
Home » Uncategorized (Page 162)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TOLVAPTAN

TOLVAPTAN
的合成
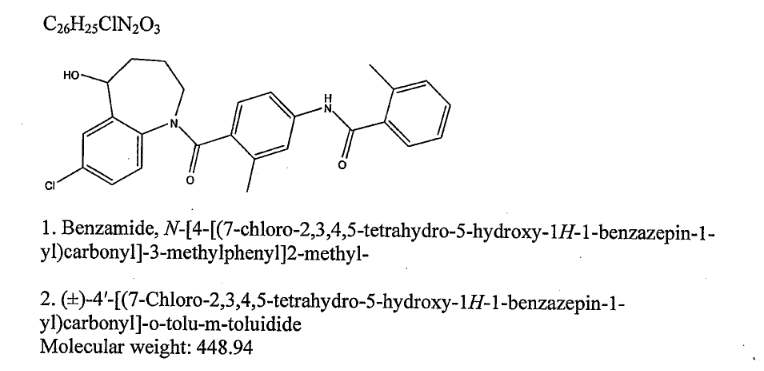
N-(4-{[(5R)-7-chloro-5-hydroxy-2,3,4,5-tetrahydro-1H-1-benzazepin-1-yl]carbonyl}-3-methylphenyl)-2-methylbenzamide
| Formula | C26H25ClN2O3 |
|---|---|
| Mol. mass | 448.941 g/mol |
150683-30-0 CAS NO
+ form 331947-66-1 Rform
OPC-41061
Otsuka…..innovator
UPDATE 2022
Tolvaptan sodium phosphate, Samtasu,
|
トルバプタンリン酸エステルナトリウム
|
| 2022/3/28 JAPAN PPROVED |
| Formula |
C26H24ClN2O6P. 2Na
|
|---|---|
| CAS |
|
| Mol weight |
572.8849
|
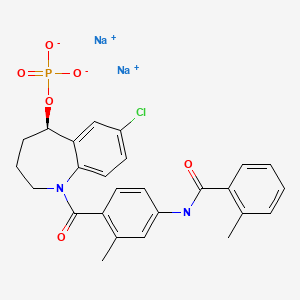
Tolvaptan sodium phosphate
disodium;[(5R)-7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-2,3,4,5-tetrahydro-1-benzazepin-5-yl] phosphate
European Medicines Agency (EMA) Accepts Otsuka’s Marketing Authorisation Application (MAA) for Tolvaptan, an Investigational Compound for Autosomal Dominant Polycystic Kidney Disease (ADPKD)
•Tolvaptan was discovered by Otsuka in Japan and, if approved by the EMA, would become the first pharmaceutical therapy in Europe for patients with ADPKD
•ADPKD is an inherited genetic disease that causes cyst growth in the kidneys, which gradually impairs their functioning. There is no current pharmaceutical treatment option
•Otsuka’s development of tolvaptan as a treatment for ADPKD illustrates the company’s commitment to address significant patient needs for diseases that traditionally have not been a priority for the pharmaceutical industry
TOKYO–(BUSINESS WIRE)–Otsuka Pharmaceutical Co., Ltd. announced today that the European Medicines Agency (EMA) has accepted the submission of a marketing authorisation application (MAA) for the potential approval of tolvaptan for the treatment of autosomal dominant polycystic kidney disease (ADPKD). Phase III clinical trial results that form the basis of the regulatory filing were published in the New England Journal of Medicine.
http://www.pharmalive.com/ema-accepts-otsukas-maa-for-tolvaptan
Tolvaptan is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP).
Tolvaptan is (±)-4′-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl) carbonyl]-otolu-m-toluidide. The empirical formula is C26H25ClN2O3. Molecular weight is 448.94. The chemical structure is:
 |
SAMSCA tablets for oral use contain 15 mg or 30 mg of tolvaptan. Inactive ingredients include corn starch, hydroxypropyl cellulose, lactose monohydrate, low-substituted hydroxypropyl cellulose, magnesium stearate and microcrystalline cellulose and FD&C Blue No. 2 Aluminum Lake as colorant.
SEE NEW UPDATE AT END OF PAGE
Tolvaptan (INN), also known as OPC-41061, is a selective, competitive vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated withcongestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone(SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca and in India is manufactured & sold by MSN laboratories Ltd. under the trade name Tolvat & Tolsama.
Tolvaptan is also in fast-track clinical trials[2] for polycystic kidney disease. In a 2004 trial, tolvaptan, when administered with traditional diuretics, was noted to increase excretion of excess fluids and improve blood sodium levels in patients with heart failure without producing side effects such as hypotension (low blood pressure) or hypokalemia(decreased blood levels of potassium) and without having an adverse effect on kidney function.[3] In a recently published trial (TEMPO 3:4 ClinicalTrials.gov number, NCT00428948) the study met its primary and secondary end points. Tolvaptan, when given at an average dose of 95 mg per day over a 3-year period, slowed the usual increase in kidney volume by 50% compared to placebo (2.80% per year versus 5.51% per year, respectively, p<0.001) and reduced the decline in kidney function when compared with that of placebo-treated patients by approximately 30% (reciprocal serum creatinine, -2.61 versus -3.81 (mg/mL)-1 per year, p <0.001)[4]
Tolvaptan was first approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, then approved by the European Medicines Agency (EMA) on August 3, 2009 and approved by Pharmaceuticals and Medical Devices Agency of Japan on Feb 4, 2013. It was developed and marketed as Samsca® by Otsuka in the US, DE and JP.
UPDATED
Tolvaptan is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP) and that is 29 times greater than for the V1a-receptor. When taken orally, 15 to 60 mg doses of tolvaptan antagonize the effect of vasopressin and cause an increase in urine water excretion that results in an increase in free water clearance (aquaresis), a decrease in urine osmolality, and a resulting increase in serum sodium concentrations. It is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia [serum sodium < 125 mEq/L or less marked hyponatremia that is symptomatic and has resisted correction with fluid restriction], including patients with heart failure, cirrhosis, and syndrome of inappropriate antidiuretic hormone (SIADH).
Samsca® is available as tablet for oral use, containing 7.5 mg/15 mg/30 mg of free Tolvaptan. The recommended starting dose is 15 mg once daily and it may be increased at intervals ≥ 24 hr to 30 mg once daily, and to a maximum of 60 mg once daily as needed to raise serum sodium.
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US5258510 | No | 1993-11-02 | 2010-11-02 |  |
| US5753677 | No | 1998-05-19 | 2020-05-19 | |
| US8501730 | No | 2013-08-06 | 2026-09-01 | |
| US5972882 | No | 1999-10-26 | 2018-12-14 | |
| US10905694 | No | 2021-02-02 | 2030-04-07 | |
Synthesis Reference
Bandi Parthasaradhi Reddy, “PROCESS FOR PREPARING TOLVAPTAN INTERMEDIATES.” U.S. Patent US20130190490, issued July 25, 2013.
US20130190490

Reference:1. US5258510A.

Reference:1. Bio. Med. Chemistry 2006, 14, 6165–6173.

Reference:1. Bio. Med. Chem. Lett. 2007, 17, 6455–6458.

Reference:1. CN102060769B.

Reference:1. Org. Lett. 2014, 16, 6041−6043.

Reference:1. Bio. Med. Chem. 1999, 7, 1743-1754.
2. WO2007026971A2.
SYN
SYN
Chemical synthesis:[5] 
Tolvaptan is chemically, N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide. Tolvaptan is represented by the following structure:
Tolvaptan, also known as OPC-41061, is a selective, competitive arginine vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.
Tolvaptan and its process for preparation were disclosed in U.S. Pat. No. 5,258,510.
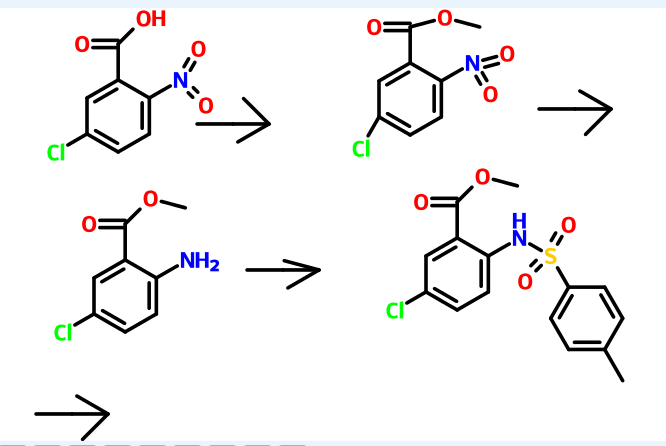
Processes for the preparation of 7-chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one, 7-chloro-1-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine and 7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine were reported in Bioorganic & medicinal chemistry 7 (1999), 1743-1754. According to the journal, 7-chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one can be prepared by reacting 7-chloro-4-ethoxycarbonyl-5-oxo-N-p-toluenesufonyl-2,3,4,5-tetrahydro-1H-1-benzazepine with acetic acid in the presence of hydrochloric acid and water to obtain 7-chloro-5-oxo-2,3,4,5-tetrahydro-1-p-toluenesulfonyl-1H-1-benzazepine, and then reacted with polyphospholic acid. According to the journal, 7-chloro-1-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine can be prepared by reacting 7-chloro-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine with 2-methyl-4-nitobenzoyl chloride in the presence of triethylamine.
According to the journal, 7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine can be prepared by reacting 1-(4-amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine with 2-methylbenzoylchloride in the presence of triethylamine.
PCT publication no. WO 2007/026971 disclosed a process for the preparation oftolvaptan can be prepared by the reduction of 7-chloro-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one with sodium borohydride.
7-Chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one is a key intermediate for the preparation of tolvaptan.
Biooganic and Medicinal Chemistry I (2007) 6455-6458, Biooganic andMedicinal Chemistry 14 (2000) 2493-2495 reported in the literature of the intermediate 2 – carboxylic acid -5 – (2 – methyl-benzoylamino) toluene synthesis method,


5-Chloro-2-nitrobenzoic acid (I) was converted into methyl ester (II) using dimethyl sulfate and K2CO3 in acetone. The nitro group of (II) was then reduced with SnCl2 to afford aniline (III), which was protected as the p-toluenesulfonamide (IV) with tosyl chloride in pyridine. Alkylation of (IV) with ethyl 4-bromobutyrate (V) yielded diester (VI). Subsequent Dieckmann cyclization of (VI) in the presence of potassium tert-butoxide provided benzazepinone (VIIa-b) as a mixture of ethyl and methyl esters, which was decarboxylated to (VIII) by heating with HCl in AcOH. Deprotection of the tosyl group of (VIII) was carried out in hot polyphosphoric acid. The resulting benzazepinone (IX) was condensed with 2-methyl-4-nitrobenzoyl chloride (X) to give amide (XI). After reduction of the nitro group of (XI) to the corresponding aniline (XII), condensation with 2-methylbenzoyl chloride (XIII) provided diamide (XIV). Finally, ketone reduction in (XIV) by means of NaBH4 led to the target compound.
……………………………………….
PATENT

……………………………………………..
PATENT

Synthesis of Intermediate III: 1.
Example
2-methyl-4-nitrobenzoic acid (available from Alfa Aesar Tianjin Chemical Co., purity> 99%, 25g,
0.14mol) was added to a 250ml reaction flask, is reacted with thionyl chloride under reflux conditions for 3h, thionyl chloride was distilled off under reduced pressure to give 2-methyl-4-nitrobenzoyl chloride (26.Sg, light yellow oily liquid), without purification, was used directly in the next step.
Intermediate II (20g, 0.1moI) and 2_ methyl _4_ nitrobenzoylchloride (22.4g, 0.llmol) was added to a 250ml reaction flask. Dichloromethane (50ml), cooled to ice bath with stirring to dissolve O~5 ° C, was slowly added dropwise N- methylmorpholine (11.2g, 0.llmol), Bi dropwise with stirring while, at room temperature the reaction 4h. TLC [developing solvent: ethyl acetate – petroleum ether (I: I), hereinafter] is displayed after completion of the reaction, saturated aqueous sodium bicarbonate (20ml), stirred for lOmin, filtered, the filter cake with dichloromethane (15ml X 2 ) washing. The filtrate and washings were combined, washed with saturated sodium chloride solution (30ml X 3), dried over anhydrous sodium sulfate and filtered. The filtrate under reduced pressure to recover the solvent, the residue was recrystallized from anhydrous methanol to give a white powder 111 (27.5g, 75.2%), mp 154.8 ~155.6 ° C. Purity 97.9% (HPLC normalization method).
Synthesis of Intermediate IV:
Intermediate III (10g, 28mmol) was added to a 250ml reaction flask, concentrated hydrochloric acid (40ml) and ethanol (50ml), with stirring, was slowly added dropwise stannous chloride (20g, 88mmol) in ethanol (40ml) . Bi room temperature drops 5h. After TLC showed completion of the reaction, ethanol was distilled off under reduced pressure to about 70ml, the residue was -10 ° C -0 ° C allowed to stand overnight to cool. Filtered, and the filter cake was washed with water poured into water (40ml) in. Plus 20% sodium hydroxide solution (approximately 60ml) was adjusted to pH 9. Filtered, washed with ethanol and recrystallized to give a pale yellow powdered solid IV (6.3g, 68.7%), mp 190.4~191.1 ° C. Purity 97.2% (HPLC normalization method).
Synthesis of intermediate V:
Intermediate IV (5g, 15mmol) and triethylamine (2.3g, 23mmol) was added followed by IOOml reaction flask was added dichloromethane (30ml), stir until dissolved. Solution of o-methylbenzoyl chloride (2.8g, 18mmol), dropwise at room temperature completion of the reaction Ih0 TLC showed the reaction was complete was poured into ice-water (about 40ml) in, (20ml X 3) and extracted with dichloromethane, the combined organic phases, and saturated sodium chloride solution successively (25ml X 3), dried over anhydrous sodium sulfate and filtered with 5% hydrochloric acid (25ml X 3). The filtrate under reduced pressure to recover the solvent (about 50ml), dried over anhydrous methanol residue – petroleum ether (2: 1) and recrystallized to give white crystals of Intermediate V (6.2g, 90.9%), mp 121.1 ~123.6 ° C. Purity 98.6% (HPLC normalization method).
Synthesis of tolvaptan: Example 4
Intermediate V (5g, Ilmmol) IOOml added to the reaction flask, was added anhydrous methanol (25ml), stirred and then added portionwise sodium borohydride (0.65g, 17mmol) to the reaction mixture, addition was complete the reaction at room temperature lh. After TLC showed the reaction was complete, the methanol recovered under reduced pressure (approximately 20ml), the residue was added methylene chloride (25ml), (25mlX3) and washed with saturated sodium chloride solution. Anhydrous sodium sulfate and filtered, and the filtrate under reduced pressure to recover the solvent, the residue with absolute methanol – petroleum ether (2: 1) and recrystallized tolvaptan white crystals (4.85g, 96.6%), mp 220.1~221.5 ° C. Purity 99.2% (HPLC normalization method). ES1-HRMS (C26H25C1N203, m / z) found (calc): 447.1476 (447.1481) [MH] – “
…………..
PATENT
http://www.google.com/patents/WO2012046244A1?cl=en
Tolvaptan is chemically, N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxylH-l- benzazepin- 1 -yl)carbonyl]-3-methylphenyl]-2-methylbenzamide. Tolvaptan is represented by the following structure:

Tolvaptan, also known as OPC-41061, is a selective, competitive arginine vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.
Tolvaptan and its process for preparation were disclosed in U.S. patent no. 5,258,510. Processes for the preparation of 7-chloro-2,3,4,5-tetrahydro-lH-l-benzazepin-5- one, 7-chloro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine and 7-chloro- 1 -[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine were reported in Bioorganic & medicinal chemistry 7 (1999), 1743-1754. According to the journal, 7-chloro-2,3,4,5-tetrahydro-lH-l- benzazepin-5-one can be prepared by reacting 7-chloro-4-ethoxycarbonyl-5-oxo-N-p- toluenesufonyl-2,3,4,5-tetrahydro-lH-l-benzazepine with acetic acid in the presence of hydrochloric acid and water to obtain 7-chloro-5-oxo-2,3,4,5-tetrahydro-l-p- toluenesulfonyl-lH-l-benzazepine, and then reacted with polyphospholic acid.
According to the journal, 7-chloro- 1 -(2 -methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine can be prepared by reacting 7-chloro-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine with 2-methyl-4-nitobenzoyl chloride in the presence of triethylamine.
According to the journal, 7-chloro- l-[2-methyl-4-[(2- methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine can be prepared by reacting l-(4-amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro- lH-l-benzazepine with 2-methylbenzoylchloride in the presence of triethylamine.
PCT publication no. WO 2007/026971 disclosed a process for the preparation of tolvaptan can be prepared by the reduction of 7-chloro- l-[2-methyl-4-(2- methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-lH-l-benzazepin-5-one with sodium borohydride.
7-Chloro-2,3,4,5-tetrahydro-lH-l-benzazepin-5-one is a key intermediate for the preparation of tolvaptan.
SYNTHESIS CONSTRUCTION

