
Home » PATENT (Page 4)
Category Archives: PATENT
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
WO 2016092561, Ivacaftor, New patent, Laurus Labs Pvt Ltd

WO-2016092561, Ivacaftor, NEW PATENT
https://www.google.com/patents/WO2016092561A2?cl=en
Novel polymorphs of ivacaftor, process for its preparation and pharmaceutical composition thereof
Laurus Labs Pvt Ltd
LAURUS LABS PRIVATE LIMITED [IN/IN]; Plot No. DS1, IKP Knowledge Park, Genome Valley Turkapally, Shameerpet Mandal, Ranga District Hyderabad 500078 (IN)

| Ram Thaimattam, Venkata Srinivasa Rao DAMA, Venkata Sunil Kumar Indukuri, Seeta Rama Anjaneyulu GORANTLA,Satyanarayana Chava, | |
| Applicant | Laurus Labs Private Limited |
THAIMATTAM, Ram; (IN).
DAMA, Venkata Srinivasa Rao; (IN).
INDUKURI, Venkata Sunil Kumar; (IN).
GORANTLA, Seeta Rama Anjaneyulu; (IN).
CHAVA, Satyanarayana; (IN)

Novel crystalline forms of ivacaftor (designated as forms L1 to L14), processes for their preparation and composition comprising them are claimed.
Vertex, in research collaboration with Cystic Fibrosis Foundation Therapeutics, had developed and launched ivacaftor.
Ivacaftor, also known as N-(2,4-di-tert-butyl-5-hydroxyphenyl)-l,4-dihydro-4-oxoquinoline-3-carboxamide, having the following Formula I:

Formula I
Ivacaftor was approved by FDA and marketed by Vertex pharma for the treatment of cystic fibrosis under the brand name KALYDECO® in the form of 150 mg oral tablets.
WO2006/002421 publication discloses modulators of ATP-binding cassette transporters such as ivacaftor. This patent generally discloses a process for the preparation of modulators of ATP-binding cassette transporters such as quinoline compounds; however, specific process for the preparation of ivacaftor and its solid state details were not specifically disclosed.
WO2007/079139 publication discloses Form A, Form B and amorphous form of ivacaftor characterized by PXRD, DSC and TGA and process for their preparation. Further this publication discloses ethanol crystalate of ivacaftor in example part.
WO2009/038683 publication discloses the solid forms of ivacaftor, which are designated as Form-I (2-methylbutyric acid), Form-II (propylene glycol), Form-HI (PEG400.KOAc), Form-IV (lactic acid), Form-V (isobutyric acid), Form-VI (propionic
acid), Form- VII (ethanol), Form- VIII (2-propanol), Form-IX (monohydrate), Form-X (besylate Form A), Form-XI (besylate Form B), Form-XII (besylate Form D), Form-XIII (besylate Form E), Form-XIV (besylate Form F), Form-XV (besylate (2: 1)), Form-XVI (besylate mono hydrate). This publication also discloses the characterization details like PXRD, DSC and TGA for the above forms and process for their preparation.
WO201 1/1 16397 publication discloses crystalline Form C of ivacaftor, process for its preparation and pharmaceutical composition comprising the same. Also discloses characterization details of Form C, such as PXRD, IR, DSC and 13CSSNMR.
WO2013/158121 publication discloses solvated forms of ivacaftor, which are designated as Form D (acetonitrile or acetonitrile/water (75/25) solvate), Form E (Methyl ethyl ketone (MEK), MEK/water (90/1), MEK/water (90/10), MEK/water (80/20) solvate), Form F (acetonitrile/water (75/25) solvate), Form G (isopropyl acetate solvate), Form H (isopropyl acetate/water (95/5) solvate), Form I (MEK solvate), Form J (MEK/water (99/1) solvate), Form K (MEK or MEK/water (99/1) or MEK/water (90/10) or MEK/water (80/20) solvate), Form L (isopropyl acetate/water (95/5) solvate), Form M (MEK or MEK/water (99/1) solvate), Form N (MEK water (90/10) or MEK/water (80/20) solvate), Form O (MEK or MEK/water (99/1) solvate), Form P (MEK water (90/10) or MEK water (80/20) solvate), Form Q (MEK/water (80/20) solvate), Form R (acetonitrile solvate), Form S (MEK/water (80/20) solvate), Form T (isopropyl acetate/water (95/5) solvate), Form W (acetonitrile/water (90/10) solvate), Form XX (from 10% water/ acetonitrile) and hydrate B (hydrated form). This patent further discloses characterization details like PXRD and TGA for the above forms and process for their preparation.
WO2014/118805 publication discloses crystalline forms of ivacaftor designated as Form D, Form E, Form El, Form G and Form G’; amorphous ivacaftor designated as Form I and Form II; crystalline ivacaftor solvates such as n-butanol solvate, methanol solvate, propylene glycol solvate, DMF solvate, THF solvate, DMF:ethylacetate solvate. This publication further discloses the process for the preparation of said forms along with their characterization details.
WO2015/070336 publication discloses polymorphic form APO-I and MIBK solvate of ivacaftor along with its characteristic PXRD details, process for its preparation and pharmaceutical composition comprising them.
CN 104725314A publication discloses ivacaftor new polymorph D, which is obtained by crystallization of ivacaftor from acetonitrile/water. This publication further discloses characteristic details such PXRD, IR and DSC of ivacaftor new polymorph D.
Polymorphism is the occurrence of different crystalline forms of a single compound and it is a property of some compounds and complexes. Thus, polymorphs are distinct solids sharing the same molecular formula, yet each polymorph may have distinct physical properties. Therefore, a single compound may give rise to a variety of polymorphic forms where each form has different and distinct physical properties, such as different solubility profiles, different melting point temperatures and/or different x-ray diffraction peaks. Since the solubility of each polymorph may vary, identifying the existence of pharmaceutical polymorphs is essential for providing pharmaceuticals with predictable solubility profiles. It is desirable to investigate all solid state forms of a drug, including all polymorphic forms and solvates, and to determine the stability, dissolution and flow properties of each polymorphic form.
Polymorphic forms and solvates of a compound can be distinguished in a laboratory by X-ray diffraction spectroscopy and by other methods such as, infrared spectrometry. Additionally, polymorphic forms and solvates of the same drug substance or active pharmaceutical ingredient, can be administered by itself or formulated as a drug product (also known as the final or finished dosage form), and are well known in the pharmaceutical art to affect, for example, the solubility, stability, flowability, tractability and compressibility of drug substances and the safety and efficacy of drug products.
The discovery of new polymorphic forms and solvates of a pharmaceutically useful compound, like ivacaftor, may provide a new opportunity to improve the performance characteristics of a pharmaceutical product. It also adds to the material that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic. New polymorphic forms of the ivacaftor have now been discovered and have been designated as ivacaftor Form-Ll, Form-L2, Form-L3, Form-L4, Form-L5, Form-L6, Form-L7, Form-L8, Form-L9, Form-LlO, Form-Ll 1, Form-Ll 2 A, Form-Ll 2B, Form-Ll 3 and Form-Ll 4.
EXAMPLE 1 : Preparation of Ivacaftor Form-Ll
A suspension of ivacaftor ethanolate (5 g) in n-heptane (200 mL) was heated to 95-100°C and stirred for 5 hrs at the same temperature. Then the reaction mixture was cooled to 25-35°C and stirred for an hour. The solid obtained was filtered, washed with n-heptane and suck dried. The wet solid was further dried at 60-65°C for 16 hrs under vacuum yielded ivacaftor Form-Ll . The XRPD is set forth in Figure- 1.
In a similar manner, ivacaftor Form-Ll was prepared from different solvates of ivacaftor in place of ivacaftor ethanolate as input using the following conditions;

Ivacaftor cyclopentyl methyl ether (0.5 g) n-heptane (20 mL) 50°C/8 hr
Ivacaftor methyltertiarybutyl ether (0.5 g) n-heptane (20 mL) 50°C/8 hr

When Dr Satyanarayana Chava started Laurus Labs in 2007, he invested nearly Rs 60 crore of his own money into it. His confidence in its success was neither bravado nor bluster, but defined by his knowledge of the pharmaceutical industry. Eight years on, the Hyderabad-based company is on track to reach revenues of Rs 2,000 crore by the end of FY2016.
Chava, now 52, has more than two decades of experience in the pharmaceutical industry; in his last job, he was chief operating officer (COO) of the successful startup, Matrix Laboratories. Of his 10 years there, he says with pride, “I never skipped a promotion and got to work in all departments.” His dedication, coupled with a sound understanding of what it takes to start a pharmaceutical company, is what makes Laurus Labs among the hottest startups in this sector.
Initially, Chava planned the business around research and development (R&D). He wanted Laurus Labs to focus on contract research and make money from royalties. “In India, companies start with manufacturing and then get into R&D,” he explains. “I did it the other way round.” He focussed his fledgling company’s resources on developing formulations for medicines, and licensed them to other pharmaceutical players. In the early months, Laurus Labs had 10 people in manufacturing and 300 in R&D.

In June 2007, Aptuit, a US-based contract research organisation (CRO), signed it on for a $20 million (then Rs 80 crore) contract. But despite this injection of funds, Chava was unable to sustain his original idea of developing technologies for other companies. At the time of the Aptuit deal, Laurus Labs’s annual revenues were not even $20,000 (Rs 8 lakh at the time). In 2008, Chava decided to start manufacturing active pharmaceutical ingredients (API), which, as the name suggests, are chemicals or key ingredients in drugs required to make the medication work. His early investment into R&D benefitted Laurus Labs; it maintains a large repository of research-based knowledge that forms the bedrock of any successful pharmaceutical business.
Today, it is a key manufacturer supplier of APIs and holds its own against better-known competitors like US generic drug giant Mylan, which, incidentally, acquired a controlling stake in Matrix around the time Chava founded Laurus Labs. It has also carved a niche for itself by supplying antiretroviral or ARVs (used to fight infections caused by retroviruses like HIV) and oncology drugs. And despite being a relatively new player, its clients include giants like Pfizer, Teva Pharmaceutical Industries and Merck.

The person behind it
A Master’s degree in chemistry was never on the cards for Chava. In the early 1980s, the best students usually studied physics, and he had planned to do the same. But when he went to his college in Amravati (Andhra Pradesh) to enroll, his elder sister’s friend suggested he study chemistry too. Chava took up the subject on a whim. He ended up liking chemistry so much so that in his final year he topped his batch despite not having written one out of the four required papers. He went on to complete his PhD in the subject in 1991.
Upon graduating, he was hired by Ranbaxy Laboratories in Delhi as a researcher. In those early years itself Chava knew he’d spend a lifetime in the industry. He enjoyed the work and gained valuable experience as a young researcher in what was then India’s finest pharmaceutical company.
But through his years in the industry, Chava was conscious of the fact that he needed to broaden his experience outside of research. His stint at Matrix Laboratories afforded him that opportunity. As it was a startup, he was able to rise through the ranks quickly and got the opportunity to work in key departments from sales and marketing to finance and accounts. Within eight years of joining Matrix, he became its COO.

This experience was to come in handy when, due to differences with the board—he refused to elaborate on this—he decided to leave Matrix and set up Laurus Labs. And though he is the company’s chief executive officer (CEO), Chava remains true to his calling as a chemist. He has strived to build an organisation that is not very hierarchical. It is not uncommon to see him interacting with the chemists in the company and discussing formulations with them—something unheard of in an industry where most CEOs are from a sales and marketing background.


Chandrakanth Chereddi
VP Synthesis Business Unit
Prior to his current assignment at Laurus Labs India, Chandra headed the Project Management division for all scientific projects at the Laurus R&D center. Chandra previously worked for McKinsey & Company in India as a member of the healthcare practice and at Google Inc. as a software engineer in Google’s Mountain View, CA office. Chandra holds a BE from the College of Engineering, Osmania University, Hyderabad, and MS from University of Illinois at Urbana-Champaign, and an MBA from Indian School of Business, Hyderabad.
///////WO 2016092561, Ivacaftor, New patent, Laurus Labs Pvt Ltd
Lurasidone hydrochloride, Jubilant Generics Ltd, WO 2016059649, New patent
 |
|
 |

Lurasidone hydrochloride, Jubilant Life Sciences Ltd, WO 2016059649, New patent
An improved process for the preparation of lurasidone hydrochloride
Jubilant Life Sciences Ltd
WO 2016059649
JUBILANT GENERICS LIMITED (FORMERLY JUBILANT LIFE SCIENCES DIVISION) [IN/IN]; Plot 1A, Sector 16 A, NOIDA Uttar Pradesh 201301 (IN)
MISHRA, Vaibhav; (IN).
DUBEY, Shailendr; (IN).
SINGH, Kumber; (IN).
CHOUDHARY, Alka Srivastava; (IN).
VIR, Dharam; (IN)

![]()
Disclosed herein is an improved process for the preparation of Lurasidone and its pharmaceutically acceptable salts via novel intermediate and use thereof for the preparation of an antipsychotic agent useful for the treatment of schizophrenia and bipolar disorder. Further, present invention provides a cost effective and eco-friendly process for producing Lurasidone hydrochloride of formula (I) substantially free of residual solvent(s) at industrial scale.
Improved process for preparing lurasidone or its hydrochloride, substantially free of residual solvent, useful for treating schizopherenia and bipolar disorder. Also claims novel intermediate of lurasidone eg ((R,R)-cyclohexane-1,2-diyl)bis((1H-imidazol-1-yl)methanone) and its preparation method.

In April 2016, Newport Premium™ reported that Jubilant Life Sciences was capable of producing commercial quantities of lurasidone and lists the drug as a molecule available under research and development on the company’s website.
This is the first patenting to be seen from Jubilant Life Sciences that focuses on lurasidone – it having been developed and launched by Sumitomo Dainippon Pharma and EU licensee Takeda, for treating schizophrenia.

