
Home » Uncategorized (Page 13)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
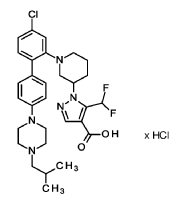
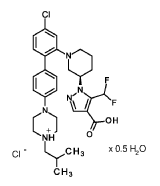
Nurandociguat
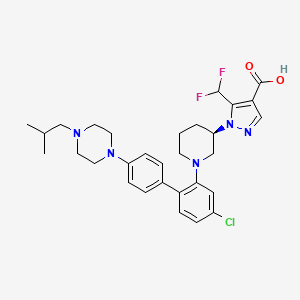
Nurandociguat
CAS 2781965-75-9
MF C30H36ClF2N5O2 MW 572.1 g/mol
1-[(3R)-1-{4-chloro-4′-[4-(2-methylpropyl)piperazin-1-yl][1,1′-biphenyl]-2-yl} piperidin-3-yl]-5-(difluoromethyl)-1H-pyrazole-4-carboxylic acid
1-[(3R)-1-[5-chloro-2-[4-[4-(2-methylpropyl)piperazin-1-yl]phenyl]phenyl]piperidin-3-yl]-5-(difluoromethyl)pyrazole-4-carboxylic acid
guanylate cyclase activator, BAY 3283142, LPU8429UK5
Nurandociguat is a small molecule drug candidate, previously known as BAY 3283142, that is a guanylate cyclase activator being developed by Bayer for cardiovascular conditions. The “ciguat” stem in its name indicates its function as a guanylate cyclase activator, a mechanism that is also being investigated for related drugs like runcaciguat. It is currently in clinical trials, including a Phase 2 program for chronic kidney disease (CKD).
- Drug class: Guanylate cyclase activator
- Developer: Bayer
- Previous name: BAY 3283142
- Indication: Investigated for cardiovascular conditions
- Current status: In clinical development, including a Phase 2 study for chronic kidney disease (CKD)
- OriginatorBayer
- ClassAntihypertensives; Cardiovascular therapies; Hepatoprotectants; Urologics
- Mechanism of ActionGuanylate cyclase stimulants
- Phase IIRenal failure
- Phase ICardiovascular disorders; Diabetic retinopathy; Hypertension; Liver disorders
- 28 Sep 2025No recent reports of development identified for phase-I development in Renal-failure in Germany (PO, Immediate release)
- 16 Sep 2025(CTIS2024-510856-11-00) (EudraCT2024-510856-11-00): Trial initiation and completion info added; updated DevT; Corrected intro to match DevT as most of the info about indication and countries missing
- 28 May 2025No recent reports of development identified for phase-I development in Renal-failure(In volunteers, In adults) in Japan (PO, Immediate release)
Nurandociguat is a small molecule drug. The usage of the INN stem ‘-ciguat’ in the name indicates that Nurandociguat is a guanylate cyclase activator and stimulator. Nurandociguat has a monoisotopic molecular weight of 571.25 Da.
PAT
- Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in womenPublication Number: WO-2023237577-A1Priority Date: 2022-06-09
- Substituted pyrazolo piperidine carboxylic acidsPublication Number: WO-2022122910-A1Priority Date: 2020-12-10
- The use of sgc activators for the treatment of ophthalmologic diseasesPublication Number: WO-2022122917-A1Priority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmic diseasesPublication Number: CN-115175681-APriority Date: 2020-12-10
- Use of sgc activators for the treatment of ophthalmologic diseasesPublication Number: US-2022241273-A1Priority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmic diseasesPublication Number: JP-2023514928-APriority Date: 2020-12-10
- The use of sgc activators for the treatment of ophthalmologic diseasesPublication Number: EP-4259140-A1Priority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmic diseasesPublication Number: KR-20230118143-APriority Date: 2020-12-10
- Substituted pyrazolo piperidine carboxylic acidsPublication Number: US-2023265072-A1Priority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmic diseasesPublication Number: JP-2024073585-APriority Date: 2020-12-10
- Use of sGC activators for the treatment of ophthalmological diseasesPublication Number: JP-7458683-B2Priority Date: 2020-12-10Grant Date: 2024-04-01
- Use of sgc activators for the treatment of ophthalmologic diseasesPublication Number: US-2023346777-A1Priority Date: 2020-12-10
- Use of sGC activators for treating ophthalmic diseasesPublication Number: CN-115175681-BPriority Date: 2020-12-10Grant Date: 2024-10-25
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022122917&_cid=P20-MHVQYD-96133-1
soluble guanylate cyclase (sGC) activators for use in the treatment and/or prophylaxis of ophthalmologic diseases, including non-proliferative diabetic retinopathy (NPDR), diabetic macular edema (DME), retinal ganglion cell/photoreceptor neurodegeneration and cataract, especially wherein the soluble guanylate cyclase (sGC) activators are compounds selected from the group consisting of
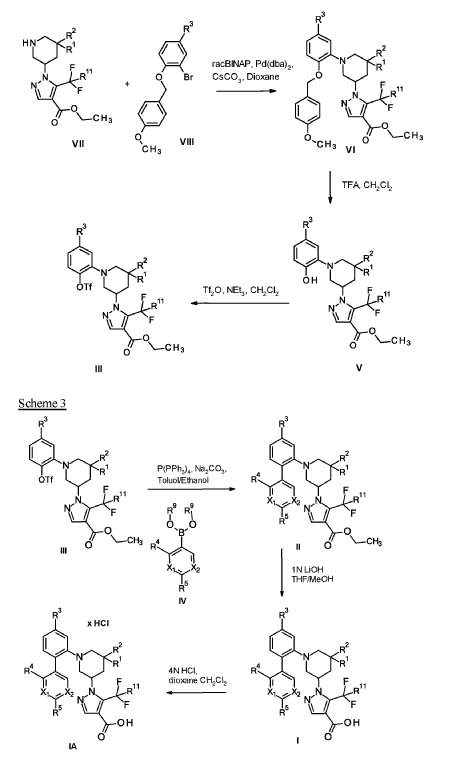
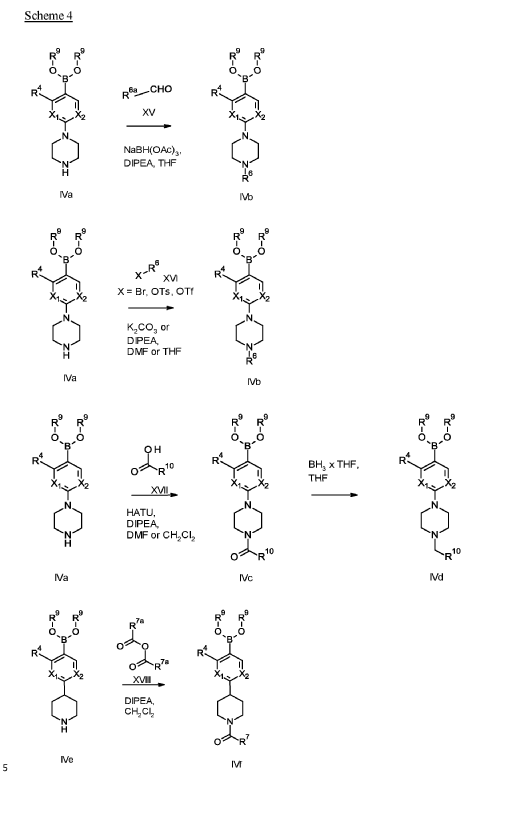
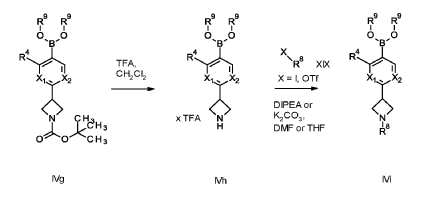
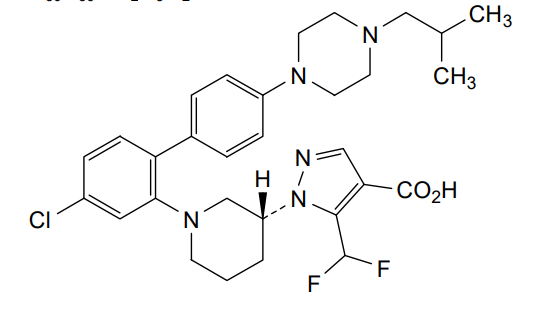
Example 1
1 – [ 1 – { 4-Chloro-4′- [4-(2-methylpropyl)piperazin- 1 -yl] [1,1 ’-biphenyl] -2-yl }piperidin-3-yl] -5- (difluoromethyl)-lH-pyrazole-4-carboxylic acid hydrochloride (Enantiomer 1)

Ethyl 1 – [ 1 – { 5-chloro-2- [(trifluoromethanesulfonyl)oxy]phenyl }piperidin-3-yl] -5-(difluoromethyl)- 1 H-pyrazole-4-carboxylate (prepared in analogy to Example 11A, Enantiomer 1, 80.0 mg, 147 pmol) and l-(2-methylpropyl)-4- [4-(4,4,5 ,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl]piperazine (Example 18 A 62.8 mg, 97 % purity, 177 pmol) were placed under argon in toluene/ethanol (820/820 pl). 2 M sodium carbonate solution (220 pl, 2.0 M, 440 pmol) and tetrakis(triphenylphosphine)palladium(0) (8.52 mg, 7.37 pmol) were added and the mixture was stirred at 100°C. overnight. The reaction mixture was diluted with ethyl acetate and 1 M hydrochloric acid was added. The aqueous phase was extracted three times with ethyl acetate. The organic phase was dried with sodium sulfate, filtered off and evaporated. The crude mixture was dissolved with THF/ethanol (2.0/0.2 ml), 1 M lithium hydroxide solution (1.5 ml, 1.5 mmol) was added and the mixture was stirred at room temperature overnight. A I M lithium hydroxide solution (740 pl, 740 pmol) was added again. After about 6 h the reaction mixture was evaporated at 50°C. The residue was dissolved in
SUBSTITUTE SHEET (RULE 26)
acetonitrile/water/0.25 ml trifluoroacetic acid and purified by preparative HPLC (RP18 column, acetonitrile/water gradient with the addition of 0.1% trifluoroacetic acid). The crude product was purified by means of thick layer chromatography (dichloromethane/methanol/formic acid: 10/1/0.1). The silica gel mixture was stirred with dichloromethane/1 M hydrochloric acid in dioxane (10/1) in ethanol, filtered off and carefully evaporated at 30°C and lyophilized. 34 mg of the target compound (36% of theory, purity 95%) were obtained.
LC-MS (Method 6): Rt = 1.23 min; MS (ESIpos): m/z = 572 [M-HC1+H]+
‘H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 1.004 (15.87), 1.015 (16.00), 1.500 (0.51), 1.521 (0.57), 1.728 (0.73), 1.750 (0.61), 1.897 (0.57), 1.917 (0.62), 1.975 (0.79), 2.122 (0.42), 2.133 (0.84), 2.144 (1.02), 2.156
(0.79), 2.571 (0.47), 2.587 (0.91), 2.610 (0.52), 3.004 (0.84), 3.022 (2.01), 3.026 (2.20), 3.038 (3.72), 3.048
(2.50), 3.065 (0.75), 3.154 (2.66), 3.161 (2.75), 3.169 (2.36), 3.177 (1.88), 3.224 (0.84), 3.237 (0.70), 3.589
(1.41), 3.602 (1.80), 3.825 (1.02), 3.841 (0.78), 3.866 (1.05), 3.882 (0.75), 4.223 (2.57), 4.445 (0.68), 4.463
(0.97), 4.481 (0.57), 7.045 (0.55), 7.055 (3.63), 7.070 (3.72), 7.084 (2.72), 7.087 (3.09), 7.110 (1.47), 7.113
(1.11), 7.123 (2.19), 7.127 (2.02), 7.163 (3.67), 7.177 (2.19), 7.215 (0.46), 7.428 (0.83), 7.495 (4.24), 7.510
(4.02), 7.515 (2.07), 7.602 (0.82), 7.959 (4.79), 9.484 (0.54).
Example 2
1 – [ 1 – { 4-Chloro-4′- [4-(2-methylpropyl)piperazin- 1 -yl] [1,1 ’-biphenyl] -2-yl }piperidin-3-yl] -5-(difluoromethyl)-lH-pyrazole-4-carboxylic acid (Enantiomer 2)
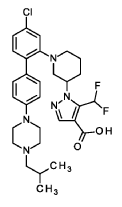
Method A
A solution of ethyl l-[l-{4-chloro-4′-[4-(2-methylpropyl)piperazin-l-yl][l,T-biphenyl]-2-yl}piperidin-3-yl]-5-(difluoromethyl)-lH-pyrazole-4-carboxylate (prepared in analogy to Example 17A, Enantiomer 2, 50.8 g, 84.6 mmol) in a THF/methanol mixture 9:1 (1.0 1) was treated with an aqueous solution of lithium hydroxide (850 ml, 1.0 M, 850 mmol) and stirred overnight at room temperature. The reaction mixture was
SUBSTITUTE SHEET (RULE 26)
concentrated, diluted with dichloromethane (1.5 1) and adjusted to pH = 2 with an aqueous solution of hydrogen chloride (2N). The resulting suspension was stirred 45 minutes at room temperature. The solid was filtered, washed with water and dried under vacuum affording 43 g (90 % yield) of the title compound.
LC-MS (Method 7): Rt = 1.27 min; MS (ESIpos): m/z = 572 [M+H]+
‘H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 1.002 (15.68), 1.013 (16.00), 1.080 (0.57), 1.092 (1.18), 1.103 (0.63), 1.498 (0.74), 1.519 (0.83), 1.719 (1.03), 1.741 (0.88), 1.902 (0.78), 1.908 (0.74), 1.922 (0.88), 1.928
(0.83), 1.943 (0.45), 1.978 (1.13), 1.994 (0.74), 2.102 (0.71), 2.112 (0.85), 2.123 (0.70), 2.571 (1.40), 2.591
(0.77), 2.882 (1.10), 3.018 (1.27), 3.035 (3.01), 3.053 (2.14), 3.239 (2.40), 3.254 (2.32), 3.368 (1.13), 3.379
(1.40), 3.391 (1.33), 3.403 (0.92), 3.493 (0.76), 4.463 (0.65), 4.482 (1.12), 4.500 (0.62), 7.033 (4.22), 7.048
(4.45), 7.074 (3.47), 7.077 (4.04), 7.100 (1.85), 7.103 (1.52), 7.113 (2.53), 7.117 (2.34), 7.162 (4.18), 7.175
(2.71), 7.439 (1.03), 7.481 (4.88), 7.495 (4.57), 7.526 (2.04), 7.613 (0.91), 7.952 (5.28).
Method B
1 – { 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)- 1 H-pyrazole-4-carboxylic acid hydrochloride (prepared in analogy to Example 3, Enantiomer 2, 31.2 mg, 51.3 pmol) were dissolved in 17 ml of dichloromethane and 1 ml of methanol. The solution was shaken once with 1.5 ml of saturated, aqueous sodium bicarbonate solution. The phases were separated. 5 ml of dichloromethane and 3 ml of methanol were added to the organic phase. The organic phase was then dried over sodium sulfate, filtered, evaporated and purified by preparative HPLC (RP18 column, acetonitrile/water gradient, neutral without acid addition). Product fractions were combined and lyophilized. 22 mg of the target compound (74% of theory) were obtained.
LC-MS (Method 3): Rt = 1.73 min; MS (ESIpos): m/z = 572 [M+H]+
‘H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 0.887 (15.60), 0.898 (16.00), 1.493 (0.64), 1.514 (0.70), 1.695 (0.89), 1.718 (0.74), 1.799 (0.48), 1.811 (0.88), 1.822 (1.12), 1.833 (0.92), 1.844 (0.48), 1.890 (0.68), 1.910
(0.74), 1.977 (0.93), 1.995 (0.62), 2.118 (3.91), 2.130 (3.66), 2.516 (5.14), 3.017 (1.09), 3.035 (2.76), 3.053
(1.94), 3.181 (5.03), 3.185 (5.02), 3.267 (1.53), 4.473 (0.55), 4.491 (0.96), 4.509 (0.54), 6.963 (3.96), 6.977
(4.06), 7.048 (3.13), 7.051 (3.31), 7.081 (1.60), 7.084 (1.26), 7.095 (2.21), 7.098 (1.89), 7.152 (3.52), 7.165
(2.42), 7.434 (4.45), 7.448 (4.50), 7.533 (1.51), 7.621 (0.67), 7.930 (4.14).
Example 3
1 – { 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)- 1 H-pyrazole-4-carboxylic acid hydrochloride (Enantiomer 2)
SUBSTITUTE SHEET (RULE 26)
Method A
A suspension of 1 – [ 1 – { 4-chloro-4′- [4-(2-methylpropyl)piperazin- 1 -yl] [1,1 ’-biphenyl] -2-yl }piperidin-3-yl] -5-(difluoromethyl)-lH-pyrazole-4-carboxylic acid (prepared in analogy to Example 2, Enantiomer 2, 43.5 g, 76.0 mmol) in diethyl ether (870 ml) was treated with a solution of hydrogen chloride in diethyl ether (84 ml, 1.0 M, 84 mmol). The resulting mixture was stirred overnight at room temperature and evaporated affording 46.1 g (quant.) of the title compound.
LC-MS (Method 3): Rt = 1.72 min; MS (ESIpos): m/z = 572 [M+H]+
‘H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 1.026 (15.64), 1.037 (16.00), 1.497 (0.56), 1.519 (0.61), 1.722 (0.78), 1.743 (0.65), 1.903 (0.59), 1.910 (0.53), 1.924 (0.66), 1.930 (0.61), 1.978 (0.82), 1.994 (0.50), 2.142
(0.45), 2.154 (0.91), 2.165 (1.11), 2.176 (0.89), 2.187 (0.45), 2.557 (0.64), 2.577 (1.02), 2.594 (0.55), 2.992
(1.81), 3.002 (2.77), 3.012 (1.87), 3.018 (1.15), 3.036 (2.40), 3.054 (1.60), 3.133 (1.12), 3.148 (1.19), 3.168
(0.53), 3.237 (0.88), 3.250 (0.76), 3.338 (0.81), 3.360 (1.42), 3.379 (0.88), 3.580 (1.61), 3.791 (0.89), 3.819
(1.25), 3.844 (0.81), 4.463 (0.89), 4.474 (0.97), 4.481 (1.26), 4.488 (0.99), 4.499 (0.88), 7.051 (3.56), 7.065
(3.77), 7.077 (2.72), 7.080 (3.14), 7.103 (1.42), 7.106 (1.13), 7.116 (2.00), 7.120 (1.84), 7.165 (3.40), 7.178
(2.22), 7.443 (0.84), 7.489 (4.04), 7.504 (3.79), 7.531 (1.66), 7.618 (0.72), 7.954 (4.33), 10.519 (0.49).
Method B
Ethyl 1 – [ 1 – { 5-chloro-2- [(trifluoromethanesulfonyl)oxy]phenyl }piperidin-3-yl] -5-(difluoromethyl)- 1 H-pyrazole-4-carboxylate (prepared in analogy to Example 14A, Enantiomer 2, 80.0 mg, 150 pmol) and l-(2-methylpropyl)-4- [4-(4,4,5 ,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl]piperazine (Example 18 A 64.1 mg, 97 % purity, 180 pmol) were dissolved under argon in toluene/ethanol (0.83/0.83 ml). Tetrakis(triphenylphosphine)palladium(0) (8.69 mg, 7.52 pmol) and 2 M sodium carbonate solution (226 pl, 452 pmol) were added and the mixture was stirred at 100°C overnight. The reaction mixture was diluted with ethyl acetate and water. The aqueous phase was acidified with 1 M hydrochloric acid. The phases were
SUBSTITUTE SHEET (RULE 26)
separated and the aqueous phase was extracted twice with ethyl acetate. The combined organic phases were dried over sodium sulfate, filtered and evaporated. The crude product was dissolved in THF/ethanol (3.9/0.39 ml), 1 M aqueous lithium hydroxide solution (1.5 ml, 1.5 mmol) was added and the mixture was stirred overnight at room temperature. The mixture was evaporated, the residue was dissolved in acetonitrile/TFA/water and purified using preparative HPLC (RP18 column, acetonitrile/water gradient with the addition of 0.1% TFA). The product fractions were combined and evaporated. The residue was mixed with 0.1 M hydrochloric acid in dioxane, carefully evaporated at 30°C (twice) and then lyophilized. 53 mg of the target compound (55% of theory, purity 95%) were obtained.