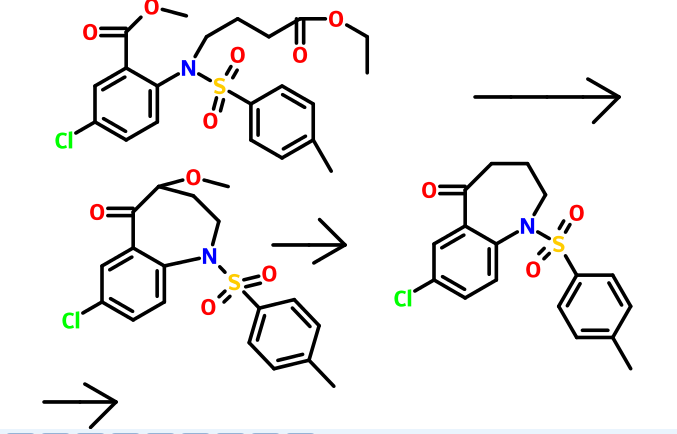


Reference example 1 :
Preparation of methyl 5-chloro-2-nitrobenzoate
Potassium carbonate (515 gm) was added to a solution of 5-chloro-2-nitro benzoic acid (500 gm) in acetone (2750 ml) at room temperature. Dimethyl sulphate (306.5 gm) was added to the reaction mixture slowly and heated to reflux for 30 minutes. The reaction mass was filtered and then concentrated to obtain a residual mass. The residual mass was poured to the ice water and extracted with methylene chloride. The solvent was distilled off under reduced pressure to obtain a residual solid of methyl 5- chloro-2-nitrobenzoate (534 gm). Reference example 2:
Preparation of methyl 2-amino-5-chlorobenzoate
A mixture of methyl 5-chloro-2-nitrobenzoate (534 gm) as obtained in reference example 1 and concentrated hydrochloric acid (2250 ml) was added to ethyl acetate (1120 ml). To the reaction mixture was added a solution of tin chloride (1680 gm) in ethyl acetate (2250 ml). The reaction mass was stirred for 16 hours at room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 8.0 to 9.0 with aqueous sodium hydroxide solution (2650 ml). The separated aqueous layer was extracted with ethyl acetate and then concentrated to obtain a residual solid of methyl 2- amino-5-chlorobenzoate (345 gm). Reference example 3:
Preparation of methyl 5-chIoro-2-(N-p-toluenesulfonyl)aminobenzoate
To a solution of methyl-2-amino-5-chloro benzoate (345 gm) as obtained in reference example 2 in pyridine (1725 ml) was added p-toluenesulfonyl chloride (425 gm). The reaction mixture was stirred for 2 hours at room temperature and poured to the ice water. The separated solid was filtered and dried to obtain 585 gm of methyl 5- chloro-2-(N-p-toluenesulfonyl)aminobenzoate.
Reference example 4:
Preparation of methyl 5-chloro-2-[N-(3-ethoxycarbonyI)propyI-N-p- toluenesulfonyl] aminobenzoate
Methyl 5-chloro-2-(N-p-toluenesulfonyl)aminobenzoate (585 gm) as obtained in reference example 3, ethyl-4-bromo butyrate (369.6 gm) and potassium carbonate (664 gm) in dimethylformamide (4400 ml) were added at room temperature. The contents were heated to 120°C and maintained for 2 hours. The reaction mass was poured into water and filtered. The solid obtained was dried to give 726 gm of methyl 5-chloro-2-[N- (3 -ethoxycarbonyl)propyl-N-p-toluenesulfonyl] aminobenzoate.
Reference example 5:
Preparation of 7-chloro-4-ethoxycarbonyI-5-oxo-N-p-toluenesufonyl-2,3,4,5- tetrahydro-lH-l-benzazepine
To a heated mixture of potassium tetrabutoxide (363 gm) in toluene (1000 ml) at 70°C was added portion wise methyl 5-chloro-2-[N-(3-ethoxycarbonyl)propyl-N-p- toluenesulfonyl]aminobenzoate (726 gm) as obtained in reference example 4. The contents were heated to reflux and maintained for 30 minutes. The reaction mass was then cooled to room temperature and then poured to the ice water. The layers were separated and the aqueous layer was extracted with toluene. The solvent was distilled off under reduced pressure to obtain a residual solid of 7-chloro-4-ethoxycarbonyl-5-oxo-N- p-toluenesufonyl-2,3,4,5-tetrahydro-lH-l-benzazepine (455 gm).
Example 1:
Preparation of 7-chIoro-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine
7-Chloro-4-ethoxycarbonyl-5-oxo-N-p-toluenesufonyl-2,3,4,5-tetrahydro- 1 H- 1 – benzazepine (455 gm) as obtained in reference example 5 was added to aqueous sulfuric acid (80%, 2275 ml). The contents heated to 75°C and maintained for 2 hours. The reaction mass was then cooled to room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 7.5 to 8.0 with sodium hydroxide solution (2575 ml). The solid obtained was collected by filtration and dried to give 160 gm of 7- chloro-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 -benzazepine.
Example 2:
Preparation of 7-chIoro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l- benzazepine
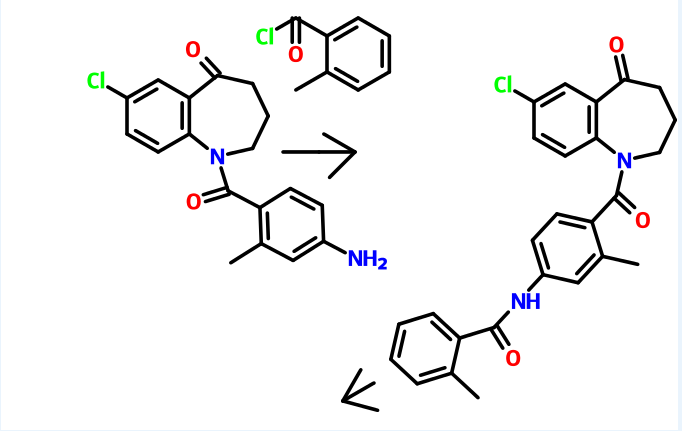
7-Chloro-5-oxo-2,3,4,5-tetrahydro-lH-l -benzazepine (160 gm) as obtained in example 1 was dissolved in methylene dichloride (480 ml) and then added aqueous sodium bicarbonate solution (20%, 68.75 gm). The reaction mixture was then cooled to 0 to 5°C and then added 2-methyl-4-nitrobenzoylchloride (180 gm) slowly. The pH of the reaction mass was adjusted to 7.0 to 8.0 with aqueous sodium bicarbonate solution (170 ml). The layers were separated and the aqueous layer was extracted with methylene chloride. The solvent was distilled off under reduced pressure to obtain a residual mass. To the residual mass was dissolved in isopropyl alcohol (7300 ml) and maintained for 2 hours at reflux temperature. The separated solid was filtered and dried to obtain 250 gm of 7-chloro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine. Example 3:
Preparation of l-(4-amino-2-methylbenzoyl)-7-chIoro-5-oxo-2,3,4,5-tetrahydro-lH- 1-benzazepine
7-Chloro- 1 -(2-methyl-4-nitrobenzoyl)-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 – benzazepine (250 gm) as obtained in example 2 was dissolved in methanol (575 ml) and then added a solution of tin chloride (630 gm) in methanol (1130 ml). The reaction mixture was stirred for 16 hours at room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 8.0 to 9.0 with sodium hydroxide solution (1250 ml). The layers were separated and the aqueous layer was extracted with ethyl acetate. The solvent was distilled off under vacuum to obtain a residual solid of l-(4- amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro- 1 H- 1 -benzazepine (185 gm).
Example 4:
Preparation of 7-chloro-l-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-dxo- 2,3,4,5-tetrahydro-lH-l-benzazepine
1 -(4-Amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 – benzazepine (185 gm) as obtained in example 3 was dissolved in methylene chloride (4000 ml) and then added sodium bicarbonate solution (10%, 47.3 gm). The reaction mass was cooled to 0 to 5°C and then added 2-methyl benzoyl chloride (95.7 gm) slowly. -The pH of the reaction mass was adjusted to 7.0 to 8.0 with aqueous sodium bicarbonate solution (120 ml). The separated aqueous layer was extracted with methylene chloride and then concentrated to obtain a residual solid of 7-chloro-l-[2- methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro- 1 H- 1 – benzazepine (185 gm). Example 5:
Preparation of tolvaptan
7-Chloro- 1 -[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5- tetrahydro-lH-1 -benzazepine (63 gm) as obtained in example 4 was dissolved in methanol (570 ml) and then added sodium borohydride (2.07 gm) at room temperature. The reaction mass was stirred for 1 hour and pH of the reaction mass was adjusted to 6.0 to 7.0 with hydrochloric acid solution (1%, 630 ml). The separated solid was filtered and dried to obtain 57 gm of tolvaptan.
……………….

Process used to prepare Tolvaptan involves condensing 7-chloro-1, 2, 3, 4-tetrahydro-benzo[b]azepin-5-one with 2-methyl, 4-nitro benzoyl chloride, followed by reduction using SnCl2/HCl catalyst resulting in amine which is then condensed with o-toluoyl chloride followed by reduction with sodium borohydride to give Tolvaptan
///////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN 1
Synthetic Reference
Cordero-Vargas, Alejandro; Quiclet-Sire, Beatrice; Zard, Samir Z. A flexible approach for the preparation of substituted benzazepines: Application to the synthesis of tolvaptan. Bioorganic & Medicinal Chemistry. Volume 14. Issue 18. Pages 6165-6173. 2006.

SYN 2
Synthetic Reference
Torisawa, Yasuhiro; Abe, Kaoru; Muguruma, Yasuaki; Fujita, Shigekazu; Ogawa, Hidenori; Utsumi, Naoto; Miyake, Masahiro. Process for preparation of benzoylaminobenzoylbenzazepinones by reaction of benzazepinones with benzoylaminophenyl halides in the presence of carbonylating agents. Assignee Otsuka Pharmaceutical Co.,

SYN 3
Synthetic Reference
Zard, Samir; Cordero Vargas, Alejandro; Sire, Beatrice. Improved process for the preparation of benzazepines and their derivatives. Assignee Centre National de la Recherche Scientifique CNRS, Fr.; Ecole Polytechnique. FR 2867187. (2005).

SYN 4
Synthetic Reference
Gao, Junlong; Li, Peng; Liu, Kai; Guo, Dapeng. Method for preparing high-purity Tolvaptan intermediate. Assignee Jiangsu Hengrui Medicine Co., Ltd., Peop. Rep. China. CN 108503586. (2018).

SYN 5
Synthetic Reference
Han, Shin; Jeon, Seong Hyeon; Lee, Shin Yoon. Improved method for preparing synthetic intermediates for tolvaptan. Assignee Hexa Pharmatec Co., Ltd., S. Korea. JP 2018012690. (2018).

SYN 6
Synthetic Reference
Guo, Xinfu; Wang, Qiang; Liu, Zhaoguo; Wang, Zhipeng. Preparation method of tolvaptan. Assignee Tianjin Taipu Pharmaceutical Co., Ltd., Peop. Rep. China. CN 106883175. (2017).

SYN 7
Synthetic Reference
Lixin, Juanzi; Li, Jianzhi; Ma, Xilai; Chi, Wangzhou; Liu, Hai; Hu, Xuhua; Zheng, Xiaoli; Zhai, Zhijun; Li, Jianxun. Process for the preparation of tolvaptan. Assignee Shanghai Tianci International Pharmaceutical Co., Ltd., Peop. Rep. China. CN 105753735. (2016).

STR8
Synthetic Reference
Patel, Dhaval J.; Shah, Tejas C.; Singh, Manoj Kumar. A process for the preparation of tolvaptan. Assignee Cadila Healthcare Limited, India. IN 2012MU01559. (2014).

STR9
Synthetic Reference
Sethi, Madhuresh Kumar; Rawat, Vijendrasingh; Thirunavukarasu, Jayaprakash; Yerramala, Raja Krishna; Kumar, Anish. Improved process for the preparation of tolvaptan. Assignee Matrix Laboratories Ltd., India. IN 2011CH01303. (2013).

/////////////////////


Title: Tolvaptan
CAS Registry Number: 150683-30-0
CAS Name: N-[4-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide
Additional Names: 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine
Manufacturers’ Codes: OPC-41061
Molecular Formula: C26H25ClN2O3
Molecular Weight: 448.94
Percent Composition: C 69.56%, H 5.61%, Cl 7.90%, N 6.24%, O 10.69%
Literature References: Nonpeptide arginine vasopressin V2 receptor antagonist. Prepn: H. Ogawa et al., WO 9105549; eidem, US 5258510 (1991, 1993 both to Otsuka); K. Kondo et al., Bioorg. Med. Chem. 7, 1743 (1999). Pharmacology: Y. Yamamura et al., J. Pharmacol. Exp. Ther. 287, 860 (1998). Clinical trial in heart failure: M. Gheorghiade et al., J. Am. Med. Assoc. 291, 1963 (2004).
Properties: Colorless prisms, mp 225.9°.
Melting point: mp 225.9°
Therap-Cat: In treatment of congestive heart failure.
Keywords: Vasopressin Receptor Antagonist.
- Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther 10 (3): 165–71. doi:10.1177/107424840501000304. PMID 16211205.
- Otsuka Maryland Research Institute, Inc.
- Gheorghiade M, Gattis W, O’Connor C, et al. (2004). “Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: a randomized controlled trial”. JAMA 291 (16): 1963–71. doi:10.1001/jama.291.16.1963. PMID 15113814.
- (2012) Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease
- Kondo, K.; Ogawa, H.; Yamashita, H.; Miyamoto, H.; Tanaka, M.; Nakaya, K.; Kitano, K.; Yamamura, Y.; Nakamura, S.; Onogawa, T.; et al.; Bioor. Med. Chem. 1999, 7, 1743.
- http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm?source=govdelivery
- Gheorghiade M, Niazi I, Ouyang J et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation 107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04.PMID 12742979.
G. R. Belum, V. R. Belum, S. K. Chaitanya Arudra, and B. S. N. Reddy, “The Jarisch-Herxheimer reaction: revisited,” Travel Medicine and Infectious Disease, vol. 11, no. 4, pp. 231–237, 2013.
H. D. Zmily, S. Daifallah, and J. K. Ghali, “Tolvaptan, hyponatremia, and heart failure,” International Journal of Nephrology and Renovascular Disease, vol. 4, pp. 57–71, 2011.
M. N. Ferguson, “Novel agents for the treatment of hyponatremia: a review of conivaptan and tolvaptan,” Cardiology in Review, vol. 18, no. 6, pp. 313–321, 2010.
H. Ogawa, H. Miyamoto, K. Kondo, et al., US5258510, 1993.
K. Kondo, H. Ogawa, H. Yamashita et al., “7-Chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5- tetrahydro-1H-1-benzazepine (OPC-41061): a potent, orally active nonpeptide arginine vasopressin V2 receptor antagonist,” Bioorganic and Medicinal Chemistry, vol. 7, no. 8, pp. 1743–1754, 1999.
| WO2012046244A1 * | Aug 23, 2011 | Apr 12, 2012 | Hetero Research Foundation | Process for preparing tolvaptan intermediates |
| CN102060769A * | Dec 20, 2010 | May 18, 2011 | 天津药物研究院 | Preparation method of tolvaptan |
| CN102060769B | Dec 20, 2010 | Sep 18, 2013 | 天津药物研究院 | Preparation method of tolvaptan |
| US9024015 | Aug 23, 2011 | May 5, 2015 | Hetero Research Foundation | Process for preparing tolvaptan intermediates |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101817783A | May 12, 2010 | Sep 1, 2010 | 天津泰普药品科技发展有限公司 | Method for preparing tolvaptan intermediate |
| WO2007026971A2 | Sep 1, 2006 | Mar 8, 2007 | Otsuka Pharma Co Ltd | Process for preparing benzazepine compounds or salts thereof |
| Reference | ||
|---|---|---|
| 1 | Cordero-Vargas, Alejandro | |
| 2 | Kondo, Kazumi et al.7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoyl-amino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine (OPC-41061): A potent, orally active nonpeptide arginine vasopressin V2 receptor antagonist.《Bioorganic & Medicinal Chemistry》.1999,1743-1757. | |
| 3 | Quiclet-Sire, Beatrice | |
| 4 | Torisawa, Yasuhiro et al.Aminocarbonylation route to tolvaptan.《Bioorganic & Medicinal Chemistry Letters》.2007,6455-6458. | |
| 5 | Zard, Samir Z.A flexible approach for the preparation of substituted benzazepines: Application to the synthesis of tolvaptan.《Bioorganic & Medicinal Chemistry》.2006,6165-6173. | |
///////////////
CC1=CC=CC=C1C(=O)NC2=CC(=C(C=C2)C(=O)N3CCCC(C4=C3C=CC(=C4)Cl)OP(=O)([O-])[O-])C.[Na+].[Na+]
////////////UPDATE 2022
-Tolvaptan_Structural_Formula_V1.svg) |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Samsca, Jinarc, Jynarque, others |
| Other names | OPC-41061 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609033 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Unknown (40% absorbed) |
| Protein binding | 99% |
| Metabolism | Liver (CYP3A4-mediated)[7] |
| Elimination half-life | 12 hours (terminal) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.219.212 |
| Chemical and physical data | |
| Formula | C26H25ClN2O3 |
| Molar mass | 448.95 g·mol−1 |
| 3D model (JSmol) | |
| |
|
Tolvaptan, sold under the brand name Samsca among others, is an aquaretic drug that functions as a selective, competitive vasopressin receptor 2 (V2) antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.[8] Tolvaptan, as Jynarque, was granted approval for medical use in the United States in April 2018.[9]
The U.S. Food and Drug Administration (FDA) granted tolvaptan a fast track designation for clinical trials investigating its use for the treatment of polycystic kidney disease.[10] The FDA granted Jynarque an orphan drug designation in April 2012, for the treatment of autosomal dominant polycystic kidney disease.[11]
Tolvaptan is available as a generic medication.[12]
Medical uses
Tolvaptan (Samsca) is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia.[13]
Tolvaptan (Jynarque) is indicated to slow kidney function decline in adults at risk of rapidly progressing autosomal dominant polycystic kidney disease (ADPKD).[14]
Side effects
The FDA has determined that tolvaptan should not be used for longer than 30 days and should not be used in patients with underlying liver disease because it can cause liver injury, potentially leading to liver failure.[15] When using to treat hyponatremia, it may cause too rapid correction of hyponatremia resulting in fatal osmotic demyelination syndrome.[16]
Pharmacology
Tolvaptan is a selective vasopressin V2 receptor antagonist.[13][14]
Chemistry
Tolvaptan is a racemate, a 1:1 mixture of the following two enantiomers:[17]
| Enantiomers of tolvaptan | |
|---|---|
-Tolvaptan_Structural_Formula_V1.svg) (R)-Tolvaptan CAS number: 331947-66-1 |
-Tolvaptan_Structural_Formula_V1.svg) (S)-Tolvaptan CAS number: 331947-44-5 |
References
- ^ “Samsca 15 mg tablets – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 14 December 2020.
- ^ “Jinarc 15 mg tablets – Summary of Product Characteristics (SmPC)”. (emc). 21 April 2020. Retrieved 14 December 2020.
- ^ “Jynarque- tolvaptan kit Jynarque- tolvaptan tablet”. DailyMed. 31 March 2020. Retrieved 14 December 2020.
- ^ “Samsca- tolvaptan tablet”. DailyMed. 26 October 2020. Retrieved 14 December 2020.
- ^ “Samsca EPAR”. European Medicines Agency (EMA). Retrieved 14 December 2020.
- ^ “Jinarc EPAR”. European Medicines Agency (EMA). Retrieved 14 December 2020.
- ^ Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther. 10 (3): 165–71. doi:10.1177/107424840501000304. PMID 16211205. S2CID 39158242.
- ^ “Drug Approval Package: Samsca (Tolvaptan) Tablets NDA #022275”. U.S. Food and Drug Administration (FDA). 21 July 2009. Retrieved 15 August 2020. Lay summary (PDF).
{{cite web}}: Cite uses deprecated parameter|lay-url=(help) - ^ “Drug Approval Package: Jynarque (tolvaptan)”. U.S. Food and Drug Administration (FDA). 8 June 2018. Retrieved 15 August 2020.
- ^ “Otsuka Maryland Research Institute, Inc. Granted Fast Track Designation For Tolvaptan In PKD”. Medical News Today. Healthline Media UK Ltd. Retrieved 6 December 2018.
- ^ “Tolvaptan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 6 April 2012. Retrieved 15 August 2020.
- ^ “Drugs@FDA: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 August 2020.
- ^ Jump up to:a b “Samsca- tolvaptan tablet”. DailyMed. 28 May 2019. Retrieved 15 August 2020.
- ^ Jump up to:a b “Jynarque- tolvaptan kit Jynarque- tolvaptan tablet”. DailyMed. 31 March 2020. Retrieved 15 August 2020.
- ^ “U.S. Food and Drug Administration.” Samsca (Tolvaptan): Drug Safety Communication. N.p., 30 Apr. 2013. Web. 1 June 2014. <http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm>[dead link]
- ^ Goodman & Gilman’s the pharmacological basis of therapeutics. Brunton, Laurence L, Knollmann, Björn C, Hilal-Dandan, Randa (Thirteenth ed.). New York. 5 December 2017. ISBN 9781259584732. OCLC 994570810.
- ^ Rote Liste Service GmbH (Hrsg.): Rote Liste 2017 – Arzneimittelverzeichnis für Deutschland (einschließlich EU-Zulassungen und bestimmter Medizinprodukte). Rote Liste Service GmbH, Frankfurt/Main, 2017, Aufl. 57, ISBN 978-3-946057-10-9, S. 222.
Further reading
- Gheorghiade M, Niazi I, Ouyang J, et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation. 107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04. PMID 12742979.
External links
- “Tolvaptan”. Drug Information Portal. U.S. National Library of Medicine.
Synthesis
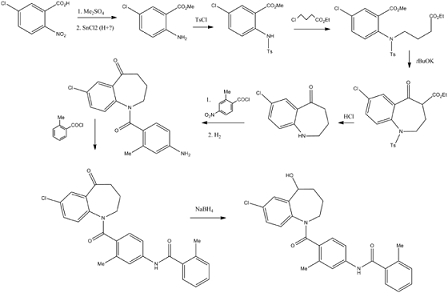
Fugure 1: Synthesis of Tolvaptan

