
Home » Uncategorized (Page 227)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
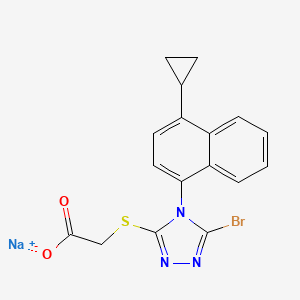
Lesinurad
Lesinurad
Acetic acid, 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]-,
sodium salt (1:1)
Sodium 2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-
yl]sulfanyl}acetate
2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid
MOLECULAR FORMULA C17H13BrN3NaO2S
MOLECULAR WEIGHT 426.3
http://clinicaltrials.gov/show/NCT01508702
http://www.ama-assn.org/resources/doc/usan/lesinurad.pdf
Ardea Biosciences, Inc.
- Lesinurad
- RDEA 594
- RDEA594
- UNII-09ERP08I3W
Gout phase 3
Gout is associated with elevated levels of uric acid that crystallize and deposit in joints, tendons, and surrounding tissues. Gout is marked by recurrent attacks of red, tender, hot, and/or swollen joints.
This study will assess the serum uric acid lowering effects and safety of lesinurad compared to placebo in patients who are intolerant or have a contraindication to allopurinol or febuxostat.
http://euroscan.org.uk/technologies/technology/view/2386
Lesinurad (RDEA-594, lesinurad sodium) is a selective urate transporter-1 (URAT-1) inhibitor, which blocks the reabsorption of urate within the renal proximal tubule. It is intended for the treatment of gout after failure of first line therapy and is administered orally at 400mg once daily
A Phase 3 Randomized, Double-Blind, Multicenter, Placebo- Controlled Study to Assess the Efficacy and Safety of Lesinurad Monotherapy Compared to Placebo in Subjects With Gout and an Intolerance or Contraindication to a Xanthine Oxidase Inhibitor
AstraZeneca’s lesinurad (formerly known as RDEA-594) is a selective oral Uric Acid Transporter URAT1 inhibitor currently in Phase III development for the treatment of of gout. The regulatory filings for lesinurad in the US and Europe are expected for the first half of 2014.

Gout (also known as podagra when it involves the big toe), while not life-threatening, is an excruciatingly painful condition caused by a buildup of a waste product in the blood called uric acid, which is normally eliminated from the body through urine. Excess Uric acid crystallizes and get deposited in the joints (usually the big toes), creating symptoms similar to an acute arthritis flare. Gout has seen a recent gradual resurgence as a result of rising obesity rates and poor diet according to a study in the journal Annals of the Rheumatic Diseases.
The current Standard treatment for gout works by inhibiting a protein called xanthine oxidase that helps in the formation of the uric acid. These therapies, some of which have been used for more than 50 years, are not effective in all patients. One is a generic drug called allopurinol that was approved in the U.S. in 1966. The other is febuxostat, marketed by Takeda Pharmaceutical Co. in the U.S. asUloric and by Ipsen SA and others in Europe as Adenuric and approved in the U.S. in 2009.
AstraZeneca’s new product Lesinurad, a selective uric acid re-absorption inhibitor (SURI), tackles gout by blocking a protein called Uric acid trasporter 1 (URAT1) that otherwise would cause the body to reabsorb the uric acid. AstraZeneca acquired lesinurad (aka RDEA-594) as part of its $1.26 billion takeouver of San Diego-based Ardea Biosciences in 2012. RDEA594 is a metabolite of RDEA806, a non-nucleoside reverse transcriptase inhibitor originally developed for HIV.

In top-line results from a Phase III LIGHT study released by AstraZeneca in December 2013 on gout patients who get no benefit from Zyloprim (allopurinol) and febuxostat, lesinurad alone significantly reduced serum levels of uric acid. The company has three other phase III studies ongoing that are testing the use of the drug alongside allopurinol and febuxostat, and these should generate results in the middle of 2014. Analysts at JPMorgan Chase forecast lesinurad alone may have peak sales of $1 billion a year. AstraZeneca also has a second, more potent drug called RDEA3179 to treat elevated levels of uric acid or hyperuricemia. Pfizer’s KUX-1151, licensed from Japan’s Kissei Phmarceuticals, is in early stage development.
Gout is not an automatic success indication of drugmakers. Savient Pharmaceuticals filed for Chapter 11 bankruptcy in October 2013 in the face of a severe cash crisis, having spent hundreds of millions of dollars on its would-be flagship — the gout-fighting drug Krystexxa (pegloticase) — with limited results. Krystexxa (pegloticase), a twice-monthly infusion designed to treat severe chronic gout that doesn’t respond to conventional therapy, was approved by the U.S. Food and Drug Administration in September 2010. Crealta Pharmaceuticals acquired Savient for $120.4 million in December 2013.
Lesinurad
RDEA-594
2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]sulfanyl}acetic acid
CAS number: 878672-00-5 (Lesinurad), 1151516-14-1 (Lesinurad sodium)
Mechanism of Action:once-daily inhibitor of URAT1, a transporter in the kidney that regulates uric acid excretion from the body
US patents:US8242154 , US8173690, US808448
Indication: Gout
Developmental Status: Phase III (US, UK, EU)
Originator: Ardea Biosciences (Acquired by AstraZeneca for $1.26 billion in 2012)
Developer: AstraZeneca
…………………………
http://www.google.co.in/patents/US8242154
Example 8 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)-N-(2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume, to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 mins and the organic layer removed. The aqueous layer was cooled to 0° C. and acidified by treatment with HCl (1N) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3×) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
Example 102 Methyl 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate


Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0° C., and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%).

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).

A solution of 1-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropylnaphthalene (3.1 g, 73%).

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of 1-amino-4-cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) at 0° C. The reaction mixture was stirred for 5 min at 0° C. and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
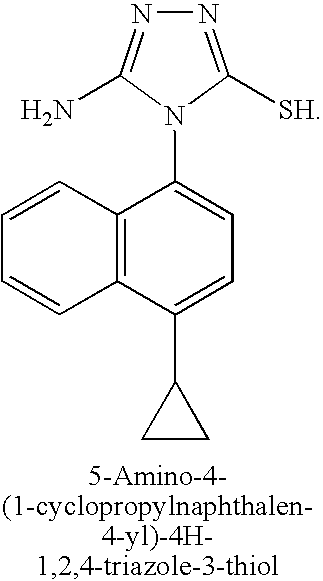
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanatonaphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50° C. for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.0 g, 49%).

