2-methoxyphenyl] -N ‘- [2- (propane -2 – to obtain a solution containing a sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine. To this solution was added 35% hydrochloric acid (39.1 kg) of mixed solution of water (217kg), and stirred for 15 hours at room temperature. To the reaction mixture, added potassium carbonate (64.8kg) and a mixed solution of water (217kg), and extracted. Activated carbon (10.8 kg) was added to the obtained organic layer and stirred for 17 hours at room temperature, filtered and washed activated carbon with tetrahydrofuran (96.0kg). The resulting filtrate was concentrated under reduced pressure until the total volume of about 380L at 40 ° C.. After the resultant mixture was added acetone (257.1Kg), as a seed crystal, 1- [3-methoxy-4 – ({4- [2- (propane-2-sulfonyl) anilino] 1,3,5 – after stirring for 1 hour was inoculated triazin-2-yl} amino) phenyl] piperidin-4-one crystals (approximately 11g), the addition of water (865Kg) was stirred for 15 hours, the precipitated crystals were collected by filtration did. The obtained crystals were washed with a mixed solution of acetone (50.9kg) and Tsunemizu (173 kg), and dried in vacuo at 50 ℃, 1- [3- methoxy-4 – ({4- [2- (propane 2-sulfonyl) anilino] -1,3,5-triazine-2-yl} amino) phenyl] piperidine-4-one (82.9kg, 89.0% yield (yield in a continuous two steps)) as crystals Obtained.
D2,343K: 1.17 (6H, d, J = 6.8Hz), 2.46-2.50 (4H, m), 3.40 (1H, sept, J = 6.8Hz), 3.61 (4H, dd, J = 6.1,6.2Hz) , 3.79 (3H, s), 6.57 (1H, dd, J = 2.6,8.7Hz), 6.70 (1H, d, J = 2.6Hz), 7.25-7.29 (1H, m), 7.38 (1H, d, J 8.7 Hz =), 7.61 (1H, br), 7.77-7.80 (1H, yd), 8.28 (1H, s), 8.50 (1H, br), 8.66 (1H, br), 9.25 (1H, br)
ESI +: 497
Seventh Step N- {2- methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] – 1,3,5-triazine-2,4-diamine (formula (1) compounds) synthesis
of 1- [3-methoxy-4 – ({4- [2- (propane-2-sulfonyl) anilino] -1 , 3,5-triazin-2-yl} amino) phenyl] piperidin-4-one (60.1kg), methylpiperazine (24.2kg), was mixed with toluene (500 kg) and acetic acid (28.4kg), 1 hour at room temperature It stirred. To this mixture solution was added sodium triacetoxyborohydride (51.4kg), and stirred at room temperature for 17 hours. To the reaction mixture, methanol (47.5kg) and water (180.1kg) was added, extraction to give an organic layer and an aqueous layer 1. The organic layer was re-extracted by adding water (60.0kg), to obtain an aqueous layer 2. After mixing the aqueous layer 1 and aqueous layer 2 was extracted by adding isopropyl acetate (523.4kg). The resulting aqueous layer methanol (522.3kg), a mixed solution of 48% sodium hydroxide (60.6kg) and water (112.7kg) was added, as a seed crystal N- {2- methoxy-4- [4- (4 – methyl-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-crystal of diamine (about 6 g) were inoculated, after stirring at room temperature for 2 hours, added water (660.2kg), further stirred for 3.5 hours at room temperature, the precipitated crystals were collected by filtration. The obtained crystals were washed with a mixed solution of methanol (104.4kg) and water (132.0kg), and dried in vacuo at 50 ℃, N- {2- methoxy-4- [4- (4-methylpiperazin- 1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (54.2kg, yield: 77.1 %) was obtained as A06-form crystals.
D1: 1.31 (6H, d, J = 6.8Hz), 1.59-1.78 (2H, m), 1.90-2.01 (2H, m), 2.24-2.80 (11H, m), 2.30 (3H, s), 3.19- 3.32 (1H, m), 3.65-3.75 (2H, m), 3.88 (3H, s), 6.50-6.59 (2H, m), 7.18-7.30 (1H, m), 7.53-7.70 (2H, m), 7.88 (1H, dd, J = 1.5,8.3Hz), 8.10 (1H, br), 8.37 (1H, br), 8.53 (1H, br), 9.29 (1H, s)
ESI +: 581
N- {2- methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3 , purification step by recrystallization 5-triazine-2,4-diamine (compound of formula (1))
N-{2-methoxy-4- [4- (4-methylpiperazin-1-yl) piperidine-1 yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (54.3kg), activated charcoal (5.4 kg), 2-butanone (1046.1 kg) were mixed, stirred for 1 hour at 75 ° C., was subjected to active carbon filtration. The filtrate activated carbon (5.4kg) in addition to, and the mixture was stirred for 1 hour at 75 ℃, was activated carbon filtration. The filtrate activated carbon (5.4kg) in addition to, and the mixture was stirred for 1 hour at 75 ℃, was activated carbon filtration. After filtrate was total volume approximately until 430L normal pressure concentrated and cooled to 70 ° C., as a seed crystal N- {2- methoxy-4- [4- (4-methylpiperazin-1-yl) piperidine-1 yl] phenyl} -N ‘- inoculated with [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-crystalline diamine (approximately 5 g), after stirring for 3 hours, It was cooled to 5 ℃ at a rate of 20 ℃ / h, and the precipitated crystals were collected by filtration. After washing with the resulting crystals were cooled in 5 of 5 ° C. 2-butanone (220L), and dried in vacuo at 50 ℃, N- {2- methoxy-4- [4- (4-methylpiperazin-1- yl) piperidin-1-yl] phenyl} -N ‘- [2- (propane-2-sulfonyl) phenyl] -1,3,5-triazine-2,4-diamine (42.6kg, 78.5% yield, purity 99.5%) was obtained as A04-form crystals (A04 type ratio 100%).


