

![Acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91758795&t=l)
![2-(4-Chlorophenyl)-N-[[2-(2,6-dioxopiperidin-3-yl)-1-oxo-3H-isoindol-5-yl]methyl]-2,2-difluoroacetamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118647211&t=l)
Candidate: TAK-981
Home » PHASE1 (Page 2)
Category Archives: PHASE1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AK 3280
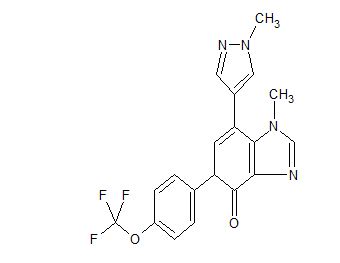
AK-3280
AK 3280; GDC3280; RG 6069
C19 H15 F3 N4 O2, 388.34
CAS 1799412-33-1
4H-Benzimidazol-4-one, 1,5-dihydro-1-methyl-7-(1-methyl-1H-pyrazol-4-yl)-5-[4-(trifluoromethoxy)phenyl]-
Ci8Hi4N502F3, mass 389.3 g/mol),
ROCHE,
Ark Biosciences , under license from Roche , is developing AK-3280, an antifibrotic agent, for the potential oral treatment of IPF. In July 2018, Ark intended to further clinical development of the drug, for IPF. In June 2019, a phase I trial was planned in Sweden.
- Originator Genentech
- Mechanism of Action Undefined mechanism
- Phase I Interstitial lung diseases
- 19 Jun 2019Ark Biosciences plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in Sweden (PO, Tablet), in August 2019 , (NCT03990688)
- 28 Sep 2018GDC 3280 is still in phase I trials for Interstitial lung diseases (Genentech pipeline, September 2018)
- 28 Jun 2018No recent reports of development identified for phase-I development in Fibrosis(In volunteers) in United Kingdom (PO)
Introduction
GDC 3280 (also known as RG 6069), an orally administered drug, is being developed by Genentech, for the treatment of interstitial lung diseases. Early stage clinical development is underway in the UK.
Company Agreements
In September 2018, Genentech licensed exclusive worldwide development and commercialisation rights of GDC 3280 to Ark Biosciences, for the treatment of idiopathic pulmonary fibrosis
Key Development Milestones
As at September 2018, GDC 3280 is still in phase I development for interstitial lung disease (Genentech pipeline, September 2018).
In December 2015, Genentech completed a phase I trial that evaluated the safety, pharmacokinetics and tolerability of GDC 3280 in healthy volunteers, compared with placebo (GB29751; EudraCT2015-000560-33; NCT02471859). The randomised, double-blind, single and multiple oral dose trial was initiated in June 2015 and enrolled eight volunteers in the UK .
PATENT
WO-2019152863
Novel crystalline salt forms of 1-methyl-7-(1-methyl-lH-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-1,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one (compound I; presumed to be AK-3280 ), processes for their preparation and compositions comprising them are claimed.
Compound I is an orally available small molecule having the structure:
[0004] Compound I has therapeutic value in several different indications that display fibrotic pathophysiology, including idiopathic pulmonary fibrosis (IPF).
[0005] Idiopathic pulmonary fibrosis is a disease of unknown etiology that occurs mainly in middle-aged and elderly patients, which is characterized by progressive fibrosis of the lung, leading to pulmonary insufficiency and death. Because fibrosis has long been considered to be a clinically irreversible process, treatments have traditionally been focused on managing the symptoms and complications, with little hope of significantly slowing progression of the condition. For many years, mainstay treatments have been typically anti inflammatory, immunosuppressive, and anti-oxidant agents. The effectiveness of these therapies in the treatment of IPF and other fibrotic conditions appears to be minimal and variable, and their side effects are often poorly tolerated by patients.
[0006] New treatment options have only recently become available. Both pirfenidone and nintedanib have been approved for use in the treatment of IPF. Current research efforts to develop new anti-fibrotic agents are targeting multiple mechanisms proposed to be linked to the underlying molecular pathogenic processes. This changing landscape has raised hopes and expectations for what might be achievable with new single agents or combination therapies targeting additional pathways.
Preparation of Compound I and its salts
[0045] A synthesis of Compound I and its tosylate salt is shown in the scheme below:
[0046] l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one (5) was synthesized in 4 steps, including a copper-catalyzed coupling reaction e.g., a Goldberg-Ullmann coupling reaction. In another aspect of the invention, intermediate (5) is synthesized using any transition metal-catalyzed coupling reaction. The skilled chemist would know that intermediate (5) could be synthesized from intermediate (4) and compounds
LG
of the general formula: OCF3 , wherein the leaving group“LG” includes but is not limited to halogen, tosylate, mesylate, triflate, etc.
[0047] Compound I was synthesized in 6 steps, using a transition metal cross-coupling reaction, e.g., a Suzuki reaction. In another aspect of the invention, Compound I is synthesized using any cross -coupling reaction. Compound I is synthesized from intermediate 6 containing any leaving group. For example, the skilled chemist would use compounds of
the general formula:
, wherein the leaving group“LG” includes but is not limited to halogen, tosylate, mesylate, triflate, etc.
An alternative synthesis of Compound I and its salts is shown in the scheme below:
Example 13 – Synthesis of Compound I Tosylate Salt
[00183] A process for the formation of mono- and di-tosylate salts of Compound I was developed and a batch was performed to successfully produce the mono-tosylate salt.
Step 1 : Synthesis of2-chloro-N-methyl-3-nitropyridin-4-amine
[00184] A reactor was charged with 2,4-dichloro-3-nitropyridine and 3.0 volumes of DMF. The solution was stirred at 20-25 °C until a clear solution was obtained. The solution was then cooled to 0-5 °C, and 2.1 equivalents of 40% methylamine in water were slowly added over at least 2 hours at 0-5 °C. The reaction mixture was stirred for at least 2 hours at 0-5 °C until conversion to the product was 95% (as measured by HPLC). The reaction mixture was diluted by slowly adding 10 volumes of water over at least 30 minutes at 0-5 °C. The obtained suspension was stirred for at least 60 minutes at 0-5 °C. The precipitate was collected by filtration, and the filter cake was rinsed via the reactor with 10 volumes of water at 0-5 °C. The damp filter cake was then dried in a flow of dry nitrogen to yield 2-chloro-A-methyl-3-nitropyridin-4-amine in 78% yield.
Step 2: Synthesis of 2-chloro-N4 -methylpyridine-3, 4-diamine
[00185] A reactor was charged with catalyst [2% Pt on charcoal, 59 %wt. water] (0.0004 equivalents Pt), damp 2-chloro-/V-methyl-3-nitropyridin-4-amine from step 1 and 9.4 volumes of THF. The solution was stirred, and then the suspension was transferred from the glass-reactor to an autoclave. The line was rinsed with 1.2 volumes of THF into the autoclave, and the autoclave was purged with nitrogen for 15 minutes at 50 rpm, followed by hydrogen for 15 minutes at 150 rpm. The autoclave was closed, and the hydrogen pressure was adjusted to 2 bar at 20-30 °C. The reaction mixture was stirred for 4-8 hours at 2 bar and 20-30 °C.
[00186] Next, the autoclave was released to atmospheric pressure and purged with nitrogen for at least 15 minutes. Conversion to the product was verified by HPLC, and then the catalyst was removed by filtration. The filtered catalyst was rinsed with 1.3 volumes of THF and the filtrates were combined. The combined filtrates were charged to a second reactor via a particle filter, and the line was rinsed with 0.5 volumes of THF. The solution was concentrated to a final volume of 2.5 volumes by distillation under reduced pressure at 40-45 °C.
[00187] The solution was then diluted with 10 volumes of THF in portions while concentrating the solution to a final volume of 2.5 volumes by distillation under reduced pressure at 45-50 °C. The reactor was purged with nitrogen to atmospheric pressure, and 5.0 volumes of heptane were added to the residue at 40-50 °C. The reaction mixture was cooled over 2 hours to 20-25 °C, and stirring was continued for 1 hour. The reaction mixture was then further cooled to 0-5 °C over 1 hour, and stirring was continued for 1 hour. The precipitated product was collected by filtration, rinsed via the reactor with 5.0 volumes of heptane, and the damp filter cake was dried in a vacuum drying oven at max. 40 °C until loss on drying was < 2 % weight, giving 2-chloro-/V4-methylpyridine-3, 4-diamine in 85% yield.
Step 3 : Synthesis of -inelhyl- 1 ,5-dihvdro-4H-iinidazoi4,5-c h yridin-4-one
[00188] A reactor was charged with 2-chloro-/V4-methylpyridine-3, 4-diamine and 4 volumes of formic acid. The reaction mixture was heated to smooth reflux within one hour, and reflux was maintained for 6 hours. The reaction mixture was then cooled to
approximately 60 °C, and conversion to the product was verified by HPLC.
[00189] The reaction mixture was then concentrated by distillation under reduced pressure at 60-80 °C to a final volume of 2 volumes. The temperature of the solution was adjusted to 60 °C, maintaining the temperature above 50 °C to avoid precipitation.
[00190] Next, a second reactor was charged with 10 volumes of acetone, and heated to gentle reflux. The product solution from the first reactor was slowly transferred to the acetone in the second reactor over 20 minutes, and the line was rinsed with approximately 0.05 volumes of formic acid. Reflux of the obtained suspension was maintained for 15 minutes. The slurry was cooled to 0 °C within 1 hour, and stirring was continued for 1 hour at that temperature. The precipitate was collected by filtration, and the filter cake was rinsed via the reactor with 3.7 volumes of cold acetone at 0-10 °C. The filter cake was dried in a flow of dry nitrogen or in a vacuum drying oven at 50 °C until loss on drying was < 2% of weight, giving 1 -methyl- 1 ,5-dihydiO-4/7-imidazo[4,5-c]pyndin-4-onc in 95% yield.
Step 4: Synthesis of l-methyl-5-(4-(trifluoromethoxy)phenyl)-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one
[00191] A first reactor (Reactor A) was charged with 1 -methyl- 1 ,5-dihydro-4/7-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalent), Cu(0Ac)2 H20 (0.1 mol equivalents), and K2C03 (1.1 mol equivalents). The reactor was closed and the atmosphere replaced with nitrogen.
[00192] Next, l-bromo-4-(trifluoromethoxy)benzene (1.5 mol equivalents) and N-methylpyrrolidinone (5.4 volume equivalents) were added, whereupon a suspension was formed. The suspension was stirred until the temperature had fallen again to approximately 20-25 °C and gas evolution had slowed. The reaction mixture was heated to approximately 130-150 °C at which time a blue/green color was observed, changing to dark brown after some time. The reaction was stirred at 130-150 °C for at least 40 hours. Stirring times of 40 hours up to 72 hours were required to reach an acceptable level of conversion. In general, higher reaction temperatures supported faster conversion.
[00193] Next, the reaction mixture was cooled to approximately 20-30 °C, and 25% aqueous NH3 (0.7 volume equivalents) was added, followed by water (3.5 volume equivalents). The resulting suspension was transferred into a second reactor (Reactor B). Additional water was added (18.1 volume equivalents) to the reaction mixture via Reactor A, followed by n-heptane (3.2 volume equivalents). The resulting suspension was cooled to approximately 0-5 °C, and stirred for approximately 2 hours.
[00194] The suspension was filtered, and the filter cake was washed with water (9.7 volume equivalents). The filter cake was then dissolved in dichloromethane (14.1 volume equivalents) and transferred back into reactor B. To this solution was added water (5.7 volume equivalents) via the filter, followed by 25% aq. NH3(1.6 volume equivalents). The mixture was stirred for approximately 1 hour at approximately 15-25 °C.
[00195] Next, the layers were separated, and dichloromethane was added (3.6 volume equivalents) to the aqueous layer. The biphasic mixture was stirred at approximately 15-25 °C for approximately 20-30 minutes. The layers were separated over a period of at least 1 hour, and to the combined organic layers was added a solution of NH4Cl (2.5 mol equivalents) in water (7.0 volume equivalents). The biphasic mixture was stirred at approximately 15-25 °C for about 20-30 minutes, then the layers were separated over the course of 1 hour.
[00196] The lower organic layer was filtered through a particle filter and diluted with toluene (7.1 volume equivalents) via the filter. The organic layer was concentrated under ambient pressure at approximately 80 °C, until no further liquid was seen to evaporate and a precipitate began to form. Toluene was added (16.6 volume equivalents), then concentrated in vacuo, followed by addition of more toluene (7.1 volume equivalents) and again concentrated in vacuo. The suspension was cooled to approximately 0-5 °C, stirred for approximately 2 hours, and filtered. The filter cake was washed with toluene (2.9 volume equivalents), and dried in vacuo at approximately 50 °C until the loss on drying was 0.5% of the weight to give l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one as a beige-colored solid in 83.1% yield.
Step 5 : Synthesis of 7-bromo- 1 -methyl-5-(4-( trifluoromethoxy Iphenyl )- l,5- 4H-
imidaz.o[4,5-clpyridin-4-one
[00197] A first reactor (Reactor A) was charged with water (1.8 volume equivalents) and cooled to approximately 0-5 °C, to which was slowly added 96% sulfuric acid (14 mol. equivalents) at approximately 0-20 °C. The temperature of the solution was adjusted to approximately 0-5 °C, and l -mcthyl-5-(4-(tnfluoromcthoxy)phcnyl)-l ,5-dihydro-4/7-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalent) was added in 3-4 portions at approximately 0-5 °C. The temperature of the mixture was adjusted to approximately 0-5 °C, and N-bromosuccinimide (1.0 mol equivalents) was slowly added in 3-4 portions, while maintaining the temperature at approximately 0-5 °C.
[00198] The reaction mixture was stirred for about 1 hour at approximately 0-5 °C, and then for an additional 4-16 hours at approximately 0-22 °C. Conversion to the product was confirmed by HPLC, then the reaction mixture was cooled to approximately 0-5 °C.
[00199] A second reactor (Reactor B) was charged with water (42.7 volume equivalents) and cooled to approximately 0-5 °C. The reaction mixture from Reactor A was transferred into the pre-cooled water in Reactor B at a temperature below 30 °C over 2 hours. The reaction was rinsed with water (1.6 volume equivalents), and 50% aqueous sodium hydroxide (25 mol. equivalents) was carefully added at approximately 0-30 °C over about 2 hours until the pH reached 2-5.
[00200] Next, MTBE (6.5 volume equivalents) was added at approximately 0-20 °C, and the mixture was stirred for about 5 minutes. Additional 50% aqueous sodium hydroxide (2 mol. equivalents) was added at approximately 0-30 °C until the pH of the solution was in the range of 10-14. The reaction was stirred for at least 1.5 hours at approximately 15-25 °C, and then the layers were allowed to separate over a period of at least 1 hour. The suspension was filtered, taking care to capture the product, which accumulated at the interface of the aqueous and organic layers. The filter cake was washed with MTBE (1.7 volume equivalents), water (3.0 volume equivalents), and then MTBE again (3.0 volume equivalents). The product was dried in vacuo at below 50 °C until the loss on drying was < 1% of the weight, giving 7-bromo-l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one as a pale beige-colored solid in 97.6% yield.
Step 6: Synthesis of 1 -methyl-7 -( 1 -methyl-lH-pyraz.ol-4-yl )-5-(4-( trifluoromethoxy )pheml )-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one (Compound /)
[00201] A reactor was charged with 7-bromo-l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalents), ( 1 -methyl- 1 //-pyrazol-4-yl)boronic acid pinacol ester (l-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l//-pyrazole, 1.6 mol equivalents), Pd[Ph3]4 (0.025 mol equivalents, and K2C03 (2.0 mol equivalents), to which were added acetonitrile (10.0 volume equivalents) and water (3.0 volume equivalents). The reaction mixture was stirred for approximately 10-20 minutes at about 20-25 °C to form a suspension.
[00202] The mixture was heated to slight reflux, whereupon a biphasic, yellow solution formed. The mixture was stirred at slight reflux for at least 10 hours. The reaction mixture was cooled to between 30-50 °C, then passed through a particle filter. The filter was washed with acetonitrile (2.6 volume equivalents), the filtrates were combined, and the solution was concentrated to a final volume of approximately 120 mL (4.8 volume equivalents) under reduced pressure at below 60 °C.
[00203] To the resulting suspension was added water (1.9 volume equivalents), methanol (26 mL, 1.0 volume equivalents), and dichloromethane (14.8 volume equivalents). The mixture was warmed to about 30-35 °C and stirred until two clear layers were observed. The layers were allowed to separate without stirring at about 30-35 °C, and additional dichloromethane (3.7 volume equivalents) was added to the aqueous layer. The mixture was warmed to approximately 30-35 °C and stirred for about 5 minutes, and then the layers were allowed to separate at approximately 30-35 °C.
[00204] To the combined organic layers was added water (1.9 volume equivalents), and the mixture was warmed to approximately 30-35 °C and stirred for about 5 minutes. The layers were separated at approximately 30-35 °C. Charcoal was added to the combined organic layers and stirred for 30-60 minutes at approximately 30-35 °C. The charcoal was removed by filtration, and the filter was washed with dichloromethane (39 mL, 1.6 volume equivalents).
[00205] The solution was concentrated to approximately 4.0 volume equivalents at ambient pressure and at below 50 °C, then diluted with methanol (5.0 volume equivalents). The solution was again concentrated to approximately 4.0 volume equivalents at ambient pressure and below 60 °C, diluted with methanol (5.0 volume equivalents), and concentrated to a final volume of approximately 3.0 volume equivalents under reduced pressure below 60 °C.
[00206] To the resulting suspension was added methanol (2.9 volume equivalents), and the suspension was warmed to approximately 45-55 °C and stirred for about 1 hour. The suspension was cooled to approximately 0-5 °C within approximately 1 hour, stirred for 1 hour at approximately 0-5 °C, and then filtered. The filter cake was washed with cold methanol (pre-cooled to approximately 0-10 °C, 2.9 volume equivalents), and the product was dried under a stream of nitrogen and in vacuo at below 60 °C until the loss on drying was < 1% by weight, giving Compound I (l-methyl-7-(l-methyl-l -pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one) as a white solid in 88.5% yield.
Step 7: Recrystallization of 1 -methyl-7 -(1 -methyl- lH-pyraz.ol-4-yl)-5-( 4-(trifluoromethoxy)phenyl)-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one (Compound /)
[00207] A reactor was charged with crude l-methyl-7-(l -methyl- l//-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one from step 6, and to this was added glacial acetic acid (1.5 volume equivalents). The suspension was warmed to approximately 50-60 °C and stirred until a clear solution was obtained, approximately 10-20 minutes. The warm solution was passed through a particle filter into a second reactor.
[00208] To this solution was added ethanol (10.0 volume equivalents) at approximately 45-55 °C over 2 hours. The suspension was stirred for approximately 30 minutes at approximately 45-55 °C, then cooled to approximately 0-5 °C over about 4 hours. The suspension was then stirred for approximately 4-16 hours at about 0-5 °C.
[00209] Next, the suspension was filtered and the filter cake was washed with cold isopropanol (4.2 volume equivalents) at approximately 0-20 °C. The product was dried under a nitrogen stream and in vacuo at below 60 °C until the loss on drying was < 1% by weight, giving Compound I ( 1 – mcthyl-7-( 1 -methyl- 1 /7-pyrazol-4-yl)-5-(4-(tnfluoromcthoxy)phcnyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one) as a white solid in 93.0% yield.
Step 8 : Synthesis of 1 -methyl-7 -( 1 -methyl- 1 H-pyrazol-4-yl )-5-(4-( trifluoromethoxy )phenyl )- 1 ,5-dihvdro-4H-imidaz.oi 4,5-clpyridin-4-one, mono – mono -tosylate
salt)
[00210] A reactor was charged with Compound I ( 1 -mcthyl-7-( 1 -methyl- 1 /7-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one, 1.00 mol equivalent), para-toluenesulfonic acid monohydrate (1.05 mol equivalents), acetone (6.75 volume equivalents), and water (0.75 volume equivalents). The mixture was stirred at 15-25 °C until a clear solution formed, and then this solution was filtered through a particle filter into a second reactor.
[00211] The filter was washed with acetone (2.5 volume equivalents), and to the combined filtrates was added MTBE (7.5 volume equivalents) at 15-25 °C and Compound I mono-tosylate seeding crystals (0.001 mol equivalents).
[00212] The resulting suspension was stirred at 15-25 °C for approximately 30-60 minutes, and MTBE was added (22.5 volume equivalents) at 15-25 °C during a period of
approximately 30 minutes. Stirring was continued at 15-25 °C for approximately 30-60 minutes, and then the suspension was filtered. The filter was washed with MTBE (2.5 volume equivalents), and the material was dried in vacuo at below 55 °C to give Compound I mono-tosylate salt (l-methyl-7-(l-methyl-l//-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one, mono-tosylate salt) as a white, crystalline solid in 93% yield.
PATENT
WO2018102323 ,
claiming use of a specific compound, orally administered, in combination with food (eg low, medium or high fat meal) for treating fibrotic, inflammatory or autoimmune disorders eg idiopathic pulmonary fibrosis IPF, assigned to Genentech Inc ,
References
-
Roche licenses IPF candidate to Ark Biosciences. Internet-Doc 2019;.
Available from: URL: https://scrip.pharmaintelligence.informa.com/deals/201820364
-
Roche Q3 2018. Internet-Doc 2018;.
Available from: URL: https://www.roche.com/dam/jcr:f9cad8fc-8655-4692-9a85-efbe1cf7a59b/en/irp181017.pdf
-
A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Ascending, Single- and Multiple-Oral-Dose, Safety, Tolerability, and Pharmacokinetic Study of GDC-3280 in Healthy Subjects
// AK-3280, AK 3280, AK3280, GDC 3280, RG 6069, PHASE 1, Idiopathic pulmonary fibrosis
SY-008


