
Home » CHINA 2024
Category Archives: CHINA 2024
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Linaprazan
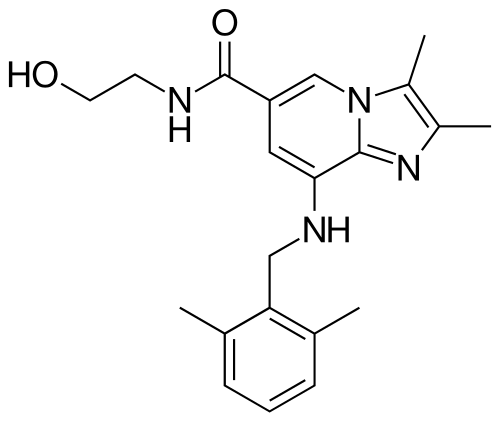
Linaprazan
CHINA 2024, APPROVALS 2024, AstraZeneca, CINCLUS, GERD, linaprazan glurate, for the treatment of moderate to severe GERD,
8-[(2,6-dimethylphenyl)methylamino]-N-(2-hydroxyethyl)-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxamide
- CAS 248919-64-4
- AZD-0865
- E0OU4SC8DP
- DTXSID90870279
366.5 g/mol, C21H26N4O2- CS-5725
- MS-25870
- DB-302288
- HY-100412
- F85407
- Q27276714
- 8-(2,6-dimethylbenzylamino)-N-(2-hydroxyethyl)-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxamide
- 8-[(2,6-dimethylphenyl)methylamino]-N-(2-hydroxyethyl)-2,3-dimethyl-imidazo[1,2-a]pyridine-6-carboxamide
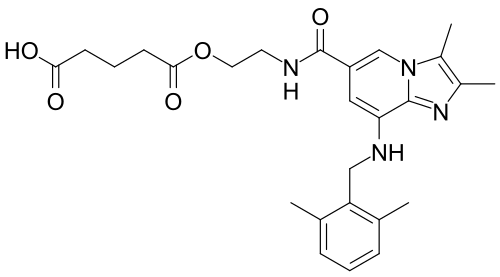
Chemical structure of linaprazan glurate CAS No.: 1228559-81-6 , X842
| Molecular formula | C26H32N4O5 |
|---|---|
| Molecular weight | 480.556086540222 |
| Accurate quality | 480.237 |
5-[2-[[8-[(2,6-dimethylphenyl)methylamino]-2,3-dimethylimidazo[1,2-a]pyridine-6-carbonyl]amino]ethoxy]-5-oxopentanoic acid
- OriginatorAstraZeneca
- DeveloperCinclus Pharma; Jiangsu Sinorda Biomedicine Co., Ltd; Shanghai Pharmaceutical Group
- Class2 ring heterocyclic compounds; Amines; Aminopyridines; Anti-inflammatories; Antibacterials; Antiulcers; Glutarates; Imidazoles; Pentanoic acids; Pyridines; Small molecules; Toluenes
- Mechanism of ActionPotassium-competitive acid blockers
- RegisteredReflux oesophagitis
- Phase IIDuodenal ulcer; Erosive oesophagitis; Helicobacter infections
- Phase IGastro-oesophageal reflux
- 28 Aug 2025No recent reports of development identified for phase-I development in Gastro-oesophageal-reflux(In volunteers) in Sweden (PO, Tablet)
- 29 Jun 2025Cinclus Pharma Holding plans a phase III trial for Gastro-oesophageal-reflux in the US, Bulgaria, Czech Republic, Georgia, Germany, Hungary, Poland (PO) (NCT07037875)
- 13 Jun 2025Cinclus Pharma secures EMA and FDA pediatric study waivers for linaprazan glurate in H. pylori infection
Linaprazan is a lipophilic, weak base with potassium-competitive acid blocking (P-CAB) activity. Linaprazan concentrates highly in the gastric parietal cell canaliculus and on entering this acidic environment is instantly protonated and binds competitively and reversibly to the potassium binding site of the proton pump hydrogen-potassium adenosine triphosphatase (H+/K+ ATPase), thereby inhibiting the pump’s activity and the parietal cell secretion of H+ ions into the gastric lumen, the final step in gastric acid production.
Linaprazan is an experimental drug for the treatment of gastroesophageal reflux disease (GERD). Unlike the proton-pump inhibitors (PPIs) which are typically used to treat GERD, linaprazan is a potassium-competitive acid blocker (P-CAB).[1][2] Linaprazan was developed by AstraZeneca, but it was not successful in clinical trials.[3]
The drug was then licensed to Cinclus Pharma,[4] which is now investigating linaprazan glurate, a prodrug of linaprazan which is expected to have a longer biological half-life than linaprazan itself.[4]
Linaprazan glurate inhibits exogenously or endogenously stimulated gastric acid secretion. Linaprazan glurate exhibits several favorable properties, such as rapid onset of action, high in vivo potency, and/or prolonged duration of action. Linaprazan glurate is useful in the research of gastrointestinal inflammatory diseases and peptic ulcer disease (disclosed in patent WO2010063876A1).
- Imidazo pyridine derivatives which inhibit gastric acid secretionPublication Number: WO-9955706-A9Priority Date: 1998-04-29
- Imidazo pyridine derivatives which inhibit gastric acid secretion.Publication Number: ZA-200005797-BPriority Date: 1998-04-29
- Imidazo pyridine derivatives which inhibit gastric acid secretionPublication Number: KR-20050121760-APriority Date: 1998-04-29
- Imidazopyridine derivatives which inhibit gastric acid secretion, pharmaceutical formulation containing such derivatives, processes for their preparation, use thereof, and intermediates.Publication Number: NO-317262-B1Priority Date: 1998-04-29
- Imidazo pyridine derivatives which inhibit gastric acid secretionPublication Number: PL-195000-B1Priority Date: 1998-04-29
- Imidazo pyridine derivatives which inhibit gastric acid secretionPublication Number: US-6313137-B1Priority Date: 1998-04-29Grant Date: 2001-11-06
- Imidazo pyridine derivatives which inhibit gastric acid secretionPublication Number: WO-9955706-A1Priority Date: 1998-04-29
SYN
WO2010063876
https://patentscope.wipo.int/search/en/WO2010063876
Examples
Example 1
Preparation of 5- {2-[( {8-[(2,6-dimethylbenzyl)amino]-2,3-dimethylimidazo[ 1 ,2-a]pyridin-6-yl}carbonyl)amino]ethoxy}-5-oxopentanoic acid
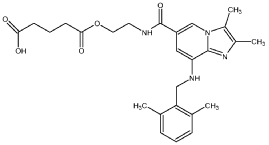
2,3-dimethyl-8-(2,6-dimethylbenzylamino)-N-hydroxyethyl-imidazo[l,2-a]pyridi-ne-6-carboxamide (obtained using the process according to WO02/20523) (2.0 g,
5.46 mmol) and glutaric anhydride (0.95 g, 8.33 mmol) was added to DMF (10 ml). The mixture was heated to 80 0C and stirred 16 h at this temperature.
Acetone (20 ml) was added to the reaction mixture whereby the product started to crystallize. The mixture was cooled to room temperature. After 4 h the product was filtered off and washed with acetone (20 ml). 2.25 g (86%) of the title compound was obtained. The structure of the compound was confirmed with 1H- NMR spectrum.
1H-NMR (300 MHz, DMSO): δ 1.73 (m, 2H), 2.2-2.4 (m, 16H), 3.52 (m,2H), 4.18 (t, 2H), 4.36 (d, 2H), 4.99 (t, IH), 6.67 (s, IH), 7.0-7.2 (m, 3H), 8.04 (s, IH), 8.56 (t, IH), 12.10 (bs, IH).
SYN
US6900324B2.
https://patentscope.wipo.int/search/en/detail.jsf?docId=US40374322&_cid=P12-MEXO1E-18626-1
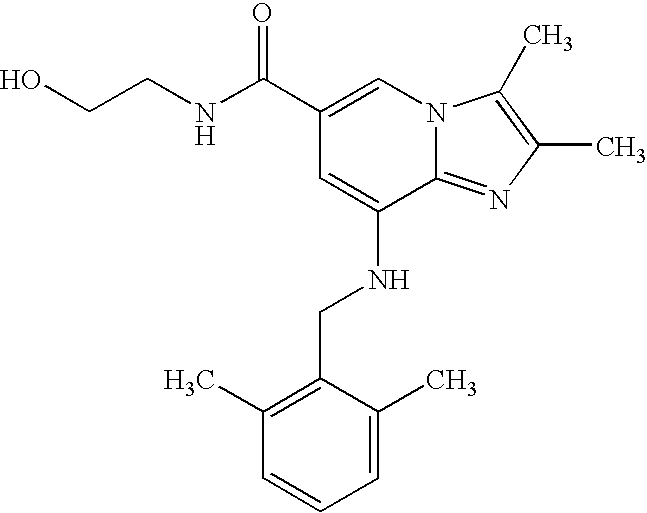
Example 1.16
| Synthesis of 8-[(216-dimethylbenzyl)amino]-N-(2-hydroxyethyl)-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxamide |
Example 2.1
Example 2.2
Example 2.3
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Linaprazan is a potassium-competitive acid blocker (P-CAB) initially developed by AstraZeneca between 2001 and 2005 for treating gastroesophageal reflux disease (GERD). Subsequently, Cinclus Pharma ac
quired the rights to linaprazan and developed linaprazan glurate. In 2024, the NMPA approved linaprazan glurate for the treatment of moderate to severe GERD, marking Cinclus Pharma’s first marketing approval in China. Linaprazan glurate is a P-CAB that inhibits gastric acid secretion by reversibly blocking the potassium-binding site of the gastric H+/K +-ATPase enzyme, leading to rapid and sustained acid suppression [94]. Clinical efficacy was demonstrated in Phase III trials NCT04567810), showing superior acid suppression and symptom relief compared to PPIs in GERD patients. Regarding toxicity, linaprazan was generally well tolerated in clinical studies. However, some issues were
noted, such as elevated liver transaminases in a few patients, which were addressed in the development of linaprazan glurate by achieving lower peak plasma concentrations (Cmax) to minimize liver load 95,96]. The synthetic route of Linaprazan, shown in Scheme 22 [97], initiates with condensative Cyclization between Lina-001 and Lina-002 to yield Lina-003. This intermediate undergoes nucleophilic substitution with Lina-004 under basic conditions to generate Lina-005. Final thermolytic amidation of Lina-005 at 100 DEG CENT affords Linaprazan
[95] C. Scarpignato, R.H. Hunt, Potassium-competitive acid blockers: current clinical use and future developments, Curr. Gastroenterol. Rep. 26 (2024) 273–293.
[96] J.F. Willart, M. Durand, L.E. Briggner, A. Marx, F. Dan`ede, M. Descamps, Solid-state amorphization of linaprazan by mechanical milling and evidence of polymorphism, J Pharm Sci 102 (2013) 2214–2220.
[97] B. Elman, S. Erback, E. Thiemermann, Process for Preparing a Substituted Imidazopyridine Compound, 2002. US6900324B2.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Rawla P, Sunkara T, Ofosu A, Gaduputi V (December 2018). “Potassium-competitive acid blockers – are they the next generation of proton pump inhibitors?”. World Journal of Gastrointestinal Pharmacology and Therapeutics. 9 (7): 63–68. doi:10.4292/wjgpt.v9.i7.63. PMC 6305499. PMID 30595950.
- “Linaprazan”. Inxight Drugs. National Center for Advancing Translational Sciences.
- Tong A (4 March 2020). “Can reformulation of an AstraZeneca castoff rival Takeda’s new heartburn drug? Here’s a $26M bet on yes”. endpts.com.
- “Linaprazan glurate”. Cinclus Pharma.
| Clinical data | |
|---|---|
| Other names | AZD-0865 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 248919-64-4 |
| PubChem CID | 9951066 |
| UNII | E0OU4SC8DP |
| Chemical and physical data | |
| Formula | C21H26N4O2 |
| Molar mass | |
////////////Linaprazan, CHINA 2024, APPROVALS 2024, AstraZeneca, CINCLUS, GERD, linaprazan glurate, moderate to severe GERD, 248919-64-4, AZD 0865, E0OU4SC8DP, DTXSID90870279, X 842
Pradefovir
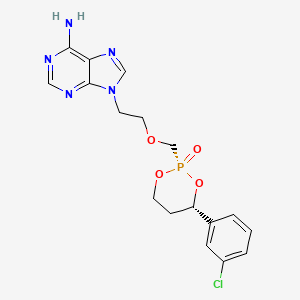
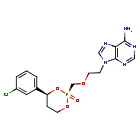
Pradefovir
WeightAverage: 423.79
Monoisotopic: 423.0863188
Chemical FormulaC17H19ClN5O4P
9-[2-[[(2R,4S)-4-(3-chlorophenyl)-2-oxo-1,3,2λ5-dioxaphosphinan-2-yl]methoxy]ethyl]purin-6-amine
2R,4S-(+)-9-(2-(4-(3-chlorophenyl)-2-oxo-1,3,2-dioxaphosphorinan-2-yl)methoxyethyl)adenine
HEPATITIS B VIRUS, APPROVALS 2024, CHINA 2024, Xi’an Xintong Pharmaceutical Research Co, Xinshumu
Pradefovir Mesylate
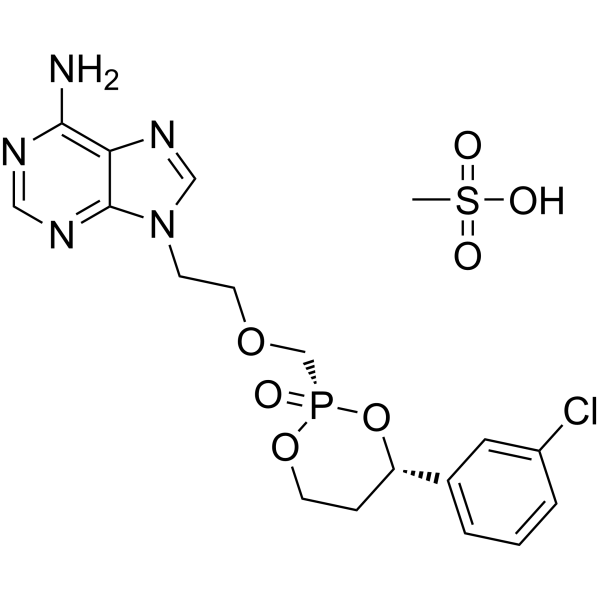
CAS No. : 625095-61-6, Remofovir mesylate
| 분자량 | 519.90 |
|---|---|
| 화학식 | C18H23ClN5O7PS |
Pradefovir is a cyclodiester antiviral prodrug with specific activity against hepatitis B virus (HBV). Pradefovir is specifically metabolized in the liver by hepatic enzymes, mainly by CYP4503A4, to adefovir. In turn, adefovir is phosphorylated by cellular kinases to its activated form adevofir diphosphate. By competing with the natural substrate dATP, the diphosphate form is incorporated into viral DNA and inhibits RNA-dependent DNA polymerase. This causes DNA chain termination and eventually results in an inhibition of HBV replication.
PAT
- Novel phosphonic acid based prodrugs of PMEA and its analoguesPublication Number: US-2003229225-A1Priority Date: 2002-05-13
- Process for Preparation of Cyclic Prodrugs of PMEA and PMPAPublication Number: US-2007203339-A1Priority Date: 2002-05-13
- Process for preparation of cyclic prodrugs of PMEA and PMPAPublication Number: US-7193081-B2Priority Date: 2002-05-13Grant Date: 2007-03-20
- Phosphonic acid based prodrugs of PMEA and its analoguesPublication Number: US-7214668-B2Priority Date: 2002-05-13Grant Date: 2007-05-08
- Lewis acid mediated synthesis of cyclic estersPublication Number: US-2005282782-A1Priority Date: 2004-06-08
- Lewis acid mediated synthesis of cyclic estersPublication Number: US-7582758-B2Priority Date: 2004-06-08Grant Date: 2009-09-01
- Process for preparation of cyclic prodrugs of pmea and pmpaPublication Number: EP-1504014-B1Priority Date: 2002-05-13Grant Date: 2015-12-02
- Salts of a phosphonic acid based prodrug of PMEAPublication Number: EP-2223927-B1Priority Date: 2002-05-13Grant Date: 2014-10-15
- Process for preparation of cyclic prodrugs of PMEA and PMPAPublication Number: US-2003225277-A1Priority Date: 2002-05-13
Syn
J. Med. Chem. 51 (2008) 666–676
SYN
https://pubs.acs.org/doi/10.1021/jm7012216
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
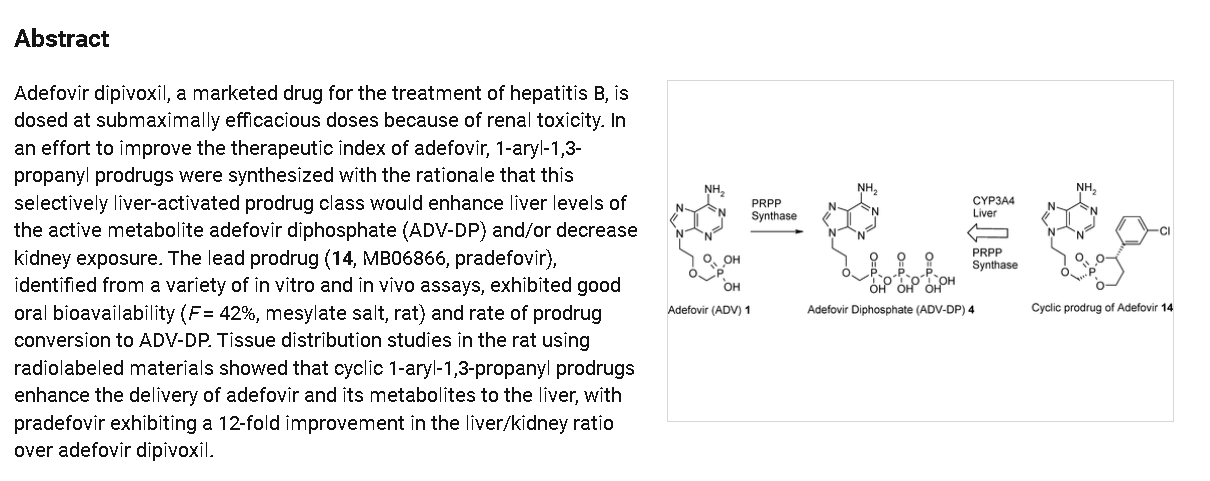
Pradefovir, developed by Xi’an Xintong Pharmaceutical Research Co., Ltd., is a liver-targeted nucleotide analog prodrug designed for the treatment of chronic hepatitis B virus (HBV) infection. It was approved
by the NMPA in 2024, under the brand name Xinshumu, for the treat ment of adult patients with chronic hepatitis B. Pradefovir utilizes HepDirect liver-targeting technology, allowing it to remain stable in the
gastrointestinal tract and bloodstream. It is specifically metabolized into its active form in the liver by the enzyme CYP3A4, leading to high hepatic concentrations and low systemic exposure. This targeted activation enhances antiviral efficacy while minimizing potential side effects on other organs. The clinical efficacy of pradefovir was demonstrated in a Phase III randomized, double-blind, positive-controlled trial (NCT04543565) involving patients with chronic hepatitis B. Participants were randomized to receive pradefovir or tenofovir disoproxil fumarate (TDF) in a 2:1 ratio, with a treatment duration of 96 weeks
followed by a 48-week open-label extension. Interim analysis conducted after 48 weeks showed that pradefovir achieved comparable reductions in HBV DNA levels to TDF, with a favorable safety profile. Regarding safety, pradefovir exhibited a favorable profile [90]. The total occurrence rate of adverse events was similar in both the pradefovir and TDF groups. Nevertheless, the incidence of drug-related adverse events was notably lower in the pradefovir group. Significantly, compared to the typical concerns associated with nucleotide analogs, pradefovir showed a diminished influence on renal function and bone mineral density. This is a crucial aspect considering the known side-effects of nucleotide an alogs in clinical applications, where issues related to kidney function and bone health often pose challenges. Pradefovir, in contrast, appears to have a more favorable safety profile in these regards, which could potentially offer significant advantages in long-term treatment scenarios
[91]. Additionally, it had minimal effects on lipid profiles, suggesting a lower potential risk for cardiovascular events during long-term therapy.
The approval of pradefovir mesylate offers a new therapeutic option for adult patients with chronic hepatitis B, providing effective antiviral activity with an improved safety profile, particularly concerning renal and bone health [92,93].