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 mins to a suspension of 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
Example 104 Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate

Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 mins to a solution of 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10° C. The mixture was stirred at 10° C. for a further 10 mins. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 103 2-(5-Bromo-4-(1-cyclopropylnapthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 mins to a solution of methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (prepared as described in example 1 above; 1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0° C. The mixture was stirred at 0° C. for a further 45 mins and then neutralized to pH 7 by the addition of 0.5N HCl solution at 0° C. The resulting mixture was concentrated in vacuo to ⅕th of its original volume, then diluted with water (˜20 mL) and acidified to pH 2-3 by the addition of 0.5N HCl to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with DCM is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (1.02 g, 93%).
 |
1H NMR (400 MHz, DMSO-d6) δ ppm 0.84-0.91 (m, 2 H) 1.12-1.19 (m, 2 H) 2.54-2.61 (m, 1 H) 3.99 (d, J = 1.45 Hz, 2 H) 7.16 (d, J = 7.88 Hz, 1 H) 7.44 (d, J = 7.46 Hz, 1 H) 7.59-7.70 (m, 2 H) 7.75 (td, J = 7.62, 1.14 Hz, 1 H) 8.59 (d, J = 8.50 Hz, 1 H) 12.94 (br. s., 1 H) | Mass found: 404.5 (M + 1) | B |
……
POLYMORPHS AND SYNTHESIS
Described herein are various polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate which decreases uric acid levels, (see for example US patent publication 2009/0197825, US patent publication 2010/0056464 and US patent publication 2010/0056465). Details of clinical studies involving sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate have been described in International patent application
PCT/US2010/052958.
Polymorph Form A
In one embodiment, sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H- l,2,4-triazol-3-ylthio)acetate polymorph Form A exhibits an x-ray powder diffraction pattern characterized by the diffraction pattern summarized in Table 1 A or Table IB. In some embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4- cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 3 peaks of (±0.1°2Θ) of Table 1A or IB. In certain embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 4 peaks of (±0.1°2Θ) of Table 1A or IB, at least 5 peaks of (±0.1°2Θ) of Table 1A or IB, at least 6 peaks of (±0.1°2Θ) of Table 1A or IB, at least 8 peaks of
(±0. Γ2Θ) of Table 1A or IB, at least 10 peaks of (±0. Γ2Θ) of Table 1A, at least 15 peaks of (±0. Γ2Θ) of Table 1A, at least 20 peaks of (±0. Γ2Θ) of Table 1A, at least 25 peaks of (±0.1 °2Θ) of Table 1A, or at least 30 peaks of (±0.1 °2Θ) of Table 1A.


Examples
I Preparation of compounds
Example 1: Preparation of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate
Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetate was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.

[00103] Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 min to a solution of 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H- l,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10 °C. The mixture was stirred at 10 °C for a further 10 min. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 2: Preparation of 2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol- 3-ylthio)acetic acid
2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetic acid was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.
[00104] Route i:

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)-N- (2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures, see US 2009/0197825; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 min and the organic layer removed. The aqueous layer was cooled to 0 °C and acidified by treatment with HCl (IN) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3x) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5- bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
[00105] Route ii:

STEP A: 1-Cyclopropylnaphthalene

Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [l,3-bis(diphenylphosphino)propane] dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0 °C, and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%). ] STEP B: l-Cyclopropyl-4-nitronaphthalene

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0 °C. The reaction mixture was stirred at 0 °C for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).
[00108] STEP C: l-Amino-4-cyclopropylnaphthalene

A solution of l-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-amino-4-cyclopropylnaphthalene (3.1 g, 73%).
STEP D: l-Cyclopropyl-4-isothiocvanatonaphthalene

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of l-amino-4- cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in
dichloromethane (50 mL) at 0 °C. The reaction mixture was stirred for 5 min at 0 °C and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield l-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%>).
[00110] STEP E: 5-Amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol

A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), l-cyclopropyl-4- isothiocyanato naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50 °C for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50 °C for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol (2.0 g, 49%). [00111] STEP F: Methyl 2-(5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- -3 -ylthio)acetate

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 min to a suspension of 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room
temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the presence of P2O5 to yield methyl 2-(5- amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).
[00112] STEP G: Methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3 -ylthio)acetate

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room
temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
[00113] STEP H: 2-(5-Bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- )acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 min to a solution of methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3-ylthio)acetate (1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0 °C. The mixture was stirred at 0 °C for a further 45 min and then neutralized to pH 7 by the addition of 0.5N HC1 solution at 0 °C. The resulting mixture was concentrated in vacuo to l/5th of its original volume, then diluted with water (~20 mL) and acidified to pH 2-3 by the addition of 0.5N HC1 to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with dichloromethane is recommended.) The tan solid was
collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the
presence of P2O5 to yield 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- ylthio)acetic acid (1.02 g, 93%).
………………………….
EXAMPLES
The following experiments are provided only by way of example, and should not be understood as limiting the scope of the invention.
COMPOUNDS OF THE INVENTION 2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method A)

1-Cyclopropyl-naphthalene
Cyclopropylmagnesium bromide (150 mL, 0.5 M in tetrahydrofuran) was slowly added to a solution of 1-bromo-naphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloronickel(II) in tetrahydrofuran (10 mL) stirred at 0° C. The reaction mixture was stirred at room temperature for 16 hours and the solvent was evaporated under reduced pressure. EtOAc and ammonium chloride in water were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-naphthalene (6.4 g, 76%).
1-Cyclopropyl-4-nitro-naphthalene
Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropyl-naphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then was slowly poured into ice. Water was added, followed by EtOAc. After extraction, the organic layer was washed with a 1% aqueous solution of NaOH, then washed with water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitro-naphthalene (5.2 g, 64%).
1-Amino-4-cyclopropyl-naphthalene
A solution of 1-cyclopropyl-4-nitro-naphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, then filtered over celite. The solvent was evaporated, and the residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropyl-naphthalene (3.1 g, 73%).
1-Cyclopropyl-4-isothiocyanato-naphthalene
Thiophosgene (1.1 g, 9.7 mmol) was added to a solution of 1-amino-4-cyclopropyl-naphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) stirred at 0° C. The reaction mixture was stirred for 5 min at this temperature, then a 1% solution of HCl in water was added and the organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent was evaporated under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanato-naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was evaporated, toluene was added, and the solvent was evaporated again. A 2.0 M aqueous solution of sodium hydroxide (30 mL) was added and the reaction mixture was heated at 50° C. for 60 hours. The reaction mixture was filtered, and the filtrate was neutralized with a 2.0 M aqueous solution of HCl. New filtration, then evaporation of solvent and purification of the residue by silica gel chromatography to yield 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (2.0 g, 49%).
2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)Acetamide
In a solution of 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (708 mg, 2.5 mmol), K2CO3 (380 mg, 2.5 mmol) in DMF (20 mL) was added 2-chloro-N-(2-chloro-4-sulfamoylphenyl)acetamide (710 mg, 2.5 mmol). The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction, the solvent was evaporated. The residue was purified by silica gel chromatography to yield 2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (1.26 g, 95%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide
Dichloroacetic acid (180 uL, 2.2 mmol) was added to a suspension of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (0.59 g, 1.1 mmol), sodium nitrite (1.5 g, 22 mmol) and BTEABr (0.91 g, 3.3 mmol) in dibromomethane (30 mL). The reaction mixture was stirred at room temperature for 4 hours, then extracted with dichloromethane and sodium bicarbonate in water. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (224 mg, 31%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method B)

