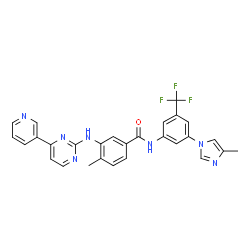
NILOTINIB
- Molecular FormulaC28H22F3N7O
- Average mass529.516 Da
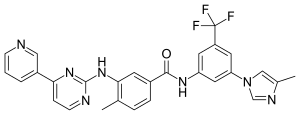
4-Methyl-N-(3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)benzamide
4-Methyl-N-(3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)benzamide
4-Methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluormethyl)phenyl]-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}benzolcarboxamid
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-{[4-(3-pyridinyl)-2-pyrimidinyl]amino}benzamide
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}benzamide
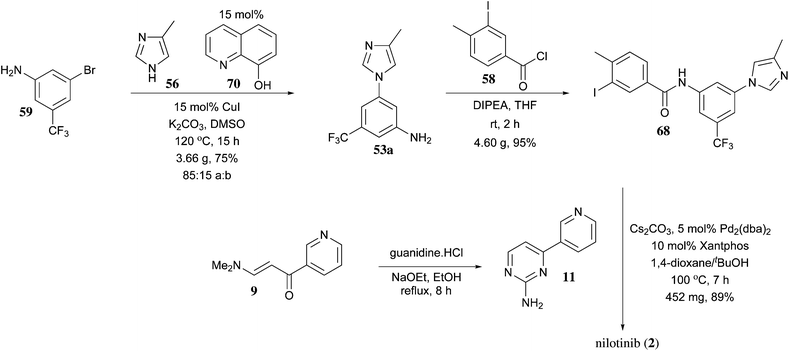
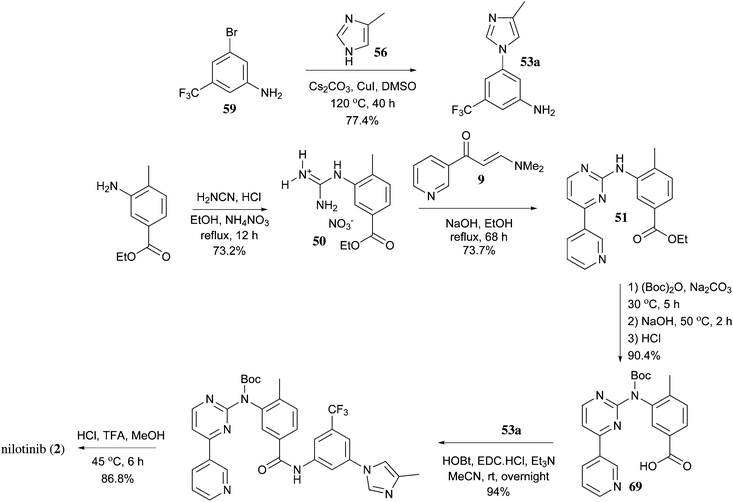
8654
Benzamide, 4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-
Nilotinib (AMN107, trade name Tasigna[2]), in the form of the hydrochloride monohydrate salt, is a small-molecule tyrosine kinase inhibitor approved for the treatment of imatinib-resistant chronic myelogenous leukemia.[3] Structurally related to imatinib,[4] it was developed based on the structure of the Abl-imatinib complex to address imatinib intolerance and resistance.[5][6][7] Nilotinib is a selective Bcr-Abl tyrosine kinase inhibitor[5][6] that is 10–30 fold more potent than imatinib in inhibiting Bcr-Abl tyrosine kinase activity and proliferation of Bcr-Abl expressing cells.[4][6][7][8] Nilotinib was developed by Novartis and is sold under the trade name Tasigna.[9]
Medical uses
It is FDA– (29 October 2007),[10] EMA– (29 September 2009),[11] MHRA– (19 November 2007)[12] and TGA– (17 January 2008)[13] approved for use as a treatment for Philadelphia chromosome (Ph+)-positive chronic myelogenous leukaemia.[1]
The drug carries a black box warning for possible heart complications.[14][15]
Clinical trials
CML
In June 2006, a phase I clinical trial found nilotinib has a relatively favorable safety profile and shows activity in cases of CML resistant to treatment with imatinib, another tyrosine kinase inhibitor currently used as a first-line treatment.[16] In that study 92% of patients (already resistant or unresponsive to imatinib) achieved normal white blood cell counts after five months of treatment.[17]
Contraindications
Contraindications include long QT syndrome, hypokalaemia, hypomagnesaemia, pregnancy, planned pregnancy, lactation and galactose/lactose intolerance.[1][13]
Cautions include:[1]
- Myelosuppression
- Tumour lysis syndrome
- Liver impairment
- History of pancreatitis
- Check serum lipase periodically in order to detect pancreatitis
- Total gastrectomy
- Avoid pregnancy or impregnating women
Dose reduction of nilotinib has been recommended in hepatically impaired population which involves recommendation of lower starting dose and monitoring of any hepatic function abnormalities.[18]
Adverse effects
Nilotinib has a number of adverse effects typical of anti-cancer drugs. These include headache, fatigue, gastrointestinal problems such as nausea, vomiting, diarrhea and constipation, muscle and joint pain, rash and other skin conditions, flu-like symptoms, and reduced blood cell count. Less typical side effects are those of the cardiovascular system, such as hypertension (high blood pressure), various types of arrhythmia, and prolonged QT interval. Nilotinib can also affect the body’s electrolyte and glucosebalance.[10] Though pulmonary-related adverse effects are rare when compared with imatinib and dasatinib, there is a case report of acute respiratory failure from diffuse alveolar hemorrhage in a patient taking nilotinib.[19]
Interactions
Nilotinib has been reported as a substrate for OATP1B1 and OATP1B3. Interaction of nilotinib with OATP1B1 and OATP1B3 may alter its hepatic disposition and can lead to transporter mediated drug-drug interactions.[18] Nilotinib is an inhibitor of OATP-1B1 transporter but not for OATP-1B3.[20]
It is a substrate for CYP3A4 and hence grapefruit juice and other CYP3A4 inhibitors[21] will increase its action and inducers like St. John’s wort[22] will decrease it. Patients report that pomegranates and starfruit may also interfere.
Food should not be eaten two hours before or one hour afterwards because it unpredictably increases its bioavailability, approximately doubling it.
Pharmacology
Nilotinib inhibits the kinases BCR-ABL,[23] KIT, LCK, EPHA3, EPHA8, DDR1, DDR2, PDGFRB, MAPK11 and ZAK.[24]
Research
Parkinson’s disease
There is weak evidence that nilotinib may be beneficial with Parkinson’s Disease (PD), with a small clinical trial suggesting it might halt progression and improve symptoms.[25]However, there were significant side effects including infection, liver function tests abnormalities, hallucinations and heart attack, and the benefit in PD disappeared at follow up after drug discontinuation, raising question as to whether it was truly a disease modifying therapy. Nilotinib is currently undergoing phase II studies for treatment of Parkinson’s.[26]Scientists and medical professionals have advised caution with over-optimistic interpretation of its effects in Parkinson’s due to the significant media hype surrounding the small and early clinical trial.[27][28]
Other
Novartis announced on April 11, 2011 that it was discontinuing a phase III trial of Tasigna (nilotinib) for investigational use in the first-line treatment of gastrointestinal stromal tumor(GIST) based on the recommendation of an independent data monitoring committee. Interim results showed Tasigna is unlikely to demonstrate superiority compared to Novartis’s Gleevec (imatinib)*, the current standard of care in this setting.[29]
Low dose nilotinib is also being investigated for use for and Alzheimer’s disease, as well as for ALS, dementia and Huntington’s disease.[30]
Patent

WO 2016024289, NILOTINIB, New Patent by SUN
SUN PHARMACEUTICAL INDUSTRIES LTD [IN/IN]; 17/B, Mahal Industrial Estate, Off Mahakali Caves Road, Andheri (east), Mumbai 400093 (IN)
THENNATI, Rajamannar; (IN).