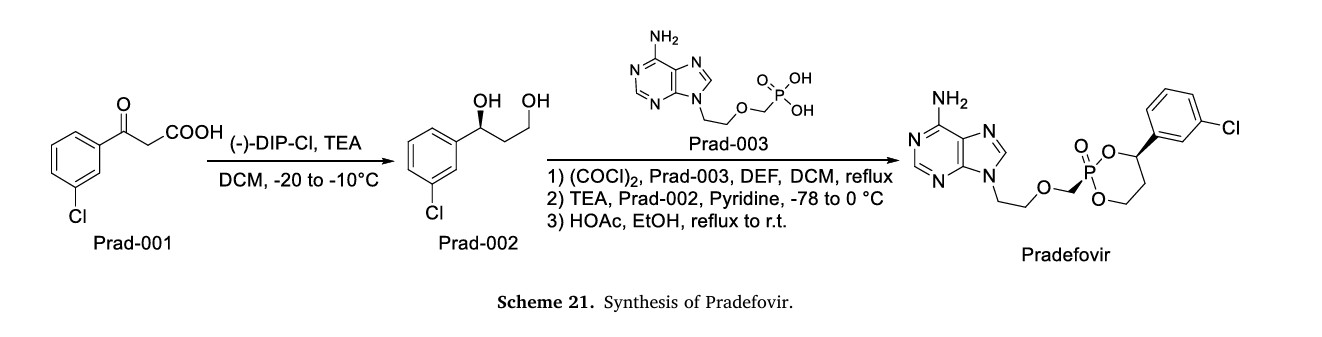
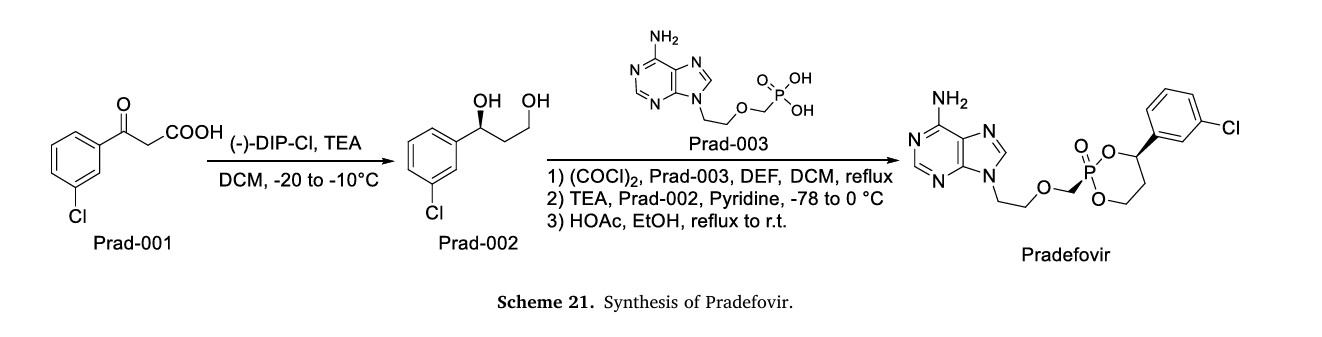
The synthetic route of Pradefovir Mesylate is shown in Scheme 21 [93]. The route commences with a stereoselective reduction of Prad-001 employing ( )-DIP-Cl, affording Prad-002. Subsequent acid-catalyzed cyclodehydration between the hydroxyl groups of Prad-002 and Prad-003 generates Pradefovir, followed by mesylate salt formation to complete the synthesis.
90-93
[90] Y. Gao, F. Kong, X. Song, J. Shang, L. Yao, J. Xia, Y. Peng, W. Liu, H. Gong, M. Mu,
H. Cui, T. Han, W. Chen, X. Wu, Y. Yang, X. Yan, Z. Jin, P. Wang, Q. Zhu, L. Chen,
C. Zhao, D. Zhang, W. Jin, D. Wang, X. Wen, C. Liu, J. Jia, Q. Mao, Y. Ding, X. Jin,
Z. Zhang, Q. Mao, G. Li, J. Niu, Pradefovir treatment in patients with chronic
hepatitis B: week 24 results from a multicenter, double-blind, randomized,
noninferiority, phase 2 trial, Clin. Infect. Dis. 74 (2022) 1925–1932.
[91] Y. Ding, H. Zhang, X. Li, C. Li, G. Chen, H. Chen, M. Wu, J. Niu, Safety,
pharmacokinetics and pharmacogenetics of a single ascending dose of pradefovir, a
novel liver-targeting, anti-hepatitis B virus drug, in healthy Chinese subjects,
Hepatol Int 11 (2017) 390–400.
[92] H. Zhang, M. Wu, X. Zhu, C. Li, X. Li, W. Jin, D. Zhang, H. Chen, C. Liu, Y. Ding,
J. Niu, J. Liu, Safety, efficacy, and pharmacokinetics of pradefovir for the
treatment of chronic hepatitis B infection, Antiviral Res 174 (2020) 104693.
[93] K.R. Reddy, M.C. Matelich, B.G. Ugarkar, J.E. G´omez-Galeno, J. DaRe, K. Ollis,
Z. Sun, W. Craigo, T.J. Colby, J.M. Fujitaki, S.H. Boyer, P.D. van Poelje, M.D. Erion,
Pradefovir: a prodrug that targets adefovir to the liver for the treatment of hepatitis
B, J. Med. Chem. 51 (2008) 666–676.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Pradefovir, HEPATITIS B VIRUS, APPROVALS 2024, CHINA 2024, Xi’an Xintong Pharmaceutical Research Co, Xinshumu, Pradefovir Mesylate, Remofovir, 625095-60-5, GZE85Q9Q61, DTXSID10870372, MB 6866, MB-06866, ICN2001-3
Bevifibatide
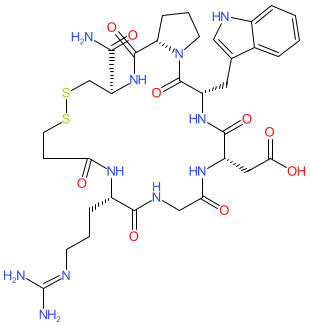
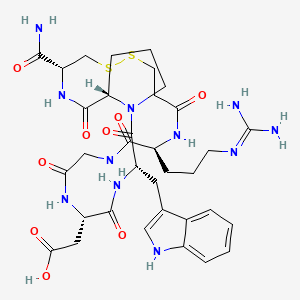
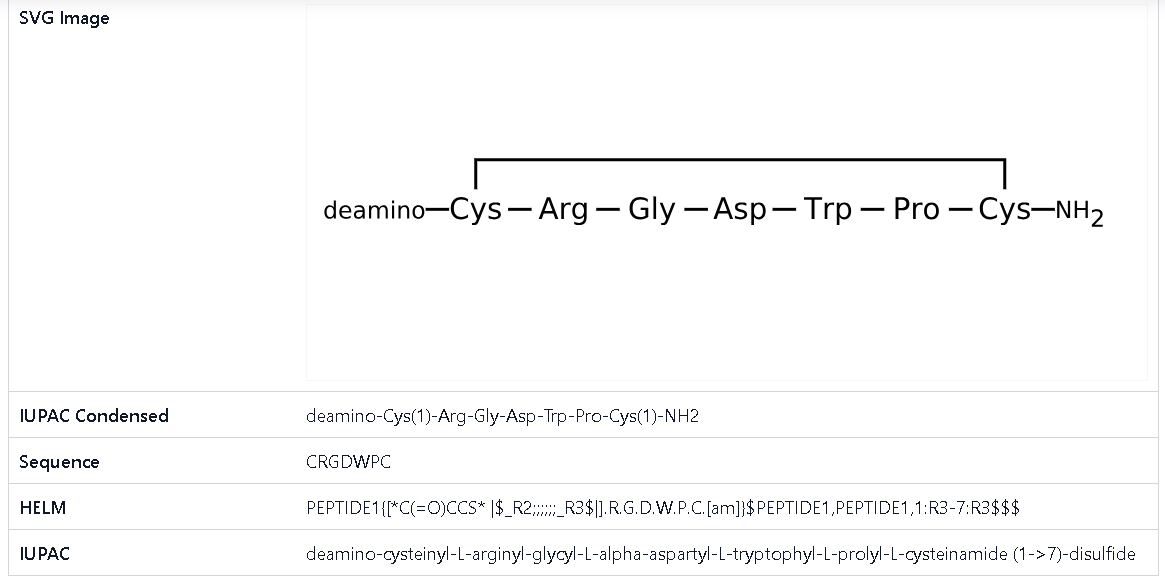
Bevifibatide
Cas 710312-77-9
817.9 g/mol, C34H47N11O9S2
2-[(3S,6S,12S,20R,23S)-20-carbamoyl-12-[3-(diaminomethylideneamino)propyl]-3-(1H-indol-3-ylmethyl)-2,5,8,11,14,22-hexaoxo-17,18-dithia-1,4,7,10,13,21-hexazabicyclo[21.3.0]hexacosan-6-yl]acetic acid
APPROVALS 2025, CHINA 2025, Bio-Thera Solutions, Beitaning RegisteredAcute coronary syndromes
BAT-2094 | batifiban | Beitaning | Betagrin® | Compound I [CN101085809A]
- OriginatorBio-Thera Solutions
- ClassAntiplatelets; Cardiovascular therapies; Cyclic peptides
- Mechanism of ActionIntegrin alphaVbeta3 antagonists; Platelet glycoprotein GPIIb-IIIa complex anatgonists
- 02 Dec 2024Zhujiang Hospital plans a phase II BCAIS-I trial for Acute ischemic stroke in China (IV) (NCT06712004)
- 07 Aug 2024Chemical structure information updated
- 28 Jun 2024Registered for Acute coronary syndromes in China (IV) – First global approval
- Correctin
- 7AKM76YKN5
Bevifibatide is a synthetic cyclic heptapeptide, and its synthesis involves several stages of peptide chemistry. The primary methods used for producing peptides of this nature are solid-phase peptide synthesis (SPPS), followed by cleavage, purification, and cyclization.
Bevifibatide is a cyclic peptide with the Peptide sequence Arg-Gly-Asp-MeAsp-Phg-Val-Nal.
Bevifibatide (Bio-Thera Solutions) is a synthetic cyclic heptapeptide that functions as a αIIbβ3 and αvβ3 integrin receptor antagonist [1]. It was designed to inhibit platelet aggregation as an antiplatelet cardiovascular therapy.
SYN
CN101085809
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN83278873&_cid=P21-MEUT2B-21989-1
| Example 1: Fmoc solid phase synthesis |
| 1: Synthesis of Fmoc-Cys(Trt)-HN-Rink Amide AM resin |
| (1) Fmoc-Rink Amide AM resin (produced by Tianjin Nankai Hecheng Technology Co., Ltd., substitution degree 0.59 mmol/g, 8.4746 g) was added to the solid phase reaction column, washed three times with DMF, and swelled with DCM for 30 minutes. |
| (2) The solution was drained and Fmoc-protection was removed with 20% piperidine in DMF at room temperature for 20 minutes. |
| (3) The solution was drained, the resin was washed five times with DMF, and the solution was drained. |
| (4) Fmoc-Cys(Trt)-OH (2.9285 g), HOBt (0.6755 g), and DIPCDI (0.8 ml) were dissolved in DMF (20 ml) and DCM (20 ml) and pre-reacted in an ice bath for 20 minutes. |
| (5) Add the above reaction solution to the solid phase reaction column, N 2 Stir with air flow to ensure full contact and reaction with the resin, and react at room temperature (31°C) for 2 hours. |
| (6) The solution was drained, and the resin was washed three times with DMF and once with DCM. Acetic anhydride (10 ml), pyridine (8 ml), and N 2 Stir with air flow to ensure full contact and reaction with the resin, and react at room temperature (31°C) for 10 hours. |
| (7) The solution was drained, and the resin was washed three times with DMF, three times with DCM, and three times with methanol. The resin was then dried under vacuum to obtain Fmoc-Cys(Trt)-HN-Rink Amide AM resin (10.2578 g). The degree of substitution was measured to be 0.5086 mmol/g. |
| 2: Synthesis of Fmoc-Pro-Cys(Trt)-HN-Rink Amide AM resin |
| (1) 10.2578 g of Fmoc-Cys(Trt)-Rink Amide AM resin (substitution degree: 0.5086 mmol/g) was added to the solid phase reaction column, washed three times with DMF, and swelled with DCM for 30 minutes. |
| (2) The solution was drained and Fmoc-protection was removed with 20% piperidine in DMF at room temperature for 20 minutes. |
| (3) The solution was drained, the resin was washed five times with DMF, and the solution was drained. |
| (4) Fmoc-Pro-OH (5.061 g), HOBt (3.04 g), and DIPCDI (7.5 ml) were dissolved in DMF (20 ml) and DCM (20 ml) and pre-reacted in an ice bath for 20 minutes. |
| (5) Add the above reaction solution to the solid phase reaction column, N 2 Stir with air flow to ensure full contact and reaction with the resin. React at room temperature (30°C) for 2 hours, and monitor the reaction progress with Kaiser test. |
| (6) The solution was drained and the resin was washed three times with DMF to obtain Fmoc-Pro-Cys(Trt)-HN-Rink Amide AM resin. |
| 3: Synthesis of Mpr-X-Gly-Asp(OtBu)-Trp(Boc)-Pro-Cys(Trt)-HN-Rink Amide AM resin, where X is Arg(Pbf), Har(Pbf) or Lys(Boc) |
| The reaction steps for coupling each protected amino acid are the same as 2, except that the protected amino acids to be coupled are: Fmoc-Trp(Boc)-OH (7.899 g); Fmoc-Asp(OtBu)-OH (6.172 g); Fmoc-Gly-OH (4.460 g); Fmoc-X-OH (9.732 g); Mpr (1.592 g). |
| 4: Linear crude peptide Mpr-X-Gly-Asp-Trp-Pro-Cys-NH 2 Preparation |
| (1) The resin obtained in step 3 was washed three times with DMF, three times with DCM, and three times with methanol, and then dried under vacuum to obtain 21.182 g of Mpr-X-Gly-Asp(OtBu)-Trp(Boc)-Pro-Cys(Trt)-HN-Rink Amide AM resin. |
| (2) The obtained resin was placed in a round-bottom flask and TFA (180 ml), H 2 A mixed solution of O (10 ml) and TIS (10 ml) was introduced into 2 , stir electromagnetically in an ice bath for 10 minutes, remove the ice bath, and react at room temperature (29°C) for 2 hours. |
| (3) After the reaction is completed, the solution is filtered, and the resin is washed twice with TFA. The filtrates are combined and ice-cold ether (2 L) is added to the filtrate. A white precipitate is precipitated, and the precipitate is collected by centrifugation and fully dried in vacuo. |
| (4) The dried white precipitate (4.237 g) was collected to obtain the crude linear peptide Mpr-X-Gly-Asp-Trp-Pro-Cys-NH 2 , sealed and stored at -20℃. |
| 5: Linear crude peptide Mpr-X-Gly-Asp-Trp-Pro-Cys-NH 2 Cyclization |
| The crude linear peptide of batifiban and its analogues obtained in Example 4 was dissolved in water, and 1 mmol/ml of I 2 The mixture was stirred for 30 minutes at room temperature, and the cyclization reaction was followed by analytical HPLC until completion, thereby obtaining Mpr-X-Gly-Asp-Trp-Pro-Cys-NH 2 (Disulfide bridge,Mpr1-Cys7)。 |
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Bevifibatide, developed by Bio-Thera Solutions, is a synthetic peptide that functions as a glycoprotein IIb/IIIa (GP IIb/IIIa) receptor antagonist. It is marketed under the brand name Beitaning. In 2024, the
NMPA approved Bevifibatide citrate injection for use in patients with acute coronary syndrome undergoing percutaneous coronary intervention (PCI), including coronary stent implantation, to reduce the risk of acute occlusion, in-stent thrombosis, no-reflow, and slow flow phenomena. Bevifibatide exerts its therapeutic effects by specifically binding to the GP IIb/IIIa receptors on platelets, thereby inhibiting the binding of fibrinogen, von Willebrand factor, and other adhesive ligands to these receptors [80]. This inhibition prevents platelet aggregation, reducing the risk of thrombotic complications during and after PCI procedures. The clinical efficacy of Bevifibatide was demonstrated in a multicenter Phase III trial involving patients with acute coronary syndrome undergoing PCI (NCT04567890). The study achieved its primary efficacy endpoint, with the composite endpoint event rate at 30 days post-procedure being significantly lower in the Bevifibatide group (4.06%) compared to the control group (6.56 %), indicating superior antithrombotic efficacy. Regarding toxicity, Bevifibatide was generally well-tolerated. The most common adverse events included bleeding complications, which are consistent with the pharmacological action of GP IIb/IIIa inhibitors. These events were manageable with appropriate clinical interventions, and the overall safety profile was comparable to other agents in its class. The approval of Bevifibatide provides a new therapeutic option for patients undergoing PCI, aiming to enhance procedural safety by mitigating thrombotic risks associated with such
interventions [81].
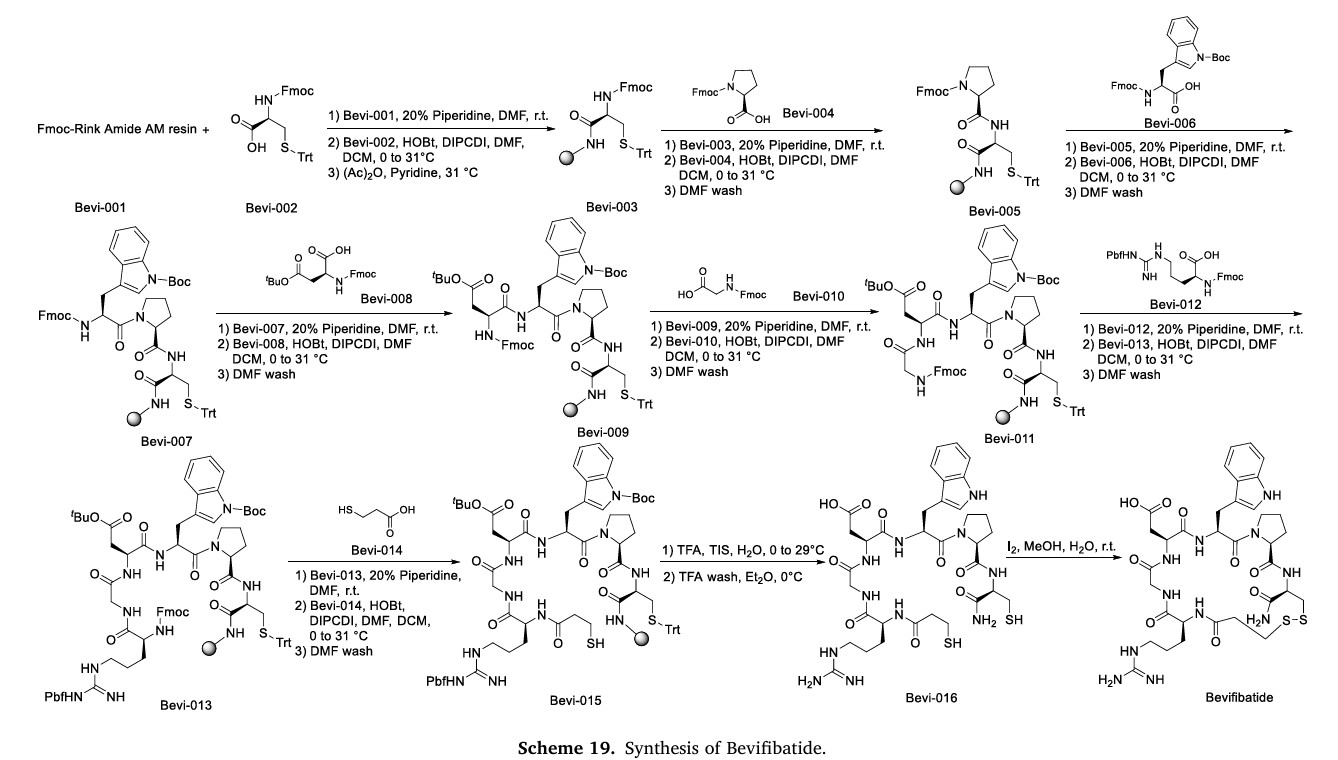
The synthetic route of Bevifibatide, shown in Scheme 19, comprises sequential amidation reactions: Bevi-001 reacts with Bevi-002 to form Bevi-003, which undergoes deprotection and subsequent coupling with
Bevi-004 to generate Bevi-005 [82]. This intermediate undergoes consecutive amidation steps with Bevi-006 and Bevi-008, producing Bevi-007 and Bevi-009 respectively. Bevi-009 then reacts with Bevi-010
to form Bevi-011, followed by coupling with Bevi-012 to yield Bevi-013. Subsequent amidation with Bevi-014 produces Bevi-015, which undergoes TFA-mediated deprotection to give Bevi-016. The final synthesis involves oxidation of the sulfhydryl group in Bevi-016 followed by iodine-mediated coupling to afford Bevifibatide.
