
Home » china pipeline
Category Archives: china pipeline
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DIMDAZENIL


DIMDAZENIL
CAS 308239-86-3
WeightAverage: 372.81
Monoisotopic: 372.1101515
Chemical FormulaC17H17ClN6O2
EVT-201, 308239-86-3, EVT201, 6J8AF7CLE4, EVT 201
7-Chloro-3-[5-[(dimethylamino)methyl]-1,2,4-oxadiazol-3-yl]-4,5-dihydro-5-methyl-6H-imidazo[1,5-a]
[1,4]benzodiazepin-6-one
7-chloro-3-[5-[(dimethylamino)methyl]-1,2,4-oxadiazol-3-yl]-5-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-6-one
EVT 201 is a novel partial positive allosteric modulator of the GABAA receptor complex which is being developed as a treatment for insomnia. It is being developed by Evotec Inc.
- OriginatorRoche
- DeveloperEvotec SE; Zhejiang Jingxin Pharmaceutical
- ClassBenzodiazepines; Chlorobenzenes; Dimethylamines; Imidazoles; Ketones; Oxadiazoles; Sleep disorder therapies; Small molecules
- Mechanism of ActionGABA A receptor modulators
- RegisteredInsomnia
- 29 Nov 2023Registered for Insomnia in China (PO) – First global approval
- 24 Oct 2023Efficacy and adverse events data from a phase III trial in Insomnia released by Zhejiang Jingxin Pharmaceutical
- 21 Oct 2023Efficacy and adverse events data from a phase II trial in Insomnia released by Zhejiang Jingxin Pharmaceutical
Dimdazenil, sold under the brand name Junoenil, is a medication used in the treatment of insomnia in China.[1] It is a benzodiazepine derivative and a partial positive allosteric modulator of the GABAA receptor[2] with two- to four-fold higher functional affinity for the α1 subunit relative to the α2, α3, and α5 subunits.
Medical use
Dimdazenil shows effectiveness in the treatment of insomnia, but has less intrinsic activity in comparison to currently-marketed benzodiazepines and the Z-drugs;[3] however, it is thought that the lower efficacy may result in fewer side effects, such as motor incoordination.[3] In China, dimdazenil is approved for short-term treatment of insomnia.[4]
History
Dimdazenil was originally developed by Roche, based on preclinical data, as a non-sedating anxiolytic, but was found to produce sedation in humans in phase I clinical trials. For this reason, it was subsequently licensed to Evotec, which is now developing it for the treatment of insomnia.[3] By 2007, dimdazenil completed phase II clinical trials for this indication, with positive findings reported.[5] In China, the drug was developed by Zhejiang Jingxin Pharmaceutical.
SCHEME

PATENT
CN111620834
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN306317338&_cid=P10-MAWAJX-84923-1
| Example 16 |
| |
| 1M lithium bis(trimethylsilyl)amide (320 mL, 0.32 mol, 3 eq, 1 Mol/liter) was added to the flask, nitrogen was passed through, the temperature was lowered to -15°C, and the compound K1 (22.6 g, 0.11 mol, 1 eq) obtained in Example 11 was added dropwise. After the addition, the mixture was kept warm at -15°C to -5°C for 2 hours. After the addition, the compound b (26 g, 0.11 mol, 1 eq) obtained by the method of Example 15 was added dropwise. The mixture was kept warm at -15°C to -5°C for 2 hours. After the addition, the mixture was naturally heated to room temperature, and glacial acetic acid was slowly added dropwise. The temperature was controlled to be below 35°C. After completion, the temperature was raised to 55-60°C, and the reaction was kept warm for 2 hours. Then, the mixture was transferred to a rotary evaporator, and the mixture was concentrated under reduced pressure at 45-50°C in batches. The temperature was lowered to 25-30°C, and water and dichloromethane were added in batches. The layers were stirred and separated, and the organic layer was collected. The aqueous layer was extracted once more with dichloromethane, and the organic layers were combined. The layers were washed with a saturated aqueous solution of sodium bicarbonate and water. After washing, the organic layers were collected and transferred to a rotary evaporator for concentration to obtain a solid. The solid was slurried with ethanol at -15°C to -5°C for 15 minutes, filtered, rinsed with cold ethanol, and dried under reduced pressure at 55-60°C to obtain a compound of formula I (36 g, 96.6%), MS: M ++ 1=373.1, HPLC purity 99.85%. |
| 1 H-NMR data: 1 H NMR (400 MHz, DMSO-d 6 δ8.57(s,1H),7.69(d,J=1.9Hz,3H),4.60(d,J=3.7Hz,2H),3.61(s,2H),3.05(s,3H),2.16(s,6H). |
| 13 C-NMR data: 13 C NMR (101 MHz, DMSO) δ 163.35, 163.25, 161.50, 138.88, 134.17, 133.15, 132.81, 130.95, 128.29, 122.67, 114.56, 110.52, 61.10, 46.6 (2), 41.77, 34.48. |
PATENT
WO2000069858
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2000069858&_cid=P10-MAWAOS-90001-1


EXAMPLE
a) 6-Chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5(lH)-dione (III).
25.0 g 6-chloro-isatoic anhydride (II) and 12.4 g sarcosine were suspended under stirring and argon atmosphere in 100 ml p-xylene and heated at reflux for two hours. The suspension was cooled to room temperature and further stirred 1 hour, then filtered off. The precipitate was washed with 25 ml p-xylene twice and dried at 50°C under vacuum. The solid so obtained (6-chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5( lH)-dione (II)) was digested in 75 ml deionized water at 0°C for 1 hour, filtered off, washed with 25 ml deionized water and dried under vacuum 18 hours at 80°C. Crude product: 25.2 g as a beige powder, m.p. 230-232°C
b) Ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[ l,5-a] [ 1,4] benzodiazepine- 3-carboxylate (V).
25.0 g 6-Chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5( lH)-dione (III) were suspended under stirring and argon atmosphere in 200 ml toluene and 32.1 ml N,N-dimethyl-p-toluidine. The suspension was heated to 100°C and 11.2 ml phosphorus oxychloride were added over 30 minutes and stirring was pursued two and an half hours at 100°C. The dark-orange solution was cooled to 40°C and toluene was removed under reduced pressure to give 82 g of a dark-orange oil.
Meanwhile, 81.2 ml hexamethyldisilazane and 265 ml tetrahydrofuran were mixed and cooled to -35°C. 229.5 ml Butyllithium were added over 45 minutes and, after stirring 30 minutes at -35°C, a solution of 35.2 g ethyl(dimethylamino-methylenamino)acetate in 70.4 ml tetrahydrofuran was added over 30 minutes. The orange solution obtained was stirred one more hour at -35°C and a solution of the crude iminochloride in 100 ml
tetrahydrofuran was added over 1 hour at -15°C. The dark red solution was stirred one hour at -15°C, then 18 hours at room temperature (r.t.). 75 ml Acetic acid were added in 10 minutes, then 75 ml deionized water were added in one portion and the orange suspension was heated at reflux for two hours. Tetrahydrofuran was removed under reduced pressure and the residue was partitioned between 200 ml dichloromethane and 100 ml deionized water. The phases were separated and the organic phase was washed with 100 ml aqueous HC1 IN twice and with 100 ml deionized water. The aqueous phases were extracted twice with 100 ml dichloromethane. The combined organic extracts were dried (Na2S04) and evaporated. The residue was digested in 200 ml n-heptane 30 minutes at r.t. and filtered off. The sticky crystals obtained were digested at reflux for 30 minutes in 213.5 ml ethanol, then stirred 3 hours to r.t. and 2 hours at -20°C. The precipitate (ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3-carboxylate (V)) was filtered off, washed three times with 20 ml ethanol and dried under reduced pressure 16 hours at 60°C. Crude product: 23.4 g as a beige powder, m.p. 225.5-226.5 °C c) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo [ 1 ,5-a] [ 1 ,4]benzodiazepine-3- carboxamide (VI).
22.8 g Ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [l,4]- benzodiazepine-3-carboxylate (V)were suspended under stirring and argon atmosphere in 91.2 ml 1 ,4-dioxane. 14.1 ml Formamide and 13.9 ml sodium methanolate were successively added to yield a clear light-orange solution, which turned to a white suspension after 10 minutes. This suspension was stirred two hours at 30°C. 200 ml deionized water were added in one portion and 1,4-dioxane was distilled off at 40°C under reduced pressure. The remaining white suspension was stirred two hours at 0°C and filtered. The precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[ l,5-a] [ l,4]benzodiazepine-3-carboxamide (VI)) was washed with 50 ml deionized water three times and dried under reduced pressure for 18 hours at 80°C. Crude product: 19.43 g as a white powder. m.p.>250°C
d) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5-a] [l,4]benzodiazepine-3- carbonitrile (VII).
19.0 g 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3- carboxamide (VI) were suspended under stirring and argon atmosphere in 95 ml 1,4- dioxane and 6.58 phosphorous oxychloride were added in one portion. The reaction mixture was heated to reflux for one hour giving a yellow solution, which was concentrated at 50°C under reduced pressure. The residue was digested in 100 ml deionized water for two hours at r.t.. The precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5- a] [ l ,4]benzodiazepine-3-carbonitrile (VII)) was filtered off, washed three times with 30 ml deionized water and dried under vacuum at 80°C for 18 hours. Crude product: 17.3 g as a light yellow powder, m.p. 238.5-239.5°C
_ e) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5-a] [l,4]benzodiazepine-3- carboxamidoxime (VIII).
16.8 g 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3- carbonitrile (VIII) were suspended under stirring and argon atmosphere in 101 ml N,N- dimethylformamide and 13.48 g hydroxylamine hydrochloride was added in one portion. 34.2 ml Sodium methanolate were then added over 60 minutes to the yellow suspension, which turned to a colorless suspension. It was stirred one more hour at r.t., then cooled to 0-2°C and 202 ml deionized water were added over 30 minutes. After stirring one more hour at 0°C, the precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[l,5-a] [l,4]benzodiazepine-3-carboxamidoxime (VIII) was filtered off, washed twice with 40 ml deionized water and dried under vacuum at 70°C for 18 hours Crude product 17.84 g as a white powder m.p.>250°C
f) 7-Chloro-3- (5-chloromethyl- [ 1 ,2,4] oxadiazol-3-yl)-5-methyl-4,5-dihydro- imidazo [ 1 ,5-a] [ 1 ,4] benzodiazepin-6-one (IX).
8.0 g 7-chloro-5,6-dιhydro-5-methyl-6-oxo-4H-ιmιdazo[ 1,5-a] [ l,4]benzodιazepιne-3-carboxamidoxime (VIII) and 1.0 g magnesium oxide were suspended under stirring and argon atmosphere in 160 ml 1,4-dioxane. 2 7 ml Chloracetyl chloride were added in one portion and the white thick gel obtained was stirred 4 hours at r.t. and then 17 hours at reflux to give a lightly orange fluid suspension 100 ml Dioxane were distilled off and the reaction mixture was cooled to room temperature. 180 ml Deionized water were added within 15 minutes and the suspension was stirred 1 hour at r.t . The precipitate was filtered off, washed with 50 ml deionized water twice and dried under vacuum at 80°C for 18 hours Crude product: 8.3 g as a light pink powder. This crude product was dissolved in 120 ml tetrahydrofuran at reflux and 0.83 g active charcoal Darco G 60 were added. The system was refluxed 1 hour, then filtered on 25 g Dicaht-Speedex and the filter cake was washed with three portions of 50 ml warm tetrahydrofuran. The filtrate was concentrated at 40°C under reduced pressure The residue was digested in 80 ml ethanol 1 hour at reflux, then stirred 16 hours at r.t. and finally 2 hours at 2°C. The precipitate (7-chloro-3-(5-chloromethyl- [ l,2,4]oxadιazol-3-yl)-5-methyl-4,5-dιhydro-ιmιdazo [ 1,5-a] [ l,4]benzo-dιazepιn-6-one (IX)) was filtered off, washed with 2 portions of 25 ml cold tert-butyl ethvl- ether and dried under vacuum 5 hours at 80°C Crude product: 7.6 g as a light beige powder, m p. 234-238°C
g) 7-Chloro-3-(5-dimethylaminomethyl-[l,2,4]oxadiazol-3-yl)-5-methyl-4,5- dιhydro-imidazo[l,5-a] [l,4]benzodιazepin-6-one (I).
7.0 g 7-Chloro-3-(5-chloromethyl- [ l,2,4]oxadιazol-3-yl)-5-methyl-4,5-dιhydro-ιmιdazo-[ 1,5-a] [ l,4]benzodιazepιn-6-one (IX) were suspended under stirring and argon
atmosphere in 70 ml 1,4-dioxane and 25.7 ml dimethylamine (33% in ethanol) were added over 60 minutes The reaction mixture was stirred one more hour at r.t. and then the solvents were removed under reduced pressure at 35°C. The residue was partitioned between 50 ml dichloromethane and 20 ml deionized water. The phases were separated and the organic phase was washed twice with 20 ml deionized water. The aqueous phases were extracted separately with the same portion of 25 ml dichloromethane, twice. The combined organic extracts were dried (Na2SO4) and the solvent was removed under reduced pressure Crude product: 8.0 g as a light yellow foam Purification
The crude product was dissolved in 40 ml ethanol at reflux and 400 mg active charcoal Darco G 60 were added. The system was stirred 1 hour at reflux, then filtered on a hot pad of Dicalit Speedex, which was washed with two portions of 40 ml hot ethanol. The filtrate was concentrated to 14 g under reduced pressure, heated to reflux and at this temperature and 40 ml terf-butyl-methylether were added over 5 minutes. The suspension was cooled slowly to r.t., stirred 16 hours, further cooled to 2°C. After stirring 1 hour at 2°C, the precipitate was filtered off, washed with 20 ml tert-butyl-methylether and dried 1 hour at 60°C under vacuum. The so obtained powder was dissolved at reflux in 26 ml ethyl acetate. 6.5 ml Ethyl acetate were then distilled off and the turbid solution obtained was slowly cooled to r.t., then to 0°C. After 1 hour stirring at 0°C, the precipitate was filtered off, washed with 10 ml cold tert-butyl-methylether and dried under vacuum at 60°C for 16 hours. The so obtained powder (7-chloro-3-(5-dimethylaminomethyl-[ 1,2,4] oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[ 1,5-a] [l,4]benzodiazepin-6-one (I)) was crystallized a second time in 24.3 ml ethyl acetate according to the procedure described above. Product: 5.5 g as a white powder, m.p. 151.5-153°C
7-Chloro-3-(5-dimethylaminomethyl-[l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo [ 1 ,5-a] [ 1 ,4] benzodiazepin-6-one maleate (1:1)
373 mg 7-Chloro-3-(5-dimethylaminomethyl-[ l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[ 1,5-a] [ l,4]benzodiazepin-6-one (I) and 116 mg maleic acid were dissloved in 3 ml hot ethanol. The salt crystalized on cooling. The suspension was stirred for 10 min at 0°C. Filtration and drying afforded 460 mg 7-Chloro-3-(5-dimethylaminomethyl-[ l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[l,5-a] [ l,4]benzodiazepin-6-one maleate (1:1) as a white solid, m.p. 182-184°C
References
- ^ Huang Z, Zhan S, Chen C, Zhang R, Zhou Y, He J, et al. (February 2024). “Efficacy and safety of Dimdazenil in adults with insomnia disorder: results from a multicenter, randomized, double-blind, placebo-controlled phase III trials”. Sleep. 47 (2). doi:10.1093/sleep/zsad272. PMC 10851846. PMID 37875349.
- ^ Guilleminault C (2010). Sleep Medicine. Elsevier Health Sciences. pp. 574–. ISBN 978-1-4377-1836-2.
- ^ Jump up to:a b c Monti JM, Pandi-Perumal SR, Möhler H (28 September 2010). GABA and Sleep: Molecular, Functional and Clinical Aspects. Springer Science & Business Media. pp. 50–51. ISBN 978-3-0346-0226-6.
- ^ Syed YY (March 2024). “Dimdazenil: First Approval”. Drugs. doi:10.1007/s40265-024-02020-9. PMID 38546956.
- ^ Plunkett JW (September 2007). Plunkett’s Biotech & Genetics Industry Almanac 2008: Biotech & Genetics Industry Market Research, Statistics, Trends & Leading Companies. Plunkett Research, Ltd. pp. 311–. ISBN 978-1-59392-087-6.
External links
| Clinical data | |
|---|---|
| Trade names | Junoenil |
| Other names | EVT-201; EVT201 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 308239-86-3 |
| PubChem CID | 9885841 |
| DrugBank | DB05721 |
| ChemSpider | 8061514 |
| UNII | 6J8AF7CLE4 |
| ChEMBL | ChEMBL5095096 |
| CompTox Dashboard (EPA) | DTXSID301032055 |
| Chemical and physical data | |
| Formula | C17H17ClN6O2 |
| Molar mass | 372.81 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////DIMDAZENIL, EVT-201, 308239-86-3, EVT201, 6J8AF7CLE4, EVT 201, CHINA 2023, INSOMNIA
SULCARDINE SULPHATE
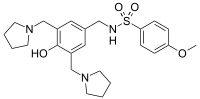
sulcardine, HBI-3000
B 87823
- Molecular FormulaC24H33N3O4S
- Average mass459.602 Da
N-[[4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)phenyl]methyl]-4-methoxybenzenesulfonamide
heart arrhythmia
.gif)
CAS No. : 343935-61-5 (Sulcardine sulfate)
| Synonyms: | B-87823; HBI-3000; B87823; HBI3000; B 87823; HBI 3000;N-(4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)benzyl)-4-methoxybenzenesulfonamide sulfate |
| Molecular Formula: | C24H35N3O8S2 |
| Molecular Weight: | 557.67 |
- Originator Jiangsu Furui Pharmaceuticals; Shanghai Institute of Materia Medica
- Developer HUYA Bioscience International; Jiangsu Furui Pharmaceuticals
- Class Antiarrhythmics; Small molecules
- Mechanism of ActionIon channel antagonists
- Phase I Atrial fibrillation
- No development reported Arrhythmias
- 13 Mar 2020 Chemical structure information added
- 28 Feb 2020 No recent reports of development identified for preclinical development in Arrhythmias in USA (IV)
- 16 Dec 2019 Adverse events data from a phase I trial in Atrial fibrillation (In volunteers) presented at the American Heart Association Scientific Sessions 2019 (AHA-2019)
HUYA Bioscience , under license from Shanghai Institute of Materia Medica (SIMM), is developing sulcardine (HBI-3000, oral, i.v, heart arrhythmia), a myocardial ion channel inhibitory compound, for the treatment of arrhythmia; In September 2016, the drug was still in phase II development, as of August 2020, the company website states that a phase II trial was pending in China.
HBI-3000 (sulcardine sulfate) is an experimental drug candidate that is currently in phase II of human clinical trials as an antiarrhythmic agent.[1][needs update] Clinical investigation will test the safety and efficacy of HBI-3000 as a treatment for both atrial and ventricular arrhythmias.[2]
The molecular problem
Anti-arrhythmic medication is taken to treat irregular beating of the heart. This irregular beating results from a deregulation of the initiation or propagation of the electrical stimulus of the heart. The most common chronic arrhythmia is atrial fibrillation.[3] There is an increased incidence of atrial fibrillation in the elderly and some examples of complications include heart failure exacerbation, hypotension and thrombembolic events.[3]
Most anti-arrhythmic medications exert their effects by decreasing the permeability of potassium ion channels (IKr) in heart cells. These potassium channel blockers delay ventricular repolarization and prolong action potential duration (APD; the prolongation of the electrical stimulus within heart cells). These changes can lower heart rate, eliminate atrial fibrillation, and ultimately sudden cardiac death.[4][5]
Mechanism of action in ventricular myocytes
Ventricular myocytes are heart muscle cells found in the lower chambers of the heart. Heart rate is dependent on the movement of an electrical stimulus through the individual heart cells. This is mediated by the opening of ion channels on cell surfaces. HBI-3000 exerts its effects on the heart by inhibiting multiple ion channels (INa-F, INa-L, ICa-L and IKr), but predominantly the INa-L ion channel . By decreasing the ion permeability of these channels, HBI-3000 slightly prolongs APD (due to IKr); however, unlike pure IKr channel blockers, it is self-limited (due to the decreased permeability of INa-L and ICa-L). This is similar to the medications ranolazine and amiodarone.[5] HBI-3000 suppresses early afterdepolarizations (EADs; a change in the normal net flow of ions during repolarization), does not produce any electrical abnormalities, and displays minimally pronounced prolongation of APD during a slow heart rate (i.e. stimulated at a slower frequency). Pronounced prolongation of APD during a slow heart rate can lead to proarrythmias. Overall, HBI-3000 seems to have a low proarrhythmic risk. The effect of HBI-3000 on contractility and cardiac conduction requires further investigation.[5]
Studies
Animal model
In a canine model, the intravenous injection of HBI-3000 demonstrated to be an effective anti-arrhythmic and anti-fribrillatory agent.[6]
Cellular isolation
The administration of HBI-3000 to isolated heart muscle cells demonstrated the potential to improve arrhythmias while having low proarrhythmic risk.[5]
Human studies
Jiangsu Furui Pharmaceuticals Co., Ltd is currently recruiting participants in their study.[1][
PAPER
http://www.simm.cas.cn/wyp/wyp_lw/201804/W020180420480084769998.pdf

