
Home » Uncategorized (Page 34)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TENAPANOR
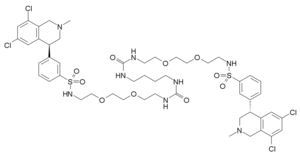
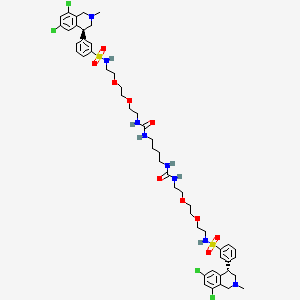


Tenapanor
Molecular FormulaC50H66Cl4N8O10S2
Average mass1145.049 Da
1234423-95-0 [RN]
1234423-95-0 (free base) 1234365-97-9 (2HCl)
9652
3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)-N-(26-((3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)phenyl)sulfonamido)-10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosyl)benzenesulfonamide
Benzenesulfonamide, N,N’-(10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosane-1,26-diyl)bis[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]-
12,15-Dioxa-2,7,9-triazaheptadecanamide, 17-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-
1-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethyl]-3-[4-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethylcarbamoylamino]butyl]urea
17-[[[3-[(4S)-6,8-Dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-12,15-dioxa-2,7,9-triazaheptadecanamide
Tenapanor, also known as AZD-1722 and RDX 5791, is an inhibitor of the sodium-proton (Na(+)/H(+)) exchanger NHE3, which plays a prominent role in sodium handling in the gastrointestinal tract and kidney. Tenapanor possesses an excellent preclinical safety profile and, as of now, there are no serious concerns about its side effects.
Tenapanor is a drug developed by Ardelyx, which acts as an inhibitor of the sodium-proton exchanger NHE3. This antiporterprotein is found in the kidney and intestines, and normally acts to regulate the levels of sodium absorbed and secreted by the body. When administered orally, tenapanor selectively inhibits sodium uptake in the intestines, limiting the amount absorbed from food, and thereby reduces levels of sodium in the body.[1] This may make it useful in the treatment of chronic kidney disease and hypertension, both of which are exacerbated by excess sodium in the diet.[2]
Ardelyx and licensees Kyowa Hakko Kirin and Fosun Pharma are developing tenapanor, an NHE3 (Na+/H+ exchange-3) inhibitor that increases fluid content in the GI tract and which also reduces GI tract pain via an unknown TRPV-1-dependent pathway, for treating constipation-predominant irritable bowel syndrome (IBS-C) and hyperphosphatemia in patients with end stage renal disease (ESRD).
Syn

PATENT
WO2010078449
PATENT
WO-2019091503
A novel crystalline form of tenapanor free base, process for its preparation, composition comprising it and its use for the preparation of tenapanor with chemical purity >98.8% is claimed. Also claimed are salt forms of tenapanor, preferably tenapanor phosphate and their use for treating irritable bowel syndrome, constipation, hyperphosphatemia, final stage renal failure, chronic kidney disease and preventing excess sodium in patients with kidney and heart conditions. Further claimed are processes for the preparation of tenapanor comprising the steps of reaction of a diamine compound with 1,4-diisocyanatobutane, followed by deprotection and condensation to obtain tenapanor. Novel intermediates of tenapanor and their use for the preparation of tenapanor are claimed. Tenapanor is known to be a sodium hydrogen exchanger 3 inhibitor and analgesic.
enapanor, having the chemical name 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazaheptadecaneamide, is a selective inhibitor of the sodium protonic NHE3 antiporter. Orally administered tenapanor selectively inhibits the absorption of sodium in the intestine. This leads to an increase of water content in the digestive tract, improved bowel flow and normalization of the frequency of bowel movement and stool consistency. At the same time it exhibits antinociceptive activity and ability to lower serum phosphate levels. Because of these properties, it is clinically tested for the treatment of irritable bowel syndrome, especially when accompanied by constipation, treatment of hyperphosphatemia, especially in patients with dialysis with final stage renal failure, treatment of chronic kidney disease, and prevention of excess sodium in patients with kidney and heart conditions. The tenapanor molecule, which was first described in the international patent application WO 2010/078449, has the following structural formula:
In this document, tenapanor was prepared as bishydrochloride salt. The bishydrochloride salt was prepared only in the form of an amorphous foam, which, after solidification, required grinding for further processing. However, the thus obtained particles are of varying sizes, while a narrow particle size distribution is required for pharmaceutical use in order to ensure uniform behavior. The amorphous foam obtained in the said document is essentially a thickened reaction mixture or a slightly purified reaction mixture containing, in addition to tenapanor, various impurities. The possibilities to purify the reaction mixtures are limited. Moreover, amorphous foams tend to adsorb solvents, and it is usually difficult to remove (or dry out) the residual solvents from the amorphous foam. This is undesirable for pharmaceutical use. A typical feature of amorphous foams is a large specific surface, resulting in a greater interaction of the substance with the surrounding environment. This significantly increases the risk of decomposition of the substance, for example through air oxygen, moisture or light. The present invention aims at overcoming these problems.
It would be advantageous to provide tenapanor solid forms (tenapanor free base or tenapanor salts) which are precipitated in solid forms, thus allowing to filter off the liquid reaction mixture containing the impurities. This results in a significantly improved purity.
The process used in WO 2010/078449 for the preparation of bishydrochloride salt of tenapanor was based on the preparation of 3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzene-l-sulfonyl chloride of formula III from 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l, 2,3,4-te
Scheme 1
The said document also discloses resolution of the starting tetrahydroisoquinoline of formula II by L-or D-dibenzoylt
(II) (S-II) (R-II)
Scheme 2
WO 2010/078449 discloses further steps of preparation of tenapanor, as shown in Scheme 3.
(V) (I)
Scheme 3
Individual synthetic steps described in Scheme 3 result in low yields: 42% for the reaction of the chloride of formula III with 2-(2-(2-aminoethoxy)ethoxy)ethylamine of formula IV, and 59% for the subsequent reaction with 1,4-diisocyanatobutane of formula V. The products of both synthetic steps are isolated by preparative chromatography which is technologically an unsuitable isolation and purification technique. The low yields and the need to use preparative chromatography for the isolation are caused by an abundance of side products and impurities and by the inability of the intermediates as well as of the product to provide a crystalline form.
Thus present invention thus further aims at providing a method of preparation of tenapanor which would be economically effective, in particular in relation to the expensive starting compound 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline, and which would also enable industrial scale production, in particular by removing steps which cannot be scaled up effectively or which cannot be scaled up at all. Furthermore, the method of preparation of tenapanor should provide tenapanor in a form which is useful for use in pharmaceutical forms and does not have the disadvantages of an amorphous foam.
Tenapanor free base in the form of an amorphous solid foam was prepared by the procedure disclosed in patent application WO 2010/078449, Example 202. The chemical purity of the tenapanor prepared by this procedure was 96.5% (HPLC). The structure of tenapanor was verified by MS and H and 13C NMR spectra.
Step A
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline
Potassium carbonate (9.30 g) and anhydrous xylene (500 ml) were added to the reaction vessel. Benzyl mercaptane (25 g) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
(S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline 50 g in anhydrous xylene (500 ml), Pd2(dba)3 (3 g) and Xantphos (3 g). The resulting solution was stirred at 25 °C for 30 minutes and then added to a solution of benzyl mercaptane. The resulting reaction mixture was maintained at 140 °C for 16 h. The mixture was then concentrated and the residue was subjected to preparative chromatography on silica gel with the mobile phase ethyl acetate / petroleum ether (1: 100-1 :50). 20 g of product are obtained as a yellow oil (36% yield).
Ste B
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochloride
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (16 g) was dissolved in the reaction vessel in acetic acid/water (160 mL: 16 mL) mixture. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the well stirred mixture. After disappearance of the starting material, the reaction mixture was purged with nitrogen and concentrated in vacuo. A product (10 g, 66.6%) was obtained as a colorless substance.
Step C
Preparation of (S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l, 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
2-(2-(2-Aminoethoxy)ethoxy)ethylamine HC1 (30 g; 0.2 mol) and triethylamine (5.2 g; 52 mmol) were dissolved in dichloromethane (500 ml) and the mixture was chilled in an ice bath. (S)-3-(6,8-Dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (10 g; 26 mmol) was added in parts during 40 minutes to the chilled reaction mixture. The ice bath was removed and the reaction mixture was stirred at laboratory temperature for additional 30 minutes.
The dichloromethane solution was extracted three times by brine (2x 250 ml), dried over sodium sulphate, and concentrated in vacuo. The residue was purified using preparative chromatography on silica gel with dichloromethane-methanol mobile phase.
Yield 7.2 g. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Step D
Preparation of 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazah
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide (5g; 10 mmol) prepared in step A was dissolved in dichloromethane (50 ml). Triethylamine (1.5 g; 14.9 mmol) and 1 ,4-diisocyanatobutane (0.48 g; 3.4 mmol) were added to the solution. The reaction mixture was cooled using ice and stirred overnight. The resulting fine suspension was filtered off, the filtrate was concentrated and the obtained product was purified by preparative chromatography on on silica gel with dichloromethane-methanol mixture as a mobile phase
Yield: 2 g of tenapanor in the form of amorphous solid foam. HPLC purity 96.5 %.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. *H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H).
Ste E
Preparation of bishydrochloride salt of tenapanor
Tenapanor free base (1 g; 0.85 mmol) prepared in step B was dissolved in a mixture of methanol (10 ml) and 4M aqueous HCl (0.5 ml; 2 mmol) under mild reflux. The solution was concentrated on rotary vacuum evaporator, and the title product was obtained in the yield of 1 g of amorphous solid foam.
Example 1
Preparation of tenapanor, crystalline form I
Tenapanor free base (200 mg, 0.17 mmol), prepared as in step D of the comparative example, was dissolved in 0.4 ml acetonitrile under mild reflux. The clear solution was cooled at the rate of 1 °C/min with stirring to laboratory temperature (i.e., range from 22 °C to 26 °C) and then stirred for additional 2 hours at this temperature. The resulting crystals were isolated by filtration on sintered glass filter and dried for 6 hours in a vacuum oven at 40 °C. Crystallization yield was 170 mg of crystalline form I of tenapanor. HPLC showed a purity of 99.5%.
Examples 4 to 9 illustrate the inventive method of preparation of crystalline tenapanor.
Example 4
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinoline
DIPEA (9.6 mL) and anhydrous dioxane (100 mL) were added to a reaction vessel. Benzyl mercaptan (8.1 ml) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
In a second reaction vessel, (S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (21.2 g) in anhydrous dioxane (140 mL), Pd2(dba)3 (835 mg)and Xantphos (835 mg) were mixed. The resulting solution was stirred at 25 °C for 30 minutes and then added to the solution of benzyl mercaptan. The resulting reaction mixture was maintained at gentle reflux for 3 hours.
After cooling, the suspension obtained was filtered through a thin layer of celite. HC1 was added to the filtrate. The precipitated hydrochloride was isolated by filtration, washed well and dried. 21 g of pinkish product were obtained (81.6% yield).
Example 5
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochlorid
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline hydrochloride (11.1 g) was stirred in DCM/2M HC1 (70 mL:6 mL) mixture in a reaction vessel. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the vigorously stirred mixture. After disappearance of the starting material, the resulting suspension was bubbled through by nitrogen and the product was filtered off and washed with DCM. 9.2 g of white product was obtained (82.7% yield).
Example 6
In the reaction vessel, t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (21.8 g) was stirred in DCM. The mixture was cooled in an ice bath under an inert atmosphere. To the cooled solution was
added 1 ,4-diisocyanatobutane (6.14 g) and TEA (0.1 mL). The cooling bath was removed and the reaction mixture was further stirred for 2 h.
35% HCl was added to the reaction mixture and the mixture was stirred under gentle reflux overnight.
After cooling, the precipitated product was filtered off and washed with DCM.
The product was recrystallized from propan-2-ol. 22.3 g of white product was obtained (80% yield).
Example 7
Preparation of (5)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l , 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
(S)-3-(6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (11.7 g) prepared in Example 2 was stirred in dichloromethane (100 ml) and the suspension was cooled in an ice bath. To the cooled suspension was added a solution of t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (6.8 g) and DIPEA (14 ml) in DCM (50 ml). The resulting solution was stirred for 2 hours in an ice bath. The reaction mixture was extracted twice with water. Concentrated HCl (15 mL) was added to the dichloromethane solution and the mixture heated at gentle reflux for 2 h.
The precipitated product, after cooling, was extracted into water. The aqueous phase was separated and basified with Na2C03. The product as the free base was extracted into DCM and the dichloromethane solution was dried over sodium sulfate and concentrated in vacuo. 12.9 g of product were obtained.
Yield 93.4%. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Example 8
Preparation of 17-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-triazaheptadecanamide (tenapanor free base)
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4- tetrahydroisoquinolin- 4-yl)benzenesulfonamide (12.9 g) prepared in Example 4 was dissolved in dichloromethane (150 ml). To the solution was added triethylamine (0.3 ml) and 1,4-diisocyanatobutane (1.7 g). The reaction mixture was stirred at 25 °C for 2 h. The resulting reaction mixture was extracted with water and aqueous Na2C03. The dichloromethane solution of the product was dried over sodium sulfate and concentrated to a solid foam. Yield 13.9 g. The crude product was taken up in acetone (100 ml) and then recrystallized from methanol (80 ml). 7.3 g of white crystalline product was obtained. Yield 49.8%.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. !H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H)
Example 9
Preparation of 17-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-
(S)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (0.81 g) prepared in Example 2 and l,l’-(butane-l,4-diyl)bis(3-(2-(2-(2-aminoethoxy)ethoxy)ethyl)urea) dihydrochloride prepared according to Example 3 (0.48 g) were stirred in anhydrous ΝΜΡ (10 ml). To the suspension was added DIPEA (2 mL) and the resulting solution was stirred at 60 °C for 1.5 h. Water (10 mL) was added dropwise to the reaction mixture and the mixture was cooled to 5 °C. The precipitated product was isolated and stirred in acetone at 5 °C overnight. The beige product was filtered off (0.67 g) and recrystallized from methanol (12 ml).
0.53 g of a colorless crystalline product was obtained.
Yield 78.7 %. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S. DSC analysis showed the melting temperature of 130.5 °C.
Example 10
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 11
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and hydrobromic acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with hydrobromic acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 12
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 13
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and citric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with citric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Other pharmaceutically acceptable acids were tested by the procedures shown in Examples 10-13, but did not yield salts which would precipitate in amorphous stable solid form from the solution. The tested acids were: methanesulfonic acid, benzenesulfonic acid, oxalic acid, maleinic acid, tartaric acid, fumaric acid, trichloroacetic acid.
Example 14
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, a solution of phosphoric acid in THF (50 μ1/5 ml) is added dropwise during 10 minutes. The resulting suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with phosph (79 %) oric is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 430 mg of colourless salt of tenapanor with phosphoric acid. XRPD showed amorphousness of the product.
Example 15
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, hydrobromic acid (48%; 100 μΐ) is added dropwise during 10 minutes. A fine precipitate forms already during the dropwise addition of HBr, and the suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with HBr is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 397 mg (69 %) of colourless salt of tenapanor with HBr (1 :2). XRPD showed amorphousness of the product.
References
- ^ Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D (2014). “Intestinal inhibition of the na+/h+ exchanger 3 prevents cardiorenal damage in rats and inhibits na+ uptake in humans”. Sci Transl Med. 6 (227): 227ra36. doi:10.1126/scitranslmed.3007790. PMID 24622516.
- ^ Salt-buster drug cuts sodium absorbed from food. New Scientist, 14 March 2014
REFERENCES
1: Johansson SA, Knutsson M, Leonsson-Zachrisson M, Rosenbaum DP. Effect of Food Intake on the Pharmacodynamics of Tenapanor: A Phase 1 Study. Clin Pharmacol Drug Dev. 2017 Mar 24. doi: 10.1002/cpdd.341. [Epub ahead of print] PubMed PMID: 28339149.
2: Johansson S, Rosenbaum DP, Ahlqvist M, Rollison H, Knutsson M, Stefansson B, Elebring M. Effects of Tenapanor on Cytochrome P450-Mediated Drug-Drug Interactions. Clin Pharmacol Drug Dev. 2017 Mar 16. doi: 10.1002/cpdd.346. [Epub ahead of print] PubMed PMID: 28301096.
3: Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor Treatment of Patients With Constipation-Predominant Irritable Bowel Syndrome: A Phase 2, Randomized, Placebo-Controlled Efficacy and Safety Trial. Am J Gastroenterol. 2017 Feb 28. doi: 10.1038/ajg.2017.41. [Epub ahead of print] PubMed PMID: 28244495.
4: Carney EF. Dialysis: Efficacy of tenapanor in hyperphosphataemia. Nat Rev Nephrol. 2017 Apr;13(4):194. doi: 10.1038/nrneph.2017.27. PubMed PMID: 28239171.
5: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Åstrand M, Johansson S, Knutsson M, Langkilde AM, Chertow GM. Effect of Tenapanor on Serum Phosphate in Patients Receiving Hemodialysis. J Am Soc Nephrol. 2017 Feb 3. pii: ASN.2016080855. doi: 10.1681/ASN.2016080855. [Epub ahead of print] PubMed PMID: 28159782.
6: Koliani-Pace J, Lacy BE. Update on the Management of Chronic Constipation. Curr Treat Options Gastroenterol. 2017 Mar;15(1):126-134. doi: 10.1007/s11938-017-0118-2. Review. PubMed PMID: 28116695.
7: Charoenphandhu N, Kraidith K, Lertsuwan K, Sripong C, Suntornsaratoon P, Svasti S, Krishnamra N, Wongdee K. Na(+)/H(+) exchanger 3 inhibitor diminishes hepcidin-enhanced duodenal calcium transport in hemizygous β-globin knockout thalassemic mice. Mol Cell Biochem. 2017 Mar;427(1-2):201-208. doi: 10.1007/s11010-016-2911-y. PubMed PMID: 27995414.
8: Thammayon N, Wongdee K, Lertsuwan K, Suntornsaratoon P, Thongbunchoo J, Krishnamra N, Charoenphandhu N. Na(+)/H(+) exchanger 3 inhibitor diminishes the amino-acid-enhanced transepithelial calcium transport across the rat duodenum. Amino Acids. 2017 Apr;49(4):725-734. doi: 10.1007/s00726-016-2374-1. PubMed PMID: 27981415.
9: Afsar B, Vaziri ND, Aslan G, Tarim K, Kanbay M. Gut hormones and gut microbiota: implications for kidney function and hypertension. J Am Soc Hypertens. 2016 Dec;10(12):954-961. doi: 10.1016/j.jash.2016.10.007. Review. PubMed PMID: 27865823.
10: Johansson S, Leonsson-Zachrisson M, Knutsson M, Spencer AG, Labonté ED, Deshpande D, Kohler J, Kozuka K, Charmot D, Rosenbaum DP. Preclinical and Healthy Volunteer Studies of Potential Drug-Drug Interactions Between Tenapanor and Phosphate Binders. Clin Pharmacol Drug Dev. 2016 Sep 22. doi: 10.1002/cpdd.307. [Epub ahead of print] PubMed PMID: 27654985.
11: Ketteler M, Liangos O, Biggar PH. Treating hyperphosphatemia – current and advancing drugs. Expert Opin Pharmacother. 2016 Oct;17(14):1873-9. doi: 10.1080/14656566.2016.1220538. Review. PubMed PMID: 27643443.
12: Johansson S, Rosenbaum DP, Knutsson M, Leonsson-Zachrisson M. A phase 1 study of the safety, tolerability, pharmacodynamics, and pharmacokinetics of tenapanor in healthy Japanese volunteers. Clin Exp Nephrol. 2016 Jul 1. [Epub ahead of print] PubMed PMID: 27368672.
13: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Stefansson BV, Rydén-Bergsten T, Greasley PJ, Johansson SA, Knutsson M, Carlsson BC. Effect of Tenapanor on Interdialytic Weight Gain in Patients on Hemodialysis. Clin J Am Soc Nephrol. 2016 Sep 7;11(9):1597-605. doi: 10.2215/CJN.09050815. PubMed PMID: 27340281; PubMed Central PMCID: PMC5012484.
14: Nusrat S, Miner PB Jr. New pharmacological treatment options for irritable bowel syndrome with constipation. Expert Opin Emerg Drugs. 2015;20(4):625-36. doi: 10.1517/14728214.2015.1105215. Review. PubMed PMID: 26548544.
15: Spencer AG, Greasley PJ. Pharmacologic inhibition of intestinal sodium uptake: a gut centric approach to sodium management. Curr Opin Nephrol Hypertens. 2015 Sep;24(5):410-6. doi: 10.1097/MNH.0000000000000154. Review. PubMed PMID: 26197202.
16: Zielińska M, Wasilewski A, Fichna J. Tenapanor hydrochloride for the treatment of constipation-predominant irritable bowel syndrome. Expert Opin Investig Drugs. 2015;24(8):1093-9. doi: 10.1517/13543784.2015.1054480. Review. PubMed PMID: 26065434.
17: Thomas RH, Luthin DR. Current and emerging treatments for irritable bowel syndrome with constipation and chronic idiopathic constipation: focus on prosecretory agents. Pharmacotherapy. 2015 Jun;35(6):613-30. doi: 10.1002/phar.1594. Review. PubMed PMID: 26016701.
18: Gerritsen KG, Boer WH, Joles JA. The importance of intake: a gut feeling. Ann Transl Med. 2015 Mar;3(4):49. doi: 10.3978/j.issn.2305-5839.2015.03.21. PubMed PMID: 25861604; PubMed Central PMCID: PMC4381464.
19: Labonté ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Dy E, Black D, Zhong Z, Langsetmo I, Spencer AG, Bell N, Deshpande D, Navre M, Lewis JG, Jacobs JW, Charmot D. Gastrointestinal Inhibition of Sodium-Hydrogen Exchanger 3 Reduces Phosphorus Absorption and Protects against Vascular Calcification in CKD. J Am Soc Nephrol. 2015 May;26(5):1138-49. doi: 10.1681/ASN.2014030317. PubMed PMID: 25404658; PubMed Central PMCID: PMC4413764.
20: Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med. 2014 Mar 12;6(227):227ra36. doi: 10.1126/scitranslmed.3007790. PubMed PMID: 24622516.
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.243.471 |
| Chemical and physical data | |
| Formula | C50H66Cl4N8O10S2 |
| Molar mass | 1145.046 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////Tenapanor, AZD 1722, RDX 5791, chronic kidney disease, hypertension
CN1CC(C2=CC(=CC(=C2C1)Cl)Cl)C3=CC(=CC=C3)S(=O)(=O)NCCOCCOCCNC(=O)NCCCCNC(=O)NCCOCCOCCNS(=O)(=O)C4=CC=CC(=C4)C5CN(CC6=C(C=C(C=C56)Cl)Cl)C
Etosalamide, этосаламид , إيتوسالاميد , 依托柳胺 ,
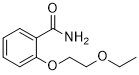
Cas 15302-15-5
Chemical Formula: C11H15NO3
Molecular Weight: 209.245
o-(2-Ethoxyethoxy)benzamide
Etosalamide, also known as Ethosalamide, is an antipyretic and analgesics agent
SYN