KILARU, Srinivasu; (IN).
VALANCE SURENDRAKUMAR, Macwan; (IN).
SHRIPRAKASH DHAR, Dwivedi; (IN)
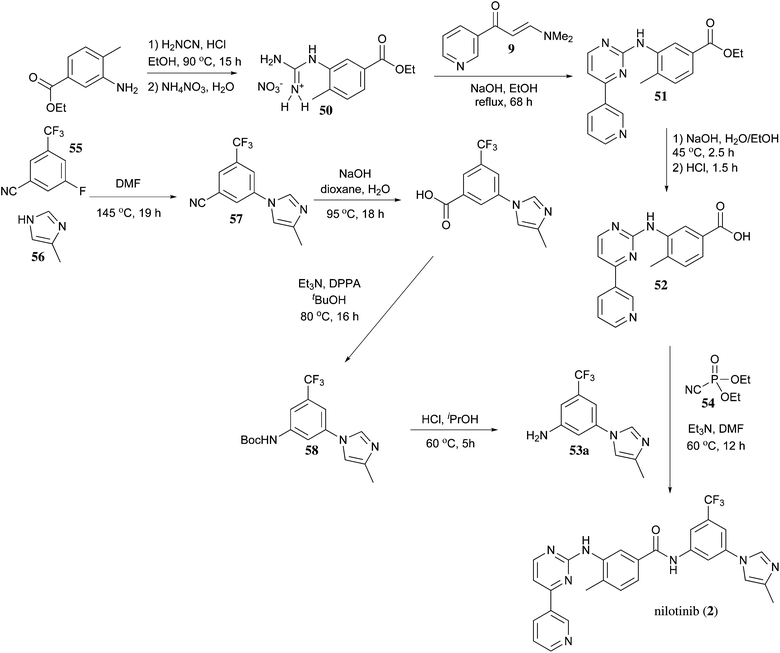
The present invention provides novel salts of nilotinib and polymorphs thereof. The acid addition salts of nilotinib with benzenesulfonic acid, butanedisulfonic acid, 1-5- naphthalenedisulfonic acid, naphthalene-1-sulfonic acid and 1-hydroxynaphthoic acid; hydrates and anhydrates thereof.
Nilotinib, 4-methyl-N-[3-(4-methyl-lH-imidazol-l-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl] amino] -benzamide, having the following formula

is marketed under the name Tasigna® in US and Europe. Tasigna contains nilotinib monohydrate monohydrochloride salt and is available as capsules for the treatment of adult patients with newly diagnosed Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase. Tasigna is also indicated for the treatment of chronic phase and accelerated phase Philadelphia chromosome positive chronic myelogenous leukemia (Ph+ CML) in adult patients resistant or intolerant to prior therapy that included imatinib.
Nilotinib is considered a low solubility/low permeability (class IV) compound in the Biopharmaceutics Classification System (BCS). Therefore, dissolution of nilotinib can potentially be rate limiting step for in-vivo absorption. It is soluble in acidic media; being practically insoluble in buffer solutions of pH 4.5 and higher.
WIPO publication 2014059518A1 discloses crystalline forms of nilotinib hydrochloride and methods of the preparation of various crystalline solvates of nilotinib hydrochloride including benzyl alcohol, acetic acid and propylene glycol.
WIPO publication 2011033307A1 discloses nilotinib dihydrochloride and its hydrates and method for their preparation.
WIPO publication 2011163222A1 discloses the preparation of nilotinib salts and crystalline forms thereof. The salts of nilotinib disclosed are hydrochloride, fumarate, 2-chloromandelate, succinate, adipate, L-tartrate, glutarate, p-toluenesulfonate, camphorsulfonate, glutamate, palmitate, quinate, citrate, maleate, acetate, L-malate, L-aspartate, formate, hydrobromide, oxalate and malonate.
WIPO publication number 2011086541A1 discloses a nilotinib monohydrochloride monohydrate salt and methods for preparing.
WIPO publication number 2010054056A2 describes several crystalline forms of nilotinib hydrochloride.
WIPO publication number 2007/015871A1 discloses the preparation of nilotinib salts and crystalline forms thereof. The salts are mixtures of nilotinib and one acid wherein the acids are selected from the group consisting of hydrochloric acid, phosphoric acid, sulfuric acid, sulfonic acid, methane sulfonic acid, ethane sulfonic acid, benzene sulfonic acid, p-toluene sul- fonic acid, citric acid, fumaric acid, gentisic acid, malonic acid, maleic acid, and tartaric acid.
WIPO publication number 2007015870A2 discloses several nilotinib salts including amorphous and crystalline forms of nilotinib free base, nilotinib HC1 and nilotinib sulfate along with their hydrate and solvates.
EXAMPLES:
Example 1: Preparation of nilotinib benzenesulfonate crystalline Form I
Nilotinib base (1 g) was suspended in water (20 ml). A solution of benzenesulfonic acid (0.4 g) in water (3ml) was added and the content was heated at 60 °C for 2-3 h. The mixture was cooled to 25-30 °C, filtered, washed with water (3 x 5 ml) and dried under vacuum for 2 h at 50-55 °C.
1H NMR (500 MHz, DMSO-d6) δ 2.40 (s,3H), 2.42 (s,3H), 7.35-7.37 (m,3H), 7.51-7.66 (m,5H),7.83 (d,lH), 7.96 (s,lH),8.08 (s,lH),8.30 (s,lH) 8.39 (s,lH),8.54 (d,lH), 8.61 (d,lH), 8.64 (s,lH), 8.75 (d,lH), 9.25 (s,lH), 9.34 (d,lH), 9.61 (s,lH), 10.84 (s,lH).
The salt provides an XRPD pattern substantially same as set forth in FIG. 1.
Example 2: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form II
Nilotinib base (100 g) was dissolved in 20 % water in THF solution (2000 ml) at 60-65 °C and insoluble matter was filtered. The filtrate was concentrated under vacuum below 60 °C. Filtered water (1000 ml) was added to the reaction mixture and it was heated at 50-55 °C, followed by addition of 1,4-butanedisulfonic acid -60% aqueous solution (28.6 ml) at same temperature. The content was stirred at 50-55 °C for 2-3h. Reaction mixture as cooled to 25-30 °C and product was filtered, washed with water (200 ml x 2) and dried in air oven at 50-55 °C (yield: 115 g).