80-82
[80] G. Tonin, J. Klen, Eptifibatide, an older therapeutic peptide with new indications:
from clinical pharmacology to everyday clinical practice, Int. J. Mol. Sci. 24 (2023)5446.
[81] H. Patel, I. Lunn, S. Hameed, M. Khan, F.M. Siddiqui, A. Borhani, A. Majid, S.M. Bell, M. Wasay, Treatment of cerebral venous thrombosis: a review, Curr. Med.Res. Opin. 40 (2024) 2223–2236.
[82] S. Tan, Y. Yang, Y. Li, Synthesis of N2-(3-mercapto-1-oxopropyl)-L-arginylglycyl-L-α-aspartyl-L
α-tryptophyl-Lα-prolyl-L Its Analogues, 2007 CN101085809A.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
1. Zhao X, Yuan L, Gong Z, Li M, Yuan Y, Geng J. (2025)
New drugs approved by the NMPA in 2024: Synthesis and clinical applications.
Eur J Med Chem, 291: 117643. [PMID:40262297]
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Pharmacokinetic and pharmacodynamic properties of batifiban coadministered with antithrombin agents in Chinese healthy volunteersPublication Name: Journal of Huazhong University of Science and Technology. Medical sciences = Hua zhong ke ji da xue xue bao. Yi xue Ying De wen ban = Huazhong keji daxue xuebao. Yixue Yingdewen banPublication Date: 2013-10-20PMID: 24142738DOI: 10.1007/s11596-013-1198-4
- Safety, pharmacokinetic and pharmacodynamic studies of batifiban injection following single- and multiple-dose administration to healthy Chinese subjectsPublication Name: Journal of Huazhong University of Science and Technology. Medical sciences = Hua zhong ke ji da xue xue bao. Yi xue Ying De wen ban = Huazhong keji daxue xuebao. Yixue Yingdewen banPublication Date: 2009-02-18PMID: 19224155DOI: 10.1007/s11596-009-0103-7
////////Bevifibatide, APPROVALS 2025, CHINA 2025, Bio-Thera Solutions, Beitaning, BAT 2094, batifiban, 710312-77-9, Correctin, 7AKM76YKN5
Tegileridine
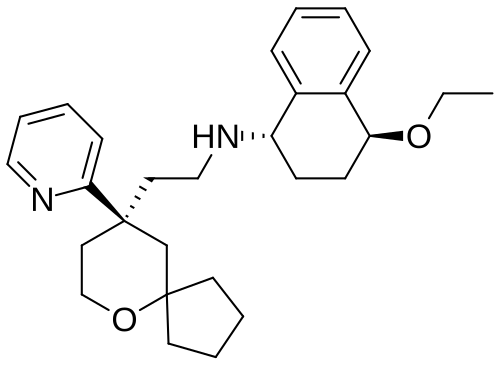
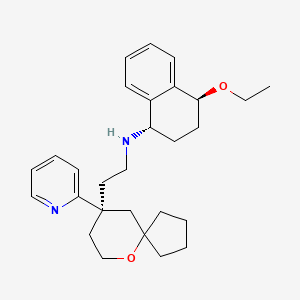
Tegileridine
- YFJS8L4TGU
- CAS 2095345-66-5
- (9R)-N-((1S,4S)-4-Ethoxy-1,2,3,4-tetrahydro-1-naphthalenyl)-9-(2-pyridinyl)-6-oxaspiro(4.5)decane-9-ethanamine
- 434.6 g/mol
WeightAverage: 434.624
Monoisotopic: 434.293328472
Chemical FormulaC28H38N2O2
(1S,4S)-4-ethoxy-N-[2-[(9R)-9-pyridin-2-yl-6-oxaspiro[4.5]decan-9-yl]ethyl]-1,2,3,4-tetrahydronaphthalen-1-amine
- (9R)-N-((1S,4S)-4-Ethoxy-1,2,3,4-tetrahydro-1-naphthalenyl)-9-(2-pyridinyl)-6-oxaspiro(4.5)decane-9-ethanamine
- (1S,4S)-4-Ethoxy-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro(4.5)decan-9-yl)ethyl)-1,2,3,4-tetrahydronaphthalen-1-amine
- (1S,4S)-4-Ethoxy-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro(4.5)decane-9-yl)ethyl)-1,2,3,4-tetrahydronaphthalen-1-amine
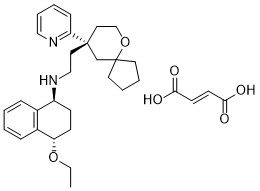
Tegileridine fumarate
CAS#2245827-85-2 (fumarate)
Chemical Formula: C32H42N2O6
Exact Mass: 550.3000
Molecular Weight: 550.70
CHINA 2025, APPROVALS 2025, AISUTE, Jiangsu Hengrui
Tegileridine is under investigation in clinical trial NCT06458400 (To Evaluate the Efficacy and Safety of Tegileridine and Oliceridine Injections in the Treatment of Postoperative Pain).
Tegileridine is a drug which acts as a μ-opioid receptor agonist. It is closely related to compounds such as oliceridine, TRV734, and SHR9352, and shares a similar profile as a biased agonist selective for activation of the G-protein signalling pathway over β-arrestin 2 recruitment.[1]
In January 2024, tegileridine was approved in China for the treatment of moderate to severe pain after abdominal surgery.[2]
SYN
CN107001347
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN203399246&_cid=P20-METU4Y-21400-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017063509&_cid=P20-METU6J-22458-1
[0184](S)-1-Ethyl-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl)ethyl)-1,2,3,4-tetrahydroquinolin-1-amine 1
[0185](R)-1-ethyl-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl)ethyl)-1,2,3,4-tetrahydroquinolin-1-amine 2
[0186]
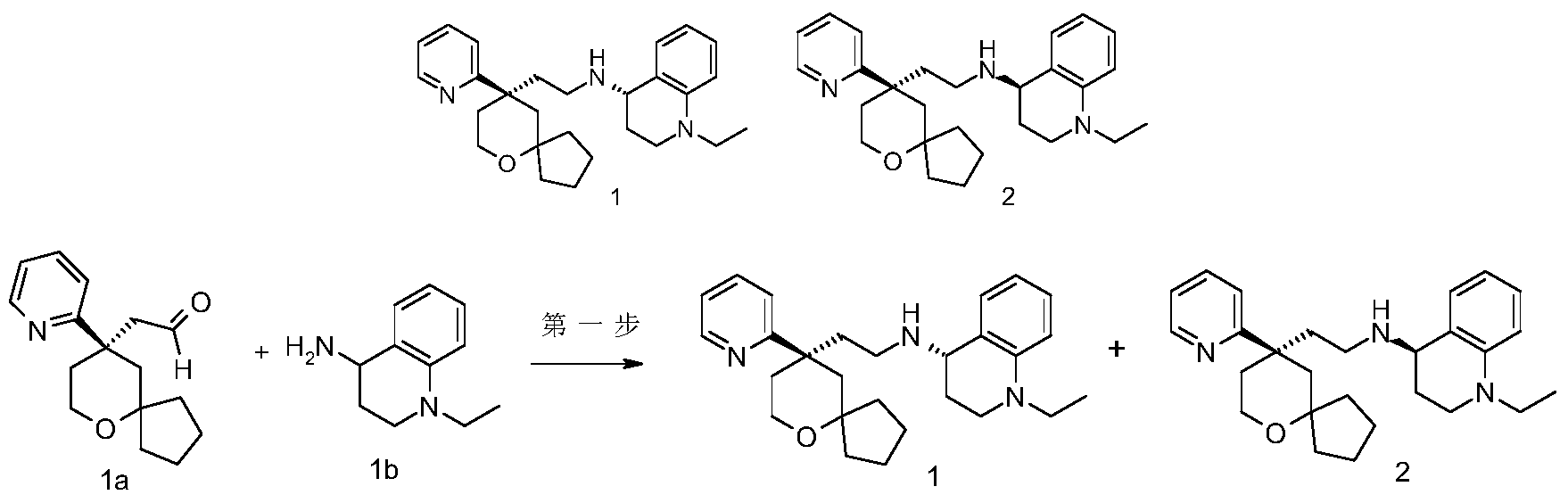
[0187](R)-2-(9-(pyridin-2-yl)-6-oxaspiro[4,5]decane-9-yl)acetaldehyde 1a (294 mg, 1.135 mmol, prepared by the method disclosed in patent application “WO2012129495”) and 1-ethyl-1,2,3,4-tetrahydroquinolin-4-amine 1b (200 mg, 1.135 mmol, prepared by the method disclosed in patent application “WO2014078454”) were dissolved in 15 mL of dichloromethane, stirred for 1 hour, and sodium triacetoxyborohydride (1.203 g, 5.675 mmol) was added and stirred for 16 hours. 20 mL of water was added, and the mixture was extracted with dichloromethane (20 mL×3). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by high performance liquid chromatography to obtain the title product, 1-ethyl-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl)ethyl)-1,2,3,4-tetrahydroquinolin-1-amine. Chiral preparation was performed (separation conditions: chiral preparative column Superchiral S-AD (Chiralway), 2 cm ID*25 cm, 5 um; mobile phase: CO
2 :methanol:diethanolamine=75:25:0.05, flow rate: 50 g/min). The corresponding fractions were collected and concentrated under reduced pressure to give the title products 1 (98 mg, brown oil) and 2 (95 mg, yellow solid).
[0190]Chiral HPLC analysis: retention time 4.028 minutes, chiral purity: 99.7% (chromatographic column: Superchiral S-AD (Chiralway), 0.46 cm ID*15 cm, 5 μm; mobile phase: CO2: methanol: diethanolamine = 75:25:0.05 (v/v/v))
[0191]
1H NMR(400MHz,DMSO-d 6)δ8.54(s,1H),7.72(s,1H),7.45(d,1H),7.20(s,1H),6.95(s,1H),6.78(d,1H),6.52(d,1H),6.37(s,1H),3.60(br,2H),3.18-3.43(m,3H),2.99(m,1H),2.33-2.45(m,3H),1.77-1.99(m,3H),1.19-1.60(m,12H),1.00-1.06(m,4H),0.63(m,1H).
[0194]Chiral HPLC analysis: retention time 3.725 minutes, chiral purity: 99.8% (chromatographic column: Superchiral S-AD (Chiralway), 0.46 cm ID*15 cm, 5 μm; mobile phase: CO2: methanol: diethanolamine = 75:25:0.05 (v/v/v))
[0195]
1H NMR(400MHz,DMSO-d 6)δ8.53(s,1H),7.72(s,1H),7.46(d,1H),7.20(s,1H),6.97(s,1H),6.85(d,1H),6.54(d,1H),6.40(s,1H),3.61(br,2H),3.17-3.25(m,3H),3.00-3.01(m,1H),2.33-2.46(m,3H),1.78-1.97(m,3H),1.24-1.65(m,12H),1.01-1.06(m,4H),0.61(m,1H).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US306969245&_cid=P20-METUA8-25189-1
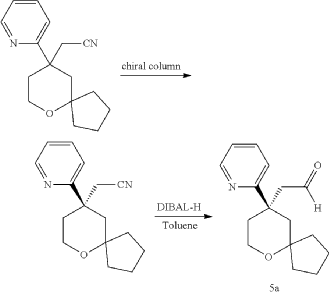
Embodiment 1: Preparation of (1S,4S)-4-ethoxy-N-(2-((R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl)ethyl)-1,2,3,4-tetrahydronaphthalen-1-amine
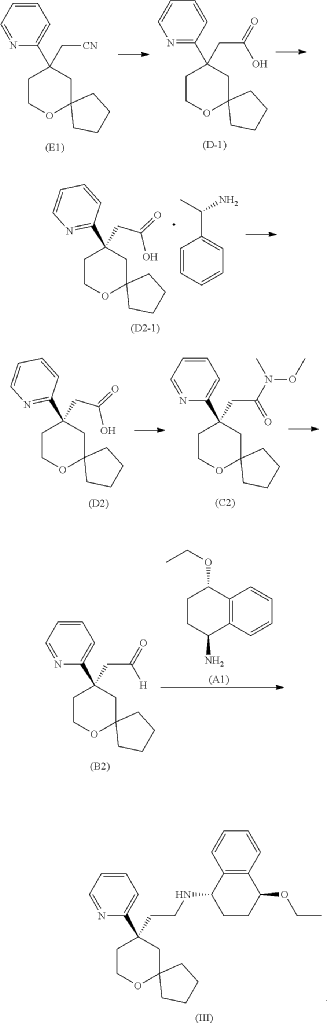
Step One: Synthesis of Intermediate (D-1)
Step Two: Synthesis of Intermediate (D2-1)
Step Three: Synthesis of Intermediate (D2)
Step Four: Synthesis of Intermediate (C2)
Step Five: Synthesis of Intermediate (B2)
Step Six: Synthesis of the Compound Represented by Formula (III)
SYN
SYN
Tegileridine fumarate, developed by Jiangsu Hengrui Pharmaceuti
cals Co., Ltd., is a novel small-molecule analgesic that functions as a
complete opioid receptor agonist with relative selectivity for -opioid
receptors (MOR). It is marketed under the brand name Aisute. In 2024,
the NMPA approved Tegileridine fumarate injection for the treatment of moderate to severe pain following abdominal surgery. Tegileridine ex
erts its analgesic effects by activating MOR, leading to inhibition of
adenylate cyclase activity, decreased intracellular cAMP levels, and
subsequent modulation of ion channel conductance. This results in hy
perpolarization of neuronal membranes and reduced neuronal excit
ability, effectively alleviating pain. The clinical efficacy of Tegileridine
was evaluated in a Phase III randomized, double-blind, placebo-
controlled trial (NCT05012516) involving patients experiencing mod
erate to severe pain after abdominal surgery. The research indicated that
Tegileridine offered substantial alleviation of pain in contrast to the
placebo. It manifested a quick-acting property, and its analgesic effects
endured throughout the period of observation. In terms of toxicity,
Tegileridine was typically well-tolerated by the subjects. The most
frequently encountered adverse reactions were nausea, vomiting, and
dizziness, all of which were of mild to moderate intensity. Importantly,
Tegileridine exhibited a favorable safety profile with a lower incidence
of gastrointestinal adverse reactions compared to traditional MOR ag
onists, potentially offering an improved therapeutic window for post
operative pain management. The approval of Tegileridine provides a
new treatment option for patients suffering from moderate to severe
postoperative pain, particularly following abdominal surgeries,
addressing a significant clinical need in pain management [72,73].
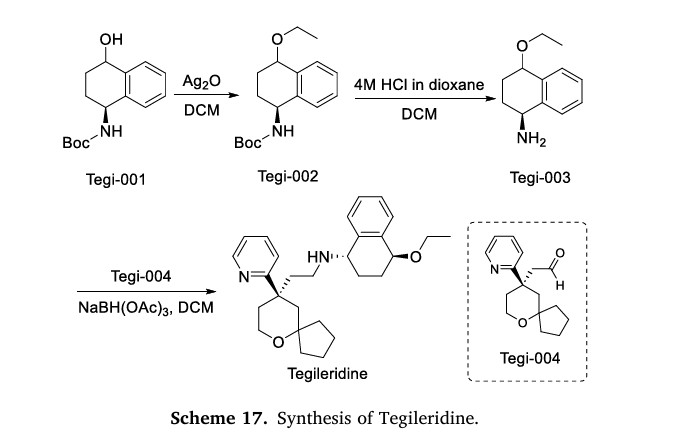
The synthesis of Tegileridine fumarate, illustrated in Scheme 17,
begins with nucleophilic substitution reaction involving Tegi-001 to
yield Tegi-002 [74]. Tegi-002 is subsequently acidified to produce
Tegi-003. Finally, Tegi-003 undergoes reductive amination with
Tegi-004 to synthesize Tegileridine.
[72] S. Dhillon, Correction: tegileridine: first approval, Drugs 84 (2024) 1011.
[73] S. Dhillon, Tegileridine: first approval, Drugs 84 (2024) 717–720.
[74] X. Li, B. Feng, Y. Chen, T. Liu, F. He, M. He, W. Tao, P. Sun, Oxa Spiro Derivative
Useful in Treatment of Pain and pain-related Disease and Its Preparation, 2017.
CN107001347A.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- WO 2017/063509, “Oxa spiro derivative, preparation method therefor, and applications thereof in medicines”, published 10 April 2018, assigned to Jiangsu Hengrui Medicine Company and Shanghai Hengrui Pharmaceutical Company Ltd .
- Dhillon S (June 2024). “Tegileridine: First Approval”. Drugs. 84 (6): 717–720. doi:10.1007/s40265-024-02033-4. PMID 38771484.