OR

CAS:592-55-2, 2-Bromoethyl ethyl ether
Cas, 611-20-1, 2-Hydroxybenzonitrile

PATENT
DE 1013643
PATENT
GB 774635
PATENT
US2822391
78 – 79 MP
PAPER
Journal of Chemical and Engineering Data (1962), 7, 265-6
70 – 71.5 MP
PATENT
WO 2004003198
US 20100226943
/////////Etosalamide, этосаламид , إيتوسالاميد , 依托柳胺 , ethosalamide
O=C(N)C1=CC=CC=C1OCCOCC
KETOROLAC
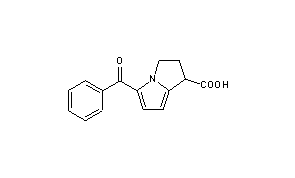

Ketorolac
- Molecular FormulaC15H13NO3
- Average mass255.269 Da
Ketorolac, sold under the brand name Toradol among others, is a nonsteroidal anti-inflammatory drug (NSAID) used to treat pain.[1]Specifically it is recommended for moderate to severe pain.[2] Recommended duration of treatment is less than six days.[1] It is used by mouth, by injection into a vein or muscle, and as eye drops.[1][2] Effects begin within an hour and last for up to eight hours.[1]
Common side effects include sleepiness, dizziness, abdominal pain, swelling, and nausea.[1] Serious side effects may include stomach bleeding, kidney failure, heart attacks, bronchospasm, heart failure, and anaphylaxis.[1] Use is not recommended during the last part of pregnancy or during breastfeeding.[1] Ketorolac works by blocking cyclooxygenase 1 and 2 (COX1 and COX2) thereby decreasing prostaglandins.[1][3]
Ketorolac was patented in 1976 and approved for medical use in 1989.[4][1] It is avaliable as a generic medication.[2] In the United Kingdom it costs the NHS less than a £ per injectable dose as of 2019.[2] In the United States the wholesale cost of this amount is about 1.50 USD.[5] In 2016 it was the 296th most prescribed medication in the United States with more than a million prescriptions.[6]
Medical uses
Ketorolac is used for short-term management of moderate to severe pain.[7]It is usually not prescribed for longer than five days.[8][9][10][11] Ketorolac is effective when administered with paracetamol to control pain in neonates because it does not depress respiration as do opioids.[12] Ketorolac is also an adjuvant to opioid medications and improves pain relief. It is also used to treat dysmenorrhea.[11] Ketorolac is used to treat idiopathic pericarditis, where it reduces inflammation.[13]
Ketorolac is used for short-term pain control not lasting longer than five days, and can be administered orally, by intramuscular injection, intravenously, and by nasal spray.[8] Ketorolac is initially administered by intramuscular injection or intravenously.[7] Oral therapy is only used as a continuation from the intramuscular or intravenous starting point.[8][12]
Ketorolac is used during eye surgery help with pain.[14] Ketorolac is effective in treating ocular itching.[15] The ketorolac ophthalmic formulation is associated with a decreased development of macular edema after cataract surgery and is more effective alone rather than as an opioid/ketorolac combination treatment.[16][17] Ketorolac has also been used to manage pain from corneal abrasions.[18]
During treatment with ketorolac, clinicians monitor for the manifestation of adverse effects and side effects. Lab tests, such as liver function tests, bleeding time, BUN, serum creatinine and electrolyte levels are often used and help to identify potential complications.[8][9]
Contraindications
Ketorolac is contraindicated in those with hypersensitivity, allergies to the medication, cross-sensitivity to other NSAIDs, prior to surgery, history of peptic ulcer disease, gastrointestinal bleeding, alcohol intolerance, renal impairment, cerebrovascular bleeding, nasal polyps, angioedema, and asthma.[8][9] Recommendations exist for cautious use of ketorolac in those who have experienced cardiovascular disease, myocardial infarction, stroke, heart failure, coagulation disorders, renal impairment, and hepatic impairment.[8][9]
Adverse effects
Though uncommon, potentially fatal adverse effects are stroke, myocardial infarction, GI bleeding, Stevens-Johnson Syndrome, toxic epidermal necrolysis and anaphylaxis. A less serious and more common (>10%) side effect is drowsiness. Infrequent (<1%) side effects are paresthesia, prolonged bleeding time, injection site pain, purpura, sweating, abnormal thinking, increased production of tears, edema, pallor, dry mouth, abnormal taste, urinary frequency, increased liver enzymes, itching and others. Ketorolac can cause premature constriction of the ductus arteriosis in an infant during the third trimester of pregnancy.[8][9] Platelet function is decreased related to the use of ketorolac.[19]
The practice of restricting treatment with ketorolac is due to its potential to cause kidney damage.[20]
Interactions
Ketorolac can interact with other medications. Probenecid can increase the probability of having an adverse reaction or experiencing a side effect when taken with ketorolac. Pentoxifylline can increase the risk of bleeding. When aspirin is taken at the same time as ketorolac, the effectiveness is decreased. Problematic GI effects are additive and become more likely if potassium supplements, aspirin, other NSAIDS, corticosteroids, or alcohol is taken at the same time. The effectiveness of antihypertensives and diuretics can be lowered. The use of ketorolac can increase serum lithium levels to the point of toxicity. Toxicity to methotrexate is more likely if ketorolac is taken at the same time. The risk of bleeding increases with the concurrent medications clopidogrel, cefoperazone, valproic acid, cefotetan, eptifibatide, tirofiban, and copidine. Anticoagulants and thrombolytic medications also increase the likelihood of bleeding. Medications used to treat cancer can interact with ketorolac along with radiation therapy. The risk of toxicity to the kidneys increases when ketorolac is taken with cyclosporine.[8][9]
Interactions with ketorolac exist with some herbal supplements. The use of Panax ginseng, clove, ginger, arnica, feverfew, dong quai, chamomile, and Ginkgo biloba increases the risk of bleeding.[8][9]
Mechanism of action
The primary mechanism of action responsible for ketorolac’s anti-inflammatory, antipyretic and analgesic effects is the inhibition of prostaglandin synthesis by competitive blocking of the enzyme cyclooxygenase (COX). Ketorolac is a non-selective COX inhibitor.[21] Ketorolac has been assessed to be a relatively higher risk NSAID when compared to aceclofenac, celecoxib, and ibuprofen.[13] It is considered a first-generation NSAID.[19]
History
In the US, ketorolac was the only widely available intravenous NSAID for many years; an IV form of paracetemol, which is not an NSAID, became available in Europe in 2009 and then in the US.[12]
The Syntex company, of Palo Alto, California developed the ophthalmic solution Acular around 2006.[citation needed]
In 2007, there were concerns about the high incidence of reported side effects. This led to restriction in its dosage and maximum duration of use. In the UK, treatment was initiated only in a hospital, although this was not designed to exclude its use in prehospital care and mountain rescue settings.[7] Dosing guidelines were published at that time.[22]
Concerns over the high incidence of reported side effects with ketorolac trometamol led to its withdrawal (apart from the ophthalmic formulation) in several countries, while in others its permitted dosage and maximum duration of treatment have been reduced. From 1990 to 1993, 97 reactions with a fatal outcome were reported worldwide.[23]
The eye-drop formulation was approved by the FDA in 1992.[24] An intranasal formulation was approved by the FDA in 2010[25] for short-term management of moderate to moderately severe pain requiring analgesia at the opioid level.
Synthesis
DOI: 10.1021/jo00348a014

1H-Pyrrolizine-1-carboxylic acid, 2,3-dihydro-5-benzoyl-, (+-)-, could be produced through many synthetic methods.
Following is one of the reaction routes:

2-Methylthiopyrrole (I) is benzoylated with N,N-dimethylbenzamide (II) to produce 5-benzoyl-2-methylthiopyrrole (III) in the presence of POCl3 in refluxing CH2Cl2, and the yielding product is condensed with spiro[2.5]-5,7-dioxa-6,6-dimethyloctane-4,8-dione (IV) in the presence of NaH in DMF giving compound (V). The oxidation of (V) with m-chloroperbenzoic acid in CH2Cl2affords the sulfone (VI), which is submitted to methanolysis with methanol and HCl giving 1-(3,3-dimethoxycarbonylpropyl)-2-methanesulfonyl-5-benzoylpyrrole (VII). The cyclization of (VII) with NaH in DMF yields dimethyl 5-benzoyl-1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1,1-dicarboxylate (VIII), which is finally hydrolyzed and decarboxylated with KOH in refluxing methanol.Compound (III) can be oxidized with m-chloroperbenzoic acid as before giving 2-methanesulfonyl-5-benzoylpyrrole (IX), which is then condensed with spiro compound (IV) as before to afford compound (VI), already obtained.
SYN
DE 2731678; ES 460706; ES 470214; FR 2358406; FR 2375234; GB 1554075
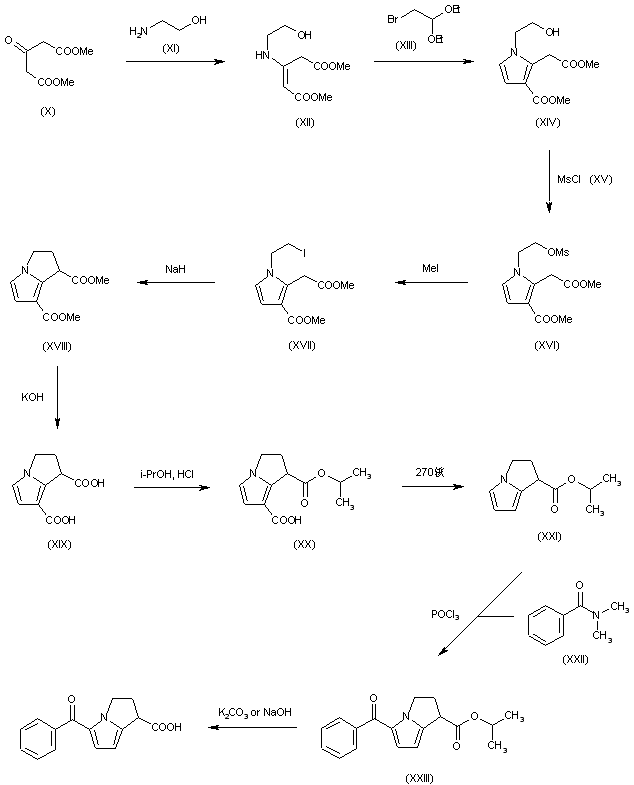
The condensation of dimethylacetone-1,3-dicarboxylate (X) with ethanolamine (XI) yields methyl 3-(methoxycarbonylmethyl)-3-(2-hydroxyethylamino)acrylate (XII), which is cyclized with bromoacetaldehyde diethylacetal (XIII) affording methyl 1-(2-hydroxyethyl)-3-methoxycarbonylpyrrol-2-acetate (XIV). Acylation of (XIV) with methanesulfonyl chloride (XV) and triethylamine in CH2Cl2 yields the corresponding mesylate (XVI), which by treatment with methyl iodide in refluxing acetonitrile is converted into methyl 1-(2-iodoethyl)-3-methoxycarbonylpyrrole-2-acetate (XVII). The cyclization of (XVII) with NaH in DMF yields dimethyl 1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1,7-dicarboxylate (XVIII), which is hydrolyzed with KOH in refluxing methanol – water to the corresponding diacid (XIX). Partial esterification of (XIX) with isopropanol and HCl gives isopropyl 1,2-dihydro-3H-7-carboxypyrrolo[1,2-a]pyrrole-1-carboxylate (XX), which is decarboxylated by heating at 270 C affording isopropyl 1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1-carboxylate (XXI). Benzoylation of (XXI) with N,N-dimethylbenzamide (XXII) and POCl3 in refluxing CH2Cl2 yields isopropyl 5-benzoyl-1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1-carboxylate (XXIII), which is finally hydrolyzed with K2CO3 or NaOH in methanol – water.
SYN2
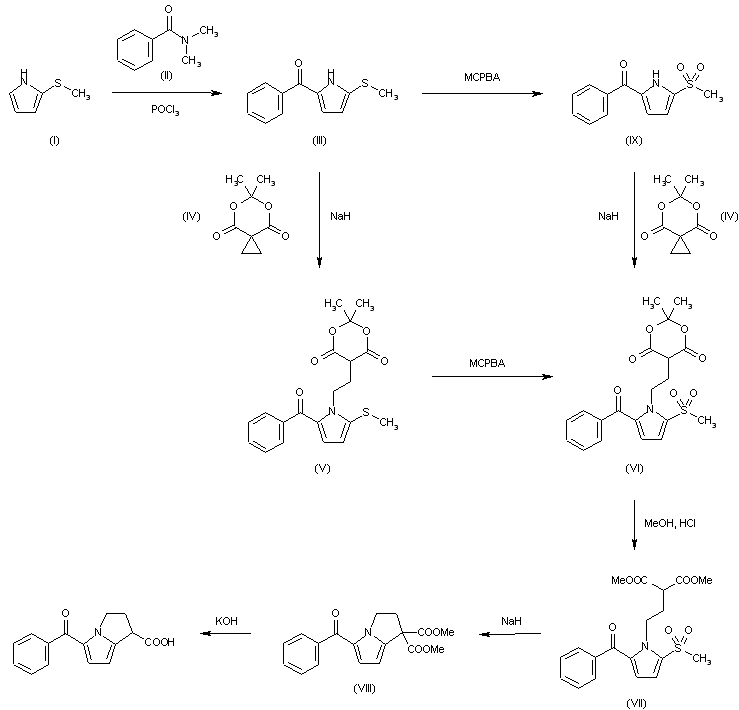
The benzoylation of 2-methylthiopyrrole (I) with N,N-dimethylbenzamide (II) by means of POCl3 in refluxing CH2Cl2 gives 5-benzoyl-2-methylthiopyrrole (III), which is condensed with spiro[2.5]-5,7-dioxa-6,6-dimethyloctane-4,8-dione (IV) by means of NaH in DMF yielding compound (V). The oxidation of (V) with m-chloroperbenzoic acid in CH2Cl2 affords the sulfone (VI), which is submitted to methanolysis with methanol and HCl giving 1-(3,3-dimethoxycarbonylpropyl)-2-methanesulfonyl-5-benzoylpyrrole (VII). The cyclization of (VII) with NaH in DMF yields dimethyl 5-benzoyl-1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1,1-dicarboxylate (VIII), which is finally hydrolyzed and decarboxylated with KOH in refluxing methanol. Compound (III) can be oxidized with m-chloroperbenzoic acid as before giving 2-methanesulfonyl-5-benzoylpyrrole (IX), which is then condensed with spiro compound (IV) as before to afford compound (VI), already obtained.
References
- ^ Jump up to:a b c d e f g h i “Ketorolac Tromethamine Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 13 April 2019.
- ^ Jump up to:a b c d British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 1144, 1302–1303. ISBN 9780857113382.
- ^ “DailyMed – ketorolac tromethamine tablet, film coated”. dailymed.nlm.nih.gov. Retrieved 14 April 2019.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 521. ISBN 9783527607495.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December 2018.
- ^ Jump up to:a b c Mallinson, Tom (2017). “A review of ketorolac as a prehospital analgesic”. Journal of Paramedic Practice. 9 (12): 522–526. doi:10.12968/jpar.2017.9.12.522. Retrieved 2 June 2018.
- ^ Jump up to:a b c d e f g h i Vallerand, April H. (2017). Davis’s Drug Guide for Nurses. Philadelphia: F.A. Davis Company. p. 730. ISBN 9780803657052.
- ^ Jump up to:a b c d e f g Physician’s Desk Reference 2017. Montvale, New Jersey: PDR, LLC. 2017. pp. S–474–5. ISBN 9781563638381.
- ^ “Ketorolac-tromethamine”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- ^ Jump up to:a b Henry, p. 291.
- ^ Jump up to:a b c Martin, Lizabeth D; Jimenez, Nathalia; Lynn, Anne M (2017). “A review of perioperative anesthesia and analgesia for infants: updates and trends to watch”. F1000Research. 6: 120. doi:10.12688/f1000research.10272.1. ISSN 2046-1402. PMC 5302152. PMID 28232869.
- ^ Jump up to:a b Schwier, Nicholas; Tran, Nicole (2016). “Non-Steroidal Anti-Inflammatory Drugs and Aspirin Therapy for the Treatment of Acute and Recurrent Idiopathic Pericarditis”. Pharmaceuticals. 9 (2): 17. doi:10.3390/ph9020017. ISSN 1424-8247. PMC 4932535. PMID 27023565.
- ^ Saenz-de-Viteri, Manuel; Gonzalez-Salinas, Roberto; Guarnieri, Adriano; Guiaro-Navarro, María Concepción (2016). “Patient considerations in cataract surgery – the role of combined therapy using phenylephrine and ketorolac”. Patient Preference and Adherence. 10: 1795–1801. doi:10.2147/PPA.S90468. ISSN 1177-889X. PMC 5029911. PMID 27695298.
- ^ Karch, Amy (2017). Focus on nursing pharmacology. Philadelphia: Wolters Kluwer. p. 272. ISBN 9781496318213.
- ^ Lim, Blanche X; Lim, Chris HL; Lim, Dawn K; Evans, Jennifer R; Bunce, Catey; Wormald, Richard; Wormald, Richard (2016). “Prophylactic non-steroidal anti-inflammatory drugs for the prevention of macular oedema after cataract surgery”. Cochrane Database Syst Rev. 11: CD006683. doi:10.1002/14651858.CD006683.pub3. PMID 27801522.
- ^ Sivaprasad, Sobha; Bunce, Catey; Crosby-Nwaobi, Roxanne; Sivaprasad, Sobha (2012). “Non-steroidal anti-inflammatory agents for treating cystoid macular oedema following cataract surgery”. Cochrane Database Syst Rev (2): CD004239. doi:10.1002/14651858.CD004239.pub3. PMID 22336801.
- ^ Wakai A, Lawrenson JG, Lawrenson AL, Wang Y, Brown MD, Quirke M, Ghandour O, McCormick R, Walsh CD, Amayem A, Lang E, Harrison N (2017). “Topical non-steroidal anti-inflammatory drugs for analgesia in traumatic corneal abrasions”. Cochrane Database Syst Rev. 5: CD009781. doi:10.1002/14651858.CD009781.pub2. PMID 28516471.
- ^ Jump up to:a b Henry, p. 279.
- ^ Henry, p. 280.
- ^ Lee, I. O.; Seo, Y. (2008). “The Effects of Intrathecal Cyclooxygenase-1, Cyclooxygenase-2, or Nonselective Inhibitors on Pain Behavior and Spinal Fos-Like Immunoreactivity”. Anesthesia & Analgesia. 106 (3): 972–977, table 977 contents. doi:10.1213/ane.0b013e318163f602. PMID 18292448.
- ^ MHRA Drug Safety Update October 2007, Volume 1, Issue 3, pp 3-4.
- ^ Committee on the Safety of Medicines, Medicines Control Agency: Ketorolac: new restrictions on dose and duration of treatment. Current Problems in Pharmacovigilance:June 1993; Volume 19 (pages 5-8).
- ^ “Ketorolac ophthalmic medical facts from”. Drugs.com. Retrieved 2013-10-06.
- ^ “Sprix Information from”. Drugs.com. Retrieved 2013-10-06.
Bibliography
- AHFS drug information. Bethesda, MD: American Society of Health-System Pharmacists. 2011. ISBN 9781585282609.
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 9. ISBN 9781284057560.
- Handley, Dean A.; Cervoni, Peter; McCray, John E.; McCullough, John R. (1998). “Preclinical enantioselective pharmacology of (R)- and (S)- ketorolac”. J Clin Pharmacol. 38 (2 Suppl): 25S–35S. doi:10.1002/j.1552-4604.1998.tb04414.x. ISSN 0091-2700. PMID 9549656.
- Henry, Norma (2016). RN pharmacology for nursing : review module. Overland Park, KS: Assessment Technologies Institute. ISBN 9781565335738.
- Kizior, Robert (2017). Saunders nursing drug handbook 2017. St. Louis, MO: Elsevier. ISBN 9780323442916.
External links
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Toradol, Acular, Sprix, others |
| Synonyms | Ketorolac tromethamine |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a693001 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth, IM, IV, eye drops |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100% (All routes) |
| Metabolism | Liver |
| Elimination half-life | 3.5 h to 9.2 h, young adults; 4.7 h to 8.6 h, elderly (mean age 72) |
| Excretion | Kidney: 91.4% (mean) Biliary: 6.1% (mean) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.110.314 |
| Chemical and physical data | |
| Formula | C15H13NO3 |
| Molar mass | 255.27 g/mol g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
//////////Ketorolac,
Cefamandole, セファマンドール ,цефамандол , سيفاماندول , 头孢孟多 ,
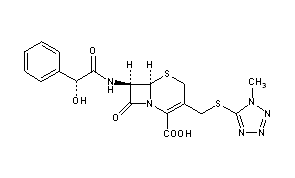
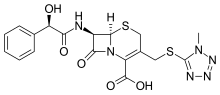