Purity (by HPLC):99.76%
1H NMR (400 MHz,DMSO-d6) δ 1.63-1.66(m,2H), 2.40(d,3H),2.42(s,3H),2.43-2.47(m,2H), 7.51-7.62(m,3H),7.85(dd,lH),7.96(s,lH),8.08(s,lH),8.34(s,lH),8.38(d,lH),8.52-8.55(m,lH), 8.60-8.62 (m,2H), 8.75(d,lH), 9.25(S,1H),9.34(S,1H),9.59(S,1H),10.86(S,1H)
Water content: 7.95 %.
The salt has a XRPD pattern substantially same as set forth in FIG. 2.
Example 3: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form II
Nilotinib base (300 g) was suspended in methanol (3000 ml) and aqueous hydrochloric acid was added to get pH less than 2. Reaction contents were heated at reflux and was filtered and washed with methanol (100 ml). 5% (w/w) NaOH (1200 ml) solution was added at 40-45 °C within 15 min, reaction mixture was stirred for 2h. Product was filtered, washed with water
(300 ml x 3) and dried for lh. Wet material was suspended in water (3000 ml), heated at 50- 55 °C followed by addition of 1,4-butanedisulfonic acid -60% aqueous solution. The reaction mixture was stirred at 50-55°C for 2hrs. Product was filtered at room temperature, washed with water (500 ml x 2) and dried in air oven at 50-55 °C (yield: 293 g).
Purity (by HPLC): 99.88 %
1H NMR (400 MHz,DMSO-d6+TFA-dl) δ 1.75-1.78(m,2H), 2.36(d,3H),2.38(s,3H),2.69- 2.72(m,2H),7.45(d,lH),7.68(d,lH),7.83(s,lH),7.88(dd,lH),7.97(s,lH),8.16-8.19(m,lH), 8.35
(s,2H), 8.63(d,lH),8.68(d,lH),9.04(d,lH),9.21(d,lH),9.53(br s,lH),9.69(d,lH)10.80 (s,lH)
Water content: 6.44 %
Example 4: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form III
Nilotinib butanedisulfonate (210g) was dissolved in acetic acid water mixture (50:50) (2520 ml) at 75-80 °C and was filtered to remove insoluble matter and washed with acetic acid water mixture (50:50) (210 ml). Water (3150ml) was added to the filtrate and stirred first at room temperature and then at 0-5 °C. Product was filtered and washed with water. Material was dried in air oven at 70-75 °C. Dried material was leached with methanol (3438 ml) at reflux temperature, filtered and dried in air oven 70-75°C (yield: 152.6 g)
Purity (by HPLC): 99.89 %
1H NMR (400 MHz,DMSO-d6+TFA-dl) δ 1.73-1.77(m,2H), 2.40(s,6H),2.67-2.70(m,2H), 7.50 (d,lH), 7.70(d,lH), 7.88-7.92(m,2H), 8.07(s,lH),8.23 (dd,lH), 8.34(s,2H), 8.67 (d,lH), 8.72 (d,lH), 9.09(d,lH), 9.23 (s,lH), 9.54(d,lH), 9.74(d,lH), 10.86(s,lH).
Water content: 0.61 %
The salt provides an XRPD pattern substantially same as set forth in FIG. 3.
Example 5: Preparation of crystalline form of nilotinib butanedisulfonate (2: 1)
Crystalline Nilotinib butanedisulfonate (1 g) of Example 2 was suspended in methanol (20 ml) and was stirred at reflux for 60 min. The mixture was cooled to room temperature. Solid was filtered, washed with methanol (2 ml x 3) and dried in air oven at 70-75°C (yield: 0.8 g)
Example 6: Preparation of nilotinib butanedisulfonate (1: 1) crystalline Form IV
Nilotinib base (20 g) was suspended in methanol (800 ml) and 1,4-butanedisulfonic acid -60
% aqueous solution (6 ml) was added at 50-55 °C, and was filtered to remove insoluble matter. Filtrate was stirred at room temperature for 2-3 h. Product formed was filtered, washed with methanol (20 ml x 2) and dried the product in air oven at 70-75 °C (yield: 18.4 g).
Purity (by HPLC):99.86 %
1H NMR (400 MHz,DMSO-d6) δ 1.64-1.68(m,4H), 2.47-2.5 l(m,4H), 2.41(s,3H), 2.42(d,3H), 7.52(d,lH), 7.83-7.89(m,2H), 7.99(s,lH), 8.15(s,lH), 8.36 (d,lH), 8.39(s,lH), 8.65-8.66(m,2H), 8.79(d,lH), 8.89(br s,lH), 9.36(s,lH), 9.41(br s,lH), 9.74(d,lH), 10.91(s,lH).
The salt has XRPD pattern substantially same as set forth in FIG. 4.
Example 7: Preparation of nilotinib 1,5-napthalenedisulfonic acid salt (2: 1) crystalline Form V
Nilotinib base (1 g) was suspended in water (20 ml). A solution of 1,5-napthalenedisulfonic acid (0.4 g; 0.6 eq.) in water (5ml) was added and the content was heated at 50-55 °C for lh. The mixture was cooled to 25-30 °C, filtered and washed with water (10 ml). The product was dried in air oven at 50-55°C (yield: 1.2 g).
1H NMR (400 MHz,DMSO-d6) δ 2.39 (s,3H), 2.42 (s,3H), 7.45-7.61 (m,4H),7.84 (d,lH), 7.97(s,2H),8.08 (m,lH),8.31 (s,lH) 8.38 (s,lH),8.55 (d,lH), 8.63 (s,2H), 8.75 (s,lH), 8.92 (d,lH), 9.26 (s, 1H), 9.34 (s,lH),9.62 (s,lH), 10.85 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 5.
Example 8: Preparation of nilotinib 1,5-napthalenedisulfonic acid salt (1: 1) crystalline Form VI
Nilotinib base (1 g) was suspended in water (20 ml). A solution of 1,5-napthalenedisulfonic acid (0.8 g; 1.2eq) in water (5 ml) was added and the content was heated at 50-55 °C for 1 h. The mixture was cooled to 25-30 °C, filtered, washed with water (10 ml) and dried in air oven at 50-55 °C (yield: 1.4g).
1H NMR(400 MHz,DMSO-d6) δ 2.40 (s,3H),2.41 (s,3H), 7.43-7.52 (m,3H),7.61 (d,lH), 7.85-7.99(m,5H),8.11 (s,lH),8.34 (s,2H), 8.64-8.67 (m,2H), 8.89-8.92 (m,4H),9.40(d,2H), 9.72 (s,lH), 10.87 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 6.
Example 9: Preparation of nilotinib napthalene-1- sulfonic acid salt crystalline Form VII Nilotinib base (1 g) was suspended in water (10 ml) and heated to 50-55 °C. A solution of napthelene-1 -sulfonic acid and methanol (10 ml) was added to it and heated at 70-75 °C for 30 min. The mixture was cooled to 25-30 °C and stirred for 10 min. The product was filtered, washed with water (2 x 2 ml) and dried under vacuum for 1-2 h at 50-55 °C.
1H NMR (400 MHz,DMSO-d6) δ 2.41 (s,3H),2.42 (s,3H), 7.46-7.58 (m,5H), 7.70-8.00 (m,7H)8.11(s,lH)8.31(s,lH),8.37(s,lH),8.63-8.66 (m,3H), 8.81-8.89 (m,2H), 9.31 (s,lH), 9.37 (d,lH), 9.71 (d,lH), 10.86 (s,lH)
The salt has a XRPD pattern substantially same as set forth in FIG. 7.
