

C=CC(=O)NC1=C(C=C2C(=C1)C(=NC=N2)NC3=CC(=C(C=C3)F)Cl)OCCCN4CCOCC4
Home » Uncategorized (Page 33)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
LENALIDOMIDE, レナリドミド, леналидомид , ليناليدوميد , 来那度胺 ,
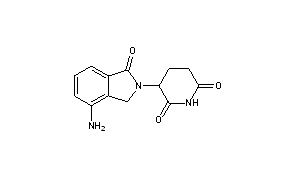
LENALIDOMIDE
- Molecular FormulaC13H13N3O3
- Average mass259.261 Da
|
レナリドミド;
|
леналидомид , ليناليدوميد , 来那度胺 ,
191732-72-6 [RN]
1-Oxo-4-amino-2-(2,6-dioxopiperidin-3-yl)isoindole
2,6-Piperidinedione, 3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-
3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-2,6-piperidinedione
3-(4-Amino-1-oxo-1,3-dihydro-2H-isoindol-2-yl)-2,6-piperidinedione
3-(4-Amino-1-oxo-1,3-dihydro-2H-isoindol-2-yl)piperidin-2,6-dion
3-(4-amino-1-oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione
3-(4-amino-1-oxoisoindolin-2-yl)piperidine-2,6-dione
3-(7-amino-3-oxo-1h-isoindol-2-yl)piperidine-2,6-dione
8505
E3 Ligase ligand
IMiD3
CAS Registry Number: 191732-72-6
CAS Name: 3-(4-Amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-2,6-piperidinedione
Additional Names: 1-oxo-2-(2,6-dioxopiperidin-3-yl)-4-aminoisoindoline
Manufacturers’ Codes: CC-5013
Trademarks: Revimid (Celgene); Revlimid (Celgene)
Molecular Formula: C13H13N3O3
Molecular Weight: 259.26
Percent Composition: C 60.22%, H 5.05%, N 16.21%, O 18.51%
Literature References: Immunomodulatory drug; analog of thalidomide, q.v. Prepn: G. W. Muller et al., US 5635517 (1997 to Celgene); and in vitro TNF-a inhibition: eidem, Bioorg. Med. Chem. Lett. 9, 1625 (1999). LC-MS determn in plasma: T. M. Tohnya et al., J. Chromatogr. B 811, 135 (2004). Clinical evaluation in multiple myeloma: P. G. Richardson et al., Blood 100, 3063 (2002); in myelodysplastic syndromes: A. List et al., N. Engl. J. Med. 352, 549 (2005). Review of development, pharmacology and therapeutic potential: J. B. Bartlett et al., Nature Rev. 4, 314-322 (2004); C. S. Mitsiades, N. Mitsiades, Curr. Opin. Invest. Drugs 5, 635-647 (2004).
Therap-Cat: Immunomodulator.
Keywords: Immunomodulator.
- 191732-72-6
- SYP-1512
- LENALIDOMIDE [VANDF]
- LENALIDOMIDE [WHO-DD]
- LENALIDOMIDE [EMA EPAR]
- LENALIDOMIDE [MI]
- LENALIDOMIDE [MART.]
- LENALIDOMIDE [ORANGE BOOK]
- LENALIDOMIDE [USAN]
- LENALIDOMIDE [INN]
- CDC-501
- REVLIMID
- LENALIDOMIDE
- 3-(4-AMINO-1-OXO-1,3-DIHYDRO-2H-ISOINDOL-2-YL)PIPERIDINE-2,6-DIONE
- 2,6-PIPERIDINEDIONE, 3-(4-AMINO-1,3-DIHYDRO-1-OXO-2H-ISOINDOL-2-YL)-
- CC-5013
Lenalidomide (trade name Revlimid) is a derivative of thalidomide approved in the United States in 2005.[1]
It was initially intended as a treatment for multiple myeloma, for which thalidomide is an accepted therapeutic treatment. Lenalidomide has also shown efficacy in the class of hematological disorders known as myelodysplastic syndromes (MDS). Along with several other drugs developed in recent years, lenalidomide has significantly improved overall survival in myeloma (which formerly carried a poor prognosis), although toxicity remains an issue for users.[2] It costs $163,381 per year for the average patient.[3]
It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[4]
Medical uses
Multiple myeloma
Multiple myeloma is a cancer of the blood, characterized by accumulation of a plasma cell clone in the bone marrow.[5] Lenalidomide is one of the novel drug agents used to treat multiple myeloma. It is a more potent molecular analog of thalidomide, which inhibits tumor angiogenesis, tumor secreted cytokines and tumor proliferation through the induction of apoptosis.[6][7][8]
Compared to placebo, lenalidomide is effective at inducing a complete or “very good partial” response as well as improving progression-free survival. Adverse events more common in people receiving lenalidomide for myeloma were neutropenia (a decrease in the white blood cell count), deep vein thrombosis, infections, and an increased risk of other hematological malignancies.[9] The risk of second primary hematological malignancies does not outweigh the benefit of using lenalidomide in relapsed or refractory multiple myeloma.[10] It may be more difficult to mobilize stem cells for autograft in people who have received lenalidomide.[6]
On 29 June 2006, lenalidomide received U.S. Food and Drug Administration (FDA) clearance for use in combination with dexamethasone in patients with multiple myeloma who have received at least one prior therapy.[11] On 22 February 2017, the FDA approved lenalidomide as standalone maintenance therapy (without dexamethasone) for patients with multiple myeloma following autologous stem cell transplant.[12]
On 23 April 2009, The National Institute for Health and Clinical Excellence (NICE) issued a Final Appraisal Determination (FAD) approving lenalidomide, in combination with dexamethasone, as an option to treat patients with multiple myeloma who have received two or more prior therapies in England and Wales.[13]
On 5 June 2013, the FDA designated lenalidomide as a specialty drug requiring a specialty pharmacy distribution for “use in mantle cell lymphoma (MCL) in patients whose disease has relapsed or progressed after two prior therapies, one of which included bortezomib.” Revlimid is only available through a specialty pharmacy, “a restricted distribution program in conjunction with a risk evaluation and mitigation strategy (REMS) due to potential for embryo-fetal risk.”[14]
Myelodysplastic syndromes
With myelodysplastic syndromes (MDS), the best results of lenalidomide were obtained in patients with the Chromosome 5q deletion syndrome (5q- syndrome).[15] The syndrome results from deletions in human chromosome 5 that remove three adjacent genes, granulocyte-macrophage colony-stimulating factor, Platelet-derived growth factor receptor B, and Colony stimulating factor 1 receptor.[16][17]
It was approved by the FDA on 27 December 2005, for patients with low or intermediate-1 risk MDS with 5q- with or without additional cytogenetic abnormalities. A completed Phase II, multi-centre, single-arm, open-label study evaluated the efficacy and safety of Revlimid monotherapy treatment for achieving haematopoietic improvement in red blood cell (RBC) transfusion dependent subjects with low- or intermediate-1-risk MDS associated with a deletion 5q cytogenetic abnormality.
63.8% of subjects had achieved RBC-transfusion independence accompanied by a median increase of 5.8 g/dL in blood Hgb concentration from baseline to the maximum value during the response period. Major cytogenetic responses were observed in 44.2% and minor cytogenetic responses were observed in 24.2% of the evaluable subjects. Improvements in bone marrow morphology were also observed. The results of this study demonstrate the efficacy of Revlimid for the treatment of subjects with Low- or Intermediate-1-risk MDS and an associated del 5 cytogenetic abnormality.[15][18][19]
Lenalidomide was approved on 17 June 2013 by the European Medicines Agency for use in low- or intermediate-1-risk myelodysplastic syndromes (MDS) patients who have the deletion 5q cytogenetic abnormality and no other cytogenetic abnormalities, are dependent on red blood cell transfusions, and for whom other treatment options have been found to be insufficient or inadequate.[20]
Mantle cell lymphoma
Lenalidomide is approved by FDA for mantle cell lymphoma in patients whose disease has relapsed or progressed after at least two prior therapies.[1] One of these previous therapies must have included bortezomib.
Other cancers
Lenalidomide is undergoing clinical trial as a treatment for Hodgkin’s lymphoma,[21] as well as non-Hodgkin’s lymphoma, chronic lymphocytic leukemia and solid tumor cancers, such as carcinoma of the pancreas.[22] One Phase 3 clinical trial being conducted by Celgene in elderly patients with B-cell chronic lymphocytic leukemia was halted in July 2013, when a disproportionate number of cancer deaths were observed during treatment with lenalidomide versus patients treated with chlorambucil.[23]
Adverse effects
In addition to embryo-fetal toxicity, lenalidomide also carries Black Box Warnings for hematologic toxicity (including significant neutropenia and thrombocytopenia) and venous/arterial thromboembolisms.[1]
Serious potential side effects are thrombosis, pulmonary embolus, and hepatotoxicity, as well as bone marrow toxicity resulting in neutropenia and thrombocytopenia. Myelosuppression is the major dose-limiting toxicity, which is contrary to experience with thalidomide.[24] Lenalidomide may also be associated with adverse effects including second primary malignancy, severe cutaneous reactions, hypersensitivity reactions, tumor lysis syndrome, tumor flare reaction, hypothyroidism, and hyperthyroidism. [1]
Teratogenicity
Lenalidomide is related to thalidomide which is known to be teratogenic. Tests in monkeys have suggested lenalidomide is also teratogenic.[25] It therefore has the pregnancy category X and cannot be prescribed for women who are pregnant or who may become pregnant during therapy. For this reason, the drug is only available in the United States(under the brand name Revlimid) through a restricted distribution system called RevAssist. Females who may become pregnant must use at least two forms of reliable contraception during treatment and for at least four weeks after discontinuing treatment with lenalidomide.[1]
Venous thromboembolism
Lenalidomide, like its parent compound thalidomide, may cause venous thromboembolism (VTE), a potentially serious complication with their use. Bennett et al. have reviewed incidents of lenalidomide-associated VTE among patients with multiple myeloma.[26] They have found that there are high rates of VTE when patients with multiple myeloma received thalidomide or lenalidomide in conjunction with dexamethasone, melphalan, or doxorubicin. When lenalidomide and dexamethasone are used to treat multiple myeloma, a median of 14% of patients had VTE (range,3-75%). In patients who took prophylaxis to treat lenalidomide-associated VTE, such as aspirin, thromboembolism rates were found to be lower than without prophylaxis, frequently lower than 10%. Clearly, thromboembolism is a serious adverse drug reaction associated with lenalidomide, as well as thalidomide. In fact, a black box warning is included in the package insert for lenalidomide, indicating that lenalidomide-dexamethasone treatment for multiple myeloma is complicated by high rates of thromboembolism.
Currently,[when?] clinical trials are under way to further test the efficacy of lenalidomide to treat multiple myeloma, and to determine how to prevent lenalidomide-associated venous thromboembolism.[citation needed]
Stevens-Johnson syndrome
In March 2008, the U.S. Food and Drug Administration (FDA) included lenalidomide on a list of 20 prescription drugs under investigation for potential safety problems. The drug is being investigated for possibly increasing the risk of developing Stevens–Johnson syndrome, a life-threatening condition affecting the skin.[27]
FDA ongoing safety review
As of 2011, the FDA has initiated an ongoing review which will focus on clinical trials which found an increased risk of developing cancers such as acute myelogenous leukemia (AML) and B-cell lymphoma,[3] though the FDA is currently advising all people to continue their treatment.[28]
Mechanism of action
Lenalidomide has been used to successfully treat both inflammatory disorders and cancers in the past ten years.[when?] There are multiple mechanisms of action, and they can be simplified by organizing them as mechanisms of action in vitro and in vivo.[29] In vitro, lenalidomide has three main activities: direct anti-tumor effect, inhibition of angiogenesis, and immunomodulation. In vivo, lenalidomide induces tumor cell apoptosis directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects, and by immunomodulatory activity. Lenalidomide has a broad range of activities that can be exploited to treat many hematologic and solid cancers.
On a molecular level, lenalidomide has been shown to interact with the ubiquitin E3 ligase cereblon[30] and target this enzyme to degrade the Ikaros transcription factors IKZF1 and IKZF3.[31] This mechanism was unexpected as it suggests that the major action of lenalidomide is to re-target the activity of an enzyme rather than block the activity of an enzyme or signaling process, and thereby represents a novel mode of drug action. A more specific implication of this mechanism is that the teratogenic and anti-neoplastic properties of lenalidomide, and perhaps other thalidomide derivatives, could be disassociated.
Research
The low level of research that continued on thalidomide, in spite of its scandalous history of teratogenicity, unexpectedly showed that the compound affected immune function. The drug was, for example, recently approved by the FDA for treatment of complications from leprosy; it has also been investigated as an adjunct for treating some malignancies. Recent research on related compounds has revealed a series of molecules which inhibit tumor necrosis factor (TNF-α).[citation needed]
Price
Lenalidomide costs $163,381 per year for the average person in the United States.[3] Lenalidomide made almost $9.7bn for Celgene in 2018.[32]
In 2013, the UK National Institute for Health and Care Excellence (NICE) rejected lenalidomide for “use in the treatment of people with a specific type of the bone marrow disorder myelodysplastic syndrome (MDS)” in England and Scotland, arguing that Celgene “did not provide enough evidence to justify the £3,780 per month (USD$5746.73) price-tag of lenalidomide for use in the treatment of people with a specific type of the bone marrow disorder myelodysplastic syndrome (MDS)”.[33]
SYN
https://link.springer.com/article/10.1007/s10593-015-1670-0

A new process for the synthesis of anticancer drug lenalidomide was developed, using platinum group metal-free and efficient reduction of nitro group with the iron powder and ammonium chloride. It was found that the bromination of the key raw material, methyl 2-methyl-3-nitrobenzoate, could be carried out in chlorine-free solvent methyl acetate without forming significant amounts of hazardous by-products. We also have compared the known synthetic methods for cyclization of methyl 2-(bromomethyl)-3-nitrobenzoate and 3-aminopiperidinedione to form lenalidomide nitro precursor.
SYN