LC-MS (Method 4): Rt = 0.91 min; MS (ESIpos): m/z = 572 [M-HC1+H]+
‘H-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1.004 (15.46), 1.020 (16.00), 1.491 (0.44), 1.522 (0.50), 1.722 (0.68), 1.753 (0.55), 1.890 (0.47), 1.920 (0.55), 1.967 (0.84), 2.129 (0.76), 2.146 (0.96), 2.163 (0.76), 2.582
(0.91), 2.613 (0.48), 2.999 (0.86), 3.010 (1.71), 3.025 (3.88), 3.041 (2.30), 3.131 (0.88), 3.161 (1.25), 3.177
(2.08), 3.213 (1.75), 3.242 (1.16), 3.467 (1.06), 3.496 (0.84), 3.503 (0.60), 3.519 (0.54), 3.525 (0.50), 3.549
(0.75), 3.555 (0.84), 3.572 (1.57), 3.582 (1.48), 3.589 (1.38), 3.601 (2.78), 3.608 (1.89), 3.633 (0.44), 3.640
(0.41), 3.811 (0.94), 3.847 (1.32), 3.878 (0.71), 4.329 (0.49), 4.439 (0.46), 4.466 (0.73), 4.477 (0.52), 4.839
(0.49), 7.047 (3.30), 7.070 (3.64), 7.082 (2.61), 7.087 (3.29), 7.104 (1.46), 7.109 (0.86), 7.124 (2.34), 7.129
(2.03), 7.160 (3.99), 7.181 (1.96), 7.388 (0.88), 7.490 (4.02), 7.512 (3.81), 7.519 (2.20), 7.650 (0.72), 7.959
(3.78), 9.708 (0.41).
[OC]D20 = -73.05°, c = 0.465g/100cm3, trichloromethane.
Enantiomer 2 has an absolute configuration of R as shown in example 3 A below.
1 – { 3(2?)- 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)- 1H-pyrazole-4-carboxylic acid hydrochloride
Example 3A
1 – { 3(7?)- 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)- 1H-pyrazole-4-carboxylic acid hydrochloride hemihydrate
SUBSTITUTE SHEET (RULE 26)
100 mg 1 – { 1 – [4-Chloro-4′-(4-isobutylpiperazin- 1 -yl) [biphenyl] -2-yl]piperidin-3-yl } -5-(difluoromethyl)-lH-pyrazole-4-carboxylic acid hydrochloride (Enantiomer 2) (example 3) were solved at 60°C in 3,5 ml 2 -propanol, wherein the 2-propanol was dosed portion wise in lOOpl -portions at 60°C until a clear solution was obtained. Afterwards the vessel was closed with a septum and placed into a slowly cooling sand bath from 60°C to roomtemperature over the weekend -> small amounts of solids were detected. Thereafter the septum was provided with a canula, in order to slowly let the solvent evaporate. After 4 weeks crystals were collected and inspected under a microscope.



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////////Nurandociguat, guanylate cyclase activator, BAY 3283142, LPU8429UK5
Muvadenant
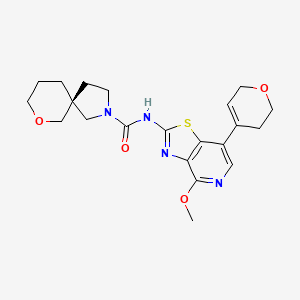
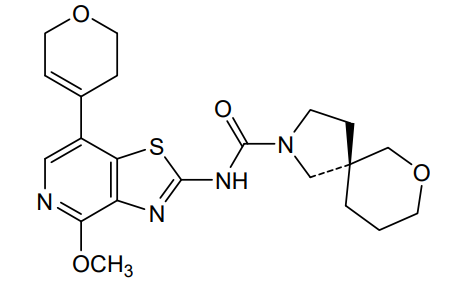
Muvadenant
CAS 2459881-03-7
MF C21H26N4O4S , 430.5 g/mol
(5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy[1,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5] decane-2-carboxamide
(5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide
adenosine receptor antagonist, antineoplastic, 6LSF69F6A8, M1069 , M 1069
Muvadenant is a small molecule drug. The usage of the INN stem ‘-adenant’ in the name indicates that Muvadenant is a adenosin receptor antagonist. Muvadenant has a monoisotopic molecular weight of 430.17 Da.
Adenosine is an ubiguitous modulator of numerous physiological activities, particularly within the cardiovascular, nervous and immune systems. Adenosine is related both structurally and metabolically to the bioactive nucleotides adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP) and cyclic adenosine monophosphate (cAMP), to the biochemical methylating agent S-adenosyl-L-methione (SAM) and structurally to the coenzymes NAD, FAD and coenzym A and to RNA.
Via cell surface receptors, adenosine modulates diverse physiological functions including induction of sedation, vasodilatation, suppression of cardiac rate and contractility, inhibition of platelet aggregability, stimulation of gluconeogenesis and inhibition of lipolysis. Studies show that adenosine is able to activate adenylate cyclases, open potassium channels, reduce flux through calcium channels, and inhibit or stimulate phosphoinositide turnover through receptor-mediated
mechanisms (Muller C. E. and Stein B., Current Pharmaceutical Design, 2: 501 , 1996; Muller C. E., Exp. Opin. Ther. Patents, 7(5): 419, 1997).
Adenosine receptors belong to the superfamily of G-protein-coupled receptors (GPCRs). Four major subtypes of adenosine receptors have been
pharmacologically, structurally and functionally characterized (Fredholm et al., Pharm. Rev., 46: 143-156, 1994) and referred to as A1, A2A, A2B and A3. Though the same adenosine receptor can couple to different G-proteins, adenosine A1 and A3 receptors usually couple to inhibitory G-proteins referred to as G, and Go which inhibit adenylate cyclase and down-regulate cellular cAMP levels. In contrast, the adenosine A2A and A2B receptors couple to stimulatory G-proteins referred to as Gs that activate adenylate cyclase and increase intracellular levels of cAMP (Linden J., Annu. Rev. Pharmacol. Toxicol., 41 : 775-87 2001).
PAT
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: US-2022119412-A1Priority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: EP-3914600-B1Priority Date: 2019-01-22Grant Date: 2024-08-07
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: CN-113939520-BPriority Date: 2019-01-22Grant Date: 2024-11-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: ES-2992081-T3Priority Date: 2019-01-22Grant Date: 2024-12-09
- Thiazolopyridine derivatives as adenosine receptor antagonists.Publication Number: JP-7600119-B2Priority Date: 2019-01-22Grant Date: 2024-12-16
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: AU-2020211697-A1Priority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: KR-20210116572-APriority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: CN-113939520-APriority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: EP-3914600-A1Priority Date: 2019-01-22
- Thiazolopyridine derivative as an adenosine receptor antagonistPublication Number: JP-2022524914-APriority Date: 2019-01-22
- Combination therapy for cancerPublication Number: CN-117858723-APriority Date: 2021-06-07
- Combination treatment of cancerPublication Number: EP-4351640-A1Priority Date: 2021-06-07
- Combination Treatment of CancerPublication Number: JP-2024520764-APriority Date: 2021-06-07
- Combination Treatment of CancerPublication Number: US-2024279338-A1Priority Date: 2021-06-07
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: WO-2020152132-A1Priority Date: 2019-01-22
- Novel crystalline forms of (s)-7-oxa-2-aza-spiro[4.5]decane-2-carboxylic acid [7-(3,6-dihydro-2h-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin-2-yl]-amide and co-crystal forms thereofPublication Number: TW-202415667-APriority Date: 2022-08-02
- Novel crystalline forms of (s)-7-oxa-2-aza-spiro[4.5]decane-2-carboxylic acid [7-(3,6-dihydro-2h-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin-2-yl]-amide and co-crystal forms thereofPublication Number: WO-2024028273-A1Priority Date: 2022-08-02
- Combination treatment of cancerPublication Number: WO-2022258622-A1Priority Date: 2021-06-07
- Combination treatment of cancerPublication Number: AU-2022288571-A1Priority Date: 2021-06-07
- Combination treatment of cancerPublication Number: CA-3220380-A1Priority Date: 2021-06-07
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020152132&_cid=P10-MHPOEV-06540-1
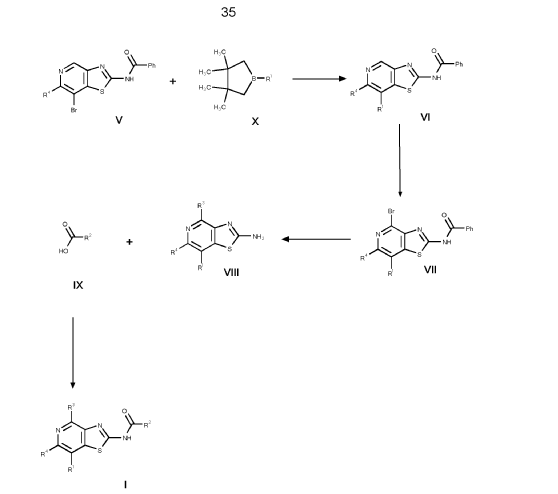
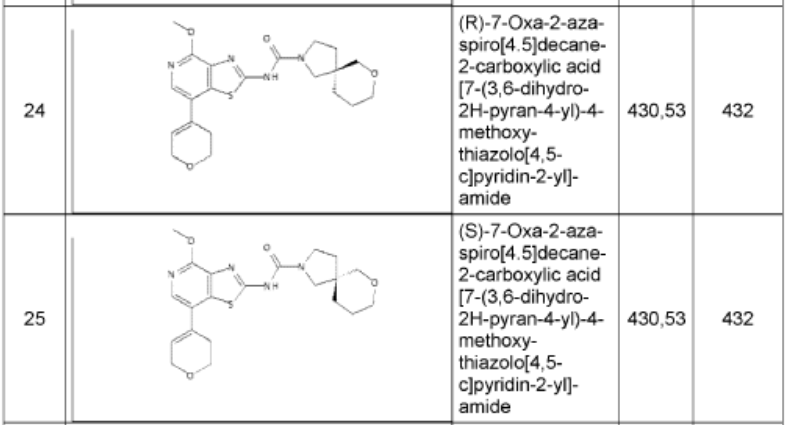
1. (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 24
and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5- c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 25
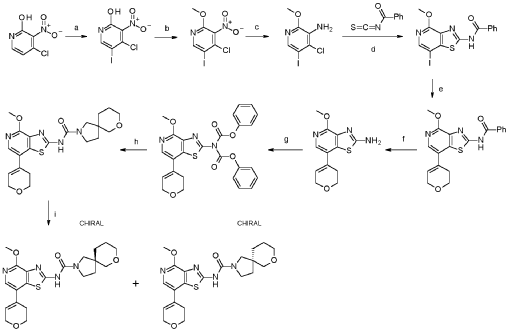
a. 4-chloro-5-iodo-3-nitropyridin-2-ol
Into a 250-mL round-bottom flask was placed 4-chloro-3-nitropyridin-2-ol (10.0 g, 54.4 mmol, 95%), N-lod-succinimid (NIS, 14.2 g, 59.9 mmol, 95%) in acetonitrile (115 mL). The solution was stirred for 1 h overnight at 80°C in an oil bath. The mixture was concentrated and the precipitate formed collected by filtration. The residue was washed with twice with petrol ether (500 mL) dried under vacuum at 60°C overnight. This resulted in 4-chloro-5-iodo-3-nitropyridin-2-ol (16.5 g, 97.9%, 97% purity) as a yellow solid. MS: m/z = 300.9 [M+H]+.
b. 4-chloro-5-iodo-2-methoxy-3-nitropyridine
Into a 500-mL round-bottom flask was placed 4-chloro-5-iodo-3-nitropyridin-2-ol (16.5 g, 53.3 mmol, 97%), Ag2CO3 (15.5 g, 53.3 mmol, 95%) in toluene (310 mL). To this suspension CH3I (15.9 g, 107 mmol, 95%) was added at 50°C and the mixture was stirred at 80°C for 4 h. The precipitate was collected by filtration and discarded. The filtrate was evaporated to dryness under vacuum and the residue purified by silica gel chromatography with ethyl acetate/petroleum ether (15:85).
This resulted in 4-chloro-5-iodo-2-methoxy-3-nitropyridine (9.90 g, 52.6%, 89% purity) as a light yellow solid. MS: m/z = 315.5 [M+H]+.
c. 4-chloro-5-iodo-2-methoxypyridin-3-amine
Into a 250-mL 3-necked round-bottom flask was placed 4-chloro-5-iodo-2-methoxy-3-nitropyridine (9.90 g, 28.0 mmol, 89%), iron (16.5 g, 281 mmol, 95%) and NH 4C (9.40 g, 174 mmol, 99%) in ethanol (152 mL) and water (30 mL). The mixture was stirred for 2 h at 80°C in an oil bath. The reaction mixture was filtered over Celite, washed with ethanol and the mother liquor was concentrated to dryness. The residue was stirred for 30 min. with 100 ml water at 60°dried in vacuo. This resulted in 4-chloro-5-iodo-2-methoxypyridin-3-amine (7.20 g, 75%, 83% purity) as an off-white solid. It was used without further purification in the next step. MS: m/z = 285.9 [M+H]+.
d. N-[7-iodo-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2-yl]benzamide
Into a 500-mL round-bottom flask was placed 4-chloro-5-iodo-2-methoxypyridin-3-amine (7.20 g, 21.0 mmol, 83%) in acetone (150 mL) and benzoyl isothiocyanate (5.21 g, 31.5 mmol, 99%) was added dropwise at room temperature. The solution was stirred for 1 h at 50 °C in an oil bath. The solids were collected by filtration, washed with acetone and dried in vacuo to give N-[7-iodo-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (8.73 g, 91 %, 90% purity) as a white solid. MS: m/z = 412.2 [M+H]+.
e. N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin- 2-yl]benzamide
To a solution of N-[7-iodo-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (6.00 g, 13.1 mmol, 90%) and 2-(3,6-dihydro-2H-pyran-4-yl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane (6.13 g, 27.7 mmol, 95%) in dioxane (200 mL) and water (40.00 mL) were added NaOH (2.90 g, 68.9 mmol, 95%) and Pd(dppf)Cl2* dichloromethane (1.20 g, 1.40 mmol, 95%). After stirring for 1 h at 100°C under a nitrogen atmosphere, the mixture was concentrated to dryness under vacuo. The residue was purified by silica gel chromatography with ethyl acetate/hexane (95:5). This resulted in 3.32 g (62%, 90% purity) of N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide as colorless solid. MS: m/z = 368.1 [M+H]+.
f. 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2- amine
To a stirred mixture of N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (3.27 g, 8.00 mmol, 90%) in water/methanol (1 :1 , 300 mL) was added NaOH (3.36 g, 80.0 mmol, 95%) at room temperature under nitrogen atmosphere. The mixture was stirred for overnight at 90°C under nitrogen atmosphere and evaporated to dryness. The residue was taken up in water and extracted 3 times with dichloromethane (100 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography, eluted with petrol ether/ethyl acetate (1 :1) to afford 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-amine (1.50 g, 68%, 96% purity) as a light brownish solid. MS: m/z = 264.1 [M+H]+.
g. phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy- [1,3]thiazolo[4,5c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate
To a stirred solution of 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-amine (600 mg, 2.19 mmol, 96%) and phenyl chloroformate (1.81 g,
11.0 mmol, 95%) in THF (50 mL) was added K2CO3 (1.59 g, 11.0 mmol, 95%) and pyridine (913 mg, 11.0 mmol, 95%) at room temperature under nitrogen
atmosphere. The mixture was stirred for 6 h at 50° and then after re-cooling to room temperature quenched by the addition of water (300 mL). The mixture was extracted 3 times with dichloromethane (200 mL), the combined organic layers were washed once with brine (200 mL), dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. This resulted in phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate (1.00 g, 69%, 76% purity) as a light yellow solid. The crude product was used in the next step directly without further purification. MS: m/z = 504.1 [M+H]+.
h. N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide
To a mixture of phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate (1.00 g, 1.52 mmol, 76.) and bis(7-oxa-2-azaspiro[4.5]decane), oxalic acid (1.19 g, 3.03 mmol, 95%) in THF (50 mL) was added diisopropylethyl amine (1.24 g, 9.09 mmol, 95%) at room temperature under nitrogen atmosphere. The mixture was stirred for 1 h at 60°. After re-cooling to room temperature, the mixture was extracted twice with dichloromethane (100 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography, eluted with petrol ether/ethyl acetate (1 :1) to afford N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (600 mg, 92%) as a white solid. HPLC: 99.9 % purity, RT = 1.17 min. MS: m/z = 431.1 [M+H]+. 1 H NMR (300 MHz, DMSO-d6) d 1 1.37 (s, 1 H), 7.95 (s, 1 H), 6.25 (s, 1 H), 4.30-4.29 (m, 2H), 3.99 (s, 3H), 3.89 (t, J=5.4Hz, 2H), 3.61-3.29 (m, 8H), 2.55-2.51 (m, 2H), 1.82-1.54 (m, 6H).
i. (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 24
and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5- c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 25
N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (450 mg, 1.044 mmol, 1 equiv, 99.9%) was purified by chiral-preparative HPLC (Preparative HPLC-032, column: ChiralPak IA, 2*25cm, 5 mm; mobile phase, dichloromethane:ethanol (20:80); detector, UV). This resulted in (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (178 mg, 39%) as a white solid. HPLC: 99.7 % purity, RT (chiral) = 3.86 min, 100% ee. MS: m/z = 431.2 [M+H]+. 1 H NMR (400 MHz, DMSO-d6) d 1 1.36 (s, 1 H), 7.94 (s, 1 H), 6.24 (s, 1 H), 4.29-4.27 (m, 2H), 3.97 (s,3H), 3.88 (t, J=5.2 Hz, 2H), 3.51-3.19 (m, 8H), 2.55-2.50 (m, 2H), 1.83-1.53 (m, 6H) and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (171 mg, 38%) as a white solid. HPLC: 99.8 % purity, RT (chiral) = 5.23 min, 99.9% ee. MS: m/z = 431.2 [M+H]+. 1 H NMR (400 MHz, DMSO-d6) d 1 1.35 (s, 1 H), 7.94 (s, 1 H), 6.24 (s, 1 H), 4.29-4.28 (m, 2H), 3.99 (s, 3H), 3.88-3.85 (m, 2H), 3.61-3.29 (m, 8H), 2.55-2.50 (m,2H), 1.83-1.53 (m,6H).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024028273&_cid=P10-MHPOFP-06905-1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////muvadenant, adenosine receptor antagonist, antineoplastic, 6LSF69F6A8, M1069 , M 1069
Matsupexole
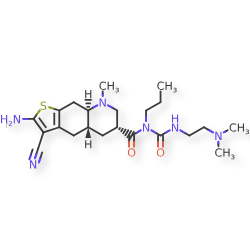
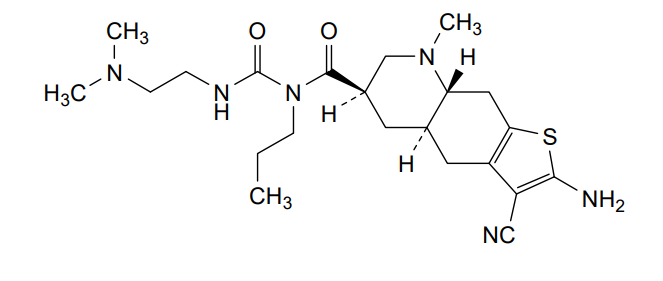
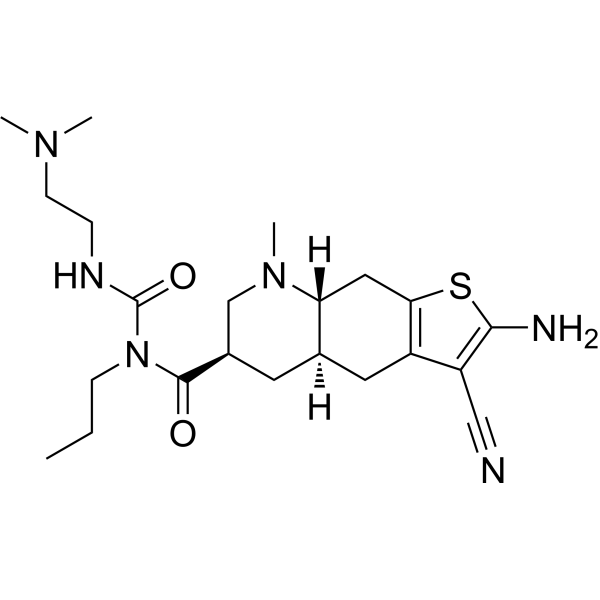

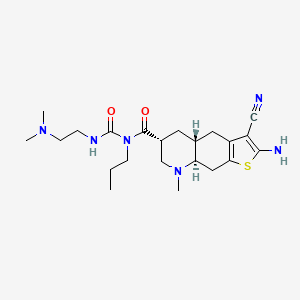
Matsupexole
CAS 1399442-97-7
MF C22H34N6O2S, Molecular Weight, 446.61
(4aR,6R,8aR)-2-amino-3-cyano-N-{[2-(dimethylamino)ethyl]carbamoyl}-8-methyl-N-propyl 4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinoline-6-carboxamide
(4aR,6R,8aR)-2-amino-3-cyano-N-[2-(dimethylamino)ethylcarbamoyl]-8-methyl-N-propyl-4a,5,6,7,8a,9-hexahydro-4H-thieno[3,2-g]quinoline-6-carboxamide
dopamine receptor agonist, Phase 2, Parkinson’s disease, K4UEG65HTX
- OriginatorKissei Pharmaceutical
- DeveloperAffaMed Therapeutics; Kissei Pharmaceutical
- ClassAmides; Amines; Antiparkinsonians; Dimethylamines; Ethylenediamines; Nitriles; Quinolines; Small molecules; Thiophenes; Urea compounds
- Mechanism of ActionDopamine receptor agonists
- Phase IIParkinson’s disease
- 28 Aug 2025Chemical structure information added.