SY-008
CAS 1878218-66-6
FREE FORM 1480443-32-0
SGLT1 inhibitor (type 2 diabetes),
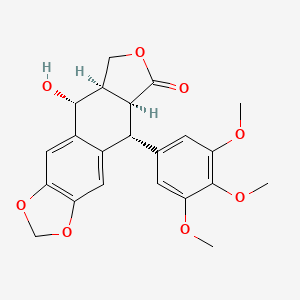
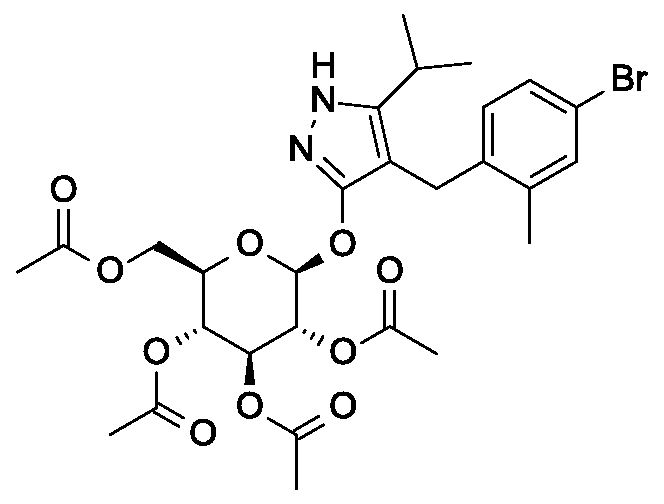
β-D-Glucopyranoside, 4-[[4-[(1E)-4-(2,9-diazaspiro[5.5]undec-2-yl)-1-buten-1-yl]-2-methylphenyl]methyl]-5-(1-methylethyl)-1H-pyrazol-3-yl, acetate (1:1)
acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol
MF H50 N4 O6 . C2 H4 O2
MW 58.8 g/mol,C35H54N4O8
Originator Eli Lilly
- Developer Eli Lilly; Yabao Pharmaceutical Group
- Class Antihyperglycaemics; Small molecules
- Mechanism of Action Sodium-glucose transporter 1 inhibitors
- Phase I Diabetes mellitus
- 28 Aug 2018 No recent reports of development identified for phase-I development in Diabetes-mellitus in Singapore (PO)
- 24 Jun 2018 Biomarkers information updated
- 12 Mar 2018 Phase-I clinical trials in Diabetes mellitus (In volunteers) in China (PO) (NCT03462589)
-
Eli Lilly is developing SY 008, a sodium glucose transporter 1 (SGLT1) inhibitor, for the treatment of diabetes mellitus. The approach of inhibiting SGLT1 could be promising because it acts independently of the beta cell and could be effective in both early and advanced stages of diabetes. Reducing both glucose and insulin may improve the metabolic state and potentially the health of beta cells, without causing weight gain or hypoglycaemia. Clinical development is underway in Singapore and China.
As at August 2018, no recent reports of development had been identified for phase-I development in Diabetes-mellitus in Singapore (PO).
Suzhou Yabao , under license from Eli Lilly , is developing SY-008 , an SGLT1 inhibitor, for the potential oral capsule treatment of type 2 diabetes in China. By April 2019, a phase Ia trial was completed
PATENT
WO 2013169546
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 201 1 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLTl is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLTl may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLTl inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 201 1/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides certain novel inhibitors of SGLTl which may be suitable for the treatment of diabetes.
Accordingly, the present invention provides a compound of Formula II:
Preparation 1
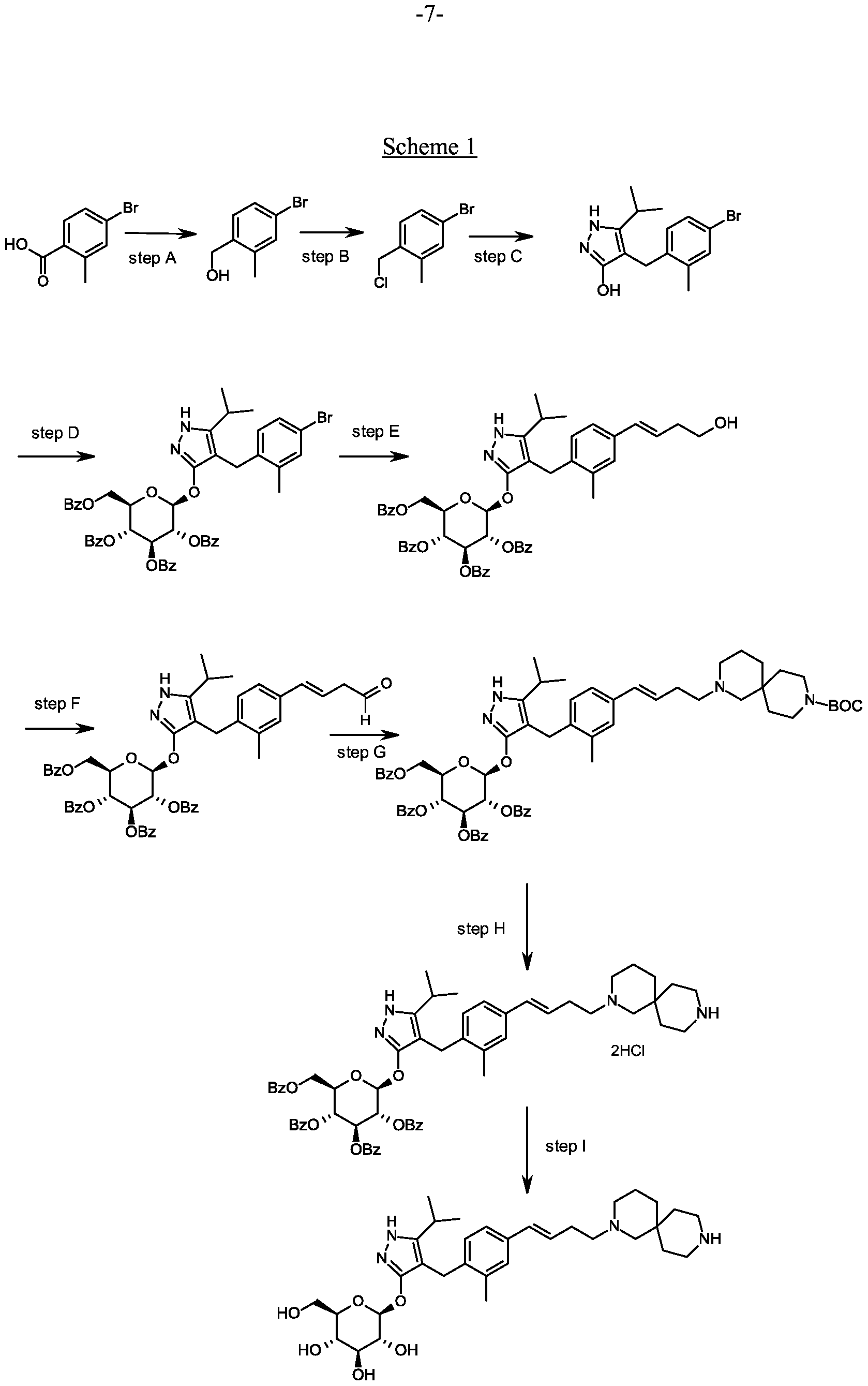
Synthesis of (4-bromo-2-methyl-phenyl)methanol.
Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). 1H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)methanol.
Borane-dimethyl sulfide complex (2M in THF; 1 16 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.1 13 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
Synthesis of 4-bromo- l-2-methyl-benzene.
Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4-bromo-2-methyl-phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and
-Cl-
dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. XH NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo- 1 -chloromethyl-2-methyl-benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). XH NMR (300.1 1 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
Synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol.
Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo- l-chloromethyl-2-methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction.
Extract with ethyl acetate (200 mL), wash extract with water (200 rnL) and brine (200 mL), dry over a2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l).
Alternative synthesis of 4-r(4-bromo-2-methyl-phenyl)methyl1-5-isopropyl- !H-pyrazol- 3-oL
A solution of 4-bromo- 1 -chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (1 1.94 g, 71.94 mmol) and methyl 4-methyl-3-oxo valerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2O5 at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/31 1 (M+l).
Preparation 4
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (ml 2): 889.2 (M+l), 887.2 (M-l).
Preparation 5
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
Synthesis of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2-dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+1).
Preparation 8
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D- glucopyranoside dihydrochloride.
Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3is)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
Example 1
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40 °C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 um C18XBridge ODB column, solvent A – 1¾0 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Preparation 9
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-acetyl-beta-D-glucopyranoside.
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammomum chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate
(32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l).
Preparation 10
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol). MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0- acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2-dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
Synthesis oftert-butyl 2-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,8- diazaspiro[4.5]decane-8-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 852.8, 853.6 (M+l), 850.8, 851.6 (M-l).
Preparation 14
Synthesis oftert-butyl 9-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-3,9- diazaspiro[5.5]undecane-3-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 866.8, 867.6 (M+l), 864.8, 865.6 (M-l).
Preparation 15
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D- glucopyranoside dihydrochloride.
Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Preparation 16
Synthesis of 4-{4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5- (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
dihydrochloride.
The title compound is prepared essentially by the method of Preparation 15. MS (m/z): 752.8, 753.8 (M+1), 750.8 (M-1).
First alternative synthesis of Example 1
First alternative synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en- 2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 urn C18XBridge ODB column, solvent A – H20 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+1), 596.8 (M-1). 1H MR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=1.3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.1 1 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Example 2
Synthesis of 4- {4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbi
(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
O H
The title compound is prepared essentially by the method of the first alternative synthesis of Example 1. MS (m/z): 584.7 (M+l), 582.8 (M-l).
Example 3
Synthesis of 4- {4-[( 1 E)-4-(3 ,9-diazaspiro[5.5]undec-3 -yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
The title compound is prepared essentially by first treating the compound of Prearation 14 with HC1 as discussed in Preparation 15 then treating the resulting hydrochloride salt with triethyl amine as discussed in the first alternative synthesis of Example 1. MS (m/z): 598.8, 599.8 (M+l), 596.8, 597.8 (M-l).
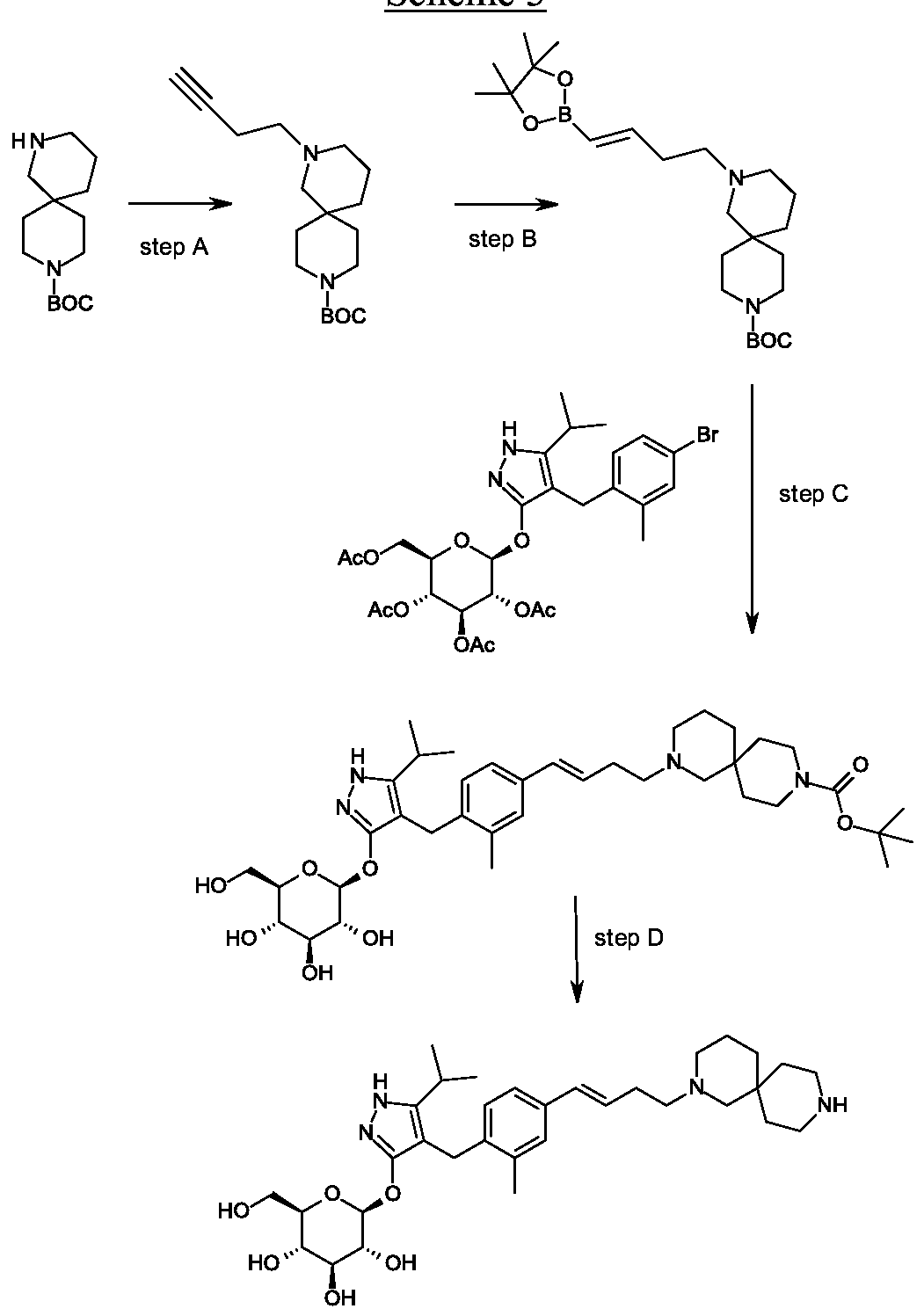
Example 1 Preparation 17
Synthesis of tert-butyl 4-but-3- nyl-4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgSC^, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). iH MR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H),
2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 18
Synthesis of tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]- 4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5, 5-tetramethyl-1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield), !H NMR (300.1 1 MHz, CDC13): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H),
3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 19
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2-dicyclohexylphosphino-2′,4′,6′-tri-i-propyl-l, r-biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).
Second alternative Synthesis of Example 1
Second alternative synthesis of 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200ml) and concentrated in vacuo. This is repeated several times give the title compound (12.22 g, yield 96%). MS (m/z): 599 (M+l). [a]D20 = -12 ° (C=0.2, MeOH).
PATENT
WO 2015069541
https://patents.google.com/patent/WO2015069541A1
4-{4-[(1 E)-4-(2,9-DIAZASPIRO[5.5]UNDEC-2-YL)BUT-1 -EN-1
-YL]-2-METHYLBENZYL}-5-(PROPAN-2-YL)-1 H-PYRAZOL-3-YL
BETA-D- GLUCOPYRANOSIDE ACETATE
The present invention relates to a novel SGLT1 inhibitor which is an acetate salt of a pyrazole compound, to pharmaceutical compositions comprising the compound, to methods of using the compound to treat physiological disorders, and to intermediates and processes useful in the synthesis of the compound.
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 2011 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLT1 is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLT1 inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 2011/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides an acetate salt of a pyrazole compound, which is an SGLT1 inhibitor, and as such, may be suitable for the treatment of certain disorders, such as diabetes. Accordingly, the present invention provides a compound of Formula I:

or hydrate thereof.
Preparation 1
(4-bromo-2-methyl-phenyl)methanol

Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). !H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)mefhanol.
Borane-dimethyl sulfide complex (2M in THF; 116 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.113 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
4-bromo- 1 -chloromethyl -2 -methyl -benzene

Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4- bromo-2 -methyl -phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. !H NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo-l-chloromethyl-2-methyl -benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). !H NMR (300.11 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
4- [(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-3 -ol

Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo-l-chloromethyl-2- methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction. Extract with ethyl acetate (200 mL), wash extract with water (200 mL) and brine (200 mL), dry over Na2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l). Alternative synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-
3-ol.
A solution of 4-bromo-l-chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (11.94 g, 71.94 mmol) and methyl 4-methyl-3-oxovalerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2Os at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/311 (M+l).
Preparation 4
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl- beta-D-glucopyranoside
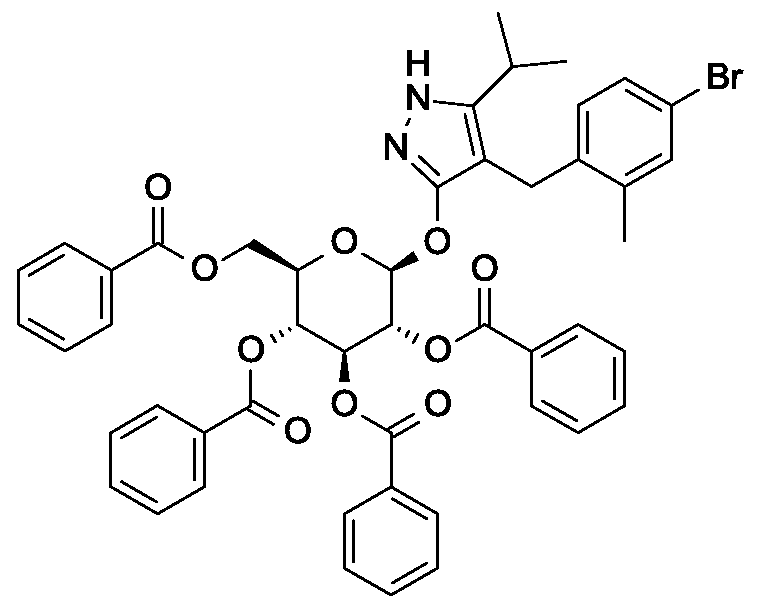
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (m/z): 889.2 (M+l), 887.2 (M-l).
Preparation 5
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
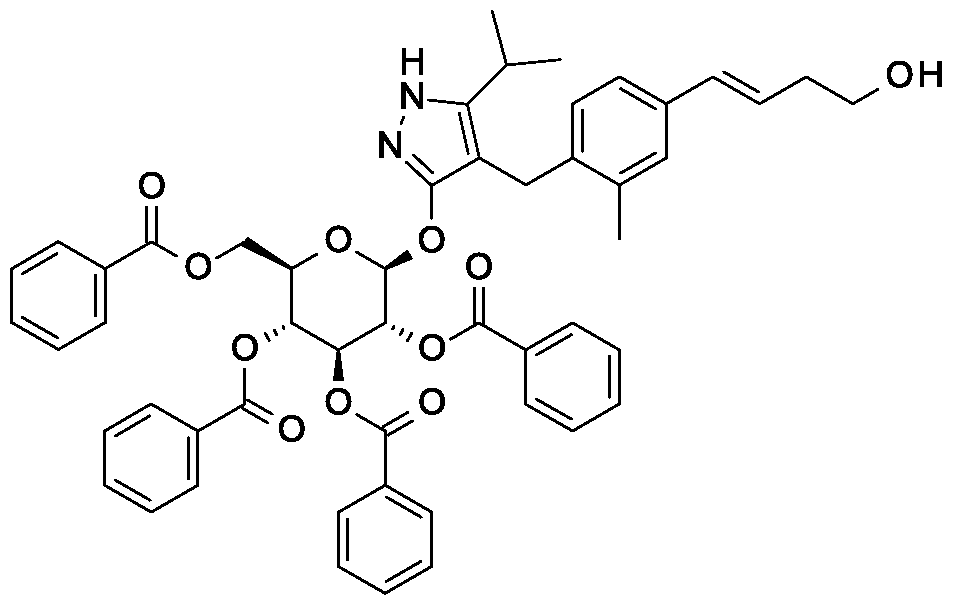
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- lH-pyrazol-3 -yl 2,3 ,4,6-tetra-O-benzoyl-beta-D- glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
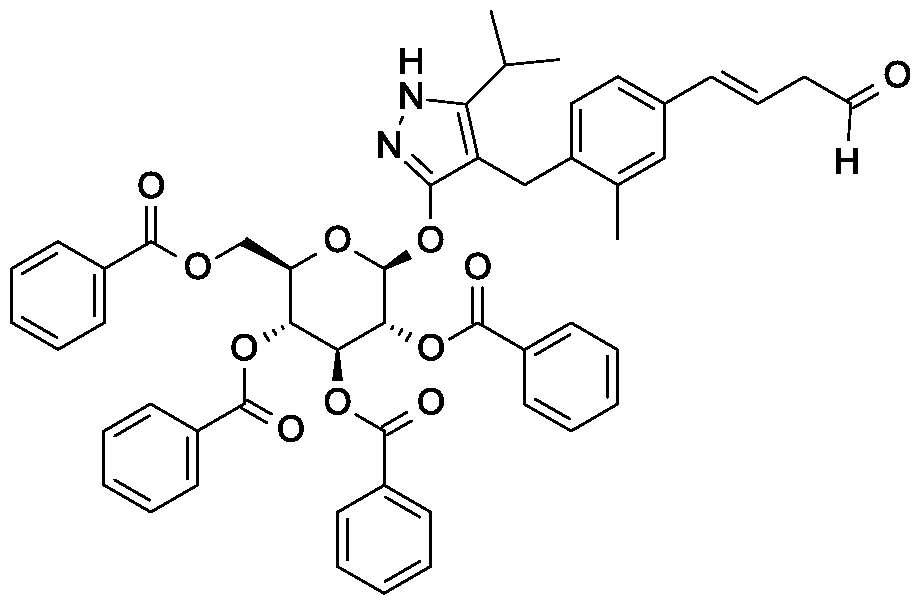
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate

Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2- dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+l).
Preparation 8
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride

Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
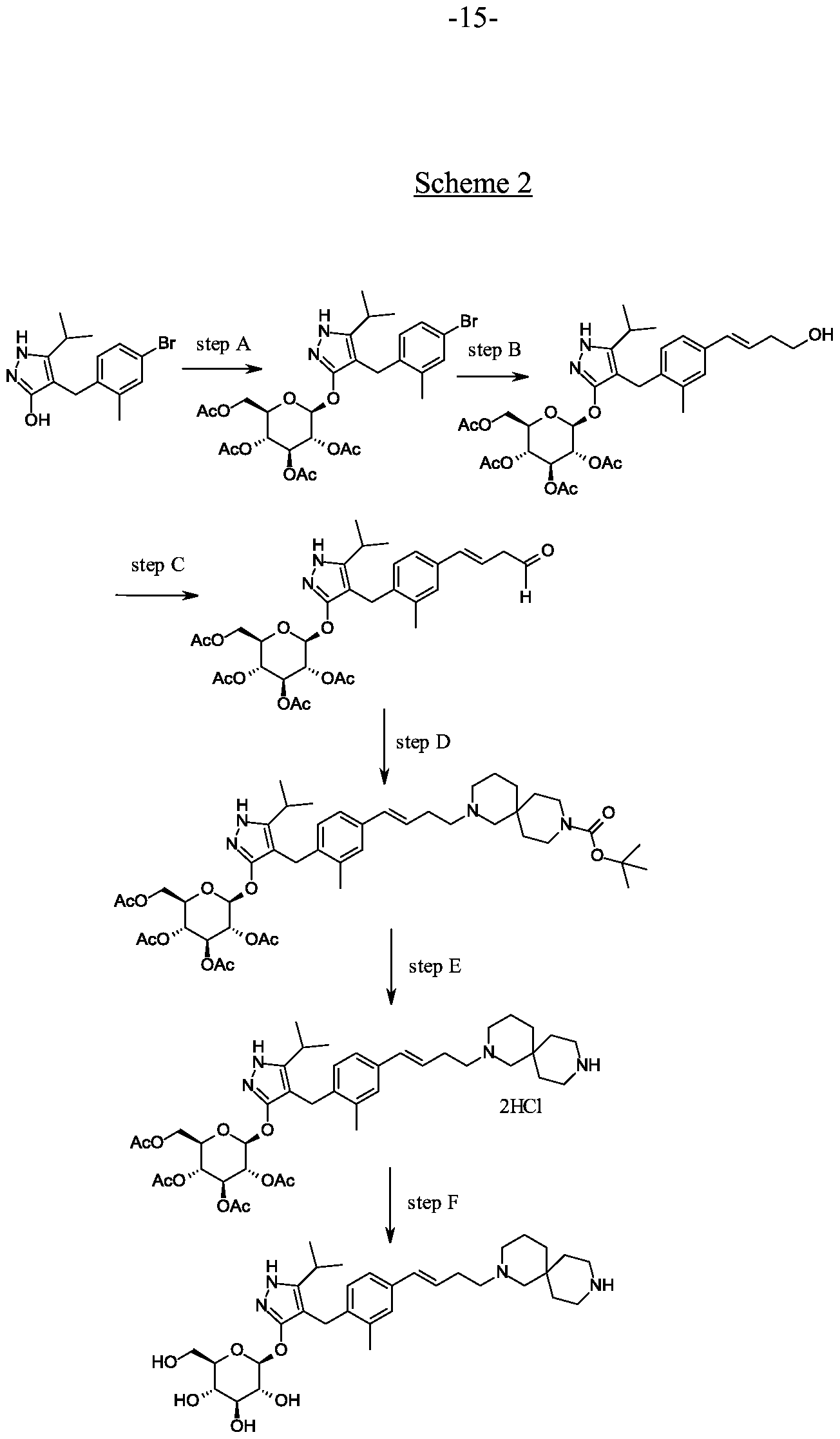
Preparation 9
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl- beta-D-glucopyranoside.
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)mefhyl]-5- isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D- glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate (32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l). Preparation 10
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- 1 H-pyrazol-3 -yl 2,3 ,4,6-tetra-O-acetyl-beta-D- glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol) MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside

Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12a
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy] -lH-pyrazol-4-yl}methyl)phenyl]but-3-en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2- dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride

Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6- tetra-0-acetyl-beta-D-glucopyranosyl)oxy] – lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 – yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Scheme 3
Preparation 14
tert-butyl 4-but-3-ynyl-4,9-diazas iro[5.5]undecane-9-carboxylate

Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgS04, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). lH NMR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H), 2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 15
tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9- diazaspiro[5.5]undecane-9-carboxylate
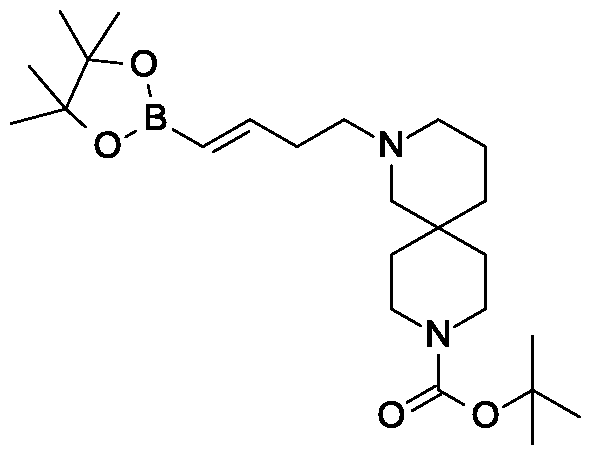
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield). 1H NMR (300.11 MHz, CDCI3): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H), 3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 16
tert-butyl 2-{(3£’)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH- pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9-diazaspiro [5.5]undecane-9-carboxylate

Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert- butyl 4-[(£)-4-(4,4,5 ,5 -tetramethyl- 1 ,3,2-dioxaborolan-2-yl)but-3 -enyl] -4,9- diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2- dicyclohexylphosphino-2′,4′,6′-tri-i -propyl- Ι, -biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).

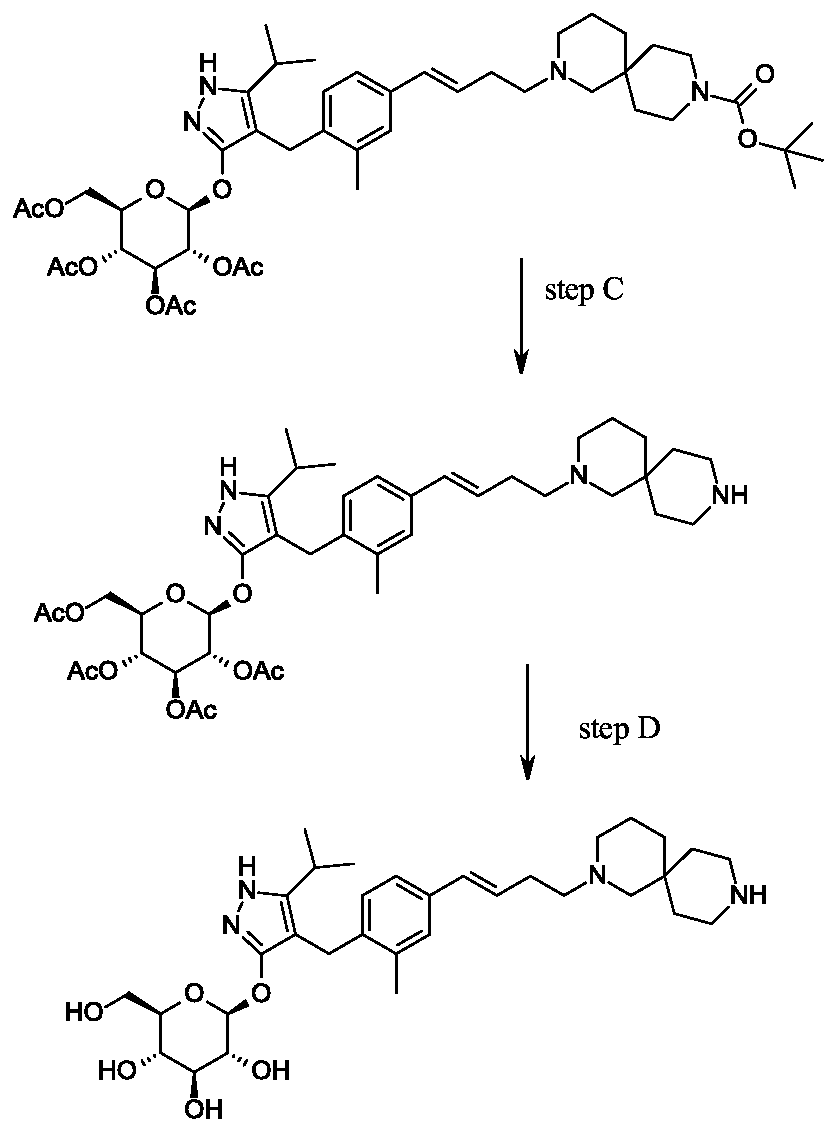
Preparation 17
tert-butyl 4- [(E)-4- [4- [(3 -hydroxy-5-isopropyl- 1 H-pyrazol-4-yl)methyl] -3 -methyl- phenyl]but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate

Scheme 4, step A: Add tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (35.8 kg, 82.4 mol) in methanol (130 L) to a solution of (4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (23.9 kg, 77.3 mol) in methanol (440 L) at room temperature. Add water (590 L) and tripotassium phosphate (100 kg, 471.7 mol) and place the reaction under nitrogen atmosphere. To the stirring solution, add a suspension of
tris(dibenzylideneacetone) dipalladium (1.42 kg, 1.55 mol) and di-tert- butylmethylphosphonium tetrafluoroborate (775 g, 3.12 mol) in methanol (15 L). The resulting mixture is heated at 75 °C for 2 hours. Cool the mixture and filter over diatomaceous earth. Rinse the the filter cake with methanol (60 L), and concentrate the filtrate under reduced pressure. Add ethyl acetate (300 L), separate the layers, and wash the organic layer with 15% brine (3 x 120 L). Concentrate the organic layer under reduced pressure, add ethyl acetate (300 L), and stir the mixture for 18 to 20 hours. Add heptane (300 L), cool the mixture to 10 °C, and stir the mixture for an additional 18 to 20 hours. Collect the resulting solids by filtration, rinse the cake with ethyl acetate/heptane (2:3, 2 x 90 L), and dry under vacuum at 40°C to give the title compound (29.3 kg, 70.6% yield) as a white solid. lH NMR (400 MHz, CD3OD): δ 7.14 (s, 1H), 7.07 (d, J= 8.0 Hz, 1H), 6.92 (d, J= 7.6 Hz, 1H), 6.39 (d, J= 16.0 Hz, 1H), 6.25-6.12 (m, 1H), 3.63 (s, 2H), 3.45-3.38 (bs, 3H), 3.34 (s, 3 H), 3.33 (s, 3H), 2.85-2.75 (m, 1H), 2.49-2.40 (m, 5 H), 2.33 (s, 3H), 1.68-1.62 (m, 2H), 1.60-1.36 (m, 15H), 1.11 (s, 3H), 1.10 (s, 3H).
Preparation 12b
Alterternative preparation of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but- 3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate.

