




, click to zoom.")
Ono Pharmaceutical Co has become the first company in the world to get an approval for a PD-1 checkpoint inhibitor, as regulators in Japan gave the green light to nivolumab, developed with Bristol-Myers Squibb, as a treatment for melanoma.
Home » Japan pipeline (Page 2)
Category Archives: Japan pipeline
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
K 912, NC 6300, Epirubicin nano

Cancer Research UK
Irish Cancer Society
Macmillan Cancer Support
NHS Evidence
 Epirubicin – Substance Summary
Epirubicin – Substance Summary
PubChem
MedlinePlus
– See more at: http://www.cancerindex.org/Epirubicin#sthash.BBioCC6K.dpuf
PHASE 1 JAPAN SOLID TUMOURS
DNA/RNA Synthesis Inhibitor
WITH Nano Carrier Co.,Ltdhttp://pdf.irpocket.com/C4571/qnwX/eFou/vG1J.pdf
KOWA COMPANY LTD
CAS FREE FORM. 56420-45-2
Smiles
- O=C2c1c(O)c5c(c(O)c1C(=O)c3cccc(OC)c23)C[C@@](O)(C(=O)CO)C[C@@H]5O[C@@H]4O[C@H]([C@H](O)[C@@H](N)C4)C
- NMR
- http://file.selleckchem.com/downloads/nmr/S122302-Epirubicin-Hydrochloride-NMR-Selleck.pdf
- http://www.medkoo.com/Product-Data/Epirubicin/Epirubicin-QC-TZC20130604web.pdf
-
Epirubicin CAS No.: 56420-45-2 Synonyms: - 4′-Epi-DX;
- Epirubicina;
- WP 697;
- Pidorubicin;
- 4′-epiadriamycin;
- Adriblastin;
- epi-dx;
- Epiadriamycin;
- farmorubicin;
- imi28;
- Farmarubicin;
Formula: C27H29NO11 Exact Mass: 543.17400
NC-6300, an epirubicin-incorporating micelle, extends the antitumor effect and reduces the cardiotoxicity of epirubicin.
Epirubicin is widely used to treat various human tumors. However, it is difficult to achieve a sufficient antitumor effect because of dosage limitation to prevent cardiotoxicity. We hypothesized that epirubicin-incorporating micelle would reduce cardiotoxicity and improve the antitumor effect. NC-6300 comprises epirubicin covalently bound to PEG polyaspartate block copolymer through an acid-labile hydrazone bond. The conjugate forms a micellar structure of 40-80 nm in diameter in an aqueous milieu. NC-6300 (10, 15 mg/kg) and epirubicin (10 mg/kg) were given i.v. three times to mice bearing s.c. or liver xenograft of human hepatocellular carcinoma Hep3B cells. Cardiotoxicity was evaluated by echocardiography in C57BL/6 mice that were given NC-6300 (10 mg/kg) or epirubicin (10 mg/kg) in nine doses over 12 weeks. NC-6300 showed a significantly potent antitumor effect against Hep3B s.c. tumors compared with epirubicin. Moreover, NC-6300 also produced a significantly longer survival rate than epirubicin against the liver orthotopic tumor of Hep3B. With respect to cardiotoxicity, epirubicin-treated mice showed significant deteriorations in fractional shortening and ejection fraction. In contrast, cardiac functions of NC-6300 treated mice were no less well maintained than in control mice. This study warrants a clinical evaluation of NC-6300 in patients with hepatocellular carcinoma or other cancers.

K-912(NC-6300)の概要 K-912(NC-6300)は、世界的に幅広く使用されているアントラサイクリン系の抗が ん剤の一つであるエピルビシンを内包したミセル化ナノ粒子製剤で、その特性により、 エピルビシンの有する心毒性の軽減が期待できます。さらに、pH 応答性システムを採 用することで、腫瘍細胞内でのエピルビシンの放出量を高め、既存のエピルビシンに比 べより強力な抗腫瘍効果が期待できます。
Epirubicin is an anthracycline drug used for chemotherapy. It can be used in combination with other medications to treat breast cancer in patients who have had surgery to remove the tumor. It is marketed by Pfizer under the trade name Ellence in the US andPharmorubicin or Epirubicin Ebewe elsewhere.
Similarly to other anthracyclines, epirubicin acts by intercalating DNA strands. Intercalation results in complex formation which inhibits DNA and RNA synthesis. It also triggers DNA cleavage by topoisomerase II, resulting in mechanisms that lead to cell death. Binding to cell membranes and plasma proteins may be involved in the compound’s cytotoxic effects. Epirubicin also generates free radicalsthat cause cell and DNA damage.
Epirubicin is favoured over doxorubicin, the most popular anthracycline, in some chemotherapy regimens as it appears to cause fewer side-effects. Epirubicin has a different spatial orientation of the hydroxyl group at the 4′ carbon of the sugar – it has the opposite chirality – which may account for its faster elimination and reduced toxicity. Epirubicin is primarily used against breast and ovarian cancer, gastric cancer, lung cancer and lymphomas.
Development history
The first trial of epirubicin in humans was published in 1980.[1] Upjohn applied for approval by the U.S. Food and Drug Administration(FDA) in node-positive breast cancer in 1984, but was turned down because of lack of data.[2] It appears to have been licensed for use in Europe from around this time however.[3] In 1999 Pharmacia (who had by then merged with Upjohn) received FDA approval for the use of epirubicin as a component of adjuvant therapy in node-positive patients.
Patent protection for epirubicin expired in August 2007.
References
- Bonfante, V; Bonadonna, G; Villani, F; Martini, A (1980). “Preliminary clinical experience with 4-epidoxorubicin in advanced human neoplasia”. Recent results in cancer research 74: 192–9. PMID 6934564. PM6934564.
- “On Target”.
- According to the proprietary database iddb.com
External links
- http://www.bccancer.bc.ca/HPI/DrugDatabase/DrugIndexPro/Epirubicin.htm
- http://www.pfizerpro.com/page_not_found?rid=/wyeth_html/home/minisites/oncology/ellence/pi/description.html
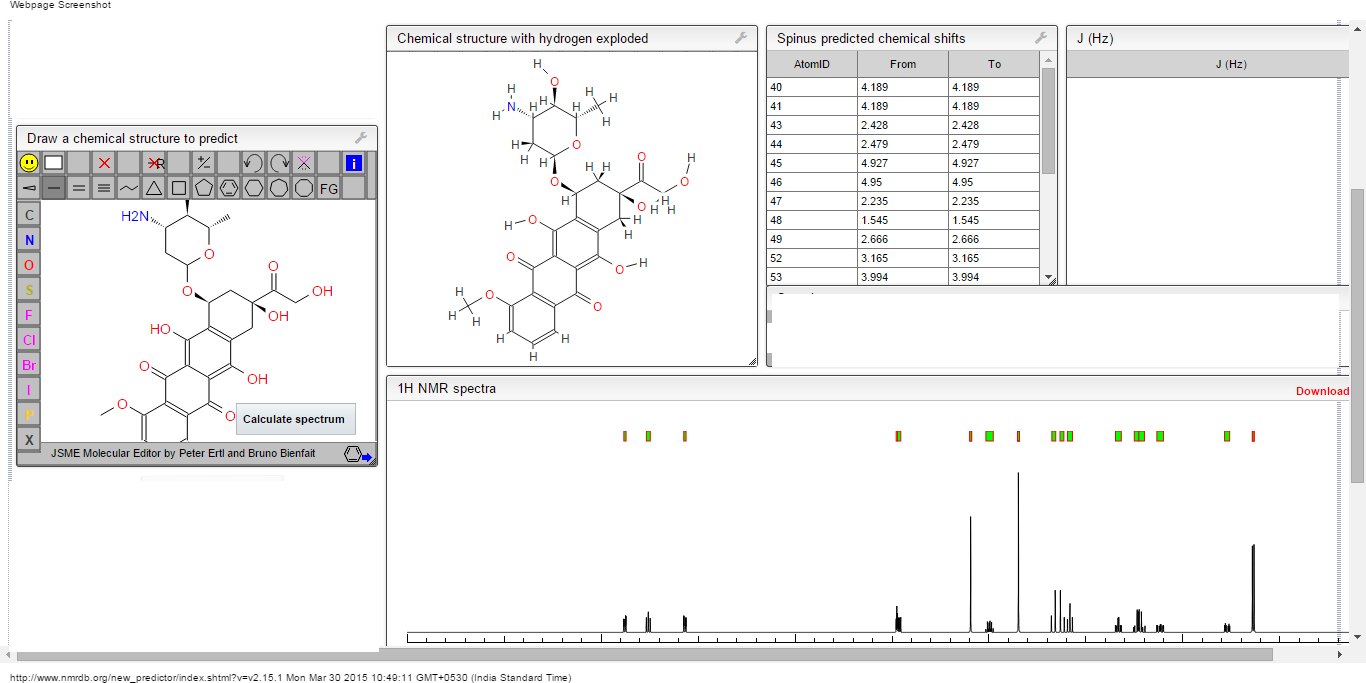
1H NMR PREDICT

13C NMR PREDICT

COSY
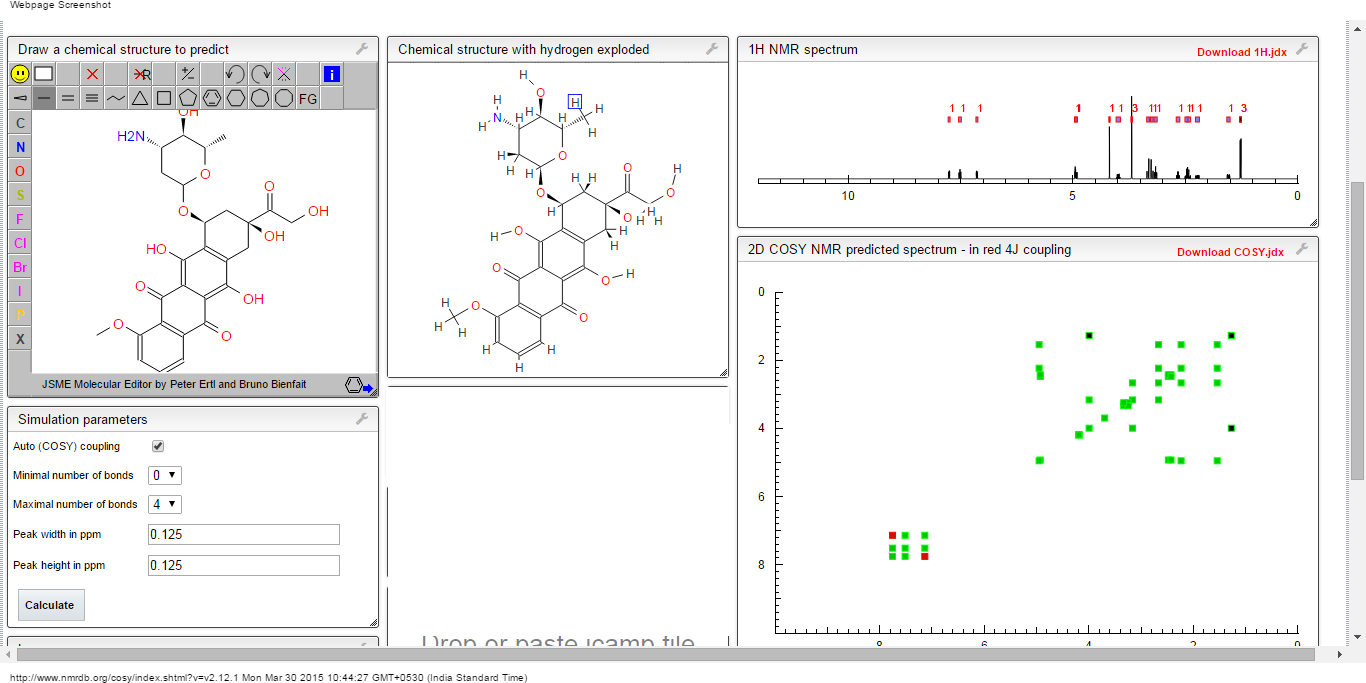
1H NMR

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (8R,10S)-10-((2S,4S,5R,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione | |
| Clinical data | |
| Trade names | Ellence |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a603003 |
| Intravenous | |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Protein binding | 77% |
| Metabolism | Hepatic glucuronidationand oxidation |
| Excretion | Biliary and renal |
| Identifiers | |
| 56420-45-2 |
|
| L01DB03 | |
| PubChem | CID 41867 |
| DrugBank | DB00445 |
| ChemSpider | 38201 |
| UNII | 3Z8479ZZ5X |
| KEGG | D07901 |
| ChEBI | CHEBI:47898 |
| ChEMBL | CHEMBL417 |
| Chemical data | |
| Formula | C27H29NO11 |
| 543.519 g/mol | |
KOWA COMPANY LTD


Nano Carrier Co


P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

PRI-724, ICG 001, What is correct structure?

PRI 724 AND ICG001 do confuse us, my efforts to unlock this confusion
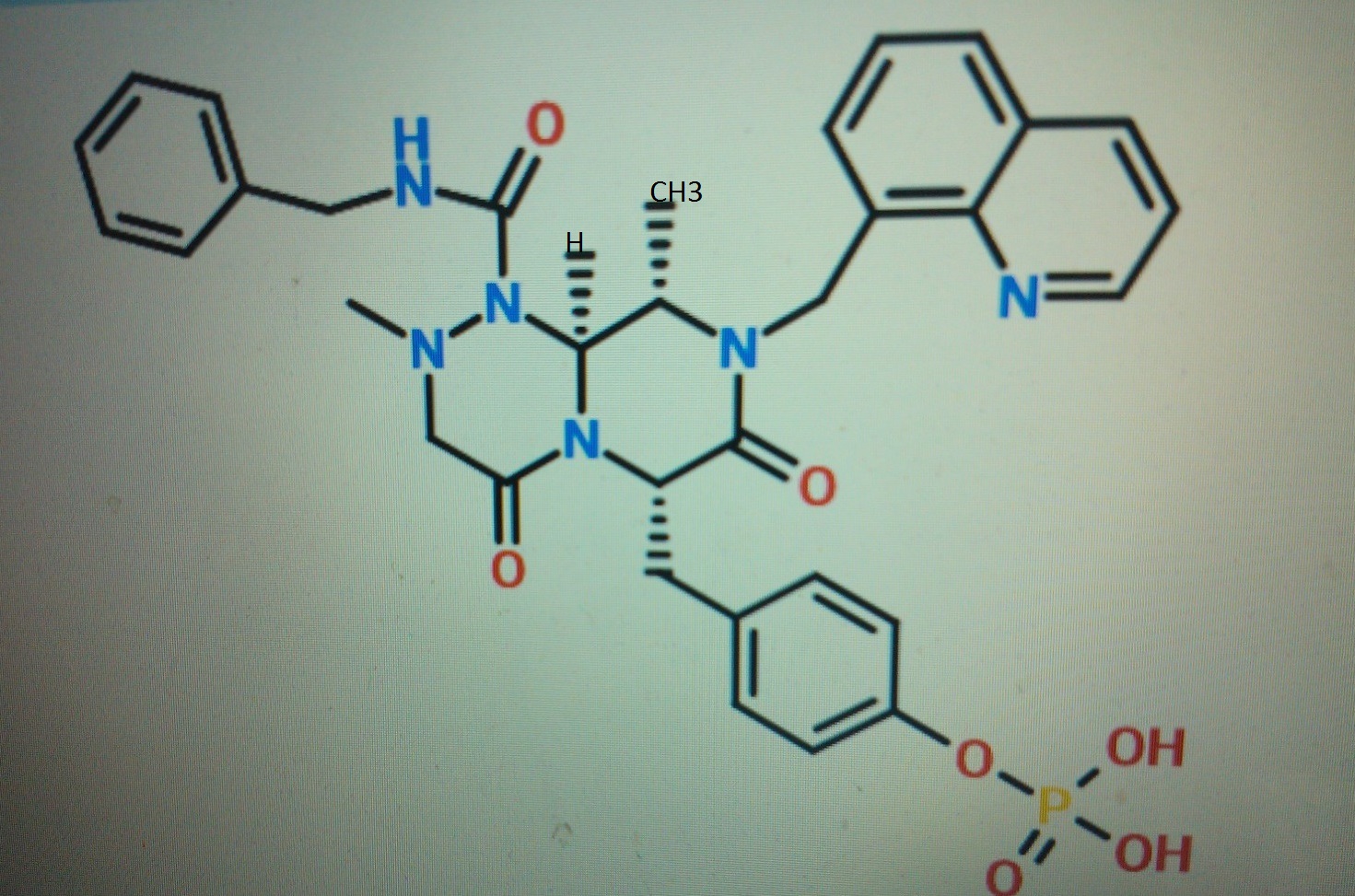
STRUCTURE 4
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate……………seems most likely PRI 724

STRUCTURE 5
Cas 1422253-37-9
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.

compd 2 and 1
OR
COMPD 3
 http://www.medkoo.com/Anticancer-trials/PRI-724.htm and similar/Same
http://www.medkoo.com/Anticancer-trials/PRI-724.htm and similar/Same
http://www.nature.com/nrc/journal/v14/n4/fig_tab/nrc3690_T1.html
compd 3.both above str are same
One of compd 1,2, 3, 4, 5 see at the end as an update , CAN BE ICG001, PRI-724,
Beta-catenin (CTNNB1) inhibitor
ICG001, also known as PRI-724, is a potent, specific inhibitor of the canonical Wnt signaling pathway in cancer stem cells with potential antineoplastic activity. Wnt signaling pathway inhibitor PRI-724 specifically inhibits the recruiting of beta-catenin with its coactivator CBP (the binding protein of the cAMP response element-binding protein CREB); together with other transcription factors beta-catenin/CBP binds to WRE (Wnt-responsive element) and activates transcription of a wide range of target genes of Wnt/beta-catenin signaling. Blocking the interaction of CBP and beta-catenin by this agent prevents gene expression of many proteins necessary for growth, thereby potentially suppressing cancer cell growth. The Wnt/beta-catenin signaling pathway regulates cell morphology, motility, and proliferation; aberrant regulation of this pathway leads to neoplastic proliferation.
JAPAN
4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate
(6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide,
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.
Compound A as in wo 2014061827……..4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyI dihydrogen phosphate in WO2014061827
4-(((6S,9S)-1-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl)octahydro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl)phenyl dihydrogen phosphate (presumed to be PRI-724; first disclosed in WO2009148192), useful for treating cancer, neurodegenerative diseases, glaucoma and idiopathic pulmonary fibrosis.
Eisai, under license from PRISM Pharma, is developing PRI-724, an inhibitor of CREB binding protein or beta-catenin complex formation, for treating cancer (phase 1, as of March 2015) and HCV-induced cirrhosis (preclinical trial).
Follows on from WO2014061827, claiming the use of PRI-724 for treating pulmonary fibrosis.
IS IT

cas 847591-62-2…………http://www.medkoo.com/Anticancer-trials/PRI-724.htm
(6S,9aS)-N-Benzyl-6-(4-hydroxybenzyl)-8-(naphthalen-1-ylmethyl)-4,7-dioxoperhydropyrazino[1,2-a]pyrimidine-1-carboxamide
COMPD 3
OR

COMPD 2
PRI724
1198780-43-6, 578.66, C33 H34 N6 O4
(6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide,

COMPD1
PRI 724
4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate
COMPD 1
SEE
http://www.google.co.in/patents/WO2009148192A1?cl=en