2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole | 2.24 | g | 282.36 | 7.9 |
| methyl chloroacetate | 0.73 | ml | 108.52 | 8.3 (1.05 eq) |
| potassium carbonate | 1.21 | g | 138.21 | 8.7 (1.1 eq) |
| dimethylformamide | 40 | ml | (5 mL/mmol) | |
Procedure:
To a suspension of thiotriazole and potassium carbonate in DMF was added methyl chloroacetate dropwise at room temperature for 5 min. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2.24 g (80%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | ||
| thiotriazole L10183-58 | 709 | mg | 354.43 | 2.0 | |
| bromoform | 10 | ml | (5 ml/mmol) | ||
| sodium nitrite | 2.76 | g | 69.00 | 40 | (20 eq) |
| benzyltriethylammonium | 1.63 | g | 272.24 | 6.0 | (3 eq) |
| bromide | |||||
| dichloroacetic acid | 0.33 | ml | 128.94 | 4.0 | (2 eq) |
Procedure:
To a solution of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester and benzyltriethylammonium chloride in bromoform was added sodium nitrite. To the mixture was added dichloroacetic acid and the reaction mixture was stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel that was packed with CH2Cl2. The column was first eluted with CH2Cl2 until all CHBr3 eluted, and was then eluted with acetone/CH2Cl2 (5:95) to give 713 mg (85%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole methyl ester | 1.14 | g | 418.31 | 2.7 |
| tetrahydrofuran | 10 | ml | (~3 ml/mmol) | |
| ethanol | 10 | ml | (~3 ml/mmol) | |
| water | 10 | ml | (~3 ml/mmol) | |
| lithium hydroxide | 98 | mg | 23.95 | 4.1 (1.5 eq) |
Procedure:
To a solution of 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester, in a mixture of THF and EtOH at 0° C., was added a solution of LiOH in H2O dropwise over 5 min. The reaction was complete after stirring at 0° C. for an additional 45 min. The reaction was neutralized to pH 7 by the addition of 0.5 N HCl solution at 0° C., and the resulting mixture was concentrated in vacuo to ⅕th of its original volume. The mixture was diluted with H2O (˜20 mL) and acidified to pH 2-3 by the addition of 0.5 N HCl to produce sticky solid. (If the product comes out as an oil during acidification, extraction with CH2Cl2 is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 1.02 g (93%) of the title compound.
REF:
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8242154 B2 ;Also published as US20100056465, US20130040907;Original Assignee: Ardea Biosciences, Inc
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8173690 B2;Also published as US20100056464;Original Assignee: Ardea Biosciences, Inc
Barry D. Quart, Jean-Luc Girardet, Esmir Gunic, Li-Tain Yeh;Compounds and compositions and methods of use;US patent number US8084483 B2; Also published as CA2706858A1, CA2706858C, CN101918377A, CN102643241A, CN103058944A, EP2217577A2, EP2217577A4, US8283369, US8357713, US8546437, US20090197825, US20110268801, US20110293719, US20120164222, US20140005136, WO2009070740A2, WO2009070740A3;Original Assignee:Ardea Biosciences, Inc.
Gunic, Esmir; Galvin, Gabriel;Manufacture of 2-[5-bromo-4-(cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio]acetic acid and related compounds;PCT Int. Appl., WO2014008295 A1
Zamansky, Irina et al;Process for preparation of polymorphic, crystalline, and mesophase forms of 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]acetic acid sodium salt; PCT Int. Appl., WO2011085009
Gunic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels; U.S. Pat. Appl. Publ., 20100056465
unic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels;U.S. Pat. Appl. Publ., 20100056464
Quart, Barry D. et al;Preparation of azole carboxylates as modulators of blood uric acid levels;PCT Int. Appl., 2009070740, 04 Jun 2009
Girardet, Jean-Luc et al;Preparation of S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase;PCT Int. Appl., WO2006026356
US20100056465 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
US20100056542 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
WO2009070740A2 * Nov 26, 2008 Jun 4, 2009 Ardea Biosciences Inc Novel compounds and compositions and methods of use
WO2011085009A2 * Jan 5, 2011 Jul 14, 2011 Ardea Biosciences, Inc. Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof
| WO2011159732A1 * | Jun 14, 2011 | Dec 22, 2011 | Ardea Biosciences,Inc. | Treatment of gout and hyperuricemia |
| WO2012092395A2 * | Dec 28, 2011 | Jul 5, 2012 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio) acetic acid and uses thereof |
| EP2560642A2 * | Mar 29, 2011 | Feb 27, 2013 | Ardea Biosciences, Inc. | Treatment of gout |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US20100056465 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20100056542 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| WO2011085009A2 * | Jan 5, 2011 | Jul 14, 2011 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof |
| US8283369 | 30 Jun 2011 | 9 Oct 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | 30 Jun 2011 | 22 Jan 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8546437 | 30 Jun 2011 | 1 Oct 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8633232 * | 4 May 2012 | 21 Jan 2014 | Ardea Biosciences, Inc. | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20130040907 * | 4 May 2012 | 14 Feb 2013 | Ardea Biosciences Inc. | Compounds, Compositions and Methods of Using Same for Modulating Uric Acid Levels |
| WO2006026356A2 * | Aug 25, 2005 | Mar 9, 2006 | La Rosa Martha De | S-TRIAZOLYL α-MERCAPTOACETANILDES AS INHIBITORS OF HIV REVERSE TRANSCRIPTASE |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| US20090197825 * | Nov 26, 2008 | Aug 6, 2009 | Ardea Biosciences, Inc. | Novel compounds and compositions and methods of use |
| US7947721 | Aug 25, 2005 | May 24, 2011 | Ardes Biosciences Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8084483 | Nov 26, 2008 | Dec 27, 2011 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8106205 | Feb 2, 2010 | Jan 31, 2012 | Ardea Biosciences, Inc. | N[S(4-aryl-triazol-3-yl)α-mercaptoacetyl]p-amino benzoic acids as HIV reverse transcriptase inhibitors |
| US8252828 | Jun 30, 2011 | Aug 28, 2012 | Ardea Biosciences, Inc. | S-triazolyl α-mercapto acetanilides as inhibitors of HIV reverse transcriptase |
| US8283369 | Jun 30, 2011 | Oct 9, 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | Jun 30, 2011 | Jan 22, 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8372807 | May 20, 2010 | Feb 12, 2013 | Ardea Biosciences, Inc. | Methods of modulating uric acid levels |
| US8481581 | Jul 18, 2012 | Jul 9, 2013 | Ardea Biosciences, Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8524754 | Jan 5, 2011 | Sep 3, 2013 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio) acetate, and uses thereof |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US8546437 | Jun 30, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |

An unexpected phenomenon concerning an otherwise common impurity of lesinurad has been observed in the context of synthetic process development. A new industrial process was designed as a chlorine-free process, but the critical chlorinated impurity 10 was surprisingly detected in the isolated product. Because of the structural similarity of the impurity and the product, no efficient separation of 10 by conventional methods (e.g., crystallization) was discovered. The formation of the impurity was explained by a chlorine impurity in a commercial brominating agent. This communication also describes control of the critical impurity.
Identification of an Unexpected Impurity in a New Improved Synthesis of Lesinurad

2-[[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]thio]acetic Acid (1)
1H NMR (DMSO-d6), δ (ppm): 0.87 (m, 2H); 1.15 (m, 2H); 2.56 (m, 1H); 4.00 (m, 2H); 7.16 (d, J = 8.5 Hz, 1H); 7.44 (d, J = 7.3, 1H); 7.64 (d, J = 7.3 Hz, 1H); 7.66 (t, J= 7.6 Hz, 1H); 7.74 (t, J = 7.6 Hz, 1H); 8.58 (d, J = 8.5 Hz, 1H); 12.99 (s, COOH). 13C NMR (DMSO-d6), δ (ppm): 7.3; 7.4; 12.9; 34.1; 121.8; 122.7; 125.2; 126.6; 126.8; 127.3; 128.1; 128.6; 131.4; 133.5; 143.2; 153.5; 169.0.
TONIX Completes Pre-Phase 3 Meeting With U.S. Food and Drug Administration for TNX-102 SL in Fibromyalgia