Example 10: Preparation of nilotinib l-hydroxy-2-napthoic acid salt crystalline Form VIII Nilotinib base (1 g) was suspended in water (20 ml) and heated to 50-55 °C. l-Hydroxy-2-napthoic acid was added to it and the content was heated at 50-55 °C for 1 h. Methanol (5 ml) was added to the mixture and stirred for 30 min. The content was filtered, washed with water (2 x 2 ml) and dried under vacuum for 1 h at 50-55 °C.
1H NMR (400 MHz, DMSO-d6) δ 2.25 (s,3H), 2.41 (s,3H), 7.40-7.92 (m,l lH), 8.23-8.73 (m,8H), 9.24 (s,lH), 9.34(s,lH), 10.70 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 8.
PATENT
https://patents.google.com/patent/WO2010009402A2/en
Nilotinib, 4-methyl-N-[3-(4-methyl-lH-imidazol-l-yl)-5-
(trifluoromethyl)phenyl] -3 – [ [4-(3 -pyridinyl)-2-pyrimidinyl] amino] -benzamide, having the following formula
is a tyrosine kinase inhibitor used for the treatment of drug-resistant chronic myelogenous leukemia (CML), and in particular, for the treatment of chronic phase and accelerated phase Philadelphia chromosome positive chronic myeloid leukemia (CML) in adult patients whose disease has progressed on or who cannot tolerate other therapies that included imatinib. Nilotinib is administered as a hydrochloride salt in forms of capsules that are marketed in the USA and the EU under the name Tasigna®.
[0004] US patent no. 7,169,791 (“US 791”) and its parallel PCT publication WO
2004/005281, the journal article in Synthesis, 2007, vol 14, pp 2121-2124, as well as PCT publication nos.: WO 2006/135640, WO 2006/135641 (“WO “641”), WO 2007/018325 and WO 2007/017734, report processes for preparing Nilotinib intermediate, 3-(trifluoromethyl)- 5-(4-methyl-lH-imidazole-l-yl)-benzeneamine of formula I
I by reacting 3-bromo-5-trifluoromethylaniline of formula II and 4-methylimidazole of formula III in the presence of a non-alkaline hydroxide inorganic base, such as potassium carbonate, cesium carbonate and sodium hydride, a copper (I) salt, such as copper iodide and a complexing amine ligand, such as ethylene diamine. The process can be illustrated by the following scheme:
Il ‘
Scheme 1
[0005] The journal article in Synthesis, 2007, VoI 14, pp 2121-2124, describes a purification process of 3-(trifluoromethyl-5-(4-methyl-lH-imidazole-l-yl)-benzeneamine of formula I.
[0006] US 791 describes processes for preparing Nilotinib and its different intermediates, using di-ethyl cyano phosphate, as described in the following scheme:
[0007] WO ‘641 further describes a process for preparing Nilotinib according to the following scheme:
Scheme 3
[0008] The present invention provides improved processes to prepare and/or purify 3-
(trifluoromethyl)-5-(4-methyl-lH-imidazole-l-yl)-benzeneamine of formula I without requiring the use of column chromatography, and thus can be easily applied to large scale manufacture, as well as new intermediates of Nilotinib, which result in higher yields in the preparation of Nilotinib.
[0009] PCT publications WO 2007/015870 (“WO ‘870”) and WO 2007/015871
(“WO ‘871”) describe several Nilotinib salts including crystalline forms of nilotinib free base, Nilotinib hydrochloride and Nilotinib Sulfate.
[0010] The present invention also relates to the solid state physical properties of
Nilotinib»3HCl, 4-methyl-N-[3-(4-methyl-lH-imidazol-l-yl)-5-(trifluoromethyl)phenyl]-3- [[4-(3-pyridinyl)-2-pyrimidinyl]amino]-benzamide trihydrochloride. These properties can be influenced by controlling the conditions under which Nilotinib-3HC1 is obtained in solid form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must necessitate the use of glidants such as colloidal silicon dioxide, talc, starch, or tribasic calcium phosphate.
[0011 ] Another important solid state property of a pharmaceutical compound is its rate of dissolution in aqueous fluid. The rate of dissolution of an active ingredient in a patient’s stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally administered active ingredient can reach the patient’s bloodstream. The rate of dissolution is also a consideration in formulation syrups, elixirs, and other liquid medicaments. The solid state form of a compound can also affect its behavior on compaction and its storage stability. [0012] These practical physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which define a particular polymorphic form of a substance. The polymorphic form can give rise to thermal behavior different from that of the amorphous material or another polymorphic form. Thermal behavior is measured in the laboratory by such techniques as capillary melting point, thermogravimetric analysis (“TGA”), and differential scanning calorimetry (“DSC”) and can be used to distinguish some polymorphic forms from others. A particular polymorphic form can also give rise to distinct spectroscopic properties that can be detectable by powder x-ray crystallography, solid state 13C NMR spectroscopy, and infrared spectrometry.
[0013] Generally, a crystalline solid has improved chemical and physical stability over the amorphous form, and forms with low crystallinity. Crystalline forms may also exhibit improved solubility, hygroscopicity, bulk properties, and/or flowability.
[0014] The discovery of new polymorphic forms of a pharmaceutically useful compound provides a new opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic.
[0015] There is a need in the art for new intermediates of Nilotinib and processes for their preparation, new processes for preparing Nilotinib and new crystalline forms of Nilotinib»3HCl salt and processes for the preparation thereof.
xample 1: Preparation of 3-(ϊrifluoromethyl)-5-(4-methyl-lH-imidazole-l-yl)- benzeneamine of formula I
[00245] 200Og of 3-bromo-5- trifluoromethylaniline of formula II, 1368g of 4- methylimidazole of formula III , 181g of 8-hydroxyquinoline, 238g of CuI, 666.6g of NaOH, 933g of CaO and 7000ml of DMSO were loaded into a 1OL of 3-neck flask. The reaction mixture was protected with nitrogen and was then stirred at 12O0C for 69 hours while monitoring for the consumption of 3-bromo-5- trifluoromethy aniline by HPLC. Heating was stopped when 3-bromo-5- trifluoromethyaniline / 4-methylimidazole is not more than 5%. The reaction mixture was cooled down to 45-5O0C and poured into a 5OL reactor. 8.4L of 14% ammonia was added dropwise and then stirred for lhour at 45-5O0C. The mixture was cooled down to room temperature.16.8L of water and 1OL of ethyl acetate were added to the extract. The upper organic layer was separated and filtered through the filter aid. The lower aqueous layer was washed with 7.5L of ethyl acetate and combined with the above filtrate.