May 2, 2014
Neeraj Agrawal: Took charge of API business for Jubilant Life Sciences at the age of 31
Position: CEO Generics, Jubilant Life Sciences
Education: IIIM-C, MBA, 1998; IIT, Bombay, Electrical Engg., 1995.
Previous Jobs: Associate-Business Strategy, Operations Improvement, McKinsey & Co.
Claim to Fame: Took charge of the API business for Jubilant when he was just 31-years-old
Management mantra: It revolves around trust, freedom and teams. I like my team to think and act like an entrepreneur – assess business risks and rewards suitably and then take decisions.
Lurasidone and its pharmaceutically acceptable salts like lurasidone hydrochloride is chemically, (3a ?,45,7 ?,7a5)-2-{ (1 ?,2 ?)-2-[4-(l,2-benzisothiazol-3-yl)piperazin-lyl-methyl] cyclohexylmethyl }hexahydro-4,7-methano-2H-isoindole- 1 ,3 -dione hydrochloride and has the structure represented by the Formula (I):

Formula-I
Lurasidone hydrochloride is marketed in the United States under the trade name Latuda®. Lurasidone and its pharmaceutically acceptable salts as well as process for their preparation was first disclosed in US patent no. 5,532,372. The patent discloses the preparation of lurasidone hydrochloride using racemic trans 1,2-cyclohexane dicarboxylic acid. Racemic trans 1,2-cyclohexane dicarboxylic acid on reduction with lithium aluminium hydride in THF at reflux temperature forms l,2-bis(hydroxymethyl)cyclohexane which is converted into racemic iran5-l,2-bis(methanesulfonyloxymethyl)cyclohexane by reaction with methane sulfonyl halide. l-(l,2-benzisothiazol-3-yl)piperazine on reaction with trans-l, 2-b (methanesulfonyloxymethyl)cyclohexane in the presence of sodium carbonate and acetonitrile forms iran5-3a,7a-octahydroisoindolium-2-spiro- -[4′-(l,2-benzisothiazol-3-yl)]piperazine methanesulfonate which on reaction with bicyclo[2.2.1]heptane-2-exo-3-exo-dicarboximide in the presence of potassium carbonate, dibenzo-18-crown-6-ether and xylene on refluxing forms racemic lurasidone free base. The compound is obtained by column chromatography and then treated the resulting lurasidone free base with IPA.HCl in acetone to obtain racemic lurasidone hydrochloride. Resolution of racemic lurasidone hydrochloride is carried out using tartaric acid as resolving agent. The process involves use of lithium aluminium hydride which is highly pyrophoric reagent and is not to utilize the same on commercial scale due to its handling problems associated with its reactivity. Also, the use of the column chromatography for purification is not viable on commercial scale. Further the process involves the usage of dibenzo-18-crown-6-ether as a phase transfer catalyst which is costly material and in turn increases the cost of production. Carrying out the resolution in the last stages is difficult due to the presence of six chiral centres in lurasidone and is also not suitable for an industrial scale preparation as it affects the overall yield and cost of the manufacturing process.

Chinese patent application no. CN102731512 discloses a process for preparation of lurasidone which comprises reaction of racemic irans-l,2-bis(methanesulfonyloxymethyl) cyclohexane and l-(l,2-benzisothiazol-3-yl)piperazine in toluene in the presence of sodium carbonate or potassium carbonate having particle size less than 200 micron and tetrabutyl ammonium bromide to give the intermediate /rans-3a,7a-octahydroisoindolium-2-spiro- -[4′-(l,2-benzisothiazol-3-yl)]piperazinemethanesulfonate which on reaction with bicyclo[2.2.1]heptane-2-exo-3-exo-dicarboximide in toluene using potassium carbonate having particle size less than 200 micron forms racemic lurasidone free base. The racemic free base is converted into racemic hydrochloride salt using acetone and cone, hydrochloric acid. Racemic lurasidone hydrochloride is resolved by following the method disclosed in US patent no. 5,532,372. The process involves resolution of product in the last stage which is not commercially viable as it affects the overall yield and cost of the manufacturing process.
Japanese patent no. JP4219696 discloses the resolution of trans 1,2-cycloheaxne dicarboxylic acid using (lS,2R)-(+)-norephedrine or (lR,2S)-(-)norephedrine to provide (R,R)-trans 1 ,2-cyclohexanedicarboxylic acid. The (R,R)-iran,sl,2-cyclohexane dicarboxylic acid obtained was esterified with ethanol and the obtained ester compound was reduced with vitride to provide (R,R)-l,2-bis(hydroxymethyl)cyclohexane followed by treatment with methane sulfonyl chloride to form (R,R)-1,2-bis(methanesulfonyloxymethyl)cyclohexane. The process requires large quantity of reducing agent viz., for reducing one lg of compound about 5g of reducing agent is required which is not conducive for industrial production.
Chinese patent application no. CN 102952001 discloses a process for the preparation of (lR,2R)cyclohexane-l,2-dimethanol by the reduction of (lR,2R)cyclohexane-l,2-
dicarboxylic acid using sodium borohydride or potassium borohydride and boron triflouoride diethyl ether in THF or diethyl ether as solvent. Boron triflouoride diethyl ether is used in large quantity and quite expensive which makes the process commercially unviable.
International publications no. WO 2012/131606 and WO 2014/037886 disclose a process for preparation of lurasidone which involves separating the racemic transl,2-cyclohexane dicarboxylic acid into its (R,R) trans and (S,S) trans isomers and then using the desired trans (R,R) isomer for the preparation of lurasidone hydrochloride using the chemistry disclosed in US patent no. 5,532,372 for preparation of racemic lurasidone hydrochloride. In these publications diisobutyl aluminium hydride (DIBAL) is used as the reducing agent for the preparation of (1R,2R) cyclohexane 1,2-dimethanol from (1R,2R) cyclohexane 1,2-dicarboxylic acid which is quite expensive. Further the process involves the usage of dibenzo-18-crown-6-ether as a phase transfer catalyst which is costly material and in turn increases the cost of production.
Some of the prior art processes disclose the process for the preparation of lurasidone hydrochloride from l,2-(lR,2R)-bis-(methanesulfonyloxymethyl)cyclohexane using different solvents and bases.
US patent no. 8,853,395 discloses a process for the preparation of lurasidone in which condensation of iran5-l,2-bis(methanesulfonyloxymethyl)cyclohexane with 1-(1,2-benz isothiazol-3-yl)piperazine and condensation of /rans-3a,7a-octahydroisoindolium-2-spiro- -[4′-(l,2-benzisothiazol-3-yl)]piperazine methanesulfonate with bicyclo[2.2.1] heptane-2-exo-3-exo-dicarboximide is carried out using organic bases with a ρ¾ higher than 10 such as l,4-diazabicycloundec-7-ene (DBU), l,5-diazabicyclo[4.3.0]non-5-ene (DBN), 1,4-diaza bicyclo[2.2.2] -octane (DABCO). These organic bases are comparatively expensive.
Indian patent application no. IN 2306/MUM/2014 and Chinese patent applications no. CN 102863437 and CN 103864774 disclose the use of dimethyl formamide (DMF), dimethyl sulphoxide (DMSO), dimethyl acetamide (DMA) and N-methyl pyrrolidine (NMP) for the condensation of iran5-3a,7a-octahydroisoindolium-2-spiro- -[4′-(l,2-benzisothiazol-3-yl)] piperazine methanesulfonate with bicyclo[2.2.1] heptane-2-exo-3-exo-dicarboximide to form lurasidone. These solvents have high boiling point so not preferred at commercial scale.
Some of the prior art processes are related to reduction of impurities or quality improvement of lurasidone hydrochloride.
International publication no. WO2011/136383 discloses a process for the preparation of lurasidone hydrochloride in which amount of by products are reduced by increasing the quantity of l-(l,2-benzisothiazol-3-yl)piperazine instead of sodium carbonate or potassium carbonate as base in the reaction mixture. Increasing the amount of l-(l,2-benzisothiazol-3-yl)piperazine causes an increase in cost of production and removal of excess compound makes the process less commercially viable.
International publication no. WO2011/136384 discloses a process for the preparation of lurasidone hydrochloride in which amount of by products are reduced by using dibasic potassium phosphate with a small amount of water as a base instead of sodium carbonate. Use of dibasic potassium phosphate as a base causes an increase in cost of production as dibasic potassium phosphate is expensive.
International publication no. WO2013/014665 discloses various processes for the preparation of lurasidone hydrochloride. In general the process is shown below:

Formula-(I)
In this process iran5-(lR,2R)-2-(aminomethyl)cyclohexyl)methanol of Formula (B) is first reacted with bicyclo[2.2.1]heptane-2-exo-3-exo-dicarboximide of Formula (A) to form (3aR,4S,7R,7aS)-2-(((lR,2R)-2-(hydroxymethyl)cyclohexyl)methyl)hexahydro-lH-4,7-methanoisoindole-l,3(2H)-dione of Formula (C) which on reaction with methane sulphonyl chloride followed by reaction with l-(l,2-benzisothiazol-3-yl)piperazine of Formula (D) forms lurasidone free base which was converted into lurasidone hydrochloride using acetone and cone, hydrochloric acid.
Some of the prior art processes disclose various combinations of hydrogen chloride and solvent for the preparation of lurasidone hydrochloride from lurasidone free base.
US 7,605,260 discloses use of acetone and aqueous HC1 having strength 1.8-14.4 % for preparing lurasidone hydrochloride. The yield of lurasidone hydrochloride is relatively low (85%) by this method. If the acid concentration during the salt formation is more than 5.0% then acetone quantity as the residual solvent in the reaction product is found to be greater than 0.5% in our hands which is above the ICH limits. If acid concentration during the salt formation is less than 1.8%, then yield is reduced drastically to 65%. Therefore, this method has limitations on the large-scale industrial production.
Chinese patent application no. CN102746289A discloses the process for the preparation of lurasidone hydrochloride by adding a mixture of acetone and aqueous HC1 to a solution of lurasidone free base in acetone. On reproducing this process in laboratory, it was observed that the XRPD of the product obtained does not match with XRPD of lurasidone hydrochloride.
Indian patent application IN 777/MUM/2013 discloses use of IPA, water and 35% Aqueous HC1 for the preparation of lurasidone hydrochloride. The IPA content in the product was found to be more than 5000ppm.
The methods described in the prior art are not suitable for large scale commercial production as the residual solvent is out of the ICH limits and thus the product obtained can’t be used as a drug. In order to keep the residual solvent(s) within ICH limits, repeated crystallization/purification are required which results in reduced yield and make the process quite expensive.
The prior art discloses various processes for the preparation of lurasidone hydrochloride and its intermediates. However, there still remains a need for alternative process for the preparation of lurasidone and its pharmaceutically acceptable salts substantially free of residual solvent(s) which can be used as a drug.
According to another embodiment of the present invention, novel process for the preparation of the compound of Formula (III), their isomers and pharmaceutically acceptable salts thereof, comprises condensing 1,2-cyclohexane dicarboxylic acid of Formula (II), their isomers with carbonyl diimidazole, optionally in a solvent.
(IV) 
Formula (III)
NaBH4 RT /H20
![]()
Formula (VII)
Scheme-1:

Example-1
Synthesis of trans(R,R)-l,2-cyclo exane dicarboxylic acid
A round bottom flask was charged with methanol (500 mL), IPA (500 mL) and trans (racemic)-l,2-cyclohexane dicarboxylic acid (100 g). In this reaction mass (R)-l-phenylethyl amine (74 mL) was added over a period of 30 minutes and stirred for 2-3 hrs at 30-40 °C. The solid obtained was filtered, washed with methanol and IPA solution (50+50 mL) and dried under reduced pressure to obtain crude salt of iran5(R,R)-l,2-cyclohexane dicarboxylic acid. The obtained salt was stirred in a solution of methanol (500 mL) and IPA (500 mL) at 65-70 °C for 2-3 hours, cooled to room temperature and filtered. The solid was washed with methanol and IPA solution (50+50 mL) and dried under reduced pressure. The solid thus obtained was dissolved in about 2N hydrochloric acid and extracted two times with ethyl acetate (1000 mL+200 mL). Organic layers were combined and washed with brine solution (100 mL). Ethyl acetate was distilled off under vacuum at 50-55 °C and cyclohexane was added to the residue. The solid separated out was filtered and washed with cyclohexane and dried under vacuum at 45-50 °C for 8-10 hours. Yield = 29.4 g
Example-2
Synthesis of ((R,R)-cyclohexane-L2-diyl)bis((lH-imidazol-l-yl)methanone)
To a solution of iran5(R,R)-l,2-cyclohexane dicarboxylic acid (25.0 g) in THF (250 mL), carbonyl diimidazole (60 g) is added and stirred for one hour at 25-30 °C . To the said solution of (R,R)2-(((lH-imidazole-lcarbonyl)oxy)carbonyl)cyclohexanecarboxylic acetic anhydride lH-imidazole (25.0 g) in THF (250 mL) is stirred for one hour at 45-50 °C. The compound obtained is isolated and is characterized by mass and NMR.
[m z = 272.75; 1H-NMR: 8.24 (s, 2H), 7.72 (d, 2H); 7.50 (d, 2H), 3.5 (m, 2H), 2.26-1.50 (m, 8H)]
Example-3
Synthesis of tra»,s(R,R)-l,2- bis(hydroxymethyl)cyclohexane
To a solution of ((R,R)-cyclohexane-l,2-diyl)bis((lH-imidazol-l-yl)methanone) (25 g) in THF (250 mL), sodium borohydride (22.0 g) followed by water (44.0 mL) are added and stirred for one hour. To this reaction mass, 10% solution of acetic acid (500 mL) and dichloromethane (500 mL) are added, stirred and layers separated. The organic layer is washed with 10% sodium bicarbonate solution followed by water. The dichloromethane is distilled off from organic layer under vacuum to give an oily mass. To the oily mass
dichloromethane (100 mL), water (100 mL) and 12.5mL cone, hydrochloric acid (35%) are added, stirred and layers obtained are separated. The dichloromethane is distilled off completely from organic layer at 40 °C to obtain oily mass (15.5 g).
Example-4
One pot process for synthesis of trans(R,R)-l,2- bis(hydroxymethyl)cyclohexane from trans(R,R)-l,2-cyclo exane dicarboxylic acid
To a solution of iran5(R,R)-l,2-cyclohexane dicarboxylic acid (25.0 g) in THF (250 mL), carbonyl diimidazole (60 g) was added and stirred for one hour at 25-30 °C. To the intermediate obtained sodium borohydride (22.0 g) and water (44.0 mL) were added and stirred for one hour. To this reaction mass, 10% solution of acetic acid (500 mL) and dichloromethane (500 mL) were added, stirred and layers separated. The aqueous layer was washed with dichloromethane (250 mL). The organic layer was washed with 10% sodium bicarbonate solution followed by water. The dichloromethane is distilled off from organic layer under vacuum to give an oily mass. To the oily mass dichloromethane (100 mL), water (100 mL) and 12.5mL cone, hydrochloric acid (35%) were added, stirred and layers obtained were separated. The dichloromethane was distilled off completely at 40 °C to obtain oily mass (15.5 g).
Example-5
Synthesis of m¾ns(R,R)- 2-bis(methanesulfonylmethyl) cyclohexane
To a suspension of irafts(R,R)-l,2-bis(hydroxymethyl)cyclohexane (15.0g) in dichloro methane (300 mL), triethyl amine (43.7 mL) followed by methane sulphonyl chloride (17.8 mL) were added over a period of 30-45 minutes. Reaction mass was stirred for 2-3 hrs. Reaction was monitored by HPLC (RI detector). After the completion of reaction, water was added, stirred and layers separated. The organic layer was washed with 10% sodium bicarbonate solution (150 mL) followed by water (150 mL). The dichloromethane was distilled off from organic layer under vacuum at 40-55 °C to give an oily mass. Methanol (30 mL) was added to the oily mass and strip off under vacuum at 40°C, added methanol (150 mL) and stirred for 1 h at 10-15°C and the solid obtained was filtered, washed with methanol (15 mL) and dried under vacuum to get the product (15.8g).
Example-6
Synthesis of ?ran (R,R)-3aJ(¾-octahvdroisoindolium-2-spiro- -r4-(L2-benzoisothiazole-3-yl)l piperazine methanesulfonate:
To a suspension of iran5(R,R)-l,2-bis(methanesulfonylmethyl)cyclohexane (15 g) in acetonitrile (150 mL) l-(l,2-benzisothiazol-3-yl)piperazine (10.95g) and sodium carbonate (7.8 g) were added, heated and stirred for 20 hrs at reflux temperature. Reaction was monitored by HPLC. After the completion of reaction, mass was cooled to 40-45 °C, filtered and washed with acetonitrile (20 mL). The acetonitrile was distilled off under vacuum at 45-50 °C. To the residue acetone (100 mL) was added, stirred for 1 hour, filtered, washed with acetone (10 mL), dried at 50-55°C for 6-8 hours to get the product (12.5 g).
Example-7
Synthesis of Lurasidone
To a suspension of iran5(R,R)-3<3,7(3-octahydroisoindolium-2-spiro- -[4-(l,2-benzo isothiazole-3-yl)]piperazinemethanesulfonate (10 g) in toluene (150 mL), bicycle[2.2.1] heptane-2-exo-3-exo-dicarboximide (5.9 g) and potassium carbonate (4.8 g) were added, heated to 110° C and stirred for 8-10 hours. Reaction was monitored by HPLC. After the completion of reaction, reaction mass was cooled to 20-30 °C, filtered and washed with toluene (10 mL). The toluene was distilled off at 55-60°C. To the residue IPA (100 mL) was added and stirred for 1-2 hours at room temperature. Lurasidone free base obtained was filtered and washed with IPA (10 mL). The solid was suck dried for 30 minutes to obtain lurasidone.
Example-8
Synthesis of Lurasidone hydrochloride
To lurasidone base (5g), acetone (75mL) and water (10 mL) were added. The mixture was heated to 55-60°C followed by the addition of IPA.HCl (10%) (lOmL) and stirred for 1-2 hours, reflux temperature. The clear solution obtained was stirred for 30 min and then 5ml IPA.HCl (10%) was added. The reaction mixture was stirred at reflux temperature for 30 min, cooled and stirred for 60 min. The solid obtained was filtered and washed with acetone (5ml) and dried under vacuum at 60°C for 8 hours.
Acetone: 542 ppm; IPA= 38ppm; Yield=93%
Example-9
Synthesis of Lurasidone hydrochloride
To lurasidone base (5g), acetone (75mL) and water (5 mL) were added. The mixture was heated to 55-60°C followed by the addition of IPA.HCl (10%) (5mL) and stirred for about 1-2 hours. The reaction mixture was stirred for 30 min. at 55-60°C, cooled and stirred for 60 min. The solid obtained was filtered and washed with acetone (5ml) and dried under vacuum at 70-80°C for 8 hours.
Jubilant Generics Limited