Regulatory Acceptance of Design of Registrational Clinical Studies; Dosing in First Safety and Efficacy Trial to Commence in the Third Quarter of 2013
NEW YORK, NY
March 11, 2013) – Tonix Pharmaceuticals Holding Corp. , a specialty pharmaceutical company developing novel treatments for challenging disorders of the central nervous system, including fibromyalgia (“FM”) and post-traumatic stress disorder (“PTSD”), announced that it recently held an End-of-Phase 2/Pre-Phase 3 meeting with the U.S. Food and Drug Administration (“FDA”) to discuss its proposed New Drug Application (“NDA”) plan for the Company’s novel sublingual tablet formulation of cyclobenzaprine for bedtime use, TNX-102 SL, for the management of FM. Official FDA meeting minutes indicate FDA acceptance of the clinical program and provide clear direction to achieve a successful NDA filing of TNX-102 SL in FM.
The registrational clinical trials will consist of two randomized, double-blind, placebo-controlled 12-week safety and efficacy studies in FM patients who will take either a TNX-102 SL (cyclobenzaprine HCl 2.8 mg) tablet or placebo at bedtime. The primary endpoint of both trials will be the change in pain from baseline to Week 12 as measured by the Numeric Rating Scale. The Company plans to conduct these trials in sequence, and expects to begin dosing in the first trial in the third quarter of 2013. This trial will enroll 100 to 200 FM patients, and top-line data are anticipated to become available in the second half of 2014.
Following the completion of the double-blind randomized portion of these studies, patients may be eligible to enroll in open-label extension studies of TNX-102 SL. The FDA agreed that the safety database needed to support a 505(b)(2) NDA submission for TNX-102 SL would contain a total exposure of at least 300 FM patients, with at least 100 patients receiving TNX-102 SL for six months and at least 50 patients for one year.
Seth Lederman, M.D., Chief Executive Officer of TONIX, said, “We view our meeting with the FDA as a major milestone for TONIX. We are pleased to have concurrence from the FDA on the design and selection of efficacy endpoints of our registrational clinical studies in FM in addition to receiving clear guidance on the remaining requirements for the TNX-102 SL NDA program. We are also pleased with the FDA’s requirements on chronic exposure, which are appropriately less than those typically needed for a new drug to be approved for a chronic use indication. We look forward to advancing TNX-102 SL towards a successful NDA filing.”
About Tonix Pharmaceuticals Holding Corp.
TONIX is developing innovative prescription medications for challenging disorders of the central nervous system. The Company targets conditions characterized by significant unmet medical need, inadequate existing treatment options, and high dissatisfaction among both patients and physicians. TONIX’s core technology improves the quality of sleep in patients with chronic pain syndromes, which is believed to translate into reductions in pain and other symptoms. An Investigational New Drug Application (“IND”) has been filed for the Company’s lead product candidate, TNX-102 SL, a novel under-the-tongue tablet formulation of cyclobenzaprine, the active ingredient in two FDA-approved muscle relaxants. TONIX expects to begin a registrational clinical study of TNX-102 SL in FM in the third quarter of 2013. TONIX expects to file an IND for TNX-102 SL in PTSD in the third quarter of 2013, and to begin a Phase 2 trial in this indication in the fourth quarter of 2013. To learn more, please visit www.tonixpharma.com.
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimated” and “intend,” among others. These forward-looking statements are based on TONIX’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, substantial competition; our ability to continue as a going concern; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payer reimbursement; limited sales and marketing efforts and dependence upon third parties; and risks related to failure to obtain FDA clearances or approvals and noncompliance with FDA regulations. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. TONIX does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K filed with the SEC on March 30, 2012 and future periodic reports filed with the Securities and Exchange Commission. All of the Company’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date hereof
Cyclobenzaprine, N,N-dimethyl-3-(dibenzo[a,d]cyclohepten-5-ylidene)propylamine, is synthesized by reacting 5H-dibenzo[a,d]cyclohepten-5-one with 3-dimethylaminopropylmagnesium chloride and subsequent dehydration of the resulting carbinol in acidic conditions into cyclobenzaprine. 
- H.La Roche, GB 858187 (1961).
- F.J. Villani, C.A. Ellis, C. Teihman, C. Biges, J. Med. Pharm. Chem., 5, 373 (1962).
- Winthrop, S. O.; Davis, M. A.; Myers, G. S.; Gavin, J. G.; Thomas, R.; Barber, R. (1962). “New Psychotropic Agents. Derivatives of Dibenzo[a,d]-1,4-cycloheptadiene”. The Journal of Organic Chemistry 27: 230. doi:10.1021/jo01048a057.
Phase 1- MERCK , Study of MK-8109 (Vintafolide) Given With Chemotherapy in Participants With Advanced Cancers

vintafolide
cas no 742092-03-1
http://www.ama-assn.org/resources/doc/usan/vintafolide.pdf
N-(4-{[(2-amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoyl)-L-γ-glutamyl-L-α- aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine disulfide with methyl (5S,7R,9S)- 5-ethyl-9-[(3aR,4R,5S,5aR,10bR,13aR)-3a-ethyl-4,5-dihydroxy-8-methoxy-6-methyl-5- ({2-[(2-sulfanylethoxy)carbonyl]hydrazinyl}carbonyl)-3a,4,5,5a,6,11,12,13a-octahydro- 1H-indolizino[8,1-cd]carbazol-9-yl]-5-hydroxy-1,4,5,6,7,8,9,10-octahydro-2H-3,7- methanoazacycloundecino[5,4-b]indol-9-carboxylate
Vincaleukoblastin-23-oic acid, O4-deacetyl-, 2-[(2-mercaptoethoxy)carbonyl]hydrazide, disulfide with N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-γ- glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine
Vintafolide is a water-soluble, folate-receptor-targeted conjugate of folate and the vinca alkaloid desacetylvinblastine monohydrazide (DAVLBH) with potential antineoplastic activity. The folate moiety of folate-vinca alkaloid conjugate EC145 binds to folic acid receptors on the tumor cell surface and the agent is internalized via folate receptor-mediated endocytosis, delivering the tubulin-binding DAVLBH moiety directly into the tumor cell; DAVLBH binding to tubulin results in the disruption of microtubule assembly-disassembly dynamics, cell cycle arrest, and tumor cell apoptosis. Folic acid receptors are frequently upregulated on the surfaces of many tumor cell types. DAVLBH is a derivative of the natural product vinblastine.
http://clinicaltrials.gov/show/NCT01688791
ClinicalTrials.gov Identifier:
Vintafolide is a derivative of the anti-mitotic chemotherapy drug vinblastine.[1] chemically linked to folic acid. The vintafolide molecule was designed to specifically target the toxic vinblastine group to cancer cellsthat overexpress the folic acid receptor.[2] Vintafolide is being studied for treatment of late-stage ovarian cancer and mid-stage non-small cell lung cancer.
Merck & Co. acquired the development and marketing rights to this experimental cancer drug from Endocyte in April 2012. Endocyte had planned to file for marketing approval for vintafolide in the third quarter of 2012. The drug received an orphan drug status in Europe in March 2012.[3] Endocyte remains responsible for the development and commercialization of etarfolatide, a non-invasive companion diagnostic imaging agent used to identify folate receptor positive tumor cells that may be susceptible to vintafolide.[4]
- Statement on a nonproprietary name adopted by the USAN Council, United States Adopted Names (USAN) Council, 6 April 2012
- Dosio F, Milla P, Cattel L. EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers. Curr Opin Investig Drugs. 2010 Dec;11(12):1424-33. Review. PubMed PMID: 21154124.
- Endocyte soars on cancer drug deal with Merck, Reuters, US Edition, Mon Apr 16, 2012.
- Merck, Endocyte in Development Deal Wed, Drug Discovery and Development. 04/25/2012