- Use:antibiotic
- Chemical name:[6R-[6α,7β(R*)]]-7-[(hydroxyphenylacetyl)amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
- Formula:C18H18N6O5S2
- MW:462.51 g/mol
- CAS-RN:34444-01-4
- InChI Key:OLVCFLKTBJRLHI-AXAPSJFSSA-N
- InChI:InChI=1S/C18H18N6O5S2/c1-23-18(20-21-22-23)31-8-10-7-30-16-11(15(27)24(16)12(10)17(28)29)19-14(26)13(25)9-5-3-2-4-6-9/h2-6,11,13,16,25H,7-8H2,1H3,(H,19,26)(H,28,29)/t11-,13-,16-/m1/s1
- EINECS:252-030-0
Derivatives
Formate monosodium salt (nafate)
- Formula:C19H17N6NaO6S2
- MW:512.50 g/mol
- CAS-RN:42540-40-9
- EINECS:255-877-4
- LD50:3915 mg/kg (M, i.v.);
2562 mg/kg (R, i.v.)
Cefamandole (INN, also known as cephamandole) is a second-generation broad-spectrumcephalosporinantibiotic. The clinically used form of cefamandole is the formateestercefamandole nafate, a prodrug which is administered parenterally. Cefamandole is no longer available in the United States.
The chemical structure of cefamandole, like that of several other cephalosporins, contains an N-methylthiotetrazole (NMTT or 1-MTT) side chain. As the antibiotic is broken down in the body, it releases free NMTT, which can cause hypoprothrombinemia (likely due to inhibition of the enzymevitamin K epoxide reductase)(vitamin K supplement is recommended during therapy) and a reaction with ethanol similar to that produced by disulfiram (Antabuse), due to inhibition of aldehyde dehydrogenase.
Cefamandole has a broad spectrum of activity and can be used to treat bacterial infections of the skin, bones and joints, urinary tract, and lower respiratory tract. The following represents cefamandole MIC susceptibility data for a few medically significant microorganisms.
- Escherichia coli: 0.12 – 400 μg/ml
- Haemophilus influenzae: 0.06 – >16 μg/ml
- Staphylococcus aureus: 0.1 – 12.5 μg/ml
CO2 is generated during the normal constitution of cefamandole and ceftazidime, potentially resulting in an explosive-like reaction in syringes.[2]
SYNTHESIS
US 3641021
US 3840531 US 3974153 US 3903278 US 2018600 US 2065621 DE 2018600 DE 2065621 DE 2730579
DE 2312997

SYN
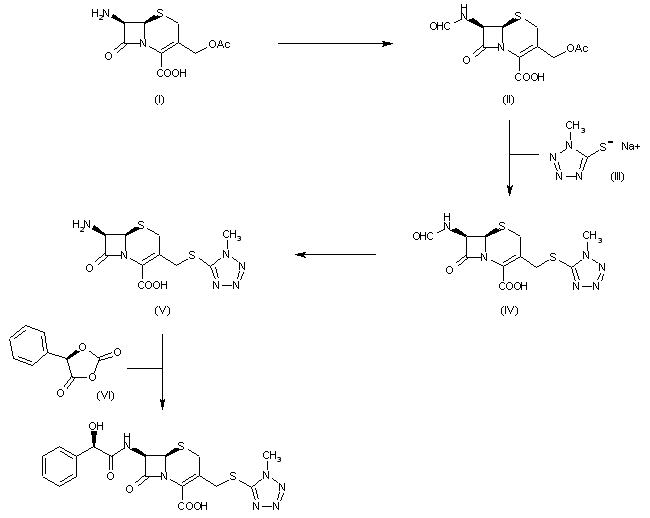
The formylation of 7-aminocephalosporanic acid (I) by the usual techniques produces 7-formamidocephalosporanic acid (II), which is then treated with the sodium salt of 1-methyl-1H-tetrazole-5-thiol (III) to yield 7-formamido-3-(1-methyl-1H-tetrazol-5-ylthio)methyl-3-cephem-4-carboxylic acid (IV). The resulting product (IV) is deformylated affording 7-amino-3-(1-methyl-1H-tetrazol-5-ylthio)methyl-3-cephem-4-carboxylic acid (V), which is finally acylated with anhydro-O-carboxymandelic acid (VI) using the usual techniques.
References
- ^ http://www.toku-e.com/Assets/MIC/Cefamandole%20sodium%20salt.pdf
- ^ Stork CM (2006). “Antibiotics, antifungals, and antivirals”. In Nelson LH, Flomenbaum N, Goldfrank LR, Hoffman RL, Howland MD, Lewin NA. Goldfrank’s toxicologic emergencies. New York: McGraw-Hill. p. 847. ISBN 0-07-143763-0. Retrieved 2009-07-03.
-
- US 3 641 021 (Lilly; 8.2.1972; appl. 18.4.1969).
- DE 2 018 600 (Lilly; appl. 17.4.1970; USA-prior. 18.4.1969).
- DAS 2 065 621 (Lilly; appl. 17.4.1970; USA-prior. 18.4.1969).
- US 3 840 531 (Lilly; 8.10.1974; appl. 21.3.1972).
- US 3 903 278 (Smith Kline Corp.; 2.9.1975; prior. 4.11.1971).
- DOS 2 730 579 (Pierrel S.p.A.; appl. 6.7.1977; GB-prior. 10.7.1976).
-
preparation and/or purification via the trimethylsilyl-derivatives:
- DOS 2 711 095 (Lilly; appl. 14.3.1977; USA-prior. 17.3.1976).
-
purification:
- US 4 115 644 (Lilly; 19.9.1978; appl. 19.9.1978).
- DOS 2 839 670 (Lilly; appl. 12.9.1978; USA-prior. 19.9.1977).
-
crystalline sodium salt:
- US 4 054 738 (Lilly; 18.10.1977; appl. 22.12.1975).
- US 4 168 376 (Lilly; 18.9.1979; appl. 5.6.1978).
-
lithium salt:
- GB 1 546 757 (Lilly; appl. 10.4.1975; valid from 7.4.1976).
-
O-formyl-derivative:
- US 3 928 592 (Lilly; 23.12.1975; appl. 21.2.1974).
- GB 1 493 676 (Lilly; appl. 20.2.1975; USA-prior. 22.2.1974).
- GB 1 546 898 (Lilly; appl. 7.4.1976; USA-prior. 11.4.1975).
- DOS 2 506 622 (Lilly; appl. 17.2.1975; USA-prior. 22.2.1974).
-
crystalline sodium salt of O-formylcefamandole:
- US 4 006 138 (Lilly; 1.2.1977; appl. 11.4.1975).
-
complex of cefamandole sodium with 1,4-dioxane and water:
- US 3 947 414 (Lilly; 30.3.1976; appl. 23.12.1974).
-
complex of cefamandole sodium with ethyl l-(–)-lactate:
- US 3 947 415 (Lilly; 30.3.1976; appl. 23.12.1974).
 |
|
| Clinical data | |
|---|---|
| Trade names | former Mandol |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a601206 |
| Pregnancy category |
|
| Routes of administration |
Intramuscular, intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 75% |
| Elimination half-life | 48 minutes |
| Excretion | Mostly renal, as unchanged drug |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.047.285 |
| Chemical and physical data | |
| Formula | C18H18N6O5S2 |
| Molar mass | 462.505 g/mol g·mol−1 |
| 3D model (JSmol) | |
Tegaserod, テガセロド
Tegaserod
- Molecular FormulaC16H23N5O
- Average mass301.387 Da
- テガセロド
Sundaram Venkataraman, Srinivasulu Gudipati, Brahmeshwararao Mandava Venkata Naga, Goverdhan Banda, Radhakrishna Singamsetty, “Process for preparing form I of tegaserod maleate.” U.S. Patent US20050272802, issued December 08, 2005.US20050272802
Tegaserod maleate [USAN]
189188-57-6
Tegaserod is a 5-HT4 agonist manufactured by Novartis and sold under the names Zelnorm and Zelmac for the management of irritable bowel syndrome and constipation.[1] Approved by the FDA in 2002, it was subsequently removed from the market in 2007 due to FDA concerns about possible adverse cardiovascular effects. Before then, it was the only drug approved by the United States Food and Drug Administration to help relieve the abdominal discomfort, bloating, and constipation associated with irritable bowel syndrome. Its use was also approved to treat chronic idiopathic constipation.[2]
In 2000, originator Novartis established an alliance with Bristol-Myers Squibb for the codevelopment and copromotion of tegaserod maleate, which is now available in more than 55 countries worldwide for the treatment of IBS with constipation. In 2015, Zelnorm was acquired by Sloan Pharma from Novartis.
Novartis’ brand name Zelnorm (tegaserod) had originally received approval from the US FDA in 2002 for the treatment of irritable bowel syndrome with constipation (IBS-C) [5, 8]. It was, however, voluntarily withdrawn from widespread use in the US market in 2007 after concerns arose over the possibility that tegaserod could potentially cause dangerous cardiovascular events in patients [5,8]. Since then, closer evaluations of the original data suggesting such cardiovascular risk have resulted in the limited reintroduction or ‘re-approval’ of tegaserod for treatment of IBS-C specifically in female patients less than 65 years of age and whom are considered to be at a lower risk of a cardiovascular event than the broader population . Zelnorm (tegaserod) by Sloan Pharma subsequently gained re-approval in April of 2019 [5]. Nevertheless, tegaserod remains un-approved in certain regions [7].
Despite the relative complications involved in its history of regulatory approval, ever since its first introduction in 2002 tegaserod remains the only therapy for IBS-C that possesses the unique mechanism of action of acting on serotonin-4 (5-HT(4)) receptors in smooth muscle cells and in the gastrointestinal wall to facilitate actions like esophageal relaxation, peristaltic gut movement, and natural secretions in the gut, among others
Mechanism of action
The drug functions as a motility stimulant, achieving its desired therapeutic effects through activation of the 5-HT4 receptors of the enteric nervous system in the gastrointestinal tract. It also stimulates gastrointestinal motility and the peristaltic reflex, and allegedly reduces abdominal pain.[3] Additionally, tegaserod is a 5-HT2B receptor antagonist.[4]
Withdrawal from market
On 30 March 2007, the United States Food and Drug Administration requested that Novartis withdraw Zelnorm from shelves.[5] The FDA alleges a relationship between prescriptions of the drug and increased risks of heart attack or stroke. An analysis of data collected on over 18,000 patients demonstrated adverse cardiovascular events in 13 of 11,614 patients treated with Zelnorm (a rate of 0.11%) as compared with 1 of 7,031 patients treated with placebo (a rate of 0.01%). Novartis alleges all of the affected patients had preexisting cardiovascular disease or risk factors for such, and further alleges that no causal relationship between tegaserod use and cardiovascular events has been demonstrated.[6] On the same day as the FDA announcement, Novartis Pharmaceuticals Canada announced that it was suspending marketing and sales of the drug in Canada in response to a request from Health Canada.[7] In a large cohort study based on a US health insurance database, no increase in the risk of cardiovascular events were found under tegaserod treatment.[8] Currently, tegaserod may only be used in emergency situations only with prior authorization from the FDA.[9]
Paper
The serotonin 5-HT4 receptor. 2. Structure-activity studies of the indole carbazimidamide class of agonists
J Med Chem 1995, 38(13): 2331
https://pubs.acs.org/doi/abs/10.1021/jm00013a010
PATENT
US 5510353
WO 2005105740
WO 2007119109
WO 2007126889
CN 103467358
WO 2006116953
Syn
PATENT
https://patents.google.com/patent/US20090306170A1/en

-
In a preferred embodiment of the first aspect of the present invention, the process of preparing tegaserod or a salt thereof comprises the steps of:
- (a) coupling S-methyl-isothiosemicarbazide or a salt thereof and 5-methoxy-indole-3-carboxaldehyde to form 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide:
-

-
and
- (b) reacting the 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide with n-pentyl amine to form tegaserod:
-

- [0013]
The skilled person will appreciate that:
-
- S-methyl-isothiosemicarbazide and salts thereof exist in two tautomeric forms:
-
-

-
- 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide exists in four tautomeric forms:
-
-