SYN
EP 0925294; US 5635517; WO 9803502
Cyclization of N-(benzyloxycarbonyl)glutamine (I) by means of CDI in refluxing THF gives 3-(benzyloxycarbonylamino)piperidine-2,6-dione (II), which is deprotected with H2 over Pd/C in ethyl acetate/4N HCl to yield 3-aminopiperidine-2,6-dione hydrochloride (III). Bromination of 2-methyl-3-nitrobenzoic acid methyl ester (IV) with NBS in CCl4 provides 2-(bromomethyl)-3-nitrobenzoic acid methyl ester (V), which is cyclized with the aminopiperidine (III) by means of triethylamine in hot DMF to afford 3-(4-nitro-1-oxoisoindolin-2-yl)piperidine-2,6-dione (VI). Finally, the nitro group of compound (VI) is reduced with H2 over Pd/C in methanol (1, 2).
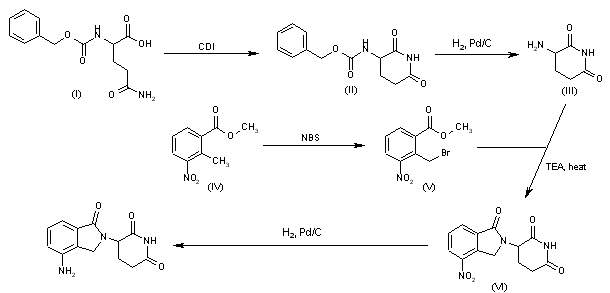
SYN
Bioorg Med Chem Lett 1999,9(11),1625
Treatment of 3-nitrophthalimide (I) with ethyl chloroformate and triethylamine produced 3-nitro-N-(ethoxycarbonyl)phthalimide (II), which was condensed with L-glutamine tert-butyl ester hydrochloride (III) to afford the phthaloyl glutamine derivative (IV). Acidic cleavage of the tert-butyl ester of (IV) provided the corresponding carboxylic acid (V). This was cyclized to the required glutarimide (VI) upon treatment with thionyl chloride and then with triethylamine. The nitro group of (VI) was finally reduced to amine by hydrogenation over Pd/C.
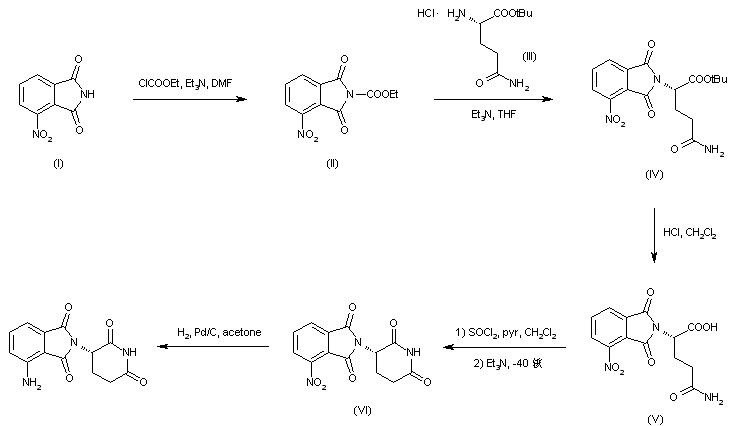
Lenalidomide
-
- Synonyms:CC-5013, CDC 501
- ATC:L04AX04
- Use:myelodysplastic syndrome (MDS)
- Chemical name:3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-2,6-piperidinedione
- Formula:C13H13N3O3
- MW:259.27 g/mol
- CAS-RN:191732-72-6
- InChI Key:GOTYRUGSSMKFNF-JTQLQIEISA-N
- InChI:InChI=1S/C13H13N3O3/c14-9-3-1-2-7-8(9)6-16(13(7)19)10-4-5-11(17)15-12(10)18/h1-3,10H,4-6,14H2,(H,15,17,18)/t10-/m0/s1
Synthesis
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Revlimid | Celgene | |
| GB | Revlimid | Celgene | |
| USA | Revlimid | Celgene ,2005 |
Formulations
- cps. 5 mg, 10 mg
References
-
- WO 9 803 502 (Celgene; 29.1.1998; USA-prior. 24.7.1996).
- WO 2 006 028 964 (Celgene; 16.3.2006; USA-prior. 3.9.2004).
- US 5 635 517 (Celgene; 3.6.1997; USA-prior. 24.7.1996).
-
medical use for treatment of certain leukemias:
- US 2 006 030 594 (Celgene; 9.2.2006; USA-prior. 4.10.2005).
-
alternative preparation of III:
- WO 2 005 005 409 (Siegfried Ltd.; 20.1.2005; CH-prior. 9.7.2003).
References
- ^ Jump up to:a b c d e REVLIMID [package insert]. Summit, NJ: Celgene Corporation; 2017. Accessed at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021880s055lbl.pdf on 14 September 2018.
- ^ McCarthy PL, Owzar K, Hofmeister CC, et al. (2012). “Lenalidomide after stem-cell transplantation for multiple myeloma”. N. Engl. J. Med. 366 (19): 1770–81. doi:10.1056/NEJMoa1114083. PMC 3744390. PMID 22571201.
- ^ Jump up to:a b c Badros AZ (10 May 2012). “Lenalidomide in Myeloma — A High-Maintenance Friend”. N Engl J Med. 366 (19): 1836–1838. doi:10.1056/NEJMe1202819. PMID 22571206.
- ^ “World Health Organization model list of essential medicines: 21st list 2019”. 2019. hdl:10665/325771.
- ^ Armoiry X, Aulagner G, Facon T (June 2008). “Lenalidomide in the treatment of multiple myeloma: a review”. Journal of Clinical Pharmacy and Therapeutics. 33 (3): 219–26. doi:10.1111/j.1365-2710.2008.00920.x. PMID 18452408.
- ^ Jump up to:a b Li S, Gill N, Lentzsch S (November 2010). “Recent advances of IMiDs in cancer therapy”. Curr Opin Oncol. 22 (6): 579–85. doi:10.1097/CCO.0b013e32833d752c. PMID 20689431.
- ^ Tageja N (March 2011). “Lenalidomide – current understanding of mechanistic properties”. Anti-Cancer Agents Med. Chem. 11 (3): 315–26. doi:10.2174/187152011795347487. PMID 21426296.
- ^ Kotla V, Goel S, Nischal S, et al. (August 2009). “Mechanism of action of lenalidomide in hematological malignancies”. J Hematol Oncol. 2: 36. doi:10.1186/1756-8722-2-36. PMC 2736171. PMID 19674465.
- ^ Yang B, Yu RL, Chi XH, et al. (2013). “Lenalidomide treatment for multiple myeloma: systematic review and meta-analysis of randomized controlled trials”. PLoS ONE. 8 (5): e64354. doi:10.1371/journal.pone.0064354. PMC 3653900. PMID 23691202.
- ^ Dimopoulos MA, Richardson PG, Brandenburg N, et al. (22 March 2012). “A review of second primary malignancy in patients with relapsed or refractory multiple myeloma treated with lenalidomide”. Blood. 119 (12): 2764–7. doi:10.1182/blood-2011-08-373514. PMID 22323483.
- ^ “FDA approves lenalidomide oral capsules (Revlimid) for use in combination with dexamethasone in patients with multiple myeloma”. Food and Drug Administration (FDA). 29 June 2006. Retrieved 15 October 2015.
- ^ “Approved Drugs – Lenalidomide (Revlimid)”. Food and Drug Administration (FDA).
- ^ “REVLIMID Receives Positive Final Appraisal Determination from National Institute for Health and Clinical Excellence (NICE) for Use in the National Health Service (NHS) in England and Wales”. Reuters. 23 April 2009.
- ^ Ness, Stacey (13 March 2014). “New Specialty Drugs”. Pharmacy Times. Retrieved 5 November 2015.
- ^ Jump up to:a b List A, Kurtin S, Roe DJ, et al. (February 2005). “Efficacy of lenalidomide in myelodysplastic syndromes”. The New England Journal of Medicine. 352 (6): 549–57. doi:10.1056/NEJMoa041668. PMID 15703420.
- ^ “PDGFRB platelet derived growth factor receptor beta [Homo sapiens (human)] – Gene – NCBI”.
- ^ Nimer SD (2006). “Clinical management of myelodysplastic syndromes with interstitial deletion of chromosome 5q”. Journal of Clinical Oncology. 24 (16): 2576–82. doi:10.1200/JCO.2005.03.6715. PMID 16735711.
- ^ List AF (August 2005). “Emerging data on IMiDs in the treatment of myelodysplastic syndromes (MDS)”. Seminars in Oncology. 32 (4 Suppl 5): S31–5. doi:10.1053/j.seminoncol.2005.06.020. PMID 16085015.
- ^ List A, Dewald G, Bennett J, et al. (October 2006). “Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion”. The New England Journal of Medicine. 355 (14): 1456–65. doi:10.1056/NEJMoa061292. PMID 17021321.
- ^ “Revlimid Approved In Europe For Use In Myelodysplastic Syndromes”. The MDS Beacon. Retrieved 17 June 2013.
- ^ “Phase II Study of Lenalidomide for the Treatment of Relapsed or Refractory Hodgkin’s Lymphoma”. ClinicalTrials.gov. US National Institutes of Health. February 2009.
- ^ “276 current clinical trials world-wide, both recruiting and fully enrolled, as of 27 February 2009”. ClinicalTrials.gov. US National Institutes of Health. February 2009.
- ^ “Celgene Discontinues Phase 3 Revlimid Study after ‘Imbalance’ of Deaths”. Nasdaq. 18 July 2013.
- ^ Rao KV (September 2007). “Lenalidomide in the treatment of multiple myeloma”. American Journal of Health-System Pharmacy. 64 (17): 1799–807. doi:10.2146/ajhp070029. PMID 17724360.
- ^ “Revlimid Summary of Product Characteristics. Annex I” (PDF). European Medicines Agency. 2012. p. 6.
- ^ Bennett CL, Angelotta C, Yarnold PR, et al. (December 2006). “Thalidomide- and lenalidomide-associated thromboembolism among patients with cancer”. JAMA: The Journal of the American Medical Association. 296 (21): 2558–60. doi:10.1001/jama.296.21.2558-c. PMID 17148721.
- ^ “Potential Signals of Serious Risks/New Safety Information Identified from the Adverse Event Reporting System (AERS) between January – March 2008”. Food and Drug Administration (FDA). March 2008.
- ^ “FDA Drug Safety Communication: Ongoing safety review of Revlimid (lenalidomide) and possible increased risk of developing new malignancies”. Food and Drug Administration(FDA). April 2011.
- ^ Vallet S, Palumbo A, Raje N, et al. (July 2008). “Thalidomide and lenalidomide: Mechanism-based potential drug combinations”. Leukemia & Lymphoma. 49 (7): 1238–45. doi:10.1080/10428190802005191. PMID 18452080.
- ^ Zhu YX, Braggio E, Shi CX, et al. (2011). “Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide”. Blood. 118 (18): 4771–9. doi:10.1182/blood-2011-05-356063. PMC 3208291. PMID 21860026.
- ^ Stewart AK (2014). “Medicine. How thalidomide works against cancer”. Science. 343(6168): 256–7. doi:10.1126/science.1249543. PMC 4084783. PMID 24436409.
- ^ “Top 10 Best-Selling Cancer Drugs of 2018”. Genetic Engineering and Biotechnology News. 22 April 2019. Retrieved 25 April 2019.
- ^ “Revlimid faces NICE rejection for use in rare blood cancer Watchdog’s draft guidance does not recommend Celgene’s drug for NHS use in England and Wales”. Pharma News. 11 July 2013. Retrieved 5 November 2015.
Further reading
- Chang DH, Liu N, Klimek V, et al. (July 2006). “Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications”. Blood. 108 (2): 618–21. doi:10.1182/blood-2005-10-4184. PMC 1895497. PMID 16569772.
- Anderson KC (October 2005). “Lenalidomide and thalidomide: mechanisms of action–similarities and differences”. Seminars in Hematology. 42 (4 Suppl 4): S3–8. doi:10.1053/j.seminhematol.2005.10.001. PMID 16344099.
External links
- Official website Includes list of adverse reactions
- Prescribing Information
- International Myeloma Foundation article on Revlimid
- multiplemyeloma.org Revlimid April 2007 Summary
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ˌlɛnəˈlɪdoʊmaɪd/ |
| Trade names | Revlimid |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a608001 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral (capsules) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Undetermined |
| Protein binding | 30% |
| Metabolism | Undetermined |
| Elimination half-life | 3 hours |
| Excretion | Renal (67% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.218.924 |
| Chemical and physical data | |
| Formula | C13H13N3O3 |
| Molar mass | 259.261 g/mol g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
//////////LENALIDOMIDE, レナリドミド ,REVLIMID, Celgene Corporation, леналидомид , ليناليدوميد , 来那度胺 ,
CANERTINIB
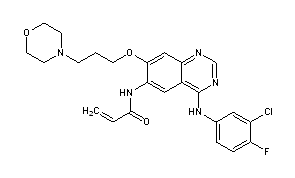
CANERTINIB
Canertinib
CAS Registry Number: 267243-28-7
CAS Name: N-[4-[(3-Chloro-4-fluorophenyl)amino]-7-[3-(4-morpholinyl)propoxy]-6-quinazolinyl]-2-propenamide
Additional Names: N-[4-(3-chloro-4-fluorophenylamino)-7-(3-morpholin-4-ylpropoxy)quinazolin-6-yl]acrylamide
Molecular Formula: C24H25ClFN5O3
Molecular Weight: 485.94
Percent Composition: C 59.32%, H 5.19%, Cl 7.30%, F 3.91%, N 14.41%, O 9.88%
Literature References: Irreversible pan-erbB tyrosine kinase inhibitor. Prepn: A. J. Bridges et al., WO 0031048; eidem, US 6344455 (2000, 2002 both to Warner-Lambert); J. B. Smaill et al., J. Med. Chem. 43, 1380 (2000). Clinical pharmacokinetics in patients with solid malignancies: E. Calvo et al., Clin. Cancer Res. 10, 7112 (2004); and tolerability in refractory cancer: J. Nemunaitis et al., ibid. 11, 3846 (2005). Review of pharmacology and mechanism of action: L. F. Allen et al., Semin. Oncol. 30, Suppl. 16, 65-78 (2003); of development and clinical experience: C. M. Galmarini, IDrugs 7, 58-63 (2004).
Properties: Crystals from methanol, mp 188-190°.
Melting point: mp 188-190°
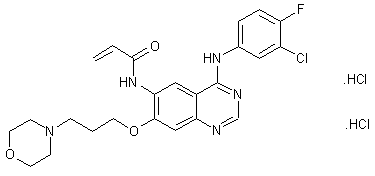
Canertinib dihydrochloride, CI-1033, PD-183805(free base)
Derivative Type: Dihydrochloride
CAS Registry Number: 289499-45-2
Manufacturers’ Codes: CI-1033
Molecular Formula: C24H25ClFN5O3.2HCl
Molecular Weight: 558.86
Percent Composition: C 51.58%, H 4.87%, Cl 19.03%, F 3.40%, N 12.53%, O 8.59%
Properties: Sol in water.
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Tyrosine Kinase Inhibitors.
267243-28-7 [RN]
2-propenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[3-(4-morpholinyl)propoxy]-6-quinazolinyl]- [ACD/Index Name]
8256
C78W1K5ASF
N-{4-[(3-Chloro-4-fluorophenyl)amino]-7-[3-(4-morpholinyl)propoxy]-6-quinazolinyl}acrylamide
Canertinib (CI-1033) is an experimental drug candidate for the treatment of cancer. It is an irreversible tyrosine-kinase inhibitor with activity against EGFR (IC50 0.8 nM), HER-2 (IC50 19 nM) and ErbB-4 (IC50 7 nM).[1][2] By 2015, Pfizer had discontinued development of the drug.[3]
Canertinib has been reported as a substrate for OATP1B3. Interaction of canertinib with OATP1B3 may alter its hepatic disposition and can lead to transporter mediated drug-drug interactions.[4] Also, canertinib is not an inhibitor of OATP-1B1 or OATP-1B3 transporter.[5]
SYN
J Med Chem 2000,43(7),1380
| EP 1131304; US 6344455; WO 0031048 |
4-Chloro-7-fluoro-6-nitroquinazoline (I) was condensed with 3-chloro-4-fluoroaniline (II) to afford the 4-anilino quinazoline (III). Displacement of the activated fluorine of (III) with the potassium alkoxide of morpholinopropanol (IV) gave the morpholinopropyl ether (V). Subsequent reduction of the nitro group of (V), either using iron dust and acetic acid or catalytic hydrogenation over Raney-Ni, furnished aminoquinazoline (VI). This was finally condensed with acrylic acid (VII), via activation as the mixed anhydride with isobutyl chloroformate or using EDC as the coupling reagent, to provide the title acrylamide.
PATENT
https://patents.google.com/patent/CN103242244A/en
canertinib (Canertinib, I), chemical name 4- (3-chloro-4-fluoroanilino) -7- [3- (4_-morpholinyl) propoxy] -6-propylene quinazoline amide group, and by the US Pfizer Warner Lambert developed jointly an irreversible epidermal growth factor receptor (pan-ErbB) selective inhibitor, which is capable of binding to the cell surface of all members of the ErbB family adenosine triphosphate binding site, thereby inhibiting the activation of these receptors and their downstream mitogenic signal transduction pathways. Clinical studies show that the product has good resistance, can be effective in treating metastatic breast cancer, ovarian cancer, cervical cancer and other tumors, and can be combined with a variety of antineoplastic agents exhibit a synergistic effect.
[0004]

[0005] China Patent No. CN1160338C, CN1438994A and No. No. CN1745073A reported the preparation of canertinib: A nucleus 4- [(3-chloro-4-fluorophenyl) amino] -6-nitro 7-fluoro-quinazoline (VIII) as a starting material, under basic conditions with 3- (4-morpholinyl) -1-propanol 7-position substitution reaction occurs to give 4- [(3-chloro – 4-fluorophenyl) amino] -6-nitro-7- [3- (4-morpholinyl) -1-propoxy] quinazoline (IX); intermediate (IX) through the 6-position nitro reduction, to give the corresponding amino compound (X); amino compound (X) to give canertinib acylation reaction (I) with acrylic acid or acryloyl chloride occurs.
[0006] In addition, “Qilu Pharmaceutical Affairs” 30, 2011, Vol. 10, page 559, and “China Industrial Medicine” 2010 Volume 41, No. 6, pp. 404 also reported an improved method of the above-prepared and studied method from 7-fluoro-quinazolin-3-one (V) via nitration, chloro and condensation reaction of the preparation of intermediate (VIII) is.
[0007]

[0008] This shows that the current Kanai prepared for Nepal is mainly the 4-position through an intermediate (VII), respectively, a functional transformation of the 6-position and 7-position achieved. Since the intermediate (VII) a fluorine-containing compounds, materials are not readily available, many steps, and many steps are required to be isolated and purified by column chromatography, which is not required for industrialization.
Example a:
[0023] at room temperature, to a three-necked flask was added diisopropyl azodicarboxylate (3mL, 15mmol) and tetrahydrofuran 5mL, dropwise addition of triphenylphosphine (4.0g, 15mmol) in tetrahydrofuran 25mL solution at room temperature, kept at room temperature for 2 hours. Under nitrogen, 3- (4-morpholinyl) -1_-propanol (0.49g, 3.4mmol) in 5mL of tetrahydrofuran was added dropwise to the reaction system after the dropwise addition is complete, 6-amino – 7-hydroxy-3,4-dihydro-quinazolin-4-one (II) (0.53g,
3.0mmol), stirred at room temperature for 4 hours. Solution of 3- (4-morpholinyl) -1-propanol (0.38g, 2.6mmol) in 5mL of tetrahydrofuran was continued at room temperature for 2 hours, the end of the reaction was monitored TLC. Recovery of the solvent by distillation under reduced pressure, the residue was treated with dilute hydrochloric acid, pH = 5-6, extracted with ethyl acetate, the organic phase was washed with saturated sodium carbonate adjusted pH = 10-11. The aqueous phase was freeze-dried in vacuo to give an off-white solid 6-amino-7- [3- (4-morpholinyl) propoxy] _3,4- dihydroquinazolin-4-one (111) 0.80g yield 87.7%.
[0024] Example II:
[0025] to a three-neck flask was added 6-amino-7- [3- (4_ morpholino) propoxy] quinazolin-dihydro _3,4_ one _4_
(III) (0.76g, 2.5mmol), triethylamine (0.25g, 2.5mmol) and dichloromethane 20mL, warmed to 40-45 ° C, stirred until homogeneous dissolution system. Dropped below 10 ° C, was slowly added dropwise acryloyl chloride (0.25g, 2.8mmol) in dichloromethane IOmL solution dropwise at room temperature after continued for 6 h, TLC detection reaction was completed. The reaction solution was respectively 10% sodium bicarbonate solution and water, dried over anhydrous sodium sulfate. Recovery of the solvent under reduced pressure, the residue was recrystallized from ethyl acetate to give a white solid 7- [3- (4-morpholinyl) propoxy] -6-acrylamido-3,4-dihydro-quinazoline – 4-one (IV) 0.81g, 90.5% yield.
[0026] Example III:
Under [0027] nitrogen, to a three-necked flask was added 7- [3- (4_-morpholinyl) propoxy] -6-acrylamido-_3,4- dihydroquinazolin-4-one (IV ) (3.58g, IOmmol), benzotriazol-1-yloxytris (dimethylamino) phosphonium iron hexafluorophosphate (BOP) (6.63g, 15mmol) and acetonitrile 100mL. Under stirring, a solution of 1,8-diazabicyclo [5.4.0] ^ a-7-ene (DBU) (2.28g, 15mmol), dropwise, at room temperature for 12 hours. Warmed to 60 ° C, the reaction was continued for 12 hours. The solvent was removed by distillation under reduced pressure, ethyl acetate was added to dissolve IOOmL, washed with 2M sodium hydroxide and 20mL. The organic phase was separated, dried and concentrated under reduced pressure. The residue was dissolved in tetrahydrofuran IOOmL, 4-chloro-3-fluoroaniline (1.89g, 13mmol) and sodium hydride (0.32g, 13mmol), was heated to 50 ° C, reaction was stirred for 5 hours, the end of the reaction was monitored TLC. Quenched with saturated brine the reaction, the organic phase was separated, dried, evaporated under reduced pressure to recover the solvent to give an off-white solid. Recrystallized from ethanol to give an off-white solid canertinib (I) 4.05g, yield 83.5%.
[0028] Example IV:
Under [0029] nitrogen, to a three-necked flask was added 7- [3- (4_-morpholinyl) propoxy] -6-acrylamido-3,4-dihydro-quinazolin-4-one (IV ) (3.58g, IOmmol), benzotriazol-1-yloxytris (dimethylamino) phosphonium iron hexafluorophosphate (BOP) (6.63g, 15mmol) and acetonitrile lOOmL. Under stirring, dropwise power port I, 5- diazabicyclo [4.3.0] – non-5-ene (DBN) (1.86g, 15mmol), dropwise, at room temperature for 12 hours. Warmed to 60 ° C, the reaction was continued for 12 hours. The solvent was removed by distillation under reduced pressure, ethyl acetate was added to dissolve IOOmL, washed with 2M sodium hydroxide and 20mL. The organic phase was separated, dried and concentrated under reduced pressure. The residue was dissolved in tetrahydrofuran IOOmL, 4-chloro-3-fluoroaniline (1.89g, 13mmol) and sodium hydride (0.32g, 13mmol), was heated to 50 ° C, reaction was stirred for 5 hours, the end of the reaction was monitored TLC. Quenched with saturated brine the reaction, the organic phase was separated, dried, evaporated under reduced pressure to recover the solvent to give an off-white solid. Recrystallized from ethanol to give an off-white solid canertinib (I) 3.85g, yield 79.4%. ·
[0030] Example Five:
Under [0031] nitrogen, to a three-necked flask was added 7- [3- (4_-morpholinyl) propoxy] -6-acrylamido-3,4-dihydro-quinazolin-4-one (IV ) (3.58g, IOmmol), benzotriazol-1-yloxytris (dimethylamino) phosphonium hexafluorophosphate gun (BOP) (6.63g, 15mmol), 4_-chloro-3-fluoroaniline ( 1.89g, 13mmol) and N, N- dimethylformamide lOOmL. Under stirring, a solution of I, 8- diazabicyclo [5.4.0] – ^ a _7_ ene (DBU) (2.28g, 15mmol), dropwise, at room temperature for 12 hours. Warmed to 60 ° C, the reaction was continued for 12 hours. The solvent was removed by distillation under reduced pressure, ethyl acetate was added to dissolve IOOmL, washed with 2M sodium hydroxide and 20mL. The organic phase was separated, dried and concentrated under reduced pressure. The residue was recrystallized from ethanol to give an off-white solid canertinib (1) 2.32g, yield 47.8%.

References
GW; Loo, JA; Greis, KD; Chan, OH; Reyner, EL; Lipka, E; Showalter, HD; et al. (2000). “Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido3,2-dpyrimidine-6-acrylamides bearing additional solubilizing functions”. Journal of Medicinal Chemistry. 43 (7): 1380–97. doi:10.1021/jm990482t. PMID 10753475.
- ^ CI-1033 (Canertinib), Selleck Chemicals
- ^ http://adisinsight.springer.com/drugs/800012072
- ^ Khurana V, Minocha M, Pal D, Mitra AK (March 2014). “Role of OATP-1B1 and/or OATP-1B3 in hepatic disposition of tyrosine kinase inhibitors”. Drug Metabol Drug Interact. 29 (3): 1–11. doi:10.1515/dmdi-2013-0062. PMC 4407685. PMID 24643910.
- ^ Khurana V, Minocha M, Pal D, Mitra AK (May 2014). “Inhibition of OATP-1B1 and OATP-1B3 by tyrosine kinase inhibitors”. Drug Metabol Drug Interact. 29 (4): 1–11. doi:10.1515/dmdi-2014-0014. PMC 4407688. PMID 24807167.
 |
|
| Names | |
|---|---|
| IUPAC name
N-{4-[(3-Chloro-4-fluorophenyl)amino]-7-[3-(morpholin-4-yl)propoxy]quinazolin-6-yl}prop-2-enamide
|
|
| Other names
CI-1033; PD-183805
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C24H25ClFN5O3 | |
| Molar mass | 485.94 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////CANERTINIB
DICYCLOPLATIN

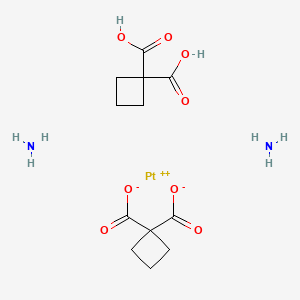
Dicycloplatin
Platinum(2+) 1-carboxycyclobutanecarboxylate ammoniate (1:2:2)
- Molecular FormulaC12H20N2O8Pt
- Average mass515.380 Da
- 287402-09-9
Has antineoplastic activity; a supramolecular complex of 1,1-cyclobutane dicarboxylic acid and cis-diammine(1,1-cyclobutane dicarboxylate)platinum (II).
1,1-Cyclobutanedicarboxylic acid, ammonium platinum(2+) salt (2:2:1) [ACD/Index Name]
Platinum(2+) 1-carboxycyclobutanecarboxylate ammoniate (1:2:2)
287402-09-9 [RN]
DICYCLOPLATIN
UNII:0KC57I4UNB
Dicycloplatin is a chemotherapy medication used to treat a number of cancers which includes the Non-small-cell lung carcinoma and prostate cancer.[1]
Some side effects which are observed from the treatment by dicycloplatin are nausea, vomiting, thrombocytopenia, neutropenia, anemia, fatigue, loss of appetite, liver enzyme elevation and alopecia. The drugs is a form of Platinum-based antineoplastic and it works by causing the mitochondrial dysfunction which leads to the cell death.[2]
Dicycloplatin was developed in China and it was used for phase I human trial clinical in 2006. The drug was approved for chemotherapy by the Chinese FDA in 2012.[3]