Scheme 4, step B: Combine tert-butyl 4-[(E)-4-[4-[(3-hydroxy-5-isopropyl-lH- pyrazol-4-yl)methyl] -3-methyl-phenyl]but-3 -enyl] -4,9-diazaspiro [5.5]undecane-9- carboxylate (17.83 kg, 33.2 moles), acetonitrile (180 L), and benzyltributylammonium chloride (1.52 kg, 4.87 moles) at room temperature. Slowly add potassium carbonate (27.6 kg, 199.7 moles) and stir the mixture for 2 hours. Add 2,3,4,6-tetra-O-acetyl-alpha- D-glucopyranosyl bromide (24.9 kg, 60.55 mol), warm the reaction mixture to 30°C and stir for 18 hours. Concentrate the mixture under reduced pressure and add ethyl acetate (180 L), followed by water (90 L). Separate the layers, wash the organic phase with 15% brine (3 x 90 L), concentrate the mixture, and purify using column chromatography over silica gel (63 kg, ethyl acetate/heptanes as eluent (1 :2→1 :0)) to provide the title compound (19.8 kg, 94% purity, 68.8% yield) as a yellow foam, !H NMR (400 MHz, CDC13): δ 7.13 (s, 1H), 7.03 (d, J= 8.0 Hz, 1H), 6.78 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 16.0,
1H), 6.25-6.13 (m, 1H), 5.64 (d, J= 8.0 Hz, 1H), 5.45-5.25 (m, 2H), 5.13-4.95 (m, 2H), 4.84-4.76 (m, 1H), 4.25-4.13 (m, 2H), 4.10-4.00 (m, 2H), 3.90-3.86 (m, 1H), 3.58-3.50 (m, 2H), 3.40-3.22 (m, 4H), 2.89-2.79 (m, 1H), 2.10-1.90 (m, 18 H), 1.82 (s, 3H), 1.62- 0.82 (m, 22H).
Preparation 18
2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane

Scheme 4, step C: Combine tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)- 3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (19.6 kg, 22.6 moles) with dichloromethane (120 L) and cool to 0°C. Slowly add trifluoroacetic acid (34.6 L, 51.6 kg, 452 moles) and stir for 9 hours. Quench the reaction with ice water (80 L), and add ammonium hydroxide (85-90 L) to adjust the reaction mixture to pH (8- 9). Add dichloromethane (120 L), warm the reaction mixture to room temperature, and separate the layers. Wash the organic layer with water (75 L), brine, and concentrate under reduced pressure to provide the title compound (16.2 kg, 95.0% purity, 93% yield) as a yellow solid. lH NMR (400 MHz, CDC13): δ 7.08 (s, IH), 6.99 (d, J= 8.0 Hz, IH),
6.76 (d, J= 7.6 Hz, IH), 6.38 (d, J=15.6 Hz, IH), 6.00-5.83 (m, IH), 5.31 (d, J= 7.6 Hz, IH), 5.25-5.13 (m, 4H), 4.32 (dd, J= 12.8, 9.2 Hz, IH), 4.14 (d, J= 11.2 Hz, IH), 3.90 (d, J= 10.0 Hz, IH), 3.75-3.50 (m, 3H), 3.30-3.00 (m, 5 H), 2.85-2.75 (m, IH), 2.70-2.48 (m, 3H), 2.25 (s, IH), 2.13-1.63 (m, 19H), 1.32-1.21 (m, IH), 1.14 (s, 3H), 1.13 (s, 3H), 1.12 (s, 3H), 1.10 (s, 3H).
Example 1
Hydrated crystalline 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside acetate
First alternative preparation of 4-{4-[(l£’)-4-(2.9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl| -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (free base).
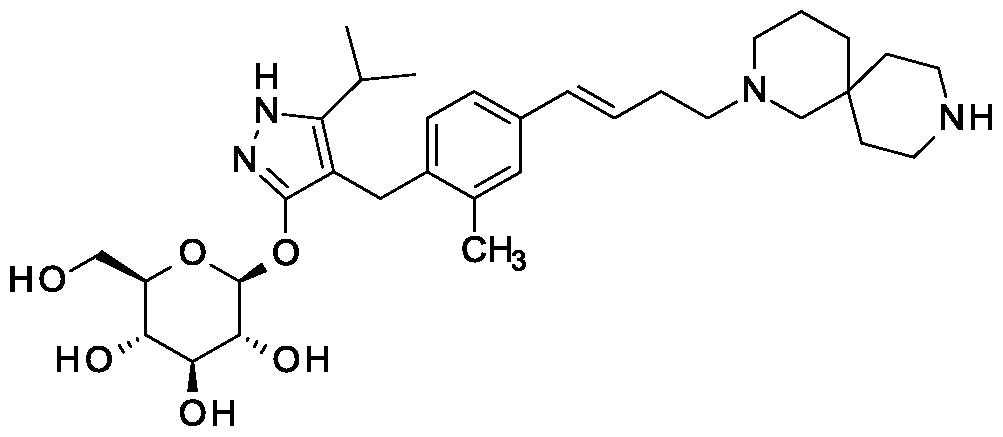
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4- {4-[( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} – 5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40°C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H.0 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Second alternative preparation of 4-{4-r(l-£’)-4-(2.9-diazaspiror5.51undec-2-yl)but-l-en- 1 -yl] -2-methylbenzyl I -5 -(propan-2-yl)- lH-pyrazol-3 -yl beta-D-glucopyranoside (free base“).

Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4- {4-[( lJE)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H20 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+l), 596.8 (M-l). 1H NMR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=l .3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.11 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Third alternative preparation of 4-{4-[(l£,)-4-(2,9-diazaspiro[5.51undec-2-yl)but-l-en-l- yll-2-methylbenzyl|-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200mL) and concentrated in vacuo. This is repeated several times to give the title compound (free base) (12.22 g, yield 96%). MS (m/z): 599 (M+l); [a]D 20 = -12 ° (C=0.2, MeOH).
Preparation of final title compound, hydrated crystalline 4-{4-|YlE)-4-(2.9- diazaspiro [5.5“|undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-vD- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.

4- {4- [(1 E)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (902 mg) is placed in a round bottom flask (100 mL) and treated with wet ethyl acetate (18 mL). [Note – wet ethyl acetate is prepared by mixing ethyl acetate (100 mL) and dionized water (100 mL). After mixing, the layers are allowed to separate, and the top wet ethyl acetate layer is removed for use. Acetic acid is a hydrolysis product of ethyl acetate and is present in wet ethyl acetate.] The compound dissolves, although not completely as wet ethyl acetate is added. After several minutes, a white precipitate forms. An additional amount of wet ethyl acetate (2 mL) is added to dissolve remaining compound. The solution is allowed to stir uncovered overnight at room temperature during which time the solvent partially evaporates. The remaining solvent from the product slurry is removed under vacuum, and the resulting solid is dried under a stream of nitrogen to provide the final title compound as a crystalline solid. A small amount of amorphous material is identified in the product by solid-state NMR. This crystalline final title compound may be used as seed crystals to prepare additional crystalline final title compound.
Alternative preparation of final title compound, hvdrated crystalline 4-{4-[(lE)-4-(2.,9- diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-yl)- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.
Under a nitrogen atmosphere combine of 4-{4-[(lE)-4-(2,9-diazaspiro[5.5]undec- 2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan-2-yl)- 1 H-pyrazol-3-yl 2,3,4,6-tetra-O- acetyl-beta-D-glucopyranoside (2.1 kg, 2.74 mol), methanol (4.4 L), tetrahydrofuran (4.2 L), and water (210 mL). Add potassium carbonate (460 g, 3.33 moles) and stir for four to six hours, then filter the reaction mixture to remove the solids. Concentrate the filtrate under reduced pressure, then add ethanol (9.0 L) followed by acetic acid (237 mL, 4.13 mol) and stir at room temperature for one hour. To the stirring solution add wet ethyl acetate (10 L, containing approx. 3 w/w% water) slowly over five hours, followed by water (500 mL). Stir the suspension for twelve hours and add wet ethyl acetate (4.95 L, containing approx. 3 w/w% water) over a period of eight hours. Stir the suspension for twelve hours and add additional wet ethyl acetate (11.5 L, containing approx. 3 w/w% water) slowly over sixteen hours. Stir the suspension for twelve hours, collect the solids by filtration and rinse the solids with wet ethyl acetate (3.3 L, containing approx. 3 w/w% water). Dry in an oven under reduced pressure below 30°C to give the title compound as an off-white crystalline solid (1.55 kg, 2.35 mol, 96.7% purity, 72.4 w/w% potency, 68.0% yield based on potency). HRMS (m/z): 599.3798 (M+l).
PATENT
CN105705509
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN175101669&tab=PCTDESCRIPTION
The present invention is in the field of treatment of diabetes and other diseases and conditions associated with hyperglycemia. Diabetes is a group of diseases characterized by high blood sugar levels. It affects approximately 25 million people in the United States, and according to the 2011 National Diabetes Bulletin, it is also the seventh leading cause of death in the United States (US Department of Health and Human Resources Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the uptake of carbohydrates such as glucose. More specifically, SGLT1 is responsible for transporting glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 can result in a decrease in glucose absorption in the small intestine, thus providing a useful method of treating diabetes.
Alternative medicines and treatments for diabetes are needed. The present invention provides an acetate salt of a pyrazole compound which is an SGLT1 inhibitor, and thus it is suitable for treating certain conditions such as diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives having human SGLT1 inhibitory activity, which are also disclosed for use in the prevention or treatment of diseases associated with hyperglycemia, such as diabetes. Moreover, WO 2011/039338 discloses certain pyrazole derivatives having SGLT1/SGLT2 inhibitor activity, which are also disclosed for use in the treatment of bone diseases such as osteoporosis.
PATENT
WO-2019141209
Diabetes is a group of lifelong metabolic diseases characterized by multiple causes of chronic hyperglycemia. Long-term increase in blood glucose can cause damage to large blood vessels and microvessels and endanger the heart, brain, kidney, peripheral nerves, eyes, feet and so on. According to the statistics of the World Health Organization, there are more than 100 complications of diabetes, which is the most common complication, and the incidence rate is also on the rise. The kidney plays a very important role in the body’s sugar metabolism. Glucose does not pass through the lipid bilayer of the cell membrane in the body, and must rely on the glucose transporter on the cell membrane. Sodium-coupled glucose co-transporters (SGLTs) are one of the transporters known to be responsible for the uptake of carbohydrates such as glucose. More specifically, SGLT1 is responsible for transporting glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 results in a decrease in glucose absorption in the small intestine and can therefore be used in the treatment of diabetes.
Ellerelli has developed a novel SGLTs inhibitor for alternative drugs and treatments for diabetes. CN105705509 discloses the SGLTs inhibitor-pyrazole compound, which has the structure shown in the following formula (1):
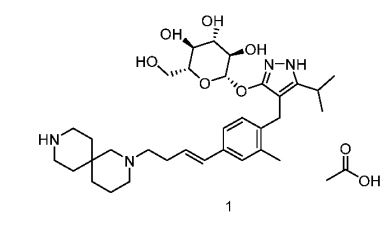
It is well known for drug production process has strict requirements, the purity of pharmaceutical active ingredients will directly affect the safety and effectiveness of drug quality. Simplified synthetic route optimization, and strictly control the purity of the intermediates has a very important role in improving drug production, quality control and optimization of the dosage form development.
CN105705509 discloses a method for synthesizing a compound of the formula (1), wherein the intermediate compound 2-{(3E)-4-[3-methyl4-({5-(propyl-2-yl)) is obtained by the step B in Scheme 4. -3-[(2,3,4,6-tetra-acetyl-β-D-glucopyranosyl)oxy]-1H-pyrazol-4-yl}methyl)phenyl]but-3- Tert-butyl-1-enyl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (Compound obtained in Preparation Example 12b) was obtained as a yellow foam, yield 68.6%, purity 94 %, this step involves silica gel column purification, low production efficiency, high cost, and poor quality controllability; the intermediate 2-{(3E)-4-[3-methyl 4-({5- (prop-2-yl)-3-[(2,3,4,6-tetra-acetyl-β-D-glucopyranosyl)oxy)-1H-pyrazol-4-yl}methyl) Phenyl]but-3-en-1-yl}-2,9-diazaspiro[5.5]undecane (Compound obtained in Preparation Example 18) as a yellow solid with a purity of 95.0%; The resulting intermediate compounds were all of low purity. Moreover, CN105705509 produces a compound of formula (1) having a purity of 96.7% as described in the publications of the publications 0141 and 0142. The resulting final compound is not of high purity and is not conducive to subsequent drug preparation.
Process for preparing pyranoglucose-substituted pyrazole compound, used as a pharmaceutical intermediate in SGLT inhibitor for treating diabetes.

Example 1
626 g of the compound of the formula (16), 6 L of acetonitrile, 840 g of cesium carbonate and 1770 g of 2,3,4,6-tetra-O-pivaloyl-α-D-glucosyl bromide (formula (17) The compound is sequentially added to the reaction vessel, heated to 40 ° C to 45 ° C, and reacted for 4 to 5 hours, then cooled to 20 to 25 ° C, filtered, and the obtained solid is rinsed once with acetonitrile; the filter cake is dissolved with 8 L of ethyl acetate and 10 L of water. After the liquid separation, the organic phase was concentrated to about 3 L, 10 L of acetonitrile was added, and the mixture was stirred for 12 h to precipitate a solid, which was filtered. The filter cake was rinsed with acetonitrile and dried under vacuum at 60 ° C for 24 h to give white crystals, 652 g of compound of formula (9c). The yield was 61%, the HPLC purity was 98.52%, and the melting point was 180.0-182.1 °C. 1 H NMR (400 MHz, MeOD) (see Figure 1): δ 7.10 (s, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.39 (d, J=15.6,1H), 6.19-6.12 (m,1H), 5.59 (d, J=8.4 Hz, 1H), 5.40-5.35 (t, J=9.6 Hz, 1H), 5.17-5.06 (m, 2H) , 4.18-4.14 (dd, J = 12.4 Hz, 4.4 Hz, 1H), 4.10-4.06 (dd, J = 12.4 Hz, 1.6 Hz, 1H), 3.92-3.89 (dd, J = 10 Hz, 2.4 Hz, 1H) , 3.64-3.54 (dd, J=20 Hz, 16.8 Hz, 2H), 3.31-3.30 (m, 4H), 2.86-2.79 (m, 1H), 2.37-2.29 (m, 11H), 1.63-1.38 (m, 17H), 1.15-1.05 (m, 42H). MS (m/z): 1035.7 (M+H).
640 g of the compound of the formula (9c) and 6.4 L of ethyl acetate were successively added to the reaction vessel, and the temperature was lowered to 15 ° C to 20 ° C. 1176 g of p-toluenesulfonic acid monohydrate was added in portions for 2 to 3 hours; after the reaction was over, 3.5 L of a 9% potassium hydroxide aqueous solution was added, and the mixture was stirred for 10 minutes, and the aqueous phase was discarded. The organic phase was washed successively with 3.5 L of 9% and 3.5 L of 3% aqueous potassium hydroxide and concentrated to 2.5 L. 21L of n-heptane was added to the residue, and the mixture was stirred for 12 hours; filtered, and the filter cake was rinsed with n-heptane; the filter cake was dried under vacuum at 60 ° C for 24 h to obtain white crystals, p-toluene of the compound of formula (10c). The sulfonate salt was 550 g, the yield was 80%, the purity was 97.59%, and the melting point was 168.0-169.2 °C. 1 H NMR (400 MHz, MeOD) (see Figure 2): δ 7.72 (d, J = 7.6 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.10 (s, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.39 (d, J = 15.6, 1H), 6.19-6.12 (m, 1H), 5.60 (d, J = 8.0 Hz, 1H) ), 5.41-5.37 (t, J = 9.6 Hz, 1H), 5.17-5.06 (m, 2H), 4.18-4.14 (dd, J = 12.4 Hz, 4.0 Hz, 1H), 4.10-4.07 (d, J = 11.6Hz, 1H), 3.94-3.91 (dd, J=7.2Hz, 2.8Hz, 1H), 3.64-3.54 (dd, J=20.0Hz, 16.8Hz, 2H), 3.31-3.30 (m, 4H), 2.86 -2.79 (m, 1H), 2.49-2.29 (m, 14H), 1.78-1.44 (m, 8H), 1.15-1.05 (m, 42H). MS (m/z): 935.7 (M+H).
82.6 g of potassium hydroxide, 5.5 L of absolute ethanol and 550 g of the p-toluenesulfonate of the compound of the formula (10c) were sequentially added to the reaction vessel, and stirred at 45 to 50 ° C for about 4 hours. The temperature was lowered to 20 to 25 ° C, filtered, and the solid was rinsed with ethanol. The filtrate and the eluent were combined, and 65 g of acetic acid was added thereto, followed by stirring for 15 min. The reaction solution was concentrated under reduced pressure to about 1.5 L, and then 52 g of acetic acid was added. After stirring for 20 min, 4.5 L of ethyl acetate containing 3% water and 160 mL of purified water were added dropwise. After the dropwise addition, continue stirring for 3 to 4 hours. Filter and filter cake was rinsed with ethyl acetate containing 3% water. The solid was transferred to a reaction kettle, 500 mL of water was added and stirred for 18 h. After filtration, the filter cake was washed successively with water and an ethanol/ethyl acetate mixed solvent. The filter cake was dried under vacuum at 35 to 40 ° C for 4 hours to obtain a white solid, 245 g of compound of formula (1), yield 75%, purity 99.55%. 1 H NMR (400 MHz, MeOD) (see Figure 3): δ 7.11 (s, 1H), 7.05 (d, J = 7.6 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 6.39 (d, J=16.0,1H), 6.20-6.13 (dt, J=15.6 Hz, 6.8 Hz, 1H), 5.03-5.01 (m, 1H), 3.83 (d, J=11.2, 1H), 3.71-3.59 (m, 3H), 3.35-3.30 (m, 4H), 3.09-3.06 (t, J = 6 Hz, 4H), 2.87-2.77 (m, 1H), 2.49-2.31 (m, 6H), 2.30 (s, 3H), 2.26(s, 2H), 1.90 (s, 3H), 1.78 (m, 2H), 1.68 (m, 2H), 1.65 (m, 2H), 1.44-1.43 (m, 2H), 1.13 (d, J = 6.8 Hz, 3H), 1.11 (d, J = 6.8 Hz, 3H), MS (m/z): 599.5 (M+H).
Example 2
5.00 kg of the maleate salt of the compound of the formula (16), 40 L of tetrahydrofuran, 5.47 kg of potassium phosphate and 11.67 kg of 2,3,4,6-tetra-O-pivaloyl-α-D-glucosyl bromide The compound (formula (17)) is sequentially added to the reaction vessel, heated to 40 to 45 ° C, and reacted for 4 to 5 hours, then cooled to 15 to 25 ° C, filtered, and the solid was rinsed once with tetrahydrofuran. The filter cake was dissolved in 36 L of ethyl acetate and 20 L of water and then separated. The organic phase was concentrated to ca. 18 L, 64 L acetonitrile was added and stirred for 15 h. Filtration, the filter cake was rinsed with acetonitrile, and dried under vacuum at 60 ° C for 24 h to give white crystals of the compound of formula (9c), 4.50 kg, yield 57%, HPLC purity 99.19%.
4.45 kg of the compound of the formula (9c) and 45 L of butyl acetate were sequentially added to the reaction vessel, and the temperature was lowered to 15 ° C to 20 ° C. 4.13 kg of methanesulfonic acid was added in portions and the reaction was carried out for 2 to 3 hours. 22 L of a 9% aqueous potassium hydroxide solution was added, stirred for 10 min, and the liquid phase was discarded. The organic phase was washed successively with 10 L of 9%, 4.5 L of 10% and 2 L of 2.5% aqueous potassium hydroxide and concentrated to 15 L. 68 L of n-heptane was added to the residue, and the mixture was stirred for further 12 h. Filtered and the filter cake was rinsed once with n-heptane. The solid was dried under vacuum at 60 ° C for 24 h to obtain white crystals. The methanesulfonic acid salt of the compound of formula (10c) was 4.37 kg, yield 99%, purity 97.94%.
0.73 kg of potassium hydroxide, 43 L of methanol and 4.30 kg of the compound of the formula (10c) were sequentially added to the reaction vessel, and stirred at 45 to 50 ° C for 4 hours. The temperature was lowered to 20 to 25 ° C, filtered, and 0.56 kg of acetic acid was added to the filtrate, and the mixture was stirred for 15 minutes. The reaction solution was concentrated to about 15 L under reduced pressure, and 0.40 g of acetic acid was added. After stirring for 10 min, 39 L of 3% water in ethyl acetate and 1.3 L of purified water were added dropwise. After the dropwise addition, stirring was continued for about 2 hours. Filter and filter cake was rinsed once with ethyl acetate containing 3% water. The solid was transferred to a reaction kettle, and 3.5 L of water was added and stirred for 18 h. After filtration, the filter cake was washed successively with water and an ethanol/ethyl acetate mixed solvent. The cake was vacuum dried at 35 to 40 ° C to give a white solid. Compound (1) (1), 1.84 g, yield 67%, purity 99.65%.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9573970 | 4–5-(PROPAN-2-YL)-1H-PYRAZOL-3-YL BETA-D GLUCOPYRANOSIDE ACETATE | 2014-10-30 | 2016-07-28 |
/////////////SY-008 , SY 008 , SY008, ELI LILY, PHASE 1, GLT1 inhibitor, type 2 diabetes, Yabao Pharmaceutical, CHINA, DIABETES
CC(=O)O.Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4O
Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4
O
CC-90009

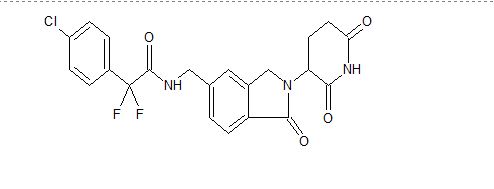
CC-90009
CC-90009-AML-001
CAS 1860875-51-9
461.8 g/mol, C22H18ClF2N3O4
2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide
- 4-Chloro-N-[[2-(2,6-dioxo-3-piperidinyl)-2,3-dihydro-1-oxo-1H-isoindol-5-yl]methyl]-α,α-difluorobenzeneacetamide
- Benzeneacetamide, 4-chloro-N-[[2-(2,6-dioxo-3-piperidinyl)-2,3-dihydro-1-oxo-1H-isoindol-5-yl]methyl]-α,α-difluoro-
Phase 1 Clinical, Acute myelogenous leukemia, Protein cereblon modulator
Useful for treating chronic lymphocytic leukemia, chronic myelocytic leukemia, acute lymphoblastic leukemia or acute myeloid leukemia.
Celgene is developing CC-90009, a cereblon E3 ligase modulator, for treating AML; in January 2019, data from a phase I trial were expected later that year.
- 0iginator Celgene Corporation
- Class Antineoplastics
- Mechanism of Action CRBN protein modulators; Ubiquitin protein ligase complex modulators
- Phase I Acute myeloid leukaemia
- 28 Mar 2019 No recent reports of development identified for clinical-Phase-Unknown development in Acute-myeloid-leukaemia in USA (IV)
- 01 Sep 2016 Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in Canada (IV) (NCT02848001)
- 04 Aug 2016 Celgene plans a phase I trial for Acute Myeloid Leukaemia in USA and Canada (NCT02848001)
In September 2016, Celgene initiated a phase I dose-finding trial of CC 90009 in patients with relapsed or refractory acute myeloid leukaemia (NCT02848001; CC-90009-AML-001). The open-label study intends to enrol 60 patients in the US and Canada
CC-90009 is a cereblon modulator. CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. .
Development Overview
cereblon modulator CC-90009A modulator of cereblon (CRBN), which is part of the cullin 4-RING E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase; CUL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and pro-apoptotic activities. Upon administration, CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. In addition, this downregulates the expression of other proteins, including interferon regulatory factor 4 (IRF4) and c-myc, which plays a key role in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins. Check for active clinical trials using this agent. (NCI Thesaurus)

WO 2017120446,
PATENT
WO2016007848
US 20170348298
WO 2017120415
WO 2017120446
WO 2017120437
PATENT
WO2017214014
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017214014&tab=PCTDESCRIPTION
Provided herein are methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy with 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of
stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof. Further provided is a compound for use in methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy, wherein the compound is 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof.
The term Compound 1 refers to”2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide” having the structure:
and its stereoisomers or mixture of stereoisomers, isotopologues, pharmaceutically acceptable salts, tautomers, solvates, hydrates, co-crystals, clathrates, or polymorphs thereof. In certain embodiments, Compound 1 refers to 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide and its tautomers. In certain embodiments, Compound 1 refers to a polymorph of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-
oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In certain embodiments, Compound 1 refers to polymorph Form C of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In one embodiment, the stereoisomer is an enantiomer.
PATENT
WO-2019136016
Novel isotopologs of the compound presumed to be CC-90009 , processes for their preparation and compositions comprising them are claimed.



| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017199193 | METHODS FOR TREATING CANCER AND THE USE OF BIOMARKERS AS A PREDICTOR OF CLINICAL SENSITIVITY TO THERAPIES | 2017-01-06 | |
| US2018224435 | METHODS FOR MEASURING SMALL MOLECULE AFFINITY TO CEREBLON | 2018-02-02 | |
| US2018353496 | FORMULATIONS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE | 2018-07-19 | |
| US2017196847 | FORMULATIONS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE | 2017-01-06 | |
| US2017348298 | TREATMENT OF A HEMATOLOGIC MALIGNANCY WITH 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE | 2017-06-05 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2018221361 | ANTIPROLIFERATIVE COMPOUNDS AND METHODS OF USE THEREOF | 2018-04-09 | |
| US9968596 | Antiproliferative compounds and methods of use thereof | 2017-10-02 | 2018-05-15 |
| US2017197934 | SOLID FORMS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE, AND THEIR PHARMACEUTICAL COMPOSITIONS AND USES | 2017-01-06 | |
| US9499514 | ANTIPROLIFERATIVE COMPOUNDS AND METHODS OF USE THEREOF | 2015-07-09 | 2016-01-14 |
| US9808451 | ANTIPROLIFERATIVE COMPOUNDS AND METHODS OF USE THEREOF | 2016-09-23 |
////////CC-90009 , CC 90009 , CC90009, chronic lymphocytic leukemia, chronic myelocytic leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, phase I, CANCER, CC-90009-AML-001
Clc1ccc(cc1)C(F)(F)C(=O)NCc2ccc3C(=O)N(Cc3c2)C4CCC(=O)NC4=O
Picropodophyllin
Picropodophyllin
Picropodophyllotoxin
CAS 477-47-4
AXL1717, NSC 36407, BRN 0099161
414.4 g/mol, C22H22O8
(5R,5aR,8aS,9R)-5-hydroxy-9-(3,4,5-trimethoxyphenyl)-5a,6,8a,9-tetrahydro-5H-[2]benzofuro[5,6-f][1,3]benzodioxol-8-one
Furo(3′,4′:6,7)naphtho(2,3-d)-1,3-dioxol-6(5aH)-one, 5,8,8a,9-tetrahydro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-, (5R-(5-alpha,5a-alpha,8a-alpha,9-alpha))-
5-19-10-00665 (Beilstein Handbook Reference)
Axelar is developing picropodophyllin, a small-molecule IGF-1 receptor antagonist for the treatment of cancer including NSCLC and malignant astrocytoma. In February 2019, a phase Ia study was planned to initiate for solid tumor in March 2019.
Picropodophyllin is a cyclolignan alkaloid found in the mayapple plant family (Podophyllum peltatum), and a small molecule inhibitor of the insulin-like growth factor 1 receptor (IGF1R) with potential antineoplastic activity. Picropodophyllin specifically inhibits the activity and downregulates the cellular expression of IGF1R without interfering with activities of other growth factor receptors, such as receptors for insulin, epidermal growth factor, platelet-derived growth factor, fibroblast growth factor and mast/stem cell growth factor (KIT). This agent shows potent activity in the suppression o f tumor cell proliferation and the induction of tumor cell apoptosis. IGF1R, a receptor tyrosine kinase overexpressed in a variety of human cancers, plays a critical role in the growth and survival of many types of cancer cells.
Picropodophyllotoxin is an organic heterotetracyclic compound that has a furonaphthodioxole skeleton bearing 3,4,5-trimethoxyphenyl and hydroxy substituents. It has a role as an antineoplastic agent, a tyrosine kinase inhibitor, an insulin-like growth factor receptor 1 antagonist and a plant metabolite. It is a lignan, a furonaphthodioxole and an organic heterotetracyclic compound.
Picropodophyllin has been investigated for the treatment of Non Small Cell Lung Cancer.
One of the largest challenges in pharmaceutical drug development is that drug compounds often are poorly soluble, or even insoluble, in aqeous media. Insufficient drug solubility means insufficient bioavailability, as well as poor plasma exposure of the drug when administered to humans and animals. Variability of plasma exposure in humans is yet a problem when developing drugs which are poorly soluble, or even insoluble, in aqeous media.
It is estimated that between 40% and 70 % of all new chemical entities identified in drug discovery programs, are insufficiently soluble in aqeous media (M. Lindenberg, S et al: European Journal of Pharmaceutics and Biopharmaceuticals, vol. 58, no.2, pp. 265-278, 2004). Scientists have investigated various ways of solving the problem with poor drug solubility in order to enhance bioavailability of poorly absorbed drugs, aiming at increasing their clinical efficacy when administered orally.
Technologies such as increase of the surface area and hence dissolution may sometimes solve solubility problems. Other techniques that may also solve bioavailability problems are addition of surfactants and polymers. However, each chemical compound has its own unique chemical and physical properties, and hence has its own unique challenges when being formulated into a pharmaceutical product that can exert its clinical efficacy.
Picropodophyllin is an insulin-like growth factor-1 receptor inhibitor fiGF-lR inhibitor) small-molecule compound belonging to the class of compounds denominated cyclolignans, having the chemical structure:
The patent applicant is presently entering clinical phase II development with its development compound picropodophyllin (AXL1717). However, picropodophyllin is poorly soluble in aqueous media. In a phase I clinical study performed by the applicant in 2012 (Ekman S et al; Acta Oncologica, 2016; 55: pp. 140-148), it was discovered that picropodophyllin, when administered as an oral suspension to lung cancer patients, resulted in unacceptable variability in drug exposure. A large variability in plasma exposure of the active drug picropodophyllin occurred not only within certain patients, but also between several patients.
Yet a problem with administering picropodophyllin as an aqeous solution, is that due to the poor solubility in aqueous media, it is difficult or even impossible to reach the required therapeutic doses.
The compound picropodophyllin is furthermore physically unstable, and transforms from amorphous picropodophyllin into crystalline picropodophyllin. Yet a stability problem with picropodophyllin is that it is chemically unstable in solution.