| Clinical data | |
|---|---|
| Trade names | 艾苏特 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2095345-66-5 |
| PubChem CID | 129049969 |
| UNII | YFJS8L4TGU |
| Chemical and physical data | |
| Formula | C28H38N2O2 |
| Molar mass | 434.624 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Tegileridine: First ApprovalPublication Name: DrugsPublication Date: 2024-05-21PMID: 38771484DOI: 10.1007/s40265-024-02033-4
- Study of the mass balance, biotransformation and safety of [14C]SHR8554, a novel μ-opioid receptor injection, in healthy Chinese subjectsPublication Name: Frontiers in PharmacologyPublication Date: 2023-09-14PMCID: PMC10538116PMID: 37781692DOI: 10.3389/fphar.2023.1231102
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: US-2018297988-A1Priority Date: 2015-10-15
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: WO-2017063509-A1Priority Date: 2015-10-15
- Oxaspiro derivatives, methods of their manufacture, and their application in pharmaceuticalsPublication Number: JP-6824502-B2Priority Date: 2015-10-15Grant Date: 2021-02-03
- Oxa spiro derivatives, their preparation, and their applications in medicinePublication Number: KR-102703513-B1Priority Date: 2015-10-15Grant Date: 2024-09-06
- Opioid Receptor (MOR) Agonist Salt, Its Fumarate Salt I Crystalline Form, and Process for Making SamePublication Number: JP-7153030-B6Priority Date: 2017-04-14Grant Date: 2023-07-24
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: EP-3354649-A1Priority Date: 2015-10-15
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: EP-3354649-B1Priority Date: 2015-10-15Grant Date: 2019-12-04
- Oxaspiro derivative, process for its production and its application in medicinePublication Number: JP-2018534257-APriority Date: 2015-10-15
- Oxa spiro derivative, preparation method therefor, and applications thereof in medicinesPublication Number: US-10442793-B2Priority Date: 2015-10-15Grant Date: 2019-10-15
////////////Tegileridine, CHINA 2025, APPROVALS 2025, AISUTE, Jiangsu Hengrui, YFJS8L4TGU, 2095345-66-5, Tegileridine FUMARATE
Cetagliptin
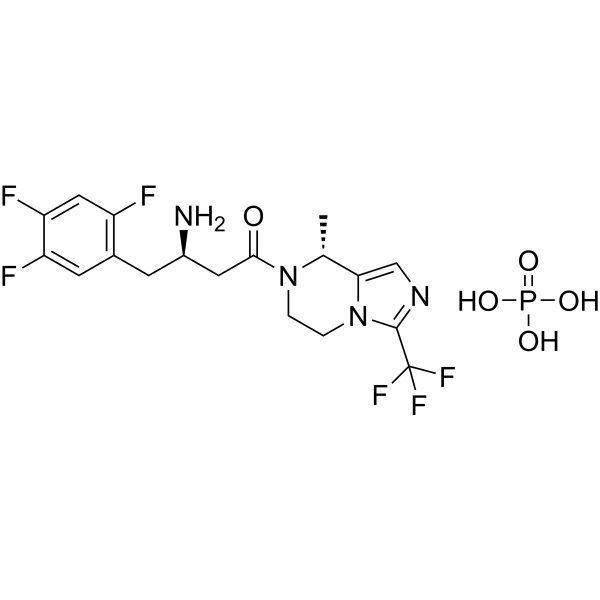
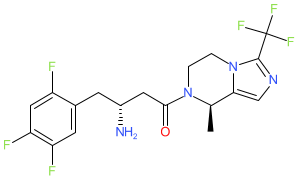
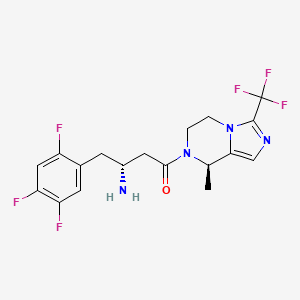
Cetagliptin
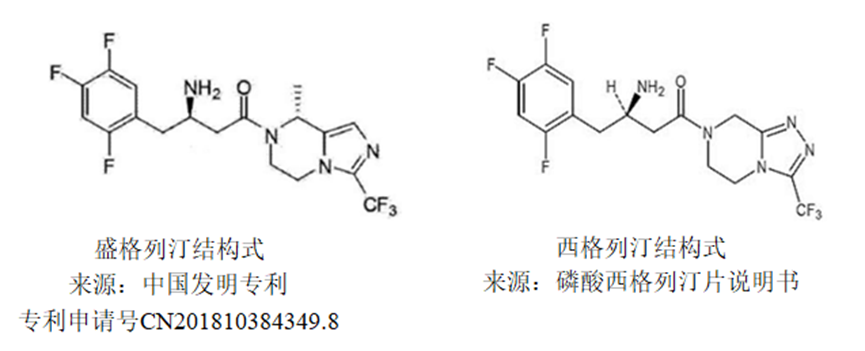
CAS No. FREE FORM : 2243737-33-7 C18H18F6N4O, 420.4 g/mol
[ Cetagliptin Phosphate 2243737-33-7 ]
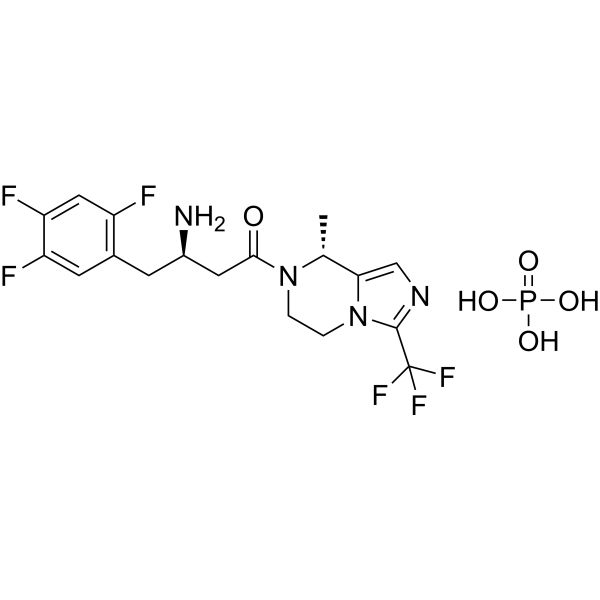
| 분자량 MW | 518.35 |
|---|---|
| 화학식 MF | C18H21F6N4O5P |
(3R)-3-amino-1-[(8R)-8-methyl-3-(trifluoromethyl)-6,8-dihydro-5H-imidazo[1,5-a]pyrazin-7-yl]-4-(2,4,5-trifluorophenyl)butan-1-one
(3R)-3-amino-1-[(8R)-8-methyl-3-(trifluoromethyl)-6,8-dihydro-5H-imidazo[1,5-a]pyrazin-7-yl]-4-(2,4,5-trifluorophenyl)butan-1-one
CHINA 2024, APPROVALS 2024, CGeneTec, DIABETES,
- GTPL13952
- CGT8012
- OriginatorCGeneTech
- Class2 ring heterocyclic compounds; Amines; Antihyperglycaemics; Fluorobenzenes; Imidazoles; Ketones; Pyrazines; Small molecules
- Mechanism of ActionDipeptidyl peptidase 4 inhibitors
RegisteredType 2 diabetes mellitus CHINA 2024
- 01 Dec 2024Registered for Type 2 diabetes mellitus in China (PO) – First global approval
- 20 Mar 2024Chemical structure information added
- 28 Jun 2023No recent reports of development identified for phase-I development in Type-2-diabetes-mellitus(In volunteers) in China (PO, Tablet)
- Cetagliptin is an orally active inhibitor for dipeptidyl peptidase 4 (DPP-4) and CYP2D6 (IC50 of 6 µM). Cetagliptin is a substrate for P-glycoprotein. Cetagliptin reduces the GLP-1 degradation, maintains the level of postprandial blood sugar, and can be used in type 2 diabetes mellitus research.
Cetagliptin (CGT-8012) is an orally bioavailable, dipeptidyl peptidase 4 enzyme (DPP-4) inhibitor (‘gliptin’) class drug. It was designed as an antihyperglycemic agent to treat type 2 diabetes mellitus (T2DM) via inhibition of DPP-4-mediated catbolism of incretin hormones including glucagon-like peptide-1 (GLP-1) [2].
- A DPP-4 inhibitor pharmaceutical composition and its preparation method and usePublication Number: CN-118557538-APriority Date: 2024-08-01
- A kind of preparation method of DPP-IV inhibitor and its key intermediatePublication Number: CN-114057751-APriority Date: 2022-01-17
- A kind of preparation method of DPP-IV inhibitor and its key intermediatePublication Number: CN-114057751-BPriority Date: 2022-01-17Grant Date: 2022-04-12
- A kind of preparation method of DPP-IV inhibitor and key intermediate thereofPublication Number: TW-202330535-APriority Date: 2022-01-17
- A preparation method of a DPP-IV inhibitor and its key intermediatePublication Number: TW-I842342-BPriority Date: 2022-01-17Grant Date: 2024-05-11
- Salt of cetagliptin, preparation method thereof, pharmaceutical composition, and use thereofPublication Number: US-2020123164-A1Priority Date: 2018-04-26
- Salt of cetagliptin, preparation method therefor, pharmaceutical composition, and use thereofPublication Number: EP-3785713-A1Priority Date: 2018-04-26
- Salt of cetagliptin, preparation method thereof, pharmaceutical composition, and use thereofPublication Number: US-11046701-B2Priority Date: 2018-04-26Grant Date: 2021-06-29
- Tetrahydro-imidaz0[1,5-a]pyrazine derivatives, preparation process and medicinal use thereofPublication Number: US-2010273786-A1Priority Date: 2007-12-26
- Tetrahydro-imidazo[1,5-α]pyrazine derivatives, preparation process and medicinal use thereofPublication Number: US-8207161-B2Priority Date: 2007-12-26Grant Date: 2012-06-26
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84092509&_cid=P20-MERZ31-36806-1
SYN
CN103351391
https://patents.google.com/patent/CN103351391A/en
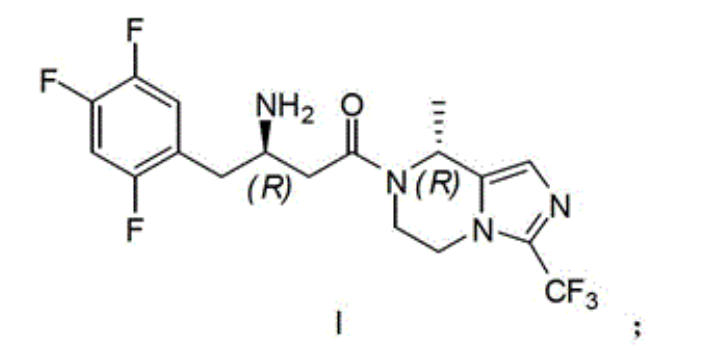
Synthetic route and the concrete steps of compound (I) are as follows:
Step 1: synthetic compound 2
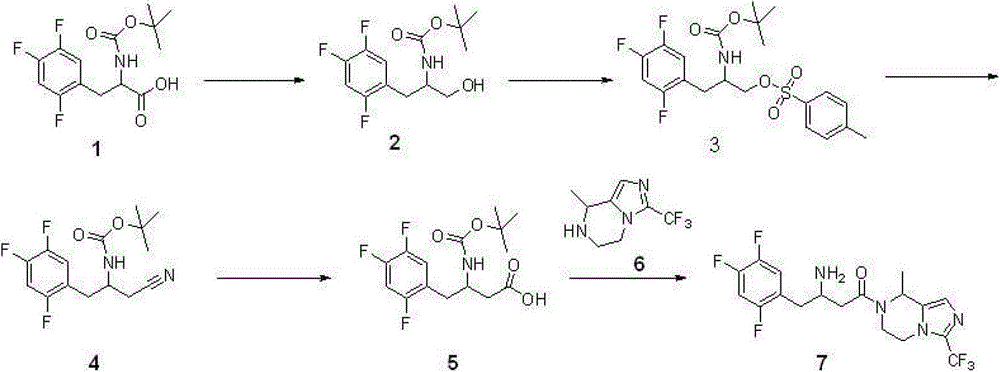
With 11.8 gram (0.037 mole) compound 1{DL N-[(1,1-dimethyl oxyethyl group) carbonyl]-2,4; 5-trifluorophenyl-L-Ala, DL N-[(1,1-dimethylethoxy) carbonyl]-2; 4; 5-trifluorophen yl-alanine, CAS:1367740-01-9, reference: synthetic chemistry; 2011; 19 (4), 557-560} is dissolved among 40 milliliters of THF, adds 5.8 milliliters of triethylamines (0.042 mole) again; reaction is cooled to 0 ℃; add 4.0 milliliters of Vinyl chloroformates (0.041 mole), 0 ℃ was reacted 1 hour under nitrogen protection, after the filtration filtered liquid was cooled to 0 ℃; slowly add sodium borohydride (1.4 grams; 0.057 the mole) mixed solution in 15 ml waters, stirring is spent the night, and adds 1N HCl acidifying; ethyl acetate extraction three times; merge organic phase, sodium hydrogen carbonate solution is washed, the saturated salt washing; anhydrous sodium sulfate drying; the concentrated 7.6 gram products that obtain, namely compound 2, yield 67%.Repeat this step, make more compound 2, use for subsequent step.
Step 2: synthetic compound 3
8.2 gram (0.027 mole) compounds 2 are dissolved in 40 milliliters of methylene dichloride; add again 4.2 milliliters of triethylamines (0.030 mole); the catalytic amount DMAP; reaction is cooled to 0 ℃; add Tosyl chloride (6.8 grams; 0.035 mole); 0 ℃ is arrived room temperature reaction 2 hours under nitrogen protection, adds 1N HCl acidifying, dichloromethane extraction three times; merge organic phase; sodium hydrogen carbonate solution is washed, saturated salt washing, anhydrous sodium sulfate drying; concentrate and obtain crude product, namely compound 3.Repeat this step, make more compound 3, use for subsequent step.
Step 3: synthetic compound 4
12.4 gram (0.027 mole) compounds 3 are dissolved in 40 milliliters of dimethyl formamides, slowly add the mixed solution of sodium cyanide (4.5 grams, 0.092 mole) in 30 milliliters of dimethyl formamides, room temperature reaction 48 hours, pour in 100 milliliters of frozen water, ethyl acetate extraction three times merges organic phase, the saturated salt washing, anhydrous sodium sulfate drying, concentrated rear column chromatography purification obtains 7.8 gram products, be compound 4, yield 92%.
Step 4: synthetic compound
5
3.1 gram (0.010 mole) compounds 4 are dissolved in 15 milliliters of 6N hydrochloric acid, and reflux is spent the night, and adds the neutralization of 2N sodium hydroxide solution, cooling drying.The gained solid is dissolved among 30 milliliters of THF, adds 20 milliliters of 0.5N sodium hydroxide solutions, adds tert-Butyl dicarbonate (2.4 grams again, 0.011 mole), room temperature reaction
16 hours, concentrated, add the neutralization of 10% sodium bisulfate, ethyl acetate extraction three times merges organic phase, the saturated salt washing, anhydrous sodium sulfate drying, the concentrated 3.3 gram products that obtain, namely, compound
5, yield 99%.
Step 5: synthetic compound 7
Compound 6{5; 6; 7; 8-tetrahydrochysene-8-methyl-3-(trifluoromethyl)-imidazo [1,5-a] pyrazine, 5; 6; 7,8-tetrahydro-8-methyl-3-(trifluoromethyl)-imidazo[1,5-a] pyrazine; synthesize and see CN103087067; 2.1 gram, 0.010 mole } be dissolved in 8 milliliters of methylene dichloride, add triethylamine 1.2 grams (0.012 mole); compound 5 (3.3 grams; 0.010 mole), EDCI2.3 restrains (0.012 mole), room temperature reaction is 24 hours under nitrogen protection; pour in 100 milliliters of frozen water; organic phase is washed saturated salt washing, anhydrous sodium sulfate drying; the concentrated crude product that obtains; be dissolved in 100 milliliters of the 2N HCl/ methanol solutions (anhydrous HCl gas is dissolved in the solution of methyl alcohol), room temperature reaction 4 hours is spin-dried for; cooling; pour in 100 milliliters of frozen water, transfer PH to 9, ethyl acetate extraction three times; merge organic phase; and wash saturated salt washing, anhydrous sodium sulfate drying; concentrated; column chromatography purification obtains 2.8 gram products, and namely compound 7, yield 66%.
Compound 7 comprises four optical isomers, and route and the concrete steps of their separation and purification are as follows:
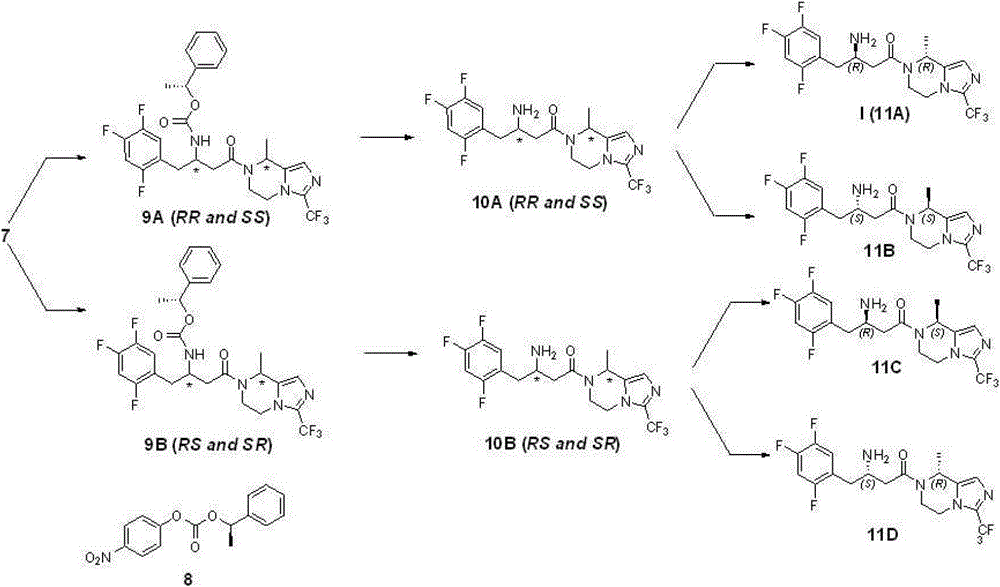
Step 6: preparation compound 9A and 9B
2.8 gram (6.67 mmole) compounds 7 are dissolved in 50 milliliters of acetonitriles; add triethylamine 1.2 grams (8.0 mmole); add again compound 8 (1.9 grams; 6.67 mmole; reference: J.Org.Chem.1995; 60 (3); 730), reflux is spent the night under nitrogen protection, and is concentrated; add ethyl acetate; the 1N sodium hydroxide solution is washed, and ethyl acetate milliliter extraction three times merges organic phase; the saturated salt washing; anhydrous sodium sulfate drying, the evaporating column chromatography purification obtains 1.6 gram 9A (43%) and 1.4 gram 9B (39%) products (de>98%); structural analysis determines that tentatively 9A is RR and SS mixture of enantiomers, and 9B is RS and SR mixture of enantiomers.Gained compound 9A and 9B give over to respectively next step and use.
Step 7: preparation compound 10A and 10B
1.5 gram (2.64 mmole) compound 9A are dissolved in 50 milliliters of methylene dichloride, reaction is cooled to 0 ℃, adds HBr solution (2M, 2.6 milliliters, 5.2 mmole), be dissolved in ethyl acetate after concentrated, sodium hydrogen carbonate solution is washed, the saturated salt washing, anhydrous sodium sulfate drying, the concentrated product that obtains, namely compound 10A (RR and SS mixture of enantiomers) gives over to next step and uses.
According to same reaction principle, condition and step, take compound 9B as starting raw material, obtain compound 10B (RS and SR mixture of enantiomers), give over to next step and use.
Step 8: preparation compound 11A, 11B and 11C, 11D
Resulting compound 10A in the step 7 (1.1 gram) is dissolved in 20 milliliters of ethanol, adds D-tartrate 0.4 gram (2.64 moles), reflux 0.5 hour, cooling, filter, obtain white solid, again with behind ten times of amount ethyl alcohol recrystallizations 2 times, obtain white solid, free with saturated sodium bicarbonate aqueous solution, obtain 0.29 and digest compound 11A, be i.e. compound (I), yield 26% is surveyed ee value>95%.
PAPER
https://www.tandfonline.com/doi/full/10.1080/00498254.2022.2091494
SYN
https://patents.google.com/patent/US11046701B2/en
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Cetagliptin phosphate, developed by CGeneTec, is a DPP-4 inhibitor designed for the treatment of T2DM. In 2024, the NMPA approved cetagliptin phosphate for managing T2DM. As a member of the DPP-4inhibitor, Cetagliptin exerts its effect on glycemic regulation by impeding the breakdown of incretin hormones. This action leads to a glucose-dependent increase in insulin secretion and a concurrent decrease in glucagon levels. Multiple clinical investigations have attested to the effectiveness and safety profile of sitagliptin. In a particular instance, a randomized, double-blind, placebo-controlled Phase 3 study was carried out to assess the use of sitagliptin as a single-agent treatment in patients diagnosed with type 2 diabetes [67]. The study found that cetagliptin significantly reduced HbA1c levels compared to placebo, with a greater proportion of patients achieving target glycemic control.
The treatment was generally well tolerated, with a safety profile comparable to placebo [68,69]. Regarding toxicity, cetagliptin was well tolerated in clinical studies, with no significant increase in adverse effects compared to placebo. No drug-related hypoglycemia was reported,
indicating a favorable safety profile [70].
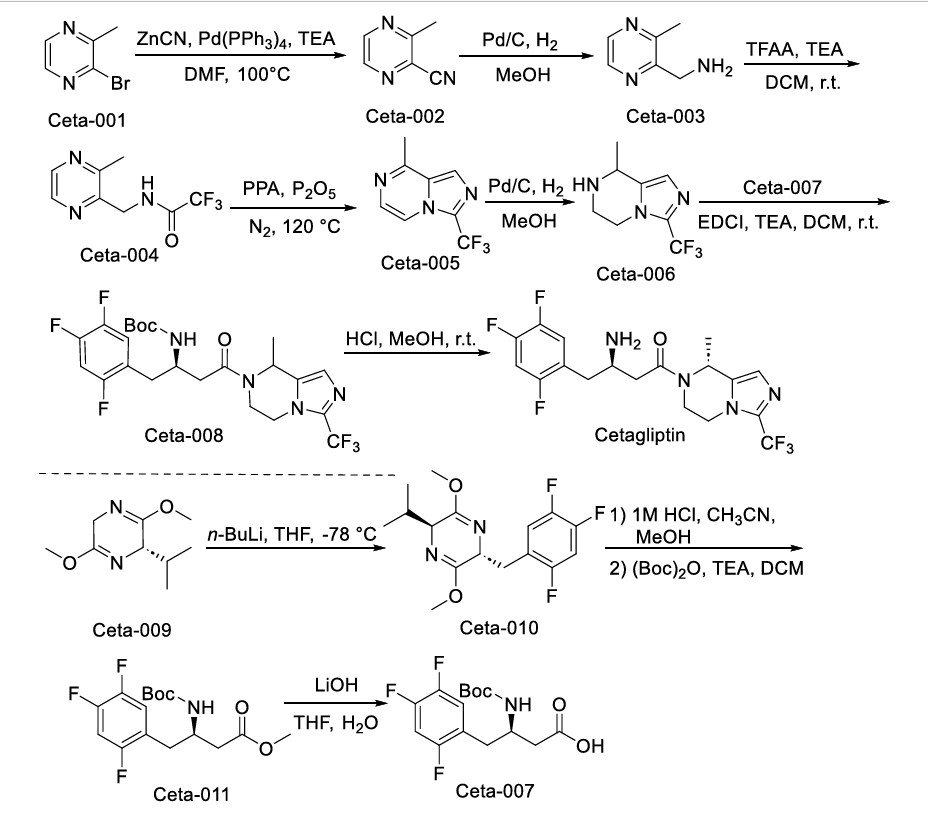
The synthesis of Cetagliptin, depicted in Scheme 16, initiates with Ceta-001 cyanidation affording Ceta-002, whose hydrogenative reduction yields Ceta-003 [71]. Subsequent amidation constructs Ceta-004,
followed by cyclization rearrangement producing Ceta-005. Hydrogenation delivers Ceta-006, which undergoes coupling with Ceta-007 assembling Ceta-008. Final TFA-mediated deprotection achieves
Cetagliptin. Concurrently, the side route involves Ceta-009 nucleophilic substitution forming Ceta-010. Sequential imine hydrolysis/protection converts Ceta-010 to Ceta-011, whose controlled hydrolysis ultimately delivers Ceta-007
67-70
[67] J. Lu, J. Zhao, D. Xie, J. Ding, Q. Yu, T. Wang, Use of a PK/PD model to select
Cetagliptin dosages for patients with type 2 diabetes in phase 3 trials, Clin.
