
Home » Phase2 drugs (Page 3)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pralnacasan
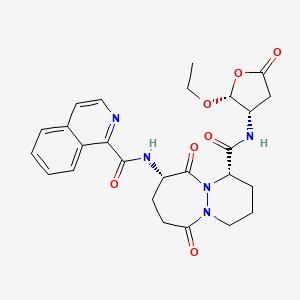
Pralnacasan
VX 740
cas 192755-52-5
(4S,7S)-N-[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]-7-(isoquinoline-1-carbonylamino)-6,10-dioxo-2,3,4,7,8,9-hexahydro-1H-pyridazino[1,2-a]diazepine-4-carboxamide
N-[(4S,7S)-4-{[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]carbamoyl}-6,10-dioxo-octahydro-1H-pyridazino[1,2-a][1,2]diazepin-7-yl]isoquinoline-1-carboxamide
(1S,9S)-N-((2R,3S)-2-Ethoxy-5-oxotetrahydrofuran-3-yl)-9-((isoquinolin-1-ylcarbonyl)amino)-6,10-dioxooctahydro-6-H-pyridazino(1,2-a)(1,2)diazepine-1-carboxamide
6H-Pyridazino(1,2-a)(1,2)diazepine-1-carboxamide, N-((2R,3S)-2-ethoxytetrahydro-5-oxo-3-furanyl)octahydro-9-((1-isoquinolinylcarbonyl)amino)-6,10-dioxo-, (1S,9S)-
- HMR 3480
- HMR3480
- HMR3480/VX-740
- Pralnacasan
- UNII-N986NI319S
- VX 470
- VX-740
C26H29N5O7, 523.543
NSAID, ICE inhibitor & metastasis inhibitor.пралнаказан [Russian] [INN]برالناكاسان [Arabic] [INN]普那卡生 [Chinese] [INN]

Pralnacasan is an orally bioavailable pro-drug of a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE).Pralnacasan is a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE, aka Caspase-1). It was originally discovered by Vertex Pharmaceuticals and licensed for development to Aventis Pharma. In 2003 Aventis and Vertex Pharmaceuticals agreed to voluntarily discontinue development based on results from a 9-month animal toxicity trial that showed liver abnormalities due to chronic high doses of pralnacasan. Pralnacasan has also been investigated for the treatment of Partial Epilepsy; advancing to Phase II clinical trials.Pralnacasan is a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE). Pralnacasan is an oral, anti-cytokine drug candidate licensed for development by Aventis Pharma from Vertex Pharmaceuticals. In November 2003, Aventis and Vertex Pharmaceuticals announced that they had voluntarily suspended the phase II clinical trials of pralnacasan due to results from an animal toxicity study that demonstrated liver abnormalities after a nine-month exposure to pralnacasan at high doses. While no similar liver toxicity has been seen to date in human trials, the companies will evaluate the animal toxicity results before proceeding with the phase II clinical program.Pralnacasan inhibits interleukin-1beta converting enzyme (ICE), an enzyme that regulates the production of IL-1 and IFN gamma – intercellular mediators that initiate and sustain the process of inflammation. Inhibiting ICE may be an effective strategy for curtailing damaging inflammatory processes common to a number of acute and chronic conditions, such as rheumatoid arthritis (RA) and osteoarthritis.
PAPERhttps://pubs.rsc.org/en/content/articlelanding/2017/ob/c7ob01403a/unauth
IDrugs (2003), 6(2), 154-158.
Chemistry (Weinheim an der Bergstrasse, Germany) (2017), 23(2), 360-369PAPER
Bioorganic & Medicinal Chemistry Letters (2006), 16(16), 4233-4236.https://www.sciencedirect.com/science/article/abs/pii/S0960894X06006184?
Abstract
Novel 1-(2-acylhydrazinocarbonyl)cycloalkyl carboxamides were designed as peptidomimetic inhibitors of interleukin-1β converting enzyme (ICE). A short synthesis was developed and moderately potent ICE inhibitors were identified (IC50 values <100 nM). Most of the synthesized examples were selective for ICE versus the related cysteine proteases caspase-3 and caspase-8, although several dual-acting inhibitors of ICE and caspase-8 were identified. Several of the more potent ICE inhibitors were also shown to inhibit IL-1β production in a whole cell assay (IC50 < 500 nM).
Graphical abstract
Novel 1-(2-acylhydrazinocarbonyl)cycloalkyl carboxamides were designed and synthesized as selective peptidomimetic inhibitors of interleukin-1β converting enzyme (ICE IC50 values <100 nM).

PAPEROrganic letters (2014), 16(13), 3488-91.https://pubs.acs.org/doi/10.1021/ol501425b
Abstract

Peptides containing N2-acyl piperazic or 1,6-dehydropiperazic acids can be formed efficiently via a novel multicomponent reaction of 1,4,5,6-tetrahydropyridazines, isocyanides, and carboxylic acids. Remarkably, the reaction’s induced intramolecularity can enable the regiospecific formation of products with N2-acyl piperazic acid, which counters the intrinsic and troublesome propensity for piperazic acids to react at N1 in acylations. The utility of the methodology is demonstrated in the synthesis of the bicyclic core of the interleukin-1β converting enzyme inhibitor, Pralnacasan.
PatentWO 9722619WO 9903852WO 9952935
PATENTWO 2000042061https://patents.google.com/patent/WO2000042061A1/enThe invention particularly relates to the process as defined above in which the compound of formula (I) is 9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) -3 ,, 7, 8, 9, 10-hexahydro-6, 10-dioxo-6H-pyridazino- [1,2- a] [1, 2] ethyl diazepine-1-carboxylate:

The invention particularly relates to the process as defined above in which the compound of formula (Iopt) is- (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8,9, 10-hexahydro-β, 10-dioxo -6H-pyridazino- [1,2- a] [1, 2] ethyl diazeρine-1-carboxylate:

The compounds of formula (I) can be generally used for the synthesis of medicaments as indicated in patent EP 94095. The compounds of formulas (II) and (III) and (F) are known and can be prepared according to the experimental method described below.The invention also relates to the application of the process as defined above as an intermediate step for the preparation of a compound of formula (V)

via the compound of formula (Iopt) as defined above, characterized in that this process comprises the steps of the process for the preparation of the compounds of formula (Iopt) from the compounds of formula (II) as defined above.The subject of the invention is also the application as defined above, characterized in that the compound of formula (Iopt) is (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo -2H- isoindol-2-yl) -3,4,7,8,9, 10-hexahydro-6, 10-dioxo-6H- pyridazino- [1,2-a] [1, 2] diazepine-1- ethyl carboxylate

The subject of the invention is also the application of the process as defined above as an intermediate step in the overall process for preparing the compounds of formula (I) and (Iopt) as defined above. Finally, the subject of the invention is, as intermediate compound, the compound of formula (IA) as defined above.Preparation 1 Preparation of bis (phenylmethyl) 1,2-hydrazinecarboxylate1.5 liters of methanol and 25 g of 80% hydrazine monohydrate are placed under nitrogen. Cooled to 0 ° C and then introduced 75 g of benzyl chloroformate and a solution of 93 g of sodium carbonate in 1100 ml of demineralized water. Maintaining the reaction mixture for 1 hour at 0 ° C, drained and washed by displacement with a mixture of 100 ml of methanol and 100 ml of water, then washed by displacement with 500 ml of water at 0 C °. Dried and obtained 107.6 g of the desired product. Preparation 2Preparation of N-phthaloyl-L-glutamic anhydride D (+) 2-tetrahydro-2,6,6-dioxo-2H-pyran-3-yl-1H-isoindole-1,3 (2H) – dione (R)Stage a: N-phthaloyl-L-glutamic acid2- (1, 3-dihydro-1,3, dioxo-2H-isoindole-2-yl) acid – pentanedioic (2S)To a solution of 14.4 g of sodium carbonate in 180 ml of water is added 10 g of L-glutamic acid then 16 g of N-carbethoxyphthalimide (nefkens reagent, commercial). The mixture is stirred at ambient temperature for 2 hours and then extracted with ethyl acetate. The organic phase is evaporated under reduced pressure until a dry extract is obtained and 2.74 g of crude product is obtained. Washing is carried out with sodium bicarbonate, then after return to the acid and extraction with ethyl acetate, 370 mg of expected product and H 2 N-C0 2 Et are isolated. Furthermore, the aqueous phase is brought to pH = 2 with 36% hydrochloric acid at a temperature below 5 ° C and then extracted with ethyl acetate, washed with a saturated chloride solution. sodium, dry, filter and concentrate under reduced pressure until 22.7 g of expected product is obtained in the form of an oil.Mass spectrum (MH) “ = 276 “ Infrared (Nujol):1775 cm “1 (m), 1720 cm ” 1 (F, complex): CO 1611 cm “1 : Aromatic Stage b:To the product obtained in stage a), 160 ml of tetrahydrofuran are added and 18.6 g of DCC (1, 3-Dicyclohexyl-carbodiimide) dissolved in 55 ml of tetrahydrofuran are added dropwise over 30 minutes. Stirred for 1 hour at 15-17 ° C, then filtered, rinsed with tetrahydrofuran, evaporated under reduced pressure until a dry extract is obtained which is taken up in isopropyl ether. After 30 minutes of stirring, the filter is washed and dried. 14.98 g of expected product are obtained. α D = -52.63 λ H NMR (DMSO) 2.12 (m, 1H); 2.61 (m, 1H); 2.98 (dm, 1H); 3.16 (ddd, 1H); 5.48 (dd, 1H); 7.82 (m,> 4H)Example 1: (IS-cis) -9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) -3,4,7,8,9,10-hexahydro-6,10 -dioxo-6H-pyridazino- [1,2- a] [1,2] diazepine-1-ethyl carboxylate. Stage a: Preparation of 2,5-dibromopentanoic acid 39 ml of bromine are added to a mixture of 106 g of 5-bromopentanoic acid and 1 ml of phosphorus tribromide. The reaction mixture is brought to 70-80 ° C for 16 h 30. The reaction medium is brought to 100 ° C for 15 minutes and allowed to return to room temperature. 147 g of sought product is obtained.Stage b: Preparation of ethyl 2,5-dibromopentanoate24.37 g of oxalyl chloride are added to a mixture containing 50 g of the acid prepared in the preceding stage, 15 drops of dimethylformamide and 300 ml of dichloromethane. The reaction mixture is kept under stirring at at room temperature, until the reaction is complete. The reaction mixture is cooled to 10 ° C and 50 ml of ethyl alcohol are added. Stirred for 30 minutes at 10 ° C, allowed to return to room temperature and stirred for 3 hours at room temperature. It is brought to dryness and the desired product is obtained. Stage c: CyclizationPreparation of (S) -tetrahydro-1,2,3-pyridazinetricarboxylate of 3-ethyl-1,2-bis (phenylmethyl) and (R) -tetrahydro-1,2,3-pyridazinetricarboxylate of 1,2 -bis (phenylmethyl). A suspension of 12.1 g of ethyl 2,5-dibromopentanoate (stage b) in 50 ml of diglyme is introduced at 20-25 ° C. in a suspension containing 10.42 g of 1,2-hydrazine carboxylate of bis (phenylmethyl) (preparation 1), 65 ml of diglyme and 8.26 g of potassium carbonate. The suspension obtained is heated to 90 ° C. and stirring is continued for 48 hours. Cooled to 20 ° C, poured into a solution containing 50 ml of 2N hydrochloric acid and 150 ml of a mixture of water and ice. Extraction is carried out with ethyl acetate, washing with water and drying. It is filtered, rinsed with ethyl acetate and dried. Finally, the crude product is purified by chromatography on silica, eluting with a heptane / ethyl acetate mixture 40/20 and 10.71 g of sought product is obtained. Stage d: Acylation and hydrogenolysisPreparation of α, (IS) – [3-oxo-3- (tetrahydro-3-ethoxycarbonyl-1 (2H) -pyridazinyl) propyl] -1,3-dihydro-1,3-dioxo-2H-isoindole acid -2-aceticThe mixture consisting of 15g of tetrahydro-1,2,3-pyridazinetricarboxylate of 3-ethyl-1,2-bis (phenylmethyl) is placed under hydrogen pressure (1.3 bar) for 24 hours. R + S mixture as prepared in stage c 150 ml of tetrahydrofuran, 2.5 g of palladium on carbon (10%) and 9.08 g of phthaloylglutamic acid anhydride as prepared according to preparation 2. After filtration, we evaporated under reduced pressure until a dry extract is obtained which is taken up in 100 ml of ethyl acetate and 150 ml of a saturated solution of sodium bicarbonate. It is extracted 3 times and the bicarbonate solution is acidified to pH = 3 with 36% hydrochloric acid. It is extracted 3 times with dichloromethane and washed with water. 13.16 g of crude product are obtained, which product is purified by chromatography on silica, eluting with a toluene / ethyl acetate / acetic acid 20/80 / 1.5 mixture to obtain 12.7 g of the expected product.NMR (250MHz, CDC1 3 ): 1.24 (d, 3H, OCH 2 CH 3 ); 4.12 (q, 2H, OCH 2 CH 3 ); 4.36-4.40 (m, 1H, Hl in alpha or beta position); 4.69-4.92 (m, 1H, H9 in the alpha position); 7.70 – 7.86 H aromatic. Stage el: cyclization with POCl 3– (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8,9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1,2- a] [1, 2] ethyl diazepine-1-carboxylate. – (lR-trans) -9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) – 3,4,7,8, 9, 10-hexahydro-6,10-dioxo -6H-pyridazino- [1,2-a] [1,2] diazepine-1-ethyl carboxylate.To a solution of 20 ml of dichloroethane heated beforehand to 75 ° C., the following solutions A and B are added over 3 hours: A: 417 mg of the ester prepared in stage d in 4 ml of dichloroethane to which 1 ml of a solution of 1.2 ml of 2,6-lutidine in 5 ml of dichloroethane. B: 1 ml of a solution of 1.9 ml of P0Cl 3 in 10 ml of dichloroethane, then the mixture is stirred for 1 hour at this temperature. Cool to 10 ° C., add demineralized water, extract with dichloromethane and evaporate under reduced pressure to obtain a crude product (0.415 g) which is purified by chromatography on silica eluting with the heptane / dichloromethane mixture. / ethyl acetate 1/1/1. 161.8 mg of the SS diastereoisomer, 126.7 mg of the SR diastereoisomer and 5.8 mg of the SS + SR mixture are isolated. Stage e2: cyclization with POBr 3– (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7, 8, 9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate. – (lR-trans) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7, 8, 9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate.To a solution of 20 ml of dichloroethane heated beforehand to 80 ° C., the following solutions A and B are added over 3 hours:A: 417 mg of the ester prepared in stage d in 4 ml of dichloroethane to which 1 ml of a solution of 2.4 ml of 2,6-lutidine in 10 ml of dichloroethane was added. B: 1 ml of a solution of 5.85 g of POBr 3 in 10 ml of dichloroethane, then the mixture is stirred for 1 hour at this temperature. Cool to 10 ° C, add demineralized water, extract with dichloromethane and evaporate under reduced pressure to obtain a crude product (0.419 g) which is purified by chromatography on silica eluting with the heptane / dichloromethane / mixture 1/1/1 ethyl acetate. 163 mg of the SS diastereoisomer, 143 mg of the SR diastereoisomer and 6.2 mg of the SS + SR mixture are isolated.Stage f: deracemization / epimerization – (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8, 9, 10-hexahydro -6,10-dioxo-6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate.Is introduced at a temperature of -45 / -48 ° C in one hour 30 minutes, a solution containing 0.029 g of potassium terbutylate and 0.3 ml of dimethylformamide in a mixture containing 0.194 g of the mixture SS + SR prepared in stage d , 1.5 ml of dimethylformamide and 0.75 ml of terbutanol. The mixture is kept stirring for 1 hour and, after cooling to -50 ° C., 0.4 g of powdered ammonium chloride is introduced. Stirred 10 minutes at -45 ° C, add 1 ml of ammonium chloride at 20 ° C and stirred again 10 minutes. 2 ml of water are added after 5 minutes demineralized. Extracted with ethyl acetate, washed with demineralized water, decanted, concentrated and dried. 0.166 g of expected SS diastereoisomer is obtained. ” D = -75.3 ° (1% in methanol) NMR (250MHz, CDC1 3 ): 1.73 (m, 3H, H-2alpha H-3alpha H-3beta; 1.24 (d, 3H, OCH 2 CH 3 ); 2.38 (m, 3H, H2beta, H7alpha, H8 alpha); 2.92 (m, 1H, H4alpha); 3.39 – 3.44 (m, 1H, H8beta); 3.62 (m, 1H, H7beta); 4.23 (m, 2H, OCH 2 CH 3 ); 4.66-4.71 (m, 1H, H4 in beta position); 5.26-5.41 (m, 2H, Hl and H9 in the alpha position); 7.72 – 7.88 H aromatics.
PATENT
WO 2000010979https://patents.google.com/patent/WO2000010979A1/en

formula II, said compound has the structure:
In the synthesis of these inhibitors, the terminal carbon of Ri adjacent the -COOH moiety contains a protecting substituent. Preferably that protecting
substituent is
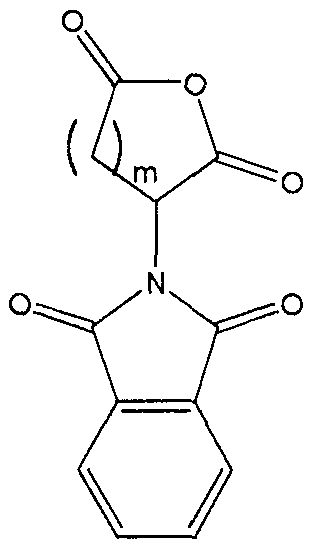
The synthesis steps from compound H to the inhibitors set forth above involve removal of the protecting substituent on Rx; coupling of the R5-NH- or R5′-NH- moiety in its place; hydrolysis of the R2 group;N .(CJ2)m.—Tand coupling of the amine ( (Ch,2)a Rs or -NH-Z)in its place. The removal of the protecting substituent on Ri is typically carried out with hydrazine. The subsequent coupling of the R5-NH- or R5′-NH- moiety is achieved with standard coupling reagents, such as EDC, DCC or acid chloride . Depending upon the nature of R2, its hydrolysis may be achieved with an acid (when R2 is t-butyl), a hydroxide (when R2 is any other alkyl, alkenyl or alkynyl or Ar) or hydrogenolysis (when R2 is an Ar-substituted alkyl, alkenyl or alkynyl) . This produces the corresponding acid from the ester.The acid is then coupled to the amine with standard coupling reagents, such as EDC, DCC or acid chloride .In order that this invention be more fully understood, the following examples are set forth. These examples are for the purpose of illustration only and are not to be construed as limiting the scope of the invention in any way. EXAMPLE 1Synthesis of a 7,6 Scaffold for a Caspase InhibitorA.

Compound A’ was dissolved m 5 equivalents of S0C12 and then heated to 80°C for 1 hour. The solution was then cooled to 50°C and 2 equivalents of bromine were added. The solution was incubated at 50°C for an additional 12 hours until the red color disappeared. We then cooled the solution to 10°C and added 4 volumes of water. The solution was then re-heated to 50°C for another hour. We then separated the organic and aqueous layer, washed the organic layer consecutively with water, Na2S0 and then brme, removing the aqueous layer after each washing. The final organic layer was then isolated, dried over Na2S0 and concentrated to produce compound B’ as an amber oil.B.

Compound B’ was treated with 1 equivalent of tert-butanol and 0.1 equivalents of 4- (dimethylammo) – pyπdme a solution of and the resulting solution cooled to 7°C. We then added a solution of 1 equivalent of DCC m toluene while maintaining reaction temperature at less than 22°C. The cooling bath was removed and the reaction was stirred at ambient temperature under a nitrogen atmosphere for 16 hours. The reaction mixture was then diluted with hexane and cooled to 9°C . The resulting solids were removed by filtration. The filtrate was washed consecutively with 0. IN HC1, water, and then sodium bicarbonate. The filtrate was then dried over sodium sulfate and concentrated in vacuo to afford compound C as a yellow oil.C.

Compound D’ was combined with 1.2 equivalents of compound C and dissolved in DMF at ambient temperature under nitrogen atmosphere. We then added granular sodium sulfate, 2.5 equivalents of LiOH monohydrate, and then 0.1 equivalents Bu4NI to the resulting solution. The reaction temperature was maintained at between 20°C and 30°C and allowed to stir for 16 hours. The reaction mixture was then diluted with ethyl acetate and water and the layers separated. The organic layer was washed with water and then brine, dried over sodium sulfate and concentrated in vacuo to produce an amber oil. This oil was then dissolved in 5 volumes of ethanol at ambient temperature. We then added 2.5 volumes of water. The resulting mixture was allowed to stir until a white solid formed (approximately 5 hours) . The crystallized product was isolated via filtration then dried in vacuo to afford compound E’ as a white solid.D.

We dissolved compound E’ in THF. We then added, at ambient temperature under a nitrogen atmosphere, 0.02 equivalents of triethylamine and 0.01 equivalents of Pd(OAc)2. A solution of 2.5 equivalents of triethylsilane (Et3SiH) in THF was then added and the resulting black solution was allowed to stir for 16 hours to complete the reaction. We then added a saturated, aqueous solution of sodium bicarbonate followed by a solution of compound F’ in THF. After 30 minutes, the layers were separated and the aqueous layer acidified to pH 4.5 with aqueous citric acid. The product in the aqueous layer was then extracted into ethyl acetate. The organic layer was isolated, washed with brine, dried over sodium sulfate and concentrated in vacuo to produce a white foam. This crude product was then recrystallized from MTBE to afford compound G’ as a white powder. E.
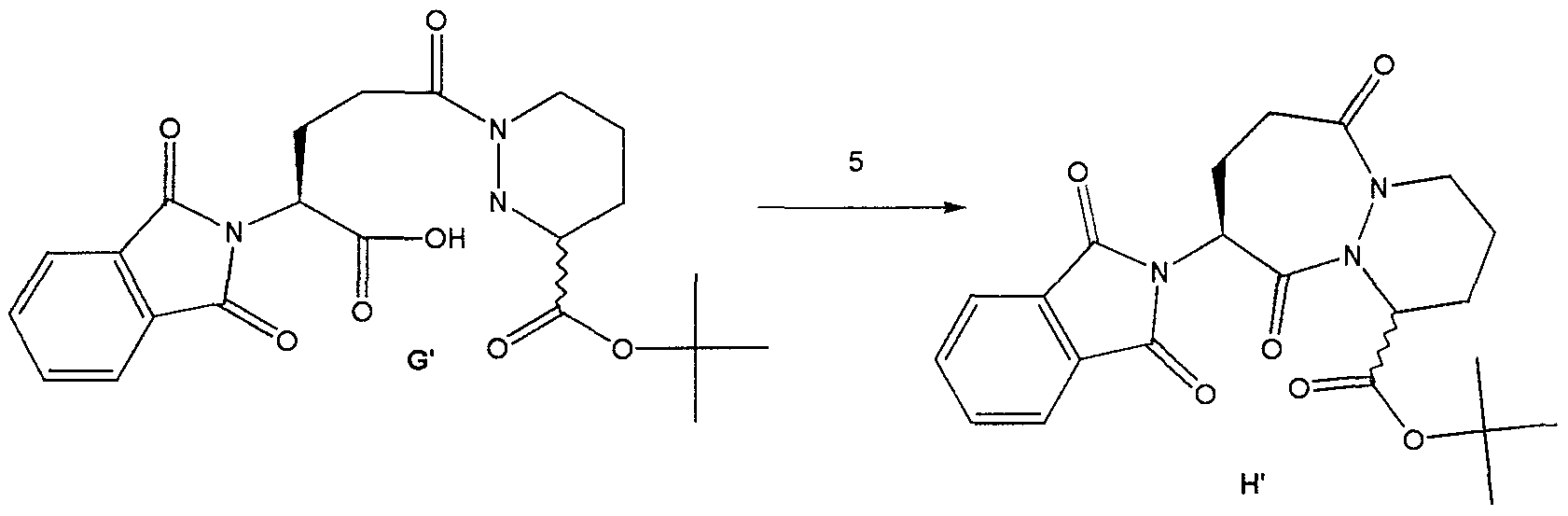
Method #1:To a suspension of compound G’ and 0.1 equivalents of DMF m dichloroethane, at 70°C we added 5 equivalents of 2, 6-lutιdme simultaneously with 2.5 equivalents of S0C12 over a period of 3 hours. The reaction was then diluted with toluene and washed consecutively with NaHC03 and br e. The solution was then dried over Na2S04 and concentrated in vacuo to afford compound H’ as a yellow solid.Method #2:To a suspension of compound G’ m dichloroethane, at 70°C, we added 4 equivalents of 2,6- lutid e followed by 2 equivalents of methanesulfonyl chloride. The resulting solution was stirred at 70°C for 12 hours. The reaction was then diluted with toluene and washed consecutively with NaHC03 and brme. The solution was then dried over Na2S04 and concentrated in vacuo to afford compound H’ as a white solid. Method #2 produced a significantly higher yield of H’ as compared to Method #1. EXAMPLE 2 Use of Intermediate H’ to Produce an Inhibitor of ICE A.

t-Butyl-9-amino-6 , 10-dioxo-l ,2,3,4,7,8,9, 10-octahydro-6- H-pyridazino [1 ,2-a] [1 ,2] diazepine-1-carboxylate (GB2,128,984) To a suspension of H’ (107 g, 0.25 mol) in ethanol (900 iriL) was added hydrazine (27 L, 0.55 mol) and the resulting mixture was allowed to stir at ambient temperature. After 4 hours, the reaction was concentrated in vacuo and the resulting white solid was suspended in acetic acid (IL of 2N) and allowed to stir at ambient temperature for 16 hours. The resulting white solid was filtered off and washed with water. The filtrate was made basic by the addition of solid sodium carbonate and the product extracted with dichloromethane. The organic layer was washed with brine, dried over magnesium sulfate and concentrated in vacuo to afford 79 mg of compound I’ as a yellow viscous oil.B.

t-Butyl-9- (isoquinolin-1-oylamino) -6, 10-dioxo- 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [ 1 , 2-a] [1,2] diazepine-1-carboxylate To a solution of the amine I’ (79 g, 0.265 mol) and isoquinolin-1-carboxylic acid (56g, 0.32 mol) in dichloromethane : DMF (400mL: 400mL) was added hydroxybenztriazole (54 g, 0.4 mol) and l-(3- dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (74 g, 0.39 mol) and the resulting mixture was allowed to stir at ambient temperature for 16 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The ethyl acetate layer was washed with 0.5N sodium bisulfate, water, sodium bicarbonate, brine, dried over sodium sulfate and concentrated in vacuo to afford 122 g of compound J’ as an orange solid-foam.C.

9- (isoquinolin-1-oylamino) -6, 10-dioxo-l ,2 ,3 ,4 , 7 , 8 , 9 , 10- octahydro-6-H-pyridazino [1 ,2-a] [1,2] diazepine-1- carboxylate A solution of the ester J’ (122 g) in dichloromethane and trifluoroacetic acid (200 mL) was allowed to stir at ambient temperature for 16 hours. The reaction mixture was concentrated to a black oil which was then triturated with acetonitrile and ether to afford 98 g of compound K’ as a pale yellow solid. D .

