

Chidamide (Epidaza)
CS055; HBI-8000
CAS 743438-44-0 CORRECT
C22 H19 F N4 O2, Benzamide, N-(2-amino-4-fluorophenyl)-4-[[[1-oxo-3-(3-pyridinyl)-2-propen-1-yl]amino]methyl]-
- Molecular Weight, 390.41
- Benzamide, N-(2-amino-4-fluorophenyl)-4-[[[1-oxo-3-(3-pyridinyl)-2-propenyl]amino]methyl]-
- N-(2-Amino-4-fluorophenyl)-4-[[[1-oxo-3-(3-pyridinyl)-2-propen-1-yl]amino]methyl]benzamide
- CS 055
- Chidamide
- Epidaza
Activity: HDAC Inhibitor; Cancer Drug; Histone Deacetylase Inhibitor; HDAC-1, 2,3,10 Inhibitor; Treatment for Peripheral T-cell Lymphomas; Treatment for PTCL
Status: Launched 2014 (China)
Originator: Shenzhen Chipscreen Biosciences Ltd
SHENZHEN CHIPSCREEN BIOSCIENCES LTD. [CN/CN]; Research Institute of Tsinghua University, Suite C301, P.O. Box 28, High-Tech Industrial Park Nanshan District, Shenzhen, Guangdong 518057
ERROR IN STRUCTURE
FLUORO IN WRONG POSITION

CAS Registry Number: 743420-02-2
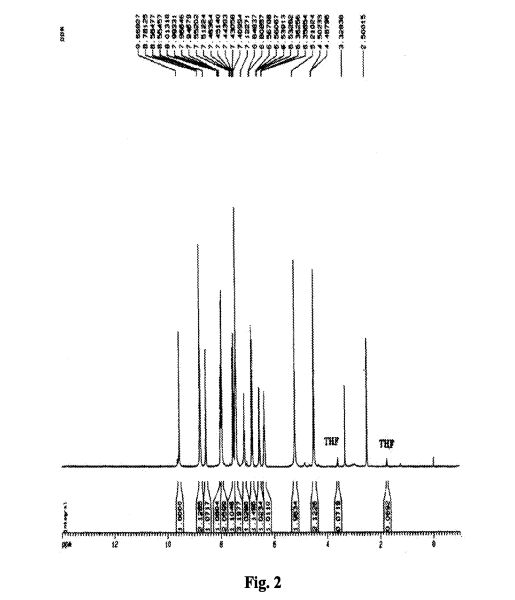
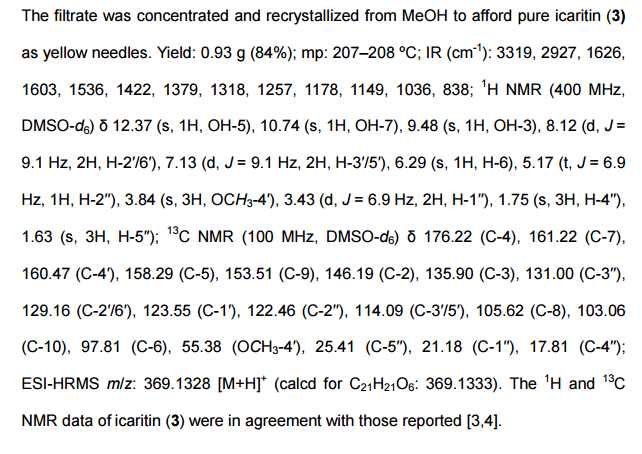
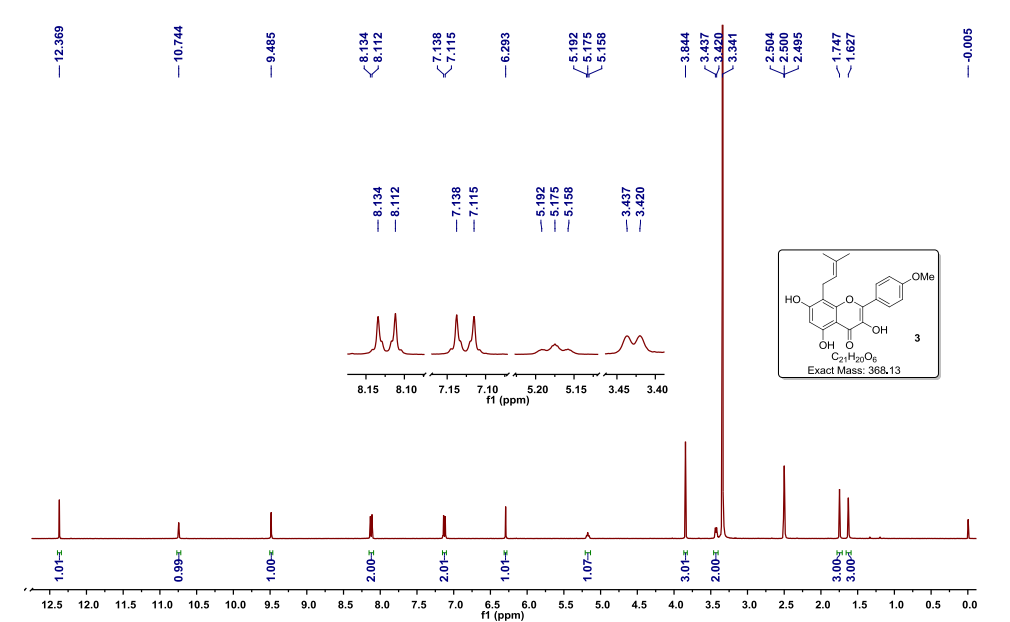
As described for Example 2 according to the patent ZL03139760.3 obtained chidamide poor purity (about 95%). LC / MS analysis results shown in Figure 1, show that the product contains N- (2- amino-5-fluorophenyl) -4- (N- (3- pyridin-acryloyl group of 4.7% of the structure shown in formula II) aminomethyl) benzamide. 1H NMR analysis of the results shown in Figure 2, show that the product contains 1.80% of tetrahydrofuran, far beyond the technical requirements for people with drug registration International Conference on Harmonization (ICH, International Conference of Harmonizition) provided 0.072% residual solvent limits. Therefore, the solid
Body not for pharmaceutical manufacturing.
 Chidamide (Epidaza) is an HDAC inhibitor (HDI) developed wholly in China.[1] It was originally known as HBI-8000.[2]
Chidamide (Epidaza) is an HDAC inhibitor (HDI) developed wholly in China.[1] It was originally known as HBI-8000.[2]
It is a benzamide HDI) and inhibits Class I HDAC1, HDAC2, HDAC3, as well as Class IIb HDAC10.[3]
It is approved by the Chinese FDA for relapsed or refractory peripheral T-cell lymphoma (PTCL), and having orphan drug status in Japan.[2]
As of April 2015 it is only approved in China.[1]
It shows potential in treating pancreatic cancer.[4][5][6]
Is NOT approved for the treatment of pancreatic cancer.

