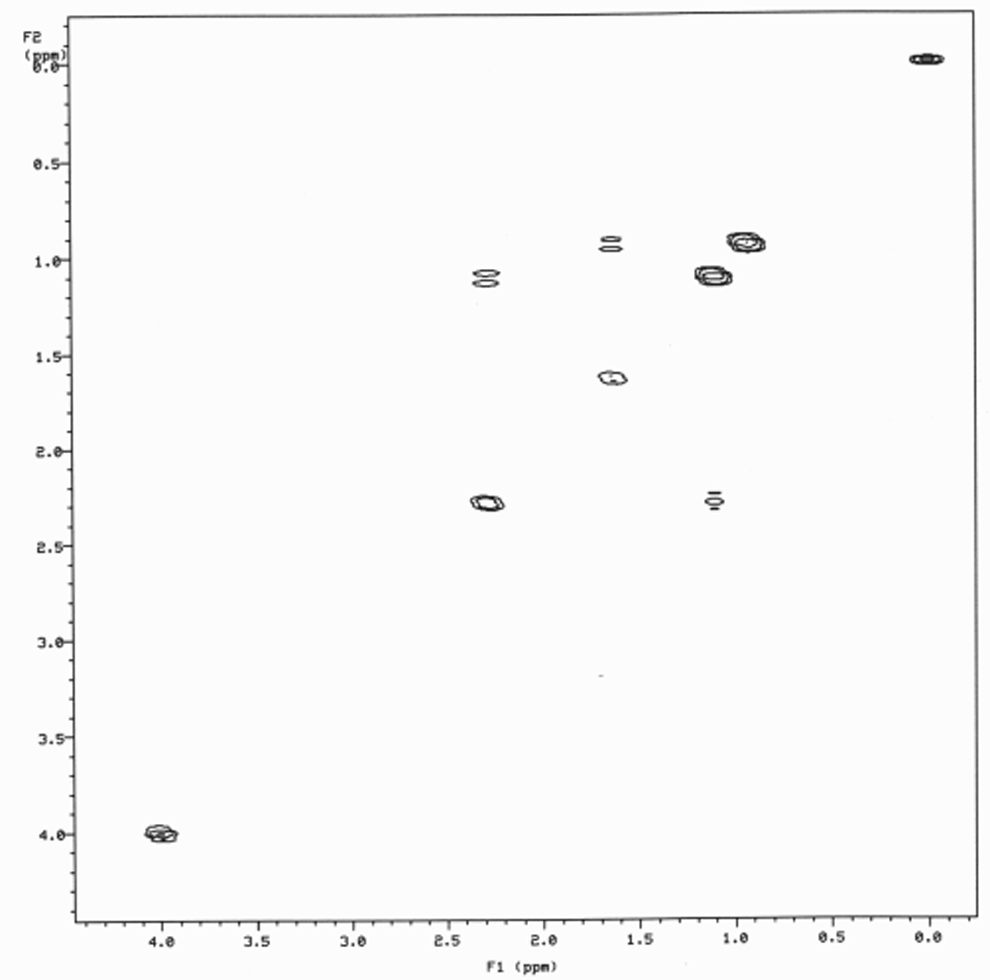
APRICOXIB
A COX-2 inhibitor.
MF; C19H20N2O3S
Mol wt: 356.439
CAS: 197904-84-0
CS-701; TG01, R-109339, TG-01 ,TP-1001
TP-2001, Capoxigem, Kymena, UNII-5X5HB3VZ3Z,
Benzenesulfonamide, 4-[2-(4-ethoxyphenyl)-4-methyl-1H-pyrrol-1-yl]-;
4-[2-(4-Ethoxyphenyl)-4-methyl-1H-pyrrol-1-yl]benzenesulfonamide
4-[2-(4-ethoxyphenyl)-4-methyl-1H-pyrrol-1-yl]benzenesulfonamide .
PHASE 2 http://clinicaltrials.gov/search/intervention=Apricoxib
Daiichi Sankyo (innovator)Daiichi Sankyo Co Ltd,
Current developer: Tragara Pharmaceuticals, Inc.

