WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Atrasentan is an experimental drug that is being studied for the treatment of various types of cancer,[1] including non-small cell lung cancer.[2] It is also being investigated as a therapy for diabetic kidney disease.
Atrasentan failed a phase 3 trial for prostate cancer in patients unresponsive to hormone therapy.[3] A second trial confirmed this finding.[4]
In April 2014, de Zeeuw et al. showed that 0.5 mg and 1.25 mg of atrasentan reduced urinary albumin by 35 and 38% respectively with modest side effects. Patients also had decreased home blood pressures (but no change in office readings) decrease total cholesterol and LDL. Patients in the 1.25 mg dose group had increased weight gain which was presumably due to increased edema and had to withdraw from the study more than the placebo or 0.5 mg dose group.[5] Reductions in proteinuria have been associated with beneficial patient outcomes in diabetic kidney disease with other interventions but is not an accepted end-point by the FDA.
The recently initiated SONAR trial[6] will determine if atrasentan reduces kidney failure in diabetic kidney disease.
Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes. For a prior filing see WO2015006219 , claiming the stable solid composition in the form of a tablet comprising atrasentan and an anti-oxidant. AbbVie (following its spin-out from Abbott), is developing atrasentan (phase III; February 2015) for treating chronic kidney disease, including diabetic nephropathy.
PAPER
European Journal of Organic Chemistry
Enantioselective Synthesis of the Pyrrolidine Core of Endothelin Antagonist ABT-627 (Atrasentan) via 1,2-Oxazines
A mixture of bromoacetyl bromide (72.3 mL) in toluene (500 mL) at 0° C. was treated with dibutylamine (280 mL) in toluene (220 mL) while keeping the solution temperature below 10° C., stirred at 0° C. for 15 minutes, treated with 2.5% aqueous phosphoric acid (500 mL) and warmed to 25° C. The organic layer was isolated, washed with water (500 mL) and concentrated to provide the product as a solution in toluene.
EXAMPLE 25-((E)-2-nitroethenyl)-1,3-benzodioxole
3,4-methylenedioxybenzaldehyde (15.55 Kg) was treated sequentially with ammonium acetate (13.4 Kg,), acetic acid (45.2 Kg) and nitromethane (18.4 Kg), warmed to 70° C., stirred for 30 minutes, warmed to 80° C., stirred for 10 hours, cooled to 10° C. and filtered. The filtrant was washed with acetic acid (2×8 Kg) and water (2×90 Kg) and dried under a nitrogen stream then in under vacuum at 50° C. for 2 days.
EXAMPLE 3ethyl 3-(4-methoxyphenyl)-3-oxopropanoate
A mixture of potassium tert-amylate (50.8 Kg) in toluene (15.2 Kg) at 5° C. was treated with 4-methoxyacetophenone (6.755 Kg) and diethyl carbonate (6.4 Kg) in toluene over 1 hour while keeping the solution temperature below 10° C., warmed to 60° C. for 8 hours, cooled to 20° C. and treated with acetic acid (8 Kg) and water (90 Kg) over 30 minutes while keeping the solution temperature below 20° C. The organic layer was isolated, washed with 5% aqueous sodium bicarbonate (41 Kg) and concentrated at 50° C. to 14.65 Kg.
EXAMPLE 4ethyl 2-(4-methoxybenzoyl)-4-nitromethyl-3-(1,3-benzodioxol-5-yl)butyrate
A mixture of EXAMPLE 3 (7.5 Kg) in THF (56 Kg) was treated with EXAMPLE 3 (8.4 Kg), cooled to 17° C., treated with sodium ethoxide (6.4 g), stirred for 30 minutes, treated with more sodium ethoxide (6.4 g), stirred at 25° C. until HPLC shows less than 1 area % ketoester remaining and concentrated to 32.2 Kg.
EXAMPLE 5ethyl cis,cis-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate
Raney nickel (20 g), from which the water had been decanted, was treated sequentially with THF (20 mL), EXAMPLE 4 (40.82 g), and acetic acid (2.75 mL). The mixture was stirred under hydrogen (60 psi) until hydrogen uptake slowed, treated with trifluoroacetic acid, stirred under hydrogen (200 psi) until HPLC shows no residual imine and less than 2% nitrone and filtered with a methanol (100 mL) wash. The filtrate, which contained 13.3 g of EXAMPLE 5, was concentrated with THF (200 mL) addition to 100 mL, neutralized with 2N aqueous NaOH (50 mL), diluted with water (200 mL), and extracted with ethyl acetate (2×100 mL). The extract was used in the next step.
EXAMPLE 6ethyl trans,trans-2-(4-methoxyphenyl)-4-(1,3 -benzodioxol-5 -yl)pyrrolidine-3-carboxylate
Example 501E (38.1 g) was concentrated with ethanol (200 mL) addition to 100 mL, treated with sodium ethoxide (3.4 g), heated to 75° C., cooled to 25° C. when HPLC showed less than 3% of EXAMPLE 1E and concentrated. The concentrate was mixed with isopropyl acetate (400 mL), washed with water (2×150 mL) and extracted with 0.25 M phosphoric acid (2×400 mL). The extract was mixed with ethyl acetate (200 mL) and neutralized to pH 7 with sodium bicarbonate (21 g), and the organic layer was isolated.
EXAMPLE 7ethyl (2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate, (S)-(+) mandelate
EXAMPLE 501F was concentrated with acetonitrile (100 mL) addition to 50 mL, treated with (S)-(+)-mandelic acid (2.06 g), stirred until a solution formed, stirred for 16 hours, cooled to 0° C., stirred for 5 hours and filtered. The filtrant was dried at 50° C. under a nitrogen stream for 1 day. The purity of the product was determined by chiral HPLC using Chiralpak AS with 95:5:0.05 hexane/ethanol/diethylamine, a flow rate of 1 mL/min. and UV detection at 227 nm. Retention times were 15.5 minutes for the (+)-enantiomer and 21.0 minutes for the (−)-enantiomer.
EXAMPLE 8(2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)pyrrolidine-3-carboxylic acid
A mixture of EXAMPLE 7 (20 g) in ethyl acetate (150 mL) and 5% aqueous sodium bicarbonate was stirred at 25° C. until the salt dissolved and gas evolution stopped. The organic layer was isolated and concentrated. The concentrate was treated with acetonitrile (200 mL), concentrated to 100 mL, cooled to 10° C., treated with diisopropylethylamine (11.8 mL) and EXAMPLE 1 (10.5 g), stirred for 12 hours and concentrated. The concentrate was treated with ethanol (200 mL), concentrated to 100 mL, treated with 40% aqueous NaOH (20 mL), stirred at 60° C. for 4 hours, cooled, poured into water (400 mL), washed with hexanes (2×50 mL then 2×20 mL), treated with ethyl acetate (400 mL) and adjusted to pH 5 with concentrated HCl (12 mL). The organic layer was isolated and concentrated.
………………….
The Michael reaction between 3,4-(methylenedioxy)-beta-nitrostyrene (I) and ethyl (4-methoxybenzoyl)acetate (II) in the presence of DBU gave adduct (III) as a mixture of isomers. Hydrogenation of this nitro ketone over Raney-Ni afforded, after spontaneous cyclization of the resulting amino ketone, the pyrroline (IV). Further reduction of the imine with NaBH3CN yielded a mixture of three pyrrolidine isomers. The desired trans-trans isomer (VI) could not be separated from the cis-trans isomer by column chromatography. However, the pure cis-cis compound (V) was isomerized to (VI) with NaOEt in refluxing EtOH. The protection of the amine as the tert-butyl carbamate with Boc2O, and saponification of the ester function provided the racemic acid (VII). Resolution of (VII) was achieved by conversion to the mixed anhydride (VIII) with pivaloyl chloride, followed by condensation with the lithium salt of (S)-4-benzyl-2-oxazolidinone (IX), and chromatographic separation of the resulting diastereomeric imides. Alternatively, racemic (VII) could be resolved by crystallization of its salt with (R)-a-methylbenzylamine. Removal of the Boc group from the appropriate isomer (X) with HCl in dioxan, followed by alkylation with N,N-dibutylbromoacetamide (XI) in the presence of i-Pr2NEt furnished the pyrrolidinylacetamide (XII). Finally, hydrolysis of the imide with lithium hydroperoxide provided the target acid.
J Med Chem1996,39,(5):1039
Cyclization of 5-(2-nitrovinyl)-1,3-benzodioxole (I) with ethyl 2-(4-methoxybenzoyl)acetate (II) by means of DBU in THF gives the 4-nitrobutyrate (III), which is reduced with H2 over Ni in ethanol to the corresponding amine, which undergoes immediate cyclization to give the pyrroline carboxylate (IV). Reduction of pyrroline (IV) with NaCNBH3 in THF affords the expected pyrrolidine as a mixture of the (trans,trans)-(V), (cis,cis)-(VI) and (cis,trans)-(VII) isomers. Using chromatography on silica gel, only the (cis,cis)-isomer (VI) is separated and completely isomerized to the (trans,trans)-isomer (V) by treatment with NaOEt in refluxing ethanol. Pure (trans,trans)-isomer (V) or the remaining mixture of (trans,trans)-(V) and (cis,trans)-(VII) is N-protected with Boc2O in dichloromethane to provide a mixture of carbamates. Then hydrolysis of the esters is performed with NaOH in ethanol/water at room temperature, and under these conditions only the (trans,trans)-isomer hydrolyzes, giving the racemic (trans,trans)-acid (VIII). Unreacted (cis,trans)-ester (VII) is easily removed by conventional methods. Condensation of the racemic acid (VIII) with the lithium salt of the chiral oxazolidinone (IX) by means of pivaloyl chloride yields the corresponding amide as a diastereomeric mixture of (X) and (XI) that are separated by chromatography. The desired isomer (XI) is deprotected with HCl in dioxane to afford the chiral pyrrolidine (XII), which is condensed with 2-bromo-N,N-dibutylacetamide (XIII) by means of diisopropylamine in acetonitrile to give the adduct (XIV). Finally, the chiral auxiliary of (XIV) is eliminated by means of LiOOH (LiOH + H2O2) in water.
EXAMPLE 95D(2R,3R,4S)-(+)-2-(4-Methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)pyrrolidine-3-carboxylic acidTo the resulting compound from Example 95C (131 mg, 0.355 mmol) was added, diisopropylethylamine (137 mg, 185 μL, 1.06 mmol), acetonitrile (2 mL), N,N-di-(n-butyl)bromoacetamide (133 mg, 0.531 mmol), and the mixture was heated at 50° C. for 1.5 hours. The reaction mixture was concentrated to a solid, dried under high vacuum, and purified by chromatography on silica gel eluting with 1:3 ethyl acetate-hexane to give pure ester as a colorless oil. 1 H NMR (CDCl3, 300MHz) δ 0.81 (t, J=7 Hz, 3H), 0.88 (t, J=7 Hz, 3H), 1.10 (t, J=7 Hz, 3H), 1.00-1.52 (m, 8H), 2.78 (d, J=14 Hz,1H), 2.89-3.10 (m, 4H), 3.23-3.61 (m, 5H), 3.71 (d, J=9 Hz, 1H), 3.80 (s, 3H), 4.04 (q, J=7 Hz, 2H), 5.94 (dd, J=1.5 Hz, 2H), 6.74 (d, J=9 Hz, 1H), 6.83-6.90 (m, 3H), 7.03 (d, J=2 Hz, 1H), 7.30 (d, J=9 Hz, 2H). MS (DCl/NH3) m/e 539 (M+H)+.To the ethyl ester dissolved in 7 mL of ethanol was added a solution of lithium hydroxide (45 mg, 1.06 mmol) in water (2.5 mL). The mixture was stirred for 1 hour at ambient temperature and then warmed slowly to 40° C. over 2.5 hours at which point all of the starting material had been consumed. The reaction mixture was concentrated to remove the ethanol, diluted with 60 mL water and extracted with ether (3×40 mL). The aqueous solution was treated with 1N aqueous hydrochloric acid until cloudy, and the pH was then adjusted to ˜4-5 with 10% aqueous citric acid. This mixture was extracted with 1:19 ethanol-methylene chloride (3×50 mL). The combined extracts were dried (Na2 SO4), filtered, concentrated and dried under high vacuum to give the title compound as a white foam (150 mg, 83%). 1 H NMR (CDCl3, 300MHz) δ 0.80 (t, J=7 Hz, 3H), 0.88 (t, J=7 Hz, 3H), 1.08 (m, 2H), 1.28 (m, 3H), 1.44 (m, 3H), 2.70-3.77 (svr br m, 12H), 3.79 (s, 3H), 5.95 (m, 2H), 6.75 (d, J=8 Hz, 1H), 6.87 (br d, J=8 Hz, 3H), 7.05 (br s,1H),7.33 (v br s, 2H). MS (DCl/NH3) m/e 511 (M+H)+. α!22 =+74.42°. Anal calcd for C29 H38 N2 O6.0.5 H2 O: C ,67.03; H, 7.56; N, 5.39. Found: C, 67.03; H, 7.59; N, 5.33.
SYN
EP 0885215; WO 9730045
Condensation of 1,3-benzodioxole-5-carbaldehyde (XV) with nitromethane by means of ammonium acetate in HOAc gives the nitrostyrene (I), which is condensed with ethyl 2-(4-methoxybenzoyl)acetate (II) [obtained by reaction of acetophenone (XVI), diethyl carbonate and potassium tert-amyloxide] by means of NaOEt in THF to yield the 4-nitrobutyrate (III). Reductive cyclization of (III) with H2 over Raney-Ni in THF affords the (cis, cis)-pyrrolidine (VI), which is isomerized to the (trans,trans)-isomer (V) by means of NaOEt in refluxing ethanol. This racemic ester (V) is submitted to optical resolution with (S)-(+)-mandelic acid to provide the pure chiral ester (XVII). This compound is condensed with 2-bromo-N,N-dibutylacetamide (XIII) [obtained by reaction of 2-bromoacetyl bromide (XVIII) with dibutylamine (XIX) in toluene] by means of DIEA in acetonitrile to give the ethyl ester (XX), which is finally hydrolyzed with NaOH in hot ethanol.
SYN
Condensation of ketoester (I) with nitrovinyl benzodioxole (II) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene gave adduct (III). Hydrogenation of the nitro group of (III) over Raney Nickel with concomitant cyclization yielded dihydropyrrole (IV). Further reduction of (IV) with sodium cyanoborohydride provided a mixture of diastereomeric pyrrolidines. Chromatographic separation removed the cis,cis isomer, affording a mixture of trans,trans and cis,trans products (V). N-Alkylation of the pyrrolidine (V) with N,N-dibutyl bromoacetamide (VI) furnished (VIIa-b). Finally, selective hydrolysis of the ester group from the trans,trans isomer produced a mixture of cis,trans ester (VIII) and the target trans,trans acid, which were readily separated by fractional extraction.
SYN
SYN
J Med Chem 1996,39(5),1039
The Michael reaction between 3,4-(methylenedioxy)-beta-nitrostyrene (I) and ethyl (4-methoxybenzoyl)acetate (II) in the presence of DBU gave adduct (III) as a mixture of isomers. Hydrogenation of this nitro ketone over Raney-Ni afforded, after spontaneous cyclization of the resulting amino ketone, the pyrroline (IV). Further reduction of the imine with NaBH3CN yielded a mixture of three pyrrolidine isomers. The desired trans-trans isomer (VI) could not be separated from the cis-trans isomer by column chromatography. However, the pure cis-cis compound (V) was isomerized to (VI) with NaOEt in refluxing EtOH. The protection of the amine as the tert-butyl carbamate with Boc2O, and saponification of the ester function provided the racemic acid (VII). Resolution of (VII) was achieved by conversion to the mixed anhydride (VIII) with pivaloyl chloride, followed by condensation with the lithium salt of (S)-4-benzyl-2-oxazolidinone (IX), and chromatographic separation of the resulting diastereomeric imides. Alternatively, racemic (VII) could be resolved by crystallization of its salt with (R)-a-methylbenzylamine. Removal of the Boc group from the appropriate isomer (X) with HCl in dioxan, followed by alkylation with N,N-dibutylbromoacetamide (XI) in the presence of i-Pr2NEt furnished the pyrrolidinylacetamide (XII). Finally, hydrolysis of the imide with lithium hydroperoxide provided the target acid.
SYN
Reaction of 2-(1,3-dioxol-5-yl)acetic acid (XXI) with pivaloyl chloride and TEA gives the corresponding anhydride (XXII), which is condensed with the chiral oxazolidinone (XXIII) by means of n-BuLi in THF to yield the amide (XXIV). Condensation of (XXIV) with 2-bromoacetic acid tert-butyl ester (XXV) by means of NaHMDS in THF affords the adduct (XXVI). Elimination of the chiral auxiliary of (XXVI) by means of LiOOH in THF/water provides the chiral succinic acid hemiester (XXVII) (93% ee), which is selectively reduced with BH3璗HF complex to give the 4-hydroxysuccinate (XXVIII). Reaction of succinate (XXVIII) with 4-chlorophenylsulfonyl chloride, TEA and DMAP in dichloromethane yields the sulfonate (XXIX), which is condensed with 4-methoxybenzaldoxime (XXX) by means of Cs2CO3 in hot acetonitrile to afford the oxime ether (XXXI). Transesterification of the tert-butyl ester of (XXXI) with trimethyl orthoformate and p-toluenesulfonic acid in hot methanol provides the methyl ester (XXXII), which is cyclized by means of trimethylsilyl triflate and tributylamine in dichloroethane to afford a 9:1 diastereomeric mixture of perhydro-1,2-oxazines (XXXIII) and (XXXIV) which is easily separated. The reductive N-O-bond cleavage of the major oxazine diastereomer (XXXIII) by means of Zn/HOAc or H2 over Pd/C gives the trisubstituted 4-aminobutanol (XXXV), which is cyclized by means of CBr4, PPh3 and TEA to yield chiral pyrrolidine (XXXVI) (4). Finally, pyrrolidine (XXXVI) is alkylated with N,N-dibutyl-2-bromoacetamide (XIII) followed by ester hydrolysis as before.
References
1
“Atrasentan”. NCI Dictionary of Cancer Terms. National Institute of Cancer.
2
Chiappori, Alberto A.; Haura, Eric; Rodriguez, Francisco A.; Boulware, David; Kapoor, Rachna; Neuger, Anthony M.; Lush, Richard; Padilla, Barbara; Burton, Michelle; Williams, Charles; Simon, George; Antonia, Scott; Sullivan, Daniel M.; Bepler, Gerold (March 2008). “Phase I/II Study of Atrasentan, an Endothelin A Receptor Antagonist, in Combination with Paclitaxel and Carboplatin as First-Line Therapy in Advanced Non–Small Cell Lung Cancer”. Clinical Cancer Research14 (5): 1464–9. doi:10.1158/1078-0432.CCR-07-1508. PMID18316570.
Quinn, David I; Tangen, Catherine M; Hussain, Maha; Lara, Primo N; Goldkorn, Amir; Moinpour, Carol M; Garzotto, Mark G; Mack, Philip C; Carducci, Michael A; Monk, J Paul; Twardowski, Przemyslaw W; Van Veldhuizen, Peter J; Agarwal, Neeraj; Higano, Celestia S; Vogelzang, Nicholas J; Thompson, Ian M (August 2013). “Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (SWOG S0421): a randomised phase 3 trial”. The Lancet Oncology14 (9): 893–900. doi:10.1016/S1470-2045(13)70294-8. PMID23871417.