Pharmacokinet. 63 (2024) 1463–1476.
[68] L. Guo, F. Tian, L. Liu, M. Chen, C. Jiang, S. Li, C. Liu, Y. Zhang, J. Qin, D. Yu,
Y. Zong, W. Dai, Retagliptin as add-on therapy to metformin in Chinese patients
with type 2 diabetes inadequately controlled with metformin: a multicentre,
randomized, double-blind, placebo-controlled, phase 3 trial, Diabetes Obes Metab
26 (2024) 2830–2838.
[69] C. Hu, J. Zheng, J. Miao, F. Liu, T.T. Hu, J.K. Gu, S.Q. Shu, Y. Wang, X.H. Zhu, M.
Z. Liang, [Pharmacokinetics of Phosphate Retagliptin Tabletin in Patients with
Renal Dysfunction], Sichuan Da Xue Xue Bao Yi Xue Ban 49 (2018) 74–80.
[70] A. Cahn, S. Cernea, I. Raz, An update on DPP-4 inhibitors in the management of
type 2 diabetes, Expert Opin Emerg Drugs 21 (2016) 409–419.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
https://en.cgenetech.com.cn/news/55.html
Since the listing application of a class of innovative drug Cetagliptin independently developed by CGeneTech was accepted by the State Food and Drug Administration, it has received great attention in the industry. Recently, the well-known industry media “Shell News Agency” also took this opportunity to comprehensively sort out the hot track and broad market prospects of domestic DPP-4 inhibitors. This article is shared with you. In the face of the high expectations given by the industry, CGeneTech will continue to run the last “one kilometer” of product launch with a scientific and rigorous attitude.
Diabetes (DM), as a chronic disease, has attracted much attention. Diabetes drugs have become the second largest drug market after tumor drugs, and it is also a place for pharmaceutical enterprises to compete.
With the development of medicine, some new drugs with different mechanism of action from traditional oral hypoglycemic drugs have emerged in recent years. Dipeptidyl peptidase-4 (DPP-4) inhibitor is one of them. At present, there are dozens of DPP-4 inhibitors, which are collectively known as “gliptin drugs”. In the future, the market size of gliptin drugs in China will exceed 30 billion yuan.
Cetagliptin seven-year long run
On February 2, CGeneTech submitted to the National Drug Administration (NMPA) the marketing application (NDA) of Cetagliptin, a DPP-4 inhibitor, which was mainly used to treat type 2 diabetes. This means that the domestic DPP-4 inhibitor market will usher in new members, and the official website of CGeneTech will also publicize the progress of Cetagliptin research and development in the product pipeline for the first time, and the listing has been confirmed.
Cetagliptin is a Class 1 innovative drug independently developed by CGeneTech, and once was its own fist product in its pipeline. It has also experienced a seven-year long run since its launch of research and development, and is about to hit the line successfully.
In 2006, the targeted hypoglycemic drug Sigliptin was approved by the FDA of the United States, which is undoubtedly a major event in the industry. Ten years later, CGeneTech completed the pre-clinical study of head-to-head comparison of Cetagliptin and Xigliptin.
At the beginning of 2018, CGeneTech launched the phase I clinical trial of head-to-head comparison of Cetagliptin and Xigliptin. Among nearly 200 patients in the Phase I clinical trial completed by Cetagliptin, the data showed that when the intake of Cetagliptin reached 50 mg, it was able to achieve the DPP-4 inhibition capacity equivalent to the intake of 100 mg of Xigliptin. Cetagliptin is administered once a day. It can reach the peak within 1 to 2 hours after administration, and has a longer half-life than Sigliptin, which can maintain stable glucose reduction for a longer time.
Diabetes requires long-term medication, and safety is the first factor to be considered when doctors choose drugs when prescribing. In the safety study, the adverse effects of the intake of Cetagliptin on the body of patients were almost undetectable, lower than that of the blank group and Sigliptin group. In addition, although Cetagliptin has a long half-life, there is no accumulation of residual drugs in the body in the phase I clinical trial, which reflects the high selectivity and strong inhibition of Cetagliptin. The beautiful phase I clinical trial data have provided the foundation for the later clinical trial research of Cetagliptin.
In 2019, Cetagliptin was officially approved by the National Drug Evaluation Center to “exempt Phase II clinical trials from Phase III trials”, becoming the first DPP-4 inhibitor in the world to pass the quantitative pharmacological model, exempt Phase II clinical trials, and directly carry out Phase III confirmatory trials, which attracted the attention of experts in the field of diabetes at home and abroad.
In October 2022, the unblinding results of Cetagliptin phase III clinical trial showed that the reduction of glycosylated hemoglobin (HbA1c) in Cetagliptin tablet 50mg group reached the main clinical end point at the end of the 24th week, which was significantly superior to the control group. After 28 weeks, the Cetagliptin 100mg dose group also showed good drug safety, and the incidence of adverse reactions was similar to that of the placebo group. The clinical trial has shown the advantages of halving the dose but the same efficacy as similar products.
In February 2023, the marketing application (NDA) of Cetagliptin has been accepted by NMPA for the treatment of type 2 diabetes.
The approval of Cetagliptin has attracted much attention, which means that CGeneTech will officially participate in the domestic hot track of DPP-4 inhibitors, and the market of 10 billion statins will usher in new members.
DPP-4 inhibitor track is hot
DPP-4 inhibitors play a hypoglycemic role mainly by inhibiting the degradation of glucagon-like peptide-1 (GLP-1) by DPP-4 enzyme, promoting insulin and glucose dependent secretion, and inhibiting glucagon secretion, which can improve β Cell dysfunction does not increase the risk of hypoglycemia and body weight of patients. Moreover, DPP-4 inhibitor is a “mild and versatile”. It is mild, versatile and safe in reducing blood sugar. It is an oral drug that can be combined with various drugs in the whole process of management.
As the current mainstream hypoglycemic drug, DPP-4 inhibitor has become a hot spot in the eyes of major pharmaceutical enterprises. At present, there are five kinds of DPP-4 inhibitors that are taken daily on the market in China: Sigliptin, Viggliptin, Shagliptin, Aggliptin and Liggliptin, and these “five golden flowers” are included in the national health insurance list.
After entering medical insurance, the sales of several products have increased significantly. It is understood that from 2016 to 2022, the annual sales of DPP-4 inhibitors showed a continuous growth trend, with the highest year-on-year growth rate in 2018. In 2021 alone, the domestic sales of DPP-4 inhibitors reached nearly 7 billion yuan.
Sigliptin
Sigliptin is the first oral DPP-4 inhibitor on the market in the world, developed by MSD. It was approved by FDA for listing in October 2006; Sigliptin was approved for listing in China in September 2009; In July 2012, its compound preparation was approved for registration in China.
According to MSD’s annual report, the global market share of Sigliptin has been stable at more than US $3 billion in the past four years, ranking first in the global sales of DPP-4 inhibitors. At present, there are 14 pharmaceutical enterprises in China, including Zhengda Tianqing, Qilu Pharmaceutical, Kelun Pharmaceutical and Zhejiang Pharmaceutical, which have been copied and approved for production.
Viggliptin is the second DPP-4 inhibitor in the world developed by Novartis. In September 2007, Viggliptin was first approved for listing by the European Commission; In August 2011, it was officially approved for listing in China.
According to Novartis annual report, the global sales volume of Vigiletin has fluctuated steadily in recent years, basically maintaining at about 1.1 billion US dollars. The imitative production of Viggliptin in the domestic market is also hot. At present, 18 pharmaceutical enterprises such as Qilu Pharmaceutical, Yangzijiang Pharmaceutical, Jiangsu Haosen Pharmaceutical, Shandong Langnuo Pharmaceutical and Nanjing Shenghe Pharmaceutical have been approved for production. They are worthy of the title of the king of domestic imitative drugs for DPP-4 inhibitors.
Shagliptin was jointly developed by Bristol-Myers Squibb and AstraZeneca. It was approved by FDA for listing in July 2009; In May 2011, Shagliptin was approved for listing in China. Shagliptin’s overseas market share exceeded 20%. At present, there are five pharmaceutical enterprises in China, including Zhengda Tianqing, Qilu Pharmaceutical and Jiangsu Aosaikang Pharmaceutical, whose generic drugs have been approved for production.
Liggliptin was developed by BI. In May 2011, it was approved for listing by the FDA of the United States, and was jointly sold by Berger Ingelheim and Lilly. In March 2013, China approved the import registration of liggliptin. Liggliptin’s overseas market share exceeds 15%. At present, there are 6 pharmaceutical enterprises in China, including Guangdong East Sunshine Pharmaceutical, Yangzijiang Pharmaceutical and Kelun Pharmaceutical, which have been approved for production.
Agiletin
Agiletin was developed by Takeda Pharmaceutical of Japan. Approved for listing in Japan in April 2010; In January 2013, it was approved by the US FDA for listing; In July of the same year, Agiletin obtained the import registration certificate of China. According to the statistics of IQVIA, the sales amount of Agiletin in the Chinese market in 2022 was 52.36 million yuan. At present, 11 pharmaceutical enterprises such as Yabao Pharmaceutical, Ruiyang Pharmaceutical and Guorui Pharmaceutical of the National Pharmaceutical Group have been approved for production.
Throughout the domestic market of DPP-4 inhibitors, the original drugs and generic drugs of the “five golden flowers” are all in the Jianghu. In order to break the competition pattern, pharmaceutical enterprises have also invested in innovative self-research teams.
At present, the research and development of innovative DPP-4 inhibitors is also advancing rapidly. According to the data, in addition to the approval of CGeneTech’s Cetagliptin, many innovative DPP-4 inhibitors (excluding compound preparations) have entered the clinical research stage in China.
TQ-F3083 of Nanjing Shunxin, Shingliptin of Chenxin Pharmaceutical, and Boggliptin of Shandong Baiji Dichang Pharmaceutical are in clinical phase II; Fugliptin of Xinritai, DBPR108 of Shiyao Group, HSK7653 of Hisco and Unigliptin of Yuandong Biological are all in clinical phase III; Hengrui Pharmaceutical’s Retagliptin has submitted its listing application.
Although there are only a few “Ting” who have been approved to market independently developed DPP-4 inhibitors in China, the approval of Cetagliptin will take the lead in ushering in the harvest period of domestic innovative DPP-4 inhibitors, break the monopoly of non-self-developed DPP-4 inhibitors again, and give great confidence to pharmaceutical enterprises engaged in the research and development of DPP-4 inhibitors.
epilogue
The huge market potential of diabetes is like a magnet, attracting pharmaceutical enterprises to participate in the hot domestic track of DPP-4 inhibitors.
As the first oral DPP-4 inhibitor launched in the world and China, Sigliptin has been in the Chinese market for more than ten years, and still dominates the market. According to the Phase I clinical trial study, Cetagliptin has obtained significantly better data than Sigliptin in terms of efficacy, safety, half-life, toxicology and pathology, which will have considerable market persuasion and is expected to help it become a similar Best-in-class product, or change the curve overtaking into a competitive pattern.
Cetagliptin is only one step away from its listing. Not only is CGeneTech full of expectations for it, but also the industry has high expectations. Cetagliptin can be expected in the future, and we also expect more home-made original new “Ting” to come out.
reference material:
1. CGeneTech official website, official account
2. New weapon for treating diabetes (I) – DPP-4 inhibitor, Department of General Medicine, Shenzhen Hospital, University of Hong Kong, December 9, 2020
3. Unique Mechanism, Multi-dimensional Benefits – Mechanism and Clinical Application of DPP-4 Inhibitor, China Medical Forum Endocrinology Today, April 9, 2020
4. DPP-4 inhibitor market may add new force. Can CGeneTech break the “five giants” pattern
5. Market | DPP-4 inhibitor market pattern seen from the withdrawal of the first generic antidiabetic drug from the network of East Sunshine, CPHI Pharmaceutical Online, November 17, 2022
- [1]. Zhou C, et al., Safety, tolerability, pharmacokinetics and pharmacokinetic-pharmacodynamic modeling of cetagliptin in patients with type 2 diabetes mellitus. Front Endocrinol (Lausanne). 2024 Mar 11;15:1359407. [Content Brief][2]. Lu J, et al., In vitro study of the drug-drug interaction potential of cetagliptin and clinical study of pharmacokinetic interaction of cetagliptin and metformin in healthy volunteers. Xenobiotica. 2021 Oct;51(10):1122-1131. [Content Brief]
/////////Cetagliptin, CHINA 2024, APPROVALS 2024, CGeneTec, DIABETES, GTPL13952, CGT 8012,
Cofrogliptin
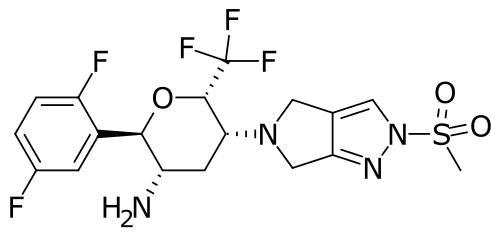
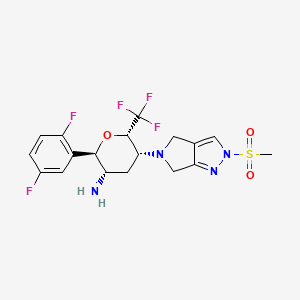
Cofrogliptin
HSK 7653
- Haisco HSK 7653
- CAS 1844874-26-5
- 466.4 g/mol
- C18H19F5N4O3S
(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)-6-(trifluoromethyl)oxan-3-amine
- (2R,3S,5R,6S)-2-(2,5-Difluorophenyl)-5-(2- (methanesulfonyl)-2,6-dihydropyrrolo(3,4-C)pyrazol- 5(4H)-yl)-6-(trifluoromethyl)oxan-3-amine
- (2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-[2- (methanesulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol- 5(4H)-yl]-6-(trifluoromethyl)oxan-3-amine
- (6R)-5-Amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-(2,6-dihydro-2-(methylsulfonyl)pyrrolo(3,4-C)pyrazol-5(4H)-yl)-1,1,1-trifluoro-D-arabino-hexitol
- (6R)-5-Amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-[2,6-dihydro-2-(methylsulfonyl)pyrrolo[3,4-c]pyrazol-5(4H)-yl]-1,1,1-trifluoro-D-arabino-hexitol
- D-Arabino-hexitol, 5-amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-(2,6-dihydro-2-(methylsulfonyl)pyrrolo(3,4-C)pyrazol-5(4H)-yl)-1,1,1-trifluoro-, (6R)-
- (2r,3s,5r,6s)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4h)-yl]-6-(trifluoromethyl)-tetrahydro-2h-pyran-3-amine
APPROVALS 2024, CHINA 2024, Haisco Pharmaceutical Group Co, Beichangping, DIABETES
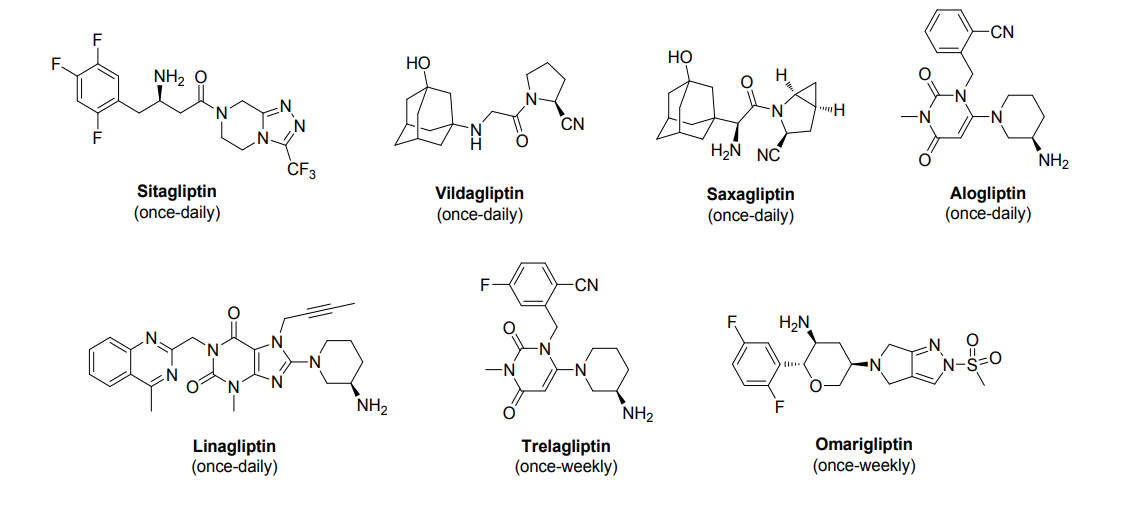
Cofrogliptin (developmental name HSK7653) is a long-acting DPP4 inhibitor dosed once every two weeks.[1][2][3][4]
Cofrogliptin (HSK7653) (compound 2), a tetrahydropyran derivative, is a potent oral dipeptidyl aminopeptidase 4 (DPP-4) inhibitor with Long-acting antidiabetic efficacy. Cofrogliptin (compound 2) has a great potential for type 2 diabetes mellitus (T2DM) .
SYN
J Med Chem. 2020 Jul 9;63(13):7108-7126
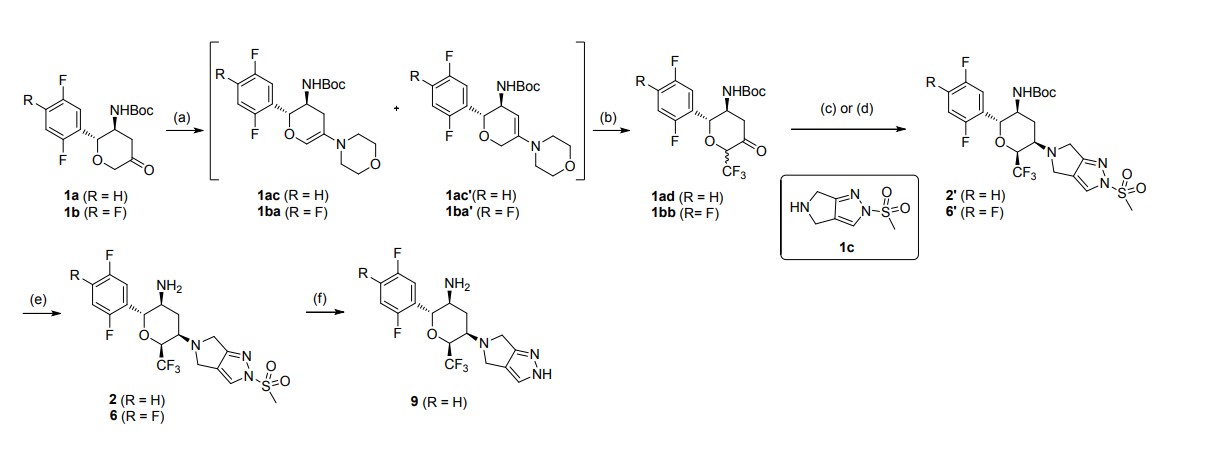
aReagents and conditions: (a) morpholine, toluene, reflux in Dean-Stark appartus; (b)
Umemoto’s reagent, DMAP, DMAc; (c) step 1: 1c, toluene, reflux; step 2: NaBH(OAc)3, CH3COOH, 1,2-DCE; (d) step 1: 1c, CHCl3, reflux in Dean-Stark apparatus; step 2:
NaBH(OAc)3, CH3COOH, 1,2-DCE; (e) TFA, DCM; (f) t-BuOK, THF
Step 2: To a stirred solution of tert-butyl N-[(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-
(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)-6-
(trifluoromethyl)tetrahydropyran-3-yl]carbamate (2′) (407.5 mg, 0.72 mmol) in DCM (6
mL) was added CF3COOH (2 mL) under nitrogen at 0 ℃. After the addition, the reaction
mixture was allowed to warm to room temperature and stirred for 2 h. The reaction mixture
was quenched with a saturated solution of Na2CO3 (15 mL), and extracted with DCM (15
mL × 2). The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo.