N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-methoxybenzenesulfamide (sulcardine, 6f) and the sulfate (sulcardine sulfate) (1) To a suspension of 4-hydroxybenzylamine (133 g, 1.08 mol) in DMF (500 mL) was added dropwise 4-methoxybenzensul-fonyl chloride (206 g, 1.00 mol) in DMF (320 mL) over a period of 30 min at 0–10 °C with stirring, followed by the addition of triethylamine (158 mL, 1.12 mol) over 30 min at the same temperature. The stirring was continued for an additional 1.5 h at room temperature. The reaction mixture was poured into ice-water (5 L). After stirring for 10 min, the suspension was allowed to stand for 2 h. The solid was filtered, washed with water (300 mL×3), and dried in a desiccator over anhydrous calcium chloride, yielding N-(4-hydroxybenzyl)-4-methoxybenzenesulfamide (11) (248 g, 85%) as a white solid, mp 160–162 °C. The authentic sample was obtained by recrystallization from ethyl acetate, mp 161–162 °C. 1 H NMR (CD3OD) δ 3.70 (s, 3H), 3.76 (s, 2H), 6.48 (d, J=8.4 Hz, 2H), 6.82(d, J=8.4 Hz, 2H), 6.86 (d, J=8.7 Hz, 2H), 7.56 (d, J=8.7 Hz, 2H). EIMS (m/z): 293 (M+ ), 254, 195, 185, 171, 155, 149, 122 (100), 107, 99, 77, 65. Anal. (C14H15NO4S) C, H, N.
(2) A mixture of 11 (230 g, 0.78 mmol), pyrrolidine (200 mL, 2.44 mol) and 36% aqueous formaldehyde (250 mL, 3.30 mol) in ethanol (800 mL) was stirred under reflux for 8 h. The reaction mixture was concentrated under vacuum to dryness. The resulting oil residue was dissolved in chloroform (350 mL), and the solution was washed with water (300 mL×3). Under stirring, the organic layer was mixed with water (300 mL), and then concentrated hydrochloric acid (approximately 165 mL) was added portionwise at 0-10 °C to adjust the pH of the aqueous phase to ~2. The aqueous phase was washed with chloroform (200 mL) and then mixed with additional chloroform (300 mL). Under stirring, the two-phase mixture was treated portionwise with 25%–28% aqueous ammonia (~300 mL) to adjust the pH of the aqueous phase to 9–10. The organic layer was separated, and the aqueous layer was further extracted with chloroform (200 mL×2). The combined organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to dryness. The oily residue was treated with acetone (45 mL) and isopropyl ether (290 mL), and the mixture was heated under reflux until the suspension became a solution. The solution was cooled to room temperature, seeded with an authentic sample, and allowed to stand at 0°C overnight. The solid was filtered and dried under vacuum, yielding product 6f (290 g, 81%) as a yellowish solid, mp 96–98 °C. The authentic sample was obtained by preparative TLC or column chromatography (silica gel; CHCl3:MeOH:25% NH4OH=92:7:1). The compound could be recrystallized from ethanol-water, mp 101–102 °C. 1 H NMR (CDCl3) δ 1.77–1.86 (m, 8H), 2.53–2.63 (m, 8H), 3.68 (s, 4H), 3.86 (s, 3H), 3.97 (s, 2H), 6.86 (s, 2H), 6.95 (d, J=8.7 Hz, 2H), 7.78 (d, J=8.6 Hz 2H). EIMS (m/z): 459 (M+ ), 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S) C, H, N.
(3) Under stirring, the Mannich base 6f (150.5 g, 0.327 mol) was mixed with 2 mol/L H2SO4 (172 mL, 0.344 mol), and the mixture was heated at 80 °C until the solid dissolved. The solution was cooled to room temperature, seeded with an authentic sample, and the sulfate of 6f was formed as crystals. To the stirred mixture was added anhydrous ethanol (520 mL), and the mixture was allowed to stand at 0°C for 24 h. The solid was filtered, washed with ethanol, and recrystallized with 80% ethanol (250 mL). The sulfate was dried over concentrated sulfuric acid in a desiccator, giving the sulfate of 6f (143 g, 71%) as a trihydrate, mp 125–140°C. 1 H NMR (D2O) δ 2.00–2.13 (m, 4H), 2.14–2.25 (m, 4H), 3.12–3.22 (m, 4H), 3.45– 3.55 (m, 4H), 3.90 (s, 3H), 4.20 (s, 2H), 4.33 (s, 4H), 7.06 (d, J=8.7 Hz, 2H), 7.28 (s, 2H), 7.66 (d, J=8.9 Hz, 2H). 13C NMR (D2O) δ 24.7, 47.6, 55.7, 56.1, 58.1, 116.6, 122.5, 131.3, 132.3, 133.3, 136.0, 155.8, 164.8. EIMS (m/z): 459, 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S∙H2SO4∙3H2O) C, H, N, S.
PATENT
Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
https://patents.google.com/patent/US6605635
Synthesis and antiarrhythmic activities of changrolin (1) have been reported (Liangquan Li, et al., Scientia Sinica, 1979, 7, 723; Weizhou Chen, et al., Acta Pharmaceutica Sinica, 1979, 14, 710). Thereafter, investigations of the chemical structural modifications and the physiological activities have successively been carried out by domestic and foreign scientists (Cunji Sun, et al., Acta Pharmaceutica Sinica, 1981, 16, 564; 1986, 21, 692; Mulan Lin, et al., ibid., 1982, 17, 212; D. M. Stout, et al. J. Med. Chem., 1983, 26, 808; 1984, 27, 1347; 1985, 28, 295; 1989, 32, 1910; R. J. Chorvat, et al., ibid., 1993, 36, 2494).