About PRI-724
PRI-724 is an antiproliferative small molecule that selectively inhibits the CBP/beta-catenin complex, which modulates the beta-catenin dependent pathway of Wnt signaling. Activation of the Wnt/beta-catenin signaling pathway is observed in various tumor cells and results in proliferation and metastasis. PRI-724 exhibits a selective antiproliferative effect, inhibiting various cancer cell lines in vitroand substantially inhibiting tumor growth in animal studies. PRI-724 is currently in clinical trials in oncology indications, partnered with Eisai Co., Ltd. PRI-724 also has potential to provide therapeutic benefit in non-oncology areas such as fibrosis and clinical trials in that indication are targeted to start in the second half of 2013.
About PRISM Pharma Co., Ltd.
PRISM Pharma Co., Ltd. has developed its platform technology to modulate inter-cellular protein-protein interactions using peptide mimetic small molecules and found various hit compounds including PRI-724.
SEE
Eisai Research Institute; PRISM Pharma Co Ltd
出願人:エ_ ザイ■ ア_ ル■ アンド■ ディ_ ■
マネジメン卜株式会社(EISAI R&D MANAGEMENT
CO., LTD.) [JP /JP ];亍1128088 東京都文京区
小石川四丁目6 番1 O 号Tokyo (JP).株式会社P
R I S M P h a r m a (PRISM PHARMA CO.,
LTD.) [JP /JP ];亍2268510神奈川県横浜市緑区長津
田町 4 2 5 9 — 3 Kanagawa (JP)
(IO) 国際公開番号
2 0 1 5 ^ ® S 3 .2 0 1 5 )
WO 2015/037587 Al
This method of producing 4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate involves a step for adding a reaction solution (I) comprising (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide, triethylamine and a solvent to a reaction solution (2) comprising a phosphorylating agent and a solvent.
1
1H-NMR (600MHz, METHAN0L-d4) δ (ppm):1.15 (d, J=6 Hz, 3H), 2.65 (s, 3H), 3.12 (d, J=18 Hz, 1H), 3.35 (d, J=7 Hz, 2H), 3.48 (d, J=18 Hz,1H), 4.15 (m,1H), 4.32 (d, J=15 Hz, 1H), 4.40 (d, J=15 Hz, 1H), 5.33(d, J=16 Hz, 1H), 5.41(d, J=16 Hz, 1H), 5.44 (d, J=7 Hz, 1H), 5.64 (d, J=10 Hz, 1H), 7.07 (dd, J=9,1 Hz, 2H), 7.15 (d, J=9 Hz, 2H), 7.24 (t, J=7 Hz, 1H), 7.27 (d, J=7 Hz, 2H), 7.34 (t, J=8 Hz, 2H), 7.55 (d d, J=8, 4 Hz, 1H), 7.60 (brd, J=6 Hz, 1H), 7.62 (dd, J=8, 7 Hz, 1H), 7.88 (dd, J=8,1 Hz, 1H), 8.38 (dd, J=8, 2 Hz, 1H), 8.90 (dd, J =4, 2 Hz, 1H).
…………………………………………………………………….
SEE
http://www.google.co.in/patents/WO2009148192A1?cl=en
SYNTHESIS OF COMPD 2
PART A
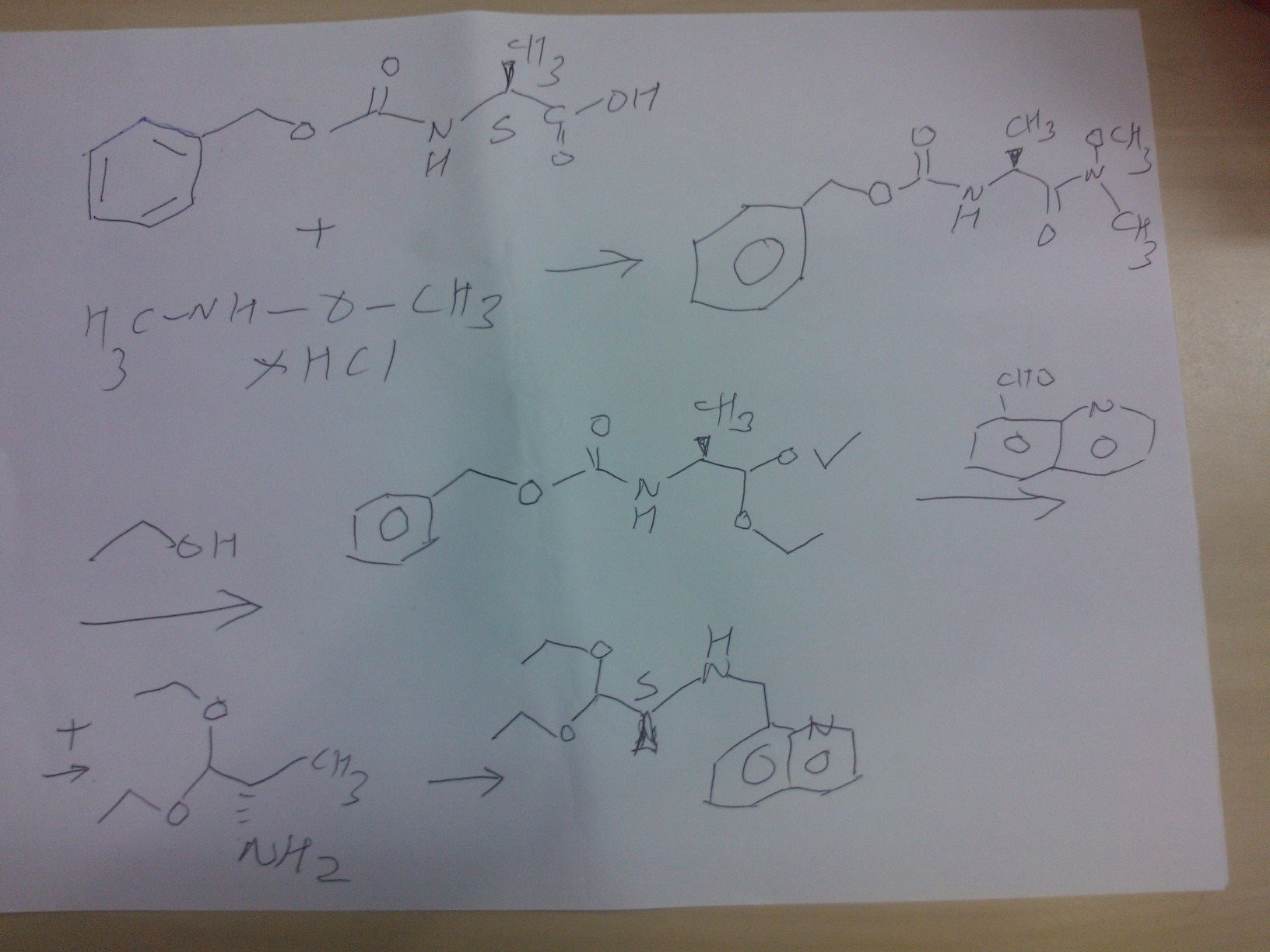
Synthesis Part A
step A
(S)-benzyl 1-(methoxy(methyl)amino)-1-oxopropan-2-ylcarbamate
Reaction of the foll……………….N-methoxy-N-methylamine hydrochloride, 1N sodium hydroxide , (S)-2-(benzyloxycarbonylamino)propanoic acidand 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride to obtain
(S)-benzyl 1-(methoxy(methyl)amino)-1-oxopropan-2-ylcarbamate.
STEP B
(S)-benzyl 1,1-diethoxypropan-2-ylcarbamate
Reaction of the foll……………….(S)-benzyl 1-(methoxy(methyl)amino)-1-oxopropan-2-ylcarbamate, 2M lithium aluminium hydride in tetrahydrofuran solution to obtain (S)-benzyl 1,1-diethoxypropan-2-ylcarbamate
STEP C
(S)-1,1-diethoxypropan-2-amine
Reaction of the foll……………….(S)-benzyl 1,1-diethoxypropan-2-ylcarbamate, 5% palladium on carbon title compound . (S)-1,1-diethoxypropan-2-amine,
STEP D
(S)-1,1-diethoxy-N-(quinolin-8-ylmethyl)propan-2-amine,
Reaction of the foll……………….(S)-1,1-diethoxypropan-2-amine,was reacted with 8-Quinolinecarboaldehyde to obtain the title
compound (S)-1,1-diethoxy-N-(quinolin-8-ylmethyl)propan-2-amine
PART B
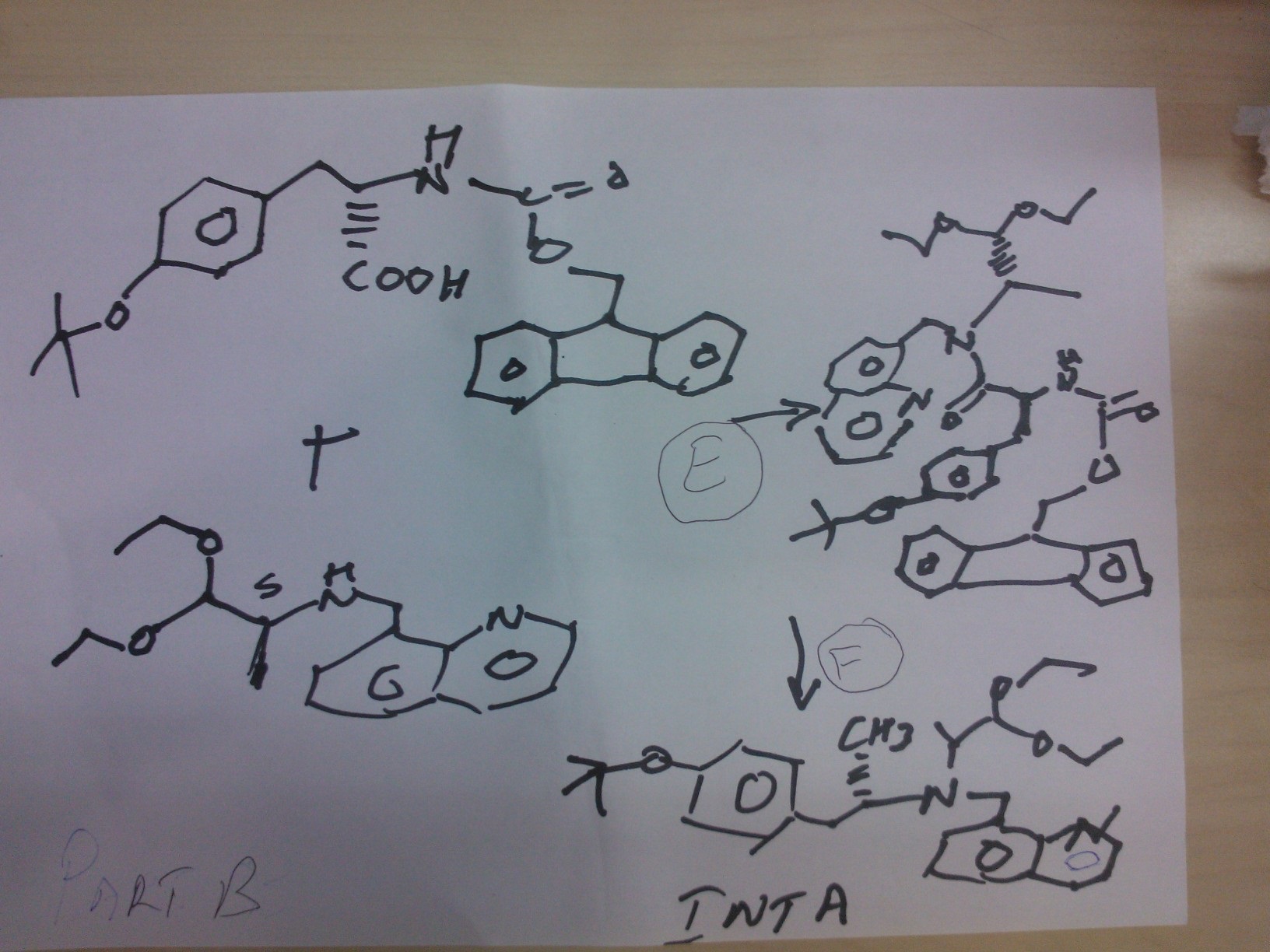
STEP E
(9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate
Reaction of the foll………………. (S)-1,1-diethoxy-N-(quinolin-8-ylmethyl)propan-2-amine, (S)-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-3-(4-tertbutoxyphenyl)propanoic acid to obtain the title compound (9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate
STEP f
(S)-2-amino-3-(4-tertbutoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide INT A
Reaction of the foll………………. (9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate and piperidine to
obtain the title compound (S)-2-amino-3-(4-tertbutoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide INT A
PART C

STEP g
ethyl 2-(1-methylhydrazinyl)acetate
Reaction of the foll……………….methylhydrazine 7 was reacted with ethyl 2-bromoacetate 1to obtain the title compound
STEP h
ethyl 2-(1-Methyl-2-(benzylcarbamoyl)hydrazinyl)acetate
Reaction of the foll………………. ethyl 2-(1-methylhydrazinyl)acetateand benzyl isocyanate to obtain the title
compound ethyl 2-(1-Methyl-2-(benzylcarbamoyl)hydrazinyl)acetate
STEP i
2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid
Reaction of the foll………………. ethyl 2-(1-allyl-2-
(benzylcarbamoyl)hydrazinyl)acetate and lithium hydroxide monohydrate to obtain the title compound 2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid
STEP j
N-benzyl-2-(2-((S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-
diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylamino)-2-oxoethyl)-2-
methylhydrazinecarboxamide……… precursor
Reaction of the foll………………. 2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid and (S)-2-amino-3-(4-tert-butoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide ( INT A )yielded the title compound ie the precursor
PART D
THIS PRECURSOR GIVES FINAL PRODUCT
Synthesis of (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-
dimethyl-8-(naphthalen-1-ylmethyl)-4,7-dioxooctahydro-1H-pyrazino[2,1-c][1,2,4]triazine-1-
carboxamide ……….final
fOLL reactants……….. N-benzyl-2-(2-((S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(naphthalen-1-ylmethyl)amino)-1-oxopropan-2-ylamino)-2-oxoethyl)-2-methylhydrazinecarboxamide, ie the precursor and 10%-water/HCOOH gave (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro-1Hpyrazino[2,1-c][1,2,4]triazine-1-carboxamide
RT 4.22; Mass 578.9
COMPD 3

(6S,9aS)-N-Benzyl-6-(4-hydroxybenzyl)-8-(naphthalen-1-ylmethyl)-4,7-dioxoperhydropyrazino[1,2-a]pyrimidine-1-carboxamide
SEE
US 6762185
……………………………..
SEE
http://www.google.com/patents/WO2012141038A1?cl=en

novel compounds, agent for inducing differentiation into hepatocytes of mesenchymal stem cells, Wnt / β- catenin signaling pathway inhibitor, method for producing hepatocytes with them on hepatocytes such as by their production.
Liver disease is said to be Japan’s national disease, a large number of patients suffering from liver disease. In addition, the annual number of deaths from hepatocellular carcinoma amounts to about 30 004 thousand people. Recently, hepatocellular cancer outcome is improved by advances in treatment, but the increase of advanced cancer, with hepatic dysfunction cirrhosis to merge, so-called hepatic failure death has increased. Liver failure therapy, although liver transplantation is ideal, it is difficult in Japan to obtain sufficient donors, it is necessary to develop a liver regeneration therapy with stem cells.
As stem cells that have the potential to differentiate into liver cells, bone marrow cells, tissue stem cells, such as umbilical cord blood cells can be expected.Therefore, a number of research institutions, for the realization of by regenerative medicine liver cell transplantation treatment of chronic liver failure patient, to differentiate human tissue stem cells into functional hepatocytes, truly clinically applicable efficient differentiation induction technology you are conducting research and development with the goal of developing a.
For example, in the laboratory of Shioda Professor of Tottori University Graduate School of Medicine, reported that the Wnt / β- catenin signaling pathway were differentiated into hepatocytes showed that suppressed by RNA interference at the time of induction of differentiation from human mesenchymal stem cells into hepatocytes you are (Non-Patent Document 1 and Non-Patent Documents 3-5).Furthermore, studies to induce differentiation of hepatocytes in other institutions have been conducted (Non-Patent Document 2, Patent Documents 1 and 2).
On the other hand, recently, from 4,000 or more screening of large compound libraries, Wnt / β- catenin signaling pathway inhibitory low molecular compound 5 types have been identified (Non-Patent Documents 6-9).
Kohyo 2009-535035 JP Patent Publication No. 2010-75631
Atsushi Yanagitani et al., ” retinoic Acid Receptor Dominant Level Negative Form Causes steatohepatitis and Liver Tumors in Transgenic Mice “, Hepatology, Vol. 40, No. 2, 2004, P. 366-375 Seoyoung Park et al.,”Hexachlorophene Inhibits Wnt / beta-catenin Pathway by Promoting Siah-Mediated beta-catenin Degradation “, Mol Pharmacol Vol. 70, No. 3, 960-966, 2006 Yoko Yoshida et al.,” A role of Wnt / beta-catenin Signals in hepatic fate Specification of human umbilical cord blood-derived mesenchymal stem cells “, Am J Physiol Gastrointest Liver Physiol 293:. G1089-G1098, 2007 Shimomura T et al,” Hepatic differentiation of human bone marrow-derived UE7T-13 cells: Effects of cytokines and CCN family Gene expression “, Hepatol Res., 37, 1068-79, 2007 Ishii K et al.,” Hepatic differentiation of human bone marrow-derived mesenchymal stem cells by tetracycline-regulated Hepatocyte Nuclear factor 3Beta “Hepatology, 48, 597- 606, 2008 Maina Lepourcelet et al., ” Small-molecule Antagonists of the oncogenic Tcf / beta-catenin protein complex “, CANCER CELL, JANUARY 2004, VOL. 5, 91-102 Emami KH et al.,” A Small molecule inhibitor of beta-catenin / CREB-binding protein Transcription “, Proc Natl Acad Sci US A. 2004 Aug 24; 101 (34):.. 12682-7 Jufang Shan et al,”Identification of a Specific Inhibitor of the Dishevelled PDZ Domain ” , Biochemistry 2005 Nov 29; 44 (47):.. 15495-503 Trosset JY et al, ” Inhibition of protein-protein Interactions: the discovery of beta-catenin Druglike Inhibitors by combining virtual and Biophysical Screening . “, Proteins 2006 Jul 1 ; 64 (1): 60-7
However, the conventional techniques described above literature, had a room for improvement in the following points.
Patent Documents 1 and 2, it has been described for proteins to induce stem cells from Hikimomiki cells, due to the use of the protein formulation as a differentiation inducing agent, a room for further improvement in terms of stability and safety and there was.
Non-Patent Document 1 and Non-Patent Document 3 to 5, and have reported that induced differentiated hepatocytes from human mesenchymal stem cells, the use of siRNA as a differentiation inducing agent, such as stability and safety there is room for further improvement in the surface. Non-Patent Document 2, 6 to 9, is not described with respect to method of inducing differentiation into hepatocytes.
The present invention has been made in view of the above circumstances, and an object thereof is to provide an effective low-molecular compounds that induce differentiation into hepatocytes from mesenchymal stem cells. Or, it is intended that the low-molecular compound was used to provide a secure differentiation inducing method is excellent from the mesenchymal stem cell differentiation efficiency of liver cells.
According to the present invention, there is provided formula (1) and one or more compounds selected from the group of compounds represented by the formula (2), a salt thereof or a solvate thereof.



<Example 1> synthetic ICG-001 of synthesis (1) ICG-001 of the IC-2 is an oligopeptide having two rings of β- turn mimic structure in central skeleton, and transcription by β-catenin / Tcf complex can function as a potent antagonist for activation has been reported (Drug Discov. Today 2005, 10, 1467-1474). Synthesis of ICG-001 in accordance with the literature (Tetrahedron 2007, 63, 12912-12916), was subjected to examination.

(1-1) of Compound 1 Synthesis 1-naphtaldehyde (Wako Pure Chemical) (1.56 g, 10 mmol) and 2,2-diethoxyethanamine (Tokyo Kasei Kogyo) (1.33 g, 10 mmol) were mixed 100 I was stirred 20 min at o C. After cooling to room temperature, diluted with EtOH (20 mL), was added portionwise NaBH 4 (0.38 g, 10 mmol), at room temperature, and stirred for 16 h. After completion of the reaction, was distilled off by concentration under reduced pressure EtOH, the product was extracted with AcOEt. The resulting product was purified by silica gel column chromatography (hexane / AcOEt = 5/1) to give the to give compound 1 (2.29 g, 8.5 mmol, 85%).

(1-2) Synthesis of Compound 3 Fmoc-L-Tyr (t-Bu) -OH (0.87 g, 1.9 mmol) in DMF (7 mL) solution of a condensing agent HATU (0.76 g, 2.0 mmol) and diisopropylethylamine (DIEA) (0.35 mL, 2.0 mmol) was added and after stirring for 20 min, compound 1 (0.54 g, a 2.0 mmol) was added, at room temperature, 16 h the mixture was stirred. After the reaction, DMF was distilled off by concentration under reduced pressure, and the resulting product was purified by column chromatography (hexane / AcOEt = 10/1), compound 2 was obtained (1.33 g, 1.9 mmol, 93%). The resulting compound 2 (1.33 g, 1.9 mmol) was dissolved in CH 2 Cl 2 (20 mL), was added diethylamine (DEA) (10 ml, excess), at room temperature, was 2 h stirring.After confirming the completion of the reaction by TLC, vacuum was distilled off CH 2 Cl 2 by concentration, the resulting product was purified by silica gel column chromatography (AcOEt), to give compound 3 (0.92 g, 1. 8 mmol, 92%).

(1-3) Synthesis Fmoc-β-Ala-OH (0.53 g, 1.7 mmol) of compound 5 in DMF (8 mL) solution of a condensing agent HATU (0.70 g, 1.8 mmol) and diisopropylethylamine (DIEA) (0.32 mL, 1.8 mmol) was added and after stirring for 20 min, compound 3 (0.92 g, 1.8 mmol) was added, at room temperature, and stirred for 14 h. After the reaction, DMF was distilled off by concentration under reduced pressure, the resulting product was purified by column chromatography (hexane / AcOEt = 1/1), compound 4 was obtained (1.2 g, 1.5 mmol, 82%). Obtained compound 4 (1.2 g, 1.5 mmol) was dissolved in CH 2 Cl 2 (20 mL), was added diethylamine (DEA) (9 mL, excess), at room temperature, and stirred for 1 h. After confirming the completion of the reaction by TLC, was distilled off CH 2 Cl 2 by concentration under reduced pressure, and the resulting product was purified by silica gel column chromatography (AcOEt / EtOH = 1/1), to give compound 5 (0 .66 g, 1.2 mmol, 80%).

(1-4) synthetic compounds 5 (0.66 g, 1.2 mmol) of compound 7 CH 2 Cl 2 of solution (8 mL) to benzylisocyanate (0.16 g, 1.2 mmol) of CH 2 Cl 2 solution (8 mL) was added, at room temperature, and stirred for 12 h. After confirming the completion of the reaction by TLC, was distilled off CH 2 Cl 2 by concentration under reduced pressure, and the resulting product was purified by column chromatography (AcOEt / EtOH = 1/1), to give compound 6 (0. 59 g, 0.85 mmol, 73%). The obtained compound 6 (0.59 g, 0.85 mmol) at room temperature in the formic acid (9 ml), I was stirred 20 h. Was evaporated formic acid by concentration under reduced pressure, the resulting product was purified by column chromatography (AcOEt), Compound 7a to (ICG-001) was obtained as a white solid (0.26 g, 0.48 mmol, 57 %).
The resulting product, MS spectra and were identified from the 1 H NMR spectrum (with the literature value) (Fig. 1).