NEW DRUG APPROVALS
HELP TO MAINTAIN THIS BLOG SUBSCRIPTION
$10.00
Title: Tolvaptan
CAS Registry Number: 150683-30-0
CAS Name: N-[4-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide
Additional Names: 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine
Manufacturers’ Codes: OPC-41061
Molecular Formula: C26H25ClN2O3
Molecular Weight: 448.94
Percent Composition: C 69.56%, H 5.61%, Cl 7.90%, N 6.24%, O 10.69%
Literature References: Nonpeptide arginine vasopressin V2 receptor antagonist. Prepn: H. Ogawa et al., WO 9105549; eidem, US 5258510 (1991, 1993 both to Otsuka); K. Kondo et al., Bioorg. Med. Chem. 7, 1743 (1999). Pharmacology: Y. Yamamura et al., J. Pharmacol. Exp. Ther. 287, 860 (1998). Clinical trial in heart failure: M. Gheorghiade et al., J. Am. Med. Assoc. 291, 1963 (2004).
Properties: Colorless prisms, mp 225.9°.
Melting point: mp 225.9°
Therap-Cat: In treatment of congestive heart failure.
Keywords: Vasopressin Receptor Antagonist.
Medicinal Chemistry International: DELDEPREVIR (NECEPREVIR)
Medicinal Chemistry International: DELDEPREVIR (NECEPREVIR)
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
DELDEPREVIR OR NECEPREVIR

CHEMICAL NAMES
1. Cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a(5H)-carboxamide, N-
(cyclopropylsulfonyl)-6-[2-(3,3-difluoro-1-piperidinyl)-2-oxoethyl]-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydro-2-[[7-methoxy-8-methyl-2-[4-(1-
methylethyl)-2-thiazolyl]-4-quinolinyl]oxy]-5,16-dioxo-, (2R,6R,12Z,13aS,14aR,16aS)-
2. (2R,6R,12Z,13aS,14aR,16aS)-N-(cyclopropylsulfonyl)-6-[2-(3,3-difluoropiperidin-1-yl)-
2-oxoethyl]-2-({7-methoxy-8-methyl-2-[4-(1-methylethyl)thiazol-2-yl]quinolin-4-yl}oxy)-
5,16-dioxo-1,2,3,6,7,8,9,10,11,13a,14,15,16,16atetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a(5H)-
carboxamide
MOLECULAR WEIGHT 911.1
SPONSOR Achillion Pharmaceuticals, Inc.
CODE DESIGNATION ACH-0142684, ACH-2684
CAS REGISTRY NUMBER 1229626-28-1
WHO NUMBER 9600
NOTE: This adoption statement replaces adoption N12/17 and the name neceprevir is hereby rescinded.
(cyclopropylsulfonyl)-6-[2-(3,3-difluoro-1-piperidinyl)-2-oxoethyl]-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydro-2-[[7-methoxy-8-methyl-2-[4-(1-
methylethyl)-2-thiazolyl]-4-quinolinyl]oxy]-5,16-dioxo-, sodium salt (1:1),
(2R,6R,12Z,13aS,14aR,16aS)-
1-yl)-2-oxoethyl]-2-({7-methoxy-8-methyl-2-[4-(1-methylethyl)thiazol-2-yl]quinolin-4-
yl}oxy)-5,16-dioxo-1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-
tetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-14a(5H)-
yl]formyl]azanide
| WO 2010068761 | ||
| US 2010152103 |

click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
Deleobuvir » All About Drugs
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
DELEOBUVIR

BI-207127 (free acid)
| Company | Boehringer Ingelheim GmbH |
| Description | Oral non-structural protein 5B (NS5B) RNA-dependent polymerase inhibitor |
| Molecular Target | HCV NS5B polymerase |
| Mechanism of Action | Viral polymerase inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase III |
| Indication | Hepatitis C virus (HCV) |
| Partner |
- Interferon-free hepatitis C treatment with faldaprevir proves safe and effective in people with cirrhosis. Alcorn, K. Aidsmap.com. 20 November 2012.
- S Zeuzem, J-F Dufour, M Buti, V Soriano, R Buynak, P Mantry, J Taunk, JO Stern, R Vinisko, J-P Gallivan, WO Bocher and FJ Mensa.“Interferon-free treatment with faldaprevir, deleobuvir (BI 207127) and ribavirin in SOUND-C3: 95% SVR12 in HCV GT-1b”. 23rd Conference of the Asian Pacific Association for the Study of the Liver (APASL) 6–9 June 2013. Retrieved 12 Sep 2013.
| WO 2013147749 |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010059667A1 | Nov 18, 2009 | May 27, 2010 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition of a potent hcv inhibitor for oral administration |
| WO2011005646A2 | Jul 1, 2010 | Jan 13, 2011 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition for a hepatitis c viral protease inhibitor |
| WO2012041771A1 * | Sep 23, 2011 | Apr 5, 2012 | Boehringer Ingelheim International Gmbh | Combination therapy for treating hcv infection |
| US4211771 | Feb 13, 1978 | Jul 8, 1980 | Robins Ronald K | Treatment of human viral diseases with 1-B-D-ribofuranosyl-1,2,4-triazole-3-carboxamide |
| US6063772 | Jun 15, 1998 | May 16, 2000 | Icn Pharmaceuticals, Inc. | Specific modulation of Th1/Th2 cytokine expression by ribavirin in activated T-lymphocytes |
| US6277830 | Jul 7, 1999 | Aug 21, 2001 | Schering Corporation | 5′-amino acid esters of ribavirin and the use of same to treat hepatitis C with interferon |
| US6323180 | Aug 5, 1999 | Nov 27, 2001 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| US6403564 | Oct 14, 1999 | Jun 11, 2002 | Schering Corporation | Ribavirin-interferon alfa combination therapy for eradicating detectable HCV-RNA in patients having chronic hepatitis C infection |
| US7141574 | Jul 18, 2002 | Nov 28, 2006 | Boehringer Ingelheim (Canada) Ltd. | Viral polymerase inhibitors |
| US7514557 | May 23, 2005 | Apr 7, 2009 | Boehringer Ingelheim International Gmbh | Process for preparing acyclic HCV protease inhibitors |
| US7582770 | Feb 18, 2005 | Sep 1, 2009 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US7585845 | May 20, 2004 | Sep 8, 2009 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US7642352 | Feb 10, 2006 | Jan 5, 2010 | Boehringer Ingelheim International Gmbh | Process for preparing 2,3-disubstituted indoles |
| US20090087409 | Nov 26, 2008 | Apr 2, 2009 | Boehringer Ingelheim (Canada) Ltd. | Viral Polymerase Inhibitors |
| US20100068182 | Sep 16, 2009 | Mar 18, 2010 | Boehringer Ingelheim International Gmbh | Combination therapy for treating hcv infection |
| US20100093792 | Sep 15, 2009 | Apr 15, 2010 | Boehringer Ingelheim International Gmbh | Crystalline forms of a potent hcv inhibitor |
| Ref | ||
|---|---|---|
| 1 | BALAGOPAL GASTROENTEROLOGY vol. 139, 2010, pages 1865 – 1876 | |
| 2 | BERG ET AL. HEPATOL vol. 52, no. S1, 2010, | |
| 3 | * | DOMINIQUE LARREY ET AL: “Rapid and strong antiviral activity of the non-nucleosidic NS5B polymerase inhibitor BI 207127 in combination with peginterferon alfa 2a and ribavirin“, JOURNAL OF HEPATOLOGY, vol. 57, no. 1, 7 March 2012 (2012-03-07), pages 39-46, XP55040240, ISSN: 0168-8278, DOI: 10.1016/j.jhep.2012.02.015 |
| 4 | G. CAIRNS GENE VARIANT THAT HELPS HEPATITIS C TREATMENT MAY HINDER HIV TREATMENT, [Online] 2011, Retrieved from the Internet: <URL:http://www.bhiva.org/Ncws.aspx?NewsID=a7503829-94b9-4d2f-bd91-ld2fbaad6c8d> | |
| 5 | GE ET AL. NATURE vol. 461, 2009, pages 399 – 401 | |
| 6 | GHANY; MARC ET AL.: ‘An Update on Treatment of Genotype 1 Chronic Hepatitis C Virus Infection: 2011 Practice Guideline by the American Association for the Study of Liver Diseases‘ HEPATOLOGY vol. 54, no. 4, 2011, pages 1433 – 44 | |
| 7 | * | LIZ HIGHLEYMAN: “AASLD: All-Oral Combination of BI 201335, BI 207127 and Ribavirin Shows Good Efficacy at 12 Weeks“, INTERNET CITATION, [Online] 1 December 2011 (2011-12-01), pages 1-3, XP002684260, Retrieved from the Internet: URL:www.hivandhepatitis.com/hepatitis-c/he patitis-c-topics/hcv-treatment/3371-aasld- all-oral-combination-of-bi-201335-bi-20712 7-and-ribavirin-shows-good-efficacy-at-12- weeks> [retrieved on 2012-09-27] |
| 8 | * | POL S ET AL: “SVR AND PHARMACOKINETICS OF THE HCV PROTEASE INHIBITOR BI201335 WITH PEGIFN/RBV IN HCV GENOTYPE-1 PATIENTS WITH COMPENSATED LIVER CIRRHOSIS AND NON-RESPONSE TO PREVIOUS PEGIFN/RBV“, JOURNAL OF HEPATOLOGY, vol. 54, no. Suppl. 1, March 2011 (2011-03), page S486, XP55038942, & 46TH ANNUAL MEETING OF THE EUROPEAN-ASSOCIATION-FOR-THE-STUDY-OF-THE- LIVER (EASL); BERLIN, GERMANY; MARCH 30 -APRIL 03, 2011 ISSN: 0168-8278 |
| 9 | S. M. BIRGE ET AL. J. PHARM. SCI. vol. 66, 1977, pages 1 – 19 | |
| 10 | * | STEFAN ZEUZEM ET AL: “Efficacy of the Protease Inhibitor BI 201335, Polymerase Inhibitor BI 207127, and Ribavirin in Patients With Chronic HCV Infection“, GASTROENTEROLOGY, ELSEVIER, PHILADELPHIA, PA, vol. 141, no. 6, 1 December 2011 (2011-12-01), pages 2047-2055, XP002664706, ISSN: 0016-5085, DOI: 10.1053/J.GASTRO.2011.08.051 |
| 11 | SULKOWSKI MS ET AL. HEPATOL vol. 50, 2009, page 2A | |
| 12 | SULKOWSKI MS ET AL. J HEPATOL vol. 52, no. 1, 2010, pages S462 – S463 | |
| 13 | WHITE PW ET AL. ANTIMICROB AGENTS CHEMOTHER vol. 54, no. 11, 2010, pages 4611 – 4618 | |
| 14 | WHO COLLABORATIVE STUDY GROUP. VOX SANG vol. 76, 1999, pages 149 – 158 | |
| 15 | * | ZEUZEM STEFAN ET AL: “STRONG ANTIVIRAL ACTIVITY AND SAFETY OF IFN-SPARING TREATMENT WITH THE PROTEASE INHIBITOR BI 201335, THE HCV POLYMERASE INHIBITOR BI 207127 AND RIBAVIRIN IN PATIENTS WITH CHRONIC HEPATITIS C“, HEPATOLOGY, WILLIAMS AND WILKINS, BALTIMORE, MD, US, vol. 52, no. Suppl, 1 October 2010 (2010-10-01), pages 876A-877A, XP009154421, ISSN: 0270-9139 |
| 16 | * | ZEUZEM STEFAN ET AL: “VIROLOGIC RESPONSE TO AN INTERFERON-FREE REGIMEN OF BI201335 AND BI207127, WITH AND WITHOUT RIBAVIRIN, IN TREATMENT-NAIVE PATIENTS WITH CHRONIC GENOTYPE-1 HCV INFECTION: WEEK 12 INTERIM RESULTS OF THE SOUND-C2 STUDY“, HEPATOLOGY, WILLIAMS AND WILKINS, BALTIMORE, MD, US, vol. 54, no. Suppl. 1, 1 November 2011 (2011-11-01), page 1436A, XP009163087, ISSN: 0270-9139, DOI: 10.1002/HEP.24666 [retrieved on 2011-09-30] |













click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
LEDIPASVIR , 来迪派韦 , Ледипасвир , ليديباسفير


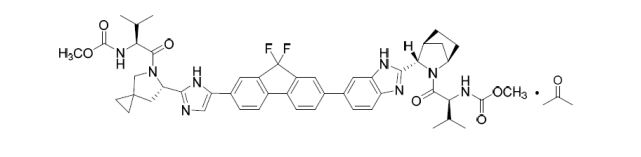
Carbamic acid, N-((1S)-1-(((6S)-6-(5-(9,9-difluoro-7-(2-((1R,3S,4S)-2-((2S)-2-((methoxycarbonyl)amino)-3-methyl-1-oxobutyl)-2-azabicyclo(2.2.1)hept-3-yl)-1H-benzimidazol-6-yl)-9H-fluoren-2-yl)-1H-imidazol-2-yl)-5-azaspiro(2.4)hept-5-yl)carbonyl)-2-me
Chemical Formula:C52H60F2N8O7
Molecular Weight:947.08