Apricoxib is an orally bioavailable nonsteroidal anti-inflammatory agent (NSAID) with potential antiangiogenic and antineoplastic activities. Apricoxib binds to and inhibits the enzyme cyclooxygenase-2 (COX-2), thereby inhibiting the conversion of arachidonic acid into prostaglandins. Apricoxib-mediated inhibition of COX-2 may induce tumor cell apoptosis and inhibit tumor cell proliferation and tumor angiogenesis. COX-related metabolic pathways may represent crucial regulators of cellular proliferation and angiogenesis.

R-109339 is a cyclooxygenase-2 (COX-2) inhibitor currently in phase II clinical development at Tragara Pharmaceuticals for the oral treatment of non-small cell lung cancer (NSCLC) and for the treatment of inflammation. Additional phase II clinical trials are ongoing in combination with gemcitabine and erlotinib for the treatment of pancreas cancer. The company had been evaluating R-109339 for the treatment of colorectal cancer, but development for this indication was discontinued for undisclosed reasons. Daiichi Sankyo and Tragara Pharmaceuticals had been conducting phase II clinical trials with the drug candidate for the oral treatment of arthritis and for the treatment of breast cancer, respectively; however, no recent development for this indication has been reported.
COX catalyzes the formation of prostaglandins and thromboxane from arachidonic acid, which is derived from the cellular phospholipid bilayer by phospholipase A2. In addition to several other functions, prostaglandins act as messenger molecules in the process of inflammation. The compound is also designed to act against a well-defined cancer pathway that affects several routes of cancer pathogenesis. In preclinical cancer models, R-109339 demonstrated superiority to compounds with similar mechanisms of action and potential for use in combination with cisplatin. Furthermore, the compound demonstrated the ability to inhibit the cachexia and weight loss seen in mouse tumor models.

Apricoxib, (CS-706, 1) 2-(4-ethoxyphenyl)-4-methyl-1-(4-sulfamoylphenyl)-pyrrole, a small-molecule, orally active, selective COX-2 inhibitor was discovered by investigators at Daiichi Sankyo in 1996. Clinical studies demonstrated potent analgesic activity and preclinical studies demonstrated good pharmacokinetics, pharmacodynamics and gastrointestinal tolerability. As an anticancer agent, preclinical studies demonstrated efficacy in biliary tract cancer models and colorectal carcinoma, and Recamp et al.
The original synthetic route is outlined below. Though the initial two steps were accomplished with decent yields, the final step of pyrrolidine formation followed by dehydration and dehydrocyanation produced only 3% of 1 as a brown powder. The yield in the last step of the synthesis of the 2-(4-methoxyphenyl) analog, 2-(4-methoxyphenyl)-4-methyl-1-(4-sulfamoylphenyl)-pyrrole, was 6%, indicating that this synthesis route is problematic.
14 Kimura T, Noguchi Y, Nakao A, Suzuki K, Ushiyama S, Kawara A, Miyamoto M. 799823. EP. 1997:A1.

……………………….
Synthesis
Published online Aug 19, 2011. doi: 10.1016/j.bmcl.2011.08.050
SEE AT
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3310163/
An efficient synthesis of apricoxib (CS-706), a selective cyclooxygenase inhibitor, was developed using copper catalysed homoallylic ketone formation from methyl 4-ethoxybenzoate followed by ozonolysis to an aldehyde, and condensation with sulphanilamide. This method provided multi-gram access of aprocoxib in good yield. Apricoxib exhibited potency equal to celecoxib at inhibition of prostaglandin E2 synthesis in two inflammatory breast cancer cell lines.