K'[IS, 9S (2RS, 3S) ] N-(2-benzyloxγ-5-oxotetrahydrofuran-3- yl) -6 , 10-dιoxo-9- (ιsoquιnolιn-1-oγlamιno) -1,2,3,4,7,8,9, 10-octahydro-6-H-pyrιdazιno [ 1 , 2-a] [1,2] dιazepιne-l-carboxamιde To a solution of (3S, 2RS) 3- allyloxycarbonylammo-2- (4-chlorobenzyl) oxy-5- oxotetrahydrofuran [Bioorg. & Med. Chem. Lett., 2, pp. 615-618 (1992)] (4.4 g, 15.1 mmol) in dichloromethane was added N, N-dimethylbarbituric acid (5.9g, 3.8 mmol) then tetrakispalladium ( 0) tπphenyl phosphme (1.7 g, 1.5 mmol) and the resulting mixture was allowed to stir at ambient temperature for 15 minutes. To the resulting mixture was added the acid, compound K’ (5.0 g, 12.6 mmol), hydroxybenztπazole (2.0 g, 14.8 mmol) then and 1- (3-dιmethylammopropyl) -3-ethylcarbodιιmιde hydrochloride (2.7g, 14 mmol) and the reaction was allowed to stir for 3 hours at ambient temperature. The reaction mixture was then poured into water and extracted with ethyl acetate. The organics were washed with 0.5M sodium bisulfate, water, sodium bicarbonate, br e, dried over magnesium sulfate and concentrated m vacuo to afford 2.6 g of the crude product as a yellow foam. The crude material was purified by column chromatography (Sι02, dichloromethane : acetone 9:1 – 3:1) to afford 1.2 g of the compound L’ . Compound L’ and related compounds that may be synthesized using the method of this invention as an intermediate step are described in WO 97/22619, the disclosure of which is herein incorporated by reference. Those related compounds may be synthesized from the product of the method of this invention, H or H’ , through modifications of the procedure set forth in Example 2. Such modifications are well known in the art.
PATENTWO 2001083458https://patents.google.com/patent/WO2001083458A2/enScheme IV

C 2 5,> R’==OH (S)-VI-a *•* 6 6., R R”==<CI

Example 1

(S) -t-butyl- bis- (1,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate (>90% ee) : To a solution of bis-Cbz hydrazine and (R) -t-butyl-2, 5- dimesylvalerate (from the diol prepared by the method of Schmidt et al., Synthesis, p. 223 (1996)) in DMF was added Na2S04 then TBAF (2.5 equivalents). The resulting reaction mixture was allowed to stir at room temperature for 24 hrs. The reaction was then diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound. The optical purity of the title compound was greater than 90% ee as determined by HPLC using a ChiralPak® AD column and eluting with ethanol at 0.7 ml per minute.Example 2

(S) -t-butyl-bis- (1 ,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate (40% ee) : To a solution of bis-Cbz hydrazine and (R) -t-butyl-2, 5-dimesylvalerate(96.5% ee) in DMF was added Na2S04 then K2C03 (5 equivalents) and TBAI (0.1 equivalents). The resulting reaction mixture was heated at 80°C for 24 hrs. The reaction was allowed to cool and diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound as a 70:30 mixture of the S:R enantiomers (40% ee, as determined by HPLC using a ChiralPak® AD column, eluting with ethanol at 0.7 ml/min) .Example 3

Racemic t-butyl- bis- (1 ,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate: To a solution of bis- Cbz hydrazine and (R) -t-butyl-2, 5-dimesylvalerate (96.5% ee) in THF was added NaH (2 equivalents) . The resulting reaction mixture was stirred at room temperature. The reaction was quenched then diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound as a racemic mixture.Example 4 A. Deprotection and salt formation

Hexahydro-pyridazine-3-carboxylic acid tert-butyl ester , L-tartaric acid salt (B) : Compound A was combined with 10% Pd/C (10% w/w) in tetrahydrofuran. The resulting suspension was stirred at 60 °C under a hydrogen atmosphere until deprotection complete. The catalyst was removed via filtration, to the filtrate was added L- tartaric acid (1 equivalent) and the resulting solution concentrated in vacuo.B. Enantiomeric Enrichment
Hθ

The concentrate (B) was taken up in n-butanol(10 volumes), heated to reflux, then allowed to slowly cool to ambient temperature while stirring. The resulting solids were collected via filtration to afford(S) -piperazic acid, t-butyl ester as the tartrate salt (C) in 33% yield.C. Chiral AnalysisCompound (C) was suspended in water and DCM and cooled. We then added NaOH to basify the aqueous layer. The layers were then separated and to the organic layer we added two equivalents of benzyl chloroformate andNaOH. After stirring for 1 hour, the layers were again separated and the organic layer was washed with water.The organic layer was then dried over MgS04 and then concentrated in vacuo to produce the bis-Cbz piperazic acid, t-butyl ester for chiral HPLC analysis. The bis-Cbz piperazic acid, t-butyl ester was applied to a Chiralpak AD HPLC column (Chiral Technologies, Exton, PA) and eluted with ethanol at 0.8 ml/minute. Fractions from the column were quantitate by absorption at 210 nm. The results demonstrated that (S)- piperazic acid, t-butyl ester accounted for 94.5% of the piperazic acid, t-butyl ester present in the preparation.
Example 5 Conversion of Intermediate IV to Intermediate Vl-a Cbzy


IV’ C02t-Bu yi-a C02t-Bu Tetrahydro-pyridazine-l,3-dicarboxylic acid 1-benzyl ester 3-tert-butyl ester (Vl-a) : Compound IV (1 mmol) is combined with toluene and sodium hydroxide (aqueous, 2M, 3 equivalents) and the resulting mixture cooled to 1 °C. A solution of benzylchloroformate (1.05 equivalents) in toluene is added while maintaining the reaction pH at 10 or higher by the addition of sodium hydroxide, as needed. After stirring an additional 1 hour, allow the mixture to warm to room temperature then extract with ethyl acetate. The organic layer is washed with brine, dried over sodium sulfate and concentrated to afford Vl-a.Example 6 Conversion of Intermediate X to an Inhibitor of ICE
A. Phthalimide removal to form IX-b

X IX-b t-Butyl-9-amino-6 , 10-dioxo-l ,2,3,4,7,8,9, 10-octa ydro-6-H-pyridazino[l,2-a] [1,2] diazepine-1-carboxylate (GB 2,128,984): To a suspension of X (107 g, 0.25 mol) in ethanol (900 mL) was added hydrazine (27 mL, 0.55 mol) and the resulting mixture was allowed to stir at ambient temperature. After 4 hours, the reaction was concentrated in va cuo and the resulting white solid was suspended in acetic acid (1L of 2N) and allowed to stir at ambient temperature for 16 hours. The resulting white solid was filtered off and washed with water. The filtrate was made basic by the addition of solid sodium carbonate and the product extracted with dichloromethane. The organic layer was washed with brine, dried over magnesium sulfate and concentrated in va cuo to afford 79g of compound IX-b as a yellow viscous oil.B. Formation of compound XII

IX-b XII t-Butyl-9- (isoquinolin-1-oylamino) -6 , 10-dioxo- 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [1 , 2-a] [1,2] diazepine-1-carboxylate (XII) : To a solution of IX-b (79 g, 0.265 mol) and isoquinolin-1-carboxylic acid (56g, 0.32 mol) in dichloromethane and DMF (400mL: 00mL) was added hydroxybenzotriazole (54 g, 0.4 mol) and l-(3- dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (74 g, 0.39 mol) and the resulting mixture was allowed to stir at ambient temperature for 16 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The ethyl acetate layer was washed with 0.5N sodium bisulfate, water, sodium bicarbonate, brine, dried over sodium sulfate and concentrated in vacuo to afford 122 g of compound XII as an orange solid-foam.t-Butyl ester hydrolysis to form compound XIII

XIII 9- (isoquinolin-1-oylamino) -6 , 10-dioxo-l ,2,3,4,7,8,9, 10- octahydro-6-H-pyridazino [1 , 2-a] [1 , 2] diazepine-1- carboxylate (XIII) : A solution of the ester XII (from step B) (122 g) in dichloromethane and trifluoroacetic acid (200 mL) was allowed to stir at ambient temperature for 16 hours. The reaction mixture was concentrated to a black oil which was then triturated with acetonitrile and ether to afford 98 g of compound XIII as a pale yellow solid.D. Formation of compound 4-b

[1S, 9S (2RS,3S) ]N- (2-benzyloxy-5-oxotetrahydrofuran-3- yl) -6,10-dioxo-9- (isoquinolin-1-oylamino) – 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [1 , 2-a] [1,2] diazepine-1-carboxamide (4-b) : To a solution of (3S, 2RS) 3-allyloxycarbonylamino-2-benzyloxy-5-oxotetrahydrofuran [Bioorq. & Med. Chem. Lett., 2, pp. 615-618 (1992)] (4.4 g, 15.1 mmol) in dichloromethane was added N,N- dimethylbarbituric acid (5.9g, 3.8 mmol) then tetrakispalladium(O) triphenyl phosphine (1.7 g, 1.5 mmol) and the resulting mixture was allowed to stir at ambient temperature for 15 minutes. To the resulting mixture was added the acid, compound XIII (from step C) (5.0 g, 12.6 mmol), hydroxybenzotriazole (2.0 g, 14.8 mmol), then 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (2.7g, 14 mmol) and the reaction was allowed to stir for 3 hours at ambient temperature. The reaction mixture was then poured into water and extracted with ethyl acetate. The organics were washed with 0.5M sodium bisulfate, water, sodium bicarbonate, brine, dried over magnesium sulfate and concentrated in vacuo to afford 2.6 g of the crude product as a yellow foam. The crude material was purified by column chromatography (Si02, dichloromethane: acetone 9:1 – 3:1) to afford 1.2 g of the compound 4-b. Compounds of formulae VII and VIII, and related compounds, that may be synthesized using the method of this invention as an intermediate step are described in WO 97/22619 and United States Patent 6,204,261 the disclosure of which is herein incorporated by reference. Those related compounds may be synthesized from the product of the method of this invention, I, IV, or V, through modifications of the procedure set forth in Examples 4 through 6. Such modifications are well known in the art.PATENTUS 6559304https://patents.google.com/patent/US6559304B1PATENTWO 2008074816https://patents.google.com/patent/WO2008074816A1/en
Patent
Publication numberPriority datePublication dateAssigneeTitleEP0094095A2 *1982-05-121983-11-16F. Hoffmann-La Roche AgBicyclic carboxylic acids and their alkyl and aralkyl estersUS4692438A *1984-08-241987-09-08Hoffmann-La Roche Inc.Pyridazo-diazepines, diazocines, and -triazepines having anti-hypertensive activityWO1993023403A1 *1992-05-151993-11-25Merrell Dow Pharmaceuticals Inc.NOVEL MERCAPTOACETYLAMIDO PYRIDAZO[1,2]PYRIDAZINE, PYRAZOLO[1,2]PYRIDAZINE, PYRIDAZO[1,2-a][1,2]DIAZEPINE AND PYRAZOLO[1,2-a][1,2]DIAZEPINE DERIVATIVES USEFUL AS INHIBITORS OF ENKEPHALINASE AND ACEWO1994011353A1 *1992-11-121994-05-26University College LondonProcess for the preparation of (3r)- and (3s)-piperazic acid derivativesWO1995035308A1 *1994-06-171995-12-28Vertex Pharmaceuticals IncorporatedINHIBITORS OF INTERLEUKIN-1β CONVERTING ENZYMEFamily To Family CitationsUS6204261B11995-12-202001-03-20Vertex Pharmaceuticals IncorporatedInhibitors of interleukin-1β Converting enzyme inhibitorsFR2777888B11998-04-272004-07-16Hoechst Marion Roussel IncNOVEL DERIVATIVES OF ACID (3,4,7,8,9,10-HEXAHYDRO-6,10- DIOXO-6H-PYRIDAZINO [1,2-A] [1,2] DIAZEPINE-1-CARBOXYLIC, THEIR PROCESS OF PREPARATION AND THEIR APPLICATION TO THE PREPARATION OF MEDICINESFR2777889B11998-04-272004-07-09Hoechst Marion Roussel IncNOVEL DERIVATIVES OF OCTAHYDRO-6,10-DIOXO-6H- PYRIDAZINO [1,2-A] [1,2] DIAZEPINE-1-CARBOXYLIC, THEIR PREPARATION PROCESS AND THEIR APPLICATION TO THE PREPARATION OF THERAPEUTICALLY ACTIVE COMPOUNDS
////////////////Pralnacasan, VX 740, VX 470, HMR 3480, пралнаказан , برالناكاسان , 普那卡生 ,
CCOC1C(CC(=O)O1)NC(=O)C2CCCN3N2C(=O)C(CCC3=O)NC(=O)C4=NC=CC5=CC=CC=C54
NEW DRUG APPROVALS
ONE TIME
$10.00
VX 148
VX 148
297730-05-3
Name: VX-148
CAS#: 297730-05-3
Chemical Formula: C23H25N5O4
Exact Mass: 435.19065
Molecular Weight: 435.48
Elemental Analysis: C, 63.44; H, 5.79; N, 16.08; O, 14.70
| Molecular Weight | 435.48 |
| Formula | C23H25N5O4 |
| CAS No. | 297730-05-3 (VX 148); |
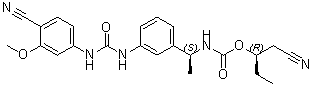
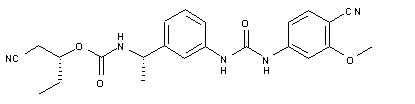
| Chemical Name | Carbamic acid, N-[(1S)-1-[3-[[[(4-cyano-3-methoxyphenyl)amino]carbonyl]amino]phenyl]ethyl]-, (1R)-1-(cyanomethyl)propyl ester |
- OriginatorVertex Pharmaceuticals
- ClassAntipsoriatics
- Mechanism of ActionInosine monophosphate dehydrogenase inhibitors
- DiscontinuedPsoriasis; Transplant rejection; Viral infections
- 13 Nov 2003Interim data from a media release have been added to the adverse events and Skin Disorders therapeutic trials sections
- 23 May 2003Vertex Pharmaceuticals has completed enrolment in a phase IIa trial for Psoriasis in Iceland
- 24 Dec 2002Phase-II clinical trials in Psoriasis in Iceland (unspecified route)
VX-148 is a second-generation, orally administered inhibitor of inosine monophosphate dehydrogenase (IMPDH). The IMPDH enzyme plays a key role in regulating immune response and proliferation of specific cell types, including lymphocytes. VX-148 is a developed for the treatment of autoimmune diseases.
Investigated for use/treatment in autoimmune diseases, psoriasis and psoriatic disorders, and viral infection.
VX-148 is a novel, uncompetitive IMPDH inhibitor with a K(i) value of 6 nM against IMPDH type II enzyme. VX-148 is slightly more potent than mycophenolic acid and VX-497 in inhibiting the proliferation of mitogen-stimulated primary human lymphocytes (IC(50) value of ~80 nM). The inhibitory activity of VX-148 is alleviated in the presence of exogenous guanosine. VX-148 does not inhibit proliferation of nonlymphoid cell types such as fibroblasts, indicating selectivity for inhibition of IMPDH activity. VX-148 is orally bioavailable in rats and mice; oral administration of VX-148 inhibits primary antibody response in mice in a dose-dependent manner with an ED(50) value of 38 mg/kg b.i.d. VX-148 significantly prolongs skin graft survival at 100 mg/kg b.i.d. in mice.
SYN
WO 0056331
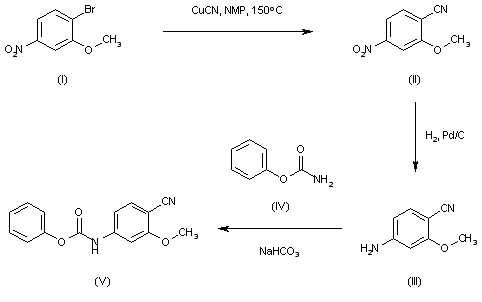
The intermediate carbamate (V) has been obtained as follows. The reaction of 4-bromo-3-methoxynitrobenzene (I) with CuCN in NMP at 150 C gives 2-methoxy-4-nitrobenzonitrile (II), which is reduced with H2 over Pd/C in ethyl acetate to yield 4-amino-2-methoxybenzonitrile (III). Finally, this compound is condensed with phenyl carbamate (IV) by means of NaHCO3 in ethyl acetate to afford the desired carbamate intermediate (V).
SYN
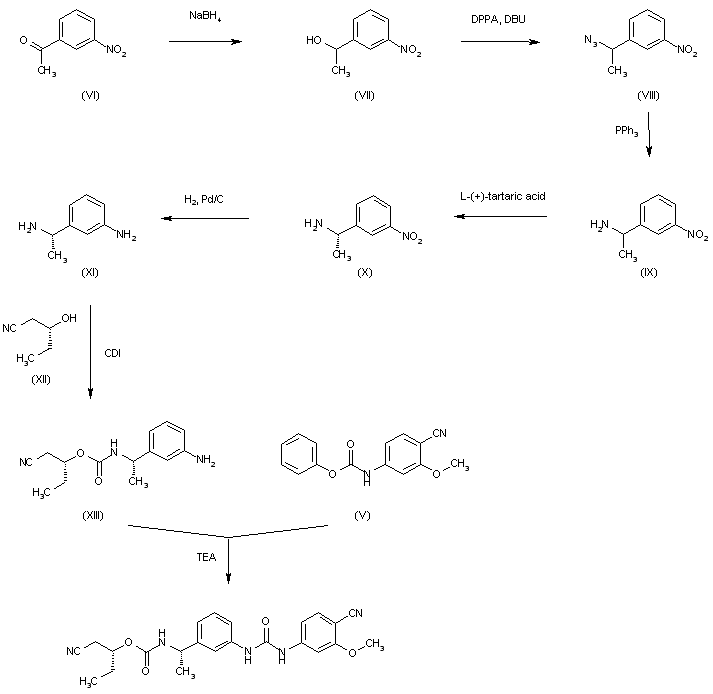
The reduction of 3-nitroacetophenone (VI) by means of NaBH4 in ethanol gives 1-(3-nitrophenyl)ethanol (VII), which is treated with DPPA and DBU in hot toluene to yield the azido derivative (VIII). The reduction of (VIII) with PPh3 in THF/water affords 1-(3-nitrophenyl)ethylamine (IX) as a racemic mixture that is submitted to optical resolution with L-(+)-tartaric acid to provide the desired (S)-isomer (X). The reduction of the nitro group of (X) by means of H2 over Pd/C in methanol gives 1(S)-(3-aminophenyl)ethylamine (XI), which is condensed with 2(R)-hydroxypentanenitrile (XII) and CDI to yield the carbamate (XIII). Finally, this compound is condensed with intermediate carbamate (V) by means of TEA in hot ethyl acetate to afford the target urea.
- Jain J, Almquist SJ, Heiser AD, Shlyakhter D, Leon E, Memmott C, Moody CS, Nimmesgern E, Decker C: Characterization of pharmacological efficacy of VX-148, a new, potent immunosuppressive inosine 5′-monophosphate dehydrogenase inhibitor. J Pharmacol Exp Ther. 2002 Sep;302(3):1272-7. [Article]
////////////VX 148, phase 2
O=C(O[C@H](CC)CC#N)N[C@H](C1=CC=CC(NC(NC2=CC=C(C#N)C(OC)=C2)=O)=C1)C

NEW DRUG APPROVALS
one time
$10.00
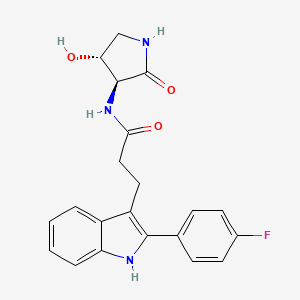
VX- ? (3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide)
VX- ?
CAS 2446817-72-5
HYDRATE 2446818-26-2
Acetic acid, 1-methylethyl ester 2446818-27-3
C21 H20 F N3 O3, 381.4
1H-Indole-3-propanamide, 2-(4-fluorophenyl)-N-[(3S,4R)-4-hydroxy-2-oxo-3-pyrrolidinyl]-
3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide
use in treating focal segmental glomerulosclerosis (FSGS) and/or non-diabetic kidney disease (NDKD).