Chidamide drug administration and clinical milestone
November 2005: China declared IND
November 2006: eligible for Phase I clinical documents of approval
November 2006: completion of the International Patent Licensing, China entered the international fray original new drug development
May 2008: completed Phase I clinical, showing international mechanism similar drugs have the potential to become the best
February 2009: eligible lymphoma indications II / III of this document
March 2009: Start of the Phase II clinical trial for the NDA to ①CTCL goal of clinical trials and ②PTCL
March 2009: IND by the FDA application is eligible to start Phase I clinical in the United States
July 2009: eligible for non-small cell lung cancer, breast cancer and prostate cancer clinical documents of approval
December 2010: of PTCL by a conventional phase II directly into Phase II clinical trial registered drug trial center and by recognition
March 2011: combination chemotherapy for non-small cell lung cancer clinical trials enter phase Ib
September 2012: of PTCL indication test deadline
December 2012: of PTCL clinical summary will be held
January 2013: Chidamide declare China NDA
December 2014: the State Food and Drug Administration (CFDA) approved the listing

Chidamide overview, location and clinical significance
Chidamide (Chidamide, love spectrum sand ® / Epidaza®) Shenzhen microchip biotechnology limited liability company developed a new subtype selective histone having a chemical structure and is eligible for a global patent licensing deacetylase inhibitor, belong to the new mechanisms of epigenetic regulation new class of targeted anticancer drugs, has now completed with relapsed or refractory peripheral T-cell lymphoma clinical trial study registered indications, was in March 2013 to the SFDA reporting new drug certificate (NDA) and the marketing authorization (MAA). While a number of Chinese Cancer clinical trials undertaken Chidamide is also China’s first approved by the US FDA clinical studies in the United States of Chinese chemical original new drug trials in the United States Phase I has been completed. Chidamide has won the national “Eleventh Five-Year” 863 major projects (project number: 2006AA020603) and the national “Eleventh Five-Year”, “significant Drug Discovery” science and technology and other major projects funded project (project number: 2009ZX09401-003), was chosen the Ministry of Science and one of the “Eleventh five-Year” major national scientific and technological achievements.
Relapsed or refractory peripheral T-cell lymphoma (PTCL) is Chidamide first approvedclinical indications, PTCL belongs to the category of rare diseases, the lack of standard drug currently recommended clinical treatment, conventional chemotherapy response rate is low, recur, 5-year overall survival rate was about 25%. The world’s first PTCL treatment Folotyn (intravenous drug use) is eligible for FDA clearance to market in 2009, the second drugs Istodax (intravenous drug use) approved by the FDA in 2011. Add a new drug information for these drugs is very expensive, and were listed in China. Chidamide album clinical trial results showed that the primary endpoint of objective response rate was 28%, reaching the intended target research and development; sustained remission rate of 24% three months; drug safety was significantly better than the international similar drugs, and oral medication.
Chidamide is a completely independent intellectual property rights China originator of innovative medicines, has been multi-national patent. In China, for patients with relapsed or refractory PTCL to carry out effective drug treatment is urgent clinical need, Chidamide expected to bring new treatment options for patients with PTCL, prolong survival and improve quality of life of patients.

In China, for the effective treatment of patients with relapsed or refractory PTCL has undertaken urgent clinical need
Chidamide is a completely independent intellectual property rights China originator of innovative medicines
Chidamide (Chidamide) has been multi-national invention patents
In October 2006, the US HUYA biological microchip company formally signed the International Patent Chidamide licensing and international clinical cooperative development agreement; the United States in the ongoing Phase I clinical
Chidamide (Epidaza), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I , as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.
Chidamide, the English called Chidamide, by the Shenzhen-core biotechnology limited liability company independent design and synthesis of a novel anti-cancer drugs with new chemical structures and global intellectual property, and its chemical name N- (2-amino-_4_ fluorophenyl) -4_ (N- (3- topiramate Li acryloyl) aminomethyl) benzamide, its chemical structure of the structural formula I
The patent ZL03139760.3 and said US7,244,751, Chidamide have histone deacetylase inhibitory activity can be used to treat the differentiation and proliferation-related diseases such as cancer and psoriasis, especially for leukemia and solid tumors with excellent results.
Patent No. ZL03139760.3 and US7,244,751 discloses a method for preparing chidamide, but did not specify whether the resulting product is a crystalline material, nor did the presence or absence of the compound polymorphism. In the above patent, the activity of the compound for evaluation is not conducted in a solid state and, therefore, does not disclose any description about characteristics of the crystal.
Chipscreen BioSciences announced that the CFDA had approved chidamide for the treatment of relapsed or refractory peripheral T-cell lymphoma (PTCL) in December 2014. The drug and Hengrui’s apatinib were the only two NCEs launched by domestic drug makers last year.
Chidamide (CS055/HBI-8000) is a HDAC1/2/3/10 inhibitor derived from entinostat (MS-27-275)[1] which was first discoved by Mitsui Pharmaceuticals in 1999. Chipscreen holds worldwide IP rights to chidamide (patents: WO2004071400, WO2014082354).