We envisioned that 7 could be prepared by ozonolysis of homoallylic ketone (8) (Route B). A recent development in the synthesis of homoallylic ketones by Dorr et al. via copper-catalyzed cascade addition of alkenylmagnesium bromide to an ester a24 was examined. Treatment of commercially available methyl 4-ethoxybenzoate with 1-propenylmagnesium bromide (4.0 equiv) in presence of CuCN (0.6 equiv) resulted in 95% yield of desired ketone8 after silica gel chromatography, along with a minor amount of unreacted ester).b25
Efficient synthesis of apricoxib (1):
The product was a mixture of cis/trans R/S stereoisomers, as detected in the 1H NMR spectrum, and was used directly in the next step without separation. Ozone was bubbled through a solution of 8 in MeOH/CH2Cl2 at −78°C, until all starting materials were consumed. The ozonide was then reduced to aldehyde 7 by treatment with Me2S overnight. Removal of volatiles and subsequent addition and evaporation of toluene gave the crude 1,4-dicarbonyl compound 7 which was sufficiently pure for the following condensation step. The 1H NMR signal at 9.78 ppm of the crude product confirmed the formation of the aldehyde. No attempt was made to characterize the enantiomeric ratio of 7 since the dehydration/aromatization reaction of the next step removes the chirality of the product. Treatment of 7 with sulfanilamide in 40% acetic acid-acetonitrile at 70°C for three hours resulted in a brown product. Purification by silica gel flash chromatography yielded 71% of pure 1 as a white solid.c26
a24. Dorr AA, Lubell WD. Can J Chem. 2007;85:1006.
b25. Synthesis of 1-(4-ethoxy-phenyl)-3-methyl-hex-4-en-1-one (8): To a stirred suspension of CuCN (1.8 g, 20.0 mmol) in 50 mL of dry THF at −78°C under argon, a solution of 1-propenylmagnesium bromide (133.2 mmol, 265 mL of 0.5 M solution in THF) was added dropwise. The slurry was stirred for an additional 30 min and then a solution of methyl 4-ethoxybenzoate (6.0 g, 33.3 mmol) in 60 mL of dry THF was added slowly. The stirred reaction mixture was allowed to warm to room temperature overnight. The reaction was quenched with ice cold saturated aqueous NaH2PO4 (100mL) and the mixture was extracted with ether (4 × 100 mL). The combined ether extracts were washed with brine (2 × 100mL), dried (MgSO4), filtered, and evaporated to dryness. The crude homoallylic ketone was purified by silica gel flash chromatography using a gradient of ethyl acetate in hexane as the eluent to give 8 (7.4 g, 95%) as a colorless oil. 1H NMR (CDCl3, 300.0 MHz) δ 1.04–1.07 (m, 3H), 1.44 (t, J = 6.9 Hz, 3H), 1.6–1.64 (m, 3H), 2.8–2.96 (m, 2.5H), 3.2 (m, 0.5H), 4.1 (q, J = 6.9 Hz, 2H), 5.25 (m, 0.5 H), 5.34–5.46 (m, 1.5H), 6.92 (d, J = 9.0 Hz, 2H), 7.92 (d, J = 9.0 Hz, 2H). 13C NMR (CDCl3, 75.0 MHz) δ 12.9, 14.6, 17.9, 20.4, 21.0, 28.4, 33.0, 45.4, 45.5, 63.7, 114.1, 123.1, 123.4, 130.2, 130.3, 135.5, 136.0, 141.9, 162.7, 198.1. M+H Calcd: 233.1542; Found, 233.2482.
c26. Synthesis of Apricoxib (1): Homoallylic ketone (8) (5.0 g, 21.53 mmol) in 180 mL of CH2Cl2/MeOH (1:5) was treated with ozone bubbles at −78°C until a blue coloration persisted. The solution was purged with argon, 8.0 mL of dimethylsulphide (21.5 mmol) was added, and the reaction mixture then warmed slowly to rt overnight. The solvent was evaporated under vacuum to give 7 which was then diluted with 100 mL of 40 % acetic acid in acetonitrile, (v/v) and sulphanilamide (4.0 g, 23.2 mmol) was added. The mixture was refluxed until complete consumption of 1,4-dicarbonyl compound was detected by TLC (ca 3 h). After cooling to room temperature, the product was concentrated under vacuum and diluted with 250 mL of ethyl acetate. The organic layer then washed with saturated Na2CO3 solution (3 × 50 mL) followed by brine (1 × 50 mL), dried (MgSO4), and evaporated to dryness. The crude brown material was purified by silica gel flash chromatography using a gradient of EtOAc in hexane to give apricoxib as white solid (5.5 g, 15.43 mmol, 71%).
m.p. 161–163°C (lit. 135–139°C14).