Product case, WO02102804
Patent
WO-2019130194
Novel amorphous forms of picropodophyllin , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating cancers, such as neurologic cancer, lung cancer, breast cancer, head and neck cancer, gastrointestinal cancer, genitourinary cancer, gynecologic cancer, hematologic cancer, musculoskeletal cancer, skin cancer, endocrine cancer, and eye cancers. , claiming picropodophyllin derivatives as modulators of insulin-like growth factor-1 receptor (IGF-1), useful for treating cancers, assigned to Axelar AB ,
CLIP

CLIP
https://pubs.rsc.org/en/content/articlelanding/2004/cc/b312245j/unauth#!divAbstract
http://www.rsc.org/suppdata/cc/b3/b312245j/b312245j.pdf
dH(CDCl3; 300 MHz; Me4Si): 2.64-2.78 (1 H, m, 3-H), 3.23 (1 H, dd, J 4.4 and 8.2, 2-H), 3.81 (6 H, s, 2 x OMe), 3.85 (3 H, s, OMe), 4.09 (1 H, d, J 4.4, 1-H), 4.38–4.59 (3 H, m, 11-H2 and 4-H), 5.91 (1 H, d, J 1.5, OCH2O), 5.93 (1 H, d, J 1.5, OCH2O), 6.35 (1 H, s, 5-H/8-H), 6.46 (1 H, s, 2’-H and 6’-H) and 7.07 (1 H, s, 5-H/8-H).
CLIP
PAPER
Organic Letters (2018), 20(6), 1651-1654
https://pubs.acs.org/doi/abs/10.1021/acs.orglett.8b00408

A nickel-catalyzed reductive cascade approach to the efficient construction of diastereodivergent cores embedded in podophyllum lignans is developed for the first time. Their gram-scale access paved the way for unified syntheses of naturally occurring podophyllotoxin and other members.
Synthesis of (−)-Podophyllotoxin (1)
https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.8b00408/suppl_file/ol8b00408_si_001.pdf
The residue was purified by flash column chromatography (petroleum ether/EtOAc = 4 : 1 → petroleum ether/EtOAc = 2 : 1) on silica gel to afford 1 (8.6 mg, 87% yield) as a white solid; Rf = 0.23 (petroleum ether/EtOAc = 1 : 1); [α]20 D = –115.00 (c = 1.00, CHCl3) [ref.13: [α]20 D = –101.7 (c = 0.55, EtOH)]; Mp. 167–168 °C; 1H NMR (400 MHz, CDCl3): δ = 7.11 (s, 1H), 6.51 (s, 1H), 6.37 (s, 2H), 5.98 (s, 1H), 5.96 (s, 1H), 4.77 (t, J = 8.4 Hz, 1H), 4.60 (t, J = 8.0 Hz, 1H), 4.59 (d, J = 4.4 Hz, 1H), 4.08 (dd, J = 9.6, 8.8 Hz, 1H), 3.81 (s, 3H), 3.75 (s, 6H), 2.84 (dd, J = 14.0, 4.4 Hz, 1H), 2.83−2.74 (m, 1H), 2.13 (d, J = 8.0 Hz, 1H, −OH) ppm; 13C NMR (100 MHz, CDCl3): δ = 174.6, 152.5 (2C), 147.7, 147.6, 137.1, 135.5, 133.3, 131.0, 109.7, 108.4 (2C), 106.3, 101.4, 72.6, 71.4, 60.7, 56.2 (2C), 45.2, 44.1, 40.6 ppm.

https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.8b00408/suppl_file/ol8b00408_si_002.pdf


PAPER
Organic Letters (2017), 19(24), 6530-6533
https://pubs.acs.org/doi/abs/10.1021/acs.orglett.7b03236

he first catalytic enantioselective total synthesis of (−)-podophyllotoxin is accomplished by a challenging organocatalytic cross-aldol Heck cyclization and distal stereocontrolled transfer hydrogenation in five steps from three aldehydes. Reversal of selectivity in hydrogenation led to the syntheses of other stereoisomers from the common precursor.
https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.7b03236/suppl_file/ol7b03236_si_001.pdf
(-)-Picropodophyllin 4. The lactone 5 (0.2 g, 0.38 mmol) was taken in 1-pentanol (5 mL) in a double neck RB flask at rt. Water (0.14 mL, 7.6 mmol) was added to above mixture and it was then degassed with argon followed by addition of Pd/C (0.04 g, 20% by wt.) and HCO2Na (0.78g, 11.4 mmol). The reaction mixture was heated at 40 °C for 12 h. On completion, the reaction mixture was diluted with EtOAc (200 mL), filtered through a celite pad and solvent was removed under vacuum. This crude mixture was dissolved in THF (3.8 mL), TBAF (1.9 mL, 1.9 mmol, 1M in THF) was added and stirred for 6 h at 27 °C. On completion, EtOAc (250 mL) was added, washed with water (100 mL), brine and dried over Na2SO4. After removal of solvent, the crude product was purified by column chromatography (hexanes-EtOAc, 3:2) to get the title compound as a white solid (0.082 g, 52%): Rf 0.32 (hexanes/EtOAc, 1:1); [α]25 D = -10.6 (c = 0.4, CHCl3) [lit. -10 (c = 0.3, CHCl3), -11 (c = 0.41, CHCl3)]3a,b;
Mp 214-216 °C; 1H NMR (600 MHz, CDCl3) δ 7.05 (s, 1H), 6.47 (s, 2H), 6.41 (s, 1H), 5.95 (d, J = 14.1 Hz, 2H), 4.5 (m, 2H), 4.44 (t, J = 8.0 Hz, 1H), 4.15 (d, J = 4.1 Hz, 1H), 3.86 (s, 3H), 3.83 (s, 6H), 3.24 (dd, J = 8.7, 5.0 Hz, 1H), 2.75 (m, 1H), 2.12 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 177.6, 153.7, 147.5, 147.1, 139.3, 137.4, 131.9, 130.6, 109.3, 105.9, 105.5, 101.2, 69.8, 69.6, 60.9, 56.3, 45.4, 44.1, 42.7; HRMS (ESI-TOF) m/z 437.1219 [(M+Na)+ ; calcd for C22H22O8Na+ : 437.1212].
PAPER
The Journal of organic chemistry (2000), 65(3), 847-60.
https://pubs.acs.org/doi/abs/10.1021/jo991582+

REF
Berichte der Deutschen Chemischen Gesellschaft [Abteilung] B: Abhandlungen (1932), 65B, 1846.
Justus Liebigs Annalen der Chemie (1932), 499, 59-76.
Justus Liebigs Annalen der Chemie (1932), 494, 126-42.
Journal of the American Chemical Society (1954), 76, 5890-1
Helvetica Chimica Acta (1954), 37, 190-202.
Journal of the American Chemical Society (1988), 110(23), 7854-8.
//////////////Picropodophyllin, AXL1717, NSC 36407, BRN 0099161, Picropodophyllotoxin, AXELAR, PHASE 1, CANCER, neurologic cancer, lung cancer, breast cancer, head and neck cancer, gastrointestinal cancer, genitourinary cancer, gynecologic cancer, hematologic cancer, musculoskeletal cancer, skin cancer, endocrine cancer, eye cancers, NSCLC, malignant astrocytoma, SOLID TUMOUR
COC1=CC(=CC(=C1OC)OC)C2C3C(COC3=O)C(C4=CC5=C(C=C24)OCO5)O
Podofilox, Podophyllotoxin, Wartec, Condyline, Condylox
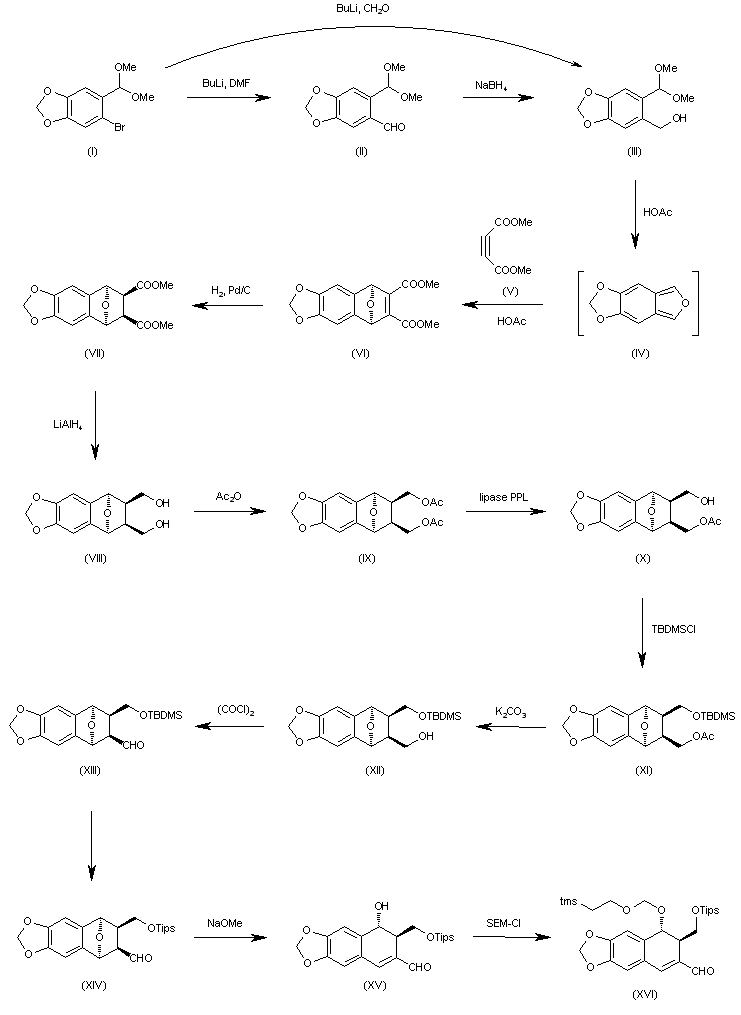
| J Org Chem 2000,65(3),847 |
The formylation of 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) with BuLi and DMF gives the 6-formyl derivative (II), which is reduced with NaBH4 in ethanol to yield the corresponding carbinol (III). The cyclization of (III) with dimethyl acetylenedicarboxylate (V) in hot acetic acid (through the nonisolated intermediate (IV)) affords dimethyl 1,4-epoxy-6,7-(methylenedioxy)naphthalene-2,3-dicarboxylate (VI), which is hydrogenated with H2 over Pd/C in ethyl acetate to give the (1R*,2S*,3R*,4S*)-tetrahydro derivative (VII). The reduction of (VII) with LiAlH4 in refluxing ethyl ether affords the corresponding bis carbinol (VIII), which is treated with acetic anhydride to afford the diacetate (IX). The enzymatic monodeacetylation of (VIII) with PPL enzyme in DMSO/buffer gives (1R,2R,3S,4S)-2-(acetoxymethyl)-1,4-epoxy-3-(hydroxymethyl)-6,7-(methylenedioxy)-1,2,3,4-tetrahydronaphthalene (X), which is silylated with TBDMS-Cl and imidazole in DMF yielding the silyl ether (XI). The hydrolysis of the acetoxy group of (XI) with K2CO3 in methanol affords the carbinol (XII), which is oxidized with oxalyl chloride in dichloromethane affording the carbaldehyde (XIII). The exchange of the silyl protecting group of (XIII) (for stability problems) provided the triisopropylsilyl ether (XIV), which is treated with sodium methoxide in methanol to open the epoxide ring yielding the hydroxy aldehyde (XV). The protection of the hydroxy group of (XV) with 2-(trimethylsilyl)ethoxymethyl chloride and DIEA in dichloromethane provides the corresponding ether (XVI). The carbinol (III) can also be obtained directly from 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) by reaction with formaldehyde and BuLi in THF.
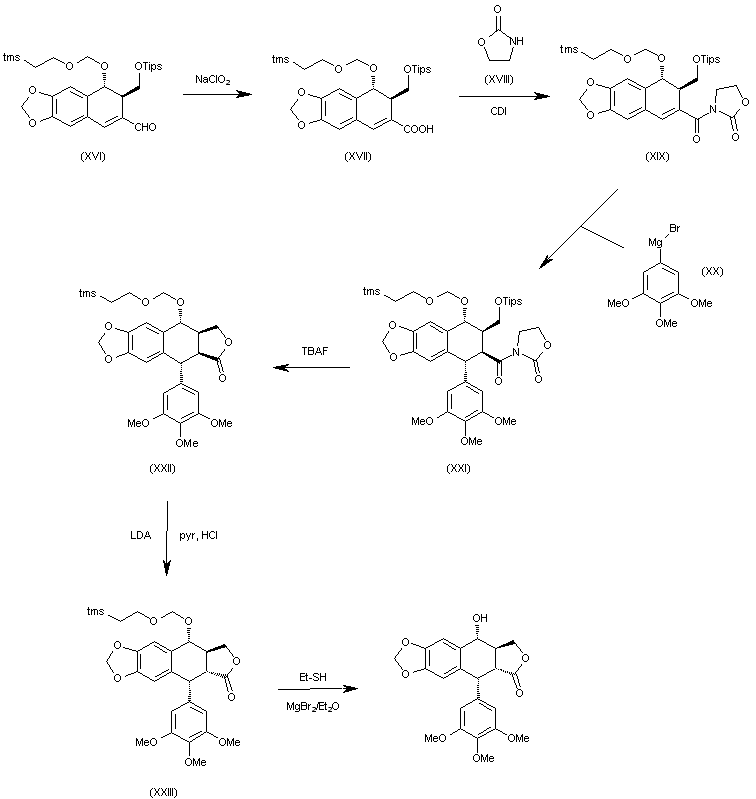
The oxidation of the aldehyde group of (XVI) with NaClO2 in tert-butanol affords the corresponding carboxylic acid (XVII), which is condensed with 2-oxazolidinone (XVIII) by means of carbonyldiimidazole (CDI) in THF to give the acyl imidazolide (XIX). The arylation of (XIX) with 3,4,5-trimethoxyphenylmagnesium bromide (XX) in THF yields the expected addition product (XXI), which is cyclized by means of TBAF in hot THF to afford the tetracyclic intermediate (XXII). Isomerization of the cis-lactone ring of (XXII) with LDA in THF affords intermediate (XXIII) with its lactone ring with the correct trans-conformation. Finally, this compound is deprotected with ethyl mercaptane and MgBr2 in ethyl ether to provide the target compound.
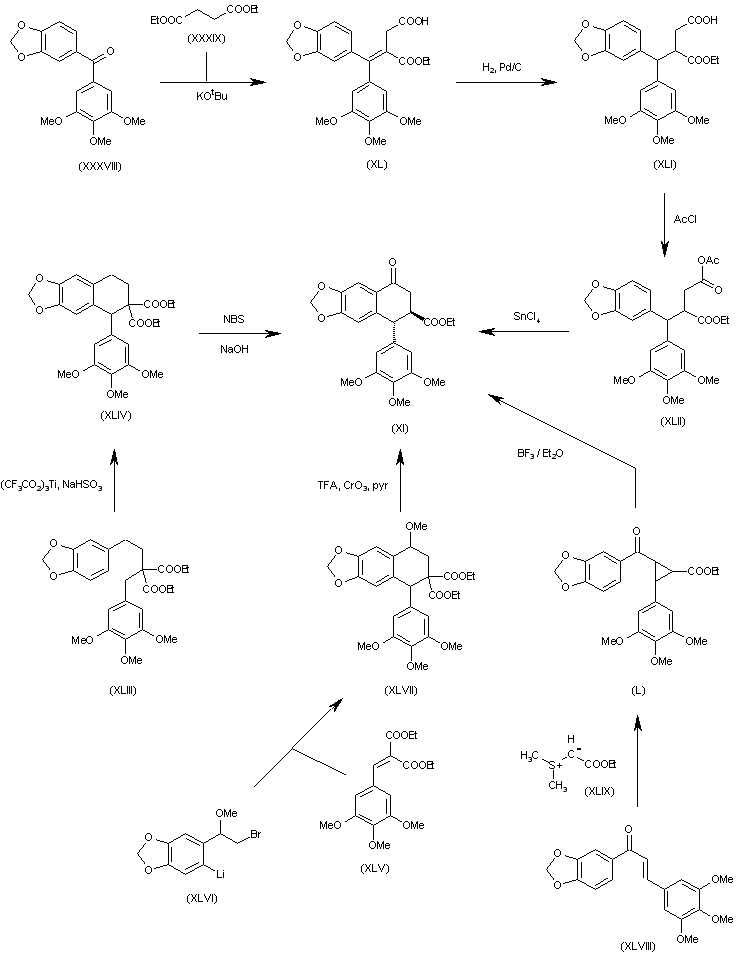
Synthesis 1992,719
The intermediate trans-8-oxo-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetra-hydronaphtho[2,3-d][1,3]benzodioxole-6-carboxylic acid ethyl ester (XI) has been obtained by several different ways: (a) The condensation of benzophenone (XXXVIII) with diethyl malonate (XXXIX) by means of t-BuOK gives the alkylidenemalonate (XL), which is hydrogenated with H2 over Pd/C to the alkylmalonate hemiester (XLI). The reaction of (XLI) with acetyl chloride affords the mixed anhydride (XLII), which is finally cyclized to the target (XI) by means of SnCl4. (b) The cyclization of the malonic ester derivative (XLIII) by means of Ti(CF3–CO2)3 gives the 5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho [2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLIV), which is finally oxidized and decarboxylated with NBS and NaOH in methanol to afford the target intermediate (XI). (c) The cyclization of the benzylidenemalonate (XLV) with the aryllithium derivative (XLVI) gives the 8-methoxy-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho[2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLVII), which is demethylated with TFA and oxidized with CrO3 and pyridine to the target compound (XI). (d) The cyclopropanation of the chalcone (XLVIII) with (ethoxycarbonyl) (dimethylsulfonium)methylide (XLIX) gives the cyclopropanecarboxylate (L), which is finally rearranged with BF3/Et2O to the target intermediate (IX).
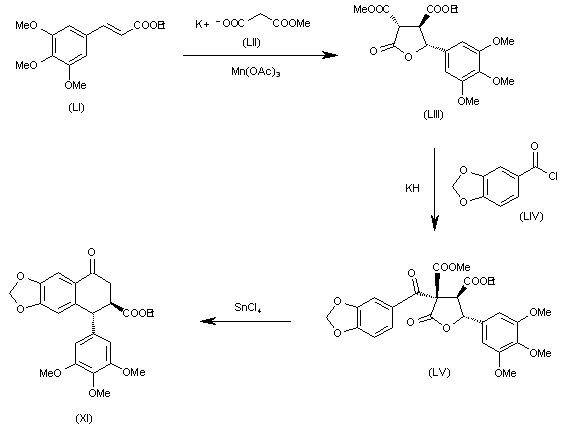
The cyclization of 3,4,5-trimethoxycinnamic acid ethyl ester (LI) with malonic acid ethyl ester potassium salt (LII) by means of Mn(OAc)3 gives the tetrahydrofuranone (LIII), which is acylated with 1,3-benzodioxol-5-ylcarbonyl chloride (LIV) yielding the tetrahydrofuranone (LV). Finally, this compound is rearranged and decarboxylated with SnCl4 to the target intermediate (XI).
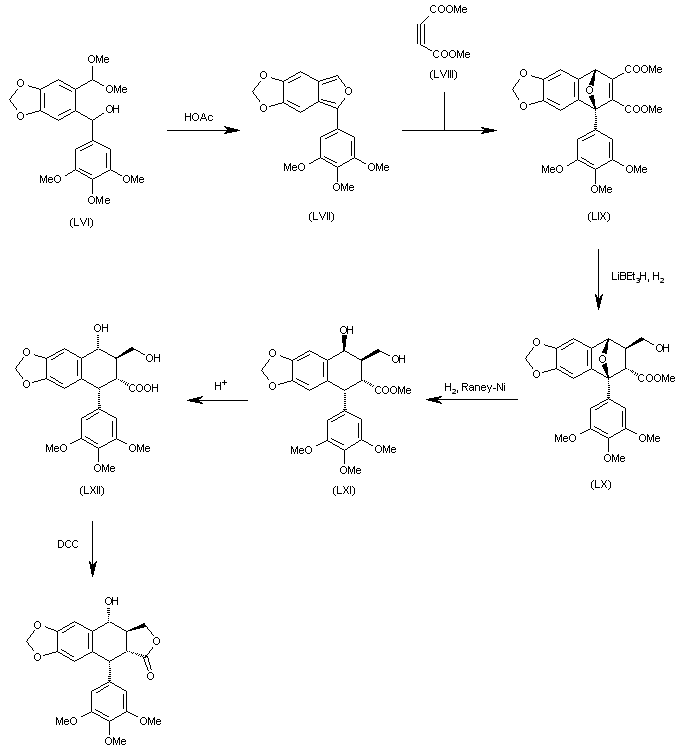
The cyclization of 6-[1-hydroxy-1-(3,4,5-trimethoxyphenyl)methyl]-1,3-benzodioxol-5-carbaldehyde dimethylacetal (LVI) by means of AcOH gives 5-(3,4,5-trimethoxyphenyl)-1,3-dioxolo[4,5-f]isobenzofuran (LVII), which is submitted to a Diels-Alder cyclization with acetylenedicarboxylic acid dimethyl ester (LVIII) yielding the epoxy derivative (LIX). The selective reduction of (LIX) with LiBEt3H and H2 affords the carbinol (LX), which is treated with H2 over RaNi in order to open the epoxide ring to give the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to (LXII) with the suitable configuration. Finally, this compound is cyclized with DCC to the desired target compound.
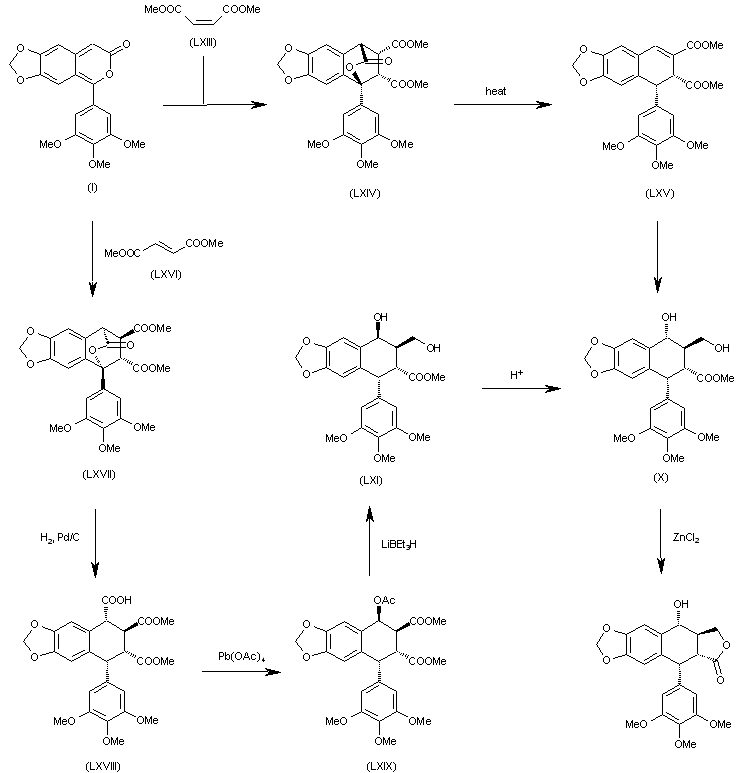
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl maleate (LXIII) gives the expected adduct (LXIV), which by thermal extrusion of CO2 yields the dihydronaphthodioxole (LXV). This compound is then converted to dihydroxycompound (X), which is finally cyclized by means of ZnCl2 to provide the target compound. The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl fumarate (LXVI) gives the expected adduct (LXVII), which by hydrogenation with H2 over Pd/C yields the tricarboxylic acid derivative (LXVIII). The reaction of (LXVIII) with Pb(OAc)4 affords the acetoxy derivative (LXIX), which is selectively reduced with LiBEt3H providing the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to give the previously described (X) with the suitable configuration.
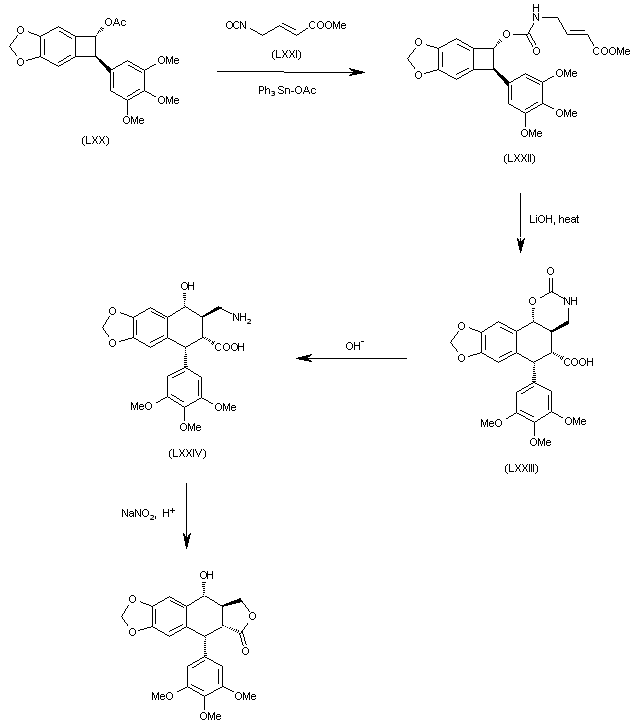
The reaction of benzocyclobutane derivative (LXX) with isocyanate (LXXI) by means of Ph3SnOAc gives the carbamate (LXXII), which is cyclized by a thermal treatment with LiOH yielding the tetracyclic carboxylic acid (LXXIII). The opening of the oxazinone ring of (LXXIII) in basic medium affords the tricyclic amino acid (LXXIV), which is finally cyclized to the target compound by reaction with sodium nitrite in acidic medium (pH = 4).
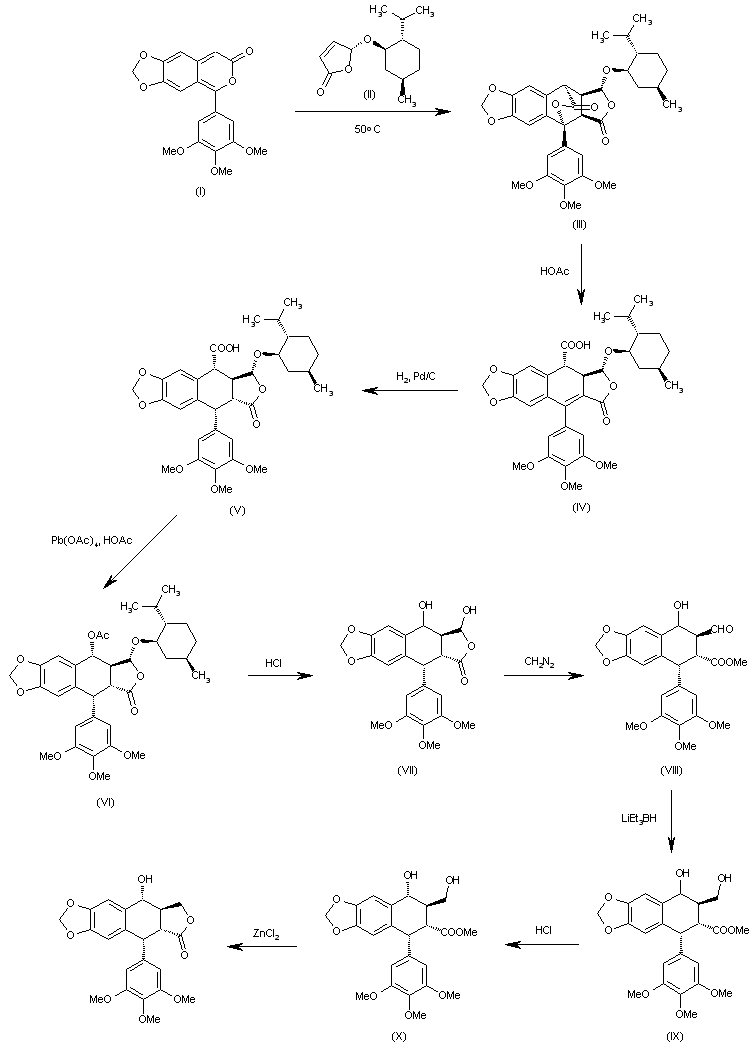
J Chem Soc Chem Commun 1993,1200
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with the chiral dihydrofuranone (II) in hot acetonitrile gives the pentacyclic anhydride (III), which is opened with warm acetic acid yielding the carboxylic acid (IV). Hydrogenation of the benzylic double bond of (IV) with H2 over Pd/C affords (V), which is treated with lead tetraacetate and acetic acid in THF to give the acetoxy compound (VI). The hydrolysis of the acetoxy group and the menthol hemiacetal group with HCl in hot dioxane yields the diol (VII), which is treated with diazomethane in ether/methanol affording the aldehyde (VIII). The reduction of the aldehyde group of (VIII) with LiEt3BH in THF gives the diol (IX) as a diastereomeric mixture, which is treated with HCl in THF to afford the diol (X) with the right conformation. Finally, this compound is lactonized to the target compound with ZnCl2 in THF.
//////////
HEC-68498
HEC-68498, CT-365
CAS 1621718-37-3
C20 H13 F2 N5 O3 S
441.41
Benzenesulfonamide, N-[5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxy-3-pyridinyl]-2,4-difluoro-
N-[5-(3-Cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxy-3-pyridinyl]-2,4-difluorobenzenesulfonamide
HEC Pharm , Calitor Sciences Llc; Sunshine Lake Pharma Co Ltd
PHASE 1, idiopathic pulmonary fibrosis and solid tumors
Phosphoinositide 3-kinase inhibitor; mTOR inhibitor