STRUCTURE 4
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate
STRUCTURE 5
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.
Cas 1422253-37-9
2H-Pyrazino[2,1-c][1,2,4]triazine-1(6H)-carboxamide, hexahydro-6-[(4-hydroxyphenyl)methyl]-2,9-dimethyl-4,7-dioxo-N-(phenylmethyl)-8-(8-quinolinylmethyl)-, (6S,9S,9aS)-
Structure can represented as

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

Vibegron ビベグロン
Vibegron, MK-4618, KRP 114V
update FDA APPROVED 12/23/2020, GEMTESA, To treat overactive bladder
UNII-M5TSE03W5U; M5TSE03W5U; D10433
Molecular Formula: C26H28N4O3 Molecular Weight: 444.52552
Merck Sharp & Dohme Corp. INNOVATOR
phase 2 for the treatment of overactive bladder
(6S)-N-[4-([(2S,5R)-5-[(R)-Hydroxy(phenyl)methyl]pyrrolidin-2-yl]methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-a]pyrimidine-6-carboxamide
(6S)-N-[4-[[(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl]methyl]phenyl]-4-oxo-7,8-dihydro-6H-pyrrolo[1,2-a]pyrimidine-6-carboxamide
Target-based Actions Beta 3 adrenoceptor agonist
Indications Overactive bladder; Urinary incontinence
UPDATE 2018/9/21 pmda Beova JAPAN 2018Kyorin Pharmaceutical, under license from Merck, is developing vibegron (phase II, September 2014) for the treating of overactive bladder. In July 2014, Merck has granted to Kyorin an exclusive license to develop, manufacture and commercialize vibegron in Japan.
MK-4618 is being developed in phase II clinical trials at Merck & Co. for the treatment of overactive bladder. The company had been developing the compound for the treatment of endocrine disorders and hypertension; however, recent progress reports are not available at present.
In 2014, Merck licensed the product to Kyorin for development and commercialization in Japan.
The function of the lower urinary tract is to store and periodically release urine. This requires the orchestration of storage and micturition reflexes which involve a variety of afferent and efferent neural pathways, leading to modulation of central and peripheral neuroeffector mechanisms, and resultant coordinated regulation of sympathetic and parasympathetic components of the autonomic nervous system as well as somatic motor pathways. These proximally regulate the contractile state of bladder (detrusor) and urethral smooth muscle, and urethral sphincter striated muscle.
β Adrenergic receptors (βAR) are present in detrusor smooth muscle of various species, including human, rat, guinea pig, rabbit, ferret, dog, cat, pig and non-human primate. However, pharmacological studies indicate there are marked species differences in the receptor subtypes mediating relaxation of the isolated detrusor; β1AR predominate in cats and guinea pig, β2AR predominate in rabbit, and β3AR contribute or predominate in dog, rat, ferret, pig, cynomolgus and human detrusor. Expression of βAR subtypes in the human and rat detrusor has been examined by a variety of techniques, and the presence of β3AR was confirmed using in situ hybridization and/or reverse transcription-polymerase chain reaction (RT-PCR). Real time quantitative PCR analyses of β1AR, β2AR and β3AR mRNAs in bladder tissue from patients undergoing radical cystectomy revealed a preponderance of β3AR mRNA (97%, cf 1.5% for β1AR mRNA and 1.4% for β2AR mRNA). Moreover, β3AR mRNA expression was equivalent in control and obstructed human bladders. These data suggest that bladder outlet obstruction does not result in downregulation of β3AR, or in alteration of β3AR-mediated detrusor relaxation. β3AR responsiveness also has been compared in bladder strips obtained during cystectomy or enterocystoplasty from patients judged to have normal bladder function, and from patients with detrusor hyporeflexia or hyperreflexia. No differences in the extent or potency of β3AR agonist mediated relaxation were observed, consistent with the concept that the β3AR activation is an effective way of relaxing the detrusor in normal and pathogenic states.
Functional evidence in support of an important role for the β3AR in urine storage emanates from studies in vivo. Following intravenous administration to rats, the rodent selective β3AR agonist CL316243 reduces bladder pressure and in cystomeric studies increases bladder capacity leading to prolongation of micturition interval without increasing residual urine volume.
Overactive bladder is characterized by the symptoms of urinary urgency, with or without urgency urinary incontinence, usually associated with frequency and nocturia. The prevalence of OAB in the United States and Europe has been estimated at 16 to 17% in both women and men over the age of 18 years. Overactive bladder is most often classified as idiopathic, but can also be secondary to neurological condition, bladder outlet obstruction, and other causes. From a pathophysiologic perspective, the overactive bladder symptom complex, especially when associated with urge incontinence, is suggestive of detrusor overactivity. Urgency with or without incontinence has been shown to negatively impact both social and medical well-being, and represents a significant burden in terms of annual direct and indirect healthcare expenditures. Importantly, current medical therapy for urgency (with or without incontinence) is suboptimal, as many patients either do not demonstrate an adequate response to current treatments, and/or are unable to tolerate current treatments (for example, dry mouth associated with anticholinergic therapy). Therefore, there is need for new, well-tolerated therapies that effectively treat urinary frequency, urgency and incontinence, either as monotherapy or in combination with available therapies. Agents that relax bladder smooth muscle, such as β3AR agonists, are expected to be effective for treating such urinary disorders.
PATENT
http://www.google.com/patents/WO2013062881A1?cl=en

EXAMPLE 3

To a three neck flask equipped with a N2 inlet, a thermo couple probe was charged pyrrolidine i-11 (10.0 g), sodium salt i-12 (7.87 g), followed by IPA (40 mL) and water (24 mL). 5 N HC1 (14.9 mL) was then slowly added over a period of 20 min to adjust pH = 3.3- 3.5, maintaining the batch temperature below 35 °C. Solid EDC hydrochloride (7.47 g) was charged in portions over 30 min. The reaction mixture was aged at RT for additional 0.5 – 1 h, aqueous ammonia (14%) was added dropwise to pH ~8.6. The batch was seeded and aged for additional 1 h to form a slurry bed. The rest aqueous ammonia (14%, 53.2 ml total) was added dropwise over 6 h. The resulting thick slurry was aged 2-3 h before filtration. The wet-cake was displacement washed with 30% IPA (30 mL), followed by 15% IPA (2 x 20mL) and water (2 X 20mL). The cake was suction dried under N2 overnight to afford 14.3 g of compound of Formula (I)-
1H NMR (DMSO) δ 10.40 (s, NH), 7.92 (d, J = 6.8, 1H), 7.50 (m, 2H), 7.32 (m, 2H), 7.29 (m, 2H), 7.21 (m, 1H), 7.16 (m, 2H), 6.24 (d, J = 6.8, 1H), 5.13 (dd, J = 9.6, 3.1, 1H), 5.08 (br s, OH), 4.22 (d, J = 7.2, 1H), 3.19 (p, J = 7.0, 1H), 3.16-3.01 (m, 3H), 2.65 (m, 1H), 2.59-2.49 (m, 2H), 2.45 (br s, NH), 2.16 (ddt, J = 13.0, 9.6, 3.1, 1H), 1.58 (m, 1H), 1.39 (m, 1H), 1.31-1.24 (m, 2H).
13C NMR (DMSO) δ 167.52, 165.85, 159.83, 154.56, 144.19, 136.48, 135.66, 129.16, 127.71, 126.78, 126.62, 119.07, 112.00, 76.71, 64.34, 61.05, 59.60, 42.22, 31.26, 30.12, 27.09, 23.82.
HPLC method – For monitoring conversion
Column: XBridge C18 cm 15 cm x 4.6 mm, 3.5 μιη particle size;
Column Temp. : 35 °C; Flow rate: 1.5 mL/min; Detection: 220 nm;
Mobile phase: A. 5 mM Na2B407.10 H20 B: Acetonitrile
Gradient:

HPLC method – For level of amide epimer detection
Column: Chiralpak AD-H 5 μηι, 250 mm x 4.6 mm.
Column Temp: 35 °C; Flow rate: 1.0 mL/min; Detection: 250 nm;
Mobile phase: Isocratic 30% Ethanol in hexanes + 0.1% isobutylamine
PATENT
WO 2009124167
http://www.google.com/patents/WO2009124167A1?cl=en
EXAMPLE 103
(6y)-N-r4-({(‘25′. 5R)-5-r(‘R)-hvdroxy(‘phenvnmethyl1pyrrolidin-2-yl}methvnphenyl1-4-oxo- 4,6J,8-tetrahydropyiτolori,2-α1pyrimidine-6-carboxamide

ter?-butyl(2R. 55f)-2-rCR)-hvdroxy(‘phenvnmethyl1-5-r4-(‘{r(‘65f)-4-oxo-4.6.7.8-
tetrahydropyrrolof 1.2-alpyrimidin-6- yl]carbonyl} amino)benzyl]pyrrolidine- 1 – carboxylate

To a solution of i-13a (21.4 g, 55.9 mmol) in N,N-dimethylformamide (100 ml) at O0C was added [(65)-4-oxo-4,6,7,8-tetrahydropyrrolo[l,2-α]pyrimidine-6-carboxylic acid (11.1 g, 61.5 mmol), followed by 1 -hydroxybenzotriazole (i-44, 7.55 g, 55.9 mmol), N-(3- dimethylaminopropyl)-Nl-ethylcarbodiimide hydrochloride (16.1 g, 84.0 mmol) and N,N- diisopropylethylamine (29.2 ml, 168 mmol). The reaction mixture was stirred from O0C to ambient temperature for 2 h. Water (600 ml) was added and it was extracted with dichloromethane (600 ml x 2). The combined organic layers were dried over Na2SO4. After removal of the volatiles, the residue was purified by using a Biotage Horizon® system (0-5% then 5% methanol with 10% ammonia/dichloromethane mixture) to afford the title compound which contained 8% of the minor diastereomer. It was further purified by supercritical fluid chromatography (chiral AS column, 40% methanol) to afford the title compound as a pale yellow solid (22.0 g, 72%). 1H NMR (CDCl3): δ 9.61 (s, IH), 7.93 (d, J = 6.6 Hz, IH), 7.49 (d, J = 8.4 Hz, 2H), 7.35-7.28 (m, 5H), 7.13 (d, J = 8.5 Hz, 2H), 6.40 (d, J = 6.7 Hz, IH), 5.36 (d, J = 8.6 Hz, IH), 4.38 (m, IH), 4.12-4.04 (m, 2H), 3.46 (m,lH), 3.15-3.06 (m, 2H), 2.91 (dd, J = 13.1, 9.0 Hz, IH), 2.55 (m, IH), 2.38 (m, IH), 1.71-1.49 (m, 13H). LC-MS 567.4 (M+23).
(6S)-N-\4-( U2S. 5R)-5-r(R)-hvdroxy(phenyl)methyl1pyrrolidin-2-
yl}methyl)phenyl1-4-oxo-4,6J,8-tetrahvdropyrrolori,2-α1pyrimidine-6- carboxamide

To a solution of the intermediate from Step A (2.50 g, 4.59 mmol) in dichloromethane (40 ml) was added trifluoroacetic acid (15 ml). The reaction mixture was stirred at ambient temperature for 1.5 h. After removal of the volatiles, saturated NaHCCh was added to make the PH value to 8-9. The mixture was then extracted with dichloromethane. The combined organic layers were dried over Na2SO4. After concentration, crystallization from methanol/acetonitrile afforded the title compound as a white solid (1.23g, 60%). 1H NMR (DMSO-Cl6): δ 10.40 (s, IH), 7.91 (d, J = 6.7 Hz, IH), 7.49 (d, J = 8.3 Hz, 2H), 7.32-7.26 (m, 4H), 7.21 (m, IH), 7.15 (d, J = 8.4 Hz, 2H), 6.23 (d, J = 6.7 Hz, IH), 5.11 (dd, J = 9.6, 2.9 Hz, IH), 5.10 (br, IH), 4.21 (d, J = 7.1 Hz, IH), 3.20-3.00 (m, 4H), 2.66-2.51 (m, 3H), 2.16 (m, IH), 1.57 (m, IH), 1.38 (m, IH), 1.29-1.23 (m, 2H). LC-MS 445.3 (M+l).
Using the Biological Assays described above, the human β3 functional activity of Example 103 was determined to be between 11 to 100 nM.
PATENT
CHECK STRUCTURE…………….CAUTION
http://www.google.com/patents/US8247415


CAUTION…………….
Example 103(6S)-N-[4-({(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl}methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxamide

Step A: tert-butyl(2R,5S)-2-[(R)-hydroxy(phenyl)methyl]-5-[4-({[(6S)-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidin-6-yl]carbonyl}amino)benzyl]pyrrolidine-1-carboxylate

To a solution of i-13a (21.4 g, 55.9 mmol) in N,N-dimethylformamide (100 ml) at 0° C. was added [(6S)-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxylic acid (11.1 g, 61.5 mmol), followed by 1-hydroxybenzotriazole (i-44, 7.55 g, 55.9 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (16.1 g, 84.0 mmol) and N,N-diisopropylethylamine (29.2 ml, 168 mmol). The reaction mixture was stirred from 0° C. to ambient temperature for 2 h. Water (600 ml) was added and it was extracted with dichloromethane (600 ml×2). The combined organic layers were dried over Na2SO4. After removal of the volatiles, the residue was purified by using a Biotage Horizon® system (0-5% then 5% methanol with 10% ammonia/dichloromethane mixture) to afford the title compound which contained 8% of the minor diastereomer. It was further purified by supercritical fluid chromatography (chiral AS column, 40% methanol) to afford the title compound as a pale yellow solid (22.0 g, 72%). 1H NMR (CDCl3): δ 9.61 (s, 1H), 7.93 (d, J=6.6 Hz, 1H), 7.49 (d, J=8.4 Hz, 2H), 7.35-7.28 (m, 5H), 7.13 (d, J=8.5 Hz, 2H), 6.40 (d, J=6.7 Hz, 1H), 5.36 (d, J=8.6 Hz, 1H), 4.38 (m, 1H), 4.12-4.04 (m, 2H), 3.46 (m, 1H), 3.15-3.06 (m, 2H), 2.91 (dd, J=13.1, 9.0 Hz, 1H), 2.55 (m, 1H), 2.38 (m, 1H), 1.71-1.49 (m, 13H). LC-MS 567.4 (M+23).
Step B: (6S)-N-[4-({(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl}methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxamide

To a solution of the intermediate from Step A (2.50 g, 4.59 mmol) in dichloromethane (40 ml) was added trifluoroacetic acid (15 ml). The reaction mixture was stirred at ambient temperature for 1.5 h. After removal of the volatiles, saturated NaHCO3 was added to make the PH value to 8-9. The mixture was then extracted with dichloromethane. The combined organic layers were dried over Na2SO4. After concentration, crystallization from methanol/acetonitrile afforded the title compound as a white solid (1.23 g, 60%). 1H NMR (DMSO-d6): δ 10.40 (s, 1H), 7.91 (d, J=6.7 Hz, 1H), 7.49 (d, J=8.3 Hz, 2H), 7.32-7.26 (m, 4H), 7.21 (m, 1H), 7.15 (d, J=8.4 Hz, 2H), 6.23 (d, J=6.7 Hz, 1H), 5.11 (dd, J=9.6, 2.9 Hz, 1H), 5.10 (br, 1H), 4.21 (d, J=7.1 Hz, 1H), 3.20-3.00 (m, 4H), 2.66-2.51 (m, 3H), 2.16 (m, 1H), 1.57 (m, 1H), 1.38 (m, 1H), 1.29-1.23 (m, 2H). LC-MS 445.3 (M+1).
Using the Biological Assays described above, the human β3 functional activity of Example 103 was determined to be between 11 to 100 nM.
PATENT
WO2014150639
Step 6. Preparation of Compound 1-7 from Compound 1-6 and Compound A-2

To a three neck flask equipped with a N2 inlet, a thermo couple probe was charged pyrrolidine hemihydrate 1-6 (10.3 g), sodium salt A-2 (7.87 g), followed by IPA (40 mL) and water (24 mL). 5 N HC1 (14.9 mL) was then slowly added over a period of 20 minutes to adjust pH = 3.3-3.5, maintaining the batch temperature below 35°C. Solid EDC hydrochloride (7.47 g) was charged in portions over 30 minutes. The reaction mixture was aged at RT for additional 0.5 – 1 hour, aqueous ammonia (14%) was added dropwise to pH -8.6. The batch was seeded and aged for additional 1 hour to form a slurry bed. The rest aqueous ammonia (14%, 53.2 ml total) was added dropwise over 6 hours. The resulting thick slurry was aged 2-3 hours before filtration. The wet-cake was displacement washed with 30% IPA (30 mL), followed by 15% IPA (2 x 20mL) and water (2 X 20mL). The cake was suction dried under N2 overnight to afford 14.3 g of compound 1-7.
1H NMR (DMSO) δ 10.40 (s, NH), 7.92 (d, J = 6.8, 1H), 7.50 (m, 2H), 7.32 (m, 2H), 7.29 (m, 2H), 7.21 (m, 1H), 7.16 (m, 2H), 6.24 (d, J = 6.8, 1H), 5.13 (dd, J = 9.6, 3.1, 1H), 5.08 (br s, OH), 4.22 (d, J = 7.2, 1H), 3.19 (p, J = 7.0, 1H), 3.16-3.01 (m, 3H), 2.65 (m, 1H), 2.59-2.49 (m, 2H), 2.45 (br s, NH), 2.16 (ddt, J = 13.0, 9.6, 3.1, 1H), 1.58 (m, 1H), 1.39 (m, 1H), 1.31-1.24 (m, 2H).
13C NMR (DMSO) δ 167.52, 165.85, 159.83, 154.56, 144.19, 136.48, 135.66, 129.16, 127.71, 126.78, 126.62, 119.07, 112.00, 76.71, 64.34, 61.05, 59.60, 42.22, 31.26, 30.12, 27.09, 23.82.
The crystalline freebase anhydrous form I of Compound 1-7 can be characterized by XRPD by

PATENT
WO-2014150633
Merck Sharp & Dohme Corp
Process for preparing stable immobilized ketoreductase comprises bonding of recombinant ketoreductase to the resin in a solvent. Useful for synthesis of vibegron intermediates. For a concurrent filling see WO2014150639, claiming the method for immobilization of ketoreductase. Picks up from WO2013062881, claiming the non enzymatic synthesis of vibegron and intermediates.
PAPER
Discovery of Vibegron: A Potent and Selective β3 Adrenergic Receptor Agonist for the Treatment of Overactive Bladder
Merck Research Laboratories, 2015 Galloping Hill Road, PO Box 539, Kenilworth, New Jersey 07033, United States
J. Med. Chem., Article ASAP
DOI: 10.1021/acs.jmedchem.5b01372
Publication Date (Web): December 27, 2015
Copyright © 2015 American Chemical Society
*Telephone: (908) 740-0287. E-mail scott.edmondson@merck.com.
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01372
http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.5b01372/suppl_file/jm5b01372_si_001.pdf

The discovery of vibegron, a potent and selective human β3-AR agonist for the treatment of overactive bladder (OAB), is described. An early-generation clinical β3-AR agonist MK-0634 (3) exhibited efficacy in humans for the treatment of OAB, but development was discontinued due to unacceptable structure-based toxicity in preclinical species. Optimization of a series of second-generation pyrrolidine-derived β3-AR agonists included reducing the risk for phospholipidosis, the risk of formation of disproportionate human metabolites, and the risk of formation of high levels of circulating metabolites in preclinical species. These efforts resulted in the discovery of vibegron, which possesses improved druglike properties and an overall superior preclinical profile compared to MK-0634. Structure–activity relationships leading to the discovery of vibegron and a summary of its preclinical profile are described.



| Reference | ||
|---|---|---|
| 1 | H.P. Kaiser, et al., “Catalytic Hydrogenation of Pyrroles at Atmospheric Pressure“, J. Org. Chem., vol. 49, No. 22, p. 4203-4209 (1984). | |
A study of the efficacy and safety of MK-4618 in patients with overactive bladder (OAB) (MK-4618-008 EXT1) (NCT01314872)
ClinicalTrials.gov Web Site 2011, April 28
ClinicalTrials.gov Web Site 2011, April 28
| WO2011043942A1 * | Sep 27, 2010 | Apr 14, 2011 | Merck Sharp & Dohme Corp. | Combination therapy using a beta 3 adrenergic receptor agonist and an antimuscarinic agent |
| US20090253705 * | Apr 2, 2009 | Oct 8, 2009 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| US20110028481 * | Apr 2, 2009 | Feb 3, 2011 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8642661 | Aug 2, 2011 | Feb 4, 2014 | Altherx, Inc. | Pharmaceutical combinations of beta-3 adrenergic receptor agonists and muscarinic receptor antagonists |
| US8653260 | Jun 20, 2012 | Feb 18, 2014 | Merck Sharp & Dohme Corp. | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| US20120202819 * | Sep 27, 2010 | Aug 9, 2012 | Merck Sharp & Dohme Corporation | Combination therapy using a beta 3 adrenergic receptor agonists and an antimuscarinic agent |
| US20020028835 | Jul 12, 2001 | Mar 7, 2002 | Baihua Hu | Cyclic amine phenyl beta-3 adrenergic receptor agonists |
| US20070185136 | Feb 2, 2007 | Aug 9, 2007 | Sanofi-Aventis | Sulphonamide derivatives, their preparation and their therapeutic application |
| US20110028481 | Apr 2, 2009 | Feb 3, 2011 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| WO2003072572A1 | Feb 17, 2003 | Sep 4, 2003 | Jennifer Anne Lafontaine | Beta3-adrenergic receptor agonists |
|
8-22-2012
|
Hydroxymethyl pyrrolidines as [beta]3 adrenergic receptor agonists
|
////////////C1CC(NC1CC2=CC=C(C=C2)NC(=O)C3CCC4=NC=CC(=O)N34)C(C5=CC=CC=C5)O
Japan approves world’s first PD-1 drug, nivolumab
http://www.pharmatimes.com/Article/14-07-07/Japan_approves_world_s_first_PD-1_drug_nivolumab.aspx
old article cut paste

NIVOLUMAB
Anti-PD-1;BMS-936558; ONO-4538
PRONUNCIATION nye vol’ ue mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL DESCRIPTION
A fully human IgG4 antibody blocking the programmed cell death-1 receptor (Medarex/Ono Pharmaceuticals/Bristol-Myers Squibb)
MOLECULAR FORMULA C6362H9862N1712O1995S42
MOLECULAR WEIGHT 143.6 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION MDX-1106, BMS-936558
CAS REGISTRY NUMBER 946414-94-4
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
Nivolumab (nye vol’ ue mab) is a fully human IgG4 monoclonal antibody designed for the treatment of cancer. Nivolumab was developed by Bristol-Myers Squibb and is also known as BMS-936558 and MDX1106.[1] Nivolumab acts as an immunomodulator by blocking ligand activation of the Programmed cell death 1 receptor.
A Phase 1 clinical trial [2] tested nivolumab at doses ranging from 0.1 to 10.0 mg per kilogram of body weight, every 2 weeks. Response was assessed after each 8-week treatment cycle, and were evaluable for 236 of 296 patients. Study authors concluded that:”Anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use.”[3]
Phase III clinical trials of nivolumab are recruiting in the US and EU.[4]
- Statement On A Nonproprietary Name Adopted By The USAN Council – Nivolumab, American Medical Association.
- A Phase 1b Study of MDX-1106 in Subjects With Advanced or Recurrent Malignancies (MDX1106-03), NIH.
- Topalian SL, et al. (June 2012). “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer”. New England Journal of Medicine 366. doi:10.1056/NEJMoa1200690. Lay summary – New York Times.
- Nivolumab at ClinicalTrials.gov, A service of the U.S. National Institutes of Health.
The PD-1 blocking antibody nivolumab continues to demonstrate sustained clinical activity in previously treated patients with advanced non-small cell lung cancer (NSCLC), according to updated long-term survival data from a phase I trial.
Survival rates at one year with nivolumab were 42% and reached 24% at two years, according to the median 20.3-month follow up. Additionally, the objective response rate (ORR) with nivolumab, defined as complete or partial responses by standard RECIST criteria, was 17% for patients with NSCLC. Results from the updated analysis will be presented during the 2013 World Conference on Lung Cancer on October 29.
“Lung cancer is very difficult to treat and there continues to be a high unmet medical need for these patients, especially those who have received multiple treatments,” David R. Spigel, MD, the program director of Lung Cancer Research at the Sarah Cannon Research Institute and one of the authors of the updated analysis, said in a statement.
“With nivolumab, we are investigating an approach to treating lung cancer that is designed to work with the body’s own immune system, and these are encouraging phase I results that support further investigation in larger scale trials.”
In the phase I trial, 306 patients received intravenous nivolumab at 0.1–10 mg/kg every-other-week for ≤12 cycles (4 doses/8 week cycle). In all, the trial enrolled patients with NSCLC, melanoma, renal cell carcinoma, colorectal cancer, and prostate cancer.
The long-term follow up focused specifically on the 129 patients with NSCLC. In this subgroup, patients treated with nivolumab showed encouraging clinical activity. The participants had a median age of 65 years and good performance status scores, and more than half had received three or more prior therapies. Across all doses of nivolumab, the median overall survival was 9.9 months, based on Kaplan-Meier estimates.
In a previous update of the full trial results presented at the 2013 ASCO Annual Meeting, drug-related adverse events of all grades occurred in 72% of patients and grade 3/4 events occurred in 15%. Grade 3/4 pneumonitis related to treatment with nivolumab emerged early in the trial, resulting in 3 deaths. As a result, a treatment algorithm for early detection and management was developed to prevent this serious side effect.
Nivolumab is a fully human monoclonal antibody that blocks the PD-1 receptor from binding to both of its known ligands, PD-L1 and PD-L2. This mechanism, along with early data, suggested an associated between PD-L1 expression and response to treatment.
In separate analysis presented at the 2013 World Conference on Lung Cancer, the association of tumor PD-L1 expression and clinical activity in patients with NSCLC treated with nivolumab was further explored. Of the 129 patients with NSCLC treated with nivolumab in the phase I trial, 63 with NSCLC were tested for PD-L1 expression by immunohistochemistry (29 squamous; 34 non-squamous).
Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.