1H NMR (CDCl3, 300.0 MHz) δ 1.32 (t, J = 6.9 Hz, 3H), 2.1 (s, 3H), 3.92 (q, J = 6.9 Hz, 2H), 4.95 (s, 2H), 6.14 (m, 1H), 6.63 (m, 1H), 6.69 (d, J = 6.6 Hz, 2H), 6.94 (d, J = 6.6 Hz, 2H), 7.13 (d, J = 6.6 Hz, 2H), 7.74 (d, J= 6.6 Hz, 2H).
13C NMR (CDCl3, 75.0 MHz) δ 11.7, 14.8, 63.4, 82.4, 113.2, 114.4, 121.0, 121.1, 124.9, 125.2, 127.4, 129.7, 133.6, 138.7, 144.2, 158.0
M+H Calcd: 357.1273; Found, 357.1252.
01
OR

……………
SYNTHESIS

synthesis
In one strategy, bromination of 4-ethoxyacetophenone (I) with Br2 yields 2-bromo-1-(4-ethoxyphenyl)ethanone (II) along with the byproduct 2-bromo-1-(3-bromo-4-ethoxyphenyl)ethanone, which are separated using HPLC. Alkylation of propionaldehyde N,Ndiisobutylenamine (III) with bromo ketone (II) and subsequent ketalization with neopentyl glycol (IV) using p-TsOH·H2O and, optionally, H2SO4 in MeCN gives monoprotected ketoaldehyde (V) (1). Finally, cyclization of ketoaldehyde derivative (V) with 4-aminobenzenesulfonamide (VI) in the presence of AcOH in PrOH/H2O at 90-100 °C furnishes apricoxib
Intermediate (V) can also be prepared by reaction of 1-(4- ethoxyphenyl)-2-buten-1-one (VII) with CH3NO2 in the presence of DBU in THF to produce nitro ketone (VIII). Subsequent treatment of nitroderivative (VIII) with neopentyl glycol (IV) and NaOMe and MeOH gives acetal (V) (2).In an alternativestrategy, condensation of 4-ethoxyacetaldehyde (IX) with 4-sulfamoylaniline (VI) in refluxing EtOH furnishesN-(4-ethoxybenzylidene)-
4-sulfamoylaniline (X), which then condenses with trimethylsilyl cyanide (XI) in the presence of ZnCl2 in THF yielding α- amino nitrile (XII). Cyclization of this compound with methacrolein (XIII) using LiHMDS in THF affords apricoxib
reference for above
- Drugs of the Future 2011, 36(7): 503-509
- Kojima, S., Ooyama, J. (Daiichi Sankyo Co., Ltd.). Process for production of brominated acetophenone. WO 2008020617.
- Fujimoto, K., Takebayashi, T., Noguchi, Y., Saitou, T. (Daiichi Sankyo Co., Ltd.). Production of 4-methyl-1,2-diarylpyrrole and intermediate for synthesizing the same. JP 2000080078
- Kimura, T., Noguchi, Y., Nakao, A., Suzuki, K., Ushiyama, S., Kawara, A., Miyamoto, M. (Daiichi Sankyo Co., Ltd.). 1,2-Diphenylpyrrole derivatives,their preparation and their therapeutic uses. CA 2201812, EP 0799823, JP 1997823971, US 5908858.