Changrolin is an effective antiarrhythmic agent. Ventricular premature beats disappear 2-3 days after oral administration of changrolin to patients suffering from arrhythmia; I.v. injection or instillaton may result in significant reduction or even disappearence of ventricular premature beats and ventricular tachycardia. However, oral administration of changrolin for a period of over one month may cause a reversible pigmentation on the skin of patients, which gradually retrogresses after ceasing the administration. This pigmentation is associated to the subcutaneous oxidation of certain structural moieties in changrolin molecule or to its instability in solution.
EXAMPLE 1N-[3,5-bis(1-Piperidinomethyl)-4-hydroxy]phenyl-1-naphthalenesulfonamide (B-87836)
(1) To a solution of 4-aminophenol (4.5 g) in dioxane (20 ml) was added dropwise a solution of 1-naphthalenesulfonyl chloride (4.4 g) in dioxane (20 ml). The mixture was further stirred at room temperatue for 4.5 hours and poured into water. The precipitate was collected by filtration, recrystallized from ethanol and decolored with activated carbon to give N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (4.2 g), mp 195-196° C.
(2) A mixture of N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (2.0 g), 37% aqueous formaldehyde (4.5 g) and piperidine (5.6 g) in ethanol (100 ml) was heated to reflux for 50 hours. The ethanol was removed by evaporation in vacuo and chloroform was added to the residue. The organic layer was washed with water then dried over anhydrous Na2SO4. Then the chloroform was removed in vacuo and the residue was triturated in water to give a solid, which was then recrystallized from ethanol to give the titled product (1.4 g), mp 197-198° C.
1HNMR(CDCl3): 1.30-1.50(m, 12H), 2.10-2.21(m, 8H), 3.28(s, 4H), 6.45(s, 2H), 7.24-8.04(m, 6H), 8.56(m, 1H). Elemental analysis (C28H35N3O3S ): Calcd. (%): C, 68.12; H, 7.15; N, 8.51. Found (%): C, 67.96; H, 7.16; N, 8.56.
PATENT
WO-2020159959
Novel crystalline forms of acid salts of sulcardine useful for treating arrhythmia and atrial fibrillation.
4-Methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)-4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), also known as sulcardine, and its salts, such as sulcardine sulfate, constitute a group of compounds with potent anti -arrhythmic activity. Sulcardine is a multi-ion channel blocker that specifically inhibits iNa-Peak, iNa-Late, Ica,L, and Ixrwith similar in vitro potencies (and Ito and IKUT to a lesser degree) in human atrial cardiomyocytes and represents what may be the sole example of a substituted sulfonamide class of anti-arrhythmic. Sulcardine salts can be used as an intravenous injectable or as oral doses for the treatment of arrhythmias, including supraventricular tachyarrhythmia, premature ventricular contractions, ventricular tachycardia, ventricular fibrillation, and atrial fibrillation. See, e.g ., U.S. Patent Nos. 8,541,464 and 8,637,566. Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
[0004] In addition, the evidence to date suggests that one advantage of sulcardine and its salts is that they lack significant pro-arrhythmic activity, as demonstrated in rigorous preclinical safety models, including a post-MI sudden-death conscious canine model and the validated rabbit ventricular wedge model. Additionally, it has been shown that they do not significantly increase defibrillation threshold, nor increase defibrillation failure risk in a post-MI canine model as was seen with flecainide. On the basis of these data, sulcardine and salts, with their very low apparent pro-arrhythmic potential, could potentially be used to treat acute and recurrent atrial fibrillation in the presence of organic heart disease, prolonged QR syndrome, and ventricular arrhythmias, including premature ventricular contractions (PVCs), ventricular tachycardia (VT), and ventricular fibrillation (VF), in either acute- or chronic-administration settings owing to their ability to be formulated into intravenous and oral dosing formulations.
Sulcardine has a chemical name of 4-methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)- 4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), and has the following structure:
[0062] Sulcardine sulfate has the following structure:
[0063] Sulcardine sulfate can exist in a hydrated form. One such form is a trihydrate.
HPLC analysis was performed on a Dionex Ultimate 3000 instrument with the following parameters:
Column: Phenomenex Luna C18, 150×4.6mm, 5pm
Column Temperature: 30°C
Mobile Phase A: 0.2% Phosphoric Acid
Mobile Phase B: Methanol
Diluent: 50:50 MeOH:H20
Runtime: 12 minutes
Flow Rate: l.OmL/min
Injection Volume: 5pL
Detection: 237 nm
Gradient:
EXAMPLE 2 – PREPARATION OF FREE BASE AND SCREENING
[00348] Sulcardine sulfate trihydrate was dissolved in ethyl acetate (16 vol.) and saturated sodium bicarbonate solution (16 vol.). The biphasic solution was transferred to a separating funnel and the layers separated. The organic layer was dried over sodium sulfate and then the solvent was removed by rotary evaporation and the resulting oil dried under vacuum at ambient temperature for ca. 3 hr. FIG. 4 is an XRPD pattern of the resulted amorphous sulcardine free base. In all cases, the initial screening work detailed below was performed on 10 mg of sulcardine free base. All XRPD diffractograms were compared with sulcardine sulfate trihydrate, sulcardine free base and relevant counterions and found to be distinct.
Patent
WO2020123824
claiming treatment of atrial fibrillation (AF) by intravenously administering sulcardine sulfate .
PATENT
References
- ^ Jump up to:a b Jiangsu Furui Pharmaceuticals (November 5, 2010). “Efficacy and safety of sulcardine sulfate tablets in patients with premature ventricular contractions”. ClinicalTrials.gov. U.S. National Library of Medicine. Retrieved 2019-12-20.
- ^ “HUYA Bioscience Int’l announces clinical trial milestones in China for promising new anti-arrhythmic compound; Data supports desirable safety profile” (Press release). San Francisco, California: HUYA Bioscience International. Retrieved 2019-12-20.
- ^ Jump up to:a b Mashal, Abdallah; Katz, Amos; Shvartzman, Pesach (2011). “Atrial fibrillation: A primary care cross-sectional study”. Israel Medical Association Journal. 13 (11): 666–671. PMID 22279699.
- ^ Farkas, András; Leprán, István; Papp, Julius Gy. (1998). “Comparison of the antiarrhythmic and the proarrhythmic effect of almokalant in anaesthetised rabbits”. European Journal of Pharmacology. 346 (2–3): 245–253. doi:10.1016/S0014-2999(98)00067-3. PMID 9652366.
- ^ Jump up to:a b c d Guo, Donglin; Liu, Que; Liu, Tengxian; Elliott, Gary; Gingras, Mireille; Kowey, Peter R.; Yan, Gan-Xin (2011). “Electrophysiological properties of HBI-3000: A new antiarrhythmic agent with multiple-channel blocking properties in human ventricular myocytes”. Journal of Cardiovascular Pharmacology. 57 (1): 79–85. doi:10.1097/FJC.0b013e3181ffe8b3. PMID 20980921.
- ^ Lee, Julia Y.; Gingras, Mireille; Lucchesi, Benedict R. (2010). “HBI-3000 prevents sudden cardiac death in a conscious canine model”. Heart Rhythm. 7 (11): 1712. doi:10.1016/j.hrthm.2010.09.028.
 |
|
| Names | |
|---|---|
| IUPAC name
N-({4-Hydroxy-3,5-bis[(pyrrolidin-1-yl)methyl]phenyl}methyl)-4-methoxybenzene-1-sulfonamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H33N3O4S | |
| Molar mass | 459.61 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
////////////////sulcardine sulfate, phase 2, china, HBI 3000, atrial fibrillation, B 87823,
COC1=CC=C(C=C1)S(=O)(=O)NCC2=CC(=C(C(=C2)CN3CCCC3)O)CN4CCCC4
GST-HG-121
GST-HG-121
mw 431.4
C23 H29 N07
Fujian Cosunter Pharmaceutical Co Ltd
Preclinical for the treatment of hepatitis B virus infection
This compound was originally claimed in WO2018214875 , and may provide the structure of GST-HG-121 , an HBsAg inhibitor which is being investigated by Fujian Cosunter for the treatment of hepatitis B virus infection; in June 2019, an IND application was planned in the US and clinical trials of the combination therapies were expected in 2020. Fujian Cosunter is also investigating GST-HG-131 , another HBsAg secretion inhibitor, although this appears to be being developed only as a part of drug combination.
WO2017013046A1
PATENT
WO2018214875
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018214875&_cid=P21-KB0QYA-12917-1
PATENT
WO-2020103924
Novel crystalline forms of 11-oxo-7,11-dihydro-6H-benzo[f]pyrido[1,2-d][1,4]azepine, a hepatitis B surface antigen and HBV replication inhibitor, useful for treating HBV infection.
Step H: Compound 9 (15.80 g, 35.95 mmol) was dissolved in dichloromethane (150.00 mL), and trifluoroacetic acid (43.91 mL, 593.12 mmol) was added. The reaction solution was stirred at 10 degrees Celsius for 3 hours. The reaction solution was concentrated under reduced pressure and spin-dried, sodium bicarbonate aqueous solution (100.00 mL) was added, and dichloromethane (100.00 mL) was extracted. The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain compound 10.
Step J: Compound 12 (875.00 mg, 1.90 mmol) was dissolved in toluene (20.00 mL) and ethylene glycol dimethyl ether (20.00 mL), and tetrachlorobenzoquinone (1.40 g, 5.69 mmol) was added. The reaction solution was stirred at 120 degrees Celsius for 12 hours. The reaction solution was cooled to room temperature, and a saturated aqueous sodium carbonate solution (50.00 ml) and ethyl acetate (60.00 ml) were added. The mixed solution was stirred at 10-15 degrees Celsius for 20 minutes, and the liquid was separated to obtain an organic phase. Add 2.00 mol/L aqueous hydrochloric acid solution (60.00 mL) to the organic phase, stir at 10-15 degrees Celsius for 20 minutes, and separate the liquid. Wash the organic phase with 2 mol/L aqueous hydrochloric acid solution (60.00 mL×2), separate the liquid, and separate the water phase A 2 mol/L aqueous sodium hydroxide solution (200.00 ml) and dichloromethane (200.00 ml) were added. The layers were separated, and the organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain compound 13.
Step K: Compound 13 (600.00 mg, 1.31 mmol) was dissolved in methanol (6.00 mL), and 4.00 mol/L aqueous sodium hydroxide solution (2.00 mL, 6.39 equiv) was added. The reaction solution was stirred at 15 degrees Celsius for 0.25 hours. The reaction solution was adjusted to pH=3-4 with a 1.00 mol/L hydrochloric acid aqueous solution, and then extracted with dichloromethane (50.00 mL×3). The organic phases were combined, washed with saturated brine (50.00 mL), and dried over anhydrous sodium sulfate , Filtered and concentrated under reduced pressure to obtain the compound of formula (I). ee value (enantiomeric excess): 100%.
////////////GST-HG-121, Fujian Cosunter, Preclinical , hepatitis B, virus infection
O=C(O)C1=CN2C(=CC1=O)c3cc(OC)c(OCCCOC)cc3OC[C@H]2C(C)(C)C
O=C(O)C1=CN2C(=CC1=O)c3cc(OC)c(OCCCOC)cc3OC[C@H]2C(C)(C)C
CK-101
![N-[3-[2-[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]anilino]quinazolin-8-yl]phenyl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117909640&t=l)

CK-101, RX-518
CAS 1660963-42-7
N-[3-[2-[[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]phenyl]amino]quinazolin-8-yl]phenyl]acrylamide
N-(3-(2-((2,3-Difluoro-4-(4-(2-hydroxyethyl)piperazin-1-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide
EGFR-IN-3
UNII-708TLB8J3Y
Suzhou NeuPharma (Originator)
Checkpoint Therapeutics
Non-Small Cell Lung Cancer Therapy
Solid Tumors Therapy
PHASE 2 Checkpoint Therapeutics, Cancer, lung (non-small cell) (NSCLC), solid tumour
RX518(CK-101) is an orally available third-generation and selective inhibitor of certain epidermal growth factor receptor (EGFR) activating mutations, including the resistance mutation T790M, and the L858R and exon 19 deletion (del 19) mutations, with potential antineoplastic activity.
In August 2019, Suzhou Neupharma and its licensee Checkpoint Therapeutics are developing CK-101 (phase II clinical trial), a novel third-generation, covalent, EGFR inhibitor, as a capsule formulation, for the treatment of cancers including NSCLC and other advanced solid tumors. In September 2017, the FDA granted Orphan Drug designation to this compound, for the treatment of EGFR mutation-positive NSCLC; in January 2018, the capsule was being developed as a class 1 chemical drug in China.
CK-101 (RX-518), a small-molecule inhibitor of epidermal growth factor receptor (EGFR), is in early clinical development at Checkpoint Therapeutics and Suzhou NeuPharma for the potential treatment of EGFR-mutated non-small cell lung cancer (NSCLC) and other advanced solid malignancies.
In 2015, Suzhou NeuPharma granted a global development and commercialization license to its EGFR inhibitor program, excluding certain Asian countries, to Coronado Biosciences (now Fortress Biotech). Subsequently, Coronado assigned the newly acquired program to its subsidiary Checkpoint Therapeutics.
In 2017, the product was granted orphan drug designation in the U.S. for the treatment of EGFR mutation-positive NSCLC.
There are at least 400 enzymes identified as protein kinases. These enzymes catalyze the phosphorylation of target protein substrates. The phosphorylation is usually a transfer reaction of a phosphate group from ATP to the protein substrate. The specific structure in the target substrate to which the phosphate is transferred is a tyrosine, serine or threonine residue. Since these amino acid residues are the target structures for the phosphoryl transfer, these protein kinase enzymes are commonly referred to as tyrosine kinases or serine/threonine kinases.
[0003] The phosphorylation reactions, and counteracting phosphatase reactions, at the tyrosine, serine and threonine residues are involved in countless cellular processes that underlie responses to diverse intracellular signals (typically mediated through cellular receptors), regulation of cellular functions, and activation or deactivation of cellular processes. A cascade of protein kinases often participate in intracellular signal transduction and are necessary for the realization of these cellular processes. Because of their ubiquity in these processes, the protein kinases can be found as an integral part of the plasma membrane or as cytoplasmic enzymes or localized in the nucleus, often as components of enzyme complexes. In many instances, these protein kinases are an essential element of enzyme and structural protein complexes that determine where and when a cellular process occurs within a cell.
[0004] The identification of effective small compounds which specifically inhibit signal transduction and cellular proliferation by modulating the activity of tyrosine and serine/threonine kinases to regulate and modulate abnormal or inappropriate cell proliferation, differentiation, or metabolism is therefore desirable. In particular, the identification of compounds that specifically inhibit the function of a kinase which is essential for processes leading to cancer would be beneficial.
[0005] While such compounds are often initially evaluated for their activity when dissolved in solution, solid state characteristics such as polymorphism are also important. Polymorphic forms of a drug substance, such as a kinase inhibitor, can have different physical properties, including melting point, apparent solubility, dissolution rate, optical and mechanical properties, vapor pressure, and density. These properties can have a direct effect on the ability to process or manufacture a drug substance and the drug product. Moreover, differences in these properties
can and often lead to different pharmacokinetics profiles for different polymorphic forms of a drug. Therefore, polymorphism is often an important factor under regulatory review of the ‘sameness’ of drug products from various manufacturers. For example, polymorphism has been evaluated in many multi-million dollar and even multi-billion dollar drugs, such as warfarin sodium, famotidine, and ranitidine. Polymorphism can affect the quality, safety, and/or efficacy of a drug product, such as a kinase inhibitor. Thus, there still remains a need for polymorphs of kinase inhibitors. The present disclosure addresses this need and provides related advantages as well.
PATENT
WO2015027222
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015027222