Chairman & Managing Director
Jubilant Bhartia Group Shyam, together with his brother Hari, is founder of Jubilant Bhartia Group (www.jubilantbhartia.com) headquartered in New Delhi, India. The Jubilant Bhartia Group, with 30,000 employees, has a strong presence in diverse sectors like Pharmaceuticals and Life Sciences, Oil and Gas (exploration and production), Agri products, Performance Polymers, Retail, Food and Consulting in Aerospace and Oilfield Services. Jubilant Bhartia Group has four flagships Companies- Jubilant Life Sciences Limited, Jubilant FoodWorks Limited and Jubilant Industries Limited, listed on Indian Stock Exchange and Jubilant Energy NV, listed at AIM market of London Exchange.Shyam, holds a bachelors’ degree in commerce from St. Xavier’s College, Calcutta University, and is a qualified cost and works accountant & a fellow member of the Institute of Cost and Works Accountants of India (ICWAI).Shyam has been associated with various institutions and has served as Member of Board of Governors, Indian Institute of Technology (IIT), Mumbai, and Indian Institute of Management (IIM), Ahmedabad. Shyam has also served as a Member of the Executive Committee of Federation of Indian Chamber of Commerce & Industry (FICCI) & Confederation of Indian Industry (CII) and was also a member of Task Force on Chemicals appointed by the Government of IndiaShyam’s immense contributions have been recognized by various awards. CHEMEXCIL has conferred Lifetime Achievement Award 2010-11 to him. He, along with his brother, was felicitated with the Entrepreneur of the Year Award at the prestigious AIMA Managing India Awards 2013, presented by the President of India. In 2010, the duo also shared the much-covetedErnst & Young Entrepreneur of the Year Award for Life Sciences & Consumer Products category.Shyam serves on the Board of several Public and Private and Foreign companies likes of Chambal Fertilizers and Chemicals Ltd, Putney Inc., CFCL Technologies Limited (Cayman Islands), Tower Promoters, BT Telecom India Pvt Ltd., American Orient Capital Partners India Pvt Ltd, IMACID, Morocco, Safe Food Corporation, etc. He was also a Director on the Board of Air India.Shyam is a regular participant at the World Economic Forum Annual Meeting in Davos and a member of the Chemical Governors Council of the World Economic Forum.Shyam is married to Shobhana, Former Member of Parliament & Chairperson, The Hindustan Times Media Ltd. They have two sons- Priyavrat and Shamit.
ISO Certification
ISO 9001:2008, 14001:2004 & OHSAS 18001:2007 certified
Code of Conduct
Code Of Conduct for Directors and Senior ManagementThis Code of Conduct highlights the standards of conduct expected from the Company’s Directors and Senior Management so as to align these with the Company’s Vision, Promise and Values.Jubilant Life Sciences Ltd. (Jubilant) has a well formulated Vision which drives the business and has the promise of Caring, Sharing, Growing to all the stakeholders–We will, with utmost care for the environment, continue to enhance value for our customers by providing innovative products and economically efficient solutions and for our shareholders through sales growth, cost effectiveness and wise investment of resources.
Director’s Desk

Co-Chairman & Managing Director
Jubilant Bhartia Group
Hari, together with his brother Shyam, is co-founder of Jubilant Bhartia Group (www.jubilantbhartia.com) headquartered in New Delhi, India.The Jubilant Bhartia Group, with 30,000 employees, has a strong presence in diverse sectors like Pharmaceuticals and Life Sciences, Oil and Gas (exploration and production), Agri products, Performance Polymers, Retail, Food and Consulting in Aerospace and Oilfield Services. Jubilant Bhartia Group has four flagships Companies- Jubilant Life Sciences Limited, Jubilant FoodWorksLimited and Jubilant Industries Limited, listed on Indian Stock Exchange and Jubilant Energy NV, listed at AIM market of London Exchange.A Chemical Engineering Graduate from the prestigious Indian Institute of Technology (IIT), Delhi, Hari was conferred the Distinguished Alumni award by his alma mater in 2000. He has been associated in various capacities with the IIT system and with the Ministry of Human Resource Development, Government of India.Hari is a past President of the Confederation of Indian Industry (CII) & a member of several educational, scientific and technological programmes of the Government of India. He is currently the Chairman of the Board of Governors of the Indian Institute of Management (IIM), Raipur and Member of the International Advisory Board of McGill University, Canada.Hari is the Co-Chairman of India-Canada CEO’s Forum appointed by the Prime Minister of India. He is also a member of CEO’s Forum for India-USA, India-France and India-Sri Lanka and Joint Task Force for India-Myanmar & India-UAE. He is a regular participant at the World Economic Forum Annual Meeting in Davos and is a member of the World Economic Forum’s International Business Council and the Health Governors.Hari’s immense contributions have been recognized by various awards. He, along with his brother, was felicitated with the Entrepreneur of the Year Award at the prestigious AIMA Managing India Awards 2013, presented by the President of India. In 2010, the duo also shared the much-coveted Ernst & Young Entrepreneur of the Year Award for Life Sciences & Consumer Products category.Hari serves on the board of several public and private companies like TV 18 Broadcast Ltd., Shriram Pistons & Rings Ltd., Export Credit Guarantee Corporation of India Ltd., BT Telecom India Pvt. Ltd & India Brand Equity Foundation.Hari is married to Kavita, a leading Fashion Designer and Retailer. They have a daughter, Aashti and a son, Arjun.

Executive Leadership Team

Shyam S Bhartia
Chairman

Hari S Bhartia
Co-Chairman & Managing Director

Shyamsundar Bang
Executive Director –Manufacturing & Supply Chain

R Sankaraiah
Executive Director – Finance

Pramod Yadav
Co-CEO
Life Science Ingredients
Rajesh Srivastava
Co-CEO
Life Science Ingredients
G. P. Singh
Fine Chemicals and CRAMS
CEO – Jubilant Pharma
Chandan Singh
President – Life Science Chemicals

Martyn Coombs
President – Jubilant DraxImage

Bryan Downey
President – Allergy Business

T. S. Parmar
President – India Branded Pharmaceuticals

Dr. Ashutosh Agarwal
Chief Scientific Officer –Chemicals and Life Science Ingredients

Ajay Khanna
Chief – Strategic & Public Affairs
///////Lurasidone hydrochloride, Jubilant Life Sciences Ltd, WO 2016059649, New patent
NEW PATENT, WOCKHARDT LIMITED, WO 2016055918, ISAVUCONAZOLE
![]()
WO2016055918) NOVEL STABLE POLYMORPHS OF ISAVUCONAZOLE OR ITS SALT THEREOF
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
KHUNT, Rupesh Chhaganbhai; (IN).
RAFEEQ, Mohammad; (IN).
MERWADE, Arvind Yekanathsa; (IN).
DEO, Keshav; (IN)
The present invention relates to novel stable novel stable polymorphs of Isavuconazole or its salt thereof, having purity more than 90 % when measured by HPLC. In particular the present invention directs process for the preparation of solid amorphous and crystalline form of Isavuconazole base. In a further embodiment present invention directs to crystalline form Isavuconazole Hydrobromide salt and oxalate salt of 2-(2,5-difluoro- phenyl)-1-[1,2,4]triazol-1-yl-butane-2,3-diol.
Isavuconazole, Isavuconazonium, Voriconazole, and Ravuconazole are azole derivatives and known as antifungal drugs for treatment of systemic mycoses as reported in US 5,648,372, US 5,792,781, US 6,300,353 and US 6,812,238.
The US patent No. 6,300,353 discloses Isavuconazole and its process. It has chemical name [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-l -(lH-l,2,4-triazol-l-yl)-2-(2,5-difluorophenyl)43utan-2-ol; and has the structural formula I:

Formula I
The ‘353 described the process for the preparation Isavuconazole, involve the use of 2-(2,5-difluoro-phenyl)-l-[l ,2,4]triazol-l-yl-butane-2,3-diol (referred herein after “diol base”) in an oil form, which is difficult to isolate and purify. The use of 2-(2,5-difluoro-phenyl)-l-[l ,2,4]triazol-l-yl-butane-2,3-diol base, without purification, reflects the purity of Isavuconazole and Isavuconazonium sulfate. However, the reported process not feasible industrially.
Thus, an object of the present invention is to provide simple, cost effective and industrially feasible processes for preparation of Isavuconazole or its salt thereof in enhanced yield as well as purity. In a particular present invention directs to novel stable polymorphs of Isavuconazole or its salt thereof.

Examples
Example-1: Preparation of Amorphous Isavuconazole
In a round bottomed flask charged ethanol (250 ml), thioamide compound (25.0 gm) and 4-cyano phenacyl bromide (18.4 gm) under stirring. The reaction mixture were heated to 70 °C. After completion of reaction the solvent was removed under vacuum distillation and water (250 ml) and Ethyl acetate (350 ml) were added to reaction mass. The reaction mixture was stirred and its pH was adjusted between 7 to 7.5 by 10 % solution of sodium bicarbonate. The layer aqueous layer was discarded and organic layer was washed with saturated sodium chloride solution (100 ml) and concentrated under vacuum to get residue. The residue was suspended in methyl tert-butyl ether (250 ml) and the reaction mixture was heated to at 40°C to make crystals uniform and finally reaction mass is cooled to room temperature filtered and washed with the methyl tert-butyl ether. The product was isolated dried to get pale yellowish solid product.
Yield: 26.5 gm
HPLC purity: 92.7%
Example-2: Preparation of crystalline Isavuconazole Base
Charged methylene dichloride (250 ml) and 25.0 gm Isavuconazole Hydrobromide compound of formula-II into 1.0 L flask and stirred. Added aqueous solution of sodium bi carbonate in to the reaction mass to obtained clear solution. The layers were separated and organic layer was washed with dilute hydrochloric acid solution followed by saturated solution of sodium chloride. Finally, Organic layer was concentrated under vacuum to get titled product.
Yield: 18.5 gm
HPLC Purity: 97%
Example-3: Preparation of crystalline Isavuconazole Hydrobromide
Charged isopropanol alcohol (250 ml) followed by thioamide compound (25.0 gm) and 4-cyano phenacyl bromide (18.4 gm) into 1.0 L flask. The reaction mixture was stirred and heated to 50 C, after completion of reaction the precipitated material was filtered and washed with isopropanol alcohol (25 ml). The wet cake is dried under vacuum for 4-5 hrs at 40 C to obtain off-white solid product.
Yield: 26.5 gm
HPLC Purity: 97.3%
Exaniple-4: Synthesis of 2-(2,5-difluoro-phenyl)-l -[l,2,4]triazol-l-yl-butane-2,3-diol oxalate
Dissolved crude 50 gm 2-(2,5-difluoro-phenyl)-l-[l ,2,4]triazol-l -yl-butane-2,3-diol base compound in 150 ml of ethyl acetate. Oxalic acid dihydrate 25 gm was added into the reaction mixture and stirred. Heat the reaction mixture for 1 hour at 50-55 °C. The reaction mixture was cooled to 25°C to 35°C. Toluene 300 ml was added into the reaction mixture to precipitate the solid. The precipitate was washed with toluene and dried under vacuum to obtain the solid crystalline form of titled compound.
Yield: 58 g
HPLC Purity: 76%
Exaniple-5: Synthesis of 2-(2,5-difluoro-phenyl)-l -[l,2,4]triazol-l-yl-butane-2,3-diol oxalate salt
Exemplified procedure in example 1 with the replacement ethyl acetate solvent with tetrahydrofuran and antisolvent toluene with petroleum ether were used to get the title compound.
Exaniple-6: Synthesis of 2-(2,5-difluoro-phenyl)-l -[l,2,4]triazol-l-yl-butane-2,3-diol oxalate
Exemplified procedure in example 1 with the replacement ethyl acetate solvent with isopropyl acetate and antisolvent toluene with diisopropyl ether were used to get the title compound.
Exaniple-7: Synthesis of 2-(2,5-difluoro-phenyl)-l -[l,2,4]triazol-l-yl-butane-2,3-diol oxalate
Exemplified procedure in example 1 wherein diethyl ether is used in place of ethyl acetate and toluene or heptane was used as antisolvent to get the title compound.
Example-8: Synthesis of 2-(2,5-difluoro-phenyl)-l -[l,2,4]triazol-l-yl-butane-2,3-diol oxalate
Exemplified procedure in example 1 wherein diethyl ether is used in place of ethyl acetate and isolation of the product were done by means of partial removal of the solvent under vacuum.
Example-9: Synthesis of 2-(2,5-difluoro-phenyl)-l -[l,2,4]triazol-l-yl-butane-2,3-diol oxalate
Exemplified procedure in example 1 wherein ethyl acetate is replaced with isopropyl acetate and further, the reaction mass was stirred at lower temperatures to about 10°C to about 15°C for 3-5 hours and subsequently precipitated product was isolated and dried.
Example-10: Synthesis of 2-(2,5-difluoro-phenyl)-l-[l ,2,4]triazol-l-yl-butane-2,3-diol base
Stirring the suspension of 260 ml water and 65 gm 2-(2,5-difluoro-phenyl)-l-[l,2,4] triazol-l-yl-butane-2,3-diol oxalate salt were added. The reaction mixture pH was adjusted by addition of 10 % aqueous sodium carbonate solution. The pH was maintained to about pH 7 to about 8, 300 ml dichloro methane was added into the reaction mixture with stirring. The layers were separated and dichloromethane layer was collected. Aqueous layer was extracted with 150 ml dichloromethane. Dichloromethane layer was combined and washed with water. Dichloromethane was distilled out to get titled compound.
Yield: 35 gm
Purity: 87%

Wockhardt Ltd chairman Habil Khorakiwala.