Sorafenib
(4-(4-(3-(4-chloro-3-(trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide)
BAY 43-9006

Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.
Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.
Nexavar’s approval in Japan is supported by data from the multicentre, placebo-controlled Phase III DECISION (‘stuDy of sorafEnib in loCally advanced or metastatIc patientS with radioactive Iodine refractory thyrOid caNcer’) study.
The international Phase III DECISION study, which randomised a total of 417 patients, met its primary endpoint of extended progression-free survival. Safety and tolerability profile of sorafenib was generally consistent with the known profile of sorafenib.
The most common treatment-emergent adverse events in the sorafenib arm were hand-foot skin reaction, diarrhea, alopecia, weight loss, fatigue, hypertension and rash.
Nexavar was awarded orphan drug status by the MHLW for thyroid carcinoma in September 2013.
Sorafenib (co-developed and co-marketed by Bayer and Onyx Pharmaceuticals as Nexavar),[1] is a drug approved for the treatment of primary kidney cancer (advanced renal cell carcinoma), advanced primary liver cancer (hepatocellular carcinoma), and radioactive iodine resistant advanced thyroid carcinoma.
Medical uses
At the current time sorafenib is indicated as a treatment for advanced renal cell carcinoma (RCC), unresectable hepatocellular carcinomas (HCC) and thyroid cancer.[2][3][4][5]
Kidney cancer
An article in The New England Journal of Medicine, published January 2007, showed compared with placebo, treatment with sorafenib prolongs progression-free survival in patients with advanced clear cell renal cell carcinoma in whom previous therapy has failed. The median progression-free survival was 5.5 months in the sorafenib group and 2.8 months in the placebo group (hazard ratio for disease progression in the sorafenib group, 0.44; 95% confidence interval [CI], 0.35 to 0.55; P<0.01).[6] A few reports described patients with stage IV renal cell carcinomas that were successfully treated with a multimodal approach including neurosurgical, radiation, and sorafenib.[7] This is one of two TGA-labelled indications for sorafenib, although it is not listed on the Pharmaceutical Benefits Scheme for this indication.[5][8]
Liver cancer
At ASCO 2007, results from the SHARP trial[9] were presented, which showed efficacy of sorafenib in hepatocellular carcinoma. The primary endpoint was median overall survival, which showed a 44% improvement in patients who received sorafenib compared to placebo (hazard ratio 0.69; 95% CI, 0.55 to 0.87; p=0.0001). Both median survival and time to progression showed 3-month improvements. There was no difference in quality of life measures, possibly attributable to toxicity of sorafenib or symptoms related to underlying progression of liver disease. Of note, this trial only included patients with Child-Pugh Class A (i.e. mildest) cirrhosis. The results of the study appear in the July 24, 2008, edition of The New England Journal of Medicine. Because of this trial Sorafenib obtained FDA approval for the treatment of advanced hepatocellular carcinoma in November 2007.[10]
In a randomized, double-blind, phase II trial combining sorafenib with doxorubicin, the median time to progression was not significantly delayed compared with doxorubicin alone in patients with advanced hepatocellular carcinoma. Median durations of overall survival and progression-free survival were significantly longer in patients receiving sorafenib plus doxorubicin than in those receiving doxorubicin alone.[10] A prospective single-centre phase II study which included the patients with unresectable hepatocellular carcinoma (HCC)concluding that the combination of sorafenib and DEB-TACE in patients with unresectable HCC is well tolerated and safe, with most toxicities related to sorafenib.[11] This is the only indication for which sorafenib is listed on the PBS and hence the only Government-subsidised indication for sorafenib in Australia.[8] Along with renal cell carcinoma, hepatocellular carcinoma is one of the TGA-labelled indications for sorafenib.[5]
Thyroid cancer
A phase 3 clinical trial has started recruiting (November 2009) to use sorafenib for non-responsive thyroid cancer.[12] The results were presented at the ASCO 13th Annual Meeting and are the base for FDA approval. The Sorafenib in locally advanced or metastatic patients with radioactive iodine-refractory differentiated thyroid cancer: The Phase 3 DECISION trial showed significant improvement in progression-free survival but not in overall survival. However, as is known, the side effects were very frequent, specially hand and foot skin reaction.[13]
Adverse effects
Adverse effects by frequency
Note: Potentially serious side effects are in bold.
Very common (>10% frequency)
- Lymphopenia
- Hypophosphataemia[Note 1]
- Haemorrhage[Note 2]
- Hypertension[Note 3]
- Diarrhea
- Rash
- Alopecia (hair loss; occurs in roughly 30% of patients receiving sorafenib)
- Hand-foot syndrome
- Pruritus (itchiness)
- Erythema
- Increased amylase
- Increased lipase
- Fatigue
- Pain[Note 4]
- Nausea
- Vomiting[Note 5][14]
Common (1-10% frequency)
- Leucopoenia[Note 6]
- Neutropoenia[Note 7]
- Anaemia[Note 8]
- Thrombocytopenia[Note 9]
- Anorexia (weight loss)
- Hypocalcaemia[Note 10]
- Hypokalaemia[Note 11]
- Depression
- Peripheral sensory neuropathy
- Tinnitus[Note 12]
- Congestive heart failure
- Myocardial infarction[Note 13]
- Myocardial ischaemia[Note 14]
- Hoarseness
- Constipation
- Stomatitis[Note 15]
- Dyspepsia[Note 16]
- Dysphagia[Note 17]
- Dry skin
- Exfoliative dermatitis
- Acne
- Skin desquamation
- Arthralgia[Note 18]
- Myalgia[Note 19]
- Renal failure[Note 20]
- Proteinuria[Note 21]
- Erectile dysfunction
- Asthenia (weakness)
- Fever
- Influenza-like illness
- Transient increase in transaminase
Uncommon (0.1-1% frequency)
- Folliculitis
- Infection
- Hypersensitivity reactions[Note 22]
- Hypothyroidism[Note 23]
- Hyperthyroidism[Note 24]
- Hyponatraemia[Note 25]
- Dehydration
- Reversible posterior leukoencephalopathy
- Hypertensive crisis
- Rhinorrhoea[Note 26]
- Interstitial lung disease-like events[Note 27]
- Gastro-oesophageal reflux disease (GORD)
- Pancreatitis[Note 28]
- Gastritis[Note 29]
- Gastrointestinal perforations[Note 30]
- Increase in bilirubin leading, potentially, to jaundice[Note 31]
- Cholecystitis[Note 32]
- Cholangitis[Note 33]
- Eczema
- Erythema multiforme[Note 34]
- Keratoacanthoma[Note 35]
- Squamous cell carcinoma
- Gynaecomastia (swelling of the breast tissue in men)
- Transient increase in blood alkaline phosphatase
- INR abnormal
- Prothrombin level abnormal
- bulbous skin reaction[15]
Rare (0.01-0.1% frequency)
Mechanism of action
Sorafenib is a small molecular inhibitor of several tyrosine protein kinases (VEGFR and PDGFR) and Raf kinases (more avidly C-Raf than B-Raf).[16][17] Sorafenib also inhibits some intracellular serine/threonine kinases (e.g. C-Raf, wild-type B-Raf and mutant B-Raf).[10] Sorafenib treatment induces autophagy,[18] which may suppress tumor growth. However, autophagy can also cause drug resistance.[19]
History
Renal cancer
Sorafenib was approved by the U.S. Food and Drug Administration (FDA) in December 2005,[20] and received European Commission marketing authorization in July 2006,[21] both for use in the treatment of advanced renal cancer.
Liver cancer
The European Commission granted marketing authorization to the drug for the treatment of patients with hepatocellular carcinoma(HCC), the most common form of liver cancer, in October 2007,[22] and FDA approval for this indication followed in November 2007.[23]
In November 2009, the UK’s National Institute of Clinical Excellence declined to approve the drug for use within the NHS in England, Wales and Northern Ireland, stating that its effectiveness (increasing survival in primary liver cancer by 6 months) did not justify its high price, at up to £3000 per patient per month.[24] In Scotland the drug had already been refused authorization by the Scottish Medicines Consortium for use within NHS Scotland, for the same reason.[24]
In March 2012, the Indian Patent Office granted a domestic company, Natco Pharma, a license to manufacture generic Sorafenib, bringing its price down by 97%. Bayer sells a month’s supply, 120 tablets, of Nexavar for![]() 280000 (US$4,700). Natco Pharma will sell 120 tablets for
280000 (US$4,700). Natco Pharma will sell 120 tablets for ![]() 8800 (US$150), while still paying a 6% royalty to Bayer.[25][26] Under Indian Patents Act, 2005 and the World Trade Organisation TRIPS Agreement, the government can issue a compulsory license when a drug is not available at an affordable price.[27]
8800 (US$150), while still paying a 6% royalty to Bayer.[25][26] Under Indian Patents Act, 2005 and the World Trade Organisation TRIPS Agreement, the government can issue a compulsory license when a drug is not available at an affordable price.[27]
Thyroid Cancer
As of November 22, 2013, sorafenib has been approved by the FDA for the treatment of locally recurrent or metastatic, progressive differentiated thyroid carcinoma (DTC) refractory to radioactive iodine treatment.[28]
Research
Lung
In some kinds of lung cancer (with squamous-cell histology) sorafenib administered in addition to paclitaxel and carboplatin may be detrimental to patients.[29]
Brain (Recurrent Glioblastoma)
There is a phase I/II study at the Mayo Clinic[30] of sorafenib and CCI-779 (temsirolimus) for recurrent glioblastoma.
Desmoid Tumor (Aggressive Fibromatosis)
A study performed in 2011 showed that Sorafenib is active against Aggressive fibromatosis. This study is being used as justification for using Sorafenib as an initial course of treatment in some patients with Aggressive fibromatosis.[31]
Nexavar Controversy
In January 2014, Bayer’s CEO stated that Nexavar was developed for “western patients who [could] afford it”. At the prevailing prices, a kidney cancer patient would pay $96,000 (£58,000) for a year’s course of the Bayer-made drug. However, the cost of the Indian version of the generic drug would be around $2,800 (£1,700).[32]
Notes
- Low blood phosphate levels
- Bleeding; including serious bleeds such as intracranial and intrapulmonary bleeds
- High blood pressure
- Including abdominal pain, headache, tumour pain, etc.
- Considered a low (~10-30%) risk chemotherapeutic agent for causing emesis)
- Low level of white blood cells in the blood
- Low level of neutrophils in the blood
- Low level of red blood cells in the blood
- Low level of plasma cells in the blood
- Low blood calcium
- Low blood potassium
- Hearing ringing in the ears
- Heart attack
- Lack of blood supply for the heart muscle
- Mouth swelling, also dry mouth and glossodynia
- Indigestion
- Not being able to swallow
- Sore joints
- Muscle aches
- Kidney failure
- Excreting protein [usually plasma proteins] in the urine. Not dangerous in itself but it is indicative kidney damage
- Including skin reactions and urticaria (hives)
- Underactive thyroid
- Overactive thyroid
- Low blood sodium
- Runny nose
- Pneumonitis, radiation pneumonitis, acute respiratory distress, etc.
- Swelling of the pancreas
- Swelling of the stomach
- Formation of a hole in the gastrointestinal tract, leading to potentially fatal bleeds
- Yellowing of the skin and eyes due to a failure of the liver to adequately cope with the amount of bilirubin produced by the day-to-day actions of the body
- Swelling of the gallbladder
- Swelling of the bile duct
- A potentially fatal skin reaction
- A fairly benign form of skin cancer
- A potentially fatal abnormality in the electrical activity of the heart
- Swelling of the skin and mucous membranes
- A potentially fatal allergic reaction
- Swelling of the liver
- A potentially fatal skin reaction
- A potentially fatal skin reaction
- The rapid breakdown of muscle tissue leading to the build-up of myoglobin in the blood and resulting in damage to the kidneys
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino] phenoxy]-N-methyl-pyridine-2-carboxamide |
|
| Clinical data | |
| Trade names | Nexavar |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a607051 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | D (AU) D (US) |
| Legal status | Prescription Only (S4) (AU) ℞-only (CA) POM (UK) ℞-only (US) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 38–49% |
| Protein binding | 99.5% |
| Metabolism | Hepatic oxidation and glucuronidation (CYP3A4 & UGT1A9-mediated) |
| Half-life | 25–48 hours |
| Excretion | Faeces (77%) and urine (19%) |
| Identifiers | |
| CAS number | 284461-73-0 |
| ATC code | L01XE05 |
| PubChem | CID 216239 |
| DrugBank | DB00398 |
| ChemSpider | 187440 |
| UNII | 9ZOQ3TZI87 |
| KEGG | D08524 |
| ChEBI | CHEBI:50924 |
| ChEMBL | CHEMBL1336 |
| Synonyms | Nexavar Sorafenib tosylate |
| PDB ligand ID | BAX (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C21H16ClF3N4O3 |
| Mol. mass | 464.825 g/mol |

4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-Λ/2-methylpyridine-2- carboxamide is commonly known as sorafenib (I). Sorafenib is prepared as its tosylate salt. Sorafenib blocks the enzyme RAF kinase, a critical component of the RAF/MEK/ERK signaling pathway that controls cell division and proliferation; in addition, sorafenib inhibits the VEGFR-2/PDGFR-beta signaling cascade, thereby blocking tumor angiogenesis.
Sorafenib, marketed as Nexavar by Bayer, is a drug approved for the treatment of advanced renal cell carcinoma (primary kidney cancer). It has also received “Fast Track” designation by the FDA for the treatment of advanced hepatocellular carcinoma (primary liver cancer). It is a small molecular inhibitor of Raf kinase, PDGF (platelet-derived growth factor), VEGF receptor 2 & 3 kinases and c Kit the receptor for Stem cell factor.

Sorafenib and pharmaceutically acceptable salts thereof is disclosed in WO0042012. Sorafenib is also disclosed in WO0041698. Both these patents disclose processes for the preparation of sorafenib.
WO0042012 and WO0041698 describe the process as given in scheme I which comprises reacting picolinic acid (II) with thionyl chloride in dimethyl formamide (DMF) to form acid chloride salt (III). This salt is then reacted with methylamine dissolved in tetrahydrofuran (THF) to give carboxamide (IV). This carboxamide when further reacted with 4- aminophenol in anhydrous DMF and potassium tert-butoxide 4-(2-(N-methylcarbamoyl)-4- pyridyloxy)aniline (V) is formed. Subsequent reaction of this aniline with 4-chloro-3- (trifluoromethyl) phenyl isocyanate (Vl) in methylene chloride yields sorafenib (I). The reaction is represented by Scheme I as given below.
Scheme I

Picolini

Sorafenib (I)
WO2006034796 also discloses a process for the preparation of sorafenib and its tosylate salt. The process comprises reacting 2-picolinic acid (II) with thionyl chloride in a solvent inert toward thionyl chloride without using dimethyl formamide to form acid chloride salt (III). This acid salt on further reaction with aqueous solution methylamine or gaseous methylamine gives compound (IV). Compound (IV) is then reacted with 4-aminophenol with addition of a carbonate salt in the presence of a base to yield compound (V).
Compound (V) can also be obtained by reacting compound (IV) with 4-aminophenol in the presence of water with addition of a phase transfer catalyst. Compound (V) when reacted with 4-chloro-3-(trifluoromethyl) phenyl isocyanate (Vl) in a non-chlorinated organic solvent, inert towards isocyanate gives sorafenib (I). Sorafenib by admixing with p- toluenesulfonic acid in a polar solvent gives sorafenib tosylate (VII). The reaction is represented by Scheme Il as given below.
Scheme Il
P

A key step in the synthesis of sorafenib is the formation of the urea bond. The processes disclosed in the prior art involve reactions of an isocyanate with an amine. These isocyanate compounds though commercially available are very expensive. Further synthesis of isocyanate is very difficult which requires careful and skillful handling of reagents.
Isocyanate is prepared by reaction of an amine with phosgene or a phosgene equivalent, such as bis(trichloromethyl) carbonate (triphosgene) or trichloromethyl chloroformate (diphosgene). Isocyanate can also be prepared by using a hazardous reagent such as an azide. Also, the process for preparation of an isocyanate requires harsh reaction conditions such as strong acid, higher temperature etc. Further, this isocyanate is reacted with an amine to give urea.
Reactions of isocyanates suffer from one or more disadvantages. For example phosgene or phosgene equivalents are hazardous and dangerous to use and handle on a large scale. These reagents are also not environment friendly. Isocyanates themselves are thermally unstable compounds and undergo decomposition on storage and they are incompatible with a number of organic compounds. Thus, the use of isocyanate is not well suited for industrial scale application.
Sorafenib and its pharmaceutically acceptable salts and solvates are reported for the first time in WO0041698 (corresponding US 03139605) by Bayer. In the literature only one route is disclosed for the preparation of sorafenib. According to this route (Scheme-I), picolinic acid of formula III is reacted with thionyl chloride to give the 4-chloro derivative which on treatment

VII
Scheme-I with methanol gave the methyl ester of formula V. Compound of formula V is reacted with methylamine to get the corresponding amide of formula VL Compound of formula VI is reacted with 4-aminophenol to get the ether derivative of formula VII. Compound of formula VII is reacted with 4-chloro-3-trifluoromethylphenylisocyante to get sorafenib base of formula I. Overall yield of sorafenib in this process is 10% from commercially available 2-picolinic acid of formula II. Main drawback in this process is chromatographic purification of the intermediates involved in the process and low yield at every step.
Donald Bankston’s (Org. Proc. Res. Dev., 2002, 6, 777-781) development of an improved synthesis of the above basic route afforded sorafenib in an overall yield of 63% without involving any chromatographic purification. Process improvements like reduction of time in thionyl chloride reaction; avoid the isolation of intermediates of formulae IV and V5 reduction of base quantity in p-aminophenol reaction, etc lead to the simplification of process and improvement in yield of final compound of formula I.
Above mentioned improvements could not reduce the number of steps in making sorafenib of formula-I. In the first step all the raw materials are charged and heated to target temperature (72°C). Such a process on commercial scale will lead to sudden evolution of gas emissions such as sulfur dioxide and hydrogen chloride. Also, in the aminophenol reaction two bases (potassium carbonate and potassium t-butoxide) were used in large excess to accomplish the required transformation.
A scalable process for the preparation of sorafenib is disclosed in WO2006034796. In this process also above mentioned chemistry is used in making sorafenib of formula I. In the first step, catalytic quantity. of DMF used in the prior art process is replaced with reagents like hydrogen bromide, thionyl bromide and sodium bromide. Yield of required product remained same without any advantages from newly introduced corrosive reagents. Process improvements like change of solvents, reagents, etc were applied in subsequent steps making the process scalable. Overall yield of sorafenib is increased to 74% from the prior art 63% yield. Purity of sorafenib is only 95% and was obtained as light brown colored solid.
Main drawbacks in this process are production of low quality sorafenib and requirement of corrosive and difficult to handle reagents such as thionyl bromide and hydrogen bromide. Also, there is no major improvement in the yield of sorafenib.
Sorafenib tosylate ( Brand name: Nexavar ®, BAY 43-9006 other name, Chinese name: Nexavar, sorafenib, Leisha Wa) was Approved by U.S. FDA for the treatment of advanced kidney cancer in 2005 and liver cancer in 2007 .
Sorafenib, co-Developed and co-marketed by Germany-based Bayer AG and South San Francisco-based Onyx Pharmaceuticals , is an Oral Multi-kinase inhibitor for VEGFR1, VEGFR2, VEGFR3, PDGFRbeta, Kit, RET and Raf-1.
In March 2012 Indian drugmaker Natco Pharma received the first compulsory license ever from Indian Patent Office to make a generic Version of Bayer’s Nexavar despite the FACT that Nexavar is still on Patent. This Decision was based on the Bayer Drug being too expensive to most patients. The Nexavar price is expected to drop from $ 5,500 per person each month to $ 175, a 97 percent decline. The drug generated $ 934 million in global sales in 2010, according to India’s Patent Office.