- Originator HEC Pharm
- Developer HEC Pharm; Sunshine Lake Pharma
- Class Anti-inflammatories; Antifibrotics; Isoenzymes
- Mechanism of Action 1 Phosphatidylinositol 3 kinase inhibitors; MTOR protein inhibitors
- Phase I Idiopathic pulmonary fibrosis
- 22 May 2018 Phase-I clinical trials in Idiopathic pulmonary fibrosis in USA (PO) (NCT03502902)
- 24 Apr 2018 Sunshine Lake Pharma in collaboration with Covance plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in China , (NCT03502902)
- 19 Apr 2018 Preclinical trials in Idiopathic pulmonary fibrosis in China (PO)
- US 20140234254
- CN 103965199
CN 103965199

CN 103965199

Sunshine Lake Pharma , a subsidiary of HEC Pharm is developing an oral capsule formulation of HEC-68498 (phase 1, in July 2019) sodium salt, a dual inhibitor of phosphoinositide-3 kinase and the mTOR pathway, for the treatment of idiopathic pulmonary fibrosis and solid tumors
HEC 68498 is an oral inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin in clinical development at HEC Pharm for the treatment of idiopathic pulmonary fibrosis. A phase I trial is under way in healthy volunteers.
The phosphoinositide 3-kinases (PI3 kinases or PI3Ks), a family of lipid kinases, have been found to play key regulatory roles in many cellular processes including cell survival, proliferation and differentiation. The PI3K enzymes consist of three classes with variable primary structure, function and substrate specificity. Class I PI3Ks consist of heterodimers of regulatory and catalytic subunits, and are subdivided into 1A and 1B based on their mode of activation. Class 1A PI3Ks are activated by various cell surface tyrosine kinases, and consist of the catalytic pl lO and regulatory p85 subunits. The three known isoforms of Class 1A pl lO are pl lOot, rΐ ΐqb, and rΐ ΐqd, which all contain an amino terminal regulatory interacting region (which interfaces with p85), a Ras binding domain, and a carboxy terminal catalytic domain. Class IB PI3Ks consist of the catalytic (pl lOy) and regulatory (p 101 ) subunits and are activated by G-protein coupled receptors. (“Small-molecule inhibitors of the PI3K signaling network” Future Med. Chem ., 2011, 3, 5, 549-565).
[0004] As major effectors downstream of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), PI3Ks transduce signals from various growth factors and cytokines into intracellular massages by generating phospholipids, which activate the serine-threonine protein kinase ART (also known as protein kinase B (PKB)) and other downstream effector pathways. The tumor suppressor or PTEN (phosphatase and tensin
homologue) is the most important negative regulator of the PI3K signaling pathway. (“Status of PBK/Akt/mTOR Pathway Inhibitors in Lymphoma.” Clin Lymphoma, Myeloma Leuk , 2014, 14(5), 335-342.)
[0005] The signaling network defined by phosphoinositide 3-kinases (PI3Ks), AKT and mammalian target of rapamycin (mTOR) controls most hallmarks of cancer, including cell cycle, survival, metabolism, motility and genomic instability. The pathway also contributes to cancer promoting aspects of the tumor environment, such as angiogenesis and inflammatory cell recruitment. The lipid second messenger produced by PI3K enzymes, phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3; also known as PIP3), is constitutively elevated in most cancer cells and recruits cytoplasmic proteins to membrane-localized‘onco’ signal osomes.
[0006] Cancer genetic studies suggest that the PI3K pathway is the most frequently altered pathway in human tumors: the PIK3CA gene (encoding the PI3K catalytic isoform pl lOa) is the second most frequently mutated oncogene, and PTEN (encoding phosphatase and tensin homolog, the major PtdIns(3,4,5)P3 phosphatase) is among the most frequently mutated tumor suppressor genes. In accord, a recent genomic study of head and neck cancer found the PI3K pathway to be the most frequently mutated. Indeed, even in cancer cells expressing normal PI3K and PTEN genes, other lesions are present that activate the PI3K signaling network (that is, activated tyrosine kinases, RAS and AKT, etc ). As a net result of these anomalies, the PI3K pathway is activated, mutated or amplified in many malignancies, including in ovarian cancer (Campbell et al., Cancer Res., 2004, 64, 7678-7681; Levine et al., Clin. Cancer Res., 2005, 11, 2875-2878; Wang et al., Hum. Mutat., 2005, 25, 322; Lee et al., Gynecol. Oncol. ,2005, 97, 26-34), cervical cancer, breast cancer (Bachman et al.,· Cancer Biol., Ther, 2004, 3, 772-775; Levine et al., supra; Li et al., Breast Cancer Res. Treat., 2006, 96, 91-95; Saal et al., Cancer Res., 2005, 65, 2554-2559; Samuels and Velculescu, Cell Cycle, 2004, 3, 1221-1224), colorectal cancer (Samuels et al., Science, 2004, 304, 554; Velho et al., Eur. J. Cancer, 2005, 41, 1649-1654), endometrial cancer (Oda et al ., Cancer Res., 2005, 65, 10669-10673), gastric carcinomas (Byun et al., M. J. Cancer, 2003 , 104, 318-327; Li et al., supra; Velho et al., supra; Lee et al., Oncogene, 2005 , 24, 1477-1480), hepatocellular carcinoma (Lee et al., id), small and non-small cell lung cancer (Tang et al., Lung Cancer 2006, 11, 181-191; Massion et al , Am. J. Respir. Crit. Care Med., 2004, 170, 1088-1094), thyroid carcinoma (Wu et al., J. Clin. Endocrinol. Metab., 2005, 90, 4688-4693),
acute myelogenous leukemia (AML) (Sujobert et al., Blood, 1997, 106, 1063-1066), chronic myelogenous leukemia (CML) (Hickey et al., J. Biol. Chem ., 2006, 281, 2441-2450), glioblastomas (Hartmann et al. Jlcta Neuropathol (Bert ), 2005, 109, 639-642; Samuels et al., supra), Hodgkin and non-Hodgkin lymphomas (“PI3K and cancer: lessons, challenges and opportunities”, Nature Reviews Drug Discovery., 2014, 13, 140).
[0007] The PI3K pathway is hyperactivated in most cancers, yet the capacity of PI3K inhibitors to induce tumor cell death is limited. The efficacy of PI3K inhibition can also derive from interference with the cancer cells’ ability to respond to stromal signals, as illustrated by the approved PI3K5 inhibitor idelalisib in B-cell malignancies. Inhibition of the leukocyte-enriched PI3K5 or RI3Kg may unleash antitumor T-cell responses by inhibiting regulatory T cells and immune-suppressive myeloid cells. Moreover, tumor angiogenesis may be targeted by PI3K inhibitors to enhance cancer therapy. (“Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy”, Cancer Discov., 2016, 6(10), 1090-1105.)
[0008] mTOR is a highly conserved serine-threonine kinase with lipid kinase activity and participitates as an effector in the PI3K/AKT pathway. mTOR exists in two distinct complexes, mTORCl and mTORC2, and plays an important role in cell proliferation by monitoring nutrient avaliability and cellular energy levels. The downstream targets of mTORCl are ribosomal protein S6 kinase 1 and eukaryotic translation initiation factor 4E-binding protein 1, both of which are crucial to the regulation of protein synthesis. (“Present and future of PI3K pathway inhibition in cancer: perspectives and limitations”, Current Med. Chem., 2011, 18, 2647-2685).
[0009] Knowledge about consequences of dysregulated mTOR signaling for tumorigenesis comes mostly from studies of pharmacologically disruption of mTOR by repamycin and its analogues such as temsirolimus (CCI-779) and everolimus (RADOOl).Rapamycin was found to inhibit mTOR and thereby induce G1 arrest and apoptosis. The mechanism of rapamycin growth inhibition was found to be related to formation of complexes of rapamycin with FK-binding protein 12 (FKBP-12). These complexes then bound with high affinity to mTOR, preventing activation and resulting in inhibition of protein translation and cell growth. Cellular effects of mTOR inhibition are even more pronounced in cells that have concomitant inactivation of PTEN. Antitumor activity of rapamycin was subsequently identified, and a number of rapamycin analogues such as temsirolimus and everolimus have been approved by the US Food and Drug
Administration for the treatment certain types of cancer.
[0010] Fibrosis is the formation of excess fibrous connective tissue in an organ or tissue in a reparative or reactive process. Examples of fibrosis include, but are not limited to pulmonary fibrosis, liver fibrosis, dermal fibrosis, and renal fibrosis. Pulmonary fibrosis, also called idiopathic pulmonary fibrosis (IPF), interstitial diffuse pulmonary fibrosis, inflammatory pulmonary fibrosis, or fibrosing alveolitis, is a lung disorder and a heterogeneous group of conditions characterized by abnormal formation of fibrous tissue between alveoli caused by alveolitis comprising cellular infiltration into the alveolar septae with resulting fibrosis. The effects of IPF are chronic, progressive, and often fatal.
[0011] The clinical course of IPF is variable and largely unpredictable. IPF is ultimately fatal, with historical data suggesting a median survival time of 2-3 years from diagnosis. A decline in forced vital capacity (FVC) is indicative of disease progression in patients with IPF and change in FVC is the most commonly used endpoint in clinical trials. A decline in FVC of 5% or 10% of the predicted value over 6-12 months has been associated with increased mortality in patients with IPF.
[0012] Our understanding of the pathogenesis of IPF has evolved from that of a predominantly inflammatory disease to one driven by a complex interplay of repeated epithelial cell damage and aberrant wound healing, involving fibroblast recruitment, proliferation and differentiation, and culminating in excess deposition of extracellular matrix. This shift in knowledge prompted a change in the type of compounds being investigated as potential therapies, with those targeted at specific pathways in the development and progression of fibrosis becoming the focus.
[0013] In patients with IPF, the mechanisms whereby PI3K/mTOR inhibitors act may involve inhibition of kinases such as PI3Ks and mTOR. This results in inactivation of cellular receptors for mediators involved in the development of pulmonary fibrosis. As a result, fibroblast proliferation is inhibited and extracellular matrix deposition is reduced. (“Update on diagnosis and treatment of idiopathic pulmonary fibrosis”, J Bras Pneumol. 2015, 41(5), 454-466.)
[0014] Accordingly, small-molecule compounds that specially inhibit, regulate and/or modulate the signal transduction of kinases, particularly including PI3K and mTOR as described above, are desirable as a means to prevent, manage, or treat proliferative disorders and fibrosis, particular idiopathic pulmonary fibrosis in a patient. One such small-molecule is A-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfon-amide, which has the chemical structure as shown in the following:
[0015] WO 2014130375A1 described the synthesis of N-(5 -(‘3 -cyanopyrazol o [l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (Example 3) and also disclosed the therapeutic activity of this molecule in inhibiting, regulating and modulating the signal transduction of protein kinases.
[0016] Different salts and solid state forms of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, improving the dissolution profile, stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also provide improvements to the final dosage form, for example, if they serve to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
Different salts and solid state forms of /V-(5-(3-cyanopyrazolo[l,5- ]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide are described herein.
PATENT
WO2014130375 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014130375
claiming new pyrazolo[1,5-a]pyridine derivatives are PI3K and mTOR inhibitors, useful for treating proliferative diseases
Example 3 N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
Step 1) 5-bromopyrazolo[1,5-a]pyridine
[196] A solution of ethyl 5-bromopyrazolo[1,5-a]pyridine-3-carboxylate (240
mmol) in 40% H2SO4 (12 mL) was stirred at 100 °C for 4 hours, then cooled to rt, and neutralized to pH=7 with aq. NaOH (6 M) in ice bath. The resulted mixture was extracted with DCM (25 mL x 2). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (175 mg, 99.5%).
MS (ESI, pos. ion) m/z: 196.9 [M+H]+.
Step 2) 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde
[197] To a solution of 5-bromopyrazolo[1,5-a]pyridine (175 mg, 0.89 mmol) in DCM (6 mL) was added (chloromethylene)dimethyliminium chloride (632 mg, 3.56 mmol). The reaction was stirred at 44 °C overnight, and concentrated in vacuo. The residue was dissolved in saturated NaHCO3 aqueous solution (25 mL) and the resulted mixture was then extracted with EtOAc (25 mL x 3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (225 mg, 100%).
MS (ESI, pos. ion) m/z: 225.0 [M+H]+.
Step 3) (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime
[198] To a suspension of 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde (225 mg, 1 mmol) in EtOH (10 mL) and H2O (5 mL) was added hydroxylamine hydrochloride (104 mg, 1.5 mmol). The reaction was stirred at 85 °C for 2 hours, then cooled to rt, and concentrated in vacuo. The residue was adjusted to pH=7 with saturated NaHCO3 aqueous solution. The resulted mixture was then filtered and the filter cake was dried in vacuo to give title compound as a yellow solid (240 mg, 99%).
MS (ESI, pos. ion) m/z: 240.0 [M+H]+.
Step 4) 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile
[199] A solution of (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime (240 mg,
1 mmol) in Ac2O (6 mL) was stirred at 140 °C for 18 hours, then cooled to rt, and concentrated in vacuo. The residue was washed with Et2O (1 mL) to give the title compound as a yellow solid (44 mg, 22.5%).
MS (ESI, pos. ion) m/z: 222.0 [M+H]+.
Step 5) N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
[200] 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (612 mg, 1.5 mmol), 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile (222 mg, 1 mmol), Pd(dppf)Cl2·CH2Cl2 (16 mg, 0.02 mmol) and Na2CO3 (85 mg, 0.8 mmol) were placed into a two-neck flask, then degassed with N2 for 3 times, and followed by adding 1,4-dioxane (5 mL) and water (1 mL). The resulted mixture was degassed with N2 for 3 times, then heated to 90 °C and stirred further for 5 hours. The mixture was cooled to rt and filtered. The filtrate was concentrated in vacuo and the residue was purified by a silica gel column chromatography (PE/EtOAc (v/v) = 1/2) to give the title compound as a light yellow solid (400 mg, 81.6%).
MS (ESI, pos. ion) m/z: 442.0 [M+H]+;
1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.37 (s, 1H), 9.02 (d, J = 7.2 Hz, 1H), 8.67 (s, 1H), 8.60 (d, J = 2.2 Hz, 1H), 8.26-8.16 (m, 2H), 7.82-7.72 (m, 1H), 7.57 (dd, J = 13.2, 5.8 Hz, 2H), 7.21 (t, J= 8.5 Hz, 1H), 3.67 (s, 3H).
PATENT
WO-2019125967
The invention relates to salts of pyrazolo[l,5-a]pyridine derivatives and use thereof, specifically relates to salt of /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl) -2,4-difluorobenzenesulfonamide (compound of formula (I)) and use thereof, further relates to composition containing said salts above. The salts or the composition can be used to inhibit/modulate protein kinases, further prevent, manage or treat proliferative disorders or pulmonary fibrosis in a patient.
Amorphous form of mono-sodium salt of HEC-68498 , useful for treating a proliferative disorder or pulmonary fibrosis.
The invention is further illustrated by the following examples, which are not be construed as limiting the invention in scope.
[00108] /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluoroben zenesulfonamide can be prepared according to the synthetic method of example 3 disclosed in WO2014130375 Al.
//////////////HEC-68498, HEC 68498, HEC68498, HEC Pharm , Calitor Sciences, Sunshine Lake Pharma, PHASE 1, proliferative disorder, pulmonary fibrosis, idiopathic pulmonary fibrosis, solid tumors, CT-365 , CT 365 , CT365
Fc1ccc(c(F)c1)S(=O)(=O)Nc2cc(cnc2OC)c3ccn4ncc(C#N)c4c3
GNE-0877