Medical uses
Dicycloplatin can inhibit the proliferation of tumor cells via the induction of apoptosis . It is used to treat a number types of cancer which are Non-small-cell lung carcinoma and prostate cancer.[4]
Side effects
Similar to cisplatin and carboplatin, dicycloplatin also contains some side effects, which are nausea, vomiting, thrombocytopenia, neutropenia, anemia, fatigue, anorexia, liver enzyme elevation, and alopecia. However, with doses up to 350 mg/m(2), there is no significant toxicity; these effects are observed only at higher doses. Furthermore, the nephrotoxicity of dicycloplatin is reported to be less than that of cisplatin, and its myelosuppressive potency is similar to that of carboplatin.[5]
Chemical structure
Dicycloplatin consists of carboplatin and cyclobutane-1,1-dicarboxylic acid (CBDC) linked by the hydrogen bond. In the structure of dicycloplatin, there are two types of bond: O-H…O is the bond between the hydroxyl group of CBDC with carboxyl oxygen atom. It creates the one-dimensional polymer chain of carboplatin and CBDC. The second one is N-H…O which links between the ammoniagroup of carboplatin and oxygen of CBDC. It forms the two-dimensional polymer chain of carboplatin and CBDC. In aqueous solution, the 2D-hydrogen bonded polymeric structure of dicycloplatin is destroyed. Firstly, the bond between ammonia group of carboplatin and oxygen of CBDC breaks, thus inducing the formation of one-dimensional dicycloplatin. After that, the strong hydrogen bond breaks and creates an intermediate state of dicycloplatin. Finally, the rearrangement of different orientation of carboplatin and CBDC leads to the formation of intramolecular hydrogen bond and a supramolecule of dicycloplatin with two O-H…O and N-H…O is created.[6]
Mechanism of action
Similar to carboplatin, dicycloplatin inhibits the proliferation of cancer cells by inducing cell apoptosis. When treated with dicycloplatin, some changes in the properties of Hep G2 cells are observed: the declination of Mitochondria Membrane Potential, the release of cytochrome c from mitocondria to cytosol, the activation of caspase-9, caspase-3 and the decrease of Bcl-2.[4] Those phenomena indicate the role of mitochondrial in the apoptosis by intrisic way.[7] Furthermore, the increase in caspase-8 activation is also observed. This can stimulate the apoptosis by activating downstream caspase-3 [8] or by cleaving Bid.[9] As a result, the cleavage of Bid (tBid) transfers to the mitochondria and induce mitochondrial dysfunction which promotes the release of cytochrome c from mitochondria to cytosol.[10] From the dicycloplatin-treated Hep G2 cell, an excessive amount of reactive oxygen species was detected,[4] which plays an important role in the release of cytochrome c. In the mitochondria, the release of hemoprotein happens through 2-step process: Firstly, the dissociation of cytochrome c from its binding to cardiolipin happens. Due to the reactive oxygen species, the cardiolipin is oxidized, thus reducing the cytochrome c binding and increase the concentration of free cytochrome c [11]
PATENT
WO2018171371
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018171371
Since the FDA approved cisplatin as an anticancer drug in 1978, the mortality rate of testicular cancer patients has been reduced from 100% to less than 10%. For patients with early detection, the cure rate can reach 100%, making cisplatin An outstanding representative of anticancer drugs. In 1986, the FDA approved the second-generation platinum anticancer drug carboplatin. Its anticancer spectrum is similar to that of cisplatin, but it has good water solubility and light toxicity. In 2002, the FDA approved the third-generation platinum anticancer drug oxaliplatin to enter clinical treatment of colorectal cancer. Its anticancer spectrum is different from cisplatin, and it does not produce cross-resistance with cisplatin.
In addition to the above three products, four products, including Nida Platinum, Shuplatin, Lobaplatin and Miplatin, have been listed in different countries and are the first in other countries.
In CN1311183A, Yang Xuqing et al. designed and prepared a new class of platinum antitumor drugs, diammonium platinum dichloride (II) derivatives, based on the abnormal changes in the spatial configuration of cancer cells DNA and RNA. A typical representative drug is bicycloplatinum. Bicycloplatinum in English is called Dicycloplatin, which is called bis(1,1-cyclobutanedicarboxylic acid) diammine platinum (II) (English name [Bis-(1,1-cyclobutane dicarboxylic acid)]diammine platinum(II) ), the structural formula is:
It is a supramolecular compound composed of carboplatin and 1,1-cyclobutanedicarboxylic acid through four hydrogen bonds. It is the first self-developed platinum antitumor drug in China with broad spectrum, low toxicity and high efficiency. It does not produce cross-resistance and good penetrability.
Bicycloplatinum is usually obtained by reacting carboplatin with 1,1-cyclobutanedicarboxylic acid. The prior art discloses various preparation methods, but both have the problems of complicated preparation process and low product purity.
CN1311183A As the earliest publication of bicycloplatin and its preparation method, it is disclosed that bicycloplatinum is prepared by the following method: carboplatin is dissolved in pure water at normal temperature, and then an equimolar amount of 1,1-cyclobutanedicarboxylic acid is added. After the reaction was completed, it was evaporated to dryness, washed with ethanol, and then recrystallized from distilled water. This method is cumbersome in operation due to the need for evaporation and recrystallization steps, and the yield of bicycloplatinum is low.
CN104693245A discloses a preparation method of bicyclo platinum, which is prepared by using carboplatin as a raw material in a ratio of 1:11 to 1,1-cyclobutanedicarboxylic acid in a molar ratio of 1:1, and is protected from light at 0-60 ° C. After -9 days, the excess water is removed by concentration under reduced pressure or freeze-drying to obtain a bicyclic platinum product. Although according to reports, the HPLC purity of the product is more than 99%, it requires a long standing process, is inefficient, and greatly increases the risk of carboplatin decomposition, especially for the process of amplification; The heating and concentration in the final process makes the bicyclic platinum product exist in the higher temperature aqueous solution for a long time, and the product has a high risk of degradation, and the quality stability is inevitably affected. In fact, bicycloplatinum with the reported yield and purity was not obtained according to this method.
CN106132408A discloses a process for the preparation of another bicyclic platinum in which carboplatin is mixed with a corresponding ratio of 1,1-cyclobutanedicarboxylic acid and a solvent to form a suspension, and the precipitated solid formed is separated from the suspension. Although the report states that the obtained product does not contain XRPD detectable amount of carboplatin, the suspension method uses a small amount of solvent, so that the product formed during the reaction is also precipitated as a solid, which is mixed with the unreacted raw material solid. This prevents the reaction from proceeding and makes the purification of the product more difficult. Especially in the case where the product is coated with carboplatin, the carboplatin can hardly be removed by purification. Therefore, the suspension method has the disadvantages of difficulty in control, poor operability, and incapability of industrial scale-up production. In fact, bicycloplatinum with the reported yield and purity cannot be obtained according to this method as well.
1 is a nuclear magnetic resonance-hydrogen spectrum of the bicyclic platinum product of Example 1.
2 is a nuclear magnetic resonance-carbon spectrum of the bicyclic platinum product of Example 1.
Drawing
[ figure 1] 
[ figure 2] 
Preparation Example 1:
Take 20.0 g of cis-diiododiammine platinum (II), add 600 ml of purified water, stir well and heat to 80 ° C in water bath, then add 14.1 g of silver 1,1-cyclobutanedicarboxylate, after reacting for 30 minutes. The AgI slag was filtered off, and the filtrate was concentrated under reduced pressure to a residue of about 50 ml, cooled to room temperature, and the precipitated product was filtered. After recrystallization, the mixture was dried at 60 ° C to obtain 11.26 g of carboplatin, and the yield was 69.88%.
Example 1
32.0 g (222.2 mmol) of 1,1-cyclobutanedicarboxylic acid was taken, and 260 ml of water was added thereto, and the mixture was heated to 80 ° C in a water bath. Add 10.0 g (26.95 mmol) of carboplatin, stir for 40 minutes, cool at 10 ° C for 8 hours, filter the precipitated solid, wash the filter cake with appropriate amount of purified water, drain the washing water, and dry at 40 ° C under reduced pressure to obtain bicyclo platinum 9.32 g. The yield is 67.15% and the content is 99.78%. The obtained products were characterized by elemental analysis, negative ion electrospray mass spectrometry, nuclear magnetic resonance-hydrogen spectroscopy, nuclear magnetic resonance-carbon spectroscopy and X-ray diffraction. The content of bicycloplatin was measured by high performance liquid chromatography.
The test results are shown in Figure 1. The attribution of each peak is as follows:
The peak of chemical shift 1.7159-1.7793ppm is H a , the actual number of hydrogen nuclei is 2, and it is divided into 5 heavy peaks by 4 H b on both sides ; the peak of chemical shift 1.8281-1.8928ppm is H c , actual hydrogen the number of cores 2, a total of four sides by H D impact crack 5 doublet; 2.3965-2.4288ppm peak chemical shift of H B , the actual number of hydrogen nuclei to 4, were subjected to unilateral 2 H a of Effect split into three doublet; 2.7140-2.7457ppm peak chemical shift of H D , the actual number of hydrogen nuclei is 4, were subjected to unilateral 2 H Caffected divided into three split doublet; chemical shifts of the peaks 4.0497ppm is H E , the actual number of hydrogen nuclei 6 as broad singlet; due to D 2 exchange interaction of O, carboxy FIG active hydrogen protons H does not appear f peaks. 4. Nuclear Magnetic Resonance – Carbon Spectrum (D 2 O, 500MHz)
The test results are shown in Figure 2, where the peaks are as follows:
The peak of chemical shift 15.25ppm is C a ; the peak of chemical shift 15.39ppm is C h ; the peak of chemical shift 28.60ppm is C b ; the peak of chemical shift 31.02ppm is C g ; the peak of chemical shift 52.93ppm is C c ; The peak of chemical shift 56.19 ppm is C f ; the peak of chemical shift 176.11 ppm is C d ; the peak of chemical shift 181.85 ppm is C e .
PATENT
WO-2019161526




One-pot method for preparing twin dicarboxylic acid diamine complex platinum (II) derivatives ( dicycloplatin ) comprising the separation of intermediate carboplatin or carboplatin analogue.
For the preparation of bicycloplatin, CN1311183A, as the earliest publication of bicycloplatin and its preparation method, discloses the preparation of bicycloplatinum by the following method: carboplatin is dissolved in pure water at normal temperature, and then an equimolar amount of 1,1-ring is added. Butane dicarboxylic acid was evaporated to dryness after completion of the reaction, washed with ethanol, and recrystallized from distilled water. The method needs to completely evaporate the solvent water, which increases the risk of degradation of the bicyclic platinum, and also introduces more impurities into the crude bicycloplatinum. Therefore, ethanol washing and recrystallization are required, and the operation is cumbersome, and the yield of the bicyclic platinum is low.
[0015]
CN104693245A discloses a preparation method of bicyclo platinum, which is prepared by using carboplatin as a raw material in a ratio of 1:11 to 1,1-cyclobutanedicarboxylic acid in a molar ratio of 1:1, and is protected from light at 0-60 ° C. After -9 days, the excess water is removed by concentration under reduced pressure or freeze-drying to obtain a bicyclic platinum product. Although according to reports, the HPLC purity of the product is more than 99%, it requires a long standing process, is inefficient, and greatly increases the risk of carboplatin decomposition, especially for the process of amplification; In the final process, the solvent water is completely evaporated to make the bicyclic platinum product exist in a relatively high temperature aqueous solution for a long time, and the product has a high risk of degradation, and the quality stability is inevitably affected. In fact, bicycloplatinum with the reported yield and purity was not obtained according to this method.
[0016]
CN106132408A also discloses a process for the preparation of another bicyclic platinum in which carboplatin is mixed with a corresponding ratio of 1,1-cyclobutanedicarboxylic acid and a solvent to form a suspension, and the precipitated solid formed is separated from the suspension. Although the report states that the obtained product does not contain XRPD detectable amount of carboplatin, the suspension method uses a small amount of solvent, so that the product formed during the reaction is also precipitated as a solid, which is mixed with the unreacted raw material solid. This prevents the reaction from proceeding and makes the purification of the product more difficult. Especially in the case where the product is coated with carboplatin, the carboplatin can hardly be removed by purification. Therefore, the suspension method has the disadvantages of difficulty in control, poor operability, and incapability of industrial scale-up production. In fact, bicycloplatinum with the reported yield and purity cannot be obtained according to this method as well.
Notes
- ^ D., Zhao; Y., Zhang; C., Xu; C., Dong; H., Lin; L., Zhang; C., Li; S., Ren; X., Wang; S., Yang; D., Han; X., Chen (February 2012). “Pharmacokinetics, Tissue Distribution, and Plasma Protein Binding Study of Platinum Originating from Dicycloplatin, a Novel Antitumor Supramolecule, in Rats and Dogs by ICP-MS”. Biological Trace Element Research. 148 (2): 203–8. doi:10.1007/s12011-012-9364-2. PMID 22367705.
- ^ G.Q., Li; X.G., Chen; X.P., Wu; J.D., Xie; Y.J., Liang; X.Q., Zhao; W.Q, Chen; L.W., Fu (November 2012). “Effect of Dicycloplatin, a Novel Platinum Chemotherapeutical Drug, on Inhibiting Cell Growth and Inducing Cell Apoptosis”. PLOS ONE. 7 (11): e48994. Bibcode:2012PLoSO…748994L. doi:10.1371/journal.pone.0048994. PMC 3495782. PMID 23152837.
- ^ J.J, Yu; X.Q, Yang; Q.H, Song; M. D., Mueller; S. C., Remick (2014). “Dicycloplatin, a Novel Platinum Analog in Chemotherapy: Synthesis of Chinese Pre-clinical and Clinical Profile and Emerging Mechanistic Studies”. Anticancer Research. 34: 455–464.
- ^ Jump up to:a b c Guang-quan, Li; Xing-gui, Chen; Xing-ping, Wu; Jing-dun, Xie; Yong-ju, Liang; Xiao-qin, Zhao; Wei-qiang, Chen; Li-wu, Fu (November 2012). “Effect of Dicycloplatin, a Novel Platinum Chemotherapeutical Drug, on Inhibiting Cell Growth and Inducing Cell Apoptosis”. PLOS ONE. 7 (11): e48994. Bibcode:2012PLoSO…748994L. doi:10.1371/journal.pone.0048994. PMC 3495782. PMID 23152837.
- ^ Li.S; Huang H; Liao H; Zhan J; Guo Y; Zou BY; Jiang WQ; Guan ZZ; Yang XQ (2015). “Phase I clinical trial of the novel platin complex dicycloplatin: clinical and pharmacokinetic results”. International Journal of Clinical Pharmacology and Therapeutics. 51 (2): 96–105. doi:10.5414/CP201761. PMID 23127487.
- ^ Y., Xu Qing; J., Xiang Lin; S., Q.; TANG, Ka Luo; Y., Zhen Yun; Z., Xiao Feng; T., You Qi (June 2010). “Structural studies of dicycloplatin, an antitumor supramolecule”. Science China Chemistry. 53 (6): 1346–1351. doi:10.1007/s11426-010-3184-z.
- ^ R., Kumar; P.E., Herbert; A.N., Warrens (September 2005). “An introduction to death receptors in apoptosis”. International Journal of Surgery. 3 (4): 268–77. doi:10.1016/j.ijsu.2005.05.002. PMID 17462297.
- ^ Yang, BF; Xiao, C; Li, H; Yang, SJ (2007). “Resistance to Fas-mediated apoptosis in malignant tumours is rescued by KN-93 and cisplatin via downregulation of cFLIP expression and phosphorylation”. Clinical and Experimental Pharmacology and Physiology. 34 (12): 1245–51. doi:10.1111/j.1440-1681.2007.04711.x. PMID 17973862.
- ^ Blomgran, R; Zheng, L; Stendahl, O (2007). “Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization”. Journal of Leukocyte Biology. 81 (5): 1213–23. doi:10.1189/jlb.0506359. PMID 17264306.
- ^ Yin, XM (2006). “Bid, a BH3-only multi-functional molecule, is at the cross road of life and death”. Gene. 369: 7–19. doi:10.1016/j.gene.2005.10.038. PMID 16446060.
- ^ Ott, M; Gogvadze, V; Orrenius, S; Zhivotovsky, B (May 2007). “Mitochondria, oxidative stress and cell death”. Apoptosis. 12 (5): 913–22. doi:10.1007/s10495-007-0756-2. PMID 17453160.

Chemical structure of Dicycloplatin
|
|
| Clinical data | |
|---|---|
| Trade names | Dicycloplatin |
| Synonyms | Platinum(2+) 1-carboxycyclobutanecarboxylate ammoniate (1:2:2), 1,1-Cyclobutanedicarboxylic acid, compd. with (sp-4-2)-diammine(1,1-cyclobutanedi(carboxylato-kappaO)(2-))platinum (1:1) |
| Routes of administration |
Intravenous |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Protein binding | < 88.7% |
| Elimination half-life | 24.49 – 108.93 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C12H20N2O8Pt |
| Molar mass | 515.382 g/mol |
| 3D model (JSmol) |
|
/////////////Dicycloplatin
C1CC(C1)(C(=O)O)C(=O)O.C1CC(C1)(C(=O)[O-])C(=O)[O-].N.N.[Pt+2]
Octamoxin, октамоксин , أوكتاموكسين , 奥他莫辛 ,
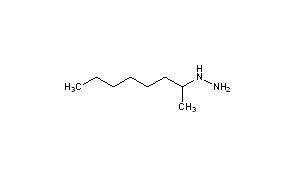
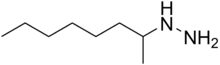
Octamoxin
- Molecular FormulaC8H20N2
- Average mass144.258 Da
Octan-2-ylhydrazine
Octomoxine
UNII:0HXY3M6S54
UNII:2NJ66SLA5C
UNII:895PL98ZMY
CAS Registry Number: 4684-87-1
CAS Name: (1-Methylheptyl)hydrazine
Additional Names: 2-hydrazinooctane; octomoxine
Trademarks: Ximaol
Molecular Formula: C8H20N2
Molecular Weight: 144.26
Percent Composition: C 66.61%, H 13.97%, N 19.42%
Literature References: Monoamine oxidase inhibitor. Prepd by condensation of methyl hexyl ketone and hydrazine hydrate followed by hydrogenation under pressure: Michel-Ber et al., GB 899385 (1962 to Soc. Civile Auguil).
Derivative Type: Sulfate
CAS Registry Number: 3845-07-6
Trademarks: Nimaol
Molecular Formula: C8H20N2.H2SO4
Molecular Weight: 242.34
Percent Composition: C 39.65%, H 9.15%, N 11.56%, S 13.23%, O 26.41%
Properties: Crystals, mp 78-80°.
Melting point: mp 78-80°
Therap-Cat: Antidepressant.
Keywords: Antidepressant; Hydrazides/Hydrazines; Monoamine Oxidase Inhibitor.
Octamoxin (trade names Ximaol, Nimaol), also known as 2-octylhydrazine, is an irreversible and nonselective monoamine oxidase inhibitor (MAOI) of the hydrazine class that was used as an antidepressant in the 1960s but is now no longer marketed.[2][3][4][5]
CLIP
OXIME TO AMINO TO PRODUCT

Zhurnal Russkago Fiziko-Khimicheskago Obshchestva1899vol. 31p. 878Ch emisches Zentralblatt 1900 vol. 71 Ip. 653
References
- ^ “Octamoxin – Compound Summary”. USA: National Center for Biotechnology Information. 26 March 2005. Identification and Related Records. Retrieved 31 May 2012.
- ^ “Dictionary of pharmacological agents – Google Books”.
- ^ “13-06781. Octamoxin [Archived]: The Merck Index”.
- ^ Levy J, Michel-Ber E (1966). “[Relations between the antidepressive effects of octamoxine revealed by 3 pharmacological tests and inhibition of cerebral monoamine oxidase in mice]”. Thérapie (in French). 21 (4): 929–45. PMID 5925088.
- ^ Gayral L, Stern H, Puyuelo R (1966). “[Indications and results of the treatment of mental depression by octamoxine (ximaol)]”. Thérapie (in French). 21 (5): 1183–90. PMID 5976767.
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
1-Methylheptylhydrazine[citation needed]
|
|
| Systematic IUPAC name
Octan-2-ylhydrazine[1]
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C8H20N2 | |
| Molar mass | 144.262 g·mol−1 |
| Density | 0.831 g/mL |
| Boiling point | 228 °C (442 °F; 501 K) |
| Pharmacology | |
| Oral | |
| Related compounds | |
|
Related compounds
|
Tuaminoheptane |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////////Octamoxin, Ximaol, Nimaol, 2-octylhydrazine, октамоксин , أوكتاموكسين , 奥他莫辛 ,
Labetalol Hydrochloride, ラベタロール ,

Labetalol
ラベタロール;
- Molecular FormulaC19H24N2O3
- Average mass328.405 Da
Labetalol hydrochloride, AH-5158A, Sch-15719W, Amipress, Trandate, Normodyne
Labetalol was granted FDA approval on 1 August 1984
Presolol; (RS)-2-Hydroxy-5-{1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl}benzamide; 5-[1-Hydroxy-2-[(1-methyl-3-phenyl propyl)amino]ethyl]salicylamide
A salicylamide derivative that is a non-cardioselective blocker of BETA-ADRENERGIC RECEPTORS and ALPHA-1 ADRENERGIC RECEPTORS.
253-258-3 [EINECS]
2-Hydroxy-5-{1-hydroxy-2-[(4-phenyl-2-butanyl)amino]ethyl}benzamide [ACD/IUPAC Name]
2-Hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide
36894-69-6 [RN]
Benzamide, 2-hydroxy-5-(1-hydroxy-2-((1-methyl-3-phenylpropyl)amino)ethyl)-
Benzamide, 2-hydroxy-5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]- [ACD/Index Name]
Dilevalol
Labetalol[Wiki]
labetolol
[32780-64-6]
[36894-69-6]
2948416[Beilstein]
2-Hydroxy-5-(1-hydroxy-2-((1-methyl-3-phenylpropyl)amino)ethyl)benzamide
- AH 5158
- Albetol
- EC 253-258-3
- EINECS 253-258-3
- HSDB 6537
- Ibidomide
- Labetalol
- Labetalolum
- Labetalolum [INN-Latin]
- Labetolol
- SCH 15719W
- UNII-R5H8897N95
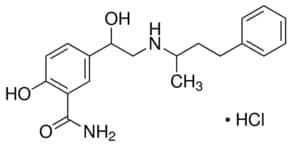
Labetalol hydrochloride
- CAS Number 32780-64-6,
- Empirical Formula (Hill Notation) C19H24N2O3 · HCl,
- Molecular Weight 364.87
REF https://www.accessdata.fda.gov/drugsatfda_docs/anda/98/74787_Labetalol%20Hydrochloride_Chemr.pdf