- 06 Sep 2021Kissei Pharmaceutical completes a phase II trial in Parkinson’s disease (In adults, In elderly) in Japan (PO) (NCT04867551)
- 04 Aug 2021Phase-II clinical trials in Parkinson’s disease in China (PO) (Kissei Pharmaceutical pipeline, August 2021)
PAT
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: WO-2012124649-A1Priority Date: 2011-03-14
- COMPOUNDS DERIVED FROM OCTAIDROTHIENOQUINOLINE, PHARMACEUTICAL COMPOSITION AND PHARMACEUTICAL AGENT COMPRISING SUCH COMPOUNDSPublication Number: BR-112013023575-B1Priority Date: 2011-03-14
- New octahydrothienoquinoline derivative, pharmaceutical composition containing derivative, and using themPublication Number: RU-2573399-C2Priority Date: 2011-03-14Grant Date: 2016-01-20
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: SG-193400-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing the same, and their usesPublication Number: TW-I537274-BPriority Date: 2011-03-14Grant Date: 2016-06-11
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: US-2014243311-A1Priority Date: 2011-03-14
- Octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: US-9138434-B2Priority Date: 2011-03-14Grant Date: 2015-09-22
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: HU-E033449-T2Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and their usePublication Number: JP-5563716-B2Priority Date: 2011-03-14Grant Date: 2014-07-30
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and their usePublication Number: JP-WO2012124649-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions containing them and uses thereofPublication Number: KR-20140010137-APriority Date: 2011-03-14
- NEW OCTAHYDROTHYENOCHINOLINE DERIVATIVE, PHARMACEUTICAL COMPOSITION CONTAINING A DERIVATIVE AND THEIR APPLICATIONPublication Number: RU-2013145799-APriority Date: 2011-03-14
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions comprising said derivatives and their usesPublication Number: CN-103443106-BPriority Date: 2011-03-14Grant Date: 2015-09-30
- New octahydrothienoquinoline derivative, pharmaceutical composition comprising the derivative, and use thereofPublication Number: DK-2687532-T3Priority Date: 2011-03-14Grant Date: 2017-02-20
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: EP-2687532-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: EP-2687532-B1Priority Date: 2011-03-14Grant Date: 2016-12-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising the derivative, and use thereofPublication Number: ES-2613658-T3Priority Date: 2011-03-14Grant Date: 2017-05-25
- Novel dopamine D2 receptor agonistPublication Number: JP-2014074013-APriority Date: 2012-09-12
- Novel dopamine D2 receptor agonistPublication Number: JP-6177061-B2Priority Date: 2012-09-12Grant Date: 2017-08-09
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: AU-2012227428-A1Priority Date: 2011-03-14
- Novel octahydrothienoquinoline derivative, pharmaceutical composition comprising derivative, and use of thesePublication Number: AU-2012227428-B2Priority Date: 2011-03-14Grant Date: 2016-05-05
- Novel octahydrothienoquinoline derivatives, pharmaceutical compositions comprising said derivatives and their usesPublication Number: CN-103443106-APriority Date: 2011-03-14
- Succinate of octahydrothienoquinoline compound, and crystals thereofPublication Number: WO-2022009815-A1Priority Date: 2020-07-06
- Succinate of octahydrothienoquinoline compound and its crystalPublication Number: CN-115803329-APriority Date: 2020-07-06
- Succinate salts of octahydrothienoquinoline compounds and crystals thereofPublication Number: KR-20230035050-APriority Date: 2020-07-06
- Succinate of octahydrothienoquinoline compound, and crystals thereofPublication Number: EP-4177257-A1Priority Date: 2020-07-06
- Succinate salts of octahydrothienoquinoline compound and crystals thereofPublication Number: US-2023286998-A1Priority Date: 2020-07-06
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022009815&_cid=P22-MHO952-66657-1
[0018]Example 11-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea sesquisuccinate monohydrate (Form I crystals of salt (A-1)) 102.8 g of acetone was added to 1-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4,4a,5,6,7,8,8a,9-octahydrothieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (22.00 g), the mixture was suspended, and the suspension was heated and stirred at an external temperature of 52°C to dissolve the suspension. Activated carbon (2.2 g) was added to this solution and stirred for 10 minutes. This suspension was hot filtered and washed with 35.2 g of acetone. 220.0 g of acetone was then added, and the reaction solution was heated to an external temperature of 52°C and stirred. Next, 44.0 g of water was added to the reaction solution. Separately, 8.73 g of succinic acid was dissolved in a mixed solution of 156.1 g of acetone and 19.8 g of water. This succinic acid solution was added dropwise to the reaction solution over approximately 10 minutes. The dropping funnel was washed with a mixed solution of 17.4 g of acetone and 2.2 g of water and then added dropwise to the reaction solution. The reaction solution was stirred at an internal temperature of 50°C for 1 hour and cooled to 15°C over 30 minutes. The reaction solution was stirred at an external temperature of 10°C for 2 hours, and the crystals were collected by filtration. The crystals were washed twice with 52.8 g of acetone. The obtained wet crystals were dried under reduced pressure at 50°C for 37 hours and then returned to room temperature under reduced pressure over 3 hours. The crystals were stored under air for 24 hours to obtain crystals (27.75 g) of the title compound.
1 H-NMR (DMSO-d6) (δ (ppm)): 0.85 (3H, t, J = 7.4Hz), 1.32 (1H, ddd, J=12.2Hz, 12.2Hz, 12.2Hz), 1.42-1.57 (2H, m), 1.57-1.70 (1H, m ), 1.89-2.00 (2H, m), 2.20-2.13 (1H, m), 2.13-2.28 (2H, m), 2.21 (3H, s), 2.24 (6H, s ), 2.35-2.48 (1H, m), 2.40 (6H, s), 2.46 (2H, t, J = 6.4Hz), 2.81-2.96 (2H, m), 3.00-3 .12 (1H, m), 3.21-3.33 (2H, m), 3.47-3.66 (2H, m), 6.99 (2H, s), 8.50-8.90 (1H, br).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012124649&_cid=P22-MHO8UB-55660-1
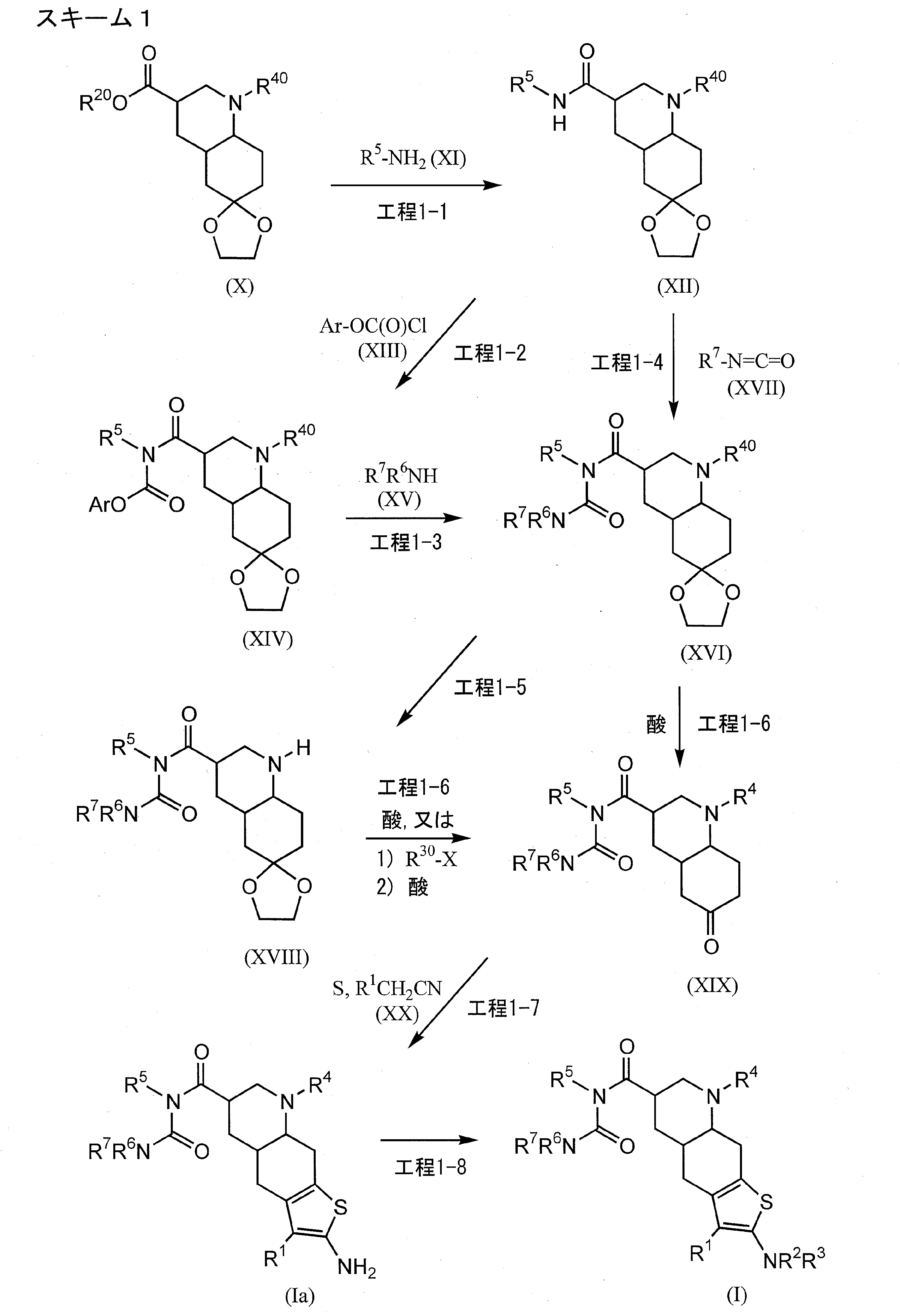
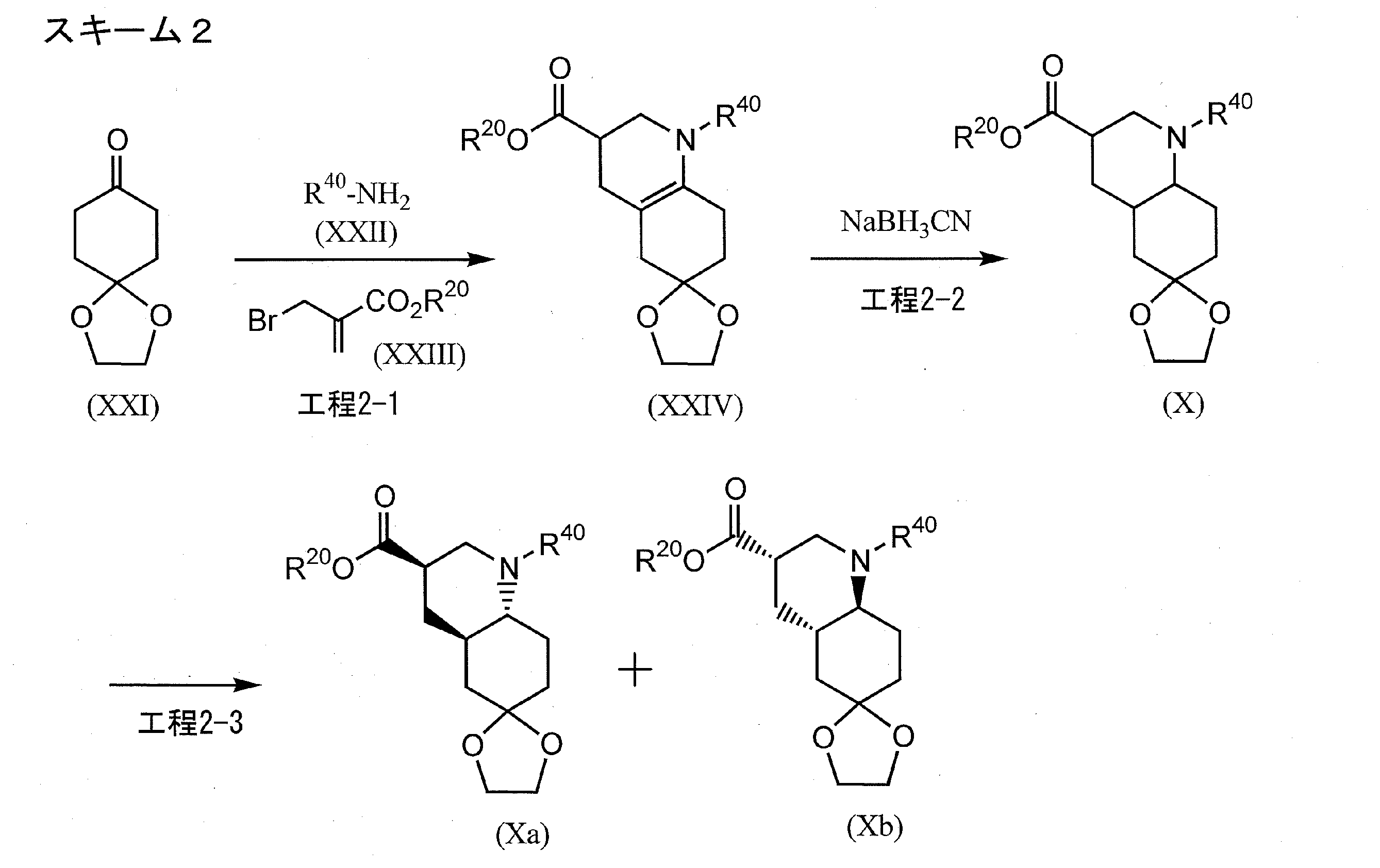
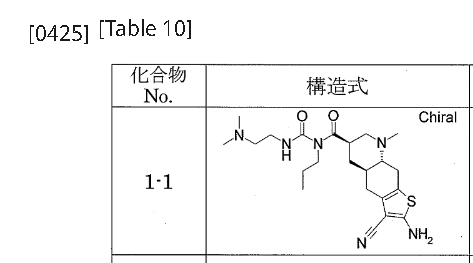
[0422]Example 1-11-{[(4aR,6R,8aR)-2-amino-3-cyano-8-methyl-4H,4aH,5H,6H,7H,8H,8aH,9H-thieno[3,2-g]quinolin-6-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Compound 1-1) To a mixture of 1-{[(3R,4aR,8aR)-1-methyl-6-oxodecahydroquinolin-3-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Reference Example 10-1) (1.602 g) and ethanol (44 mL) were added malononitrile (435 mg), morpholine (0.572 mL), and then elemental sulfur (282 mg) with stirring at room temperature, and the mixture was heated to 55°C and stirred for 1.5 hours. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure, and the residue was purified by column chromatography on aminopropyl silica gel (eluent: 0%-5% methanol/ethyl acetate, gradient elution) to give the title compound (1.479 g) as a solid.
1 H-NMR (CDCl
3 ) δ ppm: 0.94(3H, t, J=7.4Hz), 1.45-1.85(4H, m), 1.95-2.15(2H, m), 2.15-2.30(7H, m), 2.30-2.55(7H, m), 2.60-2.75(1H, m), 2.90-3.00(2H, m), 3.00-3.10(1H, m), 3.35-3.45(2H, m), 3.60-3.85(2H, m), 4.65(2H, s), 9.27(1H, br)[α]
D 29 =-105.54°(c=0.30, MeOH)
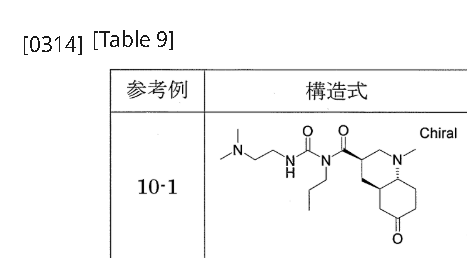
[0311]Reference Example 10-11-{[(3R,4aR,8aR)-1-methyl-6-oxodecahydroquinolin-3-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea 1-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea (Reference Example 8-1) (2.366 g) was added to 2 mol/L hydrochloric acid (30 mL), and the mixture was stirred at room temperature for 2 hours. The reaction mixture was washed with diethyl ether, and then potassium carbonate was added to the aqueous layer to make it alkaline. The mixture was extracted with a methylene chloride/methanol mixed solvent (methylene chloride:methanol = 9:1). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the title compound (1.605 g).
1 H-NMR (CDCl
3 ) δ ppm: 0.94 (3H, t, J=7.4 Hz), 1.45-1.90 (6H, m), 1.95-2.05 (1H, m), 2.10-2.55 (17H, m), 2.90-3.10 (2H, m), 3.30-3.45 (2H, m), 3.60-3.80 (2H, m), 9.22 (1H, brs).[α]
D 28 =-37.56° (c=0.38, MeOH).
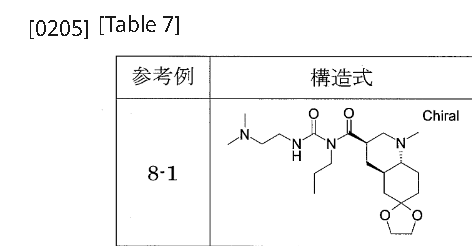
[0198]Reference Example 8-1To a mixture of phenyl 1-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-3-[2-(dimethylamino)ethyl]-1-propylurea N-{[(3’R,4’aR,8’aR)-1′-methyloctahydro-1’H-spiro[1,3-dioxolane-2,6′-quinoline]-3′-yl]carbonyl}-N-propylcarbamate (Reference Example 6-1) (2.401 g) and 2-propanol (30 mL), N,N-dimethylethylenediamine (1.26 mL) was added with stirring at room temperature, and the mixture was heated to 53°C and stirred for 13 hours. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was purified by aminopropyl silica gel column chromatography (eluent: 0%-100% ethyl acetate/hexane, gradient elution) to give the title compound (2.383 g).