GNE-0877
Maybe DNL-151 ?
CAS 1374828-69-9
Chemical Formula: C14H16F3N7
Molecular Weight: 339.31895
2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-1H-pyrazol-1-yl)propanenitrile
Denali Therapeutics Inc, useful for treating Alzheimer’s disease, breast tumor, type I diabetes mellitus and Crohn’s disease
GNE-0877 is a highly potent and selective LRRK2 inhibitor. Leucine-rich repeat kinase 2 (LRRK2) has drawn significant interest in the neuroscience research community because it is one of the most compelling targets for a potential disease-modifying Parkinson’s disease therapy.
- Developer Denali Therapeutics Inc
- Class Antiparkinsonians; Small molecules
- Mechanism of Action LRRK2 protein inhibitors
- Phase I Parkinson’s disease
- 20 Dec 2017 Denali Therapeutics plans clinical studies for Parkinson’s disease
- 13 Nov 2017 Phase-I clinical trials in Parkinson’s disease (In volunteers) in Netherlands (unspecified route)
- 13 Nov 2017 Preclinical trials in Parkinson’s disease in USA (unspecified route) before November 2017
Denali Therapeutics is developing DNL-151 (phase 1, in July 2019), a lead from a program of small-molecule inhibitors of LRRK2 originally licensed from Genentech, for the treatment of Parkinson’s disease.
Leucine-rich repeat kinase 2 (LRRK2) is a complex signaling protein that is a key therapeutic target, particularly in Parkinson’s disease (PD). Combined genetic and biochemical evidence supports a hypothesis in which the LRRK2 kinase function is causally involved in the pathogenesis of sporadic and familial forms of PD, and therefore that LRRK2 kinase inhibitors could be useful for treatment (Christensen, K.V. (2017) Progress in medicinal chemistry 56:37-80). Inhibition of the kinase activity of LRRK2 is under investigation as a possible treatment for Parkinson’s disease (Fuji, R.N. et al (2015) Science Translational Medicine 7(273):ral5;
Taymans, J.M. et al (2016) Current Neuropharmacology 14(3):214-225). A group of LRRK2 kinase inhibitors have been studied (Estrada, A.A. et al (2015) Jour. Med. Chem. 58(17): 6733-6746; Estrada, A.A. et al (2013) Jour. Med. Chem. 57:921-936; Chen, H. et al (2012) Jour. Med. Chem. 55:5536-5545; Estrada, A.A. et al (2015) Jour. Med. Chem. 58:6733-6746; US 8354420; US 8569281; US8791130; US 8796296; US 8802674; US 8809331; US 8815882; US 9145402; US 9212173; US 9212186; WO 2011/151360; WO 2012/062783; and WO 2013/079493.
PATENT
WO2012062783 , assigned to Hoffmann-La Roche , but naming inventors specifically associated with both Genentech and BioFocus (which had an agreement with Genentech for drug discovery programs); the compound was also later identified in J.Med.Chem (57(3), 921-936, 2014) in an article from these two companies, with the lab code GNE-0877. So while this represents the first application in the name of Denali Therapeutics Inc that focuses on this compound, it is likely that it provides the structure of DNL-151 , a lead from a program of small-molecule inhibitors of leucine-rich repeat kinase 2 (LRRK2) originally licensed from Genentech, being developed for the oral treatment of Parkinson’s disease, and which had begun phase I trials by December 2017 (when this application was lodged).
PATENT
WO2019104086 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019104086
claiming novel crystalline and amorphous forms of pyrimidinylamino-pyrazole compound, useful for treating Alzheimer’s disease, breast tumor, type I diabetes mellitus and Crohn’s disease.
Novel crystalline and amorphous forms of 2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-1H-pyrazol-1-yl)propanenitrile (which is substantially pure form) and their anhydrous and solvates such as cyclohexanol solvate (designated as Forms B-D), processes for their preparation and compositions comprising them are claimed. The compound is disclosed to be leucine rich serine threonine kinase 2 inhibitor, useful for treating Gaucher disease, Alzheimer’s disease, motor neurone disease, Parkinson’s disease, prostate tumor, Lewy body dementia, mild cognitive impairment, breast tumor, type I diabetes mellitus and Crohn’s disease.
The present disclosure relates to crystalline polymorph or amorphous forms of a pyrimidinylamino-pyrazole kinase inhibitor, referred to herein as the Formula I compound and having the structure:
FORMULA I COMPOUND
The present disclosure includes polymorphs and amorphous forms of Formula I compound, (CAS Registry Number 1374828-69-9), having the structure:
and named as: 2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-lH-pyrazol-l-yl)propanenitrile (WO 2012/062783; US 8815882; US 2012/0157427, each of which are incorporated by reference). As used herein, the Formula I compound includes tautomers, and pharmaceutically acceptable salts or cocrystals thereof. The Formula I compound is the API (Active Pharmaceutical Ingredient) in formulations for use in the treatment of neurodegenerative and other disorders, with pKa when protonated calculated at 6.7 and 2.1.
CRYSTALLIZATION
Initial polymorph screening experiments were performed using a variety of
crystallization or solid transition methods, including: anti-solvent addition, reverse anti-solvent addition, slow evaporation, slow cooling, slurry at room temperature (RT), slurry at 50 °C, solid vapor diffusion, liquid vapor diffusion, and polymer induced crystallization. By all these methods, the Form A crystal type was identified. Polarized light microscopy (PLM) images of Form A obtained from various polymorph screening methods were collected (Example 5).
Particles obtained via anti-solvent addition showed small size of about 20 to 50 microns (pm) diameter while slow evaporation, slow cooling (except for THF/isooctane), liquid vapor diffusion and polymer-induced crystallization resulted in particles with larger size. Adding isooctane into a dichloromethane (DCM) solution of the Formula I compound produced particles with the most uniform size. Crude Formula I compound crystallized from THF///-heptane and then was micronized. A crystallization procedure was developed to control particle size.
A total of four crystal forms (Forms A, B, C, and D) and an amorphous form E of Formula I compound were prepared, including 3 anhydrates (Form A, C, and D) and one solvate (Form B). Slurry competition experiments indicated that Form D was thermodynamically more stable when the water activity aw< 0.2 at RT, while Form C was more stable when aw> 0.5 at RT. The 24 hrs solubility evaluation showed the solubility of Form A, C and D in FLO at RT was 0.18, 0.14 and 0.11 mg/mL, respectively. DVS (dynamic vapor sorption) results indicated that Form A and D were non-hygroscopic as defined by less than 0.1% reversible water intake in DVS, while Form C was slightly hygroscopic. Certain characterization data and observations of the crystal forms are shown in Table 1.
Table 1 Characterization summary for crystal forms of Formula I compound
Differential Scanning Calorimetry (DSC) analysis of Forms A and C showed that Form C had higher melting point and higher heat of fusion (Table 1), suggesting that the two forms are monotropic with Form C being the more stable form. Competitive slurry experiments with 1 : 1 Form A and C in a variety of solvents always produced Form C confirming that Form C was
more stable than Form A. In accordance with this, Form C was produced even when the crystallization batch was seeded with seeds of Form A.
PATENT
WO-2019126383
Methods of making leucine-rich repeat kinase 2 (LRRK2)-inhibiting, pyrimidinyl-4-aminopyrazole compounds (eg 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol-1-yl)propanenitrile), useful for treating LRRK2 mediated diseases such as Parkinson’s disease.
Example 1 Preparation of 2-(4-amino-3 -methyl- liT-pyrazol-l -yl)-2-methylpropanamide 5a
4a 5a
To a 20-L reactor containing dimethyl formamide (4.5 L) was charged 5-methyl-4-nitro-lH-pyrazole la (1.5 kg, 1.0 equiv). The solution was cooled to 0 °C and charged with finely ground K2CO3 (2.45 kg, 1.5 equiv) in three portions over ~l h. Methyl 2-bromo-2-methylpropanoate (3.2 kg, 1.5 equiv) was added dropwise to the mixture and then was allowed to warm to ~25 °C. The reaction mixture was maintained for 16 h and then quenched with water (15 L) and product was extracted with ethyl acetate. The combined organic layer was washed with water, and then with a brine. The organic layer was dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure to give a light yellow solid. The crude product was purified by crystallization with petroleum ether (15 L), filtered, and dried to give methyl 2-m ethyl -2-(3 -methyl -4-nitro- l//-pyrazol- l -yl)propanoate 3a (2.25 kg, >99% purity by HPLC, 84 % yield) as an off-white solid. ¾ NMR (400 MHz, CDCb) 8.28 (s, 1H), 3.74 (s, 3H), 2.53 (s, 3H), 1.85 (s, 6H).
Methanol (23 L) and 2-methyl-2-(3-methyl-4-nitro-lif-pyrazol-l-yl)propanoate 3a (2.25 kg, 1.0 equiv) were charged into a 50-L reactor and cooled to approximately -20 °C. Ammonia gas was purged over a period of 5 h and then the reaction mixture warmed to 25 °C. After 16 h, the reaction mixture was concentrated under reduced pressure (~50 °C) to give the crude product. Ethyl acetate (23 L) was charged and the solution agitated in the presence of charcoal (0.1 w/w) and Celite® (0.1 w/w) at 45 °C. The mixture was filtered and concentrated under reduced pressure, and then the solid was slurried in methyl tert-butyl ether (MTBE, 11.3 L) at RT for 2 h. Filtration and drying at ~45 °C gave 2-m ethyl -2-(3 -m ethyl -4-ni tro- 1 //-pyrazol – 1 -yl)propanamide 4a (1.94 kg, >99% purity by HPLC, 92% yield).
Methanol (5 L) and 2-m ethyl-2-(3 -methyl -4-nitro-lif-pyrazol-l-yl)propanamide 4a (0.5 kg) were charged into a 10-L autoclave under nitrogen atmosphere, followed by slow addition of 10 % (50% wet) Pd/C (50 g). Hydrogen was charged (8.0 kg pressure/l 13 psi) and the reaction mixture agitated at 25 °C until complete. The mixture was filtered, concentrated under reduced
pressure and then slurried in MTBE (2.5 L) for 2 h at 25 °C. Filtration and drying under reduced pressure (45 °C) gave 2-(4-amino-3-methyl- l//-pyrazol- l -yl)-2-methyl propanamide 5a (0.43 kg, >99% purity by HPLC, 99% yield).
Example 2 Preparation of 2-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-3-methyl-lH-pyrazol-l-yl)-2-methylpropanamide 7a
DCM
Into a first reactor was charged /-BuOH (or alternatively 2-propanol) (15.5 vol) and 2-(4-amino-3 -methyl- li7-pyrazol-l-yl)-2-methylpropanamide 5a (15 kg), followed by zinc chloride (13.5 kg, 1.2 equiv) at room temperature and the suspension agitated ~2 h. Into a second reactor was charged dichloromethane (DCM, 26.6 vol) and 2,4-dichloro-5-trifluoromethyl pyrimidine 6a (19.6 kg, 1.1 equiv) and then cooled to 0 °C. The contents from first reactor were added portion-wise to the second reactor. After addition, the reaction mixture was agitated at 0 °C for ~l h and then Et3N (9.2 kg, 1.1 equiv) was slowly charged. After agitation for 1 h, the temperature was increased to 25 °C and monitored for consumption of starting material. The reaction mixture was quenched with 5% aqueous NaHCO, and then filtered over Celite®. The DCM layer was removed and the aqueous layer was back-extracted with DCM (3x). The combined organics were washed with water, dried (Na2S04), and concentrated. Methanol (2.5 vol) was charged and the solution was heated to reflux for 1 h, then cooled to 0 °C. After 1 h, the solids were filtered and dried under reduced pressure to give 2-(4-((4-chloro-5-(tri fluoromethyl)pyri mi din-2-yl)amino)-3 -methyl – l//-pyrazol- l -yl)-2-methyl propanamide 7a
(31.2 kg (wet weight)). 1H NMR (600 MHz, DMSO-de) 10.05 (br. s., 1H), 8.71 (d, J= 11 Hz, 1H), 7.95 (app. d, 1H), 7.18 (br. s., 1H), 6.78 (br. s., 1H), 2.14 (s, 3H), 1.67 (s, 6H).
Example 3 Preparation of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol- 1 -yl)propanamide 8a
A reactor was charged with anhydrous tetrahydrofuran (THF, 10 vol) and 2-(4-((4-chloro-5-(trifl uoromethyl )pyrimi din-2-yl)amino)-3 -methyl – l //-pyrazol- l -yl)-2-methylpropanamide 7a (21 kg) at room temperature with agitation. A solution of 2M
methylamine in THF (3.6 vol) was slowly charged to the reactor at 25 °C and maintained for ~3 h. The reaction mixture was diluted with 0.5 w/w aqueous sodium bicarbonate solution (10 w/w), and extracted with ethyl acetate (EtOAc, 4.5 w/w). The aqueous layer was extracted with EtOAc (4x), the organics were combined and then washed with H20 (7 w/w). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. «-Heptane (3 w/v) was added to the residue, agitated, filtered and dried under reduced pressure to give 2-m ethyl -2-(3 -methyl -4-((4-(methyl ami no)-5-(trifl uoromethyl )pyri mi din-2-yl)amino)- l //-pyrazol-1 -yl)propanamide 8a (19.15 kg, 93% yield). ¾ NMR (600 MHz, DMSO-d6) 8.85 (m, 1H), 8.10 (s, 1H), 8.00 (m, 1H), 7.16 (br. s., 1H), 6.94 (m, 1H), 6.61 (br. s., 1H), 2.90 (d, J = 4.3 Hz, 3H), 2.18 (br. s., 3H), 1.65 (s, 6H).
Example 4 Preparation of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol- 1 -yl)propanenitrile 9a
To a reactor was charged 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifl uoromethyl )pyri mi din-2-yl)amino)- l //-pyrazol- l -yl)propan amide 8a (15 kg, 1 equiv) at room temperature followed by EtOAc (2 vol) and 6.7 vol T3P (50% w/w in EtOAc). The reaction mixture was heated to 75 °C over 1 h and then agitated for 16 h until consumption of starting material. The reaction mixture was cooled between -10 to -15 °C then added drop-wise 5N aqueous NaOH (7 vol) resulting in pH 8-9. The layers were separated and the aqueous layer back-extracted with EtOAc (2 x 4 vol). The combined organic extracts were washed with 5 %
aqueous NaHCO, solution, and then distilled to azeotropically remove water. The organics were further concentrated, charged with «-heptane (2 vol) and agitated for 1 h at room temperature. The solids were filtered, rinsed with «-heptane (0.5 vol) and then dried under vacuum (<50 °C). The dried solids were dissolved in EtOAc (1.5 vol) at 55 °C, and then «-heptane (3 vol) was slowly added followed by 5-10% of 9a seeds. To the mixture was slowly added «-heptane (7 vol) at 55 °C, agitated for 1 h, cooled to room temperature and then maintained for 16 h. The suspension was further cooled between 0-5 °C, agitated for 1 hour, filtered, and then rinsed the filter with chilled 1 :6.5 EtOAc/«-heptane (1 vol). The product was dried under vacuum at 50 °C to give 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1 //-pyrazol – 1 -yl )propaneni tri 1 e 9a (9.5 kg, first crop), 67% yield). ‘H NMR (600 MHz, DMSO-d6) 8.14 (s, 1H), 8.13 (br. s., 1H), 7.12 (br. s., 1H), 5.72 (br. s, 1H), 3.00 (d, J= 4.6 Hz, 3H),
2.23 (s, 3H), 1.96 (s, 3H).
Example 5 Preparation of methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a
Following the procedure of Example 1, a mixture of methanol and methyl 2-methyl-2-(3-methyl-4-nitro-lH-pyrazol-l-yl)propanoate 3a (0.5 kg) was charged into an autoclave under nitrogen atmosphere, followed by slow addition of 10 % (50% wet) Pd/C. Hydrogen was charged under pressure and the reaction mixture agitated at 25 °C until complete. The mixture was filtered, concentrated under reduced pressure and then slurried in MTBE for 2 h at 25 °C. Filtration and drying under reduced pressure gave methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a (LC-MS, M+l=l98).
Example 6 Preparation of methyl 2-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-3 -methyl- lH-pyrazol- 1 -yl)-2-methylpropanoate 11a
Following the procedure of Example 2, a mixture of methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a and DIPEA (1.2 equiv) in /-BuOH was warmed to 80 °C. Then a solution of 2,4-dichloro-5-trifluoromethyl pyrimidine 6a in /-BuOH was added slowly drop wise at 80 °C. After 15 minutes, LCMS showed the reaction was complete, including later eluting 59.9% of product ester 11a, earlier eluting 31.8% of undesired regioisomer (ester), and no starting material 10a. After completion of reaction, the mixture was cooled to room temperature and a solid was precipitated. The solid precipitate was filtered and dried to give methyl 2-(4-((4-chloro-5-(trifluoromethyl)pyrimi din-2 -yl)amino)-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 11a (LC-MS, M+l=378).
PAPER
J.Med.Chem (57(3), 921-936, 2014
https://pubs.acs.org/doi/full/10.1021/jm401654j
2-Methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1H-pyrazol-1-yl)propanenitrile (11)
A solution of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1H-pyrazol-1-yl)propanamide (34, 250 mg, 0.7 mmol) in POCl3 (5 mL) was stirred at 90 °C for 1 h. The POCl3 was removed by evaporation. The mixture was then slowly poured onto ice (10 mL). The pH of the solution was adjusted to 8 with saturated sodium carbonate. The aqueous phase was extracted with EtOAc (3×). The combined organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated to give a residue that was purified by recrystallization to give 11 (100 mg, 42% yield) as a white solid. 1H NMR (300 MHz, DMSO) δ 9.18 (s, 1H), 8.29 (s, 1H), 8.14 (s, 1H), 7.10 (s, 1H), 2.91 (d, 3H), 2.22 (s, 3H), 1.94 (s, 6H). HRMS (ES) m/z: [M + H]+ calcd for C14H16F3N7H+, 340.1492; found, 340.1484.
Scheme 2

Scheme 2. Synthesis of N-Alkyl Pyrazole Analoguesa
aReagents and conditions: (a) NaH, methyl 2-bromo-2-methylpropanoate, DMF, 70%; (b) LiOH, THF-H2O, 90%; (c) (i) (COCl)2, CH2Cl2, (ii) R-NH2, THF; (d) Pd/C, H2, MeOH; (e) 26, Et3N, n-BuOH, 120 °C; (f) 26, TFA, 2-methoxyethanol, 70 °C; (g) POCl3, 90 °C, 42%.
GNE-9605
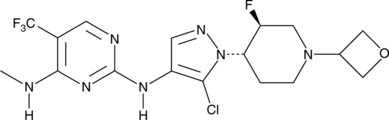
CAS № 1536200-31-3
Molecular Formula
C17H20ClF4N7O
Formula Weight
449.8
GNE-9065 is an orally bioavailable and potent inhibitor of leucine-rich repeat kinase 2 (LRRK2; IC50 = 18.7 nM).1 It is selective for LRRK2 over 178 kinases, inhibiting only TAK1-TAB1 >50% at a concentration of 0.1 μM. GNE-9065 (10 and 50 mg/kg) inhibits LRRK2 Ser1292 autophosphorylation in BAC transgenic mice expressing human LRRK2 protein with the G2019S mutation found in families with autosomal Parkinson’s disease.
CNC1=C(C(F)(F)F)C=NC(NC2=C(Cl)N([C@H]3CCN(C4COC4)C[C@@H]3F)N=C2)=N1
N2-(5-Chloro-1-((trans)-3-fluoro-1-(oxetan-3-yl)piperidin-4-yl)-1H-pyrazol-4-yl)-N4-methyl-5-(trifluoromethyl)pyrimidine-2,4-diamine (20)
A mixture of (±)-(trans)-4-(5-chloro-4-nitro-1H-pyrazol-1-yl)-3-fluoro-1-(oxetan-3-yl)piperidine (53, 2.2 g, 3.9 mmol), iron dust (1.6 g, 29 mmol), and ammonium chloride (1.5 g, 29 mmol) in ethanol (20 mL) was stirred at 90 °C for 30 min. The reaction was filtered and concentrated. The residue was sonicated with 100 mL of EtOAc for 5 min. The mixture was filtered to remove all insoluble solids. The filtrate was then concentrated to give crude (±)- (trans)-4-(5-chloro-4-amino-pyrazol-1-yl)-3-fluoro-1-(oxetan-3-yl)piperidine (1.9 g).
To a mixture of the crude (±)-(trans)-4-(5-chloro-4-amino-pyrazol-1-yl)-3-fluoro-1-(oxetan-3-yl)piperidine (1.9 g) and 2-chloro-N-methyl-5-(trifluoromethyl)pyrimidin-4-amine (26, 1.5 g, 6.9 mmol) in 2-methoxyethanol (25 mL) was added TFA (0.60 mL, 7.7 mmol). The reaction was stirred at 90 °C for 15 min. The mixture was then diluted with saturated sodium bicarbonate and extracted with EtOAc (3×). The combined extracts were washed with brine, dried over sodium sulfate, filtered, and concentrated. The crude product was purified by preparative HPLC, chiral SFC, and recrystallized in isopropanol to give 20 (0.70 g, 40% yield). 1H NMR (400 MHz, DMSO) δ 8.91 (s, 1H), 8.08 (s, 1H), 7.87 (s, 1H), 7.00 (s, 1H), 5.03 4.79 (m, 1H), 4.56 (m, 1H), 4.46 (m, 2H), 3.68–3.51 (m, 1H), 3.26–3.12 (m, 1H), 2.92–2.73 (m, 3H), 2.54 (s, 2H), 2.20–1.88 (m, 3H). HRMS (ES) m/z: [M + H]+ calcd for C17H20ClF4N7OH+, 450.1427; found, 450.1418.
Scheme 8