RR
CAS 75659-07-3
- (R,R)-Labetalol
- Dilevalol
- Dilevalolum
- Dilevalolum [Latin]
- UNII-P6629XE33T
Labetalol is a racemic mixture of 2 diastereoisomers where dilevalol, the R,R’ stereoisomer, makes up 25% of the mixture.8 Labetalol is formulated as an injection or tablets to treat hypertension
Labetalol is a medication used to treat high blood pressure and in long term management of angina.[1][2] This includes essential hypertension, hypertensive emergencies, and hypertension of pregnancy.[2] In essential hypertension it is generally less preferred than a number of other blood pressure medications.[1] It can be given by mouth or by injection into a vein.[1]
Common side effects include low blood pressure with standing, dizziness, feeling tired, and nausea.[1] Serious side effects may include low blood pressure, liver problems, heart failure, and bronchospasm.[1] Use appears safe in the latter part of pregnancy and it is not expected to cause problems during breastfeeding.[2][3] It works by blocking the activation of β-receptors and α-receptors.[1]
Labetalol was patented in 1966 and came into medical use in 1977.[4] It is available as a generic medication.[2] A month supply in the United Kingdom costs the NHS about 8 £ as of 2019.[2] In the United States the wholesale cost of this amount is about US$12.[5] In 2016 it was the 233rd most prescribed medication in the United States with more than 2
Medical uses
Labetalol is effective in the management of hypertensive emergencies, postoperative hypertension, pheochromocytoma-associated hypertension, and rebound hypertension from beta blocker withdrawal. [7]
It has a particular indication in the treatment of pregnancy-induced hypertension which is commonly associated with pre-eclampsia. [8]
It is also used as an alternative in the treatment of severe hypertension.[7]
Special populations
Pregnancy: studies in lab animals showed no harm to the baby. However, a comparable well-controlled study has not been performed in pregnant women.[9]
Nursing: breast milk has been shown to contain small amounts of labetalol (0.004% original dose). Prescribers should be cautious in the use of labetalol for nursing mothers.[9]
Pediatric: no studies have established safety or usefulness in this population.[9]
Geriatric: the elderly are more likely to experience dizziness when taking labetalol. Labetalol should be dosed with caution in the elderly and counseled on this side effect.[9]
Side effects
Common
- Neurologic: headache (2%), dizziness (11%) [9]
- Gastrointestinal: nausea (6%), dyspepsia (3%) [9]
- Cholinergic: nasal congestion (3%), ejaculation failure (2%) [9]
- Respiratory: dyspnea (2%) [9]
- Other: fatigue (5%), vertigo (2%), orthostatic hypotension [9]
Low blood pressure with standing is more severe and more common with IV formulation (58% vs 1%[9]) and is often the reason larger doses of the oral formulation cannot be used.[10]
Rare
- Fever [9]
- Muscle cramps [9]
- Dry eyes [9]
- Heart block [9]
- Hyperkalemia [9]
- Hepatotoxicity [9]
- Drug eruption similar to lichen planus[11]
- Hypersensitivity – which may result in a lethal respiratory distress[9]
Contraindications
Labetalol is contraindicated in people with overt cardiac failure, greater-than-first-degree heart block, severe bradycardia, cardiogenic shock, severe hypotension, anyone with a history of obstructive airway disease including asthma, and those with hypersensitivity to the drug.[12]
Chemistry
The minimum requirement for adrenergic agents is a primary or secondary amine separated from a substituted benzene ring by one or two carbons.[13] This configuration results in strong agonist activity. As the size of the substituent attached to the amine becomes greater, particularly with respect to a t-butyl group, then the molecule typically is found to have receptor affinity without intrinsic activity, and is, therefore, an antagonist.[13] Labetalol, with its 1-methyl-3-phenylpropyl substituted amine, is greater in size relative to a t-butyl group and therefore acts predominantly as an antagonist. The overall structure of labetalol is very polar. This was created by substituting the isopropyl group in the standard beta-blocker structure with an aralkyl group, including a carboxamide group on the meta position, and by adding a hydroxyl group on the para position.[14]
Labetalol has two chiral carbons and consequently exists as four stereoisomers.[15] Two of these isomers, the (S,S)- and (R,S)- forms are inactive. The third, the (S,R)-isomer, is a powerful α1 blocker. The fourth isomer, the (R,R)-isomer which is also known as dilevalol, is a mixed nonselective β blocker and selective α1 blocker.[14] Labetalol is typically given as a racemic mixture to achieve both alpha and beta receptor blocking activity.[16]
| Stereoisomers of labetalol | |
|---|---|
-Labetalol_Structural_Formula_V1.svg) (R,R)-Labetalol CAS number: 75659-07-3 |
-Labetalol_Structural_Formula_V1.svg) (S,S)-Labetalol CAS number: 83167-24-2 |
-Labetalol_Structural_Formula_V1.svg) (R,S)-Labetalol CAS number: 83167-32-2 |
-Labetalol_Structural_Formula_V1.svg) (S,R)-Labetalol CAS number: 83167-31-1 |
Labetalol acts by blocking alpha and beta adrenergic receptors, resulting in decreased peripheral vascular resistance without significant alteration of heart rate or cardiac output.
The β:α antagonism of labetalol is approximately 3:1.[17][18]
It is chemically designated in International Union of Pure and Applied Chemistry (IUPAC) nomenclature as 2-hydroxy-5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]benzamide monohydrochloride.[16][19]
Pharmacology
Mechanism of action
Labetalol’s dual alpha and beta adrenergic antagonism has different physiological effects in short- and long-term situations. In short-term, acute situations, labetalol decreases blood pressure by decreasing systemic vascular resistance with little effect on stroke volume, heart rate and cardiac output.[20] During long-term use, labetalol can reduce heart rate during exercise while maintaining cardiac output by an increase in stroke volume.[21]
Labetalol is a dual alpha (α1) and beta (β1/β2) adrenergic receptor blocker and competes with other Catecholamines for binding to these sites.[22] Its action on these receptors are potent and reversible.[12] Labetalol is highly selective for postsynaptic alpha1- adrenergic, and non-selective for beta-adrenergic receptors. It is about equipotent in blocking both beta1- and beta2- receptors.[14]
The amount of alpha to beta blockade depends on whether labetalol is administered orally or intravenously (IV). Orally, the ratio of alpha to β blockade is 1:3. Intravenously, alpha to β blockade ratio is 1:7.[14][12] Thus, the labetalol can be thought to be a beta-blocker with some alpha-blocking effects.[12][22][23] By comparison, labetalol is a weaker β-blocker than propranolol, and has a weaker affinity for alpha-receptors compared to Phentolamine.[14][22]
Labetalol possesses intrinsic sympathomimetic activity.[23] In particular, it is a partial agonist at beta2- receptors located in the vascular smooth muscle. Labetalol relaxes vascular smooth muscle by a combination of this partial beta2- agonism and through alpha1- blockade.[23][24] Overall, this vasodilatory effect can decrease blood pressure.[25]
Similar to local anesthetics and sodium channel blocking antiarrhythmics, labetalol also has membrane stabilizing activity.[23][26] By decreasing sodium entry, labetalol decreases action potential firing and thus has local anesthetic activity.[27]
Physiological action
The physiological effects of labetalol when administered acutely (intravenously) are not predictable solely by their receptor blocking effect, i.e. blocking beta1- receptors should decrease heart rate, but labetalol does not. When labetalol is given in acute situations, it decreases the peripheral vascular resistance and systemic blood pressure while having little effect on the heart rate, cardiac output and stroke volume, despite its alpha1-, beta1- and beta2- blocking mechanism.[20][21] These effects are mainly seen when the person is in the upright position.[25]
Long term labetalol use also has different effects from other beta-blocking drugs. Other beta-blockers, such as propranolol, persistently reduce cardiac output during exercise. The peripheral vascular resistance decreases when labetalol is first administered. Continuous labetalol use further decreases peripheral vascular resistance. However, during exercise, cardiac output remains the same due to a compensatory mechanism that increases stroke volume. Thus, labetalol is able to reduce heart rate during exercise while maintaining cardiac output by the increase in stroke volume.[21]
Pharmacokinetics
Labetalol, in animal models, was found to cross the blood-brain-barrier in only negligible amounts.[28]
History
Labetalol was the first drug created that combined both alpha- and beta- adrenergic receptor blocking properties. It was created to potentially fix the compensatory reflex issue that occurred when blocking a single receptor subtype, i.e. vasoconstriction after blocking beta-receptors or tachycardia after blocking alpha receptors. Because the reflex from blocking the single receptor subtypes acted to prevent the lowering of blood pressure, it was postulated that weak blocking of both alpha- and beta- receptors could work together to decrease blood pressure.[14][21]
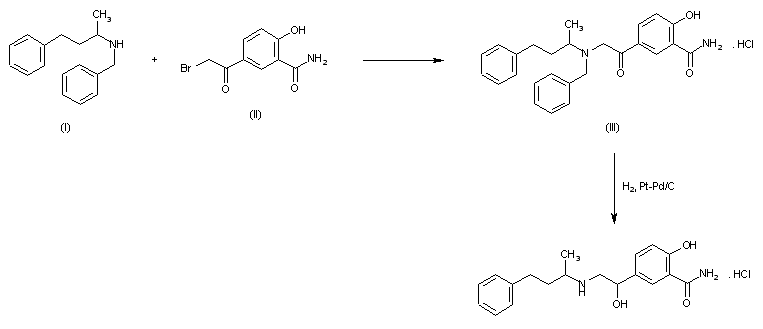
Syn 1
Drugs Fut 1976,1(3),125
DE 1643224; FR 1557677; FR 8010M; GB 1200886; US 3642896; US 3644353; US 3705233
Condensation of 5-bromoacetylsalicylamide (I) with N-benzyl-N-(1-methyl-3-phenylpropyl)amine (II) in refluxing butanone to 5-(N-benzyl-N-(1-methyl-3-phenylpropyl) glycyl)salicylamide hydrochloride (III), m.p. 139-141 C, which is reduced with H2 over Pt-Pd/C in ethanol.
SYN 2
Reductocondensation of 5-(N,N-dibenzylglycyl)salicylamide (IV) and benzylace-tone (V) with H2 over Pd-Pt/C in methanol – acetic acid.
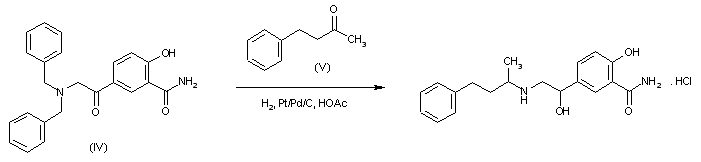
SYN 3
Reaction of methyl 5-(2-amino-1-hydroxyethyl)salicylate hydrochloride (VI) with NH3 to 5-(2-amino-1-hydroxyethyl)salicylamide hydrochloride (VII), m.p. >360 C, which is finally condensed with benzylacetone (V) and reduced with H2 over Pd-Pt/C in methanol.
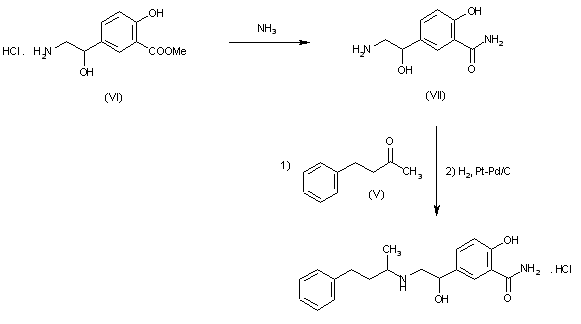
SYN 4

SYN 5
2-hydroxy-5-(1-hydroxy-2-((1-methyl-3-phenylpropyl)amino)ethyl)-, monohydrochloride, could be produced through many synthetic methods.
Following is one of the synthesis routes: 5-Bromoacetylsalicylamide (I) with N-benzyl-N-(1-methyl-3-phenylpropyl)amine (II) is condensed in the presence of refluxing butanone to produce 5-(N-benzyl-N-(1-methyl-3-phenylpropyl) glycyl)salicylamide hydrochloride (III), m.p. 139-141 C, and next the yielding compound is reduced with H2 over Pt-Pd/C in ethanol.

SYN 6
https://patents.google.com/patent/WO2017098520A1/en
aration of Labetaiol Hydrochloride of

Scheme -I illustrates the process for preparation of Labetaiol Hydrochloride of formula (I).

30% NaOH
Step – Sodium borohydride

Pure Labetaiol Hydrochloride (I)
aration of Labetaiol Hydrochloride of
Scheme -I illustrates the process for preparation of Labetaiol Hydrochloride of formula (I).
30% NaOH
Step – Sodium borohydride
Pure Labetaiol Hydrochloride (I)
SYN
https://patents.google.com/patent/EP0009702A1/en
-
The substance labetalol is known from British patent specification 1,266,058 and U.S.P. 4,012,444. Its pharmacological properties are discussed by Farmer et. al. in British Journal of Pharmacology, 45: 660-675 (1972), who designate it AH5158; it is shown to block a- and β-adrenergic receptors, suggesting that it would be useful in the treatment of arrhythmia, hypertension and angina pectoris.
- [0003]
The unique pharmacological properties of labetalol and its use as an antihypertensive agent are said to be largely a function of the exquisite balance of its a- and a-blocking activities. The file history of U.S.P. 4,012,444 indeed indicates that slight changes in the chemical structure of labetalol deleteriously affect this balance, and, even in the few analogous compounds where the balance is retained, the absolute potencies of these compounds are shown to be too low for them to be useful antihypertensive agents. Therefore, in the treatment of hypertension, labetalol is the compound of choice among those disclosed in British patent specification 1,266,058 and U.S.P. 4,012,444.
- [0004]
Labetalol has two asymmetrically substituted carbon atoms and therefore can exist as two diastereoisomers and four optical isomers. Indeed, British patent specification 1,266,058 and U.S.P. 4,012,444 disclose that compounds such as labetalol have optically active forms, but give no example of an optically active form. These patent specifications .teach that “the racemic mixtures may be resolved by conventional methods, for example by salt formation with an optically active acid, followed by fractional crystallization”, but give no method of resolution. Example 14 of each specifi– cation does indeed describe the separation of labetalol into two diastereoisomers “1” and “2”, using benzoic acid, but this is not an optical resolution. In British patent specifications 1,541,932 and 1,541,933, “isomer 1” is designated “diastereoisomer A” and is characterised as that diastereoisomer whose hydrochloride salt has the higher melting point. These two British patent specifications also disclose that diastereoisomer A is a valuable antiarrhythmic agent since it has strongly reduced β-adrenergic blocking activity and is therefore useful in the treatment of people who have suffered myocardial infarction.
- [0005]
We have now discovered that diastereoisomer A is composed of the (S,R) and (R,S) optical isomers of labetalol, whereas diastereoisomer B is composed of the (S,S) and (R,R) optical isomers. We have also-surprisingly found that the novel (R,R) optical isomer of labetalol exhibits, in comparison with labetalol itself, both an unexpectedly high increase in β-adrenergic blocking potency and a decrease in a-adrenergic blocking potency. Thus, when the (R,R) optical isomer is compared with labetalol, the ratio of the β-adrenergic blocking potency to the a-adrenergic blocking potency is found to be greatly and unexpectedly increased. In particular, animal tests have indicated that the (R,R) optical isomer has about twelve times the β-blocking potency of labetalol, but only about one third of the a-blocking potency of labetalol. These. properties could in no way have been predicted theoretically, especially as the β-blocking potency of diastereoisomer B is not significantly different from that of labetalol and the a-blocking potency of diastereoisomer B is half that of labetalol. Indeed, it is clear, when the activities of the four optical isomers of labetalol are compared, that the activities of the diastereoisomers A and B and indeed of labetalol itself cannot be calculated from the activities of their components. One can put this the other way around by saying that the α-and β-blocking activities of the four optical isomers of labetalol do not merely average to give the a- and β-blocking activites of labetalol and of its diastereoisomers A and B. Some of the activities are much greater than could ever have been expected on a simple basis of mathematical proportions, in particular the high β-blocking activity of the (R,R) optical isomer: this activity is much higher than the β-blocking activity of diastereoisomer B so that antagonism evidently exists between the (S,S) and (R,R) optical isomers with respect to the β-blocking activity. This degree of antagonism could in no way have been foreseen. In the absence of this antagonism, the (R,R) optical isomer shows a balance of properties that make it the optical isomer of choice in the treatment of hypertension. In particular, the (R,R) optical isomer possesses potent antihypertensive activity and rapid onset of activity while substantially lacking the undesirable side-effects usually associated with a-blockade, e.g. postural hypotension.
-
The following Table shows the relationships between labetalol, its diastereoisomersA and B and the four pure optical isomers; below each compound are given its potencies as an a-blocking and then as a β-blocking agent, all relative to the values for labetalol (assigned values 1.0 for each blocking activity):

This table clearly shows the unexpectedly high β-blocking activity and ratio of β-:α-blocking activities possessed by the (R,R)-optical isomer. Additionally, the (R,R)–optical isomer has been found to possess greater direct peripheral vasodilation activity than labetalol, and this also contributes to its anti-hypertensive activity. Moreover, the (R,R)-optical isomer is substantially non-toxic at therapeutic doses.
- [0007]
According to the invention therefore we provide the (R,R)-optical isomer of labetalol, namely 5- {(R)–1-hydroxy-2-[(R)-(1-methyl-3-phenylpropyl)amino]ethyl} salicylamide, which can be characterised by means of its hydrochloride salt which is dimorphic with m.pts. of about 133-134°C. and about 192-193.5°C. and an [α]D 26 of about -30.6° (conc. 1 mg./ml., ethanol), said (R,R) optical isomer being substantially free of the corresponding (R,S), (S,R) and (S,S) optical isomers
reaction scheme:

- E. (-)-5- { (R)-l-Hydroxy-2-[(R)-(l-methyl-3-phenylpropyl)-amino]ethyl} salicylamide hydrochloride salt (9)
- [0032]
Treat a solution of 3.0 g. (0.0059 mol.) of 2-0-benzyl-5-{(R) -1-hydroxy-2-[(R)-(1-methyl-3-phenylpropyl)benzylamino]ethyl} salicylamide in 30 ml. of ethyl ether with 2N ethereal hydrogen chloride until no further precipitation occurs. Wash the precipitated 2-0-benzyl-5-{(R)-1-hydroxy-2-[(R)-(1-methyl–3-phenylpropyl)benzylamino]ethyl} salicylamide hydrochloride with ether to remove excess hydrogen chloride and dissolve it in 100 ml. ethanol. To the ethanol solution add 300 mg. of a 20% palladium hydroxide on carbon catalyst and hydrogenate (3 atm.; 3.1 kg. cm.-2) in a Paar apparatus with shaking at room temperature for 3 hours. Filter off the catalyst, evaporate, and triturate the solid residue with isopropanol. Dissolve the solid in 11 ml. of 1N sodium hydroxide, adjust the pH to about 8 and precipitate the free base by bubbling in carbon dioxide. Collect the free base, wash it with water and dry it in vacuo at 40°C. Chromatograph the free base on 450 g. of silica gel and dissolve the pure product in 20 ml. of boiling acetonitrile. Cool the solution and carefully acidify with 2N ethereal HC1 to about pH2. Solidify the gum which precipitates by refluxing the mixture for 10 minutes, filter off the solid, wash it with ethyl ether and recrystallize it from ethanol to obtain analytically pure product (9), m.p. 192-193.5°C.(dec.), [α]D26 = -30.6° (c=1.0, ethanol).
Dilevalol
Synonyms:(R,R)-Labetalol
ATC:C02CB
- Use:α- and β-adrenoceptor antagonist, α- and β-blocker, isomer of labetalol, antihypertensive
- Chemical name:[R-(R*,R*)]-2-hydroxy-5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]benzamide
- Formula:C19H24N2O3
- MW:328.41 g/mol
- CAS-RN:75659-07-3
- LD50:1719 mg/kg (M, p.o.);
1228 mg/kg (R, p.o.)
Derivatives
Monohydrochloride
- Formula:C19H24N2O3 • HCl
- MW:364.87 g/mol
- CAS-RN:75659-08-4
- LD50:1079 mg/kg (M, p.o.);
82 mg/kg (R, i.v.); 1026 mg/kg (R, p.o.)
Synthesis Path
Labetalol
CAS Registry Number: 36894-69-6
CAS Name: 2-Hydroxy-5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]benzamide
Additional Names: 5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]salicylamide; ibidomide
Molecular Formula: C19H24N2O3
Molecular Weight: 328.41
Percent Composition: C 69.49%, H 7.37%, N 8.53%, O 14.62%
Literature References: Specific competitive antagonist at both a- and b-adrenergic receptor sites. Prepn: L. H. Lunts, D. T. Collin, DE2032642; eidem,US4012444 (1971, 1977 both to Allen & Hanburys). Synthesis of labetalol and enantiomers: J. E. Clifton et al.,J. Med. Chem.25, 670 (1982); and comparison of cardiovascular properties: E. H. Gold et al., ibid. 1363. Abs config of dilevalol: P. Murray-Rust et al.,Acta Crystallogr.C40, 825 (1984). Adrenoceptor blocking properties: E. J. Sybertz et al.,J. Pharmacol. Exp. Ther.218, 435 (1981). HPLC determn in serum or plasma: T. F. Woodman, B. Johnson, Ther. Drug Monit.3, 371 (1981). Metabolism in animals and man: R. Hopkins et al.,Biochem. Soc. Trans.4, 726 (1976). Toxicity: K. Shimpo et al.,Hokkaido Igaku Zasshi53, 15 (l978), C.A.90, 66465v (1974). Review of pharmacology: R. Donnelly, G. J. A. Macphee, Clin. Pharmacokinet.21, 95-109 (1991); of therapeutic applications in hypertension and ischemic heart disease: K. L. Goa et al.,Drugs37, 583-627 (1989).
Derivative Type: Hydrochloride
CAS Registry Number: 32780-64-6
Manufacturers’ Codes: AH-5158A; Sch-15719W
Trademarks: Amipress (Dox-Al); Ipolab (Finmedical); Labelol (ELEA); Labrocol (Lagap); Normodyne (Schering); Presdate (Alfa); Pressalolo (Locatelli); Trandate (Allen & Hanburys)
Molecular Formula: C19H24N2O3.HCl
Molecular Weight: 364.87
Percent Composition: C 62.54%, H 6.91%, N 7.68%, O 13.15%, Cl 9.72%
Properties: White crystalline solid from ethanol-ethyl acetate, mp 187-189°. Sol in water, ethanol. Insol in ether, chloroform. LD50in male, female mice, male, female rats (mg/kg): 114, 120, 113, 107 i.p.; 47, 54, 60, 53 i.v.; 1450, 1800, 4550, 4000 orally (Shimpo).
Melting point: mp 187-189°
Toxicity data: LD50 in male, female mice, male, female rats (mg/kg): 114, 120, 113, 107 i.p.; 47, 54, 60, 53 i.v.; 1450, 1800, 4550, 4000 orally (Shimpo)
Derivative Type: (R,R)-Form hydrochloride
CAS Registry Number: 75659-08-4; 75659-07-3 (free base)
Additional Names: Dilevalol hydrochloride
Manufacturers’ Codes: Sch-19927
Properties: Polymorphic crystals from ethanol, mp 133-134° (dec); mp 192-193.5° (dec). [a]D26 -30.6° (c = 1.0 in ethanol).
Melting point: mp 133-134° (dec); mp 192-193.5° (dec)
Optical Rotation: [a]D26 -30.6° (c = 1.0 in ethanol)
Therap-Cat: Antihypertensive.
Keywords: a-Adrenergic Blocker; ?Adrenergic Blocker; Antihypertensive; Arylethanolamine Derivatives.
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ləˈbɛtəlɔːl/ |
| Trade names | Normodyne, Trandate, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a685034 |
| Pregnancy category |
|
| Routes of administration |
By mouth, intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 25% |
| Protein binding | 50% |
| Metabolism | Liver pass metabolism, |
| Elimination half-life | Tablet: 6-8 hours; IV: 5.5 hours |
| Excretion | Excreted in urine, not removed by hemodialysis |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.048.401 |
| Chemical and physical data | |
| Formula | C19H24N2O3 |
| Molar mass | 328.412 g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
References
- ^ Jump up to:a b c d e f “Labetalol Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ Jump up to:a b c d e British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 147–148. ISBN 9780857113382.
- ^ “Labetalol Use During Pregnancy”. Drugs.com. Retrieved 11 March 2019.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 463. ISBN 9783527607495.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December 2018.
- ^ Jump up to:a b Koda-Kimble, Mary A.; Alldredge, Brian K. (2013). “21”. Koda-Kimble and Young’s Applied Therapeutic: The Clinical Use of Drugs. Philadelphia: Philadelphia: Lippincott Williams & Wilkins. ISBN 978-1-60913-713-7.
- ^ Arulkumaran, N; Lightstone, L (December 2013). “Severe pre-eclampsia and hypertensive crises”. Best Practice & Research. Clinical Obstetrics & Gynaecology. 27 (6): 877–84. doi:10.1016/j.bpobgyn.2013.07.003. PMID 23962474.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Trandate” (PDF). Prometheus Laboratories Inc. November 2010. Retrieved 3 November 2015.
- ^ “Labetalol hydrochloride” (PDF). Hospira. May 2015. Retrieved 3 November 2015.
- ^ Shiohara T, Kano Y (2007). “Lichen planus and lichenoid dermatoses”. In Bolognia JL (ed.). Dermatology. St. Louis: Mosby. p. 161. ISBN 978-1-4160-2999-1.
- ^ Jump up to:a b c d “Labetalol [package insert]. Spring Valley, NY: Par Pharmaceutical; 2011” (PDF). Retrieved 2015-11-03.
- ^ Jump up to:a b Medicinal Chemistry of Adrenergics and Cholinergics
- ^ Jump up to:a b c d e f Louis, W.J.; McNeill, JJ; Drummer, OH (1988). Doyle, AE (ed.). Labetalol and other vasodilator/Beta-blocking drugs. IN: Handbook of Hypertension. Amsterdam, Netherlands: Elsevier Sciences Publishing Co. pp. 246–273. ISBN 978-0-444-90469-0.
- ^ Riva E, Mennini T, Latini R (December 1991). “The alpha- and beta-adrenoceptor blocking activities of labetalol and its RR-SR (50:50) stereoisomers”. Br. J. Pharmacol. 104 (4): 823–8. doi:10.1111/j.1476-5381.1991.tb12513.x. PMC 1908821. PMID 1687367.
- ^ Jump up to:a b Robertson D, Biaggioni, I. Adrenoceptor Antagonist Drugs. In: Katzung BG, Masters SB, Trevor AJ, eds. Basic & Clinical Pharmacology. 12th ed. San Francisco, CA: McGraw Hill Lange Medical; 2012: 151-168. ISBN 978-0-07-176401-8.
- ^ Katzung, Bertram G. (2006). Basic and clinical pharmacology. New York: McGraw-Hill Medical. p. 170. ISBN 978-0-07-145153-6.
- ^ D A Richards; J Tuckman; B N Prichard (October 1976). “Assessment of alpha- and beta-adrenoceptor blocking actions of labetalol”. Br J Clin Pharmacol. 3 (5): 849–855. doi:10.1111/j.1365-2125.1976.tb00637.x. PMC 1428931. PMID 9968.
- ^ “labetalol | C19H24N2O3 – PubChem”. pubchem.ncbi.nlm.nih.gov. Retrieved 2015-11-04.
- ^ Jump up to:a b MacCarthy, E. P.; Bloomfield, S. S. (1983-08-01). “Labetalol: a review of its pharmacology, pharmacokinetics, clinical uses and adverse effects”. Pharmacotherapy. 3(4): 193–219. doi:10.1002/j.1875-9114.1983.tb03252.x. ISSN 0277-0008. PMID 6310529.
- ^ Jump up to:a b c d Louis, W. J.; McNeil, J. J.; Drummer, O. H. (1984-01-01). “Pharmacology of combined alpha-beta-blockade. I”. Drugs. 28 Suppl 2: 16–34. doi:10.2165/00003495-198400282-00003. ISSN 0012-6667. PMID 6151889.
- ^ Jump up to:a b c Robertson, D; Biaggioni, I (2012). Katzung, BG (ed.). Adrenoceptor Antagonist Drugs IN: Basic & Clinical Pharmacology (12 ed.). San Francisco: McGraw Hill Lange Medical. pp. 151–168. ISBN 978-0-07-176401-8.
- ^ Jump up to:a b c d Westfall, David P (2004). Craig, Charles R (ed.). Adrenoreceptor Antagonists IN: Modern Pharmacology with Clinical Applications (6th ed.). Baltimore, MD: Lippincott Williams & Wilkins. pp. 109–117. ISBN 978-0781737623.
- ^ Lund-Johansen, P. (1988-01-01). “Hemodynamic effects of beta-blocking compounds possessing vasodilating activity: a review of labetalol, prizidilol, and dilevalol”. Journal of Cardiovascular Pharmacology. 11 Suppl 2: S12–17. doi:10.1097/00005344-198800000-00004. ISSN 0160-2446. PMID 2464093.
- ^ Jump up to:a b Lund-Johansen, P. (1984-01-01). “Pharmacology of combined alpha-beta-blockade. II. Haemodynamic effects of labetalol”. Drugs. 28 Suppl 2: 35–50. doi:10.2165/00003495-198400282-00004. ISSN 0012-6667. PMID 6151890.
- ^ Mottram, Allan R.; Erickson, Timothy B. (2009). Field, John (ed.). Toxicology in Emergency Cardiovascular Care IN: The Textbook of Emergency Cardiovascular Care and CPR. Philadelphia, PA: Lippincott WIlliams & Wilkins. pp. 443–452. ISBN 978-0-7817-8899-1.
- ^ Exam Zone (1 January 2009). Elsevier Comprehensive Guide. Elsevier India. pp. 449–. ISBN 978-81-312-1620-0.
- ^ Detlev Ganten; Patrick J. Mulrow (6 December 2012). Pharmacology of Antihypertensive Therapeutics. Springer Science & Business Media. pp. 147–. ISBN 978-3-642-74209-5.
External links
References
-
- EP 9 702 (Schering Corp.; appl. 17.9.1979; USA-prior. 20.9.1978).
-
Improvement of diastereomer separation:
- DOS 2 616 403 (Scherico; appl. 14.4.1976; USA-prior. 17.4.1975).
- US 4 173 583 (Schering Corp.; 6.11.1979; appl. 21.9.1978; prior. 17.4.1975).
-
Synthesis without chromatographic purification:
- EP 92 787 (Schering Corp.; appl. 20.4.1983; USA-prior. 26.4.1982).
-
Chiral reduction of IV:
- Clifton, J.E. et al.: J. Med. Chem. (JMCMAR) 25, 670 (1982).
- Gold, E.H. et al.: J. Med. Chem. (JMCMAR) 25, 1363 (1982).
- EP 382 157 (Schering Corp.; appl. 6.2.1990; USA-prior. 10.2.1989, 26.9.1989).
- US 4 948 732 (Schering Corp.; 14.8.1990; prior. 26.9.1989, 10.2.1989).
///////////Labetalol hydrochloride, AH-5158A, Sch-15719W, Amipress, Trandate, Normodyne, ラベタロール , Dilevalol
Novobiocin, ノボビオシン;