1 H-NMR (CDCl
3 ) δ ppm: 0.92(3H, t, J=7.4Hz), 1.35-1.50(3H, m), 1.50-1.90(8H, m), 2.00-2.15(1H, m), 2.26(6H, s), 2.31(3H, s), 2.37(1H, t, J=11.2Hz), 2.46(2H, t, J=6.4Hz), 2.85-3.10(2H, m), 3.35-3.45(2H, m), 3.60-3.70(1H, m), 3.70-3.80(1H, m), 3.90-4.00(4H, m), 9.33(1H, br)[α]
D 28 =-6.62°(c=0.31, MeOH)
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////matsupexole, dopamine receptor agonist, Phase 2, Parkinson’s disease, K4UEG65HTX
Lunresertib
Lunresertib
CAS 2719793-90-3
MF C18H20N4O2 MW 324.4 g/mol
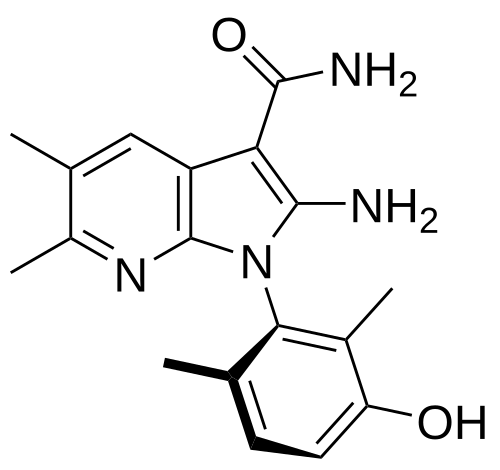
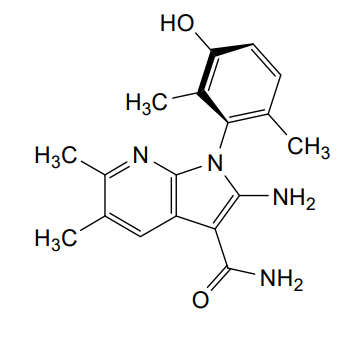
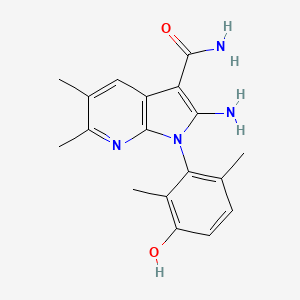
(1P)-2-amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethyl1H-pyrrolo[2,3-b]pyridine-3-carboxamide
serine/ threonine kinase inhibitor, antineoplastic, N95U3A7N57, RP-6306, RP 6306
2-Amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethylpyrrolo[2,3-b]pyridine-3-carboxamide
Lunresertib is an investigational new drug that is being evaluated for the treatment of cancer. It is an oral small molecule inhibitor of PKMYT1, developed by Repare Therapeutics.[1] This drug targets cell cycle regulation in tumors with specific genetic alterations, including CCNE1 amplifications or FBXW7 and PPP2R1A loss of function mutations. It is currently in phase 1/2 clinical trials, both as monotherapy or in combination with camonsertib, an ATR inhibitor.[2]
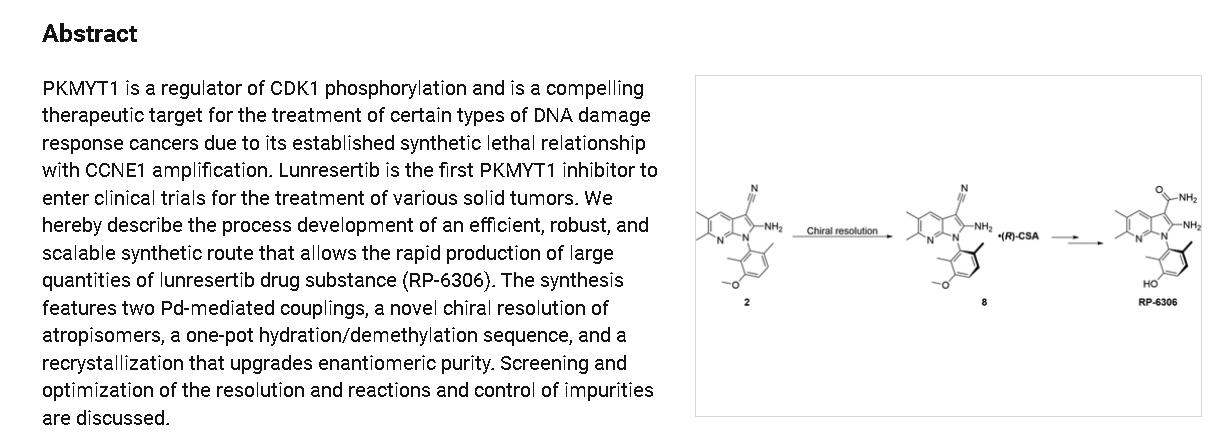
Lunresertib is an orally bioavailable inhibitor of the human membrane-associated tyrosine– and threonine-specific cdc2-inhibitory kinase (PKMYT1), with potential antineoplastic activity. Upon oral administration, lunresertib targets, binds to and inhibits the activity of PKMYT1. This results in the inhibition of CDK1 phosphorylation, which may promote both premature mitosis and a prolonged mitotic arrest, and lead to the accumulation of unrepaired DNA damage and apoptosis in susceptible tumor cells, such as CCNE1-overexpressing tumor cells. PKMYT1 phosphorylates CDK1 specifically when CDK1 is complexed to cyclins, which blocks progression from G2 into mitosis.NCI Thesaurus (NCIt)
- Study of RP-6306 With FOLFIRI in Advanced Solid TumorsCTID: NCT05147350Phase: Phase 1Status: TerminatedDate: 2025-08-20
- Study of RP-6306 Alone or in Combination With RP-3500 or Debio 0123 in Patients With Advanced Solid TumorsCTID: NCT04855656Phase: Phase 1Status: RecruitingDate: 2025-08-06
- RP-6306 in Patients With Advanced CancerCTID: NCT05605509Phase: Phase 2Status: Active, not recruitingDate: 2025-07-14
- Study of RP-6306 With Gemcitabine in Advanced Solid TumorsCTID: NCT05147272Phase: Phase 1Status: TerminatedDate: 2025-06-17
- Liquid-biopsy Informed Platform Trial to Evaluate CDK4/6-inhibitor Resistant ER+/HER2- Metastatic Breast CancerCTID: NCT05601440Phase: Phase 2Status: RecruitingDate: 2025-01-14
- Phase 1 Study of RP-6306 With Carboplatin and Paclitaxel in TP53 Ovarian and Uterine Cancer
- CTID: NCT06107868
- Phase: Phase 1
- Status: Active, not recruiting
- Date: 2024-03-22
PAT
- Compounds, Pharmaceutical Compositions, and Methods of Preparing and Using CompoundsPublication Number: JP-2023521633-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: US-2023151014-A1Priority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: US-2023158022-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: EP-4126879-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: IL-296934-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of making the compounds and methods of using themPublication Number: KR-20230011279-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions and methods of making compounds and methods of their usePublication Number: CN-115916783-APriority Date: 2020-04-01
- Methods of using MYT1 inhibitorsPublication Number: JP-2023519430-APriority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: WO-2021195782-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: AU-2021250744-A1Priority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: CA-3173955-A1Priority Date: 2020-04-01
- Methods of using MYT1 inhibitorsPublication Number: CN-115811976-APriority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: EP-4125907-A1Priority Date: 2020-04-01
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021195781&_cid=P20-MHLE6P-37080-1
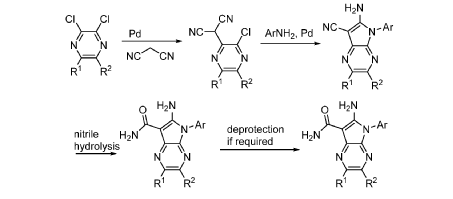
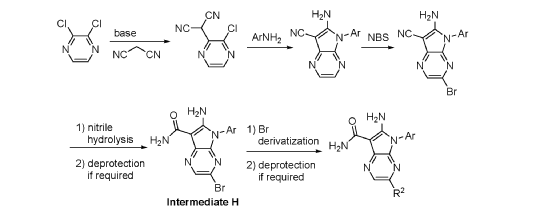
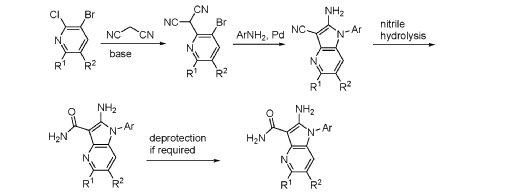
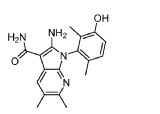
Step 9. To a suspension of 2-amino-1-(3-methoxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (2.22 g, 6.56 mmol, 77% purity) in DCM (25 mL) was added tribromoborane in DCM (1 M, 26 mmol, 26 mL) dropwise. The reaction mixture was stirred at RT for 45 min, then concentrated to dryness. The crude product was taken in DCM and placed in an ice bath and MeOH was added carefully (exotherm). The mixture was concentrated to dryness then co-evaporated twice with MeOH. The residue was triturated with saturated aqueous NaHCO3. The solids were collected by filtration on a Buchner funnel, washed with H2O and air-dried. The still wet solid was dissolved in DCM/MeOH, concentrated to dryness and triturated in 20% MeOH/DCM (50 mL). The solid was collected by filtration, washed with 20% MeOH/DCM, air-dried then dried in vacuo to afford 2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (1.60g, 75% yield) as a light beige solid. MS: [M+1]: 325.1. A different batch was purified by preparative HPLC to yield 2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (63% yield) as an off-white fluffy solid.
1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 7.82 (s, 1H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J =
8.2 Hz, 1H), 6.71 (br s, 2H), 6.64 (br s, 2H), 2.26 (s, 3H), 2.23 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
Chiral SFC separation of Compound 181 (1.60g, 4.93 mmol) (Instrument: Waters Prep 100 SFC-MS; Column: Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm; Conditions: isocratic at 55% IPA + 10mM Ammonium Formate with 45% CO2 ; Flow Rate: 70 mL/min) provided
Compound 182 and Compound 183.
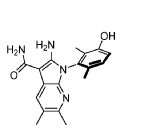
Compound 182 from SFC separation of 181. Peak 1 (retention time 3.94 min, 99.86%): (S)-2- amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (381 mg) was obtained as an off white fluffy solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1 H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.72 (s, 2H), 6.65 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
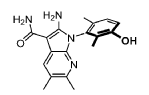
Compound 183 from SFC separation of 181. Peak 2 (retention time 4.35 min, 98.09%): (R)-2- amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (495 mg) was obtained as an off white fluffy solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1 H), 7.05 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.72 (s, 2H), 6.66 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
SYN
https://pubs.acs.org/doi/full/10.1021/acs.oprd.4c00493
REF
- The Science and Art of Structure-Based Virtual ScreeningPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2024-03-25PMCID: PMC11017385PMID: 38628791DOI: 10.1021/acsmedchemlett.4c00093
- Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306Publication Name: Journal of Medicinal ChemistryPublication Date: 2022-07-26PMCID: PMC9837800PMID: 35880755DOI: 10.1021/acs.jmedchem.2c00552
- CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibitionPublication Name: NaturePublication Date: 2022-04-20PMCID: PMC9046089PMID: 35444283DOI: 10.1038/s41586-022-04638-9
- Contributions in the domain of cancer research: Review¶Negative regulators of cyclin-dependent kinases and their roles in cancersPublication Name: Cellular and molecular life sciences : CMLSPublication Date: 2001-11PMCID: PMC11337304PMID: 11766887DOI: 10.1007/pl00000826
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | RP-6306 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2719793-90-3 |
| PubChem CID | 156869388 |
| ChemSpider | 115008046 |
| UNII | N95U3A7N57 |
| KEGG | D12736 |
| ChEMBL | ChEMBL5199076 |
| Chemical and physical data | |
| Formula | C18H20N4O2 |
| Molar mass | 324.384 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Szychowski J, Papp R, Dietrich E, Liu B, Vallée F, Leclaire ME, et al. (August 2022). “Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306”. Journal of Medicinal Chemistry. 65 (15): 10251–10284. doi:10.1021/acs.jmedchem.2c00552. PMC 9837800. PMID 35880755.
- Previtali V, Bagnolini G, Ciamarone A, Ferrandi G, Rinaldi F, Myers SH, et al. (July 2024). “New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives”. Journal of Medicinal Chemistry. 67 (14): 11488–11521. doi:10.1021/acs.jmedchem.4c00113. PMC 11284803. PMID 38955347.
///////lunresertib, Serine/ threonine kinase inhibitor, antineoplastic, N95U3A7N57, RP-6306, RP 6306
Lomedeucitinib
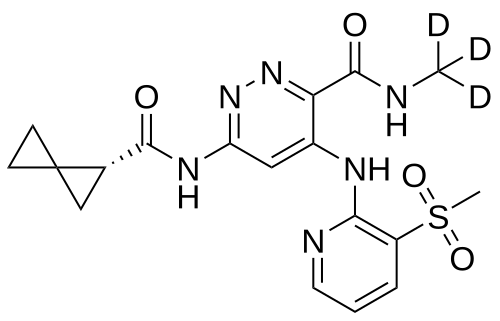
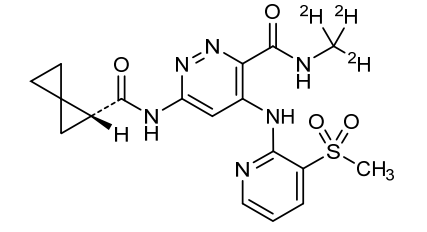
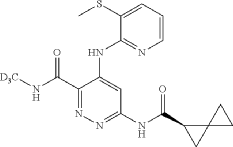
Lomedeucitinib
CAS 2328068-29-5
MF C18H172H3N6O4S
MW 419.5 g/mol

4-{[3-(methanesulfonyl)pyridin-2-yl]amino}-N-(2H3)methyl-6-[(1R)-spiro[2.2]pentane-1-carboxamido]pyridazine-3-carboxamide
4-[(3-methylsulfonyl-2-pyridinyl)amino]-6-[[(2R)-spiro[2.2]pentane-2-carbonyl]amino]-N-(trideuteriomethyl)pyridazine-3-carboxamide
Janus kinase inhibitor, anti-inflammatory, BMS-986322, BMS 986322, EYQ7KA55XA
Lomedeucitinib is an investigational new drug that is being evaluated for the treatment of psoriasis and psoriatic arthritis. It is a tyrosine kinase 2 (TYK2) inhibitor.[1]
- A Study to Evaluate Effectiveness and Safety of BMS-986322 in Participants With Moderate-to-Severe PsoriasisCTID: NCT05730725Phase: Phase 2Status: CompletedDate: 2024-09-19
- A Study to Evaluate the Drug Levels, Metabolism, and Removal of BMS-986322 in Healthy Adult Male ParticipantsCTID: NCT06088264Phase: Phase 1Status: CompletedDate: 2024-03-29
- A Study Investigating Interactions Between BMS-986322 and Rosuvastatin, Metformin and Methotrexate in Healthy ParticipantsCTID: NCT05615012Phase: Phase 1Status: CompletedDate: 2024-03-27
- A Study to Investigate the Interaction of BMS-986322 and a Combined Oral Hormonal Contraceptive (Ethinyl Estradiol [EE]/Norethindrone [NET]) in Healthy Female ParticipantsCTID: NCT05579574Phase: Phase 1Status: CompletedDate: 2023-08-18
- A Study to Assess the Safety and Tolerability of BMS-986322 in Healthy Participants of Japanese DescentCTID: NCT05546151Phase: Phase 1Status: CompletedDate: 2023-06-22
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US333829535&_cid=P10-MHIXWK-98212-1
General Scheme for Examples 252 and 253:
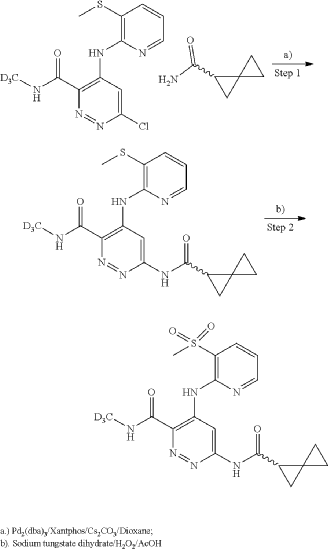
Example 252
Step 1
| A mixture of cesium carbonate (149 mg, 0.457 mmol), Xantphos (14.43 mg, 0.025 mmol), Pd 2(dba) 3 (11.42 mg, 0.012 mmol), 6-chloro-N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)pyridazine-3-carboxamide (65 mg, 0.208 mmol), and (R)-spiro[2.2]pentane-1-carboxamide (50.8 mg, 0.457 mmol) in dioxane (3 mL) was degassed using a vacuum/N2 fill cycle three times. The reaction was heated at 110° C. for 16 hours. The reaction was diluted with water and DCM. The DCM layer was separated and washed two more times with water and then dried (Na 2SO 4), filtered and concentrated. Purification via automated flash chromatography, eluting with methanol in DCM from 0 to 10%, gave the title compound (R)—N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (54 mg, 67% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 12.15 (br s, 1H), 9.88 (s, 1H), 8.68 (br s, 1H), 8.36 (br d, J=3.5 Hz, 1H), 8.25 (br s, 1H), 7.72 (br d, J=7.4 Hz, 1H), 6.97 (br dd, J=7.0, 5.1 Hz, 1H), 2.51 (s, 3H), 2.21-2.09 (m, 1H), 1.58-1.10 (m, 6H), 1.08-0.93 (m, 5H). |
| LCMS (ESI) m/e 388.1 [(M+H) +, calc’d C 18H 18D 3N 6O 2S 1, 388.1]; LC/MS retention time (method D): t R=0.80 min. |
Step 2
To a suspension of hydrogen peroxide (30% solution in water, 0.258 mL, 2.52 mmol) and (R)—N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (0.0489 g, 0.126 mmol) in AcOH (1 mL) was added sodium tungstate dihydrate (0.042 g, 0.126 mmol) at room temperature. After stirring at room temperature for 1 hour, the reaction was diluted with water, basified with Na 2CO 3 powder and extracted three times with DCM. The DCM layers were combined, washed with Na 2S 2O 3 (5% solution), dried (Na 2SO 4), filtered and concentrated. The crude product was purified using reverse phase prepHPLC to give the title compound (R)—N-(methyl-d3)-4-((3-(methylsulfonyl)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (16.2 mg, 31%) as a colorless solid. 1H NMR (500 MHz, DMSO-d 6) δ 12.07 (s, 1H), 11.22 (s, 1H), 9.49 (s, 1H), 9.16 (s, 1H), 8.63 (dd, J=4.6, 1.5 Hz, 1H), 8.29 (dd, 0.1=7.8, 1.4 Hz, 1H), 7.34 (dd, 0.1=7.8, 4.7 Hz, 1H), 2.48-2.43 (m, 1H), 1.46-1.41 (m, 1H), 1.42-1.36 (m, 1H), 0.95-0.82 (m, 3H), 0.80-0.73 (m, 1H). (3H methyl sulfone was buried under DMSO peak). LCMS (ESI) m/e 420.0 [(M+H) +, calc’d C 18H 18D 3N 6O 4S, 420.1]; LC/MS retention time (method E): t R=1.38 min; OR: −205.39 (20° C.).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US242383764&_cid=P10-MHIXVD-97150-1
PAT
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-11787779-B2Priority Date: 2017-11-21Grant Date: 2023-10-17
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2024002364-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: KR-102702228-B1Priority Date: 2017-11-21Grant Date: 2024-09-02
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: NZ-805343-APriority Date: 2017-11-21
- Sulfonepyridine alkylamide-substituted heteroaryl compoundsPublication Number: JP-2023098942-APriority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2023255024-A1Priority Date: 2017-11-21
- Heteroaryl compounds substituted with sulfone pyridinylalkylamidesPublication Number: CN-111315737-BPriority Date: 2017-11-21Grant Date: 2024-06-18
- The heteroaryl compounds are substituted with sulfone-pyridine alkyl amidesPublication Number: IL-274816-B2Priority Date: 2017-11-21
- Sulfonepyridine alkylamide substituted heteroaryl compoundsPublication Number: JP-7490107-B2Priority Date: 2017-11-21Grant Date: 2024-05-24
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2022228101-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: TW-I776994-BPriority Date: 2017-11-21Grant Date: 2022-09-11
- Sulfonepyridine alkylamide-substituted heteroaryl compoundsPublication Number: JP-7258903-B2Priority Date: 2017-11-21Grant Date: 2023-04-17
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2022228101-B2Priority Date: 2017-11-21Grant Date: 2023-08-03
- The heteroaryl compounds are substituted with sulfone-pyridine alkyl amidesPublication Number: IL-274816-B1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2019152948-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: CA-3083122-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: KR-20200089706-APriority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-11021462-B2Priority Date: 2017-11-21Grant Date: 2021-06-01
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2021253554-A1Priority Date: 2017-11-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | BMS-986322 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2328068-29-5 |
| PubChem CID | 138620496 |
| IUPHAR/BPS | 13210 |
| UNII | EYQ7KA55XA |
| KEGG | D12725 |
| ChEMBL | ChEMBL5314608 |
| Chemical and physical data | |
| Formula | C18H17D3N6O4S |
| Molar mass | 419.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Ahsan S, Degener R, Schlamp M (2024). “Non-Invasive Treatments Invade the Psoriasis Pipeline”. Drugs in Context. 13: 2024–5–6. doi:10.7573/dic.2024-5-6. PMC 11313207. PMID 39131603.