Chemical structure of EC-145
(source: THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, 2011, 336(2):336–343)
Phase 2 ORM-12741, Orion’s Experimental Alzheimer’s Drug Shows Promise, Study Finds
 ORM-12741 in WO 2003082866 ORM-12741 in WO 2003082866 |
The purpose of this study is to determine whether ORM-12741 is safe and effective in the treatment of Alzheimer’s disease.,
March 11, 2013
A small Finnish study is raising hopes for a new drug designed to help stave off memory loss among patients struggling with moderate Alzheimer’s disease.
Still in the preliminary stages of investigation, the drug — called ORM-12741 — showed promise during a three-month trial involving 100 such patients, half of whom were given the medication on top of their current drug treatment.
By the end of the study, memory scores plummeted by 33 percent among the 50 patients who were given a dummy pill (placebo) rather than the new drug, while patients who took the new drug showed a 4 percent improvement on the tests.
“The bottom line is that this was the first study investigating [effectiveness] of a drug with a novel mechanism of action in patients with Alzheimer’s disease,” said study lead author Dr. Juha Rouru, who heads the central nervous system therapy area at Orion Pharma in Turku, Finland.
“The results were clearly positive,” he said, adding they were seen particularly on important episodic memory, which involves remembering events and personal experiences. Orion, the maker of ORM-12741, funded the research.
Rouru and his colleagues are scheduled to present their work in San Diego at a meeting of the American Academy of Neurology, which starts Saturday.
By 2050, as the elderly population increases, an estimated 13.8 million Americans will have Alzheimer’s, a progressive brain disease that robs people of their memory and the ability to perform even simple everyday tasks. There is no cure for the disease, and drugs aimed at controlling the debilitating symptoms are only moderately effective, Rouru said.
With that in mind, the team set out to assess the potential of ORM-12741, the first drug to target a specific receptor in the brain, called alpha-2C. This receptor is thought to play a role in the brain’s “fight or flight” response to stress, and the authors noted that the new drug’s impact on alpha-2C had shown promise in prior animal studies.
All the patients in the study were already taking a cholinesterase drug. Some were also using memantine, another type of Alzheimer’s medication.
Fifty patients were then given a placebo on top of their current regimen, while 50 were given either a low-dose (30 to 60 milligrams) twice daily supply of ORM-12741 or a high-dose (100 to 200 milligrams) version.
Computerized memory tests highlighted an apparent memory benefit (without prompting severe side effects) among the ORM-12741 patients, and Rouru suggested that the new drug should be seen as just one more potentially effective tool in an ongoing battle to reign in “a devastating disease.”
“I am afraid that wonder drugs hardly exist,” he noted. “In the present study, our drug was used on top of existing Alzheimer’s medications. In that setting it showed clear effect, which suggests that it is giving additional clinically significant benefit for patients that are already using Alzheimer’s medications.”
Catherine Roe, an assistant professor of neurology at Washington University School of Medicine in St. Louis, described Rouru’s research as “impressive.”
“This is really a new approach, in terms of the biology that they’re targeting,” she noted. “And they showed significant results after only three months of treatment, which is exciting particularly because this drug combination was tested on people who had moderate Alzheimer’s disease.”
Many experts have thought moderate Alzheimer’s disease would be untreatable, she said. “By the time it’s that advanced, the nerves have already died and it would be too late to do anything about memory by this stage,” she explained.
Still, much more testing will need to be done, Roe cautioned. “And these results will have to be replicated with other groups of people,” she said. “But if they can do that, this would be awesome.”
The data and conclusions of research presented at medical meetings are typically considered preliminary until published in a peer-reviewed journal.
More information
For more on Alzheimer’s disease, visit the U.S. National Institutes of Health.
Phase 2, Sarepta Therapeutics, Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
s excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Clinical studies
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
References
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods.. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303.. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
Phase 2 Drug: Ustekinumab A monoclonal antibody against the p40 subunit of IL-12/23 Other Name: Stelara
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-12 and IL-23
|