Sorafenib tosylate
Chemical Name: 4-Methyl-3-((4 – (3-pyridinyl)-2-pyrimidinyl) amino)-N-(5 – (4-methyl-1H-imidazol-1-yl) -3 – (trifluoromethyl) phenyl) benzamide monomethanesulfonate, Sorafenib tosylate
CAS Number 475207-59-1 (Sorafenib tosylate ) , 284461-73-0 (Sorafenib)
References for the Preparation of Sorafenib References
1) Bernd Riedl, Jacques Dumas, Uday Khire, Timothy B. Lowinger, William J. Scott, Roger A. Smith, Jill E. Wood, Mary-Katherine Monahan, Reina Natero, Joel Renick, Robert N. Sibley; Omega-carboxyaryl Substituted diphenyl Ureas as RAF kinase inhibitors ; U.S. Patent numberUS7235576
2) Rossetto, Pierluigi; Macdonald, Peter, Lindsay; Canavesi, Augusto; Process for preparation of sorafenib and Intermediates thereof , PCT Int. Appl., WO2009111061
3) Lögers, Michael; gehring, Reinhold; Kuhn, Oliver; Matthäus, Mike; Mohrs, Klaus; müller-gliemann, Matthias; Stiehl, jürgen; berwe, Mathias; Lenz, Jana; Heilmann, Werner; Process for the preparation of 4 – {4 – [( {[4-chloro-3-(TRIFLUOROMETHYL) phenyl] amino} carbonyl) amino] phenoxy}-N-methylpyridine-2-carboxamide , PCT Int. Appl., WO2006034796
4) Shikai Xiang, Liu Qingwei, Xieyou Rong, sorafenib preparation methods, invention patent application Publication No. CN102311384 , Application No. CN201010212039
5) Zhao multiply there, Chenlin Jie, Xu Xu, MASS MEDIA Ji Yafei; sorafenib tosylate synthesis ,Chinese Journal of Pharmaceuticals , 2007 (9): 614 -616
Preparation of Sorafenib Tosylate (Nexavar) Nexavar, sorafenib Preparation of methyl sulfonate

Sorafenib (Sorafenib) chemical name 4 – {4 – [({[4 – chloro -3 – (trifluoromethyl) phenyl] amino} carbonyl) amino] phenoxy}-N-methyl-pyridine -2 – formamide by Bayer (Bayer) research and development, in 2005 the U.S. Food and Drug Administration (FDA) approval. Trade name Nexavar (Nexavar). This product is an oral multi-kinase inhibitor, for the treatment of liver cancer and kidney cancer.
Indian Patent Office in March this year for Bayer’s Nexavar in liver and kidney cancer drugs (Nexavar) has released a landmark “compulsory licensing” (compulsory license). Indian Patent Office that due to the high price Nexavar in India, the vast majority of patients can not afford the drug locally, thus requiring local Indian pharmaceutical company Natco cheap Nexavar sales. Nexavar in 2017 before patent expiry, Natco pay only Bayer’s pharmaceutical sales to 6% royalties. The move to make Nexavar patent drug prices, the supply price from $ 5,500 per month dropped to $ 175, the price reduction of 97%. Compulsory licensing in India for other life-saving drugs and patent medicines overpriced open a road, the Indian Patent Office through this decision made it clear that the patent monopoly does not guarantee that the price is too high. Nexavar is a fight against advanced renal cell carcinoma, liver cancer cure. In China, a box of 60 capsules of Nexavar price of more than 25,000 yuan. In accordance with the recommended dose, which barely enough to eat half of patients with advanced cancer. In September this year India a patent court rejected Bayer Group in India cheap drugmaker emergency appeal. Indian government to refuse patent medicine overpriced undo “compulsory licensing rules,” allowing the production of generic drugs Nexavar.
Sorafenat by Natco – Sorafenib – Nexavar – India natco Nexavar

Chemical Synthesis of Sorafenib Tosylate (Nexavar)
Sorafenib tosylate (brand name :Nexavar®, other name BAY 43-9006, was approved by US FDA for the treatment of kidney cancer in 2005 and advanced liver cancer in 2007.

US Patent US7235576, WO2006034796, WO2009111061 and Faming Zhuanli Shenqing(CN102311384) disclosed processes for preparation of sorafenib base and its salt sorafenib tosylate.
References
1)Bernd Riedl, Jacques Dumas, Uday Khire, Timothy B. Lowinger, William J. Scott, Roger A. Smith, Jill E. Wood, Mary-Katherine Monahan, Reina Natero, Joel Renick, Robert N. Sibley; Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors; US patent numberUS7235576
2)Rossetto, pierluigi; Macdonald, peter, lindsay; Canavesi, augusto; Process for preparation of sorafenib and intermediates thereof, PCT Int. Appl., WO2009111061
3)Lögers, michael; gehring, reinhold; kuhn, oliver; matthäus, mike; mohrs, klaus; müller-gliemann, matthias; stiehl, jürgen; berwe, mathias; lenz, jana; heilmann, werner; Process for the preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-n-methylpyridine-2-carboxamide, PCT Int. Appl., WO2006034796CN102311384, CN201010212039
Full Experimental Details for the preparation of Sorafenib Tosylate (Nexavar)
Synthesis of 4-(2-(N-methylcarbamoyl)-4-pyridyloxy)aniline.
A solution of 4-aminophenol (9.60 g, 88.0 mmol) in anh. DMF (150 mL) was treated with potassium tert-butoxide (10.29 g, 91.7 mmol), and the reddish-brown mixture was stirred at room temp. for 2 h. The contents were treated with 4-chloro- N -methyl-2-pyridinecarboxamide (15.0 g, 87.9mmol) and K2CO3 (6.50 g, 47.0 mmol) and then heated at 80°C. for 8 h. The mixture was cooled to room temp. and separated between EtOAc (500 mL) and a saturated NaCl solution (500 mL). The aqueous phase was back-extracted with EtOAc (300 mL). The combined organic layers were washed with a saturated NaCl solution (4×1000 mL), dried (Na2SO4) and concentrated under reduced pressure. The resulting solids were dried under reduced pressure at 35°C. for 3 h to afford 4-(2-(N-methylcarbamoyl)-4-pyridyloxy)aniline as a light-brown solid 17.9 g, 84%):. 1H-NMR (DMSO-d6) δ 2.77 (d, J = 4.8 Hz, 3H), 5.17 (br s, 2H), 6.64, 6.86 (AA’BB’ quartet, J = 8.4 Hz, 4H), 7.06 (dd, J = 5.5, 2.5 Hz, 1H), 7.33 (d, J = 2.5 Hz, 1H), 8.44 (d, J = 5.5 Hz; 1H), 8.73 (br d, 1H); HPLC ES-MS m/z 244 ((M+H)+).
Synthesis of 4-{4-[({[4-Chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-N-methylpyridine-2-carboxamide (sorafenib)
4-(4-Aminophenoxy)-N-methyl-2-pyridinecarboxamide (52.3 kg, 215 mol) is suspended in ethyl acetate (146 kg) and the suspension is heated to approx. 40° C. 4-Chloro-3-trifluoromethylphenyl isocyanate (50 kg, 226 mol), dissolved in ethyl acetate (58 kg), is then added to such a degree that the temperature is kept below 60° C. After cooling to 20° C. within 1 h, the mixture is stirred for a further 30 min and the product is filtered off. After washing with ethyl acetate (30 kg), the product is dried under reduced pressure (50° C., 80 mbar). 93 kg (93% of theory) of the title compound are obtained as colorless to slightly brownish crystals. m.p. 206-208° C. 1H-NMR (DMSO-d6, 500 MHz): δ =2.79 (d, J=4.4 Hz, 3H, NCH3); 7.16 (dd, J=2.5, 5.6 Hz, 1H, 5-H); 7.18 (d, J=8.8 Hz, 2H, 3′-H, 5′-H); 7.38 (d, J=2.4 Hz, 1H, 3-H); 7.60-7.68 (m, 4H, 2′-H, 6′-H, 5″-H, 6″-H); 8.13 (d, J=1.9 Hz, 1H, 2″-H); 8.51 (d, J=5.6 Hz, 1H, 6-H); 8.81 (d, J=4.5 Hz, 1H, NHCH3); 9.05 (br. s, 1H, NHCO); 9.25 (br. s, 1H, NHCO) MS (ESI, CH3CN/H2O): m/e=465 [M+H]+.
Synthesis of Sorafenib Tosylate (Nexavar)
4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-N2-methylpyridine-2-carboxamide (sorafenib) (50g, 0.1076 mol) is suspended in ethyl acetate (500 g) and water (10g). The mixture is heated to 69°C within 0.5 h, and a filtered solution of p-toluenesulfonic acid monohydrate (3.26 g, 0.017 mol) in a mixture of water (0.65 g) and ethyl acetate (7.2 g) is added. After filtration a filtered solution of p-toluenesulfonic acid monohydrate (22g, 0.11 mol) in a mixture of ethyl acetate (48 g) and water (4.34 g) is added. The mixture is cooled to 23°C within 2 h. The product is filtered off, washed twice with ethyl acetate (92.5 g each time) and dried under reduced pressure. The sorafenib tosylate (65.5 g, 96% of theory) is obtained as colorless to slightly brownish crystals.
…………………..
http://www.google.com/patents/EP2195286A2?cl=en
Example 22: Synthesis of Sorafenib
Phenyl 4-chloro-3-(trifluoromethyl)phenylcarbamate (100 g, 0.3174 mol) and 4-(4- aminophenoxy)-N-methylpicolinamide (77.14 g, 0.3174 mol) were dissolved in N1N- dimethyl formamide (300 ml) to obtain a clear reaction mass. The reaction mass was agitated at 40-450C for 2-3 hours, cooled to room temperature and diluted with ethyl acetate (1000 ml). The organic layer was washed with water (250 ml) followed by 1N HCI (250ml) and finally with brine (250 ml). The organic layer was separated, dried over sodium sulfate and degassed to obtain solid. This solid was stripped with ethyl acetate and finally slurried in ethyl acetate (1000 ml) at room temperature. It was then filtered and vacuum dried to give (118 g) of 4-(4-(3-(4-chloro-3- (trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide (sorafenib base).
Example 23: Synthesis of 1-(4-chloro-3-(trifluoromethyl)phenyl)urea (Compound 4)
Sodium cyanate (1.7 g, 0.02mol) was dissolved in water (17ml) at room temperature to obtain a clear solution. This solution was then charged drop wise to the clear solution of 3- trifluoromethyl-4-chloroaniline (5 g, 0.025 mol) in acetic acid (25 ml) at 40°C-45°C within 1- 2 hours. The reaction mass was agitated for whole day and cooled gradually to room temperature. The obtained solid was filtered washed with water and vacuum dried at 500C to afford the desired product (5.8 g) i.e. 1-(4-chloro-3-(trifluoromethyl)phenyl)urea.
Example 24: Synthesis of Sorafenib
1-(4-chloro-3-(trifluoromethyl) phenyl)urea (15 g, 0.0628 mol), 1 ,8- diazabicyclo[5.4.0]undec-7-ene (11.75 ml, 0.078 mol) and 4-(4-aminophenoxy)-N- methylpicolinamide (15.27 g, 0.0628 mol) were mixed with dimethyl sulfoxide (45 ml) and the reaction mass was then heated to 110-1200C for 12-18 hours. The reaction mass was cooled to room temperature and quenched in water (250 ml). The quenched mass was extracted repeatedly with ethyl acetate and the combined ethyl acetate layer was then back washed with water. It was dried over sodium sulfate and evaporated under vacuum to obtain solid. The obtained solid was slurried in acetonitrile (150 ml) at ambient temperature and filtered to give 4-(4-(3-(4-chloro-3-(trifluoromethyl) phenyl) ureido) phenoxy)-N-methylpicolinamide (sorafenib base) (17.5 g).
………………………..
http://www.google.com/patents/WO2009054004A2?cl=en


EXAMPLES
Example 1
Preparation of l-(4-chloro-3-(trifluoromethyl)phenyI)-3-(4-hydroxyphenyl)urea Into a 250 ml, four-necked RB flask was charged 1O g of 4-aminophenol and 100 ml of toluene. A solution of 4-chloro-3-(trifluoromethyl)phenyl isocyante (20.4 g) in toluene (50 ml) was added to the reaction mass at 25-300C. The reaction mass was stirred at room temperature for 16 h. The reaction mass was filtered and washed the. solid with 50 ml of toluene. The wet material was dried in the oven at 50-60°C to get 29.8 g of title compound as white solid. M.P. is 218-222°C. IR (KBr): 3306, 1673, 1625, 1590, 1560, 1517, 1482, 1435, 1404, 1328, 1261, 1182, 1160, 1146, 1125, 1095, 1032, 884, 849, 832, 812, 766, 746, 724, 683, 539 and 434 cm“1.
Example 2 Preparation of sorafenib tosylate
Into a 100 ml, three-necked RB flask was charged 2.0 g of l-(4-chloro-3- (trifluoromethyl)-phenyl)-3-(4-hydroxyphenyl)urea and 10 ml of DMF. Potassium tert- butoxide (2.3 g) was added to the reaction mass and stirred for 45 min at RT. 4-Chlro-N- methylpicolinamide (1.14 g) and potassium carbonate (0.42 g) were added to the reaction mass and heated to 80°C. The reaction mass was maintained at 80-85°C for 8 h and cooled to 30°C. The reaction mass was poured into water and extracted with ethyl acetate. Ethyl acetate layer was washed with water, brine and dried over sodium sulphate. Solvent was distilled of under reduced pressure.
The crude compound (4.7 g) was dissolved in 10 ml of IPA and added 1.9 g of p- toluenesulfonic acid. The reaction mass was stirred at RT for 15 h and filtered. The wet solid was washed with 10 ml of IPA and dried at 50-60°C to get 3.4 g of title compound as off-white crystalline solid.
…………………..
A Scaleable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer
Bayer Research Center, Pharmaceutical Division, 400 Morgan Lane, West Haven, Connecticut 06516, U.S.A.
Org. Proc. Res. Dev., 2002, 6 (6), pp 777–781
DOI: 10.1021/op020205n
http://pubs.acs.org/doi/abs/10.1021/op020205n

Urea 3 (BAY 43–9006), a potent Raf kinase inhibitor, was prepared in four steps with an overall yield of 63%. Significant process research enabled isolation of each intermediate and target without chromatographic purification, and overall yield increases >50% were observed compared to those from previous methods. This report focuses on improved synthetic strategies for production of scaled quantities of 3 for preclinical, toxicological studies. These improvements may be useful to assemble other urea targets as potential therapeutic agents to combat cancer.
Synthesis of N-[4-Chloro-3-(trifluoromethyl)phenyl]({4-[2-(N-methyl-carbamoyl)(4-pyridyloxy)]phenyl}amino)carboxamide (3, BAY 43–9006).
A suspension of 9 (67.00 g, 275.43 mmol) in methylene chloride ———————-DELETE………………………………The solids were washed with methylene chloride (2 × 50 mL) and dried under vacuum for 4 h at 35 °C to afford 3 (118.19 g, 254.27 mmol, 92%) as an off-white solid.
Mp = 210−212 °C.
1H NMR (DMSO-d6, 300 MHz):
δ 2.77 (d, J = 4.8 Hz, 3H, −NHCH3);
7.16 (m, 3H, aromatic);
7.37 (d, J = 2.5 Hz, 1H, aromatic);
7.62 (m, 4H, aromatic);
8.11 (d, J = 2.5 Hz, 1H, aromatic);
8.49 (d, J = 5.5 Hz, 1H, aromatic);
8.77 (br d, 1H, −NHCH3);
8.99 (s, 1H, −NHCO−); 9.21 (s, 1H, −NHCO−).
Mass spectrum (HPLC/ES): m/e = 465 (M + 1).
Anal. Calcd for C21H16N4ClF3O3: C, 54.26; H, 3.47; N, 12.05. Found: C, 54.11; H, 3.49; N, 12.03.
HPLC (ELS) purity >98%: tR = 3.5 min.
Synthesis of N-[4-Chloro-3-(trifluoromethyl)phenyl]({4-[2-(N-methyl-carbamoyl)(4-pyridyloxy)]phenyl}amino)carboxamide (3, BAY 43–9006): Use of CDI.
A solution of 11 (1.25 g, 6.39 mmol) in methylene chloride———————-DELETED……………………. high vacuum at 35 °C for 2 h to afford 3 (2.55 g, 5.49 mmol, 91%) as a white solid. Proton NMR and mass-spectral data were consistent with structure.
Anal. Calcd for C21H16N4ClF3O3: C, 54.26; H, 3.47; N, 12.05; Cl, 7.63. Found: C, 54.24; H, 3.31; N, 12.30; Cl, 7.84.
Mp (differential scanning calorimetry, 10 °C/min): 205.6 °C;
no polymorphs observed.
References
- “FDA Approves Nexavar for Patients with Inoperable Liver Cancer” (Press release). FDA. November 19, 2007. Retrieved November 10, 2012.
- “Nexavar (sorafenib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 26 December 2013.
- “NEXAVAR (sorafenib) tablet, film coated [Bayer HealthCare Pharmaceuticals Inc.]”. DailyMed. Bayer HealthCare Pharmaceuticals Inc. November 2013. Retrieved 26 December 2013.
- “Nexavar 200mg film-coated tablets – Summary of Product Characteristics (SPC) – (eMC)”. electronic Medicines Compendium. Bayer plc. 27 March 2013. Retrieved 26 December 2013.
- “PRODUCT INFORMATION NEXAVAR® (sorafenib tosylate)” (PDF). TGA eBusiness Services. Bayer Australia Ltd. 12 December 2012. Retrieved 26 December 2013.
- Escudier, B; Eisen, T; Stadler, WM; Szczylik, C; Oudard, S; Siebels, M; Negrier, S; Chevreau, C; Solska, E; Desai, AA; Rolland, F; Demkow, T; Hutson, TE; Gore, M; Freeman, S; Schwartz, B; Shan, M; Simantov, R; Bukowski, RM (January 2007). “Sorafenib in advanced clear-cell renal-cell carcinoma”. New England Journal of Medicine 356 (2): 125–34. doi:10.1056/NEJMoa060655. PMID 17215530.
- Walid, MS; Johnston, KW (October 2009). “Successful treatment of a brain-metastasized renal cell carcinoma”. German Medical Science 7: Doc28. doi:10.3205/000087. PMC 2775194. PMID 19911072.
- “Pharmaceutical Benefits Scheme (PBS) -SORAFENIB”. Pharmaceutical Benefits Scheme. Australian Government Department of Health. Retrieved 27 December 2013.
- Llovet, et al. (2008). “Sorafenib in Advanced Hepatocellular Carcinoma” (PDF). New England Journal of Medicine 359 (4): 378–90.
- Keating GM, Santoro A (2009). “Sorafenib: a review of its use in advanced hepatocellular carcinoma”. Drugs 69 (2): 223–40. doi:10.2165/00003495-200969020-00006. PMID 19228077.
- Pawlik TM, Reyes DK, Cosgrove D, Kamel IR, Bhagat N, Geschwind JF (October 2011). “Phase II trial of sorafenib combined with concurrent transarterial chemoembolization with drug-eluting beads for hepatocellular carcinoma”. J. Clin. Oncol. 29 (30): 3960–7. doi:10.1200/JCO.2011.37.1021. PMID 21911714.
- “Phase 3 Trial of Nexavar in Patients With Non-Responsive Thyroid Cancer”[dead link]
- [1]
- “Chemotherapy-Induced Nausea and Vomiting Treatment & Management”. Medscape Reference. WebMD. 3 July 2012. Retrieved 26 December 2013.
- Hagopian, Benjamin (August 2010). “Unusually Severe Bullous Skin Reaction to Sorafenib: A Case Report”. Journal of Medical Cases 1 (1): 1–3. doi:10.4021/jmc112e. Retrieved 11 February 2014.
- Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, Van Belle P, Elder DE, Wang Y, Nathanson KL, Herlyn M (January 2009). “CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations”. Oncogene 28 (1): 85–94. doi:10.1038/onc.2008.362. PMC 2898184. PMID 18794803.
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M (October 2008). “Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling”. Mol. Cancer Ther. 7 (10): 3129–40. doi:10.1158/1535-7163.MCT-08-0013. PMID 18852116.
- Zhang Y (Jan 2014). “Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways.”. J Mol Med Rep 9 (1): 83–90. PMID 24213221.
- Gauthier A (Feb 2013). “Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update..”. Hepatol Res 43 (2): 147–154. doi:10.1111/j.1872-034x.2012.01113.x. PMID 23145926.
- FDA Approval letter for use of sorafenib in advanced renal cancer
- European Commission – Enterprise and industry. Nexavar. Retrieved April 24, 2007.
- “Nexavar® (Sorafenib) Approved for Hepatocellular Carcinoma in Europe” (Press release). Bayer HealthCare Pharmaceuticals and Onyx Pharmaceuticals. October 30, 2007. Retrieved November 10, 2012.
- FDA Approval letter for use of sorafenib in inoperable hepatocellular carcinoma
- “Liver drug ‘too expensive‘“. BBC News. November 19, 2009. Retrieved November 10, 2012.
- http://www.ipindia.nic.in/ipoNew/compulsory_License_12032012.pdf
- “Seven days: 9–15 March 2012”. Nature 483 (7389): 250–1. 2012. doi:10.1038/483250a.
- “India Patents (Amendment) Act, 2005”. WIPO. Retrieved 16 January 2013.
- [2]
- “Addition of Sorafenib May Be Detrimental in Some Lung Cancer Patients”
- ClinicalTrials.gov NCT00329719 Sorafenib and Temsirolimus in Treating Patients With Recurrent Glioblastoma
- “Activity of sorafenib against desmoid tumor/deep fibromatosis”
- “‘We didn’t make this medicine for Indians… we made it for western patients who can afford it‘“. Daily Mail Reporter. 24 Jan 2014.
External links
- Nexavar.com – Manufacturer’s website
- Prescribing Information – includes data from the key studies justifying the use of sorafenib for the treatment of kidney cancer (particularly clear cell renal cell carcinoma, which is associated with the von Hippel-Lindau gene)
- Patient Information from FDA
- Sorafenib in Treating Patients With Soft Tissue Sarcomas
- Sorafenib Sunitinib differences – diagram
- ClinicalTrials.gov NCT00217399 – Sorafenib and Anastrozole in Treating Postmenopausal Women With Metastatic Breast Cancer
- Cipla launches Nexavar generic at 1/10 of Bayer’s price
| Reference | ||
|---|---|---|
| 1 | * | D. BANKSTON ET AL.: “A Scalable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer” ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 6, no. 6, 2002, pages 777-781, XP002523918 cited in the application |
| 2 | * | PAN W ET AL: “Pyrimido-oxazepine as a versatile template for the development of inhibitors of specific kinases” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 15, no. 24, 15 December 2005 (2005-12-15), pages 5474-5477, XP025314229 ISSN: 0960-894X [retrieved on 2005-12-15] |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2011036647A1 | Sep 24, 2010 | Mar 31, 2011 | Ranbaxy Laboratories Limited | Process for the preparation of sorafenib tosylate |
| WO2011036648A1 | Sep 24, 2010 | Mar 31, 2011 | Ranbaxy Laboratories Limited | Polymorphs of sorafenib acid addition salts |
| WO2011058522A1 | Nov 12, 2010 | May 19, 2011 | Ranbaxy Laboratories Limited | Sorafenib ethylsulfonate salt, process for preparation and use |
| WO2011092663A2 | Jan 28, 2011 | Aug 4, 2011 | Ranbaxy Laboratories Limited | 4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-n2-methylpyridine-2-carboxamide dimethyl sulphoxide solvate |
| WO2011113367A1 * | Mar 17, 2011 | Sep 22, 2011 | Suzhou Zelgen Biopharmaceutical Co., Ltd. | Method and process for preparation and production of deuterated ω-diphenylurea |
| US8552197 | Nov 12, 2010 | Oct 8, 2013 | Ranbaxy Laboratories Limited | Sorafenib ethylsulfonate salt, process for preparation and use |
| US8604208 | Sep 24, 2010 | Dec 10, 2013 | Ranbaxy Laboratories Limited | Polymorphs of sorafenib acid addition salts |
| US8609854 | Sep 24, 2010 | Dec 17, 2013 | Ranbaxy Laboratories Limited | Process for the preparation of sorafenib tosylate |
| US8618305 | Jan 28, 2011 | Dec 31, 2013 | Ranbaxy Laboratories Limited | Sorafenib dimethyl sulphoxide solvate |
| US8669369 | Mar 17, 2011 | Mar 11, 2014 | Suzhou Zelgen Biopharmaceutical Co., Ltd. | Method and process for preparation and production of deuterated Ω-diphenylurea |
Taltirelin Талтирелин for Treatment of Neurodegenerative Diseases,