Syndax Pharmaceuticals (NASDAQ: SNDX) is testing entinostat in breast cancer and NSCLC in pivotal trials. The FDA granted Breakthrough Therapy Designation to entinostat for advanced breast cancer in 2013. Eddingpharm in-licensed China rights to entinostat from Syndax in September 2013.
Chipscreen disclosed positive results from Phase II study of chidamide in relapsed or refractory PTCL at 2013 ASCO Annual Meeting[2]. Out of 79 evaluable patients in the trial, 23 patients (29.1%) had confirmed responses (8 CR, 3 CRu, and 12 PR). The most common grade 3/4 AEs were thrombocytopenia (24%), leucocytopenia (13%), neutropenia(10%).
The FDA has approved three HDAC inhibitors, known as Zolinza (vorinostat), Istodax (romidepsin) and Beleodaq (belinostat), for the treatment of PTCL. Celgene priced Istodax at $12000-18000/month and reported annual sales of $54 million in 2013. The efficacy and safety profile of chidamide compares favorably with romidepsin.
Although a dozen of companies are developing generic vorinostat and romidepsin, no chemical 3.1 NDA has been submitted to the CFDA so far. Chipscreen will be the only domestic maker of HDAC inhibitor in the coming two years. Moreover, the company is testing chidamide in NSCLC and breast cancer in early clinical studies.


CLIP
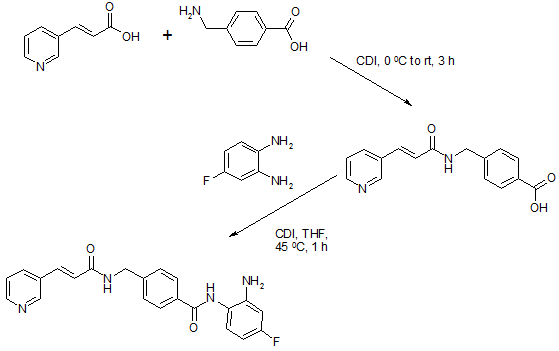
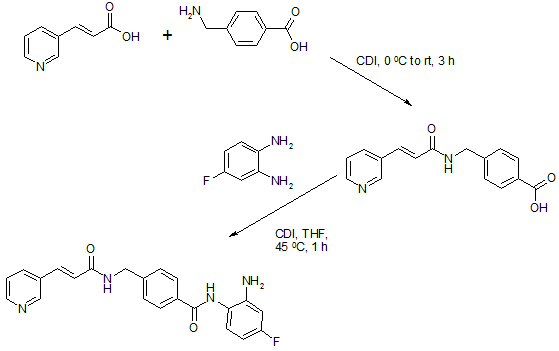
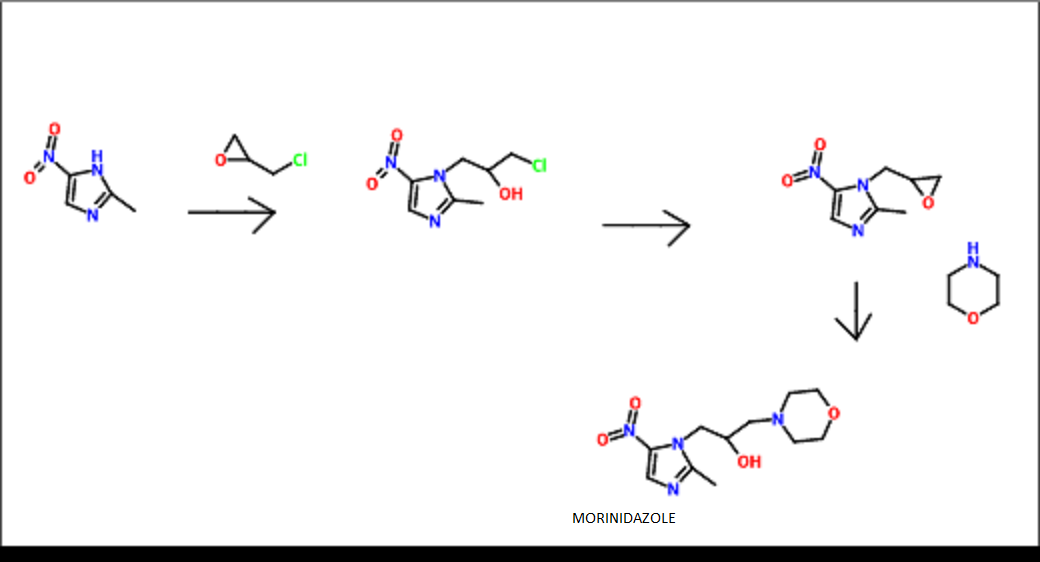
Chiamide synthesis: US7244751B2
Procedure:
Step a: To a suspension of 0.33 g (2.01 mmol) of N,N’-carbonyldiimidazole in tetrahydrofunan (10 ml) is added drop-wise a solution of 0.30 g (2.01 mmol) of 3-pyridineacrylic acid at 0 °C. Then, the mixture is stirred at room temperature for 3 hours and added drop-wise to a separately prepared 2.0 ml (2.00 mmol) of 1N aqueous sodium hydroxide solution including 0.30 g (2.00 mmol) of 4-aminomethylbenzoic acid, followed by stirring at room temperature for 8 hours. The reaction mixture is evaporated under vacuum. To the residue is added a saturated solution of sodium chloride (2 ml), then the mixture is neutralized with concentrated hydrochloric acid to pH 5. The deposited white solid is collected by filtration, washed with ice-water, and then dried to give 4-[N-(Pyridin-3-ylacryloyl)aminomethyl]benzoic acid (0.46 g, 82%). HRMS calcd for C16H14N2O3: 282.2988. Found: 282.2990. MA calcd for: C16H14N2O3: C, 68.07%; H, 5.00%; N, 9.92%. Found: C, 68.21%; H, 5.03%; N, 9.90%.
Step b: To a suspension of 0.29 g (1.78 mmol) of N,N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridin-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45 °C. for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofiman (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give N-(2-amino-4-fluorophenyl)-4-[N-(Pyridin-3-ylacryloyl)aminomethyl]benzamide (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): dppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (1H, t), 6.80 (2H, m),696 (1H, t), 7.18 (1H, d), 7.42 (2H, d), 7.52 (1H, d), 7.95 (2H, d), 8.02 (1H, d), 8.56 (1H, d), 8.72 (1H, br. t), 8.78 (1H, s), 9.60 (1H, br.s). IR (KBr) cm1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22H19N4O2F: 390.4170. Found: 390.4172. MA calcd for C22H19N4O2F: C, 67.68%; H, 4.40%; N, 14.35%. Found: C, 67.52%; H, 4.38%; N, 14.42%.
http://www.google.co.in/patents/US7244751
EXAMPLE 1
Preparation of 4-[N-(Pyridin-3-ylacryloyl)aminomethyl]benzoic acid