Scheme 8. Synthesis of Inhibitor 20a
aReagents and conditions: (a) (±)-(cis)-tert-butyl 3-fluoro-4-hydroxypiperidine-1-carboxylate, PPh3, diisopropyl azodicarboxylate, THF; (b) TFA, DCM, 58% over two steps; (c) oxetan-3-one, DIPEA, NaBH(OAc)3, acetic acid, DCE, 85%; (d) LiHMDS then C2Cl6, THF, −78 °C, 65%; (e) iron dust, NH4Cl, EtOH, 90 °C; (f) 26, TFA, 2-methoxyethanol, 90 °C, 40%, two steps.
REFERENCES
1: Estrada AA, Chan BK, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, Chen H, Dominguez SL, Dotson J, Drummond J, Flagella M, Fuji R, Gill A, Halladay J, Harris SF, Heffron TP, Kleinheinz T, Lee DW, Pichon CE, Liu X, Lyssikatos JP, Medhurst AD, Moffat JG, Nash K, Scearce-Levie K, Sheng Z, Shore DG, Wong S, Zhang S, Zhang X, Zhu H, Sweeney ZK. Discovery of Highly Potent, Selective, and Brain-Penetrant Aminopyrazole Leucine-Rich Repeat Kinase 2 (LRRK2) Small Molecule Inhibitors. J Med Chem. 2014 Jan 15. [Epub ahead of print] PubMed PMID: 24354345.
/////////////DNL-151, DNL 151, DNL151, Alzheimer’s disease, breast tumor, type I diabetes mellitus, Crohn’s disease, phase 1, Parkinson’s disease, GNE0877, GNE 0877, GNE-0877, GNE-9605, GNE 9605, GNE9605, Genentech
CC(N1N=C(C)C(NC2=NC=C(C(F)(F)F)C(NC)=N2)=C1)(C)C#N
SHR-0532
SHR-0532
CAS 2166329-09-3
C24 H26 N4 O5 . C4 H6 O6
2-Pyridinecarboxamide, 5-cyano-N-[1-[(2R)-2-(1,3-dihydro-4-methyl-1-oxo-5-isobenzofuranyl)-2-hydroxyethyl]-4-piperidinyl]-4-methoxy-, (2R,3R)-2,3-dihydroxybutanedioate (1:1)
FREE FORM
1945997-37-4
C24 H26 N4 O5
450.49
2-Pyridinecarboxamide, 5-cyano-N-[1-[(2R)-2-(1,3-dihydro-4-methyl-1-oxo-5-isobenzofuranyl)-2-hydroxyethyl]-4-piperidinyl]-4-methoxy-
5-Cyano-N-[1-[(2R)-2-(1,3-dihydro-4-methyl-1-oxo-5-isobenzofuranyl)-2-hydroxyethyl]-4-piperidinyl]-4-methoxy-2-pyridinecarboxamide
KCNJ potassium channel-1 inhibitor, Hypertension; Renal insufficiency
- Originator Jiangsu Hengrui Medicine Co.
- Class Antihypertensives
- Mechanism of Action Undefined mechanism
- Preclinical Hypertension
- 03 Jun 2019 Jiangsu Hengrui Medicine Co. plans a phase I trial for Hypertension (PO) in June 2019 (NCT03971929)
- 26 Aug 2018 Jiangsu HengRui Medicine plans a phase I trial for Hypertension (In volunteers) (PO) in August 2018 (NCT03645278)
Jiangsu Hengrui Medicine is developing an oral tablet formulation of SHR-0532, a small molecule specific inhibitor of ROMK (renal outer medullary potassium channel), for use as a diuretic to treat hypertension and renal insufficiency inducing water and sodium retention. In January 2019 a phase trial was completed, and in June 2019, another phase I trial for mild hypertension was planned.
PATENT
WO2016091042
WO 2017211271
CN 108113988
PATENT
WO2019011200
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019011200&redirectedID=true
Diuretics are widely recommended as first-line antihypertensive drugs in national hypertension guidelines for mild to moderate hypertension, especially in elderly hypertension or complicated heart failure.
Clinically, traditional diuretics have a risk of causing hypokalemia. ROMK antihypertensive diuretic development of new targets, as ROMK of inward rectifier K + channel (inwardly rectifying K channels, Kir) a family, belong Kir1 type, the maintenance of renal potassium ions play a crucial balance effect. In the rat kidney, there are at least three subtypes of ROMK channels: ROMK1, ROMK2, and ROMK3. Most of ROMK2 is distributed in the ascending limb of Henle (TALH); ROMK1 and ROMK3 are mainly expressed on the cortical collecting duct (CCD). Expressed in the TALH and ROMK of Na + / K + / 2Cl – transporter with regulating the secretion of potassium ions and sodium reabsorption, and expressed in the CCD ROMK of Na + / K + secretion was adjusted with potassium transporter. Therefore, blocking the ROMK site can be a good diuretic research direction by inhibiting the reabsorption of Na + by diuretic and reducing blood potassium and causing hypokalemia.
WO2016091042A1 (publication date 2016-06-16) discloses a class of extrarenal medulla secretory potassium channel (ROMK) inhibitors, chemical name (R)-5-cyano-N-(1-(2-hydroxy-2) a compound of (4-methyl-1-oxo-1,3-dihydroisobenzofuran-5-yl)ethyl)piperidin-4-yl)-4-methoxypyridinecarboxamide, relative In other ROMK inhibitors, the compound increases the polar group, lowers the ClogP, enhances the hERG selectivity and increases the safety based on the activity of the ROMK inhibitor, and its structure is as shown in the formula (A).
Example 1 of WO2016091042A1 discloses a preparation method of Compound A, which has a total of five steps of reaction, and the specific reaction is as follows:
The method has the problems of more reaction steps, small batch size, post-treatment method using thin layer chromatography purification, low yield, etc., wherein the yield of the second step reaction is 22.4%, and the yield of the product prepared in the last step is only 11.3. % is not conducive to industrial expansion of production, it is necessary to improve its preparation method.
Example 1. Preparation of (R)-4-methyl-5-(oxiran-2-yl)isobenzofuran-1(3H)-one
First step, preparation of compound of formula (h)
Sodium borohydride (57.8 g) was dissolved in tetrahydrofuran (2000 mL), argon-protected, cooled to 0 ° C, material i (130.0 g) was added portionwise, and stirred at 5-10 ° C for 1 hour, 5-10 ° C Add boron trifluoride diethyl ether (237 mL) dropwise, stir at room temperature for 4 hours, stop the reaction, add methanol (800 mL) to quench the reaction, stir, add 1N hydrochloric acid (1000 mL) solution, stir at 0-20 ° C for 1 hour, decompress The organic solvent was evaporated, and the residue was evaporated. mjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj Concentration gave the title product (95 g).
The second step, the preparation of the compound of formula (g)
The raw material h (120.0 g) and trifluoroacetic acid (64 mL) were dissolved in acetonitrile (1 L), stirred, and cooled to 0-5 ° C under ice bath, and solid N-bromosuccinimide (147.0 g) was added portionwise. The reaction temperature was controlled at 0-8 ° C. After the reaction was completed, the reaction was quenched by adding 200 mL of potassium carbonate aqueous solution (containing 66.0 g of potassium carbonate) under ice-cooling, and concentrated under reduced pressure, water (200 mL) and ethyl acetate (800 mL) ×1,400 mL×2), and the organic phase was combined with EtOAc EtOAc (EtOAc m. Drying gave 150.0 g of product.
The third step, the preparation of the compound of formula (f)
The cuprous cyanide (123.0 g) was added to N,N-dimethylformamide (500 mL), and the material g (150.0 g) was dissolved in N,N-dimethylformamide (250 mL), and added to the dropping funnel. Under an argon atmosphere, after heating to 140-150 ° C, the N,N-dimethylformamide solution of the raw material g was added dropwise, and the reaction was stirred at 145 ° C for 2 hours. After the reaction was completed, the temperature was lowered to 90-95 ° C, and the mixture was added dropwise. Ionized water (62 mL), reacted for 18 hours, stopped the reaction, and cooled to room temperature. The reaction solution was added to a mixed solvent of isopropyl acetate/methanol (V/V = 4:1, 1500 mL), stirred for 30 minutes, and padded with silica gel and silicon. The mixture was filtered with celite, and the filter cake was washed with isopropyl acetate/methanol (V/V = 4:1, 100 mL×3), and the filtrate was concentrated under reduced pressure. The residue was slowly added to deionized water (3 L) and stirred for 1 hour. Filtration, the filter cake was washed with ethanol (50 mL×3), and the filter cake was dried to give 133.0 g of crude product. The crude product was added to ethyl acetate/methanol (V/V=4:1, 2.0L) and heated to reflux. After filtration, the cake was washed with ethyl acetate /methanol (EtOAc/EtOAc (EtOAc)
The fourth step, the preparation of the compound of formula (e)
The starting material f (26.0 g) was dissolved in dichloromethane (520 mL), triethylamine (33 mL) was added, and the mixture was cooled to -5-0 ° C and added trifluoromethanesulfonic anhydride (29.2 mL), 0-10 After reacting at ° C for 2 hours, the reaction was stopped. Under ice-cooling conditions, water (250 mL) was added dropwise to the reaction mixture to quench the reaction, and the mixture was separated, and the aqueous phase was extracted with dichloromethane (100 mL×2). The sodium solution (300 mL) was washed with EtOAc EtOAc (mjjHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH After dissolving at 70 ° C, the supernatant liquid was separated, and the lower layer of the oil was dissolved in a mixed solution of petroleum ether and ethyl acetate (V/V = 5:1) (300 mL × 2), and the organic phases were combined and concentrated under reduced pressure. (41.0 g), EtOAc (EtOAc m.
The fifth step, the preparation of the compound of formula (d)
The starting material e (50.1 g) was dissolved in isopropanol (500 mL), and ethylene trifluoroborate (29.5 g) and 1,1′-bisdiphenylphosphinoferrocene palladium dichloride (1.25 g) were added. Further, triethylamine (71 mL) was added, and the reaction was refluxed for 1.5 hours under an argon atmosphere. The reaction was stopped, cooled to room temperature, filtered, and the filtrate was washed with ethyl acetate (20 mL×3), and the filtrate was concentrated and concentrated through silica gel column. The title product (29.0 g) was obtained (yield: ethyl acetate: petroleum ether = 1:5-1:3).
The sixth step, the preparation of the compound of formula (c)
Potassium ferricyanide (279.0 g) was added to the reaction flask, followed by potassium carbonate (116.0 g) and hydrogenated quinidine 1,4-(2,3-naphthyridinyl)diether ((DHQD) 2 PHAL , 1.1g) and potassium citrate dihydrate (103mg), add 2L of deionized water, stir for 30 minutes, add tert-butanol (1.5L) under argon atmosphere, stir for 15 minutes, 0-5 ° C raw material d ( 49.0g) was added in portions, stirred at 0-5 ° C for 4 hours, warmed to room temperature and stirred for 18 hours, the reaction was stopped, saturated sodium sulfite solution (800 mL) and ethyl acetate (1000 mL) were added, stirred until fully dissolved, layered, The aqueous layer was extracted with EtOAc (EtOAc (EtOAc) (EtOAc (EtOAc) The mixture was cooled to rt.
The seventh step, the preparation of the compound of formula (a)
The raw material c (54.0 g) was added to dichloromethane (600 mL), and the mixture was white turbid. Under argon atmosphere, b (46.9 g) was added, stirred at room temperature for 10 minutes, cooled to 0 ° C, and trimethylchlorosilane was added dropwise. (54.0g), stirring at 0 ° C for 30 minutes, the solution became clear, warmed to room temperature for 1 hour, then cooled to 0 ° C, added b (23.0g), raised to room temperature for 30 minutes, stop the reaction, the reaction solution Concentration under reduced pressure gave the crude title product which was used in the next step without purification.
The eighth step, the preparation of the compound of formula (VI)
The raw material a (69.6 g) was added to methanol (1000 mL), and potassium carbonate (90.0 g) was added, and the mixture was stirred at room temperature for 2 hours, the reaction was stopped, and the mixture was evaporated under reduced pressure. ethyl acetate (500 mL) and water (200 mL) The aqueous phase was extracted with EtOAc (EtOAc (EtOAc) (EtOAc) The title compound (35.0 g) was obtained in vacuo.
Example 2 Preparation of 5-cyano-4-methoxypyridinecarboxylic acid hydrochloride
First step, preparation of the compound of formula (p)
Raw material n (110.0g), o (150.0g), acetic anhydride (151.5g) was added to the reaction flask and refluxed for 4 hours, the reaction was stopped, concentrated under reduced pressure, and the obtained residue was controlled at a temperature of 0-10 ° C to add ammonia water and Water (V / V = 1:1, 600mL) mixed solution, when a large amount of solids were formed, add ice water (400mL), drip, stir for 30 minutes, adjust to pH 2-3 with concentrated hydrochloric acid, stir 30 After a minute, the mixture was filtered, and the filter cake was dried, and then filtered with anhydrous ethanol (500 mL) for 1 hour, filtered, filtered, washed with cold anhydrous ethanol (100 mL×3), and the filter cake was dried to give the title product (80.0 g) The yield was 59%.
The second step, the preparation of the compound of formula (q)
Sodium hydroxide (43.6 g) was added to water (800 mL) under ice bath, and the starting material p (79.8 g) was added portionwise to the above aqueous sodium hydroxide solution, the ice bath was removed, and the mixture was heated to reflux for 2 hours to terminate the reaction. The reaction solution was cooled to room temperature with ice water, 2M hydrochloric acid solution was added dropwise to adjust the pH to 2-3, stirred for 30 minutes, filtered, and the filter cake was washed with ice water (100 mL) and cold ethanol (100 mL), and the obtained solid was dried. The title product (71.2 g), yield 100%.
The third step, the preparation of the compound of formula (r)
The raw material q (70.3 g) was dissolved in phosphorus oxychloride (210 mL), stirred at 110 ° C for 2 hours under reflux, concentrated under reduced pressure to remove phosphorus oxychloride, and the residue was added to acetonitrile (350 mL). Add diisopropylethylamine (117.0 g), dilute the solution to a black suspension, add the suspension to the ammonia water (350 mL) under ice bath, drop the reaction for 30 minutes, ethyl acetate (500 mL × 3) Extraction, the organic phase was combined, washed with saturated sodium chloride (500 mL), dried over anhydrous sodium sulfate, filtered and evaporated. (44.7 g), yield 51%.
The fourth step, the preparation of the compound of formula (s)
The raw material r (44.3 g) was added to dichloromethane (440 mL) under an argon atmosphere, and the temperature was controlled to 0-5 ° C under ice-cooling, triethylamine (58.6 g) was added dropwise, and the mixture was stirred for 10 minutes. Trifluoroacetic anhydride (58.5g) was added dropwise, the addition was completed, and the reaction was carried out for 1 hour in an ice bath. The reaction was stopped, the pH of the reaction mixture was 7-8, and the reaction was quenched by adding water (400 mL), and the mixture was separated. The organic phase was extracted with EtOAc (EtOAc) (EtOAc) (HHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH g), yield 91%.
The fifth step, the preparation of the compound of formula (t)
The raw material s (25.6 g) and cesium carbonate (49.2 g) were dissolved in N,N-dimethylformamide (260 mL), cooled to 0 ° C in an ice bath, and methanol (9.5 g) was added dropwise in an ice bath, 0 ° C After reacting for 6 hours, the mixture was stirred at 20-25 ° C for 12 hours, and the reaction was stopped. The reaction mixture was quenched with water (650 mL), and extracted with ethyl acetate (200 mL×3). The title compound (16.8 g) was obtained. The title compound (16.8 g) was obtained from EtOAc (EtOAc). The rate is 67%.
The sixth step, the preparation of the compound of formula (II-1)
Add t (22.0g), palladium acetate (1.46g), 1,3-bis(diphenylphosphino)propane (2.68g), triethylamine (36mL) to the mixed solution, pressurize with carbon monoxide to 10bar, heat up The reaction was stopped at 70 ° C for 18 hours, the reaction was stopped, the organic solvent was removed by concentration, the aqueous phase was added with saturated sodium chloride solution and dichloromethane (300 mL×3), and the organic phase was combined, decolorized by activated carbon, filtered, and the organic phase was adjusted to pH with concentrated hydrochloric acid. =1, a solid precipitated, and after adding 50 mL of isopropanol, the dichloromethane was concentrated to remove the product, which was filtered and dried to give a product (23.6 g).
Example 3, (R)-5-cyano-N-(1-(2-hydroxy-2-(4-methyl-1-oxo-1,3-dihydroisobenzofuran-5-yl) Preparation of ethyl)piperidin-4-yl)-4-methoxypyridinecarboxamide (formula (I))
First step, synthesis of intermediate (IV)
Into the reaction flask, 4.0 L of absolute ethanol was added, and (R)-4-methyl-5-(oxiran-2-yl)-benzoisofuran-1(3H)-one (274.8) was added under stirring. g), 4-Boc-aminopiperidine (341.2 g), heated to 65-70 ° C, stirred for 18-20 h, and the heating was stopped. Naturally cooled to 50-55 ° C, 8.0 L of n-hexane was added under stirring, stirred until the temperature naturally dropped to 20-25 ° C, a large amount of solids were precipitated, the temperature of the ice bath was lowered to 0-5 ° C, stirred, suction filtered, filter cake It was washed twice with n-hexane (250 ml × 2) and dried to give a solid (354.3 g).
The second step, the synthesis of intermediate (III-1)
5.2 L of ethyl acetate was placed in a glass bottle, and the temperature was lowered to 0 to 5 ° C under stirring. The stirring was stopped, hydrogen chloride gas (0.48 kg) was introduced, and the temperature of the reaction liquid was controlled to be lower than 5 ° C during the passage of hydrogen chloride. The above product (349.3 g) was added to the reaction mixture with slow stirring. After the addition, the reaction was stirred for 3-4 hours, and the reaction temperature was naturally raised to 20-25 ° C, and the stirring was stopped. After suction filtration, the filter cake was washed three times with ethyl acetate (1.0 L×3), and the filter cake was dried under vacuum at 40-45 ° C for 6-8 h to give a solid (322.8 g) in a yield of 99.3%; The ratio of hydrochloric acid was determined by silver nitrate titration to be 20.5%.
The third step, the synthesis of the compound of formula (I)
Into the reaction flask, 4.0 L of N, dimethylformamide was added, and the product of the above step (317.8 g), 5-cyano-4-methoxypyridinecarboxylate II-1 (205.9 g) was sequentially added with stirring. Triethylamine (528.2 g), 1-hydroxybenzotriazole (152.7 g), N,N-diisopropylcarbodiimide (142.6 g). After the addition, the argon gas was replaced three times, and the mixture was heated to 40-45 ° C to stir the reaction for 16-18 h. The heating was stopped, and the reaction liquid was poured into ice water (30 L), and stirred for 1 hour. After suction filtration, the filter cake was washed three times with purified water, dried, and then pulverized with anhydrous ethanol (3.0 L) at 20-25 ° C for 1 h. Filtering, drying 10-12h to obtain crude (290.4g), yield 73.7%, purity: 97.76%;
N,N-dimethylformamide (2.0 L) was added to the crude product (290.4 g) with stirring. The reaction solution was heated to 70-75 ° C, 20.3 g of activated carbon (7% w/w) was added, and the mixture was stirred for 1 h. Heat filtration, wash the filter residue with hot N,N-dimethylformamide (70-75 ° C, 200 mL), combine the filtrate, heat the filtrate to 70-75 ° C, add hot (65-70 ° C, 5 L) with stirring Anhydrous ethanol to the reaction liquid in the previous step, stirring and crystallization, until the temperature naturally drops to 20-25 ° C, the reaction bottle is transferred to an ice water bath and stirring for 1 h, suction filtration, the filter cake is washed with absolute ethanol, dried to obtain a solid 219.5 g, total yield 55.7%, purity: 99.69%.
1 H-NMR (400 MHz, DMSO-d 6 ) δ 8.88 (s, 1H), 8.75 (d, 1H), 7.77 (s, 1H), 7.71-7.69 (m, 2H), 5.43-5.40 (m, 2H), 5.35 (s, 1H), 5.08 (s, 1H), 4.09 (s, 3H), 3.78 (s, 1H), 2.95 (s, 3H), 2.38 (s, 1H), 2.27 (s, 3H) ), 2.25 (s, 2H), 1.72 (s, 4H).
PATENT
WO-2019109935
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019109935&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of a renal outer medullary potassium channel inhibitor and their salts, preferably Form III, for treating hypertension or heart failure.
Strengthening the salt reabsorption of the kidneys can trigger a risk of high blood pressure. On the contrary, inhibiting the reabsorption function of the kidney can promote the excretion of urine and play a diuretic and antihypertensive effect. Common diuretics are thiazide diuretics, as the first-line antihypertensive drugs in the United States, mainly acting on Na + -Cl transport carriers; Loop diuretics are more effective in patients with impaired renal function, mainly through Na + -K + -2Cl – Transport proteins play a role. But both diuretics cause hypokalemia (symptoms: weakness, fatigue, muscle cramps, constipation and heart rhythm problems such as arrhythmia), increasing the risk of cardiovascular disease morbidity and mortality.
The renal outer medullary potassium channel (ROMK) is also called inward-rectifying potassium channel 1.1 (Kir1.1). Ion channels may ROMK thick ascending limb segment (the TAL) conductance through apical membrane of renal medullary loop, and of Na + -K + -2Cl – cotransporter NKCC2 (responsible for transport of NaCl) synergy regulation of Na + reabsorption. The study found that ROMK is directly associated with the secretory pathway of the kidney, knocking out the ROMK gene, missing the 35-pS ion channel and other TAL K + ion channels of mouse TAL and CCD . Batter syndrome is an autosomal recessive hereditary disease characterized by massive loss of salt in the kidneys, hypokalemia, and low blood pressure. Paramyelocytic hyperplasia is mainly caused by mutation of ROMK or Na + -K + -2Cl – cotransporter, except that hypokalemia caused by rotaside cell hyperplasia caused by ROMK mutation is better than Na + -K + – Parathyroid cell hyperplasia induced by 2Cl – cotransporter mutations is greatly alleviated. In summary, suppressing the function of ROMK can effectively inhibit Na without causing hypokalemia. + -K + -2Cl – The salt reabsorption function of transporters promotes the excretion of urine and acts as a diuretic and antihypertensive agent .
WO2016091042A1 (Publication Date 2016.06.16) discloses an extrarenal medullary secretory potassium channel (ROMK) inhibitor having the chemical name (R)-5-cyano-N-(1-(2-hydroxy-2-() 4-methyl-1-carbonyl-1,3-dihydroisobenzofuran-5-yl)ethyl)piperidin-4-yl)-4-methoxypyridinecarboxamide relative to other ROMK inhibitors The compound increases the polar group, reduces the ClogP on the basis of maintaining the activity of the ROMK inhibitor, improves the selectivity of hERG, and increases the safety, and the structure is as shown in formula (II):
The crystal structure of the pharmaceutically active ingredient often affects the chemical and physical stability of the drug, and the difference in crystallization conditions and storage conditions may cause changes in the crystal structure of the compound, sometimes accompanied by the formation of other forms of crystal form. In general, amorphous pharmaceutical products have no regular crystal structure and often have other defects, such as poor product stability, difficulty in filtration, easy agglomeration, and poor fluidity. Therefore, it is necessary to improve various aspects of the compound of the formula (II).
/////////////SHR-0532, SHR0532, SHR 0532, Jiangsu Hengrui Medicine Co, phase I, Antihypertensives
COc1cc(ncc1C#N)C(=O)NC2CCN(CC2)C[C@H](O)c4ccc3C(=O)OCc3c4C
TAK-981

TAK-981
C25 H28 Cl N5 O5 S2, 578.103
[(1R,2S,4R)-4-[(5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl)amino]-2-hydroxycyclopentyl]methyl sulfamate
[(1R,2S,4R)-4-[[5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methyl-thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate
Sulfamic acid, [(1R,2S,4R)-4-[[5-[[4-[(1R)-7-chloro-1,2,3,4-tetrahydro-1-isoquinolinyl]-5-methyl-2-thienyl]carbonyl]-4-pyrimidinyl]amino]-2-hydroxycyclopentyl]methyl ester
| CAS 1858276-04-6 FREE
CAS 1858279-63-6 HYDRATE |
|
| MW | 578.103 |
- Originator Takeda Oncology
- Class Antineoplastics
- Mechanism of Action Small ubiquitin-related modifier protein inhibitors
- Phase I Lymphoma; Solid tumours
- 01 Oct 2018 Phase-I clinical trials in Solid tumours (Late-stage disease, Metastatic disease) and and Lymphoma (Refractory metastatic disease, Second-line therapy or greater) in USA (IV) (NCT03648372)
- 03 Sep 2018 Takeda Oncology plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) and Lymphoma (Refractory metastatic disease, Second-line therapy or greater) in September 2018 (IV) (NCT03648372)
- 03 Sep 2018 Preclinical trials in Lymphoma in USA (IV) prior to September 2018 (NCT03648372)
Takeda is evaluating TAK-981, a SUMO-Activating Enzyme (SAE) inhibitor, in early clinical trials for the treatment of adult patients with advanced or metastatic solid tumors or with relapsed or refractory lymphomas.
Small ubiquitin-like modifier (SUMO) is a member of the ubiquitin-like protein (Ubl) family that is covalently conjugated to cellular proteins in a manner similar to Ub-conjugation (Kerscher, O., Felberbaum, R., and Hochstrasser, M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 22: 159-80). Mammalian cells express three major isoforms: SUMO l , SUM02 and SUM03. SUM02 and SUM03 share -95% amino acid sequence homology but have -45% sequence homology with SUMO l (Kamitani, T., Kito, K., Nguyen, H. P., Fukuda-Kamitani, T., and Yeh, E. T. 1998. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J Biol Chem. 273( 18): 1 1349-53). SUMO proteins can be conjugated to a single lysine residue of a protein (monosumoylation) or to a second SUMO protein that is already conjugated to a protein forming a SUMO chain (polysumoylation). Only SUM02/3 can form such chains because they possess internal consensus SUMO modification sites (Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H., Naismith, J. H., Hay, R. T. 2001. Polymeric chains of SUMO-2 and SUM 0-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 276(38):35368-74). An additional isoform, SUM04, is found in kidney, lymph node and spleen cells, but it is not known whether SUM04 can be conjugated to cellular proteins.
[0003] SUMO l , SUM02 and SUM03 are activated in an ATP-dependent manner by the SUMO-activating enzyme (SAE). SAE is a heterodimer that consists of SAE 1 (SUMO-activating enzyme subunit 1) and SAE2 (UBA2). SAE, like other El activating enzymes, uses ATP to adenylate the C-terminal glycine residue of SUMO. In a second step, a thioester intermediate is then formed between the C-terminal glycine of SUMO and a cysteine residue in SAE2. Next, SUMO is transferred from the El to the cysteine residue of the SUMO conjugating enzyme (E2), UBC9. Unlike the Ub pathway that contains many E2 enzymes, Ubc9 is currently the only known conjugating enzyme for SUMO and functions with SUMOl , SUM02 and SUM03 proteins. SUMO proteins are then conjugated to the target protein, either directly or in conjunction with an E3 ligase, through isopeptide bond formation with the epsilon amino group of a lysine side chain on a target protein. Several SUMO E3 ligases, including PIAS (protein inhibitor of activated signal transducer and activator of transcription protein) proteins and Ran-binding protein 2 (RanBP2), and polycomb 2 (Pc2), have been identified (Johnson, E. S., and Gupta, A. A. 2001. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 106(6):735-44; Pichler, A., Gast, A., Seeler, J. S., Dejean, A.; Melchior, F. 2002. The nucleoporin RanBP2 has SUMOl E3 ligase activity. Cell. 108(1): 109-20; Kagey, M. H., Melhuish, T. A., and Wotton, D. 2003. The polycomb protein Pc2 is a SUMO E3. Cell. 1 13(1): 127- 37). Once attached to cellular targets, SUMO modulates the function, subcellular localization, complex formation and/or stability of substrate proteins (Miiller, S., Hoege, C, Pyrowolakis, G., and Jentsch, S. 2001. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2(3):202-10). SUMO- conjugation is reversible through the action of de-sumoylating enzymes called SENPs (Hay, R. T. 2007. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 17(8):370-6) and the SUMO proteins can then participate in additional conjugation cycles.
[0004] SAE-initiated SUMO-conjugation plays a major role in regulating diverse cellular processes, including cell cycle regulation, transcriptional regulation, cellular protein targeting, maintenance of genome integrity, chromosome segregation, and protein stability (Hay, R. T. 2005. SUMO: a history of modification. Mol Cell. 18( 1): 1 -12; Gill, G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18(17):2046-59). For example, SUMO- conjugation causes changes in the subcellular localization of RanGAPl by targeting it to the nuclear pore complex (Mahajan, R., Delphin, C., Guan, T., Gerace, L., and Melchior, F. 1997. A small ubiquitin-related polypeptide involved in targeting RanGAPl to nuclear pore complex protein RanBP2. Cell. 88(1):97- 1070). Sumoylation counteracts ubiquitination and subsequently blocks the degradation of Ι Β, thereby negatively regulating NF-κΒ activation (Desterro, J. M., Rodriguez, M. S., Hay, R. T. 1998. SUMO- 1 modification of IkappaB alpha inhibits NF-kappaB activation. Mol Cell. 2(2):233-9). Sumoylation has been reported to play an important role in transcription exhibiting both repressive and stimulatory effects. Many of the transcriptional nodes that are modulated play important roles in cancer. For example, sumoylation stimulates the transcriptional activities of transcription factors such as p53 and HSF2 (Rodriguez, M. S., Desterro, J. M., Lain, S., Midgley, C. A., Lane, D. P., and Hay, R. T. 1999. SUMO- 1 modification activates the transcriptional response of p53. EMBO J. 18(22):6455-61 ; Goodson, M. L., Hong, Y., Rogers, R., Matunis, M. J., Park-Sarge, O. K., Sarge, K. D. 2001. Sumo- 1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J Biol Chem. 276(21 ): 18513-8). In contrast, SUMO-conjugation represses the transcriptional activities of transcription factors such as LEF (Sachdev, S., Bruhn, L., Sieber, H., Pichler, A., Melchior, F., Grosschedl, R. 2001. PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15(23):3088- 103) and c-Myb (Bies, J., Markus, J., and Wolff, L. 2002. Covalent attachment of the SUMO- 1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. / Biol Chem. 277( 1 1):8999-9009). Thus, SUMO-conjugation controls gene expression and growth control pathways that are important for cancer cell survival.
[0005] Altered expression of SAE pathway components have been noted in a variety of cancer types: (Moschos, S. J., Jukic, D. M., Athanassiou, C., Bhargava, R., Dacic, S., Wang, X., Kuan, S. F., Fayewicz, S. L., Galambos, C., Acquafondata, M., Dhir, R., and Becker, D. 2010. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum Pathol. 41(9): 1286-980); including multiple myeloma (Driscoll, J. J., Pelluru, D., Lefkimmiatis, K., Fulciniti, M., Prabhala, R. H., Greipp, P. R., Barlogie, B., Tai, Y. T., Anderson, K. C, Shaughnessy, J. D. Jr., Annunziata, C. M., and Munshi, N. C. 2010. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 1 15(14):2827-34); and breast cancer (Chen, S. F., Gong, C, Luo, M., Yao, H. R., Zeng, Y. J., and Su, F. X. 201 1. Ubc9 expression predicts chemoresistance in breast cancer. Chin J Cancer. 30(9):638-44), In addition, preclinical studies indicate that Myc-driven cancers may be especially sensitive to SAE inhibition (Kessler, J. D., Kahle, K. T., Sun, T., Meerbrey, K. L., Schlabach, M. R., Schmitt, E. M., Skinner, S. O., Xu, Q., Li, M. Z., Hartman, Z. C, Rao, M., Yu, P., Dominguez-Vidana, R., Liang, A. C, Solimini, N. L., Bernardi, R. J., Yu, B., Hsu, T., Golding, I., Luo, J., Osborne, C. K., Creighton, C. J., Hilsenbeck, S. G., Schiff, R., Shaw, C. A., Elledge, S. J., and Westbrook, T. F. 2012. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 335(6066):348-53; Hoellein, A., Fallahi, M., Schoeffmann, S., Steidle, S., Schaub, F. X., Rudelius, M., Laitinen, I., Nilsson, L., Goga, A., Peschel, C, Nilsson, J. A., Cleveland, J. L., and Keller, U. 2014. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood. 124( 13):2081 -90). Since SUMO-conjugation regulates essential cellular functions that contribute to the growth and survival of tumor cells, targeting SAE could represent an approach to treat proliferative disorders such as cancer.
[0006] SAE inhibitors may also be applicable for the treatment of other diseases and conditions outside of oncology. For example, SUMO modifies proteins that play important roles in neurodegenerative diseases (Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C, Slepko, N., Hies, K., Lukacsovich, T., Zhu, Y. Z., Cattaneo, E., Pandolfi, P. P., Thompson, L. M., Marsh, J. L. 2004. SUMO modification of Huntington and Huntington’s disease pathology. Science. 304(5667): 100-4); Dorval, V., and Fraser, P. E. 2006. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 281 ( 15):9919-24; Ballatore, C, Lee, V. M., and Trojanowski, J. Q. 2007. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 8(9):663-72). Sumoylation also has been reported to play important role in pathogenic viral infection, inflammation and cardiac function (Lee, H. R., Kim, D. J., Lee, J. M., Choi, C. Y., Ahn, B. Y., Hayward, G. S., and Ahn, J. H. 2004. Ability of the human cytomegalovirus ΓΕ1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. / Virol. 78(12):6527-42; Liu, B., and Shuai, K. 2009. Summon SUMO to wrestle with inflammation. Mol Cell. 35(6):731-2; Wang, J., and Schwartz, R. J. 2010. Sumoylation and regulation of cardiac gene expression. Circ Rei. l07( l): 19-29). [0007] It would be beneficial therefore to provide new SAE inhibitors that possess good therapeutic properties, especially for the treatment of proliferative, inflammatory, cardiovascular and neurodegenerative disorders.
PATENT
WO 2016004136

https://patents.google.com/patent/WO2016004136A1/en
Example 133: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate I-263a