////////lomedeucitinib, Janus kinase inhibitor, anti-inflammatory, BMS-986322, BMS 986322, EYQ7KA55XA
Linustedastat
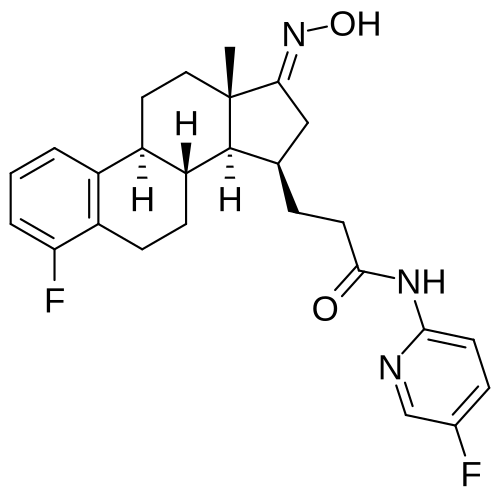
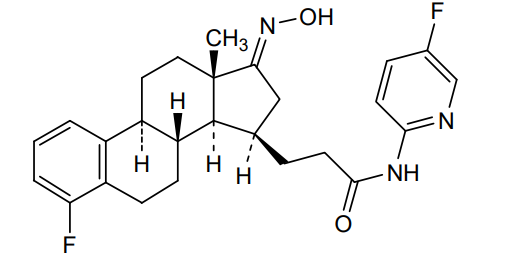
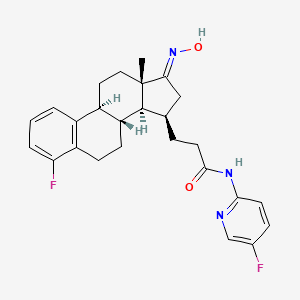
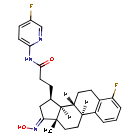
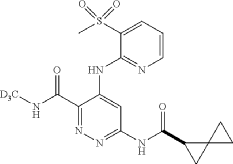
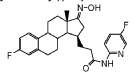
Linustedastat
CAS 2254299-48-2
MFC26H29F2N3O2 MW 453.5 g/mol
FOR-6219, OG-6219, FOR 6219, OG 6219, PP3PLL7GZY, Phase 2, Endometriosis
3-[(8R,9S,13S,14S,15R,17E)-4-fluoro-17-hydroxyimino-13-methyl-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthren-15-yl]-N-(5-fluoro-2-pyridinyl)propanamide
- (15beta,17E)-4-Fluoro-N-(5-fluoro-2-pyridinyl)-17-(hydroxyimino)estra-1,3,5(10)-triene-15-propanamide
- 3-[(17E)-4-fluoro-17-(hydroxyimino)estra-1,3,5(10)-trien-15beta-yl]-N-(5-fluoropyridin-2-yl)propanamide
- Estra-1,3,5(10)-triene-15-propanamide, 4-fluoro-N-(5-fluoro-2-pyridinyl)-17-(hydroxyimino)-, (15beta,17E)-
3-[(17E)-4-fluoro-17-(hidroxiimino)estra-1,3,5(10)-trien-15β-il]-N-(5-fluoropiridin-2-il)propanamida
inhibidor de la hidroxiesteroide 17-beta deshidrogenasa 1(HSD17B1)
- OriginatorHormos Medical; Solvay Pharmaceuticals B.V.; University of Turku
- DeveloperOrganon
- ClassSmall molecules
- Mechanism of ActionEstradiol dehydrogenase inhibitors
- Phase IIEndometriosis
- 02 Jul 2025Efficacy data from the phase II ELENA trial in Endometriosis released by Organon
- 28 May 2025Organon completes a phase-II clinical trials in Endometriosis (In adults) in Latvia, Sweden, Poland, Italy, France, Hungary, Germany, Czech Republic, Czech Republic, Bulgaria, Belgium, USA (PO) (NCT05560646)
- 28 Nov 2023No recent reports of development identified for phase-I development in Endometriosis(In volunteers) in United Kingdom (PO)
Linustedastat (developmental code names FOR-6219 and OG-6219) is a 17β-hydroxysteroid dehydrogenase 1 (17β-HSD1; HSD17B1) inhibitor which is under development for the treatment of endometriosis.[1][2][3][4][5] It is a steroidal compound derived from estrone and works by preventing the formation of the more potent estrogen estradiol from the minimally active precursor estrone.[1][2][5] This in turn results in antiestrogenic effects that may be useful in the treatment of estrogen-dependent conditions.[1][2][5] As of November 2023, the drug is in phase 2 clinical trials for endometriosis.[1][2] It is also under preclinical investigation for treatment of breast cancer and endometrial cancer.[5]
A Study to Investigate Efficacy and Safety of OG-6219 BID in 3 Dose Levels Compared With Placebo in Participants Aged 18 to 49 With Moderate to Severe Endometriosis-related Pain
CTID: NCT05560646
Phase: Phase 2
Status: Completed
Date: 2025-05-29
Pat
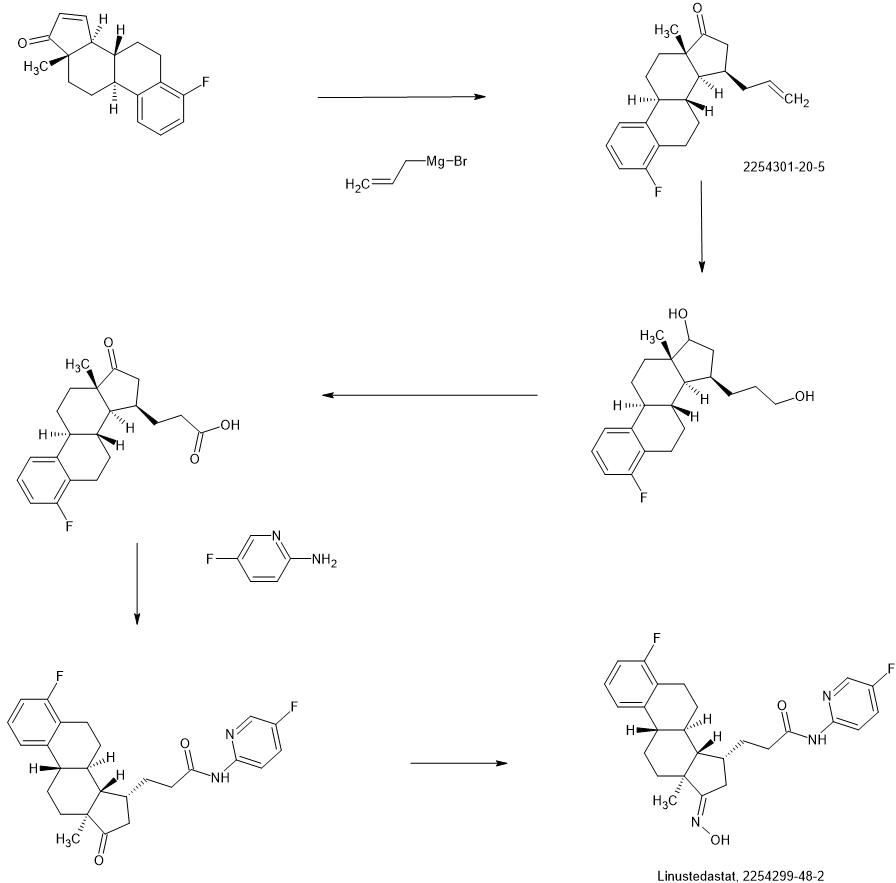
WO2018224736
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018224736&_cid=P21-MHFVBM-49409-1
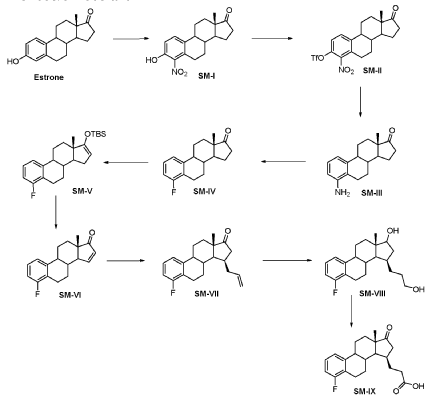
Compound 26
3-((13S,15R,E)-3-fluoro-17-(hydroxyimino)-13-methyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-15-yl)-N-(5-fluoropyridin-2-yl)propanamide
Example 26 was prepared in 94% yield from the compound 25 by the same method as with Example 2 in three hours reaction time.
1H NMR (200 MHz, DMSO-d6): 1.03 (s, 3 H), 1.12 – 2.48 (m, 15 H), 2.57 – 2.78 (m, 1 H), 2.80 – 2.95 (m, 2 H), 6.79 – 7.01 (m, 2 H), 7.18 – 7.38 (m, 1 H), 7.72 (td, 1 H), 8.15 (dd, 1 H), 8.31 (d, 1 H), 10.18 (s, 1 H), 10.64 (s, 1 H). MS m/z (TOF ES+): 454 (M+1).
SYNTHESIS
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | FOR-6219; OG-6219; 3-[(17E)-4-Fluoro-17-(hydroxyimino)estra-1,3,5(10)-trien-15β-yl]-N-(5-fluoropyridin-2-yl)propanamide |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2254299-48-2 |
| PubChem CID | 171390018 |
| UNII | PP3PLL7GZY |
| KEGG | D13078 |
| Chemical and physical data | |
| Formula | C26H29F2N3O2 |
| Molar mass | 453.534 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “FOR 6219”. AdisInsight. 28 November 2023. Retrieved 15 August 2024.
- “Delving into the Latest Updates on Linustedastat with Synapse”. Synapse. 3 August 2024. Retrieved 15 August 2024.
- Barra F, Romano A, Grandi G, Facchinetti F, Ferrero S (June 2019). “Future directions in endometriosis treatment: discovery and development of novel inhibitors of estrogen biosynthesis”. Expert Opin Investig Drugs. 28 (6): 501–504. doi:10.1080/13543784.2019.1618269. hdl:11380/1201688. PMID 31072144.
- Perrone U, Evangelisti G, Laganà AS, Bogliolo S, Ceccaroni M, Izzotti A, Gustavino C, Ferrero S, Barra F (December 2023). “A review of phase II and III drugs for the treatment and management of endometriosis”. Expert Opin Emerg Drugs. 28 (4): 333–351. doi:10.1080/14728214.2023.2296080. PMID 38099328.
- Rižner TL, Romano A (2023). “Targeting the formation of estrogens for treatment of hormone dependent diseases-current status”. Front Pharmacol. 14 1155558. doi:10.3389/fphar.2023.1155558. PMC 10175629. PMID 37188267.
Several compounds with inhibitory action on the enzyme HSD17B1 have been developed and one steroidal compound, a competitive HSD17B1 inhibitor (OG-6219) recently entered the clinical phase for endometriosis […] and it is in the preclinical phase for endometrial and breast cancer (Husen et al., 2006a; Husen et al., 2006b; Konings et al., 2018b; Jarvensivu et al., 2018; Xanthoulea et al., 2021). […] Only the C15 estrone derivative developed by Organon Finland, former Forendo pharma (compound FOR-6219/OR-6219) reached the clinical phase for endometriosis with three clinical trials registered in the database Clinical Trails (Table 2). Phase 1 and 1b trials NCT04686669 and NCT03709420 determined the bio-availability of the compound administered orally as gelatine capsule in 12 subjects (NCT04686669) and then the safety, tolerability, food interactions, the pharmacokinetics and pharmacodynamics of escalating doses of the drug in 87 subjects (NCT03709420). The phase 2 randomized, double-blind, Elena study (NCT05560646) is currently recruiting patients and aims at evaluating the efficacy and safety of OG-6219 in women with moderate to severe endometriosis […]
External links
//////////Linustedastat, FOR-6219, OG-6219, FOR 6219, OG 6219, PP3PLL7GZY, Phase 2, Endometriosis
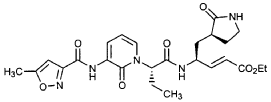
Imocitrelvir
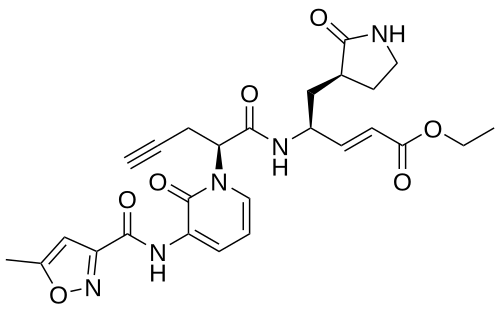
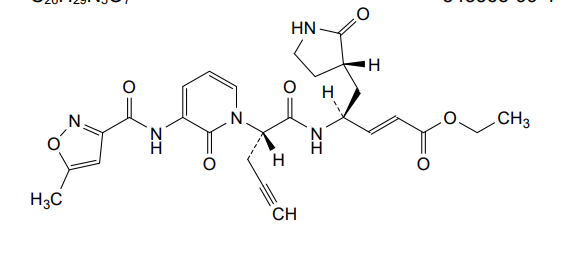
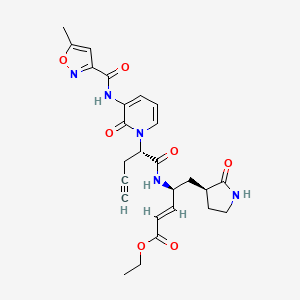
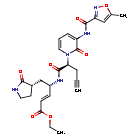
Imocitrelvir
CAS 343565-99-1
MFC26H29N5O7 MW523.5 g/mol
ethyl (2E,4S)-4-{(2S)-2-[3-(5-methyl-1,2-oxazole-3-carboxamido)-2-oxopyridin-1(2H)-yl]pent-4-ynamido}-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
ethyl (E,4S)-4-[[(2S)-2-[3-[(5-methyl-1,2-oxazole-3-carbonyl)amino]-2-oxo-1-pyridinyl]pent-4-ynoyl]amino]-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
Imocitrelvir is an investigational new drug that is being evaluated for the treatment of viral infections. It is a 3C protease inhibitor in picornaviruses. Originally developed by Pfizer for treating human rhinovirus infections,[1] this small molecule has shown promise against a broader range of viruses, including polioviruses.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2003-09-17
PMID: 14521419
DOI: 10.1021/jm030166l
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016044656&_cid=P21-MHBDH2-20719-1
PAT
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001040189&_cid=P21-MHBDI9-21481-1
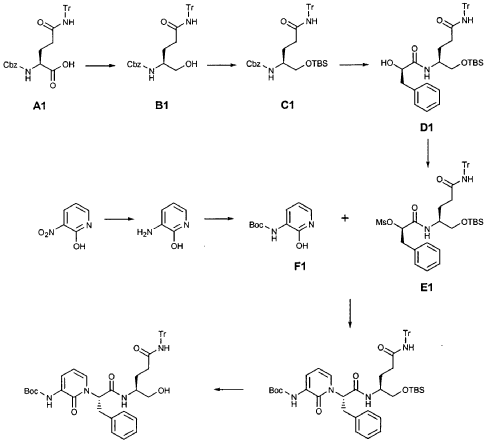
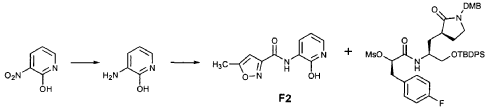
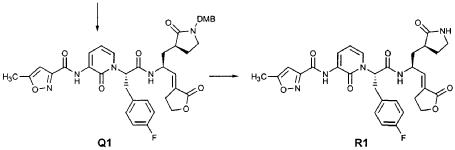
EXAMPLE 21
Preparation of Compound 22: tra«5-(4S,3″”S)-4-(2′-{3″-[(5′”-Methylisoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin- 1 “-yl} acetylamino)-5-(2″”-oxopyrrilidin-3″”-yl)pent-2-enoic Acid Ethyl Ester
Preparation of Intermediate {3-[(5′-Methylisoxazole-3′-carbonyl)amino]-2-oxo-2H-pyridin-l-yl} acetic Acid tert-Butyl Ester
To a solution of 5-methylisoxazole-3-carboxylic acid (2′-hydroxy-4′-methylpyridin-3′-yl)amide (F2, Example 19) (0.520 g, 2.37 mmol, 1 equiv) in TΗF (20 mL) at 0 °C was added NaΗ (0.095 g, 2.37 mmol, 1.0 equiv). The resulting mixture was stirred at 0 °C for 20 min, and then t-butyl bromoacetate (0.385 mL, 2.61 mmol, 1.1 equiv) was added. The reaction mixture was stirred and warmed to room temperature for 30 min, then was partitioned between 0.5 N ΗC1 (100 mL) and EtOAc (2 x 100 mL). The combined organic layers were dried over Na2SO and were concentrated. Purification of the residue by flash column chromatography (30% EtOAc in hexanes) provided the title intermediate (0.628 g, 79%) as a white solid: IR (cm-1) 3343, 1743, 1651, 1581, 1156; Η NMR (CDC13) δ 1.52 (s, 9H), 2.53 (s, 3H), 4.65 (s, 2H), 6.32 (t, 1H, 7= 7.2), 6.51 (s, IH), 7.01 (dd, 1H, 7= 6.9, 1.8), 8.50 (dd, 1H, 7= 7.5, 1.8), 9.63 (s, br. IH); Anal. C16H19N3O5: C, H, N.
Preparation of Compound 22
The preceding intermediate was transformed into Compound 22 by a process that was analogous to that described in Example 25 for the transformation of V3 to product R3: mp = 102-106 °C; IR (cm”1) 3336, 1684, 1534, 1457; JH NMR (CDCI3) δ 1.27 (t, 3H, 7= 7.2), 1.67-1.75 (m, IH), 1.98-2.09 (m, IH), 2.37-2.49 (m, IH), 2.53 (s, 3H), 2.55-2.61 (m, IH), 3.34-3.46 (m, 2H), 3.51-3.52 (m, IH), 4.17 (q, 2H, 7= 7.2), 4.61-4.78 (m, 3H), 5.98 (dd, IH, 7 = 15.6, 1.5), 6.20 (s, br. IH), 6.35 (t, 1H, 7= 7.8), 6.51 (s, IH), 6.85 (dd, IH, 7= 15.6, 5.1), 7.17 (d, IH, 7= 7.2), 8.33 (d, IH, 7= 7.2), 8.49 (d, IH, 7= 7.5), 9.57 (s, br. IH); Anal.