Novobiocin
ノボビオシン;
- Molecular FormulaC31H36N2O11
- Average mass612.624 Da
(3R,4S,5R,6R)-5-hydroxy-6-(4-hydroxy-3-(4-hydroxy-3-(3-methylbut-2-enyl)benzamido)-8-methyl-2-oxo-2H-chromen-7-yloxy)-3-methoxy-2,2-dimethyltetrahydro-2H-pyran-4-yl carbamate
(3R,4S,5R,6R)-5-Hydroxy-6-[(4-hydroxy-3-{[4-hydroxy-3-(3-methyl-2-buten-1-yl)benzoyl]amino}-8-methyl-2-oxo-2H-chromen-7-yl)oxy]-3-methoxy-2,2-dimethyltetrahydro-2H-pyran-4-yl carbamate (non-preferred name) [ACD/IUPAC Name]
(3R,4S,5R,6R)-5-Hydroxy-6-[(4-hydroxy-3-{[4-hydroxy-3-(3-methyl-2-buten-1-yl)benzoyl]amino}-8-methyl-2-oxo-2H-chromen-7-yl)oxy]-3-methoxy-2,2-dimethyltetrahydro-2H-pyran-4-yl carbamate (non-preferred name)
(3R,4S,5R,6R)-5-Hydroxy-6-[(4-hydroxy-3-{[4-hydroxy-3-(3-methylbut-2-en-1-yl)benzoyl]amino}-8-methyl-2-oxo-2H-chromen-7-yl)oxy]-3-methoxy-2,2-dimethyltetrahydro-2H-pyran-4-yl carbamate (non-preferred name)
1476-53-5 [RN]
17EC19951N
216-023-6 [EINECS]
224-321-2 [EINECS]
575
Albamycin[Trade name]
Biotexin
UNII17EC19951N
CAS number303-81-1
WeightAverage: 612.6243
Monoisotopic: 612.231910004
Monoisotopic: 612.231910004
Chemical FormulaC31H36N2O11
For the treatment of infections due to staphylococci and other susceptible organisms
Novobiocin
CAS Registry Number: 303-81-1
CAS Name: N-[7-[[3-O-(Aminocarbonyl)-6-deoxy-5-C-methyl-4-O-methyl-b-L-lyxo-hexopyranosyl]oxy]-4-hydroxy-8-methyl-2-oxo-2H-1-benzopyran-3-yl]-4-hydroxy-3-(3-methyl-2-butenyl)benzamide
Additional Names: crystallinic acid; streptonivicin
Manufacturers’ Codes: PA-93; U-6591
Molecular Formula: C31H36N2O11
Molecular Weight: 612.62
Percent Composition: C 60.78%, H 5.92%, N 4.57%, O 28.73%
Literature References: Antibiotic substance produced by Streptomyces spheroides: Kaczka et al., J. Am. Chem. Soc. 77, 6404 (1955); Wolf, US 3000873 (1961 to Merck & Co.); Stammer, Miller; Miller; Wallick, US 3049475; US 3049476; US 3049534 (all 1962 to Merck & Co.). By Streptomyces niveus: Hoeksema et al., J. Am. Chem. Soc. 77, 6710 (1955); Antibiot. Chemother. 6, 143 (1956); French, US 3068221 (1962 to Upjohn). Structure: Shunk et al., J. Am. Chem. Soc. 78, 1770 (1956); Hoeksema et al., ibid.2019; Walton et al., ibid. 82, 1489 (1960). Conformation: Golding, Richards, Chem. Ind. (London) 1963, 1081. Revised configuration: O. Achmatowicz et al., Tetrahedron 32, 1051 (1976). Synthesis: Stammer, US 2925411 (1960); Walton, Spencer, US2966484 (1960 to Merck & Co.); Vaterlaus et al., Helv. Chim. Acta 47, 390 (1964). Conversion of isonovobiocin to novobiocin: Caron et al., US 2983723 (1961 to Upjohn). Antiviral activity: Chang, Weinstein, Antimicrob. Agents Chemother. 1970, 165. Efficacy in canine respiratory infections: B. W. Maxey, Vet. Med. Small Anim. Clin. 75, 89 (1980). Mechanism of action studies: Smith, Davis, J. Bacteriol. 93, 71 (1967); H. T. Wright et al., Science 213, 455 (1981); I. W. Althaus et al., J. Antibiot. 41, 373 (1988). Review: Brock in Antibiotics vol. 1, R. Gottlieb, P. Shaw, Eds. (Springer-Verlag, New York, 1967) pp 651-665; M. J. Ryan, ibid. vol. 5(pt. 1), F. E. Hahn, Ed. (1979) pp 214-234.
Properties: Pale yellow orthorhombic crystals from ethanol. Sensitive to light. d 1.3448. Dec at 152-156° (a rarer modification dec 174-178°). Acid reaction: pKa1 4.3; pKa2 9.1. [a]D24 -63.0° (c = 1 in ethanol). uv max (0.1N NaOH; 0.1N methanolic HCl; pH 7 phosphate buffer): 307; 324; 390 nm (E1%1cm 600, 390, 350 resp.). Sol in aq soln above pH 7.5. Practically insol in more acidic solns. Sol in acetone, ethyl acetate, amyl acetate, lower alcohols, pyridine. Additional soly data: Weiss et al., Antibiot. Chemother.7, 374 (1957).
pKa: pKa1 4.3; pKa2 9.1
Optical Rotation: [a]D24 -63.0° (c = 1 in ethanol)
Absorption maximum: uv max (0.1N NaOH; 0.1N methanolic HCl; pH 7 phosphate buffer): 307; 324; 390 nm (E1%1cm 600, 390, 350 resp.)
Density: d 1.3448
Derivative Type: Monosodium salt
CAS Registry Number: 1476-53-5
Trademarks: Albamycin (Pharmacia & Upjohn)
Molecular Formula: C31H35N2NaO11
Molecular Weight: 634.61
Percent Composition: C 58.67%, H 5.56%, N 4.41%, Na 3.62%, O 27.73%
Properties: Minute crystals, dec 220°. [a]D24 -38° (c = 2.5 in 95% ethanol); [a]D24 -33° (c = 2.5 in water). Freely sol in water. A 100 mg/ml soln has a pH of 7.5 and a half-life of ~30 days at 25° and several months at 4°. Soly data: Weiss et al., loc. cit. Properties: Birlova, Traktenberg, Antibiotiki 13, 997 (1968).
Optical Rotation: [a]D24 -38° (c = 2.5 in 95% ethanol); [a]D24 -33° (c = 2.5 in water)
Therap-Cat: Antibacterial.
Therap-Cat-Vet: Antimicrobial.
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Novobiocin sodium | Q9S9NQ5YIY | 1476-53-5 | WWPRGAYLRGSOSU-RNROJPEYSA-M |
Reata Pharmaceuticals Inc
Abgentis is investigating a novobiocin analog, GYR-12 (discovery), as a re-engineered, previously-marketed-but-uncompetitive (undisclosed) antibacterial compound inhibiting ATPase activity of DNA supercoiling GyrB/ParE, for the potential broad-spectrum treatment of bacterial infections, including multi-drug resistant Gram-negative infections. In April 2017, development was underway [1924695].
Novobiocin, also known as albamycin or cathomycin, is an aminocoumarin antibiotic that is produced by the actinomycete Streptomyces niveus, which has recently been identified as a subjective synonym for S. spheroides[1] a member of the order Actinobacteria. Other aminocoumarin antibiotics include clorobiocin and coumermycin A1.[2] Novobiocin was first reported in the mid-1950s (then called streptonivicin).[3][4]
It is active against Staphylococcus epidermidis and may be used to differentiate it from the other coagulase-negative Staphylococcus saprophyticus, which is resistant to novobiocin, in culture.
Novobiocin was licensed for clinical use under the tradename Albamycin (Pharmacia And Upjohn) in the 1960s. Its efficacy has been demonstrated in preclinical and clinical trials.[5][6] The oral form of the drug has since been withdrawn from the market due to lack of efficacy.[7] Novobiocin is an effective antistaphylococcal agent used in the treatment of MRSA.[8]
Mechanism of action
The molecular basis of action of novobiocin, and other related drugs clorobiocin and coumermycin A1 has been examined.[2][9][10][11][12] Aminocoumarins are very potent inhibitors of bacterial DNA gyrase and work by targeting the GyrB subunit of the enzyme involved in energy transduction. Novobiocin as well as the other aminocoumarin antibiotics act as competitive inhibitors of the ATPase reaction catalysed by GyrB. The potency of novobiocin is considerably higher than that of the fluoroquinolones that also target DNA gyrase, but at a different site on the enzyme. The GyrA subunit is involved in the DNA nicking and ligation activity.
Novobiocin has been shown to weakly inhibit the C-terminus of the eukaryotic Hsp90 protein (high micromolar IC50). Modification of the novobiocin scaffold has led to more selective Hsp90 inhibitors.[13] Novobiocin has also been shown to bind and activate the Gram-negative lipopolysaccharide transporter LptBFGC.[14][15]
Structure
Novobiocin is an aminocoumarin. Novobiocin may be divided up into three entities; a benzoic acid derivative, a coumarin residue, and the sugar novobiose.[9] X-ray crystallographic studies have found that the drug-receptor complex of Novobiocin and DNA Gyrase shows that ATP and Novobiocin have overlapping binding sites on the gyrase molecule.[16] The overlap of the coumarin and ATP-binding sites is consistent with aminocoumarins being competitive inhibitors of the ATPase activity.[17]
Structure–activity relationship
In structure activity relationship experiments it was found that removal of the carbamoyl group located on the novobiose sugar lead to a dramatic decrease in inhibitory activity of novobiocin.[17]
Biosynthesis
This aminocoumarin antibiotic consists of three major substituents. The 3-dimethylallyl-4-hydroxybenzoic acid moiety, known as ring A, is derived from prephenate and dimethylallyl pyrophosphate. The aminocoumarin moiety, known as ring B, is derived from L-tyrosine. The final component of novobiocin is the sugar derivative L-noviose, known as ring C, which is derived from glucose-1-phosphate. The biosynthetic gene cluster for novobiocin was identified by Heide and coworkers in 1999 (published 2000) from Streptomyces spheroidesNCIB 11891.[18] They identified 23 putative open reading frames (ORFs) and more than 11 other ORFs that may play a role in novobiocin biosynthesis.
The biosynthesis of ring A (see Fig. 1) begins with prephenate which is a derived from the shikimic acid biosynthetic pathway. The enzyme NovF catalyzes the decarboxylation of prephenate while simultaneously reducing nicotinamide adenine dinucleotide phosphate (NADP+) to produce NADPH. Following this NovQ catalyzes the electrophilic substitution of the phenyl ring with dimethylallyl pyrophosphate (DMAPP) otherwise known as prenylation.[19] DMAPP can come from either the mevalonic acid pathway or the deoxyxylulose biosynthetic pathway. Next the 3-dimethylallyl-4-hydroxybenzoate molecule is subjected to two oxidative decarboxylations by NovR and molecular oxygen.[20] NovR is a non-heme iron oxygenase with a unique bifunctional catalysis. In the first stage both oxygens are incorporated from the molecular oxygen while in the second step only one is incorporated as determined by isotope labeling studies. This completes the formation of ring A.

Figure 1. Biosynthetic scheme of benzamide portion of novobiocin (4-hydroxy-3-(3-methylbut-2-en-1-yl)benzoic acid)
The biosynthesis of ring B (see Fig. 2) begins with the natural amino acid L-tyrosine. This is then adenylated and thioesterified onto the peptidyl carrier protein (PCP) of NovH by ATPand NovH itself.[21] NovI then further modifies this PCP bound molecule by oxidizing the β-position using NADPH and molecular oxygen. NovJ and NovK form a heterodimer of J2K2 which is the active form of this benzylic oxygenase.[22] This process uses NADP+ as a hydride acceptor in the oxidation of the β-alcohol. This ketone will prefer to exist in its enol tautomer in solution. Next a still unidentified protein catalyzes the selective oxidation of the benzene (as shown in Fig. 2). Upon oxidation this intermediate will spontaneously lactonize to form the aromatic ring B and lose NovH in the process.

Figure 2. Biosynthesis of 3-amino-4,7-dihydroxy-2H-chromen-2-one component of novobiocin (ring B)
The biosynthesis of L-noviose (ring C) is shown in Fig. 3. This process starts from glucose-1-phosphate where NovV takes dTTP and replaces the phosphate group with a dTDP group. NovT then oxidizes the 4-hydroxy group using NAD+. NovT also accomplishes a dehydroxylation of the 6 position of the sugar. NovW then epimerizes the 3 position of the sugar.[23] The methylation of the 5 position is accomplished by NovU and S-adenosyl methionine (SAM). Finally NovS reduces the 4 position again to achieve epimerization of that position from the starting glucose-1-phosphate using NADH.

Figure 3. Biosynthesis of L-noviose component of novobiocin (ring C)
Rings A, B, and C are coupled together and modified to give the finished novobiocin molecule. Rings A and B are coupled together by the enzyme NovL using ATP to diphosphorylate the carboxylate group of ring A so that the carbonyl can be attacked by the amine group on ring B. The resulting compound is methylated by NovO and SAM prior to glycosylation.[24] NovM adds ring C (L-noviose) to the hydroxyl group derived from tyrosine with the loss of dTDP. Another methylation is accomplished by NovP and SAM at the 4 position of the L-noviose sugar.[25] This methylation allows NovN to carbamylate the 3 position of the sugar as shown in Fig. 4 completing the biosynthesis of novobiocin.

Figure 4. Completed biosynthesis of novobiocin from ring systems A, B, and C.
CLIP
CLIP