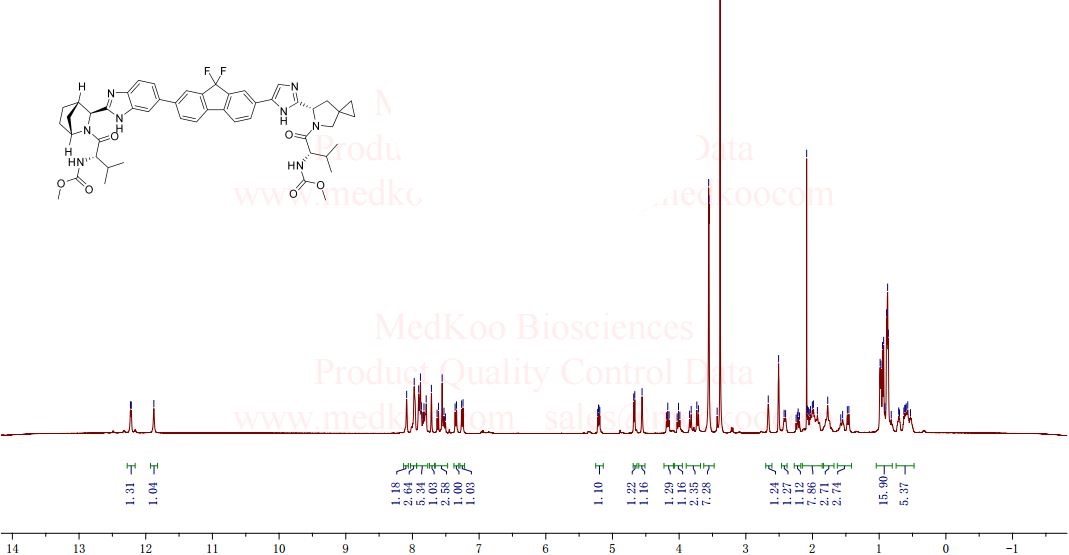
The structure of ledipasvir was unambiguously confirmed by 1 H, 13C and 19F NMR spectroscopy, UV spectroscopy, IR spectroscopy, high resolution mass spectrometry, elemental analysis and X-ray crystallography. LDV-AS is a white to tinted (off-white, tan, yellow, orange, or pink), slightly hygroscopic crystalline solid. It shows pH dependent solubility in aqueous media: it is slightly soluble in pH 2.3 buffer but practically insoluble in pH 4-7.5 buffers. It is freely soluble in ethanol and DMSO and slightly soluble in acetone. Ledipasvir is chiral and possesses 6 stereogenic centres and enantiomeric purity is controlled in starting material specifications. Three crystalline forms are known and ledipasvir acetone solvate is the designated commercial form. The first step for finished product manufacture involves the dissolution of ledipasvir in ethanol followed by spray-drying and thus precise control of morphology and particle size is not considered important. Ledipasvir is a chemical substance not previously authorised as a medicinal product in the European Union. Furthermore, it is not a salt, complex, derivative or isomer, (nor mixture of isomers), of a previously authorised substance. Whilst it contains some structural features in common with daclastavir, it is metabolically stable and the applicant presented data indicating that there are no common active metabolites. Therefore, the therapeutic moieties are not the same. Ledipasvir thus meets the definition of a New Active Substance according to the Notice to Applicants (NtA), Vol 2A, Chapter 1, Annex 3.
The mode of action of ledipasvir has not been directly established but indirect evidence is consistent with the compound targeting the NS5A molecule. In vitro resistance selection and cross-resistance studies, and the lack of HCV enzyme or kinase inhibition was taken to support the conclusion that ledipasvir targets NS5A as its mode of action. Ledipasvir has shown antiviral activity against HCV genotypes 1a and 1b with mean EC50 values of 0.031 and 0.004 nM, respectively. Antiviral activity determined as EC50 against genotypes 2 to 6 ranged from 0.15 to 530 nM. Ledipasvir showed no relevant antiviral activity at the highest concentration tested, or the highest concentration without cytotoxicity, against other virus such as bovine viral diarrhea virus (BVDV), RSV, HBV, HIV-1, HRV, influenza A and B, and a panel of flaviviruses (including West Nile virus, yellow fever virus, dengue virus, and banzai virus). Cytotoxicity of ledipasvir was characterised by CC50 of 4029 to >50000 nM using different cell lines (1b-Rluc-2, Huh-luc, 1a-HRlucp, Hep G2, SL3, Huh7, Hep-2, AD-38 and MT4 cells). Ledipasvir at 10 µM showed significant binding to 3 ion channels and 1 receptor in a radioligand binding assay screen against a panel of 68 mammalian ion channels and receptors. The IC50s of ledipasvir were 0.210 and 3.47 μM against sodium channel site 2 and calcium channel L-type (dihydropyridine), respectively. A 50% inhibition of androgen receptor was noted at 10 μM. Ledipasvir activity against 442 kinases was assessed using a quantitative polymerase chain reaction (qPCR)-based competition assay. Results showed weak competition for binding of 2 kinases, Bruton’s tyrosine kinase (BTK) and homeodomain-interacting protein kinase 1 (HIPK1) at 0.1 and 1 μM, respectively. Taking into account the high protein binding, >99.5%, of ledipasvir the large margin between unbound maximum clinical plasma levels (0.8 nM) and potential ion channel/receptor inhibition indicates limited clinical relevance.
Ledipasvir (formerly GS-5885) is a drug for the treatment of hepatitis C that was developed by Gilead Sciences.[1] After completingPhase III clinical trials, on February 10, 2014 Gilead filed for U.S. approval of a ledipasvir/sofosbuvirfixed-dose combination tablet for genotype 1 hepatitis C.[2][3] The ledipasvir/sofosbuvir combination is a direct-acting antiviral agent that interferes with HCV replication and can be used to treat patients with genotypes 1a or 1b without PEG-interferon or ribavirin.
Ledipasvir is an inhibitor of the hepatitis C virusNS5A protein.
Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of the nucleotide analog inhibitor sofosbuvir, ledipasvir, and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[4][5] The sofosbuvir/ledipasvir coformulation is being tested with and without ribavirin. In February 2014 Gilead has filed for United StatesFood and Drug Administration (FDA) approval of ledipasvir/sofosbuvir oral treatment, without interferon and ribavirin.[6]
On October 10, 2014 the FDA approved the combination product ledipasvir 90 mg/sofosbuvir 400 mg called Harvoni.[7]


https://www.google.co.in/patents/WO2013184698A1
CLIP
SYN


https://www.google.co.in/patents/WO2013184702A1









PATENT
https://www.google.co.in/patents/US8088368
Example ED Preparation of Intermediate 5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-methyl ester

4-Methylene-pyrrolidine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester
Example ED′


2,7-Dibromo-9,9-difluoro-9H-fluorene
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-[2-(7-bromo-9,9-difluoro-9H-fluoren-2-yl)-2-oxo-ethyl]ester
6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carboxylic acid benzyl ester
(1-{6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester
(1-{3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
https://www.google.co.in/patents/US8088368

2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester: A mixture of 2-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carboxylic acid tert-butyl ester (324 mg, 0.627 mmol), 3-[6-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (1.1 eq., 304 mg), [1,1′ bis(diphenylphosphino)ferrocene]dichloropalladium(II)(3%, 15 mg), tetrakis(triphenylphosphine)palladium (3%, 22 mg) and potassium carbonate (3.3 eq., 285 mg) in 10 mL DME and 3 mL water was heated to 90° C. under Argon for 3 hours. The reaction mixture was cooled and diluted with ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was dried (MgSO4), concentrated and purified by flash column chromatography (silica gel, 20 to 100% ethyl acetate/hexane) to give 2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester (361 mg, yield 77%). LCMS-ESI−: calc’d for C43H46F2N6O4: 748.86. Found: 749.2 (M+H+).
PATENTS
SEE
WO 2010132601
WO 2013040492
WO 2013059630
WO 2013059638
CLIP
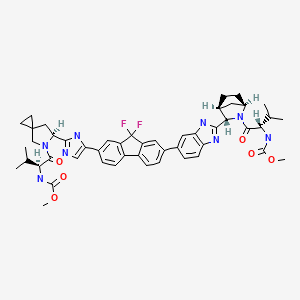
Ledipasvir (Harvoni) Ledipasvir is a potent NS5A inhibitor that is approved for use in combination with sofosbuvir, a nucleotide inhibitor of viral polymerase, for the treatment of chronic hepatitis C virus genotype 1 infection.14,130,131 This combination was discovered and developed at Gilead Sciences and is marketed as the fixed combination with brand name of Harvoni. The synthesis of ledipasvir has been reported in the literature132 and the routes shown in Schemes 22–24 below represent the most efficient and largest scale sequence reported in the patent literature.133,134 The synthesis of the spirocyclopropane proline intermediate 136 is described in Scheme 21. Bis-iodination of cyclopropane-1,1-diyldimethanol (131) in the presence of triphenylphosphine gave diiodide 132 in 70% yield. N-Boc-glycine ethyl ester (133) was then treated with sodium hydride followed by diiodide 132 to give the protected proline analog 134 in 61% yield. Saponification of the ester followed by a classical resolution with (1S,2R)-amino-indanol gave enantomerically pure salt 135. Liberation of the free acid with 1 M HCl followed by treatment with potassium tert-butoxide provided enantiopure potassium salt 136 in high yield. The synthesis of the difluoro-fluorene Suzuki coupling intermediate 143 is described in Scheme 22. Iodination of 2-bromofluorene (137) produced aryl iodide 138 in 95% yield, which was then treated with lithium hexamethyldisilazide and N-fluorobenzenesulfonimide (NFSI) to give the difluoro intermediate 139 in 82% yield. Formation of the Grignard reagent of 139 through reaction with isopropylmagnesium chloride followed by condensation with Weinreb amide 140 gave chloroketone 141 in 71% yield. The potassium salt of the cyclopropyl proline intermediate 136 (described in Scheme 21) was coupled with 141 to give keto ester 142 in high yield. Heating 142 with ammonium acetate resulted in formation of the imidazole ring in intermediate 143 in 77% yield. The completion of the synthesis of ledipasvir is described in Scheme 23. Commercially available (1R,3S,4S)-N-Boc-2-azabicyclo [2.2.1]heptane-3-carboxylic acid (144) was coupled to 4-bromo- 1,2-benzenediamine (145) using EDC/HOBt to give a mixture ofamides 146a/146b in 72% yield. Heating mixture 146a/146b with acetic acid affected cyclization to benzimidazole 147 in 94% yield. Palladium mediated coupling of bromide 147 to bis(pinacolato)diboron gave intermediate148 which was then coupled in the same reaction vessel to bromide 143 generated in Scheme 22. This was followed by formation of the oxalate salt to give the protected central core of ledipasvir (149) in good overall yield. Removal of the amine protecting groups gave diamine 150 which was coupled to two equivalents of Moc-valine (151) via EDC/HOBt to give ledipasvir XVII in 73% yield. 19. Lobeglitazone sulfate