1. Bierbach, Ulrich. Platinum acridine anti-cancer compounds and methods thereof. PCT Int. Appl. (2010), 54pp. CODEN: PIXXD2 WO 2010048499 A1 20100429 CAN 152:517954 AN 2010:529827
2. Zaknoen, Sara L.; Lawhon, Tracy. Methods and compositions for the treatment of cancer, tumors, and tumor-related disorders. PCT Int. Appl. (2009), 119 pp. CODEN: PIXXD2 WO 2009070546 A1 20090604 CAN 151:24882 AN 2009:676598
3. Zaknoen, Sara L.; Lawhon, Tracy. Cancer treatment using a 1,2-diphenylpyrrole derivative cyclooxygenase 2 (COX-2) inhibitor and antimetabolite combinations. PCT Int. Appl. (2009), 107pp. CODEN: PIXXD2 WO 2009070547 A1 20090604 CAN 151:24877 AN 2009:672256
4. Estok, Thomas M.; Zaknoen, Sara L.; Mansfield, Robert K.; Lawhon, Tracy. Therapies for treating cancer using combinations of COX-2 inhibitors and anti-HER2(ErbB2) antibodies or combinations of COX-2 inhibitors and HER2(ErbB2) receptor tyrosine kinase inhibitors. PCT Int. Appl. (2009), 121pp. CODEN: PIXXD2 WO 2009042618 A1 20090402 CAN 150:390188 AN 2009:386123
5. Estok, Thomas M.; Zaknoen, Sara L.; Mansfield, Robert K.; Lawhon, Tracy. Therapies for treating cancer using combinations of COX-2 inhibitors and aromatase inhibitors or combinations of COX-2 inhibitors and estrogen receptor antagonists. PCT Int. Appl. (2009), 88pp. CODEN: PIXXD2 WO 2009042612 A1 20090402 CAN 150:390184 AN 2009:385226
6. Estok, Thomas M.; Zaknoen, Sara L.; Mansfield, Robert K.; Lawhon, Tracy. Combination therapy for the treatment of cancer using COX-2 inhibitors and dual inhibitors of EGFR (ErbB1) and HER-2 (ErbB2). PCT Int. Appl. (2009), 87pp. CODEN: PIXXD2 WO 2009042613 A1 20090402 CAN 150:390183 AN 2009:385196
7. Lawhon, Tracy; Zaknoen, Sara; Estok, Thomas; Green, Mark. Patient selection and therapeutic methods using markers of prostaglandin metabolism. PCT Int. Appl. (2009), 121pp. CODEN: PIXXD2 WO 2009009776 A2 20090115 CAN 150:136599 AN 2009:55595
8. Estok, Thomas M.; Zaknoen, Sara L.; Mansfield, Robert K.; Lawhon, Tracy. Methods and compositions for the treatment of cancer, tumors, and tumor-related disorders using combination of a 1,2-diphenylpyrrole derivative and an EGFR inhibitor. PCT Int. Appl. (2009), 104 pp. CODEN: PIXXD2 WO 2009009778 A1 20090115 CAN 150:136628 AN 2009:54177
9. Rohatagi, Shashank; Kastrissios, Helen; Sasahara, Kunihiro; Truitt, Kenneth; Moberly, James B.; Wada, Russell; Salazar, Daniel E. Pain relief model for a COX-2 inhibitor in patients with postoperative dental pain. British Journal of Clinical Pharmacology (2008), 66(1), 60-70.
10. Senzaki, Michiyo; Ishida, Saori; Yada, Ayumi; Hanai, Masaharu; Fujiwara, Kosaku; Inoue, Shin-Ichi; Kimura, Tomio; Kurakata, Shinichi. CS-706, a novel cyclooxygenase-2 selective inhibitor, prolonged the survival of tumor-bearing mice when treated alone or in combination with anti-tumor chemotherapeutic agents. International Journal of Cancer (2008), 122(6), 1384-1390. CODEN: IJCNAW ISSN:0020-7136. CAN 148:440459 AN 2008:228248
11. Kojima, Shunshi; Ooyama, Jo. Process for production of brominated acetophenone as drug intermediate. PCT Int. Appl. (2008), 37pp. CODEN: PIXXD2 WO 2008020617 A1 20080221 CAN 148:262335 AN 2008:220659
12. Ushiyama, Shigeru; Yamada, Tomoko; Murakami, Yukiko; Kumakura, Sei-ichiro; Inoue, Shin-ichi; Suzuki, Keisuke; Nakao, Akira; Kawara, Akihiro; Kimura, Tomio. Preclinical pharmacology profile of CS-706, a novel cyclooxygenase-2 selective inhibitor, with potent antinociceptive and anti-inflammatory effects. European Journal of Pharmacology (2008), 578(1), 76-86.