The combined organic layer was washed with 5L*3 of 5% of brine for three times. The upper organic layer was separated and dried over lkg of anhydrous Na2SO4overnight. The mixture was filtered and concentrated to obtain 2.3kg of solid. The residue was dissolved in 2L of ethyl acetate at 450C. To the solution was then added 8L of petroleum ether dropwise at 450C. The mixture was cooled down slowly to 0-150C and stirred for lhour. A large amount of precipitate was formed and filtered. The filtered cake was dissolved in 2L of ethyl acetate at 450C. The solution was then added 8L of petroleum ether dropwise at 450C. The mixture was cooled down slowly to 15-O0C and stirred for lhour. A large precipitate was formed and filtered. The filter cake was dried at 450C and 954g of 3-(trifluoromethyl)-5-(4-methyl-lH- imidazole-l-yl)-benzeneamine of formula I were obtained. (Yield: 47.5%). The obtained compound of formula I had purity of 99.7% on area by HPLC and contained 0.13% on area by HPLC, of the 5 methyl isomer impurity.
Example 2: Recrystallization of 3-ftrifluoromethyl)-5-f4-methyl-lH-imidazole-l-yl)- benzeneamine of formula I from IPA/water
[00246] A 5OmL flask was charged with Ig of the compound of Formula I crude
(purity of 82.5%) and 3.5mL of IPA. The mixture was heated to 45°C under stirring until the entire solid dissolved. At 45°C, 6mL of water was added drop-wise. The mixture was stirred for lOmin and cooled slowly to 0~10°C. The mixture was stirred at 0~10°C for 10 min and filtered to obtain the recrystallized compound of Formula I having a purity of 98%.
Example 3: Recrystallization of 3-ftrifluoromethyl)-5-f4-methyl-lH-imidazole-l-yl)- benzeneamine of formula I from Ethanol/water
[00247] A 5OmL flask was charged with 2g of the compound of Formula I crude
(purity of 83.1%) and 4mL of Ethanol. The mixture was heated to reflux under stirring until the entire solid dissolved. While refluxing, 1OmL of water was added drop-wise. The mixture was cooled slowly to 25±5°C. The mixture was filtered and washed with a mixture of ethanol/water to obtain the recrystallized compound of Formula I having a purity of 86.5%.
[00248] The purification factor can be seen in the following table:
Example 4: Preparation of compound of formula IV
[00249] The compound of formula X (31.Og, 0.1 Omol) was suspended in 310ml toluene, and SOCl2 (47.6g, 0.40mol) was added to the mixture under the protection of N2. The formed mixture was reacted at 5O0C for 2 h. The solvent was evaporated completely, and a compound of formula (X-Cl) was obtained as yellow solid. The compound of formula (X- Cl) was then added to a THF solution of the compound of formula II (27.Og, 0.1 lmol), DIPEA (15.Og, 0.12mol) and DMAP (0.5g, 4.0mmol). The reaction mixture was reacted at 3O0C for 12 h, and then quenched with 8% solution of sodium bicarbonate (620ml). The mixture was filtered, and washed with H2O, then dried in vacuum. The solid was re-slurried with MTBE, and dried in vacuum again. 49.5g of the compound of formula IV were obtained as light yellow powder. The yield is about 93.7% by weight. The purity of the isolated product is 98% (% on area by HPLC).
Example 5: Preparation of compound of formula IV
[00250] To a 50ml 3-neck flask was charged compound of formula X 3. Ig and 21ml of toluene. The suspension was charged 5.1g dichlorosulfoxide (SOCl2) under nitrogen protection. The reaction mixture was heated to 5O0C and reacted for 2hrs. The reaction was then concentrated to dry. To another 100ml 3-neck flask was charged 2.7g of compound of formula II, 1.5g of DIPEA, O.lg of DMAP and 30ml of THF. To the mixture was charged the above concentrated residue. The reaction mixture was stirred at 25±5°C overnight. The mixture was charged 45ml of ethyl acetate and 20ml of water. The mixture was then stirred at 25±5°C for lOmin, filtered and the filtrate was phase separated. The organic layer was washed by water 10ml twice. Then the organic layer was concentrated to dry. The residue was combined with the filter cake and slurried in MTBE. The mixture was filtered and dried under vacuum at 5O0C. The water layer was adjusted pH to 8 with NaHCO3solution. The second crop 0.5g was thus precipitated out. Total yield was 94%.
Example 6: Preparation of compound of formula IV
[00251] To a 50ml 3-neck flask was charged compound of formula X 3. Ig, 20 mL of toluene and 18ml of dichlorosulfoxide (SOCl2) under nitrogen protection. The reaction mixture was heated to 5O0C and reacted overnight. The reaction was then concentrated to dry and co-evaporated with 20ml of toluene of once. To another 100ml 3-neck flask was charged 2.7g of compound of formula II, 1.5g OfK2CO3, O.lg of DMAP and toluene. To the mixture was charged the above concentrated residue. The reaction mixture was stirred at 5O0C overnight. The mixture was charged 30ml of half saturated NaHCO3 solution, 15ml of MTBE and stirred for lOmin. Large amount of solid was precipitated out and filtered. The filter cake was washed with MTBE and fired under vacuum at 55 0C. The resulted product was of 81% of purity. There were about 9% of the compound of formula X.
Example 7: Preparation of compound of formula IV [00252] The compound of formula X (50 g), HOBt (26.5 g)/ EDCI (37.5 g) and DMF
(500 mL) were loaded into a reactor at 25±5°C. After being reacted for 3h, the compound of formula II (39 g) was added to the reactor. The reaction mixture was stirred at 800C for about 18 hours while monitoring for the consumption of active ester by HPLC. After being cooled to 25±5°C, the mixture was dropped to a solution of half-saturated aqueous solution of sodium hydrogen carbonate, and the product was precipitated as canary yellow solid. [00253] The yield of this step was about 29.0% by weight. The purity of the isolated product was 95% (% on area by HPLC method described in Appendix 1).
Example 8: Preparation of Nilotinib
[00254] The compound of formula IV (21.Og, 39.7mmol), NaI (12.Og, 79.8mmol), CuI
(1.3g, β.Ommol) and N,N-Dimethylethylenediamine (1.Ig, 12.0mmol) were dissolved in DMF (105ml) under the protection of N2. The formed solution was reacted at 12O0C for 24h. The temperature of the above solution was decreased to 6O0C.
[00255] 8-Hydroxyquinoline (1.8g, 1 l.βmmol), CuI (1.3g, β.Ommol), the compound of formula III (4.6g, 56.3mmol) and DBU (9.Og, 59.3mmol) were added to the above solution under the protection of N2. The formed solution was reacted at 12O0C for 48h. After the reaction was competed (detected by the consumption of the compound of formula IV, HPLC), the reaction solution was dropped to a mixture of saturated solution of NaHCO3 (15ml) and water (300ml) at 25±5°C. The mixture was then filtered, and the filter cake was washed with water. 26.9g crude product was obtained as pale brown powder with 69% purity after drying in vacuum.