/////////NEW PATENT, WOCKHARDT LIMITED, WO 2016055918, ISAVUCONAZOLE
WO 2016042441, Mankind Research Centre, Silodosin, New patent

WO 2016042441, Mankind Research Centre, Silodosin, New patent


Mankind Research Centre
MANKIND RESEARCH CENTRE [IN/IN]; 191-E, Sector 4-II, IMT-Manesar, Haryana 122050 (IN)
A novel process for the preparation of considerably pure silodosin
GANGWAR, Kuldeep Singh; (IN).
KUMAR, Anil; (IN).
BHASHKAR, Bhuwan; (IN)
The present invention relates to a novel, improved, commercially viable and industrially advantageous process for the preparation of Silodosin of Formula (I), its pharmaceutically acceptable salts or solvates thereof. The invention relates to the preparation of considerably pure Silodosin with high yield.
Silodosin, l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl} amino)propyl]-2,3-dihydro-lH-indole-7-carboxamide of Formula (I) is an indoline antidysuric which has a selectively inhibitory effect against urethra smooth muscle constriction, and decreases urethra internal pressure without great influence on blood pressure. Silodosin is available under trade names RAPAFLO® or UROREC®. Silodosin was first disclosed in EP 0600675 as a therapeutic agent for the treatment of dysuria associated with benign prostatic hyperplasia, where a process for producing the compound is also disclosed.

Formula (I)
Since, Silodosin is an optically active compound having a complex chemical structure; its synthesis is relatively complex and requires a sequence of multiple steps.
US patent no. 6,310,086, discloses a process for preparing Silodosin analogue compound from reaction of (R)-3-{5-(2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl} propylbenzoate with 2-(2-ethoxyphenoxy)ethyl methanesulfonate and finally isolated as a crude compound which is purified by column chromatography. The said process has a major disadvantage of using column chromatography which is not feasible at plant scale production.
PCT application no. WO 2012147019, discloses the preparation of Silodosin as shown in scheme- 1, wherein the Ν,Ν-dialkyl impurity of Formula (Ila) formed during condensation of 3-{7-cyano-5-[(2R)-2-aminopropyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate of Formula (III) with 2-(2-(2,2,2-trifluoroethoxy)phenoxy)ethyl methanesulfonate of Formula (IV); is removed through preparation of monotartarate salt to give compound of Formula (VI). The compound of Formula (VI) is base hydrolyzed followed by cyano hydrolysis to give crude Silodosin of Formula (VIII) which is then further purified by crystallization to get desired pure Silodosin.
Scheme- 1:

Major drawback of above said reaction process is that multiple isolations and crystallizations are required to get pure Silodosin.
Similarly, US 7,834,193 discloses monooxalate salt represented by Formula Via having 0.9% of dialkyl impurity represented by Formula Ila. The oxalate salt so obtained is subjected to alkaline hydrolysis followed by transformation of the nitrile to an amide.

Formula (Ila)
Similarly, PCT application no. WO 2012147107, discloses the method wherein Silodosin is prepared by condensation of 3-{7-cyano-5-[(2R)-2-aminopropyl]-2,3-dihydro-lH-indol-l-yl} propyl benzoate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate in solvent using base and phase transfer catalyst wherein, dialkyl impurity is formed up to 11%, followed by hydroxyl deprotection in protic solvent using base and phase transfer catalyst which is then subjected to purification to remove N,N-dialkyl impurity represented by Formula (lib) up to 0.6% through the preparation of acetate salt. This process suffers from a serious drawback i.e., accountable formation of dialkyl impurity and even after purification the impurity is reduced to only up to 0.6%. Secondly, the process requires multiple isolations and purifications ensuing into time engulfing workups and purifications and hence incurring solvent wastage. This makes process lengthy, uneconomical and tedious to be performed at plant scale.
Another PCT application no. WO 2012131710, discloses the preparation of Silodosin in which the chiral compound (3-(5-((R)-2-aminopropyl)-7-cyanoindolin-l-yl)propyl benzoate) is reacted with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methane sulfonate in isopropyl alcohol using sodium carbonate as base. The reaction is completed in 40-50h and about 9-11% of dimer is formed during condensation. After completion of reaction, it is subjected to hydroxyl deprotection and the crude compound so obtained is purified to remove the Ν,Ν-dialkyl impurity of Formula (lib). The pure compound is then reacted with hydrogen peroxide in dimethyl sulfoxide to give Silodosin. The major drawback of this process is that the process is a multistep process wherein the condensation reaction is long-drawn-out resulting into countable amount of dimer formation during the process.
Thus, the prior art methods of preparing Silodosin require multiple and repeated purifications to synthesize DMF (Drug Master File) grade Silodosin. None of the prior art produces compound of Formula (VI) or (VII) with Ν,Ν-dialkyl impurity of Formula (Ila) or (lib) in an amount less than 0.6% to 0.5% even after purification. Therefore to prepare highly pure Silodosin, there is a need to explore new synthetic schemes that could be more economical and scalable. The present invention provides a novel, improved, commercially viable and industrially advantageous process for the synthesis of Silodosin and its pharmaceutically acceptable salts or solvates thereof. The present invention focus on preparation of highly pure Silodosin in appreciable yields with minimal use of solvents wherein the Silodosin is isolated with purity >99.5% having Ν,Ν-dialkyl impurity less than 0.03% and other individual impurities below 0.1%.

Ramesh Juneja (seated), founder of Mankind Pharma, with brother Rajeev, who is senior director (marketing & sales)

Mankind Pharma Chairman and Founder RC Juneja
In accordance to one embodiment of the present invention, the process of the preparation of Silodosin represented by Formula (I)

comprises the steps of:
a) condensing chiral compound represented by Formula (III)

Formula (III)
wherein, Bz represents to Benzoyl group with compound represented by Formula (IV)
![]()
Formula (IV)
wherein, Ms represents to Methanesulfonyl group in presence of base and phase transfer catalyst in an organic solvent to give intermediate represented by Formula (V)

Formula (V)
wherein, n is an integer of 1 and 2 and Bz is as defined above, wherein the compound having n=2 is formed in an amount of less than 5%;
b) optionally isolating compound of Formula (V),
c) without purification converting it to de-protected compound represented by Formula (IX) in an organic solvent;

Formula (IX)
wherein, n is as defined above;
d) optionally isolating compound of Formula (IX), and
e) without purification converting it to compound represented by Formula (X)

Formula (X)
wherein n is as defined above;
f) subjecting compound of Formula (X) to purification by converting to acid salt for removal of Ν,Ν-dialkyl impurity represented by Formula (lie);

Formula (He)
g) hydrolysis of the said acid salt to get Silodosin of Formula (I) with purity >99.5%.
Examples
The invention is explained in detail in the following examples which are given solely for the purpose of illustration only and therefore should not be construed to limit the scope of the invention.
Example 1
Preparation of Crude Silodosin:
Method A:
To the solution of lOg (0.0275 mol) of (3-(5-((R)-2-aminopropyl)-7-cyanoindolin-l-yl)propyl benzoate) in 100ml of toluene was added 14.3g (0.0826 mol) of dipotassium hydrogen phosphate and 8.20g (0.0261 mol) of 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methane sulfonate followed by addition of 2.0g (0.0055 mol) of tetrabutyl ammonium iodide and stirred the reaction mass at 85-90°C for 10-12h. Cooled the reaction mass, added de-mineralized water and separated the toluene layer followed by distillation to get crude viscous mass. Added 110ml of dimethyl sulfoxide and a solution of 1.51g (0.0415 mol) of sodium hydroxide dissolved in 8.52ml of water followed by addition of 6.42g (0.0567 mol) of 30% w/w of hydrogen peroxide. Stirred the reaction mass at 20-25°C till completion and added sodium sulfite solution. Extracted the compound in ethylacetate, washed the organic layer with brine solution and concentrated to get 10.2g of crude Silodosin.
Ν,Ν-dialkyl impurity is 3.2% as per HPLC.
Method B:
To the solution of lOg (0.0275 mol) of (3-(5-((R)-2-aminopropyl)-7-cyanoindolin-l-yl)propyl benzoate) in 100ml of toluene was added 14.3g (0.0826 mol) of dipotassium hydrogen phosphate and 8.20g (0.0261 mol) of 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methane sulfonate followed by addition of 2.0g (0.0055 mol) of tetrabutyl ammonium iodide and stirred the reaction mass at 85-90°C for 10-12h. Added solution of 4.4g of sodium hydroxide dissolved in 10ml of water and stirred the reaction at ambient temperature till completion. Quenched the reaction mass with water and separated the layers. Washed the toluene layer with brine and concentrated under reduced pressure to get crude mass. Dissolved the crude mass so obtained in 110ml of dimethyl sulfoxide and added a solution of 1.95g (0.0488 mol) of sodium hydroxide dissolved in 7.95ml of water followed by addition of 7.5g (0.066 mol) of 30% w/w of hydrogen peroxide. Stirred the reaction mass at room temperature followed by addition of 210ml of aqueous solution of sodium sulfite and extracted the compound in ethyl acetate. Washed the organic layer with brine and concentrated under reduced pressure to get 10. lg of crude Silodosin.
Ν,Ν-dialkyl impurity is 3.0% as per HPLC
Method C:
To the solution of lOg (0.0275 mol) of (3-(5-((R)-2-aminopropyl)-7-cyanoindolin-l-yl)propyl benzoate) in 100ml of dimethyl sulfoxide was added 14.3g (0.0826 mol) of dipotassium hydrogen phosphate and 8.20g (0.0261 mol) of 2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methane sulfonate followed by addition of 2.0g (0.0055 mol) of tetrabutyl ammonium iodide and stirred the reaction mass at 85-90°C for 2-3h. Added 100ml of water and 50ml of toluene and stirred the reaction mass at room temperature for half an hour. Separated the toluene layer and concentrated under reduced pressure. To the crude mass so obtained was added 110ml of dimethyl sulfoxide and a solution of 4.4g of sodium hydroxide dissolved in 10ml of water followed by addition of 7.5g (0.066 mol) of 30% w/w of hydrogen peroxide. Stirred the reaction mass at room temperature followed by addition of 210ml of aqueous solution of sodium sulfite and extracted the compound in ethyl acetate. Washed the organic layer with brine and concentrated under reduced pressure to get 9.8 g of crude Silodosin.
Ν,Ν-dialkyl impurity is 2.1% as per HPLC
Method D:
To the solution of 20g (0.055 mol) of (3-(5-((R)-2-aminopropyl)-7-cyanoindolin-l-yl)propyl benzoate) in 200ml of toluene was added 28.6g (0.165 mol) of dipotassium hydrogen phosphate and 16.4g (0.0522 mol) of 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methane sulfonate followed by addition of 4.0g (0.11 mol) of tetrabutyl ammonium iodide and stirred the reaction mass at 85-90°C for 10-12h. Added de-mineralized water and stirred at room temperature for half an hour. Separated the toluene layer to which was added a solution of 8.8g of sodium hydroxide dissolved in 20ml of water and stirred the reaction at ambient temperature till completion. Quenched the reaction mass with water and separated the layers. Washed the toluene layer with brine and concentrated under reduced pressure to get crude mass. Dissolved the crude mass so obtained in 200ml of dimethyl sulfoxide and added a solution of 3.9g (0.0976 mol) of sodium hydroxide dissolved in 16ml of water followed by addition of 15g (0.132 mol) of 30% w/w of hydrogen peroxide. Stirred the reaction mass at room temperature followed by addition of 400ml of aqueous solution of sodium sulfite and extracted the compound in ethyl acetate. Washed the organic layer with brine and concentrated under reduced pressure to get 21. Og of crude Silodosin.
Ν,Ν-dialkyl impurity is 2.8% as per HPLC
Method E:
To the solution of 2g (0.0055 mol) of (3-(5-((R)-2-aminopropyl)-7-cyanoindolin-l-yl)propyl benzoate) in 20ml of was dimethyl sulfoxide was added 2.87g (0.0165 mol) of dipotassium hydrogen phosphate and 1.64g (0.0052 mol) of 2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methane sulfonate followed by addition of 0.29g (0.0011 mol) of 16-crown ether and stirred the reaction mass at 85-90°C for 10-12h. Added a solution of 0.88g of sodium hydroxide dissolved in 2ml of water and stirred the reaction at ambient temperature till completion. Added de-mineralized water and toluene and stirred at room temperature for half an hour. Separated the toluene layer and concentrated under reduced pressure and to the solid mass so obtained were added 20ml of dimethyl sulfoxide and a solution of 0.38g (0.0231 mol) of sodium hydroxide dissolved in 1.6ml of water followed by addition of 1.5g (0.0132 mol) of 30% w/w of hydrogen peroxide. Stirred the reaction mass at room temperature followed by addition of aqueous solution of sodium sulfite and extracted the compound in ethyl acetate. Washed the organic layer with brine and concentrated under reduced pressure to get 2.1g of crude Silodosin.
Ν,Ν-dialkyl impurity is 2.2% as per HPLC
Method F:
To the solution of lOg (0.0275 mol) of (3-(5-((R)-2-aminopropyl)-7-cyanoindolin-l-yl)propyl benzoate) in 100ml of was acetonitrile was added 14.3g (0.0826 mol) of dipotassium hydrogen phosphate and 8.20g (0.0261 mol) of 2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methane sulfonate followed by addition of 2.0g (0.0055 mol) of tetra butyl ammonium iodide and stirred the reaction mass at 85-90°C for 10-12h. Added a solution of 4.4g of sodium hydroxide dissolved in 10ml of water and stirred the reaction at ambient temperature till completion. Added de-mineralized water and toluene and stirred at room temperature for half an hour. Separated the toluene layer and concentrated under reduced pressure and to the solid mass so obtained were added 110ml of dimethyl sulfoxide and a solution of 1.95g (0.0488 mol) of sodium hydroxide dissolved in 7.95ml of water followed by addition of 7.5g (0.066 mol) of 30% w/w of hydrogen peroxide. Stirred the reaction mass at room temperature followed by addition of 210ml of aqueous solution of sodium sulfite and extracted the compound in ethyl acetate. Washed the organic layer with brine and concentrated under reduced pressure to get 9.5g of crude Silodosin.
Ν,Ν-dialkyl impurity is 3.1% as per HPLC
Method G:
To the solution of lOg (0.0275 mol) of (3-(5-((R)-2-aminopropyl)-7-cyanoindolin-l-yl)propyl benzoate) in 100ml of was Dimethyl sulfoxide was added 14.3g (0.0826 mol) of dipotassium hydrogen phosphate and 8.20g (0.0261 mol) of 2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methane sulfonate followed by addition of 4.0g (0.0055 mol) of tetra butyl ammonium iodide and stirred the reaction mass at 85-90°C for 10-12h. Added a solution of 4.4g of sodium hydroxide dissolved in 10ml of water and stirred the reaction at ambient temperature till completion. Added de-mineralized water and toluene and stirred at room temperature for half an hour. Separated the toluene layer and concentrated under reduced pressure and to the solid mass so obtained were added 110ml of dimethyl sulfoxide and a solution of 1.95g (0.0488 mol) of sodium hydroxide dissolved in 7.95ml of water followed by addition of 7.5g (0.066 mol) of 30% w/w of hydrogen peroxide. Stirred the reaction mass at room temperature followed by addition of 210ml of aqueous solution of sodium sulfite and extracted the compound in ethyl acetate. Washed the organic layer with brine and concentrated under reduced pressure to get 10.4g of crude Silodosin.
Ν,Ν-dialkyl impurity is 1.83% as per HPLC
Example 2
Purification of Crude Silodosin:
To the lOg (0.0080 mol) of crude mass of Silodosin was added 110ml of isopropyl alcohol followed by addition of 1.75g of oxalic acid at ambient temperature. Stirred the solution 6-8h and filtered the precipitates. Added ethyl acetate and water in the ratio of 1: 1 to the above solid followed by addition of 5ml of liquor ammonia. Stirred the reaction mass at ambient temperature for 15 min and separated the layers. Concentrated the organic layer to ¼ of its volume and left undisturbed overnight. Filtered the precipitates and recrystallized with ethyl acetate followed by drying under reduced pressure to get 5.1g of pure Silodosin. The amount of impurities and the percent impurity of the Silodosin obtained was as follows:
Ν,Ν-dialkyl impurity: undetectable amount
Other impurities: 0.03 to 0.09%
Silodosin purity: 99.65% (HPLC)
////WO 2016042441, Mankind Research Centre, Silodosin, New patent
New patent, WO 2016042573, Acitretin, Emcure Pharmaceuticals Ltd