CLIP

CLIP

PATENT
US-20190241599
Novel co-crystal forms of novobiocin and its analogs and proline, processes for their preparation and compositions comprising them are claimed. Also claims are methods for inhibiting heat shock protein 90 and treating or preventing neurodegenerative disorders, such as diabetic peripheral neuropathy.
References
- ^ Lanoot B, Vancanneyt M, Cleenwerck I, Wang L, Li W, Liu Z, Swings J (May 2002). “The search for synonyms among streptomycetes by using SDS-PAGE of whole-cell proteins. Emendation of the species Streptomyces aurantiacus, Streptomyces cacaoi subsp. cacaoi, Streptomyces caeruleus and Streptomyces violaceus”. International Journal of Systematic and Evolutionary Microbiology. 52 (Pt 3): 823–9. doi:10.1099/ijs.0.02008-0. PMID 12054245.
- ^ Jump up to:a b Alessandra da Silva Eustáquio (2004) Biosynthesis of aminocoumarin antibiotics in Streptomyces: Generation of structural analogues by genetic engineering and insights into the regulation of antibiotic production. DISSERTATION
- ^ Hoeksema H.; Johnson J. L.; Hinman J. W. (1955). “Structural studies on streptonivicin, a new antibiotic”. J Am Chem Soc. 77 (24): 6710–6711. doi:10.1021/ja01629a129.
- ^ Smith C. G.; Dietz A.; Sokolski W. T.; Savage G. M. (1956). “Streptonivicin, a new antibiotic. I. Discovery and biologic studies”. Antibiotics & Chemotherapy. 6: 135–142.
- ^ Raad I, Darouiche R, Hachem R, Sacilowski M, Bodey GP (November 1995). “Antibiotics and prevention of microbial colonization of catheters”. Antimicrobial Agents and Chemotherapy. 39 (11): 2397–400. doi:10.1128/aac.39.11.2397. PMC 162954. PMID 8585715.
- ^ Raad II, Hachem RY, Abi-Said D, Rolston KV, Whimbey E, Buzaid AC, Legha S (January 1998). “A prospective crossover randomized trial of novobiocin and rifampin prophylaxis for the prevention of intravascular catheter infections in cancer patients treated with interleukin-2”. Cancer. 82 (2): 403–11. doi:10.1002/(SICI)1097-0142(19980115)82:2<412::AID-CNCR22>3.0.CO;2-0. PMID 9445199.
- ^ “Determination That ALBAMYCIN (Novobiocin Sodium) Capsule, 250 Milligrams, Was Withdrawn From Sale for Reasons of Safety or Effectiveness”. The Federal Register. 19 January 2011.
- ^ Walsh TJ, Standiford HC, Reboli AC, John JF, Mulligan ME, Ribner BS, Montgomerie JZ, Goetz MB, Mayhall CG, Rimland D (June 1993). “Randomized double-blinded trial of rifampin with either novobiocin or trimethoprim-sulfamethoxazole against methicillin-resistant Staphylococcus aureus colonization: prevention of antimicrobial resistance and effect of host factors on outcome”. Antimicrobial Agents and Chemotherapy. 37 (6): 1334–42. doi:10.1128/aac.37.6.1334. PMC 187962. PMID 8328783.
- ^ Jump up to:a b Maxwell A (August 1993). “The interaction between coumarin drugs and DNA gyrase”. Molecular Microbiology. 9 (4): 681–6. doi:10.1111/j.1365-2958.1993.tb01728.x. PMID 8231802.
- ^ Maxwell A (February 1999). “DNA gyrase as a drug target”. Biochemical Society Transactions. 27 (2): 48–53. doi:10.1042/bst0270048. PMID 10093705.
- ^ Lewis RJ, Tsai FT, Wigley DB (August 1996). “Molecular mechanisms of drug inhibition of DNA gyrase”. BioEssays. 18 (8): 661–71. doi:10.1002/bies.950180810. PMID 8760340.
- ^ Maxwell A, Lawson DM (2003). “The ATP-binding site of type II topoisomerases as a target for antibacterial drugs”. Current Topics in Medicinal Chemistry. 3 (3): 283–303. doi:10.2174/1568026033452500. PMID 12570764.
- ^ Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BS (September 2005). “Hsp90 inhibitors identified from a library of novobiocin analogues”. Journal of the American Chemical Society. 127 (37): 12778–9. doi:10.1021/ja0535864. PMID 16159253.
- ^ Mandler MD, Baidin V, Lee J, Pahil KS, Owens TW, Kahne D (June 2018). “Novobiocin Enhances Polymyxin Activity by Stimulating Lipopolysaccharide Transport”. Journal of the American Chemical Society. 140 (22): 6749–6753. doi:10.1021/jacs.8b02283. PMC 5990483. PMID 29746111.
- ^ May JM, Owens TW, Mandler MD, Simpson BW, Lazarus MB, Sherman DJ, Davis RM, Okuda S, Massefski W, Ruiz N, Kahne D (December 2017). “The Antibiotic Novobiocin Binds and Activates the ATPase That Powers Lipopolysaccharide Transport”. Journal of the American Chemical Society. 139 (48): 17221–17224. doi:10.1021/jacs.7b07736. PMC 5735422. PMID 29135241.
- ^ Tsai FT, Singh OM, Skarzynski T, Wonacott AJ, Weston S, Tucker A, Pauptit RA, Breeze AL, Poyser JP, O’Brien R, Ladbury JE, Wigley DB (May 1997). “The high-resolution crystal structure of a 24-kDa gyrase B fragment from E. coli complexed with one of the most potent coumarin inhibitors, clorobiocin”. Proteins. 28 (1): 41–52. doi:10.1002/(sici)1097-0134(199705)28:1<41::aid-prot4>3.3.co;2-b. PMID 9144789.
- ^ Jump up to:a b Flatman RH, Eustaquio A, Li SM, Heide L, Maxwell A (April 2006). “Structure-activity relationships of aminocoumarin-type gyrase and topoisomerase IV inhibitors obtained by combinatorial biosynthesis”. Antimicrobial Agents and Chemotherapy. 50 (4): 1136–42. doi:10.1128/AAC.50.4.1136-1142.2006. PMC 1426943. PMID 16569821.
- ^ Steffensky M, Mühlenweg A, Wang ZX, Li SM, Heide L (May 2000). “Identification of the novobiocin biosynthetic gene cluster of Streptomyces spheroides NCIB 11891”. Antimicrobial Agents and Chemotherapy. 44 (5): 1214–22. doi:10.1128/AAC.44.5.1214-1222.2000. PMC 89847. PMID 10770754.
- ^ Pojer F, Wemakor E, Kammerer B, Chen H, Walsh CT, Li SM, Heide L (March 2003). “CloQ, a prenyltransferase involved in clorobiocin biosynthesis”. Proceedings of the National Academy of Sciences of the United States of America. 100 (5): 2316–21. Bibcode:2003PNAS..100.2316P. doi:10.1073/pnas.0337708100. PMC 151338. PMID 12618544.
- ^ Pojer F, Kahlich R, Kammerer B, Li SM, Heide L (August 2003). “CloR, a bifunctional non-heme iron oxygenase involved in clorobiocin biosynthesis”. The Journal of Biological Chemistry. 278 (33): 30661–8. doi:10.1074/jbc.M303190200. PMID 12777382.
- ^ Chen H, Walsh CT (April 2001). “Coumarin formation in novobiocin biosynthesis: beta-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI”. Chemistry & Biology. 8 (4): 301–12. doi:10.1016/S1074-5521(01)00009-6. PMID 11325587.
- ^ Pacholec M, Hillson NJ, Walsh CT (September 2005). “NovJ/NovK catalyze benzylic oxidation of a beta-hydroxyl tyrosyl-S-pantetheinyl enzyme during aminocoumarin ring formation in novobiocin biosynthesis”. Biochemistry. 44 (38): 12819–26. CiteSeerX 10.1.1.569.1481. doi:10.1021/bi051297m. PMID 16171397.
- ^ Thuy TT, Lee HC, Kim CG, Heide L, Sohng JK (April 2005). “Functional characterizations of novWUS involved in novobiocin biosynthesis from Streptomyces spheroides”. Archives of Biochemistry and Biophysics. 436 (1): 161–7. doi:10.1016/j.abb.2005.01.012. PMID 15752721.
- ^ Pacholec M, Tao J, Walsh CT (November 2005). “CouO and NovO: C-methyltransferases for tailoring the aminocoumarin scaffold in coumermycin and novobiocin antibiotic biosynthesis”. Biochemistry. 44 (45): 14969–76. doi:10.1021/bi051599o. PMID 16274243.
- ^ Freel Meyers CL, Oberthür M, Xu H, Heide L, Kahne D, Walsh CT (January 2004). “Characterization of NovP and NovN: completion of novobiocin biosynthesis by sequential tailoring of the noviosyl ring”. Angewandte Chemie. 43 (1): 67–70. doi:10.1002/anie.200352626. PMID 14694473.
External links
- Novobiocin bound to proteins in the PDB
 |
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
intravenous |
| ATCvet code | |
| Pharmacokinetic data | |
| Bioavailability | negligible oral bioavailability |
| Metabolism | excreted unchanged |
| Elimination half-life | 6 hours |
| Excretion | renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.005.589 |
| Chemical and physical data | |
| Formula | C31H36N2O11 |
| Molar mass | 612.624 g·mol−1 |
| 3D model (JSmol) | |
4309-70-0 CAS
calcium;7-[(2R,3R,4S,5R)-4-carbamoyloxy-3-hydroxy-5-methoxy-6,6-dimethyloxan-2-yl]oxy-3-[[4-hydroxy-3-(3-methylbut-2-enyl)benzoyl]amino]-8-methyl-2-oxochromen-4-olate
///////// Novobiocin, ノボビオシン , Antibacterial, Antimicrobial, crystallinic acid, streptonivicin,
Manufacturers’ Codes: PA-93; U-6591
History
Novobiocin is a coumarin antibiotic obtained from Streptomyces niveus and other Streptomyces species. Novobiocin is useful primarily in infections involving staphylococci, and other gram-positive organisms. It acts by inhibiting the initiation of DNA replication in bacterial and mammanlian cells. Evidences indicated that Novobiocin blocks prokaryotic DNA gyrase and eukaryotic II topoisomerase, enzymes that relax super-coiled DNA and are crucial for DNA replication.1
Novobiocin

| UIPAC Name | 4-Hydroxy-3-4-hydroxy-3-(3-methylbut-2-enyl)benzamido-8-methylcoumarin-7-yl 3-O-carbamoyl-5,5-di-C-methyl-α-l-lyxofuranoside |
| CAS Number | 303-81-1 |
| Molecular Mass | 612.624 g / mol |
| Chemical Formular | C31H36N2O11 |
Biosynthesis
The substituted coumarin (ring B, red) and the 4-OH benzoyl moiety (ring A, aqua) in novobiocin were derived from -Tyr based on earlier labeling studies. β-OH-Tyr is proposed to be a common intermediate in these two biosynthetic pathways.2

NovH is a -Tyr specific didomain NRPS that generates the
-tyrosyl-S-NovH intermediate. NovH, isolated from E. coli is primed by a PPTase with CoA. The A domain activates
-Tyr as
-tyrosyl-AMP and then transfers the
-tyrosyl group to the HS-pant-PCP domain of NovH through thioester formation.3

-tyrosyl-S-NovH is then function as a cytochrome P450 monooxygenase that hydroxylates the β-carbon of the tethered
-tyrosyl group on NovH. While the substrate
-tyrosyl-S-NovH provides two electrons for a single round of the hydroxylation reaction, the other two electrons needed to reduce the oxygen atom are provided by NADPH via two-electron transfer effected by electron transfer proteins ferrodoxin (Fd) and ferrodoxin reductase (Fd Red).3 The electron transfer route is from NADPH→FAD in Fd Red→Fe–S center in Fd→Heme in NovI→oxygen.

Both NovJ and NovK are similar to 3-keto-ACP reductase and they may form a heterodimer and operate in the reverse direction to oxidize 3-OH to 3-keto. NovO is similar to some quinone C-methyltransferases 3 but the timing of methylation is not clear. NovC resembles flavin-dependent monooxygenases (35 and 32% similarity to dimethylaniline and cyclohexanone monooxygenases, respectively) 3 and is proposed to hydroxylate the ortho position of the phenyl ring. The nucleophilic attack of the ortho hydroxyl group on the thioester carbonyl center would release the coumarin ring and regenerate NovH. Ring B is then synthesized.

Synthesis





Mechanism of action
E.Coli DNA gyrase utilizes ATP to catalyze the negative supercoiling, or under-twisting, of duplex DNA. The energy coupling components of the supercoiling reaction includes 1) the DNA-dependent hydrolysis that converts ATP to ADP and Pi, and 2) the gyrase cleavage reaction that targets the specified DNA site. The two activities are induced by treating the stable gyrase-DNA complex trapped by the inihibitor oxolinic acid with sodium dodecyl sulfate (SDS or Sulphate). 4 Novobiocin competes with ATP in the ATPase and supercoiling assays, hence Novobiocin prevents the ATP from shifting the primary cleavage site on ColE1 DNA by places the site of action of the antibiotics at a reaction step prior to ATP hydrolysis and blocks the binding of ATP. 4 Such a simple mechanism of action represents for all effects of the drugs on DNA gyrase.
Clinical Use
Due to factors as low solubility, poor pharmacokinetics, and limited activity agasinst Gram-negative bacteria, the clinical usage of Novobiocin is not achieved. 5 Therefore, it is of interest to study the novobiocin biosynthetic pathway in order to generate analogs with enhanced solubility and pharmacokinetic properties while maintaining the gyrase inhibitory properties.
References
1 J.C. D’Halluin, M. Milleville, and P. Boulanger. “Effect of Novobiocin on adenovirus DNA synthesis and encapsidation”. Nucleic Acids Research 1980; 8: 1625-1641
2 M. Steffensky, S.M. Li and L. Heide, “Cloning, overexpression, and purification of novobiocic acid synthetase from Streptomyces spheroides ” NCIB 11891. J. Biol. Chem. 275 (2000), pp. 21754–21760.
3 Huawei Chen and Christopher T. Walsh, “Coumarin formation in novobiocin biosynthesis: β-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI” Chemistry and Biology; 2001; 8: 301-312
4 K. Scheirer and N. P. Higgins. “The DAN Cleavage Reaction of DNA Gyrase ” The Journal of Biological Chemistry; 1997; 272 (43): 27202-27209
5 N Pi, C. L. F. Meyers, M. Pacholec, C. T. Walsh, and J. A. Leary. “Mass spectrometric characterization of a three-enzyme tandem reacton for assembly and modification of the novobiocin skeleton” PNAS 2004;101;10036-10041
Imipenem, イミペネム水和物
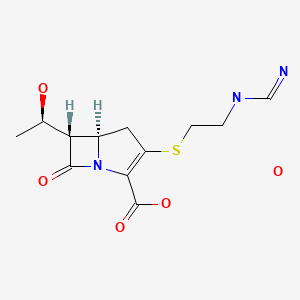
Imipenem
イミペネム水和物
Cas 74431-23-5
- Molecular FormulaC12H19N3O5S
- Average mass317.361 Da
(5R,6S)-3-((2-(Formimidoylamino)ethyl)thio)-6-((R)-1-hydroxyethyl)-7-oxo-1-azabicyclo(3.2.0)hept-2-ene-2-carboxylic acid monohydrate
1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid, 6-[(1R)-1-hydroxyethyl]-3-[[2-[(iminomethyl)amino]ethyl]thio]-7-oxo-, (5R,6S)-, monohydrate
264-734-5 [EINECS]
74431-23-5 [RN]
N-Formimidoylthienamycin Monohydrate
Primaxin monohydrate
Tienam monohydrate
(5R,6S)-3-((2-Formimidamidoethyl)thio)-6-((R)-1-hydroxyethyl)-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid hydrate
(5R,6S)-3-[2-(aminomethylideneamino)ethylsulfanyl]-6-[(1R)-1-hydroxyethyl]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid and hydrate
8174596 [Beilstein]
imipemide monohydrate
Antibacterial, Cell wall biosynthesis inhibitor
Imipenem
CAS Registry Number: 74431-23-5; 64221-86-9 (anhydrous)
CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-3-[[2-[(iminomethyl)amino]ethyl]thio]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid monohydrate
Additional Names: N-formimidoylthienamycin monohydrate; imipemide
Manufacturers’ Codes: MK-787
Molecular Formula: C12H17N3O4S.H2O
Molecular Weight: 317.36
Percent Composition: C 45.41%, H 6.03%, N 13.24%, O 25.21%, S 10.10%
Literature References: Extremely broad-spectrum semi-synthetic antibiotic; first stable derivative of thienamycin, q.v. Prepn: W. J. Leanza et al., J. Med. Chem. 22, 1435 (1979); T. W. Miller, EP 6639 (1980 to Merck & Co.), C.A. 93, 155845y (1980); B. G. Christensen et al., US 4194047 (1980 to Merck & Co.). Totally synthetic prepn without formation of thienamycin: I. Shinkai et al.,Tetrahedron Lett. 23, 4903 (1982). HPLC determn in serum: C. M. Myers, J. L. Blumer, Antimicrob. Agents Chemother. 26, 78 (1984). Series of articles on in vitro activity, pharmacokinetics, clinical efficacy of combination with cilastatin sodium, q.v., a renal dehydropeptidase I inhibitor: J. Antimicrob. Chemother. 12, Suppl. D, 1-155 (1983); Rev. Infect. Dis. 7, Suppl. 3, S389-S536 (1985); Am. J. Med. 78, Suppl. 6A, 1-167 (1985); Infection 14, Suppl. 2, S111-S180 (1986). Comprehensive description: E. R. Oberholtzer, Anal. Profiles Drug Subs. 17, 73-114 (1988).
Properties: Crystals from water-ethanol. [a]D25 +86.8° (c = 0.05 in 0.1M phosphate, pH 7). pKa1 ~3.2, pKa2 ~9.9. uv max (water): 299 nm (e 9670, 98% NH2OH ext). Soly (mg/ml): water 10, methanol 5, ethanol 0.2, acetone <0.1, dimethylformamide <0.1, dimethylsulfoxide 0.3.
pKa: pKa1 ~3.2, pKa2 ~9.9
Optical Rotation: [a]D25 +86.8° (c = 0.05 in 0.1M phosphate, pH 7)
Absorption maximum: uv max (water): 299 nm (e 9670, 98% NH2OH ext)
Derivative Type: Combination with cilastatin sodium
CAS Registry Number: 85960-17-4
Trademarks: Imipem (Neopharmed); Primaxin (Merck & Co.); Tenacid (Sigma-Tau); Tienam (Merck & Co.); Zienam (Merck & Co.)
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Carbapenems.
Imipenem (Primaxin among others) is an intravenous β-lactam antibiotic discovered by Merck scientists Burton Christensen, William Leanza, and Kenneth Wildonger in the mid-1970s.[1] Carbapenems are highly resistant to the β-lactamase enzymes produced by many multiple drug-resistant Gram-negative bacteria,[2] thus play a key role in the treatment of infections not readily treated with other antibiotics.[3]
Imipenem was patented in 1975 and approved for medical use in 1985.[4] It was discovered via a lengthy trial-and-error search for a more stable version of the natural product thienamycin, which is produced by the bacterium Streptomyces cattleya. Thienamycin has antibacterial activity, but is unstable in aqueous solution, so impractical to administer to patients.[5] Imipenem has a broad spectrum of activity against aerobic and anaerobic, Gram-positive and Gram-negative bacteria.[6] It is particularly important for its activity against Pseudomonas aeruginosa and the Enterococcus species. It is not active against MRSA, however.
Medical uses
Spectrum of bacterial susceptibility and resistance
Acinetobacter anitratus, Acinetobacter calcoaceticus, Actinomyces odontolyticus, Aeromonas hydrophila, Bacteroides distasonis, Bacteroides uniformis, and Clostridium perfringens are generally susceptible to imipenem, while Acinetobacter baumannii, some Acinetobacter spp., Bacteroides fragilis, and Enterococcus faecalis have developed resistance to imipenem to varying degrees. Not many species are resistant to imipenem except Pseudomonas aeruginosa (Oman) and Stenotrophomonas maltophilia.[7]
Coadministration with cilastatin
Imipenem is rapidly degraded by the renal enzyme dehydropeptidase 1 when administered alone, and is almost always coadministered with cilastatin to prevent this inactivation[8]
Adverse effects
Common adverse drug reactions are nausea and vomiting. People who are allergic to penicillin and other β-lactam antibiotics should take caution if taking imipenem, as cross-reactivity rates are high. At high doses, imipenem is seizurogenic.[9]
Mechanism of action
Imipenem acts as an antimicrobial through inhibiting cell wall synthesis of various Gram-positive and Gram-negative bacteria. It remains very stable in the presence of β-lactamase (both penicillinase and cephalosporinase) produced by some bacteria, and is a strong inhibitor of β-lactamases from some Gram-negative bacteria that are resistant to most β-lactam antibiotics.
SYM
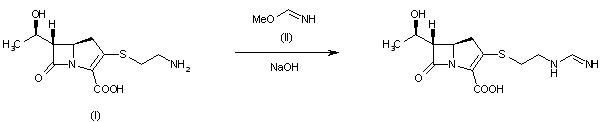
By reaction of thienamycin (I) with methyl formimidate (II) by means of NaOH in water.
| DE 2652679; FR 2332012; GB 1570990; NL 7612939 |
SYN 2
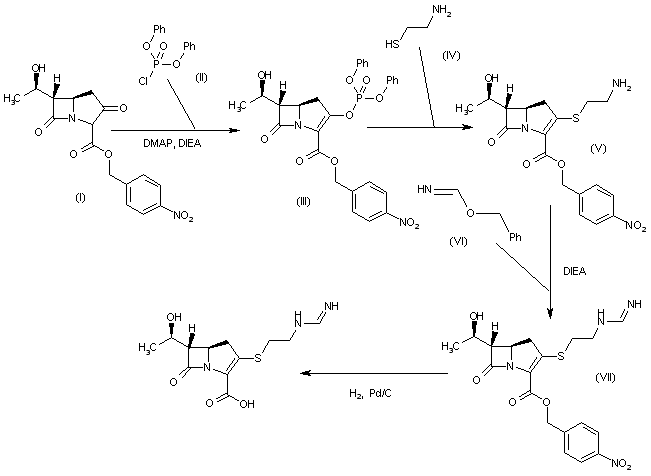
WO 0294828
The reaction of (3R,5R,6S)-6-(1(R)-hydroxyethyl)-2-oxo-1-carbapenem-3-carboxylic acid p-nitrobenzyl ester (I) with diphenyl chlorophosphate by (II) means of DMAP and DIEA in DMA/dichloromethane gives the enol phosphate (III), which is condensed with 2-aminoethanethiol (IV) in DMA to yield the 2-aminoethylsulfanyl derivative (V). The reaction of (V) with benzyl formimidate (VI) by means of DIEA in DMA affords the intermediate p-nitrobenzyl ester (VII), which is finally hydrogenated with H2 over Pd/C in water/isopropanol/N-methylmorpholine to provide the target Imipemide.
SYN 3
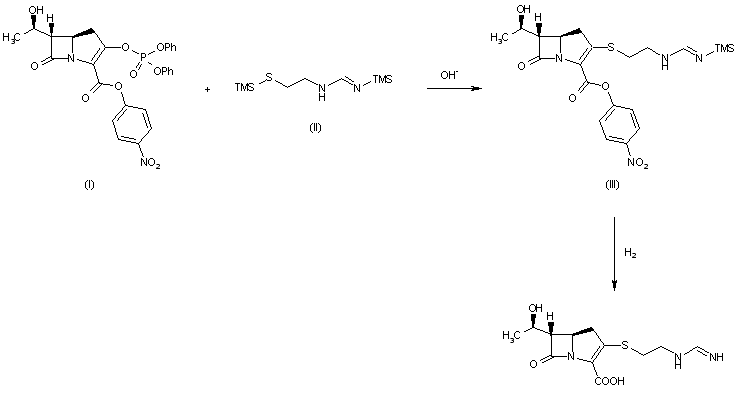
Tetrahedron Lett 1982,23(47),4903
The condensation of 7-oxo-6-(1-hydroxyethyl)-3-(diphenoxyphosphate)-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid p-nitrophenyl ester (I) with the bis(trimethylsilyl) derivative of 2-(iminomethylamino)ethanethiol (II) in the presence of base gives p-nitrophenyl ester of MK-0787, protected with a trimethylsilyl group (III), which is finally deprotected by hydrogenolysis.
CLIP