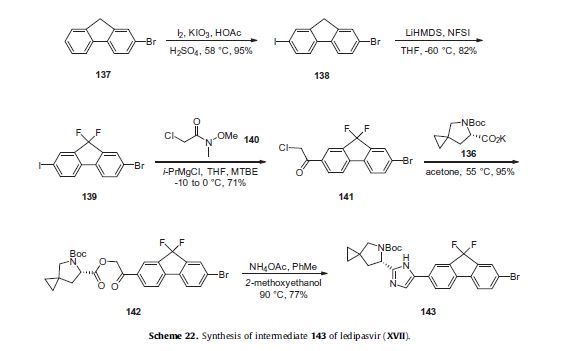


130. Gentile, I.; Buonomo, A. R.; Borgia, F.; Castaldo, G.; Borgia, G. Expert Opin.Invest. Drugs 2014, 23, 561.
131. Smith, M. A.; Chan, J.; Mohammad, R. A. Ann. Pharmacother. 2015, 49, 343.132. Link, J. O.; Taylor, J. G.; Xu, L.; Mitchell, M.; Guo, H.; Liu, H.; Kato, D.;Kirschberg, T.; Sun, J.; Squires, N.; Parrish, J.; Keller, T.; Yang, Z. Y.; Yang, C.;Matles, M.; Wang, Y.; Wang, K.; Cheng, G.; Tian, Y.; Mogalian, E.; Mondou, E.;Cornpropst, M.; Perry, J.; Desai, M. C. J. Med. Chem. 2014, 57, 2033.
133. Guo, H.; Kato, D.; Kirschberg, T. A.; Liu, H.; Link, J. O.; Mitchell, M. L.; Parrish, J.P.; Squires, N.; Sun, J.; Taylor, J.; Bacon, E. M.; Canales, E.; Cho, A.; Cottell, J. J.;Desai, M. C.; Halcomb, R. L.; Krygowski, E. S.; Lazerwith, S. E.; Liu, Q.;Mackman, R.; Pyun, H. J.; Saugier, J. H.; Trenkle, J. D.; Tse, W. C.; Vivian, R. W.;Schroeder, S. D.; Watkins, W. J.; Xu, L.; Yang, Z. Y.; Kellar, T.; Sheng, X.; Clarke,M. O. N. H.; Chou, C. H.; Graupe, M.; Jin, H.; McFadden, R.; Mish, M. R.;Metobo, S. E.; Phillips, B. W.; Venkataramani, C. WO Patent 2010132601A1,2010.
134. Scott, R. W.; Vitale, J. P.; Matthews, K. S.; Teresk, M. G.; Formella, A.; Evans, J.W. US Patent 2013324740A1, 2013.
135. Jin, S. M.; Park, C. Y.; Cho, Y. M.; Ku, B. J.; Ahn, C. W.; Cha, B.-S.; Min, K. W.;Sung, Y. A.; Baik, S. H.; Lee, K. W.; Yoon, K.-H.; Lee, M.-K.; Park, S. W. Diab.Obes. Metab. 2015, 17, 599.
136. Lee, H. W.; Ahn, J. B.; Kang, S. K.; Ahn, S. K.; Ha, D.-C. Org. Process Res. Dev.2007, 11, 190.
137. Lee, H. W.; Kim, B. Y.; Ahn, J. B.; Kang, S. K.; Lee, J. H.; Shin, J. S.; Ahn, S. K.; Lee,S. J.; Yoon, S. S. Eur. J. Med. Chem. 2005,
PAPER
The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection
http://pubs.acs.org/doi/abs/10.1021/jm401499g?prevSearch=LEDIPASVIR&searchHistoryKey=
http://pubs.acs.org/doi/pdf/10.1021/jm401499g
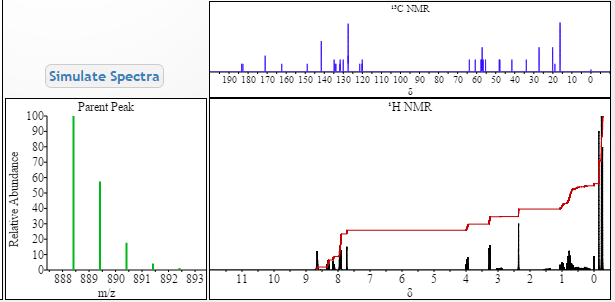
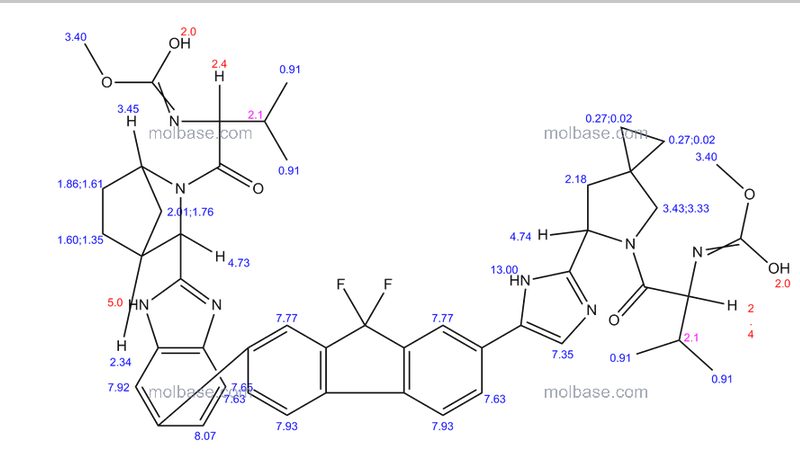
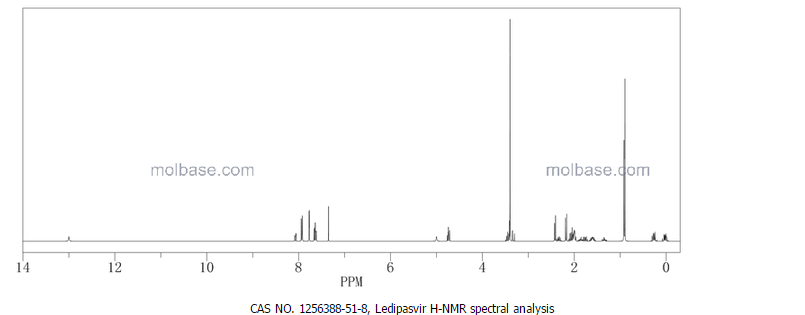
1H-NMR: 300 MHz, (dmso-d6) δ: 8.20-7.99 (m, 8H), 7.73 (s, 2H), 7.37 – 7.27
(m, 2H), 5.25 (dd, J = 7.2 Hz, 1H), 4.78 (s, 1H) 4.54 (s, 1H), 4.16 (m, 1H), 4.02 (m,
1H), 3.87 (m,1H), 3.74 (m, 1H), 3.55 (s, 3H), 3.53 (s, 3H), 2.75 (m, 1H), 2.25 (m,
2H), 2.09 – 2.04 (m, 2H), 1.88 – 1.79 (m, 2H), 1.54 (m, 1H), 0.94 – 0.77 (m, 15H)
0.63 (m, 4H) ppm.
19F-NMR: 282 MHz, (dmso-d6) δ: -109.1 ppm [-74.8 ppm TFA].
HRMS (ESI-TOF) m/z: [M + H]+
calc’d for C49H55F2N8O6: 889.4207; Found: 889.4214.
methyl [(2S)-1-{(6S)-6-[5-(9,9-difluoro-7-{2-[(1R,3S,4S)-2-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-azabicyclo[2.2.1]hept-3-yl]-1H-benzimidazol-6-yl}-9H-fluoren-2-yl)-1H-imidazol-2-
yl]-5-azaspiro[2.4]hept-5-yl}-3-methyl-1-oxobutan-2-yl]carbamate (39 NOS IS LEDISPAVIR
PATENT
Synthesis of 25
25
B. Synthesis of 26 and 27
25 26 27
[0186] To a flask was charged 25 (20.00 g, 0.083 mol), 4-bromo-l,2-benzenediamine (16.74 g, 0.089 mol, 1.08 equiv.), hydroxybenzotriazole (HOBt) (13.96 g, 0.091 mol, 1.1 equiv.), and l-ethyl-3-(3-dimethylaminopropyl) carbodiimide HC1 (EDC.HC1) (17.48 g, 0.091 mol, 1.1 equiv.). The flask was cooled in an ice bath, and was charged with N,N- dimethylacetamide (DMAc, 80 mL). The reaction was allowed to cool to ca. 10 °C with stirring. N-methylmorpholine (NMM) (27.34 mL, 0.249 mol, 3 equiv.) was added over 5 minutes keeping the internal temperature below 20 °C. The reaction was stirred at rt for 20 h. Upon reaction completion, the reaction mixture was added to MTBE (200 mL) and water (600 mL) in a separatory funnel and was gently shaken. The layers were allowed to separate, and the aqueous layer was removed. The aqueous layer was extracted twice with MTBE (50 mL), and the organic extracts were combined. The combined organic extracts were then extracted with water (500 mL), forming a mixture that did not separate well. The mixture was filtered over an appropriate solid support and the layers were separated. The organic phase was concentrated under vacuum, and the resulting residue was dissolved in diisopropyl ether (100 mL). The solution was cooled to ca. 5 °C with stirring. Acetic acid (5.22 mL, 0.091 mol, 1.1 equiv.) was added slowly keeping the internal temperature below 10 °C, and the resulting suspension was stirred 2 h at 5 °C. The thick suspension was then filtered, and the solid was rinsed with diisopropyl ether (100 mL), followed by heptane (100 mL). The cake was dried under vacuum to give the product as a light-beige solid as a mixture of regioisomers 26 and 27 (28.19 g, 72%, >99% AN). 1H NMR (400 MHz, DMSO) mixture of 26 & 27 (data is for the two rotamers of the major regioisomer): δ 9.25 (s, 0.5H), 9.13 (s, 0.5H), 7.08 (d, J= 8.3 Hz, 0.5H); 7.06 (d, J= 8.2 Hz, 0.5H), 6.92 (d, J= 2.2 Hz, 0.5H), 6.89 (d, J= 2.1 Hz, 0.5H), 6.71 (dd, J= 8.4, 2.2, 0.5H), 6.66 (dd, J= 8.4, 2.2, 0.5H), 5.10 (br s, 1H), 5.05 (br s, 1H), 4.15 (br s, 0.5H), 4.10 (br s, 0.5H), 3.76 (s, 1H), 2.64 (br s, 1H), 1.96- 1.88 (m, 1H), 1.77-1.67 (m, 1H), 1.67-1.19 (m, 4H), 1.41 (s, 4.5H), 1.33 (s, 4.5H). MS-ESI+: [M + H]+ calcd for Ci8H25Br03N3, 410.1, 412.1; found, 410.0, 412.0
[0187] The disclosure provides in some embodiments the use of other coupling reagents. These include but are not limited to N,N”-dicyclohexylcarbodiimide (DCC), NJV- diisopropylcarbodiimide (DIC), 6-chloro-2,4-dimethoxy-s-triazine (CDMT), O- benzotriazole-N^N^A^-tetramethyl-uronium-hexafluoro-phosphate (HBTU), and 2-(7-Aza- 1H- benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU).
[0188] The amine base also can be varied or omitted completely. For instance the amine is selected from tertiary amines (R3N), 2,6-lutidine, pyridine, dicyclohexylmethylamme, and N- methylmorpholine (NMM).
[0189] Suitable solvent alternatives are selected from DMF, NMP, dialkyl and cyclic ethers R20, THF, 2-MeTHF, DCM, DCE, toluene, EtOAc, IP Ac, acetone, MIBK, and MEK.
[0190] Suitable temperatures for the reaction range from about -20 °C to 80 °C.
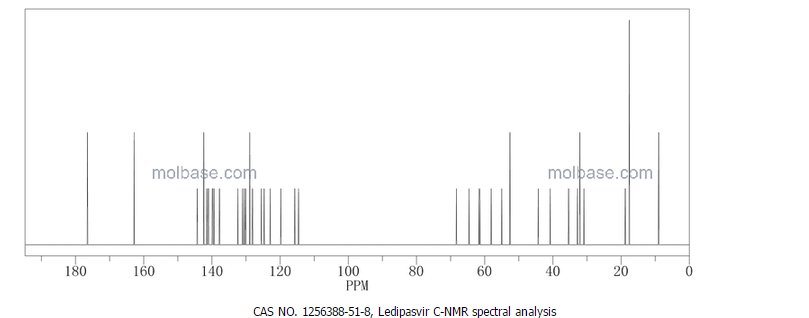
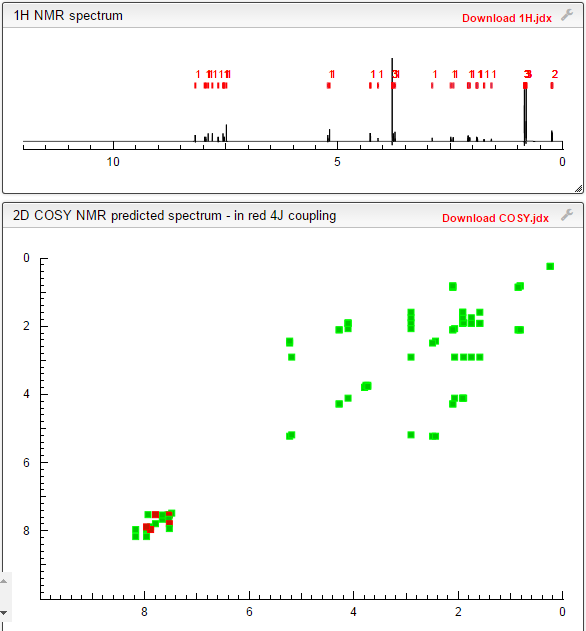
NMR PREDICT
1H/13C NMR PREDICT


COSY
Links
1)Link, John O.et al; The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection; Journal of Medicinal Chemistry (2013), Ahead of Print.DOI:10.1021/jm401499g
2)Ray, Adrian S. et al; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., WO2013040492
3) Delaney, William E. et al ; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., wo2012087596
4) Delaney, William E., IV et al; Preparation of quinoline derivatives and analogs for use in the treatment of hepatitis C virus infection in combination with ribavirin; PCT Int. Appl., wo2011156757
5) Guo, Hongyan et al; Preparation of biaryls, arylheteroaryls, heteroaryls, biarylacetylenes and related compounds end-capped with amino acid or peptide derivatives as antiviral agents; PCT Int. Appl., WO2010132601
6)Phase III (Sofosbuvir + Ledipasvir) ION-1 study: (Clinical Trial number: NCT01701401):
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection
7) Phase III (Sofosbuvir + Ledipasvir) ION-2 study: (Clinical Trial number: NCT01768286)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/GS-5885 Fixed-Dose Combination ± Ribavirin for 12 and 24 Weeks in Treatment-Experienced Subjects With Chronic Genotype 1 HCV Infection
8) Phase III (Sofosbuvir + Ledipasvir) ION-3 study: (Clinical trial number: NCT01851330)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection

References
- “Ledipasvir” (PDF). United States Adopted Name.
- “Ledipasvir-submitted-to-FDA”.
- “GS-5885”. Gilead Sciences.
- ELECTRON: 100% Suppression of Viral Load through 4 Weeks’ Post-treatment for Sofosbuvir + Ledipasvir (GS-5885) + Ribavirin for 12 Weeks in Treatment-naïve and -experienced Hepatitis C Virus GT 1 Patients. Gane, Edward et al. 20th Conference on Retroviruses and Opportunistic Infections. March 3–6, 2013. Abstract 41LB.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, Liz. HIVandHepatitis.com. 4 March 2013.
- “Gilead Files for U.S. Approval of Ledipasvir/Sofosbuvir Fixed-Dose Combination Tablet for Genotype 1 Hepatitis C”. Gilead Sciences. 10 February 2014.
- “U.S. Food and Drug Administration Approves Gilead’s Harvoni® (Ledipasvir/Sofosbuvir), the First Once-Daily Single Tablet Regimen for the Treatment of Genotype 1 Chronic Hepatitis C”. 10 October 2014. Retrieved 10 October 2014.
- Afdhal, N; Zeuzem, S; Kwo, P; Chojkier, M; Gitlin, N; Puoti, M; Romero-Gomez, M; Zarski, J. P.; Agarwal, K; Buggisch, P; Foster, G. R.; Bräu, N; Buti, M; Jacobson, I. M.; Subramanian, G. M.; Ding, X; Mo, H; Yang, J. C.; Pang, P. S.; Symonds, W. T.; McHutchison, J. G.; Muir, A. J.; Mangia, A; Marcellin, P; Ion-1, Investigators (2014). “Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection”. New England Journal of Medicine 370 (20): 1889–98. doi:10.1056/NEJMoa1402454. PMID 24725239.
- http://www.gilead.com/~/media/Files/pdfs/medicines/liver-disease/harvoni/harvoni_pi.pdf
- http://www.hepatitisc.uw.edu/page/treatment/drugs/ledipasvir-sofosbuvir
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Methyl N-[(2S)-1-[(6S)-6-[5-[9,9-Difluoro-7-[2-[(1S,2S,4R)-3-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]-3-azabicyclo[2.2.1]heptan-2-yl]-3H-benzimidazol-5-yl]fluoren-2-yl]-1H-imidazol-2-yl]-5-azaspiro[2.4]heptan-5-yl]-3-methyl-1-oxobutan-2-yl]carbamate
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 76% |
| Protein binding | >99% |
| Metabolism | No cytochromemetabolism |
| Biological half-life | 47 hrs |
| Identifiers | |
| CAS Registry Number | 1256388-51-8 |
| ATC code | None |
| ChemSpider | 29271894 |
| ChEBI | CHEBI:85089 |
| Chemical data | |
| Formula | C49H54F2N8O6 |
| Molecular mass | 889.00 g/mol |
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT
////////////////GS-5885, LEDIPASVIR
PATENT 1
Patent application WO2010132601A1 (primary patent) discloses the base compound of ledipasvir. The application claims a general structural formula (Markush) of new amide compounds useful for treating disorders associated with HCV. This patent, if granted, serves as a blocking patent preventing competitors from making the product. The claims are very broad, using a Markush structure of antiviral agents. As per the WIPO ISR, claims 1-19 are novel and inventive. However, according to the ISR, all remaining claims (claims 20 to 173), covering a large number of compounds, lack both novelty and inventive step, due to lack of support from the patent specification and in the light of prior art. Prosecution at the USPTO Three patents have been granted in the United States: US8088368B2, claiming the base compound by general structural formula; US8273341B2 (a division of US8088368B2), claiming a method of inhibiting HCV; and US8575118B2 (a continuation of US8273341B2 and a division of US8088368B2), claiming specific amide compounds not covered in the other two related patents. The examination report of US8088368B2 reveals that the application was allowed after the applicant cancelled and amended claims on Markush substuents. The examination report of US8273341B2 reveals that the application was allowed after the applicant amended a claim ‘A method of treating HCV’ to ‘A method of inhibiting HCV´. The examination report of US8575118B2 reveals that the application was allowed after the applicant cancelled claims already covered by the related patents, and limited claims to four specific compounds. Patent 1 has been filed in various jurisdictions: The patent has been granted by the ARIPO, in South Africa, and the United States. The patent (or a related patent) is pending in Argentina, Australia, Canada, China, as well as China, Hong Kong SAR, the EAPO, the EPO, Israel, India, Japan, New Zealand, Singapore, and Ukraine. Legal status is not available for Colombia, Ecuador, Mexico, Peru, Uruguay, and Viet Nam. 13 Litigation / Opposition on Patent 1 In December 2013, Gilead Sciences filed apatent infringement lawsuit against Abbott Laboratories and AbbVie Inc., in the United States District Court for the District of Delaware (case Number: 1:13cv02034). The case involves Gilead Sciences patents US8088368B2, US8273341B2, and US8575118B2.
PATENT 2 Patent application WO2013184698A1 is a product and process patent, claiming new crystalline solvate forms of ledipasvir useful for treating a subject suffering from HCV infection. The application also claims processes of manufacture of such amorphous and crystalline forms with specific X-ray diffraction peaks, and compositions and combinations comprising them. The application has just recently been published and no written opinion on patentability is available at this stage. As per the available information (details available in the Annex): The patent is pending at the EPO and the United States. There are no litigation or opposition procedures reported.
PATENT 3 Patent application WO2013184702A1 is a process patent, claiming processes for the preparation of ledipasvir. The disclosure also provides compounds that are synthetic intermediates to compounds of ledipasvir. The claims are moderately narrow covering crystalline and amorphous forms of ledipasvir with specific X-ray diffraction peaks. The application has just recently been published and no written opinion on patentability is available at this stage. As per the available information (details available in the Annex): The patent is pending at the EPO and the United States. There are no litigation or opposition procedures reported.
PATENT 4 Patent application WO2012087596A1 is a formulation patent, claiming various formulations comprising a combination of ledipasvir with GS-9256, or tegobuvir or with other compounds. The application also claims methods of treatment with the said combinations for reducing viral load in a person infected with HCV. 14 As per the WIPO ISR, the application is novel but not inventive in comparison to the closest prior art retrieved during the search. The combinations claimed in the instant application are not disclosed in the prior art, thus the combinations are novel. However, the prior art discloses various combinations, therefore, the problem to be solved through the invention should be new combinations with fewer side effects. Further, no experimental data of synergism has been provided to support double, triple, or quadruple combinations. Thus, according to the ISR, the instant invention cannot be regarded as inventive. As per the available information (details available in the Annex): The patent has been granted in Argentina. The patent is pending in Australia, Canada, the EPO, and the United States. Legal status is not available for Japan and Uruguay. There are no litigation or opposition procedures reported.
PATENT 5 Patent application WO2013040492A2 is a formulation and method of use patent, claiming compositions and a method of using the combination for the treatment of HCV. Drug combinations are used, and the compositions include sofosbuvir, PSI-7851 and ledipasvir. Since the application claims a group of compounds of Markush structure, it gives the claims a broad scope. As per the WIPO ISR the application is novel but lacks the inventive step in light of prior art. The invention lacks an inventive step as it would be obvious to a person skilled in the art to combine the diastereoisomer of the present invention, disclosed in the prior art, with other antiviral agents to provide an alternative HCV therapy. As per the available information (details available in the Annex): The patent is pending in Australia, Canada, the EPO, and the United States. There are no litigation or opposition procedures reported. This patent is listed in the sofosbuvir report as Patent No. 7
http://www.who.int/phi/implementation/ip_trade/ledipasvir_report_2014-09-02.pdf
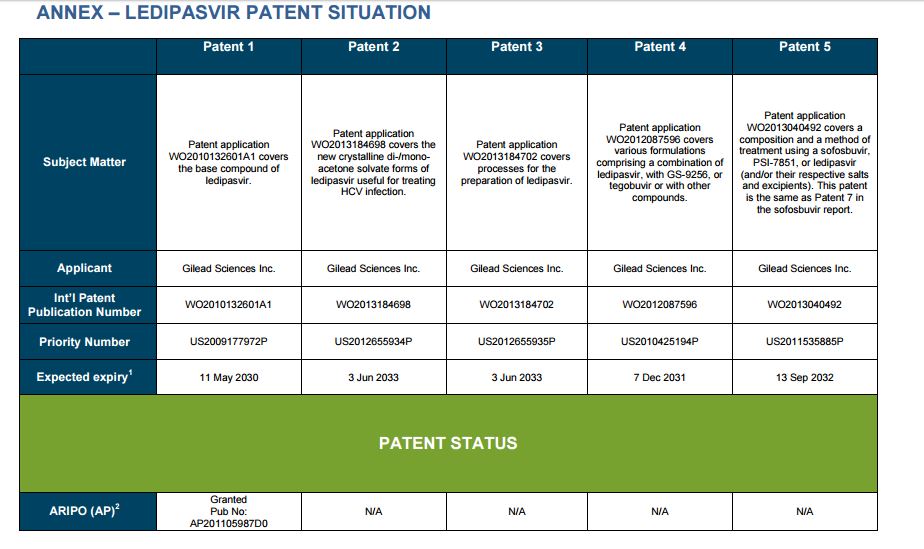
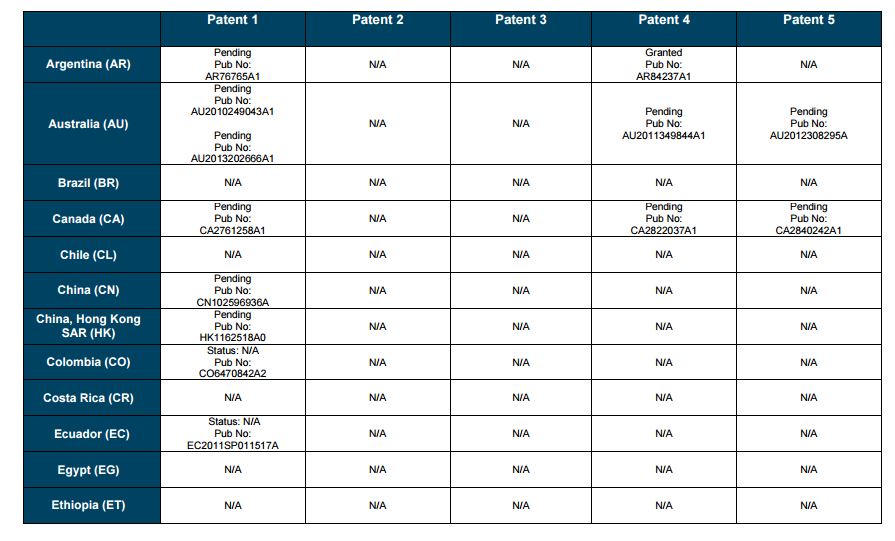
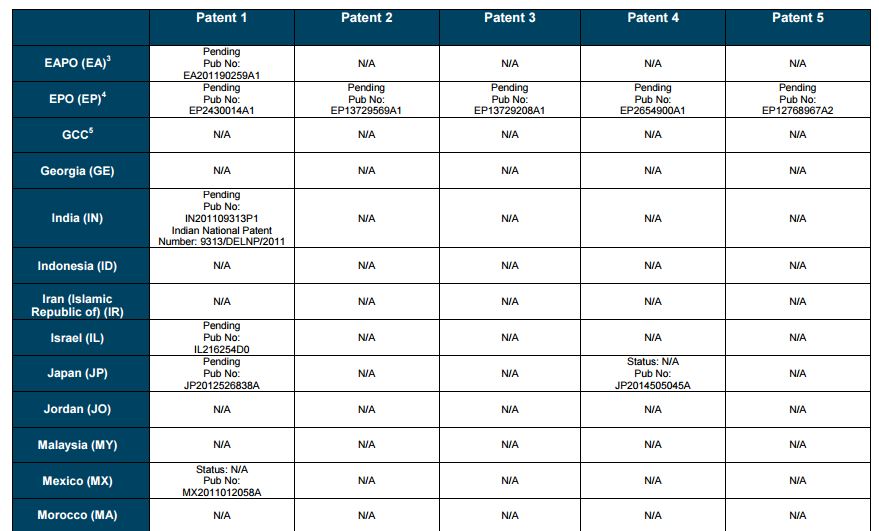
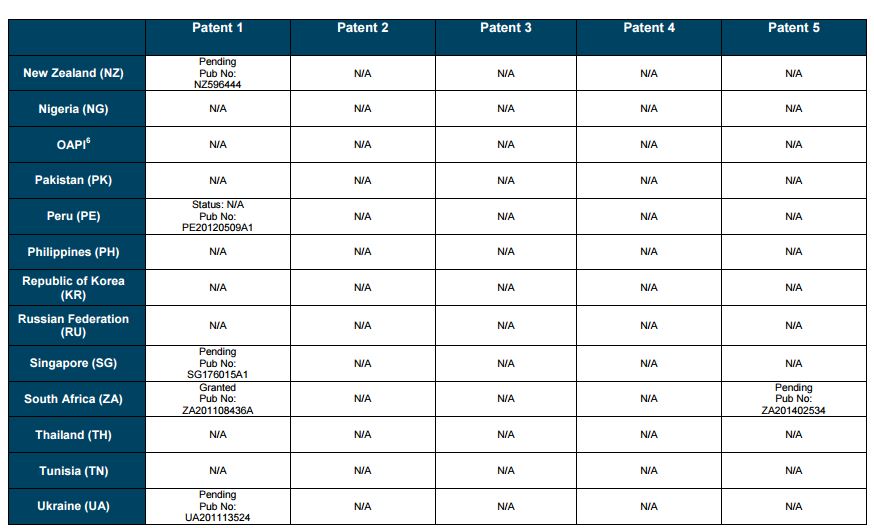
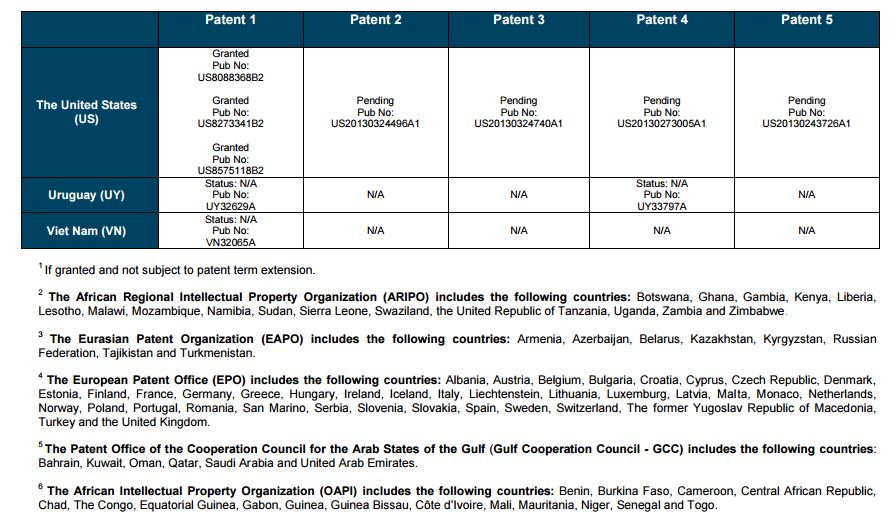
SUMMARY The search revealed patents filed with respect to ledipasvir by the Sponsor as well as a nonSponsor. The ledipasvir Sponsor patent collection comprises 5 different patents (patent families) with 47 family members published in 23 jurisdictions. The majority of these patent applications are still pending in the respective patent offices (see Patents 1 to 5 in the Annex). Patent 1 is the primary patent, claiming the base compound through a Markush claim, along with various substituents. Where granted, this patent can prevent competitors from making ledipasvir. Patents 2 and 3 claim processes to make ledipasvir and thus if granted will require competitors to design around these patents and use other production processes. The chemical product itself is not protected. Patents 4 and 5 claim combinations of different HCV drugs with ledipasvir, and their formulations. There is competition in the field by AbbVie, Inc., which filed formulation patents. Note: The search also revealed two patents that are relevant for all seven reports. Patent applications WO2013059630A1 and WO2013059638A1 inter alia claim the use of combinations of unnamed direct-acting antiviral agents for treating HCV, where the treatment does not include administration of interferon or ribavirin, and the treatment lasts between 8-12 weeks. The description and the dataset for these two patents can be found in the Working Paper on ombitasvir (Patents No 3 and 4). These patents are in litigation. Detailed information can be found in the Working Paper on sofosbuvir under Patent No 2.
World Drug Tracker: LEDIPASVIR
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
LEDIPASVIR
Biological Activity of Ledipasvir
Ledipasvir(GS5885) is an inhibitor of the hepatitis C virus NS5A protein. Ledipasvir is an experimental drug for the treatment of hepatitis C.
IC50 Value: 141 nM (EC50, JFH1/3a-NS5A hybrid replicon) [1]
Target: HCV NS5A
in vitro: Against JFH1/3a-NS5A, DCV was more potent (EC(50) = 0.52 nM) than GS-5885 (EC(50) = 141 nM). DCV sensitivity was increased against JFH1/3a-NS5A-M28V (EC50 = 0.006 nM), A30V (EC(50) = 0.012 nM), and E92A (EC(50) = 0.004 nM) while the NS5A-A30K and -Y93H variants exhibited reduced sensitivity to DCV (EC50 values of 23 nM and 1120 nM, respectively) and to GS-5885 (EC50 values of 1770 nM and 4300 nM, respectively) [1].
in vivo: GS-5885 was well tolerated and resulted in median maximal reductions in HCV RNA ranging from 2.3 log(10) IU/ml (1 mg QD) to 3.3 log(10) IU/ml (10 mg QD in genotype 1b and 30 mg QD). E(max) modeling indicated GS-5885 30 mg was associated with>95% of maximal antiviral response to HCV genotype 1a. HCV RNA reductions were generally more sustained among patients with genotype 1b vs. 1a. Three of 60 patients had a reduced response and harbored NS5A-resistant virus at baseline. NS5A sequencing identified residues 30 and 31 in genotype 1a, and 93 in genotype 1b as the predominant sites of mutation following GS-5885 dosing. Plasma pharmacokinetics was consistent with QD dosing [2].
Toxicity:
Clinical trial: Combination Therapy for Chronic Hepatitis C Infection. Phase 2
Clinical Information of Ledipasvir
| Product Name | Sponsor Only | Condition | Start Date | End Date | Phase | Last Change Date |
|---|---|---|---|---|---|---|
| Ledipasvir | Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-12 | 31-DEC-14 | Phase 3 | 12-SEP-13 |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-13 | 31-JAN-15 | Phase 3b | 11-NOV-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-MAY-13 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-DEC-10 | 30-APR-14 | Phase 2b | 28-AUG-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-JUN-13 | Phase 2 | 22-AUG-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-APR-13 | Phase 2b | 03-OCT-12 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-13 | 31-JAN-15 | Phase 3 | 11-NOV-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-MAY-13 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-12 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-APR-13 | Phase 2 | 03-OCT-12 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-JUN-13 | Phase 2b | 22-AUG-13 |
update………..
![]()

WO 2016145990, Ledipasvir, New patent, SHANGHAI FOREFRONT PHARMACEUTICAL CO., LTD
(WO2016145990) METHOD OF PREPARATION FOR LEDIPASVIR AND DERIVATIVE THEREOF, AND INTERMEDIATE COMPOUND FOR PREPARATION OF LEDIPASVIR
SHANGHAI FOREFRONT PHARMCEUTICAL CO., LTD [CN/CN]; Room 1306, No.781 Cailun Road China (Shanghai) Pilot Free Trade Zone, Pudong New Area Shanghai 201203 (CN)
HUANG, Chengjun; (CN).
FU, Gang; (CN).
FU, Shaojun; (CN).
WEI, Zhewen; (CN).
LI, Wei; (CN).
ZHANG, Xixuan; (CN)
chinese machine translation please bear………..
SMILES COC(=O)N[C@@H](C(C)C)C(=O)N1CC2(CC2)C[C@H]1c3ncc([nH]3)c4ccc5c6ccc(cc6C(F)(F)c5c4)c7ccc8nc([nH]c8c7)[C@@H]9[C@H]%10CC[C@H](C%10)N9C(=O)[C@@H](NC(=O)OC)C(C)C
DARUNAVIR


DARUNAVIR
206361-99-1 CAS NO
[(1S,2R)-3-[[(4-Aminophenyl)sulfonyl] (2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester
M. P.:- 72-74 °C (dec)
MW: 547.66
Darunavir and processes for its preparation are disclosed in EP0715618, W09967417, EP1725566 and Bioorganic & Medicinal Chemistry Letters (2004), 14(4), 959-963.
J Med Chem. 2013 May 23;56(10):4017-27. doi: 10.1021/jm400231v
US20050250845 discloses various pseudopolymorphs of darunavir and processes for their preparation. According to this application, “pseudopolymorph” is defined as a crystalline form of a compound in which solvent molecules are incorporated in the lattice structure. The Form B disclosed in the patent application is a pseudopolymorph wherein water is used as solvent. The thermogravimetric experiments of the Form B shows weight loss of 3.4% in the temperature range 25-78°C (water), 5.1% in the temperature range 25-1 10°C (ethanol and water) and further 1.1% weight loss (ethanol) in temperature range 110-200° C. Further at the drying step the Form B showed about 5.6% weight loss. The obtained dried product was hygroscopic and it adsorbed up to 6.8% water at high relative humidity. Amorphous form of darunavir is disclosed in US20050250845 and the publication in J.Org. Chem. 2004, 69, 7822 – 7829.
US 7700645 patent disclosed amorphous Darunavir, various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. Journal of Organic Chemistry 2004, 69, 7822-7829 disclosed amorphous Darunavir is obtained by purification with column chromatography in 2% methanol in chloroform as eluent. PCT publication WO2010086844A1 disclosed crystalline dimethylsulfoxide solvate and crystalline tetrahydrofuran solvate of darunavir. The publication also disclosed the amorphous darunavir having the IR spectrum with characteristic peaks at about 1454 and 1365 cm“1
PCT publication WO201 1083287A2 disclosed crystalline darunavir hydrate substantially free of any non aqueous solvent.
Drug information:- Darunavir is an Anti-microbial drug further classified as anti-viral agent of the class protease inhibitor. It is used either single or in combination with other drugs for the treatment of human immunodeficiency virus.
Darunavir (brand name Prezista, formerly known as TMC114) is a drug used to treat HIV infection. It is in the protease inhibitor class. Prezista is an OARAC recommended treatment option for treatment-naïve and treatment-experienced adults and adolescents.Developed by pharmaceutical company Tibotec, darunavir is named after Arun K. Ghosh, the chemist who discovered the molecule at the University of Illinois at Chicago. It was approved by the Food and Drug Administration (FDA) on June 23, 2006.[2]
Darunavir is a second-generation protease inhibitor (PIs), designed specifically to overcome problems with the older agents in this class, such as indinavir. Early PIs often have severe side effects and drug toxicities, require a high therapeutic dose, are costly to manufacture, and show a disturbing susceptibility to drug resistant mutations. Such mutations can develop in as little as a year of use, and effectively render the drugs useless.
Darunavir was designed to form robust interactions with the protease enzyme from many strains of HIV, including strains from treatment-experienced patients with multiple resistance mutations to PIs.
Darunavir received much attention at the time of its release, as it represents an important treatment option for patients with drug-resistant HIV. Patient advocacy groups pressured developer Tibotec not to follow the previous trend of releasing new drugs at prices higher than existing drugs in the same class. Darunavir was priced to match other common PIs already in use, such as the fixed-dose combination drug lopinavir/ritonavir.
PREZISTA (darunavir) is an inhibitor of the human immunodeficiency virus (HIV-1) protease.
PREZISTA (darunavir), in the form of darunavir ethanolate, has the following chemical name: [(1S,2R)-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]-carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester monoethanolate. Its molecular formula is C27H37N3O7S • C2H5OH and its molecular weight is 593.73. Darunavir ethanolate has the following structural formula:
 |
Darunavir ethanolate is a white to off-white powder with a solubility of approximately 0.15 mg/mL in water at 20°C.
|
|
4-11-2012
|
METHODS FOR THE PREPARATION OF HEXAHYDROFURO[2,3-b]FURAN-3-OL
|
|
|
12-28-2011
|
Substituted Aminophenylsulfonamide Compounds as Hiv Protease Inhibitor
|
|
|
12-23-2011
|
POLYMORPHS OF DARUNAVIR
|
|
|
12-14-2011
|
METHODS FOR THE PREPARATION OF N-ISOBUTYL-N-(2-HYDROXY-3-AMINO-4-PHENYLBUTYL)-P-NITROBENZENESULFONYLAMIDE DERIVATIVES
|
|
|
11-30-2011
|
Protease inhibitor precursor synthesis
|
|
|
6-31-2011
|
PROCESS FOR THE PREPARATION OF (3R,3AS,6AR)-HEXAHYDROFURO [2,3-B] FURAN-3-YL (1S,2R)-3-[[(4-AMINOPHENYL) SULFONYL] (ISOBUTYL) AMINO]-1-BENZYL-2-HYDROXYPROPYLCARBAMATE
|
|
|
9-29-2010
|
Aminophenylsulfonamide Derivatives as Hiv Protease Inhibitor
|
|
|
8-11-2010
|
Process for the preparation of (3R,3aS,6aR)-hexahydrofuro [2,3-b] furan-3-yl (1S,2R)-3[[(4-aminophenyl) sulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate
|
|
|
7-30-2010
|
RELATING TO ANTI-HIV TABLET FORMULATIONS
|
|
|
7-30-2010
|
COMBINATION FORMULATIONS
|
|
7-2-2010
|
METHODS AND INTERMEDIATES USEFUL IN THE SYNTHESIS OF HEXAHYDROFURO [2,3-B]FURAN-3-OL
|
|
|
5-7-2010
|
METHODS AND COMPOSITIONS FOR TREATING HIV INFECTIONS
|
|
|
4-21-2010
|
Pseudopolymorphic forms of a hiv protease inhibitor
|
|
|
9-21-2007
|
Immunoassays, Haptens, Immunogens and Antibodies for Anti-HIV Therapeutics
|
|
|
6-23-2006
|
Method for treating HIV infection through co-administration of tipranavir and darunavir
|
|
|
6-3-2005
|
Combination of cytochome p450 dependent protease inhibitors
|
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010086844A1 | Dec 8, 2009 | Aug 5, 2010 | Mapi Pharma Hk Limited | Polymorphs of darunavir |
| WO2011048604A2 * | Sep 16, 2010 | Apr 28, 2011 | Matrix Laboratories Limited | An improved process for the preparation of darunavir |
| WO2011083287A2 | Oct 6, 2010 | Jul 14, 2011 | Cipla Limited | Darunavir polymorph and process for preparation thereof |
| CN102584844A * | Jan 11, 2011 | Jul 18, 2012 | 浙江九洲药业股份有限公司 | Darunavir crystal form and method for preparing same |
| US6248775 | Apr 8, 1999 | Jun 19, 2001 | G. D. Searle & Co. | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| US7700645 | May 16, 2003 | Apr 20, 2010 | Tibotec Pharmaceuticals Ltd. | Pseudopolymorphic forms of a HIV protease inhibitor |
| Reference | ||
|---|---|---|
| 1 | JOURNAL OF ORGANIC CHEMISTRY vol. 69, 2004, pages 7822 – 7829 | |
| 2 | * | VAN GYSEGHEM E ET AL: “Solid state characterization of the anti-HIV drug TMC114: Interconversion of amorphous TMC114, TMC114 ethanolate and hydrate“, EUROPEAN JOURNAL OF PHARMACEUTICAL SCIENCES, ELSEVIER, AMSTERDAM, NL, vol. 38, no. 5, 8 December 2009 (2009-12-08), pages 489-497, XP026764329, ISSN: 0928-0987, DOI: 10.1016/J.EJPS.2009.09.013 [retrieved on 2009-09-24] |
Virus-encoded proteases, which are essential for viral replication, are required for the processing of viral protein precursors. Interference with the processing of protein precursors inhibits the formation of infectious virions. Accordingly, inhibitors of viral proteases may be used to prevent or treat chronic and acute viral infections. Darunavir has HIV protease inhibitory activity and is particularly well suited for inhibiting HIV-I and HIV -2 viruses. Darunavir, chemically (1 S^R.S’R.S’aS.e’aRJ-fS’he ahydrofuro^.S-b ]furanyl-[3-( 4-aminobenzenesulfonyl)isobutylamino [- 1-benzyl-zhydroxypropyl]carbamate. Darunavir is represented by the following structure:
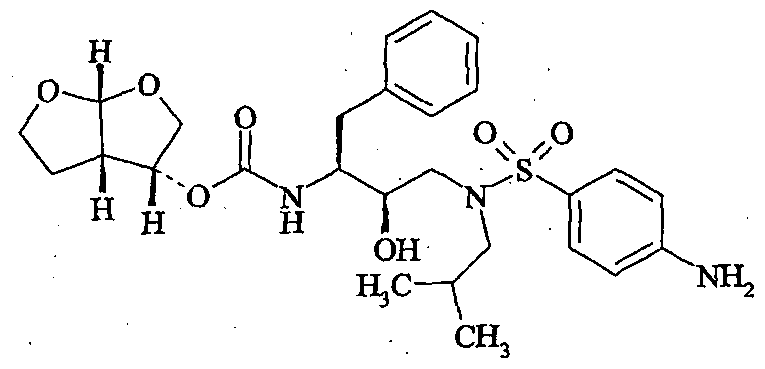
Darunavir and its pharmaceutically acceptable salts were disclosed in US 6248775 patent, wherein Darunavir is prepared by condensing 2R-hydroxy-3-[[(4-aminophenyl)sulfonyl](2- methylpropyl)amino]-1S(phenylmethyl)propylamine with hexahydro-furo[2,3-b]furan-3-ol in anhydrous acetonitrile in the presence of anhydrous pyridine and Ν,Ν’-disuccinimidyl carbonate at ambient temperature.
US 7700645 patent disclosed amorphous Darunavir, various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. Journal of Organic Chemistry 2004, 69, 7822-7829 disclosed amorphous Darunavir is obtained by purification with column chromatography in 2% methanol in chloroform as eluent. PCT publication WO2010086844A1 disclosed crystalline dimethylsulfoxide solvate and crystalline tetrahydrofuran solvate of darunavir. The publication also disclosed the amorphous darunavir having the IR spectrum with characteristic peaks at about 1454 and 1365 cm“1
PCT publication WO201 1083287A2 disclosed crystalline darunavir hydrate substantially free of any non aqueous solvent.
Darunavir Ethanolate, has the chemical name: [(1 S, 2R)-3-[[(4-aminophenyl) sulfonyl](2- methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3/?, 3aS, 6a/?)- hexahydrofuro[2,3-i>]furan-3-yl ester monoethanolate and has the following structural formula:

Darunavir and its process are first disclosed in US 6248775, wherein 2 ?-hydroxy-3-[[(4- aminophenyl)sulfonyl](2-methylpropyl)amino]-1 S(phenylmethyl) propylamine (4) is reacted with (3R, 3aS, 6aR)-hexahydrofuro[2,3- >]furan-3-ol in anhydrous acetonitrile in the presence of N, W-disuccinimidyl carbonate, anhydrous pyridine at ambient temperature followed by workup to get Darunavir (Scheme A).
Scheme A

Darunavir
US 20050250845 disclosed the various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. The same application disclosed the amorphous Darunavir by Raman spectra without process details.
WO 2005063770 discloses process for the preparation of Darunavir ethanolate, wherein 2R-hydroxy-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-1 S-(phenylmethyl)propyl amine (4) is reacted with (3R, 3aS, 6a ?)-hexahydrofuro[2,3-b]furan-3-ol in the presence of N, /V-disuccinimidyl carbonate, triethylamine, 41% methylamine in ethanol in a mixture of ethyl acetate and acetonitrile followed by workup and crystallization from ethanol to get Darunavir ethanolate (Scheme B).
Scheme B

In the prior art process, compound of formula 4 condensed with (3/?, 3aS, 6aR)- hexahydrofuro[2,3-6]furan-3-ol in large excess of solvent or solvent mixture containing large excess of base or mixture of bases to get Darunavir. Further, the obtained products by the processes described in the prior art are not satisfactory, from purity point of view. We have repeated the Darunavir synthetic procedures as described in the prior art and found that relatively large amounts of impurities were obtained along with Darunavir (Table-1) which need repeated crystallizations in different solvents to get desired quality of the final product resulting in poor yields. Among other impurities, the carbonic acid [(1/?,2S)-1-{((4-amino-benzenesulfonyl)-isobutyl-amino)-methyl}-2-((3R,3aSI6aR)- hexahydro-furot2,3-/3]furan-3-yloxycarbonylamino)-3-phenyl-propylester (3R,3aS,6aR)- hexahydro-furo[2,3-ft]furan-3-yl ester (difuranyl impurity of formula 1) is identified.


Conditions:-
i. Phenyl magnesium bromide, Cuprous cyanide, tetrahydrofuran, 23 °C, 1 h,
ii. t-Butyl hydroperoxide, titanium tetraisopropoxide, diethyl D-tartrate, dichloromethane, -22 °C, 24 h,
iii. Azidotrimethylsilane, titanium tetraisopropoxide, Benzene, reflux, 25 min,
iv. 2-Acetoxyisobutyryl chloride, Chloroform, 23 °C, 8 h,
v. Isobutyl amine, isopropanol, 80 °C, 12 h,
vi 4-aminobenzenesulfonyl chloride, aq. Sodium bicarbonate, dichloromethane, 23 °C, 12 h,
vii. 10% palladium on carbon, hydrogen gas (50 psi), methanol, acetic acid, tetrahydrofuran, room temperature, 2 h,
viii. [3R, 3aS,6aS]-3-hydroxyhexahydrofuro[2,3-b]-furan, disuccanamidyl carbonate, triethylamine, acetonitrile, 23 °C, 12 h
Schematic Representation for Synthesis of Darunavir
Preparation of Darunavir is described in US patent 05,158,713, and also in WO9967417 and WO9967254. Accordingly, 2-vinyloxirane 1 on reacting with phenyl magnesium bromide in presence of tetrahydrofuran solvent and cuprous cyanide catalyst give 4-phenylbut-2-ene-1-ol 2. Oxidizing 2 with t-Butyl hydroperoxide in presence of titanium tetraisopropoxide and diethyl D-tartrate using dichloromethane as solvent give [(3S)-3-benzyloxiran-2-yl]methanol 3.
Heating 3 with azidotrimethylsilane in presence of titanium tetraisopropoxide using benzene as solvent give (2S,3S)-3-azido-4-phenyl-butane-1,2-diol 4. The 1,2-dipl compound 4 underwent cyclization when treated with 2-acetoxyisobutyryl chloride in chloroform give (2S)-2-[(1S)-1-azido-2-phenyl-ethyl]oxirane 5, which was further heating with isobutylamine and isopropanol at higher temperature give (2R,3S)-3-azido-1-(isobutylamino)-4-phenyl-butan-2-ol 6. Compound 6 was reacted with 4-aminobenzenesulfonyl chloride in presence of aq. Sodium bicarbonate as base and dichloromethane as solvent resulting in to 4-amino-N-[(2R,3S)-3-azido-2-hydroxy-4-phenyl-butyl]-N-isobutyl-benzenesulfonamide 7.
Hydrogenating 7 with 10% palladium on carbon catalyst using hydrogen gas (50 psi) in methanol and tetrahydrofuran solvent in presence of small amount of acetic acid at ambient temperature resulted in to 4-amino-N-[(2R,3S)-3-amino-2-hydroxy-4-phenyl-butyl]-N-isobutyl-benzenesulfonamide 8. The final step involves reacting 8 with [3R,3aS,6aS]-3-hydroxyhexahydrofuro[2,3-b]-furan and disuccanamidyl carbonate in presence of triethylamine base and acetonitrile as solvent afford [(1S,2R)-3-[[(4-Aminophenyl)sulfonyl] (2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester also called Darunavir 9.
…………………………
http://www.google.com/patents/WO2013114382A1?cl=en
process for the preparation of amorphous Darunavir is as

Process for the preparation of intermediate 2 is as shown in below scheme.

Examples
Example -1 : Preparation of [(1S, 2S)-3-chloro-2-hydroxy-1-(phenyl methyl) propyl] carbamic acid tert-butyl ester (5).
The solution of (3S)-3-(tert-butoxycarbonyl) amino-1-chloro-4-phenyl-2-butanone (Chloromethyl ketone 6,100 g) and aluminium isopropoxide (35 g) in isoprpylalcohol was heated to mild reflux and maintained for 3 hours. After completion of reaction distilled off isopropyl alcohol up to 50 % under vacuum and the resultant mass was cooled to 25-35°C. Water was added to the distillate, pH was adjusted to 3.0-4.0 with acetic acid and maintained the stirring for 2 hours at 25-35°C. The obtained solid was filtered and washed with water. The wet cake was taken into isopropyl alcohol (400mL) and heated to reflux for 60minutes, the mass was cooled to 25-35°C again maintain the stirring for 60minutes, the obtained solid was filtered and washed with isopropyl alcohol. The wet product was dried under normal drying to get title compound 5 (yield 80 g). Example -2: Preparation of [(1 S, 2R)-3-[(2-methylpropyl) amino]-2-hydroxy-1- (phenylmethyl) propyl] carbamic acid tert-butyl ester (4).
The mixture of [(1S, 2S)-3-chloro-2-hydroxy-1-(phenylmethyl) propyl] carbamic acid tert-butyl ester (5,100 g), isobutyl amine (294 g), sodium carbonate (31.3 g) and water was heated to 60 – 65°C and maintained for 3hours. After completion of reaction water (200 mL) was added and distilled out excess isobutyl amine under vacuum at below 75°C. Water (800 mL) was added to the distillate, cooled to 25-35°C and stirred for 2 hours. The obtained solid was filtered and washed with water to get title compound 4 (yield 105 g).
Example -3: Preparation of [(1S, 2R)-3-[[(4-nitrophenyl) sulfonyl] (2-methylpropyl) amino]- 2-hydroxy-1-(phenylmethyl) propyl] carbmic acid tert-butylester (3).
[(1 S, 2R)-3-[(2-methylpropyl) amino]-2-hydroxy-1 -(phenyl methyl) propyl] carbamic acid tert-butyl ester (4, 100 gm) and triethylamine (39.04 g) was added to methylenedichloride (1200 mL) and the temperature was raised to 40°C. p-nitro benzene sulfonyl chloride solution (72.3g of p-NBSC dissolve in 300mL methylenedichloride) was added slowly at 40-45°C for 2-3 hrs. The reaction was maintained for 3hours at 40 – 45°C. After completion of the reaction, water (500 mL) was added, separated the organic layer and distilled out methylene dichloride at atmospheric pressure. Finally, strip out the methylene dichloride by using isopropyl alcohol (200 mL). Isopropyl alcohol (1000 mL) was added to the distillate and maintained the stirring for 60 minutes at 70- 80°C. Cooled the mass to 30 – 35°C, filtered and washed with Isopropyl alcohol to get title compound 3 (yield 145 g). Example – 4: Preparation of 4-Amino-N-(2R, 3S) (3-amino-2-hydroxy-4-phenylbutyl)-N- isobutyl-benzene sulfonamide (1).
(1S, 2R)-{1-benzyl-2-hydroxy-3-[isobutyl-(4-nitro-benzenesulfonyl)-amino]-propyl}-carbamic acid tert-butyl ester (3, 100g), 10% palladium carbon (10gm) and triethanolamine (2gm) were suspended in isopropyl alcohol. The reaction was heated to 40 – 45°C and maintained under 4 – 6kg/cm2 of hydrogen pressure for 3 hours. After completion of reaction, the mass was filtered and hydrochloric acid (70mL) was added to the filtered mass. The solution was heated to reflux and maintained for 2-3hours. After completion of reaction the mass was cooled to 25-35°C, the reaction mass pH was adjusted to 6.0 – 7.0 with 20% sodium hydroxide solution and distilled out isopropyl alcohol under vacuum at below 55°C. Ethanol (200mL) and water (400mL) was added to the distillate, the mass pH was adjusted to 9.0 – 10.0 with 20% sodium hydroxide solution at 25-35°C and maintained the stirring for 2 hours at 25-35°C. The mass was cooled to 0 – 5°C, filtered and wash with water. The wet product was taken into ethanol (350mL), maintained the stirring for 30minutes at reflux temperature. The mass was cooled to 2 – 4°C, stirred for 2 hours, filtered and washed with ethanol (50 mL). The wet product was dried under normal drying to get title compound 1 (Yield 60 g).
Example-5: Preparation of ethyl-2-(4,5-dihydrofuran-3-yl)-2-oxoacetate (VI).
2, 3-Dihydrofuran (250 g) was taken in toluene (2000 mL) and triethyl amine (505 g) was added to above solution. Ethyl oxalyl chloride (536.5 g) was slowly added to the above mixture by maintaining temperature at 25-30°C and maintained the stirring for 5 hours. After completion of reaction separated the organic layer, washed the organic layer with 8% sodium bicarbonate solution (2x500mL). Organic layer was distilled completely under vacuum to get title compound VI (Yield 560g).
1 H NMR : 1.38 (t, 3H), 2.93 (t, 2H), 4.34 (q, 2H), 4.63 (t, 2H), 8.02 (s, 1 H).
Example-6: Preparation of ethyl-2-(3-bromo-2-ethoxytetrahydrofuran-3-yl)-2-oxoacetate (V).
Ethyl-2-(4,5-dihydrofuran-3-yl)-2-oxoacetate (Vl, 100g) was dissolved in dichloromethane (500ml) and Ethanol (150mL) was added. The reaction mass was cooled to 5 to 10°C. N- bromosuccinimide (1 15 g) was added lot wise by maintaining temp below 10°C. Reaction mass was then stirred at 20-30°C till completion of reaction. Reaction mass was washed with sodium bicarbonate solution (2%, 3x400mL) and the organic layer was used for the next step.
Example-7: Preparation of hexahydrofuro [2, 3-b] furan-3-ol (IV).
To the solution of Ethyl-2-(3-bromo-2-ethoxy tetra hydrofuran-3-yl)-2-oxoacetate in dichloromethane (V, 500mL) as prepared in above example, sodium sulphite solution (225g was dissolved in 1700mL of water) was added at 25-35°C. Reaction mass was stirred for 5-8hours at the same temperature and separated the organic and aqueous layers. Organic layer was washed with water (340mL). Distilled out the solvent completely get ethyl-2-(2-ethoxy tetra hydrofuran-3- yl)-2-oxoacetate. Sodium borohydride (35.5g)was dissolved in ethanol (400mL) under nitrogen atmosphere, ethyl-2-(2-ethoxytetra hydrofuran-3-yl)-2-oxoacetate was dissolved in ethanol (100mL) and slowly added to above solution at 15-30°C. Reaction mass was heated to 30-45X, maintained for 5-8 hours, the reaction mass temperature was raised to 55°C and stirred for 8 hours. The reaction mass was cooled to 20-30°C, ammonium chloride solution (1 5g in 200mL water) was slowly added and stirred for 1-2hours. The reaction mass was filtered and filtrate was distilled out under vacuum to get residue. Dichloromethane (600mL) was added to residue and cooled to -10°C. Hydrochloric acid (85mL) was added slowly drop wise in 2 hours by maintaining temp -5 to 0°C, reaction mass was stirred for 60minutes at -5 to 0°C and distilled the solvent completely. The obtained residue was stripped out with isopropyl alcohol (2x200mL, 1x100mL), ethyl acetate (500mL) was added to the resultant residue, stirred for 30-60minutes and cooled to 10-15°C. The solution was filtered and filtrate was concentrated to get title compound IV (yield 56 g).
Example-8: Preparation of Hexahydrofuro [2, 3-b] furan-3-yl acetate (III).
Hexahydrofuro [2, 3-b] furan-3-ol (IV, 60g) was dissolved in dichloromethane (300mL) and cooled to 0-5°C. To the cooled solution triethylamine (58.2 g), N, N-dimethylaminopyridine (1.12g) was added, acetic anhydride (56.5g) was added for 30-60 minutes at the same temperature, the mass temperature was raised to 25-35°C and stirred for 2-4hours. After completion of reaction the mass was cooled to 10-20°C, water (120mL) was added, stirred for 30minutes, separated the organic layer, washed with 10% sodium chloride solution (120mL) and distilled out dichloromethane to get title compound (yield 72g). Further, the product was purified by fractional distillation to get pure Hexahydrofuro [2, 3-b] furan-3-yl acetate III (yield 54g).
1 H NMR : 1.9-2.09(m, 2H), 2.10(s, 3H), 3.0-3.1 (m, 1 H), 3.86-4.03(m, 2H), 3.73(dd, 1 H), 4.10(dd, 1 H), 5.19(m, 1 H), 5.72 (d, 1 H)
Example-9: Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-yl acetate (II). To the buffer solution (104.3g of sodium dihydrogen orthophosphate dissolved in 530mL of water & pH adjusted to 6.0-6.5 with saturated sodium bicarbonate solution(68g in 680 mL water) solution) hexahydrofuro [2, 3-b] furan-3-yl acetate (111,115g) and CAL-B (17.25g) was added at 25-35°C, heated to 38-45°C and stirred for 24 hours. CAL-B (17.25g) was added stirred for 16 hours, again CAL- B (11.5g) was added at 38-45°C and stirred for 16 hours (pH should maintain 6.0-6.5). The reaction mass was cooled to 20-30°C, methylenedichloride (1 150mL) was added to the mass and stirred for 30 minutes. The reaction mass was filtered through hyflowbed then separated the organic layer and washed with 10%sodiumchloride solution (575mL). Organic layer was distilled completely under vacuum to get title compound II (yield 40. Og). Example-10: Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-ol (I).
(3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-yl acetate (II, 14.0g) was dissolved in methanol (42mL). Potassium carbonate (0.34g) was added and stirred at 25-35°C for 6-8hours. Methanol was distilled out completely under vacuum, to the distillate methylenedichloride (28mL) was added, stirred the mass for 30 minutes and again distilled the solvent to get residue. Dissolved the residue in dichloromethane (56mL), the resultant solution was treated with carbon and the solvent was completely distilled out get title compound I (yield 10.5g). Example-11 : Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b]-furan-3-yl-4-nitrophenyl carbonate (2).
To the solution of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-ol (l,100g) and Bis-nitrophenyl carbonate (257.2g) in methylene dichloride (1200mL), triethylamine solution (132 g in 300 mL of methylene dichloride) was added slowly at 20-30°C for 2-3hours. Maintained the reaction at the same temperature for 8-10hours, after completion of reaction water (500mL) was added for 30- 60minut.es and settled the reaction mass then separated the organic layer. Organic layer was washed with 10% acetic acid (100mL) and 10% sodium chloride solution (500mL), distilled the organic layer and co distilled with ethyl acetate (100mL). Ethyl acetate (300mL) was added to the distillate and heated to 50-55°C for 30-45minut.es to get clear solution, the solution was cooled to 5-10°C and maintained at the same temperature for 60 minutes. The obtained solid was filtered, washed with ethanol (100mL) and dried the wet material at 40-45°C for 10-14 hours to get title compound 2 (yield 160g). Example-12: Preparation of dimethylformamide solvate of Darunavir.
To a mixture of 4-amino-N-(2r,3S)(3-amino-2-hydroxy-4-phenylbutyl)-N-lsobutyl- benzenesulfonamide (1 ,25g) and N-methyl-2-pyrrolidinone (NMPO, 50mL), a solution of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]-furan-3-yl-4-nitrophenyl carbonate (2, 8.85g) and N-methyl- 2-pyrrolidinone (75mL) was added at -5 to 0°C for 2 to 3 hours under nitrogen atmosphere. The mass temperature was slowly raised to 25 to 30°C and stirred for 6 to 8 hours. The reaction mass was quenched in to the solution of methylene chloride (125mL) and water (250mL) at 25-35°C for 30 to 45 minutes. Separated the organic layer followed by washed with 10% sodium carbonate solution (150mL), 10% sodium chloride solution (150mL) and with water (6x150mL). Organic layer was dried over sodium sulphate and distill off the solvent under vacuum at below 50°C to obtain darunavir as a residue. To the residue Ν,Ν-dimethyl formamide (50mL) was added and cooled to 0 to -5°C, water (25mL) was added to the solution and maintained for 12hours at 0 to -5 °C, the obtained solid was filtered and washed with pre-cooled mixture of N,N-dimethyl formamide & water (25mL+25mL) to get dimethylformamide solvate of darunavir.
Example-13: Preparation of non-solvated crystalline Darunavir.
To a mixture of 4-amino-N-(2r,3S)(3-amino-2-hydroxy-4-phenylbutyl)-N-lsobutyl- benzenesulfonamide (1, 25g) and N-methyl-2-pyrrolidinone (NMPO, 50mL), a solution of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]-furan-3-yl-4-nitrophenyl carbonate (2, 18.85g) and N- methyl-2-pyrrolidinone (75mL) was added at -5 – 0°C for 2 to 3 hours under nitrogen atmosphere. The mass temperature was slowly raised to 25 – 30°C and stirred for 6 to 8 hours. The reaction mass was quenched in to the solution of methylene chloride (250mL) and water (250mL) at 25- 35°C for 30 – 45 minutes. Separated the organic layer followed by washed with 10% potassium carbonate solution (5x125mL), water (5x125mL), 20% sodium chloride solution (25mL), finally washed with 20% citric acid solution (125mL). The organic layer was treated with carbon and distilled off the solvent under vacuum at below 50°C to obtain darunavir as a residue. To the residue ethylacetate (250mL) was added and cooled to 0 to -5°C, to the cooled solution hexane (225mL) was added and maintained for 12hours at 0 to -5 °C, the obtained solid was filtered, washed with pre-cooled mixture of ethylacetate and hexane (25mL+25mL) and dried the compound to get non-solvated crystalline darunavir(yield 25g).
Example -14: Preparation of Amorphous Darunavir.
Darunavir (200g) as obtained in above example was dissolved in methylene dichloride (10L) and washed with water (3×1000 mL). Organic layer was taken into agitated thin film dryer (ATFD) feed tank. Applied initial temperature about 36 – 40°C and high vacuum (580mm/Hg) to the vessel. Slowly feed the solution to the Vessel (feed rate 5L r) over 1hour finally given the methylene chloride (3L) flushing. The material is collected in the material collecter. Dried at 58 -62°C for 40 hours to get amorphous darunavir (yield 160g).
………………..
http://www.google.com/patents/WO2011048604A2?cl=en