To a suspension of 0.33 g (2.01 mmol) of N,N′-carbonyldiimidazole in tetrahydrofunan (10 ml) is added drop-wise a solution of 0.30 g (2.01 mmol) of 3-pyridineacrylic acid at 0° C. Then, the mixture is stirred at room temperature for 3 hours and added drop-wise to a separately prepared 2.0 ml (2.00 mmol) of 1N aqueous sodium hydroxide solution including 0.30 g (2.00 mmol) of 4-aminomethylbenzoic acid, followed by stirring at room temperature for 8 hours. The reaction mixture is evaporated under vacuum. To the residue is added a saturated solution of sodium chloride (2 ml), then the mixture is neutralized with concentrated hydrochloric acid to pH 5. The deposited white solid is collected by filtration, washed with ice-water, and then dried to give the title compound (0.46 g, 82%). HRMS calcd for C16H14N2O3: 282.2988. Found: 282.2990. MA calcd for: C16H14N2O3: C, 68.07%; H, 5.00%; N, 9.92%. Found: C, 68.21%; H, 5.03%; N, 9.90%.EXAMPLE 2
Preparation of N-(2-amino-4-fluorophenyl)-4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzamide

To a suspension of 0.29 g (1.78 mmol) of N,N′-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45° C. for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofiman (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (1H, t), 6.80 (2H, m),696 (1H, t), 7.18 (1H, d), 7.42 (2H, d), 7.52 (1H, d), 7.95 (2H, d), 8.02 (1H, d), 8.56 (1H, d), 8.72 (1H, br. t), 8.78 (1H, s), 9.60 (1H, br.s). IR (KBr) cm1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22H19N4O2F: 390.4170. Found: 390.4172. MA calcd for C22H19N4O2F: C, 67.68%; H, 4.40%; N, 14.35%. Found: C, 67.52%; H, 4.38%; N, 14.42%.EXAMPLE 3
Preparation of 4-[N-cinnamoylaminomethyl]benzoic acid
To a suspension of 0.33 g (2.01 mmol) of N,N′-carbonyldiimidazole in tetrahydrofunan (10 ml) is added drop-wise a solution of 0.30 g (2.01 mmol) of cinnamic acid at 0° C. Then, the mixture is stirred at room temperature for 3 hours and added drop-wise to a separately prepared 2.0 ml (2.00 mmol) of 1N aqueous sodium hydroxide solution including 0.30 g (2.00 mmol) of 4-aminomethylbenzoic acid, followed by stirring at room temperature for 8 hours. The reaction mixture is evaporated under vacuum. To the residue is added a saturated solution of sodium chloride (2 ml), then the mixture is neutralized with concentrated hydrochloric acid to pH 7. The deposited white solid is collected by filtration, washed with ice-water, and then dried to give the title compound (0.51 g, 91%). HRMS calcd for C17H15NO3: 281.3242. Found: 281.3240. MA calcd for C17H15NO3: C, 72.58%; H, 5.38%; N, 4.98. Found: C, 72.42%; H, 5.37%; N, 4.98%.
EXAMPLE 4
Preparation of N-(2-amino-4-fluorophenyl)-4-[N-cinnamoylaminomethyl]benzamide

To a suspension of 0.29 g (1.78 mmol) of N,N′-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-cinnamoylaminomethyl]benzoic acid, followed by stirring at 45° C. for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 16 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.45 g, 64%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.42 (2H, d), 4.92 (2H, br.s), 6.62 (1H, t), 6.78 (2H, m), 7.01 (1H, t), 7.32 (5H, m), 7.54 (5H, m), 8.76 (1H, br.t), 9.58 (1H, br.s). IR (KBr) cm−1: 3306, 1618, 1517, 1308, 745. HRMS calcd for C23H20N3O2F: 389.4292. Found: 389.4294. MA calcd for C23H20N3O2F: C, 70.94%; H, 5.18%; N, 10.79%. Found: C, 70.72%; H, 5.18%; N, 10.88%.
PATENT
https://www.google.com/patents/US20150299126