Acitretin
PDT PATENT US4105681

Process for preparation of acitretin
Emcure Pharmaceuticals Ltd
EMCURE PHARMACEUTICALS LIMITED [IN/IN]; an Indian company at EMCURE HOUSE, T-184, MIDC., Bhosari, Pune – 411 026 Maharashtra (IN)
GURJAR MUKUND KESHAV; (IN).
JOSHI SHASHIKANT GANGARAM; (IN).
BADHE SACHIN ARVIND; (IN).
KAMBLE MANGESH GORAKHANATH; (IN).
MEHTA SAMIT SATISH; (IN)
The present invention Provides a process for preparation of {(2E, 4E, 6E, 8E) -9- (4-methoxy-2,3,6-trimethyl) phenyl-3,7-dimethyl-nona-2,4,6 , 8} tetraenoate, acitretin year intermediate of formula (VI) with trans isomer ≥97%, comprenant of Reacting 3-formyl-Crotonic acid butyl ester of formula (V) Substantially free of impurities, with 5- (4-methoxy- 2,3,6-trimethylphenyl) -3-methyl-penta-2,4-diene-l-triphenyl phosphonium bromide of formula (IV) and isolating resulting compound of formula (VI) Treating the filtrate with iodine for isomerization of the Undesired cis intermediate and finally Obtaining acitretin (I), with trans isomer Desired ≥97%.


Samit Satish Mehta holds the position of the President – Research & Development
Acitretin of formula (I), chemically known as (2E,4E,6E,8E)-9-(4-methoxy-2,3,6- trimethyl)phenyl-3,7-dimethyl-nona-2,4,6,8-tetraenoic acid, is a second generation retinoid a roved by USFDA in 1996, for the treatment of psoriasis.

Acitretin (I)
The process for preparation of acitretin (I) was first disclosed in US 4,105,681 wherein the intermediate, 5-(4-methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene-l-triphenyl phosphonium bromide was reacted with 3-formyl-crotonic acid butyl ester in presence of sodium hydride as base and dimethylformamide as solvent. The resultant ester derivative was obtained with a trans is (E/Z) ratio of around 55:45 which was subjected to hydrolysis in presence of potassium hydroxide and ethyl alcohol to obtain acitretin.
Use of hazardous, highly pyrophoric and moisture sensitive reagent like sodium hydride, along with cumbersome work-up and successive crystallizations to obtain the desired isomer rendered the process unviable for commercial scale.
Indian patent application 729/MUM/2012 discloses use of organic bases such as triethyl amine or pyridine for the reaction of 3-formyl-crotonic acid butyl ester and 5-(4-methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene-l -triphenyl phosphonium bromide for the synthesis of acitretin. The process utilizes a large excess of the organic base (2.85:1.0) with respect to the reactant phosphonium bromide derivative. Further, there is no mention of the ratio of cis and trans geometric isomers of the product thus obtained either at the intermediate or final stage. The trans: cis (E/Z) ratio of the intermediate significantly impacts the final yield and purity of the product as several purifications and crystallizations are required to obtain the desired trans isomer.
The present inventors have experimentally observed that use of organic base in such large quantities severely hampers the removal of the undesired side product triphenyl phosphonium oxide formed in significant amounts. Also, the intermediate is obtained with a very modest trans: cis (E/Z) ratio.
WO2012/155796 discloses another method wherein alkali metal alkoxides are used as bases in the reaction of 5-(4-methoxy-2,3,6-trimethylphenyl)-3 -methyl -penta-2,4-diene-l -triphenyl phosphonium bromide with 3-formyl-crotonic acid. The obtained reaction mass, after adjusting pH to 7-8 with acid, is directly subjected to catalytic isomerization using catalysts such as Pd(OAc)2 or Pd(NH3)2Cl2. The reaction mixture so obtained is quenched with water, neutralized and filtered to get the desired product, which is further recrystallized from ethyl acetate. Although this procedure avoids the hydrolysis step and attempts in-situ isomerization, however the use of expensive, soluble palladium catalyst which cannot be recycled from the reaction mass coupled with lengthy reaction time of 25-30 hours and large solvent volumes make the process unviable.
It may be noted that in the synthesis of acitretin, the key reaction of 5-(4-methoxy-2,3,6-trimethylphenyl)-3 -methyl-penta-2 ,4-diene- 1 -triphenylphosphoniumbromide with 3 -formyl crotonic acid or its ester in presence of either strong inorganic bases such as sodium hydride, alkali metal alkoxides or organic bases like triethylamine is common to almost all synthetic routes disclosed in the prior art. Hence, all these routes suffer from the inherent problems of formation of undesired impurities including cis-isomeric compounds and their separation from the desired all-trans product which necessitates various purification methods ranging from column chromatography, multiple crystallizations etc.
Thus, there still exists a need for a convenient, easy-to-scale up process for synthesis of acitretin (I) which avoids use of pyrophoric strong bases and provides a robust method which affords acitretin having desired isomeric purity in high yield.

5-(4-methoxy,2,3,6 trimethylphenyl)- 3-formyl crotonic acid butyl glyoxalate L(+) tartaric acid
3-methyl-penta-2,4-dien-1-triphenyl butyl ester (V) dibutyl ester
phosphonium bromide (IV)

Acitretin (I)


Satish Mehta,CEO, Above and here Inspiring the participants
EXAMPLES
Example 1: Preparation of 4-(4-methoxy-2,3,6-trimethylphenyl)-but-3-en-2-one (II)
Acetone (6000 ml) was added to 4-methoxy-2,3,6 trimethyl benzaldehyde (500.3 g) and the mixture was stirred at 20-30°C. Aqueous solution of sodium hydroxide (134.8 g in 500 ml water) was gradually added to it and the resulting mixture was heated to 45-50°C with continued stirring. After completion of the reaction, as monitored by HPLC, the reaction mass was cooled and acetic acid was added till pH 4.5 to 5.5. Distillation of acetone, followed by addition of cyclohexane to the residue, followed by washing with water, separation and concentration of the organic layer gave 4-(4-methoxy-2,3,6 trimethylphenyl)-but-3-en-2-one of formula (II).
Yield: 80-84%
Example 2: Preparation of 5-(4-methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene- 1-triphenyl phosphonium bromide (IV)
4-(4-Methoxy-2,3,6-trimethylphenyl)-but-3-en-2-one (II; 500 g) dissolved in toluene (2000 ml) was gradually added to a mixture of vinyl magnesium bromide (3500 ml; 1 molar solution in THF) and lithium chloride (4.8 g) and stirred at 20-30 C till completion of the reaction as monitored by HPLC. The reaction mixture was quenched with water and concentrated hydrochloric acid was added till the pH was between 3 and 4. The organic layer was separated and concentrated to give residue containing 5-(4-methoxy-2,3,6 trimethylphenyl)-3 -methyl -penta l,4-dien-3-ol (III). Methyl isobutyl ketone (3500 ml) was added to the residue, followed by gradual addition of triphenyl phosphine hydrobromide (745.3 g) at room temperature. The reaction mixture was heated to 50-60°C till completion of the reaction. The reaction mixture was cooled and filtered to give 5-(4-methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene-l-triphenyl phosphonium bromide of formula (IV).
Yield: 1000 g (76%)
Example 3: Preparation of 3-formyl crotonic acid butyl ester (V)
Dibutyl-L- tartrate (500 g) was dissolved in isopropanol (3500 ml) at room temperature, and water (750 ml) was added to it. The reaction mixture was cooled to 15-25°C and sodium metaperiodate (448.5 g) was gradually added to it with stirring. The reaction was continued at 20-30°C till completion of the reaction based on GC analysis. The reaction mixture was filtered and the filtrate was concentrated. The resulting residue was dissolved in toluene (1000 ml), stirred and filtered to obtain the filtrate containing butyl glyoxylate. Propionaldehyde (221.0 g) was added to the filtrate and heated to around 60°C, followed by gradual addition of piperidine (26.4 g, dissolved in toluene). The reaction mixture was further heated and stirred at 110-120°C till completion of the reaction, as monitored by GC. After completion, the reaction mass was cooled, washed with aqueous sulfuric acid, water and finally with aqueous sodium bicarbonate solution. The organic layer was concentrated and the residue was distilled to give 3-formyl crotonic acid butyl ester (V)
Yield: 230-280 g (35-43%)
Example 4. Preparation of butyI{(2E,4E,6E,8E)-9-(4-methoxy-2,3,6-trimethyl) phenyl-3,7-dimethyl-nona-2,4,6,8}tetraenoate (VI)
Sodium carbonate (297. lg), was added to the mixture of 5-(4-Methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene-l-triphenyl-phosphoniumbromide (IV; 1000 g) in toluene (5000 ml) followed by gradual addition of 3-formyl crotonic acid butyl ester (330 g) at room temperature. The stirred reaction mixture was heated to 60-70°C till completion of the reaction as monitored by HPLC. The reaction mass was cooled, quenched with water. The organic layer was separated, concentrated and n-heptane was added to the residue. The mass was stirred, filtered and 40% aqueous methanol (2000 ml) was added to it with stirring. Layer separation, concentration of the organic layer, and crystallization of the resulting residue from isopropyl alcohol, optionally with seeding followed by filtration gave crop I of butyl {{(2E,4E,6E,8E)— 9-(4-methoxy-2,3,6 trimethyl)phenyl-3,7 dimethyl -nona-2,4,6,8} tetraenoate (VI),.
Yield: 45-50%;
Cis: Trans isomer ratio (2.0:98.0)
The filtrate was concentrated, the residue was dissolved in toluene (2000 ml) and treated with iodine (4.5 g) at room temperature. After completion of the reaction, as monitored by HPLC, the reaction mixture was stirred with aqueous sodium thiosulfate solution. Separation and concentration of the organic layer and crystallization of the resulting residue from isopropyl alcohol, optionally with seeding, gave crop II of butyl {{(2E,4E,6E,8E)-9-(4-methoxy-2,3,6-trimethyl)phenyl-3,7-dimethyl-nona-2,4,6,8} tetraenoate (VI).
Yield (crop II): 15 to 20%.
Cis: Trans isomer ratio (2.0:98.0)
Total yield (crop I+II): 60-70%.
Example 5: Preparation of acitretin (I)
Aqueous solution of potassium hydroxide (155.2 g in 600 ml water) was added to a solution of butyl {(2E,4E,6E,8E)-9-(4-methoxy-2,3 ,6-trimethyl) phenyl-3 ,7-dimethyl-nona- 2,4,6,8}tetraenoate, VI (300.0 g) in ethanol (1800 ml) at 25-30°C and the reaction mixture was stirred at reflux temperature till completion of the reaction. After completion, as monitored by HPLC, the reaction mixture was quenched with water, and hydrochloric acid was added till pH was between 2.5 and 3.5. The mass was heated at 70°C, stirred, cooled to 40-50°C and filtered. Recrystallization of the resulting solid from tetrahydrofuran gave acitretin (I).
Yield: 154.0 g (60%)
Desired trans isomer: > 98%

India’s hockey stars Sardara Singh and Sandeep Singh with Emcure Pharmaceuticals COO, Arun Khanna

HE Dr. Kenneth Kaunda, First President of Zambia interacting with Mr. A. K. Khanna, COO & ED, Emcure at Emcure booth at AIDS 2012 conference, Washington
 Mr. Sunil Mehta is an Executive Director and Senior Director (Projects)
Mr. Sunil Mehta is an Executive Director and Senior Director (Projects)

Arun Khanna is the Chief Operating Officer and Executive Director on the Board of Emcure Pharmaceuticals Limited.
//////New patent, WO 2016042573, Acitretin, Emcure Pharmaceuticals Ltd