Preparation of Durumvir ethanolate
A solution of (3R,3aS,6a ?)-hexahydrofuro[2,3-D]furan-3-yl 4-nitrophenyl carbonate (5b, 75.4 g) in A -methyl-2-pyrrolidinone (300 mL) was added to a pre-cooled (-2 ± 2°C) solution of the compound of formula 4 (100 g) in W-methyl-2-pyrrolidinone (200 mL) at -4 to 0°C over a period of 2 h. The temperature of the reaction mass was slowly raised to 25 – 30°C and maintained for 8 h. After completion of the reaction (TLC monitoring), ethyl acetate (1000 mL) and purified water (500 mL) were added to the reaction mass. The layers were separated; organic layer was washed with sodium carbonate solution (2 X 500 mL) followed by sodium chloride solution. The organic layer was concentrated; ethanol (300 mL) was added, heated to 45 – 50°C, maintained for 1 h, filtered and washed with ethanol. The wet compound was taken into a mixture of ethyl acetate- ethanol (7:93, 600 mL), heated to reflux, charcoal was added and filtered. The resultant filtrate was cooled to 0 – 5°C, filtered the separated solid and washed with ethanol. The wet compound was dried at 45°C to obtain the in 124.3 g (yield-82.5%). The obtained Darunavir ethanolate had purity of 99.79% on area by HPLC and contained 0.08% on area by HPLC of the difuranyl impurity. Preparation of Amorphous Darunavir
Example – 4
A solution of Darunavir ethanolate (200 g) in dichloromethane (10 L) was taken into ATFD Feed tank. The solvent was evaporated by fed the solution slowly to the ATFD Vessel (feed rate 5 L /h) at 36 – 40°C and high vacuum (580 mm/Hg) over 2 h and then flushed with dichloromethane (3 L). The material is collected in the material collector in 160g with the HPLC Purity of 99.60% and particle size D50 of approximately 50 micrometers and Dgo of approximately 100 to 180 micrometers. Example-5
Darunavir Ethanolate (200 gm) was dissolved in Methylene chloride (1000 ml) and solvent was evaporated by applying vacuum followed by isolation of amorphous Darunavir as a solid as such or by charging n-Heptane or Isopropyl ether. Example – 6
Darunavir Ethanolate (10 g) was dissolved in ethyl acetate (50 mL). The solution was heated to 40 – 45°C and maintained for 30 min. Ethyl acetate was distilled off under vacuum completely to get residue in the form of semisolid. n-Heptane (50 mL) was added to the residue and stirred for 30 min. at ambient temperature. The separated solid was filtered, washed the wet cake with n-heptane (5 mL) and dried at 40 – 45°C under vacuum to get 8.0 g of amorphous Darunavir.
Example – 7
Darunavir Ethanolate (10 g) was placed into a dry round bottom flask and heated to 110 – 120°C to melt and maintained under vacuum for 4 h. The reaction mass was slowly cooled to 25 – 35°C. The obtained glass type crystal was broken into powder to afford 8.5 g of amorphous Darunavir.
Example – 8
Darunavir Ethanolate (5.0 g) was suspended into glycerol (25 g), heated to 110 – 120°C under vacuum and maintained for 30min. Water (50 mL) was added to the cooled reaction mass at 25 – 35°C under stirring and the obtained suspension was stirred for 30 min at 25 – 35°C. The separated solid was filtered and dried at 40 – 45°C under vacuum to yield 3.5 g of amorphous Darunavir. Example – 9
Carbonic acid [(1 R,2S)-1-{((4-amino-benzenesulfonyl)-isobutyl-amino)-methyl}-2- ((3/?,3aS,6aR)-hexahydro-furo[2,3-ft]furan-3-yloxycarbonylamino)-3-phenyl-propylester (3R,3aS,6a ?)-hexahydro-furo[2,3- )]furan-3-yl ester (difuranyl impurity, 1).
The difuranyl impurity (1) isolated from the mother liquor by preparative HPLC using a mixture of formic acid-water (1 :99) as eluent. The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.

1H-NMR (DMSO-cfe, 300 MHz, ppm) – δ 0.79 (d, J=6.6 Hz, 6H, 15 & 15′), 1.14-1.20 (m, 1 H, 20Ha), 1.34-1.42 (m, 1 H, 20Hb), 1.75-1.85 (m, 2H, 20’Ha & 14), 1.94-2.01(m, 1 H, 20’Hb), 2.54-2.64 (m, 2H, 8Ha & 13Ha), 2.74-2.89 (m, 3H, 8Hb, 13Hb & 19), 3.00-3.11 (m, 2H, 5Ha & 19′), 3.34-3.39 (m, 1H, 5Hb), 3.54-2.63 (m, 3H, 21 Ha & 17Ha), 3.65-3.74 (m, 3H, 21’Ha, 21 Hb &17Hb), 3.81-3.89 (m, 2H, 21’Hb & 17’Ha), 3.94-4.04 (m, 2H, 7 & 17’Hb), 4.81-4.88 (m, 1 H, 6), 4.92-4.96 (m, 1 H, 18′), 5.03-5.10 (m, 1 H, 18), 5.11 (d, J=5.4 Hz, 1 H, 22′), 5.61 (d, J=5.1 Hz, 1 H, 22), 6.03 (brs, 2H, NH2, D20 exchangeable), 6.63 (d, J=8.7 Hz, 2H, 2 & 2″), 7.15-7.28 (m, 5H, 10H, 10Ή, 11 H, 11′ & 12), 7.40 (d, J=8.7 Hz, 2H, 3 & 3′), 7.55 (d, J=9.3 Hz, 1 H, NH, D20 exchangeable).
“H-NMR (DMSO-d6, 75 MHz, ppm)- δ 19.56 & 19.81 (15C & 15’C), 25.42 (20 ), 25.47 (20C), 26.28 (14C), 35.14 (8C), 44.45(19’C), 45.01 (19C), 49.21 (5C), 53.39 (7C), 57.55 (13C), 68.70 (21 ‘C), 68.74 (21C), 69.95 (17’C), 70.20(17C), 72.65 (6C), 76.27 (18C), 79.59 (18’C), 108.70 (22’C), 108.75 (22C), 112.69 (2C), 122.56 (4C), 126.12 (12C), 128.04 (11 C & 11’C), 129.03 (10C & 10’C), 129.08 (3C), 138.03 (9C), 152.99 (1C), 153.55 (16’C), 155.32 (16C).
DIP MS: m/z (%) 1108 [M+Hf, 1131 [M+Naf
……………
http://www.google.com/patents/US20130244297

According to the present invention Darunavir having the below impurity not more than 0.1, preferably 0.05%.

………….
DARUNAVIR

CHYAVAN PRASH DABUR ; AN EVALUATION OF AYURVEDIC REMEDY IN K.P.C.A.R.C. LABORATORY TEST
CHYAVAN PRASH is an AYURVEDIC REMEDY used as RASAYANA in Ayurveda. The PRASH is also used as a Food or Food Supplement for maintaining GENERAL HEALTH CONDITION [GHC].
Above DABUR CHYAVAN PRASH container, which is tested at our Laboratory for evaluation puurposes.
Dabur branded CHYAVAN PRASH is taken randomised examination and test for evaluation of AYURVEDIC FUNDAMENTALS.The batch number of the test material container is given above.
5 gramms DABUR CHYAVAN PRASH is taken for test and examination purposes and absorbed in 100 ml solvent, used for the liquification level for laboratory test.
For Physical test and texture of the CHYAVAN PRASH, as for as prepared by me few years ago, on the similar lines , which was laid down and instructed by CHARAK SAMHITA. Although I prepared several years CHYAVAN PRASH for my patient, therefore I know well about the taste and texture of the Chyavan Prash.
A well…
View original post 330 more words
Medicinal Chemistry International: ASUNAPREVIR
Medicinal Chemistry International: ASUNAPREVIR
CLICK ABOVE
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
ASUNAPREVIR

- “A Phase 3 Study in Combination With BMS-790052 and BMS-650032 in Japanese Hepatitis C Virus (HCV) Patients”. ClinicalTrials.gov.
- C. Reviriego (2012). Drugs of the Future 37 (4): 247–254.doi:10.1358/dof.2012.37.4.1789350.
- Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- “Bristol-Myers’ Daclatasvir, Asunaprevir Cured 77%: Study”. Bloomberg. Apr 19, 2012.
- AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Bioorganic and Medicinal Chemistry Letters, 2011 , vol. 21, 7 pg. 2048 – 2054
patents
WO 2003099274, WO 2003099274, WO 2009085659
| US8202996 | 6-20-2012 | Crystalline forms of N-(tert-butoxycarbonyl)-3-methyl-L-valyl-(4R)-4-((7-chloro-4-methoxy-1-isoquinolinyl)oxy)-N- ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-L-prolinamide |
| US8163921 | 4-25-2012 | Hepatitis C Virus Inhibitors |
| US7915291 | 3-30-2011 | HEPATITIS C VIRUS INHIBITORS |
| US7449479 | 11-12-2008 | Hepatitis C virus inhibitors |
| US6995174 | 2-8-2006 | Hepatitis C virus inhibitors |










REFERENCES AND NOTES
-
- Rev. Med. Virol., 13 (2003), p. 57
-
- Hepatology, 36 (2002), p. S237
-
- Am. J. Med., 117 (2004), p. 344
-
- For a recent review on HCV anti-viral agents, see: Expert Opin. Invest. Drugs, 19 (2010), p. 63
- Curr. Opin. Pharmacol., 8 (2008), p. 522
-
- For a recent review on HCV NS3/4A protease inhibitors, see: Curr. Opin. Invest. Drugs, 10 (2009), p. 821
- Expert Rev. Anti. Infect. Ther., 7 (2009), p. 537
-
- Infect. Disord. Drug Targets, 6 (2006), p. 3
-
- Acc. Chem. Res., 41 (2008), p. 50
-
- J. Med. Chem., 47 (2004), p. 1605
-
- Antimicrob. Agents Chemother., 52 (2008), p. 4432
-
- Bioorg. Med. Chem. Lett., 18 (2008), p. 4853
-
- J. Med. Chem., 53 (2010), p. 2443
-
- Gastroenterology, 127 (2004), p. 1347
-
- J. Med. Chem., 53 (2010), p. 6466
-
- (a)Chemical and Engineering News (April 12, 2010 issue), 88, pp 30–33.
- (b)Perrone, R.K.; Wang, C.; Ying, W.; Song, A.I. WO 2009085659
-
- Sci. Transl. Med., 2 (2010), p. 30ra32
…………………………………………….
CONTD ONhttp://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
…………………………………………………………..
DR ANTHONY MELVIN CRASTO Ph.D