-
- tegaserod exists in four tautomeric forms:
-
-

- [0017]
It is to be understood that where tautomeric forms occur, the present invention embraces all tautomeric forms and their mixtures, i.e. although S-methyl-isothio-semicarbazide and 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemi-carbazide are mostly defined for convenience by reference to one isothiosemicarbazide form only, and although tegaserod is mostly defined for convenience by reference to one guanidino form only, the invention is not to be understood as being in any way limited by the particular nomenclature or graphical representation employed.
- [0018]
When an S-methyl-isothiosemicarbazide salt is used in the process of the present invention, this may be an acid addition salt with acids, including but not limited to inorganic acids such as hydrohalogenic acids (for example, hydrofluoric, hydrochloric, hydrobromic or hydroiodic acid) or other inorganic acids (for example, nitric, perchloric, sulfuric or phosphoric acid), or organic acids such as organic carboxylic acids (for example, propionic, butyric, glycolic, lactic, mandelic, citric, acetic, benzoic, salicylic, succinic, malic or hydroxysuccinic, tartaric, fumaric, maleic, hydroxymaleic, mucic or galactaric, gluconic, pantothenic or pamoic acid), organic sulfonic acids (for example, methanesulfonic, trifluoromethanesulfonic, ethanesulfonic, 2-hydroxyethanesulfonic, benzenesulfonic, p-toluenesulfonic, naphthalene-2-sulfonic or camphorsulfonic acid) or amino acids (for example, ornithinic, glutamic or aspartic acid). Preferably the S-methyl-isothiosemicarbazide salt is a hydrohalide (such as the hydrofluoride, hydrochloride, hydrobromide, or hydroiodide) or a sulfonate (such as the methanesulfonate, benzenesulfonate, or p-toluenesulfonate). Preferably the S-methyl-isothiosemicarbazide salt is S-methyl-isothiosemicarbazide hydroiodide.
-
-
The following synthetic scheme demonstrates a preferred process of the present invention.
-
- [0032]
The invention is now demonstrated by the following non-limiting illustrative example.
-
EXAMPLE Step 1: Schiff’s Base Formation of 5-methoxy-indole-3-carboxaldehyde and S-methyl-isothiosemi-carbazide hydroiodide
-
- [0033]
5-Methoxy-indole-3-carboxaldehyde (1.5 g, 1 eq) and S-methyl-isothiosemicarbazide hydroiodide (3.99 g, 2 eq) in methanol (15 ml, 10 vol) were stirred in the presence of triethylamine (3 ml, 2 vol) at 25-30° C. for 2 hours. After completion of the reaction, the methanol was removed by distillation under reduced pressure at 45-50° C. and ethyl acetate (10.5 ml, 7 vol) was added to the residue to precipitate out the product. The product, 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemi-carbazide, was separated by filtration, washed with ethyl acetate (3 ml, 2 vol) and dried under vacuum at 45-50° C. The yield was almost quantitative (˜100%).
- [0033]
Step 2: Conversion of 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide to 1-((5-methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (Tegaserod)
-
- [0034]
A solution of 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide (8.0 g, 1 eq) and n-pentyl amine (2.65 g, 1 eq) was refluxed in methanol (8 ml, 1 vol) at 66° C. for 4 hours. After completion of the reaction, the methanol was removed by distillation under reduced pressure at 45-50° C. to obtain tegaserod free base as a yellowish brown solid. Yield=97%. HPLC purity=95%.
- [0034]
Step 3: Conversion of 1-((5-methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (Tegaserod) to Tegaserod Maleate
- [0035]
1-((5-Methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (55 g, 1 eq) was taken in methanol (357.5 ml, 6.5 vol) and stirred. To this reaction mixture was added at room temperature a solution of maleic acid (74.15 g, 3.5 eq) in water (137.5 ml, 2.5 vol) and the reaction mixture stirred for one hour at room temperature. The solid obtained was then filtered through a Buchner funnel and dried at 700 mmHg and 500° C. Yield=36.8 g, 48.42%. HPLC purity=99.45%.
Polymorphs
WO 2007084697
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007084697
EXAMPLES
PXRD:
EV 320 251 655 US Powder X-ray diffraction (“PXRD”) analysis using a SCINTAG powder X-ray diffϊactometer model X’TRA equipped with a solid-state detector. Copper radiation of λ=1.5418 A was used. The sample was introduced using a round standard aluminum sample holder with round zero background quartz plate in the bottom.
Thermal Gravimetric Analysis TTGA):
TGA/SDTA 85 r, Mettler Toledo , Sample weight 7-15 mg.
Heating rate: 100C/ min., in N2 stream: flow rate: 50 ml/min
Example 1 : Preparation of Tegaserod maleate Form B
To a mixture of 90 g MICHO and 63 g NaOH [47 %] was added a solution of 212 g AGPΗI dissolved in 566 mL of water at room temperature. The resultant reaction mixture was heated to 400C. After 3 hours, 522 mL of ethyl acetate was added and the reaction mixture was stirred for an additional hour. The organic phase was washed with water (3 x 450 mL), and vacuum filtered. After addition of 211 mL ethyl acetate and 870 mL of n-propanol, the mixture was heated to 600C and a solution of maleic acid (86.5 g in 180 mL water), at the same temperature, was added to the reaction mixture and stirred at the same temperature. After 2 hours the reaction mixture was cooled to about 100C and stirred for an additional hour. The resulting solid was filtered off, washed with n-propanol, and dried in a vacuum oven over night to give 195.8 g of tegaserod maleate Form B.
6
EV 320251 655 US
PATENT
Tegaserod maleate is an aminoguanidine indole 5HT4 agonist for the treatment of irritable bowel syndrome (IBS). Tegaserod maleate has the following structure:
According to the prescribing information (Physician’s Desk Reference, 57th Ed., at Page 2339), tegaserod as the maleate salt is a white to off-white crystalline powder and is slightly soluble in ethanol and very slightly soluble in water. Tegaserod maleate is available commercially as ZELNORM®, in which it is present as crystalline form.
Tegaserod maleate is disclosed in US patent No. 5,510,353 and in its equivalent EP 0 505 322 (example 13), and is reported to have a melting point of 1900C (table 1 example 13).
The literature (Buchheit K.H, et al., J.Med.Chem., 1995, 38, 2331) describes a general method for the condensation of amino guanidines with indole-3-carbadehydes in methanol in the presence of HCl (pH 3-4). The product obtained after solvent evaporation maybe converted to its hydrochloride salt by treatment of the methanolic solution with diethylether/HCl followed by recrystallization from
methanol/diethylether. Tegaserod base prepared according to this general method is characterized solely by a melting point of 155 0C (table 3 compound 5b). Additional Tegaserod maleate characterization was done by 1H and 13C-NMR according to the literature (Jing J. et. al., Guangdong Weiliang Yuansu Kexue, 2002, 9/2, 51).
WO 04/085393 discloses four crystalline forms of tegaserod maleate. The search report for WO 04/085393 further identifies WO 00/10526, and Drugs Fut. 1999, 24(1) which provides an overview for tegaserod maleate. Additional crystalline forms of tegaserod maleate are provided in WO 2005/058819, one of which is characterized by an X-ray Diffraction pattern having peaks at 15.7, 16.9, 17.2, 24.1, 24.6 and 25.2±0.2 two theta (designated as Form B in that PCT publication).
The solid state physical properties of tegaserod salt may be influenced by controlling the conditions under which tegaserod salt is obtained in solid Form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must take that fact into account in developing a tablet or capsule formulation, which may necessitate the use of glidants such as colloidal silicon dioxide, talc, starch or tribasic calcium phosphate.
Another important solid state property of a pharmaceutical compound is its rate of dissolution in aqueous fluid. The rate of dissolution of an active ingredient in a patient’s stomach fluid may have therapeutic consequences since it imposes an upper limit on the rate at which an orally- administered active ingredient may reach the patient’s bloodstream. The rate of dissolution is also a consideration in
formulating syrups, elixirs and other liquid medicaments. The solid state Form of a compound may also affect its behavior on compaction and its storage stability.
These practical physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which defines a particular polymorphic Form of a substance. The polymorphic form may give rise to thermal behavior different from that of the amorphous material or another polymorphic Form. Thermal behavior is measured in the laboratory by such techniques as capillary melting point, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) and may be used to distinguish some polymorphic forms from others. A particular polymorphic Form may also give rise to distinct spectroscopic properties that may be detectable by powder X-ray crystallography, solid state C NMR spectrometry and infrared spectrometry.
The discovery of new polymorphic forms of a pharmaceutically useful compound provides a new opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic.
The polymorphic forms may further help in purification of tegaserod, particularly if they possess high crystallinity. In the event of metastability, a metastable polymorphic form may be used to prepare a more stable polymorph.
Hence, discovery of new polymorphic forms and new processes help in advancing a formulation scientist in preparation of tegaserod as an active pharmaceutical ingredient in a formulation.
The present invention provides an additional polymorphic form of a maleate salt of tegaserod.
Example 1 : Preparation of sesqui-tefiaserod maleate Foπn H2 through tegaserod base
To a mixture of AGPΗI (112.7 g) in 283 mL of water was added 5-MICHO (45 g) followed by NaOH (52.8 g, 47%) and stirred at room temperature. After three hours, 522 mL of ethyl acetate were added and the mixture stirred for an additional four hours. After phase separation at 400C the organic phase was washed with water (3 x 218 ml), and filtrated under vacuum. The resulting solution was heated to 60 0C and a solution of maleic acid (14.4 g) in 45 mL water was dropped during half hour, and the reaction mixture stirred at the same temperature for an additional two hours. The mixture was cooled to 100C during one hour, kept under stirring at the same temperature for 12 hrs and then filtered under vacuum. The wet product was washed twice with 65 ml of ethyl acetate and dried in a vacuum oven at 45°C for 16 hours to give 85% of the product.
Example 2: Preparation of sesqui-tegaserod maleate Form H2
45 gr MICHO were added to a 1 L reactor at RT. A solution of 112.7 gr of AGP HI and 283 ml water was added to the reactor. 52.8 gr of NaOH 47% were added to the mixture while stirring. The mixture was heated to 400C and stirred for 12 hrs. 522 ml of Ethyl Acetate were added and the mixture was stirred for 4 hrs.
After phase separation at 400C the organic phase was washed with water (3 x 218 ml), and filtrated under vacuum.
The mixture was heated to 600C and a mixture o 14.4 gr of Maleic Acid in 45 ml water was dropped during 5 min.
The mixture was stirred at 600C for 2 hrs.
The mixture was cooled to 100C during 1 hour, stirred at 100C for 13 hrs and then filtered under vacuum. The wet product was washed twice with 65 ml of n-Propanol. The wet product was dried in a vacuum oven at 45°C.
Yield: 71.2%
Example 3: Preparation of Tegaserod maleate Form B from Sesqui-tegaserod maleate Form H2
6.9 g of maleic acid were added to a slurry of Sesqui-Tegaserod maleate Form H2 (41.5 g) in 208 ml n-propanol at room temperature. The mixture was stirred for 5 hours at the same temperature, filtered and washed with n-propanol. After drying on vacuum oven at 450C for 15 hours the product was analyzed by XRD and found to be Form B (89% yield).
PATENT
https://patents.google.com/patent/WO2005058819A2/en


The formation of hydrazones is catalyzed by both general acids and general bases. General base catalysis of dehydration of the tetrahedral intermediate involves nitrogen deprotonation concerted with elimination of hydroxide ion as shown in the Scheme (Sayer J.M., et al. J. Am. Chem. Soc. 1973, 95, 4277). R fast O I H h° NH2R’ R- -NHR’ R R

In many cases, the equilibrium constant for their formation in aqueous solution is high. The additional stability may be attributed to the participation of the atom adjacent to the nitrogen in delocalized bonding. – + RRC = N – NH2 ~*→- RRC – N = NH2
In order to obtain only the maleic salt, the product when using an acid halide (HA) or other acids has to first be converted into the free base, before the addition of maleic acid (Path a), which results in an additional step to the synthesis. On the other hand, the reaction of the present invention in the presence of organic or inorganic base results in the formation of tegaserod free base which gives only the maleate salt after the addition of maleic acid (Path b).


TGS

TGS-MA
EXAMPLES
HPLC method for detecting the level of the impurities:
Column: Atlantis dcl8(150*4.6),
Mobile phase: A.80% KH2PO4(0.02M) pH=5, 20% acetonitrile(ACN), B.100% ACN. Gradient: time 0= A: 100 B: 0, time 25 min= A:50%, B:50%, time 30 min= A:50%, B:50%, + 10 minutes of equilibration time. Wavelength= 225 nm
Sample concentration: 0.5 mg/mL
Temperature = 25°C
Example 1- Preparation of Tegaserod maleate in water with HCl.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by HCl (37%) until pH 4. The mixture was heated to reflux for 1 hour and then cooled to room temperature. To the resulting slurry was added a solution of NaHCO3 (10%) until pH 9, and heated to 65°C for 20 minutes. After cooling, 100 mL of EtOAc were added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.27 g of crude tegaserod maleate with a purity of 99.70% (by HPLC).
Example 2- Preparation of Tegaserod maleate in water with HCl in two steps. a. Preparation of Tegaserod free base.
To a mixture of AGP-HI (163.3 g, 0.6 mol) in 375 mL water was added 5-MICHO (52.5 g, 0.3 mol) followed by HCl (37%) until pH 4. The mixture was heated to reflux for 1 hour and then cooled to room temperature. To the resulting slurry was added a liter of a solution of NaHCO (10%) until pH 9, and heated to 65 °C for one hour. After cooling, 1500 mL of EtOAc were added, and the organic phase washed with water. The remaining organic phase was evaporated to dryness to give tegaserod free base with a purity of 87.42 % (by HPLC). b. Preparation of Tegaserod maleate. To a solution of 2 g of tegaserod free base in MeOH was added a solution of maleic acid (1.28 g, 0.011 mol) in 10 mL MeOH. The resulting solid was filtered off and washed with MeOH to give 1.09 g of crude tegaserod maleate with a purity of 96.81 % (by HPLC).
Example 3- Preparation of Tegaserod maleate in water with TEA.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by TEA (11.0 mL, 0.08 mol) and stirred at room temperature. After one hour, 25 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 7.92 g of crude tegaserod maleate with a purity of 94 % (by HPLC).
Example 4- Preparation of Tegaserod maleate in water with NaHCO3. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaHCO3 (6.72 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, 50 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.71 g of crude tegaserod maleate with a purity of 98 % (by HPLC) .
Example 5- Preparation of Tegaserod maleate in water with NaHCO3 in two steps. a. Preparation of Tegaserod free base. To a mixture of AGP-HI (32.66 g, 0.12 mol) in 300 mL water was added 5-MICHO (10.51 g, 0.06 mol) followed by NaHCO3(20.16 g, 0.24 mol) and heated to reflux for 1 hour. After cooling, 150 mL of EtOAc was added, and the organic phase washed with water and evaporated to dryness to give 20.4 g of tegaserod free base (91.55%) purity by HPLC). b. Preparation of Tegaserod maleate.
To a solution of 2g of the resulting tegaserod free base in 8 mL MeOH was added a solution of maleic acid (1.28 g, 0.011 mol) in 5 mL MeOH. The resulting solid was filtered off and washed with MeOH to give 2.1 g of crude tegaserod maleate with a purity of 99.63 % (by HPLC).
Example 6- Preparation of Tegaserod maleate in water with Na2CO3. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by Na2CO3 (4.24 g, 0.04 mol) and heated to reflux for 1 hour. After cooling, 50 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.48 g of crude tegaserod maleate with a purity of 98.2 % (by HPLC).
Example 7- Preparation of Tegaserod maleate in MeOH with TEA in two steps. a. Preparation of tegaserod free base
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL MeOH was added 5-MICHO (3.50 g, 0.02 mol) followed by triethylamine (11.0 mL, 0.08 mol). After 1 h at room temperature the mixture was evaporated to dryness, and washed with water, giving 5.79 g of tegaserod free base (86.90 % purity by HPLC). b. Preparation of tegaserod maleate
To a solution of 2 g of the resulting tegaserod free base in 10 mL MeOH was added a solution of maleic acid (1.16 g, 0.01 mol) in water. The resulting solid was filtrated and washed with water to give 1.45 g of crude tegaserod maleate as a white solid (94.60 % purity by HPLC). Crystallization in MeOH improved the purity to 98.94% by HPLC.
Example 8- Preparation of Tegaserod maleate in IPA with K2CO3.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL IPA was added 5-MICHO (3.50 g, 0.02 mol) followed by K2CO3 (5.53g, 0.04 mol). After 22 h at room temperature the mixture was washed with brine. The organic phase was treated with a solution of maleic acid (3.48 g, 0.03 mol) in IPA. The resulting solid was filtrated and washed with IPA to give 3.26 g of a white solid (98.97% purity by HPLC).
Example 9- Preparation of Tegaserod maleate in TEA.
To a mixture of AGP-HI (10.88 g, 0.04 mol) and 5-MICHO (3.50 g, 0.02 mol) was added 11 mL of TEA (0.08 mol). After 2 h at room temperature 25 mL of EtOAc were added and the mixture was stirred for 1 h. The resulting solid was filtrated and washed with 25 mL EtOAc, to give 5.7 g of crude.
2 g of the residue was dissolved in 13 mL MeOH and treated with 7 mL of a solution of maleic acid (2.7 g, 0.023 mol) in water. The resulting solid was filtered and washed with water to give 1.5 g of tegaserod maleate (99.26 % purity by HPLC). Crystallization of the solid in MeOH improved the purity to 99.89%) by HPLC.
Example 10- Preparation of Tegaserod maleate in toluene/water with NaHCO3. a. Preparation of tegaserod free base To a mixture of AGP-HI (10.88 g, 0.04 mol) in 200 mL of water/toluene 1:1 was added 5-MICHO (3.50 g, 0.02 mol) followed by NaHCO3 (6.72 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.25 g of tegaserod free base was obtained (93.8 % purity by HPLC). b. Preparation of tegaserod maleate To a solution of 3 g of the product in 10 mL MeOH was added a solution of maleic acid (2.31 g, 0.02 mol) in 10 mL water. The resulting solid was filtered off and washed with a solution of MeOH / water to give 2.50 g of crude tegaserod maleate with a purity of 96.6 % (by HPLC).
Example 11- Preparation of Tegaserod maleate in water with NaOH. a. Preparation of tegaserod free base
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaOH (2 g, 0.05 mol) and stirred at room temperature. After 3 hours 50 mL of EtOAc was added, and the organic phase washed with water and evaporated to dryness to give 5.6 g of tegaserod free base (98.80% purity by HPLC). b. Preparation of Tegaserod maleate.
To a solution of 1.6 g of tegaserod free base in 15 mL ethyl acetate was added a solution of maleic acid (0.7 g, 0.006 mol) in 5 mL ethyl acetate. The resulting solid was filtered off and washed with ethyl acetate to give 1.65 g of crude tegaserod maleate, with a purity of 99.87 % (by HPLC)
Example 12- Preparation of Tegaserod maleate in water with maleic acid. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by maleic acid (9.3 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.92 g of tegaserod maleate crude was obtained (92.4 % purity by HPLC).
Example 13- Preparation of Tegaserod maleate in methanol with maleic acid.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of methanol was added 5- MICHO (3.50 g, 0.02 mol) followed by maleic acid (9.29 g, 0.08 mol) and heated to reflux for 2 hours. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.51 g of tegaserod maleate crude was obtained (97.4 % purity by HPLC).
Example 14- Preparation of Tegaserod maleate in water with NaOH in one pot. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaOH (2 g, 0.05 mol) and stirred at room temperature. After 4 hours a solution of maleic acid (4.35 g, 0.0375 mol) in 25 mL water was added, and the reaction mixture was stirred overnight. The resulting solid was filtered off and washed with water to give 7.87 g of crude tegaserod maleate (99.16% purity by HPLC).
Example 15- Preparation of Tegaserod maleate in water with NaOH in one pot.
To a mixture of AGP-HI (174.2 g, 0.64 mol) in 362 mL of water was added 5-MICHO (56.2 g, 0.32 mol) followed by NaOH (68.1 g, 47%) and stirred at room temperature. After 4.5 hours, 640 mL of EtOAc was added, and the organic phase washed with water, treated with active carbon and filtrated through hyper flow bed. A solution of maleic acid (44.57 g, 0.38 mol) in 415 mL ethyl acetate / water 97:3 was added, and the reaction mixture was heating to 65 °C and stirrer overnight. The resulting solid was filtered off and washed with water and ethyl acetate to give 121.4 g of crude tegaserod maleate (up to 99.88 % purity by HPLC).
Example 16- Preparation of Tegaserod maleate (from Tegaserod acetate).
To a solution of 8.2 g of tegaserod acetate in 15 mL ethyl acetate heated to 65 °C was added a solution of 3.3 g maleic acid in 5 ml ethyl acetate/water 95:5, and the mixture was stirred at the same temperature for an additional 2 hours, followed by cooling to room temperature and stirring overnight. The resulting solid was filtered off and washed with ethyl acetate/water 95:5. After drying on vacuum oven at 45 °C for 15 hours, 9.18 g of tegaserod maleate were obtained. Tegaserod acetate is prepared according to Examples 19, 20 and 21 of U.S. Appl. No. 11/015,875 and PCT/US04/42822.
Example 19 of U.S. Appl. No. 11/015,875 reads as follows: A slurry of tegaserod base amorphous (6 g) in 50 mL ethyl acetate was stirred at 20- 30 °C for 24 hours. The solid was filtrated and washed with 15 mL of same solvent and dried in a vacuum oven at 40 °C for 16 hours.
Example 20 of U.S. Appl. No. 11/015,875 reads as follows:
A slurry of tegaserod base amorphous (6 g) in 50 mL ethyl acetate was stirred at reflux for 24 hours. The solid was filtrated and washed with 15 mL of same solvent and dried in a vacuum oven at 40 °C for 16 hours.
Example 21 of U.S. Appl. No. 11/015,875 reads as follows:
To a slurry of tegaserod maleate Form A (15 g) in EtOAc (210 mL) and water (210 mL) was added 38.4 g of NaOH 47%. The mixture was stirred overnight and the resulting white solid was isolated by filtration and washed with 100 mL of water. Drying in vacuum oven at 40 °C for 16 hours gives 12.38 g (90% yield). Tegaserod acetate was characterized by H and C-NMR.
Example 17: General method for the preparation of Tegaserod maleate Form A from crystallization.
Tegaserod maleate (1 g) was combined with the appropriate solvent (5 mL), and heated to reflux. Then, additional solvent was added until complete dissolution. After the compound was dissolved, the oil bath was removed and the solution was cooled to room temperature. The solid was filtrated and washed with 5 mL of the same solvent and dried in a vacuum oven at 40 C for 16 hours.