Taltirelin Талтирелин
N-{[(4S)-1-methyl-2,6-dioxohexahydropyrimidin-4-yl]carbonyl}-L-histidyl-L-prolinamide
(S)-1-Methyl-4,5-dihydroorotyl-L-histidyl-L-prolinamide
(S)-N-(1-Methyl-2,6-dioxohexahydropyrimidin-4-ylcarbonyl)-L-histidyl-L-prolinamide
launched 2000 by Mitsubishi Tanabe Pharma
| Tanabe Seiyaku Co., Ltd. |
Taltirelin tetrahydrate, Taltirelin hydrate, 201677-75-0, TA 0910
Molecular Formula: C17H31N7O9 Molecular Weight: 477.46954
Taltirelin (marketed under the tradename Ceredist) is a thyrotropin-releasing hormone (TRH) analog, which mimics the physiological actions of TRH, but with a much longer half-life and duration of effects,[1] and little development of tolerance following prolonged dosing.[2] It has nootropic,[3] neuroprotective[4] and analgesic effects.[5]
Taltirelin is primarily being researched for the treatment of spinocerebellar ataxia; limited research has also been carried out with regard to other neurodegenerative disorders, e.g., spinal muscular atrophy.[6][7][8]
Taltirelin is a thyrotropin-releasing hormone (TRH) analog that was first commercialized by Tanabe Seiyaku (now Mitsubishi Tanabe Pharma) in Japan in 2000 for the oral treatment of ataxia due to spinocerebellar degeneration.
In 2008, the company filed a regulatory application seeking approval of taltirelin orally disintegrating tablets for the treatment of spinocerebellar degeneration, and in 2009 the approval was received for this formulation.
TRH is a tripeptide hormone that stimulates the release of thyroid-stimulating hormone and prolactin by the anterior pituitary. TRH is produced by the hypothalamus and travels across the median eminence to the pituitary via the hypophyseal portal system.
Taltirelin (TAL) is a thyrotropin-releasing hormone (TRH) analog that is approved for use in humans in Japan. In this study, we characterized TAL binding to and signaling by the human TRH receptor (TRH-R) in a model cell system. We found that TAL exhibited lower binding affinities than TRH and lower signaling potency via the inositol-1,4,5-trisphosphate/calcium pathway than TRH. However, TAL exhibited higher intrinsic efficacy than TRH in stimulating inositol-1,4,5-trisphosphate second messenger generation. This is the first study that elucidates the pharmacology of TAL at TRH-R and shows that TAL is a superagonist at TRH-R



……………………………
Synthesis and central nervous system actions of thyrotropin-releasing hormone analogues containing a dihydroorotic acid moiety
J Med Chem 1990, 33(8): 2130\
http://pubs.acs.org/doi/abs/10.1021/jm00170a013
………………
http://www.google.com/patents/US4665056
EXAMPLE 2
(1) 1.56 g of 1-methyl-L-4,5-dihydroorotic acid and 1.15 g of N-hydroxysuccinimide are dissolved in 30 ml of dimethylformamide, and 2.06 g of dicyclohexylcarbodiimide are added thereto at 0° C. The mixture is stirred at room temperature for 2 hours. The solution thus obtained is hereinafter referred to as “Solution A”. On the other hand, 3.43 g of benzyl L-histidyl-L-prolinate.2HCl are dissolved in dimethylformamide, and 1.67 g of triethylamine are added thereto. The mixture is stirred at 0° C. for 20 minutes, and insoluble materials are filtered off. The filtrate is added to “Solution A”, and the mixture is stirred at 0° C. for 4 hours and then at 10° C. for one day. Insoluble materials are filtered off, and the filtrate is concentrated under reduced pressure at 40° C. to remove dimethylformamide. The residue is dissolved in water, and insoluble materials are filtered off. The filtrate is adjusted to pH 8 with sodium bicarbonate and then passed through a column packed with CHP-20P resin. The column is washed with 500 ml of water, 500 ml of 20% methanol and 300 ml of 50% methanol, successively. Then, the desired product is eluted with 70% methanol. The fractions which are positive to the Pauly’s reaction are collected from the eluate and concentrated under reduced pressure, whereby 3.65 g of benzyl (1-methyl-L-4,5-dihydroorotyl)-L-histidyl-L-prolinate are obtained as an oil.
IRνmax chloroform (cm-1) 3300, 1725, 1680.
650 mg of the product obtained above are dissolved in 1 N-HCl and then lyophilized to give 690 mg of benzyl (1-methyl-L-4,5-dihydroorotyl)-L-histidyl-L-prolinate.HCl.H2 O as powder.
[α]D 22 : -39.8° (C=0.5, H2 O).
IRνmax nujol (cm-1): 1720, 1640-1680.
NMR (DMSO-d6, δ): 1.7-2.4 (m, 4H), 2.90 (s, 3H), 2.4-3.9 (m, 6H), 3.9-4.2 (m, 1H), 4.3-4.5 (m, 1H), 4.6-5.0 (m, 1H), 5.09 (s, 2H), 7.2-7.5 (m, 5H), 8.96 (s, 1H).
Mass (m/e): 496 (M+).
(2) 700 mg of benzyl (1-methyl-L-4,5-dihydroorotyl)-L-histidyl-L-prolinate are dissolved in 20 ml of methanol, and 20 mg of palladium-black are added thereto. The mixture is stirred at room temperature for 3 hours in hydrogen gas. 20 ml of water are added to the reaction mixture, and the catalyst is filtered off. The filtrate is evaporated to remove solvent. The residue is crystallized with methanol, whereby 290 mg of (1-methyl-L-4,5-dihydroorotyl)-L-histidyl-L-proline.5/4 H2 O are obtained.
M.p.: 233°-236° C. (decomp.).
[α]D 20 : -17.2° (C=0.5, H2 O).
IRνmax nujol (cm-1): 1715, 1680, 1630.
NMR (D2 O, δ): 1.7-2.4 (m, 4H), 2.6-3.9 (m, 6H), 3.03 (s, 3H), 4.0-4.45 (m, 2H), 4.95 (t, 1H), 7.27 (s, 1H), 8.57 (s, 1H).
(3) A mixture of 4.29 g of (1-methyl-L-4,5-dihydroorotyl)-L-histidyl-L-proline, 1.15 g of N-hydroxysuccinimide, 2.26 g of dicyclohexylcarbodiimide and 30 ml of dimethylformamide is stirred at 0° C. for 40 minutes and at room temperature for 80 minutes. 30 ml of 15% ammonia-methanol are then added to the mixture at 0° C., and the mixture is stirred at 0° C. for 30 minutes and at room temperature for one hour. Insoluble materials are filtered off, and the filtrate is evaporated to remove dimethylformamide. The residue is dissolved in 20 ml of water, and insoluble materials are again filtered off. The filtrate is adjusted to pH 8 with sodium bicarbonate and then passed through a column packed with CHP-20P resin. After the column is washed with 2 liters of water, the desired product is eluted with 10% methanol. The fractions which are positive to the Pauly’s reaction are collected and concentrated under reduced pressure. The residue is dissolved in 10 ml of water, and allowed to stand in a refrigerator. Crystalline precipitates are collected by filtration, washed with water, and then dried at 25° C. for one day, whereby 3.3 g of (1-methyl-L-4,5-dihydroorotyl)-L-histidyl-L-prolinamide.7/2 H2 O are obtained.
M.p.: 72°-75° C.
[α]D 25 : -13.6° (C=1, H2 O).
IRνmax nujol (cm-1): 3400, 3250, 1710, 1660, 1610, 1540.
References
- Fukuchi, I.; Asahi, T.; Kawashima, K.; Kawashima, Y.; Yamamura, M.; Matsuoka, Y.; Kinoshita, K. (1998). “Effects of taltirelin hydrate (TA-0910), a novel thyrotropin-releasing hormone analog, on in vivo dopamine release and turnover in rat brain”. Arzneimittel-Forschung 48 (4): 353–359. PMID 9608876.
- Asai, H.; Asahi, T.; Yamamura, M.; Yamauchi-Kohno, R.; Saito, A. (2005). “Lack of behavioral tolerance by repeated treatment with taltirelin hydrate, a thyrotropin-releasing hormone analog, in rats”. Pharmacology Biochemistry and Behavior 82 (4): 646–651. doi:10.1016/j.pbb.2005.11.004. PMID 16368129.
- Yamamura, M.; Suzuki, M.; Matsumoto, K. (1997). “Synthesis and pharmacological action of TRH analog peptide (Taltirelin)”. Nihon yakurigaku zasshi. Folia pharmacologica Japonica. 110 Suppl 1: 33P–38P. PMID 9503402.
- Urayama, A.; Yamada, S.; Kimura, R.; Zhang, J.; Watanabe, Y. (2002). “Neuroprotective effect and brain receptor binding of taltirelin, a novel thyrotropin-releasing hormone (TRH) analogue, in transient forebrain ischemia of C57BL/6J mice”. Life Sciences 72 (4–5): 601–607. doi:10.1016/S0024-3205(02)02268-3. PMID 12467901.
- Tanabe, M.; Tokuda, Y.; Takasu, K.; Ono, K.; Honda, M.; Ono, H. (2009). “The synthetic TRH analogue taltirelin exerts modality-specific antinociceptive effects via distinct descending monoaminergic systems”. British Journal of Pharmacology 150 (4): 403–414. doi:10.1038/sj.bjp.0707125. PMC 2189720. PMID 17220907.
- Takeuchi, Y.; Miyanomae, Y.; Komatsu, H.; Oomizono, Y.; Nishimura, A.; Okano, S.; Nishiki, T.; Sawada, T. (1994). “Efficacy of Thyrotropin-Releasing Hormone in the Treatment of Spinal Muscular Atrophy”. Journal of Child Neurology 9 (3): 287–289. doi:10.1177/088307389400900313. PMID 7930408.
- Tzeng, A. C.; Cheng, J.; Fryczynski, H.; Niranjan, V.; Stitik, T.; Sial, A.; Takeuchi, Y.; Foye, P.; Deprince, M.; Bach, J. R. (2000). “A study of thyrotropin-releasing hormone for the treatment of spinal muscular atrophy: A preliminary report”. American journal of physical medicine & rehabilitation / Association of Academic Physiatrists 79 (5): 435–440. doi:10.1097/00002060-200009000-00005. PMID 10994885.
- Kato, Z.; Okuda, M.; Okumura, Y.; Arai, T.; Teramoto, T.; Nishimura, M.; Kaneko, H.; Kondo, N. (2009). “Oral Administration of the Thyrotropin-Releasing Hormone (TRH) Analogue, Taltireline Hydrate, in Spinal Muscular Atrophy”. Journal of Child Neurology 24 (8): 1010–1012. doi:10.1177/0883073809333535. PMID 19666885.
-
-
EP 168 042 (Tanabe Seiyaku; appl. 10.7.1985; GB-prior. 10.7.1984).
-
JP 62 234 029 (Tanabe Seiyaku; J-prior. 27.12.1985).
-
Suzuki, M. et al.: J. Med. Chem. (JMCMAR) 33 (8), 2130-2137 (1990).
-
External links
- (Japanese) Ceredist セレジスト錠 (PDF) Mitsubishi Tanabe Pharma. October 2007.
SITAFLOXACIN …………Antibacterial [DNA-gyrase inhibitor]

7-[(4S)-4-Amino-6-azaspiro[2.4]heptan-6-yl]-8-chloro-6-fluoro-1-[(2S)-2-fluorocyclopropyl]-4-oxoquinoline-3-carboxylic acid
(1R-(1a(S*),2a))-7-(7-Amino-5-azaspiro[2.4]hept-5-yl)-8-chloro-6-fluoro-1-(2-fluorocyclopropyl)-1,4-dihydro-4-oxo-3-quinolinecarboxylic Acid
SYNTHESIS……….http://www.drugfuture.com/synth/syndata.aspx?ID=176447
127254-10-8 [RN]
127254-10-8(ACETATE)
Sitafloxacin isomer II, DU-6859a, STFX, 127254-12-0, 127254-10-8, 163253-35-8
Molecular Formula: C19H18ClF2N3O3 Molecular Weight: 409.814326
- DU 6859A
- DU-6859a
- Sitafloxacin
- UNII-9TD681796G
Sitafloxacin (INN; also called DU-6859a) is a fluoroquinolone antibiotic[1] that shows promise in the treatment of Buruli ulcer. The molecule was identified by Daiichi Sankyo Co., which brought ofloxacin and levofloxacin to the market. Sitafloxacin is currently marketed in Japan by Daiichi Sankyo under the tradename Gracevit.
Sitafloxacin is a new-generation, broad-spectrum oral fluoroquinolone antibiotic.It is very active against many Gram-positive, Gram-negative and anaerobic clinical isolates, including strains resistant to other fluoroquinolones, was recently approved in Japan for the treatment of respiratory and urinary tract infections. Sitafloxacin is active against methicillin-resistant staphylococci, Streptococcus pneumoniae and other streptococci with reduced susceptibility to levofloxacin and other quinolones and enterococci

163253-35-8
-
C19-H18-Cl-F2-N3-O3.3/2H2-O
- 427.833

AU 8933702; EP 0341493; JP 1990231475; JP 1995300416; JP 1999124367; JP 1999124380; US 5587386; US 5767127
The condensation of 3-chloro-2,4,5-trifluorobenzoylacetic acid ethyl ester (I) with (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) and ethyl orthoformate (II) in hot acetic anhydride gives (1R,2S)-2-(3-chloro-2,4,5-trifluorobenzoyl)-3-(2-fluorocyclopropylamino)acrylic acid ethyl ester (IV). The cyclization of (IV) by means of NaH yields the quinolone (V), which is hydrolyzed with HCl to the free acid (VI). The condensation of (VI) with 7(S)-(tert-butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) by means of triethylamine in refluxing acetonitrile affords the protected final product (VIII), which is finally deprotected with trifluoroacetic acid and anisole.

The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) is obtained as follows: 1) The cyclization of butadiene (IX) with dibromofluoromethane by means of BuONa, followed by oxidation with KMnO4, esterification with ethanol – sulfuric acid and reduction with tributyltin hydride gives 2-fluorocyclopropanecarboxylic acid ethyl ester as a cis/trans mixture (X), which is separated by crystallization. The cis-racemic-isomer (XI) is hydrolyzed with NaOH to the corresponding acid (XII), which is condensed with (R)-alpha-methylbenzylamine (XIII) by means of diphenyl chlorophosphate to give the mixture of diastereomers (XIV). This mixture is separated by crystallization, yielding pure (1S,2S)-2-fluoro-N-[alpha(R)-methylbenzyl]cyclopropanecarboxamide (XV), which is hydrolyzed with HCl to the corresponding free acid (XVI). Finally, this compound is converted into (III) by treatment with diphenylphosphoryl azide in refluxing tert-butanol.

b) The intermediate 7(S)-(tert-Butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) can also be obtained as follows: 1) The cyclopropanation of ethyl acetoacetate (XXXI) with 1,2-dibromoethane (XXXII) by means of K2CO3 in DMF gives 1-acetylcyclopropane-1-carboxylic acid ethyl ester (XXXIII), which is brominated with Br2 in ethanol yielding the bromoacetyl derivative (XXXIV). The cyclization of (XXXI) with (R)-alpha-methylbenzylamine (XIII) by means of triethylamine affords 5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane-4,7-dione (XXXV), which by reaction with hydroxylamine is converted into the monooxime (XXXVI). The reduction of (XXXVI) with H2 over RaNi in methanol affords 7-amino-5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptan-4-one as a diastereomeric mixture (XXXVII) + (XXXVIII), which is separated by column chromatography. The reduction of the (7S)-isomer (XXXVIII) with LiAlH4 in THF gives 7(S)-amino-5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane (XXXIX), which is protected in the usual way to the tert-butoxycarbonyl derivative (XL). Finally, this compound is debenzylated to (VII) by hydrogenation with H2 over Pd/C in ethanol.
The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) is obtained as follows: 1) The cyclization of butadiene (IX) with dibromofluoromethane by means of BuONa, followed by oxidation with KMnO4, esterification with ethanol – sulfuric acid and reduction with tributyltin hydride gives 2-fluorocyclopropanecarboxylic acid ethyl ester as a cis/trans mixture (X), which is separated by crystallization. The cis-racemic-isomer (XI) is hydrolyzed with NaOH to the corresponding acid (XII), which is condensed with (R)-alpha-methylbenzylamine (XIII) by means of diphenyl chlorophosphate to give the mixture of diastereomers (XIV). This mixture is separated by crystallization, yielding pure (1S,2S)-2-fluoro-N-[alpha(R)-methylbenzyl]cyclopropanecarboxamide (XV), which is hydrolyzed with HCl to the corresponding free acid (XVI). Finally, this compound is converted into (III) by treatment with diphenylphosphoryl azide in refluxing tert-butanol.

b) The intermediate 7(S)-(tert-Butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) can also be obtained as follows: 2) The reaction of 1-acetylcyclopropane-1-carboxylic acid ethyl ester (XXXIII) with (R)-alpha-methylbenzylamine (XIII) by means of NaOH and ethyl chloroformate gives the corresponding amide (XLI), which by reaction with ethylene glycol and p-toluenesulfonic acid is converted into the ethylene ketal (XLII). The bromination of (XLII) with Br2 in dioxane affords the bromomethyl dioxolane (XLIII), which is finally cyclized to 5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane-4,7-dione (XXXV), already obtained as an intermediate in the preceding synthesis.

The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) can also be obtained as follows: 3) A study of the influence of different substituents in the cis/trans ratio of the cyclopropanation process has been performed. The general method is as follows: the reaction of benzylamine (XXIII) with acetaldehyde and trichloromethyl chloroformate gives the N-benzyl-N-vinylcarbamoyl chloride (XXIV), which by treatment with alcohol yields the N-vinylcarbamate (XXV). The cyclopropanation of (XXV) with fluorodiiodomethane and diethyl zinc as before preferentially affords the cis-N-(2-fluorocyclopropyl)carbamate (XXVI), which is purified by crystallization. The hydrogenolysis of (XXVI) with H2 over Pd/C in acetic acid gives cis-racemic-2-fluorocyclopropylamine (XXVII), which is submitted to optical resolution with L-menthyl chloroformate to afford pure (1R,2S)-isomer (XXII). Finally, this compound is converted into (III) with tert-butoxycarbonyl anhydride as before.
References
- Anderson, DL. (Jul 2008). “Sitafloxacin hydrate for bacterial infections.”. Drugs Today (Barc) 44 (7): 489–501. doi:10.1358/dot.2008.44.7.1219561.PMID 18806900.
- Chem Pharm Bull 1998,46(4),587
- J Med Chem 1994,37(20),3344
- Drugs Fut 1994,19(9),827
- 33rd Intersci Conf Antimicrob Agents Chemother (Oct 17-20, New Orleans) 1993,Abst 975
- Tetrahedron Lett 1992,33(24),3487-90
- Keating GM (April 2011). “Sitafloxacin: in bacterial infections”. Drugs 71 (6): 731–44. doi:10.2165/11207380-000000000-00000.PMID 21504249.
- (Japanese) Gracevit グレースビット (PDF) Daiichi Sankyo Co. January 2008.
|
3-7-2012
|
Method for Production of Quinolone-Containing Lyophilized Preparation
|
|
|
12-5-2007
|
Stabilized liquid preparation
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
3-2-2005
|
Highly absorptive solid preparation
|
|
|
7-9-2004
|
Highly absorbable solid preparation
|
|
|
2-6-2004
|
Medicinal composition
|
|
|
12-17-1999
|
NOVEL THERAPEUTIC AGENTS THAT MODULATE ENZYMATIC PROCESSES
|
Japanese Pharmacopoeia and Japanese GMP Regulations available online
Japanese Pharmacopoeia and Japanese GMP Regulations available online
On Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) website, you can download documents on GMP as well as on marketing authorisations for medicinal products. An English version of the Japanese Pharmacopoeia (JP) is also available. You will find the direct links in the News.
On Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) website, you can find in the section “Regulations and Procedures” under the heading “GMP” requirements regarding the inspection of manufacturers of medicinal products and APIs who want to introduce their products into Japan.
Now, a document was supplemented in January 2014 which describes which documents have to be submitted to the Japanese Agency within a pre-approval inspection and/ or a periodical post-approval inspection.
Go to the PMDA webpage to get more information.
There, you can also access the current Japanese Pharmacopoeia Sixteenth Edition in English.
Source: PMDA, Japan
Topiroxostat 托匹司他 for gout and hyperuricemia


Topiroxostat
托匹司他
FUJI YAKUHIN ……..INNOVATOR
Approved in japan PMDA JUNE 28 2013
Xanthine oxidase inhibitor
FOR GOUT AND HYPERURICEMIA
Launched – 2013, Fuji YakuhinSanwa, Topiloric Uriadec
IUPAC Name: 4-(5-pyridin-4-yl-1H-1,2,4-triazol-3-yl)pyridine-2-carbonitrile
CAS Registry Number: 577778-58-6
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1)
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
3-(3-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole
Synonyms: 4-(5-PYRIDIN-4-YL-1H-1,2,4-TRIAZOL-3-YL)PYRIDINE-2-CARBONITRILE,
AC1NRB9T, Topiroxostat (JAN/INN), DB01685, D09786, FYX-051
SK-0910
4-[5-PYRIDIN-4-YL-1H-[1,2,4]TRIAZOL-3-YL]-PYRIDINE-2-CARBONITRILE,
C13H8N6 MF,248.2482 MW
 TOPIROXOSTAT
TOPIROXOSTAT
托匹司他
A xanthine oxidase inhibitor used to treat gout and hyperuricemia.