5
de Zeeuw, Dick; Coll, Blai; Andress, Dennis; Brennan, John J.; Tang, Hui; Houser, Mark; Correa-Rotter, Ricardo; Kohan, Donald; Lambers Heerspink, Hiddo J.; Makino, Hirofumi; Perkovic, Vlado; Pritchett, Yili; Remuzzi, Giuseppe; Tobe, Sheldon W.; Toto, Robert; Viberti, Giancarlo; Parving, Hans-Henrik (May 2014). “The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy”. Journal of the American Society of Nephrology25 (5): 1083–93. doi:10.1681/ASN.2013080830. PMID24722445.
Granted in February 2015, this patent claims novel crystalline anhydrous S-mandelate salt of atrasentan. Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes.
Szczepankiewicz BG, Bal RB, von Geldern TW, Wu-Wong JR, Chiou WJ, Dixon DB, Opgenorth TJ, Hoffman DJ, Borre AJ, Marsh KC, Nguyen BN: The effects of diminishing albumin binding to some Endothelin receptor antagonists. Life Sci. 1998;63(21):1905-12. doi: 10.1016/s0024-3205(98)00466-4. [Article]
Rajasekaran A, Julian BA, Rizk DV: IgA Nephropathy: An Interesting Autoimmune Kidney Disease. Am J Med Sci. 2021 Feb;361(2):176-194. doi: 10.1016/j.amjms.2020.10.003. Epub 2020 Oct 8. [Article]
FDA Approved Drug Products: Vanrafia (atrasentan) tablets for oral use (April 2025) [Link]
Novartis Media Release: Novartis receives FDA accelerated approval for Vanrafia® (atrasentan), the first and only selective endothelin A receptor antagonist for proteinuria reduction in primary IgA nephropathy (IgAN) [Link]
StatPearls [Internet]: IgA Nephropathy (Berger Disease) [Link]
ResearchGate: Total Synthesis of Atrasentan (Craig S. Harris, Reims Symposium, October 2002) [Link]
//////////ATRASENTAN, FDA 2025, APPROVALS 2025, Vanrafia, A 147627, (+)-A-127722, ABT 627, UNII-V6D7VK2215
Bremelanotide is a compound that is currently under investigation for its potential uses in managing reperfusion injury, female sexual dysfunction or hemorrhagic shock. The chemical may also see success in managing modulate inflammation or limiting the effects of ischemia.
Palatin, in collaboration with European licensee Gedeon Richter, is developing an sc formulation of the synthetic peptide bremelanotide (PT-141; BMT), a melanocortin MCR-4 agonist and a synthetically modified analog of PT-14, also analogous to alpha-melanocyte-stimulating hormone (alpha-MSH), for the potential treatment of female sexual dysfunction (FSD) including hypoactive sexual desire disorder (HSDD)
The Bremelanotide or PT-141 is a mean that explains the revolution caused by the medical world in a silent but attractive manner in the human health related study. Bremelanotide is the latest arrival from the company called Palatin Technologies which forms the basic treatment for the hemorrhagic shock and reperfusion injury.( In short about the company, the Palatin Technologies is the owner of this research and is located in New Jersey. Hence this medicine is a Jersey based Product. And regarding the product under research, is waiting for the approval from the Food and Drug Association. Once this is done, the company has targeted to reach those customers, whom the Viagra has approached. This has the effect of helping the male patients suffering with an erectile dysfunction syndrome. Also if it gets the approval as a treatment measure for the female sexual dysfunction, then this medicine is expected to bring a relief to the post-menopausal and also supports or provides their sexual happiness and also they are checking regarding thehyposexual desire disorder. This is expected to be a blockbuster, if released. So this medicine is waiting for a confirmation as well as an approval.
In February 2015, a randomized, double-blind, placebo-controlled, open-label extension, phase III trial (NCT02338960; BMT-302, Reconnect Study) was initiated in the US in premenopausal women (expected n = 550) with hypoactive sexual desire disorder to evaluate the efficacy and safety of bremelanotide. At that time, the trial was expected to complete in July 2017
Study – Potential Use Erectile Dysfunction
One study has explored the potential use of bremelanotide as a replacement for natural peptide melanocyte stimulating hormones for the sake of treating erectile dysfunction.
The goal of this study was to determine if the effects of bremelanotide stimulating sexual desire that was shown in male rats could be replicated in the brains of female rats. To do this, hormone primed female rats in a control group and a test group that were treated with bremelanotide and known to have consummatory sexual disorders was introduced to a group of male rats and the reactions were measured.
Heart racing, hops and darts, pacing and customary sexual behaviors were assessed while the brain was stimulated. The stimulation of specific molecular markers within the brain was examined to determine arousal in the female subjects.
Results indicated that the females saw an increase in sexual behavior when bremelanotide was applied to the limbic and hypothalamic regions of their brains. It is suggested that this was because the chemical that stimulated the mPOA terminals, leading to activated dopamine in the brain.
Additional study is necessary to determine the extent of the effects bremelanotide has on the brain and natural stimulating chemicals.
Bremelanotide and Ongoing Research
This is an advanced research involved even now. This functions by activating the Melanocortin, which is a group of peptide hormones which includes the adrenocorticotropic hormone and also the different forms of the melanocyte stimulating hormones. These melanocortins are produced or prepared from the proopiomelanocortin in the pituitary glands. The melanocortin releases or exert their effects by making a bind with the melanocortin and thereby activating it).The Bremelanotide functions by activating the melanocortin receptors and thereby makes a modulation in the inflammation. This is actually produced for making use in treating the sexual dysfunction. Due to certain reasons; the process of researching was kept under hold in recently, since it created some adverse side effects of increased blood pressure. In the chemistry of the preparation of the bremelanotide, the Peptide Melanaton II forms the basic compound. This compound is tested using a sunless tanning agent.
The actual information about the peptide melanaton has the effect of making sexual arousal and speed as well as sudden erections and some other side effects. However, there are several other measures taken to test the property of the same under several other health situations to make a detailed study about the chemical compound structure to make a change in the combination of the chemical structure. This medicine has made a revolution in the field of science of the human structure. When made a deep verification of the compound structure of the chemical study showed the following information. The structural design has an appearance of white colored powder like material, which has an accurate purity of nearly 98%. The actual molecular weight of the compound formed is around 1025.2. This compound has the collective share of Amino acids in the composition, peptide and acetate contents also.
The study of the compound structure PT-141 has an enhanced support of making a recombination that produces a different profile of the same medicine but in a different standard with different properties that may support the human requirement.
Bremelanotide PT-141 is known for its aphrodisiac properties
Development
Bremelanotide was developed from the peptide hormoneMelanotan II which underwent testing as a sunless tanningagent. In initial testing, Melanotan II did induce tanning but additionally caused sexual arousal and spontaneous erections as unexpected side effects in nine out of the ten original male volunteer test subjects.[4]
A Phase IIIclinical trial was scheduled to begin in the first half of 2007, but was delayed until August 2007. On August 30, Palatin announced that the U.S. Food and Drug Administration had expressed serious concerns regarding therisk/benefit ratio of bremelanotide with regards to the side effect of increased blood pressure. The FDA stated that it would consider alternate uses for bremelanotide, including as a treatment for individuals who do not respond to more established ED treatments. However, On May 13, 2008, Palatin Technologies announced it had “discontinued development of Bremelanotide for the treatment of male and female sexual dysfunction” while concurrently announcing plans to develop it as a treatment for hemorrhagic shock instead.[7] The company additionally announced intentions to focus its attention on another compound, PL-6983, that causes lower blood pressure in animal models.[8]Palatin has since re-initiated Bremelanotide studies for ED and FSD using a subcutaneous delivery method. On August 12, 2009, the company announced that in a double-blind study of 54 volunteers bremelanotide failed to evoke the hypertensive side effects seen with the nasal delivery system used in prior studies, concluding that “variability of uptake” inherent in intranasal administration of the drug resulted in “increases in blood pressure and gastrointestinal events…primarily related to high plasma levels in [only] a subset of patients” and that subcutaneous administration of the drug circumvented the potential for this side effect.[8] Palatin has completed a human Phase 2B study utilizing subcutaneous administration and reported positive results.[9]
Sexual dysfunction, including both penile erectile dysfunction or impotence and female sexual dysfunction, are common medical problems. Significant effort has been devoted over the last twenty or more years to develop methods, devices and compounds for treatment of sexual dysfunction. While more effort has been undertaken for treatment of penile erectile dysfunction, female sexual dysfunction is also an area to which significant research and effort has been devoted.
At present, one commonly used orally administered drug for treatment of sexual dysfunction in the male is Viagra®, a brand of sildenafil, which is a phosphodiesterase 5 inhibitor, increasing the persistence of cyclic guanosine monophosphate and thereby enhancing erectile response. There are several other medical treatment alternatives currently available depending on the nature and cause of the impotence problem. Some men have abnormally low levels of the male hormone testosterone, and treatment with testosterone injections or pills may be beneficial. However, comparatively few impotent men have low testosterone levels. For many forms of erectile dysfunction, treatment may be undertaken with drugs injected directly into the penis, including drugs such as papaverin, prostaglandin E1, phenoxybenzamine or phentolamine. These all work primarily by dilating the arterial blood vessels and decreasing the venous drainage. Urethral inserts, such as with suppositories containing prostaglandin, may also be employed. In addition, a variety of mechanical aids are employed, including constriction devices and penile implants.
A variety of treatments have also been explored for female sexual dysfunction, including use of sildenafil, although the Food and Drug Administration has not specifically approved such use. Testosterone propionate has also been employed to increase or augment female libido.
Melanocortin receptor-specific compounds have been explored for use of treatment of sexual dysfunction. In one report, a cyclic α-melanocyte-stimulating hormone (“α-MSH”) analog, called Melanotan-II, was evaluated for erectogenic properties for treatment of men with psychogenic erectile dysfunction. Wessells H. et al., J Urology 160:389-393 (1998); see also U.S. Pat. No. 5,576,290, issued Nov. 19, 1996 to M. E. Hadley, entitled Compositions and Methods for the Diagnosis and Treatment of Psychogenic Erectile Dysfunction and U.S. Pat. No. 6,051,555, issued Apr. 18, 2000, also to M. E. Hadley, entitled Stimulating Sexual Response in Females. The peptides used in U.S. Pat. Nos. 5,576,290 and 6,051,555 are also described in U.S. Pat. No. 5,674,839, issued Oct. 7, 1997, to V. J. Hruby, M. E. Hadley and F. Al-Obeidi, entitled Cyclic Analogs of Alpha–MSH Fragments, and in U.S. Pat. No. 5,714,576, issued Feb. 3, 1998, to V. J. Hruby, M. E. Hadley and F. Al-Obeidi, entitled Linear Analogs of Alpha–MSH Fragments. Melanotan-II is a peptide of the following formula:
Additional related peptides are disclosed in U.S. Pat. Nos. 5,576,290, 5,674,839, 5,714,576 and 6,051,555. These peptides are described as being useful for both the diagnosis and treatment of psychogenic sexual dysfunction in males and females. These peptides are related to the structure of melanocortins.
In use of Melanotan-II, significant erectile responses were observed, with 8 of 10 treated men developing clinically apparent erections, and with a mean duration of tip rigidity greater than 80% for 38 minutes with Melanotan-II compared to 3.0 minutes with a placebo (p=0.0045). The drug was administered by subcutaneous abdominal wall injection, at doses ranging from 0.025 to 0.157 mg/kg body weight. Transient side effects were observed, including nausea, stretching and yawning, and decreased appetite.
The minimum peptide fragment of native α-MSH needed for erectile response is the central tetrapeptide sequence, His6-Phe7-Arg8-Trp9 (SEQ ID NO:1). In general, all melanocortin peptides share the same active core sequence, His-Phe-Arg-Trp (SEQ ID NO:1), including melanotropin neuropeptides and adrenocorticotropin. Five distinct melanocortin receptor subtypes have been identified, called MC1-R through MC5-R, and of these MC3-R and MC4-R are believed to be expressed in the human brain. MC3-R has the highest expression in the arcuate nucleus of the hypothalamus, while MC4-R is more widely expressed in the thalamus, hypothalamus and hippocampus. A central nervous system mechanism for melanocortins in the induction of penile erection has been suggested by experiments demonstrating penile erection resulting from central intracerebroventricular administration of melanocortins in rats. While the mechanism of His-Phe-Arg-Trp (SEQ ID NO:1) induction of erectile response has not been fully elucidated, it has been hypothesized that it involves the central nervous system, and probably binding to MC3-R and/or MC4-R.
Other peptides and constructs have been proposed which are ligands that alter or regulate the activity of one or more melanocortin receptors. For example, International Patent Application No. PCT/US99/09216, entitled Isoquinoline Compound Melanocortin Receptor Ligands and Methods of Using Same, discloses two compounds that induce penile erections in rats. However, these compounds were administered by injection at doses of 1.8 mg/kg and 3.6 mg/kg, respectively, and at least one compound resulted in observable side effects, including yawning and stretching. Other melanocortin receptor-specific compounds with claimed application for treatment of sexual dysfunction are disclosed in International Patent Application No. PCT/US99/13252, entitled Spiropiperidine Derivatives as Melanocortin Receptor Agonists.
Both cyclic and linear α-MSH peptides have been studied; however, the peptides heretofore evaluated have had an amide or —NH2 group at the carboxyl terminus. See, for example, Wessells H. et al., J Urology, cited above; Haskell-Luevano C. et al., J Med Chem 40:2133-39 (1997); Schiöth H. B. et al., Brit J Pharmacol 124:75-82 (1998); Schiöth H. B. et al., Eur J Pharmacol 349:359-66 (1998); Hadley M. E. et al., Pigment Cell Res 9:213-34 (1996); Bednarek M. A. et al., Peptides20:401-09 (1999); U.S. Pat. Nos. 6,054,556, 6,051,555 and 5,576,290; and, International Patent Applications PCT/US99/04111 and PCT/US98/03298. While significant research has been conducted in an effort to determine the optimal structure of α-MSH peptides, including a variety of structure-function, agonist-antagonist, molecular modeling and pharmacophore studies, such studies have relied upon peptides with an art conventional —NH2 group at the carboxyl terminus. Further, it has long been believed that biologically active neuropeptides, including α-MSH peptides, are amidated, with an —NH2 group at the carboxyl terminus, and that such amidation is required both for biological activity and stability. See, for example, Metabolism of Brain Peptides, Ed. G. O’Cuinn, CRC Press, New York, 1995, pp. 1-9 and 99-101.
The peptide of Compound 1 has a formula of C50H68N14O10, and a net molecular weight of 1025.18. This peptide may be synthesized by solid-phase means and purified to greater than 96% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of Compound 1 is:
In general, the peptide compounds of this invention may be synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of well-known procedures utilizing a variety of resins and reagents may be used to prepare the compounds of this invention.
The peptides of this invention may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, succinic or methanesulfonic. The acetate salt form is especially useful. Where the compounds of this invention include an acidic moiety, suitable pharmaceutically acceptable salts may include alkali metal salts, such as sodium or potassium salts, or alkaline earth metal salts, such as calcium or magnesium salts.
The invention provides a pharmaceutical composition that includes a peptide of this invention and a pharmaceutically acceptable carrier. The carrier may be a liquid formulation, and is preferably a buffered, isotonic, aqueous solution. Pharmaceutically acceptable carriers also include excipients, such as diluents, carriers and the like, and additives, such as stabilizing agents, preservatives, solubilizing agents, buffers and the like, as hereafter described.
EXAMPLE 1
Peptide Synthesis
The peptide Ac-Nle-cyclo(-Asp-His-D-Phe-Arg-Trp-Lys)-OH was synthesized by standard solid phase peptide synthesis methods, and is a cyclic heptapeptide melanocortin peptide analog with a free acid at the carboxyl terminus and an acetylated amino group at the amino terminus, with the structure:
The peptide has a net molecular weight of 1025.18, and is supplied in an acetate salt form. The peptide is a white, odorless amorphous hygroscopic powder, soluble in 0.9% saline, composed of C50H68N14O10. For synthesis, an Fmoc-Lys(R3)-p-alkoxybenzyl alcohol resin was transferred to a solid phase peptide synthesizer reactor with a mechanical stirrer. The R3group, such as 1-(1′-adamantyl)-1-methyl-ethoxycarbonyl (Adpoc), allyloxycarbonyl (Aloc) or 4-methyltrityl (Mtt), was removed and the next Fmoc-protected amino acid (Fmoc-Trp(Boc)-OH) was added to the resin through standard coupling procedures. The Fmoc protective group was removed and the remaining amino acids added individually in the correct sequence, by repeating coupling and deprotection procedures until the amino acid sequence was completed. After completion of coupling with the last Fmoc-amino acid derivative, Fmoc-Nle-OH, and cleavage of the Fmoc protective group, the exposed terminal amino group was acetylated with acetic anhydride and pyridine in N,N-dimethylformamide (DMF). The peptide-resin was dried and the Lys and Asp protective groups cleaved. The Lys and Asp deprotected peptide resin was suspended in a suitable solvent, such as DMF, dichloromethane (DCM) or 1-methyl-2-pyrrolidone (NMP), a suitable cyclic coupling reagent, such as 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU), 2-(7-aza-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TATU), 2-(2-oxo-1(2H)-pyridyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) or N,N′-dicyclohexylcarbodiimide/1-hydroxybenzotriazole (DCCl/HOBt) was added, and coupling initiated by use of a suitable base, such as N,N-diispropylethylamine (DIPEA), sym-collidine or N-methylmorpholine (NMM). After cyclization, the peptide-resin was washed and the peptide cleaved from the resin and any remaining protective groups using trifluoroacetic acid (TFA) in the presence of water and 1,2-ethanedithiol (EDT). The final product was precipitated by adding cold ether and collected by filtration. Final purification was by reversed phase HPLC using a C18 column. The purified peptide was converted to acetate salt by passage through an ion-exchange column.
The peptide of bremeianotide has a formula of CsaHesN< C½, and a net mofecufar weight of 1025.18, This peptide may be synthesized by conventional means, including either solid-phase or Squid-phase techniques, and purified to greater than 99% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of bremeianotide is:
in one embodiment of the invention, bremeianotide is synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of ‘well-known procedures utilizing a variety of resins and reagents may be used to prepare bremeianotide.
Bremeianotide may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacefie, maieic, citric, tartaric, oxalic, succinic or methanesu!fonic acid. The acetate salt form is especially useful.
in a preferred embodiment, bremelanotide is an acetate salt form, and is formulated in a buffered aqueous solution including giycerin, and prepackaged in a syringe and auto-injector device. In alternative embodiments, bremelanotide is any pharmaceutically acceptable salt form, and is formulated in any pharmaceutically acceptable aqueous solution, the aqueous solution optionally including one or more salts, such as sodium chloride, one or more acids, such as citric acid, and one or more additional ingredients, including cellulose or derivatives thereof, saccharides o
polysaccharides such as dextrose, and any of a wide variety of surfactants, chelating agents and preservatives.
In yet another embodiment of the present invention, the melanocortin receptor agonist is: Ac–Nle-cyclo(-Asp–His–D–Phe–Arg–Trp–Lys)-OH PT-141
The peptide of PT-141 has a formula of C50H68N14O10, and a net molecular weight of 1025.18. This peptide may be synthesized by conventional means, including either solid-phase or liquid-phase techniques, and purified to greater than 99% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of PT-141 is:
In one embodiment of the invention, PT-141 is synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of well-known procedures utilizing a variety of resins and reagents may be used to prepare PT-141.