Example 18: Preparation of Tegaserod maleate in water with p-TSOH.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by para-toluenesulfonic acid monohydrate (0.45 g, 0.0024 mol). The mixture was heated to reflux for 4 hour and then cooled to room temperature. The resulting solid was filtered off and washed with water to give 8.32 g of a white solid (84.74 % purity by HPLC).
Example 19: Preparation of Tegaserod maleate from Tegaserod Hemi-maleate hemihydrate
To a solution of 1.72 g of Tegaserod Hemi-maleate hemihydrate in 20 mL ethyl acetate at room temperature was added a solution of 0.134 g maleic acid in 5 ml ethyl acetate/water 95:5, and the mixture was stirred at the same temperature for overnight. The resulting solid was filtered off and washed with ethyl acetate/water 95:5. After drying on vacuum oven at 45°C for 15 hours, 1.68 g of tegaserod maleate were obtained. Tegaserod Hemi-maleate hemihydrate was prepared according to Example 23 of U.S. Appl. No. 11/015,875 and PCT/US04/42822. Example 23 of U.S. Appl. No. 11/015,875 and PCT/US04/42822 reads as follows: A solution of maleic acid (2.32 g in 22 mL ethyl acetate/water 97:3) was added to a mixture of tegaserod base in ethyl acetate, and the reaction mixture was heated to 65 °C and stirrer overnight. The resulting solid was filtered off and washed with water and ethyl acetate. Drying in vacuum oven at 40 °C for 16 hours gives 12.19 g of Tegaserod hemi-maleate hemihydrate. Depending on the base polymorph used a solution or slurry is obtained. When using amorphous tegaserod base, a solution is obtained, while when using any other base polymorph of tegaserod, a slurry is obtained.
PATENT
https://patents.google.com/patent/WO2009063247A1/en
Tegaserod, chemically named 2-[(5-methoxy-liϊ-indol-3-yl)methylene]-IV-pentylhydrazine- carboximidamide, is a selective serotonin 4 (5-HT4) receptor agonist, which can be used to treat gastrointestinal disorders such as heartburn, bloating, postoperative ileus, abdominal pain and discomfort, epigastric pain, nausea, vomiting, regurgitation, intestinal pseudoobstruction, irritable bowel syndrome and gastro-oesophageal reflux. Tegaserod as the maleate salt is marketed for the short-term treatment of irritable bowel syndrome in women whose primary bowel symptom is constipation.
Tegaserod, represented by the formula (I), was first described in US 5 510 353 as well as processes for its preparation. The maleate salt of tegaserod is also disclosed, but interestingly a method of manufacturing tegaserod maleate is not disclosed. The only characterizing data is the melting point which is disclosed as 1900C for the maleate salt and 124°C for the tegaserod base.

WO 2006/116953 describes crystalline forms of the hydrobromide, dihydrogen phosphate and oxalate salts of tegaserod. Also claimed is a process for preparing the hydrochloride, hydrobromide, dihydrogen phosphate, tartrate, citrate, lactate, mesylate, oxalate, succinate, glutarate, adipate, salicylate, sulfate, mandelate, camphor sulfonate and hydrogen sulfate salts of tegaserod from a specific crystalline form of tegaserod base. Another process described is a method of preparing the dihydrogen phosphate, maleate, tartrate, citrate, mesylate, lactate, succinate, oxalate, hydrochloride, salicylate, glutarate, adipate, hydrobromide, sulfate and hydrogen sulfate from a hydrogen halide salt of tegaserod.
There are often major hurdles to overcome before an active pharmaceutical ingredient (API) can be formulated into a composition that can be marketed. For example, the rate of dissolution of an API that has poor aqueous solubility is often problematic. The aqueous solubility is a major influence on the bioavailability of the API such that a poorly soluble API can mean the API is not available to have a pharmaceutical effect on the body. The API can also cause problems during manufacture of a pharmaceutical composition. For example, flowability, compactability and stickiness are all factors affected by the solid state properties of an API.
It has thus always been an aim of the pharmaceutical industry to provide many forms of an API in order to mitigate the problems described above. Different salts, crystalline forms also known as polymorphs, solvates and amorphous forms are all forms of an API that can have different physiochemical and biological characteristics. Indeed, it has been discovered that the tegaserod maleate product on the market, Zelnorm , has been linked to an increase in heart problems in a proportion of individuals. One possible reason is that the maleate moiety reacts with the tegaserod, resulting over time in the production of a toxic impurity.
This impurity could be a contributor to the heart problems seen in some patients.
PATENT
https://patents.google.com/patent/WO2004085393A1/en
Figure 1 is a x-ray powder diffraction pattern of tegaserod maleate Form I. Figure 2 is a x-ray powder diffraction pattern of tegaserod maleate Form II. Figure 3 is a x-ray powder diffraction pattern of tegaserod maleate Form III. Figure 4 is a x-ray powder diffraction pattern of tegaserod maleate Form IV. x-Ray powder diffraction spectrum was measured on a Siemens D5000 x- ray powder diffractometer having a copper-Kα radiation.
The following examples further illustrate the invention.
Example 1 Tegaserod free base (10 gm) is dissolved in acetone (100 ml). Maleic acid (4 gm) is added to the solution and the contents are maintained for 1 hour at 25°C. The separated solid is filtered to give 12.5 gm of tegaserod maleate Form I.
Example 2 Tegaserod maleate Form II (5 gm) and acetone (70 ml) are mixed and refluxed for 1 hour and cooled to 25°C and filtered to give 4.8 gm of tegaserod maleate Form I.
Example 3 Tegaserod maleate Form I (10 gm) is dissolved in methanol (100 ml). Acetonitrile (150 ml) is added to the solution and the contents are heated to reflux. The contents are then cooled to 25°C and maintained for 30 minutes. The separated crystals are collected by filtration to give 9 gm of tegaserod maleate Form II.
Example 4 Tegaserod free base (10 gm) is dissolved in methanol (100 ml) and maleic acid (4 gm) is added to the solution. Then the contents are maintained for 30 minutes at 25°C. Then the separated solid is filtered to give 13 gm of tegaserod maleate Form III.
Example 5
Tegaserod maleate (5 gm) is dissolved in methanol (50 ml) and the solution is maintained at 25°C for 30 minutes. The separated crystals are collected by filtration to give 4.8 gm of tegaserod maleate Form III. Example 6 Tegaserod free base (10 gm) is dissolved in methanol (50 ml), maleic acid (4 gm) is added and the contents are refluxed for 30 minutes and then the resulting solution is cooled to 25°C. Methylene dichloride (200 ml) is added and the contents are maintained for 30 minutes at 25°C. The separated solid is collected by filtration to give 13 gm of tegaserod maleate Form IV.
Example 7 Maleic acid (4 gm) is added to a solution of tegaserod free base (10 gm) in methanol (50 ml). The contents are maintained for 30 minutes at 25°C and isopropyl alcohol (150 ml) is mixed and contents are maintained for 30 minutes at 25°C. The separated solid is collected by filtration to give 12.5 gm of tegaserod maleate Form IV
CLIP
References
- ^ “New Data for Zelnorm”. Archived from the original on December 9, 2007. Retrieved March 30, 2007.
- ^ “FDA approves first treatment for women with irritable-bowel syndrome”. Archived from the original on February 5, 2007. Retrieved March 30, 2007.
- ^ Rossi, S. (2004). Australian Medicines Handbook. Adelaide: Health Communication Network. ISBN 0-9578521-4-2.
- ^ Beattie DT, Smith JA, Marquess D, et al. (November 2004). “The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo”. Br. J. Pharmacol. 143 (5): 549–60. doi:10.1038/sj.bjp.0705929. PMC 1575425. PMID 15466450.
- ^ “FDA Announces Discontinued Marketing of GI Drug, Zelnorm, for Safety Reasons”. FDA Press Release. 30 March 2007.
- ^ “Zelnorm” (PDF). Novartis. Archived from the original (PDF) on 2007-04-10. Retrieved 2007-03-30.
- ^ “Novartis suspends Canadian marketing and sales of Zelnorm in response to request from Health Canada”. Retrieved 2007-03-30.
- ^ Loughlin J, Quinn S, Rivero E, Wong J, Huang J, Kralstein J, Earnest DL, Seeger JD (2010). “Tegaserod and the Risk of Cardiovascular Ischemic Events: An Observational Cohort Study”. J Cardiovasc Pharmacol Ther. 15 (2): 151–7. doi:10.1177/1074248409360357. PMID 20200325.
- ^ http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm103223.htm
- Beattie DT, Smith JA, Marquess D, Vickery RG, Armstrong SR, Pulido-Rios T, McCullough JL, Sandlund C, Richardson C, Mai N, Humphrey PP: The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo. Br J Pharmacol. 2004 Nov;143(5):549-60. Epub 2004 Oct 4. [PubMed:15466450]
- Talley NJ: Irritable bowel syndrome. Intern Med J. 2006 Nov;36(11):724-8. doi: 10.1111/j.1445-5994.2006.01217.x. [PubMed:17040359]
- Borman RA, Tilford NS, Harmer DW, Day N, Ellis ES, Sheldrick RL, Carey J, Coleman RA, Baxter GS: 5-HT(2B) receptors play a key role in mediating the excitatory effects of 5-HT in human colon in vitro. Br J Pharmacol. 2002 Mar;135(5):1144-51. doi: 10.1038/sj.bjp.0704571. [PubMed:11877320]
- Vickers AE, Zollinger M, Dannecker R, Tynes R, Heitz F, Fischer V: In vitro metabolism of tegaserod in human liver and intestine: assessment of drug interactions. Drug Metab Dispos. 2001 Oct;29(10):1269-76. [PubMed:11560869]
- FDA approves the reintroduction of Zelnorm™ (tegaserod) for Irritable Bowel Syndrome with Constipation (IBS-C) in women under 65 [Link]
- Tegaserod 2019 FDA Label [File]
- EMA Refusal Assessment Report for Zelnorm (Tegaserod) [File]
- FDA Joint Meeting of the Gastrointestinal Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee Briefing Document for Zelnorm (tegaserod maleate) [File]
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Zelnorm, Zelmac |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 10% |
| Protein binding | 98% |
| Metabolism | Gastric and hepatic |
| Elimination half-life | 11 ± 5 hours |
| Excretion | Fecal and renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C16H23N5O |
| Molar mass | 301.39 g/mol g·mol−1 |
| 3D model (JSmol) | |
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US5510353 | No | 1996-04-23 | 2013-04-26 |  |
References
-
- Buchheit, K.-H. et al.: J. Med. Chem. (JMCMAR) 38, 2331 (1995).
- US 5 510 353 (Novartis; 23.4.1996; GB-prior. 22.3.1991).
- EP 505 322 (Sandoz; GB-prior. 22.3.1991).
-
Preparation of 5-methoxyindole:
- Tsuji, Y. et al.: J. Org. Chem. (JOCEAH) 55 (2), 580 (1990).
- Jones, G.B. et al.: J. Org. Chem. (JOCEAH) 58 (20), 5558 (1993).
- Kondo, Y. et al.: J. Org. Chem. (JOCEAH) 62 (19), 6507 (1997).
- JP 3 024 055 (Kawaken Fine Chemicals; 1.2.1991; J-prior. 21.6.1989).
/////////Tegaserod, HTF 919, HTF-919, SDZ HTF 919, SDZ-HTF-919, テガセロド , Sloan Pharma, Novartis,
CCCCCNC(=N)N\N=C\C1=CNC2=C1C=C(OC)C=C2
BMS 986236
BMS-986236
CAS 2058035-15-5
MW C22 H25 N9 O
MF 431.49
1-(5-(4-(3-Hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile
1H-Pyrazolo[3,4-b]pyridine-5-carbonitrile, 1-[5-[4-(3-hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl]-4-[(1-methylethyl)amino]-2-pyridinyl]-
1-[5-[4-(3-hydroxy-3-methylbutyl)triazol-1-yl]-4-(propan-2-ylamino)pyridin-2-yl]pyrazolo[3,4-b]pyridine-5-carbonitrile
|
The present invention generally relates to heteroaryl substituted aminopyridine compounds useful as kinase inhibitors, including the modulation of IRAK-4. Provided herein are heteroaryl substituted aminopyridine compounds, compositions comprising such compounds, and methods of their use. The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to kinase modulation and methods of inhibiting the activity of kinases, including IRAK-4 in a mammal.
|
PATENT
US2018186799
https://patentscope.wipo.int/search/en/detail.jsf?docId=US222843237&tab=PCTDESCRIPTION&maxRec=1000
PATENT
Gardner, D. S.; Santella, J. B.; Paidi, V. R.; Wu, H.; Duncia, J. V.; Nair, S. K.; Hynes, J. (BMS, USA). Heteroaryl Substituted Aminopyridine Compounds. PCT Int. Appl. WO/2016/210034 A1, 2016.
https://patents.google.com/patent/WO2016210034A1/en
Clip
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00023
Development of a Scalable Synthesis for the Potent Kinase Inhibitor BMS-986236; 1-(5-(4-(3-Hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile
 , Richard Rampulla‡, Muthalagu Vetrichelvan*† , Anuradha Gupta† , Arun Kumar Gupta†, and Arvind Mathur‡
, Richard Rampulla‡, Muthalagu Vetrichelvan*† , Anuradha Gupta† , Arun Kumar Gupta†, and Arvind Mathur‡
A scalable route to 1-(5-(4-(3-hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile (1, BMS-986236) was developed by incorporating an alternate azide intermediate following safety-driven processes. The newly developed process involved mitigating safety hazards and eliminating the column chromatography purification. The issue of trace metal contamination in the final API observed in the first-generation synthesis has been overcome.
1 (92.5 g, 73% yield, 99.5% purity by HPLC) as a cream-colored solid.
1H NMR (400 MHz, DMSO-d6) δ = 9.21–8.86 (m, 2H), 8.66 (s, 1H), 8.45–8.24 (m, 2H), 7.49 (s, 1H), 6.57 (d, J = 7.5 Hz, 1H), 4.33 (s, 1H), 3.83 (d, J = 7.0 Hz, 1H), 2.91–2.72 (m, 2H), 1.97–1.68 (m, 2H), 1.24 (d, J = 6.5 Hz, 12H).
13C NMR (100 MHz, DMSO) δ = 151.7, 150.8, 149.8, 147.9, 147.7, 143.7, 136.8, 136.3, 122.9, 118.9, 117.6, 116.0, 102.8, 99.4, 68.4, 43.6, 42.7, 29.2, 21.7, 20.2.
HRMS [M + H]+ calcd for C22H25N9O 432.2255, found 432.2259.



//////// BMS-986236, BMS 986236
CC(C)(O)CCc1cn(nn1)c2cnc(cc2NC(C)C)n4ncc3cc(cnc34)C#N
Acefylline
Acefylline
- Molecular FormulaC9H10N4O4
- Average mass238.200 Da
Acefylline (INN),[1] also known as acetyloxytheophylline, is a stimulant drug of the xanthine chemical class. It acts as an adenosine receptor antagonist. It is combined with diphenhydramine in the pharmaceutical preparation etanautine to help offset diphenhydramine induced drowsiness.[2]
Synthesis
DE 352980 (1922 to E. Merck); Frdl. 14, 1320; S. M. Ride et al., Pharmazie 32, 672 (1977).
Acefylline
- Use:cardiotonic, diuretic, antispasmodic, bronchodilator
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid
- Formula:C9H10N4O4
- MW:238.20 g/mol
- CAS-RN:652-37-9
- EINECS:211-490-2
- LD50:1180 mg/kg (M, i.p.); 2733 mg/kg (M, p.o.)
Acepifylline
- Use:
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid compd. with piperazine
- Formula:C9H10N4O4 • xC4H10N2
- MW:unspecified
- CAS-RN:18833-13-1
- EINECS:242-614-3
Acefylline heptaminol
- Use:
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid compd. with 6-amino-2-methyl-2-heptaminol (1:1)
- Formula:C9H10N4O3 • C8H19NO
- MW:367.45 g/mol
- CAS-RN:59989-20-7
- EINECS:262-012-4
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names (Rec. INN): List 21” (PDF). World Health Organization. Retrieved 29 December 2016.
- ^ Zuidema, Jan. (1978). “Biofarmaceutische en farmacokinetische aspecten van theofylline en acefylline”. Thesis (doctoral)–Universiteit van Amsterdam. References
Baisse, J.: Bull. Soc. Chim. Fr. (BSCFAS) 1949, 769.
DE 352 980 (E. Merck; 1922).
 |
|
 |
|
| Clinical data | |
|---|---|
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| ECHA InfoCard | 100.010.447 |
| Chemical and physical data | |
| Formula | C9H10N4O4 |
| Molar mass | 238.20 g/mol g·mol−1 |
| 3D model (JSmol) | |
////////Acefylline
DESLORATADINE, デスロラタジン
Desloratadine
- Molecular FormulaC19H19ClN2
- Average mass310.821 Da
Desloratadine (trade name Clarinex and Aerius) is a tricyclic H1-antihistamine that is used to treat allergies. It is an active metaboliteof loratadine.
It was patented in 1984 and came into medical use in 2001.[1]
Medical uses
Desloratadine is used to treat allergic rhinitis, nasal congestion and chronic idiopathic urticaria (hives).[2] It is the major metabolite of loratadine and the two drugs are similar in safety and effectiveness.[2] Desloratadine is available in many dosage forms and under many trade names worldwide.[3]
An emerging indication for desloratadine is in the treatment of acne, as an inexpensive adjuvant to isotretinoin and possibly as maintenance therapy or monotherapy.[4][5]
Side effects
The most common side-effects are fatigue, dry mouth, and headache.[2]
Interactions
A number of drugs and other substances that are prone to interactions, such as ketoconazole, erythromycin and grapefruit juice, have shown no influence on desloratadine concentrations in the body. Desloratadine is judged to have a low potential for interactions.[6]
Pharmacology
Pharmacodynamics
Desloratadine is a selective H1–antihistamine which functions as an inverse agonist at the histamine H1 receptor.[7]
At very high doses, is also an antagonist at various subtypes of the muscarinic acetylcholine receptors. This effect is not relevant for the drug’s action at therapeutic doses.[8]
Pharmacokinetics
Desloratadine is well absorbed from the gut and reaches highest blood plasma concentrations after about three hours. In the bloodstream, 83 to 87% of the substance are bound to plasma proteins.[6]
Desloratadine is metabolized to 3-hydroxydesloratadine in a three-step sequence in normal metabolizers. First, n-glucuronidation of desloratadine by UGT2B10; then, 3-hydroxylation of desloratadine N-glucuronide by CYP2C8; and finally, a non-enzymatic deconjugation of 3-hydroxydesloratadine N-glucuronide.[9] Both desloratadine and 3-hydroxydesloratadine are eliminated via urine and feces with a half-life of 27 hours in normal metabolizers.[6][10]

3-Hydroxydesloratadine, the main metabolite
It exhibits only peripheral activity since it does not readily cross the blood-brain barrier; hence, it does not normally cause drowsiness because it does not readily enter the central nervous system.[11]
Desloratadine does not have a strong effect on a number of tested enzymes in the cytochrome P450 system. It was found to weakly inhibit CYP2B6, CYP2D6, and CYP3A4/CYP3A5, and not to inhibit CYP1A2, CYP2C8, CYP2C9, or CYP2C19. Desloratadine was found to be a potent and relatively selective inhibitor of UGT2B10, a weak to moderate inhibitor of UGT2B17, UGT1A10, and UGT2B4, and not to inhibit UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, UGT2B15, UGT1A7, and UGT1A8.[9]
Pharmacogenomics
2% of Caucasian people and 18% of people from African descent are desloratadine poor metabolizers. In these people, the drug reaches threefold highest plasma concentrations six to seven hours after intake, and has a half-life of about 89 hours. However, the safety profile for these subjects is not worse than for extensive (normal) metabolizers.[6][10]
Clip
https://www.beilstein-journals.org/bjoc/articles/9/265
The value of substituted 3-picoline precursors is illustrated in the synthesis of clarinex (1.22, Desloratadine, Scheme 5), a dual antagonist of platelet activating factor (PAF) and of histamine used in the treatment of allergies. This compound consists of a highly functional tricyclic core with an unsaturated linkage to a pendant piperidine ring. The picoline derivative 1.23 is first treated with two equivalents of n-butyllithium (n-BuLi) followed by alkylation with benzyl chloride to give the chain elongated adduct [27]. The tert-butylamide 1.24 is then dehydrated with phosphorous oxychloride at elevated temperatures to yield the nitrile derivative 1.25. Introduction of the piperidine ring is achieved by utilisation of the appropriately substituted Grignard reagent 1.26. A Friedel–Crafts type acylation promoted by either triflic acid or polyphosphoric acid (PPA) furnishes the tricyclic structure 1.28 which upon N-demethylation affords clarinex (1.22).