C23H27N5O7: C, H, N.
EXAMPLE 24
Preparation of Compound 25: trans-(2’S,3″”‘S,4S)-4-(3,-(4″-Fluorophenyl)-2′-{3″‘-[(5″”-methylisoxazole-3″”-carbonyl)amino]-2′”-oxo-2′”H-pyridin- “-yl}propionylamino)-5-(2″ oxopyrrolidin-3′””-yl)pent-2-enoic Acid Ethyl Ester
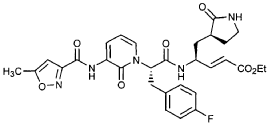
The title compound was prepared from F2 (Example 19) in a manner analogous to that described for the conversion of U2 to 13 in Example 23 utilizing intermediate Y2 (Example 25) where appropriate: IR (cm-1) 3331, 1690, 1590, 1531, 1455; !H NMR (CDCI3) δ 1.30 (t, 3H, 7= 7.0), 1.45-1.55 (m, IH), 1.64-1.75 (m, IH), 2.03-2.31 (m, 3H), 2.49 (s, 3H), 3.10 (dd, IH, 7= 13.7, 7.9), 3.20-3.46 (m, 3H), 4.20 (q, 2H, 7= 7.0), 4.36-4.47 (m, IH), 5.67 (dd, IH, 7 = 15.7, 1.4), 5.85-5.92 (m, IH), 6.29 (t, 1H, 7= 7.2), 6.45 (s, IH), 6.70 (dd, IH, 7= 15.7, 5.7), 6.86 (s, IH), 6.90-6.97 (m, 2H), 7.10-7.16 (m, 2H), 7.60 (dd, IH, 7= 7.2, 1.6), 8.37 (dd, IH, 7 = 7.2, 1.6), 8.51 (d, IH, 7= 6.6), 9.47 (s, IH).
EXAMPLE 25
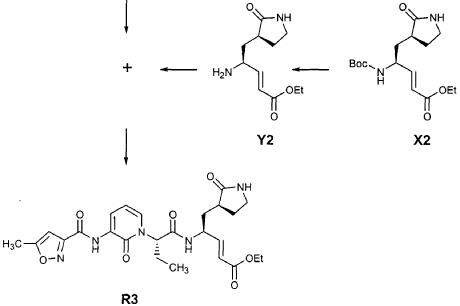
Preparation of Compound 26: tr_.«5-(2’S,3″”S,4S)-4-(2′-{3″-[(5″‘-Methyl-isoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin-l”-yl}butyrylamino)-5-(2″”-oxopyrrolidin-3″”-yl)pent-2-enoic Acid Ethyl Ester (R3)
Preparation of Intermediate (2R)-2-Trifluoromethanesulfonyl-oxybutyric acid tert-butyl ester (U3)
Commercially available T3 (0.575 g, 3.59 mmol, 1 equiv) was dissolved in CH2CI2 (25 mL) and cooled in an ice bath. 2,6-Lutidine (0.836 mL, 7.18 mmol, 2 equiv) and trifluoromethanesulfonic anhydride (1.15 mL, 6.84 mmol, 1.9 equiv) were added and the reaction mixture was stirred 30 min. It was then diluted with MTBE (400 mL), washed with a mixture of brine and 1 N HCl (2:1, 100 mL) and brine (100 mL), dried over Na2SO4 and evaporated to provide the title intermediate which was used without further purification.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyri din- l’-yl} butyric Acid tert-Butyl Ester (V3)
Intermediate F2 from above (0.200 g, 0.912 mmol, 1.1 equiv) was suspended in TΗF (6 mL). Sodium hydride (60% dispersion in mineral oil, 0.0332 g, 0.830 mmol, 1 equiv) was added in one portion. After stirring 30 min, a solution of intermediate U3 (0.830 mmol, 1 equiv, based on T3) in TΗF (7 mL) was added dropwise. The resulting mixture was stirred 2 hours, then diluted with EtOAc (200 mL) and washed with brine (2 x 50 mL). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (25% EtOAc in hexanes) to provide the title intermediate (0.178 g, 59%) as an oil: R/= 0.30 (25% EtOAc in hexanes); IR (cm”1) 3331, 1731, 1690, 1649, 1602, 1531 ; *Η NMR (CDCI3) δ 0.93 (t, 3H, 7= 7.3), 1.45 (s, 9H), 1.83-2.01 (m, IH), 2.17-2.31 (m, IH), 2.50 (s, 3H), 5.44-5.51 (m, IH), 6.32 (t, IH, 7= 7.2), 6.48 (s, IH), 7.10 (dd, IH, 7= 7.2, 1.8), 8.45 (dd, 1H, 7= 7.2, 1.8), 9.64 (s, IH); Anal. C18H23N3O5: C, H, N.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyridin-l’-yl}butyric Acid (W3)
Intermediate V3 from above (0.143 g, 0.397 mmol, 1 equiv) was stirred for 1 h in a solution of TFA (2 mL) in CΗ2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Intermediate trα«5-(3’S,4S)-4-Amino-5-(2′-oxopyrrolidin-3′-yl)pent-2-enoic Acid Ethyl Ester (Y2)
Intermediate X2, prepared according to the method disclosed in the co-pending application, U.S. Provisional Patent Application No. 60/150,358, filed August 24, 1999(0.130 g, 0.398 mmol, 1 equiv), was stirred for 30 min in a solution of TFA (2 mL) in CH2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
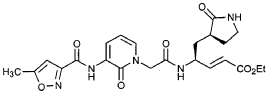
Preparation of Product R3 (Compound 26)
Intermediates W3 and Y2 (as prepared above) were combined in CH2CI2 (7 mL) and cooled in an ice bath. HOBt (0.064 g, 0.47 mmol, 1.2 equiv), iP^NEt (0.484 mL, 2.78 mmol, 7 equiv) and EDC (0.084 g, 0.44 mmol, 1.1 equiv) were added sequentially. The reaction mixture was allowed to warm to 23 °C overnight, then diluted with EtOAc (500 mL) and washed with 5% KHSO4 , half saturated NaHCO3, and brine (100 mL each). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (gradient elution, 2→3% CH3OH in CH2CI2) to provide the title intermediate (0.119 g, 58%) as a white foam: IR (cm”1) 3331, 1684, 1649, 1590, 1531; JH NMR (CDCI3) δ 0.92 (t, 3H, J = 7.3), 1.29 (t, 3H, J = 7.1), 1.47-1.58 (m, IH), 1.62-1.77 (m, IH), 1.85-2.00 (m, IH), 2.08-2.33 (m, 4H), 2.49 (s, 3H), 3.25-3.42 (m, 2H), 4.19 (q, 2H, J = 7.1), 4.39-4.50 (m, IH), 5.73 (dd, IH, J = 8.8, 6.8), 5.97 (dd, IH, J = 15.7, 1.4), 6.34 (t, IH, J = 7.2), 6.46 (s, IH), 6.86 (dd, IH, J = 15.7, 5.9), 7.18 (s, IH), 7.59 (dd, IH, J = 7.2, 1.8), 8.42 (dd, IH, J = 7.2, 1.8), 8.58-8.62 (m, IH), 9.56 (s, 1); Anal. C25H31N5O7O.5OH2O: C, H, N.
PAT
- Treatment of infection by human enterovirus d68Publication Number: US-2020016243-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: WO-2016044656-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: US-2021052708-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus D68Publication Number: US-11191817-B2Priority Date: 2014-09-17Grant Date: 2021-12-07
- Therapeutic compounds and methodsPublication Number: US-2025051283-A1
- Protease Inhibitors for Treatment or Prevention of Coronavirus DiseasePublication Number: US-2023192660-A1Priority Date: 2020-05-08
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-2019030027-A1Priority Date: 2016-01-29
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-10864210-B2Priority Date: 2016-01-29Grant Date: 2020-12-15
- Treatment of infection by human enterovirus D68Publication Number: US-10328128-B2Priority Date: 2014-09-17Grant Date: 2019-06-25
- Treatment of infection by human enterovirus d68Publication Number: US-2017290893-A1Priority Date: 2014-09-17
- Nucleotide and nucleoside therapeutic compositions, combinations and related uses thereofPublication Number: CN-117881402-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: EP-4333859-A1Priority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations, and related usesPublication Number: JP-2024517807-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: WO-2022235874-A1Priority Date: 2021-05-05
- Protease inhibitors for treatment or prevention of coronavirus diseasePublication Number: EP-4146267-A1Priority Date: 2020-05-08
- 4′-substituted nucleosides and nucleotides as antiviral agentsPublication Number: WO-2024227159-A2Priority Date: 2023-04-28
- Therapeutic compoundsPublication Number: WO-2024206284-A2Priority Date: 2023-03-27
- Antibody molecules binding to sars-cov-2Publication Number: WO-2024168061-A2Priority Date: 2023-02-07
- Predictive model for variants associated with drug resistance and theranostic applications thereofPublication Number: WO-2023172635-A1Priority Date: 2022-03-08
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: CA-3216679-A1Priority Date: 2021-05-05
LIT
- Structure and inhibition of SARS-CoV-1 and SARS-CoV-2 main proteases by oral antiviral compound AG7404Publication Name: Antiviral ResearchPublication Date: 2022-12PMCID: PMC9632241PMID: 36336176DOI: 10.1016/j.antiviral.2022.105458
- Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug DesignPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-09-30PMID: 34591488DOI: 10.1021/acs.jmedchem.1c01215
- In Vitro Antiviral Activity of New Oxazoline Derivatives as Potent Poliovirus InhibitorsPublication Name: Journal of Medicinal ChemistryPublication Date: 2018-12-04PMCID: PMC9169555PMID: 30512950DOI: 10.1021/acs.jmedchem.8b01482
- A Novel Series of Highly Potent Small Molecule Inhibitors of Rhinovirus ReplicationPublication Name: Journal of Medicinal ChemistryPublication Date: 2017-06-15PMID: 28581749DOI: 10.1021/acs.jmedchem.7b00175
- Anti-poliovirus activity of protease inhibitor AG-7404, and assessment of in vitro activity in combination with antiviral capsid inhibitor compoundsPublication Name: Antiviral ResearchPublication Date: 2013-05PMID: 23499651DOI: 10.1016/j.antiviral.2013.03.003

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | AG-7404, V-7404 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 343565-99-1 |
| PubChem CID | 5280053 |
| IUPHAR/BPS | 13223 |
| UNII | VQ1AN3OO42 |
| ChEMBL | ChEMBL141157 |
| Chemical and physical data | |
| Formula | C26H29N5O7 |
| Molar mass | 523.546 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Imocitrelvir”. PatSnap.
- Xie H, Rhoden EE, Liu HM, Ogunsemowo F, Mainou BA, Burke RM, et al. (November 2024). “Antiviral Development for the Polio Endgame: Current Progress and Future Directions”. Pathogens. 13 (11). Basel, Switzerland: 969. doi:10.3390/pathogens13110969. PMC 11597170. PMID 39599522.
- Bandyopadhyay AS, Burke RM, Hawes KM (June 2024). “Polio Eradication: Status, Struggles and Strategies”. The Pediatric Infectious Disease Journal. 43 (6): e207-211. doi:10.1097/INF.0000000000004330. PMID 38564755.
////////Imocitrelvir, protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
Ilantimod
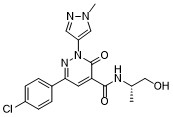
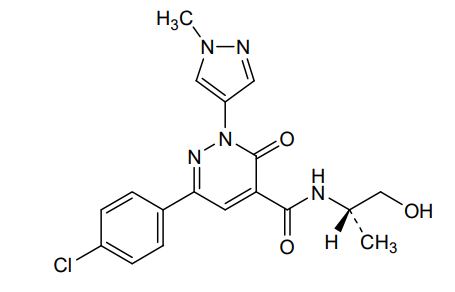
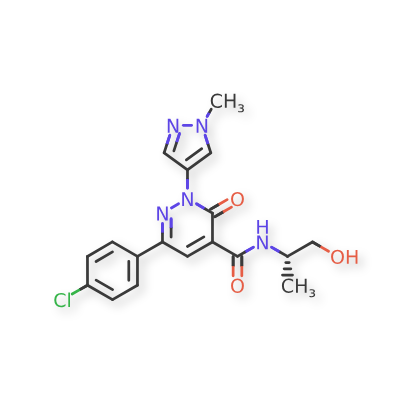
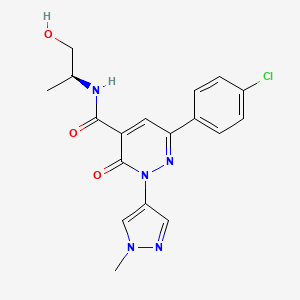
Ilantimod
CAS 2242464-44-2
MF C18H18ClN5O3 MW 387.82
6-(4-chlorophenyl)-N-[(2S)-1-hydroxypropan-2-yl]-2-(1-methyl-1H-pyrazol-4-yl)-3-oxo-2,3-dihydropyridazine-4-carboxamide
(S)-6-(4-chlorophenyl)-N-(1-hydroxypropan-2-yl)-2-(1-methyl-1H-pyrazol-4-yl)-3-oxo-2,3-dihydropyridazine-4-carboxamide
immunomodulator, BAY-2416964, BAY 2416964, Y87V4WXQ4Z
Ilantimod is an orally available formulation containing a small molecule antagonist of the aryl hydrocarbon receptor (AhR; class E basic helix-loop-helix protein 76; bHLHe76) with potential immunomodulating and antineoplastic activities. Upon oral administration, ilantimod specifically binds to AhR, inhibits AhR activation, and prevents AhR-mediated signaling. Abrogation of AhR activation prevents the activation of immune-tolerant dendritic cells (DCs) and regulatory T-cells (Tregs) in the tumor microenvironment (TME). This may restore the immune response against tumor cells. AhR, a member of the basic helix-loop-helix/Per-Arnt-Sim (bHLH/PAS) family of transcription factors, has important roles in regulating immunity and cellular differentiation. AhR can exhibit both pro-oncogenic and tumor suppressor-like functions depending on the tumor type; therefore, its expression may serve as a negative or positive prognostic factor.
- A Study to Learn How Safe the Study Drug BAY 2416964 (AhR Inhibitor) in Combination With the Treatment Pembrolizumab is, How This Combination Affects the Body, the Maximum Amount That Can be Given, How it Moves Into, Through and Out of the Body and Its Action Against Advanced Solid Cancers in AdultsCTID: NCT04999202Phase: Phase 1Status: TerminatedDate: 2025-02-10
- A First-in-Humans Dose Finding Study for an Aryl Hydrocarbon Receptor Inhibitor (AhRi) in Patients With Advanced CancerCTID: NCT04069026Phase: Phase 1Status: CompletedDate: 2024-03-06
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018146010&_cid=P11-MHAFJG-41587-1
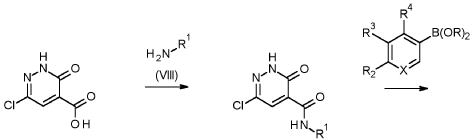
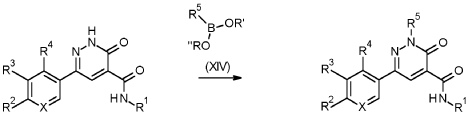
Example 17
6-(4-Chlorophenyl)-/V-[(2S)-1 -hydroxypropan-2-yl]-2-(1 -methyl-1 H-pyrazol-4-yl)-3-oxo-2,3-dihydropyridazine-4-carboxamide
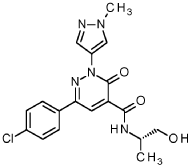

A solution of 80 mg intermediate 1 1 , 29.1 mg (2S)-2-aminopropan-1 -ol, 1 10 mg HATU and 0.1 mL ethyldiisopropylamine in 5 mL of DMF was stirred at room temperature for 14 hours. Then the reaction was quenched by water, and the mixture was extracted with dichloromethane two times. The combined organic phases were dried over sodium sulfate and evaporated to dryness. The residue was subjected to RP-HPLC ((column: X-Bridge C18 5μηι 100x30mm, mobile phase: acetonitrile / water (0.1 vol% formic acid)-gradient)) to yield 50 mg 6-(4-chlorophenyl)-/V-[(2S)-1 -hydroxypropan-2-yl]-2-(1 -methyl-1 H-pyrazol-4-yl)-3-oxo-2,3-dihydropyridazine-4-carboxamide
1H-NMR (400 MHz, CDC ): δ [ppm] = 1.34 (d, 3H); 2.73-2.82 (m, 1 H); 3.66-3.73 (m, 1 H); 3.77-3.84 (m, 1 H); 3.98 (s, 3H); 4.26-4.36 (m, 1 H); 7.49 (d, 2H); 7.87 (d, 2H); 8.12 (s, 1 H); 8.33 (s, 1 H); 8.69 (s, 1 H); 9.82 (bd, 1 H).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US438191125&_cid=P11-MHAFQQ-47913-1
SEE EX 17
PAT
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamide for the treatment of cancerPublication Number: KR-102627266-B1Priority Date: 2017-02-09Grant Date: 2024-01-24
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: US-11795164-B2Priority Date: 2017-02-09Grant Date: 2023-10-24
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: US-2024294505-A1Priority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamidesPublication Number: TW-I770113-BPriority Date: 2017-02-09Grant Date: 2022-07-11
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: CN-110678459-BPriority Date: 2017-02-09Grant Date: 2023-04-04
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: US-2023121195-A1Priority Date: 2017-02-09
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: CN-116531380-APriority Date: 2017-02-09
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: CN-116554152-APriority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: WO-2018146010-A1Priority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: EP-3580211-B1Priority Date: 2017-02-09Grant Date: 2020-12-02
- 2-HETEROARYL-3-OXO-2,3-DIHYDROPYRIDAZINE-4-CARBOXAMIDES FOR THE TREATMENT OF CANCERPublication Number: HR-P20210143-T1Priority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: AU-2018217860-B2Priority Date: 2017-02-09Grant Date: 2021-07-08
- 2-Troaril-3-oxo-3,2-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: IL-268469-APriority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: CA-3052718-A1Priority Date: 2017-02-09
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamide for the treatment of cancerPublication Number: CN-110678459-APriority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: EP-3580211-A1Priority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamide for the treatment of cancerPublication Number: KR-20190115460-APriority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamidePublication Number: TW-201840549-APriority Date: 2017-02-09
- Combination of an ahr-inhibitor and an pd1-inhibitor antibody and its use in the treatment of cancerPublication Number: EP-4076462-A1Priority Date: 2019-12-16
- Combinations of AHR inhibitors and PD1 inhibitor antibodies and their use in the treatment of cancerPublication Number: JP-2023505907-APriority Date: 2019-12-16
- Combinations of AHR-inhibitors and PD1-inhibitor antibodies and their use in the treatment of cancerPublication Number: KR-20220128622-APriority Date: 2019-12-16
- Combination of an ahr-inhibitor and an pd1-inhibitor antibody and its use in the treatment of cancerPublication Number: US-2023084899-A1Priority Date: 2019-12-16
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: AU-2018217860-A1Priority Date: 2017-02-09
- Methods and compositions for treating inflammatory and fibrotic pulmonary disordersPublication Number: US-2021401987-A1Priority Date: 2020-03-20
- Methods and compositions for treating inflammatory and fibrotic pulmonary disordersPublication Number: EP-4121111-A1Priority Date: 2020-03-20
- Combination of an ahr-inhibitor and an pd1-inhibitor antibody and its use in the treatment of cancerPublication Number: WO-2021122434-A1Priority Date: 2019-12-16
- Combination of an AhR-inhibitor and an PD1-inhibitor antibody and its use in the treatment of cancerPublication Number: AU-2020403801-A1Priority Date: 2019-12-16
- Combinations of AHR inhibitor and PD1 inhibitor antibodies and their use in cancer therapyPublication Number: CN-114786674-APriority Date: 2019-12-16
- Compositions and methods for treating myelin deficiency by rejuvenating glial progenitor cellsPublication Number: US-2023190961-A1Priority Date: 2021-10-20
- Prophylactic or therapeutic agent for severe pulmonary hypertension, refractory pulmonary hypertension, or drug-induced pulmonary hypertensionPublication Number: WO-2022149605-A1Priority Date: 2021-01-08
- Deuterated 2-arylheterocycle-3-oxo-2,3-dihydropyridazine-4-carboxamide inhibitor and preparation method therefor and application thereofPublication Number: EP-4253374-A1Priority Date: 2020-11-27
- Heteroaromatic ahr inhibitorPublication Number: WO-2022078356-A1Priority Date: 2020-10-15
- Methods and compositions for treating inflammatory and fibrotic pulmonary disordersPublication Number: WO-2021188849-A1Priority Date: 2020-03-20
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Ilantimod, immunomodulator, BAY-2416964, BAY 2416964, Y87V4WXQ4Z
Ibrilatazar
Ibrilatazar
CAS 57818-44-7
MF C18H32O3 MW 296.4 g/mol
rac-(2R)-(9Z,12Z)-2-hydroxyoctadeca-9,12-dienoic acid
(9Z,12Z)-2-hydroxyoctadeca-9,12-dienoic acid
peroxisome proliferator activated receptor (PPAR) alpha and gamma agonist, antineoplastic, ABILITY PHARMA, ABTL 0812, alpha-Hydroxylinoleic acid, ABTL0812
- alpha-Hydroxylinoleic acid
- ABTL0812
- 2-hydroxylinoleic acid
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| ABTL-0812 Sodium | X1840C8161 | Not Available | VFXKYDDSDQXKLC-NBTZWHCOSA-M |
Ibrilatazar also known as α-hydroxylinoleic acid is a small-molecule, experimental cancer drug being developed by Ability Pharmaceuticals.[1]
Ibrilatazar is an orally bioavailable, lipid analogue and inhibitor of raptor-mammalian target of rapamycin (mTOR) (mTOR complex 1; mTORC1), rictor-mTOR (mTOR complex 2; mTORC2) and dihydrofolate reductase (DHFR) with potential antineoplastic activity. Upon oral administration, ibrilatazar binds to and inhibits both mTORC1 and mTORC2, which may result in apoptosis and a decrease in proliferation in mTORC1/2-expressing tumor cells. mTOR is a serine/threonine kinase that is upregulated in some tumors; it plays an important role in the PI3K/Akt/mTOR signaling pathway which is often deregulated in cancer cells. In addition, ibrilatazar inhibits DHFR, an enzyme that reduces dihydrofolic acid to tetrahydrofolic acid, thereby blocking tetrahydrofolate synthesis, and resulting in both the depletion of nucleotide precursors and the inhibition of DNA, RNA and protein synthesis. This induces autophagy-induced cell death and further inhibition of cell proliferation.