PT-141 may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, citric, tartaric, oxalic, succinic or methanesulfonic acid. The acetate salt form is especially useful. Where the compounds of this invention include an acidic moiety, suitable pharmaceutically acceptable salts may include alkali metal salts, such as sodium or potassium salts, or alkaline earth metal salts, such as calcium or magnesium salts.
In a preferred embodiment, PT-141 is an acetate salt form, and is formulated in a buffered aqueous solution including glycerin, prepackaged in a metered unit dose intranasal delivery device. In alternative embodiments, PT-141 is any pharmaceutically acceptable salt form, and is formulated in any pharmaceutically acceptable aqueous solution, the aqueous solution optionally including one or more salts, such as sodium chloride, one or more acids, such as citric acid, and one or more additional ingredients, including cellulose or derivatives thereof, saccharides or polysaccharides such as dextrose, and any of a wide variety of surfactants, chelating agents and preservatives. In one preferred embodiment, PT-141 is administered to patients in volumes of 100 μL, with the quantity of PT-141 delivered determined by the concentration thereof. As described hereafter, in one preferred embodiment a metered unit dose contains 7.5 mg of PT-141.
While certain embodiments of the present invention are described primarily in the context of PT-141, it is to be understood that other melanocortin receptor agonists may be employed. For example, the metallopeptide melanocortin receptor agonists disclosed in WO 02/064091, filed on Feb. 13, 2001, and U.S. Ser. No. 10/640,755, filed on Aug. 13, 2003, both entitled Melanocortin Metallopeptides for Treatment of Sexual Dysfunction; and WO 01/13112, filed on Jun. 14, 2000, entitled Melanocortin Metallopeptide Constructs, Combinatorial Libraries and Applications, may be employed. In addition, the peptidomimetic melanocortin receptor agonists disclosed in U.S. Ser. No. 10/776,419, filed on Feb. 10, 2004, entitled Peptidomimetics of Biologically Active Metallopeptides; the pyrrolidine melanocortin receptor agonists disclosed in U.S. Ser. No. 10/766,657, filed on Feb. 10, 2004, entitled Pyrrolidine Melanocortin-Specific Compounds; and the bicyclic melanocortin receptor agonists disclosed in PCT/US04/01505, filed on Jan. 20, 2004, entitled Bicyclic Melanocortin-Specific Compounds, may also be employed. Also particular preferred are the piperazine melanocortin agonists disclosed in PCT/US04/01462, filed on Jan. 20, 2004 and U.S. Ser. No. 10/762,079, filed on Jan. 20, 2004, both entitled piperazine Melanocortin-Specific Compounds; the melanocortin agonists disclosed in WO 03/006620, filed on Jul. 11, 2002, entitled Linear and Cyclic Melanocortin Receptor-Specific Peptides; WO 04/005324, filed on Jul. 9, 2003, entitled Peptide Compositions for Treatment of Sexual Dysfunction; PCT/US00/18217, filed on Jun. 29, 2000 and U.S. Ser. No. 10/040,547, filed on Jan. 4, 2002, entitled Compositions and Methods for Treatment of Sexual Dysfunction; and U.S. Ser. No. 10/638,071, filed on Aug. 8, 2003, entitled Cyclic Peptide Compositions and Methods for Treatment of Sexual Dysfunction. The entire disclosure of each of the foregoing are incorporated here by reference. It is to be understood that the foregoing listing of patent applications disclosing melanocortin receptor agonists is intended to only be exemplary, and that other melanocortin receptor agonists, whether heretofore known or hereafter developed, may similarly be used in the practice of this invention.
The Khajuraho Group of Monuments are a group of Hindu and Jain temples in Madhya Pradesh, India. About 620 kilometres (385 mi) southeast of New Delhi, …
his Is The Famous ‘Ganj-Golai’ As The Central Place Of The Latur City. There Are 16 Roads Connecting To This Place And Seperate Markets i.e. Jewellers …
Vilasrao Deshmukh’s ancestral home at Babhalgaon village in Latur. Machindra Amle
UDGIR: Udgir is one of the most important towns of Latur district. Udgir has a great historical significance. It has witnessed the war between the Marathas …
The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
One of the most popular CDK inhibitor in clinical trials in the recent years was dinaciclib (MK-7965, SCH 727965) (Figure 3), the inhibitor of CDK1, CDK2, CDK5, and CDK9. A Phase I trial on the effect of dinaciclib in combination with aprepitant was performed in patients with advanced malignancies [44]. Aprepitant is used for the prevention of chemotherapy-induced nausea and vomiting, is known as an inhibitor and inducer of CYP3A4, which metabolizes dinaciclib.
Coadministration of dinaciclib with aprepitant resulted in no clinically significant effect on the pharmacokinetics and did not alter the safety profile of dinaciclib. The first Phase I clinical trial on dinaciclib as a single agent was performed on patients with advanced malignancies [68]. Forty-eight patients with various solid tumors were treated and 10 of them achieved prolonged stable disease for at least four treatment cycles. Adverse effects were mild, the most common being nausea, anemia, decreased appetite and fatigue.
A phase II multi-center study of dinaciclib for relapsed and/or refractory AML was performed on 20 patients [69]. Temporary decrease in peripheral blood and/or bone marrow blasts was observed in 60% of patients. Four of 13 (31%) patients with circulating blasts had >50% decrease and 6 (46%) >80% decrease in the absolute blast count within 1–8 days of the first dinaciclib dose. Toxicities included diarrhea, fatigue, transaminitis, and manifestations of tumor lysis syndrome, with one patient who deceased of acute renal failure. Another Phase II study was performed of dinaciclib versus erlotinib in patients with non-small cell lung cancer [70].
Unfortunately, it was found that dinaciclib was not successful as monotherapy in non-small cell lung cancer. Most common toxicities included neutropenia, leukopenia, vomiting, and diarrhea. Yet another Phase II study was performed on dinaciclib versus capecitabine in patients with advanced breast cancer [71]. Dinaciclib treatment demonstrated antitumor activity in two of seven patients with ER-positive and ERBB 2-negative metastatic breast cancer, however efficacy was not superior to capecitabine (p = 0.991).
Toxicities included neutropenia, leukopenia, increase in aspartate aminotransferase, and febrile neutropenia. Phase I nonrandomized dose-escalation trial was performed, where patients with relapsed or refractory chronic lymphocytic leukemia were treated with dinaciclib and rituximab [72]. Four out of six patients achieved stable disease, and one patient achieved complete response. Drug-related adverse events were mostly hematological, digestive and metabolic and no dose-limiting toxicities were observed. Dinaciclib was also moved into Phase III development for refractory chronic lymphocytic leukemia [73]. Phase I/II clinical trial Dinaciclib in patients with relapsed multiple myeloma showed promise as single agent [74]. The overall confirmed response rate was 3 of 27 (11%). Adverse effects included leukopenia, thrombocytopenia, gastrointestinal symptoms, alopecia, and fatigue. –
Dinaciclib (SCH-727965) is an experimental drug that inhibits cyclin-dependent kinases (CDKs.[1] It is being evaluated in clinical trials for various cancer indications.[2]
Mechanisms of action
Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains.[3]
Dinacliclib induces the apoptosis of osteosarcoma cells.[8]
Apoptosis of osteosarcoma cultures can be induced by the combination of the cyclin-dependent kinase inhibitor SCH727965 and a heat shock protein 90 inhibitor.[9]
As described in the ‘878 publication, Synthetic Scheme II leading to the compound of Formula II has several disadvantages from the standpoint of commercial scale synthesis. In step 1, the starting material (compound “C”) used in the formation of compound “D” is a sticky, viscous oil which is difficult to process (weigh, transfer, and blend). Moreover, step 1, as described in the ‘878 publication, requires isolation and chromatographic purification of compounds C and D prior to carrying out each subsequent derivatization reaction. In addition, as described in the ‘878 publication, the reaction of compound C with malonate diester is carried out using the diester as a solvent. After isolation and purification of the resultant malonate adduct, compound D, ring closure to form diketone compound E is carried out in methanol. In accordance with the procedure described in the ‘878 publication, compound E is isolated and dried, then converted to the corresponding dichloride in N,N-dimethyl aniline by treatment with phosphorous oxychloride (POCl3). The dichloride thus formed was isolated and purified by chromatography prior to the sequential amination reactions. Additionally, the compounds of Formula G and of Formula II require chromatography purification and isolations, as described in the ‘878 publication.
As further described in the ‘878 publication, each of the amination reactions were run separately with isolation and chromatographic purification between amination reactions. Accordingly, the ‘878 publication describes the preparation of the compound of Formula II utilizing a scheme consisting of five separate reaction steps with intervening isolation and purification of the products, each sequential step being carried out in a different solvent system. The overall yield of the compound of Formula II reported for this synthesis, based on starting compound C (Scheme II) is about 20%.
Example 1Preparation of Diketone Compound E (Scheme VI) 3-Ethylpyrazolo[1,5-a]pyrimidine-5,7(4H,6H)-dione
To a 250 ml, three-necked flask equipped with a thermometer, a reflux condenser and mechanical stirrer was charged 3-amino-4-ethylpyrazole oxalate (10 g, 50 mmole), dimethylmalonate (10 ml, 88 mmole), methyl alcohol (80 ml) and sodium methoxide (50 ml, 245 mmole, 25% in methyl alcohol). The batch was heated at reflux for 16 hours then cooled to room temperature. Celite (5 g) and water (60 ml) were added to the batch and agitated for 10 minutes. The batch was filtered to remove the solid residue. The filtrate was pH adjusted to pH˜3 with aqueous HCl (10 ml) to effect precipitation. The precipitate (compound “E”) was filtered and washed with water (40 ml). The wet cake was dried for 18 hours in vacuum oven maintained in the range of oven at 45° C. to 55° C., to give a solid product (84.3%, 7.5 g). C8H9N3O3, Mp: 200-205° C.; NMR in DMSO-d6: 1.05 (t, 3H), 2.23 (q, 2H), 3.26 (bs, 1H), 3.89 (bs, 1H), 7.61 (s, 1H), 11.50(bs, 1H).
Example 2Preparation of Dichloride Compound F (Scheme VI) 5,7-Dichloro-3-Ethylpyrazolo[1,5-a]pyrimidine
Into a 3-neck flask fitted with an inert gas inlet, a reflux condenser and a mechanical stirring apparatus and containing 83 liters of acetonitrile was placed 3-Ethylpyrazolo[1,5-a]pyrimidine-5,7(4H,6H)-dione (E) prepared as described in Step 1 (11.0 kg, 61.5 mole), N,N-dimethylaniline (8.0 L, 63 mole) and POCl3 (7 kg, 430 mole). With stirring the mixture was brought to reflux and maintained under refluxing conditions for 15 hours. The reaction mixture was sampled periodically to monitor the amount of compound “E” present. After the conversion was complete, the solution was cooled to 15° C. Into the cooled reaction mixture was added water which had been cooled to a temperature of less than 20° C. The product is filtered and washed with 4 aliquots of acetonitrile-water (1:3) which had been cooled to a temperature of 20° C. followed by a wash with 10× water. The wet cake is dried in a vacuum oven maintained at 40° C. for at least 15 hours to yield the compound “F” (86.7%); 1H NMR (CDCl3): 1.32(t, 3H), 2.81 (q, 2H), 6.92 (s, 1H), 8.10 (s, 1H)
mp: 90-95° C.
Example 3Preparation of Compound G (Scheme VI) 5-Chloro-3-Ethyl-N-[(1-oxido-pyridinyl)methyl]pyrazolo-[1,5-a]pyrimidine-5.7(4H,6H)-dion-7-amine
Into a 3-liter, three-necked flask equipped with a thermometer, a reflux condenser and mechanical stirrer was charged an aliquot of the dichloride compound “F” prepared in Step 2 (150 g, 0.69 mole), potassium phosphate tribasic monohydrate (338.0 g, 1.47 mole), the dihydrochloride salt of N-oxide-pyridin-3-yl-methylamine, compound F1a (142.5 g, 0.72 mole), water (1500 ml) and acetonitrile (300 ml). The batch was heated at reflux for 6 hours. At the end of the refluxing period the batch was cooled to room temperature over 2 hours and then held at room temperature for 4 hours. The resulting precipitate was filtered and washed with water (600 ml). The wet cake was returned to the flask with water (1500 ml) and acetonitrile (300 ml), and heated to reflux. Reflux was maintained for 6 hours additional. At the end of the second reflux period the reaction mixture was cooled to room temperature over a 2 hour period and left to stand at room temperature for 4 hours. The resulting precipitate was filtered and washed with water (600 ml). The wet cake was dried in an air draft oven at 50° C. for 18 hours to give the first amine adduct “G” material (179 g, 84.9%). mp: 187-189C; NMR in CDCl3, 1.26(t, 3H), 2.73(q, 2H), 4.60(d, 2H), 5.87(s, 1H), 6.83(bs, 1H), 7.33(t, 1H), 7.70(d, 1H), 7.84(s, 1H), 8.58(d, 1H), 8.64(d, 1H).
Example 4
Preparation of the Compound of Formula II (Scheme VI) 1-[3-Ethyl-7-[(1-oxido-3-pyridinyl)methyl]amino]pyrazolo[1,5-a]pyrimidin-5-yl]-2(s)-piperidinemethanol
Into a three-neck flask fitted with a mechanical stirrer and a reflux condenser were placed the first amine adduct prepared in Step 3, compound “G”, (7 kg, 23 mole), amino-alcohol compound G1a (5.6 kg, 43.3 mole), sodium carbonate (3.5 kg, 33.0 mole), 110 ml of water and 1-methyl-2-pyrrolidinone (NMP) (11 L). The reaction mixture was heated to 150° C. for 4 days. After chromatography indicated that the reaction was complete (90-95% substrate consumed), the reaction mixture was cooled to room temperature and quenched by adding water. The mixture was then extracted with ethyl acetate. The batch was dried by distillation of the water azeotrope under atmospheric pressure and concentrated to about 28 L volume. THF was added and the solution was heated to reflux until all the solids dissolve. Ethyl acetate and trietylamine are added to the hot solution. The batch was cooled to ambient and then agitated with the temperature maintained in the range of from 20° C. to 25° C. for 12 hours. The solids were collected by filtration, washed first with ethyl acetate then water, and dried in the filter under vacuum for 24 hours with the temperature maintained at from 40° C. to 50° C., yielding 4.9 kg, 51.3% of the compound of Formula II.
DSC, 168.6° C.; Specific Rotation (10 mg/ml in MeOH, 20° C.), −117.8 °;
Nguyen, T. K.; Grant, S (2013). “Dinaciclib (SCH727665) inhibits the unfolded protein response (UPR) through a CDK1 and CDK5-dependent mechanism”. Molecular Cancer Therapeutics13(3): 662–74. doi:10.1158/1535-7163.MCT-13-0714. PMID24362465.edit
Jump up^Fu, W; Ma, L; Chu, B; Wang, X; Bui, M. M.; Gemmer, J; Altiok, S; Pledger, W. J. (2011). “The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells”. Molecular Cancer Therapeutics10 (6): 1018–27. doi:10.1158/1535-7163.MCT-11-0167. PMID21490307.edit
Jump up^Mita, M; Joy, A. A.; Mita, A; Sankhala, K; Jou, Y. M.; Zhang, D; Statkevich, P; Zhu, Y; Yao, S. L.; Small, K; Bannerji, R; Shapiro, C. L. (2013). “Randomized Phase II Trial of the Cyclin-Dependent Kinase Inhibitor Dinaciclib (MK-7965) Versus Capecitabine in Patients with Advanced Breast Cancer”. Clinical Breast Cancer14 (3): 169–76. doi:10.1016/j.clbc.2013.10.016.PMID24393852.edit
Jump up^Stephenson, J. J.; Nemunaitis, J; Joy, A. A.; Martin, J. C.; Jou, Y. M.; Zhang, D; Statkevich, P; Yao, S. L.; Zhu, Y; Zhou, H; Small, K; Bannerji, R; Edelman, M. J. (2014). “Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer”. Lung Cancer83 (2): 219–23.doi:10.1016/j.lungcan.2013.11.020. PMID24388167.edit
Process and intermediates for the synthesis of (3-alkyl-5-piperidin-1-yl-3,3a-dihydro-pyrazolo[1,5-a]pyrimidin-7-yl)-amino derivatives and intermediates [US8076479]2008-03-06 GRANT2011-12-13
Process for resolving chiral piperidine alcohol and process for synthesis of pyrazolo[1,5-a] pyrimidine derivatives using same [US7786306]2008-02-28 GRANT2010-08-31
Sequential Administration of Chemotherapeutic Agents for Treatment of Cancer [US2011129456]2011-06-02
TARGETING CDK4 AND CDK6 IN CANCER THERAPY [US2011009353]2011-01-13
Pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2007225270]2007-09-27
Takeda’s flagship experimental cancer drug ixazomib is a giant leap closer to being filed with regulatory authorities around the globe for multiple myeloma, after turning in a solid performance in late-stage trials.
Takeda’s ixazomib soon to be filed for multiple myeloma
Entinostat, developed by Syndax Pharmaceuticals, is an oral selective histone deacetylase (HDAC) inhibitor primarily targeting class IHDACs (HDAC1, HDAC2, and HDAC3) . It was later licensed to Jiangsu Hengrui Medicine Co., Ltd., for development and commercialization in China. In 2024, Entinostat has been approved by the NMPA for use in combination with exemestane to treat advanced breast cancer that is HR-positive and HER2-negative.
TOKYO and WALTHAM, Mass., Jan. 7, 2015 /PRNewswire/ — Kyowa Hakko Kirin Co., Ltd., (Headquarters: Chiyoda-ku, Tokyo; president and CEO: Nobuo Hanai, “Kyowa Hakko Kirin”) and Syndax Pharmaceuticals, Inc., (Waltham, Mass.; president and CEO:Arlene M. Morris, “Syndax”) today jointly announced that the companies have entered into a license agreement for the exclusive rights to develop and commercialize entinostat in Japan and Korea. Entinostat is a Class I selective histone deacetylase (HDAC) inhibitor being developed by Syndax in the United States and Europe in combination with hormone therapy for advanced breast cancer and immune therapy combinations in solid tumors.
Entinostat inhibits class I HDAC1 and HDAC3 with IC50 of 0.51 μM and 1.7 μM, respectively.[2]
Entinostat (formerly known as MS-275) is a histone deacetylase (HDAC) inhibitor in phase III clincal trials at Syndax in combination with exemestane for the treatment of advanced HR-positive breast cancer.
Entinostat (MS-275) preferentially inhibits HDAC1 (IC50=300nM) over HDAC3 (IC50=8µM) and has no inhibitory activity towards HDAC8 (IC50>100µM). MS-275 induces cyclin-dependent kinase inhibitor 1A (p21/CIP1/WAF1), slowing cell growth, differentiation, and tumor development in vivo. Recent studies suggest that MS-275 may be particularly useful as an antineoplastic agent when combined with other drugs, like adriamycin.
In September 2013, Syndax Pharmaceuticals entered into a licensing, development and commercialization agreement with Eddingpharm in China and other asian countries. In 2013, a Breakthrough Therapy Designation was assigned to the compound for the treatment of locally recurrent or metastatic estrogen receptor-positive (ER+) breast cancer when added to exemestane in postmenopausal women whose disease has progressed following non-steroidal aromatase inhibitor therapy.