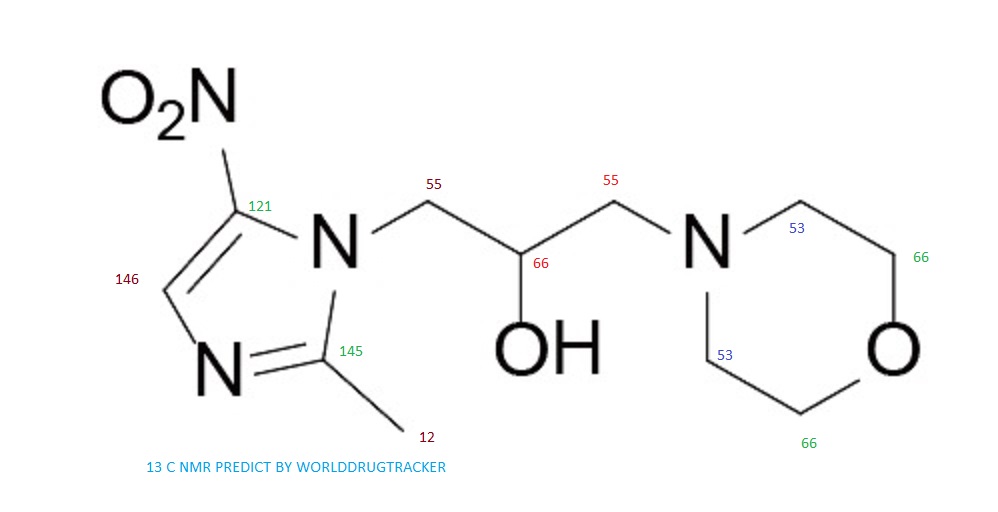
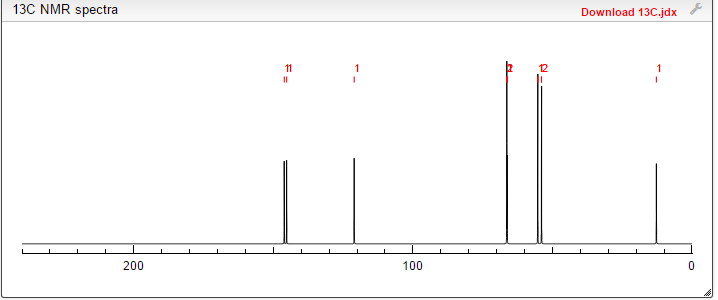
NMR, MS ETC CLICK TO VIEW
C-NMR
CLIP
Chidamide (Epidaza), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I 1–3, as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.74
This agent also inhibits the expression of signaling kinases in the PI3K/ Akt and MAPK/Ras pathways and may result in cell cycle arrest and the induction of tumor cell apoptosis.75
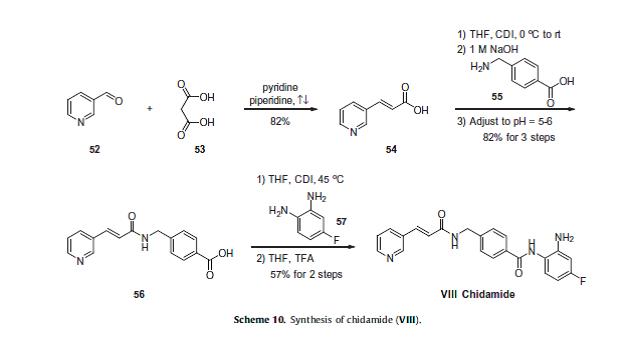
Currently, phases I and II clinical trials are underway for the treatment of non-small cell lung cancer and for the treatment of breast cancer, respectively.76 The scalable synthetic approach to chidamide very closely follows the discovery route,77–79 and is described in Scheme 10. The sequence began with the condensation of commercial nicotinaldehyde (52) and malonic acid (53) in a mixture of pyridine and piperidine. Next, activation of acid 54 with N,N0-carbonyldiimidazole (CDI) and subsequent reaction with 4-aminomethyl benzoic acid (55) under basic conditions afforded amide 56 in 82% yield.
Finally, activation of 56 with CDI prior to treatment with 4-fluorobenzene- 1,2-diamine (57) and subsequent treatment with TFA and THF yielded chidamide (VIII) in 38% overall yield from 52. However, no publication reported that mono-N-Boc-protected bis-aniline was used to approach Chidamide.