The residue was purified by flash column chromatography (Eluent: DCM/MeOH = 80:1–
30:1) to afford the desired product 2 (301.9 mg, yield: 90%). White solid. Mp: 150.1–152.0
℃. [α]D20 = +17.6 (c = 2.000 in MeOH). Rf= 0.40 (1:15 MeOH/CH2Cl2, TLC).
1H NMR
(400 MHz, CDCl3) δ = 7.71 (s, 1H), 7.20 – 7.12 (m, 1H), 7.10 – 6.97 (m, 2H), 4.63 (d, J =
10.0 Hz, 1H), 4.49 – 4.38 (m, 1H), 4.07 – 3.97 (m, 2H), 3.93 – 3.81 (m, 2H), 3.53 – 3.42
(m, 1H), 3.29 (s, 3H), 3.01 – 2.91 (m, 1H), 2.45 – 2.35 (m, 1H), 2.07 – 1.93 (m, 1H), 1.19
(br. s, 2H). 13C NMR (100 MHz, CDCl3) δ = 163.6, 159.1 (dd, J = 2.3 Hz, 235.8 Hz), 156.6
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0223523424003441
SYN
WO2015192701
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015192701&_cid=P20-MEQV3M-18104-1
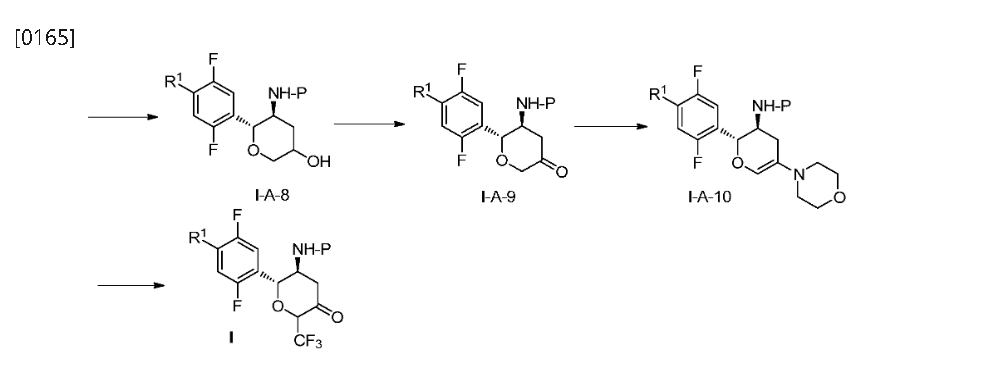
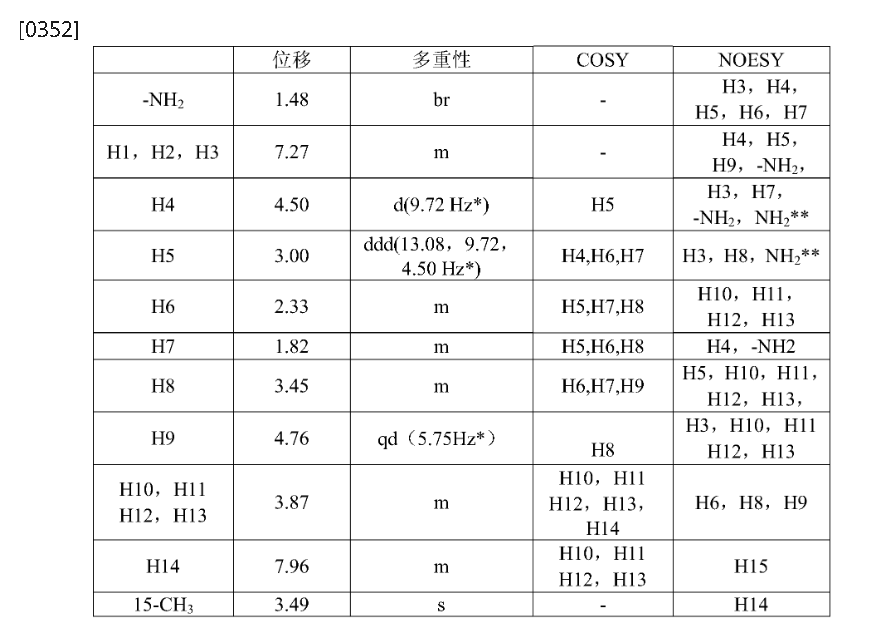
Step 4: (2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-(methylsulfonyl)-pyrrolo[3,4]pyrazol-5(2H,4H,6H)-yl)-6-(trifluoromethyl)tetrahydro-2H-pyran-3-amine (Compound 3)
[0345]
(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-(methylsulfonyl)pyrrolo[3,4-c]pyrazol-5(2H,4H,6H)-yl)-6-(trifluoromethyl)tetrahydro-2H-pyran-3-amine
[0346]3c (410 mg, 0.72 mmol) was dissolved in 6 mL of dichloromethane and 2 mL of trifluoroacetic acid and stirred at room temperature for 1 hour. After completion, saturated aqueous sodium bicarbonate (30 mL) was added to quench the reaction. After separation, the aqueous phase was extracted with ethyl acetate (30 mL x 2). The combined organic phases were dried over anhydrous sodium sulfate, and concentrated. Purification by silica gel column chromatography (dichloromethane/methanol (v/v) = 30:1) afforded compound 3 (250 mg, 75% yield) as a white powder.
[0347]MS m/z(ESI): 467.1[M+1];
[0348]
1H NMR(400MHz,DMSO-d 6):δ7.96(m,1H),7.35–7.04(m,3H),4.86–4.63(qd,1H),4.50(d,1H),3.95(dd,2H),3.78(dd,2H),3.49(s,3H),3.45(m,1H),3.00(ddd,1H),2.33(m,1H),1.82(m,1H),1.48(br,2H)。
SYN
Cofrogliptin, developed by Haisco Pharmaceutical Group Co., Ltd., is a novel, ultra-long-acting dipeptidyl peptidase-4 (DPP-4) inhibitor designed for the treatment of T2DM. It is marketed under the brand name (Beichangping). In 2024, the NMPA approved Cofrogliptin for improving blood glucose control in adult patients with T2DM [59].Cofrogliptin acts pharmacologically by inhibiting DPP-4, an enzyme tasked with degrading incretin hormones like glucagon-like peptide-1(GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). By obstructing the degradation of these hormones, it amplifies their activity. This leads to a glucose-dependent rise in insulin secretion and a
corresponding decrease in glucagon release, which in turn improves glycemic control. The clinical efficacy of Cofrogliptin was demonstrated in Phase III, randomized, double-blind, non-inferiority trial
(NCT04556851), where its efficacy and safety were compared to those of daily linagliptin in patients with T2DM whose blood sugar was not well-controlled by metformin. The study reported that Cofrogliptin
administered once every two weeks achieved a reduction in HbA1c comparable to that of daily linagliptin, with a mean decrease of approximately 0.96 % over 24 weeks. Regarding toxicity, Cofrogliptin
was generally well-tolerated [60,61]. The incidence of hypoglycemia was low, and no severe hypoglycemic events directly attributed to the drug were reported.
The synthesis of Cofrogliptin, illustrated in Scheme 14, initiates with trifluoromethylation of Cofr-001 via oxidation, affording Cofr-002 [62]. Nucleophilic addition of Cofr-003 to Cofr-002 yields Cofr-004, followed by NaBH(OAc)3 reduction to Cofr-005. TFA-mediated deprotection of Cofr-005 ultimately delivers Cofrogliptin. Concurrently, Cofr-006 undergoes nucleophilic substitution with Cofr-007 to form Cofr-008, whose deprotection regenerates Cofr-003
[59] L. Gao, F. Bian, T. Pan, H. Jiang, B. Feng, C. Jiang, J. Sun, J. Xiao, P. Yan, L. Ji,
Efficacy and safety of cofrogliptin once every 2 weeks in Chinese patients with type
2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial, Diabetes
Obes Metab 27 (2025) 280–290.
[60] C. Cui, F. Cao, I.I. Kong, Q. Wu, F. Li, H. Li, D. Liu, A model-informed approach to
accelerate the clinical development of cofrogliptin (HSK7653), a novel ultralong-
acting dipeptidyl peptidase-4 inhibitor, Diabetes Obes Metab 26 (2024) 592–601.
[61] Q. Ren, L. Li, X. Su, X. Hu, G. Qin, J. Han, Y. Liu, J. Wang, L. Ji, Cofrogliptin once
every 2 weeks as add-on therapy to metformin versus daily linagliptin in patients
with type 2 diabetes in China: a randomized, double-blind, non-inferiority trial,
Diabetes Obes Metab 26 (2024) 5013–5024.
[62] C. Zhang, J. Wang, C. Li, Y. Wei, Amino Pyranoid Ring Derivative as DPP-IV
Inhibitor and Its Preparation, 2015. WO2015192701A1.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ji, Linong; Bian, Fang; Pan, Tianrong; Jiang, Hongwei; Jiang, Chengxia; Ren, Qian (20 June 2023). “55-OR: HSK7653, a Novel Ultralong-Acting DPP-4 Inhibitor, as Monotherapy in Patients With Type 2 Diabetes—A Randomized, Double-Blind, Placebo-Controlled Phase III Trial”. Diabetes. 72 (Supplement_1). doi:10.2337/db23-55-OR. S2CID 259433641.
- Zhang, Miao; Zhang, Shudong; Yu, Zhiheng; Yao, Xueting; Lei, Zihan; Yan, Pangke; Wu, Nan; Wang, Xu; Hu, Qin; Liu, Dongyang (October 2023). “Dose decision of HSK7653 oral immediate release tablets in specific populations clinical trials based on mechanistic physiologically-based pharmacokinetic model”. European Journal of Pharmaceutical Sciences. 189 106553. doi:10.1016/j.ejps.2023.106553. PMC 10485820. PMID 37532063.
- Liu, Yang; Yan, Shuai; Liu, Jie; Liu, Hongzhong; Song, Ling; Yao, Xueting; Jiang, Ji; Li, Fangqiong; Du, Ke; Liu, Dongyang; Hu, Pei (May 2023). “Development and validation of an HPLC coupled with tandem mass spectrometry method for the determination of HSK7653, a novel super long-acting dipeptidyl peptidase-4 inhibitor, in human plasma and urine and its application to a pharmacokinetic study”. Biomedical Chromatography. 37 (5): e5607. doi:10.1002/bmc.5607. PMID 36802077. S2CID 257048524.
- Bai, Nan; Wang, Jin; Liang, Wenxin; Gao, Leili; Cui, Wei; Wu, Qinghe; Li, Fangqiong; Ji, Linong; Cai, Yun (6 November 2023). “A Multicenter, Randomized, Double-Blind, Placebo-Controlled, and Dose-Increasing Study on the Safety, Tolerability and PK/PD of Multiple Doses of HSK7653 by Oral Administration in Patients with Type 2 Diabetes Mellitus in China”. Diabetes Therapy. 15 (1): 183–199. doi:10.1007/s13300-023-01496-0. PMC 10786778. PMID 37930584.
| Clinical data | |
|---|---|
| Other names | HSK7653 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1844874-26-5 |
| PubChem CID | 118613788 |
| ChemSpider | 115037226 |
| UNII | LH4G6K6NKP |
| ChEMBL | ChEMBL4646510 |
| Chemical and physical data | |
| Formula | C18H19F5N4O3S |
| Molar mass | 466.43 g·mol−1 |
- [1]. International Nonproprietary Names for Pharmaceutical Substances (INN)[2]. Chen Zhang, et al. Design, Synthesis, and Evaluation of a Series of Novel Super Long-Acting DPP-4 Inhibitors for the Treatment of Type 2 Diabetes. J Med Chem. 2020 Jul 9;63(13):7108-7126. [Content Brief]
///////Cofrogliptin, APPROVALS 2024, CHINA 2024, Haisco Pharmaceutical Group Co, Beichangping, DIABETES, HSK 7653, Haisco HSK 7653, 1844874-26-5
Janagliflozin
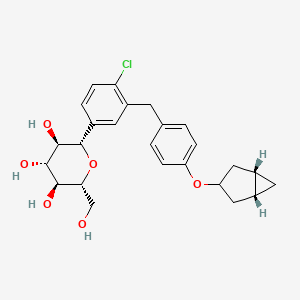
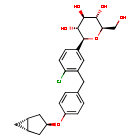
Janagliflozin
WeightAverage: 460.95
Monoisotopic: 460.1652664
Chemical FormulaC25H29ClO6
- WK4RT85HCA
- XZP-5695
- UNII-WK4RT85HCA
- 1800115-22-3
- (2S,3R,4R,5S,6R)-2-[3-[[4-[[(1S,5R)-3-bicyclo[3.1.0]hexanyl]oxy]phenyl]methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
- D-Glucitol, 1,5-anhydro-1-C-(3-((4-((1alpha,3alpha,5alpha)-bicyclo(3.1.0)hex-3-yloxy)phenyl)methyl)-4-chlorophenyl)-, (1S)-
- (2S,3R,4R,5S,6R)-2-[3-[[4-[[(1S,5R)-3-bicyclo[3.1.0]hexanyl]oxy]phenyl]methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
- D-GLUCITOL, 1,5-ANHYDRO-1-C-(3-((4-((1.ALPHA.,3.ALPHA.,5.ALPHA.)-BICYCLO(3.1.0)HEX-3-YLOXY)PHENYL)METHYL)-4-CHLOROPHENYL)-, (1S)-
- (2S,3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
China 2024, approvals 2024, Jilin Huisheng Biopharmaceutical Co, sihuan, SGLT2 inhibitors, Huiyoujing
Janagliflozin is an SGLT2 inhibitor developed by Sihuan Pharmaceutical.[1][2][3][4][5][6] It is approved in China for the treatment of type 2 diabetes.[7]
PAPER
https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-0042-1751524
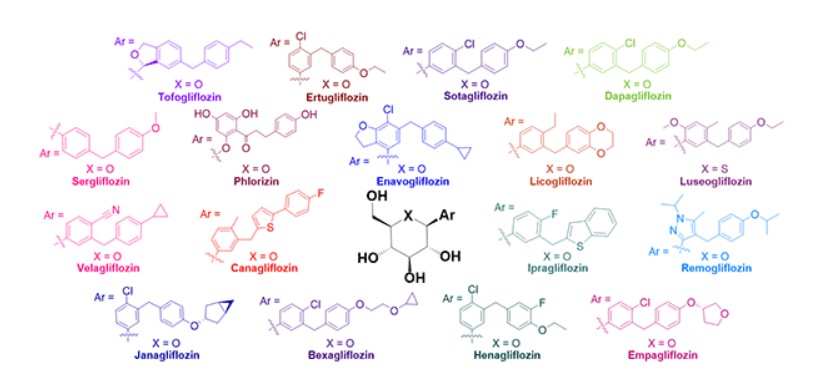
(71) (a) Wu, F. US9315438B2, 2016. (b) Wu, F. EP2891654A1, 2014.
Initially, the two advanced intermediates were synthesized and then coupled under cryogenic conditions using nBuLi. The construction of 242 commences with the reaction of 5-bromo-2-chlorobenzoic acid (26c) with oxalyl chloride and a catalytic amount of DMF in DCM, yielding the acid chloride derivative 26c′. This intermediate is then subjected to Friedel–Crafts acylation with anisole to produce 240 in
71% yield. Subsequent reduction of 240 was carried out using boron trifluoride–diethyl etherate and triethylsilane in a DCM/acetonitrile mixture, leading to the formation of 241 in an excellent yield. Demethylation of compound 241 is accomplished using boron tribromide at low temperature, resulting in 242 with a yield of 97%. On the other hand, the synthesis of 245 involves two steps starting from commercially available cyclopent-3-en-1-ol (243). The Simmons Smith cyclopropanation of 243 is performed using a mixture of trifluoroacetic acid, diiodomethane, and diethylzinc in DCM, providing 244 with a yield of 48%. Compound 244 is then further treated with methanesulfonyl chloride to give the mesylated compound 245 in a yield of 68%. Subse quently, 4-(5-bromo-2-chlorobenzyl)phenol (242) is allowed to react with 245 in the presence of NMP, cesium carbonate, and BTEAC (benzyltriethylammonium chloride) to give 246. The next step involves a lithium–halogen exchange on
246 using n-butyllithium, with addition to 22 at –78 °C affording the hydroxy intermediate. Methylation of this hydroxy intermediate using methanesulfonic acid and methanol provides 247 in 98% yield. Reduction of 247 using borontrifluoride–diethyl etherate and triethylsilane at –78 °C furnishes 248. To achieve the desired isomer, all of the hydroxy groups of compound 248 were protected using acetic anhydride, DMAP, and pyridine in DCM at 0 °C to give the O-acylated compound 249. In the final step, 249 is hydrolyzed us ing lithium hydroxide monohydrate in a mixed solvent consisting of methanol, THF, and water to provide the desired compound janagliflozin (14) in a yield of 91%. This truncated synthetic route is protection-group-free, and is well suited for scale-up. The drawback of the synthetic route is
the late-stage enrichment of the desired isomer in the final product via acylated derivative 249. The poor isolated yield of 249 is not commercially favored due to low throughput and an increase in raw material and production costs.