NEW DRUG APPROVALS
one time
$10.00
PATENT
SOLID FORMS OF APOL1 INHIBITOR AND METHODS OF USING SAME
Compound I is disclosed as Compound 87 in U.S. Provisional Application No.62/780,667 filed on December 17, 2018, U.S. Application No. 16/717,099 filed onDecember 17, 2019, and PCT International Application No. PCT/US2019/066746 filed on December 17, 2019, the entire contents of each of which are incorporated herein by reference.
Compound I, which can be employed in the treatment of diseases mediated by APOLl, such as FSGS and NDKD
Example 1. Synthesis of Compound
Preparation of Compound I and Forms Thereof
Compound I Compound I /‘– PrOAc solvate Form A
n-pentanol/
n-heptane
Compound I
Form B
Step 1. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00156] To a mixture of C104 (100.0 g, 1.0 equiv) and phenyl hydrazine hydrochloride (72.2 g, 1.05 eqiv) was charged AcOH (800 mL, 8 vol). The mixture was agitated and heated to 85 °C for 16 hours. The batch was cooled to 22 °C. A vacuum was applied and the batch distill at <70 °C to ~3 total volumes. The batch was cooled to 19- 25 °C. The reactor was charged with iPrOAc (800 mL, 8 vol) and then charged with water (800 mL, 8 vol). The internal temperature was adjusted to 20 – 25 °C and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. 1 N HC1 (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the
biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The reactor was charged with 1 N HC1 (500 mL, 5 vol). The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The organic phase was distilled under vacuum at <75 °C to 3 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The resulting slurry was heated to an internal temperature of 85 °C until complete dissolution of solids was achieved. The mixture was allowed to stir for 0.5 h at 85 °C and then cooled to an internal temperature of 19 – 25 °C over 5 h. The mixture was allowed to stir at 25 °C for no less than 2 h. The slurry was filtered. The filter cake was washed with toluene (1 x 2 vol (200 mL) and 1 x 1.5 vol (150 mL)). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford product C101 (95.03 g, 70%).
Step 2. Synthesis of Compound I
[00157] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (50 g, 1.0 equiv), S2 hydrochloride (28.3 g, 1.05 equiv), and CDMT (34.1 g, 1.1 equiv) was charged with 2-MeTHF (200 mL, 4 vol) and DMF (50 mL, 1 vol) and the mixture was agitated. The internal temperature adjusted to <13 °C. The reactor was charged with NMM (64.5 g, 3.5 equiv) over 1 h, while maintaining internal temperature <20 °C. The internal temperature was adjusted to 25 °C and the batch was stirred at that temperature for 14 h. The batch was cooled to 10 °C and charged with water (250 mL, 5 vol) while keeping the internal temperature <20 °C. The batch was then warmed to 20 – 25 °C. Stirring was stopped, and the phases allowed to separate for 10 min. The lower aqueous phase was removed. The aqueous layer was back extracted with 2-MeTHF (2 x 200 mL, 2 x 4 vol) at
20 – 25 °C. The combined organic phases were washed with 1 N HC1 (500 mL, 10 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The lower aqueous phase was removed. The organic phases were washed with 0.25 N HC1 (2 x 250 mL, 2 x 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min for each wash. Lower aqueous phases were removed after each wash. The organic phase was washed with water (250 mL, 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The reactor was charged with 20 wt % Nuchar RGC® and stirred for 4 h. The reaction mixture was filtered through a pad of celite®. The reactor and celite® pad were rinsed with 2-MeTHF. The combined organics were distilled under vacuum at <50 °C to 5 total volumes. The reactor was charged with iPrOAc (500 mL, 10 vol). The organic phase was distilled under vacuum at <50 °C to 5 total volumes. The mixture was charged with additional iPrOAc (400 mL, 8 vol) and distillation under vacuum was repeated. The mixture was charged with additional iPrOAc (250 mL, 5 vol), heated to an internal temperature of 75 °C and stirred for 5 h. The slurry was cooled to 25 °C, over 5 h and stirred for no less than 12 h. The slurry was filtered and the filter cake washed with iPrOAc (2 x 50 mL, 2 x 1 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C to afford Compound I as an iPrOAc solvate (60.38 g including 9.9% w/w iPrOAc, 80.8% yield).
Recrystallization to Form A of Compound I
[00158] Compound I as an iPrOAc solvate (17.16 g after correction for iPrOAc content, 1.0 equiv) was charged to a reactor. A mixture of IP A (77 mL, 4.5 vol) and water (137 mL, 8 vol) were charged to the reactor. The slurry was heated to an internal temperature of 75 °C. The batch was cooled to an internal temperature of 25 °C over 10 h and then stirred at 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 52 mL, 2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford Compound I as a neat, crystalline form (Form A, 15.35 g, 89%).
[00159] The X-ray powder diffractogram of Compound I Form A (FIG. 50) was acquired at room temperature using a PANalytical Empyrean diffractometer equipped with PIXcel ID detector. The peaks are listed in Table A below.
Table A. XRPD of Form A of Compound I
|
I
PATENT
- WO2020131807
Alternative Preparation I of Compound 87 (Indole preparation route C)
Step 1. Synthesis of 2-(4-fluorophenyl)-lH-indole (C98)
[00401] To a stirred suspension of indole (5 g, 42.7 mmol) and (4- fluorophenyl)boronic acid (8.96 g, 64.0 mmol) in AcOH (200 mL) was
added Pd(OAc)2.Trimer (1.44 g, 6.4 mmol) and the mixture stirred at room temperature for 16 h under 02-balloon pressure. Then the reaction mixture was filtered through a Celite® pad, washed with EtOAc (500 mL). The filtrates were washed with water, sat. NaHC03 solution, brine solution, then dried over Na2S04 and concentrated under reduced pressure. Purification by silica gel chromatography (Gradient: 0-10 % EtOAc in heptane) yielded the product afforded 2-(4-fluorophenyl)-lH-indole (5.5 g, 61 %). ‘H NMR (300 MHz, DMSO-de) 5 11.51 (s, 1H), 7.9 (t, J = 5.4 Hz, 2H), 7.52 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.30 (t, J = 8.7 Hz, 2H), 7.09 (t, J = 12 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.86 (s, 1H). LCMS m/z 212.4 [M+H]+.
Step 2. Synthesis of methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (C99)
[00402] 2-(4-fluorophenyl)-lH-indole (1.0 g, 4.76 mmol) and methyl 3,3-dimethoxypropanoate (0.81 mL, 5.7 mmol) were suspended in dichloromethane (15 mL). Trifluoroacetic acid (2.00 mL, 26 mmol) was added rapidly via syringe, resulting in a clear brown solution. The reaction mixture was heated to 40 °C for three hours. The reaction was diluted with dichloromethane (15 mL) to give an amber solution which was washed with saturated aqueous NaHCCh (25 mL) to yield a bright yellow/light amber biphasic mixture. The phases were separated and the organic layer was washed with saturated NaHCCh (30 mL), then dried (MgSCh) and filtered. The mixture was concentrated under a nitrogen stream overnight. The crude product was obtained as a yellow powder. The product was dissolved in minimum 2-MeTHF and pentane added until the suspension became lightly cloudy. The suspension was allowed to stand overnight, and the precipitate was filtered off. The filter cake was washed with heptane (2 x 15 mL), and dried in vacuo at 40 °C to afford the product as a yellow powder. Methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (1.30 g, 86 %). ¾ NMR (300 MHz, Chloroform -if) d 8.41 (s, 1H), 8.01 – 7.95 (m, 1H), 7.92 (d, J = 16.0 Hz,
1H), 7.58 – 7.50 (m, 2H), 7.46 – 7.41 (m, 1H), 7.33 – 7.27 (m, 2H), 7.22 (t, J = 8.6 Hz, 2H), 6.59 (d, J = 16.0 Hz, 1H), 3.79 (s, 3H). LCMS m/z 295.97 [M+H]+.
Step 3. Synthesis of methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (CIOO)
[00403] To a solution of methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (7 g, 0.02 mol) in EtOAc (350 mL) was added Palladium on carbon (4 g, 10 %w/w, 0.004 mol) and stirred at room temperature for 2 h under an atmosphere of H2 (bladder pressure). The reaction mixture was filtered through a pad of Celite® and washed with EtOAc (400 mL). The filtrates was concentrated to afford methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (7.1 g, 100 %). 1H MR (300 MHz, DMSO-<fc) 5 11.2 (s, 1H), 7.65 (q, J = 5.4 Hz, 2H), 7.54 (d, J = 8.1 Hz, 1H), 7.36 (t, J = 9.0 Hz, 3H), 7.10 (t, J = 8.1 Hz, 1H), 7.02 (t, J = 7.8 Hz, 1H), 3.53 (s, 3H), 3.10 (t, J = 15.9 Hz, 2H), 2.63 (t, J = 15.9 Hz, 2H). LCMS m/z 298.21 [M+H]+. The product was used directly in the subsequent step without further purification.
Step 4. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00404] To stirred solution of methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (14.4 g, 0.05mol) in THF (300 mL), MeOH (300 mL) and H2O (250 mL) was cooled to -10°C. LiOH.H20 (10.1 g, 0.24 mol) was slowly added in a portion-wise manner. The reaction mixture was allowed to stir at room temperature for 16 h. The mixture was
evaporated and ice cold water (200 mL) was added, pH was adjusted to pH- 2 with 1M HC1 (400 mL, Cold solution). The mixture was stirred for 10 minutes, filtered and dried to afford 3-[2-(4-fhiorophenyl)-lH-indol-3-yl]propanoic acid (12.9 g, 94 %). ‘H NMR (400 MHz, DMSCMJ) 5 12.11 (s, 1H), 11.18 (s, 1H), 7.65 (q, J = 5.2 Hz, 2H), 7.56 (d, J = 7.6 Hz, 1H), 7.36 (t, J = 8.8 Hz, 3H), 7.10 (t, J = 8 Hz, 1H), 7.01 (t, J = 8 Hz, 1H), 3.06 (t, J = 16.4 Hz, 2H), 2.55 (t, J = 16 Hz, 2H). LCMS m/z 284.21 [M+H]+.
Step 5. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide (87)
[00405] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (40 g, 120.0 mmol) and (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one (Hydrochloride salt) S2 (23.8 g, 156.0 mmol) in DMF (270 mL) was stirred at room temperature for 5 minutes. CDMT (27.2 g, 154.9 mmol) and NMM (53 mL, 482.1 mmol) were added and the mixture was stirred at room temperature for 2 h. The mixture was poured into water (140 mL) and then stirred for 1 h at room temperature, then filtered and washing the solids with water (50 mL). The solids were dissolved in 1 : 1 IP A/water (-400 mL, until all solids dissolved) with heating (reflux) and stirring. The mixture was allowed to cool slowly to room temperature overnight. The mixture was cooled to 0 oC and stirred to break up crystals for filtration. The crystals were then filtered off, rinsed with cold 1 : 1 IP A/water to afford a tan solid (45 g). The solid was dissolved in IPA (200 mL) and heated to 80 °C to dissolve the solid. Activated charcoal (10 g) was added and the mixture was heated with stirring for 30 minutes. The mixture was filtered through Celite ® and solvent removed under reduced pressure. A mixture of 40:60 IP A/water (350 mL) was added to the solid and the mixture was heated until all solids dissolved. The mixture was cooled to room temperature over 5 h. Solids precipitated within the mixture. The mixture was then cooled to 0 °C and stirred for 1 h. The solids were filtered off and air dried on funnel for 1 h, then in a vacuum at 55 °C overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (36.6 g, 79 %). ¾ NMR (300 MHz, Methanol-i¾) d 7.63 (ddt, J= 8.6, 5.1, 2.7 Hz, 3H), 7.35 (dt, J= 8.1, 1.0 Hz, 1H), 7.25 – 7.16 (m, 2H), 7.11 (ddd, J= 8.1, 7.0, 1.3 Hz, 1H), 7.03 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H), 4.34 (td, J= 7.6, 6.8 Hz, 1H), 4.22 (d, J= 7.7 Hz, 1H), 3.55 (dd, J= 9.9, 7.5 Hz, 1H), 3.26 – 3.18 (m, 2H), 3.10 (dd, J= 9.9, 6.8 Hz, 1H), 2.69 – 2.59 (m, 2H). LCMS m/z 382.05 [M+H]+. The
product contained 0.23 % IPA by weight by NMR (1439 ppm IPA by residual solvent analysis). Purity is 99.5 % by (qNMR).
Alternative Preparation II of Compound 87 ( Indole Preparation route D)
Step 1. Synthesis of 5-(4-fluorophenyl)-5-oxo-pentanoic acid (Cl 04)
[00406] To a stirred suspension of AlCb(13.9 g, 0.10 mol) in dichloromethane (50 mL) was added a solution of tetrahydropyran-2,6-dione (5.93 g, 0.05
mol) in dichloromethane (100 mL) at 0 °C over a period of 15 minutes and stirred for 30 min. Then to the reaction mixture was added fluorobenzene (5 g, 0.05 mol) at 0 °C over a period of 15 min, gradually allowed to room temperature and stirred for 16 h. Then the reaction mixture was added to ice water (50 mL) under stirring. The resulting solid was filtered to afford a light yellow solid. The solid was diluted with 3 % NaOH solution (50 mL) and dichloromethane (50 mL). The aqueous layer was separated and acidified with IN HC1 at 0 °C. The mixture was then extracted with EtOAc (100 mL), dried over Na2SC>4, and concentrated under reduced pressure. The solid was then washed with pentane and dried to afford 5-(4-fluorophenyl)-5-oxo-pentanoic acid as an off white solid. (6 g, 53 %). ¾ NMR (300 MHz, DMSO-^) d 12.07 (s, 1H), 8.06 (d, J = 6 Hz, 1H), 8.02 (d, J = 5.4 Hz, 1H), 7.36 (t, J = 8.7 Hz, 2H), 3.06 (t, J = 12 Hz,
2H), 2.31 (t, J = 7.2 Hz, 2H), 1.86-1.78 (m, 2H). LCMS m/z 211.18 [M+H]+.
Step 2. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (Cl 01) [00407] Phenylhydrazine (Hydrochloride salt) (375.7 g, 2.6 mol) was combined with the 5-(4-fluorophenyl)-5-oxo-pentanoic acid (507.7 g, 2.4 mol) in a 12 L three-necked round-bottomed flask equipped with an overhead stirrer, temperature probe, and reflux condenser. AcOH (5 L) was added. The stirring was initiated and ZnCk (605 g, 4.44 mol) was added. The white suspension rapidly thickened after a few minutes (due to formation of the hydrazine intermediate). Approx. 500 mL of extra AcOH was added to aid stirring. The reaction was then heated to 100 °C for three hours. The reaction was cooled to room temperature and poured into water (approx. 6 L). The mixture was extracted with EtOAc (approx 8 L). The extract was washed with water, dried
(MgS04), filtered, and evaporated in vacuo to afford a golden yellow solid. The solid was triturated with approx. 4 L of 10 % EtOAc/DCM and filtered. The filter cake was washed with 50 % dichloromethane/heptane (approx 1 L). The filter cake was dissolved in 40 % EtOAc/dichloromethane (approx. 2L) and filtered over a plug of silica gel. The plug was eluted with 40 % EtOAc/ dichloromethane until the product had been eluted (checked by TLC (25 % EtOAc/ dichloromethane)). The filtrate was evaporated in vacuo to afford 382.6 g of an off-white solid (Crop 1). All filtrates were combined and evaporated in vacuo. The remaining solid was dissolved in 10 %
EtOAc/dichloromethane (approx. 1 L) and chromatographed on a 3 kg silica gel cartridge on the ISCO Torrent (isocratic gradient of 10 % EtOAc/dichloromethane). Product fractions were combined and evaporated in vacuo to afford a yellow solid that was slurried with dichloromethane, cooled under a stream of nitrogen, and filtered. The filter cake was washed with 50 % dichloromethane/heptane and dried in vacuo to afford 244.2 g of product (Crop 2). Altogether, both crops afforded 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (626.8 g, 93 %). ¾ NMR (300 MHz, DMSO-i/e) d 12.15 (s, 1H), 11.20 (s, 1H), 7.74 – 7.62 (m, 2H), 7.57 (d, J = 7.8 Hz, 1H), 7.47 – 7.28 (m, 3H), 7.11 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.02 (ddd, J = 7.9, 7.0, 1.1 Hz, 1H), 3.17 – 2.85 (m, 2H), 2.61 – 2.52 (m, 2H) ppm. 19F NMR (282 MHz, DMSO-i/e) d -114.53 ppm. LCMS m/z 284.15 [M+H]+.
Step 3. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide (87)
[00408] A 3-L three neck RBF under nitrogen was equipped with a 150 mL addition funnel and thermocouple, then loaded with 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (77.2 g, 228.6 mmol), (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one
(Hydrochloride salt) (36.6 g, 239.9 mmol) and CDMT (44.2 g, 251.7 mmol). DMF (320 mL) was added and the orange slurry was cooled to -5 °C (acetone/brine/dry ice). NMM (88 mL, 800.4 mmol) was added via a funnel over 75 minutes to keep the internal temp <0 °C. The slurry was stirred at between -10 and 0 °C for 1 hour, then allowed to warm to ambient temperature progressively over 2 hours. Additional reagents were added (10 % of the initial quantities), and the mixture was stirred overnight at ambient temperature. Water (850 mL) was added over 60 minutes, maintaining the internal temperature at <25 °C (ice bath). This slow water addition allows for complete dissolution of any visible salt before precipitation of the product. The resulting thick slurry was stirred at ambient temperature overnight. The solid was recovered by filtration and washed with water (3 x 500 mL). The solid was dried under a stream of air at ambient temperature, then purified by crystallization.
Crystallization of 3- [2-( 4-fluorophenyl)-lH-indol-3-yl ]-N-[ ( 3S, 4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl ] propanamide (87)
[00409] Under nitrogen atmosphere, a 2-L, 3 -neck flask equipped with addition funnel and thermocouple was charged with a light brown suspension of the crude 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yljpropanamide (89.5 g) in IPA (225 mL, 2.5 vol). The slurry was heated to 50 °C and water (675 mL, 7.5 vol) was added until near-complete dissolution of solid was observed. The temperature was adjusted to 70 °C-to achieve full dissolution, yielding a clear amber solution. After 30 minutes, the heat source was removed and the mixture was cooled to ambient temperature over the weekend, stirring gently while maintaining the nitrogen atmosphere. The solid was recovered by filtration, washed with IPA:H20 = 1 :2 (2 x 300 mL, 2 x 3.3 vol) dried under a stream of air overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (84.8 g, 92 %). ¾ NMR (300 MHz, DMSO-^) d 11.19 (s, 1H), 8.23 (d, J= 7.5 Hz, 1H), 7.77 (s, 1H), 7.72 – 7.63 (m, 2H), 7.60 (d, J= 7.8 Hz, 1H), 7.41 -7.31 (m, 3H), 7.12 (ddd, J= 8.1, 7.0, 1.2 Hz, 1H), 7.03 (ddd, J= 8.0, 7.0, 1.1 Hz, 1H), 5.49 (d, J= 5.0 Hz, 1H), 4.20 – 4.06 (m, 2H), 3.38 (s, 1H), 3.11 – 3.00 (m, 2H), 2.92 (dd, J= 9.4, 6.6 Hz, 1H). LCMS m/z 382.15 [M+H]+.
Crystallization of 3- [2-( 4-fluorophenyl)-lH-indol-3-yl J-N-[ ( 3S, 4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl ] propanamide (87)
[00410] A 2-L, 3-neck flask equipped with addition funnel and thermocouple was charged with a light brown suspension of the crude 3-[2-(4-fluorophenyl)-lH-indol-3- yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide in IPA (225 mL, 1 vol). The slurry was heated to 50 °C and water (675 mL, 3 vol) was added until near- complete dissolution of solid observed (mL). Temperature was increased to 70 °C under nitrogen (full dissolution, yielding a clear amber solution). After 30 minutes, the heat was removed and the mixture cooled to ambient temperature over the weekend, stirring gently under nitrogen atmosphere. The solid was recovered by filtration and washed with IPAiLLO = 1 :2 (2 x 300 mL).The solid was dried under a stream of air overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl]propanamide (84.8 g, 92 %). ¾ NMR (300 MHz, DMSO-i/e) d 11.19 (s, 1H), 8.23 (d, J= 7.5 Hz, 1H), 7.77 (s, 1H), 7.72 – 7.63 (m, 2H), 7.60 (d, J= 7.8 Hz,
1H), 7.41 – 7.31 (m, 3H), 7.12 (ddd, J= 8.1, 7.0, 1.2 Hz, 1H), 7.03 (ddd, 7= 8.0, 7.0,
1.1 Hz, 1H), 5.49 (d, J= 5.0 Hz, 1H), 4.20 – 4.06 (m, 2H), 3.38 (s, 1H), 3.11 – 3.00 (m, 2H), 2.92 (dd, J= 9.4, 6.6 Hz, 1H). LCMS m/z 382.15 [M+H]+.
Large Scale Preparation of Compound 87
/- PrOAc solvate Form A
Step 1. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00411] To a mixture of C104 (100.0 g, 1.0 equiv) and phenyl hydrazine hydrochloride (72.2 g, 1.05 eqiv) was charged AcOH (800 mL, 8 vol). The mixture was agitated and heated to 85 °C for 16 hours. The batch was cooled to 22 °C. A vacuum was applied and the batch distill at <70°C to ~3 total volumes. The batch was cooled to 19- 25 °C. The reactor was charged with iPrOAc (800 mL, 8 vol) and then charged with water (800 mL, 8 vol). The internal temperature was adjusted to 20 – 25 °C and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. 1 N HC1 (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The reactor was charged with 1 N HC1 (500 mL, 5 vol). The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h.
Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor.
The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The organic phase was distilled under vacuum at <75 °C to 3 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The resulting slurry was heated to an internal temperature of 85 °C until complete dissolution of solids was achieved. The mixture was allowed to stir for 0.5 h at 85 °C and then cooled to an internal temperature of 19 – 25 °C over 5 h. The mixture was allowed to stir at 25 °C for no less than 2 h. The slurry was filtered. The filter cake was washed with toluene (1 x 2 vol (200 mL) and 1 x 1.5 vol (150 mL)). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford product C101 (95.03 g, 70%).
Purification of Compound 87 by Recrystallization to Form A
[00412] Compound 87 as an iPrOAc solvate (17.16 g after correction for iPrOAc content, 1.0 equiv) was charged to a reactor. A mixture of IP A (77 mL, 4.5 vol) and water (137 mL, 8 vol) were charged to the reactor. The slurry was heated to an internal temperature of 75 °C. The batch was cooled to an internal temperature of 25 °C over 10 h and then stirred at 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 52 mL, 2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford Compound 87 as a neat, crystalline form (Form A, 15.35 g, 89%).
Synthetic Procedure
[00413] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (50 g, 1.0 equiv), S2 hydrochloride (28.3 g, 1.05 equiv), and CDMT (34.1 g, 1.1 equiv) was charged with 2-MeTHF (200 mL, 4 vol) and DMF (50 mL, 1 vol) and the mixture was agitated. The internal temperature adjusted to <13 °C. The reactor was charged with NMM (64.5 g, 3.5 equiv) over 1 h, while maintaining internal temperature <20 °C. The internal temperature was adjusted to 25 °C and the batch was stirred at that temperature for 14 h. The batch was cooled to 10 °C and charged with water (250 mL, 5 vol) while keeping the internal temperature <20 °C. The batch was then warmed to 20 – 25 °C. Stirring was stopped, and the phases allowed to separate for 10 min. The lower aqueous phase was removed. The aqueous layer was back extracted with 2-MeTHF (2 x 200 mL, 2 x 4 vol) at 20 – 25 °C. The combined organic phases were washed with 1 N HC1 (500 mL, 10 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The lower aqueous phase was removed. The organic phases were washed with 0.25 N HC1 (2 x 250 mL, 2 x 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min for each wash. Lower aqueous phases were removed after each wash. The organic phase was washed with water (250 mL, 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The reactor was charged with 20 wt % Nuchar RGC® and stirred for 4 h. The reaction mixture was filtered through a pad of celite®. The reactor and celite® pad were rinsed with 2-MeTHF. The combined organics were distilled under vacuum at <50 °C to 5 total volumes. The reactor was charged with iPrOAc (500 mL, 10 vol). The organic phase was distilled under vacuum at <50 °C to 5 total volumes. The mixture was charged with additional iPrOAc (400 mL, 8 vol) and distillation under vacuum was repeated. The mixture was charged with additional iPrOAc (250 mL, 5 vol), heated to an internal
temperature of 75 °C and stirred for 5 h. The slurry was cooled to 25 °C, over 5 h and stirred for no less than 12 h. The slurry was filtered and the filter cake washed with iPrOAc (2 x 50 mL, 2 x 1 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C to afford Compound 87 as an iPrOAc solvate (60.38 g including 9.9% w/w iPrOAc, 80.8% yield).
Form A of Compound 87
[00414] Compound 87 hydrate form was converted to the dehydrated, neat crystalline form (Form A) after drying.
Hydrate Form A of Compound 87
[00415] A mixture of IP A (4.5 vol) and water (8 vol) was added to compound 87
(iPrOAc solvate containing ~2.5 – 11 wt% iPrOAc, 1.0 equiv). The slurry was heated to an internal temperature of 75 °C and filtered hot. The filtrate was cooled to 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C. The product was isolated as Hydrate form.
IPAC Solvate of Compound 87:
[00416] The large scale synthesis described above provided an iPrOAc solvate containing ~2.5 – 11 wt% iPrOAc after drying.
Amorphous Form of Compound 87
[00417] ~lg of compound 87 was dissolved in 22mL of acetone. The solution was evaporated using a Genevac. The resulted solid was dried at 60C under vacuum overnight. The dried solid was amorphous form.
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2020131807-A1 | Inhibitors of apol1 and methods of using same | 2018-12-17 | |
| US-2020377479-A1 | Inhibitors of apol1 and methods of using same | 2018-12-17 |
///////////
O=C(N[C@@H]1C(=O)NC[C@H]1O)CCc1c2ccccc2[NH]c1c1ccc(F)cc1
SIMILAR

predicted
VX 147
cas 2446816-88-0 predicted
O=C(N[C@@H]1C(=O)NC[C@H]1O)CCc1c2cc(F)cc(F)c2[NH]c1c1ccc(F)cc1
- OriginatorVertex Pharmaceuticals
- ClassSmall molecules; Urologics
- Mechanism of ActionApolipoprotein L1 inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- Phase IIFocal segmental glomerulosclerosis
- Phase IKidney disorders
Most Recent Events
- 14 Apr 2020Phase-II clinical trials in Focal segmental glomerulosclerosis in USA (PO) (EudraCT2020-000185-42) (NCT04340362)
- 31 Dec 2019Vertex Pharmaceuticals completes phase I clinical trial in Focal segmental glomerulosclerosis and Kidney disorders (In volunteers) in USA (PO)
- 05 Aug 2019Vertex Pharmaceuticals plans a phase II proof-of-concept trial for focal segmental glomerulosclerosis in 2020
| NCT Number ICMJE | NCT04340362 |
|---|---|
| Other Study ID Numbers ICMJE | VX19-147-101 2020-000185-42 ( EudraCT Number ) |
ONO-2910

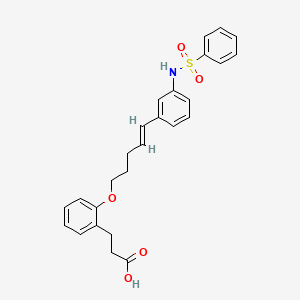
ONO-2910
CAS 2410177-35-2
3- [2-[(E) -5- [3- (benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid
3- [2-[(E) -5- [3- (benzenesulfonamido) phenyl] penta-4-enoxy] phenyl] propanoic acidC26 H27 N O5 S465.56Benzenepropanoic acid, 2-[[(4E)-5-[3-[(phenylsulfonyl)amino]phenyl]-4-penten-1-yl]oxy]-
ONO Pharmaceuticals is developing ONO-2910 , the lead from a program of novel transient receptor potential cation channel 4/5 inhibitors, for treating peripheral neuropathy. In April 2021, a phase II trial in patients with diabetic polyneuropathy was initiated.
PATENT
CN112513011-BENZENE DERIVATIVE
| Example 84: 3-[2-[(E)-5-[3-(Benzenesulfonamido)phenyl]pent-4-enyloxy]phenyl]propionic acid |
| [Chemical formula 52] |
| |
| To a solution of the compound (146 mg) produced in Example 83 in THF (0.5 mL) and methanol (0.1 mL), 1M aqueous lithium hydroxide solution (0.5 mL) was added, and the mixture was stirred at 50°C for 8 hours. 1M hydrochloric acid was added to make it acidic, and it was extracted with ethyl acetate. After drying the organic layer over sodium sulfate, it was concentrated under reduced pressure to obtain the title compound (105 mg) having the following physical properties. |
| HPLC retention time (min): 1.10 |
| 1 H-NMR(CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61,2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45-7.49, 7.55, 7.75 -7.78. |

NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT
WO-2021153690
Novel crystalline forms of 3-[2-[(E)-5-[3-(benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid act as neuroprotective, useful for treating neurological disorders eg chronic inflammatory demyelinating polyneuritis, Guillain-Barre syndrome and allergic angiitis.Example 1:
Sulfuric acid (0.26 mL) is added to a solution of isopropyl 3- (2-hydroxyphenyl) propanoate 3,4-dihydrocoumarin (50.0 g) in isopropyl alcohol (500 mL), and the reaction mixture is mixed at room temperature for 2 hours. Stirred. The reaction mixture was concentrated under reduced pressure, and the obtained residue was diluted with ethyl acetate. The mixture was washed with saturated aqueous sodium hydrogen carbonate solution, water and saturated brine, dried over sodium sulfate, and concentrated under reduced pressure to give the title compound (73.2 g) having the following physical properties.
1 1 H-NMR (CDCl 3 ): δ 1.20, 2.66-2.70, 2.87-2.91, 4.95-5.08, 6.86-6.91, 7.06-7.15, 7.35.
Example 2: Isopropyl 3- (2- (pent-4-in-1-yloxy) phenyl) propanoate In a solution of the compound (3.00 g) prepared in Example 1 in N, N-dimethylacetamide (25 mL) at room temperature. Cesium carbonate (9.39 g) was added at the same temperature, and the mixture was stirred at the same temperature for 15 minutes. 5-Chloro-1-pentyne (CAS Registry Number: 14267-92-6) (1.63 g) was added to the reaction solution at room temperature, and the mixture was stirred at 60 ° C. for 3 hours. Water was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 1: 0 → 5: 1) to give the title compound (2.40 g) having the following physical property values.
HPLC retention time (minutes): 1.13.Example 3: Isopropyl (E) -3- (2-((5- (4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) penta-4-en-1-yl) Il) Oxy) Phenyl) Propanoate In
a heptane (2 mL) solution of the compound (1.00 g) prepared in Example 2, 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1. 17 g) and 4-dimethylaminobenzoic acid (60.2 mg) were added, and the mixture was stirred at 100 ° C. for 4 hours. The reaction solution was cooled to room temperature and then concentrated. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 20: 1 → 4: 1) to give the title compound (503 mg) having the following physical characteristics.
HPLC retention time (minutes): 1.38.Example 3 (1):
Pyridine (0.95 mL), N, N-dimethyl in a solution of N- (3-bromophenyl) benzenesulfonamide 3-bromoaniline (1.02 g) in dichloromethane (20 mL) at 0 ° C. Aminopyridine (hereinafter abbreviated as DMAP) (72.4 mg) and benzenesulfonyl chloride (1.10 g) were added, and the mixture was stirred at room temperature for 2 hours. After concentrating the reaction solution, the obtained residue is purified by silica gel column chromatography (hexane: ethyl acetate = 9: 1 → 2: 1) to give the title compound (1.96 g) having the following physical properties. rice field.
HPLC retention time (minutes): 0.98.
Example 4: Isopropyl (E) -3-(2-((5- (3- (phenylsulfonamide) phenyl) penta-4-en-1-yl) oxy) phenyl) propanoate The
compound prepared in Example 3. In a solution of (180 mg) in THF (3 mL), the compound (168 mg) prepared in Example 3 (1), chloro (2-dicyclohexylphosphino-2′, 4′, 6′-triisopropyl-1,1′- Biphenyl) [2- (2′-amino-1,1′-biphenyl)] palladium (II) (0.035 g) and a 2M tripotassium phosphate aqueous solution (0.67 mL) were added, and the mixture was stirred at 60 ° C. for 1 hour. .. The reaction solution was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 7: 1 → 2: 1) to give the title compound (113 mg) having the following physical characteristics.
HPLC retention time (minutes): 1.24
Example 5: 3- [2-[(E) -5- [3- (benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid
[Chemical 2]
A 1 M aqueous lithium hydroxide solution (0.5 mL) was added to a solution of the compound (146 mg) prepared in Example 4 in THF (0.5 mL) and methanol (0.1 mL), and the mixture was stirred at 50 ° C. for 8 hours. It was acidified by adding 1M hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to give the title compound (105 mg) having the following physical characteristics.
Form: Amorphous
HPLC retention time (minutes): 1.101
1 H-NMR (CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61, 2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45-7.49, 7.55, 7.75-7.78.
PATENT
WO2020027150
https://patents.google.com/patent/WO2020027150A1/en
Example 83: Isopropyl (E) -3- (2-((5- (3- (phenylsulfonamido) phenyl) penta-4-en-1-yl) oxy) phenyl) propanoate The compound prepared in Example 82 Compound (168 mg) prepared in Example 9 and chloro (2-dicyclohexylphosphino-2 ′, 4 ′, 6′-triisopropyl-1,1′-biphenyl) [180 mg) in THF (3 mL) solution were added. 2- (2′-Amino-1,1′-biphenyl)] palladium (II) (0.035 g) and a 2M aqueous solution of tripotassium phosphate (0.67 mL) were added, and the mixture was stirred at 60 ° C. for 1 hour. After cooling the reaction solution to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 7: 1 → 2: 1) to give the title compound (113 mg) having the following physical data.
HPLC retention time (min): 1.24.Example 84: 3- [2-[(E) -5- [3- (benzenesulfonamido) phenyl] penta-4-enoxy] phenyl] propanoic acid
To a solution of the compound prepared in Example 83 (146 mg) in THF (0.5 mL) and methanol (0.1 mL) was added a 1 M aqueous lithium hydroxide solution (0.5 mL), and the mixture was stirred at 50 ° C. for 8 hours. The mixture was acidified with 1M hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to give the title compound (105 mg) having the following physical data.
HPLC retention time (min): 1.10
1 H-NMR (CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61, 2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45 -7.49, 7.55, 7.75-7.78.