CLIP
FTIR

SYN
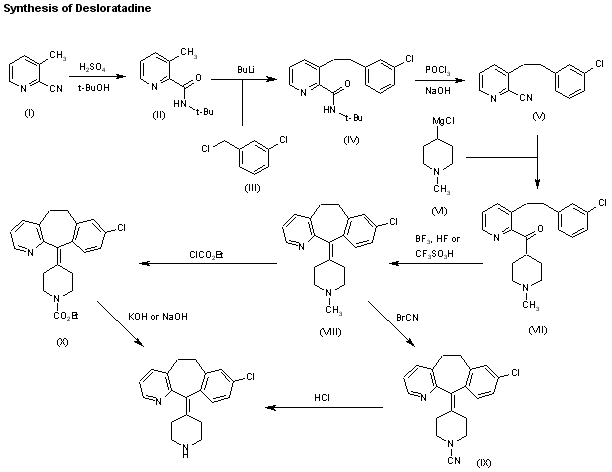
Alcoholysis of 3-methylpyridine-2-carbonitrile (I) with hot tert-butanol and H2SO4 gives the N-tert-butylcarboxamide (II), which is alkylated with 3-chlorobenzyl chloride (III) and BuLi in THF, yielding N-tert-butyl-3-[2-(3-chlorophenyl)ethyl]pyridine-2-carboxamide (IV). The reaction of (IV) with refluxing POCl3 and then with NaOH affords the corresponding nitrile (V), which is condensed with 1-methylpiperidin-4-ylmagnesium chloride (VI) in THF to give the ketone (VII). Cyclization of (VII) by means of either BF3 in HF or trifluoromethanesulfonic acid yields 8-chloro-11-(1-methylpiperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine (VIII), which is reacted with cyanogen bromide in benzene to give the N-cyano compound (IX). Finally, this compound is treated with HCl in refluxing acetic acid/water. Alternatively, 8-chloro-11-(1-methylpiperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine (VIII) is treated with ethyl chloroformate in hot toluene, affording the carbamate (X) (2), which is finally decarboxylated with KOH or NaOH in refluxing ethanol/water.
SYN
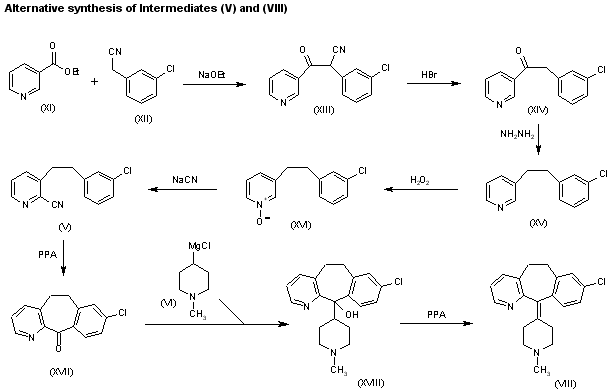
Condensation of ethyl nicotinate (XI) with 3-chlorophenylacetonitrile (XII) by means of sodium ethoxide in ethanol gives 2-(3-chlorophenyl)-3-oxo-3-(3-pyridyl)propionitrile (XIII), which by refluxing with concentrated HBr yields 2-(3-chlorophenyl)-1-(3-pyridyl)ethanone (XIV). The reduction of (XIV) with hydrazine hydrate and NaOH in diethylene glycol at 235-40 C affords 3-(2-phenylethyl) pyridine (XV), which is oxidized with H2O2 in hot acetic acid to provide the corresponding N-oxide (XVI). Reaction of (XVI) with NaCN and dimethyl sulfate in water affords the previously described 3-(2-phenylethyl)pyridine-2-carbonitrile (V), which can be worked up as previously described or cyclized with polyphosphoric acid (PPA) at 180 C to give 8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-one (XVII). The condensation of (XVII) with 1-methylpiperidin-4-ylmagnesium chloride (VI) in THF yields the corresponding carbinol (XVIII), which is dehydrated with PPA at 170 C to afford the previously reported 8-chloro-11-(1-methylpiperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine (VIII).
SYN
|
|
| Condensation of ethyl nicotinate (XI) with 3-chlorophenylacetonitrile (XII) by means of sodium ethoxide in ethanol gives 2-(3-chlorophenyl)-3-oxo-3-(3-pyridyl)propionitrile (XIII), which by refluxing with concentrated HBr yields 2-(3-chlorophenyl)-1-(3-pyridyl)ethanone (XIV). The reduction of (XIV) with hydrazine hydrate and NaOH in diethylene glycol at 235-40 C affords 3-(2-phenylethyl) pyridine (XV), which is oxidized with H2O2 in hot acetic acid to provide the corresponding N-oxide (XVI). Reaction of (XVI) with NaCN and dimethyl sulfate in water affords the previously described 3-(2-phenylethyl)pyridine-2-carbonitrile (V), which can be worked up as previously described or cyclized with polyphosphoric acid (PPA) at 180 C to give 8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-one (XVII). The condensation of (XVII) with 1-methylpiperidin-4-ylmagnesium chloride (VI) in THF yields the corresponding carbinol (XVIII), which is dehydrated with PPA at 170 C to afford the previously reported 8-chloro-11-(1-methylpiperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine (VIII). |
Syn
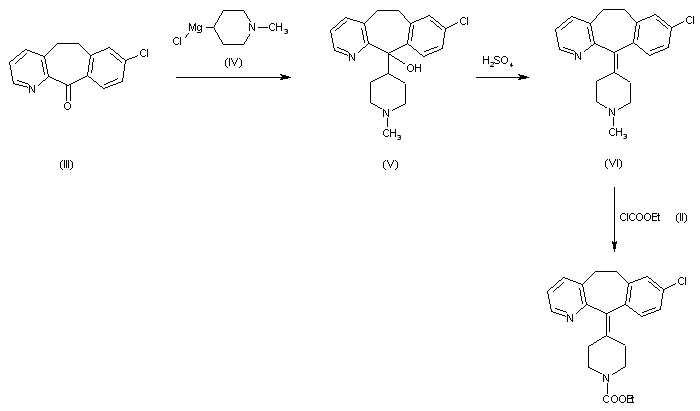
2) By reaction of 8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-one (III) with the Grignard reagent (IV) to give the tertiary carbinol (V), which is dehydrated with 85% H2SO4 affording 8-chloro-11-piperidinylidene derivative (VI). Finally, cornpound (VI) is treated with ethyl chloroformate (II) in toluene.
SYN
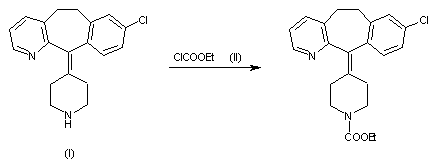
1) By carboxylation of 8-chloro-6,11-dihydro-11-(4-piperidylidene)-5H-benzo[5,6]cyctohepta[1,2-b]pyridine (I) with ethyl chloroformate (II) in refluxing benzene.
SYN
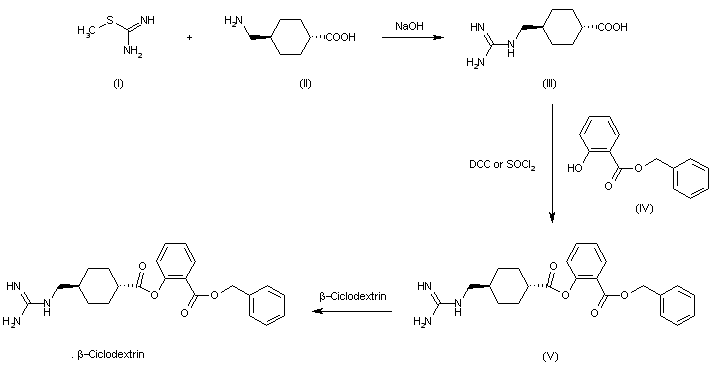
The condensation of S-methylisothiourea (I) with trans-4-(aminomethyl)cyclohexanecarboxylic acid (II) by means of NaOH in water gives trans-4-(guanidinomethyl)cyclohexanecarboxylic acid (III) (I), which is esterified with benzyl salicylate (IV) by means of dicyclohexylcarbodiimide (DCC) or SOCl2 yielding 2-benzyloxycarbonylphenyl trans-4-(guanidinomethyl)cyclohexanecarboxylate (V). Finally, this compound is treated with cyclodextrin in aqueous solution to afford the corresponding complex.
SPECTROSCOPY
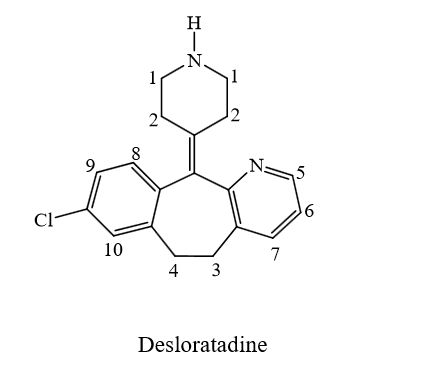
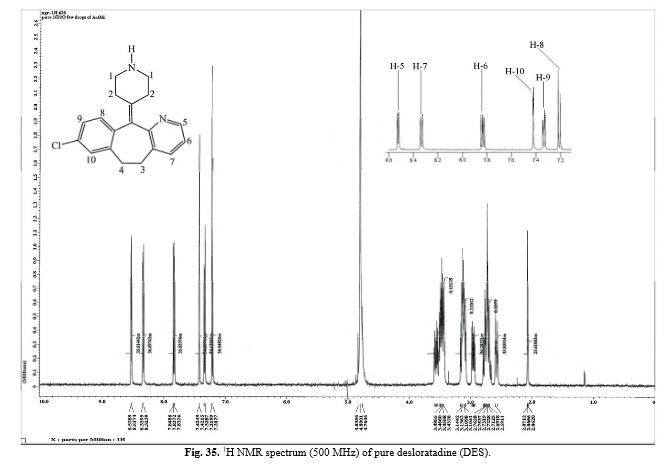
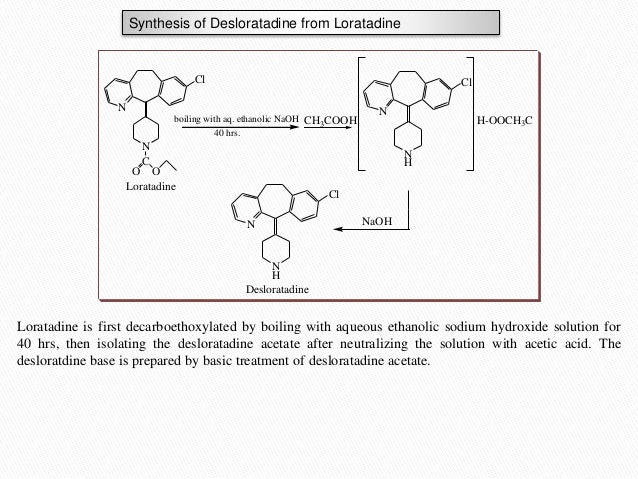
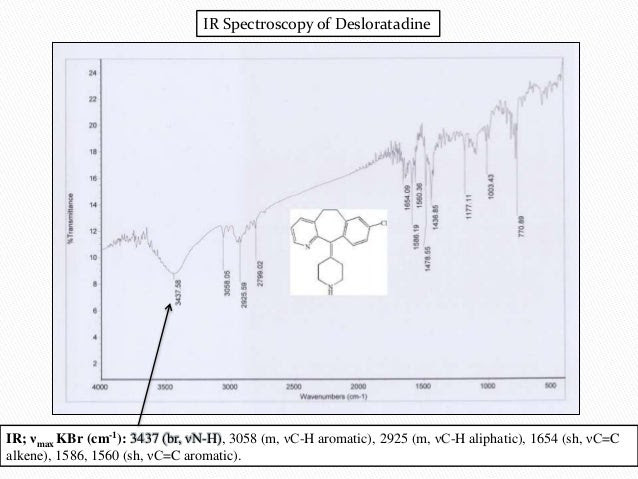


[0052] Table 1, desloratadine sample IH-NMR data of the DMS0_d6

[0055] The desloratadine 1H spectra of the samples were assigned:
[0056] (I) 1H spectra show that there are 10 groups of hydrogen from low field to high field integral hydrogen ratio was 1: 1: 1: 1: 1: 1: 2: 4:
2: 4, and desloratadine structure match.
[0057] (2) δ 8.334 处 hydrogen as a set of double doublet, number of protons is I, attributed to two hydrogen;
[0058] (3) δ 7.560 处 hydrogen as a set of double doublet, number of protons is I, attributed to four hydrogen;
[0059] (4) δ 7.282 处 doublet hydrogen as a group, the number of protons is I, 12 attributed to hydrogen.
[0060] (5) δ 7.198 处 hydrogen as a set of double doublet, number of protons is I, 14 attributed to hydrogen;
[0061] (6) δ 7.174 处 hydrogen as a set of double doublet, number of protons is I, attributed to three hydrogen;
[0062] (7) δ 7.064 处 doublet hydrogen as a group, the number of protons is I, 15 attributed to hydrogen;
[0063] (8) δ 3.314 处 hydrogen as a group multiplet, 2 protons attributable to 10 hydrogen;
[0064] (9) δ 2.831,2.554 hydrogen groups at multiplet, protons of 4, 18, 20, the home position is hydrogen;
[0065] (10) δ 2.819 处 hydrogen as a group multiplet, 2 protons attributable to 11 hydrogen;
[0066] (11) δ 2.108 处 hydrogen as a single peak, the number of protons is I, home to 19 active hydrogen;
[0067] (12) δ 2.205, 2.002 处 two hydrogen multiplet, protons of 4, 17, 21 bits attributed to hydrogen; [0068] From the foregoing, 1H-NMR spectrum data and the resulting product in this embodiment is of he will be loratadine same structure as the target product.
http://www.google.com/patents/CN103755682A?cl=en
References
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 549. ISBN 9783527607495.
- ^ Jump up to:a b c See S (2003). “Desloratadine for allergic rhinitis”. Am Fam Physician. 68 (10): 2015–6. PMID 14655812.
- ^ Drugs.com Desloratadine entry at drugs.com international Page accessed May 4, 2015
- ^ Lee HE, Chang IK, Lee Y, Kim CD, Seo YJ, Lee JH, Im M (2014). “Effect of antihistamine as an adjuvant treatment of isotretinoin in acne: a randomized, controlled comparative study”. J Eur Acad Dermatol Venereol. 28 (12): 1654–60. doi:10.1111/jdv.12403. PMID 25081735.
- ^ Layton AM (2016). “Top Ten List of Clinical Pearls in the Treatment of Acne Vulgaris”. Dermatol Clin. 34 (2): 147–57. doi:10.1016/j.det.2015.11.008. PMID 27015774.
- ^ Jump up to:a b c d “Aerius: EPAR – Product Information” (PDF). European Medicines Agency. 2017-06-07.
- ^ Canonica GW, Blaiss M (2011). “Antihistaminic, anti-inflammatory, and antiallergic properties of the nonsedating second-generation antihistamine desloratadine: a review of the evidence”. World Allergy Organ J. 4 (2): 47–53. doi:10.1097/WOX.0b013e3182093e19. PMC 3500039. PMID 23268457.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Clarinex (US), Aerius, Dasselta, Deslordis (EU), others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a602002 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral (tablets, solution) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Rapidly absorbed |
| Protein binding | 83 to 87% |
| Metabolism | UGT2B10, CYP2C8 |
| Metabolites | 3-Hydroxydesloratadine |
| Onset of action | within 1 hour |
| Elimination half-life | 27 hours |
| Duration of action | up to 24 hours |
| Excretion | 40% as conjugated metabolites into urine Similar amount into the feces |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.166.554 |
| Chemical and physical data | |
| Formula | C19H19ClN2 |
| Molar mass | 310.82 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////Desloratadine, Descarboethoxyloratadine, Sch-34117, DCL, Denosin, Clarinex RediTabs, Allex, Desalex, Opulis, Clarinex, Neoclarityn, Aerius, MK-4117
E 2212
C25 H23 F3 N6 O, 480.48
CAS 1123197-68-1
(+) -2-{(E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine
- (+)-5,6,7,8-Tetrahydro-2-[(1E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)-3-pyridinyl]ethenyl]-8-[2-(trifluoromethyl)phenyl][1,2,4]triazolo[1,5-a]pyridine
- (+)-2-[(E)-2-[5-Methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]ethenyl]-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine

E2212
CAS 1123197-82-9
- C25 H23 F3 N6 O . 3/2 C4 H6 O6
- [1,2,4]Triazolo[1,5-a]pyridine, 5,6,7,8-tetrahydro-2-[(1E)-2-[6-methoxy-5-(4-methyl-1H-imidazol-1-yl)-2-pyridinyl]ethenyl]-8-[2-(trifluoromethyl)phenyl]-, (8S)-, (2S,3S)-2,3-dihydroxybutanedioate (2:3)
PATENT
https://patents.google.com/patent/US9453000B2/en
Examples 394 and 395 Synthesis of (+) and (−)-2-{(E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine

230 mg of the racemic title compound was obtained from 1-amino-3-(2-trifluoromethylphenyl)piperidin-2-one (343 mg) and (E)-3-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]acrylic acid (500 mg) by the same method as in Examples 194 and 195. The racemic title compound (220 mg) was separated by CHIRALPAK™ IC manufactured by Daicel Chemical Industries, Ltd. (2 cm×25 cm; mobile phase: methanol) to obtain the title optically active compound with positive optical rotation and a retention time of 16 minutes (92 mg) and the title optically active compound with negative optical rotation and a retention time of 19 minutes (79 mg).
The property value of the title optically active compound with a retention time of 16 minutes is as follows.
ESI-MS; m/z 481 [M++H].
The property values of the title optically active compound with a retention time of 19 minutes are as follows.
ESI-MS; m/z 481 [M++H]. 1H-NMR (CDCl3) δ (ppm): 1.90-2.01 (m, 1H), 2.10-2.35 (m, 2H), 2.29 (s, 3H), 2.43-2.52 (m, 1H), 3.95 (s, 3H), 4.27-4.41 (m, 2H), 4.69 (dd, J=6.0, 8.4 Hz, 1H), 7.02 (d, J=8.0 Hz, 1H), 7.08 (d, J=16.4 Hz, 1H), 7.40 (dd, J=7.6, 7.6 Hz, 1H), 7.44-7.53 (m, 4H), 7.73 (d, J=8.0 Hz, 1H), 8.13 (d, J=1.6 Hz, 1H), 8.34 (s, 1H).
PATENT
WO2009028588
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=D6AD22B6CC7302560AE1ADCED305CDCE.wapp2nC?docId=WO2009028588&tab=FULLTEXT&queryString=%28PA%2Feisai%29%2520&recNum=93&maxRec=725
(+)および(-)-2-{(E)-2-[5-メトキシ-6-(4-メチル-1H-イミダゾール-1-イル)ピリジン-3-イル]ビニル}-8-(2-トリフルオロメチルフェニル)-5,6,7,8-テトラヒドロ-[1,2,4]トリアゾロ[1,5-a]ピリジンの合成
[化221]
実施例194および実施例195と同様の方法により、1-アミノ-3-(2-トリフルオロメチルフェニル)ピペリジン-2-オン(343mg)および(E)-3-[5-メトキシ-6-(4-メチル-1H-イミダゾール-1-イル)ピリジン-3-イル]アクリル酸(500mg)から、ラセミ体の表題化合物を230mg得た。ラセミ体の表題化合物(220mg)をダイセル製CHIRALPAK TM IC(2cm×25cm:移動相;メタノール)にて分取し、(+)の旋光性を有する保持時間16分の表題光学活性化合物(92mg)および(-)の旋光性を有する保持時間19分の表題光学活性化合物(79mg)を得た。
保持時間16分の表題光学活性体の物性値は以下の通りである。
ESI-MS;m/z 481[M ++H].
保持時間19分の表題光学活性体の物性値は以下の通りである。
ESI-MS;m/z 481[M ++H]. 1H-NMR(CDCl 3)δ(ppm):1.90-2.01(m,1H),2.10-2.35(m,2H),2.29(s,3H),2.43-2.52(m,1H),3.95(s,3H),4.27-4.41(m,2H),4.69(dd,J=6.0,8.4Hz,1H),7.02(d,J=8.0Hz,1H),7.08(d,J=16.4Hz,1H),7.40(dd,J=7.6,7.6Hz,1H),7.44-7.53(m,4H),7.73(d,J=8.0Hz,1H),8.13(d,J=1.6Hz,1H),8.34(s,1H).
Example 394 and Example 395
(+) and (−)-2-{(E) -2- [5-methoxy-6- (4-methyl-1H-imidazol-1-yl) pyridin-3-yl] Synthesis of vinyl} -8- (2-trifluoromethylphenyl) -5,6,7,8-tetrahydro- [1,2,4] triazolo [1,5-a] pyridine [Formula
221]
Example 194 and By a method similar to Example 195, 1-amino-3- (2-trifluoromethylphenyl) piperidin-2-one (343 mg) and (E) -3- [5-methoxy-6- (4-methyl-) 1 H-Imidazol-1-yl) pyridin-3-yl] acrylic acid (500 mg) gave 230 mg of the racemic title compound. Racemic title compound (220 mg) a Daicel CHIRALPAK TM IC (2 cm × 25 cm: mobile phase; methanol) was collected by min (+) title optically active compound of the retention time of 16 minutes with a optical rotation of (92 mg) The title optically active compound (79 mg) having a polarizability of (−) and a retention time of 19 minutes was obtained.
The physical property values of the title optically active substance with a retention time of 16 minutes are as follows.
ESI-MS; m / z 481 [M + + H].
The physical property values of the title optically active substance with a retention time of 19 minutes are as follows.
ESI-MS; m / z 481 [M + + H]. 1 H-NMR (CDCl 3)) Δ (ppm): 1.90 to 2.01 (m, 1 H), 2.10 to 2.35 (m, 2 H), 2.29 (s, 3 H), 2.43 to 2.52 (m) , 1 H), 3.95 (s, 3 H), 4.27-4. 41 (m, 2 H), 4.69 (dd, J = 6.0, 8.4 Hz, 1 H), 7.02 (d , J = 8.0 Hz, 1 H), 7.08 (d, J = 16.4 Hz, 1 H), 7.40 (dd, J = 7.6, 7.6 Hz, 1 H), 7.44-7. 53 (m, 4H), 7.73 (d, J = 8.0 Hz, 1 H), 8.13 (d, J = 1.6 Hz, 1 H), 8.34 (s, 1 H).
PATENT
https://patents.google.com/patent/WO2010098490A1/it


As a novel compound that has an effect of reducing the production of Aβ40 and
42 and is expected as a therapeutic or prophylactic agent for Alzheimer’s disease or the like, the present inventors have found a compound represented by the following formula (1) (compound
(D): [Formula 1]
and filed a patent application for the invention (PCT/JP08/065365).
Generally, properties of salts of compounds and those crystals that are useful as pharmaceuticals are highly important for the development of pharmaceuticals, because the properties greatly affect bioavailability of drugs, purity of drug substances, formulation of preparations, and the like. Therefore, it is necessary to research which salts and crystal forms of the compound of the formula (1) are most excellent as pharmaceuticals. Specifically, since their properties depend on the character of the individual compounds, it is generally difficult to estimate salts and crystal forms for drug substances having excellent properties and it is demanded to actually make various studies for each compound.
EXAMPLES [0023] The present invention will be described in detail below with reference to reference examples and examples; however, the present invention is not limited to these reference examples and examples. [0024]
The following abbreviations are used in the following reference examples and examples.
DMF: N,N’-dimethylformamide
THF: Tetrahydrofuran
EDC: lrEmyl-S-β-dimemylammopropytycarbodiimide hydrochloride HOBT: 1-Hydroxybenzotriazole IPEA: Diisopropylethylamine [0025]
In powder X-ray diffractometry of the crystals produced in the following examples, the resulting crystals were placed on a sample stage of a powder X-ray diffractometer and analyzed under the following conditions. [0026] Measurement conditions
Sample holder: Aluminum Target: Copper
Detector: Scintillation counter Tube voltage: 50 kV Tube current: 300 mA
Slit: DS 0.5 mm (Height limiting slit 2 mm), SS Open, RS Open Scanning rate : 5 °/min
Sampling interval: 0.02° Scan range: 5 to 35° Goniometer: Horizontal goniometer [0027] Reference Example 1
Svnmesis ofr8SV2-(fE)-246-memoxy-5-(4-memyl-lH-imidazol-l-vnpyridin-2-yllvmvU-8-(2-trifluoromethylphenyl‘)-5,6J,8-tetrahvdro-[1.2,41triazolo[l.,5-a]pyridine
[Formula 2]
Synthesis of l-amino-3-(2-trifluoromemylphenyl)piperidin-2-one Thionyl chloride (2.72 mL) was added to a solution of 2-trifluoromethylphenylacetic acid (1.9 g) in methanol (38 mL), followed by stirring at room temperature for three hours. The reaction solution was concentrated under reduced pressure. The resulting residue was diluted with DMF. Sodium hydride (containing 40% mineral oil, 410 mg) was added under ice-cooling, followed by stirring for 10 minutes. The reaction solution was further stirred for 30 minutes and then ice-cooled again. l-Chloro-3-iodopropane (1.02 mL) was added to the reaction mixture, and the reaction solution was stirred at room temperature overnight. Water and ethyl acetate were added to the reaction mixture, and the organic layer was separated. The resulting organic layer was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was diluted with ethanol (26.6 mL). Hydrazine monohydrate (7.6 mL) was added, and the reaction solution was stirred at room temperature for two hours and then at 60°C for further three hours. The reaction mixture was concentrated under reduced pressure. Saturated aqueous sodium bicarbonate and ethyl acetate and were added to the residue, and the organic layer was separated. The resulting organic layer was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: heptane-ethyl acetate system) to obtain 1.68 g of the title compound. The property values of the compound are as follows.
ESI-MS; m/z 259 [M+H-H]. 1H-NMR (CDCl3) δ (ppm): 1.82-2.10 (m, 3H), 2.18-2.26 (m, IH), 3.58-3.76 (m, 2H), 4.07 (dd, J = 10.0, 5.6 Hz, IH), 4.60 (s, 2H), 7.24 (d, J = 7.6 Hz, IH), 7.35 (t, J = 7.6 Hz, IH), 7.51 (t, J = 7.6 Hz, IH)5 7.66 (d, J = 7.6 Hz, IH). [0028] Synthesis of (EV3-[6-methoxy-5-(4-methyl- 1 H-imidazol- 1 -yl)pyridin-2-yl]-N-f2-oxo-3 -(2-trifluoromethylphenyl)piperidin- 1 -yl]acrylamide
EDC (834 mg), HOBT (588 mg) and IPEA (2.03 mL) were added to a suspension of (E)-3-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridm-2-yl]acrylic acid trifluoroacetate (1.42 g) and l-amήio-3-(2-trifluoromethylphenyl)piperidin-2-one (750 mg) in DMF (30 mL). After stirring at room temperature for 14 hours, a saturated sodium bicarbonate solution and ethyl acetate were added to the reaction solution, and the organic layer was separated. The resulting organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: ethyl acetate-methanol system) to obtain 1.23 g of the title compound. The property values of the compound are as follows. ESI-MS; m/z 500 [M1H-HJ. [0029]
Synthesis of r8S>-2-(fEV2-r6-methoxy-5-r4-methyl-lH-imidazol-l-vnpyridm’2-vnvinvU-8-(2-trifluoromethvlphenvD-5.6.7.8-tetrahvdro-ri.2.41triazoloπ.5-a1pvridine Phosphorus oxychloride (24.2 mL) was added to (E)-3~[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]-N-[2-oxo-3-(2-trifluoromethylphenyl)piperidin-l-yl]acrylamide (1.2 g). The reaction solution was stirred at 1000C for one hour and then concentrated under reduced pressure. Subsequently, the residue was diluted with acetic acid (24.2 mL) and then ammonium acetate (1.9 g) was added, followed by stirring at 1500C for two hours. The reaction solution was left to cool to room temperature and then concentrated under reduced pressure. A saturated sodium bicarbonate solution and ethyl acetate were added to the resulting residue, and the organic layer was separated. The resulting organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: heptane-ethyl acetate system) to obtain a racemate of the title compound (750 mg). The resulting racemate (410 mg) was separated by CHIRALP AK™ IA manufactured by Daicel Chemical Industries, Ltd. (2 cm x 25 cm, mobile phase: hexane:ethanol = 8:2, flow rate: 10 mL/min) to obtain the title compound with a retention time of 33 minutes and negative optical rotation (170 mg) as crystals. The property values of the title compound are as follows.
1H-NMR (CDCl3) δ (ppm): 1.90-2.01 (m, IH), 2.10-2.35 (m, 2H), 2.29 (d, J = 1.2 Hz, 3H), 2.42-2.51 (m, IH), 4.03 (s, 3H), 4.28-4.41 (m, 2H), 4.70 (dd, J = 8.4, 6.0 Hz, IH), 6.92 (d, J = 8.0 Hz, IH), 6.95 (t, J = 1.2 Hz, IH), 7.01 (d, J = 7.6 Hz, IH), 7.39 (t, J = 7.6 Hz5 IH), 7.44 (d, J = 16.0 Hz, IH), 7.45 (d, J = 8.0 Hz, IH), 7.49 (t, J = 7.6 Hz, IH), 7.63 (d, J = 16.0 Hz5 IH), 7.72 (d, J = 7.6 Hz, IH), 7.76 (d, J = 1.2 Hz, IH). [0030]
(8S)-2-{(E)-2-[6-Methoxy-5-(4-methyl-lH-imidazol-l-yl)ρyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine synthesized according to the above reference example was used for the following synthesis of salts. [0031] Example 1
Synthesis of r8SV2-{rEV2-[6-methoxy-5-(4-methyl-lH-imidazol-l-vπpyridin-2-vnvinvU-8-f2-trifluoromethylphenyl)-5.6.7.8-tetrahvdro-fl,2,4]triazolo[l.,5-a]pyridine 1.5 D-tartrate
(8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine (33.70 mg) was dissolved in 285 μL of a D-tartaric acid-ethanol solution (110.92 mg/3 mL) with stirring at room temperature. The oil was precipitated when 1 mL of heptane was added. Accordingly, the oily substance was dissolved by adding 1 mL of ethanol. Further, 0.5 mL of heptane was added, and the mixture was transferred to a low temperature laboratory at about 50C (under shading) and continuously stirred for 24 hours. Thus, partial gelation occurred. Thereafter, the mixture was brought back to room temperature and continuously stirred, resulting in precipitation of a solid. The solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 21.25 mg of the title compound as white solid crystals. 1H-NMR (600 MHz, DMSOd6) δ (ppm): 1.96 (m, IH), 2.14 (s, 3H), 2.16 (m, 2H), 2.29 (m, IH), 3.98 (s, 3H), 4.28 (m, 2H), 4.29 (s, 3H), 4.51 (dd, J = 9, 6 Hz, IH), 7.22 (s, IH), 7.25 (brd, J = 8 Hz, IH), 7.27 (d, J = 8 Hz, IH), 7.32 (d, J = 16 Hz, IH)5 7.46 (d, J = 16 Hz, IH), 7.49 (brdd, J = 8 Hz, IH), 7.61 (brdd, J = 8 Hz5 IH), 7.77 (brd, J = 8 Hz, IH), 7.78 (d, J = 8 Hz, IH), 7.91 (s, IH). [0032] Example 2
Synthesis of (‘8SV2-l(Ε)-2-f6-methoxy-5-(4-methyl-lH-imidazol-l-vnpyridm-2-yllvinyl>-8-f2-trifluoromethylphenylV5,6J,8-tetrahvdro-[l ,2,4]triazolo[l ,5-a]pyridine di-D-tartrate
(8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,657,8-tetrahydro-[l ,2,4]triazolo[l ,5-a]ρyridine (810.18 mg) was dissolved in 8 mL of a D-tartaric acid-ethanol solution (751.13 mg/10 mL) with stirring at room temperature. The oil was precipitated when 2 mL of heptane was added. Accordingly, the oily substance was dissolved by ultrasonic treatment to prepare a clear solution. Several mg of crystals of the 1.5 D-tartrate prepared according to Example 1 were added, followed by stirring at room temperature. Stirring for about one hour resulted in gelation and subsequent precipitation of a solid. Further, stirring was continued while gradually adding 14 mL of heptane. A part of the suspension (2 mL) was separated and the solid was collected by filtration through a glass filter. The solid was dried under reduced pressure at room temperature to obtain 71.14 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.15 (s, 3H), 2.16 (m, 2H), 2.30 (m, IH), 3.98 (s, 3H), 4.28 (m, 2H), 4.29 (s, 4H), 4.51 (dd, J = 9, 6 Hz, IH), 7.22 (brs, IH), 7.25 (brd, J = 8 Hz, IH), 7.27 (d, J = 8 Hz, IH), 7.32 (d, J = 16 Hz, IH), 7.46 (d, J = 16 Hz, IH), 7.49 (brdd, J – 8 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.77 (brd, J = 8 Hz, IH), 7.78 (d, J = 8 Hz, IH), 7.91 (brs, IH). [0033] Example 3
Synthesis of r8SV2-(rE)-2-r6-methoxy-5-r4-methyl-lH-imidazol-l-vnpyridin-2-yl1vinvU-8-α-trifluoromethylphenyl)-5,6J,8-tetrahydro-[1.2,4]triazolo[l,5-a]pyridine disulfate
Concentrated sulfuric acid (11.5 μL) was added to a solution of (8S)-2-{(E)-2-[6- methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-txifluoromethylphenyl)-5,6,7,8-tetrahydro-[l52,4]triazolo[l55-a]pyridine (98.09 mg) in ethanol (1 mL), and 1 mL of ethyl acetate was added with stirring at room temperature. Since the oily portion was confirmed on the bottom of the recovery flask, the oily substance was dissolved by ultrasonic treatment. Stirring at room temperature under shading for about 30 minutes resulted in precipitation of a solid. The solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 127.94 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.17 (m, 2H), 2.30 (m, IH), 2.34 (brd, J = 1 Hz, 3H), 4.01 (s, 3H), 4.29 (m, 2H), 4.52 (dd, J = 9, 6 Hz, IH)5 7.25 (brd, J = 8 Hz, IH), 7.37 (d, J = 16 Hz, IH), 7.40 (d, J = 8 Hz, IH), 7.50 (brdd, J = 8 Hz, IH), 7.55 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.77 (m, IH), 7.78 (m, IH), 8.00 (d, J = 8 Hz, IH), 9.36 (d, J = 2 Hz, IH). [0034] Example 4 Synthesis of (8SV2-((E)-2-[“6-methoxy-5-(4-methyl-lH-imidazol-l-ylN)ρyridin-2-yllvinvU-8-(‘2-trifluoromethylphenyl)-5,6,7,8-tetrahvdiO-[1.2,41triazolo[l,5-a]pyridine dihydrobromide
Concentrated hydrobromic acid (24.8 μL) was added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,84etrahydro-[l,254]triazolo[l55-a]pyridine (51.42 mg) m ethanol (1 mL), and 1 mL of heptane was added with stirring at room temperature. After several minutes, 1 mL of heptane was further added to the solution and stirring was continued. The solution was stirred at room temperature for one hour and then further stirred at about 50C for 20 minutes. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 49.24 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 1.99 (m, IH), 2.17 (m, 2H), 2.30 (m, IH), 2.34 (brd, J = 1 Hz5 3H), 4.01 (s, 3H), 4.30 (m, 2H), 4.52 (dd, J = 9, 6 Hz5 IH), 7.25 (brd, J = 8 Hz5 IH), 7.37 (d, J = 16 Hz, IH), 7.40 (d, J = 7 Hz, IH)57.50 (brdd, J = 8 Hz, IH), 7.55 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz5 IH), 7.77 (m, IH)5 7.78 (m, IH), 8.00 (d, J = 7 Hz, IH), 9.37 (d, J = 2 Hz, IH). [0035] Example 5
Synthesis of r8SV2-((Ε)-2-r6-methoxy-5-r4-methyl-lH-imidazol-l-yl)ρyridin-2-vnvinyl}-8-r2-trifluoromethylphenyl)-5,6J,8-tetrahvdro-[1.2,41triazolo[1.5-alpyridine hydrochloride
Concentrated hydrochloric acid (3.6 μL) was added to a solution of (8S)-2-{(E)- 2-[6-methoxy-5-(4-metiiyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylplienyl)-5,6,7,8-te1xahydro-[l,2,4]triazolo[l,5-a]pyridme (19.80 mg) in 2-propanol (1 mL), and a total of 4 mL of heptane was added in 1 mL portions with stirring at room temperature. The solution was stirred at room temperature under shading for five days. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 7.45 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.17 (m, 2H)5 2.30 (m, IH), 2.30 (s, 3H), 4.00 (s, 3H), 4.30 (m, 2H)5 4.52 (dd, J = 9, 6 Hz5 IH), 7.25 (brd, J – 8 Hz5 IH), 7.36 (d, J = 16 Hz5 IH), 7.37 (d5 J = 8 Hz, IH), 7.50 (brt, J = 8 Hz5 IH)5 7.53 (d, J = 16 Hz5 IH)5 7.61 (brt, J = 8 Hz5 IH)5 7.66 (brs, IH), 7.77 (brd, J = 8 Hz, IH)5 7.96 (d, J = 8 Hz5 IH), 9.06 (brs, IH). [0036] Example 6
Synthesis of (8S)-2-((ΕV2-r6-methoxy-5-(‘4-methyl-lH-imidazol-l-yl)pyridin-2-yl1vinvU-8-(2-trifluoromethylphenyl)-5.6,7,8-tetrahvdro-[l,2,4]triazolo[L5-a1pyridine hydrochloride Concentrated hydrochloric acid (14.3 μL) and heptane (7 mL) were added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,657,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine (79.77 mg) in 2-propanol (3 mL). A small amount of the crystals obtained in Example 5 were added as seed crystals with stirring at room temperature. The mixture was transferred to a low temperature laboratory at about 50C and stirred for one hour. Thereafter, 1 mL of heptane was further added, followed by stirring for several minutes. When the precipitated solid was collected by filtration through a glass filter, the solid was precipitated in the filtrate. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 38.02 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 1.97 (m, IH), 2.17 (m5 2H), 2.29 (m, IH), 2.32 (brd, J = 1 Hz, 3H), 4.00 (s, 3H), 4.30 (m, 2H), 4.52 (dd, J = 9, 6 Hz, IH), 7.25 (brd, J = 8 Hz, IH), 7.37 (d, J = 16 Hz5 IH), 7.38 (d, J = 8 Hz, IH), 7.50 (brdd, J = 8 Hz, IH)5 7.54 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.72 (brs, IH), 7.77 (brd, J = 8 Hz, IH), 7.98 (d, J = 8 Hz5 IH)5 9.24 (brs, IH). [0037] Example 7
SvnJhesis off8SV2-f(E>2-r6-memoxy-5-(4-mefovπ trifluoromethylt>henylV5,6,7,8-tetrahvdro-[l,2,4]triazolo[l,5-a]pyridine mesylate
Mesylic acid (0.8 μL) was added to a mixed solution of (8S)-2-{(E)-2-[6- methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphen^ tetrahydro-[l52,4]triazolo[l,5-a]pyridine (50 mg) in t-butyl methyl ether (0.8 mL)-ethaαol (0.1 mL). The mixture was solidified as a result of stirring at room temperature for two hours. The solid was collected by filtration through a glass filter. The solid was washed with t-butyl methyl ether-ethanol (8:1) and then dried under reduced pressure at room temperature to obtain 51.9 mg of the title compound as pale yellow solid crystals.
1H-NMR (DMSO-d6) δ (ppm): 1.90-2.05 (m, IH)3 2.10-2.22 (m, 2H), 2.28-2.40 (m, IH), 2.31 (s, 3H), 2.35 (s, 3H)5 4.02 (s, 3H)5 4.25-4.39 (m, 2H), 4.50-4.55 (m, IH), 7.27 (d5 J = 8.0 Hz5 IH)5 7.38 (d, J = 16.0 Hz5 IH)5 7.41 (d, J = 8.0 Hz, IH)5 7.51 (t5 J = 8.0 Hz5 IH)5 7.55 (d, J = 16.0 Hz5 IH), 7.63 (t, J = 8.0 Hz5 IH)5 7.78 (d, J = 8.0 Hz5 IH)5 7.79 (s, IH), 8.01 (d, J = 8.0 Hz5 IH), 9.37 (s, IH). [0038] Example 8 Synthesis of (8S)-2-((ΕV2-r6-methoxy-5-(4-methyl-lH-imidazol-l-vnpyridin-2-vnvinvn-8-r2-trifluoromethylphenyl)-5.6,7,8-tetrahydro-[l.,2,4‘|triazolo[l,5-a]pyridine diphosphate
A solution of phosphoric acid (52.8 mg) in acetonitrile (0.2 mL) was added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-mτidazol-l-yl)ρyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5565758-tetrahydro-[l5254]triazolo[l,5-a]pyridine (100 mg) in acetonitrile (0.8 mL) at room temperature. The precipitated oil was solidified as a result of stirring with spatula. The solid was collected by filtration through a glass filter. The solid was washed with ice-cold acetonitrile, air-dried at room temperature for 10 minutes and then dried under reduced pressure at room temperature to obtain 120 mg of the title compound as white solid crystals. 1H-NMR (DMSO-d6) δ (ppm): 1.90-2.05 (m, IH), 2.11-2.20 (m, 2H), 2.15 (s, 3H), 2.25-2.35 (m, IH), 3.99 (s, 3H)5 4.24-4.39 (m, 2H), 4.50-4.55 (m, IH)5 7.23 (s, IH), 7.26 (d, J = 7.0 Hz, IH), 7.28 (d, J = 8.0 Hz, IH), 7.33 (d, J = 16.0 Hz5 IH), 7.47 (d, J = 16.0 Hz5 IH), 7.51 (t, J = 7.0 Hz, IH), 7.63 (t, J = 7.0 Hz, IH), 7.78 (d, J = 7.0 Hz, IH), 7.79 (d, J = 8.0 Hz, IH), 7.90 (s, IH). [0039] Example 9 Svnmesis of(8SV2-{(E)-2-[6-memoxy-5-(4-methyl-lH-irnidazol-l-yl)pyridin-2-yl1vinvU-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahvdro-[l .2.41triazolo[l .5-a]pyridine diphosphate
A solution of phosphoric acid (13.2 mg) in ethanol (0.05 mL) was added to a mixed solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2!,4]triazolo[l,5-a]pyridme (50 mg) in heptane (0.6 mL)-ethanol (0.15 mL) at room temperature. The reaction solution was stirred at room temperature, and the precipitated solid was collected by filtration through a glass filter. The solid was washed with heptane-ethanol (3:1) and then dried under reduced pressure at room temperature to obtain 37.6 mg of the title compound as white solid crystals. 1H-NMR (DMSOd6) δ (ppm): 1.90-2.05 (m, IH), 2.11-2.20 (m, 2H), 2.15 (s, 3H), 2.25-2.35 (m, IH), 3.99 (s, 3H), 4.24-4.39 (m, 2H), 4.50-4.55 (m, IH), 7.23 (s, IH), 7.26 (d, J = 7.0 Hz, IH), 7.28 (d, J = 8.0 Hz, IH), 7.33 (d, J = 16.0 Hz, IH), 7.47 (d, J = 16.0 Hz, IH), 7.51 (t, J = 7.0 Hz, IH), 7.63 (t, J = 7.0 Hz, IH), 7.78 (d, J = 7.0 Hz, IH), 7.79 (d, J = 8.0 Hz, IH), 7.90 (s, IH).
CLIP
Development of an Efficient Manufacturing Process for E2212 toward Rapid Clinical Introduction
, Taiju Nakamura‡, Atsushi Kamada†, Takeo Sasaki§, Toshiyuki Uemura§, Yorihisa Hoshino†, Masaaki Matsuda†, Yongbo Hu∥, Daiju Hasegawa§, Kazato Inanaga‡, Nobuaki Sato§, Kazuhiro Yoshizawa‡, George A. Moniz∥, Gordon D. Wilkie∥, Francis G. Fang∥, Yoshihiro Nishikawa‡, and Katsuya Tagami*‡
Process studies of E2212 (1) toward rapid clinical introduction are described. Through comprehensive route-finding studies and optimization of key condensation and cyclization steps, a racemate-based manufacturing route was established and successfully scaled-up to the hundred kilogram scale. For the rapid delivery of a drug substance containing the Z isomer for preclinical safety studies, the successful scale-up of the photoisomerization of an olefin in a flow system is also presented.
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00444
E2212 (1) (18.0 kg, 92.5% yield) as a white solid. Mother liquor 3 were recycled according to the procedure described below. FTIR (cm–1, KBr) 3461, 3173, 2956, 1734, 1584, 1536, 1476, 1309, 1130, 835, 765, 752; 1H NMR (600 MHz, DMSO-d6) δ 7.91 (s, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.77 (br d, J = 8.4 Hz, 1H), 7.61 (br dd, J = 7.8, 7.8 Hz, 1H), 7.49 (br dd, J = 7.8, 7.8 Hz, 1H), 7.46 (d, J= 15.6 Hz, 1H), 7.32 (d, J = 15.6 Hz, 1H), 7.27 (d, J = 7.8 Hz, 1H), 7.25 (br d, J = 7.8 Hz, 1H), 7.22 (s, 1H), 4.51 (dd, J = 9.0, 6.0 Hz, 1H), 4.29 (s, 3H), 4.28 (m, 2H), 3.98 (s, 3H), 2.29 (m, 1H), 2.14 (s, 3H), 2.16 (m, 2H), 1.96 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 173.3, 159.3, 155.4, 155.0, 150.1, 141.1, 137.1, 136.9, 133.6, 132.9, 131.0, 130.5, 127.6, 127.1 (q, JC–F = 30 Hz), 125.8 (q, JC–F = 5.6 Hz), 124.7 (q, JC–F = 270 Hz), 122.2, 120.7, 117.2, 116.5, 72.3, 53.7, 47.0, 37.6, 30.7, 21.3, 13.6; HRMS (ESI+) calcd for C25H23F3N6O ([M + H]+) 481.1958, found 481.1953.