PATENT
WO-2019157225
Crystalline form II-VIII of the compound presumed to be CK-101 (first disclosed in WO2015027222 ), for treating a disorder mediated by epidermal growth factor receptor (EGFR) eg cancer.
SCHEME A
Scheme B
General Procedures
Example 1: Preparation of the compound of Formula I (N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide)
[0253] To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2C03 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
[0254] To a solution of 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 9.0 mmol) in MeOH (30 mL) was added Pd/C (270 mg) and the resulting mixture was stirred at r.t.
overnight. The Pd/C was removed by filtration and the filtrate was concentrated to afford 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (2.39 g, 99% yield) as off-white solid.
[0255] To a solution of 8-bromo-2-chloroquinazoline (15.4 g, 63.6 mmol, 1 eq. ) and (3-aminophenyl)boronic acid (8.7 g, 63.6 mmol, 1 eq.) in dioxane/H20 (200 mL/20 mL) was added Na2C03 (13.5 g, 127.2 mmol, 2 eq.), followed by Pd(dppf)Cl2 (2.6 g, 3.2 mmol, 0.05 eq.) under N2, then the mixture was stirred at 80 °C for 12 h. Then the solution was cooled to r.t.,
concentrated and the residue was purified via column chromatography (PE/EA=3 :2, v/v) to afford 3-(2-chloroquinazolin-8-yl)aniline as yellow solid (8.7 g, 53.7% yield).
[0256] To a solution of 3-(2-chloroquinazolin-8-yl)aniline (8.7 g, 34 mmol, 1 eq.) in DCM ( 200 mL ) cooled in ice-bath was added TEA (9.5 mL, 68 mmol, 2 eq. ), followed by acryloyl chloride (4.1 mL, 51 mmol, 1.5 eq.) dropwise. The resulting mixture was stirred at r.t. for 1 h, then washed with brine, dried over anhydrous N2S04 concentrated and the residue was purified via column chromatography (PE/EA=l : 1, v:v) to afford N-(3-(2-chloroquinazolin-8-yl)phenyl)acryl amide as yellow solid(6.6 g, 65% yield).
[0257] To a suspension of 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (83 mg,
0.32 mmol, 1 eq.) and N-(3-(2-chloroquinazolin-8-yl)phenyl)acrylamide (100 mg, 0.32 mmol, 1 eq.) in n-BuOH (5 mL) was added TFA (68 mg, 0.64 mmol, 2 eq.) and the resulting mixture was stirred at 90 °C overnight. The mixture was concentrated, diluted with DCM (20 mL) , washed with Na2C03 solution (20 mL), dried over anhydrous Na2S04, concentrated and the residue was purified via column chromatography (MeOH/DCM=l/30, v:v) to afford N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide as a yellow solid(l6.3 mg, 9.5% yield). LRMS (M+H+) m/z calculated 531.2, found 531.2. 1H NMR
(CD3OD, 400 MHz) d 9.21 (s, 1 H), 7.19-8.01 (m, 10 H), 8.90 (s, 1 H), 6.41-6.49 (m, 3 H), 5.86 (m, 1 H), 3.98-4.01 (m, 3 H), 3.70-3.76 (m, 3 H), 3.40-3.49 (m, 2 H), 3.37-3.39 (m, 4 H), 3.18 (m, 2H).
Example 2. Preparation of Form I of the compound of Formula I
[0258] Crude compound of Formula I (~30 g, 75% of weight based assay) was dissolved in ethyl acetate (3 L) at 55-65 °C under nitrogen. The resulting solution was filtered via silica gel pad and washed with ethyl acetate (3 L><2) at 55-65 °C. The filtrate was concentrated via vacuum at 30-40 °C to ~2.4 L. The mixture was heated up to 75-85 °C and maintained about 1 hour.
Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and the mixture was then cooled down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with ethyl acetate (60 mL><2). The wet cake was dried via vacuum at 30-40 °C to get (about 16 g) of the purified Form I of the compound of Formula I.
Example 3. Preparation of Form III of the compound of Formula I
[0259] The compound of Formula I (2 g) was dissolved in EtOH (40 mL) at 75-85 °C under nitrogen. n-Heptane (40 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 75-85 °C for 1 hour. Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with EtOH/n-Heptane (1/1, 5 mL><2). The wet cake was dried via vacuum at 30-40 °C to get the purified Form III of the compound of Formula I (1.7 g).
Example 4. Preparation of Form IV of the compound of Formula I The crude compound of Formula I (15 g) was dissolved in ethyl acetate (600 mL) at 75-85 °C under nitrogen and treated with anhydrous Na2S04, activated carbon, silica metal scavenger for 1 hour. The resulting mixture was filtered via neutral Al203 and washed with ethyl acetate (300 mL><2) at 75-85 °C. The filtrate was concentrated under vacuum at 30-40 °C and swapped with DCM (150 mL). n-Heptane (75 mL) was added into this DCM solution at 35-45 °C, and then the mixture was cooled down to 20-30 °C slowly. The resulting mixture was filtered and washed with DCM/n-Heptane (2/1, 10 mL><3). The wet cake was dried via vacuum at 35-40 °C to get the purified Form IV of the compound of Formula I (9.6 g).
Example 5. Preparation of Form V of the compound of Formula I
[0260] Polymorph Form III of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form V.
Example 6. Preparation of Form VI of the compound of Formula I
[0261] The compound of Formula I (1 g) was dissolved in IPA (20 mL) at 75-85 °C under nitrogen. n-Heptane (20 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 45-55 °C for 16 hours. Then heated up to 75-85 °C and maintained about 0.5 hour.
Then cooled down to 45-55 °C for 0.5 hour and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. Filtered and washed with IPA/n-Heptane (1/1, 3 mL><2). The wet cake was dried via vacuum at 75-80 °C for 2 hours to get the purified Form VI of the compound of Formula I.
Example 7. Preparation of Form VIII of the compound of Formula I
[0262] The polymorph Form VI of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form VIII.
Example 8. X-ray powder diffraction (XRD)
[0263] X-ray powder diffraction (XRD) patterns were obtained on a Bruker D8 Advance. A CuK source (=1.54056 angstrom) operating minimally at 40 kV and 40 mA scans each sample between 4 and 40 degrees 2-theta. The step size is 0.05°C and scan speed is 0.5 second per step.
Example 9. Thermogravimetric Analyses (TGA)
[0264] Thermogravimetric analyses were carried out on a TA Instrument TGA unit (Model TGA 500). Samples were heated in platinum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 60mL/min (sample purge) and 40mL/min (balance purge). The TGA temperature was calibrated with nickel standard, MP=354.4 °C. The weight calibration was performed with manufacturer-supplied standards and verified against sodium citrate dihydrate desolvation.
Example 10. Differential scanning calorimetry (DSC)
[0265] Differential scanning calorimetry analyses were carried out on a TA Instrument DSC unit (Model DSC 1000 or 2000). Samples were heated in non-hermetic aluminum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 50mL/min. The DSC temperature was calibrated with indium standard, onset of l56-l58°C, enthalpy of 25-29J/g.
Example 11. Hygroscopicity (DVS)
[0266] The moisture sorption profile was generated at 25°C using a DVS Moisture Balance Flow System (Model Advantage) with the following conditions: sample size approximately 5 to 10 mg, drying 25°C for 60 minutes, adsorption range 0% to 95% RH, desorption range 95% to 0% RH, and step interval 5%. The equilibrium criterion was <0.01% weight change in 5 minutes for a maximum of 120 minutes.
Example 12: Microscopy
[0267] Microscopy was performed using a Leica DMLP polarized light microscope equipped with 2.5X, 10X and 20X objectives and a digital camera to capture images showing particle shape, size, and crystallinity. Crossed polars were used to show birefringence and crystal habit for the samples dispersed in immersion oil.
Example 13: HPLC
[0256] HPLCs were preformed using the following instrument and/or conditions.
///////////////CK-101 , CK 101 , CK101 , phase II , Suzhou Neupharma, Checkpoint Therapeutics , Orphan Drug designation, EGFR mutation-positive NSCLC, NSCLC, CANCER, SOLID TUMOUR, China, RX-518, AK543910
OCCN1CCN(CC1)c5ccc(Nc2nc3c(cccc3cn2)c4cccc(NC(=O)C=C)c4)c(F)c5F
SY-008


![Acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91758795&t=l)
SY-008
CAS 1878218-66-6
FREE FORM 1480443-32-0
SGLT1 inhibitor (type 2 diabetes),
β-D-Glucopyranoside, 4-[[4-[(1E)-4-(2,9-diazaspiro[5.5]undec-2-yl)-1-buten-1-yl]-2-methylphenyl]methyl]-5-(1-methylethyl)-1H-pyrazol-3-yl, acetate (1:1)
acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol
MF H50 N4 O6 . C2 H4 O2
MW 58.8 g/mol,C35H54N4O8
Originator Eli Lilly
- Developer Eli Lilly; Yabao Pharmaceutical Group
- Class Antihyperglycaemics; Small molecules
- Mechanism of Action Sodium-glucose transporter 1 inhibitors
- Phase I Diabetes mellitus
- 28 Aug 2018 No recent reports of development identified for phase-I development in Diabetes-mellitus in Singapore (PO)
- 24 Jun 2018 Biomarkers information updated
- 12 Mar 2018 Phase-I clinical trials in Diabetes mellitus (In volunteers) in China (PO) (NCT03462589)
-
Eli Lilly is developing SY 008, a sodium glucose transporter 1 (SGLT1) inhibitor, for the treatment of diabetes mellitus. The approach of inhibiting SGLT1 could be promising because it acts independently of the beta cell and could be effective in both early and advanced stages of diabetes. Reducing both glucose and insulin may improve the metabolic state and potentially the health of beta cells, without causing weight gain or hypoglycaemia. Clinical development is underway in Singapore and China.
As at August 2018, no recent reports of development had been identified for phase-I development in Diabetes-mellitus in Singapore (PO).
Suzhou Yabao , under license from Eli Lilly , is developing SY-008 , an SGLT1 inhibitor, for the potential oral capsule treatment of type 2 diabetes in China. By April 2019, a phase Ia trial was completed
PATENT
WO 2013169546
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 201 1 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLTl is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLTl may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLTl inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 201 1/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides certain novel inhibitors of SGLTl which may be suitable for the treatment of diabetes.
Accordingly, the present invention provides a compound of Formula II:
Preparation 1
Synthesis of (4-bromo-2-methyl-phenyl)methanol.
Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). 1H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)methanol.
Borane-dimethyl sulfide complex (2M in THF; 1 16 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.1 13 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
Synthesis of 4-bromo- l-2-methyl-benzene.
Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4-bromo-2-methyl-phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and
-Cl-
dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. XH NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo- 1 -chloromethyl-2-methyl-benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). XH NMR (300.1 1 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
Synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol.
Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo- l-chloromethyl-2-methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction.
Extract with ethyl acetate (200 mL), wash extract with water (200 rnL) and brine (200 mL), dry over a2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l).
Alternative synthesis of 4-r(4-bromo-2-methyl-phenyl)methyl1-5-isopropyl- !H-pyrazol- 3-oL
A solution of 4-bromo- 1 -chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (1 1.94 g, 71.94 mmol) and methyl 4-methyl-3-oxo valerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2O5 at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/31 1 (M+l).
Preparation 4
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (ml 2): 889.2 (M+l), 887.2 (M-l).
Preparation 5
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
Synthesis of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2-dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+1).
Preparation 8
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D- glucopyranoside dihydrochloride.
Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3is)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
Example 1
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40 °C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 um C18XBridge ODB column, solvent A – 1¾0 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Preparation 9
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-acetyl-beta-D-glucopyranoside.
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammomum chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate
(32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l).
Preparation 10
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol). MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0- acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2-dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
Synthesis oftert-butyl 2-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,8- diazaspiro[4.5]decane-8-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 852.8, 853.6 (M+l), 850.8, 851.6 (M-l).
Preparation 14
Synthesis oftert-butyl 9-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-3,9- diazaspiro[5.5]undecane-3-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 866.8, 867.6 (M+l), 864.8, 865.6 (M-l).
Preparation 15
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D- glucopyranoside dihydrochloride.
Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Preparation 16
Synthesis of 4-{4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5- (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
dihydrochloride.
The title compound is prepared essentially by the method of Preparation 15. MS (m/z): 752.8, 753.8 (M+1), 750.8 (M-1).
First alternative synthesis of Example 1
First alternative synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en- 2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 urn C18XBridge ODB column, solvent A – H20 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+1), 596.8 (M-1). 1H MR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=1.3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.1 1 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Example 2
Synthesis of 4- {4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbi
(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
O H
The title compound is prepared essentially by the method of the first alternative synthesis of Example 1. MS (m/z): 584.7 (M+l), 582.8 (M-l).
Example 3
Synthesis of 4- {4-[( 1 E)-4-(3 ,9-diazaspiro[5.5]undec-3 -yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
The title compound is prepared essentially by first treating the compound of Prearation 14 with HC1 as discussed in Preparation 15 then treating the resulting hydrochloride salt with triethyl amine as discussed in the first alternative synthesis of Example 1. MS (m/z): 598.8, 599.8 (M+l), 596.8, 597.8 (M-l).
Example 1 Preparation 17
Synthesis of tert-butyl 4-but-3- nyl-4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgSC^, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). iH MR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H),
2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 18
Synthesis of tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]- 4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5, 5-tetramethyl-1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield), !H NMR (300.1 1 MHz, CDC13): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H),
3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 19
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2-dicyclohexylphosphino-2′,4′,6′-tri-i-propyl-l, r-biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).
Second alternative Synthesis of Example 1
Second alternative synthesis of 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200ml) and concentrated in vacuo. This is repeated several times give the title compound (12.22 g, yield 96%). MS (m/z): 599 (M+l). [a]D20 = -12 ° (C=0.2, MeOH).
PATENT
WO 2015069541
https://patents.google.com/patent/WO2015069541A1
4-{4-[(1 E)-4-(2,9-DIAZASPIRO[5.5]UNDEC-2-YL)BUT-1 -EN-1
-YL]-2-METHYLBENZYL}-5-(PROPAN-2-YL)-1 H-PYRAZOL-3-YL
BETA-D- GLUCOPYRANOSIDE ACETATE
The present invention relates to a novel SGLT1 inhibitor which is an acetate salt of a pyrazole compound, to pharmaceutical compositions comprising the compound, to methods of using the compound to treat physiological disorders, and to intermediates and processes useful in the synthesis of the compound.
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 2011 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLT1 is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLT1 inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 2011/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides an acetate salt of a pyrazole compound, which is an SGLT1 inhibitor, and as such, may be suitable for the treatment of certain disorders, such as diabetes. Accordingly, the present invention provides a compound of Formula I:

or hydrate thereof.
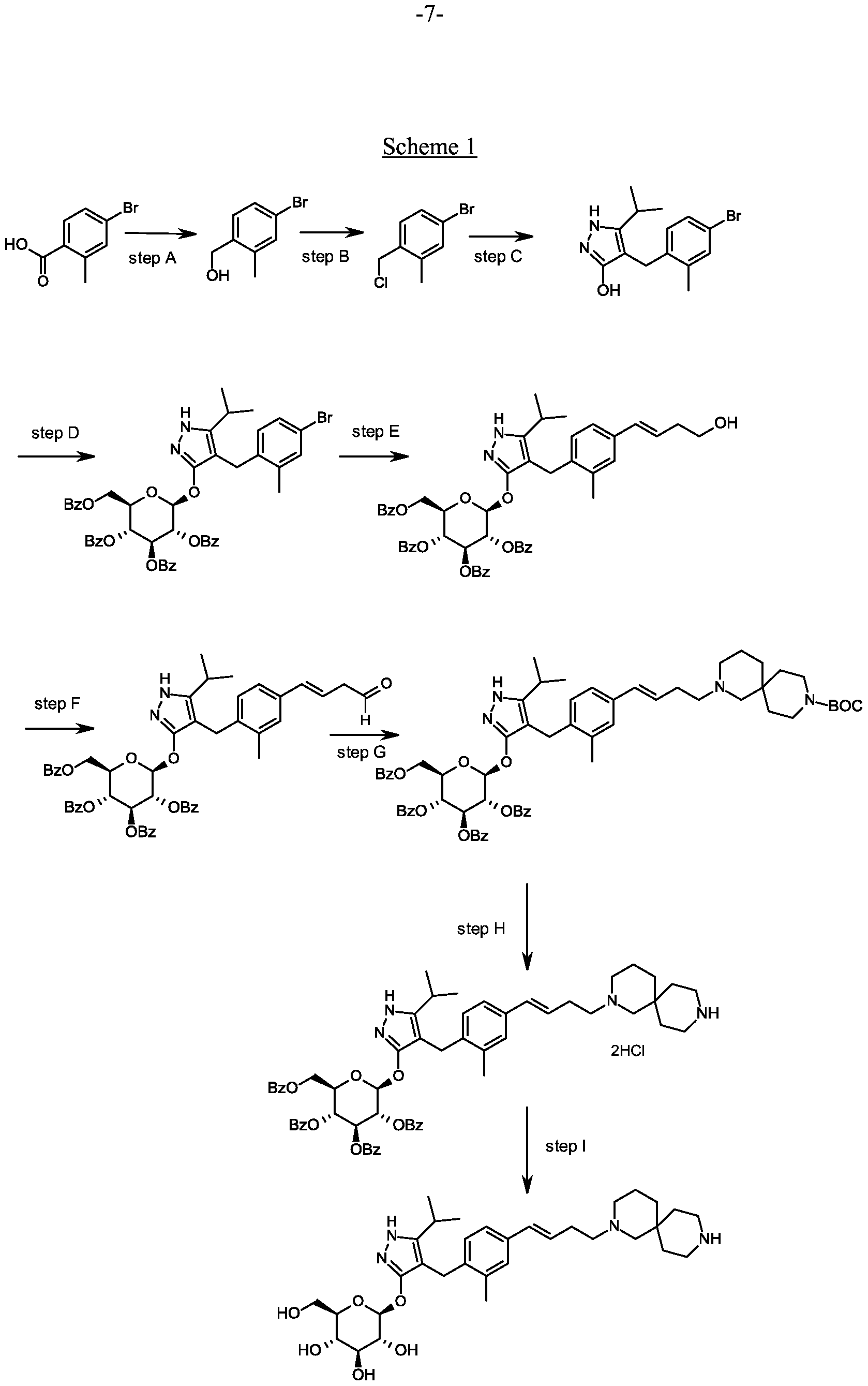
Preparation 1
(4-bromo-2-methyl-phenyl)methanol

Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). !H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)mefhanol.
Borane-dimethyl sulfide complex (2M in THF; 116 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.113 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
4-bromo- 1 -chloromethyl -2 -methyl -benzene

Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4- bromo-2 -methyl -phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. !H NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo-l-chloromethyl-2-methyl -benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). !H NMR (300.11 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
4- [(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-3 -ol

Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo-l-chloromethyl-2- methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction. Extract with ethyl acetate (200 mL), wash extract with water (200 mL) and brine (200 mL), dry over Na2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l). Alternative synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-
3-ol.
A solution of 4-bromo-l-chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (11.94 g, 71.94 mmol) and methyl 4-methyl-3-oxovalerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2Os at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/311 (M+l).
Preparation 4
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl- beta-D-glucopyranoside
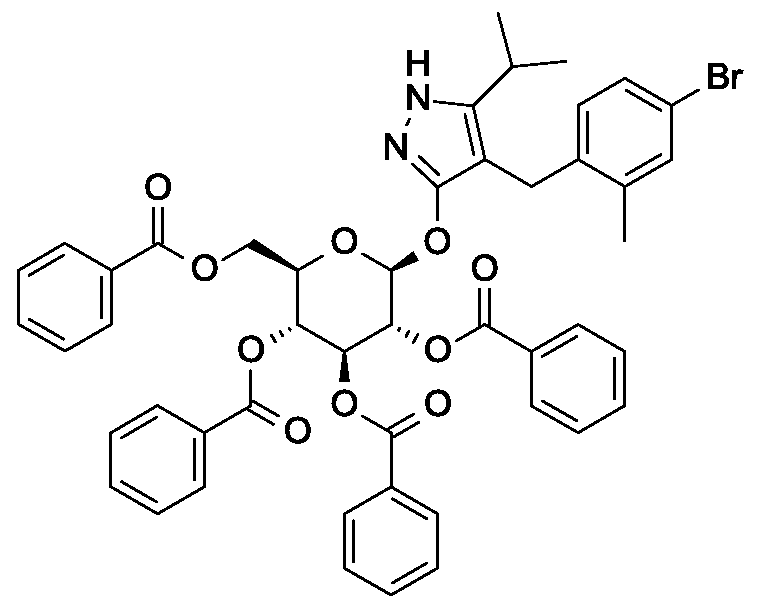
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (m/z): 889.2 (M+l), 887.2 (M-l).
Preparation 5
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- lH-pyrazol-3 -yl 2,3 ,4,6-tetra-O-benzoyl-beta-D- glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
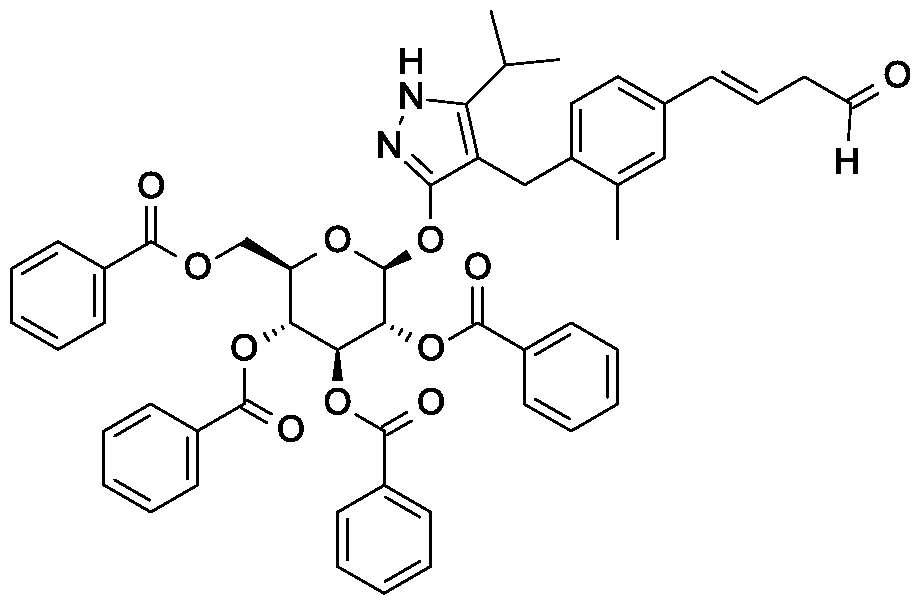
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate

Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2- dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+l).
Preparation 8
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride

Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
Preparation 9
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl- beta-D-glucopyranoside.
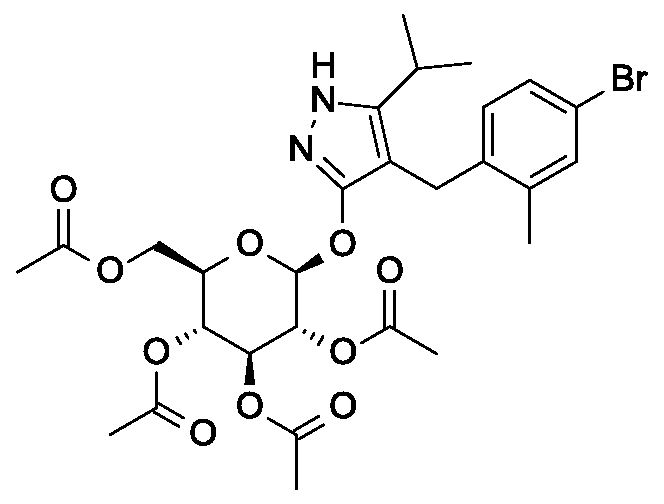
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)mefhyl]-5- isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D- glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate (32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l). Preparation 10
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
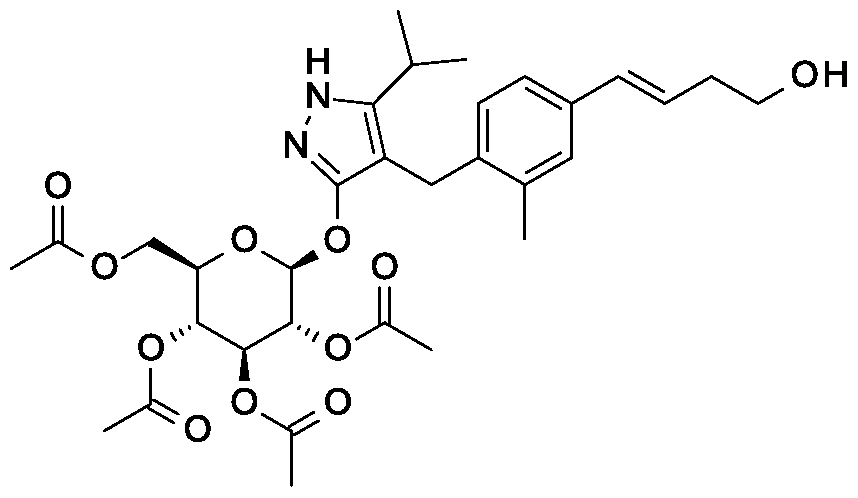
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- 1 H-pyrazol-3 -yl 2,3 ,4,6-tetra-O-acetyl-beta-D- glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol) MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside

Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12a
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy] -lH-pyrazol-4-yl}methyl)phenyl]but-3-en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate
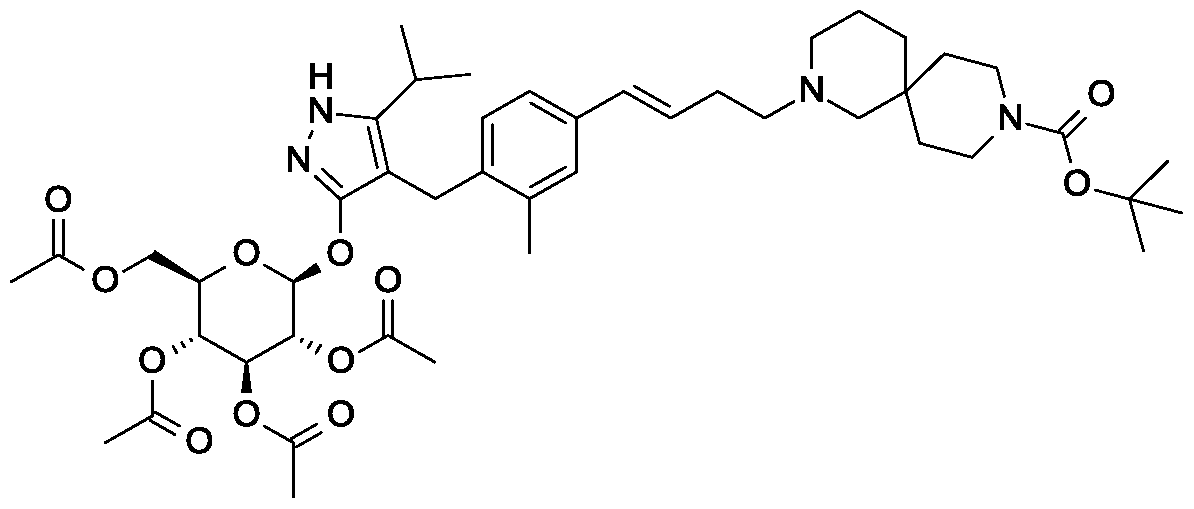
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2- dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride

Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6- tetra-0-acetyl-beta-D-glucopyranosyl)oxy] – lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 – yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Scheme 3
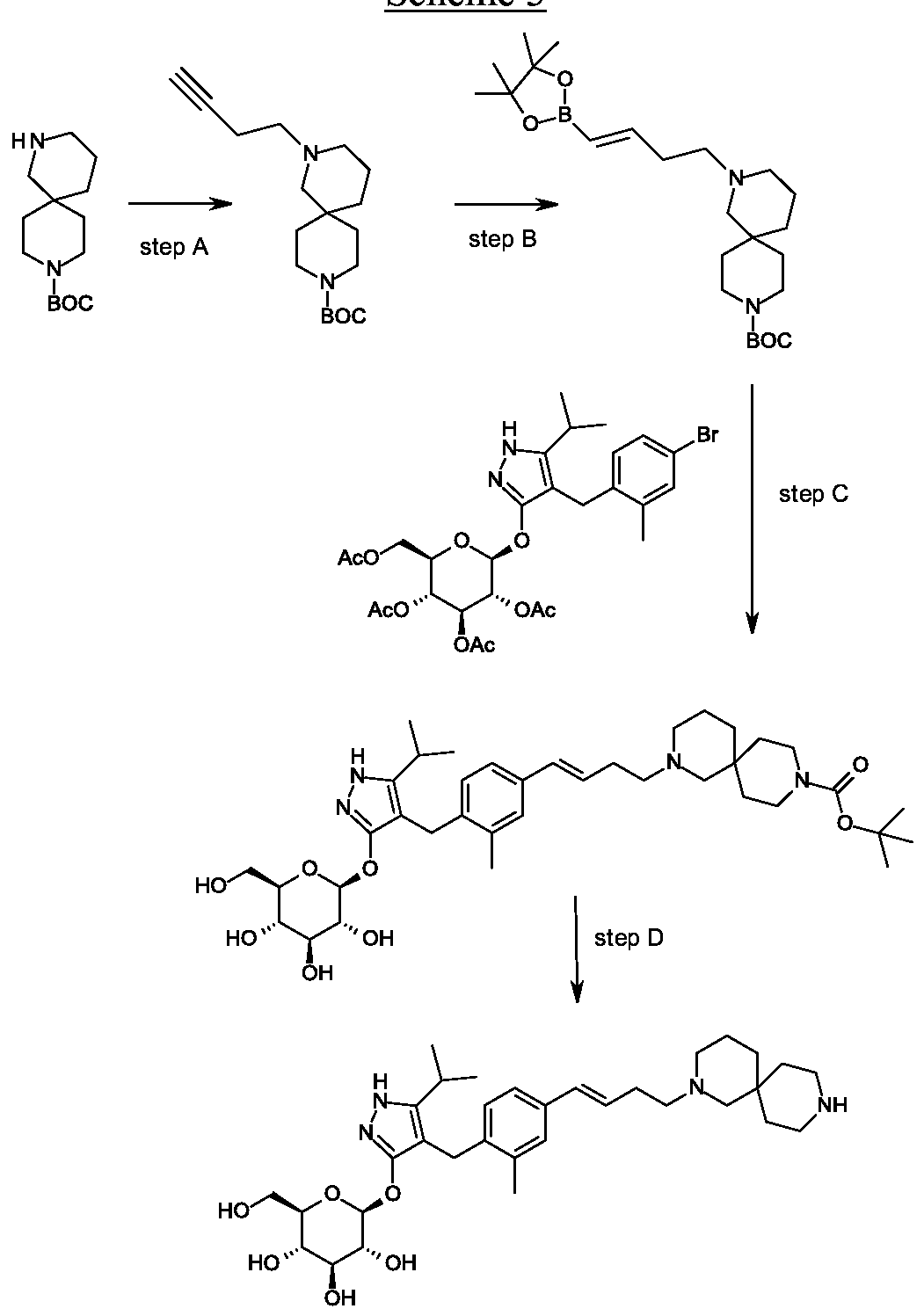
Preparation 14
tert-butyl 4-but-3-ynyl-4,9-diazas iro[5.5]undecane-9-carboxylate

Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgS04, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). lH NMR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H), 2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 15
tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9- diazaspiro[5.5]undecane-9-carboxylate
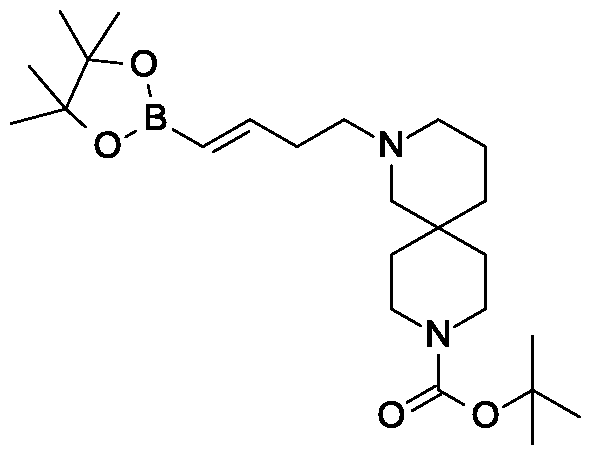
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield). 1H NMR (300.11 MHz, CDCI3): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H), 3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 16
tert-butyl 2-{(3£’)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH- pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9-diazaspiro [5.5]undecane-9-carboxylate

Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert- butyl 4-[(£)-4-(4,4,5 ,5 -tetramethyl- 1 ,3,2-dioxaborolan-2-yl)but-3 -enyl] -4,9- diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2- dicyclohexylphosphino-2′,4′,6′-tri-i -propyl- Ι, -biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).

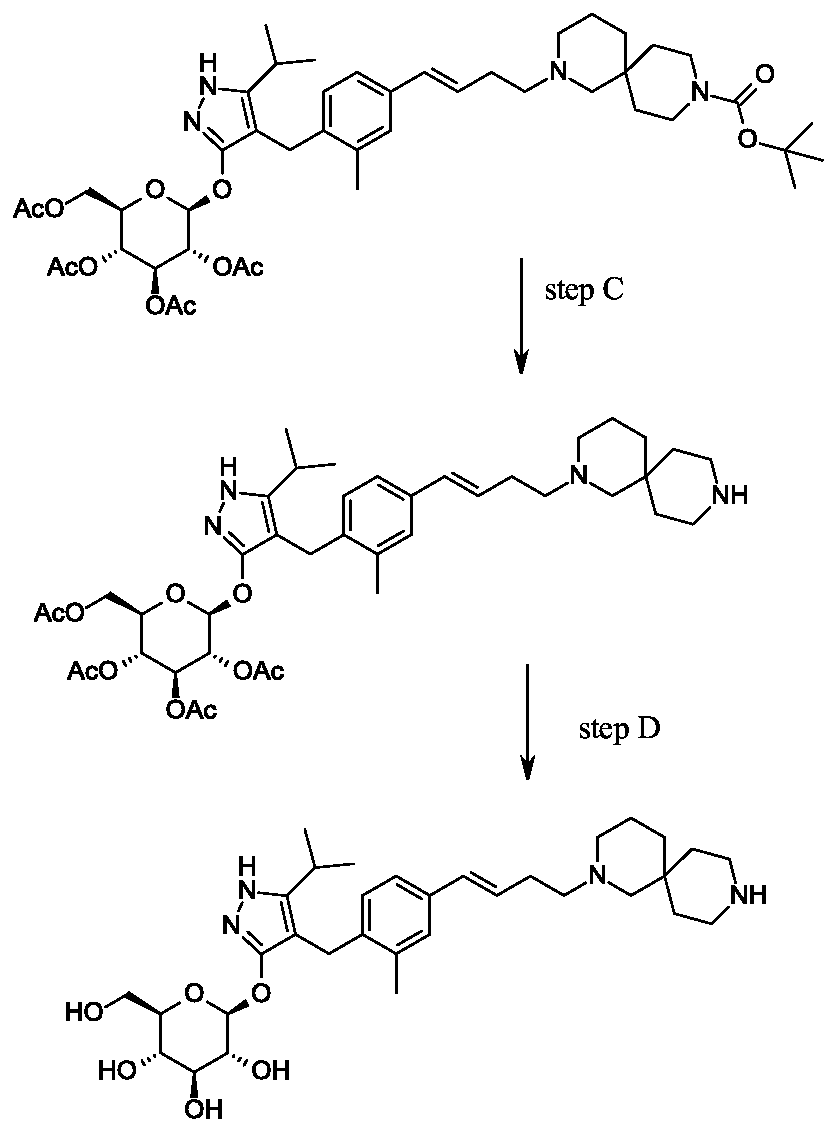
Preparation 17
tert-butyl 4- [(E)-4- [4- [(3 -hydroxy-5-isopropyl- 1 H-pyrazol-4-yl)methyl] -3 -methyl- phenyl]but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate

Scheme 4, step A: Add tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (35.8 kg, 82.4 mol) in methanol (130 L) to a solution of (4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (23.9 kg, 77.3 mol) in methanol (440 L) at room temperature. Add water (590 L) and tripotassium phosphate (100 kg, 471.7 mol) and place the reaction under nitrogen atmosphere. To the stirring solution, add a suspension of
tris(dibenzylideneacetone) dipalladium (1.42 kg, 1.55 mol) and di-tert- butylmethylphosphonium tetrafluoroborate (775 g, 3.12 mol) in methanol (15 L). The resulting mixture is heated at 75 °C for 2 hours. Cool the mixture and filter over diatomaceous earth. Rinse the the filter cake with methanol (60 L), and concentrate the filtrate under reduced pressure. Add ethyl acetate (300 L), separate the layers, and wash the organic layer with 15% brine (3 x 120 L). Concentrate the organic layer under reduced pressure, add ethyl acetate (300 L), and stir the mixture for 18 to 20 hours. Add heptane (300 L), cool the mixture to 10 °C, and stir the mixture for an additional 18 to 20 hours. Collect the resulting solids by filtration, rinse the cake with ethyl acetate/heptane (2:3, 2 x 90 L), and dry under vacuum at 40°C to give the title compound (29.3 kg, 70.6% yield) as a white solid. lH NMR (400 MHz, CD3OD): δ 7.14 (s, 1H), 7.07 (d, J= 8.0 Hz, 1H), 6.92 (d, J= 7.6 Hz, 1H), 6.39 (d, J= 16.0 Hz, 1H), 6.25-6.12 (m, 1H), 3.63 (s, 2H), 3.45-3.38 (bs, 3H), 3.34 (s, 3 H), 3.33 (s, 3H), 2.85-2.75 (m, 1H), 2.49-2.40 (m, 5 H), 2.33 (s, 3H), 1.68-1.62 (m, 2H), 1.60-1.36 (m, 15H), 1.11 (s, 3H), 1.10 (s, 3H).
Preparation 12b
Alterternative preparation of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but- 3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate.