Ken Jones, president and chief executive officer, Astellas Pharma Europe
Paper
Organic Process Research & Development (2015), 19(12), 1966-1972
† Technology Process Chemistry Laboratories, Astellas Pharma Inc., 160-2 Akahama, Takahagi, Ibaraki 318-0001,Japan
‡ Astellas Pharma Tech Co., Ltd., 160-2 Akahama, Takahagi, Ibaraki 318-0001, Japan
§ Department of Chemical Engineering, Tokyo University of Agriculture and Technology, 2-24-16, Naka-cho, Koganei, Tokyo 184-8588, Japan
Org. Process Res. Dev., 2015, 19 (12), pp 1966–1972
DOI: 10.1021/acs.oprd.5b00208
Publication Date (Web): October 23, 2015
Copyright © 2015 American Chemical Society
Abstract

ASP3026(N-{2-Methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl}-N′-[2-(propane-2-sulfonyl)phenyl]-1,3,5-triazine-2,4-diamine) was developed as a novel and selective inhibitor of the fusion protein EML4-ALK. Five polymorphs of ASP3026 (A01, A02, A03, A04, and A05) as well as a hydrate have been identified to date, and the most stable polymorph (A04) was selected for designing solid formulations. The influence of crystallization process parameters on nucleation of A03 and A04 was clarified for process development. A04 was obtained at relatively high temperatures and A03 at relatively low temperatures, regardless of the superaturation ratio. A03 and A04 were therefore able to be selectively obtained via temperature control, possibly due to temperature-dependent variations in the concentrations of conformers in solution. The relationship between polymorphs and solution structures before nucleation was investigated using in situ Raman spectroscopy. The relationship with the intensity ratios of nine Raman bands of both polymorphs and ASP3026 solution structures was investigated in detail. Our findings suggest that the solution structure shifted from a structure similar to that of A04 to one similar to that of A03 with decreasing temperature.