PATENT EXP 3/12/22, US /EU/CN

FYX-051, TOPIROXOSTAT is a xanthine oxidase inhibitor. This agent was approved in Japan by Fuji Yakuhin and Sanwa for the treatment of gout and hyperuricemia in 2013 and launched at the same year. In 2009, the compound was licensed to Sanwa by Fuji Yakuhin in Japan for the codevelopment and commercialization of gout.
The number of patients with hyperuricemia in Japan is reported to be 1.25 million and the number suffering from asymptomatic hyperuricemia is estimated to reach several millions. Hyperuricemia is becoming a popular disease.
Presently, hyperuricemia and gout due to hyperuricemia are treated by improving the living environment and administering various drug therapies for each period when an attack of gout is predicted to occur (presymptomatic period), when an attack of gout occurs, or when an attack of gout subsides. That is, preventive therapy is conducted in the presymptomatic period by administering colchicines as well as controlling the daily living environment. When an attack occurs, drug therapy using non-steroidal or steroidal anti-inflammatory agents is mainly conducted. After the attack subsides, patients are given guidance to improve their lifestyle. When improvement is judged insufficient, an assessment is made as to whether hyperuricemia is caused by reduced excretion of uric acid or by increased production of uric acid followed by treatment with drugs, which exhibit a uricosuric effect, such as probenecid and benzbromarone, those which inhibit resorption of uric acid, such as sulfinpyrazone, those which improve acidurea conditions, such as citrates, and xanthine oxidase inhibitors which inhibit production of uric acid, such as allopurinol. Colchicine is said to be able to prevent about 90% of attacks through inhibiting chemotaxis and phagocytosis of leukocytes, such as neutrophils, if administration thereof has been completed within a few hours before the attack. Since colchicine has various adverse effects, however, the use thereof is limited to the minimum and it is therefore difficult to timely administer it.
Accordingly, drug therapies are mainly adopted, but only allopurinol is available for the treatment of a disease caused by increased production of uric acid. However, a metabolite of allopurinol, oxypurinol, tends to accumulate and may cause calculi formation. Furthermore, this drug has been reported to induce adverse events such as rash, a decreased renal function and hepatitis, and it is not easy to administer.
Examples of compounds having xanthine oxidase inhibiting activity that can be used for treating gout caused by increased production of uric acid and that are effective for hyperuricemia and gout due to hyperuricemia have been described in J. Medicinal Chemistry, 1975, Vol. 18, No. 9, pp. 895–900, Japanese Patent Publication No. 49-46622 and Japanese Patent Publication No. 50-24315, which disclose some 1,3,5-substituted or 3,5-substituted 1,2,4-triazole compounds.
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1) has a xanthine oxidase inhibitory activity and serum uric acid level known as the agent that reduces (Patent Document 1).

The method for producing the compound (1), for example, 2 by Reissert Henze reaction isonicotinic acid methyl N-oxide – is a cyano isonicotinate, and the hydrazide which is then, 4 – this condensed cyanopyridine After obtaining a hydrazide of isonicotinic acid N-oxide (Patent Document 1, Example 12) and method, a cyano group after introduction, 4 by Reissert Henze reaction – method of condensing a cyano pyridine is known (Patent Document 1, Example 39).Further, 4 – as a starting material cyano-N-oxide, a triazole ring after construction (Patent Document 3), Reissert Henze unprotected or (Patent Document 2) to protect the ring condensed with isonicotinic acid hydrazide method of obtaining the compound (1) by introducing a cyano group by the reaction have also been reported.
The crystalline polymorph, yet the same molecule with the same chemical composition, the molecular arrangement in the crystal are different, and are different crystalline states. The pharmaceutical compounds having crystal polymorphism such the differences in physicochemical properties, affect pharmacological activity, solubility, bioavailability, stability and the like are known.Therefore, when the crystal polymorphism is present in a pharmaceutically useful compound, producing compounds of the crystalline form highly useful from polymorphs thereof is desirable.
WO 2003/064410 discloses WO 2005/009991 discloses Japanese Patent Publication No. 2005-41802
However, 4 of the above Patent Document – no description about the presence of crystalline polymorph on carbonitrile – pyridine-2-[yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol] It has not been, to these manufacturing methods, it is disclosed a method for the purpose of improving the chemical purity and yield, there is no description of the crystallographic plane.
Method of producing topiroxostat, useful for preventing or treating gout; and its intermediates. Picks up from WO2012060308, claiming the use of this topiroxostat for treating renal dysfunction. Along with the concurrently published WO2014017515, claiming crystalline Forms I and II of this compound, which, Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017516
Crystalline Forms I and II of topiroxostat, useful for preventing or treating gout. Along with the concurrently published WO2014017516, claiming a method of producing this compound. Picks up from WO2012060308, claiming a method of treating renal dysfunction using topiroxostat, which Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017515
novel 1,2,4-triazole compounds having an optionally substituted 2-cyanopyridin-4-yl group at 3-position and an optionally substituted aromatic group at 5-position inhibit a xanthine oxidase and are useful for treatment of gout and hyperuricemia, and have previously filed a patent application (Patent Document 1). The compounds can be prepared according to a method shown by the following reaction scheme:
-
 wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
Patent Document 1: JP-A-2002-017825 -
-
A compound represented by formula (1) which is a starting material may be prepared by a method described in, for example, JP-A-47-7120, JP-A-61-152661A, JP-A-62-149673, JP-A-2002-528447, or European Patent Application No. 559363 specification. However, it is preferable to prepare compound (1) according to the following reaction scheme:
-

-


SYNTHESIS



PATENT
- Example 2
-
To the toluene solution obtained in Example 1 (2) was added 2-propanol (700 mL), and the mixture was stirred. To the resulting solution was added p-toluenesulfonic acid monohydrate (151.16 g) and the resulting mixture was stirred for 8 hours at an internal temperature of 80°C. The mixture was brought to room temperature, and the precipitated crystals were taken out and washed with 2-propanol (210 mL×2). The white crystals were dried under reduced pressure at 60°C for 15 hours to give 106.0 g of the captioned compound as white crystals. Subsequently, 90.0 g of the crystals was suspended in a mixture of 2-butanol (49 mL) and water (491 mL) and heated to an internal temperature of 80°C for 1 hour. The internal temperature was brought to room temperature, and the crystals were filtered and washed with a mixture of 2-butanol and water (1:10) (270 mL×3). The resulting crystals were dried under reduced pressure at 60°C for 15 hours to give 75.7 g of the captioned compound in a high purity.
-
1H―NMR(DMSO-d6)δppm:2.29(s,3H), 7.11 (m,2H), 7.48 (dd, 2H, J=6.48, 1.62Hz) , 8.32-8.35(m, 3H) , 8.57(dd, 1H, J=1.62, 0.81Hz) , 8.94-8.98(m, 3H)
- Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonate
Example 3
Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
-
To the white crystals (50.5g) obtained in Example 2 was added 2-propanol (937.5 mL) and water (312.5 mL), and the resulting mixture was heated and dissolved at an internal temperature of 80°C. Immediately thereafter, the solution was filtered and the filtrate was cooled to an internal temperature of 20°C. To the resulting suspension was added dropwise 0.52 mol/l of an aqueous sodium hydrogen carbonate solution (250 mL), and the mixture was stirred at room temperature for 2 hours. Then the crystals were filtered and washed with water (150 mL×3) and 2-butanol (150 mL×2). The crystals were dried under reduced pressure at 80°C for 15 hours to give 29.4 g of the captioned compound as pale yellow crystals.
-
1H―NMR(DMSO-d6)δppm:8.02(dd, 2H, J=4.59, 1.62Hz),8.32(dd, 1H, J=5.13, 1.62Hz), 8.55(dd, 1H, J=1.62, 1.08Hz), 8.80(dd, 2H, J=4.59, 1.62Hz), 8.93 (dd, 1H, J=5.13, 1.08Hz)
SYNTHESIS
Example 12
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of methyl isonicotinate N-oxide
13.9 g of isonicotinic acid N-oxide was added to 209 ml of methylene chloride, 29.7 g of 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline was further added thereto, and the mixture was stirred under argon atmosphere at room temperature for one hour. 32.1 g of methanol was added to this mixture, which was stirred at room temperature for 17 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (3:1) was used as an eluent to yield 11.1 g of a white powder.
1H-NMR (CDCl3) δppm: 3.95 (3H, s), 7.88 (2H, d, J=7.25 Hz), 8.22 (2H, J=7.25 Hz)
2) Production of Methyl 2-cyanoisonicotinate
11.1 g of the crystal obtained in 1) was dissolved in 170 ml of acetonitrile, 14.6 g of triethylamine and 21.5 g of trimethylsilylnitrile were added thereto, and the mixture was refluxed under argon atmosphere for 16 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (95:5) was used as an eluent to yield 8.44 g of a pale yellow powder.
1H-NMR (CDCl3) δppm: 4.01 (3H, s), 8.08 (1H, d, J=5.45 Hz), 8.24 (1H, s), 8.90 (1H, d, J=5.45 Hz)
3) Production of 2-cyanoisonicotinic acid hydrazide
8.44 g of the crystal obtained in 2) was added to 85 ml of methanol, 1.84 g of hydrazine was further added thereto, and the mixture was stirred under argon temperature for 2 hours. After the solvent was evaporated under reduced pressure, chloroform was added to the residue, which was stirred at room temperature for one hour. The precipitated crystal was filtered, washed with chloroform and dried with a vacuum pump to yield 4.15 g of a pale yellow powder.
1H-NMR (DMSO-d6) δppm: 4.72 (2H, s), 8.05 (1H, d, J=5.12 Hz), 8.31 (1H, s),8.90 (1H, d, J=5.12 Hz), 10.23 (1H, s)
4) Production of the Object Compound
2.67 g of 4-cyanopyridine was dissolved in 40 ml of methanol, 0.83 g of sodium methoxide was added thereto, and the mixture was stirred at room temperature for one hour. Then 4.15 g of the crystal obtained in 3) was added and the mixture was refluxed for 37 hours. After the reaction completed, the precipitated solid was filtered, washed with methanol and dried with a vacuum pump to yield 3.66 g of the object compound as a yellow powder.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54, 1.57 Hz), 8.31 (1H, dd, J=5.11, 1.65 Hz), 8.53 (1H, dd, J=1.65, 0.50 Hz), 8.80 (2H, dd, J=4.54, 1.57 Hz), 8.93 (1H, dd, J=5.11, 0.50 Hz)
Example 39
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of isonicotinic acid (N-2-tert-butoxycarbonyl)hydrazide-1-oxide
585 ml of methylene chloride was added to 39.0 g of isonicotinic acid N-oxide, and after 34.0 g of triethylamine was further added thereto, the mixture was cooled under argon atmosphere to −15° C. 33.5 g of ethyl chlorocarbonate in 117 ml of methylene chloride was added dropwise to this mixture, which was stirred at a temperature from −5 to −10° C. for one hour. Then 44.4 g of tert-butyl ester of carbamic acid in 117 ml of methylene chloride was added dropwise to this mixture and it was allowed to slowly rise to room temperature while it was stirred. The precipitated solid was filtered after 15 hours, washed with methylene chloride, and dried with a vacuum pump to yield 49.7 g of white crystal.
1H-NMR (DMSO-d6) δppm: 1.42 (9H, s), 7.82 (2H, d, J=7.09 Hz), 8.33 (2H, d, J=7.09 Hz), 9.02 (1H, s), 10.44 (1H, s)
Production of 2-cyanoisonicotinic acid hydrazine 1½ P-Toluenesulfonic acid salt
228 ml of dioxane was added to 30.4 g of the crystal obtained in 1), and after 13.1 g of trimethylsilyl cyanide and 38.8 g of N,N-dimethylcarbamoyl chloride were further added thereto, the mixture was stirred under argon atmosphere at 60° C. for 5 hours. After the solvent was evaporated under reduced pressure, the residue was dissolved in ethyl acetate and subsequently washed with 1.5 M sodium carbonate aqueous solution and a saturated saline solution and dried over magnesium sulfate. After the magnesium sulfate was filtered off, the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue, 68.5 g of p-toluenesulfonic acid monohydrate was added thereto, and the mixture was stirred at room temperature for 22 hours. The precipitated crystal was filtered, washed with ethyl acetate, and dried with a vacuum pump to yield 40.3 g of white crystal 2).
1H-NMR (DMSO-d6) δppm: 2.28 (4.5H, s), 7.12 (3H, dd, J=7.92 & 0.66 Hz), 7.48 (3H, dd, J=7.92 & 0.66 Hz), 8.10 (1H, dd, J=5.11 & 1.81 Hz), 8.39 (1H, dd, J=1.81 & 0.33 Hz), 8.99 (1H, dd, J=5.11 & 0.33 Hz)
3) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
9.98 g of 4-cyanopyridine was dissolved in 250 ml of methanol, and after 7.77 g of sodium methoxide was added thereto, the mixture was stirred at room temperature for one hour. Then 40.3 g of the crystal obtained in 2) was added and the mixture was refluxed for 24 hours. After the reaction completed, the precipitated crystal was filtered, washed with methanol, and dried with a vacuum pump to yield 16.3 g of yellow crystal.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54 & 1.57 Hz), 8.31 (1H, dd, J=5.11 & 1.65 Hz), 8.53 (1H, dd, J=1.65 & 0.50 Hz), 8.80 (2H, dd, J=4.54 & 1.57 Hz), 8.93 (1H, dd, J=5.11 & 0.50 Hz)
4) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
45 ml of ethanol and 15 ml of 1-methyl-2-pyrrolidone were added to 3.0 g of the crystal obtained in 3), and the mixture was heated and stirred at 80° C. for 19 hours. The crystal was filtered, subsequently washed with a mixture of ethanol and 1-methyl-2-pyrrolidone (3:1) and ethanol, and dried with a vacuum pump to yield 2.71 g of yellow crystal.
5) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonic acid salt
5 ml of ethanol and 30 ml of water were added to 2.48 g of the crystal obtained in 4), and after 3.8 g of p-toluenesulfonic acid monohydrate was further added thereto, the mixture was stirred at room temperature for 5 hours. The precipitated crystal was filtered, subsequently washed with a mixture of ethanol and water (1:6), water and then ethanol, and dried with a vacuum pump to yield 3.5 g of white crystal.
1H-NMR (DMSO-d6) δppm: 2.28 (3H, s), 7.12 (2H, dd, J=7.75 & 0.50 Hz), 7.48 (2H, dd, J=7.75 & 0.50 Hz), 8.33 (1H, dd, J=5.12 & 1.65 Hz), 8.45 (2H, d, J=6.11 Hz), 8.57 (1H, dd, J=1.65 & 0.66 Hz), 8.96˜9.02 (3H, m)
6) Production of the object compound
17 ml of ethanol and 17 ml of water were added to 3.36 g of the crystal obtained in 5), and the mixture was stirred at room temperature for 30 minutes. A solution of sodium carbonate (0.74 g of sodium carbonate in 17 ml of water) was further added, and the mixture was stirred at room temperature for 2 hours. The precipitated crystal was filtered, subsequently washed with water and ethanol, and dried with a vacuum pump to yield 1.89 g of the object compound as a pale yellow crystal.
TOPIROXOSTAT
SYNTHESIS

(First step)
The first step, 4 – is a step of obtaining a compound (3) is reacted in the presence of an alkali metal alkoxide, cyano-N-oxide and (2), and isonicotinic acid hydrazide.
4 used in this reaction – isonicotinic acid hydrazide and (2) a cyano-N-oxide is a known compound both, I can be prepared by known means.
The alkali metal alkoxide is used, 6 alkoxide alkali metal C 1-C are preferred, sodium methylate, sodium ethylate and the like can be given as specific examples. The reaction is preferably carried out in a solvent, as the solvent, alcohol solvents such as methanol, ethanol and the like are preferable.
The reaction is preferably first in a solvent, is treated with an alkali metal alkoxide compound (2) and then to react the isonicotinic acid hydrazide. First, heated to reflux under cooling, at 80 ℃ from 15 ℃ preferably, 30 minutes and 12 hours in general, the reaction temperature in the reaction with an alkali metal alkoxide (2) with the compound is reacted 1-4 hours, preferably about. Under the temperature conditions, using an excess amount or one equivalent of 30 minutes to 12 hours usually, reaction with isonicotinic acid hydrazide Subsequent to reaction for 1 to 5 hours, preferably.
Example 1:
Synthesis 4 oxide (3) – – – (4 – pyridin-carbonyl) -4 – N “pyridine hydrazide imide -1 was suspended in 40mL of methanol cyanopyridine-N-oxide and (2) 5.00g, sodium was added to methylate 22.4mg, and the mixture was stirred for 2 hours under 40 ℃ nitrogen atmosphere. was cooled to room temperature. reaction solution was stirred for 4 hours at 40 ℃ was added isonicotinic acid hydrazide 5.71g at the same temperature, precipitated The filtrated crystals were, washed with methanol 15mL, and dried 15 hours at 80 ℃, N “- to give (3) 9.60g oxide – (4 – pyridin) -4 – pyridine-hydrazide imide -1.
1 H-NMR (DMSO-d 6) δ (ppm): 6.98 (br, 2H), 7.81 (d, 2H, J = 5.77Hz), 7.85 (d, 2H, J = 7 .09 Hz), 8.29 (d, 2H, J = 7.09Hz), 8.73 (d, 2H, J = 5.77Hz), 10.37 (br, 1H)
MS m / z: 256 [M-H] –
(Second step)
The second step is a step of obtaining compound (4) by cyanation agent cyano compound (3).
As the cyanation agent used, trialkyl cyanide alkali metal cyanide, sodium cyanide, potassium cyanide and the like, zinc cyanide, trimethylsilyl cyanide and the like.
The cyanation reaction is preferably, for example, be carried out (Heterocycles, Vol.22, No.5, 1994) by Reissert Henze reaction. This reaction, for example, to give compound (4) by an organic solvent in the compound (3), and after activation with carbamoyl halide, and reacting the cyano agent. The alkylcarbamoyl halide used in the carbamoylation is a first step in Reissert Henze reaction, 6 alkylcarbamoyl halide di C 1-C dimethylcarbamoyl chloride, and di-propyl carbamoyl chloride can be used, preferably, dimethylcarbamoyl is chloride. The solvent used in this reaction, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, tetrahydrofuran and acetonitrile can be used, however, N, N-dimethylformamide is preferred. Further, 15 ~ 60 ℃, more preferably 30 ~ 50 ℃ reaction temperature. The reaction time is preferably 1 to 24 hours, more preferably 1 to 3 hours. As the cyanation agent used in the cyanation reaction followed, cyano agents above can be used, sodium cyanide, potassium cyanide, zinc cyanide, and trimethylsilyl cyanide, and more preferably, it is sodium cyanide . -20 ~ 60 ℃ is preferred, more preferably -10 ~ 40 ℃, reaction temperature is 1-4 hours.
Is a novel compound (4) The compound obtained in this second step, it is useful as an intermediate for the production of compound (1). If through Compound (4) can be synthesized in good yield and easily without the need for purification in the second step is also possible, and can be produced (1) Compound industrially efficiently compound (4).
Synthetic N “hydrazide (4) – (4 – pyridine carbonyl) -4 – pyridine carboxylic acid N’-(carboxylic imidoyloxy – 2 – – cyano-4)
Example 2
4 pyridine hydrazide imide -1 – oxide ( was suspended in N, N-dimethylformamide 48mL and 3) 10.0g, under nitrogen atmosphere, followed by stirring for 1 hour was added dimethylcarbamoyl chloride 9.20g at 40 ℃. was added sodium cyanide 2.48g at the same temperature, After cooling to 5 ℃ below. reaction mixture was stirred for 1 hour, the crystals were collected by filtration. precipitate was successively added dropwise a 5% aqueous sodium bicarbonate solution 100mL, and 100mL water, and washed with water 100mL, at 80 ℃ for 15 h and dried under reduced pressure to give 4 – hydrazide (4) 9.28g of pyridine-carboxylic acid N’-(carboxylic imide yl – 2 – cyano-4).
1 H-NMR (DMSO-d 6) δ (ppm): 7.15 (br, 2H), 7.82 (d, 2H, J = 5.61Hz), 8.14 (d, 1H, J = 5 .11 Hz), 8.37 (s, 1H), 8.75 (d, 2H, J = 5.61Hz), 8.86 (d, 1H, J = 5.11Hz), 10.47 (br, 1H )
MS m / z: 265 [M-H] –