Scheme 4, step B: Combine tert-butyl 4-[(E)-4-[4-[(3-hydroxy-5-isopropyl-lH- pyrazol-4-yl)methyl] -3-methyl-phenyl]but-3 -enyl] -4,9-diazaspiro [5.5]undecane-9- carboxylate (17.83 kg, 33.2 moles), acetonitrile (180 L), and benzyltributylammonium chloride (1.52 kg, 4.87 moles) at room temperature. Slowly add potassium carbonate (27.6 kg, 199.7 moles) and stir the mixture for 2 hours. Add 2,3,4,6-tetra-O-acetyl-alpha- D-glucopyranosyl bromide (24.9 kg, 60.55 mol), warm the reaction mixture to 30°C and stir for 18 hours. Concentrate the mixture under reduced pressure and add ethyl acetate (180 L), followed by water (90 L). Separate the layers, wash the organic phase with 15% brine (3 x 90 L), concentrate the mixture, and purify using column chromatography over silica gel (63 kg, ethyl acetate/heptanes as eluent (1 :2→1 :0)) to provide the title compound (19.8 kg, 94% purity, 68.8% yield) as a yellow foam, !H NMR (400 MHz, CDC13): δ 7.13 (s, 1H), 7.03 (d, J= 8.0 Hz, 1H), 6.78 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 16.0,
1H), 6.25-6.13 (m, 1H), 5.64 (d, J= 8.0 Hz, 1H), 5.45-5.25 (m, 2H), 5.13-4.95 (m, 2H), 4.84-4.76 (m, 1H), 4.25-4.13 (m, 2H), 4.10-4.00 (m, 2H), 3.90-3.86 (m, 1H), 3.58-3.50 (m, 2H), 3.40-3.22 (m, 4H), 2.89-2.79 (m, 1H), 2.10-1.90 (m, 18 H), 1.82 (s, 3H), 1.62- 0.82 (m, 22H).
Preparation 18
2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane

Scheme 4, step C: Combine tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)- 3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (19.6 kg, 22.6 moles) with dichloromethane (120 L) and cool to 0°C. Slowly add trifluoroacetic acid (34.6 L, 51.6 kg, 452 moles) and stir for 9 hours. Quench the reaction with ice water (80 L), and add ammonium hydroxide (85-90 L) to adjust the reaction mixture to pH (8- 9). Add dichloromethane (120 L), warm the reaction mixture to room temperature, and separate the layers. Wash the organic layer with water (75 L), brine, and concentrate under reduced pressure to provide the title compound (16.2 kg, 95.0% purity, 93% yield) as a yellow solid. lH NMR (400 MHz, CDC13): δ 7.08 (s, IH), 6.99 (d, J= 8.0 Hz, IH),
6.76 (d, J= 7.6 Hz, IH), 6.38 (d, J=15.6 Hz, IH), 6.00-5.83 (m, IH), 5.31 (d, J= 7.6 Hz, IH), 5.25-5.13 (m, 4H), 4.32 (dd, J= 12.8, 9.2 Hz, IH), 4.14 (d, J= 11.2 Hz, IH), 3.90 (d, J= 10.0 Hz, IH), 3.75-3.50 (m, 3H), 3.30-3.00 (m, 5 H), 2.85-2.75 (m, IH), 2.70-2.48 (m, 3H), 2.25 (s, IH), 2.13-1.63 (m, 19H), 1.32-1.21 (m, IH), 1.14 (s, 3H), 1.13 (s, 3H), 1.12 (s, 3H), 1.10 (s, 3H).
Example 1
Hydrated crystalline 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside acetate
First alternative preparation of 4-{4-[(l£’)-4-(2.9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl| -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (free base).
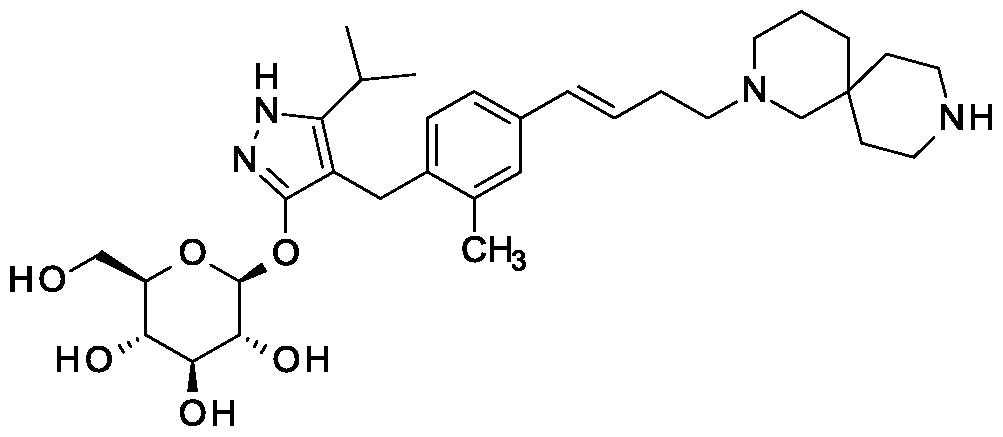
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4- {4-[( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} – 5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40°C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H.0 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Second alternative preparation of 4-{4-r(l-£’)-4-(2.9-diazaspiror5.51undec-2-yl)but-l-en- 1 -yl] -2-methylbenzyl I -5 -(propan-2-yl)- lH-pyrazol-3 -yl beta-D-glucopyranoside (free base“).

Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4- {4-[( lJE)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H20 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+l), 596.8 (M-l). 1H NMR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=l .3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.11 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Third alternative preparation of 4-{4-[(l£,)-4-(2,9-diazaspiro[5.51undec-2-yl)but-l-en-l- yll-2-methylbenzyl|-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200mL) and concentrated in vacuo. This is repeated several times to give the title compound (free base) (12.22 g, yield 96%). MS (m/z): 599 (M+l); [a]D 20 = -12 ° (C=0.2, MeOH).
Preparation of final title compound, hydrated crystalline 4-{4-|YlE)-4-(2.9- diazaspiro [5.5“|undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-vD- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.

4- {4- [(1 E)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (902 mg) is placed in a round bottom flask (100 mL) and treated with wet ethyl acetate (18 mL). [Note – wet ethyl acetate is prepared by mixing ethyl acetate (100 mL) and dionized water (100 mL). After mixing, the layers are allowed to separate, and the top wet ethyl acetate layer is removed for use. Acetic acid is a hydrolysis product of ethyl acetate and is present in wet ethyl acetate.] The compound dissolves, although not completely as wet ethyl acetate is added. After several minutes, a white precipitate forms. An additional amount of wet ethyl acetate (2 mL) is added to dissolve remaining compound. The solution is allowed to stir uncovered overnight at room temperature during which time the solvent partially evaporates. The remaining solvent from the product slurry is removed under vacuum, and the resulting solid is dried under a stream of nitrogen to provide the final title compound as a crystalline solid. A small amount of amorphous material is identified in the product by solid-state NMR. This crystalline final title compound may be used as seed crystals to prepare additional crystalline final title compound.
Alternative preparation of final title compound, hvdrated crystalline 4-{4-[(lE)-4-(2.,9- diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-yl)- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.
Under a nitrogen atmosphere combine of 4-{4-[(lE)-4-(2,9-diazaspiro[5.5]undec- 2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan-2-yl)- 1 H-pyrazol-3-yl 2,3,4,6-tetra-O- acetyl-beta-D-glucopyranoside (2.1 kg, 2.74 mol), methanol (4.4 L), tetrahydrofuran (4.2 L), and water (210 mL). Add potassium carbonate (460 g, 3.33 moles) and stir for four to six hours, then filter the reaction mixture to remove the solids. Concentrate the filtrate under reduced pressure, then add ethanol (9.0 L) followed by acetic acid (237 mL, 4.13 mol) and stir at room temperature for one hour. To the stirring solution add wet ethyl acetate (10 L, containing approx. 3 w/w% water) slowly over five hours, followed by water (500 mL). Stir the suspension for twelve hours and add wet ethyl acetate (4.95 L, containing approx. 3 w/w% water) over a period of eight hours. Stir the suspension for twelve hours and add additional wet ethyl acetate (11.5 L, containing approx. 3 w/w% water) slowly over sixteen hours. Stir the suspension for twelve hours, collect the solids by filtration and rinse the solids with wet ethyl acetate (3.3 L, containing approx. 3 w/w% water). Dry in an oven under reduced pressure below 30°C to give the title compound as an off-white crystalline solid (1.55 kg, 2.35 mol, 96.7% purity, 72.4 w/w% potency, 68.0% yield based on potency). HRMS (m/z): 599.3798 (M+l).
PATENT
CN105705509
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN175101669&tab=PCTDESCRIPTION
The present invention is in the field of treatment of diabetes and other diseases and conditions associated with hyperglycemia. Diabetes is a group of diseases characterized by high blood sugar levels. It affects approximately 25 million people in the United States, and according to the 2011 National Diabetes Bulletin, it is also the seventh leading cause of death in the United States (US Department of Health and Human Resources Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the uptake of carbohydrates such as glucose. More specifically, SGLT1 is responsible for transporting glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 can result in a decrease in glucose absorption in the small intestine, thus providing a useful method of treating diabetes.
Alternative medicines and treatments for diabetes are needed. The present invention provides an acetate salt of a pyrazole compound which is an SGLT1 inhibitor, and thus it is suitable for treating certain conditions such as diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives having human SGLT1 inhibitory activity, which are also disclosed for use in the prevention or treatment of diseases associated with hyperglycemia, such as diabetes. Moreover, WO 2011/039338 discloses certain pyrazole derivatives having SGLT1/SGLT2 inhibitor activity, which are also disclosed for use in the treatment of bone diseases such as osteoporosis.
PATENT
WO-2019141209
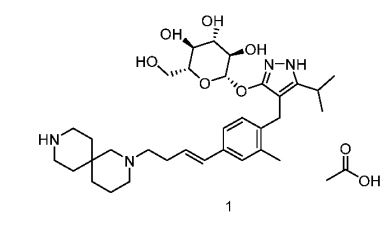
Process for preparing pyranoglucose-substituted pyrazole compound, used as a pharmaceutical intermediate in SGLT inhibitor for treating diabetes.

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9573970 | 4–5-(PROPAN-2-YL)-1H-PYRAZOL-3-YL BETA-D GLUCOPYRANOSIDE ACETATE | 2014-10-30 | 2016-07-28 |
/////////////SY-008 , SY 008 , SY008, ELI LILY, PHASE 1, GLT1 inhibitor, type 2 diabetes, Yabao Pharmaceutical, CHINA, DIABETES
CC(=O)O.Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4O
Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4
O
Fluazolepali, 氟唑帕利 , Fluzoparib

Fluazolepali
CAS 2170504-09-1
Fluzoparib; SHR-3162, (HS10160)
- HS 10160
- SHR 3162
An orally available inhibitor of poly(ADP-ribose) polymerase 1 and 2 (PARP-1/2) for treatment of solid tumors (Jiangsu Hengrui Medicine Co. Ltd., Lianyungang, China)
Fluazolepali, developed by Hengrui and Howson, is intended for the treatment of recurrent ovarian cancer, triple-negative breast cancer, advanced gastric cancer and other advanced solid tumors. Currently, the drug has been introduced into China for recurrent ovarian cancer. Clinical stage.
In February 2019, a randomized, double-blind, controlled, multicenter, phase III clinical study (CTR20190294) of flazopril capsule versus placebo for maintenance of recurrent ovarian cancer was initiated in China and was sponsored by Hengrui Medicine.
Jiangsu Hansoh Pharmaceutical , in collaboration with Jiangsu Hengrui Medicine , is developing an oral capsule formulation of fluazolepali (fluzoparib; SHR-3162), a small molecule inhibitor to PARP-1 and PARP-2, for the treatment of solid tumors including epithelial ovarian, fallopian tube or primary peritoneal, breast and gastric cancer.
- Originator Jiangsu Hengrui Medicine Co.
- Class Antineoplastics
- Mechanism of Action Poly(ADP-ribose) polymerase 1 inhibitors; Poly(ADP-ribose) polymerase 2 inhibitors
- Phase II Ovarian cancer
- Phase I Breast cancer; Fallopian tube cancer; Gastric cancer; Peritoneal cancer; Solid tumours
- 09 Jul 2019 Jiangsu HengRui Medicine initiates a phase I trial in Solid tumors in China (NCT04013048) [14C]-Fluzoparib
- 01 Jul 2019 Jiangsu HengRui Medicine plans a phase I drug-drug interaction trial (In volunteers) in China (PO) (NCT04011124)
- 12 Jun 2019 Jiangsu HengRui Medicine completes a phase I trial in Gastric cancer (Combination therapy, Recurrent, Metastatic disease, Second-line therapy or greater, Late-stage disease) in China (PO) (NCT03026881)
Fluzoparib (SHR 3162) is a selective poly [ADP-ribose] polymerase 1 (PARP1) and poly [ADP-ribose] polymerase 2 inhibitor (PARP2), being developed by Jiangsu HengRui Medicine, for the treatment of cancer. PARP enzymes play a vital role in repair of DNA damage and maintaining genomic stability. Fluzoparib inhibits PARP enzymes and induces DNA-double strands breaks, G2/M arrest and apoptosis in homologous recombination repair (HR)-deficient cells. Clinical development for ovarian cancer, breast cancer, fallopian tube cancer, peritoneal cancer, gastric cancer and solid tumours is underway in China and Australia.
An orally available inhibitor of poly (ADP-ribose) polymerase (PARP) types 1 and 2, with potential antineoplastic activity. Upon oral administration, fluzoparib inhibits PARP 1 and 2 activity, which inhibits PARP-mediated repair of damaged DNA via the base excision repair (BER) pathway, enhances the accumulation of DNA strand breaks, promotes genomic instability, and leads to an induction of apoptosis. The PARP family of proteins catalyze post-translational ADP-ribosylation of nuclear proteins, which then transduce signals to recruit other proteins to repair damaged DNA. PARP inhibition may enhance the cytotoxicity of DNA-damaging agents and may reverse tumor cell chemoresistance and radioresistance. Check for active clinical trials using this agent. (NCI Thesaurus)
PATENT
WO-2019137358
Process for preparing heterocyclic compounds (presumed to be fluazolepali ) and its intermediates as PARP inhibitors useful for treating cancer.