[00256] The crude product was added to 3.8 vol. DMF, and heated to dissolution. The solution was filtered through Celite, and the filter cake was washed with 0.5 vol. DMF. 3.5 vol. of methanol/H2O (3:1) was added to the above solution at 6O0C. The formed solution was stirred at 25±5°C overnight and at ice bath for 2h. The mixture was filtered, and the filter cake was washed with methanol (0.05 volχ3). The first round re-crystallization solid was obtained after drying in vacuum. The above solid was added to 2.9 vol DMF, and heated to dissolution. Then filtered, and the filter cake was washed with 0.1vol. DMF. The resulting solution was stirred at 25±5°C for 0.5 h, and at ice bath for 2 h. The mixture was filtered, and the cake was washed with methanol (0.05volχ3). 9.1g solid was obtained with 99.1% purity after drying in vacuum. The total yield was about 43.5% by weight. The purity of the isolated product is 99.1% (% on area by HPLC). Example 9: Preparation of Nilotinib
[00257] The compound of formula IV, the compound of formula III, CS2CO3, CuI , 8- hydroxyquinoline and CaO were loaded into a reactor at 25±5°C under the protection of N2. The reaction mixture was then stirred at 1200C for about 24 hours while monitoring for the consumption of the compound of formula IV by HPLC. After cooled to 25±5°C, the mixture was treated with a half-saturated aqueous solution of sodium hydrogen carbonate and extracted three times with ethyl acetate, then dried by Na2SO4. After concentration, the crude product was obtained as yellow solid. Then the solid was dissolved by CH2CVMeOH (10 equ., 3:2), and the mixture was washed three times with water. After a period of time, the product would be crystallized from the organic solvent (purity: 95%, detected by HPLC). Few minutes later, the product would precipitate as yellow solid. Then the product was stirred in the solvent of CH2Cl2/Me0H (5 equ., 5:1) at 400C for 1 hour. After that, the mixture would be filtered. The solid we got was dried in vacuum, and the product with 98% purity was obtained by this means.
[00258] The yield of this step was about 31.1% by weight. The purity of the isolated product was 98% (% on area by HPLC method described in Appendix 1).
Example 10: Preparation of Nilotinib:
[00259] To 250 mL glass reactor was added the compound 4-methyl-3-{[4-(pyridin-3- yl)pyrimidin-2-yl] amino} benzoic acid of formula X (10.0 g, 0.032 mol), a compound of formula I (8.7 g, 0.036 mol), SOCl2 (7.5 mL, 0.103 mol) and N-Methyl-pyrrolidone (100 mL). The reaction mixture was stirred and heated to 900C for 5 h. The reaction was then cooled to 500C and an aqueous NaOH solution was added (12 g in 72 mL H2O) until pH 10- 11. Then, the suspension was cooled to room-temperature, stirred for 30 minutes at this temperature, filtered under reduced pressure and washed with 30 mL H2O to yield a beige solid. This material was dried under vacuum at 500C and 8.2 g of Nilotinib base was obtained. To the mother-liquor was added H2O (300 mL), and the mixture was stirred for 15 hours at room-temperature. A precipitate was formed and filtered under vacuum. The solid so-obtained was washed with H2O (20 mL), and dried in vacuum oven at 500C to yield additional 5.9 g of Nilotinib base. The total amount of Nilotinib base was 14.1 g in 81% yield. Example 11: Preparation of Nilotinib:
[00260] To 250 mL glass reactor was added the compound of formula 4-methyl-3- {[4-
(pyridin-3-yl)pyrimidin-2-yl]amino}benzoic acid of formula X (20.0 g, 0.065 mol), a compound of formula I (17.3 g, 0.072 mol), SOCl2 (15 mL, 0.206 mol) and N-Methyl- pyrrolidone (100 mL). The reaction mixture was stirred and heated to 900C for 3 h. The reaction was filtered under reduced pressure and washed with NMP (10 mL) and H2O (10 mL). The filtrate was then cooled to 700C and a 47% NaOH solution (30 mL) was added and stirred for 30 minutes until pH 11-12. Then, the suspension was cooled to 5°C during 3 hours, stirred at this temperature for 10 hours room-temperature, filtered under reduced pressure and washed with 100 mL H2O to yield a beige solid. This material was dried under vacuum at 500C and 27.1 g of Nilotinib base was obtained with 76% yield. (97.2% assay, 99.17% purity).
Example 12: Preparation of Nilotinib:
[00261] To IL glass reactor was added the compound of formula 4-methyl-3-{[4-
(pyridin-3-yl)pyrimidin-2-yl]amino}benzoic acid of formula X (80.0 g, 0.26 mol), and N- Methyl-pyrrolidone (400 mL). The mixture was heated to 600C, then SOCl2 (24 mL, 0.33 mol) was added during 15 minutes. The resulted mixture was stirred at 600C for 1 h. A compound of formula I (69.2 g, 0.29 mol) was added and the reaction mixture was stirred and heated to 900C for 3 h. Water (500 mL) was added and the solution was heated to 800C. NaOH 47% solution (65 mL) was added until pH 11-12. Then, the suspension was cooled to 400C and stirred at this temperature for 2 hours, filtered under reduced pressure at 400C, and washed with 500 mL H2O to yield a beige solid. This material was slurried in water (1 L) at 400C for 1 h, filtered, washed with water (500 mL), and dried under vacuum at 500C to obtain 135.25 g of Nilotinib base with 94% yield. (95.8% assay, 99.46% purity).
Example 13: Preparation of 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-4-methylbenzoyl chloride, dihydrochloride of the formula (X-C1)*2HC1:
[00262] Thionyl chloride (1400ML) was added to 3-(4-(pyridin-3-yl)pyrimidin-2- ylamino)-4-methylbenzoic acid of formula X (39 gms). This mixture was heated to 60-700C and stirred for 10-12 hours. The reaction mixture was then cooled to 30-270C. The obtained slurry was filtered and the solid was washed with dichloromethane. The wet product was dried at 55-600C under reduced pressure.
Dry wt: 140gm
Yield: 95.4
Purity: above 98% by HPLC
Hydrochloride content (by Argentometry titration): 27.48%
Example 14: Preparation of 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-4-methylbenzoyl chloride, dihydrochloride of the formula (X-ClWHCl:
[00263] Thionyl chloride (1000ML) was added to 3-(4-(pyridin-3-yl)pyrimidin-2- ylamino)-4-methylbenzoic acid of formula X (100 gms). This mixture was heated to 60-700C and stirred for 5-6 hours. The reaction mixture was then cooled to 30-350C. Dichloromethane
(1000ML) was then added to the recation mixture and stirred for 10-15 minutes. The obtained slurry was filtered and the solid was washed with dichloromethane. The wet product was dried at 55-600C under reduced pressure.
Dry wt: 100-106gm
Purity: above 98% by HPLC
Example 15: Preparation of Nilotinib*3HCl (crude):
[00264] 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-4-methylbenzoyl chloride dihydrochloride of formula (X-C1)-2HC1 (105 gms) was added to dichloromethane (1000ml) and 3-(trifluoromethyl)-5-(4-methyl-lH-imidazol-l-yl)benzenamine of formula I (71 gms) at
25-400C. The temperature was raised to reflux point and was stirred at this temperature for
10-12 Hours. The reaction mixture was then cooled to 30-200C. The obtained slurry was filtered and the solid was washed with dichloromethane (200ml). The wet product was dried at 40-60 0C under reduced pressure.
[00265] The X-ray powder diffraction of the obtained product is shown in Figure 3.
The X-ray powder diffraction of the obtained product after exposure to 100% humidity for
96% is shown in Figure 4.
Yield: 90-92%
Purity: 85-90%
Hydrochloride content (by Argentometry titration): 16.8%.