74. Ning, Z. Q.; Li, Z. B.; Newman, M. J.; Shan, S.; Wang, X. H.; Pan, D. S.; Zhang, J.;
Dong, M.; Du, X.; Lu, X. P. Cancer Chemother. Pharmacol. 2012, 69, 901.
75. Liu, L.; Chen, B.; Qin, S.; Li, S.; He, X.; Qiu, S.; Zhao, W.; Zhao, H. Biochem.
Biophys. Res. Commun. 2010, 392, 190.
76. Gong, K.; Xie, J.; Yi, H.; Li, W. Bio. Chem. J. 2012, 443, 735.
77. Lu, X. P.; Li, Z. B.; Xie, A. H.; Shi, L. M.; Li, B. Y.; Ning, Z. Q.; Shan, S.; Deng, T.;
Hu, W. M. US Patent 2004224991A1, 2004.
78. Lu, X. P.; Li, Z. B.; Xie, A. H.; Shi, L. M.; Li, B. Y.; Ning, Z. Q.; Shan, S.; Deng, T.;
Hu, W. M. CN Patent 1513839A, 2003.
79. Yin, Z. H.; Wu, Z. W.; Lan, Y. K.; Liao, C. Z.; Shan, S.; Li, Z. L.; Ning, Z. Q.; Lu, X.
P.; Li, Z. B. Chin. J. New Drugs 2004, 13, 536.
see CN 105457038
CN 1513839
WRONG COMPD
WO2004071400
Example 2. Preparation of
N-(2-amino-5-fluorophenyl)-4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzamide
 To a suspension of 0.29 g (1.78 mmol) of N, N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45°C for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (IH, t), 6.80 (2H, m), 6.96 (IH, t), 7.18 (IH, d), 7.42 (2H, d), 7.52 (IH, d), 7.95 (2H, d), 8.02 (IH, d), 8.56 (IH, d), 8.72 (IH, br. t), 8.78 (IH, s), 9.60 (IH, br.s). IR (KBr) cm“1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22Hι9N4O2F: 390.4170. Found: 390.4172. MA calcd for C22Hι9N4O2F: C, 67.68%; H, 4.40%; N, 14.35. Found: C, 67.52%; H, 4.38%; N, 14.42%.
To a suspension of 0.29 g (1.78 mmol) of N, N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45°C for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (IH, t), 6.80 (2H, m), 6.96 (IH, t), 7.18 (IH, d), 7.42 (2H, d), 7.52 (IH, d), 7.95 (2H, d), 8.02 (IH, d), 8.56 (IH, d), 8.72 (IH, br. t), 8.78 (IH, s), 9.60 (IH, br.s). IR (KBr) cm“1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22Hι9N4O2F: 390.4170. Found: 390.4172. MA calcd for C22Hι9N4O2F: C, 67.68%; H, 4.40%; N, 14.35. Found: C, 67.52%; H, 4.38%; N, 14.42%.

Photo taken on May 22, 2015 shows a box of Chidamide in Shenzhen, south China’s Guangdong Province. Chidamide is the world’s first oral HDAC inhibitor …
A New Cancer Drug, Made in China
After 14 years, Shenzhen biotech’s medicine is one of the few locally developed from start to finish
HONG KONG— Xian-Ping Lu left his job as director of research at drug maker Galderma R&D in Princeton, N.J., to co-found a biotech company to develop new medicines in his native China.
It took more than 14 years but the bet could be paying off. In February, Shenzhen Chipscreen Biosciences’ first therapy, a medication for a rare type of lymph-node cancer, hit the market in China.
The willingness of veterans like Dr. Lu and others to leave multinational drug companies for Chinese startups reflects a growing optimism in the industry here. The goal, encouraged by the government, is to move the Chinese drug industry beyond generic medicines and drugs based on ones developed in the West.
Chipscreen’s drug, called chidamide, or Epidaza, was developed from start to finish in China. The medicine is the first of its kind approved for sale in China, and just the fourth in a new class globally. Dr. Lu estimates the research cost of chidamide was about $70 million, or about one-tenth what it would have cost to develop in the U.S.
“They are a good example of the potential for innovation in China,” said Angus Cole, director at Monitor Deloitte and pharmaceuticals and biotechnology lead in China.
China’s spending on pharmaceuticals is expected to top $107 billion in 2015, up from $26 billion in 2007, according to Deloitte China. It will become the world’s second-largest drug market, after the U.S., by 2020, according to an analysis published last year in the Journal of Pharmaceutical Policy and Practice.
China has on-the-ground infrastructure labs, a critical mass of leading scientists and interested investors, according to Franck Le Deu, head of consultancy McKinsey & Co.’s pharmaceuticals and medical-products practice in China. “There’re all the elements for the recipe for potential in China,” he said.
But there are obstacles to an industry where companies want big payoffs for a decade or more of work and tremendous costs it takes to develop a drug.
While the protection of intellectual property has improved, China’s cumbersome rules for drug approval and a government effort to cut health-care costs, particularly spending on drugs, could hurt the Chinese drug companies’ efforts, said Mr. Cole of Deloitte.
“Will you start to see success? Of course you will,” said Mr. Cole. However, “I’ve yet to see convincing or compelling evidence that it’s imminent.”
To date, many of the Chinese companies that are flourishing in the life sciences are contract research organizations that help carry out clinical trials, as well as providers of related services.
Some companies, like Shanghai-based Hua Medicine, are buying the rights to develop new compounds in China from multinational drug companies, what some experts consider more akin to an intermediate step to innovation.
Late last year, Hua Medicine completed an early-stage human clinical trial of a diabetes drug in China and in March filed an application to the Food and Drug Administration to develop it in the U.S. as well. The company has raised $45 million in venture funding to date.
Li Chen, who left an 18-year career at Roche Holding AG as head of research and development in China to help start Hua Medicine, said the company’s goal is to “create a game-changer of drug discovery.”
At Chipscreen Biosciences, Dr. Lu and his co-founders set up the company in 2001 in Shenzhen, a city that was quickly growing into a technology and research hub, just over the border from Hong Kong. They created a lab of 10 scientists to use a new analytic technique known as “chemical genomics” to examine the relationships between molecular structures of the existing and failed drugs, how they act on different targets in the body and what genes were being activated or repressed. Now they have more than 60 scientists.
By better predicting how chemicals would act on the body before entering human testing, they hoped they would be more likely get a drug to market.
“How can a small company compete with a multinational?” said Dr. Lu. “The only thing we can compete with is the scientific brain.”
The biggest challenges for the company have been financing and the Chinese regulatory system, said Dr. Lu. The company has raised a total of 300 million yuan ($48 million) over five rounds of venture funding, said Dr. Lu. Chipscreen also receives grant money from the Chinese government.
The company filed its application for approval of chidamide to the Chinese Food and Drug Administration, or CFDA, in early 2013. It had to wait nearly two years for approval, receiving the OK only in December.
Chidamide now is on the market in China for 26,500 yuan ($4,275) a month, a price far lower than patients in the U.S. pay for some of the newest cancer medicines but much more than the typical Chinese patient pays for drugs. Dr. Lu said the price reflects a balance between affordability for patients and return for shareholders. Some investors wanted to price the drug higher.
PAPER
Discovery of an orally active subtype-selective HDAC inhibitor, chidamide, as an epigenetic modulator for cancer treatment
Tumorigenesis is maintained through a complex interplay of multiple cellular biological processes and is regulated to some extent by epigenetic control of gene expression. Targeting one signaling pathway or biological function in cancer treatment often results in compensatory modulation of others, such as off-target drivers of cell survival. As a result, overall survival of cancer patients is still far from satisfactory. Epigenetic-modulating agents can concurrently target multiple aberrant or compensatory signaling pathways found in cancer cells. However, existing epigenetic-modulating agents in cancer treatment have not yet fully translated into survival benefits beyond hematological tumors. In this article, we present a historical rationale for use of chidamide (CS055/Epidaza), an orally active and subtype-selective histone deacetylase (HDAC) inhibitor of the benzamide chemical class. This compound was discovered and successfully developed as mono-therapy for relapsed and refractory peripheral T cell lymphoma (PTCL) in China. We discuss the evidence supporting chidamide as a durable epigenetic modulator that allows cellular reprogramming with little cytotoxicity in cancer treatments.