///////////ONO-2910, ONO 2910, PHASE 2,
O=S(=O)(Nc1cc(\C=C\CCCOc2ccccc2CCC(=O)O)ccc1)c1ccccc1
Rilzabrutinib
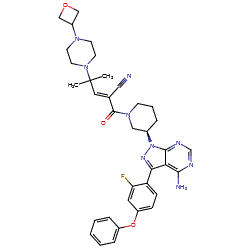
![(R)-2-(3-(4-Amino-3-(2-fluoro-4-phenoxyphenyl)-1H-pyrazolo[3,4-d]-pyrimidin-1-yl)piperidine-1-carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin-1-yl)pent-2-enenitrile.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118325989&t=l)


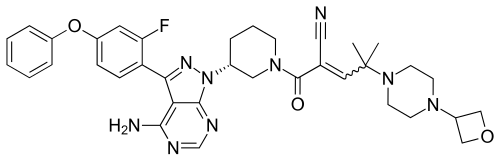
PRN 1008, Rilzabrutinib
CAS 1575591-66-0
| リルザブルチニブ; |
C36H40FN9O3,
| MW 665.7597 |
2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidine-1-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-1-yl]pent-2-enenitrile
Anti-inflammatory disease, Autoimmune disease treatment
Fda 2025, approvals 2025 8/29/2025, Wayrilz, To treat persistent or chronic immune thrombocytopenia that has not sufficiently responded to immunoglobulins, anti-D therapy, or corticosteroids
- OriginatorPrincipia Biopharma
- Class2 ring heterocyclic compounds; Amines; Anti-inflammatories; Fluorobenzenes; Nitriles; Phenyl ethers; Piperazines; Piperidines; Pyrazoles; Pyrimidines; Skin disorder therapies; Small molecules
- Mechanism of ActionAgammaglobulinaemia tyrosine kinase inhibitors
- Orphan Drug StatusYes – Idiopathic thrombocytopenic purpura; Pemphigus vulgaris
- Phase IIIIdiopathic thrombocytopenic purpura; Pemphigus vulgaris
- Phase IIAutoimmune disorders
- 02 Jun 2021Efficacy data from a phase IIa trial in Ankylosing spondylitis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
- 07 Apr 2021Sanofi initiates enrollment in a phase I pharmacokinetics trial in healthy volunteers in Australia (PO, Tablet, Capsule) (NCT04748926)
- 31 Mar 2021Sanofi announces intention to seek regulatory approval for Idiopathic thrombocytopenic purpura in 2023 (Sanofi pipeline, May 2021)
Rilzabrutinib, sold under the brand name Wayrilz, is an anti-cancer medication used for the treatment of immune thrombocytopenia.[1] Rilzabrutinib is a tyrosine kinase inhibitor.[1] It is taken by mouth.[1]
Rilzabrutinib may increase the risk of serious infections (including bacterial, viral, or fungal).[2] The most common side effects include diarrhea, nausea, headache, abdominal pain, and COVID-19.[2]
Rilzabrutinib was approved for medical use in the United States in August 2025.[2]
CLIP
Sanofi to acquire BTK inhibitor firm Principia for $3.7 billion
Principia is testing its small-molecule compounds in multiple sclerosis and immune system diseases
Sanofi will pay $3.7 billion to acquire Principia Biopharma, a San Francisco-based biotech firm developing small molecules that inhibit Bruton tyrosine kinase (BTK). The price represents about a 75% premium over Principia’s stock market value in early July, before reports surfaced that Sanofi was interested in buying the firm.
BTK is a protein important for both normal B cell development and the proliferation of lymphomas, which are B cell cancers. AbbVie, AstraZeneca, and BeiGene all market BTK inhibitors for treating specific kinds of lymphomas. Sales of AbbVie’s inhibitor, Imbruvica, approached $4.7 billion in 2019.
Other drug firms have been eager to get in on the action as well. In January, Merck & Co. spent $2.7 billion to acquire ArQule, whose experimental noncovalent BTK inhibitor is designed to overcome resistance that some cancers develop after treatment with current covalent BTK inhibitors. Eli Lilly and Company’s $8 billion acquisition of Loxo Oncology in 2019 also included a noncovalent BTK inhibitor.
BTK is also linked to inflammation, and Principia focuses on developing BTK inhibitors for immune system diseases and multiple sclerosis. Its compound rilzabrutinib is currently in clinical trials for pemphigus and immune thrombocytopenia. In 2017, Sanofi struck a deal to develop Principia’s brain-penetrant BTK inhibitor, SAR442168, for multiple sclerosis.
Sanofi announced in April of this year that the inhibitor reduced formation of new lesions—the scarred nervous tissue that gives multiple sclerosis its name—by 85% in a Phase II clinical trial. A Phase III trial of the compound began in June.
Upon announcing its deal to acquire Principia, Sanofi said that both rilzabrutinib and SAR442168 have the potential to become a “pipeline in a product,” indicating they can be used for many immune-related and neurological diseases, respectively.
The anti-inflammatory effects of BTK inhibitors have raised interest in the drugs as treatments for people hospitalized with COVID-19. Notably, the US National Cancer Institute conducted a small study suggesting acalabrutinib may help reduce the respiratory distress and inflammation in people with COVID-19. Based on that preliminary study, AstraZeneca—which markets acalabrutinib as Calquence—is conducting a 60-person randomized trial of the drug for COVID-19.
Sanofi has not indicated interest in investigating Principia’s BTK inhibitors as COVID-19 treatments.Chemical & Engineering NewsISSN 0009-2347
PATENT
WO 2021127231https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021127231&tab=PCTDESCRIPTION&_cid=P20-KRA0I9-18818-1
SOLID FORMS OF 2-[3-[4-AMTNO-3-(2-FT,TTORO-4-PHENOXY- PHEN¥L)PYRAZOLO[3,4 D]PYRIMIDIN l~YL]PIPERIDINE~l~CARBON¥L] 4~
METHYL-4-[4-(OXETAN-3-YL)PIPERAZIN-l-YLjPENT-2-ENENITRILE
[11 This application claims the benefit of priority to U.S. Provisional Application
No 62/951,958, filed December 20, 2019, and U.S Provisional Application No. 63/122,309, filed December 7, 2020, the contents of each of which are incorporated by reference herein in their entirety.
[2] Disclosed herein are solid forms of 2-[3-[4~amino-3~(2~fluoro-4-phenoxy-plienyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l Carbonyl]~4-nietliyl-4~[4-(oxetaii~3-yl)piperazin-!~yi]pent-2~enenitriie (Compound (I)), methods of using the same, and processes for making Compound (I), including its solid forms. The solid forms of Compound (I) may be inhibitors of Bruton’s tyrosine kinase (BTK) comprising low residual solvent content.
[3| The enzyme BTK is a member of the Tec family non-receptor tyrosine kinases.
BTK is expressed in most hematopoietic cells, including B cells, mast cells, and macrophages BTK plays a role in the development and activation of B cells. BTK activity has been implicated in the pathogenesis of several disorders and conditions, such as B cell-related hematological cancers (e.g., non-Hodgkin lymphoma and B cell chronic lymphocytic leukemia) and autoimmune diseases (e.g., rheumatoid arthritis, Sjogren’s syndrome, pemphigus, IBD, lupus, and asthma).
[4] Compound (I), pharmaceutically acceptable salts thereof, and solid forms of any of the foregoing may inhibit BTK and be useful in the treatment of disorders and conditions mediated by BTK activity. Compound (I) is disclosed in Example 31 of WO 2014/039899 and has the following structure:
where *C is a stereochemical center. An alternative procedure for producing Compound (!) is described in Example 1 of WO 2015/127310.
[5] Compound (I) obtained by the procedures described in WO 2014/039899 and WO 2015/127310 comprises residual solvent levels well above the limits described in the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (“ICH”) guidelines. In general, manufacturing processes producing residual solvent levels near or above the ICH limits are not desirable for preparing active pharmaceutical ingredients (APIs).
Example 1: Spray Drying Process A
[311] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was washed with pH 3 phosphate buffer to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. The dichloromethane solution was then washed with pH 7 buffer and solvent exchanged into isopropyl acetate. The isopropyl acetate solution was then washed with pH 3 phosphate buffer, bringing Compound (I) into the aqueous layer and removing non-basic impurities. The pH of the aqueous layer was adjusted to pH 9 with 10% sodium hydroxide, and the aqueous layer was extracted with isopropyl acetate. Upon concentration under vacuum, Compound (I) was precipitated from heptane at 0 °C, filtered and dried to give a white amorphous solid as a mixture of the (E) and (Z) isomers, as wet Compound (I). Wet Compound (I) was dissolved in methanol and spray dried at dryer inlet temperature of 125 °C to 155 °C and dryer outlet temperature of 48 to 58 °C to obtain the stable amorphous Compound (I) free base with levels of isopropyl acetate and heptane below 0.5% and 0.05%, respectively.
Example 2: Spray Drying Process B
intermediate A
Compound (!)
[241] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe, and recirculating fluid chiller/heater was charged with Intermediate A (20.2 kg) and Intermediate B (13.6 kg, 1.5 equiv). DCM (361.3 kg, 14.5 vol) was charged to the reactor. The mixture was agitated, and the batch cooled to 0 °C to 5 °C. The reactor was charged with pyrrolidine (18.3 kg, 6 equiv) and then charged with TMSC1 (18.6 kg, 4 eq). Stirring was continued at 0 °C to 5 °C for 0.5 to 1 hour
[242] At 0 °C to 5 °C, acetic acid (2.0 equiv) was charged to the reactor followed by water (5 equiv). Stirring was continued at 0 °C to 5 °C for 1 to 1.5 hours. Water (10 equiv) was charged to the reactor, and the solution was adjusted to 20 °C to 25 °C. The internal temperature was adjusted to 20 °C to 25 °C and the biphasic mixture was stirred for 15 to 20 mins. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed.
[243] Water (7 vol) was charged to the reactor. The pH was adjusted to 2.8-3.3 with a 10 wt. % solution of citric acid. Stirring was continued at 0 to 5 °C for 1 to 1.5 hours. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed.
[244] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe, and recirculating fluid chiller/heater was charged with an approximately 9% solution of NaHCCri (1 vol) and the organic layer. The internal temperature was adjusted to 20 °C to 25 °C, and the biphasic mixture was stirred for 15 to 20 mins. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed. The aqueous layer was measured to have a pH greater than 7.
[245] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe and recirculating fluid chiller/heater was charged with the organic layer. The organic phase ¾s distilled under vacuum at less than 25 °C to 4 total volumes. IP AC (15 vol) was charged to the reactor. The organic phase was distilled under vacuum at less than 25 °C to 10 total volumes. Water (15 vol) followed by pH 2.3 phosphate buffer were charged to the reactor at an internal temperature of 20 °C to 25 °C. The pH adjusted to 3 Stirring was stopped and phases allowed to separate for at least 0.5 h. The organic phase was removed.
[246] The following steps were repeated twice: IP AC (5 vol) was charged to the reactor containing the aqueous layer. Stirring was continued for 0.25 to 0.5 hours. Stirring was stopped and phases allowed to separate for at least 0.5 h. The organic phase was removed. [247] IP AC (15 vol) was charged to the reactor containing the aqueous layer. A pH 10 phosphate buffer was charged to the reactor and the pH adjusted to 10 with 14% NaOH solution. Stirring was continued for 1.5 to 2 hours. Stirring was stopped and phases allowed to separate for at. least 0.5 h. The aqueous layer was discarded. The organic layer was dried over brine.
[248] The organic solution was distilled under vacuum at less than 25 °C to 5 total volumes.
[249] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe and recirculating fluid chiller/heater was charged with n-heptane (20 vol). The internal temperature was adjusted to 0 to 5 °C, and the IP AC solution was added.
[250] The suspension was filtered. The filter cake was washed with n-heptane and the tray was dried at 35 °C. Compound (I) (24.6 kg) was isolated in 86% yield.
[251] Compound (1) was dissolved in methanol (6 kg) and spray dried to remove residual IP AC and n-heptane.
Example 3: Precipitation Process A
[252] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing with pH 3 aqueous solution to remove basic impurities that are more soluble than Compound (1) in the aqueous layer. Washing was repeated as needed to reduce impurities. Methanesulfonic acid was added to the dichloromethane solution, and the dichloromethane solution was concentrated by distillation under reduced pressure, followed by addition of 1% NaCi aqueous solution and isopropyl acetate before adjustment of pH to approximately 3 with potassium hydroxide. The isopropyl acetate layer was removed and discarded. The aqueous layer containing Compound (I) was washed with isopropyl acetate to remove hydrophobic impurities. Washing was repeated as needed to reduce related substance impurities. Residual isopropyl acetate was removed by distillation under reduced pressure. The aqueous solution containing Compound (I) was cooled to 0 to 5°C before adjusting the pH to approximately 9 with potassium hydroxide. The free base of Compound (I) was allowed to precipitate and maturate at 20 °C for 20 hours. The mixture temperature was then adjusted to 20 °C to 25 °C, and the hydrate impurity was verified to be less than 0.3% (< 0.3%). The cake of the free base of Compound (I) was filtered and washed as needed to reduce conductivity. The cake was then allowed to dry on the filter under vacuum and nitrogen swept to reduce water content by Karl-Fischer (KF < 50%) before transferring to the oven for drying. The wet cake of the free base of Compound (1) was dried under vacuum at 25 °C until water content by Karl -Fischer was less than 1.5% (KF < 1.5%), and then dehmiped by milling to yield a uniform white amorphous solid as a mixture of the (E) and (Z) isomers, with no detectible levels of isopropyl acetate or heptane.
Example 4: Precipitation Process 3B
[253] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing with pH 3 aqueous solution to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. The washing was repeated as needed to reduce residual solvents and impurities. The dichloromethane solution was then washed with saturated sodium bicarbonate (pH > 7). Dichloromethane was removed by distillation under reduced pressure, followed by addition of water and isopropyl acetate. The pH of the aqueous layer was adjusted to pH to 2.8 – 3.3 with 2 M aqueous sulfuric acid (H2SQ4) at 0 – 5 °C, and the mixture rvas stirred and settled. After phase separation removal of the organic layer, the aqueous layer was washed with isopropyl acetate three times and the residual isopropyl acetate in aqueous layer was distilled out under vacuum at a temperature below 25 °C and the solution was basitied with 5% aqueous KOFI to pH 9 – 10 to a slurry . The resulting suspension was stirred and warmed up to 20 °C to 25 °C and aged for 20 h. The product was filtered and washed with water and dried to give white solid in 86% yield.
Example 5: Precipitation Process C
[254] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. Washing was repeated as needed to reduce impurities. Methanesulfonic acid was added to the d chloromethane solution, and the dichloromethane solution was concentrated under reduced pressure to obtain a thin oil. The concentrated oil was cooled to approximately 5°C before washing with an aqueous solution of sodium chloride. The organic phase was discarded. Washing of the aqueous layer was repeated as needed with dichloromethane to remove low level impurities. The pH of the aqueous solution was adjusted to approximately 3 with an aqueous solution of potassium hydroxide. Residual dichloromethane was removed
under reduced pressure. The level of residual acetic acid was determined by, for example, titration. The aqueous solution containing Compound (I) was cooled to a temperature between 0°C and 5°C. Acetic acid was present at 0 wt % to 8 wt. %. Acetic acid level was 0 wt % if the aqueous acid solution was washed with aqueous sodium bicarbonate or another aqueous inorganic base. Optionally, additional acetic acid was added to achieve a 0 wt.% to 8 wt. % acetic acid level. An aqueous solution of potassium hydroxide was constantly charged to the aqueous solution to obtain a pH to approximately 9.5. The free base of Compound (I) was allowed to precipitate and maturate at approximately 20 °C for least 3 hours. The cake (wet solid) of the free base of Compound (I) was filtered and washed with water. The wet cake was then dried under reduced vacuum with slight heat. Alternatively, instead of washing the wet cake with water, the wet cake was reslurried with water at approximately 15 °C for at least 1 hour before filtering. The free base of Compound (I) in the fomi of a wet cake was dried under vacuum with slight heat at 25°C.
[255] FIGs. 12-15 are example SEM images showing the variable morphologies of particles of Compound (I) during the filtration step to isolate Compound (I) based on the amount acetic acid added during the initial step in the precipitation of Compound (Ϊ) (FIG. 12: at 0 wt. % acetic acid; FIG 13: at 3 wt. % acetic acid; FIG. 14: at 5 wt. % acetic acid; FIG 15: at 8 wt. % acetic acid). Filtration speed depended on the morphology and was the fastest for 0 wt. % acetic acid. At 1 wt. % acetic acid, the filtration speed diminished considerably, improving at 2 wt. % to 3 wt. % acetic acid. Morphologies with more open holes (such as, e.g., more porous particles) resulted in improved filtration speeds, whereas more compact particles resulted in decreased filtration speed.
Example 6: Conversion of a Crystalline Form of Compound (Ϊ) to an Amorphous Form
[256] 9.8 grams of a crystalline form of Compound (I) were dissolved in approximately 20 mL of dichloromethane and approximately 120 ml. of brine solution. Then, approximately 1 equivalent of methanesulfonic acid was added. The pH w¾s approximately 2. The layers were separated. The aqueous layer was concentrated at a temperature between 0°C and 5°C to remove residual dichloromethane before slowly adding aqueous KOI I solution (approximately 5%) to adjust the pH to a value between 9 and 10. During aqueous KOH addition, an amorphous form of Compound (I) precipitated out. The slurry was slowly warmed to room temperature and then was stirred for approximately 24 hours before filtering and rinsing the wet cake with water. The wet cake was dried under vacuum with slight heat at approximately 30°C to provide 7 grams of a white to an off-white solid (87% yield and 98 4% purity). XRPD showed that the product was an amorphous solid form of Compound (I).
Example 7: Micronization of Compound (I) Particles Obtained by Precipitation Processes
[257] A fluid jet mill equipment was used during lab scale jet milling trials. The fluid jet mill equipment includes a flat cylindrical chamber with 1.5” diameter, fitted with four symmetric jet nozzles winch are tangentially positioned in the inner wall. Prior to feeding material to the fluid jet mill in each trial, the material was sieved in a 355 iim screen to remove any agglomerates and avoid blocking of the nozzles during the feed of material to the micronization chamber. The material to be processed was drawn into the grinding chamber through a vacuum created by the venturi (P vent ~ 0 5 – 1 0 bar above P grind). The feed flow rate of solids (F_feed) was controlled by a manual valve and an infinite screw volumetric feeder. Compressed nitrogen was used to inject the feed material; compressed nitrogen was also used for the jet nozzles in the walls of the milling chamber. Compressed fluid issuing from the nozzles expands from P grind and imparts very’ high rotational speeds in the chamber. Accordingly, material is accelerated by rotating and expanding gases and subjected to centrifugal forces. Particles move outward and are impacted by high velocity jets, directing the particles radially inward at very high speeds. Rapidly moving particles impact the slower moving path of particles circulating near the periphery of the chamber. Attrition takes place due to the violent impacts of particles against each other. Particles with reduced size resulting from this sequence of impacts are entrained in the circulating stream of gas and swept against the action of centrifugal force toward the outlet at the center. Larger particles in the gas stream are subjected to a centrifugal force and returned to the grinding zone. Fine particles are carried by the exhaust gas to the outlet and pass from the grinding chamber into a collector.
[258] The feeder has continuous feed rate control; however, to more precisely control the feed rate, the full scale of feed rates was arbitrary divided in 10 positions. To calibrate F feed, the feeder was disconnected from milling chamber and 10 g of Compound (I) powder was fed through the feeder operating at various feed rate positions. The mass of powder flowing through the feeder over 6 minutes was marked. The resulting feed rate was directly proportional to feeder position. After processing each of the four trials, the jet mill was stopped, micronized product removed from the container, and the milling chamber checked for any powder accumulation.
Variables/Parameters
F_feed Feed flow rate of solids [kg/h]
P grind Grinding pressure inside the
drying chamber [bar]
P vent Feed pressure in the venturi [bar]
Example 8: Residual Solvent Levels
[251] Retention of process solvents (/.<?., res dual solvents) depends on van der Waal s’ forces that are unique to and an inherent property of each molecule. Additionally, solvent retention depends how the API solid is formed, isolated, washed, and dried (i.e., during the manufacturing process). Because residual solvents may pose safety risks, pharmaceutical processes should be designed to minimize residual solvent levels (e.g , to result in residual solvent levels below the limits established in the ICH guidelines).
[252] Residual solvent analysis was performed using gas chromatography-mass spectrometry. The residual solvent levels in solid forms of Compound (I) prepared by spray drying processes described herein and precipitation processes described herein are provided in Table 2. The residual solvent levels in crude Compound (I) listed in Table 2 are comparable to the residual solvent levels in crude Compound (I) prepared according to the procedures detailed in Example 31 of WO 2014/039899 and Example 1 of WO 2015/127310.
Table 2: Residual solvent levels in solid forms of Compound (I)
PATENT
WO 2015127310
https://patents.google.com/patent/WO2015127310A1/enExample 1Synthesis of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l- yl]-piperidine-l-carbonyl]-4-m iperazin-l-yl]pent-2-enenitrile

Step 1To a solution of 3-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin-l -yl]-l-piperidyl]-3-oxo-propanenitrile (15 g, 3.12mmol), 2-methyl-2-[4- (oxetan-3-yl)piperazin-l-yl]propanal (794.25mg, 3.74mmol) in DCM (40mL), pyrrolidine (1.54mL,18.71mmol) at 0-5 °C was added, which is followed by TMS-Cl (1.58mL,12.47mmol). The reaction mixture was stirred at 0-5 °C for 3 h and was quenched with 1 M potassium phosphate buffer (pH 3). Layers were separated and the organic layer was washed once more with 1 M potassium phosphate buffer (pH 3). The organic layer was extracted withl M potassium Phosphate buffer at pH 1.5. Layers were separated. The aqueous phase contained the desired product while the impurities stayed in the organic phase. The aqueous phase was neutralized with 1 M potassium phosphate (pH 7) and was extracted with isopropylacetate (10 volumes). Upon concentration 2-[(3R)-3-[4-amino-3-(2-fluoro-4- phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2-enenitrile was obtained as a foam having >99% HPLC purity. MS (pos. ion) m/z: 666 (M+l ).The foam containing high levels of residual solvent was dissolved in 2 M HC1 and the resulting solution was placed under vacuum to remove residual organic solvents. pH of the solution was then adjusted to ~ 7 and the resulting paste was filtered and dried in vacuum without heat. This resulted in isolation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3- yl)piperazin- l-yl]pent-2-enenitrile containing residual water up to 10%. Drying under vacuum without heat reduces the water level but lead to generation of impurities.Step 1AAlternatively, the isopropylacetate solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4- phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4- (oxetan-3-yl)piperazin-l -yl]pent-2-enenitrile can be concentrated to 4 vol and added to heptane (20 volume) at 0 °C. The resulting suspension was stirred at 0 °C overnight and the product was filtered, washed twice with heptane and dried at 45 °C for 2 days under vacuum to give 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l – yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2-enenitrile in 85 – 90 % yield as a free flowing solid. However, the solids obtained by this method contained high residual solvents (3.9 wt% isopropylacetate and 1.7 wt% heptane). In addition, the free base form was not very stable as degradation products were observed during the drying process at less than 45 °C.Salt formationExample 2Preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4-d]pyrimidin- l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2-enenitrile hemisulfate and sulfate saltHemisulfate: To the solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin-l-yl]-piperidine -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2- enenitrile (4.2 g) in EtOAc (60 mL, 15 vol) was added sulfuric acid (0.31 g, 0.17 mL, 0.5 eq) in EtOAc (20 mL, 5 vol) at ambient temperature. The suspension was stirred at ambient temperature for ~ 2 hr and then 40 °C for 4 hr and then at ambient temperature for at least 1 hr. After filtration and drying at ambient temperature under vacuum, 1.5 g of white powder was obtained. Solubility of the hemi-sulfate at ambient temperature was > 100 mg/mL in water.Sulfate saltTo the solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2- enenitrile (810 mg) in EtOAc (8 mL, 10 vol) was added sulfuric acid (0.06 mL, 1.0 equiv.) in EtOAc (2.5 mL, 5 vol) at ambient temperature. The resulting suspension was stirred at 40 °C for 2 hr and then cooled to ambient temperature for at least 1 hr. After filtration, solids were dried by suction under Argon for 1 h to give a white powder (0.68 g) in 69% yield.
Example 3Preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin- 1 -yl]-piperidine- 1 -carbonyl] -4-methyl-4-[4-(oxetan-3-yl)-piperazin- 1 -yl]pent-2- enenitrile hydrochlorideTo a solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile (100 mg, 0.15 mmol) in CH2CI2 (1ml) at ambient temperature was added 2 equivalent of HC1 (0.3 mmol, 0.15 ml of 2M HC1 in 1 : 1 dioaxane:CH2Cl2). The resulting homogeneous solution was stirred at ambient temperature for 1 h and was added dropwise to 15 volumes of ethylacetate (as compared to CH2C12) resulting in formation of a white solid. The mixtures was aged at ambient temperature for lh and placed at 2-8 C for 19 h. Upon filtration and washing of the filter cake with ethylacetate and drying a white solid was obtained. Analysis by XRPD indicated formation of an amorphous solid. Both Ή-NMR and IC analysis indicated formation of the salt. IC indicated formation mono-HCl salt.

Example 4General procedure for preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)- piperazin-l-yl]pent-2-enenitrile mono- and di-mesylate saltsTo a solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2- enenitrile (100 mg, 0.15 mmol) in CH2C12 (1 ml) at ambient temperature was added either 1 equivalent of methanesulfonic acid (0.15 mmol, 0.2 ml of 74 mg/ml solution in CH2C12) or 2 equivalent of methanesulfonic acid (0.3 mmol, 0.4 ml of 74 mg/ml solution in CH2C12). The resulting homogeneous solution was stirred at ambient temperature for 1 h and was added dropwise to 10 volumes of antisolvents (ethylacetate, methyl tert-butylether (MTBE), or cyclohexane) (10 ml as compared to CH2C12) resulting in formation of a white solid. The mixture was aged at ambient temperature for lh and placed at 2-8 °C for 19 h. Upon filtration and washing of the filter cake with the antisolvent and drying, a white solid was obtained. Analysis by XRPD indicated formation of an amorphous solid. Both Ή-NMR and IC analysis indicated formation of the salt as well as counterion ratio.Alternatively 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]- pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile can be dissolved in 4 volumes of isopropylacetate and added to 2 equivalent of methanesulfonic acid in 6 volumes of isopropylacetate at 0 °C to generate the dimesylate salt.