Chairman of Astellas Pharma Inc. Mr. Masafumi Nogimori is conferred with Netherlands Honor – ‘Officer in the Order of Oranje-Nassau’
PAPER
Effect of Temperature and Solvent of Solvent-Mediated Polymorph Transformation on ASP3026 Polymorphs and Scale-up
† Technology Process Chemistry Laboratories, Astellas Pharma Inc., 160-2 Akahama, Takahagi, Ibaraki 318-0001,Japan
‡ Astellas Pharma Tech Co., Ltd., 160-2 Akahama, Takahagi, Ibaraki 318-0001, Japan
§ Department of Chemical Engineering, Tokyo University of Agriculture and Technology, 2-24-16, Naka-cho, Koganei, Tokyo 184-8588, Japan
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00068
Publication Date (Web): April 28, 2016
Copyright © 2016 American Chemical Society
Abstract

ASP3026 (N-{2-methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl}-N′-[2-(propane-2-sulfonyl)phenyl]-1,3,5-triazine-2,4-diamine) was developed as a novel and selective inhibitor of the fusion protein EML4-ALK. Five polymorphs of ASP3026 (A01, A02, A03, A04, and A05) as well as a hydrate have been identified to date. Process development was conducted for large-scale pilot plant manufacturing, and obtaining the desired polymorph A04 was key after a synthetic route of ASP3026 was selected for scale-up. The effects of temperature and solvent species on induction time of polymorph transformation were investigated using in situ Raman spectroscopy, and selective transformation conditions of A02 to A03 and A04 were examined in detail. A04 was obtained at high temperatures using highly polar non-hydrogen-bond-donating solvents, while A03 was obtained at low temperatures using low-polarity or hydrogen-bond-donating solvents. Further, the desired polymorph A04 was successfully obtained in high purity in first stage scale-up manufacturing. Given these findings, this method of solvent-mediated polymorph transformation may aid in process development for obtaining desired polymorphs.
http://pubs.acs.org/doi/full/10.1021/acs.oprd.6b00068