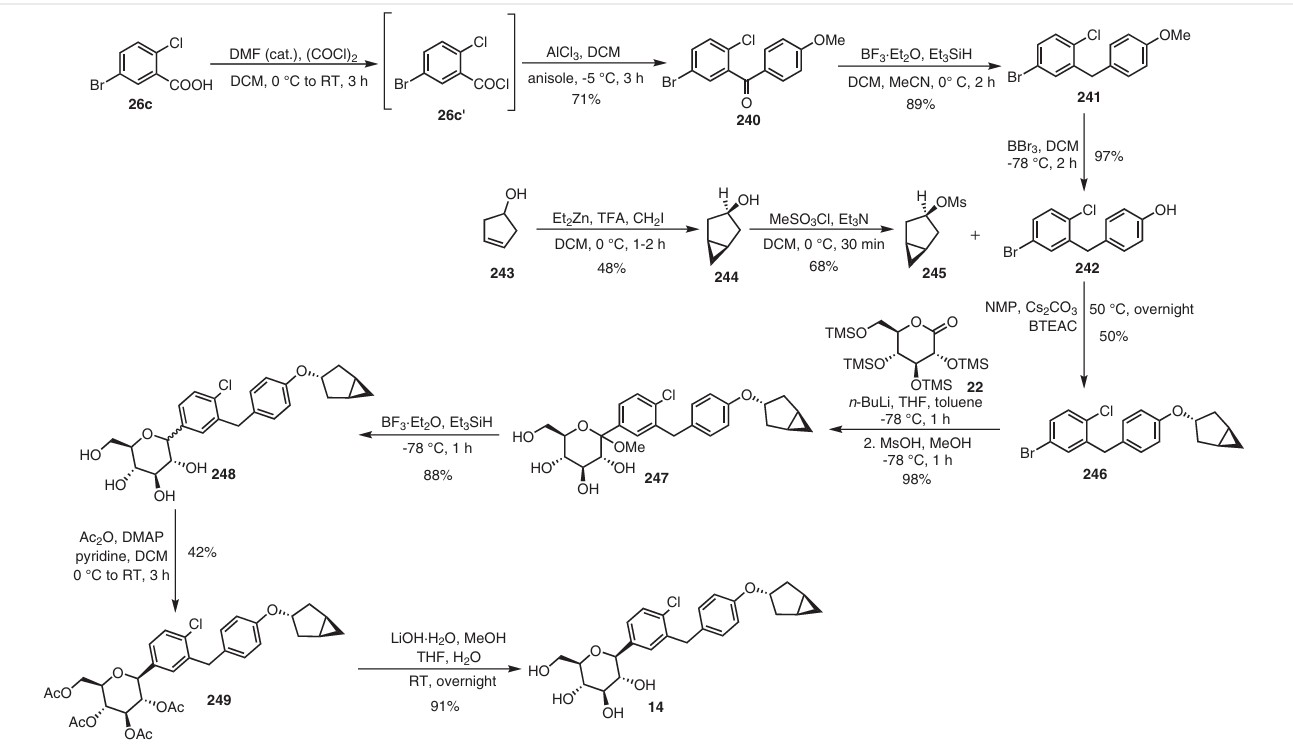
PAPER
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00017
SYN
https://www.sciencedirect.com/science/article/abs/pii/S022352342400223X
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US142552820&_cid=P11-MEPJES-88258-1
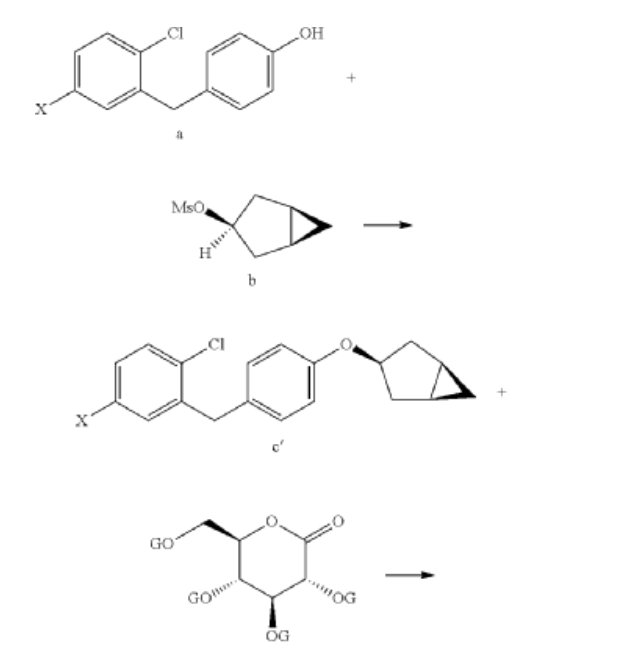
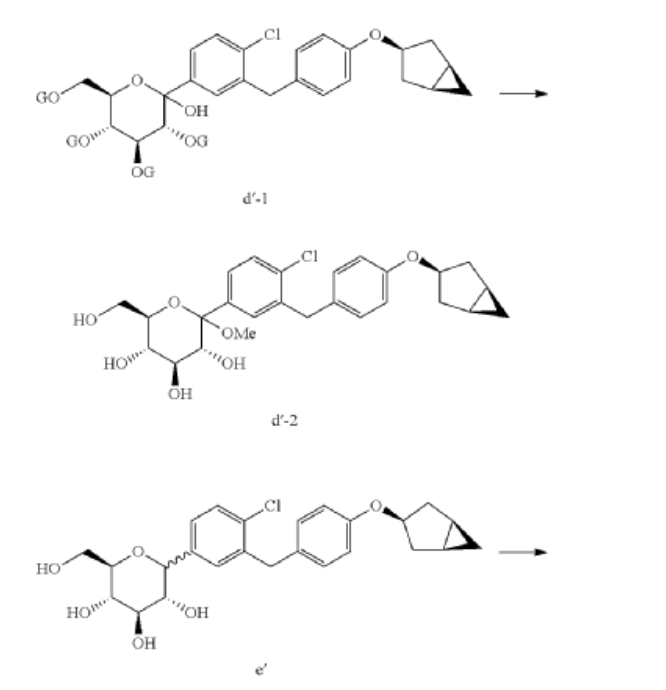
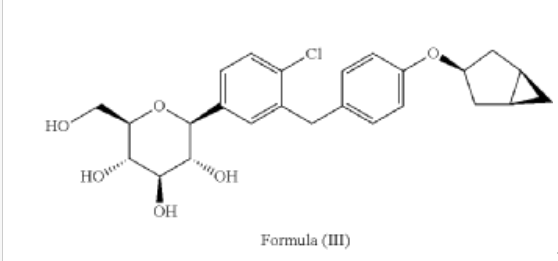
Example 1
Preparation of (2S,3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Formula II)
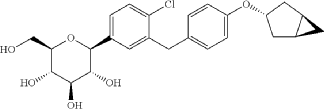
(1) Preparation of 5-bromo-2-chlorobenzoyl chloride
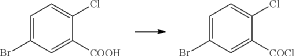
(2) Preparation of (5-bromo-2-chlorophenyl)(4-methoxyphenyl)methanone
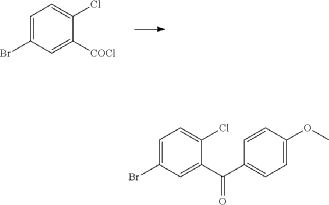
(3) Preparation of 4-bromo-1-chloro-2-(4-methoxybenzyl)benzene
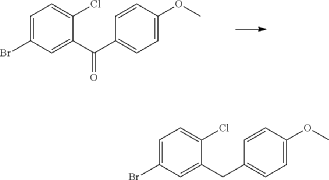
(4) Preparation of 4-(5-bromo-2-chlorobenzyl)phenol
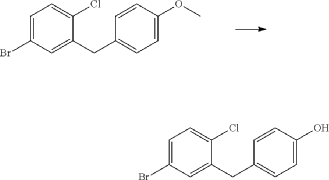
(5) Preparation of (1R,3r,5S)-bicyclo[3.1.0]hexan-3-ol
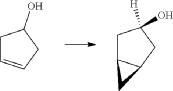
(6) Preparation of (1R,3r,5S)-bicyclo[3.1.0]hexan-3-yl methanesulfonate
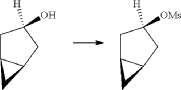
(7) Preparation of (1R,3s,5S)-3-(4-(5-bromo-2-chlorobenzyl)phenyloxy)bicyclo[3.1.0]hexane
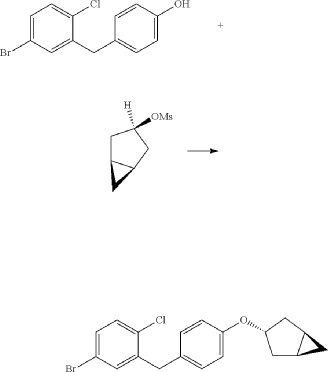
(8) Preparation of (3R,4S,5R,6R)-3,4,5-tri((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one
(9) Preparation of (3R,4S,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
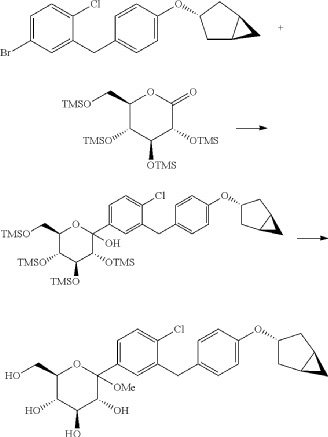
(10) Preparation of (3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
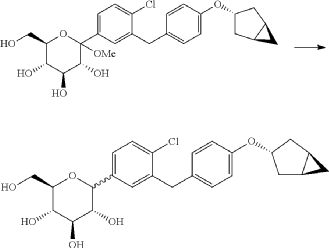
(11) Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
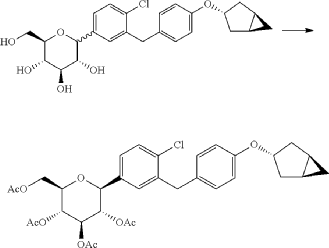
(12) Preparation of (2S,3R,4R,5 S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
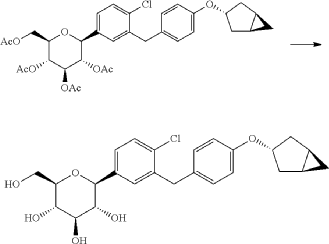
PAT
EP2891654
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP142501978&_cid=P20-MEQIAN-96633-1
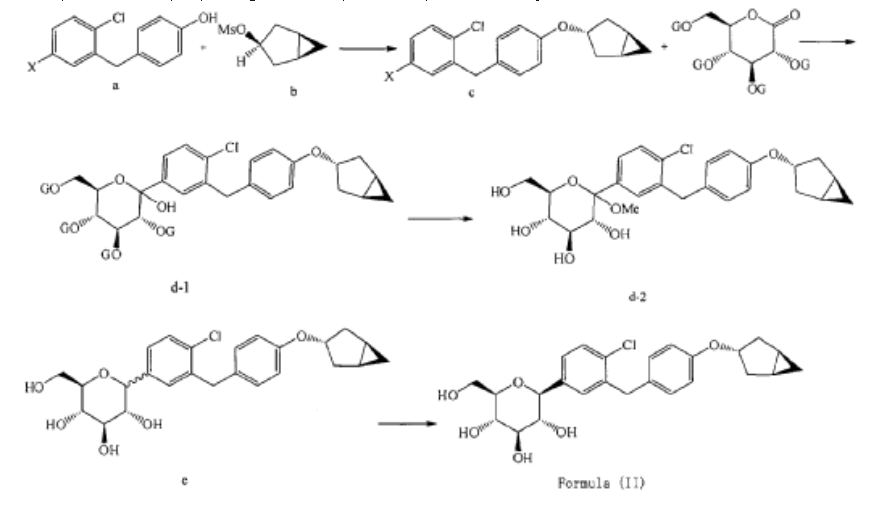
[0027] The compound represented by formula (II) as defined hereinbefore, lab-made, its chemical name and preparation process are described in the following Example 1.
Reference compound 1: Compound 4 as described in the PCT application WO2013/000275A1, lab-made (with reference to the PCT application WO2013/000275A1), its structure is as follows:
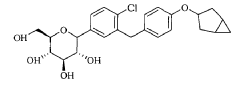
Compound 4, i.e. the compound represented by formula (I).
Reference compound 2: Compound 22 as described in the PCT application WO2013/000275A1, lab-made (with reference to the PCT application WO2013/000275A1), its structure is as follows:
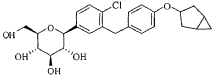
Compound 22.
(12) Preparation of
[0057] (2 S,3 R,4 R,5 S,6 R)-2-(3-(4-(((1 R,3 s,5 S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydr oxymethyl)tetrahydro-2 H-pyran-3,4,5-triol
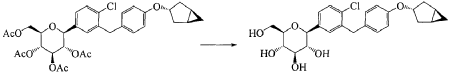
[0058] (2 R,3 R,4 R,5 S,6 S)-2-(acetoxymethyl)-6-(3-(4-(((1 R,3 s,5 S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlo rophenyl)tetrahydro-2 H-pyran-3,4,5-triyl triacetate (81g, 0.129mol) was dissolved in a mixed solvent of tetrahydrofuran (313mL), methanol (470mL) and water (156mL). To the resulting mixture was added lithium hydroxide monohydrate (6.32g, 150mmol). The mixture was stirred at room temperature overnight. TLC indicated the completion of reaction. The solvent was removed from the reaction mixture by rotary evaporation. The residual reaction mixture was dissolved with ethyl acetate (400mL). The organic phase was washed with an aqueous saturated sodium chloride solution, with an aqueous KHSO 4 solution, and with water twice, dried over anhydrous sodium sulphate, and evaporated by rotation. The residue was purified with C18 reverse phase preparative chromatography to produce 54.2g of a final product in a yield of 91%.
Formula: C 25H 29ClO 6 Mw: 460.95 LC-MS( m/ z): 478.3 [M+NH 4] +
1H-NMR (400MHz, MeOD) δ: 7.35-7.26 (m, 3H), 7.08-7.06 (d, 2H), 6.76-6.74 (d, 2H), 4.45-4.41 (m, 1H), 4.10-4.00 (m, 3H), 3.89-3.88 (d, 1H), 3.71-3.69 (m, 1H), 3.45-3.38 (m, 3H), 3.31-3.26 (m, 1H), 2.34-2.29 (m, 2H), 1.87-1.81 (m, 2H), 1.37-1.33 (m, 2H), 0.43-0.42 (m, 1H), 0.11-0.10 (m, 1H).
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Janagliflozin, engineered by Jilin Huisheng Biopharmaceutical Co., Ltd., a subsidiary under the umbrella of Sihuan Pharmaceutical Holdings Group, falls within the category of oral sodium-glucose co-transporter 2(SGLT2) inhibitors. This agent has been specifically designed with the aim of optimizing glycemic regulation in the adult population grappling with type 2 diabetes mellitus (T2DM) [54]. It is marketed under the brand name Huiyoujing. In 2024, the NMPA gave its approval for Janagliflozin, indicated for adult patients with T2DM, where it can be employed either as a standalone treatment (monotherapy) or in combination with metformin to optimize blood glucose regulation [55]. The clinical effectiveness of Janagliflozin was substantiated through a Phase III clinical trial (NCT03811548). This trial specifically assessed its application as a monotherapy in Chinese patients suffering from T2DM
whose blood glucose was not well – managed via diet and exercise alone. The findings of the study indicated notable decreases in glycated hemoglobin levels. Concurrently, improvements were observed in both body weight and blood pressure. Collectively, these outcomes serve as evidence of the drug’s ability to enhance glycemic regulation [56]. Regarding safety, Janagliflozin was generally well-tolerated. In line with the well-established safety characteristics of SGLT2 inhibitors, the frequently encountered adverse events associated with this treatment were urinary tract infections and genital mycotic infections. No serious adverse events were reported during the trial [57].
The synthesis of Janagliflozin, depicted in Scheme 13, commences with the acylation of 5-bromo-2-chlorobenzoic acid (Jana-001) using oxalyl chloride, yielding the acyl chloride intermediate Jana-002 [58]. Friedel-Crafts acylation of Jana-002 with anisole (Jana-003) affords ketone Jana-004. Subsequent reduction of the carbonyl group in Jana-004 produces Jana-005. Demethylation of Jana-005 with BBr3
generates phenol Jana-006, which undergoes substitution with intermediate Jana-007 to form ether Jana-008. Addition of gluconolactone (Jana-009) to Jana-008 affords Jana-010, where concurrent TMS
deprotection during etherification yields Jana-011. Reduction of Jana-011 using Et3SiH/BF3.ET2Oproduces Jana-012which is sequentially esterified with Ac2O , and hydrolyzed under LiOH conditions, ultimately yielding Janagliflozin
[54] L. Gao, Z. Cheng, B. Su, X. Su, W. Song, Y. Guo, L. Liao, X. Chen, J. Li, X. Tan, F. Xu,
S. Pang, K. Wang, J. Ye, Y. Wang, L. Chen, J. Sun, L. Ji, Efficacy and safety of
janagliflozin as add-on therapy to metformin in Chinese patients with type 2
diabetes inadequately controlled with metformin alone: a multicentre,
randomized, double-blind, placebo-controlled, phase 3 trial, Diabetes Obes Metab
25 (2023) 785–795.
[55] L. Ji, X. Jiang, Q. Hao, Z. Cheng, K. Wang, S. Pang, M. Liu, Y. Guo, X. Chen, X. Su,
T. Ning, J. Liu, F. Bian, Y. Li, Z. Zhang, W. Song, J. Sun, Efficacy and safety of
janagliflozin monotherapy in Chinese patients with type 2 diabetes mellitus
inadequately controlled on diet and exercise: a multicentre, randomized, double-
blind, placebo-controlled, phase 3 trial, Diabetes Obes Metab 25 (2023)
1229–1240.
[56] L. Song, X. Wang, J. Sun, X. Hu, H. Li, P. Hu, D. Liu, A model-informed approach to
accelerate the clinical development of janagliflozin, an innovative SGLT2 inhibitor,
Clin. Pharmacokinet. 62 (2023) 505–518.
[57] Canagliflozin, Drugs and Lactation Database (Lactmed®), National Institute of
Child Health and Human Development, Bethesda (MD), 2006.
[58] F. Wu, Optically Pure benzyl-4-chlorophenyl-C-glucoside Derivatives as SGLT
Inhibitors (Diabetes Mellitus), 2015. EP2891654.
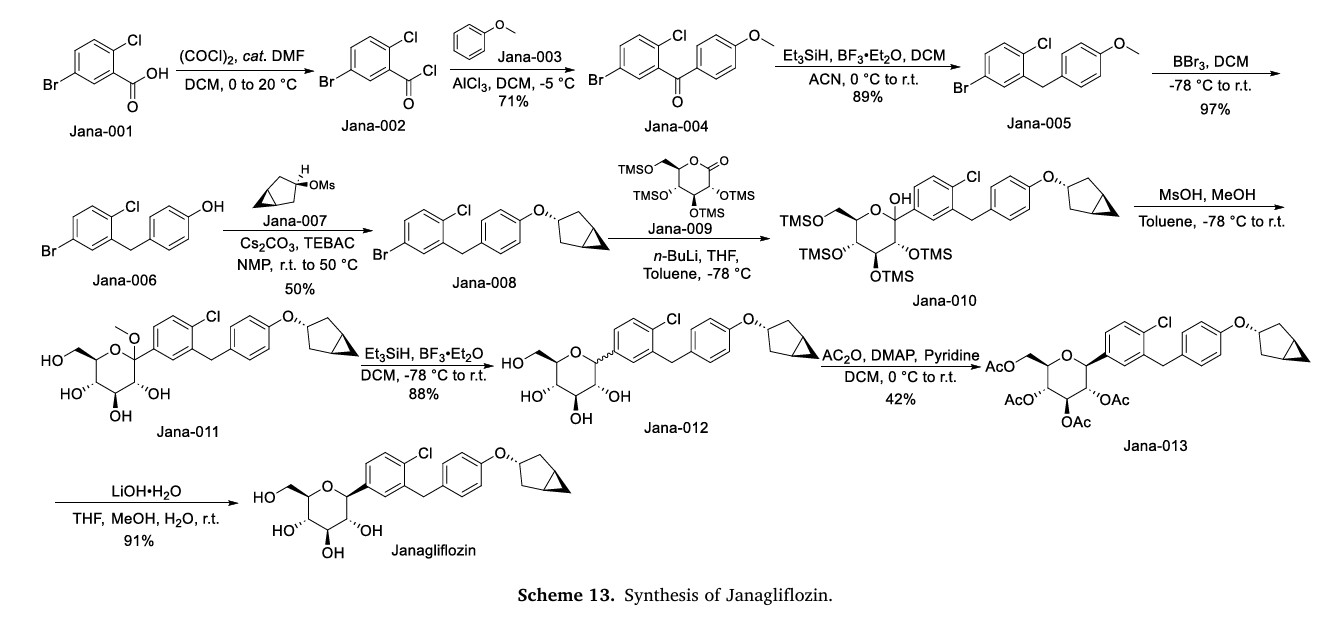
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Song, Ling; Yao, Xueting; Liu, Yang; Zhong, Wen; Jiang, Ji; Liu, Hongzhong; Zhou, Huimin; Shi, Chongtie; Zong, Kaiqi; Wang, Chong; Ma, Chuanxiang; Liu, Dongyang; Hu, Pei (April 2020). “Translational prediction of first-in-human pharmacokinetics and pharmacodynamics of janagliflozin, a selective SGLT2 inhibitor, using allometric scaling, dedrick and PK/PD modeling methods”. European Journal of Pharmaceutical Sciences. 147: 105281. doi:10.1016/j.ejps.2020.105281. S2CID 212405270.
- Liu, Dongyang; Song, Ling; Wang, Xiaoxu; Liu, Xu; Cao, Fangrui; Liu, Hongzhong; Ding, Yanhua; Xiao, Xinhua; Jiang, Ji; Hu, Pei (1 June 2019). “154-LB: Accelerating Clinical Development of Janagliflozin, a Novel Antidiabetic Drug, Using Model-Informed Drug Development Strategy”. Diabetes. 68 (Supplement_1). doi:10.2337/db19-154-LB. S2CID 195440798.
- Zhao, Hengli; Wei, Yilin; He, Kun; Zhao, Xiaoyu; Mu, Hongli; Wen, Qing (December 2022). “Prediction of janagliflozin pharmacokinetics in type 2 diabetes mellitus patients with liver cirrhosis or renal impairment using a physiologically based pharmacokinetic model”. European Journal of Pharmaceutical Sciences. 179: 106298. doi:10.1016/j.ejps.2022.106298. PMID 36162752. S2CID 252505056.
- Zhao, Hengli; Zhao, Zhirui; He, Kun; Mi, Nianrong; Lou, Kai; Dong, Xiaolin; Zhang, Wenyu; Sun, Jingfang; Hu, Xinyu; Pang, Shuguang; Cheng, Hong; Wen, Qing (August 2023). “Pharmacokinetics, Pharmacodynamics and Safety of Janagliflozin in Chinese Type 2 Diabetes Mellitus Patients with Renal Impairment”. Clinical Pharmacokinetics. 62 (8): 1093–1103. doi:10.1007/s40262-023-01256-0. PMID 37284974. S2CID 259097798.