Ustekinumab, CAS number 815610-63-0, is also known by it’s brand name Stelara, which is marketed by Janssen Biotech, Inc. Developed as a treatment for adults with moderate to severe plaque psoriasis
Rockefeller University, MAR 2013
http://clinicaltrials.gov/ct2/show/NCT01806662
Atopic dermatitis (AD) is a chronic disease associated with intense itching, which affects most aspects of everyday life in the majority of patients. Acute inflammation and extensor/facial involvement is common in infants, whereas chronic inflammation increases in prevalence with age, as do localization to flexures. AD has a complex background characterized by immune activation, increased epidermal thickness in chronic diseased skin, and defective barrier function. In normal, healthy skin, the outer layer of the epidermis, the stratum corneum is made up flattened dead cells called corneocytes held together by a mixture of lipids and proteins. The stratum corneum and, in particular, the lipid layer are vital in providing a natural barrier function that locks water inside the skin and keeps allergens and irritants out. In people with AD, the barrier function is defective, which leads to dry skin. As the skin dries out, it cracks allowing allergens and irritants to penetrate.
Mild AD can be controlled with emollients and topical medications. However, moderate to severe AD is extremely difficult to control and requires systemic treatment that is often unsatisfactory due to impracticality and lack of effectiveness. Only three therapeutic options exist for moderate to severe AD, including: 1) oral steroids 2) cyclosporine A (CsA), that is not widely used in the US as it is not FDA approved for AD and 3) ultraviolet phototherapy. Oral steroids and CsA treatments have major side effects and UV radiation therapy is highly inconvenient for patients. Several biologic medications, such as TNF-alpha inhibitors, are effective, convenient, and relatively safe therapies for psoriasis, but have thus far not shown efficacy in AD. Ustekinumab is a unique biologic medication that may specifically target AD.
The investigators study will determine whether there is a reversal of the skin thickness and the immune pathways involved in the disease during treatment with Ustekinimab and what specific immune cells are involved. The investigators are also interested to understand how the clinical reversal of the disease will correlate with tissue reversal of the disease.
Detailed Description:
In psoriasis, epidermal hyperplasia is driven by underlying immune activation, whether as a direct response to IL-20 family cytokines that induces hyperplasia and inhibits keratinocyte terminal differentiation or as an indirect response to immune-mediated injury to keratinocytes. The epidermal reaction in psoriasis is largely restored to normal with selective immune suppression. Hence, one might hypothesize that similar epidermal responses should occur in the presence of “generalized” cellular immune activation, in diseases with similar inflammatory infiltrate and epidermal hyperplasia, such as AD. In fact, psoriasis and AD share features of dense T-cells and dentritic cell infiltrates, as well as over-expression of IL-22 in skin lesions. These diseases also share similar epidermal hyperplasia in their chronic phases.
Work from the investigators group showed that IL-22 is a key cytokine in the pathogenesis of both AD and psoriasis. The investigators have demonstrated that in psoriasis, ustekinumab suppresses the production of IL-12, IL-23, and IL-22. Additionally, by RT-PCR the investigators demonstrated that the mRNA expression of p40 cytokine and the IL23R is up-regulated in AD as compared to both normal skin and psoriasis. The investigators therefore hypothesize that ustekinumab will suppress IL-22 and possibly also p40 production in AD lesions and reverse both the epidermal growth/differentiation defects and the underlying immune activation, and hence will suppress disease activity. Interestingly, p40 was also found to be significantly up-regulated in non-lesional AD skin as compared with normal skin.
Although AD is thought to be predominately a disease of Th2-type cells, in the chronic stage, there is large Th1 component. To date, the precise mechanism by which sequential activation of Th2 and Th1 cells in AD is achieved remains unknown. IL-12 induces the differentiation and maturation of human Th cells into Th1-type cells. Recent circumstantial evidence suggests that in AD patients IL-12 may facilitate a change from the Th2-type to a Th1 cytokine profile. IL-12 was recently shown to be highly elevated in pediatric AD and its levels were strongly associated with disease severity.
Expression of IL-12 p40 mRNA is significantly enhanced in lesional skin from AD, suggesting that the enhanced local production of IL-12 in dendritic cells and macrophages may be responsible for the increased production of IFN-γ in chronic lesions potentially suggesting that IL-12 may have a pivotal role in promoting inflammation in atopic dermatitis. Topical steroids which constitute a mainstay of therapy in AD are known to strongly down-regulate IL-12 expression, possibly also indicating that targeted anti IL-12 therapy might important role in treating AD.
Recently, the Th1/Th2 paradigm in autoimmunity and allergy has been revisited to include a role for a new population of IL-17-producing Th cells known as Th17. Th17 cells are characterized by the production of inflammatory cytokines such as IL-17A, IL-17F, IL-22, and IL-26. One of the key factors involved in naive Th-cell commitment to a Th17 phenotype is IL-23.
Patients with acute AD were found to have increased Th17 T-cells in peripheral blood by flow cytometry and intracellular cytokine staining 26 as well as by immunohistochemistry (IHC) in lesions. Since IL-23 is the major inducer of Th17 T-cells, as well as “T22” T-cells, neutralization of IL-23 could potentially result in both decreased Th17 signal in acute AD as well as decreased “T22/IL22″ signal. Therefore the investigators postulate that ustekinumab in AD will act both inhibiting the IL-12-dependent Th1 shift in chronic AD stage as well as the pathogenic IL-22/”T22” axis in this disease.
Ustekinumab [1] (INN, experimental name CNTO 1275, proprietary commercial name Stelara,[2] Centocor) is a human monoclonal antibody. It is directed against interleukin 12 and interleukin 23, naturally occurring proteins that regulate the immune system and immune-mediated inflammatory disorders.[3]
In two Phase III trials for moderate to severe psoriasis, the longest >76 weeks, ustekinumab was safe and effective.[4][5]
A third Phase III trial, ACCEPT, compared the efficacy and safety of ustekinumab with etanercept in the treatment of moderate to severe plaque psoriasis.[6] This trial found a significantly higher clinical response with ustekinumab over the 12-week study period compared to high-dose etanercept.[6] It also demonstrated the clinical benefit of ustekinumab among patients who failed to respond to etanercept.[6]
Ustekinumab is approved in Canada, Europe and the United States to treat moderate to severe plaque psoriasis.[7]
As of November 2009, the drug is being investigated for the treatment of psoriatic arthritis.[8][9] It has also been tested in Phase II studies for multiple sclerosis[10] and sarcoidosis, the latter versus golimumab (Simponi).[11]
- Cingoz, Oya (2009). “Ustekinumab”. MAbs 1 (3): 216–221. doi:10.4161/mabs.1.3.8593. PMC 2726595. PMID 20069753.
- ^ European Medicines Agency, 20 November 2008, http://www.emea.europa.eu/pdfs/human/opinion/Stelara_58227008en.pdf
- ^ Reddy M, Davis C, Wong J, Marsters P, Pendley C, Prabhakar U (May 2007). “Modulation of CLA, IL-12R, CD40L, and IL-2Ralpha expression and inhibition of IL-12- and IL-23-induced cytokine secretion by CNTO 1275”. Cell. Immunol. 247 (1): 1–11. doi:10.1016/j.cellimm.2007.06.006. PMID 17761156.
- ^ Leonardi CL, Kimball AB, Papp KA, et al. (May 2008). “Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1)”. Lancet 371 (9625): 1665–74. doi:10.1016/S0140-6736(08)60725-4. PMID 18486739.
- ^ Papp KA, Langley RG, Lebwohl M, et al. (May 2008). “Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2)”. Lancet 371 (9625): 1675–84. doi:10.1016/S0140-6736(08)60726-6. PMID 18486740.
- ^ a b c Griffiths C, Strober B, van de Kerkhof P et al. (2010). “Comparison of Ustekinumab and Etanercept for Moderate-to-Severe Psoriasis”. N Engl J Med 362 (2): 118–28. doi:10.1056/NEJMoa0810652. PMID 20071701.
- ^ Medarex to Receive Milestone Payment for Approval of STELARA(TM) (Ustekinumab) for the Treatment of Moderate to Severe Plaque Psoriasis
- ^ ClinicalTrials.gov NCT00267956 A Study of the Safety and Efficacy of CNTO 1275 in Patients With Active Psoriatic Arthritis
- ^ ClinicalTrials.gov NCT01009086 A Study of the Safety and Efficacy of Ustekinumab in Patients With Psoriatic Arthritis
- ^ ClinicalTrials.gov NCT00207727 A Safety and Efficacy Study of CNTO1275 in Patients With Multiple Sclerosis
- ^ ClinicalTrials.gov NCT00955279 A Study to Evaluate the Safety and Effectiveness of Ustekinumab or Golimumab Administered Subcutaneously (SC) in Patients With Sarcoidosis
- ^ http://www.empr.com/stelara-approved-for-moderate-to-severe-psoriasis/article/149760/
- ^ a b Centocor 12/19/08 Press Release, http://www.centocor.com/centocor/i/press_releases/FDA_ISSUES_COMPLETE_RESPONSE_LETTER_TO_CENTOCOR_FOR_USTEKINUMAB_BIOLOGIC_LICENSE_APPLICATION_
- ^ Johnson LL. “Study: Drug for serious psoriasis tops competition” The Associated Press. 18 Sept 2008.[dead link]
- ^ Wild, David (November 2011), “Novel IL-12/23 Antagonist Shows Potential in Severe Crohn’s”, Gastroenterology & Endoscopy News 62 (11), retrieved 2011-12-04
- ^ a b c Weber J, Keam SJ (2009). “Ustekinumab”. BioDrugs 23 (1): 53–61. doi:10.2165/00063030-200923010-00006. PMID 19344192.
- ^ Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH (September 2008). “Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study”. Lancet Neurol 7 (9): 796–804. doi:10.1016/S1474-4422(08)70173-X. PMID 18703004.
- ^ “Important Safety Information”. STELARA® (ustekinumab). Janssen Biotech.
External links
- Centocor Ortho Biotech official site
- CNTO 1275 research studies registered with U.S. National Institutes of Health:
- ClinicalTrials.gov NCT00207727 Phase II Study on Multiple Sclerosis
- ClinicalTrials.gov NCT00320216 Phase II Study on Psoriasis
- ClinicalTrials.gov NCT00267969 Phase III Study on Psoriasis
- ClinicalTrials.gov NCT00307437 Phase III Study on Psoriasis
- ClinicalTrials.gov NCT00267956 Phase II Study on Psoriatic Arthritis
- Sylvester, Bruce (2006-03-06). “CNTO 1275 Shows Efficacy for Psoriasis: Presented at AAD”. Doctor’s Guide Publishing. Retrieved 2007-01-25.
Phase 3-Gilead’s newly-acquired Sofosbuvir, GS-7977 shines in Hepatitis C trial