1. Theoretical mesylate content, monomesylate=12.6% and dimesylate=22.4%, NO- not determinedExample 5 General procedure for the preparation of carboxylate salt Approximately 20 mg of the compound (I) was dissolved in minimum amount of the allocated solvent system. These were then mixed with the appropriate number of equivalents of counterion dissolved or slurried in the allocated solvent.If compound (I) was insoluble in the selected solvent, slurry of the sample was used after adding 300 μί.If the acid was insoluble in the selected solvent, slurry of the acid was used after adding 300 xL.If the acid was a liquid, the acid was added to the dissolved/slurried compound (I) from a stock solution in the allocated solvent.The suspensions/ precipitates resulting from the mixtures of compound (I) were temperature cycled between ambient (ca. 22°C) and 40°C in 4 hour cycles for ca. 48 hrs (the cooling/heating rate after each 4 hour period was ca. 1 °C/min). The mixtures were visually checked and any solids present were isolated and allowed to dry at ambient conditions prior to analysis. Where no solid was present, samples were allowed to evaporate at ambient. Samples which produced amorphous material, after the treatment outlined above, were re- dissolved and precipitated using anti-solvent (ter/-butylmethylether) addition methods at ambient conditions (ca. 22°C). i.e. the selected anti-solvent was added to each solution, until no further precipitation could be observed visually or until no more anti-solvent could be added. The solvents used in this preparation were acetonitrile, acetone, isopropyl acetate, THF and MTBE. The acid used were oxalic acid, L-aspartic acid, maleic acid, malonic acid, L-tartaric acid, and fumaric acid.Example 6General procedure for preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)- piperazin-l-yl]pent-2-enenitrile hemicitrate saltTo a solution 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]- pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile (5 g, 7.5 mmol) in ethanol (50 ml) was added citric acid (720.5 mg, 3.76 mmol) dissolved in 2 ml of water. Mixture was stirred at ambient temperature for 15 min, additional 0.5 ml of water was added and the mixture was stirred for 1 h, concentrated in vacuo to a gum. Ethanol was added and the mixture was concentrated. This process was repeated twice more and then CH2CI2 was added to the mixture. Upon concentration a white solid was obtained which was tumble dried under reduced pressure at 40 C for 4 h, then in a vacuum oven for 19h to give 5.4 g of a solid. Analysis by XRD indicated formation of an amorphous solid
PATENT
WO2014039899, Example 31
Rilzabrutinib (PRN1008) is an oral, reversible covalent inhibitor of Bruton’s tyrosine kinase (BTK) [1].
https://patents.google.com/patent/WO2014039899A1/enExample 31Synthesis of (R)-2-(3-(4-amino-3-(2-fluoro-4-phenoxyphenyl)- 1 H-pyrazolo[3,4-d]pyrimidin- 1 -yl)piperidine- 1 -carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin- 1 -yl)pent-2-enenitrile

Step 1A solution of 2-bromo-2-methyl-propanal (696.6 mg, 4.61 mmol) in DCM (10 mL) was cooled with an ice bath and l -(oxetan-3-yl)piperazine (328 mg, 2.31 mmol), diluted with 5-10 mL of DCM, was slowly added via addition funnel over a 15 min period. Next, Hunig’s base (0.4 mL, 2.31 mmol) was added and then the cooling bath was removed. The reaction mixture was stirred at room temperature overnight and the DCM layer was washed three times with 0.5N HC1. The combined aqueous layer was neutralized with NaOH to pH 10-11 and extracted with DCM. The combined organic layer was washed with brine and dried over Na?S04. Filtration and removal of solvent afforded 2-methyl-2-[4-(oxetan-3-yl)piperazin-l- yl]propanal as a light yellow liquid, which was used directly in the next step without further purification.Step 2To a cooled (0 °C) solution of 3-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)- pyrazolo[3,4-d]pyrimidin-l-yl]-l-piperidyl]-3-oxo-propanenitrile (80 mg, 0.17 mmol), was added 2-methyl-2-[4-(oxetan-3-yl)piperazin-l-yl]propanal (-108 mg, 0.51 mmol) in DCM (10 mL) followed by pyrrolidine (0.08 mL, 1.02 mmol) and TMS-C1 (0.09 raL, 0.68 mmol.) The ice bath was removed, and the reaction stirred 1 hour. Most of the solvent was removed and the residues were purified by chromatography, using 95:5 CH2Cl2:MeOH to obtain 79 mg of (R)-2-(3-(4-amino-3-(2-fluoro-4-phenoxyphenyl)-lH-pyrazolo[3,4-d]-pyrimidin-l- yl)piperidine- 1 -carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin- 1 -yl)pent-2-enenitrile as a white solid. MS (pos. ion) m/z: 666 (M+l).
PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0223523421001781?dgcid=rss_sd_all
Therapy based on Bruton’s tyrosine kinase (BTK) inhibitors one of the major treatment options currently recommended for lymphoma patients. The first generation of BTK inhibitor, Ibrutinib, achieved remarkable progress in the treatment of B-cell malignancies, but still has problems with drug-resistance or off-target induced serious side effects. Therefore, numerous new BTK inhibitors were developed to address this unmet medical need. In parallel, the effect of BTK inhibitors against immune-related diseases has been evaluated in clinical trials. This review summarizes recent progress in the research and development of BTK inhibitors, with a focus on structural characteristics and structure-activity relationships. The structure-refinement process of representative pharmacophores as well as their effects on binding affinity, biological activity and pharmacokinetics profiles were analyzed. The advantages and disadvantages of reversible/irreversible BTK inhibitors and their potential implications were discussed to provide a reference for the rational design and development of novel potent BTK inhibitors.

Research
Rilzabrutinib is an oral, reversible covalent inhibitor of Bruton’s tyrosine kinase, that may increase platelet counts in people with immune thrombocytopenia by means of dual mechanisms of action: decreased macrophage (Fcγ receptor)–mediated platelet destruction and reduced production of pathogenic autoantibodies.[5]
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219685s000lbl.pdf
- “FDA Approves Drug to Treat Adults with Persistent or Chronic Immune Thrombocytopenia”. U.S. Food and Drug Administration. 2 September 2025. Retrieved 5 September 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “Press Release: Sanofi’s Wayrilz approved in US as first BTK inhibitor for immune thrombocytopenia” (Press release). Sanofi. 29 August 2025. Retrieved 5 September 2025 – via GlobeNewswire.
- World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 83”. WHO Drug Information. 34 (1). hdl:10665/339768.
- Kuter DJ, Efraim M, Mayer J, Trněný M, McDonald V, Bird R, et al. (April 2022). “Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia”. The New England Journal of Medicine. 386 (15): 1421–1431. doi:10.1056/NEJMoa2110297. PMID 35417637.
External links
- “Rilzabrutinib ( Code – C174769 )”. EVS Explore.
- Clinical trial number NCT04562766 for “Study to Evaluate Rilzabrutinib in Adults and Adolescents With Persistent or Chronic Immune Thrombocytopenia (ITP) (LUNA 3)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Wayrilz |
| Other names | PRN-1008 |
| AHFS/Drugs.com | Wayrilz |
| License data | US DailyMed: Rilzabrutinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1575591-66-0 |
| PubChem CID | 73388818 |
| DrugBank | DB17709 |
| ChemSpider | 58893525 |
| UNII | NWN58M4F5T |
| KEGG | D11873 |
| ChEMBL | ChEMBL3702854 |
| Chemical and physical data | |
| Formula | C36H40FN9O3 |
| Molar mass | 665.774 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
///////////////PRN-1008, PRN 1008, Rilzabrutinib, リルザブルチニブ, Fda 2025, approvals 2025 8/29/2025, Wayrilz,
N#CC(=CC(N(C1COC1)C)(C)C)C(=O)N1CCCC1Cn1nc(c2c1ncnc2N)c1ccc(cc1F)Oc1ccccc1
PAT
PAT



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Nanatinostat
Nanatinostat
Tractinostat
CHR-3996, CHR 3996, VRx 3996,
C20H19FN6O2, 394.41
CAS 1256448-47-1
2-[(1α,5α,6α)-6-[[(6-Fluoro-2-q
2-[(1R,5S,6R)-6-{[(6-fluoroquinolin-2-yl)methyl]amino}-3-azabicyclo[3.1.0]hexan-3-yl]-N-hydroxypyrimidine-5-carboxamide2-[(1R,5S,6s)-6-{[(6-Fluoro-2-quinolinyl)methyl]amino}-3-azabicyclo[3.1.0]hex-3-yl]-N-hydroxy-5-pyrimidinecarboxamide5-Pyrimidinecarboxamide, 2-[(1R,5S)-6-[[(6-fluoro-2-quinolinyl)methyl]amino]-3-azabicyclo[3.1.0]hex-3-yl]-N-hydroxy-Chroma Therapeutics Ltd. (Originator)
- OriginatorChroma Therapeutics
- DeveloperChroma Therapeutics; Viracta Therapeutics
- ClassAmides; Antineoplastics; Pyrimidines; Quinolines; Small molecules
- Mechanism of ActionHistone deacetylase inhibitors
- Orphan Drug StatusYes – Post-transplant lymphoproliferative disorder; Plasmablastic lymphoma; T-cell lymphoma
- Phase IILymphoma
- Phase I/IIMultiple myeloma
- Phase ISolid tumours
- No development reportedGastric cancer; Nasopharyngeal cancer; Post-transplant lymphoproliferative disorder
- 01 Jun 2021Phase-II clinical trials in Lymphoma (Combination therapy, Second-line therapy or greater) in North America, Europe, Asia (PO)
- 18 May 2021Ninatinostat is still in phase I trials for Solid tumour in United Kingdom and Netherlands (Viracta Therapeutics pipeline, May 2021)
- 18 May 2021Virata Therapeutics has patent protection for dose regimen in NAVAL-1 trial in USA
Nanatinostat is under investigation in clinical trial NCT00697879 (Safety Study of the Histone Deacetylase Inhibitor, CHR-3996, in Patients With Advanced Solid Tumours).
Nanatinostat is an orally bioavailable, second-generation hydroxamic acid-based inhibitor of histone deacetylase (HDAC), with potential antineoplastic activity. Nanatinostat targets and inhibits HDAC, resulting in an accumulation of highly acetylated histones, the induction of chromatin remodeling, and the selective transcription of tumor suppressor genes; these events result in the inhibition of tumor cell division and the induction of tumor cell apoptosis. This agent may upregulate HSP70 and downregulate anti-apoptotic Bcl-2 proteins more substantially than some first-generation HDAC inhibitors. HDACs, upregulated in many tumor cell types, are a family of metalloenzymes responsible for the deacetylation of chromatin histone proteins.

Patent
WO2006123121
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2006123121
Example 44: N-Hvdroxy 2-(6-fr(6-fluoroαuinolin-2-yl)methvnamino)-3-azabicvclorS.I.OIhex-S-vDpyrimidine-δ-carboxamide
LCMS purity >98%, m/z 395 [M+H]+, 1H NMR (300 MHz, c/6-DMSO) δ: 2.30 (2H, s), 2.75 (1 H, s), 3.60 (2H, dm, J = 11.7 Hz), 3.88 (2H, d, J = 11.7 Hz), 4.69 (2H, br s), 7.66 (1 H, d, J = 8.4 Hz), 7.75 (1 H, td, J = 8.7, 3.0 Hz), 7.88 (1 H, dd, J = 9.3, 2.7 Hz), 8.48 (1 H, d, J = 8.4 Hz), 8.67 (2H, s), 9.01 (1 H, br s), 9.61 (1 H, br s), 11.09 (1 H, br s).
PATENT
WO-2021113694
Crystalline hydrate form A of N-hydroxy 2-{6-[(6-fluoro-quinolin-2-ylmethyl)-amino]-3-aza-bicyclo[3.1.0]hex-3-yl}pyrimidine-5-carboxamide ( nanatinostat ) .
Compound 1 is also known as nanatinostat, VRx-3996, or CHR-3996. It has been previously described in patents and patent applications, e.g. US patent 7,932,246 and US patent application 15/959,482, each of which is incorporated by reference in their entirety.
Compound 1
PATENT
WO2021071809 , claiming dosages for HDAC treatment with reduced side effects.
/////////Nanatinostat, CHR-3996, CHR 3996, VRx 3996, CHROMA, ORPHAN DRUG, Tractinostat, PHASE 2
| FC1=CC=C2N=C(CN[C@H]3[C@]4([H])CN(C5=NC=CC(C(NO)=O)=N5)C[C@]34[H])C=CC2=C1 |

NEW DRUG APPROVALS
ONE TIME
$10.00
TROPIFEXOR

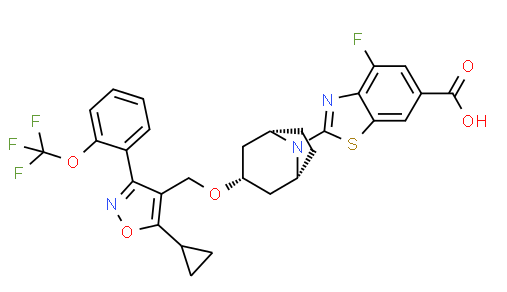
TROPIFEXOR
トロピフェクサー;
PHASE 2, NASH, PBC, liver fibrosis, bile acid diarrhea, non-alcoholic fatty liver disease
| Formula | C29H25F4N3O5S |
|---|---|
| CAS | 1383816-29-2 |
| Mol weight | 603.5845 |
TROPIFEXORLJN 452;LJN-452;LJN452;CS-2712;CPD1549;Tropifexor;Tropifexor (LJN452);LJN452;LJN452,Tropifexor;2-[(1R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]octan-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylic acidтропифексор [Russian] [INN]
تروبيفيكسور [Arabic] [INN]
曲匹法索 [Chinese] [INN]2-[(3-endo)-3-({5-Cyclopropyl-3-[2-(trifluormethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluor-1,3-benzothiazol-6-carbonsäure [German] [ACD/IUPAC Name]
2-[(3-endo)-3-({5-Cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylic acid [ACD/IUPAC Name]
6-Benzothiazolecarboxylic acid, 2-[(3-endo)-3-[[5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-4-isoxazolyl]methoxy]-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro- [ACD/Index Name]
Acide 2-[(3-endo)-3-({5-cyclopropyl-3-[2-(trifluorométhoxy)phényl]-1,2-oxazol-4-yl}méthoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylique [French] [ACD/IUPAC Name]
NMZ08KM76Z
Tropifexor fast facts
| CAS Reg. No. | 1383816-29-2 |
| Molar mass | 603.58 g/mol |
| Empirical formula | C29H25F4N3O5S |
| Appearance | White crystals |
| Melting point | 221 ºC |
| Water solubility | 6 mg/L |
| Efficacy | Anti-inflammatory, Farnesoid X receptor (FXR) agonist |
|---|---|
| Comment | Treatment of non-alcoholic steatohepatitis |
Novartis is developing tropifexor, a non-bile acid farnesoid X receptor agonist, and its analog LJP-305, for treating NASH, PBC, liver fibrosis, bile acid diarrhea and non-alcoholic fatty liver disease. In June 2021, this drug was reported to be in phase 2 clinical development.
Nonalcoholic steatohepatitis (NASH) is a liver disease that is becoming more prevalent as worldwide obesity and type 2 diabetes increase. It is characterized by accumulation of fat in the liver, inflammation, hepatocyte ballooning, and fibrosis.
Another liver disease, primary biliary cholangitis (PBC), is a cholestatic condition in which bile flow from the liver to the intestine is reduced or interrupted. It is thought to be autoimmune.
PBC is associated with decreased expression of the farnesoid X receptor (FXR), a ligand-activated nuclear receptor that is highly expressed in the liver and other organs. FXR is a key regulator of bile acid production, conjugation, and transport. FXR activation also suppresses lipogenesis; thus, it has been proposed as a treatment for NASH.
Recently, David C. Tully and colleagues at the Genomics Institute of the Novartis Research Foundation (San Diego) and the Novartis Institutes for Biomedical Research (Emeryville, CA) discovered tropifexor, a highly potent FXR agonist. They began by replacing an indole group in an existing partial FXR agonist with a 2-substituted benzothiazole-6-carboxylic acid, a change that resulted in a dramatic increase in potency. Further changes, including optimization of the benzothiazole substituent, resulted in more potent, orally bioavailable tropifexor.
Tropifexor is an investigational drug which acts as an agonist of the farnesoid X receptor (FXR). It was discovered by researchers from Novartis and Genomics Institute of the Novartis Research Foundation. Its synthesis and pharmacological properties were published in 2017.[1] It was developed for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). In combination with cenicriviroc, a CCR2 and CCR5 receptor inhibitor, it is undergoing a phase II clinical trial for NASH and liver fibrosis.[2]
Rats treated orally with tropifexor (0.03 to 1 mg/kg) showed an upregulation of the FXR target genes, BSEP and SHP, and a down-regulation of CYP8B1. Its EC50 for FXR is between 0.2 and 0.26 nM depending on the biochemical assay.
The patent which covers tropifexor and related compounds was published in 2010.[3]
PATENT
WO-2021104022
Novel, stable crystalline polymorphic form II of tropifexor , useful for treating non-alcoholic steatohepatitis (NASH), fatty liver and primary biliary cholangitis (PBC).Tropifexor was originally developed by Novartis and then licensed to Pfizer for cooperative development. It is a non-steroidal FXR (farnesoid receptor) agonist, currently in clinical phase II of indications for NASH (non-alcoholic steatohepatitis), fatty liver and primary biliary cholangitis.
The structure of Tropifexor is shown in the following formula (1):
Drug polymorphism is a common phenomenon in drug development and an important factor affecting drug quality. Different crystal forms of the same drug may have significant differences in physical and chemical properties such as appearance, fluidity, solubility, storage stability, bioavailability, etc., and there may be great differences, which will affect the storage transfer, application, stability, and efficacy of the drug In order to obtain an effective crystal form that is conducive to production or pharmaceutical preparations, it is necessary to conduct a comprehensive investigation of the crystallization behavior of the drug to obtain a crystal form that meets the production requirements.
At present, there is no literature that discloses the crystal form of Tropifexor, and there is no related literature report.
The present invention obtains a new crystal form of the compound through a large number of experimental studies on the Tropifexor compound. The new crystal form has the advantages of high solubility, good stability, low moisture absorption, simple preparation process and easy operation, etc., and has excellent properties in industrial production. Superiority.Example 1 Preparation method of Tropifexor crystal form II[0049]After mixing 60.3 mg of Tropifexor and p-aminobenzoic acid (13.7 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 51.3 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0050]Example 2 Preparation method of Tropifexor crystal form II[0051]After mixing 60.3 mg of Tropifexor and p-hydroxybenzoic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 48.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0052]Example 3 Preparation method of Tropifexor crystal form II[0053]After mixing 60.3 mg of Tropifexor and salicylic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. Filter with suction and place in a drying box at 50°C and vacuum dry to constant weight to obtain 50.0 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0054]Example 4 Preparation method of Tropifexor crystal form II[0055]After mixing 60.3 mg of Tropifexor and 2,4-dihydroxybenzoic acid (15.4 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 49.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.
PATENT
WO2021104021 ,
claiming crystalline polymorphic form I of tropifexor,Example 1 Preparation method of Tropifexor crystal form I
50.0 mg of Tropifexor was added to ethanol (1.0 ml), heated to 60° C. and stirred to obtain a clear solution, and then water (3 ml) was added dropwise to the Tropifexor solution. Stir and precipitate solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 38.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form I; its X-ray powder diffraction pattern was basically consistent with Figure 1, its DSC pattern was basically consistent with Figure 2, and its TGA pattern was basically consistent with Figure 3
PATENT
product pat, WO2012087519 , https://patents.google.com/patent/WO2012087519A1/en
has protection in the EU until November 2031, and expire in US in February 2032 with US154 extension.
PATENT
WO 2016097933
Example 1
2-r(1 R,3r,5S)-3-(f5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4-yl)methoxy)-8- azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B) and
-r(1 R,3r,5S)-3-(f5-cvclopropyl-3-r2-(trifluoromethyl)phenyll-1 ,2-oxazol-4-yl)methoxy)-8-
R1a = OCF3 (1 -1A, 1 -1 B)
R a = CF3 (1-2A, 1-2B)
Methyl 2-[(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4- yl}methoxy)-8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1 -1 A). Into a 25-mL round-bottom flask equipped with a stir bar was added sequentially 4-(((1 R,3r,5S)- 8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethoxy)phenyl)isoxazole (1 .29 mmol), N,N-dimethylacetamide (3.6 mL), cesium carbonate (3.31 mmol), and methyl 2- bromo-4-fluorobenzo[d]thiazole-6-carboxylate (3.87 mmol). After stirring the resulting slurry at room temperature for 10 minutes, the mixture was then warmed to 60 °C and stirred for 1 h. The reaction slurry was allowed to cool to room temperature, and was diluted with 200 mL of ethyl acetate and washed with water (3 χ 30 mL). The organic extracts were concentrated under vacuum and directly purified using normal phase silica gel chromatography (40 g silica column) with a 15 min gradient of 10 % to 60 % ethyl acetate/hexanes. Desired fractions were concentrated in vacuo, and the resulting residue crystallized upon standing to give methyl 2- [(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8- azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1-1 A) as a white crystalline solid. MS (m/z) : 618.2 (M+1 ).
2-r(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4-yl}methoxy)- 8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). To a 25-mL round-bottom flask equipped with a stir bar was added the ester (0.89 mmol), THF (4 mL),
MeOH (2 mL), and 3 N aqueous KOH solution (1 mL, 3 mmol). The resulting homogenous solution was stirred for 1 hour at 70 °C, cooled to room temperature, and then quenched with AcOH (roughly 0.2 mL of glacial acetic, 3 mmol) until pH=6 was achieved (Whatman class pH strip paper). At this time the reaction was diluted with ethyl acetate (40 mL) and washed with water (3 5 mL). The ethyl acetate fraction was concentrated under vacuum to give to an oily residue. To the resulting oil was then added MeOH (6 mL). The oil quickly dissolved, then immediately began to crystallize. Upon standing for 2.5 hrs, the mother liquor was withdrawn and crystals washed (3 x 2 mL of ice cold MeOH). The crystals were dried via vacuum (10 mm Hg pressure at 45 °C overnight) and then recrystallized from acetonitrile, filtered, and dried under vacuum to give 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethyl)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -2B).
Examples 1 -2A and the corresponding acid 1 -2B can be prepared following the same procedures, from the reaction of intermediate 4-((8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethyl)phenyl)isoxazole.
PAPER
European journal of medicinal chemistry (2021), 209, 112910
https://www.sciencedirect.com/science/article/abs/pii/S0223523420308825

Abstract
Farnesoid X receptor (FXR) agonists are emerging as potential therapeutics for the treatment of various metabolic diseases, as they display multiple effects on bile acid, lipid, and glucose homeostasis. Although the steroidal obeticholic acid, a full FXR agonist, was recently approved, several side effects probably due to insufficient pharmacological selectivity impede its further clinical application. Activating FXR in a partial manner is therefore crucial in the development of novel FXR modulators. Our efforts focusing on isoxazole-type FXR agonists, common nonsteroidal agonists for FXR, led to the discovery a series of novel FXR agonists bearing aryl urea moieties through structural simplification of LJN452 (phase 2). Encouragingly, compound 11k was discovered as a potent FXR agonist which exhibited similar FXR agonism potency but lower maximum efficacy compared to full agonists GW4064 and LJN452 in cell-based FXR transactivation assay. Extensive in vitro evaluation further confirmed partial efficacy of 11k in cellular FXR-dependent gene modulation, and revealed its lipid-reducing activity. More importantly, orally administration of 11k in mice exhibited desirable pharmacokinetic characters resulting in promising in vivo FXR agonistic activity.
References
- ^ Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, et al. (December 2017). “Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH)”. Journal of Medicinal Chemistry. 60 (24): 9960–9973. doi:10.1021/acs.jmedchem.7b00907. PMID 29148806.
- ^ Clinical trial number NCT03517540 for “Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and Cenicriviroc (CVC) in Adult Patients With Nonalcoholic Steatohepatitis (NASH) and Liver Fibrosis. (TANDEM)” at ClinicalTrials.gov
- ^ WO Application Filing 2012087519, Alper PB, Chianelli D, Mutnick D, Vincent P, Tully DC, “Compositions and methods for modulating fxr”, published 2012-06-28, assigned to Genomics Institute of the Novartis Research Foundation. Retrieved 17 May 2019.
| Clinical data | |
|---|---|
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1383816-29-2 |
| PubChem CID | 121418176 |
| UNII | NMZ08KM76Z |
| KEGG | D11548 |
| Chemical and physical data | |
| Formula | C29H25F4N3O5S |
| Molar mass | 603.59 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
{kind=link}
///////////TROPIFEXOR, トロピフェクサー, NOVARTIS, PHASE 2, тропифексор , تروبيفيكسور , 曲匹法索 , LJN 452, LJN-452, LJN452, CS-2712, CPD1549, Tropifexor, Tropifexor (LJN452), LJN452, LJN452, PHASE 2, NASH, PBC, liver fibrosis, bile acid diarrhea, non-alcoholic fatty liver disease
1ccc(c(c1)c2c(c(on2)C3CC3)CO[C@H]4C[C@H]5CC[C@@H](C4)N5c6nc7c(cc(cc7s6)C(=O)O)F)OC(F)(F)F