CLIPS
Chinese scientists develop world’s 1st oral HDAC inhibitor
|

Lu Xianping works in a lab at Shenzhen Chipscreen Biosciences Ltd. in Shenzhen, south China’s Guangdong Province, May 20, 2015. Lu Xianping, together with other four returned overseas scientists, spent 14 years to develop Chidamide, the world’s first oral HDAC inhibitor, which was given regulatory approval in January. (Xinhua/Mao Siqian) |

GNT Biotech and Medicals Corporation Licenses Novel Cancer Molecule from Shenzhen Chipscreen Biosciences Ltd.
PR Newswire
SHENZHEN, China, Oct. 10, 2013
SHENZHEN, China, Oct. 10, 2013 /PRNewswire/ — GNT Biotech and Medicals Corporation announces the grant of an exclusive license from Shenzhen Chipscreen Biosciences Ltd.for the development and commercialization of Chidamide in Taiwan. Chidamide, an oral, selective histone deacetylase (HDAC) inhibitor, is currently being evaluated in Phase II trials by Chipscreen Biosciences in Peripheral T-Cell Lymphoma (PTCL), Cutaneous T-Cell Lymphoma (CTCL) and Non-Small Cell Lung Cancer patients (NSCLC). GNTbm will develop and commercialize Chidamide primarily in PTCL, NSCLC and will also retain the rights to develop and commercialize Chidamide in other oncology indications in Taiwan.
About Chidamide
Chidamide is a selective HDAC inhibitor against subtype 1, 2, 3 and 10, and being studied in multiple clinical trials as a single agent or in combination with chemotherapeutic agents for the treatment of various hematological and solid cancers. Its anticancer effects are thought to be mediated through epigenetic modulation via multiple mechanisms of action, including the inhibition of cell proliferation and induction of apoptosis in blood derived cells, inhibition of epithelial to mesenchymal transition (EMT, a process that is highly relevant to tumor cell metastasis and drug resistance), induction of tumor specific antigen and antigen-specific T cell cytotoxicity, enhancement of NK cell anti-tumor activity, induction of cancer stem cell differentiation, and resensitization of tumor cells that have become resistant to anticancer agents such as platinums, taxanes and topoisomerase II inhibitors. Chidamide has demonstrated clinical efficacy in pivotal phase II trials on Cutaneous T-Cell Lymphoma (CTCL) and Peripheral T-Cell Lymphoma (PTCL) conducted in China, and is currently undergoing phase II trial in NSCLC together with first line PC therapeutic treatment. Due to its superior pharmacokinetic properties and selectivity, Chidamide may offer better clinical profile over the other HDAC inhibitors currently under development or being marketed.
About GNTbm
GNTbm is a subsidiary of GNT Inc, a Taiwanese company focused on the manufacture of nano-scale metallic particles for food and medical purposes. Founded in 1992 by a team of electronic professionals, GNT has successfully developed the innovative technology of physical metal miniaturization based on the patent of MBE (Molecular Beam Epitaxy). Further information about GNT Inc is available at www.gnt.com.tw.
GNTbm was established in August 2013, and housed in the Nankang Biotech Incubation Center, (NBIC), in Nankang, Taipei. Lead by Dr. Chia-Nan Chenalong with an experienced team of scientists, GNTbm will explore development and commercialization of novel drug delivery systems, Innovative biomedical and diagnostic tools based on gold nanoparticles.
About Shenzhen Chipscreen Biosciences Ltd.
Chipscreen is a leading integrated biotech company in China specialized in discovery and development of novel small molecule pharmaceuticals. The company has utilized its proprietary chemical genomics-based discovery platform to successfully develop a portfolio of clinical and preclinical stage programs in a number of therapeutic areas. Chipscreen’s business strategy is to generate differentiated drug candidates across multiple therapeutic areas. Drug candidates are either developed by Chipscreen or co-developed and commercialized in a partnership at the research, preclinical and clinical stages. The company was established as Sino-foreign joint venture in 2001. Further details about Chipscreen Bioscience is available atwww.chipscreen.com.
|
GNT Biotech and Medicals Corporation
Ekambaranellore Prakash, PhD
Director of International Department
GNT Biotech and Medicals Corporation
TEL: +886-2-7722-0388 #303
E-mail: prakash@gntbm.com.tw
Web site: www.gnt.com.tw
|
Shenzhen Chipscreen Biosciences Ltd.