Synthesis Path
References
- ^ U.S. Patent 4,194,047
- ^ Clissold, SP; Todd, PA; Campoli-Richards, DM (Mar 1987). “Imipenem/cilastatin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 33 (3): 183–241. doi:10.2165/00003495-198733030-00001. PMID 3552595.
- ^ Vardakas, KZ; Tansarli, GS; Rafailidis, PI; Falagas, ME (Dec 2012). “Carbapenems versus alternative antibiotics for the treatment of bacteraemia due to Enterobacteriaceae producing extended-spectrum β-lactamases: a systematic review and meta-analysis”. The Journal of Antimicrobial Chemotherapy. 67 (12): 2793–803. doi:10.1093/jac/dks301. PMID 22915465.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 497. ISBN 9783527607495.
- ^ Kahan, FM; Kropp, H; Sundelof, JG; Birnbaum, J (Dec 1983). “Thienamycin: development of imipenen-cilastatin”. The Journal of Antimicrobial Chemotherapy. 12 Suppl D: 1–35. doi:10.1093/jac/12.suppl_d.1. PMID 6365872.
- ^ Kesado, Tadataka; Hashizume, Terutaka; Asahi, Yoshinari (1980). “Antibacterial activities of a new stabilized thienamycin, N-formimidoyl thienamycin, in comparison with other antibiotics”. Antimicrobial Agents and Chemotherapy. 17 (6): 912–7. doi:10.1128/aac.17.6.912. PMC 283902. PMID 6931548.
- ^ “Imipenem spectrum of bacterial susceptibility and Resistance” (PDF). Retrieved 4 May 2012.
- ^ “IMIPENEM/CILASTATIN”. livertox.nih.gov. Retrieved 2019-03-08.
- ^ Cannon, Joan P.; Lee, Todd A.; Clark, Nina M.; Setlak, Paul; Grim, Shellee A. (2014-08-01). “The risk of seizures among the carbapenems: a meta-analysis”. Journal of Antimicrobial Chemotherapy. 69 (8): 2043–2055. doi:10.1093/jac/dku111. ISSN 0305-7453.
Further reading
- Clissold, SP; Todd, PA; Campoli-Richards, DM (1987). “Imipenem/cilastatin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 33(3): 183–241. doi:10.2165/00003495-198733030-00001. PMID 3552595.
- Buckley, MM; Brogden, RN; Barradell, LB; Goa, KL (1992). “Imipenem/cilastatin. A reappraisal of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 44 (3): 408–44. doi:10.2165/00003495-199244030-00008. PMID 1382937.
External links
- Imipenem bound to proteins in the PDB
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Primaxin |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686013 |
| Pregnancy category |
|
| Routes of administration |
IM, IV |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | 20% |
| Metabolism | Renal |
| Elimination half-life | 38 minutes (children), 60 minutes (adults) |
| Excretion | Urine (70%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.058.831 |
| Chemical and physical data | |
| Formula | C12H17N3O4S |
| Molar mass | 299.347 g/mol g·mol−1 |
| 3D model (JSmol) | |
-
- Synonyms:Imipemide
- ATC:J01DH51
- Use:carbapenem antibiotic
- Chemical name:[5R-[5α,6α(R*)]]-6-(1-hydroxyethyl)-3-[[2-[(iminomethyl)amino]ethyl]thio]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
- Formula:C12H17N3O4S
- MW:299.35 g/mol
- CAS-RN:64221-86-9
- InChI Key:ZSKVGTPCRGIANV-ZXFLCMHBSA-N
- InChI:InChI=1S/C12H17N3O4S/c1-6(16)9-7-4-8(20-3-2-14-5-13)10(12(18)19)15(7)11(9)17/h5-7,9,16H,2-4H2,1H3,(H2,13,14)(H,18,19)/t6-,7-,9-/m1/s1
- EINECS:264-734-5
- LD50:1660 mg/kg (M, i.v.); >5 g/kg (M, p.o.);
1972 mg/kg (R, i.v.); >5 g/kg (R, p.o.)
Derivatives, monohydrate
- Formula:C12H17N3O4S • H2O
- MW:317.37 g/mol
- CAS-RN:74431-23-5
References
-
-
Leanza, W.J. et al.: J. Med. Chem. (JMCMAR) 22, 1435 (1979).
-
a Salzmann, T.L. et al.: J. Am. Chem. Soc. (JACSAT) 102, 6161-6163 (1980).
-
Reider, P.J.; Grabowski, E.J.J.: Tetrahedron Lett. (TELEAY) 23, 2293-2296 (1982).
-
Grabowski, E.J.J.: Chirality (CHRLEP) 17, 249-259 (2005).
-
US 4 194 047 (Merck & Co.; 18.3.1980; prior. 21.11.1975).
-
DOS 2 652 679 (Merck & Co.; appl. 19.11.1976; USA-prior. 21.11.1975).
-
b US 5 998 612 (Merck & Co.; 7.12.1999; appl. 12.6.1992; prior. 23.10.1981).
-
c US 4 981 992 (Takasago; 27.1.1998; appl. 13.5.1996; J-prior. 11.5.1995).
-
US 5 204 460 (Takasago; 20.4.1993; appl. 8.11.1991; J-prior. 8.11.1990).
-
US 5 204 462 (Takasago; 20.4.1993; appl. 8.11.1991; J-prior. 8.11.1990).
-
US 5 712 388 (Takasago; 27.1.1998; appl. 13.5.1996; J-prior. 11.5.1995).
-
US 5 081 239 (Takasago; 14.1.1992; appl. 29.11.1989; J-prior. 29.11.1988).
-
-
Acetoxylation of 2-azetidinones in 4-position:
-
Noyori, R. et al.: J. Am. Chem. Soc. (JACSAT) 111, 9134-9135 (1989).
-
Noyori, R. et al.: Angew. Chem. (ANCEAD) 114, 2108-2123 (2002).
-
US 5 288 862 (Takasago; 22.2.1994; appl. 16.4.1992; J-prior. 18.4.1991).
-
US 5 606 052 (Takasago; 25.2.1997; appl. 16.4.1992; J-prior. 18.4.1991).
-
-
Noyori-catalyst:
-
US 4 739 084 (Takasago; 19.4.1988; appl. 15.4.1987; J-prior. 13.5.1986).
-
-
d process of Nippon Soda (Nisso):
-
US 5 026 844 (Suntory & Nippon Soda; 25.6.1991; appl. 13.10.1989; J-prior. 19.10.1988).
-
US 5 792 861 (Tanabe Seiyaku & Nippon Soda; 11.8.1998; appl. 29.6.1994, 4.11.1996; J-prior. 30.6.1993).
-
US 5 808 055 (Suntory & Nippon Soda; 15.9.1998; appl. 30.3.1993, 5.7.1995; J-prior. 30.3.1993).
-
e US 4 791 198 (Kanegafuchi; 13.12.1988; appl. 1.7.1985, 6.1.1987; J-prior. 5.7.1984, 14.1.1986).
-
US 4 861 877 (Kanegafuchi; 29.8.1989; appl. 1.7.1985, 6.1.1987; J-prior. 5.7.1984, 14.1.1985, 14.1.1986).
-
US 5 061 817 (Kanegafuchi; 29.10.1991; appl. 1.7.1985, 6.1.1987, 31.5.1988; J-prior. 5.7.1984, 14.1.1986).
-
US 4 914 200 (Kanegafuchi; 3.4.1990; appl. 28.4.1987, 14.2.1989; J-prior. 30.4.1986, 13.11.1986, 9.2.1987).
-
-
Enzymatic reduction of alkyl-2-(N-benzoylamino)methyl-3-oxobutyrates with bakers yeast:
-
US 5 463 047 (Ciba-Geigy; 31.10.1995; appl. 15.9.1994; CH-prior. 4.5.1987).
-
-
Further synthesis processes of Merck & Co. for thienamycin:
-
Johnston, D.B.R. et al.: J. Am. Chem. Soc. (JACSAT) 100, 313-315 (1978).
-
Mellilo, D.G. et al.: Tetrahedron Lett. (TELEAY) 21, 2783 (1980).
-
Melillo, D.G. et al.: J. Org. Chem. (JOCEAH) 51, 1498-1504 (1986).
-
Karady, S. et al.: J. Am. Chem. Soc. (JACSAT) 103, 6765-6767 (1981).
-
US 4 269 772 (Merck & Co.; 26.5.1981; appl. 14.1.1980).
-
US 4 282 148 (Merck & Co.; 4.8.1981; appl. 14.1.1980).
-
US 4 287 123 (Merck & Co.; 1.9.1981; appl. 14.1.1980).
-
US 4 290 947 (Merck & Co.; 22.9.1981; appl. 29.5.1980).
-
US 4 360 684 (Merck & Co.; 23.11.1982; appl. 8.4.1981).
-
US 4 206 219 (Merck & Co.; 3.6.1980; appl. 24.10.1978).
-
US 4 348 320 (Merck & Co.; 7.9.1982; appl. 20.8.1980; USA-prior. 19.11.1976).
-
US 4 460 507 (Merck & Co.; 17.7.1984; appl. 29.4.1982; USA-prior. 10.10.1980).
-
US 5 055 573 (Merck & Co.; 8.10.1991, appl. 24.8.1990; USA-prior. 19.11.1976).
-
US 5 037 974 (Merck & Co.; 6.8.1991; appl. 14.8.1990; prior. 23.5.1988, 10.4.1990).
-
-
Review of thienamycin syntheses:
-
Nicolaou, K.C.; Sorensen, E.J.: Classics in Total Synthesis, VCH 1996, Weinheim & New York, chapter 16, p. 249-263.
-
Berks, A.H.: Tetrahedron (TETRAB) 52, 331-375 (1996).
-
-
Alternative 2-azetidinone ring closure with chlorosulfonyl isocyanate:
-
US 4 350 631 (Merck & Co.; 21.9.1982; appl. 18.3.1981; prior. 18.12.1980).
-
-
Thienamycin (by fermentation of S. cattleya):
-
US 3 950 357 (Merck & Co.; 13.4.1976; appl. 25.11.1974).
-
DOS 2 552 638 (Merck & Co.; appl. 24.11.1975; USA-prior. 25.11.1974).
-
-
Combination with cilastatin:
-
EP 48 301 (Merck & Co.; appl. 24.9.1980).
-
/////////////Imipenem, イミペネム水和物 , MK-787,
Quinacillin
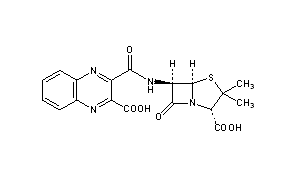
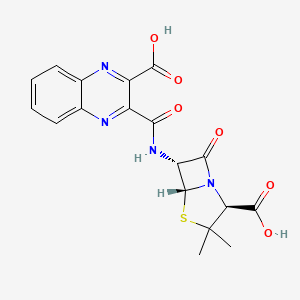
Quinacillin
UNII-83NB50X92M
Cas 1596-63-0
83NB50X92M
Quinacilina
MW 416.4 g/mol, MF C18H16N4O6S
(2S,5R,6R)-6-[(3-carboxyquinoxaline-2-carbonyl)amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
- 4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid, 6-(3-carboxy-2-quinoxalinecarboxamido)-3,3-dimethyl-7-oxo- (7CI,8CI)
- 4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid, 6-[[(3-carboxy-2-quinoxalinyl)carbonyl]amino]-3,3-dimethyl-7-oxo-, [2S-(2α,5α,6β)]-
- (2S,5R,6R)-6-[[(3-Carboxy-2-quinoxalinyl)carbonyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
- 3-Carboxy-2-quinoxalinylpenicillanic acid
- 3-Carboxy-2-quinoxalinylpenicillin
- 6-(3-Carboxy-2-quinoxalinecarboxamido)-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
- Penicillin, (3-carboxy-2-quinoxalinyl)-
CAS Registry Number: 1596-63-0
CAS Name: (2S,5R,6R)-6-[[(3-Carboxy-2-quinoxalinyl)carbonyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
Additional Names: 3-carboxy-2-quinoxalinylpenicillin
Molecular Formula: C18H16N4O6S
Molecular Weight: 416.41
Percent Composition: C 51.92%, H 3.87%, N 13.45%, O 23.05%, S 7.70%
Literature References: Semi-synthetic antibiotic related to penicillin. Prepd by condensation of quinoxaline-2,3-dicarboxylic anhydride with 6-aminopenicillanic acid: Richards et al., Nature 199, 354 (1963).
Derivative Type: Disodium salt
CAS Registry Number: 985-32-0
Molecular Formula: C18H14N4Na2O6S
Molecular Weight: 460.37
Percent Composition: C 46.96%, H 3.07%, N 12.17%, Na 9.99%, O 20.85%, S 6.97%
Properties: Crystals, dec 261-262°. [a]D23 +183.5° (water). Very hygroscopic. uv max (containing 9.2% water): 242, 326 nm (e32,100; 7280). Acquires a bright yellow color on exposure to strong sunlight but is stable at 100° for at least 3 months. Freely sol in water; a 25% aq soln is stable for 2 months at 0°. Antimicrobial activity is highest against Staphylococcus aureus.
Optical Rotation: [a]D23 +183.5° (water)
Absorption maximum: uv max (containing 9.2% water): 242, 326 nm (e 32,100; 7280)
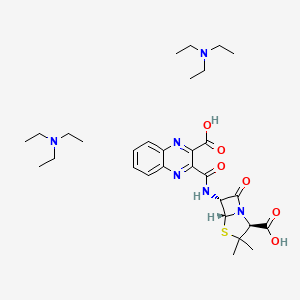
cas 13549-27-4
Derivative Type: Bistriethylammonium salt monohydrate
Molecular Formula: C30H46N6O6S.H2O
Molecular Weight: 636.80
Percent Composition: C 56.58%, H 7.60%, N 13.20%, O 17.59%, S 5.04%
Properties: Crystals from acetone, dec 135-137°. [a]D20 +142° (c = 0.376 in water).
Optical Rotation: [a]D20 +142° (c = 0.376 in water)
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Penicillins.
Quinacillin is a semisynthetic penicillase-resistant penicillin with antibacterial activity. Quinacillin binds to and inactivates penicillin-binding proteins (PBPs) located on the inner membrane of the bacterial cell wall. Inactivation of PBPs interferes with the cross-linkage of peptidoglycan chains necessary for bacterial cell wall strength and rigidity. This interrupts bacterial cell wall synthesis and results in the weakening of the bacterial cell wall, eventually causing cell lysis.
PATENTS
GB 867890
GB 967890
JP 47026513
DE 2161659
WO 2014111957
IN 2013MU00181
US 20150328323
PAPER
DOI: 10.1038/199354a0
//////Quinacillin
CC1(C(N2C(S1)C(C2=O)NC(=O)C3=NC4=CC=CC=C4N=C3C(=O)O)C(=O)O)C
Nicotinamide riboside chloride

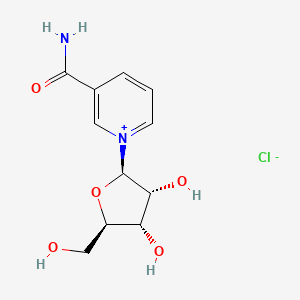

1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-methylol-tetrahydrofuran-2-yl]pyridin-1-ium-3-carboxamide
CAS 1341-23-7 [RN]
3-(aminocarbonyl)-1-β-D-ribofuranosyl-Pyridinium
Nicotinamide riboside chloride
CAS 23111-00-4 CHLORIDE
CAS : 1341-23-7 (cation) 23111-00-4 (chloride) 445489-49-6 (Triflate)
3-Carbamoyl-1-((2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyridin-1-ium chloride
Nicotinamide ribose chloride
UNII-8XM2XT8VWI
MW 290.7 g/mol
1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyridin-1-ium-3-carboxamide;chloride
C1=CC(=C[N+](=C1)C2C(C(C(O2)CO)O)O)C(=O)N.[Cl-]
Nicotinamide riboside; SRT647; SRT-647; SRT 647; Nicotinamide Riboside Triflate, α/β mixture
EH-301, nicotinamide riboside chloride,AND pterostilbene,, BY Elysium Health Inc
Nicotinamide riboside, also known as NR and SRT647, is a pyridine-nucleoside form of vitamin B3 that functions as a precursor to nicotinamide adenine dinucleotide or NAD+. NR blocks degeneration of surgically severed dorsal root ganglion neurons ex vivo and protects against noise-induced hearing loss in living mice. Nicotinamide riboside prevents muscle, neural and melanocyte stem cell senescence. Increased muscular regeneration in mice has been observed after treatment with nicotinamide riboside, leading to speculation that it might improve regeneration of organs such as the liver, kidney, and heart. Nicotinamide riboside also lowers blood glucose and fatty liver in prediabetic and type 2 diabetic models while preventing the development of diabetic peripheral neuropathy. Note: Nicotinamide Riboside chloride is a α/β mixture
Nicotinamide riboside (NR) is a pyridine–nucleoside form of vitamin B3 that functions as a precursor to nicotinamide adenine dinucleotide or NAD+.[1][2]
Chemistry
While the molecular weight of nicotinamide riboside is 255.25 g/mol,[3] that of its chloride salt is 290.70 g/mol.[4][5] As such, 100 mg of nicotinamide riboside chloride provides 88 mg of nicotinamide riboside.
History
Nicotinamide riboside (NR) was first described in 1944 as a growth factor, termed Factor V, for Haemophilus influenza, a bacterium that lives in and depends on blood. Factor V, purified from blood, was shown to exist in three forms: NAD+, NMN and NR. NR was the compound that led to the most rapid growth of this bacterium.[6] Notably, H. influenza cannot grow on nicotinic acid, nicotinamide, tryptophan or aspartic acid, which were the previously known precursors of NAD+.[7]
In 2000, yeast Sir2 was shown to be an NAD+-dependent protein lysine deacetylase,[8] which led several research groups to probe yeast NAD+ metabolism for genes and enzymes that might regulate lifespan. Biosynthesis of NAD+ in yeast was thought to flow exclusively through NAMN (nicotinic acid mononucleotide).[9][10][11][12][13]
When NAD+ synthase (glutamine-hydrolysing) was deleted from yeast cells, NR permitted yeast cells to grow. Thus, these Dartmouth College investigators proceeded to clone yeast and human nicotinamide riboside kinases and demonstrate the conversion of NR to NMN by nicotinamide riboside kinases in vitro and in vivo. They also demonstrated that NR is a natural product found in cow’s milk.[14][15]
Properties
Although it is a form of vitamin B3, NR exhibits unique properties that distinguish it from the other B3 vitamins—niacin and nicotinamide. In a head-to-head experiment conducted on mice, each of these vitamins exhibited unique effects on the hepatic NAD+ metabolome with unique kinetics, and with NR as the form of B3 that produced the greatest increase in NAD+ at a single timepoint.[16]
Different biosynthetic pathways are responsible for converting the different B3 vitamins into NAD+. The enzyme nicotinamide phosphoribosyltransferase (Nampt) catalyzes the rate-limiting step of the two-step pathway converting nicotinamide to NAD+. Two nicotinamide riboside kinases (NRK1 and NRK2) convert NR to NAD+ via a pathway that does not require Nampt.[14]
Animal studies have demonstrated that these enzymes respond differently to age and stress. In a mouse model of dilated cardiomyopathy, NRK2 mRNA expression increased, while Nampt mRNA expression decreased.[17] A similar increase in NRK1 and NRK2 expression has been observed in injured central and peripheral neurons.[18][19][20][21][22]
Niacin is known for its tendency to cause an uncomfortable flushing of the skin. This flushing is triggered by the activation of the GPR109A G-protein coupled receptor. NR does not activate this receptor,[23] and has not been shown to cause flushing in humans—even at doses as high as 2,000 mg/day.[16][24][25][26]
Despite being an NAD+ precursor, nicotinamide acts as an inhibitor of the NAD+-consuming sirtuin enzymes.[10] When sirtuins consume NAD+, they create nicotinamide and O-acetyl-ADP-ribose as products of the deacetylation reaction. Consistent with high-dose nicotinamide as a sirtuin inhibitor, NR and niacin, but not nicotinamide, have been shown to increase hepatic levels of O-acetyl-ADP-ribose.[16]
Commercialization
In 2004, Dartmouth Medical School researcher Dr. Charles Brenner discovered that NR could be converted to NAD+ via the eukaryotic nicotinamide riboside kinase biosynthetic pathway[14] Dartmouth was subsequently issued patents for nutritional and therapeutic uses of NR, in 2006.[27] ChromaDex licensed these patents in July 2012, and began to develop a commercially viable, full-scale process to bring NR to market.[28]
Human Clinical Testing
There have been five published clinical trials on groups of both men and women testing for safety. One of these trials studied NR in combination with pterostilbene,[29] while the other four examined the effects of NR alone.[16][24][25][26]
The first published clinical trial established the safety and characterized the pharmacokinetics of single doses of NR.[16] Since then, doses as high as 2,000 mg/day have been administered over periods as long as 12 weeks.[25] These studies show that NR can significantly increase levels of NAD+ and some of its associated metabolites in both whole blood and peripheral blood mononuclear cells.[16][24][26]
In a 12 week clinical trial of obese insulin-resistant men using 2000 mg/day, NR appeared safe, but did not improve insulin sensitivity or whole-body glucose metabolism.[26] In a trial of NR 250 mg plus 50 mg of pterostilbene, as well as with double this dose, the combined supplement raised NAD+ levels in a trial of older adults.[29]
PATENT
WO-2019126482
Crystalline form of nicotinamide riboside chloride, useful for treating motor neuron disease or ALS, infertility, kidney damage, and liver damage or fatty liver. Elysium Health in collaboration with Mayo Clinic , is developing EH-301 (clinical, in July 2019), a combination of nicotinamide riboside chloride and pterostilbene for the treatment of amyotrophic lateral sclerosis. See WO2019108878 , claiming use of composition comprising nicotinamide riboside and pterostilbene, for treating obesity.
Nicotinamide riboside is a pyridine-nucleoside form of niacin ( i.e ., vitamin B3) that serves as a precursor to nicotinamide adenine dinucleotide (NAD+). NAD+promotes cellular metabolism, mitochondrial function, and energy production. Currently, nicotinamide riboside is made through synthetic methods or fermentation processes. Because of its significant potential to confer health benefits when used as a dietary supplement, there exists a need to develop highly efficient and scalable processes for the manufacture and purification of nicotinamide riboside.
SUMMARY OF THE INVENTION
In certain aspects, the present invention provides a crystalline form of a compound having the structure of formula (I)
Example 1. Scale-Up Synthesis and Crystallization of Nicotinamide Riboside Chloride
900 kg of nicotinamide riboside triacetate and 2133 kg of methanol were charged to a reactor and mixed, then cooled to 0 °C. 747 kg of 7M mmmonia in methanol (i.e.,“methanolic NH3”) was slowly charged to the reactor at 0 °C. The reaction mixture was passed through a polish filter, then the reaction mixture was stirred for 14 hours. A sample from the reaction mixture was taken to assess reaction progress. Upon completion of the reaction, the reaction mixture was
placed under vacuum, then warmed to 20 °C to 25 °C for 4 hours. Vacuum was applied until solids formed. Once solids were formed, the resultant slurry was filtered on a Nutsche filter dryer. Solids were washed with 1422 kg of ethanol, then 1422 kg of acetone, then 1322 kg of methyl tert butyl ether (MTBE). The resultant solids were then dried at 40 °C. Product was formed with 60% yield. The process flow diagram for this reaction is shown in FIG. 6.
Example 2. Optional Secondary Isolation
The crystalline form may optionally undergo a second isolation process according to the following steps: The solids obtained in Example 1 were dissolved in purified water at 30 °C to 40 °C. Ethanol was slowly added to the solution and mixed for 10 hours, over which time the solids began to precipitate. MTBE was then added and mixed for 2 hours. The mixture was then filtered on a Buchner funnel, and the solids were washed with ethanol, then acetone, then MTBE. Solids were dried at 40 °C.
Example 3. Spectroscopic Data.
The crystalline form made by the process described in Examples 1 and 2 has an XRD spectrum substantially as shown in FIG. 1. The instrument utilized in collecting the XRD data is a Rigaku Smart Lab X-Ray diffraction system.
Specifically, in order to collect the XRD data, The Rigaku Smart-Lab X-ray diffraction system was configured for reflection Bragg-Brentano geometry using a line source X-ray beam. The X-ray source is a Cu Long Fine Focus tube that was operated at 40 kV and 44 mA. That source provides an incident beam profile at the sample that changes from a narrow line at high angles to a broad rectangle at low angles. Beam conditioning slits are used on the line X-ray source to ensure that the maximum beam size is less than 10 mm both along the line and normal to the line. The Bragg-Brentano geometry is a para-focusing geometry controlled by passive divergence and receiving slits with the sample itself acting as the focusing component for the optics. The inherent resolution of Bragg-Brentano geometry is governed in part by the diffractometer radius and the width of the receiving slit used. Typically, the Rigaku Smart-Lab is operated to give peak widths of 0.1 °2Q or less. The axial divergence of the X-ray beam is controlled by 5.0-degree Sober slits in both the incident and diffracted beam paths.
The samples were prepared in a low background Si holder using light manual pressure to keep the sample surface flat and level with the reference surface of the sample holder. The single crystal Si low background holder has a small circular recess (10 mm diameter and about 0.2 mm depth) that held between 20 and 25 mg of the sample. The samples were analyzed from 2 to 40
°2Q using a continuous scan of 6 °20 per minute with an effective step size of 0.02 °20. The data collection procedure used to analyze these samples was not validated. The peak lists were generated using PDXL2 v.2.3.1.0. The figures were created using PlotMon VI.00.
PATENT
WO2019108878 , claiming use of composition comprising nicotinamide riboside and pterostilbene, for treating obesity.
CLIP
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0186459
CLIP
Syntheses and chemical properties of β-nicotinamide riboside and its analogues and derivatives