13. Oitate, Masataka; Hirota, Takashi; Murai, Takahiro; Miura, Shin-ichi; Ikeda, Toshihiko. Covalent binding of rofecoxib, but not other cyclooxygenase-2 inhibitors, to allysine aldehyde in elastin of human aorta. Drug Metabolism and Disposition (2007), 35(10), 1846-1852. CODEN: DMDSAI ISSN:0090-9556. CAN 147:439860 AN 2007:1124386
14. Kiguchi, Kaoru; Ruffino, Lynnsie; Kawamoto, Toru; Franco, Eugenia; Kurakata, Shin-ichi; Fujiwara, Kosaku; Hanai, Masaharu; Rumi, Mohammad; DiGiovanni, John. Therapeutic effect of CS-706, a specific cyclooxygenase-2 inhibitor, on gallbladder carcinoma in BK5.ErbB-2 mice. Molecular Cancer Therapeutics (2007), 6(6), 1709-1717.
15. Moberly, James B.; Xu, Jianbo; Desjardins, Paul J.; Daniels, Stephen E.; Bandy, Donald P.; Lawson, Janet E.; Link, Allison J.; Truitt, Kenneth E. A randomized, double-blind, celecoxib- and placebo-controlled study of the effectiveness of CS-706 in acute postoperative dental pain. Clinical Therapeutics (2007), 29(3), 399-412.
16. Rohatagi, S.; Kastrissios, H.; Gao, Y.; Zhang, N.; Xu, J.; Moberly, J.; Wada, R.; Yoshihara, K.; Takahashi, M.; Truitt, K.; Salazar, D. Predictive population pharmacokinetic/pharmacodynamic model for a novel COX-2 inhibitor. Journal of Clinical Pharmacology (2007), 47(3), 358-370.
17. Moberly, James B.; Harris, Stuart I.; Riff, Dennis S.; Dale, James Craig; Breese, Tara; McLaughlin, Patrick; Lawson, Janet; Wan, Yaping; Xu, Jianbo; Truitt, Kenneth E. A Randomized, Double-Blind, One-Week Study Comparing Effects of a Novel COX-2 Inhibitor and Naproxen on the Gastric Mucosa. Digestive Diseases and Sciences (2007), 52(2), 442-450.
18. Oitate, Masataka; Hirota, Takashi; Koyama, Kumiko; Inoue, Shin-ichi; Kawai, Kenji; Ikeda, Toshihiko. Covalent binding of radioactivity from [14C] rofecoxib, but not [14C] celecoxib or [14C] CS-706, to the arterial elastin of rats. Drug Metabolism and Disposition (2006), 34(8), 1417-1422.
19. Kastrissios, H.; Rohatagi, S.; Moberly, J.; Truitt, K.; Gao, Y.; Wada, R.; Takahashi, M.; Kawabata, K.; Salazar, D. Development of a predictive pharmacokinetics model for a novel cyclooxygenase-2 inhibitor. Journal of Clinical Pharmacology (2006), 46(5), 537-548. CODEN: JCPCBR ISSN:0091-2700. CAN 145:327959 AN 2006:479516
20. Denis, Louis J.; Compton, Linda D. Method using camptothecin compounds, pyrimidine derivatives, and antitumor agents for treating abnormal cell growth. U.S. Pat. Appl. Publ. (2005), 32 pp. CODEN: USXXCO US 2005272755 A1 20051208 CAN 144:17160 AN 2005:1294044
21. Wajszczuk, Charles Paul; Gans, Hendrik J. Dekoning; Di Salle, Enrico; Piscitelli, Gabriella; Massimini, Giorgio; Purandare, Dinesh. Methods using exemestane, alone or with other therapeutic agents, for treating estrogen-dependent disorders. U.S. Pat. Appl. Publ. (2004), 21 pp., Cont.-in-part of WO 2002 72,106. CODEN: USXXCO US 2004082557 A1 20040429 CAN 140:368700 AN 2004:353144