Step 1: 7-Chloro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-l,2,3,4-tetrahydroisoquinoline
[00714] An oven-dried 2-neck 250 mL round bottom flask under nitrogen was charged with THF (40 mL) and cooled to -74 °C . Added 2.50 M ra-BuLi in hexane (6.92 mL, 17.3 mmol). Added a solution of Int-1 (4.00 g, 16.0 mmol) in THF (60 mL) slowly keeping the internal temperature less than -70 °C . Stirred with cooling 5 min. A second oven-dried 250 mL round bottom flask under nitrogen was charged with THF (60 mL) and Int-50 (2.04 g, 12.4 mmol) and the resulting solution was cooled to 0 °C . Added boron trifluoride diethyl ether complex ( 1.71 mL, 13.6 mmol) slowly and cooled to -30 °C . The contents of the first flask were transferred via cannula to the second flask. Reaction was quenched with saturated aqueous NaHC03 and warmed to rt. Water was added, and the mixture was extracted three times with EtOAc. Combined organic portions were washed with brine, dried over anhydrous Na2S04, filtered, and concentrated in vacuo. Residue was purified via flash column chromatography eluting with a hexane / EtOAc gradient (0 to 100% EtOAc) to afford the title compound as a white solid ( 1.88g, 45%). Ή NMR (400 MHz, Chloroform-d) δ 7.17 – 7.01 (m, 2H), 6.83 – 6.61 (m, 2H), 5.92 (s, 1H), 5.09 (s, 1H), 4.17 – 4.04 (m, 2H), 4.03 – 3.92 (m, 2H), 3.37 – 3.25 (m, 1H), 3.13 – 2.91 (m, 2H), 2.82 – 2.69 (m, 1H), 2.46 (s, 3H). LCMS: (AA) M+l 336.1
Step 2: ieri-Butyl 7-chIoro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3,4-dihydroisoquinoIine -2(lH)-carboxyIate [00715] A 50 mL round bottom flask under nitrogen was charged with 7-chloro-l -[5-(l ,3-dioxolan-2- yl)-2-methyl-3-thienyl]- l ,2,3,4-tetrahydroisoquinoline (5.67 g, 16.9 mmol) and DCM ( 100 mL), to which was added triethylamine (4.71 mL, 33.8 mmol), di-ieri-butyldicarbonate (4.61 g, 21.1 mmol), and N,N-dimethylaminopyridine (23 mg, 0.18 mmol). Reaction was stirred for 1 h at rt and then poured into saturated NaHC03 solution. Mixture was extracted three times with DCM, and the combined organic portions were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford 6.96g (95%) of the title compound. LCMS: (AA) M+ l 436.1
Step 3: tert-Butyl 7-chloro-l-(5-formyl-2-methyl-3-thienyl)-3,4-dihydroisoquinoline -2(1H)- carboxylate
[00716] A 1 L round bottom flask was charged with ferf-butyl 7-chloro-
1 -[5-( 1 ,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3 ,4-dihydroisoquinoline-2( 1 H)-carboxylate (7.30 g, 16.7 mmol), methanol (200 mL), and water (20 mL), to which was added a solution of 12M HC1 (4.00 mL, 130 mmol) in methanol (200 mL), and the reaction was stirred at rt for 1 h. Reaction was quenched via addition of 50mL of saturated NaHC03 and stirred for 5 min. Methanol was removed in vacuo, and the resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.55g, 70%). Ή NMR (400 MHz, Chloroform-d) δ 9.67 (s, 1 H), 7.27 – 7.15 (m, 2H), 7.12 (s, 1 H), 6.98 – 6.94 (m, 1 H), 6.34 (m, l H), 4.15 (s, 1 H), 3.18 – 3.06 (m, 1 H), 3.05 – 2.93 (m, 1H), 2.82 – 2.73 (m, 1 H), 2.69 (s, 3H), 1.50 (s, 9H). LCMS: (AA) M+Na 414.2
Step 4: tert-Butyl 7-chIoro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyI]-2-methyl-3-thienyl}- 3,4-dihydroisoquinoline-2(lH)-carboxylate
[00717] An oven-dried 500 mL 3-neck round bottom flask under nitrogen was charged with 4-chloro- 5-iodopyrimidine (4.08 g, 17.0 mmol) and 2-methyltetrahydrofuran ( 150 mL). An addition funnel containing a solution of rert-butyl 7-chloro- l -(5-formyl-2-methyl-3-thienyl)-3,4- dihydroisoquinoline-2(l H)-carboxylate (4.75 g, 12.1 mmol) in 2-methyltetrahydrofuran (50 mL) was attached, and the contents of the reaction flask were cooled to -75 °C . 2.50 M n-BuLi in hexane ( 14.1 mL, 35.2 mmol) was added in small portions keeping the internal temperature less than -70 °C , at which point the contents of addtion funnel were added in a single portion. Upon completion of addition, the reaction was quenched by adding 20 mL of saturated NaHC03 in small portions and warmed to rt. The aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.85g, 79%). LCMS: (AA) M+Na 528.1
Step 5: tert-Butyl 7-chloro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyl]-2-methyl-3-thienyl}- 3,4- dihydroisoquinoline-2(lH)-carboxylate
[00718] A 1 L round bottom flask was charged with fe/Y-butyl 7-chloro- l – { 5-[(4-chloropyrimidin-5- yl)(hydroxy)methyl]-2-methyl-3-thienyl}-3,4-dihydroisoquinoline-2(l H)-carboxylate (4.85 g, 9.58 mmol) and DCM (300 mL). Manganese (IV) oxide (14.2 g, 163 mmol) was added and the reaction was stirred at rt for 18 h. Mixture was filtered through Celite, and the filter cake was rinsed with hot EtOAc. Filtrate was concentrated in vacuo to afford the title compound (4.47g , 93%). Ή NMR (400 MHz, Chloroform-d) δ 9.09 (s, 1 H), 8.70 (s, 1 H), 7.24 – 7.16 (m, 1 H), 7.16
– 7.07 (m, 1 H), 7.00 – 6.90 (m, 2H), 6.32 (s, 1 H), 4.28 – 3.97 (m, 1H), 3.14 – 2.89 (m, 2H), 2.78
– 2.65 (m, 4H), 1 .53 – 1.43 (m, 9H).
Step 6: tert-Butyl (lR)-7-chloro-l-[5-[4-[[(lR,3R,4S)-3-(hydroxymethyl)-4-triisopropylsiIyloxy- cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dihydro-lH-isoquinoline-2- carboxylate
[00719] A 1 L round bottom flask under nitrogen was charged with iert-butyl 7-chloro- l – { 5-[(4- chloropyrimidin-5-yl)carbonyI]-2-methyl-3-thienyl }-3,4-dihydroisoquinoline-2( l H)-carboxylate (4.47 g, 8.86 mmol), DMF (20.0 mL, 258 mmol), Int-259 (3.06 g, 10.6 mmol), and triethylamine (3.09 mL, 22.2 mmol) and the mixture was stirred at rt for 18 h. Reaction mixture was poured into water and saturated NaHC03, and then extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a 70/30 to 60/40 hexane/EtOAc gradient to afford 0.56g of first-eluting diastereomer 1 (not pictured), 4.3 l g of a mixture of diastereomers, and 1.1 lg ( 17%) of second-eluting diastereomer 2 (the title compound). The mixture of diastereomers thus obtained was resubjected to the described chromatography conditions two additional times to afford a total of 2.62 g of the desired diastereomer. Ή NMR (400 MHz, Methanol-d4) δ 8.54 – 8.46 (m, 2H), 7.27 – 7.19 (m, 2H), 7.09 – 6.99 (m, 2H), 6.37 (s, 1H), 4.87 – 4.75 (m, 1H), 4.38 – 4.29 (m, 1H), 4.20 – 4.09 (m, 1H), 3.66 – 3.52 (m, 2H), 3.28- 3.14 (m, 2H), 3.02 – 2.89 (m, 1 H), 2.89 – 2.78 (m, 1 H), 2.68 (s, 3H), 2.54 – 2.41 (m, 1 H), 2.22 – 2.09 (m, 2H), 1.86 – 1.73 (m, 1H), 1.50 (s, 8H), 1.39 – 1.23 (m, 2H), 1.15 – 1.04 (m, 20H).
LCMS: (AA) M+ 1 755.3
Step 7: tert-Butyl (lR)-7-chloro-l-[2-methyl-5-[4-[[(lR,3R,4S)-3-(sulfamoyloxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3-thienyl]-3,4-dihydro-lH- isoquinoline-2-carboxylate [00720] A solution of ie/t-butyl (lR)-7-chloro-l-[5-[4-[[( lR,3R,4S)-3-(hydroxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dih lH-isoquinoline-2-carboxylate (2.46 g, 3.26 mmol) in 2-methyltetrahydrofuran (25 mL), and DMF (25 mL) was cooled to 0 °C. Triethylamine ( 1.82 mL, 13.0 mmol) and chlorosulfonamide (1.50 g, 13.0 mmol) were added and the reaction was stirred for 10 min. Added methanol (0.53 mL, 13.0 mmol) and stirred for 15 min. Reaction mixture was poured into saturated NaHC03, extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (2.41g, 89%). Ή NMR (400 MHz, Methanol-d4) δ 8.58 – 8.45 (m, 2H), 7.29 – 7.17 (m, 2H), 7.1 1 – 6.98 (m, 2H), 6.36 (s, 1 H), 4.84 – 4.73 (m, 1H), 4.44 – 4.33 (m, 1H), 4.21 – 4.08 (m, 4H), 3.27- 3.17 (m, 1 H),3.02 – 2.89 (m, 1 H), 2.88 – 2.78 (m, 1 H), 2.67 (s, 3H), 2.57 – 2.47 (m, 1 H), 2.41 – 2.30 (m, 1 H), 2.23 – 2.13 (m, 1 H), 1.87- 1.78 (m, 1 H), 1.50 (s, 9H), 1.43 – 1 .33 (m, 1 H), 1 .17 – 1.04 (m, 20H). LCMS: (AA) M+l 834.3
Step 8: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yI]aniino]-2-hydroxy-cyclopentyl]methyl sulfamate
[00721] A solution of f«?r/-butyl ( l R)-7-chloro- l -[2-methyl-5-[4-[[( l R,3R,4S)-3-
(sulfamoyloxymethyl)-4-triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3- thienyl]-3,4-dihydro- l H-isoquinoline-2-carboxylate (2.41 g, 2.89 mmol) in CH3CN ( 10 mL) was cooled in an ice bath to + 1 °C . Phosphoric acid ( 10 mL, 200 mmol) was added dropwise and the reaction was stirred with ice bath cooling for 60 min. The mixture was warmed to rt and stirred for an additional 3 h. Reaction was poured into a stirring mixture of 50 mL water and 50 mL EtOAc, and the the pH was adjusted to ~9 by slowly adding 200 mL of saturated NaHC03 with stirring. Resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a gradient that began with 100% DCM and increased in polarity to 80% DCM / 20% methanol / 2% ammonium hydroxide gradient to afford the title compound (1.50 g, 90%). Ή NMR (400 MHz, Methanol-d4) δ 8.61 (s, 1H), 8.52 (s, 1 H), 7.27 (s, 1 H), 7.18 – 7.13 (m, 2H), 6.73 – 6.68 (m, 1 H), 5.23 (s, 1H), 4.81 – 4.70 (m, 1 H), 4.26 – 4.10 (m, 3H), 3.29 – 3.23 (m, 2H), 3.1 1 – 2.96 (m, 2H), 2.87 – 2.76 (m, 1H), 2.60 (s, 3H), 2.55 – 2.42 (m, 1 H), 2.33 – 2.19 (m, 1H), 2.18 – 2.07 (m, 1H), 1.95 – 1.81 (m, 1H), 1.47 – 1.35 (m, 1 H). LCMS: (AA) M+l 580.0
CLIP

Credit: Tien Nguyen/C&EN
Presenter: Steven Paul Langston, associate director at Takeda Pharmaceuticals International
Target: Sumo activating enzyme
Disease: Solid tumors
Reporter’s notes: Langston gave the last talk of the morning session, placing him in the “precarious position of being between you and lunch,” he said. Takeda acquired this drug development program, falling under the umbrella of immuno-oncology, along with Millenium Pharmaceuticals in 2008. The team targeted a pathway known as SUMOylation, a protein post translation modification that is implicated in a number of cellular processes including immune response. In SUMOylation, enzymes attach a small protein to another protein. They found that inhibiting this pathway activates a type I interferon response in immune cells. How the molecule, TAK-981, inhibits this pathway is quite complicated, Langston said. TAK-981 forms an adduct with a small ubiquitin like modifier (SUMO) to inhibit a SUMO activating enzyme that catalyzes SUMOylation. While the synthesis of TAK-981 is fairly short, it requires a nonideal chiral chromatography separation after the first step. TAK-981 is in Phase I clinical trials as an intravenous infusion for patients with metastatic solid tumors or lymphomas.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2018311239 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2018-03-16 | |
| US9962386 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2017-04-17 | |
| US9683003 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2015-06-30 | 2016-01-14 |
//////////TAK-981, TAK 981, Phase I, Lymphoma, Solid tumours, TAKEDA,
Cc3sc(cc3[C@@H]1NCCc2ccc(Cl)cc12)C(=O)c5cncnc5N[C@@H]4C[C@H](COS(N)(=O)=O)[C@@H](O)C4
BIIB-095

BIIB-095
ROTATION (+)
1493790-64-9 CAS free form,
1493772-48-7 cas Hcl salt
cas 1493790-65-0, 1496563-32-6 ,SULPHATE ???
cas 1496563-31-5 SULFATE 1;1
cas 1496563-32-6 SULFATE HYDRATE 1;1;1
(2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one
1,7-Diazaspiro[4.4]nonan-6-one, 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)phenyl]-2-pyrimidinyl]-, (2R,5S)-
C20 H21 F3 N4 O, 390.40
- Originator Biogen
- Class Analgesics
- Mechanism of Action Nav1.7 voltage-gated sodium channel inhibitors
- Phase I Neuropathic pain
- 29 Mar 2018 Phase-I clinical trials in Neuropathic pain (In volunteers) in United Kingdom (PO) (NCT03454126)
- 05 Mar 2018 Biogen plans a phase I trial for Pain, including Neuropathic pain (In volunteers) in USA (PO) (NCT03454126)
- 05 Mar 2018 Preclinical trials in Neuropathic Pain in USA (PO), before March 2018
In March 2018, a randomized, double blind, placebo controlled, single and multiple-ascending dose, dose-escalation phase I study ( NCT03454126; 255HV101; 2017-003982-90) was initiated in the UK in healthy subjects (expected n = 80) to evaluate the safety, tolerability and pharmacokinetics of BIIB-095. At that time, the trial was expected to complete in December 2018
Biogen is developing BIIB-095, a voltage-gated sodium channel 1.7 inhibitor, for the potential oral treatment of neuropathic pain [2027279], [2027426]. In March 2018, a phase I trial was initiated in healthy subjects
Biogen is developing oral agent BIIB-095 for the treatment of chronic pain, including neuropathic pain. A phase I clinical trial is under way in healthy volunteers.
The compound was first claimed in WO2013175205 , for treating schizophrenia, assigned to subsidiary Convergence Pharmaceuticals Limited , naming some of the inventors. This might present the structure of BIIB-095 , a voltage-gated sodium channel 1.7 inhibitor, being developed by Biogen for the oral treatment of neuropathic pain; in March 2018, a phase I trial was initiated in healthy subjects.
PATENT
WO2013175205

CONTD………………

INTERMEDIATE

WO 2013175206
US 20150119404
https://patents.google.com/patent/US20150119404
Patent
WO-2019067961
Novel salts (citrate, mesylate, hydrosulfate, saccharinate and oxalate) forms of 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one, processes for their preparation and compositions comprising them are claimed. Also claimed are their use for treating diseases and conditions mediated by modulation of voltage-gated sodium channels.
Voltage-gated sodium channels are responsible for the initial phase of the action potential, which is a wave of electrical depolarisation usually initiated at the soma of the neuron and propagated along the axon to the terminals. At the terminals, the action potential triggers the influx of calcium and the release of neurotransmitter. Drugs, such as lidocaine, that block voltage-gated sodium channels are used as local anaesthetics. Other sodium channel blockers, such as lamotrigine and carbamazepine are used to treat epilepsy. In the latter case, partial inhibition of voltage-gated sodium channels reduces neuronal excitability and reduces seizure propagation. In the case of local anaesthetics, regional block of sodium channels on sensory neurons prevents the conduction of painful stimuli. A key feature of these drugs is their state-dependent mechanism of action. The drugs are thought to stabilise an inactivated conformation of the channel that is adopted rapidly after the channel opens. This inactivated state provides a refractory period before the channel returns to its resting (closed) state ready to be reactivated. As a result, state-dependent sodium channel blockers inhibit the firing of neurons at high frequency, for example in response to painful stimuli, and will help to prevent repetitive firing during periods of prolonged neuronal depolarisation that might occur, for example, during a seizure. Action potentials triggered at lower frequencies, for example in the heart, will not be significantly affected by these drugs, although the safety margin differs in each case, since at high enough concentrations each of these drugs is capable of blocking the resting or open states of the channels.
The voltage-gated sodium channel family is made up of 9 subtypes, four of which are found in the brain, NaV1.1 , 1.2, 1.3 and 1.6. Of the other subtypes, NaV1.4 is found only in skeletal muscle, NaV1.5 is specific to cardiac muscle, and NaV1.7, 1.8, and 1.9 are found
predominantly in sensory neurons. The hypothesised binding site for state-dependent sodium channel blockers is the local anaesthetic (LA) binding site in the inner vestibule of the pore on transmembrane S6 of domain IV. Critical residues are located in a highly conserved region among the different subtypes, thus presenting a challenge for the design of new subtype selective drugs. Drugs such as lidocaine, lamotrigine and carbamazepine do not distinguish between the subtypes. However, selectivity can be achieved, and can be further enhanced functionally, as a result of the different frequencies at which the channels operate.
Drugs that block voltage-gated sodium channels in a state-dependent manner are also used in the treatment of bipolar disorder, either to reduce symptoms of mania or depression, or as mood stabilisers to prevent the emergence of mood episodes. Clinical and preclinical evidence also suggests that state-dependent sodium channel blockers may help to reduce the symptoms of schizophrenia. For example, lamotrigine has been shown to reduce symptoms of psychosis induced by ketamine in healthy human volunteers, and furthermore, studies in patients suggest that the drug can augment the antipsychotic efficacy of some atypical antipsychotic drugs, such as clozapine or olanzapine. It is hypothesised that efficacy in these psychiatric disorders may result in part from a reduction of excessive glutamate release. The reduction in glutamate release is thought to be a consequence of sodium channel inhibition in key brain areas, such as the frontal cortex. However, interaction with voltage-gated calcium channels may also contribute to the efficacy of these drugs.
WO 2013/175205 (Convergence Pharmaceuticals Limited) describes (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one hydrochloride, sulfuric acid salt and sulfuric acid salt hydrate which are claimed to be modulators of voltage-gated sodium channels. The object of the invention is to identify alternative salts of said compound which have advantageous properties.
Example 1
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of citric acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 30 minutes a seed of citrate salt (0.1 wt%) was added and the mixture allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a very thick suspension (white) that required mobilisation with 20 ml additional ethanol and a further maturation period of 2 hours at ambient temperature. Filtration was carried out under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 6.0 g of crystalline white solid (91 % yield).
H NMR (400MHz, DMSO-D6): δΗ 1.90-2.05 (2H, m), 2.10-2.20 (2H, m,), 2.20-2.30 (1 H, m), -2.50 (1 H, m, partially masked by solvent)), 2.55-2.68 (4H, m), 2.56 (3H, s), 2.79 (3H, s),
3.28-3.40 (2H, m), 4.79 (1 H, t, J= 8.0 Hz), 7.92 (2H, d, J = 8.4 Hz), 8.03 (1 H, s), 8.45 (2H, d, J= 8.8Hz) ppm, (exchangeables not reported)
Characterisation of Example 1
The XRPD of Example 1 is presented in FIG. 1 and the DSC/TGA of Example 1 is presented in FIG. 2. The citrate salt of Example 1 displayed the following characteristics:
1 endotherm onset: 171.82°C
peak maximum: 174.55°C
There was an endotherm post the main endotherm.
There was no weight reduction until ca 168°C had been reached. The weight reduction commenced with the start of the main endotherm and coincided with the endotherm post the main endotherm which indicated that this thermal event was the onset of compound decomposition and loss of citric acid. Thermal events >220°C were due to compound decomposition.
The XPRD data in FIG. 1 demonstrated that under different extremes of humidity indicate a stable crystalline form of the citrate salt of Example 1 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 2 which show clear transitions and no evidence of solvates.
Aqueous solubility of the citrate salt (Example 1) = 22mg/ml (25°C).
Example 2
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one ) salt (E2)
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of methanesulfonic acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 10 minutes nucleation and gradual crystallisation was noted to afford a thick mixture. Additional ethanol was added (10 ml) to mobilise the suspension that was then allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a thin, mobile suspension (white) that was filtered under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 4.0 g of crystalline white solid (72% yield).
H NMR (400MHz, DMSO-D6): δΗ 2.1-2.45 (4H, m), 2.27 (3H, s), 2.50-2.75 (2H, m), 2.61 (3H, s), 2.86 (3H, s), 3.35-3.50 (2H, m), 5.20 (1 H, t, J = 8 Hz), 7.96 (2H, d, J = 8.8 Hz), 8.17 (1 H, s), 8.51 (2H, d, J = 8.4Hz), 9.45 (1 H, br), 10.16 (1 H, br) ppm.
Characterisation of Example 2
The XRPD of Example 2 is presented in FIG. 3 and the DSC/TGA of Example 2 is presented in FIG. 4. The DSC thermograph of the methanesulfonate (mesylate) (Example 2) displayed the following characteristics:
One distinct endotherm onset: 247.34°C
peak maximum: 250.34°C
The TGA thermograph showed no weight reduction until ca 250°C had been reached. The weight reduction commenced with the start of the main endotherm and indicated that this thermal event was the onset of compound decomposition. There is no evidence of entrapped solvents or water.
The XPRD data in FIG. 3 demonstrated that under different extremes of humidity indicate a stable crystalline form of the mesylate salt of Example 2 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 4 which show clear transitions and no evidence of solvates.
Aqueous solubility of the mesylate salt (Example 2) = 65mg/ml (25°C).
Example 3
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one hydrosulfate single crystals: 25.0 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluorome
one hydrosulfate was added to 4 mL vial. 1.000 mL of anhydrous EtOH was added, and the sample was filtered. Anhydrous hexanes were added dropwise until the solution neared the precipitation point. The vial was sealed and left undisturbed for 24 hr, after which time a crop of single crystals was evident. The sample was sent for single crystal analysis and confirmed as the anhydrous (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate form (FIGs. 5A-5B).
Example 4
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one freebase: 8.00 g of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate (JM Lot R-2017-4323 D 301) was added to a 1 L Erlenmeyer flask and suspended and stirred vigorously in 400 mL of THF. 20% K2C03 (250 mL) was added and dissolved. The mixture was transferred to 1 L sep. funnel. 100 mL EtOAc was added and the aqueous and organic layers were separated. The aqueous layer was re-extracted with 50 mL of EtOAc and the combined organics were back-extracted with brine (100 mL) and water (100 mL). Due to fairly poor separation, a significant quantity of MgSCU was required to dry the solution. The solution was reduced via Rotavap (45 °C) to -50 mL, transferred to a 100 mL RB flask, reduced down to -10 mL, transferred to 20 mL scintillation vial and continued to be reduced to a thick oil. The oil was left on the Rotavap for another hour and a “wet” solid was obtained. Loosened solids on the bottom of the vial were left on the Rotavap for 1 hr with no heat applied to obtain a chunky solid. The contents was transferred to a mortar and pestle, ground to powder and fine granules, placed back in a 20 mL scintillation vial and left on a Rotavap overnight to obtain a dry solid (5.1 g). The XRPD pattern of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase is shown in FIG. 6.
Example 5
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one saccharinate: 199.7 mg of (2R,5S)-7-Methyl-2-(4-
methyl-6-(4-(trifluoromethyl)phenyl)pyrirnidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one free base (0.5115 mmol) was dissolved in 4.2 mL of 2-Me-THF. 98.1 mg of saccharin (0.5106 mmol) was dissolved in 4.2 mL of 2-Me-THF. Saccharin was added to the freebase, and after 15 seconds the mixture began to precipitate and solidify. 10 mL of 2-Me-THF was added and stirred at max rpm as to provide a thick white suspension in 10 min. The suspension was filtered, air dried under vacuum for 10 min on frit, then dried under a stream of nitrogen for 30 min resulting in 215 mg of white solid product. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one saccharinate is shown in FIG. 7.
Example 6
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one oxalate: 403 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase was dissolved in 4.03 mL EtOH. 1.000 mL of this solution was added to a 4 mL vial. 23.8 mg of oxalic acid was dissolved in 1.000 mL of EtOH and added dropwise to the stirring (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase solution. After 5 min, a white precipitate was evident and 2.000 mL of EtOH was added to the slurry to aid stirring. The resulting suspension was stirred overnight. The following day the suspension was filtered and dried on a frit under vacuum for 10 min yielding 106 mg of white solid. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one oxalate is shown in FIG. 8.
Example 7
The single crystal structural information and refinement parameters for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate are shown in Table 1.
Table 1.
Largest peak, hole / e A-3 0.363, -0.264
The most prominent XRPD diffraction peaks were (2Θ): 7.8±0.2°, 8.1±0.2°, 12.6±0.2°, 14.3±0.2°, 16.5±0.2°, 18.5±0.2°, 19.6±0.2°, 24.8±0.2° and 25.3±0.2°.
PATENTS
US2018360833NOVEL PYRIMIDINYL-DIAZOSPIRO COMPOUNDS2018-06-27
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017304303 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2017-07-11 | |
| US9737536 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-05-25 | 2016-09-15 |
| US2016184306 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-02-15 | 2016-06-30 |
| US9309254 | NOVEL COMPOUNDS | 2013-05-22 | 2015-04-30 |
| US9376445 | NOVEL COMPOUNDS | 2013-05-22 | 2015-06-18 |
////////////////BIIB-095, BIIB095, BIIB 095, PHASE 1
CC1=NC(=NC(=C1)C2=CC=C(C=C2)C(F)(F)F)C3CCC4(N3)CCN(C4=O)C
VNRX-7145
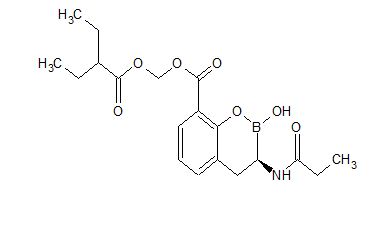

CAS 1842399-68-1
MF C19 H26 B N O7
MW 391.22
2H-1,2-Benzoxaborin-8-carboxylic acid, 3,4-dihydro-2-hydroxy-3-[(1-oxopropyl)amino]-, (2-ethyl-1-oxobutoxy)methyl ester, (3R)-
The VNRX-7145 combination is now in Phase I studies to treat resistant urinary tract infections.
VNRX-7145
PATENT
WO 2015191907
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015191907
ntibiotics are the most effective drugs for curing bacteria-infectious diseases clinically. They have a wide market due to their advantages of good antibacterial effect with limited side effects. Among them, the beta-lactam class of antibiotics (for example, penicillins, cephalosporins, and carbapenems) is widely used because they have a strong bactericidal effect and low toxicity.
[0005] To counter the efficacy of the various beta-lactams, bacteria have evolved to produce variants of beta-lactam deactivating enzymes called beta-lactamases, and in the ability to share this tool inter- and intra-species. These beta-lactamases are categorized as“serine” or“metallo” based, respectively, on presence of a key serine or zinc in the enzyme active site. The rapid spread of this mechanism of bacterial resistance can severely limit beta-lactam treatment options in the hospital and in the community.
SCHEME 1
SCHEME 2
SCHEME 3
[00390] Alternatively, (II) can be obtained by treatment of (I) with hydrochloric acid (around 3-5 Molar in dioxane) in an alcohol solvent such as methanol, ethanol, or n-butanol at a temperature between room temperature and 120 ºC (SCHEME 4).
SCHEME 4
SCHEME 5
EXAMPLE 62: (R)-2-Hydroxy-3-propionylamino-3,4-dihydro-2H-benzo[e][1,2]oxaborinine-8-carboxylic acid
Step 1. Synthesis of 2-Methoxy-3-[2-propionylamino-2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02,6]dec-4-yl)-ethyl]-benzoic acid tert-butyl ester.
[00540] Prepared from [(1S)-2-(3-tert-butoxycarbonyl-2-methoxy-phenyl)-1-chloro-ethyl]boronic acid (+) pinanediol ester and propionic acid following the procedure in Step 2 of Example 1. The crude product was purified by flash chromatography on silica gel (25-100% EtOAc/Hexane). ESI-MS m/z 486 (MH)+.
Step 2. Synthesis of (R)-2-Hydroxy-3-propionylamino-3,4-dihydro-2H-benzo[e][1,2]oxaborinine-8-carboxylic acid.
[00541] Prepared from 2-Methoxy-3-[2-propionylamino-2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02,6]dec-4-yl)-ethyl]-benzoic acid tert-butyl ester following the procedure described in Step 3 of Example 1. The crude product was purified by reverse phase preparative HPLC and dried using lyophilization. ESI-MS m/z 264 (MH)+
CLIP
Candidate: VNRX-7145

Credit: Tien Nguyen/C&EN
Presenter: Christopher John Burns, president and chief executive officer of VenatoRx Pharmaceuticals
Target: β-lactamases
Disease: Resistant urinary tract infections
Reporter’s notes: Having unveiled an antibacterial candidate at last spring’s first time disclosures session, Burns was back with another, this time the molecule can be taken orally. Both VenatoRx (pronounced Ven-a-tor-ix) compounds resuscitate the activity of β-lactam drugs, which make up more than 60% of all antibiotics prescribed. Unfortunately, many bacteria have grown resistant to these antibiotics. The new compounds rescue the old antibacterials by inhibiting β-lactamases, enzymes that chew up the antibiotics. To test the activity of new β-lactamase-targeting compounds, the researchers settled on several “sentinel” bacteria strains. Then to find a candidate with oral bioavailability, the team focused on molecules with low polarity and low molecular weight. They found VNRX-7145, developed as a prodrug in which esterases in the liver clip off the tips of the molecule to reveal the active drug. VNRX-5133, disclosed at last year’s meeting, had to be delivered intravenously along with another IV-antibiotic Cefepime, and targeted serine and metallo β-lactamases. The new oral candidate VNRX-7145 inhibits serine β-lactamases with Ceftibuten as its partner. The VNRX-7145 combination is now in Phase I studies to treat resistant urinary tract infections.
////////////VNRX-7145, VNRX7145, VNRX 7145, Phase I, VenatoRx
CCC(CC)C(=O)OCOC(=O)c1cccc2C[C@H](NC(=O)CC)B(O)Oc12
CCC(CC)C(=O)OCOC(=O)c1cccc2C[C@H](NC(=O)CC)B(O)Oc12