NEW DRUG APPROVALS
ONE TIME
$10.00
TROFINETIDE

Trofinetide
- Molecular FormulaC13H21N3O6
- Average mass315.322 Da
Tofinetide , NNZ-256610076853400-76-7[RN]
glycyl-2-methyl-L-prolyl-L-glutamic acid
H-Gly-PMe-Glu-OHL-Glutamic acid, glycyl-2-methyl-L-prolyl-UNII-Z2ME8F52QLZ2ME8F52QLтрофинетид [Russian] [INN]تروفينيتيد [Arabic] [INN]曲非奈肽 [Chinese] [INN]
| IUPAC Condensed | H-Gly-aMePro-Glu-OH |
|---|---|
| Sequence | GXE |
| HELM | PEPTIDE1{G.[*C(=O)[C@@]1(CCCN1*)C |$_R2;;;;;;;;_R1;$|].E}$$$$ |
| IUPAC | glycyl-alpha-methyl-L-prolyl-L-glutamic acid |
An (1-3) IGF-1 analog with neuroprotective activity.
OPTICAL ROT; -52.4 ° Conc: 0.19 g/100mL; water ; 589.3 nm; Temp: 20 °C; Len: 1.0 dm…Tetrahedron 2005, V61(42), P10018-10035
EU Customs Code CN, 29339980
Harmonized Tariff Code, 293399
- L-Glutamic acid, glycyl-2-methyl-L-prolyl-
- glycyl-2-methyl-L-prolyl-L-glutamic acid
- Glycyl-L-2-methylprolyl-L-glutamic acid
FDA APPROVED 2023/3/10, Daybue
853400-76-7 CAS
トロフィネチド;
Trofinetide (NNZ-2566) is a drug developed by Neuren Pharmaceuticals that acts as an analogue of the neuropeptide (1-3) IGF-1, which is a simple tripeptide with sequence Gly–Pro–Glu formed by enzymatic cleavage of the growth factor IGF-1 within the brain. Trofinetide has anti-inflammatory properties and was originally developed as a potential treatment for stroke,[1][2] but has subsequently been developed for other applications and is now in Phase II clinical trials against Fragile X syndrome and Rett syndrome.[3][4][5]
Trofinetide (NNZ-2566), a neuroprotective analogue of glypromate, is a novel molecule that has a profile suitable for both intravenous infusion and chronic oral delivery. It is currently in development to treat traumatic brain injury.
In February 2021, Neuren is developing trofinetide (NNZ-2566, phase 2 clinical ), a small-molecule analog of the naturally occurring neuroprotectant and N-terminus IGF-1 tripeptide Glypromate (glycine-proline-glutamate), for intravenous infusion treatment of various neurological conditions, including moderate to severe traumatic brain injury (TBI), stroke, chronic neurodegenerative disorders and peripheral neuropathies. At the same time, Neuren is also investigating an oral formulation of trofinetide (phase 3 clinical) for similar neurological indications, including mild TBI.
Autism Spectrum Disorders and neurodevelopment disorders (NDDs) are becoming increasingly diagnosed. According to the fourth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-4), Autism spectrum disorders (ASD) are a collection of linked developmental disorders, characterized by abnormalities in social interaction and communication, restricted interests and repetitive behaviours. Current classification of ASD according to the DSM-4 recognises five distinct forms: classical autism or Autistic Disorder, Asperger syndrome, Rett syndrome, childhood disintegrative disorder and pervasive developmental disorder not otherwise specified (PDD-NOS). A sixth syndrome, pathological demand avoidance (PDA), is a further specific pervasive developmental disorder.
More recently, the fifth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-5) recognizes recognises Asperger syndrome, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified (PDD-NOS) as ASDs.
This invention applies to treatment of disorders, regardless of their classification as either DSM-4 or DSM-5.
Neurodevelopment Disorders (NDDs) include Fragile X Syndrome (FXS), Angelman Syndrome, Tuberous Sclerosis Complex, Phelan McDermid Syndrome, Rett Syndrome, CDKL5 mutations (which also are associated with Rett Syndrome and X-Linked Infantile Spasm Disorder) and others. Many but not all NDDs are caused by genetic mutations and, as such, are sometimes referred to as monogenic disorders. Some patients with NDDs exhibit behaviors and symptoms of autism.
As an example of a NDD, Fragile X Syndrome is an X-linked genetic disorder in which affected individuals are intellectually handicapped to varying degrees and display a variety of associated psychiatric symptoms. Clinically, Fragile X Syndrome is characterized by intellectual handicap, hyperactivity and attentional problems, autism spectrum symptoms, emotional lability and epilepsy (Hagerman, 1997a). The epilepsy seen in Fragile X Syndrome is most commonly present in childhood, but then gradually remits towards adulthood. Hyperactivity is present in approximately 80 percent of affected males (Hagerman, 1997b). Physical features such as prominent ears and jaw and hyper-extensibility of joints are frequently present but are not diagnostic. Intellectual handicap is the most common feature defining the phenotype. Generally, males are more severely affected than females. Early impressions that females are unaffected have been replaced by an understanding of the presence of specific learning difficulties and other neuropsychiatric features in females. The learning disability present in males becomes more defined with age, although this longitudinal effect is more likely a reflection of a flattening of developmental trajectories rather than an explicit neurodegenerative process.
The compromise of brain function seen in Fragile X Syndrome is paralleled by changes in brain structure in humans. MRI scanning studies reveal that Fragile X Syndrome is associated with larger brain volumes than would be expected in matched controls and that this change correlates with trinucleotide expansion in the FMRP promoter region (Jakala et al, 1997). At the microscopic level, humans with Fragile X Syndrome show abnormalities of neuronal dendritic structure, in particular, an abnormally high number of immature dendritic spines (Irwin et al, , 2000).
Currently available treatments for NDDs are symptomatic – focusing on the management of symptoms – and supportive, requiring a multidisciplinary approach. Educational and social skills training and therapies are implemented early to address core issues of learning delay and social impairments. Special academic, social, vocational, and support services are often required. Medication, psychotherapy or behavioral therapy may be used for management of co-occurring anxiety, ADHD, depression, maladaptive behaviors (such as aggression) and sleep issues, Antiepileptic drugs may be used to control seizures.
Patent
WO 2014085480,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014085480

EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro-Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
WO95/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS to increase TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses certain GPE analogs having amino acid substitutions and certain other modification that are capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including injury or disease in the CNS.
EXAMPLES
The following examples are intended to illustrate embodiments of this invention, and are not intended to limit the scope to these specific examples. Persons of ordinary skill in the art can apply the disclosures and teachings presented herein to develop other embodiments without undue experimentation and with a likelihood of success. All such embodiments are considered part of this invention.
Example 1: Synthesis of N,N-Dimethylglycyl-L-prolyl)-L-glutamic acid
The following non-limiting example illustrates the synthesis of a compound of the invention, N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
All starting materials and other reagents were purchased from Aldrich; BOC=tert-butoxycarbonyl; Bn=benzyl.
BOC-L-proline-(P-benzyl)-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem. Soc: 79, 6810, 1994] (10 mmol) in dichloromethane (50 mi), cooled to 0°C, was added triethylamine (1 .39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl-L-glutamate (10 mmol) was then added and the mixture stirred at 0° C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol 1-1) then dried (MgSO4) and concentrated at reduced pressure to give BOC-L-proline-L-glutamic acid dibenzyl ester (5.0 g, 95%).
L-proline-L-glutamic acid dibenzyl ester
A solution of BOC-L-glutamyl-L-proline dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 h. at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give L-proline-L-glutamic acid dibenzyl ester.
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of L-proline-L-glutamic acid dibenzyl ester (10 mmol), N,N-dimethylglycine (10 mmol) and triethylamine ( 10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0°C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallised from ethyl acetate to yield the tripeptide derivative.
It can be appreciated that following the method of the Examples, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Eample 2: Synthesis of Glycyl-L-2-Methyl-L-Prolyl-L-Glutamate
L-2-Methylproline and L-glutamic acid dibenzyl ester p-toluenesulphonate were purchased from Bachem, N-benzyloxycarbonyl-glycine from Acros Organics and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BoPCl, 97%) from Aldrich Chem. Co.
Methyl L-2-methylprolinate hydrochloride 2
Thionyl chloride (5.84 cm3, 80.1 mmol) was cautiously added dropwise to a stirred solution of (L)-2-methylproline 1 (0.43 g, 3.33 mmol) in anhydrous methanol (30 cm3) at -5 °C under an atmosphere of nitrogen. The reaction mixture was heated under reflux for 24 h, and the resultant pale yellow-coloured solution was. concentrated to dryness in vacuo. The residue was dissolved in a 1 : 1 mixture of methanol and toluene (30 cm3) then concentrated to dryness to remove residual thionyl chloride. This procedure was repeated twice more, yielding hydrochloride 2 (0.62 g, 104%) as an hygroscopic, spectroscopically pure, off-white solid: mp 127- 131 °C; [α]D -59.8 (c 0.24 in CH2Cl2); vmax (film)/cm-1 3579, 3398 br, 2885, 2717, 2681 , 2623, 2507, 1743, 1584, 1447, 1432, 1374, 1317, 1294, 1237, 1212, 1172, 1123, 981 , 894, 861 and 764; δH (300 MHz; CDCl3; Me4Si) 1.88 (3H, s, Proα-CH3), 1 .70-2.30 (3H, br m, Proβ-HAΗΒ and Proγ-H2), 2.30-2.60 (1H, br m, Proβ-HAΗΒ), 3.40-3.84 (2H, br m, Proδ-H2), 3.87 (3H, s, CO2CH3), 9.43 (1H, br s, NH) and 10.49 ( 1H, br s, HCl); δC (75 MHz; CDCl3) 21.1 (CH3, Proα-CH3), 22.4 (CH2, Proγ-C), 35.6 (CH2, Proβ-C), 45.2 (CH2, Proδ-C), 53.7 (CH3, CO2CH3), 68.4 (quat., Proα-C) and 170.7 (quat, CO); m/z (FAB+) 323.1745 [M2.H35Cl.H+: (C7H13NO2)2. H35Cl.H requires 323.1738] and 325.1718 [M2.H37Cl.H+: (C7H13NOz)2. H37Cl.H requires 325.1708],
N-Benxyloxycarbonyl-glycyl-L-2-methylproline 5
Anhydrous triethylamine (0.45 cm3, 3.23 mmol) was added dropwise to a mixture of methyl L-2-methylprolinate hydrochloride 2 (0.42 g, 2.34 mmol) and N-benzyloxycarbonyl-glycine (98.5%) 3 (0.52 g, 2.45 mmol) in methylene chloride (16 cm3), at 0 °C, under an atmosphere of nitrogen. The resultant solution was stirred for 20 min and a solution of 1 ,3-dicyclohexylcarbodiimide (0.56 g, 2.71 mmol) in methylene chloride (8 cm3) at 0 °C was added dropwise and the reaction mixture was warmed to room temperature and stirred for a further 20 h. The resultant white mixture was filtered through a Celite™ pad to partially remove 1 ,3-dicyclohexylurea, and the pad was washed with methylene chloride (50 cm3). The filtrate was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and concentrated to dryness in vacuo. Further purification of the residue by flash column chromatography (35 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) afforded tentatively methyl N-benzyloxycarbonyl-glycyl-L-2-methylprolinate 4 (0.56 g), containing 1 ,3-dicyclohexylurea, as a white semi-solid: Rf 0.65 (EtOAc); m/z (ΕI+) 334.1534 (M+. C17H22N2O5 requires 334.1529) and 224 ( 1 ,3-dicyclohexylurea).
To a solution of impure prolinate 4 (0.56 g, ca. 1.67 mmol) in 1,4-dioxane (33 cm3) was added dropwise 1 M aqueous sodium hydroxide (10 cm3, 10 mmol) and the mixture was stirred for 19 h at room temperature. Methylene chloride ( 100 cm3) was then added and the organic layer extracted with saturated aqueous sodium hydrogen carbonate (2 x 100 cm3). The combined aqueous layers were carefully acidified with hydrochloric acid (32%), extracted with methylene chloride (2 x 100 cm3), and the combined organic layers dried (MgSO4), filtered, and
concentrated to dryness in vacuo. Purification of the ensuing residue (0.47 g) by flash column chromatography ( 17 g SiO2; 50% ethyl acetate – hexane to 30% methanol – dichloromethane; gradient elution) gave N-protected dipeptide 5 (0.45 g, 60%) as a white foam in two steps from hydrochloride 2. Dipeptide 5 was shown to be exclusively the frafw-orientated conformer by NMR analysis: Rf 0.50 (20% MeOH – CH2Cl2); [α]D -62.3 (c 0.20 in CH2Cl2); vmax (film)/cm-1 3583, 3324 br, 2980, 2942, 1722, 1649, 1529, 1454, 1432, 1373, 1337, 1251 , 1219, 1179, 1053, 1027, 965, 912, 735 and 698; δH (300 MHz; CDCl3; Me4Si) 1.59 (3H, s, Proα-CH3), 1 .89 (1H, 6 lines, J 18.8, 6.2 and 6.2, Proβ-HAHB), 2.01 (2H, dtt, J 18.7, 6.2 and 6.2, Proγ-H2), 2.25-2.40 (1H, m, Proβ-HAΗΒ), 3.54 (2H, t, J 6.6, Proδ-H2), 3.89 (1H, dd, J 17.1 and 3.9, Glyα-HAHB), 4.04 (1H, dd, J 17.2 and 5.3, Glyα-HAΗΒ), 5.11 (2H, s, OCH2Ph), 5.84 (I H, br t, J 4.2, N-H), 7.22-7.43 (5H, m, Ph) and 7.89 (1 H, br s, -COOH); δC (75 MHz; CDCl3) 21.3 (CH3, Proα-CH3), 23.8 (CH2, Proγ-C), 38.2 (CH2, Proβ-C), 43.6 (CH2, Glyα-C), 47.2 (CH2, Proδ-C), 66.7 (quat, Proα-C), 66.8 (CH2, OCH2Ph), 127.9 (CH, Ph), 127.9 (CH, Ph), 128.4, (CH, Ph), 136.4 (quat., Ph), 156.4 (quat., NCO2), 167.5 (quat., Gly-CON) and 176.7 (quat., CO); m/z (EI+) 320.1368 (M+. C16Η20Ν2Ο5 requires 320.1372).
Dibenzyl N-benzyloxycarbonyl-glycyl-L-2-methylprolyl-L-glutamate 7
Triethylamine (0.50 cm3, 3.59 mmol) was added dropwise to a solution of dipeptide 5 (0.36 g, 1.12 mmol) and L-glutamic acid dibenzyl ester /Moluenesulphonate 6 (0.73 g, 1.46 mmol) in methylene chloride (60 cm3) under nitrogen at room temperature, and the reaction mixture stirred for 10 min. Bis(2-oxo-3-oxazoIidinyl)phosphinic chloride (BoPCl, 97%) (0.37 g, 1.41 mmol) was added and the colourless solution stirred for 17 h. The methylene chloride solution was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and evaporated to dryness in vacuo. Purification of the resultant residue by repeated (2x) flash column chromatography (24 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) yielded ƒully protected tripeptide 7 (0.63 g, 89%) as a colourless oil. Tripeptide 7 was shown to be exclusively the trans-orientated conformer by NMR analysis: Rf 0.55 (EtOAc); [α]D -41.9 (c 0.29 in CH2Cl2); vmax (film)/cm-1 3583, 3353 br, 2950, 1734, 1660, 1521, 1499, 1454, 1429, 1257, 1214, 1188, 1166, 1051, 911, 737 and 697; δH (400 MHz; CDCl3; Me4Si) 1.64 (3H, s, Proot-CH3), 1.72 (1H, dt, J 12.8, 7.6 and 7.6, Proβ-HAHB), 1.92 (2H, 5 lines, J 6.7, Proγ-H2), 2.04 (1H, 6 lines, J 7.3 Gluβ-HAHB), 2.17-2.27 (1H, m, Gluβ-HAΗΒ), 2.35-2.51 (3H, m, Proβ-HAΗΒ and Gluγ-H2), 3.37-3.57 (2H, m, Proδ-H2), 3.90 (1 H, dd, J 17.0 and 3.6, Glyα-HAHB), 4.00 (1H, dd, J 17.1 and 5.1, Glyα-HAΗΒ), 4.56 (1H, td, J 7.7 and 4.9, Glyα-H), 5.05-5.20 (6H, m, 3 x OCH2Ph), 5.66-5.72 (1H, br m, Gly-NH), 7.26-7.37 (15H, m, 3 x Ph) and 7.44 (1H, d, J 7.2, Glu-NH); δC (100 MHz; CDCl3) 21.9 (CH3, Proα-CH3), 23.4 (CH2, Proγ-C), 26.6 (CH2, Gluβ-C), 30.1 (CH2, Gluγ-C), 38.3 (CH2, Proβ-C),
43.9 (CH2, Glyα-C), 47.6 (CH2, Proδ-C), 52.2 (CH, Glua-C), 66.4 (CH2, OCH2Ph), 66.8 (CH2, OCH2Ph), 67.1 (CH2, OCH2Ph), 68.2 (quat, Proα-C), 127.9 (CH, Ph), 128.0 (CH, Ph), 128.1, (CH, Ph), 128.2, (CH, Ph), 128.2, (CH, Ph), 128.3, (CH, Ph), 128.4, (CH, Ph), 128.5, (CH, Ph), 128.5, (CH, Ph), 135.2 (quat., Ph), 135.7 (quat., Ph), 136.4 (quat, Ph), 156.1 (quat, NCO2), 167.3 (quat., Gly-CO), 171.4 (quat., CO), 172.9 (quat., CO) and 173.4 (quat., CO); m/z (FAB+) 630.2809 (MH+. C35H40N3O8 requires 630.2815).
Glycyl-L-2-methylprolyl-L-glutamic acid (G-2-MePE)
A mixture of the protected tripeptide 7 (0.63 g, 1.00 mmol) and 10 wt % palladium on activated carbon (0.32 g, 0.30 mmol) in 91 :9 methanol – water (22 cm3) was stirred under an atmosphere of hydrogen at room temperature, protected from light, for 23 h. The reaction mixture was filtered through a Celite™ pad and the pad washed with 75 :25 methanol – water (200 cm3). The filtrate was concentrated to dryness under reduced pressure and the residue triturated with anhydrous diethyl ether to afford a 38: 1 mixture of G-2-MePE and tentatively methylamine 8 (0.27 g, 86%) as an extremely hygroscopic white solid. Analytical reverse-phase HPLC studies on the mixture [Altech Econosphere C 18 Si column, 150 x 4.6 mm, 5 ☐m; 5 min flush with H2O (0.05% TFA) then steady gradient over 25 min to MeCN as eluent at flow rate of 1 ml/min; detection using diode array] indicated it was a 38: 1 mixture of two eluting peaks with retention times of 13.64 and 14.44 min at 207 and 197 nm, respectively. G-2-MePE was shown to be a 73 :27 trans:cis mixture of conformers by 1H NMR analysis (the ratio was estimated from the relative intensities of the double doublet and triplet at δ 4.18 and 3.71 , assigned to the Gluα-H protons of the major and minor conformers, respectively):
mp 144 °Cɸ;
[ α]D -52.4 (c 0.19 in H2O);
δα (300 MHz; D2O; internal MeOH) 1.52 (3H, s, Proα-CH3), 1.81-2.21 (6H, m, Proβ-H2, Proγ-H, and Gluβ-H2), 2.34 (1.46H, t, J 7.2, Gluy-H2), 2.42* (0.54H, t, 77.3, Gluγ-H2), 3.50-3.66 (2H, m, Pro6-H2), 3.71 * (0.27H, t, J 6.2, Gluoc-H), 3.85 (1H, d, J 16.6, Glyα-HAHB), 3.92 (1H, d, J 16.6, Glyα-HAΗΒ) and 4.18 (0.73H, dd, J 8.4 and 4.7, Glua-H);
δC (75 MHz; D2O; internal MeOH) 21.8 (CH3, Proα-CH3), 25.0 (CH2, Proγ-C), 27.8* (CH2: Gluβ-C), 28.8 (CH2, Gluβ-C), 32.9 (CH2, Gluγ-C), 40.8 (CH2, Proβ-C), 42.7 (CH2, Glyα-C), 49.5 (CH2, Proδ-C), 56.0* (CH, Gluα-C), 56.4 (CH, Gluα-C), 69.8 (quat, Proα-C), 166.5 (quat., Gly-CO), 177.3 (quat., Pro-CON), 179.2 (quat., Gluα-CO), 180.2* (quat., Gluγ-CO) and 180.6 (quat., Gluγ-CO);
m/z (FAB+) 3 16.1508 (MH+. C13H22N3O6 requires 316.1509).
PATENT
WO02094856
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Testing; Material and Methods
The following experimental protocol followed guidelines approved by the
University of Auckland animal ethics committee.
Preparation of cortical astrocyte cultures for harvest of metabolised cell culture supernatant
One cortical hemisphere from a postnatal day 1 rat was used and collected into
4ml of DMEM. Trituration was done with a 5ml glass pipette and subsequently through an 18 gauge needle. Afterwards, the cell suspension was sieved through a lOOμm cell strainer and washed in 50ml DMEM (centrifugation for 5min at 250g). The sediment was resuspended into 20ml DMEM+10% fetal calf serum. 10 Milliliters of suspension was added into each of two 25cm3 flasks and cultivated at 37°C in the presence of 10% C02, with a medium change twice weekly. After cells reached confluence, they were washed three times with PBS and adjusted to Neurobasal/B27 and incubated for another 3 days. This supernatant was frozen for transient storage until usage at -80°C.
Preparation of striatal and cortical tissue from rat E18/E19 embryos
A dam was sacrificed by C02-treatment in a chamber for up to 4 minutes and was prepared then for cesarean section. After surgery, the embryos were removed from their amniotic sacs, decapitated and the heads put on ice in DMEM/F12 medium for striatum and PBS + 0.65% D(+)-glucose for cortex.
Striatal tissue extraction procedure and preparation of cells
Whole brain was removed from the skull with the ventral side facing upside in DMEM/F12 medium. The striatum was dissected out from both hemispheres under a stereomicroscope and the striatal tissue was placed into the Falcon tube on ice.
The collected striatal tissue was triturated by using a PI 000 pipettor in 1ml of volume. The tissue was triturated by gently pipetting the solution up and down into the pipette tip about 15 times, using shearing force on alternate outflows. The tissue pieces settled to the bottom of the Falcon tube within 30 seconds, subsequently the supernatant was transferred to a new sterile Falcon tube on ice. The supernatant contained a suspension of dissociated single cells. The tissue pieces underwent a second trituration to avoid excessively damaging cells already dissociated by over triturating them. 1 Milliliter of ice-cold DMEM/F12 medium was added to the tissue pieces in the first tube and triturated as before. The tissue pieces were allowed to settle and the supernatant was removed to a new sterile Falcon tube on ice. The cells were centrifuged at 250g for 5 minutes at 4°C. The resuspended cell pellet was ready for cell counting.
Plating and cultivation of striatal cells
Striatal cells were plated into Poly-L-Lysine (O.lmg/ml) coated 96-well plates (the inner 60 wells only) at a density of 200,000 cells /cm2 in Neurobasal/B27 medium (Invitrogen). The cells were cultivated in the presence of 5% C02 at 37°C under 100% humidity. Complete medium was changed on days 1, 3 and 6.
Cortical tissue extraction procedure and preparation of cells
The two cortical hemispheres were carefully removed by a spatula from the whole brain with the ventral side facing upside into a PBS +0.65% D(+)-glucose containing petri dish. Forcips were put into the rostral part (near B. olfactorius) of the cortex for fixing the tissue and two lateral – sagittal oriented cuttings were done to remove the paraform and entorhinal cortices. The next cut involved a frontal oriented cut at the posterior end to remove the hippocampal formation. A final frontal cut was done a few millimeters away from the last cut in order to get hold of area 17/18 of the visual cortex.
The collected cortices on ice in PBS+0.65% D(+)-glucose were centrifuged at 350g for 5min. The supernatant was removed and trypsin/EDTA (0.05%/0.53mM) was added for 8min at 37°C. The reaction was stopped by adding an equal amount of DMEM+10%) fetal calf serum. The supernatant was removed by centrifugation followed by two subsequent washes in Neurobasal/B27 medium.
The cells were triturated once with a glass Pasteur pipette in 1 ml of
Neurobasal/B27 medium and subsequently twice by using a 1ml insulin syringe with a 22 gauge needle. The cell suspension was passed through a lOOμm cell strainer and subsequently rinsed by 1ml of Neurobasal B27 medium. Cells were counted and adjusted to 50,000 cells per 60μl.
Plating and cultivation of cortical cells
96-well plates were coated with 0.2mg/ml Poly-L-Lysine and subsequently coated with 2μg/ml laminin in PBS, after which 60μl of cortical astrocyte-conditioned medium was added to each well. Subsequently, 60μl of cortical cell suspension was added. The cells were cultivated in the presence of 10% C02 at 37°C under 100%) humidity. At day 1, there was a complete medium change (1:1- Neurobasal/B27 and astrocyte-conditioned medium) with addition of lμM cytosine-β-D-arabino-furanoside (mitosis inhibitor). On the second day, 2/3 of medium was changed. On day 5, 2/3 of the medium was changed again.
Cerebellar microexplants from P8 animals: preparation, cultivation and fixation
The laminated cerebellar cortices of the two hemispheres were explanted from a P8 rat, cut into small pieces in PBS + 0.65% D(+)glucose solution and triturated by a 23gauge needle and subsequently pressed through a 125 μm pore size sieve. The microexplants that were obtained were centrifuged (60 g) twice (media exchange) into serum-free BSA-supplemented START V-medium (Biochrom). Finally, the
microexplants were reconstituted in 1500 μl STARTV-medium (Biochrom). For cultivation, 40μl of cell suspension was adhered for 3 hours on a Poly-D-Lysine
(O.lmg/ml) coated cover slip placed in 35mm sized 6-well plates in the presence of 5% C02 under 100% humidity at 34°C. Subsequently, 1ml of STARTV-medium was added together with the toxins and drugs. The cultures were monitored (evaluated) after 2-3 days of cultivation in the presence of 5% C02 under 100% humidity. For cell counting analysis, the cultures were fixed in rising concentrations of paraformaldehyde (0.4%, 1.2%, 3% and 4% for 3min each) followed by a wash in PBS.
Toxin and drug administration for cerebellar, cortical and striatal cells: analysis
All toxin and drug administration experiments were designed that 1/100 parts of okadaic acid (30nM and lOOnM concentration and 0.5mM 3-nitropropionic acid for cerebellar microexplants only), GPE (InM -ImM) and G-2Methyl-PE (InM-lmM) were used respectively at 8DIV for cortical cultures and 9DIV for striatal cultures. The incubation time was 24hrs. The survival rate was determined by a colorimetric end-point MTT-assay at 595nm in a multi-well plate reader. For the cerebellar microexplants four windows (field of 0.65 mm2) with highest cell density were chosen and cells displaying neurite outgrowth were counted.
Results
The GPE analogue G-2Methyl-PE exhibited comparable neuroprotective capabilities within all three tested in vitro systems (Figures 12-15).
The cortical cultures responded to higher concentrations of GPE (Figure 12) /or
G-2Methyl-PE (lOμM, Figure 13) with 64% and 59% neuroprotection, respectively.
Whereas the other 2 types of cultures demonstrated neuroprotection at lower doses of G-2Methyl-PE (Figures 14 and 15). The striatal cells demonstrated
neuroprotection within the range of InM to ImM of G-2Methyl-PE (Figure 15) while the postnatal cerebellar microexplants demonstrated neuroprotection with G-2Methyl-PE in the dose range between InM and lOOnM (Figure 14).
While this invention has been described in terms of certain preferred embodiments, it will be apparent to a person of ordinary skill in the art having regard to that knowledge and this disclosure that equivalents of the compounds of this invention may be prepared and administered for the conditions described in this application, and all such equivalents are intended to be included within the claims of this application.
PATENT
WO-2021026066
Composition and kits comprising trofinetide and other related substances. Also claims a process for preparing trofinetide and the dosage form comprising the same. Disclosed to be useful in treating neurodegenerative conditions, autism spectrum disorders and neurodevelopmental disorders.
Trofinetide is a synthetic compound, having a similar core structure to Glycyl-Prolyl-Glutamic acid (or “GPE”). Trofinetide has been found to be useful in treating neurodegenerative conditions and recently has been found to be effective in treating Autism Spectrum disorders and Neurodevelopmental disorders.
Formula (Ila),
Example 1: Trofinetide Manufacturing Process
In general, trofinetide and related compounds can be manufactured from a precursor peptide or amino acid reacted with a silylating or persilylating agent at one or more steps. In the present invention, one can use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group.
Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
Step 1: Preparation of Z-Gly-OSu
Several alternative procedures can be used for this step.
Procedure 1A
One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 27 eq of iPrOH and 4 eq of CH2Cl2 at 21 °C. The mixture was cooled and when the temperature reached -4 °C, 1.1 eq of EDC.HCl was added gradually, keeping the temperature below 10 °C. During the reaction a dense solid appeared. After addition of EDC.HCl, the mixture was allowed to warm to 20 °C. The suspension was cooled to 11 °C and filtered. The cake was washed with 4.9 eq of cold iPrOH and 11 eq of IPE before drying at 34 °C (Z-Gly-OSu dried product -Purity: 99.5%; NMR assay: 96%; Yield: 84%).
Procedure 1B
This Procedure is for a variant of Procedure 1A, and differs by replacing iPrOH with ACN. One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 22 eq of ACN at 35 °C. The mixture was cooled in an ice bath. When the temperature reached 1 °C, 0.9 eq of DCC in 5.5 eq of ACN was added gradually to keep the temperature below 5 °C. The coupling reaction took about 20 hrs. During the reaction, DCU precipitated and was removed by filtration at the end of the coupling. After filtration, DCU was washed with ACN to recover the product. The mixture of Z-Gly-OSu was then concentrated to reach 60% by weight. iPrOH (17 eq) was added to initiate the crystallization. Quickly after iPrOH addition a dense solid appeared. An additional 17 eq of iPrOH was needed to liquify the suspension. The suspension was cooled in an ice bath and filtered. The solid was washed with 9 eq of iPrOH before drying at 45 °C (Z-Gly-OSu dried product – Purity: 99.2%; HPLC assay: 99.6%; Yield: 71%).
Step 2: Preparation of Z-Gly-MePro-OH
Several alternative procedures can be used for this step.
Procedure 2A
One (1) eq of MePro.HCl was partially solubilized in 29 eq of CH2Cl2 at 35 °C with 1.04 eq of TEA and 1.6 eq of TMA. The mixture was heated at 35 °C for 2 hrs to perform the silylation. Then 1.02 eq of Z-Gly-OSu was added to the mixture. The mixture was kept at 35 °C for 3 hrs and then 0.075 eq of butylamine was added to quench the reaction. The mixture was allowed to return to room temperature and mixed for at least 15 min. The Z-Gly-MePro-OH was extracted once with 5% w/w NaHCO3 in 186 eq of water, then three times successively with 5% w/w NaHCO3 in 62 eq of water. The aqueous layers were pooled and the pH was brought to 2.2 by addition of 34 eq of HCl as 12N HCl at room temperature. At this pH, Z-Gly-MePro-OH formed a sticky solid that was solubilized at 45 °C with approximately 33 eq of EtOAc and 2.3 eq of iButOH. Z-Gly-MePro-OH was extracted into the organic layer and washed with 62 eq of demineralized water. The organic layer was then dried by azeotropic distillation with 11.5 eq of EtOAc until the peptide began to precipitate. Cyclohexane (12 eq) was added to the mixture to complete the precipitation. The suspension was cooled at 5 °C for 2 hrs and filtered. The solid was washed with 10 eq of cyclohexane before drying at 45 °C (Z-Gly-MePro-OH dried product – Purity: 100%; HPLC assay: 100%; Yield 79%).
Procedure 2B
This Procedure is for a variant of Procedure 2A. One (1) eq of MePro.HCl was partially solubilized in 36.6 eq of CH2Cl2 at 34 °C with 1.01 eq of TEA and 0.1 eq of TMA. Then 1.05 eq of Z-Gly-OSu was added to the mixture, followed by 1.0 eq of TEA. The mixture was maintained at 35 °C for approximately 1 hr, cooled to 25 to 30 °C and 0.075 eq of DMAPA was added to stop the reaction. One hundred (100) eq of water, 8.6 eq of HCl as 12N HCl and 0.3 eq of KHSO4 were added to the mixture (no precipitation was observed, pH=1.7). Z-Gly-MePro-OH was extracted into the organic layer and washed twice with 97 eq of demineralized water with 0.3 eq of KHSO4, then 100 eq of demineralized water, respectively. EtOAc (23 eq) was added to the mixture and CH2Cl2 was removed by distillation until the peptide began to precipitate. Cyclohexane (25 eq) was added to the mixture to complete the precipitation. The suspension was cooled at -2 °C overnight and filtered. The solid was washed with 21 eq of cyclohexane before drying at 39 °C (Z-Gly-MePro-OH dried product – Purity: 98.7%; NMR assay: 98%; Yield 86%).
Procedure 2C
In reactor 1, MePro.HCl (1 eq) was suspended in EtOAc (about 7 eq). DIPEA (1 eq) and TMA (2 eq) were added, and the mixture heated to dissolve solids. After dissolution, the solution was cooled to 0 °C. In reactor 2, Z-Gly-OH (1 eq) was suspended in EtOAc (about 15 eq). DIPEA (1 eq), and pyridine (1 eq) were added. After mixing, a solution was obtained, and cooled to -5 °C. Piv-Cl (1 eq) was added to reactor 2, and the contents of reactor 1 added to reactor 2. Upon completed addition, the contents of reactor 2 were taken to room temperature. The conversion from Z-Gly-OH to Z-Gly-MePro-OH was monitored by HPLC. When the reaction was complete, the reaction mixture was quenched with DMAPA (0.1 eq), and washed with an aqueous solution comprised of KHSO4, (about 2.5 wt%), NaCl (about 4 wt%), and conc. HCl (about 6 wt%) in 100 eq H2O. The aqueous layer was re-extracted with EtOAc, and the combined organic layers washed with an aqueous solution comprised of KHSO4 (about 2.5 wt%) and NaCl (about 2.5 wt%) in 100 eq H2O, and then with water (100 eq). Residual water was removed from the organic solution of Z-Gly-MePro-OH by vacuum distillation with EtOAc. The resulting suspension was diluted with heptane (about 15 eq) and cooled to 0 °C. The product was isolated by filtration, washed with cold heptane (about 7 eq), and dried under vacuum at 45 °C. Z-Gly-MePro-OH (85% yield) was obtained.
Step 3: Preparation of Z-Gly-MePro-Glu-OH
Several alternative procedures can be used in this step.
Procedure 3A
H-Glu-OH (1.05 eq) was silylated in 2 eq of CH2Cl2 with 3.5 eq of TMA at 65 °C. Silylation was completed after 2 hrs. While the silylation was ongoing, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 24 eq of CH2Cl2 and 1.0 eq of DMA at room temperature in another reactor. EDC.HCl (1.0 eq.) was added. The activation rate reached 97% after 15 min. The activated Oxyma Pure solution, was then added to silylated H-Glu-OH at 40 °C and cooled at room temperature. Coupling duration was approximately 15 min, with a coupling rate of 97%. Addition of 8.2% w/w NaHCO3 in 156 eq of water to the mixture at room temperature (with the emission of CO2) was performed to reach pH 8. Z-Gly-MePro-Glu-OH was extracted in water. The aqueous layer was washed twice with 29 eq of CH2Cl2. Residual CH2Cl2 was removed by concentration. The pH was brought to 2.5 with 2.5N HCl, followed by 1.4 eq of solid KHSO4 to precipitate Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 3 x 52 eq of water. The filtered solid was added to 311 eq of demineralized water and heated to 55-60 °C. iPrOH (29 eq) was added gradually until total solubilization of the product. The mixture was slowly cooled to 10 °C under moderate mixing during 40 min to initiate the crystallization. The peptide was filtered and washed with 2 x 52 eq of water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.5%; NMR assay: 96%; Yield 74%).
Procedure 3B
One (1) eq of Z-Gly-MePro-OH and 1.05 eq of Suc-OH were solubilized in 40 eq of ACN and 30 eq of CH2Cl2 at room temperature. The mixture was cooled in an ice bath, and when the temperature was near 0 °C, 1.05 eq of DCC dissolved in 8 eq of ACN was added gradually, keeping the temperature below 5 °C. After addition of DCC, the mixture was progressively heated from 0 °C to 5 °C over 1 hr, then to 20 °C between 1 to 2 hrs and then to 45 °C between 2 to 5 hrs. After 5 hrs, the mixture was cooled to 5 °C and maintained overnight. The activation rate reached 98% after approximately 24 hrs. DCU was removed by filtration and washed with 13.5 eq of ACN. During the activation step, 1.1 eq of H-Glu-OH was silylated in 30 eq of ACN with 2.64 eq of TMA at 65 °C. Silylation was completed after 2 hrs. Z-Gly-MePro-OSu was then added gradually to the silylated H-Glu-OH at room temperature, with 0.4 eq of TMA added to maintain the solubility of the H-Glu-OH. The mixture was heated to 45 °C and 0.7 eq of TMA was added if precipitation occurred. The coupling duration was about 24 hrs to achieve a coupling rate of approximately 91%. The reaction was quenched by addition of 0.15 eq of butylamine and 2.0 eq of TEA. Water (233 eq) was added and the mixture concentrated until gelation occurred. Z-Gly-MePro-Glu-OH was extracted in water by addition of 5% w/w NaHCO3 in 233 eq of water and 132 eq of CH2Cl2. The aqueous layer was washed twice with 44 eq of CH2Cl2. Residual CH2Cl2 was removed by distillation. The pH was brought to 2.0 with 24 eq of HCl as 12N HCl followed by 75 eq of HCl as 4N HCl. At this pH, Z-Gly-MePro-Glu-OH precipitated. The mixture was cooled in an ice bath over 1 hr and filtered. The solid was washed with 186 eq of cold water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – HPLC Purity: 98.4%; NMR assay: 100%; Yield 55%).
Procedure 3C
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq) was silylated in 3.7 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after approximately 1.5 to 2 hrs, as evidenced by solubilization. During the silylation step, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 31.5 eq of CH2Cl2 at 22 °C. One (1.06) eq of EDC.HCl was added to complete the activation. The silylated H-Glu-OH was then added to the activated Oxyma Pure solution. The temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 153 eq of water and 9 eq of iPrOH to reach a pH of 1.65. Residual CH2Cl2 was removed by concentration. The mixture was cooled to 12 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 90 eq of water before drying at 36 °C.
Procedure 3D
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq.) was silylated in 3.9 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after 2 hrs, as evidenced by Solubilization. During the silylation step, 1 eq of Z-Gly-MePro-OH and 1 eq of Oxyma Pure were solubilized in 25 eq of CH2Cl2 at 23 °C. One (1) eq of EDC.HCl was added. To complete the activation, an additional 0.07 eq of EDC. HCl was added. Silylated H-Glu-OH was then added to the activated Oxyma Pure solution. Temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 160 eq of water and 9.6 eq of iPrOH to reach pH 1.63.
Residual CH2Cl2 was removed by concentration. The mixture was cooled to 20 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 192 eq of water before drying at about 25 °C for 2.5 days. The solid was then solubilized at 64 °C by addition of 55 eq of water and 31 eq of iPrOH. After solubilization, the mixture was diluted with 275 eq of water and cooled to 10 °C for crystallization. The mixture was filtered and the solid was washed with 60 eq of water before drying at 27 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.6%; NMR assay: 98%; Yield 74%).
Procedure 3E
In reactor 1, H-Glu-OH (1.05 eq) was suspended in ACN (about 2.2 eq). TMA (about 3.5 eq) added, and the mixture was heated to dissolve solids. After dissolution, the solution was cooled to room temperature. In reactor 2, Z-Gly-MePro-OH (1 eq) was suspended in ACN (14 eq). Oxyma Pure (1 eq) and EDC.HCl (1 eq) were added. The mixture was stirred at room temperature until the solids dissolved. The contents of reactor 2 were added to reactor 1. The conversion from Z-Gly-MePro-OH to Z-Gly-MePro-Glu-OH was monitored by HPLC. Upon completion the reaction mixture was added to an aqueous solution comprised of KHSO4 (about 2.5 wt%) dissolved in about 100 eq H2O. ACN was removed from the aqueous suspension of Z-Gly-MePro-Glu-OH by vacuum distillation with H2O. After stirring at room temperature, the product in the resulting suspension was isolated by filtration and washed with water. The solid obtained was dissolved in an aqueous solution comprised of NaHCO3 (about 5 wt%) in 110 eq H2O, and recrystallized by addition of an aqueous solution comprised of KHSO4 (about 10 wt%) in 90 eq H2O. The product was isolated by filtration, washed with water, and dried under vacuum at 45 °C. Z-Gly-MePro-Glu-OH (75% yield) was obtained.
Step 4: Deprotection and Isolation of Trofinetide
Several alternative procedures can be used in this step.
Procedure 4A
Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 25 eq) and EtOAc (about 15 eq). Pd/C (0.025 eq by weight and containing 10% Pd by weight) was added, and the reaction mixture hydrogenated by bubbling hydrogen through the reaction mixture at room temperature. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration, and the layers separated. Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained. Alternatively, deprotection can be accomplished using MeOH only, or a combination of iPrOH and MeOH, or by use of ethyl acetate in water.
Procedure 4B
This Procedure is for a variant of Procedure 4A, excluding EtOAc. Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 50 eq). Pd/C (0.05 eq, 5% Pd by weight) was added, and the reaction mixture hydrogenated at room temperature with a pressure of 5 bar. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon
reaction completion the catalyst was removed by filtration, and the aqueous layer washed with EtOAc (about 5 eq). Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained.
Procedure 4C
This Procedure is for a variant of Procedure 4A, replacing EtOAc with MeOH. Z-Gly-MePro-Glu-OH (1 eq) was suspended in MeOH (100 eq) and water (12 eq). Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration and the layers were washed with MeOH and iPrOH. The solvents were concentrated under vacuum at 45 °C, and trofinetide precipitated. The precipitate was filtered and dried at 45 °C to provide trofinetide.
Procedure 4D
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 105 eq of MeOH and 12 eq of water. Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate approximately 99% after 1 hr), the catalyst was filtered off and washed with 20-30 eq of MeOH. iPrOH (93 eq) was added and MeOH was replaced by iPrOH by concentration at 45 °C under vacuum. The peptide was concentrated until it began to precipitate. The peptide was filtered and dried at 45 °C (H-Gly-MePro-Glu-OH dried product: Purity: 98.1%; NMR assay: 90%; Yield 81%).
Procedure 4E
This Procedure is for a variant of Procedure 4A, removing H2O and replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 44 eq of MeOH. Pd/Si type 340 (0.02 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 99.9%, after 3-3.5 hrs), the catalyst was filtered off and washed with 8 eq of MeOH. Deprotected peptide was then precipitated in 56 eq of iPrOH. After 30 min at 5 °C, the peptide was filtered and washed with three times with 11 eq of iPrOH before drying at 25 °C (H-Gly-MePro-Glu-OH dried product: Purity: 99.4%; HPLC assay: ~98%; Yield: 81%).
Procedure 4F
This Procedure is for a variant of Procedure 4A. One (1) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 14 eq of EtOAc and 25 eq of water. Pd/C (0.01 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 100%, after about 3.5 hrs), the catalyst was filtered off and washed with a mixture of 3.5 eq of EtOAc and 6 eq of water. The aqueous layer was then ready for spray-drying (Aqueous H-Gly-MePro-Glu-OH peptide solution: Purity: 98.6%; Yield: ~95%).
Procedure 4G
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si, EtOAc with MeOH, and removing H2O. Pd/Si type 340 (0.02 eq by weight) was added to 2.9 vols of MeOH for pre-reduction during 30 min. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 34 eq of MeOH. The reduced palladium was then transferred to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. Pd/C type 39 (0.007 eq by weight) was added to the mixture to increase reaction kinetics. At the end of the deprotection, the catalyst was filtered off and washed with 13.6 eq of MeOH. The deprotected peptide was then precipitated in 71 eq of iPrOH. After about 40 min, the peptide was filtered and washed with 35 eq of iPrOH. The peptide was dried below 20 °C and was then ready for solubilization in water and spray-drying.
Procedure 4H
This Procedure is for a variant of Procedure 4A. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 24.8 eq of water and 13.6 eq of EtOAc. Pd/C type 39 (0.025 eq by weight) was added to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (19 hrs), the catalyst was removed by filtration and washed with 5.3 eq of water and 2.9 eq of EtOAc. The biphasic mixture was then decanted to remove the upper organic layer. The aqueous layer was diluted with water to reach an H-Gly-MePro-Glu-OH concentration suitable for spray-drying the solution.
Example 2: Alternative Trofinetide Manufacturing Process
An alternative method for synthesis of Trofinetide is based on U.S. Patent No.
8,546,530 adapted for a tripeptide as follows.
The persilylated compounds used to synthesis Formula (Ia) (trofinetide) are obtained by silylating a corresponding peptide or amino acid by reaction with a silylating agent, optionally in an organic solvent. The persilylated peptide or amino acid can be isolated and purified if desired. One can use the persilylated peptide or amino acid in situ, e.g. by combining a solution containing persilylated peptide or amino acid with a solution containing, optionally activated, peptide or amino acid.
In step 2, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-MePro-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid can be isolated and purified if desired. One can use the persilylated amino acid in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated, amino acid (for example, Z-Gly-OH).
In step 3, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-Glu-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid or peptide can be isolated and purified if desired. It is however useful to use the persilylated amino acid or peptide in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated (for example, by using EDC.HCl and Oxyma Pure), peptide (for example, Z-Gly-MePro-OH).
In the present invention, it is useful to use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group. Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
The reaction of step 2 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 40 °C, and optionally from 15 °C to 30 °C.
The reaction of step 3 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 60 °C, optionally from 15 °C to 50 °C.
In the reaction of step 2, generally 0.5 to 5 equivalents, optionally 1 to 3 equivalents, optionally about 1.5 to 2.5 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2 to 4 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible. “Functional groups to be silylated” means particular groups having an active hydrogen atom that can react with the silylating agent such as amino, hydroxyl, mercapto or carboxyl groups.
In the reaction of step 3, generally 0.5 to 5 equivalents, optionally 2 to 4.5 equivalents, optionally about 3 to 4 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2.5 to 4.5 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible.
It is understood that “persilylated” means an amino acid or peptide or amino acid analogue or peptide analogue in which the groups having an active hydrogen atom that can react with the silylating agent are sufficiently silylated to ensure that a homogeneous reaction medium for a coupling step is obtained.
In the process according to the invention, the reaction between the amino acid or peptide and the persilylated amino acid or peptide is often carried out in the presence of a carboxyl group activating agent. In that case the carboxylic activating reagent is suitably selected from carbodiimides, acyl halides, phosphonium salts and uronium or guanidinium salts. More optionally, the carboxylic activating agent is an acyl halide, such as isobutyl chloroformate or pivaloyl chloride or a carbodiimide, such as EDC.HC1 or DCC.
Good results are often obtained when using additional carboxylic activating reagents which reduce side reactions and/or increase reaction efficiency. For example, phosphonium and uronium salts can, in the presence of a tertiary base, for example, N,N-diisopropylethylamine (DIPEA) and triethylamine (TEA), convert protected amino acids into activated species. Other reagents help prevent racemization by providing a protecting reagent. These reagents include carbodiimides (for example, DCC) with an added auxiliary nucleophile (for example, 1-hydroxy-benzo triazole (HOBt), 1-hydroxy-azabenzotriazole (HOAt), or Suc-OH) or derivatives thereof. Another reagent that can be utilized is TBTU. The mixed anhydride method, using isobutyl chloroformate, with or without an added auxiliary nucleophile, is also used, as is the azide method, due to the low racemization associated with it. These types of compounds can also increase the rate of carbodiimide-mediated couplings. Typical additional reagents include also bases such as N,N-diisopropylethylamine (DIPEA), triethylamine (TEA) or N-methylmorpholine (NMM).
When the silylation is carried out in the presence of a solvent, said solvent is optionally a polar organic solvent, more optionally a polar aprotic organic solvent. An amide type solvent such as N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMAC)
can be used. In the present invention for step 2, one can use an alkyl acetate solvent, in particular ethyl acetate is more particularly optional.
In the present invention for step 3, one can use a chlorinated hydrocarbon solvent or alkyl cyanide solvent, in particular dichloromethane or acetonitrile are more particularly optional.
In another embodiment, silylation is carried out in a liquid silylation medium consisting essentially of silylating agent and amino acid or peptide.
In the present invention, amino acid or peptide is understood to denote in particular an amino acid or peptide or amino acid analogue or peptide analogue which is bonded at its N-terminus or optionally another position, to a carboxylic group of an amino protected amino acid or peptide.
Example 3: Specifications for Compositions Containing Compounds of Formula (I)
1 ICH guideline Q3C on impurities: guideline for residual solvents
Example 4: Alternative Manufacturing of Trofinetide Example 1, Step 4, Procedure 4B
This Procedure is for a variant of Step 4, Procedure 4B. Z-Gly-MePro-Glu-OH (1 eq) was added in portions to Pd/C (0.027 eq by weight and containing 5% Pd by weight) in about 50 eq of water. The reaction mixture was hydrogenated at 20 °C at a pressure of 5 bar for at least 4 cycles of 4 hrs each. Pd/C (0.0027 eq by weight) was charged between cycles, as needed, to speed up the reaction. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon reaction completion the catalyst was removed by filtration, washed with water (12.5 eq) and the aqueous layer washed with EtOAc (about 14 eq). After phase separation, residual EtOAc was removed from the aqueous solution containing
trofinetide by sparging with nitrogen under vacuum at 20 °C for about 3 hrs. The aqueous solution was filtered. The final concentration of trofinetide was about 25 wt% and the solution was then ready for spray-drying to isolate the product.
Example 5: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (I)
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (I) on an anhydrous basis.
Example 6: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (Ia) on an anhydrous basis.
Example 7: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, wherein the product comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101
wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 8: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, and additionally comprising one or more compounds selected from the group consisting of Formula (III), Formula (IIIa), Formula (IV), Formula (V), Formula (VI), Formula (VII), Formula (VIII), and Formula (IX), wherein the composition comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101 wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 9: Analysis of Products and Compositions
The products and compositions disclosed herein may be analyzed by liquid chromatography, a suitable chromatographic method using UPLC, e.g. using materials and conditions such as Waters Acquity CSH C18, 1.7 µm, 150 x 2.1 mm column, water with 0.1 % TFA (mobile phase A), and water/ACN 70/30 + 0.1 % TFA (mobile phase B), ranging from (4% phase A/6% phase B to 100% phase B and flushed with 4% phase A/6% phase B).
Flow rate: 0.35 ml/min, Column temperature: 40 °C, autosampler temperature: 4 °C, injection volume: 4 ml (e.g. prepared by weighing about 10 mg of powder in a 10 ml volumetric flask and diluted to volume with water). Examples of detectors are UV (ultraviolet, UV 220 nm) and MS (mass spectrometry).
INDUSTRIAL APPLICABILITY
This invention finds use in the pharmaceutical, medical, and other health care fields.
PATENT
WO2014085480 ,
claiming use of trofinetide for treating autism spectrum disorders including autism, Fragile X Syndrome or Rett Syndrome.
EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro- Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
W095/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS for increasing TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses GPE analogs capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including but not limited to, injury or disease in the CNS.
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
PAPER
Tetrahedron (2005), 61(42), 10018-10035. (CLICK HERE)
The synthesis of ten proline-modified analogues of the neuroprotective tripeptide GPE is described. Five of the analogues incorporate a proline residue with a hydrophobic group at C-2 and two further analogues have this side chain locked into a spirolactam ring system. The pyrrolidine ring was also modified by replacing the γ-CH2 group with sulfur and/or incorporation of two methyl groups at C-5.
Graphical Abstract