(2E,4E,6E,8E)-9-(4-methoxy-2,3,6-trimethylphenyl)-3,7-dimethylnona-2,4,6,8-tetraenoic acid
Acitretin is an oral retinoid effective in the treatment of psoriasis. It is the major metabolite of ETRETINATE with the advantage of a much shorter half-life when compared with etretinate.
| Molecular Formula: | C21H26O3 |
|---|---|
| Molecular Weight: | 326.42934 g/mol |
Acitretin (trade names Soriatane and Neotigason) is a second-generation retinoid. It is taken orally, and is typically used for psoriasis.
It is a metabolite of etretinate, which was used prior to the introduction of acitretin. Etretinate was discontinued because it had a narrow therapeutic index as well as a long elimination half-life (t1/2=120 days), making dosing difficult. In contrast, acitretin’s half-life is approximately 2 days. However, because acitretin can be reverse metabolised into etretinate which has an extremely long half-life, women must avoid becoming pregnant for at least 3 years[1] after discontinuing acitretin. Therefore, acitretin is generally not recommended for women of child-bearing age with a risk of becoming pregnant.
Acitretin is an oral retinoid used in the treatment of severe resistant psoriasis. Because of the potential for problems and severe side effects it is generally used in only very severe cases of psoriasis that have been unresponsive to other treatments. It binds to nuclear receptors that regulates gene transcription. They induce keratinocyte differentiation and reduce epidermal hyperplasia, leading to the slowing of cell reproduction. Acitretin is readily absorbed and widely distributed after oral administration. A therapeutic effect occurs after 2 to 4 weeks or longer.
Patients that have received the medication are advised against giving blood for at least 3 years due to the risk of birth defects.[2]
- 1 “Important Safety Information for SORIATANE”. soriatane.com. Retrieved 31 October 2015.
- 2
- AABB Technical Manual, American Association of Blood Banks
- 3
- “Soriatane”. WebMD. Retrieved 15 August 2015.
- 4
- “Soriatane Side Effects”. Drugs.com. Retrieved 15 August 2015.
- 5
“Soriatane (Acitretin) Drug Information: Description, User Reviews, Drug Side Effects, Interactions – Prescribing Information at RxList”. RxList. Retrieved 15 August 2015.
Literature References:
Synthetic retinoid; free acid form and major metabolite of etretinate, q.v. Prepn: W. Bollag et al., DE 2414619; eidem, US 4105681 (1974, 1978 both to Hoffmann-La Roche).
Teratogenicity study: A. Kistler, H. Hummler, Arch. Toxicol. 58, 50 (1985).
HPLC determn in plasma: N. R. Al-Mallah et al., Anal. Lett. 21, 1603 (1988).
Pharmacokinetics in humans: F. G. Larsen et al., Pharmacol. Toxicol. 62, 159 (1988).
Clinical evaluation in cutaneous lupus erythematosus: T. Ruzicka et al., Arch. Dermatol. 124, 897 (1988).
Review of clinical pharmacology: A. Vahlquist, O. Rollman, Dermatologica 175, Suppl. 1, 20-27 (1987).
Review of clinical studies in psoriatic and nonpsoriatic dermatoses: J.-M. Geiger, B. M. Czarnetzki, ibid. 176, 182-190 (1988).
SORIATANE (acitretin), a retinoid, is available in 10-mg, 17.5-mg, and 25-mg gelatin capsules for oral administration. Chemically, acitretin is all-trans-9-(4-methoxy-2,3,6trimethylphenyl)-3,7-dimethyl-2,4,6,8-nonatetraenoic acid. It is a metabolite of etretinate and is related to both retinoic acid and retinol (vitamin A). It is a yellow to greenish-yellow powder with a molecular weight of 326.44. The structural formula is:
so
 |
Each capsule contains acitretin, black monogramming ink, gelatin, maltodextrin (a mixture of polysaccharides), microcrystalline cellulose, and sodium ascorbate.
Gelatin capsule shells contain gelatin, iron oxide (yellow, black, and red), and titanium dioxide. They may also contain benzyl alcohol, carboxymethylcellulose sodium, edetate calcium disodium.
SYNTHESIS

Synthetic retinoid; free acid form and major metabolite of etretinate, q.v. Prepn: W. Bollag et al., DE 2414619; eidem, US 4105681 (1974, 1978 both to Hoffmann-La Roche).
NMR, IR SEE………….https://www.iarc.fr/en/publications/pdfs-online/prev/handbook4/Handbook4_Retinoids-6.pdf
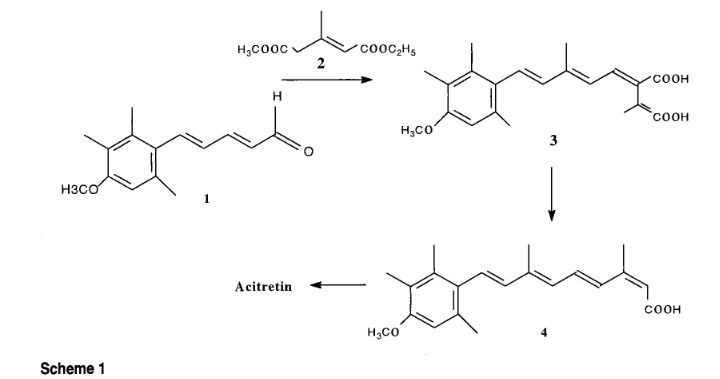
https://www.iarc.fr/en/publications/pdfs-online/prev/handbook4/Handbook4_Retinoids-6.pdf
http://nopr.niscair.res.in/bitstream/123456789/19745/1/IJBB%2039%281%29%2022-27.pdf

 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2E,4E,6E,8E)-9-(4-methoxy-2,3,6-trimethylphenyl)-3,7-dimethylnona-2,4,6,8-tetraenoic acid
|
|
| Clinical data | |
| Trade names | Soriatane, Neotigason |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a601010 |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 60% |
| Protein binding | >99.9% |
| Metabolism | Hepatic |
| Biological half-life | 49 hours |
| Excretion | Faeces & urine |
| Identifiers | |
| CAS Number | 55079-83-9 |
| ATC code | D05BB02 (WHO) |
| PubChem | CID 5284513 |
| IUPHAR/BPS | 7598 |
| DrugBank | DB00459 |
| ChemSpider | 4447573 |
| UNII | LCH760E9T7 |
| KEGG | D02754 |
| ChEBI | CHEBI:50173 |
| ChEMBL | CHEMBL1131 |
| Chemical data | |
| Formula | C21H26O3 |
| Molar mass | 326.429 g/mol |
////////////CC1=CC(=C(C(=C1C=CC(=CC=CC(=CC(=O)O)C)C)C)C)OC
New patent, Lomitapide mesylate , Zydus Cadila Healthcare Ltd, US 20160083345,


Was developed and launched by Aegerion, under license from the University of Pennsylvania (which acquired rights from BMS).

Sanjay Jagdish DESAI
Brij KHERA
Jagdish Maganlal PATEL
Harshita Bharatkumar SHAH
Arunkumar Shyam Narayan UPADHYAY
Sureshkumar Narbheram AGRAVAT
Polymorphic forms of lomitapide and its salts and processes for their preparation
Zydus Cadila Healthcare Ltd
The present invention relates to various polymorphic forms of lomitapide or its salts and processes for preparation thereof. The present invention provides Lomitapide mesylate in solid amorphous form and process for preparation thereof. The invention also provides an amorphous solid dispersion of lomitapide mesylate. Further, various crystalline forms of lomitapide mesylate like A, B and C and process for preparation thereof are provided. The invention also provides crystalline forms of lomitapide free base, in particular Form I and Form-II and their preparation. The invention further provides compositions comprising various forms of lomitapide and its salts.
A novel amorphous form of lomitapide mesylate (having >98% of purity and 0.5% of residual solvent and particles size D90 of >250 µm, D50 of >100 µm and D10 of >50 µm), a process for it preparation and a composition comprising it is claimed. Also claimed is an amorphous solid dispersion of lomitapide mesylate and a carrier (eg hydroxypropylmethyl cellulose acetate succinate). Further claimed are crystalline forms of lomitapide mesylate (designated ad Forms A, B, C, I, II and free base of lomitapide in amorphous form), processes for their preparation and compositions comprising them. Lomitapide is known to act as a microsomal triglyceride transfer protein inhibitor, useful for treating familial hypercholesterolemia.
Lomitapide is a synthetic lipid-lowering agent for oral administration. It is a microsomal triglyceride transfer protein inhibitor approved as Juxtapid® in US and as Lojuxta® in Europe as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis where available, to reduce low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), apolipoprotein B (apo B), and non-highdensity lipoprotein cholesterol (non-HDL-C) in patients with homozygous familial hypercholesterolemia (HoFH). The approved drug product is a mesylate salt of lomitapide, chemically known as N-(2,2,2-trifluoroethyl)-9-[4-[4-[[[4′(trifluoromethyl)[1,1′-biphenyl]-2-yl]carbonyl]amino]-1-piperidinyl]butyl]-9H-fluorene-9carboxamide methanesulfonate [“lomitapide mesylate” herein after] and has the structural formula

As per the approved label for Juxtapid® (US) “Lomitapide mesylate is a white to off-white powder that is slightly soluble in aqueous solutions of pH 2 to 5. Lomitapide mesylate is freely soluble in acetone, ethanol, and methanol; soluble in 2-butanol, methylene chloride, and acetonitrile; sparingly soluble in 1-octanol and 2-propanol; slightly soluble in ethyl acetate; and insoluble in heptane”.
As per Public Assessment Report for Lojuxta® (Europe) “Polymorphism has been observed for lomitapide mesylate. Of the different solid-state forms, hydrates, and solvates identified in the polymorph studies, only 2 desolvated solid-state forms, Form I and Form II, were identified in batches after drying to final drug substance.” The report further states, under the heading Manufacture, that “The final particle size distribution is controlled during the crystallisation step” (emphasis added) suggesting that the approved drug product lomitapide mesylate is a crystalline compound
U.S. Pat. No. 5,712,279 A discloses the lomitapide compound and a process for its preparation. It also discloses a process for preparation of lomitapide monohydrochloride.
U.S. Pat. No. 5,883,109 A discloses lomitapide mesylate specifically but no solid form was disclosed.
The reference article Synthesis and Applications of Isotopically Labelled Compounds, Vol. 8, Pg. 227-230 (2004) discloses the preparation of Deuterium labelled [d4]BMS-201038, [3H]BMS-201038, [14C]BMS-201038 wherein BMS-201038 is lomitapide mesylate.
International (PCT) Publication No. WO 2015/121877 A2 discloses lomitapide crystalline Form I and Form II as well as amorphous form of Lomitapide mesylate and processes for their preparation.
There is still a need to provide a novel polymorph of lomitapide or its salts which is suitable for pharmaceutical preparations. Therefore, the present invention provides new crystalline forms of lomitapide free base and lomitapide mesylate. The present invention also provides amorphous form of lomitapide free base and lomitapide mesylate, which is stable and useful for pharmaceutical preparations.
EXAMPLES
Example-1
Preparation of Lomitapide Mesylate
In a 250 mL round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 10 g lomitapide and 20 mL methanol were added and stirred to obtain a solution. 1.5 g methane sulfonic acid dissolved in 20 mL water was added slowly to the above solution under stirring. The reaction mixture was stirred till maximum salt formation was achieved. 50 mL water was added to the mixture, stirred for 15-20 min, filtered and washed with water. The product was dried further to obtain lomitapide mesylate.
EXAMPLE 2
Preparation of Amorphous Form of Lomitapide Mesylate
10 g lomitapide mesylate, 50 mL acetone and 150 mL ethyl acetate were heated in a 500 mL round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel at 50-55° C. and stirred to obtain clear solution. The solution was subjected to spray drying in JISL Mini spray drier LSD-48 with feed pump running at 30-35 rpm, inlet temperature 50-55° C., out let temperature 45-50° C., aspiration rate 1200-1300 rpm, hot air supply 1.8-2.2 Kg/cm2 and vacuum for conveying the dry product 80 mmHg. The product was collected from cyclone and characterized to an amorphous form by x-ray powder diffraction. The product was further dried to obtain the amorphous form of lomitapide mesylate
/////////////New patent, Lomitapide mesylate , Zydus Cadila Healthcare Ltd, US 20160083345, Amorphous
Glaxo……..Will help the world’s poorest people access copycat versions of its medicines at affordable prices.
Glaxo to Stop Seeking Drug Patents in Low-Income Countries
Drugmaker says move could help poor nations access cheaper copycat versions of its medicines


LONDON— GlaxoSmithKline PLC said it would stop seeking patents for its drugs in low-income countries, a move the drugmaker said could help the world’s poorest people access copycat versions of its medicines at affordable prices.
The U.K.-based company said it would take this approach in low-income and least-developed countries, a group totaling around 85 nations. In so-called lower-middle-income countries, a group of 51 nations that includes Vietnam, Cameroon and Sri Lanka, it said it would file patents but aim to grant licenses to generic manufacturers to supply low-cost versions of its drugs in those markets in return for a small royalty.
Glaxo previously filed patents in most lower-middle-income countries, and in low-income nations where a patent office exists. But that “patchwork” approach meant that generic drugmakers held back from manufacturing copycat medicines for these markets owing to the risk of being sued by pharmaceutical companies, according to Glaxo Chief Executive Andrew Witty.,,,,,,,,,continue reading
/////////Glaxo Chief Executive, Andrew Witty, filed patents, low income,poor nations, cheaper, copycat versions, medicines, GlaxoSmithKline
Mehta Api Pvt Ltd, Cinacalcet hydrochloride, New patent, WO 2016027211
![]()

Mehta Api Pvt Ltd, Cinacalcet hydrochloride, New patent, WO 2016027211
Mehta Api had cinacalcet hydrochloride under development and holds US DMF and European DMF as listed on the company’s website. Amgen and Kyowa Hakko Kirin, under license from NPS Pharmaceuticals, have developed and launched cinacalcet.
The present filing represents the first PCT filing from the assignee, which focuses on developing (using green chemistry) manufacturing and marketing of API’s- multi step, highly complex, potent, chiral and semi-synthetic, advance intermediates, specialty chemicals and building blocks.
PROCESS FOR THE PREPARATION OF CINACALCET AND ITS PHARMACEUTICALLY ACCEPTABLE SALTS
MEHTA API PVT. LTD. [IN/IN]; 203, Centre Point, 2nd Floor, Near Hotel Kohinoor, J.B. Nagar, Andheri-Kurla Road, Andheri (East), Maharashtra, Mumbai 400 059 (IN)
KHAN, Rao, Uwais, Ahmad; (IN).
PATHAK, Rajesh, Harshnath; (IN).
PATIL, Chetan, Vinesh; (IN).
GAIKWAD, Sanjay, Ramrao; (IN).
APAR, Shrikrishna, Motiram; (IN).
LINGE, Govind, Udhavrao; (IN).
SHAIKH, Mohammad, Umar; (IN)
Cinacalcet (N-[l-(R)-(-)-(l-naphthyl) ethyl]-3-[3-(trifluoromethyl) phenyl]-l-aminopropane) of Formula II, belongs to a category of calcimimetics class of compounds. It is useful for the treatment of hyperparathyroidism and the preservation of bone density in patients with kidney failure or hypercalcemia due to cancer. It is marketed under the trade name of Senipar in United States and under the trade name of Mimpara in Europe.
US6211244 and Drugs of the future (2002) 27 (9): 831, discloses a synthesis of Cinacalcet by reductive amination which implies the reaction of (R)-(l-naphthyl) ethylamine of formula (IV) with 3 -[3- (trifluoromethyl) phenyl] propionaldehyde of formula (V) in the presence of titaniumisopropoxide to afford the corresponding cinacalcet imine of formula (III), which is reduced to cinacalcet of formula (II) with NaBH4CN in ethanol.
WO2012007954 A 1 discloses process for Cinacalcet by reductive amination in presence of titanium Isopropoxide using NaBH4CN, wherein an ether solvent is used instead of ethanol. Indian patent applications 2268/DEL/2008 and 87/MUM/2011 disclose preparation of Cinacalcet wherein reaction of (R)-(I-naphthyl)ethylamine of formula (IV) with 3-[3-(trifluoromethyl)phenyl] propionaldehyde of formula (V) is carried out in the presence of titaniumisopropoxide to afford the corresponding cinacalcet imine, which is further reduced to cinacalcet with NaBH4.
The above disclosed processes require the use of reagents such as NaBH4CN, titanium isopropoxide, which are extremely toxic and flammable as well as not being environmentally sound. These reagents therefore make the industrial application of the process difficult.
US20110124917A1 and WO2008068625A2 both disclose preparation of Cinacalcet by reductive amination wherein reduction is performed by using sodiumtriacetoxyborohydride as a selective reducing agent for imines.
Sodiumtriacetoxyborohydride is hygroscopic in nature hence demands anhydrous conditions to be maintained rendering it not suitable for use on industrial scale.
WO2012007954 A 1 discloses reaction and work-up in THF followed by salt formation in Di-isopropyl ether and further purification in two solvent system consisting of Water and Methanol or Water and Acetonitrile. US20110124917 discloses reaction in Methanol, Workup in toluene, Salt formation in Ethyl Acetate and purification in Isopropanol. WO2008068625A2 discloses reaction, salt formation and Purification in two solvent system consisting of isobutyl Acetate and n-Heptane. 2268/DEL/2008 discloses reaction in MDC, Salt formation in Ethyl Acetate and Purification in Ethyl Acetate and Di-isopropyl ether. 87/MUM/2011 discloses reaction in THF, work-up in toluene. Salt formation in two solvent system consisting of cyclohexane and MTBE.
All the above prior-art process employs use of different solvents for each unit operation or a two-solvent system for purification, thereby rendering the processes not easily scalable on industrial scale.
1367/MUM/2009 discloses reductive amination using sodium borohydride with 67.6% yield reported. 3068/MUM/2012 discloses reductive amination using sodium borohydride with 86% yield but with less purity. Further 3068/MUM/2012 requires the usage of sulphuric acid for the reaction of (R)-(I-naphthyl)ethylamine of formula (II) with 3-[3-(trifluoromethyl)phenyl] propionaldehyde of formula (III).
Thus the processes disclosed above have one or other drawbacks, ranging from poor yield, purity, use of difficult to handle and toxic reagents or use of different solvents for each unit operation.
In view of the problems occurred in above methods, there remains a need for more economical and efficient industrially scalable process for the preparation of Cinacalcet and its pharmaceutically acceptable salts, which overcomes the drawbacks as disclosed in the prior art.
The present inventors have surprisingly found that when the condensation of [3-(trifluoromethyl)phenyl]propionaldehyde of formula – (V) with (R)-(l- naphthyl)ethylamine formula – (IV) is carried out in the absence of any reagent and water is removed under vacuum by azeotropic distillation at low temperatures in the optional presence of water scavengers, than Cinacalcet.hydrochloride with high purity and yield is obtained. Further the process is also industrially feasible due to the non-usage of hazardous reagents as also due to the reduction in isolation and purification steps.