- A Study of ABTL0812 in Pancreatic CancerCTID: NCT03417921Phase: Phase 1/Phase 2Status: SuspendedDate: 2024-07-31
- ABTL0812 in Combination With FOLFIRINOX for First-line Treatment of Metastatic Pancreatic StudyCTID: NCT04431258Phase: Phase 1/Phase 2Status: CompletedDate: 2024-03-18
- Phase I/Ib Clinical Trial of ABTL0812 in Advanced Cancer PatientsCTID: NCT02201823Phase: Phase 1Status: CompletedDate: 2015-07-02
- Microbiological production method of γ- and δ-lactonesPublication Number: JP-H03187387-APriority Date: 1989-08-04
- Process for the microbiological production of gamma- and delta-lactonesPublication Number: US-5168054-APriority Date: 1989-08-04Grant Date: 1992-12-01
- Degradation control of environmentally degradable disposable materialsPublication Number: US-2002123546-A1Priority Date: 1988-08-08
- Degradation control of environmentally degradable disposable materialsPublication Number: US-6323307-B1Priority Date: 1988-08-08Grant Date: 2001-11-27
- Degradation control of environmentally degradable disposable materialsPublication Number: US-6740731-B2Priority Date: 1988-08-08Grant Date: 2004-05-25
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US38087288&_cid=P12-MH8IQK-97634-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
History
In 2015, Ability announced that it had received orphan drug designation (ODD) for pediatric cancer neuroblastoma from the European Medical Agency (EMA) and the US Food and Drug Administration (FDA).[1] Also in 2016 a preclinical study confirmed that ABTL0812 was well tolerated.[2] In December 2016 the company announced Ibrilatazar has received an Orphan Drug Designation for the treatment of pancreatic cancer.[1]
Mechanism of action
One mechanism of action is the activation of the PPAR-alpha and PPAR-gamma receptors which in turn up-regulate the expression of the TRIB3 gene, leading to inhibition of the PI3K/AKT/mTOR pathway. This pathway is excessively activated in most human cancers, supporting tumor growth. It is a principal target of various new anti-tumour drugs. Tumor cells are killed via autophagic cell death, rather than apoptosis.[3][4]
ABTL0812 activates the PPAR receptors, inducing TRIB3 over-expression. TRIB3 binds to the Akt oncogene and inhibits the Akt/mTOR axis.[3]
Clinical trials
ABTL0812 showed efficacy in Phase I clinical trials in patients with advanced cancer, with low toxicity and high tolerability.[3]
References
- “Ability Pharmaceuticals Announces Orphan Drug Designation in the US for ABTL0812 in Pancreatic Cancer”. Ability Pharmaceuticals SL.
- “Ability Pharmaceuticals Announces Positive Phase 1 1b Study Results Of ABTL0812 In Cancer Patients With Advanced Solid Tumors”. http://www.biospace.com.
- “New mechanism of antitumor action identified”. Medical Xpress. 25 January 2016.
- Erazo T, Lorente M, López-Plana A, Muñoz-Guardiola P, Fernández-Nogueira P, García-Martínez JA, et al. (May 2016). “The New Antitumor Drug ABTL0812 Inhibits the Akt/mTORC1 Axis by Upregulating Tribbles-3 Pseudokinase”. Clinical Cancer Research. 22 (10): 2508–19. doi:10.1158/1078-0432.ccr-15-1808. hdl:2445/207600. PMID 26671995.
| Clinical data | |
|---|---|
| Other names | α-Hydroxylinoleic acid; 2-Hydroxylinoleic acid; ABTL-0812 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 57818-44-7 |
| PubChem CID | 21158511 |
| ChemSpider | 20118100 |
| UNII | 0DE74TJ7EZ |
| ChEBI | CHEBI:136927 |
| CompTox Dashboard (EPA) | DTXSID301258077 |
| Chemical and physical data | |
| Formula | C18H32O3 |
| Molar mass | 296.451 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Ibrilatazar, peroxisome proliferator activated receptor (PPAR) alpha and gamma agonist, antineoplastic, ABILITY PHARMA, ABTL 0812, alpha-Hydroxylinoleic acid, ABTL0812
Glovadalen



Glovadalen
CAS 2576359-31-2
MF C24H27Cl2N3O3 MW 476.4 g/mol
2-(3,5-dichloro-1-methyl-1H-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(2-hydroxypropan-2-yl)-1-methyl-3,4-dihydroisoquinolin-2(1H)-yl]ethan-1-one,
2-(3,5-dichloro-1-methylindazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(2-hydroxypropan-2-yl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanone
dopamine D1 receptor positive allosteric modulator, Phase 2, Parkinson’s disease, UCB-0022, UCB 0022, H8T5VKH4CZ
- OriginatorUCB Biopharma
- ClassAlcohols; Antiparkinsonians; Benzene derivatives; Chlorinated hydrocarbons; Isoquinolines; Ketones; Neuroprotectants; Propanols; Pyrazoles; Small molecules
- Mechanism of ActionDopamine D1 receptor modulators
- Phase IIParkinson’s disease
- 27 Aug 2025Chemical structure information added.
- 21 May 2025UCB Biopharma SRL initiate a phase I trial in healthy volunteers (PO) (NCT06970301)
- 11 Apr 2025UCB Pharma completes a phase-II ATLANTIS trial in Parkinson’s disease (In adults, In the elderly, Adjunctive treatment) in USA (PO) (NCT06055985)
Glovadalen (developmental code name UCB-0022) is a dopamine D1 receptor positive allosteric modulator which is under development for the treatment of Parkinson’s disease.[1][2][3][4][5][6] It has been found to potentiate the capacity of dopamine to activate the D1 receptor by 10-fold in vitro with no actions on other dopamine receptors.[5][6] As of May 2024, glovadalen is in phase 2 clinical trials for this indication.[1][2][5] The drug is under development by UCB Biopharma.[1][4][5] It is described as an orally active, centrally penetrant small molecule.[1][5][6]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021001288&_cid=P21-MH738G-96748-1

1. Preparation of intermediate of formula (ID- 2-(3,5-dichloro-1-methyl-indazol-4- vDacetic acid

1.1. Preparation of intermediate (Xlb) -1-methyl-5-nitro-indazole
5-Nitro-1H-indazole (Xla) (3.00 kg, 18.4 mol) and DMF (30.0 L) are charged into a 50 L three-neck round-bottom flask at 15-30°C. KOH (2.06 Kg, 36.7 mol) is added in one portion into the reactor at 0-5°C. The mixture is stirred at 0-50°C for 1h. Methyl iodide (2.87 kg, 20.2 mol) is then added at 0-5°C and the mixture is stirred for 3h at 15-30°C. The reaction mixture is added into water (30 L) at 0-10°C and the mixture is stirred for 10 min then filtered. The filter cake is washed with water (5 L) and dried. This overall procedure is carried out on 4 batches of the same size in parallel. The solids obtained from the four batches are combined to give 1-methyl-5-nitro-indazole (Xlb) as a brown solid (10.0 kg, 42.3 mol, 75% purity (LC/MS), 57.5% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCIs) d 8.65 (s, 1H), 8.21 (d, J = 9.17 Hz, 1 H), 8.13 (s, 1 H), 7.39 (d, J = 9.17 Hz, 1 H), 4.08 (s, 3 H).
1.2. Preparation of intermediate (Xa)- tert-butyl 2-(1-methyl-5-nitro-indazol-4- yl)acetate
t-BuOK (4.43 kg, 39.5 mol) and THF (30 L) are charged into a 50 L three-neck round-bottom flask and the mixture is cooled to -45 / -35°C under nitrogen and stirring. 1-Methyl-5-nitro-indazole (Xlb) (3.50 kg, 19.7 mol) is then added in portions at -45 / -35°C. Tert-butyl 2-chloroacetate (3.57 kg, 23.7 mol) is added dropwise at the same temperature and the mixture is stirred at 1h. The mixture is warmed up to 15-30°C and stirred for 5h. The reaction is quenched by the addition of a saturated ammonium chloride solution (9 L) and water (2 L) is added. The organic layer is separated and the aqueous layer is extracted with ethyl acetate (2 x 5 L). The organic phases are combined, washed with brine (2 L), dried over Na2SC>4, filtered and concentrated under vacuum. The crude product is purified by recrystallization with ethyl acetate (5 L). This overall procedure is carried out on 2 batches of the same size in parallel. The solids obtained from the two batches are combined and dried together to give tert-butyl 2-(1-methyl-5-nitro-indazol-4-yl)acetate as a yellow solid (Xa) (5.30 kg, 17.7 mol, 97.6% purity (LC/MS), 44.9% yield).
1H NMR (400 MHz, CDCIs) d 8.18-8.20 (m, 2H), 7.37 (d, J = 9.21 Hz, 1 H), 4.27 (s, 2 H), 4.14 (s, 3 H), 1.44 (s, 9 H).
1.3. Preparation of intermediate (Xb) – tert-butyl 2-(5-amino-1-methyl-indazol-4- yl)acetate
Tert-butyl 2-(1-methyl-5-nitro-indazol-4-yl)acetate (Xa) (7.30 kg, 25.0 mol) and MeOH (76 L) are charged into a reactor. Argon is purged and Pd/C (50%, 760 g) is added. Hydrogen is added three times and the mixture is stirred at 50°C under hydrogen atmosphere (50 psi) for 3h. The reaction mixture is filtered and the solid is washed with MeOH (5 L). The mixture is concentrated to give tert-butyl 2-(5-amino-1-methyl-indazol-4-yl)acetate (Xb) as a brown oil (6.50 kg, 23.9 mol, 96.2% purity (LC/MS), 95.4% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCI3) d 7.72 (s, 1H), 7.27 (d, J = 8.80 Hz, 1 H), 6.91 (d, J = 8.80 Hz, 1 H), 4.60 (s, 2 H), 3.93 (s, 3 H), 3.68 (s, 2H), 1.38 (s, 9 H).
1.4. Preparation of intermediate (Xc)- 2-(5-chloro-1-methyl-indazol-4-yl)acetic acid
Tert-butyl 2-(5-amino-1-methyl-indazol-4-yl)acetate (Xb) (2.00 kg, 7.65 mol) and concentrated HCI (10.0 L, 12M) are charged into a 50 L three-neck round bottom flask and the mixture is cooled to -10/-5°C and stirred. A water solution (5 L) of sodium nitrite (686 g, 9.95 mol) is added dropwise at -10/-5°C and stirred for 30 min. CuCI (833 g, 8.42 mol) and concentrated HCI (10.0 L, 12M) are charged into a 20 L three-neck round bottom flask and the mixture is stirred for 30 min. at -10/-5°C, then added into the other reactor. The mixture is stirred at -10/-5°C for 1 h, then at 10-30°C for 16h. The reaction mixture is filtered and the solid washed with water. This overall procedure is carried out on 3 batches of the same size in parallel. The solids obtained from the three batches are combined and dried together to give 2-(5-chloro-1-methyl-indazol-4-yl)acetic acid (Xc) as a yellow solid (4.00 kg, 16.3 mol, 92% purity (LC/MS), 71.3% yield) which is used in the next step without further purification.
1.5. Preparation of 2-(3,5-dichloro-1-methyl-indazol-4-yl)acetic acid (II)
2-(5-Chloro-1-methyl-indazol-4-yl)acetic acid (Xc) (1.30 kg, 5.79 mol) and DMF (6.5 L) are charged into a 50 L three-neck round bottom flask at 20°C. N-Chlorosuccinimide (772 g, 5.79 mol) is added portionwise at 20°C and the mixture is stirred at 20°C for 2h. The reaction mixture is poured into water (25 L) and filtered. The crude product is triturated with isopropyl etherethyl acetate (3:1) (7.0 L) at 20°C for 2h then filtered and dried. This overall procedure is carried out on 3 batches of the same size in parallel. The solids obtained from the three batches are combined to give 2-(3,5-dichloro-1-methyl-indazol-4-yl)acetic acid (II) (2.1 kg, 7.9 mol, 97.5% purity (LC/MS), 46% yield).
1H NMR (400 MHz, CDCI3) d 12.67 (s, 1 H), 7.68 (d, J = 9.05 Hz, 1 H), 7.53 (d, J = 9.05 Hz, 1 H), 4.20 (s, 2 H), 4.02 (s, 3 H).
2. Preparation of compound of formula (I)
2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1- methyl-ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanone

2.1. Preparation of intermediate (IX).
(2R)-2-amino-3-(2-bromophenyl)propan-1-ol – a6
(2R)-2-amino-3-(2-bromophenyl)propanoic acid a5 (34.0 kg, 139 mol) and THF (238 L) are charged into a reactor. Sodium borohydride (15.6 kg, 413 mol) is added slowly at 20-30°C. A solution of iodine (35.3 kg, 139 mol) in dry THF (20.0 L) is added slowly at 0-10°C and the reaction mixture is stirred at 70°C for 12h. The reaction was quenched with methanol (70.0 L) at 0°C and heated to 80°C for 30 min. The mixture was cooled down, concentrated under vacuum and the residue was suspended in NaOH (30.0 L, 2N), then filtered. The filter cake was dried under vacuum to give (2R)-2-amino-3-(2-bromophenyl)propan-1-ol a6 as a white solid (31.0 kg, 135 mol, 96.7% yield) which is used in the next step without further purification. 1H NMR (400 MHz, CDCIs) d 7.57 (d, J = 7.7 Hz, 1H), 7.21 – 7.29 (m, 2H), 7.07 – 7.15 (m, 1H), 3.66 (dd, J = 10.5, 3.6 Hz, 1 H), 3.41 (dd, J = 10.5, 7.2 Hz, 1 H), 3.18 – 3.29 (m, 1 H), 2.95 (dd, J = 13.5, 5.5 Hz, 1 H), 2.70 (dd, J = 13.5, 8.2 Hz, 1H), 1.51 – 1.91 (m, 3H).
2.2. Preparation of intermediate of formula (VIII).
(4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one – a7
(2R)-2-amino-3-(2-bromophenyl)propan-1-ol a6 (31.0 kg, 135 mol) and dichloromethane (220 L) are charged into a reactor. Triphosgene (13.9 kg, 47.1 mol) is added at room temperature then N,N-diisopropylethylamine (39.1 kg, 303 mol) is slowly added at 0-10°C. The reaction mixture is stirred at 0-10°C for 1h then washed with water (50.0 L) twice, dried with anhydrous sodium sulfate and filtered to give (4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one a7 as a solution in dichloromethane which is used directly in the next step.
2.3. Preparation of intermediate (VII).
(10aR)-9-bromo-1 ,5, 10, 10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8
A solution of (4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one a7 (135 mol) in dichloromethane (220 L) is charged into a reactor and cooled down to 0-5°C. Trimethylsilyl triflate (35.9 kg, 162 mol) and paraformaldehyde (13.3 kg, 148 mol) are added at 0-5°C, then stirred for 2h at 15-20°C. Water (170 L) is added into the mixture which is then extracted twice with dichloromethane (50.0 L). the organic layer is dried with anhydrous sodium sulfate, filtered and concentrated under vacuum. A mixture of petroleum etherethyl acetate (1 :1, 45.0 L) is added and the mixture is stirred at room temperature for 6h and filtered. The solid was dried to get (10aR)-9-bromo-1,5,10,10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8 as an off-white solid (29.0 kg, 80.2% yield).
1H NMR (400 MHz, CDCI3) d 7.45 – 7.52 (m, 1H), 7.08 – 7.14 (m, 2H), 4.83 (d, J = 17.0 Hz, 1H), 4.62 (t, J = 8.4 Hz, 1H), 4.36 (d, J = 17.0 Hz, 1H), 4.21 (dd, J = 8.6, 4.9 Hz, 1 H), 3.91 -3.99 (m, 1H), 3.25 (dd, J= 16.3, 4.2 Hz, 1 H), 2.67 (dd, J = 16.1 , 11.0 Hz, 1H).
2.4. Preparation of intermediates (VI)
2.4.1. [(3R)-5-bromo-1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9
Ethanol (120 L) and water (60.0 L) are mixed into a reactor. (10aR)-9-bromo-1,5,10,10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8 (29.7 kg, 111 mol) is added then sodium hydroxide (13.3 kg, 332 mol) is slowly added at 15-20°C. The reaction mixture is stirred at 90°C for 2h then cooled down to room temperature. Water (300 L) is added into the mixture which is centrifugated. The centrifugal cake is dried in circulation oven to give [(3R)-5-bromo- 1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9 as a white solid (23.7 kg, 88.3% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCIs) d 7.37 – 7.47 (m, 1H), 6.95 – 7.08 (m, 2H), 4.00 – 4.10 (m, 2H), 3.85 (dd, J = 10.9, 3.7 Hz, 1 H), 3.57 (dd, J = 10.9, 7.9 Hz, 1 H), 3.06 (ddt, J = 11.3, 7.6, 4.1 , 4.1 Hz, 1H), 2.79 (dd, J= 17.1, 4.4 Hz, 1H), 2.40 (dd, J= 17.1, 10.9 Hz, 1H), 1.93 (br s, 2H).