22. Di Salle, Enrico; Piscitelli, Gabriella; Massimini, Giorgio; Purandare, Dinesh; Dekoning, Gans Hendrik. Combined method for treating hormone-dependent disorders with aromatase inactivator exemestane and other therapeutic agents. PCT Int. Appl. (2002), 49 pp. CODEN: PIXXD2 WO 2002072106 A2 20020919 CAN 137:226651 AN 2002:716096
23. McKearn, John P.; Gordon, Gary; Cunningham, James J.; Gately, Stephen T.; Koki, Alane T.; Masferrer, Jaime L. Method of using a cyclooxygenase-2 inhibitor and an integrin antagonist as a combination therapy in the treatment of neoplasia. PCT Int. Appl. (2000), 348 pp. CODEN: PIXXD2 WO 2000038786 A2 20000706 CAN 133:84244 AN 2000:456950
24. McKearn, John P.; Gordon, Gary; Cunningham, James J.; Gately, Stephen T.; Koki, Alane T.; Masferrer, Jaime L. Method of using a cyclooxygenase-2 inhibitor and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia. PCT Int. Appl. (2000), 236 pp. CODEN: PIXXD2 WO 2000038730 A2 20000706 CAN 133:84243 AN 2000:456927
25. McKearn, John P.; Masferrer, Jaime L.; Milas, Luka. Combination therapy of radiation and a cyclooxygenase 2 (COX-2) inhibitor for the treatment of neoplasia. PCT Int. Appl. (2000), 96 pp. CODEN: PIXXD2 WO 2000038716 A1 20000706 CAN 133:84241 AN 2000:456913
26. McKearn, John P.; Gordon, Gary; Cunningham, James J.; Gately, Stephen T.; Koki, Alane T.; Masferrer, Jaime L. Method of using a cyclooxygenase-2 inhibitor and a matrix metalloproteinase inhibitor as a combination therapy in the treatment of neoplasia. PCT Int. Appl. (2000), 437 pp. CODEN: PIXXD2 WO 2000037107 A2 20000629 CAN 133:68922 AN 2000:441655
27. Noguchi, Yasuo; Saito, Toshinori; Fujimoto, Katsuhiko; Takebayashi, Toyonori. Preparation of 4-methyl-1,2-diarylpyrroles and and their intermediates. Jpn. Kokai Tokkyo Koho (2000), 14 pp. CODEN: JKXXAF JP 2000080078 A 20000321 CAN 132:207760 AN 2000:181022
28. Kurakata, Shinichi; Hanai, Masaharu; Kanai, Saori; Kimura, Tomio. Use of cyclooxygenase-2 inhibitors for the treatment and prevention of tumors, tumor-related disorders and cachexia. Eur. Pat. Appl. (1999), 49 pp. CODEN: EPXXDW EP 927555 A1 19990707 CAN 131:82985 AN 1999:440003
29. Kimura, Fumio; Noguchi, Yasuo; Nakao, Akira; Suzuki, Keisuke; Ushiyama, Shigeru; Kawahara, Akihiro; Miyamoto, Masaaki. Diphenylpyrrole derivatives as cyclooxygenase-2 inhibitors. Jpn. Kokai Tokkyo Koho (1999), 69 pp.
30. Kimura, Tomio; Noguchi, Yasuo; Nakao, Akira; Suzuki, Keisuke; Ushiyama, Shigeru; Kawara, Akihiro; Miyamoto, Masaaki. Preparation of 1,2-diphenylpyrroles as cyclooxygenase-2 inhibitors. Eur. Pat. Appl. (1997), 140 pp. CODEN: EPXXDW EP 799823 A1 19971008 CAN 127:331392 AN 1997:678926
31. Rao P N Praveen; Grover Rajesh K Apricoxib, a COX-2 inhibitor for the potential treatment of pain and cancer. IDrugs : the investigational drugs journal (2009), 12(11), 711-22.