(Third step)
The third step is a step of obtaining a compound (1) by the presence of an acid catalyst, the cyclization reaction of the compound (4).
As the acid, organic phosphoric acid, p-toluenesulfonic acid, such as hydrochloric acid, inorganic acids can be used, inorganic acids, phosphoric acid is particularly preferable. As the reaction solvent, water, 2 – butanol, 2 – mixed solvent of alcohol and water or alcohol, propanol, ethanol and the like can be used, but water and 2 – I was mixed 5:1 to 10:1 butanol solvent. The reaction temperature and time, 60 ~ 100 ℃, preferably 2 to 12 hours at 70 ~ 90 ℃, I want to 8-10 hours, preferably.
Intermediates and compounds of the present invention the method (1) can be isolated and purified from the washed reaction mixture, recrystallization, by means of various conventional chromatography.
Example 3:
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile 4 Synthesis of (1) – pyridine-carboxylic acid N’- (2 – cyano-4 – carboxylic imide yl) water 82mL, 2 hydrazide (4) 9.25g – butanol was added 8.2mL, phosphate 4.00g, was stirred for 8 h at 80 ℃. After cooling to room temperature, the reaction mixture was precipitated crystals were collected by filtration, water: 2 – were washed with a mixed solution of 92.5mL butanol = 10:1. The 13 h and dried under reduced pressure at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl) – 1 H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1 I got a) 7.89g.
Topiroxostat
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
MS m / z: 247 [M-H] –
PATENT
Synthetic water-carbonitrile p-toluenesulfonate – pyridine Example 1: 4 – [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol]: 2 – butanol = was added monohydrate 6.62g p-toluenesulfonic acid in a mixed solution of 55mL of 10:1, 4 at 80 ℃ – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl] pyridine-2 – – triazol-3 was added carbonitrile 7.85g, and the mixture was stirred at the same temperature for 1 hour. After cooling to room temperature, the reaction mixture, and the precipitated crystals were collected by filtration, and water: 2 – were washed with a mixed solution of 40mL of butanol = 10:1. The dried under reduced pressure for 10 hours at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile p-toluene I got a sulfonate 12.6g.
1 H-NMR (DMSO-d 6) δ (ppm): 2.29 (s, 3H), 7.11 (m, 2H), 7.48 (dd, 2H, J = 6.48,1.62 Hz ) ,8.32-8 .35 (m, 3H), 8.57 (dd, 1H, J = 1.62,0.81 Hz) ,8.94-8 .98 (m, 3H)
– [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazole and potassium carbonate 8.22g, 4 in a mixed solution of 80mL of ethanol = 9:1: preparation water of crystal form I: Example 2 I was dissolved carbonitrile p-toluenesulfonate 10.0g – -3 – yl] pyridine-2. After stirring for 5 hours plus 15mL 6M hydrochloric acid at 20 ℃, was the precipitated crystals were collected by filtration, and washed with water 100mL. The 23 h and dried under reduced pressure at 80 ℃, 4 – to obtain carbonitrile 5.78g – pyridin-2 [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazole. Having a DSC as shown in FIG 4 and the powder X-ray diffraction pattern shown in FIG 1, the resulting crystals were type-I crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
N, N carbonitrile 40.0g – preparation of 4 Form II – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl – triazol-3]-2: Example 3 – dimethylformamide was added 300mL, and stirred for 25 min at 150 ℃. After cooling to room temperature the solution, and the precipitated crystals were collected by filtration, and washed twice with water 200mL, 4 and dried under reduced pressure overnight at 80 ℃ the crystal – [5 – (pyridin-4 – yl)-1H-1 , 2,4 – I got carbonitrile 30.4g – yl] pyridine-2 – triazole-3. Having a DSC as shown in FIG 5 and powder X-ray diffraction pattern shown in FIG 2, the resulting crystals were type II crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
The 25 ℃, about 2g carbonitrile, – preparation of the hydrate 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2: Example 4 I was stored for 14 days under conditions of relative humidity 97%. Having a DSC as shown in FIG 7 and the powder X-ray diffraction pattern shown in FIG 3, the obtained crystal was a hydrate.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
Test Example: solubility test Type I crystal by crystal form, II-type crystal, and water solubility of the hydrate was calculated by absorbance measurement method, a saturated solution concentration of each sample. I Figure 8 shows the results.Whereas the 6.2μg/mL water solubility of crystalline Form I, II type crystal 4.2μg/mL, hydrate was 1.9μg/mL.
From Figure 8, the water solubility of Form II and Form I crystals is good, water-soluble type I crystal is particularly good.
NMR
BMCL Volume 19, Issue 21, 1 November 2009, Pages 6225–6229
http://www.sciencedirect.com/science/article/pii/S0960894X09012372?np=y
view compd 39 and ignore rest
 TOPIROXOSTAT, FYX O51
TOPIROXOSTAT, FYX O51
view compd 39 and ignore rest
| 1 | * | Baldwin, J.J., J. Med. Chem.; 1975; 18(9); 895-900, especially p. 898, lines 3-5. |
| 2 | * | Geldard, J.F. et al., J. Org. Chem.; 1965; 30(1); 318-319, especially p. 319, starting line 33. |
| 3 | * | Lever, A.B.P., Inorg. Chem; 1990; 29; 1271-1285, especially p. 1275, line 18 and 19. |
Nucleosides, Nucleotides and Nucleic Acids, 2008 , vol. 27, 6-7 pg. 888 – 893
Inoue, Tsutomu; Sato, Takahiro; Ashizawa, Naoki; Iwanaga, Takashi; Matsumoto, Koji; Nagata, Osamu; Nakamura, Hiroshi
Bioorganic and Medicinal Chemistry Letters, 2009 , vol. 19, 21 pg. 6225 – 6229
WO 2012060308
WO 2007148835
WO 2005009991
| WO2003064410A1 * | Dec 3, 2002 | Aug 7, 2003 | Naoki Ashizawa | Novel 1,2,4-triazole compound |
| US3882134 * | May 21, 1973 | May 6, 1975 | Merck & Co Inc | 1-Substituted-3,5-dipyridyl-1,2,4-triazoles |
| US3947577 * | Jan 8, 1975 | Mar 30, 1976 | Merck & Co., Inc. | Anti-hyperuricemia composition |
| US3984558 * | Nov 29, 1974 | Oct 5, 1976 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds used as bronchodilators |
| US4011218 * | Dec 3, 1974 | Mar 8, 1977 | Merck & Co., Inc. | 1,2,4-triazoles |
| US4104393 * | Sep 2, 1977 | Aug 1, 1978 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds |
| US5571897 * | Dec 5, 1991 | Nov 5, 1996 | Wallac Oy | Luminescent lanthanide chelates |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
US-9199970-B2 |
2015-12-01 |
|
|||
|
2.
US-20150322006-A1 |
2015-11-12 |
|
|||
|
3.
US-20150309021-A1 |
2015-10-29 |
|
|||
|
4.
US-20150291543-A1 |
2015-10-15 |
|
|||
|
5.
EP-2927219-A1 |
2015-10-07 |
EN
|
|
||
|
6.
US-20150274680-A1 |
2015-10-01 |
|
|||
|
7.
EP-2913053-A1 |
2015-09-02 |
EN
|
|
||
|
8.
EP-2511844-B1 |
2015-08-12 |
EN
|
|
||
|
9.
EP-2712861-B1 |
2015-07-29 |
EN
|
|
||
|
10.
US-20150203490-A1 |
2015-07-23 |
|
|||
|
11.
US-20150191463-A1 |
2015-07-09 |
|
|||
|
12.
US-20150166510-A1 |
2015-06-18 |
|
|||
|
13.
EP-2878594-A1 |
2015-06-03 |
EN
|
|
||
|
14.
EP-2878598-A1 |
2015-06-03 |
E
|
|
||
|
15.
EP-2878595-A1 |
2015-06-03 |
EN
|
|
||
|
16.
US-20150126558-A1 |
2015-05-07 |
|
|||
|
17.
US-8987473-B2 |
2015-03-24 |
|
|||
|
18.
EP-2842948-A1 |
2015-03-04 |
EN
|
|
||
|
19.
EP-2776028-A1 |
2014-09-17 |
EN
|
|
||
|
20.
US-20140256748-A1 |
2014-09-11 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-[5-(4-Pyridinyl)-1H-1,2,4-triazol-3-yl]-2-pyridinecarbonitrile
|
|
| Clinical data | |
| Trade names | Topiloric, Uriadec |
| Legal status |
|
| Identifiers | |
| CAS Number | 577778-58-6 |
| ATC code | None |
| PubChem | CID: 5288320 |
| ChemSpider | 4450517 |
| Chemical data | |
| Formula | C13H8N6 |
| Molecular mass | 248.24 g/mol |
/////////////
C1=CN=CC=C1C2=NC(=NN2)C3=CC(=NC=C3)C#N
TAKEDA PHARMACEUTICALS 武田薬品工業株式会社 ON THE RISE
Tadataka Yamada, M.D., Chief Medical & Scientific Officer of Takeda

TAKEDA US CHICAGO OFFICE
TAKEDA PIPELINE SEE LINKS BELOW
1 https://www.takeda.com/investor-information/annual/files/ar2013_10_en.pdf
2. http://www.takeda.com/research/files/pipeline_20131031_en.pdf
3 http://www.takeda.com/research/pipeline/
- 2012 Download Entire File
 PDF 0.4MB 34P
PDF 0.4MB 34P
Takeda’s top executives had frequently pointed to TAK-875 as one of their best shots at coming up with an important new approach to treating diabetes. The drug is designed to spur insulin secretion in the pancreas and Takeda had confidently projected an approval in Japan in 2015 with a follow-up approval in the big U.S. market a year or two later.
The termination of the high-profile program caused some anxiety among investors. Takeda’s shares plunged 8% on the loss as analysts wondered how the pharma company could counter the loss of Actos, a $3.7 billion drug that accounted for about a quarter of its revenue in 2011.
Takeda won an approval on a trio of DPP-4 diabetes drugs–Nesina (alogliptin) and two combos with alogliptin, dubbed Oseni and Kazano–at the beginning of the year. But Takeda suffered some big delays in gaining acceptance, a common fate in this field, where regulators are particularly cautious about new drugs. And Merck had already solidified its lead in the DPP-4 market with Januvia whileOnglyza trailed closely behind it. Takeda had hoped that a combination of TAK-875 and Januvia could help regain some lost market territory–but that dream has clearly vanished as well.
| January 27, 2014 | |
| January 22, 2014 | |
| January 17, 2014 | |
| January 14, 2014 | |
| January 10, 2014 |
2013
| December 27, 2013 | ||
| December 25, 2013 | ||
| December 25, 2013 | ||
| December 24, 2013 | ||
| December 20, 2013 | ||
| December 20, 2013 | ||
| December 19, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 9, 2013 | ||
| December 5, 2013 | ||
| December 4, 2013 | ||
| November 30, 2013 | ||
| November 21, 2013 | ||
| November 19, 2013 | ||
| November 14, 2013 | ||
| November 12, 2013 | ||
| November 12, 2013 | ||
| October 21, 2013 | ||
| October 7, 2013 | ||
| October 2, 2013 | ||
| October 1, 2013 |
| September 26, 2013 | ||
| September 26, 2013 | ||
| September 24, 2013 | ||
| September 20, 2013 | ||
| September 20, 2013 | ||
| September 13, 2013 | ||
| September 13, 2013 | ||
| September 5, 2013 | ||
| September 2, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 22, 2013 | ||
| August 13, 2013 | ||
| August 1, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 30, 2013 | ||
| July 29, 2013 | ||
| July 26, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 18, 2013 | ||
| July 10, 2013 | ||
| July 1, 2013 |
CLIPPED
Takeda isn’t quite in the top 10 among global drugmakers, but the company boasts the 7th-largest pipeline in the industry, according to its presentation at the conference. Yamada noted that 31% of the pipeline assets are in late-stage trials. Millennium is leading development of three late-stage contenders, TAK-700 for prostate cancer, MLN9708 for multiple myeloma and MLN0002 for ulcerative colitis andCrohn’s disease.
In an effort to revive its diabetes franchise, Takeda is in the final stage of development for a first-of-a-kind GPR40 agonist called TAK-875, designed to provide glucose-dependent insulin secretion.
With a rich late-stage pipeline at Takeda, Yamada wants the company to focus on growing its ranks of earlier-stage drug candidates. To do this the company has landed a variety of deals, including the purchase of Intellikine for $310 million to acquire anti-cancer drugs and more recently the acquisition of Envoy Therapeutics last year for $140 million.
Takeda has formed a New Frontier Science group to scout out the hottest research in academia and elsewhere and form collaborations with scientists behind those innovations. At the J.P. Morgan conference, Yamada said, he was attending many meetings with members of the biotech community.

Takeda Pharmaceutical Company Limited (武田薬品工業株式会社 Takeda Yakuhin Kōgyō Kabushiki-gaisha?) is the largest pharmaceutical company in Japan and Asia and a top 15 pharmaceutical company. The company has over 30,000 employees worldwide and achieved $16.2 billion USD in revenue during the 2012 fiscal year.[1] The company is focused on metabolic disorders, gastroenterology, neurology, inflammation, as well asoncology through its independent subsidiary, Millennium: The Takeda Oncology Company.[2] Its headquarters is located in Chuo-ku, Osaka, and it has an office in Nihonbashi, Chuo, Tokyo.[3][4] In January 2012, Fortune Magazine ranked the Takeda Oncology Company as one the 100 best companies to work for in the United States.
Takeda Pharmaceuticals was founded on June 12, 1781 and was incorporated on January 29, 1925.
Takeda’s Japanese logo
In 1977, Takeda first entered the U.S. pharmaceutical market by developing a joint venture with Abbott Laboratories called TAP Pharmaceuticals.[5]Through TAP Pharmaceuticals, Takeda and Abbott launched the blockbusters Lupron (leuprolide) in 1985 and Prevacid (lansoprazole) in 1995.
One of the firm’s mainstay drugs is Actos, a compound in the thiazolidinedione class of drugs used in the treatment of type 2 diabetes. Launched in 1999, Actos has become the best-selling diabetes drug in the world with $4 billion USD in sales during the 2008 fiscal year.[6]
In February 2005, Takeda announced its acquisition of San Diego, California-based Syrrx, a company specializing in high-throughput X-ray crystallography, for $270 million.[7]
In February 2008, Takeda acquired the Japanese operations of Amgen and rights to a dozen of the California biotechnology company’s pipeline candidates for the Japanese market.[8]
In March 2008, Takeda and Abbott Laboratories announced plans to conclude their 30-year old joint venture, TAP Pharmaceuticals, that had over $3 billion in sales in its final year. The split resulted in Abbott acquiring U.S. rights to Lupron and the drug’s support staff. On the other hand, Takeda received rights to Prevacid and TAP’s pipeline candidates. The move also increased Takeda’s headcount by 3,000 employees.[9]
In April 2008, Takeda announced that it was acquiring Millennium Pharmaceuticals of Cambridge, Massachusetts, a company specializing in cancerdrug research, for $8.8 billion. The acquisition brought in Velcade, a drug indicated for hematological malignancies, as well as a portfolio of pipeline candidates in the oncology, inflammation, and cardiovascular therapeutic areas. Millennium now operates as an independent subsidiary, serving as the global center of excellence in oncology under its new name: “Millennium: The Takeda Oncology Company.” [10]
In May 2008, the company licensed non-exclusively the RNAi technology platform developed by Alnylam Pharmaceuticals, creating a potentially long-term partnership between the companies.[11]
On May 19, 2011, Takeda Pharmaceutical and Nycomed announced that Takeda will acquire Nycomed for € 9.6 billion. The acquisition was completed by September 30, 2011.[12]
On April 11, 2012, Takeda Pharmaceutical and URL Pharma announced that Takeda will acquire URL Pharma for $800 million. The acquisition is expected to be completed within 60 days.
On 25 May 2012, Takeda announced the purchase of Brazilian pharmaceutical company Multilab by R$ 540 million.[13]

Takeda Midosuji Building, headquarters of Takeda Pharmaceutical Company, inChuo-ku, Osaka, Japan
Takeda operates two primary bases in Japan in Osaka and Tokyo. Its United States subsidiary is based in Deerfield, Illinois, and all Global Operations outside of Japan and U.S. are based in Opfikon (Zurich), Switzerland. The company maintains research & development sites in Osaka and Tsukuba, Japan; San Diego andSan Francisco, United States; Cambridge, United Kingdom; and Singapore.[14]
The company has manufacturing facilities in Japan, China, Indonesia, Italy, and Ireland.[15] Following the Nycomed acquisition, the Takeda manufacturing sites have been extended with facilities in Argentina,Austria,Belgium,Brazil,Denmark, Estonia,Germany,Mexico,Norway and Poland. Takeda has overseas marketing presences in the U.S., UK, France, Italy, Germany, Austria, Switzerland, Spain, China, Taiwan, Philippines, Thailand, Indonesia, and Singapore. It has recently[when?] announced its first foray into Canada, Portugal, Spain, Mexico, and Ireland.[15]

AT INDONESIA
Products
Some of the key products that Takeda produces on behalf of partners include:[16]
- Actos (pioglitazone) – Type 2 Diabetes
- Amitiza (lubiprostone) – Chronic idiopathic constipation
- Basen (voglibose) – Type 2 Diabetes
- Benet (risedronic acid) – Osteoporosis (Japan)
- Blopress (candesartan) – Hypertension
- Enbrel (etanercept) – Inflammatory diseases (Japan)
- Dexilant (dexlansoprazole) – Gastroesophageal reflux disease – name changed to Dexilant in U.S.
- Lupron/Leuplin (leuprorelin) – GnRH agonist for prostate cancer and endometriosis
- Prevacid/Takepron (lansoprazole) – Gastroesophageal reflux disease
- Rozerem (ramelteon) – Insomnia
- Uloric (febuxostat) – Gout
- Velcade (bortezomib) – Multiple myeloma and mantle cell lymphoma (Millennium Pharmaceuticals)
![]() AT UK
AT UK
References
- “Financial Results for Fiscal 2012” (PDF). Takeda Pharmaceutical Company Limited. May 9, 2013. Retrieved June 13, 2013.
- “Takeda Initiates Cardiovascular Outcomes Trial for Alogliptin, An Investigational Treatment for Type 2 Diabetes”. Newsblaze.com. 2009-08-28. Retrieved 2010-09-18.
- “FAQ.” Takeda Pharmaceutical Company. Retrieved on February 2, 2011. “Q : Where is Takeda located? A : The Head Office is located in Osaka, Japan, and the Tokyo Head Office is located in Tokyo, Japan.”
- “Overview.” Takeda Pharmaceutical Company. Retrieved on February 2, 2011. “Headquarters Head Office 1-1, Doshomachi 4-chome, Chuo-ku, Osaka 540-8645” and “Tokyo Head Office 12-10, Nihonbashi 2-chome, Chuo-ku, Tokyo 103-8668”
- “TAP Pharmaceutical Products, Inc.: Private Company Information – BusinessWeek”. Investing.businessweek.com. 2008-04-30. Retrieved 2010-09-18.
- Decker, Susan (2009-07-06). “Takeda Sues Torrent to Stop Generic Copy of Actos Diabetes Pill”. Bloomberg. Retrieved 2010-09-18.
- Somers, Terri (2005-02-08). “Japanese drug giant taking over Syrrx here | The San Diego Union-Tribune”. Signonsandiego.com. Retrieved 2010-09-18.
- “Takeda, Amgen in exclusive tie-up for Japanese market”. MarketWatch. 2008-02-04. Retrieved 2010-09-18.
- Marrazzo, Amanda (2008-05-15). “Featured Articles From The Chicago Tribune”. Archives.chicagotribune.com. Retrieved 2010-09-18.
- “MILLENNIUM: The Takeda Oncology Company | About Millennium | Our History”. Mlnm.com. Retrieved 2010-09-18.
- staff (2008-06-15). “Takeda Signs On as Alnylam’s Asian Partner for $150M Upfront”. Genetic Engineering & Biotechnology News (print) (Mary Ann Liebert, Inc.). p. 14.
- http://www.takeda.com/press/article_43116.html
- Hirschler, Ben (May 25, 2012). “Farmacêutica Takeda comprará Multilab por até R$ 540 mi”. Grupo Abril (in portuguese). Exame. Retrieved January 27, 2013.
- “Locations | Worldwide | Takeda Pharmaceutical Company Limited”. Takedaism.com. Retrieved 2010-09-18.
- “By Business | Worldwide | Takeda Pharmaceutical Company Limited”. Takedaism.com. Retrieved 2010-09-18.
- “Annual Reports | Investor Information | Takeda Pharmaceutical Company Limited”. Takeda.com. Retrieved 2010-09-18.
 |
|
| Native name | 武田薬品工業株式会社 |
|---|---|
| Type | Public KK |
| Traded as | |
| Industry | Pharmaceuticals |
| Founded | Doshomachi, Osaka, Japan (June 12, 1781) |
| Headquarters | 1-1, Doshomachi Yonchome,Chuo-ku, Osaka, Japan |
| Key people | Yasuchika Hasegawa (President & CEO) |
| Revenue | |
| Operating income | |
| Net income | |
| Total assets | |
| Total equity | |
| Employees | 30,481 (2012) |
| Website | takeda.com (Global website) |
References:
|
|

CMC CENTRE
The Chemistry, Manufacturing and Controls (CMC) Center is a global organization responsible for overall R&D activities ranging from chemical information on development candidates to the processes leading to “manufacturing” of pharmaceutical products.
The main sites are located in Osaka and consist of the following laboratories: the Chemical Development Laboratories in charge of R&D for developing the manufacturing methods of active pharmaceutical ingredients and the manufacturing of drug substances for clinical samples; the Pharmaceutical Technology R&D Laboratories in charge of R&D for dosage forms, manufacturing and packaging, as well as manufacturing of clinical samples; and the Analytical Development Laboratories in charge of R&D for the development of analytical methods and stability studies of clinical samples. In addition, Hikari Bio-Manufacturing Technology Laboratories is located in Hikari (Yamaguchi) and this is where antibody drug substances are manufactured.
As for overseas sites, the Cambridge Biologics CMC Group (Massachusetts) and the Chicago Pharmaceutical Science Group (Illinois) are located in the USA, while the CMC Center Europe is mainly located in Roskilde, Denmark. All research and development activities at Takeda are promoted with the cooperation of these sites.