PATENT
WO2019109938
claiming synergistic combination comprising PARP inhibitor fluazolepali and apatinib mesylate .
PATENT
WO 2018005818
WO 2018129553
WO 2018129559
WO 2018208968
WO 2018213732
WO 2018191277
WO 2018201096
WO 2018085469
WO 2018085468
WO 2019090227
WO 2019133697
WO 2019067978
WO 2019071123
WO 2019090141
///////////Fluazolepali, Jiangsu Hansoh Pharmaceutical, Jiangsu Hengrui Medicine, fluzoparib, SHR-3162, 氟唑帕利 , Phase II, Ovarian cancer, HS10160, CHINA, HS 10160
HS 10340

HS-10340
CAS 2156639-66-4

CAS 2307670-65-9
Jiangsu Hansoh Pharmaceutical Group Co Ltd
Being investigated by Jiangsu Hansoh, Shanghai Hansoh Biomedical and Changzhou Hengbang Pharmaceutical ; in June 2018, the product was being developed as a class 1 chemical drug in China.
Useful for treating liver cancer, gastric cancer and prostate cancer.
Use for treating cancers, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma and rhabdomyosarcoma


PATENT
WO2017198149
where it is claimed to be an FGFR-4 inhibitor for treating liver and prostate cancers, assigned to Jiangsu Hansoh Pharmaceutical Group Co Ltd and Shanghai Hansoh Biomedical Co Ltd .
PATENT
WO2019085860
PATENT
WO-2019085927
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019085927&tab=FULLTEXT
Novel crystalline salt (such as hydrochloride, sulfate, methane sulfonate, mesylate, besylate, ethanesulfonate, oxalate, maleate, p-toluenesulfonate) forms of FGFR4 inhibitor, particularly N-[5-cyano-4-[[(1R)-2-methoxy-1-methyl-ethyl]amino]-2-pyridyl]-7-formyl-6-[(2-oxo-1,3-oxazepan-3-yl)methyl]-3,4-dihydro-2H-1,8-naphthyridine-1-carboxamide (designated as Forms I- IX), compositions comprising them and their use as an FGFR4 inhibitor for the treatment of cancer such as liver cancer, gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and glioma or rhabdomyosarcoma are claimed.
///////////HS-10340 , HS 10340 , HS10340, CANCER, Jiangsu Hansoh, Shanghai Hansoh Biomedical, Changzhou Hengbang, CHINA, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma, rhabdomyosarcoma
C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CCS(=O)(=O)O.C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CS 3001
CS-3001
BB 7, VX 033
- Molecular Weight, 478.37
C17 H18 Br F2 N3 O2 S2
CStone Pharmaceuticals Co Ltd, JUNE 2018 IND FILED CHINA
URAT1 inhibitor – useful for treating hyperuricemia and gout.
The compound was originally claimed in WO2017202291 , covering thiophene derivative URAT1 inhibitors, useful for treating hyperuricemia and gouty arthritis, assigned to Medshine Discovery Inc , but naming the inventors.and has been reported in some instances to be a URAT1 modulator. In June 2018, an IND application was filed in



WO-2019101058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019101058&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of URAT1 inhibitor (designated as Forms A and B) are claimed. The compounds are disclosed to be useful for treating hyperuricemia and gouty arthritis.
Novel crystalline forms of a URAT1 inhibitor, designated as Forms A and B, and their preparation.
For example, remove 2.0 mL of phosphoric acid into 2000 mL of water, sonicate for 10 minutes, mix, and let cool to room temperature as mobile phase A.
////////////CS-3001, BB 7, VX 033, CHINA, PRECLINICAL, CStone Pharmaceuticals, URAT1 inhibitor, hyperuricemia, gout
O=C(O)C(C)(C)Sc4nnc(Br)n4c2sc(c1CC(F)(F)CCc12)C3CC3
TL 487
TL-487
- Molecular Weight, 507.58, MF C30 H29 N5 O3
Teligene Inc(2E)-N-[3-Cyano-7-ethoxy-4-[(4-phenoxyphenyl)amino]-6-quinolinyl]-4-(dimethylamino)-2-butenamide
(E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide
Maleate in anhydrous or monohydrate CAS, 2326561-36-6, AND 2326561-38-8 form are BTK and HER-2 kinase inhibitor useful for treating cancer
Useful for treating breast cancer, ovary cancer and colon cancer. are BTK and HER-2 kinase inhibitor useful for treating cancer.
Anticancer protein kinase inhibitor
The compound was originally claimed in WO2013152135 , and may provide the structure of TL-487 , a small molecule inhibitor to HERs, being investigated by Teligene for the treatment of breast cancer; in July 2016, the company intended to develop the product as a class 1.1 chemical drug in China.
PATENT
US 20150057312
PATENT
WO2013152135

PATENT
WO-2019096327
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019096327&redirectedID=true
Novel crystalline maleate salt of (E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide (first disclosed in WO2013152135) and its hydrates (monohydrate) and anhydrates, process for its preparation, composition comprising it and its use for treating cancers such as breast cancer, ovary cancer, colon cancer, prostate cancer, kidney cancer, bladder cancer, stomach cancer, lung cancer, mantle cell lymphoma and multiple myeloma are claimed. The compound is disclosed to be an irreversible inhibitor to BTK and Her-2 (also known as Erb-2 or neu).
///////////////TL-487, PRECLINICAL, CHINA, breast cancer, ovary cancer, olon cancer, BTK, HER-2 kinase inhibitor,
CN(C)C\C=C\C(=O)Nc3cc4c(Nc2ccc(Oc1ccccc1)cc2)c(cnc4cc3OCC)C#N
HAO 472
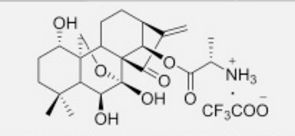
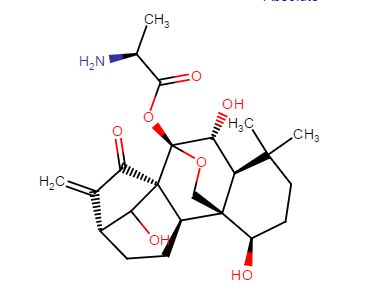 .CF3COOH
.CF3COOH
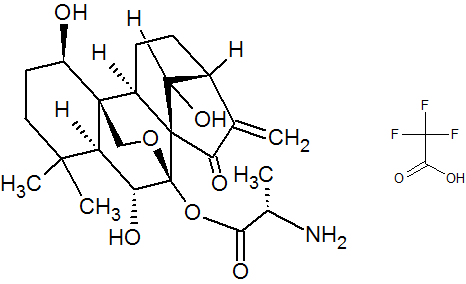
HAO 472
PHASE 1 CHINA



PRoject Name: HAO472 treatment Phase I clinical trial in relapsed / refractory AML, M2b type of AML
The main purpose: to determine HAO472 treatment of relapsed / refractory C the maximum tolerated dose (MTD). Secondary objectives: 1) evaluation of drug safety and tolerability; 2) study HAO472 in pharmacokinetic characteristics of the human body; 3) the effectiveness of HAO472 treatment of relapsed / refractory M2b type of AML.
Introduction Test
Acute myelogenous leukemia
HAO472
Phase I
Test Number: CTR20150246
Sponsor Name:
Jiangsu Hengrui Medicine Co., Ltd. 1/
2 Ruijin Hospital, Shanghai Jiaotong University School of Medicine /
3 Jiangsu Hengrui Medicine Co., Ltd. /
4 Shanghai Hengrui Medicine Co., Ltd. /

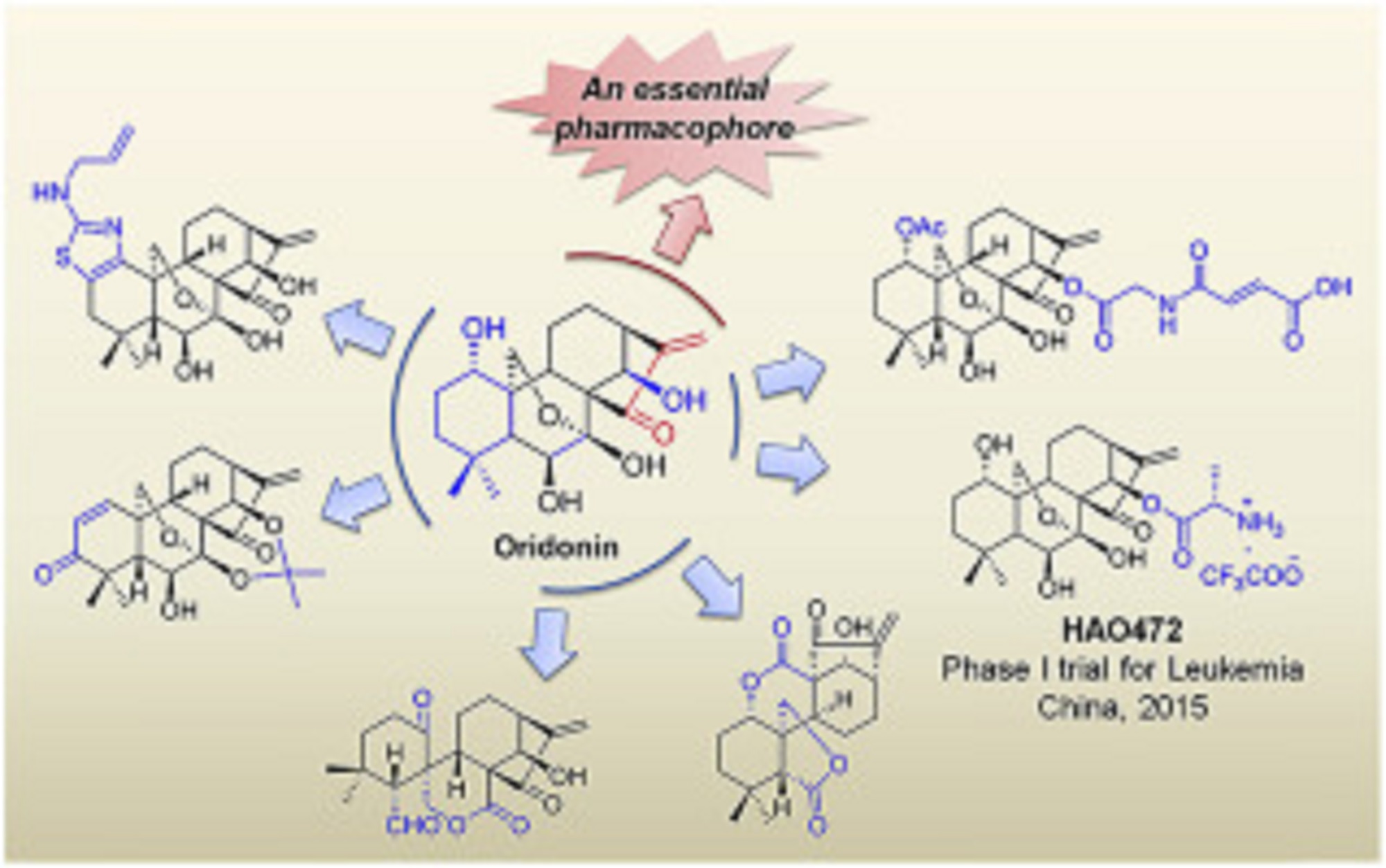
Natural products have historically been, and continue to be, an invaluable source for the discovery of various therapeutic agents. Oridonin, a natural diterpenoid widely applied in traditional Chinese medicines, exhibits a broad range of biological effects including anticancer and anti-inflammatory activities. To further improve its potency, aqueous solubility and bioavailability, the oridonin template serves as an exciting platform for drug discovery to yield better candidates with unique targets and enhanced drug properties. A number of oridonin derivatives (e.g. HAO472) have been designed and synthesized, and have contributed to substantial progress in the identification of new agents and relevant molecular mechanistic studies toward the treatment of human cancers and other diseases. This review summarizes the recent advances in medicinal chemistry on the explorations of novel oridonin analogues as potential anticancer therapeutics, and provides a detailed discussion of future directions for the development and progression of this class of molecules into the clinic.
Highlights
Oridonin displays significant anticancer activities via multi-signaling pathways.
Recent advances in medicinal chemistry of oridonin-like compounds are presented.
The article summarizes the SAR and mechanism studies of relevant drug candidates.
The milestones and future direction of oridonin-based drug discovery are discussed.
Volume 122, 21 October 2016, Pages 102–117

Discovery and development of natural product oridonin-inspired anticancer agents
- a Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, TX, 77555, United States
- b Department of Clinical Cancer Prevention, Division of Cancer Prevention and Population Sciences, The University of Texas MD Anderson Cancer Center, Houston, TX, 77030, United States

////////Natural product, Oridonin, Diterpenoids, Anticancer agents, Drug discovery, Chemical biology, AML, HAO 472, relapsed / refractory AML. Jiangsu Hengrui Medicine Co., Ltd, PHASE1, LEUKEMIA
C[C@H](N)C(=O)O[C@]15OC[C@@]2([C@H](O)CCC(C)(C)[C@@H]2[C@H]1O)[C@H]3CC[C@@H]4C(=C)C(=O)[C@@]35C4O

{kind=link}