Clinical trials
There is an ongoing phase II trial studying the effect of entinostat on Hodgkin’s lymphoma.[3] It is in other phase II trials for advanced breast cancer (in combination with aromatase inhibitors)[4] and for metastatic lung cancer (in combination with erlotinib).[5] As of September 2013, the Food and Drug Administration is working with the industry to design phase III clinical trials. They seek to evaluate the application of Entinostat for the reduction, or prevention of, treatment resistance to aromatase inhibitors in hormone receptor positive breast cancer.[6] Syndax pharmaceuticals currently holds the rights to Entinostat and recently received $26.6 million in funds to advance treatments of resistant cancers using epigenetic tools.[7]
PHASE 3………..SYNDAX, BREAST CANCER
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Entinostat, developed by Syndax Pharmaceuticals, is an oral selec tive histone deacetylase (HDAC) inhibitor primarily targeting class I HDACs (HDAC1, HDAC2, and HDAC3) [7]. It was later licensed to Jiangsu Hengrui Medicine Co., Ltd., for development and commercial ization in China. In 2024, Entinostat has been approved by the NMPA for use in combination with exemestane to treat advanced breast cancer that is HR-positive and HER2-negative. This approval is specifically for pa tients whose disease has progressed following prior endocrine therapy [8]. Entinostat inhibits HDACs, increasing histone acetylation and reactivating tumor suppressor genes. This mechanism restores sensi tivity to endocrine therapy and prevents cancer cell proliferation [9]. The therapeutic agent exerts its effects by modulating the tumor microenvironment through the suppression of immune regulatory cells, thereby augmenting the immune response. Its clinical efficacy was confirmed in the E2112 trial (NCT02115282), a global Phase III study. When used in combination with exemestane, Entinostat demonstrated the ability to extend PFS in patients with HR-positive, HER2-negative breast cancer [10]. The median PFS was significantly extended to 6.32 months, contrasting with the 3.72 months observed in the control cohort. In terms of safety profile, Entinostat demonstrated favorable tolerability. The frequently encountered adverse events were primarily neutropenia, fatigue, and nausea. Severe neutropenia occurred in 43 % of patients but was manageable with supportive care. Liver function abnormalities were reported but manageable with dose adjustments [11]. The synthetic route of Entinostat is shown in Scheme 2 [12]. Enti-001 is first treated with trifluoroacetic anhydride to afford Enti-002. Reaction of Enti-002 with oxalyl chloride yields the acyl chloride intermediate, which undergoes condensation with Enti-003 to form Enti-004. Subsequent alkaline hydrolysis of Enti-004 produces Enti-005. This compound is activated with CDI followed by reaction with Enti-006 to generate Enti-007. The synthesis concludes with acidic removal of the Boc protecting group from Enti-007, yielding Entinostat
[8] W. Li, Z. Sun, Mechanism of action for HDAC inhibitors-insights from omics approaches, Int. J. Mol. Sci. 20 (2019) 1616. [9] N. Bharathy, N.E. Berlow, E. Wang, J. Abraham, T.P. Settelmeyer, J.E. Hooper, M. N. Svalina, Z. Bajwa, M.W. Goros, B.S. Hernandez, J.E. Wolff, R. Pal, A.M. Davies, A. Ashok, D. Bushby, M. Mancini, C. Noakes, N.C. Goodwin, P. Ordentlich, J. Keck, D.S. Hawkins, E.R. Rudzinski, A. Mansoor, T.J. Perkins, C.R. Vakoc, J.E. Michalek, C. Keller, Preclinical rationale for entinostat in embryonal rhabdomyosarcoma, Skelet Muscle 9 (2019) 12. [10] B. Xu, Q. Zhang, X. Hu, Q. Li, T. Sun, W. Li, Q. Ouyang, J. Wang, Z. Tong, M. Yan, H. Li, X. Zeng, C. Shan, X. Wang, X. Yan, J. Zhang, Y. Zhang, J. Wang, L. Zhang, Y. Lin, J. Feng, Q. Chen, J. Huang, L. Zhang, L. Yang, Y. Tian, H. Shang, Entinostat, a class I selective histone deacetylase inhibitor, plus exemestane for Chinese patients with hormone receptor-positive advanced breast cancer: a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial, Acta Pharm. Sin. B 13 (2023) 2250–2258. [11] E.T. Roussos Torres, W.J. Ho, L. Danilova, J.A. Tandurella, J. Leatherman, C. Rafie, C. Wang, A. Brufsky, P. LoRusso, V. Chung, Y. Yuan, M. Downs, A. O’Connor, S. M. Shin, A. Hernandez, E.L. Engle, R. Piekarz, H. Streicher, Z. Talebi, M.A. Rudek, Q. Zhu, R.A. Anders, A. Cimino-Mathews, E.J. Fertig, E.M. Jaffee, V. Stearns, R. M. Connolly, Entinostat, nivolumab and ipilimumab for women with advanced HER2-negative breast cancer: a phase Ib trial, Nat Cancer 5 (2024) 866–879. [12] T. Suzuki, T. Ando, K. Tsuchiya, T. Nakanishi, A. Saito, S. Yamashita, G. Shiraishi, E. Tanaka, Preparation of Benzamide Derivatives as Anticancer Agents, 1998 JP10152462
In EP 0 847 992 A1 (which co-patent is US 6,794,392) benzamide derivatives as medicament for the treatment of malignant tumors, autoimmune diseases, de- rmatological diseases and parasitism are described. In particular, these derivatives are highly effective as anticancer drugs, preferred for the haematological malignancy and solid tumors. The preparation of N-(2-aminophenyl)-4-[N- (pyridine-3-yl)methoxycarbonylaminomethyl]-benzamide is described on page 57, Example 48. The compound is neither purified by chromatography nor purified by treatment with charcoal. The final step of the process comprises the re- crystallization from ethanol.
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm“1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
In EP 0 974 576 B1 a method for the production of monoacylated phenylenediamine derivatives is described. The preparation of N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl] benzamide is described on pages 12 to 13, Example 6. The final step of the process comprises the purification of the compound via silica gel column chromatography.
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm‘1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form. In J. Med. Chem. 1999, 42, 3001-3003, the synthesis of new benzamide derivatives and the inhibition of histone deacetylase (HDAC) is described. The process for the production of N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth- oxycarbonylaminomethyl] benzamide is described. The final step of the process comprises the purification of the compound via silica gel column chromatography (ethyl acetate).
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm‘1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
In WO 01/12193 A1 a pharmaceutical formulation comprising N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl]benzamide is described.
In WO 01/16106 a formulation comprising N-(2-aminophenyl)-4-[N-(pyridine-3- yl)methoxycarbonylamino-methyl]benzamide, having an increased solubility and an improved oral absorption for benzamide derivatives, and pharmaceutically acceptable salts thereof are described.
In WO 2004/103369 a pharmaceutical composition is described which comprises histone deacetylase inhibitors. That application concerns the combined use of N-(2-aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino- methyl]benzamide together with different cancer active compounds. In fact that application is a later application, which is based on the above mentioned matter and thus concerns the Polymorph A form. Finally, JP 2001-131130 (11-317580) describes a process for the purification of monoacylphenylenediamine derivatives. In Reference Example 2, the process for the production of crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide is described. Said compound has a melting point (mp) of 159 – 160 0C,
The IR spectrum shows the following bands: IR(KBr) cm“1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
Moreover, Working Example 1 describes the purification of crude N-(2- aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide in aqueous acid medium together with carbon The final crystallization is done under aqueous conditions at 40-500C.
Following the description to that example it can be seen from the Comparative Examples 1 – 3 that the crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth- oxycarbonylaminomethyl] benzamide is not purified by dissolution under reflux conditions in either ethanol, methanol or acetonithle followed by a recrystalliza- tion at 2°C. As a result, these recrystallisations do not yield any pure compound.
In addition a “purification” of crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide in ethanol under reflux conditions to- gether with carbon is dechbed. After filtering off the carbon the compound is re- crystallized at 2°C. The purification effect of this method is very limited. 1 ,1 % of an impurity remain in the N-(2-aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide. As a result, this procedure does not yield any pure compound.
None of the state of the art documents refer to a polymorph B of N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl]benzamide and no physicochemical features of said compound are known. Several biological and clinical studies have been done with N-(2-aminophenyl)- 4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide. For example, Kummar et al., Clin Cancer Res. 13 (18), 2007, pp 5411-5417 describe a phase I trial of N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide in refractory solid tumors. The compound was applied orally.
The crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylaminomethyl]- benzamide of step a) can be produced according to the method described in example 6 of EP 0974 576 B1.
Example 6Synthesis of N-(2-aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide (an example in which after activation with N,N’-carbonyldiimidazole, an acid was added to carry out reaction)
[0082]
7.78 g (48 mmole) of N,N’-carbonyldiimidazole were added to a 1,3-dimethyl-2-imidazolidinone (50 g) suspension including 11.45 g (40 mmole) of 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic acid. After stirring at room temperature for 2 hours, 17.30 g (0.16 mole) of 1,2-phenylenediamine were added to the solution. After cooling to 2°C, 9.60 g (0.1 mole) of methanesulfonic acid were added dropwise. After stirring for 2 hours, water was added, and the deposited solid was collected by filtration. Purification was then carried out through silica gel column chromatography to obtain 10.83 g (yield: 72%) of N-(2-aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide. Reaction selectivity based on the result in HPLC
Suzuki et al (Suzuki et al Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives, J Med Chem 1999, 42, (15), 3001-3) discloses benzamide derivatives having histone deacetylase inhibitory activity and methods of making benzamide derivatives having histone deacetylase inhibitory activity. Suzuki et al is hereby incorporated herein by reference in its entirety.
[18] An example of the synthesis method of Suzuki et al to produce MS-275 via a three- step procedure in 50.96% overall yield is outlined in Scheme 3 below.
Scheme 3: Previous Procedure for Synthesis of MS-275 en rt, 4h
(used without purification)
[Overall yield: 0.91 x 0.56 x 100 = 50.96%;
MS-275 [19] In addition to the modest overall yield, the procedure of Suzuki et al has other disadvantages, such as a tedious method for the preparation of an acid chloride using oxalyl chloride and requiring the use of column chromatography for purification.
The synthesis of MS-275 is shown below in Scheme 4 as an example of Applicants invention of a two-step procedure: [37] Scheme 4: Preparation of MS-275
Scheme 4: New Synthesis of MS-275 (4)
Condensation of 3-(hydroxymethyl)pyridine (7) and 4-(aminomethyl)benzoic in the presence of CDI gave 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic Acid (8) in 91.0% yield. In the previous method of Suzuki et ah, the carboxylic acid derivative 8 was first converted into acyl chloride hydrochloride by treatment of oxalyl chloride in toluene and then reacted with imidazole to form the acylimidazole intermediate. (Suzuki et al., Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem 1999, 42, (15), 3001-3.). However, Applicants synthesized the imidazolide of intermediate 8 by treatment with CDI at about 55-60 0C in THF. The imidazolide was cooled to ambient and further reacted in situ with 1,2-phenylenediamine in the presence of TFA to afford MS-275
[63] To a suspension of 4-[N-(Pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic
Acid (5.0 g, 0.017 mol) in THF (100 mL) was added CDI (3.12 g, 0.019 mol), and the mixture stirred for 3 h at 60 0C. After formation of acylimidazole the clear solution was cooled to room temperature (rt). To this was added 1,2-phenylenediamine (15.11 g, 0.14 mmol) and trifluoroacetic acid (1.2 mL, 0.015 mol) and then stirred for 16 h. The reaction mixture was evaporated to remove THF and crude product was stirred in a mixture of hexane and water (2:5, v/v) for 1 h and filtered and dried. The residue was stirred in dichloromethane twice to afford pure MS-275 (4) as off white powder 5.25 g, 80% yield:
HRMS: calcd 376.1560 (C2iH2oN4θ3), found 376.1558. These spectral and analytical data are as previously reported in J Med Chem 1999, 42, (15), 3001-3.
[64] 4-[7V-(Pyridin-3-ylmethoxycarbonyI)aminomethyl] benzoic Acid (8) may be prepared as follows. To a suspension of l, l’-carbonyldiimidazole (CDI, 25.6 g, 158 mmol) in THF (120 mL) was added 3-pyridinemethanol (7, 17.3 g, 158 mmol) in THF (50 mL) at 10 0C, and the mixture stirred for 1 h at rt. The resulting solution was added to a suspension of 4-(aminomethyl)benzoic acid (22.6 g, 158 mmol), DBU (24.3 g, 158 mmol), and triethylamine (22.2 mL, 158 mmol) in THF (250 mL). After stirring for 5 h at rt, the mixture was evaporated to remove THF and then dissolved in water (300 mL). The solution was acidified with HCl (pH 5) to precipitate a white solid which was collected by filtration, washed with water (300 mL) and methanol (50 mL), respectively, and dried to yield pure 8 (41.1 g, 91% yield):
mp 207-208 0 C;
IR (KBr) 3043, 1718, 1568, 1434, 1266, 1 108, 1037, 984, 756 cm4; 1H NMR (DMSO-^6) δ 4.28 (d, 2H, J= 5.9 Hz), 5.10 (s, 2H), 7.3-7.5 (m, 3H), 7.7-8.1 (m, 4H), 8.5-8.7 (m, 2H). These spectral and analytical data are as previously reported in Suzuki et al, J Med Chem 1999, 42, (15), 3001-3.
N-(2-Aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide (1, MS-275). To a solution of imidazole (0.63 g, 9.2 mmol) in THF (20 mL) was added 3 (1 g, 2.9 mmol), and the mixture stirred for 1 h at room temperature. After imidazole hydrochloride was removed by filtration, 1,2-phenylenediamine (2.52 g, 23.2 mmol) and trifluoroacetic acid (0.2 mL, 2.6 mmol) were added to the filtrate and stirred for 15 h. The reaction mixture was evaporated to remove THF and partitioned between ethyl acetate (500 mL) and water (400 mL). The organic layer was washed with water and dried and then purified by silica gel column chromatography (ethyl acetate) to give 1 (0.62 g, 56% yield):
References: 1. Saito, A. et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA 96 4592-4597 (1999). 2. Jaboin, J., et al. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res 62 6108-6115 (2002). 3. Rosato RR, et al. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res 2003; 63: 3637–3645.
NEWS………….DUBLIN and BUDAPEST, Hungary, Jan. 6, 2015 /PRNewswire/ — Actavis plcand Gedeon Richter Plc. today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of Actavis’ New Drug Application (NDA) resubmission for its atypical antipsychotic cariprazine, a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. The Prescription Drug User Fee Act (PDUFA) date is expected to be in the second quarter of 2015…….
The most prevalent side effects for cariprazine include akathisia, insomnia, and weight gain. Cariprazine does not appear to impact metabolic variables or prolactin levles, and unlike many other antipsychotics, does not increase the electrocardiogram (ECG) QT interval. In short term clinical trials extrapyramidal effects, sedation, akathisia, nausea, dizziness, vomiting, anxiety, and constipation were observed. One review characterized the frequency of these events as “not greatly different from that seen in patient treated with placebo”[5] but a second called the incidence of movement-related disorders “rather high”[6][7] .
Pharmacodynamics
Cariprazine acts as an antipsychotic that is effective against the positive and negative symptoms of schizophrenia.[8] Unlike many antipsychotics that are D2 and 5-HT2Areceptor antagonists, cariprazine is a D2 and D3partial agonist. It also has a higher affinity for D3 receptors. The D2 and D3 receptors are important targets for the treatment of schizophrenia, because the overstimulation of dopamine receptors has been implicated as a possible cause of schizophrenia.[9] Cariprazine acts to inhibit overstimulated dopamine receptors (acting as an antagonist) and stimulate the same receptors when the endogenous dopamine levels are low. Cariprazine’s high selectivity towards D3 receptors could prove to reduce side effects associated with the other antipsychotic drugs, because D3receptors are mainly located in the ventral striatum and would not incur the same motor side effects (extrapyramidal symptoms) as drugs that act on dorsal striatum dopamine receptors.[8] Cariprazine also acts on 5-HT1A receptors, though the affinity is considerably lower than the affinity to dopamine receptors (seen in monkey and rat brain studies).[8][10] In the same studies, cariprazine has been noted to produce pro-cognitive effects, the mechanisms of which are currently under investigation. An example of pro-cognitive effects occurred in pre-clinical trials with rats: rats with cariprazine performed better in a scopolamine-induced learning impairment paradigm in a water labyrinth test. This may be due to the selective antagonist nature of D3 receptors, though further studies need to be conducted.[8] This result could be very useful for schizophrenia, as one of the symptoms includes cognitive deficits.
Cariprazine has partial agonist as well as antagonist properties depending on the endogenous dopamine levels. When endogenous dopamine levels are high (as is hypothesized in schizophrenic patients), cariprazine acts as an antagonist by blocking dopamine receptors. When endogenous dopamine levels are low, cariprazine acts more as an agonist, increasing dopamine receptor activity.[11] In monkey studies, the administration of increasing does of cariprazine resulted in a dose-dependent and saturable reduction of specific binding. At the highest dose (300 μg/kg), the D2/D3 receptors were 94 % occupied, while at the lowest dose (1 μg/kg), receptors were 5 % occupied.[10]
Cariprazine has high oral bioavailability and can cross the blood brain barrier easily in humans because it is lipophilic.[2] In rats, the oral bioavailability was 52 % (with a dose of 1 mg/kg).[7]
Example 1 1-(2,3-dichlorophenyl)-[1,4]diazepine (starting material)
2.25 g (10 mmol) 1-bromo-2,3-dichloro-benzene was dissolved in dry toluene (50 ml), 2.3 (11 mmol) of [1 ,4]diazepine-1 -carboxylic acid tert-butylester was added followed by 0.2 g BINAP (2,2-bis(diphenylρhosphino)-1 ,1′-binaphtyl), 85 mg tris(dibenzylideneacetone)dipalladium(0) and 1.2 g (12mmol) sodium-tert-butoxyde. The reaction mixture was refluxed for eight hours and filtered. The organic layer was washed with water, dried and evaporated in vacuo. The residue was purified by chromatography and deprotected at 10 °C using 20 ml ethylacetate saturated with gaseous hydrochloric acid, the precipitate was filtered giving 2.1 g (yield: 75 %) hydrochloride salt of the title compound, melting at 182-3 °C. Example 2 Trans-N-{4-[2-[4-(2,3-dichloro-phenyl)-hexahydro-[1 ,4]diazepin-1-yl]-ethyl]- cyclohexyl}-carbamic acid tert-butylester (intermediate) 0.7 g (2.5 mmol) of 1 -(2,3-dichlorophenyl)-[1 ,4]diazepine hydrochloride and
0.6 g (2.5 mmol) of frat?s-2-{1 -[4-(N-tert-butyloxycarbonyl)amino]cyclohexyl}- acetaldehyde were dissolved in dichloroethane (35 ml), 0.35 ml (2.5 mmol) triethylamine was added, then 0.79 g (3.7 mmol) sodium triacetoxyborohydride was added portionswise and the reaction mixture was stirred for 20 hours at ambient temperature, then 20 % potassium carbonate solution in water (20 ml) was added. The organic layer was separated, dried and evaporated to dryness in vacuo. The precipitate was recrystallized from acetonitrile to give the title compound 1 .0 g (yield: 85.8 %), m.p.: 95-8 °C. Example 3
0.93 g (2.1 mmol) frarjs-N-{4-[2-[4-(2,3-dichloro-phenyl)-hexahydro- [1 ,4]diazepin-1 -yl]-ethyl]-cyclohexyl}-carbamic acid tert-butylester was deprotected at
10 °C using 15 ml ethylacetate saturated with gaseous hydrochloric acid, after 4 hours the precipitate was filtered giving 0.91 g (yield: 98 %) dihydrochloride salt of the title compound, melting at 260-6 °C. Method A
Trans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yi]-ethyl]-cyclohexyl}-3,3- dimethyl-urea (compound 1 ) 1 .39g (3 mmol) trans-4-{2-[4-(2,3-dichlorophenyl)-ρiperazin-1 -yl]-ethyl}- cyclohexyl-amine trihydrochloride was suspended in dichloromethane (100 ml), triethylamine (2.1 ml, 15 mmol) was added followed by 0.30 ml (3.3 mmol) N,N- dimethylcarbamoylchloride. The reaction mixture was stirred for 48 hours at room temperature, filtered. The filtrate was washed with water (2 x 20 ml), dried and evaporated in vacuo. Recrystallizing from methanol gave the title compound (0.83 g, 65 %), melting at 212-4 °C.