Example I:
Preparation of Cinacalcet Hydrochloride, Formula (la)
To (1000 ml) toluene in a 4Neck Round Bottom flask along with dean-stark apparatus coupled to a condenser, charge (80gms) (R)-(l- naphthyl) ethylamine of formula – (IV). Cool to 10-15°C. Charge (lOOgms) 3-[(3-Trifluoromethyl)phenyl] propionaldehyde of formula (V). Apply vacuum to the reaction mass through condenser and maintain for 8 hrs simultaneously azeotroping out water generated in the reaction till the reaction complies by thin layer chromatography to give Cinacalcet imine of formula (III) in-situ. Release vacuum after the reaction complies. Water collected after Azeotropic distillation: 7-7.5 ml. Cool the reaction mass to 5-10°C. Charge (35 gms) sodium borohydride in two lots to the reaction mass and raise the temperature to 25-30°C. Maintain the reaction mass for 8 hrs to give Cinacalcet of formula (II) in-situ. After the reaction complies by thin layer chromatography adjust the pH of the reaction mass to about pH 6 using acetic acid. Charge (200 ml) water to the reaction mass and stir for 30 mins. Separate Layers the organic layer and treat with 15% HC1 (150 ml). Stirr the Reaction mass at 40 – 50°C for one hour and separate the layer. Heat the toluene at same temperature. Adjust pH of toluene layer to below pH-2 by treating with 15% HC1 (150 ml) at 40-45 °C. Distill out 500 ml toluene under vacuum below 45 °C. Gradually charge 500 ml water to the reaction mass along with simultaneously distilling out 500 ml toluene approximately. Filter the reaction mass to give crude Cinacalcet Hydrochloride. Dry at 45-50°C for 8 hrs.
Weight: 182 gms
% Yield on theoretical basis: 98.9%
Purity: 97.54%
To (182 gms) of Crude cinacalcet Hydrochloride charge (800 ml) Methyl tert butyl ether and stirr for 60°C for 3 hrs. Cool gradually at 25-30°C and further chill the reaction mass to 0°C -5°C. Maintain the reaction mass at 0-5°C for 2 hrs and filter under vacuum followed by washing to the wet-cake with (100 ml) chilled Methyl tert butyl ether.
Wet cake is dried under vacuum at 40°C.
Weight: 163 gms
Yield on theoretical basis: 88.58%
Purity: 99.54%
To (163 gms) of MTBE pure Cinacalcet Hydrochloride is charged (400 ml) Isopropanol and heated to 70-75°C to get a clear solution which is then gradually cooled to 25-30°C and further chilled to 0-5 °C. The reaction mass is maintained for 2 hrs at same temperature and filtered under vacuum followed by washing with chilled isopropanol. Wet cake is dried under vacuum at 40°C.
Weight: 157 gms
Yield on theoretical basis: 85.32%
Purity: 99.91%
Example II:
Preparation of Crude Cinacalcet Hydrochloride, Formula (la)
To (1000 ml) toluene in a 4Neck Round Bottom flask, is charged (80gms) (R)-(l-naphthyl)ethylamine of formula (IV). Cooled to 10-15°C. Charged (lOOgms) 3-[(3-Trifluoromethyl)phenyl] propionaldehyde of formula (V) slowly. Charged (1 gm) Calcium Chloride and maintained for 8 hrs till the reaction complies by thin layer chromatography to give Cinacalcet imine of formula (III) in-situ. After the reaction complies, the reaction mass is cooled to 5-10°C. Charged (35 gms) sodium borohydride in two lots to the reaction mass and raised the temperature to 25-30°C.The reaction mass is maintained for 8 hrs to give Cinacalcet Free base of formula (II) in-situ. After the reaction complies by thin layer chromatography pH of the reaction mass is adjusted to about pH 6 using acetic acid. Charged (200 ml) water to the reaction mass and stirred for 30 mins. Layers separated and the organic layer is treated with 15% HC1 (150 ml). Reaction mass is stirred at 40 – 50°C for one hour and layer separated. Toluene layer is water washed at same temperature. pH of toluene layer adjusted to below pH-2 by treating with 15% HC1 (150 ml) at 40-45°C. Distill out 500 ml toluene under vacuum below 45 °C. Gradually charge 500 ml water to the reaction mass along with simultaneously distilling out 500 ml toluene approximately. Filter the reaction mass to give crude Cinacalcet Hydrochloride. Dry at 45-50°C for 8 hrs
Weight: 178 gms
Yield on theoretical basis: 96.73%
Purity: 94.88%
To (178 gms) of Crude cinacalcet Hydrochloride charged (800 ml) Methyl tert butyl ether and stirr for 60°C for 3 hrs. Allowed to cool gradually at 25-30°C and further chilled the reaction mass to 0-5°C. Maintained the reaction mass at 0-5°C for 2 hrs and filtered under vacuum followed by washing to the wet-cake with (100 ml) chilled Methyl tert butyl ether. Wet cake is dried under vacuum at 40°C.
Weight: 159 gms,
% Yield on theoretical basis: 86.40%
Purity: 99.77%
To (159 gms) of MTBE pure Cinacalcet Hydrochloride is charged (400 ml) Isopropanol and heated to 70-75°C to get a clear solution. Gradually cool to 25-30°C and further chill to 0-5 °C. Maintain the reaction mass is for 2 hrs at same temperature and filte under vacuum followed by washing with chilled isopropanol. Wet cake is dried under vacuum at 40°C. Weight: 150 gms
% Yield on theoretical basis: 81.51 %
Purity: 99.91 %
Example III:
Preparation of Cinacalcet Hydrochloride, Formula (la)
To (1000 ml) toluene in a 4Neck Round Bottom flask, charge (80gms) (R)-(l-naphthyl)ethylamine of formula (IV). Cool to 10-15°C. Charge (lOOgms) 3-[(3-Trifluoromethyl)phenyl] propionaldehyde of formula (V). Charge ( 1 gm) Molecular Sieves and maintain the reaction mass for 8 hrs till the reaction complies by thin layer chromatography to give Cinacalcet imine of formula (III) in-situ. After the reaction complies, cool the reaction mass to 5-10°C. Charge (35 gms) sodium borohydride in two lots to the reaction mass and raise the temperature to 25-30°C. Maintain the reaction mass for 8 hrs to give Cinacalcet of formula (II) in-situ. After the reaction complies by thin layer chromatography adjust the pH of the reaction mass to about pH 6 using acetic acid. Charge (200 ml) water to the reaction mass and stir for 30 mins. Separate the layers and treat organic layer with 15% HC1 (150 ml).Stirr Reaction mass is at 40 – 50°C for one hour and separate layers. Water wash toluene layer at same temperature. Adjust pH of toluene layer pH-2 by treating with 15% HC1 (150 ml) at 40-45 °C. Distill and degasse under vacuum below 70°C to give Cinacalcet Hydrochloride
Weight: 172 gms
Yield on theoretical basis: 93.47%
Purity: 97.29%
To (172 gms) of Crude cinacalcet Hydrochloride charge (800 ml) Methyl tert butyl ether and stirr for 60°C for 3 hrs. Cool gradually at 25-30°C and further chill the reaction mass to 0-5 °C. Maintain the reaction mass at 0-5 °C for 2 hrs and filter under vacuum followed by washing to the wet-cake with (100 ml) chilled Methyl tert butyl ether.
Wet cake is dried under vacuum at 40°C.
Weight: 155 gms
% Yield on theoretical basis: 84.23%
Purity: 99.57%
To (155 gms) of MTBE pure Cinacalcet Hydrochloride charge (400 ml) Isopropanol and heat to 70-75°C to get a clear solution which is then gradually cooled to 25-30°C and further chill to 0-5 °C. Maintain the reaction mass i for 2 hrs at same temperature and filter under vacuum followed by washing with chilled isopropanol. Wet cake is dried under vacuum at 40°C.
Weight: 146 gms
% Yield on theoretical basis: 79.34%
Purity: 99.83%
Mehta API Pvt. Ltd.

MR HARSHADRAI P MEHTA
Chairman & Managing Director

Devendra Mehta
Chief Executive Officer at MEHTA API PVT LTD
////////Mehta Api Pvt Ltd, Cinacalcet hydrochloride, New patent, WO-2016027211, WO 2016027211
Afatinib dimaleate, Dr Reddy’s, New patent, WO 2016027243
![]()
Afatinib dimaleate, Dr Reddy’s, New patent, WO-2016027243,
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills, Hyderabad, Telangana, India – 500034. Hyderabad 500034 (IN)
RAMAKRISHNAN, Srividya; (IN).
PEDDY, Vishweshwar; (IN).
MAHAPATRA, Sudarshan; (IN).
KANNIAH, Sundara Lakshmi; (IN).
CHENNURU, Ramanaiah; (IN).
JOSE, Jithin; (IN).
DHAGE, Yogesh Mohanrao; (IN).
PEDDIREDDY, Subba Reddy; (IN).
YARRAGUNTLA, Sesha Reddy; (IN).
RAGHUVEER, Sherial; (IN).
KOLLA, Srinivasa Rao; (IN).
ANIL KSHIRSAGAR, Shivani; (IN).
JAFAR SHAIKH, Latif; (IN).
BANDARU, Srinivasulu; (IN)
The drug compound having the adopted name afatinib dimaleate, has a chemical name N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-,(2E)-, (2Z)-2-butenedioate (1 :2), and is represented by structure of formula I

Formula I
Afatinib dimaleate is an anticancer protein kinase inhibitor indicated for treatment of non-small-cell lung cancer. Process for preparation of afatinib, afatinib dimaleate and intermediates useful in preparation of afatinib dimaleate are described in US Patent Nos. 7,019,012; 8,426,586 and 7,960,546.
US Patent No. 8,426,586 discloses crystalline Form A of afatinib dimaleate salt and processes for preparation thereof. US Patent Application Publication No. 20140051713 discloses crystalline Form B of afatinib dimaleate salt and processes for preparation thereof. PCT Application Publication No. 2013052157 discloses crystalline Form C, Form D and Form E of afatinib dimaleate salt and processes for preparation thereof. The PCT publication also discloses crystalline Form A, B, C and Form D of afatinib base.
Polymorphism, the occurrence of different crystal forms, is a phenomenon of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties. Polymorphs in general will have different melting points, thermal behaviors (e.g. measured by thermogravimetric analysis – “TGA”, or differential scanning calorimetry – “DSC”), X-ray powder diffraction (XRPD or powder XRD) pattern, infrared absorption fingerprint, and solid state nuclear magnetic resonance (NMR) spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms, hydrates and solvates of a pharmaceutical product can provide materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other polymorphic forms. New polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof can also provide an opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, e.g., better processing or handling characteristics, improved dissolution profile, or improved shelf-life. For at least these reasons, there is a need for additional solid state forms of Afatinib di-maleate.
SUMMARY
The present application provides novel solid state forms of Afatinib di-maleate, processes for preparing them, and pharmaceutical compositions containing them.
The present application also encompasses the use of novel solid state forms of Afatinib di-maleate provided herein, for the preparation of other afatinib salts, other solid state forms of afatinib dimaleate, and formulations thereof.
The present application also encompasses the use of any one of the novel solid state forms of Afatinib di-maleate disclosed herein for the preparation of a medicament, preferably for the treatment of cancer, particularly for the treatment of cancers mediated by epidermal growth factor receptor (EGFR) and human epidermal receptor 2 (HER2) tyrosine kinases, e.g., solid tumors including NSCLC, breast, head and neck cancer, and a variety of other cancers mediated by EGFR or HER2 tyrosine kinases. The present invention further provides a pharmaceutical composition comprising any one of the Afatinib di-maleate crystalline forms of the present invention and at least one pharmaceutically acceptable excipient.
The present application also provides a method of treating cancer, comprising administering a therapeutically effective amount of at least one of the Afatinib di-
maleate novel solid state forms of the present application, or at least one of the above pharmaceutical compositions to a person suffering from cancer, particularly a person suffering from a cancer mediated by epidermal growth factor receptor (EGFR) and human epidermal receptor 2 (HER2) tyrosine kinases, e.g., solid tumors including but not limited to NSCLC, breast, head and neck cancer, and a variety of other cancers mediated by EGFR or HER2 tyrosine kinases.
Example 1 : Preparation of amorphous form of afatinib dimaleate.
2.0 g of afatinib dimaleate was dissolved in 80 mL of a mixture of methanol and acetone (3:1 ) at 26°C and stirred for 15 min. The solution was filtered to remove the undissolved particles and the filtrate was distilled under reduced pressure at 50°C. After distillation the solid was dried under vacuum at 45°C to get 1 .29 g of amorphous afatinib dimaleate. PXRD pattern: Fig. 1 .
///////Afatinib dimaleate, Dr Reddy’s, New patent, WO-2016027243, WO 2016027243
WO 2016027283, New patent, Indacaterol, Reddy-Cheminor Inc
Beta 2 adrenoceptor agonist
Chronic obstructive pulmonary disease
WO 2016027283, New patent, Indacaterol, Reddy-Cheminor Inc
A process for preparing indacaterol and salts thereof
REDDY, G Pratap; (IN).
SUNKU, Venkataiah; (IN).
BABU, Sunkaraneni Suresh; (IN)
The present invention relates to a process for preparing indacaterol or salts thereof. The process comprises of forming compound of Formula 1 by reacting compound of Formula 2 and compound of Formula 3 in the presence of a solvent to Form compound of Formula 4, 5 which on removal of the protecting groups forms compound of Formula 1.
Indacaterol maleate is a beta-selective adrenoceptor agonist with potent bronchodilator activity. Indacaterol is chemically known as 5-[(R)-2-(5, 6-diethyl-indan-2- yl amino)-l-hydroxy-ethyl ]-8-hydroxy-(lH)-quinolin-2-one.
US7534890 claims a process to prepare 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)- 1 -hydroxy-ethyl] -8-hydroxy-(l H)-quinolin-2-one salt. One of the key steps in the process is reacting an epoxide, such as 8-substituted oxy-5-(R)- oxiranyl-(lH)-quinoline-2-one [Formula (I)] with an amine, such as 2-amino-(5,6-diethyl)-indan to form an intermediate 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l -hydroxy-ethyl]-8- substituted oxy-(lH)-quinolin-2-one [Formula (Ha)].