PAPER
Bioorganic & Medicinal Chemistry Letters (2005), 15(9), 2279-2283
A series of GPE analogues, including modifications at the Pro and/or Glu residues, was prepared and evaluated for their NMDA binding and neuroprotective effects. Main results suggest that the pyrrolidine ring puckering of the Pro residue plays a key role in the biological responses, while the preference for cis or trans rotamers around the Gly-Pro peptide bond is not important.
Graphical abstract
A series of Pro and/or Glu modified GPE analogues is described. Compounds incorporating PMe and dmP showed higher affinity for glutamate receptors than GPE and neuroprotective effects similar to those of this endogenous tripeptide in culture hippocampal neurons exposed to NMDA.

PATENT
US 20060251649
WO 2006127702
US 20070004641
US 20080145335
WO 2012102832
WO 2014085480
US 20140147491
References
- ^ Bickerdike MJ, Thomas GB, Batchelor DC, Sirimanne ES, Leong W, Lin H, et al. (March 2009). “NNZ-2566: a Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke”. Journal of the Neurological Sciences. 278 (1–2): 85–90. doi:10.1016/j.jns.2008.12.003. PMID 19157421. S2CID 7789415.
- ^ Cartagena CM, Phillips KL, Williams GL, Konopko M, Tortella FC, Dave JR, Schmid KE (September 2013). “Mechanism of action for NNZ-2566 anti-inflammatory effects following PBBI involves upregulation of immunomodulator ATF3”. Neuromolecular Medicine. 15 (3): 504–14. doi:10.1007/s12017-013-8236-z. PMID 23765588. S2CID 12522580.
- ^ Deacon RM, Glass L, Snape M, Hurley MJ, Altimiras FJ, Biekofsky RR, Cogram P (March 2015). “NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome”. Neuromolecular Medicine. 17 (1): 71–82. doi:10.1007/s12017-015-8341-2. PMID 25613838. S2CID 11964380.
- ^ Study Details – Rett Syndrome Study
- ^ Neuren’s trofinetide successful in Phase 2 clinical trial in Fragile X
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 3 | Enrolling by Invitation | Treatment | Rett’s Syndrome | 1 |
| 3 | Recruiting | Treatment | Rett’s Syndrome | 1 |
| 2 | Completed | Supportive Care | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Fragile X Syndrome (FXS) | 1 |
| 2 | Completed | Treatment | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Rett’s Syndrome | 2 |
| 2 | Terminated | Treatment | Concussions | 1 |
| 1 | Completed | Treatment | Brain Injuries,Traumatic | 2 |
| Legal status | |
|---|---|
| Legal status | US: Investigational New Drug |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 853400-76-7 |
| PubChem CID | 11318905 |
| ChemSpider | 9493869 |
| UNII | Z2ME8F52QL |
| Chemical and physical data | |
| Formula | C13H21N3O6 |
| Molar mass | 315.322 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@]1(CCCN1C(=O)CN)C(=O)N[C@@H](CCC(=O)O)C(=O)O | |
| InChI[hide]InChI=1S/C13H21N3O6/c1-13(5-2-6-16(13)9(17)7-14)12(22)15-8(11(20)21)3-4-10(18)19/h8H,2-7,14H2,1H3,(H,15,22)(H,18,19)(H,20,21)/t8-,13-/m0/s1Key:BUSXWGRAOZQTEY-SDBXPKJASA-N |
{kind=link}
////////////Tofinetide , NNZ 2566, PHASE 2, PHASE 3. NEUREN, Amino Acids, Peptides, Proteins,
CC1(CCCN1C(=O)CN)C(=O)NC(CCC(=O)O)C(=O)O
J. Med. Chem. 2025, 68, 2147−2182
Trofinetide (Daybue). Trofinetide (8) was developed by Neuren Pharmaceuticals and Acadia Pharmaceuticals for the treatment of rare childhood neurodevelopmental disorders and
was approved by the USFDA in March 2023 for adults and pediatric patients two years of age or older with Rett syndrome. 59 In most cases, Rett syndrome is caused by loss of-function mutations in the X-linked gene that encodes methyl CpG-binding protein 2 (MeCP2). 60,61 MeCP2 is a critical transcriptional regulator required for normal neurological development. In the past, treatment of Rett syndrome has
been limited to symptom management based on knowledge from treating other conditions, 62
but new research has focused on targeting the underlying genetic cause and finding agents to
restore MeCP2 function. Trofinetide is an orally available synthetic analog of glycine-proline-glutamate (GPE), the Nterminal tripeptide metabolite of insulin like growth factor-1 (IGF-1). GPE has been shown to partially reverse Rett-like symptoms in MeCP2 deficient mouse models and trofenitide was developed to have an improved pharmakokinetic profile to GPE. 64 Its use has shown significant improvement over placebo in clinical trials.
Anumberofaccountsrelated to the preparation of trofinetide have been reported with various protecting group strategies, but they are moreamenabletosmall-scaleproductionduetoreagent selection and challenging isolations requiring column chromatography.65−67 A commercially viable synthesis of the drug has been described by researchers at Neuren Pharmaceuticals and is depicted in Scheme 12.68
This synthesis takes advantage of in situ silyl protection/deprotection during its amidation steps to
avoid lengthy protecting group manipulations. Activation of Cbz protected glycine 8.1 with hydroxysuccinimide 8.2 using EDCI provided 8.3 in 84% yield as a direct drop crystallization
(isolated by crystallization directly from the reaction mixture). This activated ester was then coupled with commercially available methyl proline 8.4 69 by first silylation of the carboxylic acid of the proline analog in situ with 8.5, then adding 8.3 to couple with the amine. Deprotection of the silyl ester during the
workup provided amide 8.6 in 79% yield. In a similar sequence, 8.6 was activated with Oxyma Pure and EDCI while in a second vessel 8.7 was silyl protected using 8.5. Subsequent combination of these streams followed by workup and crystallization gave amide 8.8 in good yield. Finally, Cbz deprotection was
accomplished using Pd/C and hydrogen. Trofinetide (8) was extracted into the aqueous layer and isolated by spray drying in 90% yield.
(60) Kyle, S. M.; Vashi, N.; Justice, M. J. Rett syndrome: a
neurological disorder with metabolic components. Open Biol. 2018,
8, No. 170216.
(61) Collins, B. E.; Neul, J. L. Rett syndrome and MECP2 duplication
syndrome: disorders of MeCP2 dosage. Neuropsychiatr. Dis. Treat.
2022, 18, 2813−2835.
(62) Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt,
R.; Standridge, S.; Nues, P.; Lane, J.; Dinkel, T.; et al. Consensus
guidelines on managing Rett syndrome across the lifespan. BMJ
Paediatr. Open 2020, 4, No. e000717.
(63) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D.G.;Marsh,E.D.;Lin,T.;Stankovic,S.;Bishop,K. M.;Youakim,J.M.
Trofinetide for the treatment of Rett syndrome: a randomized phase 3
study. Nat. Med. 2023, 29, 1468−1475.
(64) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D. G.; Peters, S. U.; Jones, N. E.; Youakim, J. M. Design and outcome
measures of LAVENDER, a phase 3 study of trofinetide for Rett
syndrome. Contemp. Clin. Trials 2022, 114, No. 106704.
(65) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl-l-2-methylprolyl-l-glutamic acid. US
20140147491, 2014.
(66) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl L2012102832, 2012.-2-methylprolyl
L-glutamic acid. WO 2012102832, 2012
(67) Brimble, M.A.;Harris, P. W.R.;Sieg, F. Preparation of analogsof
glycyl-prolyl-glutamate as neuroprotective agents. US 20080145335,
2008
(68) Blower, C.; Peterson, M.; Shaw, J. M.; Bonnar, J. A.; Moniotte, E.
D. F. P.; Bousmanne, M. B. C.; Betti, C.; Decroos, K. W. L.; Ayoub, M.
Compositions of trofinetide. WO 2021026066, 2021.
(69) Beck, A. K.; Blank, S.; Job, K.; Seebach, D.; Sommerfeld, T.
Synthesis of (S)-2-methylproline: a general method for the preparation
of α-branched amino acids (L.-proline, 2-methyl-). Org. Synth. 1995, 72, 62−73