2.4.2. [(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl- silane a10
[(3R)-5-bromo-1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9 (23.7 kg, 97.8 mol) and dichloromethane (240 L) are charged into a reactor. DMAP (120 g, 0.98 mol) and imidazole (13.3 kg, 196 mol) are added. Tert-butyldimethylsilyl chloride (TBSCI) (17.7 kg, 117 mol) is slowly added at 15-20°C and the mixture is stirred for 12h. Ammonium chloride (100 L) is added into the mixture. The organic phase was separated, washed with water (50.0 L), dried with anhydrous sodium sulfate, filtered and concentrated under vacuum to give [(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a10 as a yellow oil (37.6 kg, 86% purity, 93% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCI3) d 7.36 – 7.45 (m, 1H), 7.01 (d, J = 4.6 Hz, 1H), 4.01 – 4.13 (m, 2H), 3.84 (dd, J = 9.9, 3.7 Hz, 1 H), 3.64 (dd, J = 9.8, 7.2 Hz, 1 H), 2.96 – 3.08 (m, 1 H), 2.75 (dd, J = 17.0, 4.2 Hz, 1 H), 2.44 (dd, J = 17.0, 10.8 Hz, 1H), 1.76 – 2.20 (m, 2H), 0.89 – 0.97 (m, 9H), 0.08 – 0.14 (m, 6H).
2.5. Preparation of intermediate (V).
[(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11
[(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a10 (3.42 kg, 8.31 mol) and THF (30.0 L) are charged into a reactor. N-Chlorosuccinimide (NCS) (1.17 kg, 8.73 mol) is slowly added at room temperature and the mixture is stirred at 25°C for 30 min. A solution of KOH (1.52 kg, 27.1 mol) in dry methanol (7.00 L) is slowly added at room temperature and the reaction is stirred at 25°C for 1h. The reaction is quenched with water (10.0 L) and extracted with petroleum etherethyl acetate (1:2, 5.00 L). The organic layer is separated, washed with brine (10.0 L), dried with anhydrous sodium sulfate and filtered. This overall procedure is carried out on 10 batches of the same size in parallel and the 10 reaction filtrates are combined and concentrated under vacuum to give [(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11 as a brown oil (28.0 kg, crude) which is used in the next step without further purification.
1H NMR (400 MHz, CDC ) d 8.24 (d, J = 2.6 Hz, 1H), 7.58 (dd, J = 7.8, 1.2 Hz, 1 H), 7.12 -7.25 (m, 2H), 4.03 (dd, J = 9.5, 4.0 Hz, 1 H), 3.67 – 3.77 (m, 2H), 3.07 (dd, J = 17.0, 6.2 Hz, 1H), 2.68 (dd, J = 17.1, 10.9 Hz, 1 H), 0.88 – 0.91 (m, 9H), 0.07 (d, J= 1.5 Hz, 6H).
2.6. Preparation of intermediates of formula (IV)
2.6.1. [(1S,3R)-5-bromo-1-methyl-1,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl- dimethyl-silane (IVa)
[(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11 (3.10 kg, 8.75 mol) and THF (20.0 L) are charged into a reactor. The mixture is cooled down to 0°C and methylmagnesium chloride (3M, 11.6 L) is added. The mixture is stirred at 20°C for 12h. The reaction is quenched with a saturated solution of ammonium chloride. The phases are separated and the aqueous layer is extracted twice with petroleum ether: ethyl acetate (3:1, 5.00 L). The combined organic phases are washed with brine (10.0 L), dried over anhydrous sodium sulfate and filtered. This overall procedure is carried out on 9 batches of the same size in parallel and the nine reaction filtrates are combined and concentrated under vacuum. The crude mixture is purified by silica gel chromatography with petroleum ether : ethyl acetate (10:1) to give [(1S,3R)-5-bromo-1 -methyl-1, 2, 3, 4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane (IVa) as a brown oil (4.60 kg, 99.7% purity, 15.7% yield).
1H NMR (400 MHz, DMSO-de) d 7.41 (dd, J=7.7, 0.9 Hz, 1H), 7.12 – 7.18 (m, 1H), 7.03 – 7.11 (m, 1H), 4.12 (q, J= 6.8 Hz, 1H), 3.62 (d, J= 5.7 Hz, 2H), 3.07 – 3.17 (m, 1H), 2.67 – 2.76 (m, 1H), 2.26 (dd, J=16.9, 10.0 Hz, 1H), 2.12 (br s, 1 H), 1.32 (d, J= 6.8 Hz, 3H), 0.84 – 0.93 (m, 9H), 0.07 (d, J=0.9 Hz, 6H).
2.6.2. tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1-methyl-3,4- dihydro-1 H-isoquinoline-2-carboxylate (IVb)
[(1S,3R)-5-bromo-1-methyl-1,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane (IVa) (1.85 kg, 4.99 mol) and dichloromethane (13.0 L) are charged in a reactor. N,N-diisopropylethylamine (1.94 kg, 14.9 mol) and di-tert-butyl dicarbonate (1.14 kg, 5.24 mol) are added at room temperature and the mixture is stirred for 12h. The reaction mixture is washed twice with a saturated ammonium chloride solution (10.0 L), the organic layer is dried with anhydrous sodium sulfate and filtered. This overall procedure is carried out on 2 batches of the same size in parallel and the two reaction filtrates are combined and concentrated under vacuum. The crude mixture is purified by silica gel chromatography with petroleum ether ethyl acetate (30:1) to give tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1 -methyl-3, 4-dihydro-1 H-isoquinoline-2-carboxylate (IVb) as a yellow oil (4.00 kg, 99.5% purity, 85.2% yield).
1H NMR (400 MHz, DMSO-de) d 7.50 (d, J = 7.9 Hz, 1 H), 7.22 (br d, J = 6.7 Hz, 1 H), 7.06 -7.18 (m, 1 H), 4.84 (br s, 1 H), 4.12 (br s, 1H), 3.46 (br d, J = 15.4 Hz, 2H), 2.94 (br dd, J = 15.8, 5.2 Hz, 1H), 2.71 (br t, J = 9.5 Hz, 1 H), 1.45 (s, 9 H), 1.28 (br s, 3H), 0.81 (s, 9H), -0.08 (s, 6H).
2.6.3. tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1 -methyl- ethyl)-1 -methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc)
A solution of tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1-methyl-3,4-dihydro-1 H-isoquinoline-2-carboxylate (IVb) (42.5 g, 90.3 mmol) in dry THF (0.5 M solution) and a commercial solution of n-Buthylithium in Hexanes (1.6 M solution) were pumped at respectively 6.0 ml/min (1.0 equiv) and 2.46 mL/min (1.3 equiv.) and were mixed in a glass microchip cooled at -40°C. The mixed flow stream was pumped through the reaction zone 1 of the microchip (0.3 ml_) and was then combined with a solution of dry acetone (13.5 M) pumped at 6.0 mL/min (27 equiv.). The resulting stream was then passed through the reaction zone 2 of the microchip (0.7 ml_) at -40 °C. Finally, the global flow stream exiting the reactor was collected and quenched at room temperature in a saturated solution of aqueous ammonium chloride. When all the feed solutions were consumed, a bilayer reaction mixture was obtained. The aqueous layer was separated from the organic layer, and then extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under vacuum. A yellow oil was obtained (46.5 g) and was purified by SFC chromatography on a GreenSep Nitro column (10m, 5×22.3 using CO298 %/EtOH 2% eluent). The solvent was removed under vacuum to yield to a white solid, tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc) (25 g, 56 mmol, 62 % yield).
UPLC_MS basic 1 pic @ 3.83 min (ES+): 350 (M-Boc+H)+, 332 (M-Boc-H20+H)+, 100 % purity.
1H NMR (400 MHz, DMSO-de) d 7.44 (d, J = 7.9 Hz, 1H), 7.19 (dt, J = 8.1 , 5.2 Hz, 1 H), 7.09 (t, J = 9.0 Hz, 1H), 4.99 (s, 1 H), 4.87 (dq, J = 13.4, 6.4 Hz, 1 H), 4.11 (s, 1H), 3.96 (t, J = 14.9 Hz, 1 H), 3.48 (dd, J = 9.4, 4.1 Hz, 1H), 2.98 (dd, J = 16.5, 5.0 Hz, 1H), 2.89 (t, J = 9.6 Hz, 1H), 1.65 (s, 3H), 1.58 (s, 3H), 1.55 (d, J = 2.5 Hz, 9H), 1.34 (dd, J = 20.5, 6.6 Hz, 3H), 0.90 (s, 9H), 0.08 (d, J = 7.2 Hz, 3H), -0.00 (s, 3H).
2.7. Preparation of intermediate (III) 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl- 1.2.3.4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride
2.7.1. tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)- 1.2.3.4-tetrahydroisoquinolin-3-yl]methoxy]silane- a15
Tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc) (148 g, 87% purity, 287 mmol) is dissolved in 1000 ml_ dichloromethane and transferred to a 2 liter double walled reactor. 2,6-Lutidine (100 ml_, 860 mmol) is added and the jacket temperature is set at-2°C. Trimethylsilyl trifluoromethanesulfonate (154 g, 129 ml_, 692 mmol) is added over 40 min via an addition funnel. Two hours after the start of addition, the reaction is quenched by adding 650 ml_ of an aqueous citric acid solution (1M) and the temperature of the mixture is brought back to 20°C. One hour after the start of the quench, the layers are separated. The organic layer is washed twice with 350 ml_ of an aqueous solution of citric acid (1M). The organic layer is stirred with 750 ml_ of aqueous sodium carbonate (10% w/w) for 10 min before separation of the layers. The organic layer is dried over anhydrous sodium sulfate. The organic layer is then filtered and the filtrate is concentrated under vacuum at 40°C providing a yellow oil (128 g) of tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy]silane a15 which is used in the next step without further purification.
1H NMR (400 MHz, CDC ) d 7.19 (d, J = 7.7 Hz, 1 H), 7.07 (t, J = 7.7 Hz, 1 H), 7.00 (d, J = 7.6 Hz, 1 H), 4.24 (q, J = 6.8 Hz, 1 H), 3.75 (dd, J = 9.7, 4.4 Hz, 1H), 3.60 (dd, J = 9.7, 7.0 Hz, 1H), 3.54 (dd, J = 16.3, 3.5 Hz, 1H), 3.15 (ddt, J = 10.9, 7.4, 4.0 Hz, 1 H), 2.52 (dd, J = 16.3, 10.9 Hz, 1H), 1.66 (d, J = 14.6 Hz, 6H), 1.52 – 1.43 (m, 3H), 0.92 (q, J = 1.2 Hz, 9H), 0.14 (q, J = 1.2 Hz, 2H), 0.09 (d, J = 1.1 Hz, 6H), 0.00 (q, J = 1.2, 0.8 Hz, 9H).
2.7.2. 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl-1,2,3,4-tetrahydroisoquinolin-2-ium-5- yl]propan-2-ol chloride Intermediate (III)
In a three-neck round bottom flask equipped with a mechanical stirrer, tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)-1,2,3,4-tetrahydroisoquinolin-3-yljmethoxyjsilane a15 (20.0 g, 47.4 mmol) is dissolved in 220 ml_ of isopropanol. To this solution, 42.3 ml_ of hydrochloric acid in iso-propanol (5-6 M, around 5 eq.) are added. 45 min after addition of hydrochloric acid, a 100 mg of seeds of the desired product are introduced. After 7 hours at room temperature, the reaction mixture is filtered over a sintered glass filter. The filtercake is washed with 40 ml_ isopropanol and dried under vacuum at room temperature overnight. 11.1 g of 2-[(1S,3R)-3-(hydroxymethyl)-1 -methyl-1 , 2,3,4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride (III) are obtained as a pinkish solid. The yield over the two deprotection steps is 91%.
1H NMR (400 MHz, CD3OD) d 7.46 (dd, J = 7.8, 1.3 Hz, 1H), 7.28 (t, J = 7.8 Hz, 1H), 7.21 (dd, J = 7.8, 1.3 Hz, 1H), 4.63 (q, J = 6.9 Hz, 1H), 3.97 (dd, J = 11.7, 3.8 Hz, 1 H), 3.88 (dd, J = 17.2, 4.3 Hz, 1H), 3.78 (dd, J = 11.8, 6.1 Hz, 1H), 3.66 – 3.56 (m, 1 H), 3.14 (dd, J = 17.2, 11 .4 Hz, 1 H), 1 .73 (d, J = 6.8 Hz, 3H), 1 .64 (d, J = 4.8 Hz, 6H). OH and NH protons are not observed.
2.8. Preparation of compound of formula (I).
2-(3,5-dichloro-1 -methyl-indazol-4-yl)-1 -[(1 S,3R)-3-(hydroxymethyl)-5-(1 – hydroxy-1 -methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone
In a 100 ml. Easymax reactor equipped with a mechanical stirrer, 2-(3,5-dichloro-1 -methyl-indazol-4-yl)acetic acid (II) (4.00 g, 15.4 mmol), 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl-1 ,2,3,4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride (III) (4.46 g, 16.4 mmol) and 48 mL of DMF are charged. The suspension is stirred at 20°C and then cooled by setting the jacket temperature to -2°C. Once the temperature of the mixture is below 3°C, N,N-diisopropylethylamine (9.5 mL, 54 mmol) is added. (2-(1 H-benzotriazol-1 -yl)-1 , 1 ,3,3-tetramethyluronium hexafluorophosphate (6.4 g, 17 mmol) is added in four portions over 1 hour. The mixture is stirred for 1 h 45 before setting the jacket temperature at 15°C. 16 mL of water are then added over the course of a few minutes. 15 min later, 30 mg of solid product are added as seeds to initiate the crystallization. The jacket temperature is set at 20°C. Half an hour later, 16 mL of water are added over 17 min. Stirring of the suspension is pursued for 2 h 15 at 20°C before being filtered on sintered glass. The filtercake is washed with two portions of 20 mL of water and then dried at 50°C overnight under vacuum yielding 6.03 g of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1 S,3R)-3-(hydroxymethyl)-5-(1 -hydroxy-1 -methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone (I) (crude material).
A recristallization is carried out on 5.00 g of the crude material obtained by first suspending in 50 mL acetonitrile. The jacket temperature is set to 70°C. Once the solid has dissolved and the mass temperature has reached 66°C, 720 mI of water are added. The mass temperature is then cooled to 59°C and 125 mg of solid product is added as seeding material. The mass temperature is then decreased to 55°C over 25 min at which stage crystallization is occurring. The jacket temperature is then decreased over two hours from 58°C down to 20°C. After 50 min, the suspension is filtered and the filtercake is washed with 7.5 mL acetonitrile. The filtercake is then dried under vacuum at 45°C overnight and 2 hours at 50°C providing 4.04 g of 2-(3,5-dichloro-1 -methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone (I) as an off-white powder (hydrate form) Yield = 64%.
1H NMR (400 MHz, DMSO-cfe) d 7.65 (dd, J = 9.0, 2.2 Hz, 1H), 7.52 (dd, J = 9.0, 2.1 Hz, 1 H), 7.37 (ddd, J = 19.6, 7.6, 1 .7 Hz, 1 H), 7.25 – 7.03 (m, 2H), 5.30 (q, J = 6.5 Hz, 0.3H), 5.16 -4.99 (m, 1 .7H), 4.99 – 4.84 (m, 0.7H), 4.63 – 4.30 (m, 3.3H), 4.17 – 3.93 (m, 4H), 3.28 (dt, J = 10.5, 5.1 Hz, 1.3H), 3.10 – 2.85 (m, 1.7H), 1.56 (dd, J = 13.2, 6.9 Hz, 6.7H), 1.24 (d, J = 6.5 Hz, 2.3H).
PAT
- A Substituted Tetrahydroisoquinoiline Derivative as a D1 Positive Allosteric ModulatorPublication Number: US-2022259179-A1Priority Date: 2019-07-01
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: CN-113993857-BPriority Date: 2019-07-01Grant Date: 2024-01-02
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: CN-117700395-APriority Date: 2019-07-01
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: JP-7510444-B2Priority Date: 2019-07-01Grant Date: 2024-07-03
- Substituted Tetrahydroisoquinoline Derivatives as Positive Allosteric Modulators of D1Publication Number: CN-113993857-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-3993794-A1Priority Date: 2019-07-01
- D1 Substituted tetrahydroisoquinoline derivatives as positive allosteric modulatorsPublication Number: KR-20220029686-APriority Date: 2019-07-01
- A SUBSTITUTED TETRAHYDROISOQUINOLINE DERIVATIVE AS A POSITIVE ALOSTERIC MODULATOR OF D1Publication Number: PE-20221020-A1Priority Date: 2019-07-01
- Substituted Tetrahydroisoquinoline Derivatives as D1 Positive Allosteric ModulatorsPublication Number: JP-2022539152-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2021001288-A1Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: TW-202115010-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2021001288-A9Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a D1 positive allosteric modulatorPublication Number: AU-2020299953-A1Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: CA-3139571-A1Priority Date: 2019-07-01
- A Substituted Tetrahydroisoquinoline Derivative As A D1 Positive Allosteric ModulatorPublication Number: US-2024059665-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-Dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: US-2024083925-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: AU-2021403603-A9Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-4263517-B1Priority Date: 2020-12-18Grant Date: 2024-10-02
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: EP-4263519-B1Priority Date: 2020-12-18Grant Date: 2024-10-02
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: EP-4263519-A1Priority Date: 2020-12-18
- amorphous solid dispersionPublication Number: JP-2023553457-APriority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives as D1-positive allosteric modulatorsPublication Number: JP-2023553671-APriority Date: 2020-12-18
- 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)- Prodrug of 1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: JP-2024500391-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: US-2024000769-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: IL-303693-APriority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: KR-20230121849-APriority Date: 2020-12-18
- amorphous solid dispersionPublication Number: KR-20230121867-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: EP-4262756-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-4263517-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: CA-3203281-A1Priority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives useful as D1 positive allosteric modulatorsPublication Number: CN-116583280-APriority Date: 2020-12-18
- 2-(3,5-Dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl Prodrug of -ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: CN-116601161-APriority Date: 2020-12-18
- Amorphous Solid DispersionPublication Number: CN-116685308-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: IL-303688-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: WO-2022129267-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2022129268-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: WO-2022129356-A1Priority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: AU-2021401128-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: AU-2021403603-A1Priority Date: 2020-12-18
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | UCB-0022; UCB0022 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2576359-31-2 |
| PubChem CID | 155460962 |
| IUPHAR/BPS | 13232 |
| UNII | H8T5VKH4CZ |
| Chemical and physical data | |
| Formula | C24H27Cl2N3O3 |
| Molar mass | 476.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “UCB 0022”. AdisInsight. Springer Nature Switzerland AG. 28 May 2024. Retrieved 10 August 2024.
- “Delving into the Latest Updates on Glovadalen with Synapse”. Synapse. 8 August 2024. Retrieved 10 August 2024.
- McFarthing K, Buff S, Rafaloff G, Fiske B, Mursaleen L, Fuest R, et al. (2023). “Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2023 Update”. Journal of Parkinson’s Disease. 13 (4): 427–439. doi:10.3233/JPD-239901. PMC 10357160. PMID 37302040.
Our analysis of dopaminergic therapies shows a continued emphasis on DA agonists and levodopa reformulation. These include Cerevel’s tavapadon, a D1/D5 receptor partial agonist and UCB0022, a positive allosteric modulator of the D1 receptor, as well as approaches to sub-cutaneously deliver levodopa/carbidopa such as Abbvie’s ABBV-951 and Neuroderm’s ND0612.
- “Glovadalen”. IUPHAR/BPS Guide to PHARMACOLOGY. Retrieved 10 August 2024.
- “UCB0022”. ALZFORUM. 3 May 2024. Retrieved 10 August 2024.
- Vermeiren C, Ates A, Bouzom F, Delaunois A, Gillard M, Kenda B, et al. (7 September 2022). “Preclinical characterization of UCB0022, an oral, brain penetrant, selective, clinical-stage positive allosteric modulator of the dopamine 1 receptor (D1 PAM)”. Movement Disorders. 37 (Suppl 2 [2022 International Congress September 15-18, 2022. Madrid, Spain]). Retrieved 10 August 2024.
////////Glovadalen, dopamine D1 receptor positive allosteric modulator, Phase 2, Parkinson’s disease, UCB-0022, UCB 0022, H8T5VKH4CZ

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}