The drawback of this process is opening of epoxide ring is not regioselective and thereby resulting, in formation of substantial quantities of impurities as by products, Formula (lib) and Formula (lie) resulting in overall lower yields. The quantity of 2- amino-(5,6-diethyl)-indan used in this step is also large excess than theoretical amounts. Subsequent improvements also did not address this problem effectively.
WO 2013/132514 discloses a process to prepare Indacaterol involving the steps of treating a compound of Formula (III), wherein L is a leaving group, with the amine, 2-amino-(5,6-diethyl)-indan or its acid addition salts to obtain a compound of Formula (IV) or its acid addition salts.

Though higher yields have been claimed, the process has not overcome completely all the problems mentioned earlier.
There is a need for developing a more efficient process for preparing Indacaterol or salts thereof especially for large scale production with higher yields.
The reaction scheme of synthesis of compound of Formula 3 is represented below.

Formula 3 Formula 13 Formula 12
xample 1
Process to prepare 5-[ (R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-hydroxy-( lH)-quinolin-2-one
2-Chloro-5,6-diethylindan (4.2g) was added to a solution of 5-[(R)-(2-amino-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one (6g) in dimethylformamide (20ml) followed by addition of N,N-diisopropyl-N-ethylamine (3.6 g) and sodium iodide (lg) at room temperature and stirred for 10 minutes. The reaction mixture was heated to 90° C and the temperature was maintained at 90 °C till the completion of reaction. The reaction mass was cooled to room temperature and diluted with dichloromethane (100ml) and water (100 ml) and stirred for 30 minutes. The organic phase was separated and the aqueous layer was extracted with dichloromethane. Combined organic layer was washed with water, dried and concentrated. The resulting residue was dissolved in isopropyl alcohol under reflux and cooled slowly to obtain 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-phenylmethoxy -(lH)-quinolin-2-one, which was isolated by filtration and dried under vacuum (7.4 g). Yield: 79.3 %. Purity of the product is >95 % (HPLC).
Example 2
Process to prepare 5-[(R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-hydroxy-( lH)-quinolin-2-one
Solution of 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-phenylmethoxy-(lH)-quinolin-2-one (lOg) in methanol (100ml) and acetic acid (20ml) was hydrogenated using palladium on charcoal 5% (1.5g) until completion of the reaction. The mixture was filtered over celite and the filtrate was concentrated at 55°C under vacuum. The residue obtained was dissolved in hot methanol to precipitate 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-! -hydroxy-ethyl]-8-hydroxy-(lH)-quinolin-2-one.
Example 3
Process to prepare 5-[(R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-hydroxy-(lH)-quinolin-2-one maleate
Crude 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l -hydroxy-ethyl]-8-hydroxy-(lH)-quinolin-2-one prepared by the process of Example 2 was added to a solution of maleic acid (2.6g) in methanol and the resulting clear solution was slowly cooled to 5° C and stirred for 2 hours at the same temperature. The slurry was filtered, washed with cold methanol and dried to obtain 5-[(R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-hydroxy-(lH)-quinolin-2-one maleate (8.8g). Yield: 83.5 %. Purity of the product is >99%. E.e. >99 %.
Example 4
Process for preparing 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one
Diisopropylethylamine (6g) was added to a solution of phthalimide (6g) in dimethylformamide (30 ml) at room temperature. To this solution, 8-(phenylmethoxy)-5-[(R)-2-bromo-l-hydroxy-ethyl]-(lH)-quinoline-2-one (11 gm) was added slowly followed by sodium iodide (1 g). The resulting mass was heated to 90°C and stirred till the completion of reaction as monitored by TLC. The reaction mass was diluted with water (200 ml) and the crude product was isolated by filtration. The wet filter cake was suspended in water (60 ml), stirred for 1 hour, filtered, washed with water to obtain 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one (10.4 gm) after drying. Yield: 80.7 %.
Method A- Process for preparing 5-[(R)-(2-amino-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one
To a solution of 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one ( 13.2 g) in a mixture of isopropanol (86 ml) and water (14 ml) sodium borohydride (4.6 g) was added slowly at room temperature and stirred overnight. Thereafter, the pH of the reaction mass was lowered to 5.5 with acetic acid, and then the reaction mass was heated to reflux for two hours. Isopropanol was distilled out under reduced pressure. The residue was diluted with ethyl acetate (120 ml) and concentrated hydrochloric acid (8 ml) was added and stirred for 15 minutes for the salts to precipitate out. The reaction mass was filtered and the salt was washed with ethyl acetate. To the clear filtrate concentrated hydrochloric acid (10 ml) was added and stirred at 5° C for 30 minutes for 5-[(R)-(2-amino-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one to separate out as hydrochloride salt. The product was isolated by filtration and dried under vacuum (8.2 g). The hydrochloride salt was dissolved in minimum amount of water and basified with sodium hydroxide solution. The product was isolated as free amine by concentrating the solution under reduced pressure and extracting the residue with isopropyl alcohol and distilling out the solvent (7.45 g). Yield 80 %.
1H-NMR (CDC13) ppm: 2.56-2.70 (m, 2H), 3.35 (s, br, 2H, exchangeable), 4.89 (m, 1H), 5.29 (s, 2H), 5.76 (s, 1H, exchangeable), 6.53 (d, 1H), 7.11-7.19 (dd, 2H), 7.29-7.36 (dd, 1H), 7.39 (d, 2H), 7.57 (d, 2H), 8.21 (d, 1H), 10.7 (s, br, 1H, exchangeable).
Method B- Process for preparing 5-[(R)-(2-amino-l-hydroxy-ethyl)-8-phenylmethoxy-( lH)-quinolin-2-one
To a solution of 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one ( 10 g) in ethanol (60 ml) hydrazine hydrate (4.8 g) was added and refluxed the mixture for about 6 hours. The solvent was distilled out under reduced pressure. To the residue, concentrated hydrochloric acid (16 ml) was added and heated to about 80°C and maintained till the completion of the reaction. The reaction mass was cooled to room temperature and filtered. The clear filtrate was basified and concentrated under reduced pressure. The product was isolated as free amine (5.8 g) by extracting with isopropyl alcohol and distilling out the solvent. Yield: 83%.
Method C
Preparation of 5-(2-benzylamino-l-hydroxy-ethyl)-8-phenylmethoxy-( lH)-quinolin-2-one 5-Acetyl-8-phenylmethoxy-(lH)-quinolin-2-one (30 g) was refluxed with selenium dioxide
(11.5 g) in a mixture of dioxane (350 ml) and water (30 ml) for 16 hours. The reaction mixture was diluted with dioxane (150 ml) and precipitated inorganic salts were removed by filtration. Clear filtrate was concentrated to about 60 ml under vacuum and diluted with methanol (100 ml). The reaction mass was cooled to 15° C and benzylamine (7.5 g) was added slowly over a period of 45 minutes and stirred at the same temperature for two hours.
The reaction mass was further cooled to 0°C and sodium borohydride (2.8 g) was added slowly over a period of one hour. Thereafter, the reaction mass was stirred at room temperature for 12 hours. The reaction mixture was concentrated under vacuum and diluted with 300 ml water and stirred at 20° C for three hours. The precipitated product was collected by filtration, washed with water followed by isopropyl ether and then dried (28.2 g) to obtain 5-(2-benzylamino-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one.
Example 5
Preparation of 5-acetyl-8-phenylmethoxy-(lH)-quinolin-2-one
To a solution of 5-acetyl-8-hydroxy-(lH)-quinolin-2-one (35 g) in dimethylformamide (175 ml) potassium carbonate (35 g) was added at room temperature and stirred for 10 minutes. To the suspension, benzylbromide (32 g) was slowly added over a period of 30 minutes and stirred for 2 hours at the same temperature for completion of reaction (monitored by TLC). The reaction mass was diluted with water (800 ml) and stirred for 20 minutes for the product to precipitate out. The product was filtered, washed with water and dried under vacuum to get the title product (48 g).
Example 6
Preparation of 5-(2-bromoacetyl)-8-phenylmethoxy-( lH)-quinolin-2-one
Boron trifluoride-diethyletherate (29 ml) was slowly added to a solution of 5-acetyl-8-phenylmethoxy-(lH)-quinolin-2-one (50 g) in dichloromethane (500 ml) at 0° C and stirred for 10 minutes at the same temperature to get a thick precipitate. The reaction mass was heated to reflux temperature and bromine solution was added (29 g in 190 ml dichloromethane) slowly over a period of 2 hours under reflux (the HBr fumes coming from the condenser was scrubbed). Thereafter, the reaction mass was refluxed for further 45 minutes. The solvent was distilled out completely under vacuum and the mass was triturated with 10% aqueous sodium carbonate solution (100 ml). The suspension was filtered, washed with water and the crude product was taken for the next stage reaction.
Example 7
Preparation of 5-(2-phthalimido-l-oxo-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one
Potassium carbonate (33.4 g) was added to a solution of phthalimide (21.73 g) in dimethylformamide (80 ml) at room temperature and stirred for 10 minutes. To this suspension, crude 5-(2-bromoacetyl)-8-phenylmethoxy-(lH)-quinolin-2-one of example 6, dissolved in dimethylformamide (120 ml), was added slowly over a period of 20 minutes. The resulting suspension was stirred at 50° C for about 1 hour for the completion of reaction as monitored by TLC. The mixture was diluted with water (800 ml) and the crude product was isolated by filtration. The wet filter cake was suspended in water (600 ml), stirred for 1 hour, filtered, washed with water and dried under vacuum to get 5-(2- phthalimido-l-oxo-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one (67.4 g). Over all yield
(after two steps): 90%.
Example 8
Preparation of 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-q inolin-2-one
To a solution of (R)-2-methyl-CBS-oxazaborolidine (1M in toluene, 4.2 ml) in dry
tetrahydrofuran (THF, 50 ml) Borane-diethylaniline (19 ml) was added slowly at – 10° C
and the contents were stirred at the same temperature for 15 minutes. A solution of 5-(2-
Phthalimido-l-oxo-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one ( 8.3 g), of example 7, in
a mixture of dry THF (50 ml) and dichloromethane (50 ml), was added slowly to the
reaction mass at – 10° C. The reaction mass was further stirred for 2 hours and then
methanol was added and the temperature was slowly raised to room temperature. Dilute
sulfuric acid (6N, 10 ml) was added to the reaction mixture and stirred for 15 minutes. The
reaction mixture was concentrated under vacuum and the crude mass was extracted with
ethyl acetate. The organic phase was washed with dilute sulfuric acid and then water. The
solvent was distilled out completely under vacuum and triturated with hexane. The
compound was isolated by filtration and dried (7.6 g). Yield: 91.1%. e.e.. >97%.
Example 9
Process of preparing 2-chloroindan
2-hydroxy indan (lOOg) was dissolved in 1, 2-dichloroethane (400 ml) and added to thionyl
chloride (125 g) slowly over a period of an hour. Temperature was maintained at less than
10° C. Thereafter, the reaction mass was slowly heated and refluxed till the completion of the reaction. The reaction was monitored by TLC. The reaction mass was cooled to room temperature and poured in to ice water, stirred for 1 hour and organic layer was separated. The aqueous layer was extracted with dichloroethane. Organic layers were combined and washed with water, sodium bicarbonate solution and dried over anhydrous sodium sulphate. Solvent was distilled out completely and the crude product was distilled under vacuum to obtain 2-chloroindan as a colorless liquid (118 g).
Example 10
Process for preparing 5-acetyl-2-chloroindan
Aluminium chloride (146 g) was added in small lots to nitromethane (500 ml) and the solution was cooled to 5° C under inert atmosphere while stirring. Acetyl chloride (84 g) was slowly added keeping the temperature at 5° C. Solution of 2-chloroindan (118 g) was slowly added in acetyl chloride (84 g) keeping temperature at 5° C. After completion of reaction, monitored by TLC, the reaction mass was poured into cold IN HC1 (2000 ml) solution and stirred for 30 minutes. The product was extracted into di-isopropyl ether. The combined organic layer was washed with water, bicarbonate solution, brine and dried over anhydrous sodium sulphate. The solvent was completely distilled out to obtain 5-acetyl-2-chloroindan as yellow waxy solid (130 g).
Example 11
Process for preparing 2-chloro-5-ethylindan
1 Liter hydrogenation vessel was charged with 50 grams of 5-acetyl-2-chloroindan, 400 ml of methanol and 10 ml of acetic acid. Palladium on charcoal 5% (5 g) was added and the reaction mass was hydrogenated until complete conversion to 2-chloro-5-ethylindan. The mixture was filtered over a bed of celite. The filtrate was concentrated under reduced pressure to obtain 2-chloro-5-ethylindan as an oily mass (42 g).
Example 12
Process for preparing 5-acetyl-2-chloro-6-ethylindan
5-acetyl-2-chloro-6-ethylindan was prepared from 2-chloro-5-ethylindan (20 g) in accordance with the procedure followed in Example 10.
Example 13
Process for preparing 2-chloro-5, 6-diethylindan
Hydrogenation of 5-acetyl-2-chloro-6-ethylindan using Palladium on charcoal adopting the procedure as reported in Example 11, gave 2-chloro-5, 6-diethylindan as a liquid. The crude product was distilled under vacuum to get colorless liquid.
1H-NMR (CDC13) ppm: 1.19-1.29 (t, 6H), 2.61-2.66 (q, 4H), 3.13-3.18 (dd, 2H), 3.36-3.41 (dd, 2H), 4.66-4.72 (m, 1H), 7.05 (s, 2H).
////////////WO 2016027283, New patent, Indacaterol, Reddy-Cheminor Inc