{kind=link}
Sofosbuvir
Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate

The Foster City, CA-based Gilead said that its experimental drug GS-7977, originally known as PSI-7977 before the acquisition, when combined with ribavirin, cured a group of genotype 1 hepatitis C patients after four weeks of treatment. The clinical study involved hepatitis C patients who either failed to respond to previous therapies or had not been treated before. The genotype 1 is the most common form of HCV in the United States. It affects 70 to 90 percent of the people in this country who have hepatitis C.
Norbert Bischofberger, chief scientific officer at Gilead said patients with genotype 1 hepatitis C had no detectable signs of the virus after treated with GS-7977 combination therapy for a course of close to a month. Previous study showed the drug candidate could also cure patients with genotype 2 and 3 HCV.
Gilead gained rights to GS-7977 through the $11 billion Pharmasset acquisition deal, which enable the company to be in an advanced position to compete with a few pharma companies seeking to develop an all-oral regimen for hepatitis C. The 100 percent cure rate data suggested that GS-7977 may be one of the most promising therapies for hepatitis C.
Last year, GS-7977, an oral uridine nucleotide analog polymerase inhibitor of HCV, received fast track designation from the U.S. FDA for the treatment of HCV infection.
The World Health Organization estimated that 3–4 million people are infected with HCV each year. Some 130–170 million people are chronically infected with HCV and at risk of developing liver cirrhosis and/or liver cancer, and more than 350,000 people die yearly from hepatitis C-related diseases.
Sofosbuvir (formerly PSI-7977 or GS-7977) is an experimental drug candidate for the treatment of hepatitis C.[1] It was discovered at Pharmasset and then acquired for development by Gilead Sciences. It is currently in Phase III clinical trials.[2]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-monophosphate.[3]
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) polymerase.[4] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[5] It has shown excellent clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[6]
- Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S. et al. (2010). “Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus”. Journal of Medicinal Chemistry 53 (19): 7202–7218. doi:10.1021/jm100863x. PMID 20845908. edit
- “PSI-7977”. Gilead Sciences.
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H. M. M.; Bao, D.; Chang, W.; Espiritu, C. et al. (2010). “Mechanism of Activation of PSI-7851 and Its Diastereoisomer PSI-7977”. Journal of Biological Chemistry 285 (45): 34337–34347. doi:10.1074/jbc.M110.161802. PMC 2966047. PMID 20801890. edit
- Alejandro Soza (November 11, 2012). “Sofosbuvir”. Hepaton.
- Tom Murphy (November 21, 2011). “Gilead Sciences to buy Pharmasset for $11 billion”. Bloomberg Businessweek.
- http://www.gilead.com/pr_1757156
- AASLD: PSI-7977 plus Ribavirin Can Cure Hepatitis C in 12 Weeks without Interferon. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Nucleotide Polymerase Inhibitor Sofosbuvir plus Ribavirin for Hepatitis C. Gane, E et al. New England Journal of Medicine 368:3444. January 3, 2013.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, L. HIVandHepatitis.com. 4 March 2013.
MAA EU -GSK submits diabetes drug Eperzan, albiglutide in EU

MAA EU =marketing authorisation application EU
MAR 08 2013
GlaxoSmithKline has announced the submission of a marketing authorisation application for albiglutide, which will have the brand name Eperzan, to the European Medicines Agency.
The filing of albiglutide, a once-weekly treatment for type 2 diabetes, comes almost two months after it was filed in the USA. The drug is a GLP-1 receptor agonist, the same class of injectable treatments dominated by Novo Nordisk’s once-a-day Victoza (liraglutide), twice-daily Byetta (exenatide) and an extended-release formulation of the latter, Bydureon. They were developed and sold by Amylin, which was then acquired by Bristol-Myers Squibb and AstraZeneca.
The filing is based in part on a study which assessed albiglutide against Merck & Co’s DPP-4 inhibitor Januvia (sitagliptin) which showed that GSK’s drug showed clinically and statistically significant reductions in HbA1c from baseline and superiority versus the US firm’s diabetes blockbuster. However in data from a late-stage study released in November 2011, albiglutide failed to show non-inferiority to Victoza and a number of analysts believe GSK will have its work cut out to grab a decent share of the GLP-1 market.
Albiglutide is a glucagon-like peptide-1 agonist (GLP-1 agonist) drug under investigation by GlaxoSmithKline for treatment of type 2 diabetes. It is a dipeptidyl peptidase-4-resistant glucagon-like peptide-1 dimer fused to human albumin.
Albiglutide has a half-life of four to seven days, which is considerably longer than the other two GLP-1 analogs approved for market use, exenatide (Byetta) and liraglutide (Victoza).[1] [2] GLP-1 drugs are currently only available for subcutaneous administration on a daily basis, so a GLP-1 drug with a longer half-life is desirable. Such a drug would only need to be injected biweekly or weekly instead of daily, reducing the discomfort and inconvenience of GLP-1 administration considerably.
It has not yet been determined whether albiglutide is as effective an antidiabetic agent as GLP-1 drugs currently on the market, and final data remain to be published regarding the incidence of adverse effects related to the drug. To evaluate the efficacy and safety of the drug, albiglutide is undergoing eight Phase III clinical trials. Four of these trials should report useful data by end 2010.[3]
- Matthews JE, Stewart MW, De Boever EH, et al. (December 2008). “Pharmacodynamics, Pharmacokinetics, Safety, and Tolerability of Albiglutide, a Long-Acting Glucagon-Like Peptide-1 Mimetic, in Patients with Type 2 Diabetes”. J. Clin. Endocrinol. Metab. 93 (12): 4810–4817. doi:10.1210/jc.2008-1518. PMID 18812476.
- Baggio et al. (2008). “Glucagon-like Peptide-1 Analogs Other Than Exenatide”.
- “Phase III clinical trials of Albiglutide”.
ViiV Healthcare presents phase III SAILING study data of dolutegravir vs raltegravir in treatment-experienced adults with HIV-1