REFERENCES
1: Awad MM, Shaw AT. ALK Inhibitors in Non-Small Cell Lung Cancer: Crizotinib and Beyond. Clin Adv Hematol Oncol. 2014 Jul;12(7):429-39. PubMed PMID: 25322323.
2: George SK, Vishwamitra D, Manshouri R, Shi P, Amin HM. The ALK inhibitor ASP3026 eradicates NPM-ALK⁺ T-cell anaplastic large-cell lymphoma in vitro and in a systemic xenograft lymphoma model. Oncotarget. 2014 Jul 30;5(14):5750-63. PubMed PMID: 25026277; PubMed Central PMCID: PMC4170597.
3: Mori M, Ueno Y, Konagai S, Fushiki H, Shimada I, Kondoh Y, Saito R, Mori K, Shindou N, Soga T, Sakagami H, Furutani T, Doihara H, Kudoh M, Kuromitsu S. The selective anaplastic lymphoma receptor tyrosine kinase inhibitor ASP3026 induces tumor regression and prolongs survival in non-small cell lung cancer model mice. Mol Cancer Ther. 2014 Feb;13(2):329-40. doi: 10.1158/1535-7163.MCT-13-0395. Epub 2014 Jan 13. PubMed PMID: 24419060.
| Patent ID |
Date |
Patent Title |
| US2015150850 |
2015-06-04 |
TREATING CANCER WITH HSP90 INHIBITORY COMPOUNDS |
| US8906885 |
2014-12-09 |
Treating cancer with HSP90 inhibitory compounds |
| US2013338358 |
2013-12-19 |
METHOD FOR PRODUCING DI(ARYLAMINO)ARYL COMPOUND AND SYNTHETIC INTERMEDIATE THEREFOR |
| US2013096100 |
2013-04-18 |
DI(ARYLAMINO)ARYL COMPOUND |
| US2013059855 |
2013-03-07 |
CRYSTAL OF DI(ARYLAMINO)ARYL COMPOUND |
| US2010099658 |
2010-04-22 |
DI(ARYLAMINO)ARYL COMPOUND |
////ASP3026, EML4-ALK, ASP 3026, ASTELLAS
CC(C)S(=O)(=O)C1=CC=CC=C1NC2=NC=NC(=N2)NC3=C(C=C(C=C3)N4CCC(CC4)N5CCN(CC5)C)OC
Anaplastic lymphoma kinase (ALK) is a validated therapeutic target for treating echinoderm microtubule-associated protein-like 4 (EML4)-ALK positive non-small cell lung cancer (NSCLC). ASP3026 (1) is a potent and selective ALK inhibitor that Astellas designed and synthesized through detailed structure–activity relationship (SAR) studies
Our effort toward the process improvement of anaplastic lymphoma kinase (ALK) inhibitor ASP3026 (1) is described. A cost-effective and practical synthesis of 1 was accomplished as a result of the change of starting material from 2,4-dichloro-1,3,5-triazine (6) to cyanuric chloride (9) and late-stage introduction of a highly reactive N-methyl piperazine moiety by reductive amination of intermediate ketone 13. The modified process avoided the challenges with the original synthesis and furnished the several hundred kilograms of high-quality API with high economic efficiency, operability, and reproducibility. Furthermore, a sequence of investigation of polymorphic control in the second-generation synthetic route to obtain the thermodynamically desired, most stable polymorph Form A04 is also discussed.
Improved Manufacturing Route and Polymorphic Control of a Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor ASP3026
Yuji Takahama*  , Kazuyoshi Obitsu, Kazuhiro Takeguchi, Shun Hirasawa, Koji Kobayashi, Takahiro Akiba, Norihiro Ueda, Ryoki Orii, Atsushi Ohigashi, Takumi Takahashi, Minoru Okada, and Shigeru Ieda
, Kazuyoshi Obitsu, Kazuhiro Takeguchi, Shun Hirasawa, Koji Kobayashi, Takahiro Akiba, Norihiro Ueda, Ryoki Orii, Atsushi Ohigashi, Takumi Takahashi, Minoru Okada, and Shigeru Ieda
Process Chemistry Laboratories, Astellas Pharma Inc., 160-2 Akahama, Takahagi-shi, Ibaraki 318-0001, Japan
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.8b00427
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00427
crude 1 (25.0 kg, 43.0 mol, 48% yield) as a pale yellow solid in Form A02. HPLC purity: 97.9A%. 1H NMR (600 MHz, chloroform-d) δ 9.28 (s, 1H), 8.47–8.61 (bs, 1H), 8.30–8.48 (m, 1H), 8.10 (bs, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.49–7.72 (m, 2H), 7.23 (t, J = 8.2 Hz, 1H), 6.45–6.60 (m, 2H), 3.88 (s, 3H), 3.63–3.75 (m, 2H), 3.18–3.32 (m, 1H), 2.31–2.78 (m, 11H), 2.30 (s, 3H), 1.92–2.01 (m, 2H), 1.65–1.79 (m, 2H), 1.31 (d, J = 6.9 Hz, 6H). 13C NMR (150 MHz, chloroform-d) δ (ppm) 166.3, 163.6, 149.6, 148.6, 138.4, 134.5, 131.1, 124.4, 123.6, 123.0, 122.1, 121.4, 119.6, 108.0, 100.4, 61.6, 55.6, 55.5, 55.4, 50.0, 49.0, 46.0, 28.1, 15.3. LC–MS (ESI): calcd for C29H41N8O3S ([M + H]+) 581.3; found 581.2.


/////////

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....












![CAS # 13103-34-9, Boldenone undecylenate, Boldenone undec-10-enoate, 17b-[(1-Oxo-10-undecenyl)oxy]-androsta-1,4-dien-3-one, 17b-Hydroxyandrosta-1,4-dien-3-one 10-undecenoate](https://i0.wp.com/www.chemblink.com/structures/13103-34-9.gif)























.jpg)

























{kind=link}