Method B
7rans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3-ethyl- urea (compound 2) 0.56g (1.2 mmol) trans-4-{2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl}- cyclohexyl-amine was dissolved in dry dichloromethane (20 ml), ethylisocyanate (0.1 ml, 1.3 mmol) was added and the reaction mixture was stirred at room temperature for 4 hours. The solvent was removed in vacuo. The residue was stirred with water, the precipitate was filtered, giving the title compound (0.33 g, 65 %). Melting point:
235-8 °C.
Method C rrans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3- dimethyl-urea (compound 1 )
0.56g (1.2 mmol) trans-4-{2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl}- cyclohexyl-amine trihydrochloride was suspended in dry dichloromethane (50 ml), triethylamine 0.77 ml, 6 mmol) was added and 0.13g (0.44 mmol) triphosgene dissolved in dichloromethane was dropped in. After one hour stirring at room temperature dimetilamine hydrochloride (0.49 g, 6 mmol) followed by triethylamine (0.84 ml, 6 mmol) was added and the stirring was continued for 20 hours. The mixture was filtered, the filtrate washed with water, dried and evaporated in vacuo. Recrystallizing the product from methanol gave the title compound (0.27 g, 52 %). Melting point: 212-4 °C.
U.S. Patent Publication No. 2006/0229297 discloses (thio)-carbamoyl-cyclohexane derivatives that are D3 and D2 dopamine receptor subtype preferring ligands, having the formula (I):
(I)
wherein R1, R2, X, and n are as defined therein. One particular compound disclosed therein is trans-1{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea, which is also known as trans-4-{2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl}-N,N-dimethylcarbamoyl-cyclohexylamine, the structural formula for which is shown below:
Compounds of formula (I) act as a dopamine receptor antagonists, particularly D3/D2 receptor antagonists, and are useful in the treatment and prevention of pathological conditions which require modulation of dopamine receptors.
In some cases, an appropriate salt of an active may improve certain properties suitable for pharmaceutical compounds (i.e., stability, handling properties, ease of large scale synthesis, etc.). However, selection of a suitable salt for a particular active agent is not always straightforward, since the properties of salts of different compounds formed with the same salt forming agent may differ greatly. Moreover, formation of particular salts of a compound possessing more than one basic centre may be difficult to achieve in high yield due to formation of multiple products.
We have surprisingly found that by reacting trans 4-{2-[4-(2,3-dichlorophenyl)- piperazine-l-yl]-ethyl}-cyclohexylamine of formula (III)
with a carbonic acid derivative of general formula (VI)R-O-CO-Z (VI)
then reacting the compound of general formula (IV) obtained
with an amine derivative of general formula (V)
get the compounds of general formula (I)
EXAMPLES
The invention is illustrated by the following non-limiting examples.
Example 1
Trans N-(4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-cyclohexyl)-carbamic acid methylester 6.45 g (0.015 mol) of dihydrochloride of compound of formula (III) was added to a mixture of 125 ml dichloromethane and 12.25 ml triethylamine and the thick suspension obtained was stirred at a temperature between 20-25°C for one hour. The so obtained suspension was added to a solution of 2.3 ml (0.03 mol) methyl chloroformate in 25 ml of dichloromethane at a temperature between 5-10°C. The reaction mixture obtained was stirred at a temperature between 20-25°C for 3 hours then extracted with 3×150 ml (150 g) of distilled water. The organic phase was evaporated in vacuum and the residue was recrystallized from methanol. In this manner 4.5 g of the title product was obtained.
Yield: 72 %.
Melting point: 143-147 °C
Example 2
Trans N-(4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-cyclohexyl)-carbamic acid isopropylester
6.45 g (0.015 mol) of dihydrochloride of compound of formula (III) was added to a mixture of 125 ml dichloromethane and 12.25 ml of triethylamine and the thick suspension obtained was stirred at a temperature between 20-25°C-on for one hour. The suspension was added to a solution of 3.7 g (0.03 mol) of isopropyl chloroformate in 30 ml of toluene at a temperature between 5-10°C. The reaction mixture was stirred at a temperature between 20-25°C for 3 hours and then extracted with 3×150 ml (150 g) of distilled water. The organic phase was evaporated in vacuum and the residue obtained was recrystallized from isopropanole.
In this manner 4,4 g of title compound was obtained. Yield: 67 %.
Melting point: 128-131°C
Example 3
Trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-N,N-dimethylcarbamoyl- cyclohexylamine
6.45 g (0.015 mol) of dihydrochloride of compound of formula (III) was added to a mixture of 125 ml of dichloromethane and 12.25 ml of triethylamine and the thick suspension obtained was stirred at a temperature between 20-25°C for one hour. The suspension was added to a solution of 4.9 g of bis(trichloromethyl)carbonate in 50 ml of dichloromethane at a temperature between -5-(-10)°C for one hour. The reaction mixture obtained was added to a solution of 13 g dimethylamine in 100 ml isopropyl alcohol (IP A) (40 ml, 0.12 mol) cooled at a temperature between 0-(-10)°C during which the temperature of the reaction mixture was kept under 0°C. After stirring at a temperature between 0-(-5)°C for 30 minutes to the reaction mixture 100 ml of distilled water was added under stirring. Then the pH of the aqueous phase was adjusted to 7-8 by adding concentrated hydrochloric acid and volume of the reaction mixture was concentrated to 130 ml under vacuum. To the reaction mixture obtained additional 70 ml of distilled water was added and the mixture was concentrated to 170 ml under vacuum. The suspension was stirred at 20-25°C for one hour and the product obtained was isolated by filtration.
In this manner 6.6 g of title compound was obtained.
Yield: 95 %
Melting point: 208-211 °C Example 4
Trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyI}-N,N-dimethylcarbamoyl- cyclohexylamine 4.4 g (0.011 mol) of trans N-(4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}- cyclohexyl)-carbamic acid methylester was dissolved in 120 ml of dichloromethane. The solution obtained was added to a solution of 13 g dimethylamine in 100 ml isopropyl alcohol (IP A) (100 ml, 0.3 mol) cooled at a temperature between 0-(-10)°C during which the temperature of the reaction mixture was kept under 0°C. After stirring at a temperature between 0-(-5)°C for 30 minutes to the reaction mixture 100 ml of distilled water was added under stirring. Then the pH of the aqueous phase was adjusted to 7-8 by adding concentrated hydrochloric acid and volume of the reaction mixture was concentrated to 100 ml under vacuum. To the reaction mixture obtained additional 70 ml of distilled water was added and the mixture was concentrated to 120 ml under vacuum. The suspension was stirred at 20-25°C for one hour and the product obtained was isolated by filtration.
In this manner 4.3 g of title compound was obtained.
Yield: 95 %
Melting point: 208-211 °C
Example 5
Trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-N,N-dimethylcarbamoyl- cyclohexylamine hydrochloride 6.45 g (0.015 mol) dihydrochloride of formula (III) was added to a mixture of 125 ml of dichloromethane and 12.25 ml of triethylamine and the thick suspension obtained was stirred at a temperature between 20-25°C for one hour. The suspension was added to the solution of 4.9 g of bis(trichloromethyl)carbonate in 50 ml of dichloromethane at a temperature between -5-(-10)°C for one hour. The reaction mixture obtained was added to a solution of 13 g dimethylamine in 100 ml isopropyl alcohol (IP A) (40 ml, 0.12 mol) cooled at a temperature between 0-(-10)°C during which the temperature of the reaction mixture was kept under 0°C. After stirring at a temperature between 0-(-5)°C for 30 minutes 100 ml of distilled water was added to the reaction mixture under stirring. Then the pH of the aqueous phase is adjusted to 2-3 by adding concentrated hydrochloric acid and the reaction mixture was concentrated to 130 ml, additional 70 ml of distilled water was added and the mixture was concentrated to 170 ml. The suspension was stirred at 20-25°C for one hour and the product obtained was isolated by filtration.
In this manner 6.7 g of title compound was obtained.
Yield: 96 %
Melting point: 221-224 °C
Example 6
Trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-N,N-dimethylcarbamoil- cyclohexylamine hydrochloride 6.72 g (0.015 mol) dihydrochloride monohydrate of compound of formula (III) was added to a mixture of 125 ml of dichloromethane and 12.25 ml of triethylamine and the thick suspension obtained was stirred at a temperature between 20-25 °C for one hour. The suspension was added to the solution of 4.9 g of bis(trichloromethyl)carbonate in 50 ml of dichloromethane at a temperature between -5-(-10)°C for one hour. The reaction mixture obtained was added to a solution of 13 g dimethylamine in 100 ml isopropyl alcohol (IP A) (40 ml, 0,12 mol) cooled at a temperature between 0-(-10)°C during which the temperature of the reaction mixture was kept under 0°C. After stirring at a temperature between 0-(-5)°C for 30 minutes to the reaction mixture 100 ml of distilled water was added and the pH of the aqueous phase was adjusted to 2-3 by adding concentrated hydrochloric acid. The reaction mixture was concentrated to 130 ml under vacuum then additional 70 ml of water was added and the mixture was concentrated to 170 ml. The suspension was stirred at a temperature between 20-25°C for one hour and the product obtained was isolated by filtration.
In this manner 6.7 g of title compound was obtained.
Éva Ágai-Csongor, György Domány, Katalin Nógrádi, János Galambos, István Vágó, György Miklós Keserű, István Greiner, István Laszlovszky, Anikó Gere, Éva Schmidt, Béla Kiss, Mónika Vastag, Károly Tihanyi, Katalin Sághy, Judit Laszy, István Gyertyán, Mária Zájer-Balázs, Larisza Gémesi, Margit Kapás, Zsolt Szombathelyi
Cariprazine, a potential atypical antipsychotic agent has been identified during the optimization of novel series of 4-aryl-piperazine derivatives. The recently available top line results from pivotal clinical trials demonstrated the safety and efficacy of cariprazine in bipolar mania and schizophrenia indications.
Biased agonism offers an opportunity for the medicinal chemist to discover pathway-selective ligands for GPCRs. A number of studies have suggested that biased agonism at the dopamine D2 receptor (D2R) may be advantageous for the treatment of neuropsychiatric disorders, including schizophrenia. As such, it is of great importance to gain insight into the SAR of biased agonism at this receptor. We have generated SAR based on a novel D2R partial agonist, tert-butyl (trans-4-(2-(3,4-dihydroisoquinolin-2(1H)-yl)ethyl)cyclohexyl)carbamate (4). This ligand shares structural similarity to cariprazine (2), a drug awaiting FDA approval for the treatment of schizophrenia, yet displays a distinct bias toward two different signaling end points. We synthesized a number of derivatives of 4 with subtle structural modifications, including incorporation of cariprazine fragments. By combining pharmacological profiling with analytical methodology to identify and to quantify bias, we have demonstrated that efficacy and biased agonism can be finely tuned by minor structural modifications to the head group containing the tertiary amine, a tail group that extends away from this moiety, and the orientation and length of a spacer region between these two moieties.
Using 50 (40 mg, 112 μmol) as the amine, following general procedure F the product was eluted (CHCl3/CH3OH, 20:1 to 10:1) to give the title compound as a white solid (27 mg, 56%).
Following an adapted literature procedure,(38) 10% Pd/C (881 mg, 828 μmol) was carefully added to an orange suspension of 5 (5.00 g, 27.6 mmol) in H2O (150 mL). The reaction mixture was hydrogenated on a Parr shaker at 60 psi at rt for 3 days until the uptake of hydrogen was complete and no starting materials remained by TLC (CHCl3/CH3OH, 1:1). The mixture was filtered through a Celite pad and washed with water (30 mL), and the filtrate evaporated to dryness in vacuo to reveal a white solid. The material was taken up in absolute EtOH (70 mL) to which concentrated HCl (10 mL) was addedm and the mixture was heated at reflux for 2 h. TLC confirmed ethyl ester formation, and the solvents were concentrated in vacuo. The material was basified with a 1 M NaOH solution to pH 14, and a white precipitate emerged. The product was then extracted from the mixture with EtOAc (3 × 30 mL), and the combined organic extracts were washed with brine and then dried over anhydrous Na2SO4. The product was then converted to the HCl salt by the addition of 1 M HCl in Et2O (27.6 mL, 27.6 mmol), and the solvents were concentrated to half volume in vacuo. The solution was then cooled to 0 °C, resulting in fractional crystallization of the trans stereoisomer as a white solid, which was then collected by filtration and washed with cold CH3CN (1.34 g, 22%). mp: 164–166 °C (lit.(J. Med. Chem.1998, 41, 760– 77)
the metabolite of the present invention is selected from:
EXAMPLESThe metabolites of the present invention were synthetized according to the following procedures:Example 1Trans-1-{4-[2-[4-(2,3-dichlorophenyl)-1-oxo-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea (compound D)
0.8 g (1.6 mmol) trans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea was dissolved in dichloromethane (60 ml). A solution of 0.54 g (2.4 mmol) 3-chloro-perbenzoic acid in dichloromethane (10 ml) was dropped in and the reaction mixture stirred for 24 hours at room temperature. The reaction was monitored by TLC. The solution was washed twice with saturated NaHCO3 solution, the organic layer dried and evaporated in vacuo. Flash chromatography gave 0.45 g (63.3%) of the title compound melting at 175-8° C.
Example 2Trans-1-{4-[2-[4-(2,3-dichloro-4-hydroxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea (compound C)
0.92 g (2 mmol) trans-4-{2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl}-cyclohexyl-amine dihydrochloride was suspended in dichloromethane (60 ml), triethylamine (1.26 ml, 9 mmol) was added followed by 0.21 ml (2.3 mmol) N,N-dimethylcarbamoylchloride. The reaction mixture was stirred for 48 hours at room temperature. The solution was washed with water (2×10 ml), dried and evaporated in vacuo. Purification with flash chromatography gave 0.66 g trans-1-{4-[2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea, melting at 196-8° C. This product was dissolved in dichloromethane (60 ml), then 6.4 ml (6.4 mmol) borontribromid solution (1M in CH2Cl2) was dropped in at 5° C. and the mixture stirred at room temperature for 24 hours. The reaction was monitored by TLC. 4 ml methanol was added, followed by 25 ml saturated NaHCO3 solution. After separation the organic layer was dried and evaporated in vacuo. Purification with flash chromatography gave 0.4 g of the title compound, melting at 278-80° C.
Example 3Trans-1-{4-[2-[4-(2,3-dichloro-4-hydroxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3-methyl-urea (compound B)
1.38 g (3 mmol) trans-4-{2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl}-cyclohexyl-amine dihydrochloride was suspended in dry dichloromethane (100 ml), triethylamine (1.72 ml, 12.4 mmol) was added and 0.34 g (1.14 mmol) triphosgene dissolved in dichloromethane was dropped in. After one hour stirring at room temperature methylamine (33% solution in ethanol) was added and the stirring was continued for 20 hours. The mixture was evaporated. 20 ml water was added, the precipitate filtered, washed with water, dried. Recrystallizing the product from methanol gave trans-1-{4-[2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3-methyl-urea (0.86 g, 65%) melting above 250° C. This product was dissolved in dichloromethane (60 ml), then 10 ml (10 mmol) borontribromid solution (1M in CH2Cl2) was dropped in at 5° C. and the mixture stirred at room temperature for 24 hours. The reaction was monitored by TLC. 4 ml methanol was added and the mixture evaporated. 35 ml saturated NaHCO3 solution was added. The precipitate was filtered, washed with water and dried, recrystallized from methanol giving 0.34 g of title compound, melting at 237-41° C.
Example 4Trans-1-{4-[2-[4-(2,3-dichloro-4-hydroxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-urea (compound A)
1.38 g (3 mmol) trans-4-{2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl}-cyclohexyl-amine dihydrochloride was suspended in dry dichloromethane (100 ml), triethylamine 1.72 ml, 12.4 mmol) was added and 0.34 g (1.14 mmol) triphosgene dissolved in dichloromethane was dropped in. After one hour stirring at room temperature ammonia (20% solution in methanol) was added and the stirring was continued for 20 hours. The mixture was evaporated. 20 ml water was added, the precipitate filtered, washed with water, dried. Recrystallizing the product from methanol gave 0.86 g trans-1-{4-[2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-urea melting above 250° C. This product was dissolved in dichloromethane (60 ml), then 10 ml (10 mmol) borontribromid solution (1M in CH2Cl2) was dropped in at 5° C. and the mixture stirred at room temperature for 24 hours. The reaction was monitored by TLC. 4 ml methanol was added and the mixture evaporated. 35 ml saturated NaHCO3 solution was added. The precipitate was filtered, washed with water and dried, recrystallized from methanol giving 0.37 g of title compound, melting at 195-8° C.
Kiss B; Horváth A; Némethy Z; Schmidt E; Laszlovszky I; Bugovics G; Fazekas K; Hornok K; Orosz S; Gyertyán I; Agai-Csongor E; Domány G; Tihanyi K; Adham N; Szombathelyi Z (2010). “Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile”. The Journal of Pharmacology and Experimental Therapeutics333 (1): 328–340. doi:10.1124/jpet.109.160432. PMID20093397.
Gründer G (2010). “Cariprazine, an orally active D2/D3 receptor antagonist, for the potential treatment of schizophrenia, bipolar mania and depression”. Current Opinion in Investigational Drugs11 (7): 823–832. PMID20571978.
Citrome, L (February 2013). “Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability”. Expert Opinion on Drug Metabolism and Toxicology9 (2): 193–206. doi:10.1517/17425255.2013.759211. PMID23320989.
Citrome L (February 2013). “Cariprazine in schizophrenia: clinical efficacy, tolerability, and place in therapy”. Adv Ther30 (2): 114–26. doi:10.1007/s12325-013-0006-7. PMID23361833.
Veselinović T, Paulzen M, Gründer G (November 2013). “Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression”. Expert Rev Neurother13 (11): 1141–59. doi:10.1586/14737175.2013.853448. PMID24175719.
Citrome, L (February 2013). “Cariprazine in Schizophrenia: Clinical Efficacy, Tolerability, and Place in Therapy”. Advances in Therapy30 (2): 114–126. doi:10.1007/s12325-013-0006-7.PMID23361833.
Domany, G.