Rebecca Hai
Investor Relations
Shenzhen Chipscreen Biosciences Ltd.
TEL: +86-755-26957317
E-mail: rebeccai_hai@chipscreen.com
Web site: www.chipscreen.com
|
SOURCE GNT Biotech and Medicals Corporation
| CN101397295B |
Nov 12, 2008 |
Apr 25, 2012 |
深圳微芯生物科技有限责任公司 |
2-dihydroindolemanone derivates as histone deacetylase inhibitor, preparation method and use thereof |
| CN101648920B |
Aug 20, 2009 |
Feb 8, 2012 |
苏州东南药物研发有限责任公司 |
用作组蛋白去乙酰酶抑制剂的三氟甲基酮类化合物及其用途 |
| CN101648921B |
Aug 20, 2009 |
Nov 2, 2011 |
苏州东南药物研发有限责任公司 |
Benzamide compound used as histone deacetylase inhibitor and application thereof |
| CN103833626A * |
Nov 27, 2012 |
Jun 4, 2014 |
深圳微芯生物科技有限责任公司 |
Crystal form of chidamide and preparation method and application thereof |
| CN103833626B * |
Nov 27, 2012 |
Nov 25, 2015 |
深圳微芯生物科技有限责任公司 |
西达本胺的晶型及其制备方法与应用 |
| CN104876857A * |
May 12, 2015 |
Sep 2, 2015 |
亿腾药业(泰州)有限公司 |
Preparation of benzamide histone deacetylase inhibitor with differentiation and anti-proliferation activity |
| EP2205563A2 * |
Oct 8, 2008 |
Jul 14, 2010 |
Orchid Research Laboratories Limited |
Novel histone deacetylase inhibitors |
| WO2009152735A1 * |
Jun 9, 2009 |
Dec 23, 2009 |
Jiangsu Goworth Investment Co. Ltd |
Histone deacetylase inhibitors and uses thereof |
| WO2010135908A1 * |
May 20, 2010 |
Dec 2, 2010 |
Jiangsu Goworth Investment Co. Ltd. |
N-(2-amino-4-pyridyl) benzamide derivatives and uses thereof |
| WO2014082354A1 * |
Dec 18, 2012 |
Jun 5, 2014 |
Shenzhen Chipscreen Biosciences, Ltd. |
Crystal form of chidamide, preparation method and use thereof |
| Patent ID |
Date |
Patent Title |
| US2015299126 |
2015-10-22 |
CRYSTAL FORM OF CHIDAMIDE, PREPARATION METHOD AND USE THEREOF |
| US2010222379 |
2010-09-02 |
NOVEL HISTONE DEACETYLASE INHIBITORS |
| US7244751 |
2007-07-17 |
Histone deacetylase inhibitors of novel benzamide derivatives with potent differentiation and anti-proliferation activity |
References
- “China’s First Homegrown Pharma.”. April 2015.
- ^ Jump up to:a b [1]
- HUYA Bioscience International Grants An Exclusive License For HBI-8000 In Japan And Other Asian Countries To Eisai. Feb 2016
- Qiao, Z (2013-04-26). “Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells.”. Biochem Biophys Res Commun. 434 (1): 95–101. doi:10.1016/j.bbrc.2013.03.059. PMID 23541946.
- Guha, Malini (2015-04-01). “HDAC inhibitors still need a home run, despite recent approval”. Nature Reviews Drug Discovery 14: 225–226. doi:10.1038/nrd4583.
- Wang, Shirley S. (2015-04-02). “A New Cancer Drug, Made in China”. The Wall Street Journal. Retrieved 13 April 2015.
- References:
1. Ning, Z. Q.; et. al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol2012, 69(4), 901-909. (activity)
2. Gong, K.; et. al. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J 2012, 443(3), 735-746. (activity)
3. Hu, W.; et. al. N-(2-amino-5-fluorophenyl)-4-[N-(Pyridin-3-ylacryloyl) aminomethyl ]benzamide or other derivatives for treating cancer and psoriasis. US7244751B2
4. Lu, X.; et. al. Crystal form of chidamide, preparation method and use thereof. WO2014082354A1
5. Yin, Z.-H.; et. al. Synthesis of chidamide,a new histone deacetylase (HDAC) inhibitor. Chin J New Drugs 2004, 13(6), 536-538. (starts with basic raw materials)
- Zhongguo Xinyao Zazhi (2004), 13(6), 536-538.
/////////Chidamide, Epidaza, CS055, HBI-8000, orally active subtype-selective HDAC inhibitor, epigenetic modulator, cancer treatment, CFDA, CHINA, CANCER
Fc3ccc(NC(=O)c1ccc(cc1)CNC(=O)/C=C/c2cccnc2)c(N)c3

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
























 CHINA FLAG
CHINA FLAG


 .
.







![[1860-5397-11-135-1]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-11-135-1.png?scale=2.0&max-width=1024&background=FFFFFF)

![[1860-5397-11-135-i1]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-135-i2]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-135-i3]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-135-i4]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i4.png?scale=2.0&max-width=1024&background=FFFFFF)














 AMITIZA (lubiprostone)
AMITIZA (lubiprostone)
















![Molecular Structure of 105979-17-7 (3,5-Pyridinedicarboxylicacid, 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)-, 3-methyl5-[(3R)-1-(phenylmethyl)-3-piperidinyl] ester, (4R)-rel-)](https://i0.wp.com/www.lookchem.com/300w/2010/072/105979-17-7.jpg)


