.
Telacebec

Telacebec
- Molecular FormulaC29H28ClF3N4O2
- Average mass557.006 Da
Telacebec, IAP6, CAS No. 1334719-95-7телацебек [Russian] [INN]تيلاسيبيك [Arabic] [INN]特雷贝克105731334719-95-7[RN]55G92WGH3X
6-Chloro-2-ethyl-N-(4-{4-[4-(trifluoromethoxy)phenyl]-1-piperidinyl}benzyl)imidazo[1,2-a]pyridine-3-carboxamide
Imidazo[1,2-a]pyridine-3-carboxamide, 6-chloro-2-ethyl-N-[[4-[4-[4-(trifluoromethoxy)phenyl]-1-piperidinyl]phenyl]methyl]-Q203Q-203T56 AN DNJ C2 HG BVM1R D- AT6NTJ DR DOXFFF
Qurient Therapeutics and Russia licensee Infectex are developing telacebec, an oral formulation which targets QcrB subunit of the cytochrome bc1 complex, for treating multi drug resistant or extensively drug resistant Mycobacterium tuberculosis infection. Qurient is also investigating telacebec for treating buruli ulcer (an infection caused by Mycobacterium ulcerans ). In January 2021, a global phase II trial was expected to begin by December 2021 for the treatment of buruli ulcer.
syn
Angewandte Chemie, International Edition, 57(4), 1108-1111; 2018

PATENT
WO-2021018387
Novel crystalline forms of telacebec , processes for their preparation and compositions comprising them are claimed. Also claimed is their use for treating bacterial infection.
Different forms of 6-chloro-2-ethyl-AT-(4-(4-(4- (trifluoromethoxy)phenvDpiperidine-i-vDbenzvDimidazolT.2-alpyridine- 3-carboxamide
The present invention relates to different forms of the compound 6-chloro-2-ethyl-lV-(4-(4-(4-(trifhioromethoxy)phenyl)piperidine-i-yl)benzyl)imidazo[i,2-a]pyridine-3-carboxamide and to methods of making such forms/compounds. The present invention furthermore relates to mono-acid addition salts thereof, to methods of making such mono-acid addition salts and to pharmaceutical compositions comprising any of the aforementioned compounds. Furthermore, the present invention relates to uses of any of these compounds.
Tuberculosis as a disease continues to result in millions of deaths each year. Inadequate use of chemotherapy has led to an increasing number of drug resistant cases. This situation is likely to worsen with the emergence of extremely resistant strains to all currently known drugs. Current chemotherapy consists of compounds that directly target Mycobacterium tuberculosis, either by neutralizing general information pathways and critical processes such as RNA polymerization and protein synthesis inhibition or by interfering with mycobacterial specific cell envelop synthesis. The most widely used dedicated anti-tubercular drugs isoniazid, ethionamide, and pyriazin amide are pro-drugs that first require activation. They are administered to a patient for a course of several months. Patients infected with multi-drug resistant strains of M. tuberculosis may have to undergo combination therapies for extended periods of time.
WO 2011/113606 describes various anti-tubercular compounds and their use in the treatment of bacterial infections, including compound“Q203” which chemically is 6-chloro-2-ethyl-!V-(4-(4-(4-(trifluoromethoxy)phenyl)piperidine-i-yl)benzyl)imidazo[i,2-a]pyridine-3-carboxamide. In a publication by Pethe et al. (Nature Medicine, 19, 1157-1160 (2013), this compound is reported to be active against tuberculosis by interfering with the bacterial energy metabolism, inhibiting cytochrome bci activity which is an essential component of the electron transport chain required for synthesis of ATP.
Whilst the compound shows promise for future therapy of tuberculosis and related infections, there continues to be a need for forms thereof that are particularly suitable for pharmaceutical administration. In particular there is a need to provide forms that are showing an improved solubility in comparison to the free base of this compound. Furthermore, there is a need in the art to provide for forms that show an improved stability.
In a first aspect the present invention relates to a compound 6-chloro-2-ethyl-N-(4-(4-(4-(trifluoromethoxy)phenyl)piperidine-i-yl)benzyl)imidazo[i,2-a]pyridine-3-carboxamide ditosylate having the structure
PATENT
WO2011113606 .
WO 2017049321
WO 2012143796
PAPER
Scientific reports (2019), 9(1), 8608.
Angewandte Chemie, International Edition (2018), 57(4), 1108-1111.
European journal of medicinal chemistry (2017), 136, 420-427.
European Journal of Medicinal Chemistry (2017), 136, 420-427.
European journal of medicinal chemistry (2017), 125, 807-815.
Nature communications (2016), 7, 12393.
Nature medicine (2013), 19(9), 1157-60
PAPER
Journal of Medicinal Chemistry (2014), 57(12), 5293-5305.
https://pubs.acs.org/doi/10.1021/jm5003606J. Med. Chem. 2014, 57, 12, 5293–5305
Publication Date:May 28, 2014
https://doi.org/10.1021/jm5003606

A critical unmet clinical need to combat the global tuberculosis epidemic is the development of potent agents capable of reducing the time of multi-drug-resistant (MDR) and extensively-drug-resistant (XDR) tuberculosis therapy. In this paper, we report on the optimization of imidazo[1,2-a]pyridine amide (IPA) lead compound 1, which led to the design and synthesis of Q203 (50). We found that the amide linker with IPA core is very important for activity against Mycobacterium tuberculosis H37Rv. Linearity and lipophilicity of the amine part in the IPA series play a critical role in improving in vitro and in vivo efficacy and pharmacokinetic profile. The optimized IPAs 49 and 50 showed not only excellent oral bioavailability (80.2% and 90.7%, respectively) with high exposure of the area under curve (AUC) but also displayed significant colony-forming unit (CFU) reduction (1.52 and 3.13 log10 reduction at 10 mg/kg dosing level, respectively) in mouse lung.
6-Chloro-2-ethyl-N-(4-{4-[4-(trifluoromethoxy)phenyl]piperidin-1-yl}benzyl)imidazo[1,2-a]pyridine-3-carboxamide (50)
Mp = 164.0 °C; 1H NMR (400 MHz, CDCl3) δ 1.37 (t, J = 7.6 Hz, 3H), 1.82–1.97 (m, 4H), 2.64–2.70 (m, 1H), 2.80–2.87 (m, 2H), 2.93 (q, J = 7.6 Hz, 2H), 3.80–3.83 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 6.00 (br t, J = 5.2 Hz, 1H), 6.96–6.99 (m, 2H), 7.15 (d, J = 8.0 Hz, 2H), 7.24–7.30 (m, 5H), 7.52 (dd, J = 9.6, 0.8 Hz, 1H), 9.53 (dd, J = 2.0, 0.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.3, 23.6, 33.4, 42.0, 43.3, 50.4, 115.4, 117.0, 121.2, 121.6, 121.9, 126.3, 128.2, 128.3, 128.7, 128.9, 144.5, 144.7, 147.7, 151.4, 151.5, 161.2; 19F NMR (376 MHz, CDCl3) δ 58.31 (s, 3F); LC/MS (ESI) m/z 557 [M + H]+; HRESIMS calcd for C29H29ClF3N4O2 [M + H]+ 557.1926, found 557.1918.



19F NMR (376 MHz, CDCl3) δ 58.31 (s, 3F);

13C NMR (100 MHz, CDCl3) δ 13.3, 23.6, 33.4, 42.0, 43.3, 50.4, 115.4, 117.0, 121.2, 121.6, 121.9, 126.3, 128.2, 128.3, 128.7, 128.9, 144.5, 144.7, 147.7, 151.4, 151.5, 161.2;

1H NMR (400 MHz, CDCl3) δ 1.37 (t, J = 7.6 Hz, 3H), 1.82–1.97 (m, 4H), 2.64–2.70 (m, 1H), 2.80–2.87 (m, 2H), 2.93 (q, J = 7.6 Hz, 2H), 3.80–3.83 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 6.00 (br t, J = 5.2 Hz, 1H), 6.96–6.99 (m, 2H), 7.15 (d, J = 8.0 Hz, 2H), 7.24–7.30 (m, 5H), 7.52 (dd, J = 9.6, 0.8 Hz, 1H), 9.53 (dd, J = 2.0, 0.8 Hz, 1H);
CLIP
June 3, 2019. Qurient press release:
SEONGNAM-SI, South Korea–(BUSINESS WIRE)– Qurient Co. Ltd. today announced positive results from the Phase 2a EBA (early bactericidal activity) clinical trial for telacebec (Q203), a first-in-class, orally-available antibiotic for the treatment of tuberculosis (TB). Telacebec is a selective inhibitor with high specificity for the cytochrome bc1 complex of Mycobacterium tuberculosis. This complex is a critical component of the electron transport chain, and inhibition disrupts the bacterium’s ability to generate energy.
The EBA trial assessed the pharmacokinetics, safety, and activity of telacebec in three dose strength (100 mg, 200 mg and 300 mg) in the treatment of adult patients with pulmonary TB. Telacebec met the primary objective of rate of change in the time to positivity (TTP) in sputum over days 0 to 14. Telacebec was safe and well tolerated throughout the different dose strengths. Full results from EBA trial are expected to be presented at future scientific meetings.
Phase 2. EBA began July 2018 in South Africa. As of March 2019, study is active, not enrolling.
June 2018. Q203 has a non-proprietary name assigned: telacebec. USAN: -cebec Cytochrome bc1 complex inhibitors in Mycobacterium tuberculosis.
Phase 1. Description from clinicaltrials.gov: Randomized, double-blind, placebo-controlled, dose-escalation study in healthy male and female volunteers. Subjects randomly assigned to 1 of 7 treatment cohorts (Cohorts 1 – 7) of 8 subjects each, receiving either Q203 or placebo (6 active treatment : 2 placebo) in a fasting state. Dose escalation to the next cohort may be considered when at least 6 out of 8 subjects, in a cohort, completes all procedures and none of the subjects has a clinically significant adverse event (AE) that is being followed, or at the discretion of the PI if no drug-related serious adverse events (SAEs) have occurred. A food effect cohort will be enrolled to test administration of Q203 in a fed state, at 100 mg dose level (this dose level may change based on PK analysis results). Subjects who received 100mg dose in a fasting state will return and receive the second dose, with food. Subjects will be followed up for AEs, SAE or pregnancy for 30 days postdrug administration.
Related Links
Qurient Press Release. June 2019.Kalia NP et al. 2017. Exploiting the synthetic lethality between terminal respiratory oxidases to kill M. tuberculosis and clear host infection.. PNAS.114.7426
Related Links
- Qurient website
- 2013 Pethe K–Nature Medicine–Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis
- 2014 Kang S — JMC – Lead Optimization of a Novel Series of Imidazo[1,2-a]pyridine Amides Leading to a Clinical Candidate (Q203) as a Multi- and Extensively-Drug-Resistant Anti-tuberculosis Agent
//////////////Telacebec, IAP6, 1334719-95-7, PHASE 2, QURIENT, TUBERCULOSIS, телацебек , تيلاسيبيك , 特雷贝克 , Q 203
BMS 262084
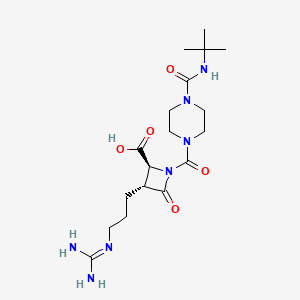
BMS-262084
CAS 253174-92-4
- Molecular FormulaC18H31N7O5
- Average mass425.483 Da
NII-I0IR71971G
I0IR71971G
(2S,3R)-1-[4-(tert-butylcarbamoyl)piperazine-1-carbonyl]-3-[3-(diaminomethylideneamino)propyl]-4-oxoazetidine-2-carboxylic acid(2S,3R)-1-{[4-(tert-butylcarbamoyl)piperazin-1-yl]carbonyl}-3-{3-[(diaminomethylidene)amino]propyl}-4-oxoazetidine-2-carboxylic acid
(2S,3R)-3-{3-[(Diaminomethylene)amino]propyl}-1-({4-[(2-methyl-2-propanyl)carbamoyl]-1-piperazinyl}carbonyl)-4-oxo-2-azetidinecarboxylic acid253174-92-4[RN]2-Azetidinecarboxylic acid, 3-[3-[(diaminomethylene)amino]propyl]-1-[[4-[[(1,1-dimethylethyl)amino]carbonyl]-1-piperazinyl]carbonyl]-4-oxo-, (2S,3R)-
Factor XIa inhibitors (thrombosis), BMS; Factor XIa inhibitors (thrombosis), Bristol-Myers Squibb; BMS-654457; Factor XIa inhibitors (cardiovascular diseases), BMS; BMS-724296
Novel crystalline forms of BMS-262084 as Factor XIa antagonist useful for treating cardiovascular diseases.
PHASE 2

PAPER
Bioorganic & Medicinal Chemistry Letters (2002), 12(21), 3229-3233.
https://www.sciencedirect.com/science/article/pii/S0960894X02006881
Abstract
A series of N1-activated C4-carboxy azetidinones was prepared and tested as inhibitors of human tryptase. The key stereochemical and functional features required for potency, serine protease specificity and aqueous stability were determined. From these studies compound 2, BMS-262084, was identified as a potent and selective tryptase inhibitor which, when dosed intratracheally in ovalbumin-sensitized guinea pigs, reduced allergen-induced bronchoconstriction and inflammatory cell infiltration into the lung.
BMS-262084 was identified as a potent and selective tryptase inhibitor that, when dosed intratracheally in ovalbumin-sensitized guinea pigs, reduced allergen-induced bronchoconstriction and inflammatory cell infiltration into the lung.

PAPER
https://pubs.acs.org/doi/10.1021/jo010757o
Journal of Organic Chemistry (2002), 67(11), 3595-3600.

A highly stereoselective synthesis of the novel tryptase inhibitor BMS-262084 was developed. Key to this synthesis was the discovery and development of a highly diastereoselective demethoxycarbonylation of diester 12 to form the trans-azetidinone 13. BMS-262084 was prepared in 10 steps from d-ornithine in 30% overall yield.
1 as a white powder (3.18 g, 99% yield). Mp: 213-215 °C dec. [α]25D = −65.9 (c 0.99, MeOH). 1H NMR (CD3OD): δ 4.17 (d, J = 3.29 Hz, 1H), 3.61−3.11 (m, 11H), 1.94−1.75 (m, 4H), 1.32 (s, 9H). 13C NMR (CD3OD): δ 176.6, 168.7, 159.4, 158.7, 152.3, 58.7, 53.2, 51.8, 46.5, 45.0, 41.8, 29.6, 27.4, 26.3. HRMS: calcd for C18H32N7O5(M+ + H) 426.2465, found 426.2470. IR (KBr): 3385, 3184, 1775, 1657, 1535, 1395, 1259, 1207, 996, 763 cm–1. Anal. Calcd for C18H31N7O5: C, 50.81, H, 7.34, N, 23.04. Found: C, 50.65, H, 7.42, N, 22.72. Chiral HPLC: ee 99.6%; Chiralpak OD column, 250 × 4.6 mm, 10 μm; mobile phase hexane/EtOH (85:15, v/v); isocratic at ambient temperature, 1.0 mL/min, 220 nm; concentration 0.25 mg/mL, 10 μL injection; RT = 18.6 min (enantiomer, RT = 15.7 min).


PATENT
WO2018133793
claiming macrocyclic compounds.
PATENT
WO-2020259366
Novel crystalline and solid forms of BMS-262084 (designates as monohydrate or 1.5 hydrate), processes for their preparation and compositions comprising them are claimed. BMS-262084 is disclosed to be Factor XIa antagonist, useful for treating cardiovascular diseases.MS-262084 (CAS number: 253174-92-4), the chemical name is (2S,3R)-1-[4-(tert-butylcarbamoyl)piperazine-1-carbonoyl]-3-[3- (Diaminomethylamino)propyl]-4-cyclopropanamide-2-carboxylic acid, also called compound (1) in the present invention, is developed by BMS (Bristol-Myers-Squibb) to treat cardiovascular diseases The drug, as an oral coagulation factor XIa inhibitor for thrombus, has the advantage of significantly reducing the risk of bleeding, and its structure is shown in formula (1):
Patent application WO 9967215A1 discloses the BMS-262084 compound, but the specific molecular formula of the solid substance obtained by the disclosed preparation process is C 18 H 31 N 7 O 5 ·1.56H 2 O, which is similar to the crystal of BMS-262084 described in this application. Type and amorphous water have different molecular weights.
“A stereoselective synthesis of BMS-262084 an azetidinone-based tryptase inhibitor” (Source: Journal of Organic Chemistry, 2002,67(11):3595-3600; Journal of Organic Chemistry,2002,67(11):3595-3600) It is mentioned that the preparation method of BMS-262084 is that hydrogenolysis under neutral conditions eliminates the benzene and Cbz protection groups, and obtains BMS-262084 (melting point 213-215℃). The inventors conducted experiments based on part of the contents disclosed in the document, and the test results obtained crystal form A and crystal form B. The X-ray powder diffraction patterns are shown in Figure 1 and Figure 2 respectively.Example 1
“A stereoselective synthesis of BMS-262084 an azetidinone-based tryptase inhibitor” (Source: Journal of Organic Chemistry, 2002,67(11):3595-3600; Journal of Organic Chemistry,2002,67(11):3595-3600) Only ethanol solvents are mentioned in the literature. Since no specific crystal refining process was provided, only part of the experiment was performed using ethanol solvent.
1) Ethanol solvent volatilization at room temperature: 50mg of BMS-262084 (amorphous) was added to 1.0 mL of ethanol solvent and completely dissolved at room temperature (about 25°C). After volatilizing at room temperature for two days, the solid product was obtained and its crystal form was tested. It is crystal form A, as shown in Figure 1. It is considered that it contains a small amount of amorphous form; but it is unstable and will undergo crystal transformation at room temperature. After standing for one day, the XRPD was tested, and it was found that it was converted to a mixture containing crystal form A, other crystal forms and amorphous forms.
2) Ethanol solvent high-temperature volatilization: 50mg BMS-262084 is added to 1.0mL ethanol solvent, completely dissolved at high temperature (about 60℃), and high-temperature volatilization is carried out in the open to obtain a solid product. The crystal form of the solid product is detected, and the crystal form is B (contains a lot of amorphous), see Figure 2.
SYN1
WO 9967215
The condensation of N-(tert-butyldimethylsilyl)-4-oxoazetidine-2(S)-carboxylic acid (I) with 1-chloro-3-iodopropane (II) by means of BuLi and triisopropylamine (TIA) in THF, followed by treatment with HCl, gives the 3(R)-(3-chloropropyl) derivative (III), which is treated with tetrabutylammonium azide and tetrabutylammonium iodide in DMF to yield the 3-azidopropyl derivative (IV). The reduction of (IV) with H2 over Pd/C in DMF affords the 3-aminopropyl compound (V), which is treated with 1-[N,N’-bis(benzyloxycarbonyl)-1H-pyrazole] (VI) in the same solvent to provide the protected 3-guanidinopropyl compound (VII). The esterification of (VII) with NaHCO3, tetrabutylammonium iodide and Bn-Br in DMF gives the benzyl ester (VIII), which is condensed with N-tert-butylpiperazine-1-carboxamide (IX) and phosgene by means of TEA in toluene to yield the protected precursor (X). Finally, this compound is debenzylated by hydrogenation with H2 over Pd/C in dioxane to give the target azetidine-carboxylic acid.
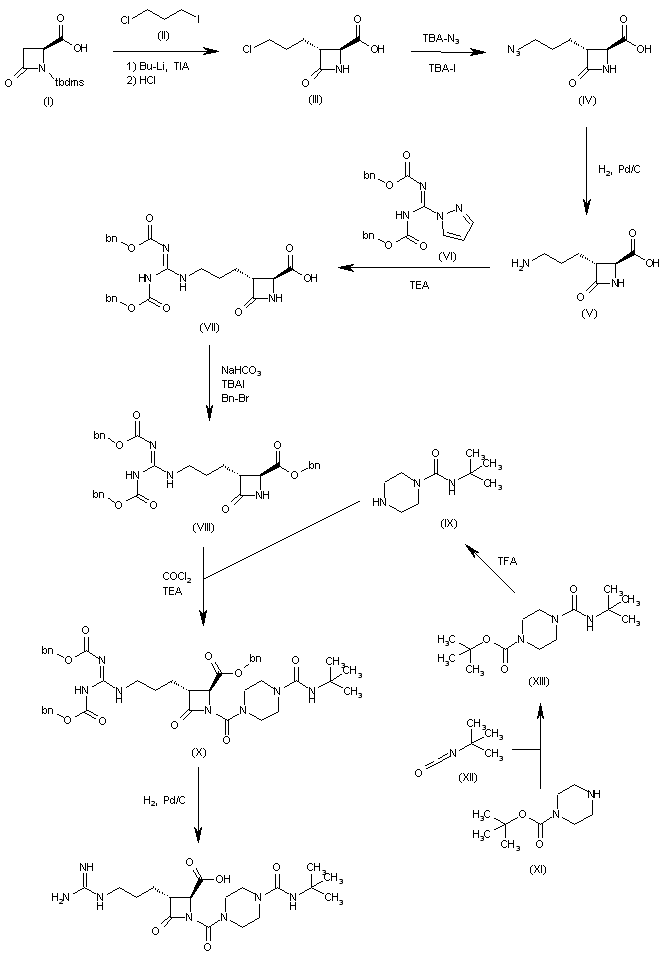
SYN 2
Ethyl nipecotate (I) was protected as the N-Boc derivative (II) and subsequently reduced to alcohol (III) by means of LiAlH4. Conversion of alcohol (III) into iodide (IV) was achieved by treatment with iodine and triphenylphosphine. The dianion of the chiral azetidinecarboxylic acid (V) was alkylated with iodide (IV) to furnish adduct (VI) as a diastereomeric mixture that was desilylated to (VII) using tetrabutylammonium fluoride. Benzyl ester (VIII) was then obtained by reaction of carboxylic acid (VII) with benzyl bromide and NaHCO3.
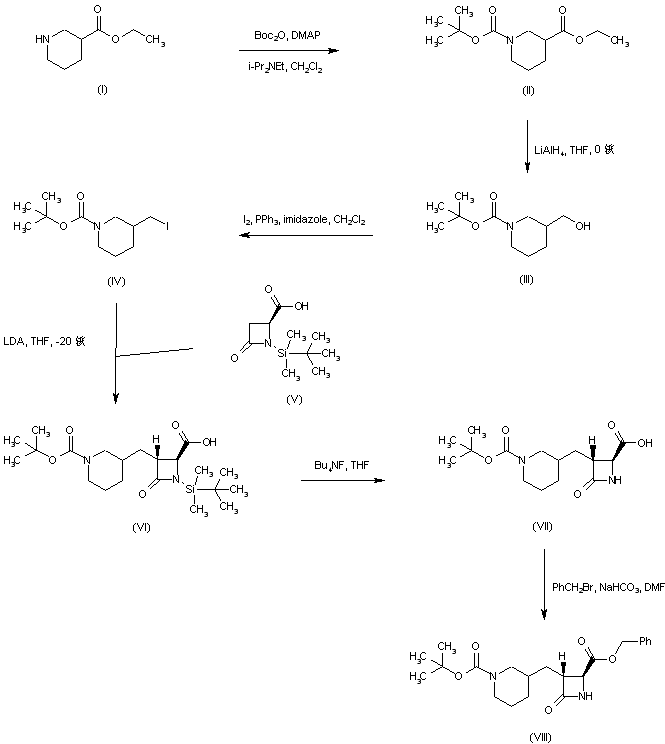
SYN 3
Coupling of 6-phenylhexanoic acid (X) with N-Boc-piperazine (IX) to give (XI), followed by acid deprotection of the Boc group of (XI), provided (6-phenylhexanoyl)piperazine (XII). This was converted to the carbamoyl chloride (XIII) upon treatment with phosgene. The condensation of carbamoyl chloride (XIII) with azetidinone (VIII) gave rise to the urea derivative (XIV). After acid cleavage of the Boc protecting group of (XIV), the resulting piperidine (XV) was condensed with N,N’-dicarbobenzoxy-S-methylisothiourea (XVI) in the presence of HgCl2, yielding the protected guanidine (XVII). This was finally deprotected by catalytic hydrogenolysis over Pd/C.
////////////////////////BMS-262084, BMS 262084, BMS 724296, Factor XIa inhibitors, thrombosis, Bristol-Myers Squibb, BMS 654457, PHASE 2
CC(C)(C)NC(=O)N1CCN(CC1)C(=O)N2C(C(C2=O)CCCN=C(N)N)C(=O)O