References
- ^ Bogan, K.L., Brenner, C. (2008). “Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition”. Annu. Rev. Nutr. 28: 115–130. doi:10.1146/annurev.nutr.28.061807.155443. PMID 18429699.
- ^ Chi Y, Sauve AA (November 2013). “Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection”. Curr Opin Clin Nutr Metab Care. 16 (6): 657–61. doi:10.1097/MCO.0b013e32836510c0. PMID 24071780.
- ^ “Nicotinamide riboside”. pubchem.ncbi.nlm.nih.gov.
- ^ “GRAS Notices, GRN No. 635”. http://www.accessdata.fda.gov. Retrieved 18 February 2019.
- ^ “Spherix/ChromaDex GRAS submission” (PDF). FDA.gov. Retrieved 18 February2019.
- ^ Gingrich, W; Schlenk, F (June 1944). “Codehydrogenase I and Other Pyridinium Compounds as V-Factor for Hemophilus influenzae and H. parainfluenzae”. Journal of Bacteriology. 47 (6): 535–50. PMC 373952. PMID 16560803.
- ^ Belenky, P.; et al. (2007). “NAD+ Metabolism in Health and Disease”. Trends in Biochemical Sciences. 32 (1): 12–19. doi:10.1016/j.tibs.2006.11.006. PMID 17161604.
- ^ Imai, S.; et al. (2000). “Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase”. Nature. 403 (6771): 795–800. doi:10.1038/35001622. PMID 10693811.
- ^ Anderson; et al. (2003). “Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae”. Nature. 423 (6936): 181–185. doi:10.1038/nature01578. PMC 4802858. PMID 12736687.
- ^ Jump up to:a b Bitterman; et al. (2002). “Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1”. J. Biol. Chem. 277 (47): 45099–45107. doi:10.1074/jbc.m205670200. PMID 12297502.
- ^ Gallo; et al. (2004). “Nicotinamide clearance by pnc1 directly regulates sir2-mediated silencing and longevity”. Mol. Cell. Biol. 24 (3): 1301–1312. doi:10.1128/mcb.24.3.1301-1312.2004.
- ^ Panozzo, C.; et al. (2002). “Aerobic and anaerobic NAD+ metabolism in Saccharomyces cerevisiae”. FEBS Lett. 517 (1–3): 97–102. doi:10.1016/s0014-5793(02)02585-1. PMID 12062417.
- ^ Sandmeier, JJ; Celic, I; Boeke, JD; Smith, JS (March 2002). “Telomeric and rDNA silencing in Saccharomyces cerevisiae are dependent on a nuclear NAD(+) salvage pathway”. Genetics. 160 (3): 877–89. PMC 1462005. PMID 11901108.
- ^ Jump up to:a b c Bieganowki, P. & Brenner, C. (2004). “Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans”. Cell. 117 (4): 495–502. doi:10.1016/s0092-8674(04)00416-7. PMID 15137942.
- ^ Hautkooper, R.H.; et al. (2012). “Sirtuins as regulators of metabolism and healthspan”. Nat. Rev. Mol. Cell Biol. 13 (4): 225–238. doi:10.1038/nrm3293. PMC 4872805. PMID 22395773.
- ^ Jump up to:a b c d e f Trammell, Samuel A. J.; Schmidt, Mark S.; Weidemann, Benjamin J.; Redpath, Philip; Jaksch, Frank; Dellinger, Ryan W.; Li, Zhonggang; Abel, E. Dale; Migaud, Marie E.; Brenner, Charles (10 October 2016). “Nicotinamide riboside is uniquely and orally bioavailable in mice and humans”. Nature Communications. 7 (1): 12948. doi:10.1038/ncomms12948. PMC 5062546. PMID 27721479.
- ^ Diguet, Nicolas; Trammell, Samuel A.J.; Tannous, Cynthia; Deloux, Robin; Piquereau, Jérôme; Mougenot, Nathalie; Gouge, Anne; Gressette, Mélanie; Manoury, Boris; Blanc, Jocelyne; Breton, Marie; Decaux, Jean-François; Lavery, Gareth G.; Baczkó, István; Zoll, Joffrey; Garnier, Anne; Li, Zhenlin; Brenner, Charles; Mericskay, Mathias (22 May 2018). “Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy”. Circulation. 137 (21): 2256–2273. doi:10.1161/CIRCULATIONAHA.116.026099. PMID 29217642.
- ^ Vaur, Pauline; Brugg, Bernard; Mericskay, Mathias; Li, Zhenlin; Schmidt, Mark S.; Vivien, Denis; Orset, Cyrille; Jacotot, Etienne; Brenner, Charles; Duplus, Eric (December 2017). “Nicotinamide riboside, a form of vitamin B , protects against excitotoxicity-induced axonal degeneration”. The FASEB Journal. 31 (12): 5440–5452. doi:10.1096/fj.201700221RR. PMID 28842432.
- ^ Sasaki, Y.; Araki, T.; Milbrandt, J. (16 August 2006). “Stimulation of Nicotinamide Adenine Dinucleotide Biosynthetic Pathways Delays Axonal Degeneration after Axotomy”. Journal of Neuroscience. 26 (33): 8484–8491. doi:10.1523/JNEUROSCI.2320-06.2006. PMID 16914673.
- ^ Frederick, David W.; Loro, Emanuele; Liu, Ling; Davila, Antonio; Chellappa, Karthikeyani; Silverman, Ian M.; Quinn, William J.; Gosai, Sager J.; Tichy, Elisia D.; Davis, James G.; Mourkioti, Foteini; Gregory, Brian D.; Dellinger, Ryan W.; Redpath, Philip; Migaud, Marie E.; Nakamaru-Ogiso, Eiko; Rabinowitz, Joshua D.; Khurana, Tejvir S.; Baur, Joseph A. (August 2016). “Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle”. Cell Metabolism. 24 (2): 269–282. doi:10.1016/j.cmet.2016.07.005. PMC 4985182. PMID 27508874.
- ^ Cantó, Carles; Jiang, Lake Q.; Deshmukh, Atul S.; Mataki, Chikage; Coste, Agnes; Lagouge, Marie; Zierath, Juleen R.; Auwerx, Johan (March 2010). “Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle”. Cell Metabolism. 11 (3): 213–219. doi:10.1016/j.cmet.2010.02.006. PMC 3616265. PMID 20197054.
- ^ Rappou, Elisabeth; Jukarainen, Sakari; Rinnankoski-Tuikka, Rita; Kaye, Sanna; Heinonen, Sini; Hakkarainen, Antti; Lundbom, Jesper; Lundbom, Nina; Saunavaara, Virva; Rissanen, Aila; Virtanen, Kirsi A.; Pirinen, Eija; Pietiläinen, Kirsi H. (March 2016). “Weight Loss Is Associated With Increased NAD /SIRT1 Expression But Reduced PARP Activity in White Adipose Tissue”. The Journal of Clinical Endocrinology & Metabolism. 101 (3): 1263–1273. doi:10.1210/jc.2015-3054. PMID 26760174.
- ^ Cantó, Carles; Houtkooper, Riekelt H.; Pirinen, Eija; Youn, Dou Y.; Oosterveer, Maaike H.; Cen, Yana; Fernandez-Marcos, Pablo J.; Yamamoto, Hiroyasu; Andreux, Pénélope A.; Cettour-Rose, Philippe; Gademann, Karl; Rinsch, Chris; Schoonjans, Kristina; Sauve, Anthony A.; Auwerx, Johan (June 2012). “The NAD+ Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity”. Cell Metabolism. 15 (6): 838–847. doi:10.1016/j.cmet.2012.04.022. PMC 3616313. PMID 22682224.
- ^ Jump up to:a b c Airhart, Sophia E.; Shireman, Laura M.; Risler, Linda J.; Anderson, Gail D.; Nagana Gowda, G. A.; Raftery, Daniel; Tian, Rong; Shen, Danny D.; O’Brien, Kevin D.; Sinclair, David A. (6 December 2017). “An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers”. PLOS ONE. 12 (12): e0186459. doi:10.1371/journal.pone.0186459. PMC 5718430. PMID 29211728.
- ^ Jump up to:a b c Dollerup, Ole L; Christensen, Britt; Svart, Mads; Schmidt, Mark S; Sulek, Karolina; Ringgaard, Steffen; Stødkilde-Jørgensen, Hans; Møller, Niels; Brenner, Charles; Treebak, Jonas T; Jessen, Niels (August 2018). “A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects”. The American Journal of Clinical Nutrition. 108 (2): 343–353. doi:10.1093/ajcn/nqy132. PMID 29992272.
- ^ Jump up to:a b c d Martens, Christopher R.; Denman, Blair A.; Mazzo, Melissa R.; Armstrong, Michael L.; Reisdorph, Nichole; McQueen, Matthew B.; Chonchol, Michel; Seals, Douglas R. (29 March 2018). “Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults”. Nature Communications. 9 (1): 1286. doi:10.1038/s41467-018-03421-7. PMC 5876407. PMID 29599478.
- ^ Brenner, Charles (20 April 2006). “Nicotinamide riboside kinase compositions and methods for using the same”. Google Patents. Dartmouth College. Retrieved 19 February2019.
- ^ “ChromaDex Licenses Exclusive Patent Rights for Nicotinamide Riboside (NR) Vitamin Technologies”. 2012-07-16. Retrieved 15 February 2019.
- ^ Jump up to:a b Dellinger, Ryan W.; Santos, Santiago Roel; Morris, Mark; Evans, Mal; Alminana, Dan; Guarente, Leonard; Marcotulli, Eric (24 November 2017). “Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD+ levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study”. NPJ Aging and Mechanisms of Disease. 3 (1): 17. doi:10.1038/s41514-017-0016-9. PMC 5701244. PMID 29184669.
Further reading
- “Press Release: NIH researchers find potential target for reducing obesity-related inflammation”. National Institutes of Health (NIH). 16 November 2015.
- Stipp, David (March 11, 2015). “Guest Blog: Beyond Resveratrol: The Anti-Aging NAD Fad”. Scientific American Blog Network.
- Zhang, H; Ryu, D; Wu, Y; Gariani, K; Wang, X; Luan, P; D’Amico, D; Ropelle, ER; Lutolf, MP; Aebersold, R; Schoonjans, K; Menzies, KJ; Auwerx, J (17 June 2016). “NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice”. Science. 352 (6292): 1436–43. doi:10.1126/science.aaf2693. PMID 27127236.
- Dolopikou CF, Kourtzidis IA, Margaritelis NV, Vrabas IS, Koidou I, Kyparos A, Theodorou AA, Paschalis V, Nikolaidis MG. (2019 Feb 6). Acute nicotinamide riboside supplementation improves redox homeostasis and exercise performance in old individuals: a double-blind cross-over study. doi:10.1007/s00394-019-01919-4.
ADDITIONAL INFORMATION
High dose nicotinic acid is used as an agent that elevates high-density lipoprotein cholesterol, lowers low-density lipoprotein cholesterol and lower free fatty acids through a mechanism that is not completely understood. It was suggested that nicotinamide riboside might possess such an activity by elevating NAD in the cells responsible for reverse cholesterol transport. The discovery that the Wallerian degeneration slow gene encodes a protein fusion with NMN adenylyltransferase 1 indicated that increased NAD+ precursor supplementation might oppose neurodegenerative processes.
ChromaDex acquired intellectual property on uses and synthesis of NR from Dartmouth College, Cornell University, and Washington University and began distributing NR as Niagen in 2013. In November 2015 ChromaDex received New Dietary Ingredient (NDI) status for Niagen from the U.S. Food and Drug Administration (FDA) and the FDA issued a generally recognized as safe (GRAS) No Objection Letter for Nicotinamide Riboside Chloride (NR) on August 3, 2016.
REFERENCES
1: Chi Y, Sauve AA. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Curr Opin Clin Nutr Metab Care. 2013 Nov;16(6):657-61. doi: 10.1097/MCO.0b013e32836510c0. Review. PubMed PMID: 24071780.
2: Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115-30. doi: 10.1146/annurev.nutr.28.061807.155443. Review. PubMed PMID: 18429699.
3: Ghanta S, Grossmann RE, Brenner C. Mitochondrial protein acetylation as a cell-intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl-lysine modifications. Crit Rev Biochem Mol Biol. 2013 Nov-Dec;48(6):561-74. doi: 10.3109/10409238.2013.838204. Review. PubMed PMID: 24050258; PubMed Central PMCID: PMC4113336.
4: Yang Y, Sauve AA. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta. 2016 Dec;1864(12):1787-1800. doi: 10.1016/j.bbapap.2016.06.014. Review. PubMed PMID: 27374990.
5: Sauve AA. NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther. 2008 Mar;324(3):883-93. doi: 10.1124/jpet.107.120758. Review. PubMed PMID: 18165311.
6: Kato M, Lin SJ. Regulation of NAD+ metabolism, signaling and compartmentalization in the yeast Saccharomyces cerevisiae. DNA Repair (Amst). 2014 Nov;23:49-58. doi: 10.1016/j.dnarep.2014.07.009. Review. PubMed PMID: 25096760; PubMed Central PMCID: PMC4254062.
7: Gerlach G, Reidl J. NAD+ utilization in Pasteurellaceae: simplification of a complex pathway. J Bacteriol. 2006 Oct;188(19):6719-27. Review. PubMed PMID: 16980474; PubMed Central PMCID: PMC1595515.
8: Srivastava S. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin Transl Med. 2016 Dec;5(1):25. doi: 10.1186/s40169-016-0104-7. Review. PubMed PMID: 27465020; PubMed Central PMCID: PMC4963347.
9: Handschin C. Caloric restriction and exercise “mimetics”: Ready for prime time? Pharmacol Res. 2016 Jan;103:158-66. doi: 10.1016/j.phrs.2015.11.009. Review. PubMed PMID: 26658171; PubMed Central PMCID: PMC4970791.
10: Ruggieri S, Orsomando G, Sorci L, Raffaelli N. Regulation of NAD biosynthetic enzymes modulates NAD-sensing processes to shape mammalian cell physiology under varying biological cues. Biochim Biophys Acta. 2015 Sep;1854(9):1138-49. doi: 10.1016/j.bbapap.2015.02.021. Review. PubMed PMID: 25770681.
11: Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014 Aug;24(8):464-71. doi: 10.1016/j.tcb.2014.04.002. Review. PubMed PMID: 24786309; PubMed Central PMCID: PMC4112140.
12: Jaehme M, Slotboom DJ. Structure, function, evolution, and application of bacterial Pnu-type vitamin transporters. Biol Chem. 2015 Sep;396(9-10):955-66. doi: 10.1515/hsz-2015-0113. Review. PubMed PMID: 26352203.
13: Magni G, Di Stefano M, Orsomando G, Raffaelli N, Ruggieri S. NAD(P) biosynthesis enzymes as potential targets for selective drug design. Curr Med Chem. 2009;16(11):1372-90. Review. PubMed PMID: 19355893.
14: Mendelsohn AR, Larrick JW. Partial reversal of skeletal muscle aging by restoration of normal NAD⁺ levels. Rejuvenation Res. 2014 Feb;17(1):62-9. doi: 10.1089/rej.2014.1546. Review. PubMed PMID: 24410488.
15: Penberthy WT. Pharmacological targeting of IDO-mediated tolerance for treating autoimmune disease. Curr Drug Metab. 2007 Apr;8(3):245-66. Review. PubMed PMID: 17430113.
16: Gazzaniga F, Stebbins R, Chang SZ, McPeek MA, Brenner C. Microbial NAD metabolism: lessons from comparative genomics. Microbiol Mol Biol Rev. 2009 Sep;73(3):529-41, Table of Contents. doi: 10.1128/MMBR.00042-08. Review. PubMed PMID: 19721089; PubMed Central PMCID: PMC2738131.
17: Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004 Jan;61(1):19-34. Review. PubMed PMID: 14704851.
18: Magni G, Orsomando G, Raffelli N, Ruggieri S. Enzymology of mammalian NAD metabolism in health and disease. Front Biosci. 2008 May 1;13:6135-54. Review. PubMed PMID: 18508649.
19: Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007 Jan;32(1):12-9. Review. Erratum in: Trends Biochem Sci. 2008 Jan;33(1):1. PubMed PMID: 17161604.
20: Niven DF, O’Reilly T. Significance of V-factor dependency in the taxonomy of Haemophilus species and related organisms. Int J Syst Bacteriol. 1990 Jan;40(1):1-4. Review. PubMed PMID: 2145965.
|
|
 |
|
| Names | |
|---|---|
| Other names
1-(β-D-Ribofuranosyl)nicotinamide; N-Ribosylnicotinamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChemSpider | |
| KEGG | |
|
PubChem CID
|
|
| Properties | |
| C11H15N2O5+ | |
| Molar mass | 255.25 g/mol |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////////// EH-301, EH 301, EH301, Nicotinamide riboside, SRT647, SRT-647, SRT 647, Nicotinamide Riboside Triflate, α/β mixture
C1=CC(=C[N+](=C1)C2C(C(C(O2)CO)O)O)C(=O)N.[Cl-]
Quinupramine, キヌプラミン
Quinupramine
キヌプラミン
- 5-(1-azabicyclo[2.2.2]oct-3-yl)-10,11-dihydro-5H-dibenz[b,f]azepine
- Formula:C21H24N2
- MW:304.44 g/mol
- CAS:31721-17-2
Quinupramine
CAS Registry Number: 31721-17-2
CAS Name: 5-(1-Azabicyclo[2.2.2]oct-3-yl)-10,11-dihydro-5H-dibenz[b,f]azepine
Additional Names: 10,11-dihydro-5-(3-quinuclidinyl)-5H-dibenz[b,f]azepine
Manufacturers’ Codes: LM-208
Trademarks: Kinupril (Bellon); Kevopril (Rhone-Poulenc)
Molecular Formula: C21H24N2
Molecular Weight: 304.43
Percent Composition: C 82.85%, H 7.95%, N 9.20%
Literature References: Analog of imipramine, q.v. Prepn: C. Gueremy, P. C. Wirth, DE 2030492 (1971 to Sogeras), C.A. 74,141581e (1971). Animal studies: W. Van Dorsser, A. Dresse, Arch. Int. Pharmacodyn. Ther. 208, 373 (1974); eidem, ibid. 220, 164 (1976). Clinical study: R. Volmat et al., Clin. Neurol. Psychiat. 239, 445 (1978).
Properties: Crystals, mp 150°.
Melting point: mp 150°
Therap-Cat: Antidepressant.
Keywords: Antidepressant; Tricyclics.
Quinupramine (brand names Kevopril, Kinupril, Adeprim, Quinuprine) is a tricyclic antidepressant (TCA) used in Europe for the treatment of depression.[1][2]
Pharmacologically, quinupramine acts in vitro as a strong muscarinic acetylcholine receptor antagonist (anticholinergic) and H1 receptorantagonist (antihistamine), moderate 5-HT2 receptor antagonist, and weak serotonin and norepinephrine reuptake inhibitor.[3] It has negligible affinity for the α1-adrenergic, α2-adrenergic, β-adrenergic, or D2 receptor.[3]
Clinically, quinupramine is reported to be stimulating similarly to imipramine, desipramine, and demexiptiline.[4] It can be inferred that its in vivo metabolites may have stronger effects on the reuptake of norepinephrine and/or serotonin than quinupramine itself
SYN
References
- ^ Swiss Pharmaceutical Society (2000). Index Nominum 2000: International Drug Directory (Book with CD-ROM). Boca Raton: Medpharm Scientific Publishers. p. 908. ISBN 3-88763-075-0.
- ^ José Miguel Vela; Helmut Buschmann; Jörg Holenz; Antonio Párraga; Antoni Torrens (2007). Antidepressants, Antipsychotics, Anxiolytics: From Chemistry and Pharmacology to Clinical Application. Weinheim: Wiley-VCH. p. 248. ISBN 978-3-527-31058-6.
- ^ Jump up to:a b Sakamoto H, Yokoyama N, Kohno S, Ohata K (December 1984). “Receptor binding profile of quinupramine, a new tricyclic antidepressant”. Japanese Journal of Pharmacology. 36 (4): 455–60. doi:10.1254/jjp.36.455. PMID 6098759.
- ^ Kent, Angela; M. Billiard (2003). Sleep: physiology, investigations, and medicine. New York: Kluwer Academic/Plenum. p. 233. ISBN 0-306-47406-9.
-
- DOS 2 030 492 (Sogeras; appl. 20.6.1970; GB-prior. 20.6.1969).
- GB 1 252 320 (Sogeras; valid from 29.5.1970; prior. 20.6.1969).
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | 33 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.046.149 |
| Chemical and physical data | |
| Formula | C21H24N2 |
| Molar mass | 304.43 g/mol g·mol−1 |
//////////////Quinupramine, キヌプラミン