|
|
9-13-2002
|
Method of using COX-2 inhibitors in the treatment and prevention of ocular COX-2 mediated disorders
|
|
|
6-2-1999
|
1,2-diphenylpyrrole derivatives, their preparation and their therapeutic uses
|
|
|
7-14-2006
|
Use of MEK inhibitors in treating abnormal cell growth
|
|
|
4-7-2006
|
Therapeutic combinations comprising poly (ADP-ribose) polymerases inhibitor
|
|
|
12-9-2005
|
Method for treating abnormal cell growth
|
|
|
6-31-2005
|
Method of using a cyclooxygenase-2 inhibitor and sex steroids as a combination therapy for the treatment and prevention of dismenorrhea
|
|
|
5-4-2005
|
Methods and compositions for treatment and prevention of tumors, tumor-related disorders and cachexia
|
|
|
4-30-2004
|
Compositions of cyclooxygenase-2 selective inhibitors and NMDA receptor antagonists for the treatment or prevention of neuropathic pain
|
|
|
4-30-2004
|
Methods for treating estrogen-dependent disorders
|
|
|
4-16-2004
|
Method of using a COX-2 inhibitor and an alkylating-type antineoplastic agent as a combination therapy in the treatment of neoplasia
|
|
|
3-26-2004
|
Method of using cox-2 inhibitors in the treatment and prevention of ocular cox-2 mediated disorders
|
|
|
3-19-2004
|
Method of using a COX-2 inhibitor and an aromatase inhibitor as a combination therapy
|
|
|
8-22-2012
|
Methods and Compositions for the Treatment of Cancer, Tumors, and Tumor-Related Disorders
|
|
|
12-21-2011
|
HUMAN MONOCLONAL ANTIBODIES TO ACTIVIN RECEPTOR-LIKE KINASE-1
|
|
|
10-6-2011
|
Use of cyclooxygenase-2 inhibitors for the treatment and prevention of tumours, tumour-related disorders and cachexia
|
|
|
6-30-2010
|
Methods and compositions for the treatment and prevention of tumors, tumor-related disorders and cachexia
|
|
|
11-13-2009
|
HETEROAROMATIC DERIVATIVES USEFUL AS ANTICANCER AGENTS
|
|
|
5-27-2009
|
Human monoclonal antibodies to activin receptor-like kinase-1
|
|
|
4-31-2009
|
BICYCLIC HETEROAROMATIC DERIVATIVES USEFUL AS ANTICANCER AGENTS
|
|
|
11-7-2008
|
Pharmaceutical Compositions Comprising an Amorphous Form of a Vegf-R-Inhibitor
|
|
|
10-24-2008
|
Compositions for the Treatment of Inflammation and Pain Using a Combination of a Cox-2 Selective Inhibitor and a Ltb4 Receptor Antagonist
|
|
|
10-32-2007
|
1,2-Diphenylpyrrole derivatives, their preparation and their therapeutic uses
|

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

















 Dedicated to all moms in the world
Dedicated to all moms in the world
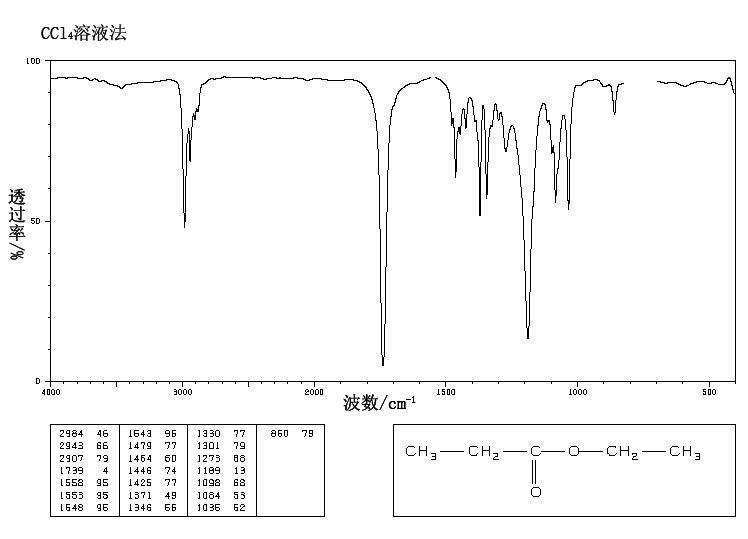



 This is the structure. See if you can assign the peaks on your own.
This is the structure. See if you can assign the peaks on your own. C has a higher chemical shift than D because it’s closer to a more electron-withdrawing functional group.
C has a higher chemical shift than D because it’s closer to a more electron-withdrawing functional group.




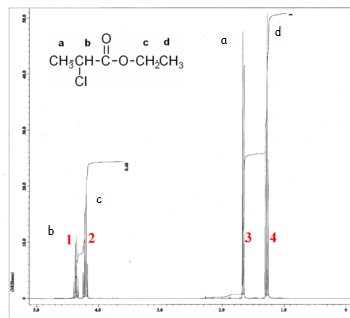






 see interpretation
see interpretation