Dolutegravir
| Identifiers | |
|---|---|
| CAS number | 1051375-16-6 |
8 TH MATCH 2013
ViiV Healthcare, a global specialist HIV company established in November 2009 by GSK and Pfizer dedicated to delivering advances in treatment and care for people living with HIV, has announced 24-week data from the phase III SAILING (ING111762) study evaluating the investigational integrase inhibitor dolutegravir in patients with HIV-1 who are failing on current therapy, but had not been treated with an integrase inhibitor.
At 24 weeks, 79% of study participants receiving the once-daily dolutegravir regimen were virologically suppressed (HIV-1 RNA <50 c/mL) vs. 70% of participants on the twice-daily raltegravir regimen. This difference in response was statistically significant with a 95% confidence interval for the difference of 3.4% to 15.9% (p=0.003).
The SAILING study was designed to demonstrate non-inferiority of a regimen containing dolutegravir versus raltegravir (both with up to two background agents) and the analysis met this criterion; statistical superiority was concluded as part of a pre-specified testing procedure. These data were presented at the 20th Conference on Retroviruses and Opportunistic Infections (CROI) in Atlanta, Georgia.
Differences in treatment outcome in favour of the dolutegravir arm were driven by greater virologic response: at Week 24, 15% of patients receiving the dolutegravir regimen had virologic non-response vs. 24% of patients receiving the raltegravir regimen. In addition, fewer subjects failed therapy with integrase inhibitor resistance on dolutegravir (n=2) than on raltegravir (n=10, p=0.016).
Overall, the tolerability of dolutegravir (DTG) was similar to that of raltegravir (RAL). At 24 weeks, 2% of subjects on the dolutegravir regimen discontinued due to adverse events (AEs) vs. 4% of subjects on the raltegravir regimen. The rate of drug-related AEs was similar for both arms (DTG 20%, RAL 23%) and commonly reported AEs (defined as events that occurred in more than 10% of subjects) were similar on both arms, namely diarrhoea (20% DTG, 17% RAL) and upper respiratory tract infection (11% DTG, 8% RAL).
“People living with HIV who have developed resistance to more than one antiretroviral drug class face increasingly narrow treatment options and clinical decisions become increasingly complex. We welcome these initial results supporting the efficacy and tolerability of dolutegravir as a potentially useful addition in the management of HIV in treatment-experienced patients.” said John Pottage, chief scientific and medical officer, ViiV Healthcare. “These encouraging data were included as part of the comprehensive clinical data package supporting recent regulatory submissions for dolutegravir and we look forward to receiving the primary analysis at 48 weeks in due course.”
The primary objective of the ongoing double-blind, double-dummy phase III SAILING study is to demonstrate the antiviral activity of once-daily dolutegravir 50mg compared to twice-daily raltegravir 400mg over 48 weeks in HIV-1 infected, antiretroviral-experienced, integrase inhibitor-naïve adults. At baseline, 715 study participants were randomised 1:1 to receive either dolutegravir or raltegravir plus investigator-selected background regimen of no more than 2 agents, one of which was fully active. All subjects had documented genotypic or phenotypic resistance to agents from at least two antiretroviral therapy drug classes, and ongoing virologic replication. Median baseline HIV-1 RNA levels were 4.18 log10 c/mL and median baseline CD4+ cell counts were 200 cells/mm3. The study population included 32% women, 42% were of African American/African heritage, and 46% of study participants were classified as CDC Class C (patients who have one or more AIDS-defining illness). The 48-week primary analysis of this study will be presented at a future scientific meeting.
S/GSK1349572 (dolutegravir, DTG) is an investigational integrase inhibitor currently in development for the treatment of HIV; it does not require an additional pharmacokinetic boosting drug to be added to the regimen. Integrase inhibitors block HIV replication by preventing the viral DNA from integrating into the genetic material of human immune cells (T-cells). This step is essential in the HIV replication cycle and is also responsible for establishing chronic infection.
SAILING is the fourth phase III dolutegravir study reporting in 2012 and 2013. Data from the two studies in treatment-naïve populations, SPRING-2 (ING113086) and SINGLE (ING114467), were announced in April and July of 2012 respectively. Data from VIKING-3 (ING112574) in integrase inhibitor-resistant patients were announced in November 2012. Dolutegravir is not yet approved as a treatment for HIV or any other indication anywhere in the world.
Dolutegravir[1] is an experimental new drug under investigation for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Also known as S/GSK1349572 or just “572”, the drug is under development by GlaxoSmithKline (GSK). Studies have shown dolutegravir to be effective in patients with resistance to the integrase inhibitor, raltegravir.[2] Clinical trials are underway to support dolutegravir in combination with abacavir and lamivudine, in a new new fixed dose combination called 572-Trii.[3] In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir’s approval process.[4]
Results from the 96-week comparison with efavirenz, SPRING-1, showed dolutegravir 50mg orally to be effective at reducing HIV viral load and raising CD4 counts in integrase-naive patients. [5]
References
- [1] American Medical Association (AMA), STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL (Dolutegravir) Accessed 3 December 2011.
- Dolutegravir (“572”) Holds Up in Heavily Raltegravir-Resistant Patients, Phase 2B Study Finds Nelson Vergel. The Body PRO. Accessed 23 April 2011.
- Shionogi-ViiV Healthcare Starts Phase 3 Trial for “572-Trii” Test positive airwave. The Body PRO. Accessed 23 April 2011.
- “GSK wins priority status for new HIV drug in U.S”. Reuters. 16 February 2013. Retrieved 18 February 2013.
- Horn, Tim. ViiV’s Dolutegravir Continues to Show Well After 96 Weeks, Versus Sustiva, for First-Time Treatment. AIDSmeds.com 7 Mar 2012. Accessed 14 Mar 2012.
NDA FDA-Nuvo reports FDA response to PENNSAID 2% , diclofenac sodium topical solution, 2% w/w

DICLOFENAC

PENNSAID 2%
7 MAR 2013
The US Food and Drug Administration (FDA) has issued a Complete Response Letter (CRL) to Nuvo Research’s US licensing partner, Mallinckrodt, following the review of Mallinckrodt’s New Drug Application (NDA) for diclofenac sodium topical solution, 2% w/w (PENNSAID 2%).
FDA in the letter mentioned that it requires Mallinckrodt’s complete pharmacokinetic study comparing PENNSAID 2% to original PENNSAID 1.5%.
FDA denied to review the similar pharmacokinetic studies submitted by Mallinckrodt with the NDA, as the reserve samples were not retained at the clinical site.
Pharmacokinetic studies are standard studies conducted during a drug development program to identify the total exposure or the amount of drug that reaches the blood stream after a patient receives both single and multiple doses of the product.
Mallinckrodt has suggested Nuvo that it expects to complete the study and submit the results to the FDA in the third quarter of 2013, and that it anticipates the FDA will provide a formal response to the filing within 6 months thereafter.
Nuvo’s Pain Group president Dr. Bradley Galer said with the new FDA’s letter the firm was disappointed that PENNSAID 2% will not be approved in this review cycle.
“We are pleased that the FDA has outlined a clear pathway to approval that we believe can be completed in a relatively short time frame,” Galer added.
“Upon approval, PENNSAID 2% will be the first and only topical NSAID in the U.S. featuring twice per day dosing and a metered dose pump bottle.”