Discovery of novel dopamine D3/D2 ligands for the treatment of schizophrenia
234th ACS Natl Meet (August 19-23, Boston) 2007, Abst MEDI 383
crystalline form of trans-1 {4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea hydrochloride (Form III) Cariprazine {RGH-188); Dysfunction of the dopaminergic neurotransmitter system is involved in the pathology of several neuropsychiatric and neurodegenerative disorders
SB-207499 is a potent second-generation inhibitor of PDE4 (phosphodiesterase-4) with decreased side effects versus those of the well-known first-generation inhibitor, (R)-rolipram. SB-207499 is in clinical development both for asthma and chronic obstructive pulmonary disease (COPD)……..J. Med. Chem.1998, 41, 821
Cilomilast (Ariflo™, SB 207499) is an orally active, second-generation phosphodiesterase (PDE) 4 inhibitor that is being developed by GlaxoSmithkline for the treatment of chronic obstructive pulmonary disease (COPD). The results of Phase I and Phase II studies have demonstrated that cilomilast significantly improves lung function and quality of life to a clinically meaningful extent, which has led to a comprehensive Phase III programme of research evaluating efficacy, safety and mechanism of action. However, the results of those Phase III studies are unremarkable and disappointing, raising doubt over the future of cilomilast as a novel therapy for COPD. This review summarizes data obtained from the Phase III clinical development programme, highlights some of the potential concerns both specific to cilomilast and to PDE4 inhibitors in general and assesses the likelihood that cilomilast will reach the market.
Cilomilast is GlaxoSmithKline’s selective phosphodiesterase type 4 (PDE4) inhibitor. The drug candidate had been preregistered in the U.S. for the maintenance of lung function in patients with chronic obstructive pulmonary disease (COPD) who are poorly responsive to albuterol. GlaxoSmithKline received an approval letter from the FDA in October 2003, however, in 2007, the company discontinued development of the compound. In 2008, the product was licensed to Alcon by GlaxoSmithKline for the treatment of eye disorders.
Phosphodiesterase (PDE) inhibitors, such as theophylline, have been used to treat Chronic Obstructive Pulmonary Disease (COPD) for centuries; however, the clinical benefits of these agents have never been shown to out-weigh the risks of their numerous adverse effects. Four clinical trials were identified evaluating the efficacy of cilomilast, the usual randomized, double-blind, and placebo-controlled protocols were used. It showed reasonable efficacy for treating COPD, but side effects were problematic and it is unclear whether cilomalast will be marketed, or merely used in the development of newer drugs.[2][3]
Christensen, Siegfried B.; Guider, Aimee; Forster, Cornelia J.; Gleason, John G.; Bender, Paul E.; Karpinski, Joseph M.; Dewolf,, Walter E.; Barnette, Mary S. et al. (1998). “1,4-Cyclohexanecarboxylates: Potent and Selective Inhibitors of Phosophodiesterase 4 for the Treatment of Asthma”. Journal of Medicinal Chemistry41 (6): 821–35. doi:10.1021/jm970090r. PMID9526558.
The reaction of 3-cyclopentyloxy-4-methoxybenzaldehyde (I) with LiBr, trimethylsilyl chloride (TMS-Cl) and 1,1,3,3-tetramethyldisiloxane in acetonitrile gives the corresponding benzyl bromide (II), which by reaction with NaCN in DMF affords 2-(3-cyclopentyloxy-4-methoxyphenyl)acetonitrile (III).
The condensation of (III) with methyl acrylate (IV) by means of Triton B in refluxing acetonitrile yields the 4-cyanopimelate (V), which is cyclized by means of NaH in refluxing DME, giving the 2-oxocyclohexanecarboxylic ester (VI). The decarboxylation of (VI) by means of NaCl in DMSO/water at 150 C yields the cyclohexanone (VII), which is condensed with 2-(trimethylsilyl)-1,3-dithiane (VIII) by means of BuLi in THF, affording the cyclohexylidene-dithiane (IX).
The methanolysis of (IX) catalyzed by HgCl2 and HClO4 in refluxing methanol gives a mixture of the cis- and trans-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexanecarboxylic acid methyl ester which is submitted to flash chromatography to obtain the cis-isomer (XII). Finally, this compound is hydrolyzed with KOH in methanol/THF/water.
The synthesis of SB-207499 is described. Investigation and development of new strategies for the homologation of ketone, 4-cyano-4-[3-(cyclopentyloxy)-4-(methoxyphenyl)]-cyclohexan-1-one 2 are described which produce SB-207499. Our ultimate route of synthesis to SB-207499 is robust and operationally simple and produces the final drug substance in good yield and purity.
cis-{-4-cyano-4-[3- (trans-3-hydroxycyclopentyloxy)-4-methoxyphenyl]cyclohexane-l -carboxylic acid} or the corresponding compounds as defined by Formula I. The preparation of any remaining compounds of the Formula (I) not described therein may be prepared by the analogous processes disclosed herein which comprise:
Example 1
Preparation of cis-r4-cvano-4-(3-cyclopentyloxy-4-methoxyphenyl)cvclohexane- 1 – carboxylic acid]
1 fa (3-Cyclopentyloxy-4-methoxyphenv acetonitrile
To a solution of 3-cyclopentyloxy-4-methoxybenzaldehyde (20 g, 90.8 mmol) in acetonitrile (100 mL) was added lithium bromide (15 g, 173 mmol) followed by the dropwise addition of trimethylsilylchloride (17.4 mL, 137 mmol). After 15 min, the reaction mixture was cooled to 0° C, 1,1,3,3-tetramethyldisiloxane (26.7 mL, 151 mmol) was added dropwise and the resulting mixture was allowed to warm to room temperature. After stirring for 3 h, the mixture was separated into two layers. The lower layer was removed, diluted with methylene chloride and filtered through Celite®. The filtrate was concentrated under reduced pressure, dissolved in methylene chloride and refiltered. The solvent was removed in vacuo to provide a light tan oil. To a solution of this crude a- bromo-3-cyclopentyloxy-4-methoxy toluene in dimethylformamide (160 mL) under an argon atmosphere was added sodium cyanide (10.1 g, 206 mmol) and the resulting mixture was stirred at room temperature for 18 h, then poured into cold water (600 mL) and extracted three times with ether. The organic extract was washed three times with water, once with brine and was dried (K2CO3). The solvent was removed in vacuo and the residue was purified by flash chromatography (silica gel, 10% ethyl acetate/hexanes) to provide an off-white solid ( m.p. 32-34g C); an additional quantity of slightly impure material also was isolated. Kb Dimethyl 4-cvano-4-(‘3-cvclopentyloxy-4-methoxyphenv pimelate
To a solution of (3-cyclopentyloxy-4-methoxyphenyl)acetonitrile (7 g, 30.3 mmol) in acetonitrile (200 mL) under an argon atmosphere was added a 40% solution of Triton-B in methanol (1.4 mL, 3.03 mmol) and the mixture was heated to reflux. Methyl acrylate (27 mL, 303 mmol) was added carefully, the reaction mixture was maintained at reflux for 5 h and then cooled. The mixture was diluted with ether, was washed once with IN hydrochloric acid and once with brine, was dried (MgSO4) and the solvent was removed in vacuo. The solid residue was triturated with 5% ethanol/hexane to provide a white solid (m.p. 81-82° C); an additional quantity was also obtained from the filtrate. Anal. (C22H29NO6) calcd: C 65.49, H 7.25, N 3.47. found: C 65.47, H 7.11, N 3.49. 1. c) 2-Caf bomethoxy-4-cvano-4-(3-cyclopentyloxy-4-methoxyphen vDcvclohexan- 1 -one To a suspension of sodium methoxide (350 mL, 1.55 mol, 25% w/w in methanol) in toluene (2.45 L) heated to 80° C under a nitrogen atmosphere was added a solution of dimethyl 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)pimelate (350.0 g, 0.87 mol) in toluene (1.05 L) over 10 min. The reaction was heated to 85° C by distilling away 250 mL of solvent and was vigorously stirred under nitrogen for 2 hours. The reaction was cooled to 50° C and was quenched with 3N (aq) HC1 (700 mL, 2.1 mol). The organic layer was isolated, was washed once with deionized water (700 mL) and once with brine (700 mL). The organic layer was concentrated via low vacuum distillation to afford crude 2- carbomethoxy-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexane- 1 -one in toluene. This was dissolved in 4.2 L of dimethyl sulfoxide and used in the next step. 1 (d) 4-Cvano-4-f3-cyclopentyloxy-4-methoxyphenyl cvclohexan- 1-one
To a suspension of sodium chloride (315 g, 5.39 mol) and deionized water ( 315 mL) was added the dimethyl sulfoxide (4.2 L) solution of 2-carbomethoxy-4-cyano-4-(3- cyclopentyloxy-4-methoxyphenyl)cyclohexane-l-one ( 323 g, 0.87 mol) and the resulting suspension was heated to 155° C for 1.75 h. The reaction was cooled to 40° C, was quenched into 8 L of iced water (22 C) and was extracted with ethyl acetate (3.5 L). The aqueous layer was isolated and re-extracted with 2.5 L of ethyl acetate. The combined organic extract (6 L) was washed two times with deionized water (2 x 1 L) and once with brine (1 L). The organic layer was isolated and concentrated in vacuo to afford a residue. This residue was dissolved in refluxing isopropanol (500 mL), was cooled to 0° C and held at this temperature for 1 hour. The crystals were isolated by filtration, were washed with 250 mL of isopropanol (0° C), and were dried in a vacuum oven (45° C at 20 inches) to produce 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-l -one . m.p. 111-112° C; Anal. (C19H23NO ) calcd: C 72.82, H 7.40, N 4.47; found: C 72.72, H 7.39, N 4.48. 1 (e) 2-r4-Cyano-4-G-cyclopentyloxy-4-methoxyphenyl)cvclohexylidenel- 1.3-dithiane To a solution of 2-trimethylsilyl-l,3-dithiane (9.25 mL, 48.7 mmol) in dry tetrahydrofuran (80 mL) at 0° C under an argon atmosphere was added rapidly n- butyllithium (2.5M in hexanes, 19.2 mL, 48 mmol). After 10 min, the mixture was cooled to -78° C and a solution of 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-l- one (7.53 g, 23 mmol) in tetrahydrofuran (40 mL) was added. After 10 min, aqueous sodium chloride was added, the mixture was allowed to warm to room temperature and was diluted with water. This mixture was combined with the product of three substantially similar reactions conducted on ketone (3.04, 6.01 and 6.1 g, 48.3 mmol total), the combined mixture was extracted three times with methylene chloride, the extract was dried (MgSO4) and evaporated. Purification by flash chromatography (silica gel, 10% ethyl acetate/hexanes) provided a white solid, m.p. 115-116° C. \(f) cis-r4-Cyano-4-(3-cyclopentyloxy-4-methoxyphenyl‘)cyclohexane- 1 -carboxylic acidl
To a suspension of 2-[4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclo- hexylidene]-l,3-dithiane ( 140.0 g, 0.34 mol) in acetonitrile (500 mL) and deioinized water (140 mL) under nitrogen was added trifluoroacetic acid (136 g, 1.19 mol). The suspension was heated to 652 C for 1.25 h followed by the addition of 20% sodium hydroxide (420 g, 2.1 mol). The solution was heated at 70 to 75° C for an additional 1.25 h, was cooled to 45° C, deionized water (420 mL)was added followed by 3N (aq) HC1 (392 mL, 1.18 mol). The suspension was cooled to 5° C and held for 1 h. The suspension was filtered, was washed with cold (5e C) deionized water ( 200 mL), and was dried in a vacuum oven (40°C at 20 inches) to obtain crude cis-[4-cyano-4-(3-cyclopentyloxy-4- methoxyphenyl)cyclohexane-l -carboxylic acid]. This material was assayed at 98.5% and was found to a 98.8:1.2 mixture of cis-to-trans isomers, which was contaminated with 0.1% of residual 1,3-propanedithiol. This material was purified via an oxidative workup as follows.
To a hot solution (65° C) of crude cis-[4-cyano-4-(3-cyclopentyloxy-4- methoxyphenyl)cyclohexane-l -carboxylic acid] (85 g, 0.247 mol) in acetonitrile (425 mL) was added 1M sodium hydroxide ( 425 mL, 0.425 mol). To the solution (60° C) was added 4.25 g of calcium hypochlorite and the suspension was vigorously stirred for 2 h. The reaction was concentrated by distilling out 320 mL of solvent, followed by the addition of ethyl acetate ( 425 mL). The reaction was again concentrated by distilling out 445 mL of solvent, was cooled to 55° C followed by the addition of ethyl acetate (1.0 L) and 6N (aq.) HC1 (100 mL). The organic layer was isolated, was washed three times with deionized water (3 x 300 mL), was filtered and was concentrated by distilling out 530 mL of solvent. To the solution was added ethyl acetate (635 mL) with continued distillation to remove 750 mL of solvent. The solution was cooled to 65° C followed by the addition of hexane ( 340 mL). The suspension was cooled to 5° C, held at this temperature for 1 hour, was filtered and was washed with cold (5° C) 10% ethyl acetate/ hexane ( 200 mL). The solid was collected and was dried in a vacuum oven (40° C at 20 inches) to obtain cis- [4- cyano-4- (3-cyclopentyloxy-4-methoxyphenyl)cyclohexane- 1 -carboxylic acid] . This material was found to contain no trans isomer. Anal.(C2θH25-Nθ4) calcd: C 69.95, H 7.34, N 4.08; found: C 69.90, H 7.35, N 4.02. Example 2
Preparation of cis-f 4-cvano-4-r3-(trans-3-hydroxycyclopentyloxy)-4-methoxyphenyll- cyclohexane-1 -carboxylic acid)
To a solution of boron tribromide in dichlorormethane (0.1M, 335 mL, 33.5 mmol) under an argon atmosphere at -78° C was slowly added a solution of cis-[4-cyano-4-(3- cyclopentyloxy-4-methoxyphenyl)cyclohexane-l -carboxylic acid] (4.03 g, 11.7 mmol) in dichloromethane (180 mL). The mixture was stirred for 5 min, 15% sodium methoxide in methanol was added to pH 8-9 and the reaction was warmed to RT. Water (lOOmL) was added and the mixture was acidified with 3N aqueous hydrochloric acid to pH 1-2. The organic layer was separated, was dried (MgSO4/Na2SO4), was filtered and was evaporated. The residue was twice dissolved in chloroform and the solution was evaporated to yield a white solid. -1H NMR(400 MHz, CDCI3) δ 7.01 (d, J=2.4 Hz, 1H), 6.96 (d of d, J=2.4, 8.5 Hz, 1H), 3.89 (s, 3H), 2.31 (m, 1H), 2.21 (br t, J=13.6 Hz, 4H), 1.98 (m,2H), 1.77 (m, 2H); mp 190-193° C. Kb) Methyl cis- r-4-cvano-4-(3-hvdroxy-4-methoxyphenyl‘)cvclohexane-l-carboxylatel -Toluenesulfonic acid monohydrate (0.015 g, 0.08 mmol) was added to a solution of the compound of Example 2(a) (0.70 g, 2.54 mmol) in dry methanol (20 mL) under an argon atmosphere and the reaction was stirred for 6 h at 45-509 C. The reaction was cooled to RT and was stirred for an additional 16 h. The solution was evaporated and the residue was purified by flash chromatography (silica gel, 50% hexane/ethyl acetate) to yield the tide compound as a white solid. -1H NMR(400 MHz, CDC13) δ 7.01 (m, 2H), 6.85 (d, J=9.1 Hz, IH), 3.90 (s, 3H), 3.72 (s, 3H), 2.35 (t of t, J=3.6, 12.2 Hz, IH), 2.14-2.25 (m, 4H), 2.00 (app q, J=13.4 Hz, IH), 1.99 (app q, J=13.4 Hz, IH), 1.77 (app t, J=13.4 Hz, IH), 1.76 (app t, J=13.4 Hz, IH); mp 106-107° C.
The compound of Example 2(b) (0.69 g, 2.37 mmol) was dissolved in tetrahydrofuran (20 mL) under an argon atmosphere and was treated with triphenylphosphine (1.24 g, 4.74 mmol) and cis-l,3-cyclopentanediol (0.49 g, 4.74 mmol). Diethyl azodicarboxylate (0.83 g, 4.74 mmol) was added and the mixture was stirred at RT for 16 h. The solution was evaporated, the residue was diluted with ether and the white solid was removed by filtration. The filtrate was concentrated and the residue was purified by flash chromatography (silica gel, 50% hexane/ethyl acetate) to yield a mixture of the title compound and triphenylphosphine oxide. The mixture was diluted with ether and the white solid triphenylphosphine oxide was removed by filtration. Evaporation of the filtrate yielded the title compound as a sticky, colorless semi-solid. 1H NMR(400 MHz, CDCI3) δ 7.07 (d, J=2.4 Hz, IH), 7.02 (d of d, J=2.4, 8.8 Hz, IH), 6.87 (d, J=8.8 Hz, IH), 4.99 (m, IH), 4.37 (m, IH), 3.85 (s, 3H), 3.74 (s, 3H), 3.16 (d, J=9.1 Hz, IH), 2.39 (m, IH), 1.88-2.25 (m, 12H), 1.80 (br t, J=13.5 Hz, 2H).
The compound of Example 2(c) (0.10 g, 0.27 mmol) was dissolved in 5:5:2 tetrahydrofuran methanol/water (5 mL), sodium hydroxide (0.035 g, 0.88 mmol) was added and the mixture was stirred at RT for 3 h. The solvent was evaporated, the residue was partitioned between 5% aqueous NaOH and dichloromethane and the layers were separated. The aqueous layer was acidified to pH 3 with 3N aqueous hydrochloric acid and was extracted three times with 5% methanol in chloroform. The organic extracts were combined, were dried (MgSO4), filtered and evaporated. The residue was purified by flash chromatography (silica gel, 90:10:1 chloroform/methanol water) to yield a solid which was slurried in ether, was collected by filtration and was dried in vacuo to afford the title compound. MS(d/NH3) m e 377 [M + NH ]+; 1H NMR(400 MHz, CDCI3) δ 7.08 (br s, IH), 7.03 (br d, J=8.5Hz, IH), 6.88 (d, J=8.5 Hz, IH), 4.98 (m, IH), 4.38 (m, IH), 3.84 (s, IH), 2.41 (m, IH), 1.77-2.29 (m, 16H); Anal. (C2oH25NO5-»0.9 H2O) calcd: C, 63.95; H,7.19; N,3.73. found: C, 64.06; H, 6.88; N, 3.77; mp 161-163° C.
Example 3 Preparation of cis- f 4-cvano-4-r3-(cis-3-hvdroxycvclopentyloxy)-4-methoxyphenyll- cyclohexane-1 -carboxylic acid) 3(a) Methyl cis-(-4-cvano-4-r3-(cis-3-formyloxycvclopentyloxy)-4-methoxyphenyll- cvclohexane- 1 -carboxylate ) The compound of Example 2(c) (0.68 g, 1.83 mmol) was dissolved in tetrahyrofuran (20 mL) under an argon atmosphere and was treated with triphenylphosphine ( 0.96 g, 3.66 mmol) and formic acid (0.17 g, 3.66 mmol). Diethyl azodicarboxylate (0.64 g, 3.66 mmol) was added and d e mixture was stirred at RT for 16 h. The solution was evaporated, ether was added and the white solid was removed by filtration. The filtrate was concentrated and die residue was purified by flash chromatography (silica gel, 65% hexane/ethyl acetate) to yield the title compound as a clear colorless oil. **-H NMR(400 MHz, CDC13) δ 8.02 (s,lH), 7.0 (d of d, J=2.4, 8.2 Hz, IH), 6.99 (d, J=2.4 Hz, 1 H), 6.87 (d, J=8.2 Hz, IH), 5.48 (m, IH), 4.95 (m, IH), 3.84 (s, 3H), 3.72 (s, 3H), 2.31-2.40 (m, 2H), 2.13-2.28 (m, 7H), 1.96-2.06 (m, 3H), 1.74-1.87 (m, 3H).