Example 16: Preparation of Nilotinib«3HCl: [00266] Methanol (50ml) was cooled to 0-50C and acetyl chloride (2.29gm) was slowly added to it. To this mixture, 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-N-(3- (trifluoromethyl)-5-(4-methyl-lH-imidazol-l-yl)phenyl)-4-methyl benzamide (Nilotinib free base) (5.00 gms) was added slowly and mixture was stirred for 2 hours. Acetone (50ml) was then added and mixture was stirred for 60 minutes. Reaction mass was filtered and washed with acetone (10ml). The obtained product was dried at 55-600C. Dry wt: 4.5gm Yield: 75% Purity: 95-98%
Example 17: Purification of Nilotinib«3HCl (Pure):
[00267] 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-N-(3-(trifluoromethyl)-5-(4-methyl- lH-imidazol-l-yl)phenyl)-4-methylbenzamide tri hydrochloride (5gm) and water (25ml) were added and the mass was heated to 60-700C. The mass was charcoalized (0.5gm carbon) and filtered through celite bed. Methanol (50ml) was added to the filtrate. The mixture was heated to 50-600C and acetone (100ml) was added. It was then cooled to 30-270C and stirred for 2hours. The obtained product was filtered and dried at 50-550C for 12 hours under vacuum. The X-ray powder diffraction of the obtained product is shown in Figure 5. Dry wt 3.5gm Yield 0.7w/w Purity: 95-98%
Example 18: Preparation of Nilotinib:
[00268] 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-N-(3-(trifluoromethyl)-5-(4-methyl- lH-imidazol-l-yl)phenyl)-4-methylbenzamide tri hydrochloride (185gms) was dissolved in 825ml water and heated to 45-55°C. A methanolic solution of sodium hydroxide (35.9gm Sodium hydroxide dissolve in 1800 ml methanol) was added to the reaction mixture over a period of 1-2 hours. The suspension was heated to 65-700C for 5-6 hours and the slurry was cooled to 35-300C. The solid was filtered and washed with equal amount of water: methanol mixture 200ml. The wet product was dried at 45-55°C under reduced pressure. Yield: 90% Purity: 99.5% Example 19: Purification of Nilotinib:
[00269] 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-N-(3-(trifluoromethyl)-5-(4-methyl- lH-imidazol-l-yl)phenyl)-4-methylbenzamide (140gm) was taken into methanol (1.41it) and sodium hydroxide (14gm). The mixture was heated to reflux and stirred for 3-4 hours. The mixture was the cooled to 40-350C and filtered. The product was washed with methanol (2X50ml) and dried at 50-600C for 12 hours under vacuum. Dry wt. 120gm Yield: 0.85w/w

PAPER
https://pubs.rsc.org/en/content/articlelanding/2013/ob/c2ob27003j/unauth#!divAbstract

References
- ^ Jump up to:a b c d e f g h “Tasigna (nilotinib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 25 January 2014.
- ^ Official Manufacturer Website http://www.tasigna.com
- ^ “Cancer Drug Information: Nilotinib”.
- ^ Jump up to:a b Manley, P.; Cowan-Jacob, S.; Mestan, J. (2005). “Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia”. Biochimica et Biophysica Acta. 1754 (1–2): 3–13. doi:10.1016/j.bbapap.2005.07.040. PMID 16172030.
- ^ Jump up to:a b Manley, P.; Stiefl, N.; Cowan-Jacob, S.; Kaufman, S.; Mestan, J.; Wartmann, M.; Wiesmann, M.; Woodman, R.; Gallagher, N. (2010). “Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib”. Bioorganic & Medicinal Chemistry. 18 (19): 6977–6986. doi:10.1016/j.bmc.2010.08.026. PMID 20817538.
- ^ Jump up to:a b c Jabbour, E.; Cortes, J.; Kantarjian, H. (2009). “Nilotinib for the treatment of chronic myeloid leukemia: An evidence-based review”. Core Evidence. 4: 207–213. doi:10.2147/CE.S6003. PMC 2899790.
- ^ Jump up to:a b Olivieri, A.; Manzione, L. (2007). “Dasatinib: a new step in molecular target therapy”. Annals of Oncology. 18 Suppl 6: vi42–vi46. doi:10.1093/annonc/mdm223. PMID 17591830.
- ^ Breccia, M.; Alimena, G. (2010). “Nilotinib: a second-generation tyrosine kinase inhibitor for chronic myeloid leukemia”. Leukemia Research. 34 (2): 129–134. doi:10.1016/j.leukres.2009.08.031. PMID 19783301.
- ^ https://www.cancer.gov/about-cancer/treatment/drugs/fda-nilotinib
- ^ Jump up to:a b “Complete Nilotinib information from Drugs.com”. Drugs.com. Retrieved 25 January2014.
- ^ “Tasigna : EPAR – Product Information” (PDF). European Medicines Agency. Novartis Europharm Ltd. 18 October 2013. Retrieved 25 January 2014.
- ^ “Tasigna 150mg Hard Capsules – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Novartis Pharmaceuticals UK Ltd. 9 September 2013. Retrieved 25 January 2014.
- ^ Jump up to:a b “TASIGNA® nilotinib” (PDF). TGA eBusiness Services. 21 October 2013. Retrieved 25 January 2014.
- ^ “FDA Approves Tasigna for Treatment of Philadelphia Chromosome Positive Chronic Myeloid Leukemia”. U.S. Food and Drug Administration. 2007-10-30. Retrieved 2009-08-04.
- ^ “Prescribing information for Tasigna (nilotinib) Capsules” (PDF). NDA 022068. U.S. FDA. 2007-10-29. Retrieved 2009-08-04.
- ^ Kantarjian H; Giles, Francis; Wunderle, Lydia; Bhalla, Kapil; O’Brien, Susan; Wassmann, Barbara; Tanaka, Chiaki; Manley, Paul; Rae, Patricia; Mietlowski, William; Bochinski, Kathy; Hochhaus, Andreas; Griffin, James D.; Hoelzer, Dieter; Albitar, Maher; Dugan, Margaret; Cortes, Jorge; Alland, Leila; Ottmann, Oliver G.; et al. (2006). “Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL”. N Engl J Med. 354 (24): 2542–51. doi:10.1056/NEJMoa055104. PMID 16775235.
- ^ “Patients with treatment-resistant leukemia achieve high responses to Tasigna (nilotinib) in first published clinical trial results”. MediaReleases. Novartis. 2006-06-14. Retrieved 2009-08-04.
- ^ Jump up to:a b Khurana V, Minocha M, Pal D, Mitra AK (March 2014). “Role of OATP-1B1 and/or OATP-1B3 in hepatic disposition of tyrosine kinase inhibitors”. Drug Metabol Drug Interact. 29 (3): 179–90. doi:10.1515/dmdi-2013-0062. PMC 4407685. PMID 24643910.
- ^ Donatelli, Christopher; Chongnarungsin, Daych; Ashton, Rendell (2014). “Acute respiratory failure from nilotinib-associated diffuse alveolar hemorrhage”. Leukemia & Lymphoma. 55 (10): 1–6. doi:10.3109/10428194.2014.887714. PMID 24467220.
- ^ Khurana V, Minocha M, Pal D, Mitra AK (May 2014). “Inhibition of OATP-1B1 and OATP-1B3 by tyrosine kinase inhibitors”. Drug Metabol Drug Interact. 29 (4): 249–59. doi:10.1515/dmdi-2014-0014. PMC 4407688. PMID 24807167.