///////////E2212, E 2212
Certolizumab pegol, セルトリズマブペゴル (遺伝子組換え)

>Amino acid sequence of the light chain DIQMTQSPSSLSASVGDRVTITCKASQNVGTNVAWYQQKPGKAPKALIYSASFLYSGVPY RFSGSGSGTDFTLTISSLQPEDFATYYCQQYNIYPLTFGQGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
>Amino acid sequence of the heavy chain EVQLVESGGGLVQPGGSLRLSCAASGYVFTDYGMNWVRQAPGKGLEWMGWINTYIGEPIY ADSVKGRFTFSLDTSKSTAYLQMNSLRAEDTAVYYCARGYRSYAMDYWGQGTLVTVSSAS TKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGL YSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCAA
Certolizumab pegol
CAS: 428863-50-7
セルトリズマブペゴル (遺伝子組換え)
CDP 870 / CDP-870 / CDP870 / PHA-738144
| Formula |
C2115H3252N556O673S16
|
|---|---|
| Cas |
428863-50-7
|
| Mol weight |
47748.8128
|
Reducing signs and symptoms of Crohn’s disease and treatment of moderately to severely active rheumatoid arthritis (RA).
Certolizumab pegol is a recombinant Fab’ antibody fragment against tumor necrosis factor alpha which is conjugated to an approximately 40kDa polyethylene glycol (PEG2MAL40K). Polyethylene glycol helps to delay the metabolism and elimination of the drugs. Chemically, the light chain is made up of 214 amino acid residues while the heavy chain is composed of 229 amino acid residues. The molecular mass of the Fab’ antibody fragment itself is 47.8 kDa. It is used for the treatment of rheumatoid arthritis and Crohn’s disease. FDA approved on April 22, 2008
Certolizumab pegol (CDP870, tradename Cimzia) is a biologic medication for the treatment of Crohn’s disease,[1][2] rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis. It is a fragment of a monoclonal antibody specific to tumor necrosis factor alpha(TNF-α) and is manufactured by UCB.[3][4][5]
Medical uses
- Crohn’s Disease
- On April 22, 2008, the U.S. FDA approved Cimzia for the treatment of Crohn’s disease in people who did not respond sufficiently or adequately to standard therapy.[4][6][7]
- Rheumatoid arthritis
- On June 26, 2009, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued a positive opinion recommending that the European Commission grant a marketing authorisation for Cimzia for the treatment of rheumatoid arthritis only – the CHMP refused approval for the treatment of Crohn’s disease. The marketing authorisation was granted to UCB Pharma SA on October 1, 2009.[8]
- Psoriatic arthritis
- On September 27, 2013, the U.S. FDA approved Cimzia for the treatment of adult patients with active psoriatic arthritis.[9]
Method of action
Certolizumab pegol is a monoclonal antibody directed against tumor necrosis factor alpha. More precisely, it is a PEGylated Fabfragment of a humanized TNF inhibitor monoclonal antibody.[10]
Clinical trials
- Crohn’s disease
- Positive results have been demonstrated in two phase III trials (PRECiSE 1 and 2) of certolizumab pegol versus placebo in moderate to severe active Crohn’s disease.[1][10][11][12]
- Axial spondyloarthritis
- In 2013, a phase 3 double blind randomized placebo-controlled study found significantly positive results in patient self-reported questionnaires, with rapid improvement of function and pain reduction, in patients with axial spondyloarthritis.[13]
- Rheumatoid arthritis
- Certolizumab appears beneficial in those with rheumatoid arthritis.[14]
Side effects
Significant side effects occur in 2% of people who take the medication.[14]
References
- ^ Jump up to:a b Sandborn WJ, Feagan BG, Stoinov S, et al. (July 2007). “Certolizumab pegol for the treatment of Crohn’s disease”. N. Engl. J. Med. 357 (3): 228–38. doi:10.1056/NEJMoa067594. PMC 3187683. PMID 17634458.
- ^ Goel, Niti; Sue Stephens (2010). “Certolizumab pegol”. mAbs. 2 (2): 137–147. doi:10.4161/mabs.2.2.11271. PMC 2840232. PMID 20190560.
- ^ Kaushik VV, Moots RJ (April 2005). “CDP-870 (certolizumab) in rheumatoid arthritis”. Expert Opinion on Biological Therapy. 5 (4): 601–6. doi:10.1517/14712598.5.4.601. PMID 15934837.
- ^ Jump up to:a b index.cfm?fuseaction=Search.Label_ApprovalHistory “Cimzia Label and Approval History” Check
|url=value (help). Drugs@FDA. U.S. Food and Drug Administration(FDA). Retrieved 2009-11-15. - ^ “Cimzia Prescribing Information” (PDF). US Food and Drug Administration (FDA). April 2016. Retrieved 2016-08-21.
- ^ UCB press release – Cimzia Approved in the US for the Treatment of Moderate to Severe Crohn’s Disease. Retrieved April 22, 2008.
- ^ Waknine, Yael (May 1, 2008). “FDA Approvals: Patanase, Actonel, Cimzia”. Medscape. Retrieved 2008-05-01.
- ^ “Cimzia European Public Assessment Report”. European Medicines Agency. Retrieved November 15, 2009.
- ^ “Cimzia (certolizumab pegol) approved by the U.S. FDA for treatment of adult patients with active psoriatic arthritis”. Archived from the original on October 1, 2013. Retrieved October 1, 2013.
- ^ Jump up to:a b Schreiber S. et al., Certolizumab pegol, a humanised anti-TNF pegylated FAb’ fragment, is safe and effective in the maintenance of response and remission following induction in active Crohn’s disease: a phase 3 study (precise), Gut, 2005, 54, suppl7, A82
- ^ Sandborn et al., Certolizumab pegol administered subcutaneously is effective and well tolerated in patients with active Crohn’s disease: results from a 26-week, placebo-controlled Phase 3 study (PRECiSE 1), Gastroenterology, 2006, 130, A107
- ^ “New Analysis Shows Cimzia (Certolizumab Pegol) Maintained Remission and Response in Recent Onset Crohn’s Disease” (Press release). UCB. October 23, 2006. Retrieved 2009-11-15.
- ^ Sieper J, Tubergen A, Coteur G, Woltering F, Landewe R (May 2013). “PMS50 – Rapid Improvements In Patient-Reported Outcomes With Certolizumab Pegol In Patients With Axial Spondyloarthritis, Including Ankylosing Spondylitis And Non-Radiographic Axial Spondyloarthritis: 24-Week Results Of A Phase 3 Double Blind Randomized Placebo-Controlled Study”. Value in Health. 16 (3): A227. doi:10.1016/j.jval.2013.03.1150.
- ^ Jump up to:a b Ruiz Garcia, V; Jobanputra, P; Burls, A; Vela Casasempere, P; Bort-Marti, S; Bernal, JA (Sep 8, 2017). “Certolizumab pegol (CDP870) for rheumatoid arthritis in adults”(PDF). The Cochrane Database of Systematic Reviews. 9: CD007649. doi:10.1002/14651858.CD007649.pub4. PMID 28884785.
External links
- certolizumab+pegol at the US National Library of Medicine Medical Subject Headings (MeSH)
- Cimzia Website
FDA approves treatment Cimzia (certolizumab pegol) for patients with a type of inflammatory arthritis
March 28, 2019
Release
The U.S. Food and Drug Administration today approved Cimzia (certolizumab pegol) injection for treatment of adults with a certain type of inflammatory arthritis called non-radiographic axial spondyloarthritis (nr-axSpA), with objective signs of inflammation. This is the first time that the FDA has approved a treatment for nr-axSpA.
“Today’s approval of Cimzia fulfills an unmet need for patients suffering from non-radiographic axial spondyloarthritis as there has been no FDA-approved treatments until now,” said Nikolay Nikolov, M.D., associate director for rheumatology of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Nr-axSpA is a type of inflammatory arthritis that causes inflammation in the spine and other symptoms. There is no visible damage seen on x-rays, so it is referred to as non-radiographic.
The efficacy of Cimzia for the treatment of nr-axSpA was studied in a randomized clinical trial in 317 adult patients with nr-axSpA with objective signs of inflammation, indicated by elevated C-reactive protein (CRP) levels and/or sacroiliitis (inflammation of the sacroiliac joints) on MRI. The trial measured the improvement response on the Ankylosing Spondylitis Disease Activity Score, a composite scoring system that assesses disease activity including patient-reported outcomes and CRP levels. Responses were greater for patients treated with Cimzia compared to patients treated with placebo. The overall safety profile observed in the Cimzia treatment group was consistent with the known safety profile of Cimzia.
The prescribing information for Cimzia includes a Boxed Warning to advise health care professionals and patients about the increased risk of serious infections leading to hospitalization or death including tuberculosis (TB), bacterial sepsis (infection in the blood steam), invasive fungal infections (such as histoplasmosis, an infection that affects the lungs), and other infections. Cimzia should be discontinued if a patient develops a serious infection or sepsis. Health care providers are advised to perform testing for latent TB and, if positive, to start treatment for TB prior to starting Cimzia. All patients should be monitored for active TB during treatment, even if the initial latent TB test is negative. The Boxed Warning also advises that lymphoma (cancer in blood cells) and other malignancies, some fatal, have been reported in children and adolescent patients treated with tumor necrosis factor (TNF) blockers, of which Cimzia is a member. Cimzia is not indicated for use in pediatric patients. Cimzia must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Cimzia was originally approved in 2008 and is also indicated for adult patients with Crohn’s disease, moderate-to-severe rheumatoid arthritis, active ankylosing spondylitis (AS) and moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
The FDA granted the approval of Cimzia to UCB.

Syringe with 200mg Certolizumab pegol
|
|
| Monoclonal antibody | |
|---|---|
| Type | Fab’ fragment |
| Source | Humanized (from mouse) |
| Target | TNF alpha |
| Clinical data | |
| Trade names | Cimzia |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a608041 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Subcutaneous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | about 11 days |
| Excretion | Renal (PEG only) |
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C2115H3252N556O673S16 |
| Molar mass | 47,750 g/mol g·mol−1 |
///////////////FDA 2019, Cimzia, certolizumab pegol, inflammatory arthritis, UCB

{kind=link}