The compound of Example 3(a) (0.52 g, 1.31 mmol) was dissolved in 5:5:2 tetrahydrofuran/methanol/water (20mL), sodium hydroxide (0.32 g, 8.0 mmol) was added and die mixture was stirred at RT for 2.5 h. The solvent was evaporated and the aqueous residue was acidified to pH 1-2 with 3N aqueous hydrochloric acid. The white solid product was collected, was washed with water and was dried in vacuo to afford the title compound as a white solid. MS(CI/NH3) m/e 377 [M + NH3]+;
Torphy TJ, Barnette MS, Underwood DC, Griswold DE, Christensen SB, Murdoch RD, Nieman RB, Compton CH. Ariflo (SB 207499), a second generation phosphodiesterase 4 inhibitor for the treatment of asthma and COPD: from concept to clinic. Pulmonary Pharmacology and Therapeutics. 1999;12(2):131-5. PMID 10373396
Ochiai H, Ohtani T, Ishida A, Kusumi K, Kato M, Kohno H, Kishikawa K, Obata T, Nakai H, Toda M. Highly potent PDE4 inhibitors with therapeutic potential. Bioorganic and Medicinal Chemistry Letters. 2004 Jan 5;14(1):207-10. PMID 14684329
WALTHAM, Mass., Dec. 21, 2014 (GLOBE NEWSWIRE) — Radius Health, Inc. today announced positive top-line 18-month fracture results from the Company’s Phase 3 clinical trial (ACTIVE) evaluating the investigational drug abaloparatide-SC for potential use in the reduction of fractures in postmenopausal osteoporosis.
BIM-44058 is a 34 amino acid analog of native human PTHrP currently in phase III clinical trials at Radius Health for the treatment of postmenopausal osteoporosis. Radius is also developing a microneedle transdermal patch using a 3M drug delivery system in phase II clinical trials. The drug candidate was originally developed at Biomeasure (a subsidiary of Ipsen), and was subsequently licensed to Radius and Teijin Pharma.
BMS-663068 is an HIV-1 attachment inhibitor in development for the treatment of HIV-1 infection. BMS-663068 is a prodrug for BMS-626529 which binds to the viral envelope glycoprotein gp120 and interferes with attachment of the virus to the cellular CD4 receptor. Administration of BMS-663068 for 8 days with or without ritonavir resulted in substantial declines in plasma HIV-1 RNA levels and was generally well tolerated. Longer-term clinical trials of BMS-663068 as part of combination antiretroviral therapy are warranted.
Fostemsavir (GSK3684934/BMS-663068) is an experimental HIVentry inhibitor and a prodrug of temsavir (BMS-626529). It is under development by [ViiV Healthcare / GlaxoSmithKline]] for use in the treatment of HIV infection. By blocking the gp120 receptor of the virus, it prevents initial viral attachment to the host CD4+ T cell and entry into the host immune cell; its method of action is a first for HIV drugs.[1] Because it targets a different step of the viral lifecycle, it offers promise for individuals with virus that has become highly resistant to other HIV drugs.[2] Since gp120 is a highly conserved area of the virus, the drug is unlikely to promote resistance to itself via generation of CD4-independent virus.[3]
Example 6Preparation of Compound I from Compound D′ (Example 5)
N-Benzoylpiperazine HCl, Compound Db, (11.73 g, 51.74 mmol) was added to a mixture of Compound D′ (14.83 g, 47.03 mmol) (prepared in Example 5) in dry THF (265 mL) and dry DMF (29.5 mL). NaOt-Bu, 30% w/w (52.3 mL, 147 mmol) was added dropwise (30 min.) keeping the temperature at 17-21° C. The resulting yellow slurry was stirred at 17-20° for 1 h more, then cooled to about 5° C. The mixture was slowly poured into cold water (90 mL) and the flask rinsed with additional water (10 mL). The pH of the resulting yellow solution was adjusted to 6-7 with slow addition (˜20 min., 5-12° C.) of 1 N HCl (105 mL). The resulting slurry was warmed and stirred at room temperature for 1.5 h. The slurry was filtered and the cake washed with water (2×60 mL) then dried in vacuo at 65-70° C. for 5 h giving 18.4 g Compound I as a white solid (82.6%), HPLC AP 99.4. 1H NMR (400 MHz, d6-DMSO): δ 2.48 (s, 3H), 3.43 (b, 4H), 3.67 (b, 4H), 3.99 (s, 3H), 7.45 (s, 5H), 7.88 (s, 1H), 8.24 (s, 1H), 9.22 (s, 1H), 12.39 (s, 1H). 13C NMR (100 MHz, d6-DMSO): 13.85, 40.65, 45.22, 56.85, 114.19, 121.02, 122.78, 123.65, 127.06, 128.42, 129.61, 129.70, 135.51, 138.59, 142.18, 149.23, 161.38, 166.25, 169.30, 185.51.
If necessary, the product could be further purified by recrystallization from acetic acid-water-ethanol, ethanol-water, or acetone-water. For example: A mixture of Compound I (25.0 g), glacial acetic acid (260 mL) and DI water (13.8 mL) was heated to 80° C. and held with stirring (overhead) until a solution was obtained (40 min.). The batch was cooled to 70° C. and seeded (0.5 g). With slow agitation (100 rpm), EtOH (300 mL) was added slowly (1 h), keeping the temperature at 70° C. The resulting slurry was kept at 70° C. for 1 h more with very slow stirring. The slurry was cooled to 20° C. over 2 hours and held at 20° C. for over 4 hours. The slurry was filtered, the wet cake washed with EtOH (125 mL), and the solid dried in vacuo at 70° C. (≧16 h), giving 22.6 g Compound I as a white solid (88.4%).
The development of a short and efficient synthesis of a complex 6-azaindole, BMS-663068, is described. Construction of the 6-azaindole core is quickly accomplished starting from a simple pyrrole, via a regioselective Friedel–Crafts acylation, Pictet–Spengler cyclization, and a radical-mediated aromatization. The synthesis leverages an unusual heterocyclic N-oxide α-bromination to functionalize a critical C–H bond, enabling a highly regioselective copper-mediated Ullmann–Goldberg–Buchwald coupling to install a challenging triazole substituent. This strategy resulted in an efficient 11 step linear synthesis of this complex clinical candidate
Attachment inhibitor BMS-663068 is currently in clinical development for the treatment of HIV infection. Key steps in the synthesis depicted are (1) a radical-mediated redox-aromatization to generate the 6-azaindole (B → C) and (2) the regioselective bromination of an N-oxide using PyBroP (D → E).
High regioselectivity was observed in the copper(I)-mediated Ullmann–Goldberg–Buchwald coupling (H → K) using the diamine ligand J (N1/N2 = 22:1), whereas a thermal SNAr reaction gave N1/N2 = 1:1. Alternative conditions for the bromination of the N-oxide D led mainly to deoxygenation.
Procedure: To a solution of the acid 6-81 (3.01 g, 10 mmol) and benzoylpiperazine hydrochloride (3.39 g, 15 mmol) in DMF (50 mL) was added triethylamine (10.1 g, 100 mmol, 10 eq.), followed by 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC; 5.75 g, 30 mmol) under N2 and the mixture stirred at room temperature for 22 h after sonication and at 40° C. for 2 h. The mixture was concentrated in vacuo to remove DMF and TEA, and to the residual solution was added water (200 mL) under stirring and sonication. The precipitates formed were collected, washed with water and dried in vacuo to obtain 2.8 g (5.9 mmol, Y. 59%) of the title compound IVc as off-white solid. The filtrate was extracted with CH2Cl2 (x2). The CH2Cl2 extracts were dried (Na2SO4), filtered and concentrated to gum which was triturated with Et2O to obtain a solid. This solid was suspended and triturated with MeOH to obtain 400 mg of the title compound IVc as off-white solid. Total yield: 3.2 g (6.8 mmol, Y. 68%): MS m/z 474 (MH); HRMS (ESI) m/z calcd for C24H24N7O4 (M+H) 474.1890, found 474.1884 (Δ-1.2 ppm); 1H NMR (DMSO-d6) δ ppm 2.50 (3H, s, overlapped with DMSO peaks), 3.43 (4H, br, CH2N), 3.68 (4H, br, CH2N), 3.99 (3H, s, CH3O), 7.46 (5H, br. s, Ar—Hs), 7.88 (1H, s, indole-H-5), 8.25 (1H, s, indole-H-2), 9.25 (1H, s, triazole-H-5), 12.40 (1H, s, NH); 13C-NMR (DMSO-d6) δ ppm 13.78 ,40.58, 45.11, 56.78, 114.11, 120.95, 122.71, 123.60, 126.98, 128.34, 129.6, 135.43, 138.52, 142.10, 149.15, 161.29, 166.17, 169.22, 185.42; UV (MeOH) λ max 233.6 nm (ε 3.43×104), 314.9 nm (ε 1.73×104); Anal: Calc for C24H24N7O4.1/5H2O; C, 60.42; H, 4.94; N, 20.55, Found; C 60.42, H 5.03, N 20.65; KF (H2O) 0.75%.
This reaction can also be performed by use of HATU and DMAP to provide more consistent yield of the title compound: To a suspension of the acid 6-81 (15.6 mmol) and HATU [O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophos phonate] (8.90 g, 23.4 mmol; 1.5 eq.) in DMF (60 mL) and CH2Cl2 (60 mL) was added a mixture of DMAP (5.72 g, 46.8 mmol, 3 eq.) and benzoylpiperazine hydrochloride (5.30 g, 23.4 mmol; 1.5 eq.) in DMF (60 mL) at room temperature and the mixture was stirred under nitrogen atmosphere for 4 hrs. The mixture was concentrated in vacuo to remove CH2Cl2 and most of DMF, and to the residual solution was added water under stirring and sonication. The precipitates formed were collected, washed with water and dried in vacuo to obtain 5.38 g (11.4 mmol, Y. 72.8%) of the title compound IVc as off-white solid: HPLC >95% (AP, uv at 254 nm)
EXAMPLE 5Preparation of Ica, (Disodium Salt)
General Procedure: A suspension of IVc (0.24 g, 0.5 mmol) in anhydrous THF (4 mL) under nitrogen atmosphere was treated with sodium hydride (60% oil dispersion, 0.08 g, 2.0 mmol), and stirred until gas evolution ceased (approximately 5 minutes). The reaction mixture was treated with iodine (0.13 g, 0.5 mmol) and stirred for 2-3 minutes followed by addition of di-tert-butyl chloromethyl phosphate (1.6 g, 6.0 mmol, crude). A stream of nitrogen was allowed to pass over the reaction to facilitate the removal of much or all of the THF. The reaction mixture was stirred overnight. HPLC analysis of crude indicated starting IVc (ca. 56%) and desired adduct (ca. 32%).
Several crude reaction mixtures (a total of 6.7 mmol based on starting material IVc) were re-dissolved in dichloromethane, combined, concentrated in vacuo to remove any remaining THF. The residue was suspended in dichloromethane and TFA (1:1, approximately 40 mL total volume). The mixture was stirred for 1.5-2 hours and then solvent was removed in vacuo. The residue was suspended in dichloromethane and extracted into water (approximately 60 mL) made weakly basic with solid or aqueous sodium bicarbonate. The aqueous layer was reduced in volume by rotary evaporator if required and the solution was loaded onto a C-18 reverse phase column (approximately 80 g of C-18, YMC ODS-Aq, 50 micron) and eluted with water, followed by water containing 2.5% acetonitrile. Fractions containing pure product were pooled and organic solvent was removed by rotary evaporator. Purified product was recovered after lyophilization to give 1.00 g (1.30 mmol, 19% over 2 steps) of the title compound Ica (disodium salt) as an off-white powder: HPLC purity>99% AP at 254 nm (gradient 0-100% B/A; A 10% CH3CN-90% H2O-0.1% TFA, B 90% CH3CN-10% H2O-0.1 % TFA, gradient time 4 min, column YMC ODS-Aq 4.6×50 mm 3 micron); MS-ESI— m/z 482 (M−H minus 2Na)−; HRMS (ESI) m/z calcd for C25H27N7O8P (M+H minus 2Na)+584.1659, found 584.1651 (Δ-1.3 ppm); 1H NMR (D2O, 500 MHz) δ ppm 2.53, 2.54 (3H, 2s), 3.56 (2H, s, CH2N), 3.72 (2H, br.s, CH2N), 3.78, 3.83 (2H, 2br.s, CH2N), 3.94, 3.96 (2H, 2br.s, CH2N), 4.14 (3H, s, CH3O), 5.38, 5.40 (2H, 2d, J=11 Hz), 7.45-7.59 (5H, m, Ar—Hs), 8.07, 8.09 (1H, 2s, indole-H-5), 8.64, 8.67 (1H, 2s, indole-H-2), 8.87, 8.89 (1H, 2s, triazole-H-5); 13C NMR (125.7 MHz, D2O) δ ppm 15.43 (N-Me), 44.03, 44.47, 44.66, 45.05, 48.20, 48.82, 49.60, 50.23, 59.78 (OMe), 75.81 (NCH2O), 115.6, 126.0, 127.2, 129.6, 131.0, 131.7, 132.1, 133.5, 136.8, 147.6, 150.1, 154.2, 164.8, 170.4, 175.8, 189.2; UV (H2O) λmax 220 nm (ε 3.91×104), 249 nm (ε 2.00×104), 303 nm (ε 1.60×104); Anal: Calc for C25H24N7O8PNa2. 8H2O. 0.2NaHCO3; C, 38.39; H, 5.14; N, 12.44, P, 3.93, Na, 6.42 Found; C, 38.16; H, 4.81; N, 12.43, P, 3.72, Na, 6.05; KF (H2O) 17.3%. A less pure fractions were collected to obtain 0.22 g (0.29 mmol, Y. 4%) of the title compound Ica (disodium salt): HPLC purity>95% (AP at 254 nm).
EXAMPLE 7Preparation of Crystalline Ic (Free Acid Mono-Hydrate)
To a mixture of IVc (600 mg, 1.27 mmol) in anhydrous THF (10 ml) in an oven-dried round bottle flask under nitrogen at r.t. was added NaH (153 mg, 6.38 mmol, dry powder, 95%), and the white suspension stirred until no gas evolution was observed. The mixture was then added I2 (375 mg, 1.48 mmol), and stirred at r.t. for 3 h. To the reaction mixture was added NaH (153 mg, 6.38 mmol, dry powder, 95%), and the mixture stirred for about 5 to 10 min. The crude chloromethyl di-tert-butylphosphate (2.0 g, about 1.6 ml, 7.79 mmol) was added to the mixture, which was then stirred at r.t. for 15 h. LCMS analysis of the reaction showed a >97% conversion of the starting material. After evaporation of the volatiles, the residue was added CH2Cl2 (10 ml), cooled in an ice-water bath, slowly added TFA (10 ml) and stirred at r.t. for 3 h. The reaction mixture was then evaporated, and the residue partitioned between CH2Cl2 (50 ml) and H2O (50 ml). The CH2Cl2 layer was poured into the reaction flask that contained some undissolved brownish solid, and this mixture was extracted with a dilute aqueous NaHCO3 solution (50 ml). The aqueous mixture was purified by reverse phase preparative HPLC (solvent A: 10% MeOH-90% H2O-0.1% TFA; solvent B: 90% MeOH-10% H2O-0.1% TFA; start % B=0, final % B=100; gradient time=6 min; flow rate=45 ml/min; column: phenomenex-Luna 30×50 mm, S5; fraction collected: 3.65 to 4.05 min). The fractions collected were evaporated to dryness, and the residue dried under high vacuum to obtain the acid Ic as a pale yellow solid (356.6 mg); 1H NMR: (500 MHz, CD3OD) δ 9.05 (s, 1H), 8.46 (s, 1H), 8.04 (s, 1H), 7.47 (b s, 5H), 5.93 (d, J=12, 2H), 4.10 (s, 3H), 4.00-3.40 (b s, 8H), 2.53 (s, 3H); 19F NMR analysis showed that the material contained residual TFA, (the percentage was not quantified); Analytical HPLC method: Start % B=0, Final % B=100, Gradient time=2 min, Flow Rate=5 mL/min, Column: Xterra MS C18 7u 3.0×50 mm, LC/MS: (ES+) m/z (M+H)+=584, HPLC Rt=0.983.
172.2 mg of the purified acid Ic was dissolved in 1 ml of H2O and then about 0.3 ml of absolute EtOH (200 proof) was added. The mixture was left standing in a refrigerator (temperature about 3° C.) overnight, after which time, crystalline material was observed. The mixture was then warmed to ambient temperature, diluted with H2O to a volumn of 3 mL, and then 20 mL of MeCN was added slowly. Following the completion of addition, the mixture was stirred at r.t. for 2 h and then filtered. The solid collected (90 mg) was dried in vacuo, and then under high vacuum. This material was shown by powder x-ray studies to be crystalline; Elemental Analysis calculated for C25H26N7O8P.H2O: C 49.92; H 4.69; N 16.30; observed: C 49.66; H 4.62; N 15.99; mp=205° C. (measured by differential scanning calorimetry). The 1H NMR pattern for crystalline material was compared with that from the purified acid and both were consistent with the structure.
EXAMPLE 10Preparation of Icb (mono tromethamine salt): [3-[(4-benzoylpiperazin-1-yl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2, 3-c]pyridin-1-yl]methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)propane-1,3-diol salt (1:1). The sequence of reactions is described in Scheme for Example 10.
Scheme for Example 10
Preparation of di-tert-butyl chloromethyl phosphate
A mixture of tetrabutylammonium di-tert-butyl phosphate (57 g, 0.126 mol, Digital Specialty Chemicals) and chloroiodomethane (221 g, 1.26 mol) was stirred at room temperature for four hours before the volatiles were removed under vacuum. 500 ml of ethyl ether was added to the residue and insoluble solid was filtered away. Concentration of the filtrate in vacuo and removal of remaining volatiles using a vacuum pump provided di-tert-butyl chloromethyl phosphate as a light brown or yellow oil, which was utilized in the next step without further purification.
Preparation of IIc: (3-(2-(4-benzoylpiperazin-1-yl)-2-oxoacetyl)-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl)methyl di-tert-butyl phosphate
NaH (2.6 g, 10.3 mmol, 95% in oil, Seq.) was added slowly into a suspension of IVc (10.0 g, 21.1 mmol) in dry THF (100 ml) and the mixture was allowed to stir for 0.5 hour at room temperature. A solution of iodine (5.27 g, 20.8 mmol) dissolved in dry THF (10 ml) was added slowly into the stirring solution at a rate which prevented foaming or a violent reaction. The resultant mixture was stirred for an additional 3 hours before a second 2.6 g portion of NaH was introduced. After 15 minutes at ambient temperature di-tert-butyl chloromethyl phosphate, the entire batch of di-tert-butyl chloromethyl phosphate, obtained from step one, was added. After stirring for 16 hours, the reaction mixture was poured into iced NH4OAc (30%) (120 ml), followed by extraction with EtOAc (3×300 ml). The combined organic extracts were washed with water (100 ml) and then brine (100 ml), dried over Na2SO4, and concentrated under vacuum to afford a residue, which was purified by silica gel chromatography (elution with EtOAc/Et3N (50/1) and then EtOAc/MeOH (100/1)) to give 8.0 g (˜75% AP, ˜41% yield) of diester IIc as a light yellow solid.