- ^ Bailey, David G; Malcolm, J; Arnold, O; David Spence, J (1998-08-01). “Grapefruit juice–drug interactions”. British Journal of Clinical Pharmacology. 46 (2): 101–110. doi:10.1046/j.1365-2125.1998.00764.x. ISSN 0306-5251. PMC 1873672. PMID 9723817.
- ^ Komoroski, Bernard J.; Zhang, Shimin; Cai, Hongbo; Hutzler, J. Matthew; Frye, Reginald; Tracy, Timothy S.; Strom, Stephen C.; Lehmann, Thomas; Ang, Catharina Y. W. (2004-05-01). “Induction and inhibition of cytochromes P450 by the St. John’s wort constituent hyperforin in human hepatocyte cultures”. Drug Metabolism and Disposition. 32 (5): 512–518. doi:10.1124/dmd.32.5.512. ISSN 0090-9556. PMID 15100173.
- ^ Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD (June 2006). “AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL”. Br. J. Cancer. 94 (12): 1765–9. doi:10.1038/sj.bjc.6603170. PMC 2361347. PMID 16721371.
- ^ Manley, PW; Drueckes, P; Fendrich, G; Furet, P; Liebetanz, J; Martiny-Baron, G; Mestan, J; Trappe, J; et al. (2010). “Extended kinase profile and properties of the protein kinase inhibitor nilotinib”. Biochimica et Biophysica Acta. 1804 (3): 445–53. doi:10.1016/j.bbapap.2009.11.008. PMID 19922818.
- ^ Pagan, F.; Hebron, M.; Valadez, E. H.; Torres-Yaghi, Y.; Huang, X.; Mills, R. R.; Wilmarth, B. M.; Howard, H.; Dunn, C.; Carlson, A.; Lawler, A.; Rogers, S. L.; Falconer, R. A.; Ahn, J.; Li, Z.; Moussa, C. (2016). “Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies”. Journal of Parkinson’s Disease. 6 (3): 503–17. doi:10.3233/JPD-160867. PMC 5008228. PMID 27434297.
- ^ Dash, Deepa (2019). “Anticancer Drugs for Parkinson’s Disease: Is It a Ray of Hope or Only Hype?”. Annals of Indian Academy of Neurology. 22 (1): 13–16. doi:10.4103/aian.AIAN_177_18. PMC 6327695. PMID 30692753.
- ^ Robledo, I.; Jankovic, J. (2017). “Media hype: Patient and scientific perspectives on misleading medical news”. Movement Disorders. 32 (9): 1319–1323. doi:10.1002/mds.26993. PMID 28370445.
- ^ Wyse, R. K.; Brundin, P.; Sherer, T. B. (2016). “Nilotinib – Differentiating the Hope from the Hype”. Journal of Parkinson’s Disease. 6 (3): 519–22. doi:10.3233/JPD-160904. PMC 5044778. PMID 27434298.
- ^ “Global Novartis News Archive”.
- ^ “Cancer drug prevents build-up of toxic brain protein”. MedicalXpress.com. 10 May 2013. Retrieved 11 April 2017.
External links
Nilotinib
-
- Synonyms:AMN-107
- ATC:L01XE08
- Use:antineoplastic, kinase inhibitor
- Chemical name:4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]benzamide
- Formula:C28H22F3N7O
- MW:529.53 g/mol
- CAS-RN:641571-10-0
- InChI Key:HHZIURLSWUIHRB-UHFFFAOYSA-N
- InChI:InChI=1S/C28H22F3N7O/c1-17-5-6-19(10-25(17)37-27-33-9-7-24(36-27)20-4-3-8-32-14-20)26(39)35-22-11-21(28(29,30)31)12-23(13-22)38-15-18(2)34-16-38/h3-16H,1-2H3,(H,35,39)(H,33,36,37)
Derivatives
Hydrochloride monohydrate
- Formula:C28H22F3N7O • HCl • H2O
- MW:586.01 g/mol
- CAS-RN:923288-90-8
Trade Names
| Country |
Trade Name |
Vendor |
Annotation |
| D |
Tasigna |
Novartis ,2008 |
|
| F |
Tasigna |
Novartis |
|
| GB |
Tasigna |
Novartis |
|
| I |
Tasigna |
Novartis |
|
| USA |
Tasigna |
Novartis ,2007 |
|
| J |
Tasigna |
Novartis ,2010 |
|
Formulations
- cps. 150 and 200 mg as hydrochloride monohydrate
References
-
- a WO 2004 005281 (Novartis; 15.1.2004; GB-prior. 5.7.2002).
- US 7 169 791 (Novartis; 30.1.2007; appl. 4.7.2003; GB-prior. 5.7.2002).
- US 7 569 566 (Novartis; 4.8.2009; GB-prior. 5.7.2002, 20.12.2002).
- WO 2006 135641 (Novartis; 21.12.2006; USA-prior. 4.8.2005).
- US 7 956 053 (Novartis; 7.6.2011; appl. 22.6.2009; GB-prior. 5.7.2002).
-
Preparation of III:
- b Huang, W.-S., Shakesperare, W.C., Synthesis (SYNTBF) (2007) 14, 2121.
- c WO 2010 060074 (Teva Pharms.; 27.5.2010; appl. 24.11.2009; USA-prior. 24.11.2008).
- d Ueda, S. et al., J. Med. Chem. Soc., (2012) 134(1), 700-706.
- US 8 017 621 (Novartis; 13.9.2011; appl. 17.11.2007; USA-prior. 18.11.2003).
- WO 2006 135619 (Novartis; 21.12.2006; USA-prior. 6.9.2005).
- e EP 2 626 355 (Natco Pharma; 14.8.2013; appl. 9.2.2012).
-
Inhibitors of mutant form of KIT:
- US 8 017 621 (Novartis; 13.9.2011; appl. 17.11.2004; USA-prior. 18.11.2003).
-
Salts of Nilotinib:
- US 8 163 904 (Novartis; 24.4.2012; appl. 18.7.2006; USA-prior. 20.7.2005).
- US 8 389 537 (Novartis; 5.3.2013; appl. 13.3.2012; USA-prior. 20.7.2005).
-
Pharmaceutical compositions:
- US 8 293 756 (Novartis; 23.10.2012; appl. 25.9.2007; EP-prior. 27.9.2006).
- US 8 501 760 (Novartis; 6.8.2013; appl. 21.9.2012; EP-prior. 27.9.2006).
-
Crystalline forms:
- US 8 343 984 (Novartis; 1.1.2013; appl. 18.7.2006; USA-prior. 20.7.2005).
- US 8 415 363 (Novartis; 9.4.2013; appl. 3.8.2012; USA-prior. 20.7.2005).
//////////Nilotinib, AMN107, Tasigna, ニロチニブ,

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
















![Graphical abstract: One-pot synthesis of high molar activity 6-[18F]fluoro-l-DOPA by Cu-mediated fluorination of a BPin precursor](https://pubs.rsc.org/en/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=C9OB01758E&imageInfo.ImageIdentifier.Year=2019 "Graphical abstract")