Preparation of Icb (mono L tromethamine salt): [3-[(4-benzoylpiperazin-1-yl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl]methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)propane-1,3-diol salt (1:1)
500 mg (˜p75 AP, 0.54 mmol) of diester IIc was dissolved in a mixture of water (2.5 ml) and acetone (2.5 ml). The resulting mixture was stirred at 40° C. for 16 hours to complete the solvolysis. To this reaction mixture was added 3.0M aqueous TRIS (mono tromethamine) solution to adjust pH to 3.32. Acetone (30 ml) was slowly added to the reaction mixture in 1 hour.* After complete addition of acetone, the solution was stirred overnight to complete the crystallization of Icb. The solid was collected by filtration and rinsed with 20:1 acetone-water (2×5 mL). The white crystalline solid was dried under house vacuum under nitrogen atomosphere at 50° C. for 24 h to afford 290 mg of Icb (>98.5 AP).
*After adding about 15 and 20 ml of acetone, the reaction mixture was seeded with crystalline Icb.
Obtained via other process (hydrolysis with TFA in methylene chloride), salt Icb is ˜1 molar mono tromethamine salt with 0.47% of water, 0.1% of acetone and 0.05% of methanol. 1H NMR (500 MHz, d6-DMSO, 30° C.) δ8.77 (s, 1H), 8.48 (s, 1H), 8.00 (s, 1H) 7.44 (b, 5H), 5.42 (d, 2H, J=15 Hz), 4.02 (s, 3H), 3.70-3.30 (m, 8H), 3.41 (s, 6H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3, 30° C.) δ184.8, 169.0, 165.8, 160.3, 150.4, 146.2, 143.2, 135.4, 129.4, 128.9, 128.2, 127.7, 126.9, 123.2, 122.2, 112.9, 72.3, 60.7, 59.0, 56.7, 13.4. MS m/z: (M-trisamine+H)+ calcd for C25H27N7O8P 584.2, found 584.0. Anal. Calcd. C, 49.11; H, 5.37; N, 15.76; P, 4.32; found: C, 48.88; H, 5.28; N, 15.71; P, 4.16. M.P. 201-205° C.
EXAMPLE 13Alternate preparation of Icb (Pro-drug of IVc)
To a 10 L reactor equipped with an overhead stirrer, thermocouple, distillation apparatus, and nitrogen inlet was charged IVc (200.00 g, 422.39 mmol), Cs2CO3 (344.06 g, 1.06 mol), KI (140.24 g, 844.81 mmol) and NMP (1.00 L, 10.38 mol). The reaction was stirred at room temperature resulting in a light brown heterogeneous suspension. Di-tert-butyl chloromethyl phosphate (273.16 g, 1.06 mol) was added via addition funnel and the reaction mixture was heated to 30° C. for 16-24 hours with stirring after which time the reaction was cooled to 5° C. To the reaction was added DCM (1.5 L) then the reaction was slowly quenched with water (3.5 L) maintaining the reaction temperature under 20° C. resulting in a biphasic mixture. The product rich bottom layer was separated, washed with water (3.5 L×3), then transferred back to the reactor. The solution was concentrated under vacuum to a volume of 1 L keeping the temperature below 25° C. IPA was added (2 L) then the reaction was concentrated under vacuum to a volume of 2 L keeping the temperature below 25° C. The reaction was then seeded with IIc (0.200 g), stirred overnight at room temperature resulting in a slurry. The slurry was filtered and the wet cake was washed with MTBE (1 L), dried in a vacuum oven at 50° C. overnight resulting in a yellow/white powder (207.1 g, 70%). 1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 8.18 (s, 1H), 7.91 (s, 1H), 7.42 (s, 5H), 5.95 (d, J=14.2 Hz, 2H), 4.06 (s, 3H), 3.97-3.36 (m, 8H), 2.50 (s, 3H), 1.27 (s, 18H); 3C NMR (100 MHz, CDCl3) δ 184.64, 170.65, 165.91, 161.60, 150.82, 145.38, 141.89, 134.96, 130.20, 129.59, 128.68, 127.58, 127.10, 124.77, 122.64, 115.22, 83.90, 83.83, 73.69, 73.63, 56.95, 46.04, 41.66, 29.61, 29.56, 13.90; ES+ MS m/z (rel. intensity) 696 (MH+,10), 640 (MH+-isobutylene, 30), 584 (MH+-2 isobutylene, 100).
To a 10 L 4 neck reactor equipped with a thermocouple, overhead stirrer, condenser and nitrogen inlet was added IIc (200.24 g, 287.82 mmol), acetone (800.00 ml, 10.88 mol) and water (800.00 ml, 44.41 mol). The reaction was heated to 40° C. and stirred for 18-24 hours. The reaction was cooled to 20° C. then tromethamine (33.62 g, 277.54 mmol) was added. The reaction was heated to 40° C. then stirred for an additional hour until all solids were dissolved. The reaction was cooled to 20° C. then filtered through a 10 micron cuno filter into a 10 L 4 neck reactor equipped with a thermocouple, overhead stirrer, and nitrogen inlet. Acetone (3 L) was added rapidly, followed by seeding with Icb (0.500 g), then additional acetone (3 L) was added. The reaction was stirred at room temperature overnight resulting in a slurry then filtered. The wet cake was washed with acetone (800 ml) then dried in a vacuum oven at 50° C. overnight resulting in a fluffy white powder (165.91 g, 82%).
Supplementary Information:
Isolation of the Free-Acid Intermediate IC:
In a 250 mL 3 neck reactor equipped with a thermocouple, overhead stirrer, condenser and nitrogen inlet was added IIc (10.0 g, 14.37 mmol), acetone (40.00 ml, 544.15 mmol) and water (40.00 ml, 2.22 mol). The reaction was heated to 40° C. and stirred for 14-24 hours. The reaction was cooled to 20° C. then stirred for three hours, resulting in a slurry. The slurry was filtered, then the wet cake washed with acetone (40.00 ml) then dried in a vacuum oven at 50° C. overnight resulting in a fluffy white powder (7.00 g, 83%). NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.47 (s, 1H), 8.06 (s, 1H), 7.45 (s, 5H), 5.81 (d, J=12.3 Hz, 2H), 4.03 (s, 3H), 3.91-3.19 (m, 8H), 2.39 (s, 3H); 13C NMR (500 MHz, DMSO-d6) δ 185.20, 169.32, 165.85, 160.75, 150.51, 146.30, 143.24, 135.53, 129.74, 129.22, 128.46, 127.34, 127.09, 123.67, 122.73, 113.94, 72.90 (d, 2JC-P=5 Hz), 57.01, 45.2 (bs), 40.8 (bs), 13.66. ES+ MS m/z (rel. intensity) 486 (MH+−H3PO4, 100).

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

















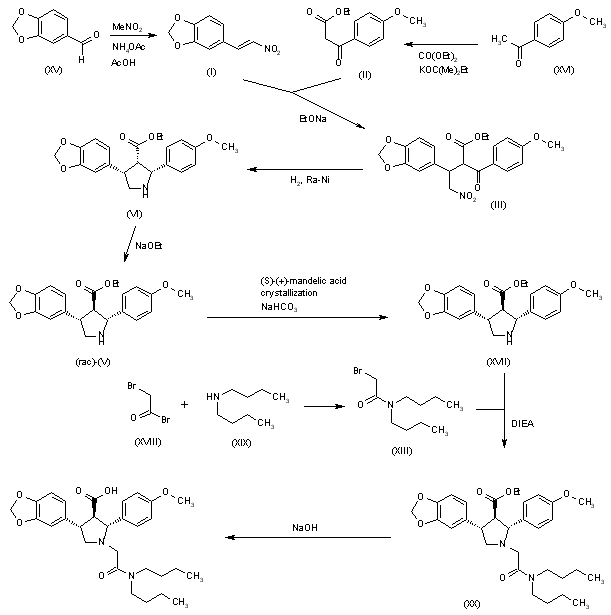
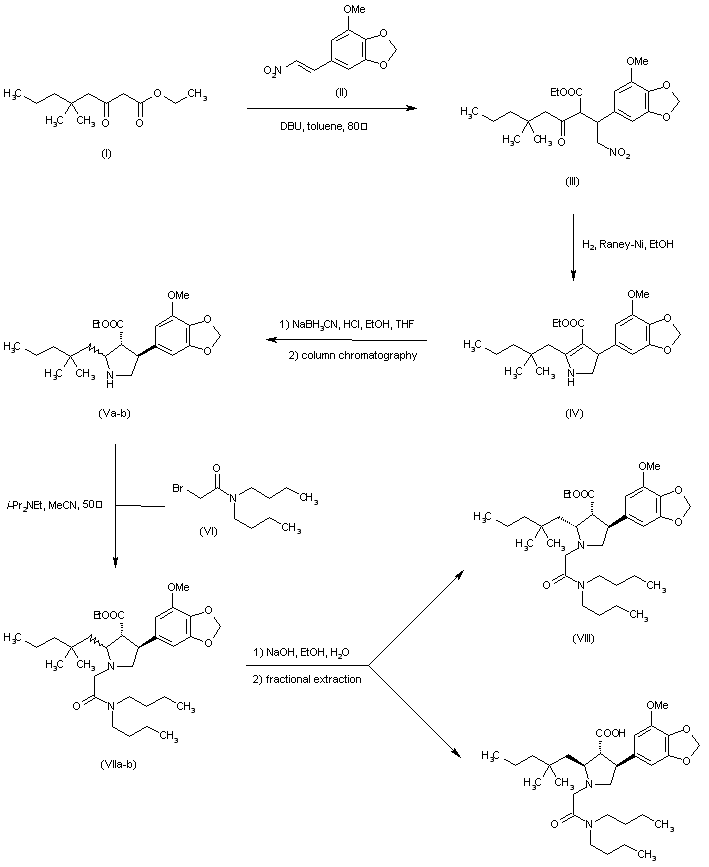
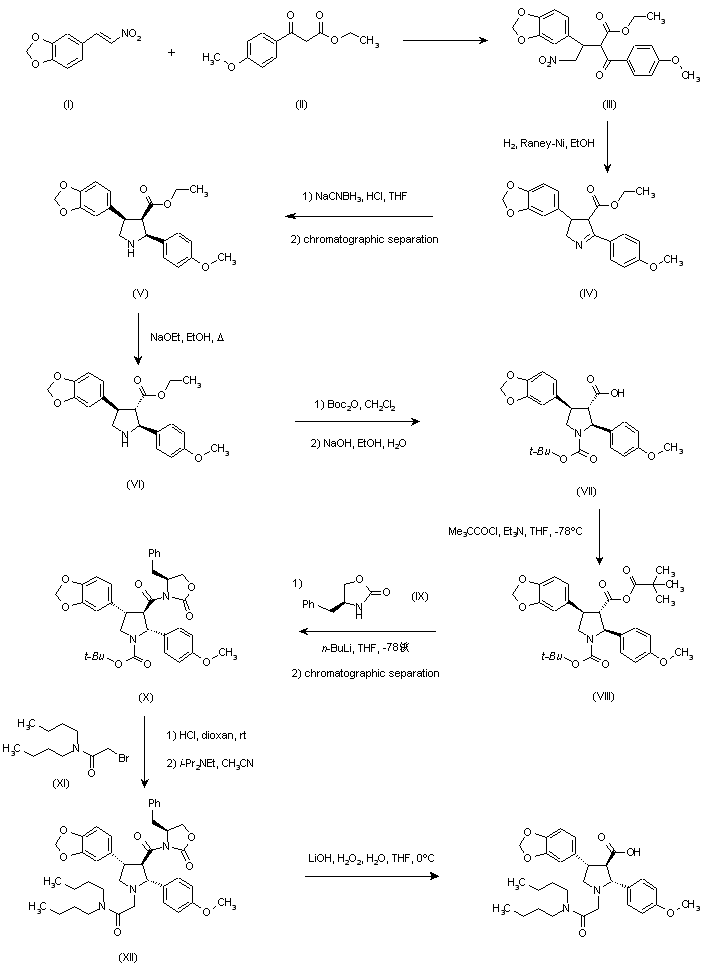
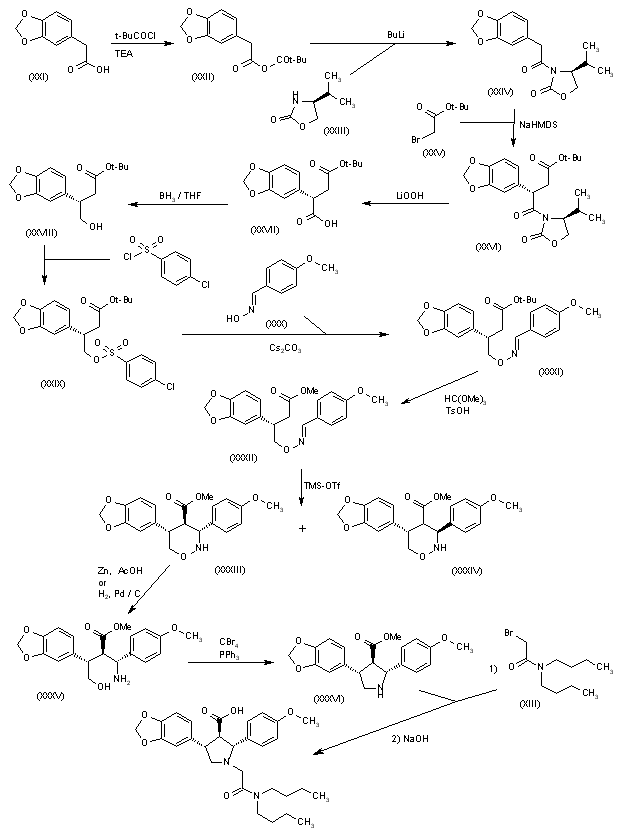






















 Location in Madhya Pradesh
Location in Madhya Pradesh















 LATUR AIRPORT
LATUR AIRPORT











 The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the











 DINACICLIB
DINACICLIB

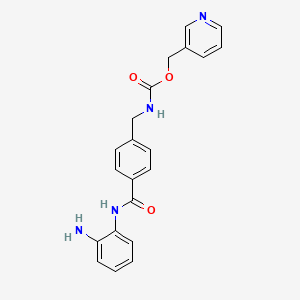





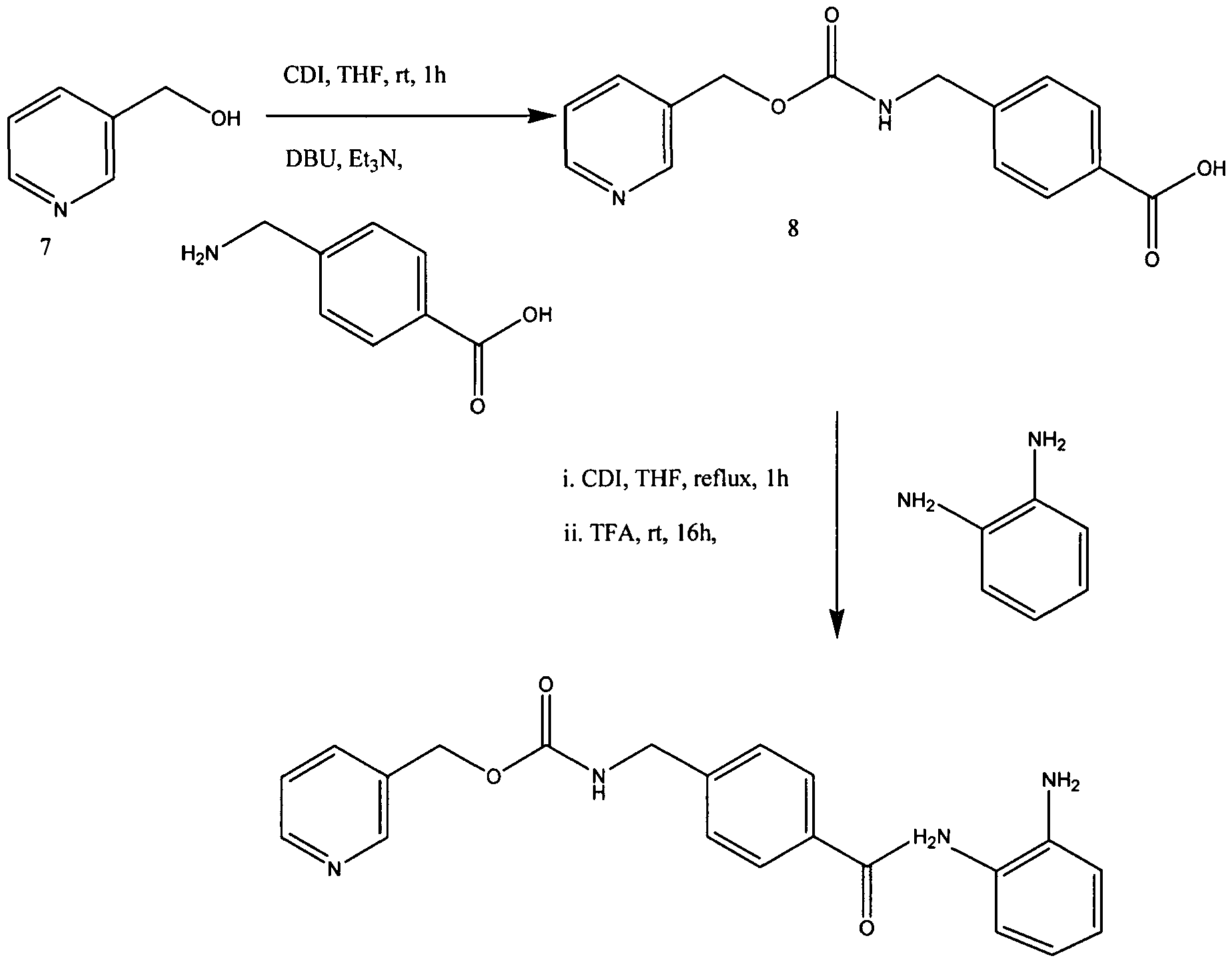










 NEWS………….DUBLIN and BUDAPEST, Hungary, Jan. 6, 2015 /PRNewswire/ — Actavis plcand Gedeon Richter Plc. today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of Actavis’ New Drug Application (NDA) resubmission for its atypical antipsychotic cariprazine, a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. The Prescription Drug User Fee Act (PDUFA) date is expected to be in the second quarter of 2015…….
NEWS………….DUBLIN and BUDAPEST, Hungary, Jan. 6, 2015 /PRNewswire/ — Actavis plcand Gedeon Richter Plc. today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of Actavis’ New Drug Application (NDA) resubmission for its atypical antipsychotic cariprazine, a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. The Prescription Drug User Fee Act (PDUFA) date is expected to be in the second quarter of 2015…….



















































{kind=link}