- Gao, Leili; Cheng, Zhifeng; Su, Benli; Su, Xiuhai; Song, Weihong; Guo, Yushan; Liao, Lin; Chen, Xiaowen; Li, Jiarui; Tan, Xingrong; Xu, Fangjiang; Pang, Shuguang; Wang, Kun; Ye, Jun; Wang, Yuan; Chen, Lili; Sun, Jingfang; Ji, Linong (March 2023). “Efficacy and safety of janagliflozin as add‐on therapy to metformin in Chinese patients with type 2 diabetes inadequately controlled with metformin alone: A multicentre, randomized, double‐blind, placebo‐controlled, phase 3 trial”. Diabetes, Obesity and Metabolism. 25 (3): 785–795. doi:10.1111/dom.14926. PMID 36433709. S2CID 253967474.
- Ji, Linong; Jiang, Xiaozhen; Hao, Qingshun; Cheng, Zhifeng; Wang, Kun; Pang, Shuguang; Liu, Meiying; Guo, Yushan; Chen, Xiaowen; Su, Xiuhai; Ning, Tao; Liu, Jie; Bian, Fang; Li, Yulan; Zhang, Zhinong; Song, Weihong; Sun, Jingfang (May 2023). “Efficacy and safety of janagliflozin monotherapy in Chinese patients with type 2 diabetes mellitus inadequately controlled on diet and exercise: A multicentre, randomized, double‐blind, placebo‐controlled, Phase 3 trial”. Diabetes, Obesity and Metabolism. 25 (5): 1229–1240. doi:10.1111/dom.14971. PMID 36594724. S2CID 255474211.
- “NMPA approves China’s second homegrown SGLT2 inhibitor janagliflozin”. bioworld.com. January 23, 2024.
| Legal status | |
|---|---|
| Legal status | Rx in China; investigational elsewhere |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1800115-22-3 |
| PubChem CID | 91820686 |
| DrugBank | DB16209 |
| UNII | WK4RT85HCA |
| Chemical and physical data | |
| Formula | C25H29ClO6 |
| Molar mass | 460.95 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Janagliflozin, china 2024, approvals 2024, Jilin Huisheng Biopharmaceutical Co, sihuan, SGLT2 inhibitors, Huiyoujing, WK4RT85HCA, XZP 5695, UNII-WK4RT85HCA, 1800115-22-3
SYN
SYNTHESIS 2024, 56, 906–943
synthesis of janagliflozin (14) was achieved through an eleven-step process in an overall yield of 3% (Scheme 45).71 Initially, the two advanced intermediates were synthesized and then coupled under cryogenic conditions using nBuLi. The construction of 242 commences with the reaction of 5-bromo-2-chlorobenzoic acid (26c) with oxalyl chloride and a catalytic amount of DMF in DCM, yielding the acid
chloride derivative 26c′. This intermediate is then subjected to Friedel–Crafts acylation with anisole to produce 240 in 71% yield. Subsequent reduction of 240 was carried out using boron trifluoride–diethyl etherate and triethylsilane in a DCM/acetonitrile mixture, leading to the formation of 241 in an excellent yield. Demethylation of compound 241 is accomplished using boron tribromide at low temperature, re
sulting in 242 with a yield of 97%. On the other hand, the synthesis of 245 involves two steps starting from commercially available cyclopent-3-en-1-ol (243). The Simmons Smith cyclopropanation of 243 is performed using a mixture of trifluoroacetic acid, diiodomethane, and diethylzinc in DCM, providing 244 with a yield of 48%. Compound 244 is then further treated with methanesulfonyl chloride to
give the mesylated compound 245 in a yield of 68%. Subsequently, 4-(5-bromo-2-chlorobenzyl)phenol (242) is allowed to react with 245 in the presence of NMP, cesium carbonate, and BTEAC (benzyltriethylammonium chloride) to give 246. The next step involves a lithium–halogen exchange on
246 using n-butyllithium, with addition to 22 at –78 °C affording the hydroxy intermediate. Methylation of this hydroxy intermediate using methanesulfonic acid and methanol provides 247 in 98% yield. Reduction of 247 using boron trifluoride–diethyl etherate and triethylsilane at –78 °C furnishes 248. To achieve the desired isomer, all of the hydroxy groups of compound 248 were protected using acetic anhydride, DMAP, and pyridine in DCM at 0 °C to give the O-acylated compound 249. In the final step, 249 is hydrolyzed us ing lithium hydroxide monohydrate in a mixed solvent consisting of methanol, THF, and water to provide the desired compound janagliflozin (14) in a yield of 91%. This truncated synthetic route is protection-group-free, and is well suited for scale-up. The drawback of the synthetic route is
the late-stage enrichment of the desired isomer in the final product via acylated derivative 249. The poor isolated yield of 249 is not commercially favored due to low throughput and an increase in raw material and production costs

(71) (a) Wu, F. US9315438B2, 2016. (b) Wu, F. EP2891654A1, 2014.
Zorifertinib
Zorifertinib
AZD 3759
CAS 1626387-80-1, 67SX9H68W2
WeightAverage: 459.91
Monoisotopic: 459.1473455
Chemical FormulaC22H23ClFN5O3
[4-(3-chloro-2-fluoroanilino)-7-methoxyquinazolin-6-yl] (2R)-2,4-dimethylpiperazine-1-carboxylate
- [4-(3-chloro-2-fluoroanilino)-7-methoxyquinazolin-6-yl] (2R)-2,4-dimethylpiperazine-1-carboxylate
- (4-(3-chloro-2-fluoroanilino)-7-methoxyquinazolin-6-yl) (2R)-2,4-dimethylpiperazine-1-carboxylate
- (R)-4-((3-chloro-2-fluorophenyl)amino)-7-methoxyquinazolin-6-yl 2,4-dimethylpiperazine-1-carboxylate
- 4-[(3-CHLORO-2-FLUOROPHENYL)AMINO]-7-METHOXYQUINAZOLIN-6-YL (2R)-2,4-DIMETHYLPIPERAZINE-1-CARBOXYLATE
China 2024, APPROVALS 2024, Alpha Biopharma, ASTRA ZENECA, Zorifer,
Zorifertinib (AZD3759) is a drug for the treatment of cancer.[1] In China, it was approved in 2024 for locally advanced or metastatic non-small-cell lung cancer (NSCLC) that has epidermal growth factor receptor exon 19 deletion or exon 21 L858R substitution mutations and central nervous system (CNS) metastases.[2]
Zorifertinib is an orally available inhibitor of the epidermal growth factor receptor (EGFR), with potential antineoplastic activity. Upon oral administration, zorifertinib binds to and inhibits the activity of EGFR as well as certain mutant forms of EGFR. This prevents EGFR-mediated signaling, and may lead to both induction of cell death and inhibition of tumor growth in EGFR-overexpressing cells. EGFR, a receptor tyrosine kinase mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization.
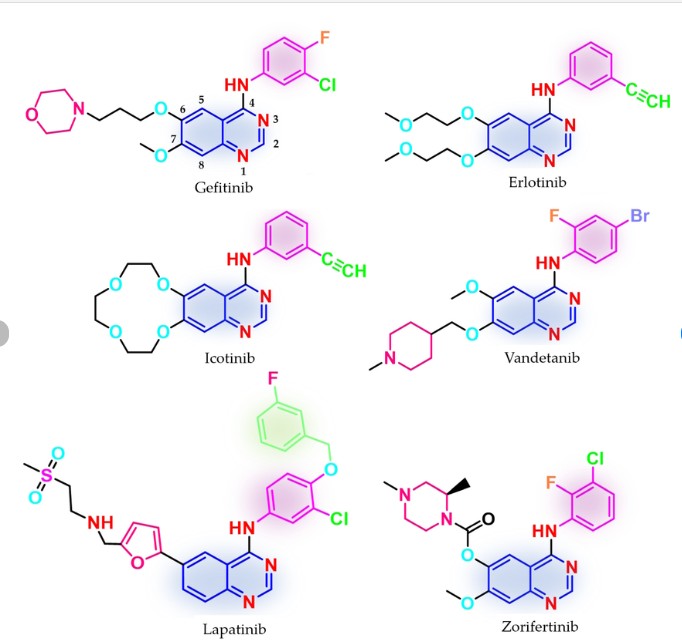
SYN
J. Med. Chem. 58 (2015) 8200–8215.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.5b01073

SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Zorifertinib, developed by AstraZeneca as AZD3759, is a novel EGFR TKI designed to effectively penetrate the blood-brain barrier (BBB) [44,45]. In 2018, Alpha Biopharma, in collaboration with AstraZeneca, advanced its development. In 2024, the NMPA gave its approval to zorifertinib hydrochloride tablets, which are sold under the brand name Zorifer. This approval is for the use of these tablets in the first-line treatment of adult patients who have the following conditions: they
have locally advanced or metastatic NSCLC with either EGFR exon 19 deletion or exon 21 L858R substitution mutations, and also have CNSmetastases [45]. Zorifertinib exerts its pharmacological action through the selective inhibition of EGFR tyrosine kinase activity, with a particular focus on mutational forms such as L858R and exon 19 deletions. In contrast to several other tyrosine kinase inhibitors (TKIs), it does not serve as a substrate for BBB efflux transporters, namely P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). This unique property enables zorifertinib to reach elevated concentrations within brain tissue and cerebrospinal fluid. As a result, it can effectively target and
act against CNS metastases [44,45]. The clinical efficacy of zorifertinib was demonstrated in the EVEREST study (NCT03653546), a random ized, open-label, international multicenter Phase II/III trial. The study
enrolled 492 patients with EGFR-mutant NSCLC and CNS metastases. Results showed that zorifertinib significantly improved systemic PFS to 9.6 months compared to 6.9 months with first-generation EGFR-TKIs, reducing the risk of disease progression or death by 28 %. Intracranial PFS was notably extended to 15.2 months versus 8.3 months in the control group. The ORR was 68.6 % for zorifertinib compared to 58.4 % for the control. Regarding toxicity, zorifertinib exhibited a manageable safety profile. The incidence of treatment-related adverse events (TRAEs) was similar between the zorifertinib and control groups (97.7 %vs. 94.0 %), with grade ≥3 TRAEs occurring in 65.9 % of patients receiving zorifertinib compared to 18.3 % in the control group. No new safety signals were identified, indicating an acceptable tolerability for patients. The approval of zorifertinib offers a significant advancement in
the treatment of EGFR-mutant NSCLC patients with CNS metastases,providing an effective therapeutic option capable of addressing both systemic and intracranial disease [44].
The synthesis of Zorifertinib, depicted in Scheme 11, initiates with nucleophilic substitution between Zori-001 and Zori-002 in MeCN, affording Zori-003 [46]. Hydrolysis of the ester moiety in Zori-003
yields Zori-004, which is subsequently esterified with Zori-005 in DMF to form Zori-006. Acidic deprotection of Zori-006 generates Zori-007, followed by methylation to deliver Zorifertinib. Concurrently, Zori-005 is prepared via amidation of Zori-008
[44] M. Roy-O’Reilly, D. Rogawski, The climb toward intracranial efficacy: Zorifertinib
in EGFR-mutant NSCLC with CNS metastases in the EVEREST trial, Med 6 (2025)
100525.
[45] Q. Zhou, Y. Yu, L. Xing, Y. Cheng, Y. Wang, Y. Pan, Y. Fan, J. Shi, G. Zhang, J. Cui,
J. Zhou, Y. Song, W. Zhuang, Z. Ma, Y. Hu, G. Li, X. Dong, J. Feng, S. Lu, J. Wu,
J. Li, L. Zhang, D. Wang, X. Xu, T.Y. Yang, N. Yang, Y. Guo, J. Zhao, Y. Yao,
D. Zhong, B. Xia, C.T. Yang, B. Zhu, P. Sun, B.Y. Shim, Y. Chen, Z. Wang, M.J. Ahn,
J. Wang, Y.L. Wu, First-line zorifertinib for EGFR-Mutant non-small cell lung
cancer with central nervous system metastases: the phase 3 EVEREST trial, Med 6
(2025) 100513.
[46] Q. Zeng, J. Wang, Z. Cheng, K. Chen, P. Johnstr¨om, K. Varn¨as, D.Y. Li, Z.F. Yang,
X. Zhang, Discovery and evaluation of clinical candidate AZD3759, a potent, oral
active, central nervous system-penetrant, epidermal growth factor receptor
tyrosine kinase inhibitor, J. Med. Chem. 58 (2015) 8200–8215.
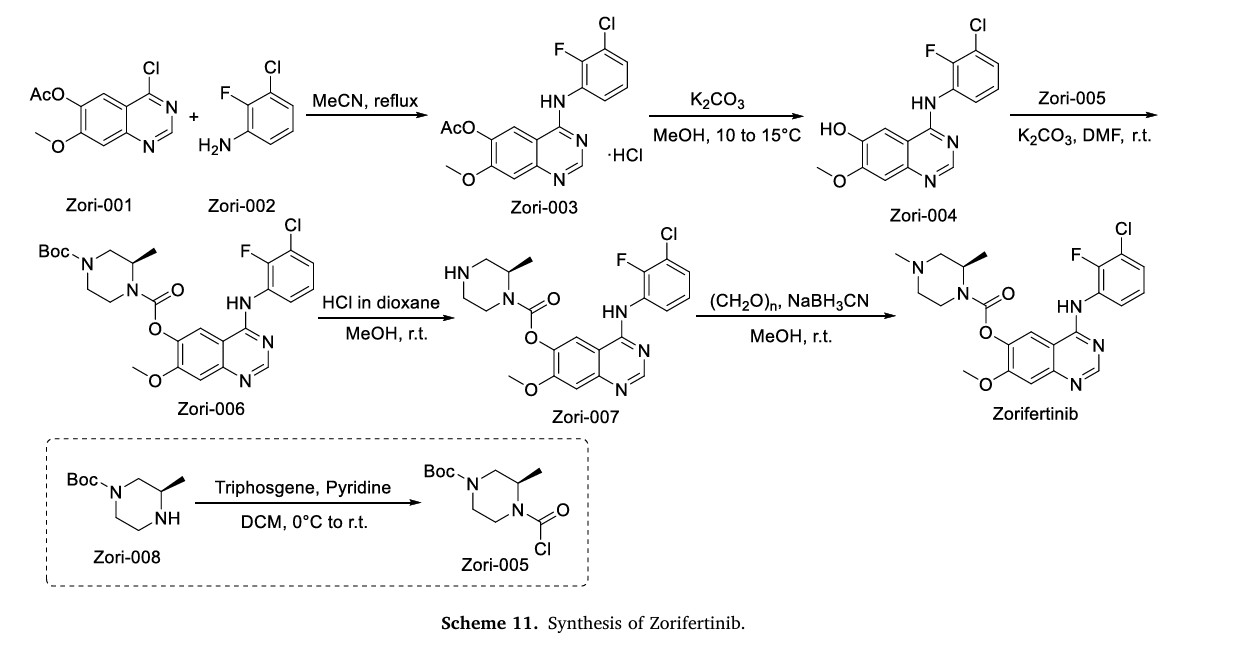
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Zhou Q, Yu Y, Xing L, Cheng Y, Wang Y, Pan Y, et al. (January 2025). “First-line zorifertinib for EGFR-mutant non-small cell lung cancer with central nervous system metastases: The phase 3 EVEREST trial”. Med. 6 (1) 100513. doi:10.1016/j.medj.2024.09.002. PMID 39389055.
- “Zorifertinib Receives NMPA Approval for EGFR+ NSCLC With CNS Metastases”. November 20, 2024.
| Clinical data | |
|---|---|
| Other names | AZD3759 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1626387-80-1 |
| PubChem CID | 78209992 |
| IUPHAR/BPS | 10456 |
| DrugBank | DB14795 |
| ChemSpider | 38772332 |
| UNII | 67SX9H68W2 |
| ChEMBL | ChEMBL3623290 |
| Chemical and physical data | |
| Formula | C22H23ClFN5O3 |
| Molar mass | 459.91 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////Zorifertinib, china 2024, APPROVALS 2024, Alpha Biopharma, ASTRA ZENECA, Zorifer, AZD 3759, 67SX9H68W2
Garsorasib
Garsorasib
Chemical Formula: C32H32F2N8O2
Exact Mass: 598.2616
Molecular Weight: 598.66
- CAS 2559761-14-5
- P491NE9G6Z
- D-1553
- 7-(2-amino-6-fluorophenyl)-1-(4,6-dicyclopropylpyrimidin-5-yl)-4-[(2S,5R)-2,5-dimethyl-4-prop-2-enoylpiperazin-1-yl]-6-fluoropyrido[2,3-d]pyrimidin-2-one
- 4-((2S,5R)-4-Acryloyl-2,5-dimethylpiperazin-1-yl)-7-(2-amino-6-fluorophenyl)-1-(4,6-dicyclopropylpyrimidin-5-yl)-6-fluoropyrido(2,3-d)pyrimidin-2(1H)-one
- Pyrido(2,3-d)pyrimidin-2(1H)-one, 7-(2-amino-6-fluorophenyl)-1-(4,6-dicyclopropyl-5-pyrimidinyl)-4-((2S,5R)-2,5-dimethyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl)-6-fluoro-
D 1553, Chia Tai Tianqing, CHINA 2024, APPROVALS 2024, Anfangning,
Garsorasib is an orally available inhibitor of the oncogenic KRAS substitution mutation, G12C, with potential antineoplastic activity. Upon oral administration, garsorasib selectively targets the KRAS G12C mutant and inhibits KRAS G12C mutant-dependent signaling. KRAS, a member of the RAS family of oncogenes, serves an important role in cell signaling, division and differentiation. Mutations of KRAS may induce constitutive signal transduction leading to tumor cell growth, proliferation, invasion, and metastasis.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021120045&_cid=P11-MEJTS8-41135-1
Example 5. Preparation and Solid state characterization of Compound 2
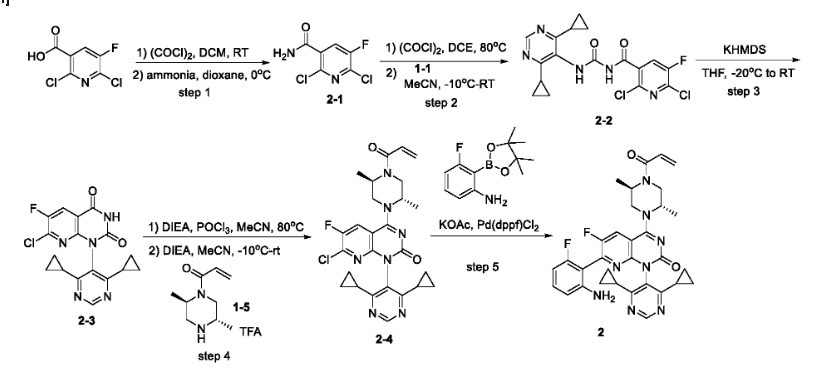
Step 1: To a mixture of 2, 6-dichloro-5-fluoronicotinic acid (23 g, 0.11 mol) in dichloromethane (300 mL) was added dimethylformamide (0.2 mL) . Oxalyl chloride (33 g, 0.26 mol) was then added slowly over 30 minutes at room temperature. The mixture was stirred at room temperature for an hour and then concentrated to give an oil which was dissolved in dioxane (50 mL) . The solution was added to ammonium hydroxide (150 mL) at 0℃ over 30 minutes. The resulting mixture was stirred at 0℃ for 30 minutes and then filtered. The filter cake was washed with cooled water (50 mL) and dried to afford 2-1.
[0183]
Step 2: A solution of 2-1 (11 g, 52.6 mmol) in 1, 2-dichloroethane (80 mL) was treated with oxalyl chloride (8.68 g, 68.4 mmol) . The mixture was stirred at 80℃ for 45 minutes and the reaction was concentrated. The residue was dissolved in acetonitrile (100 mL) , cooled to -10℃, and a solution of 1-1 (9.6 g, 55.2 mmol) in THF (30 mL) was added. The resulting mixture was stirred at room temperature for 2 hours. The solution was diluted with a sat. aqueous NaHCO 3solution and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether to petroleum ether/ethyl acetate = 4/1) to afford 2-2.
[0184]
Step 3: To a stirred solution of 2-2 (7.9 g, 19.3 mmol) in THF (100 mL) at -20℃ was added KHMDS (38.6 mL, 1 M in THF, 38.6 mmol) . The resulting mixture was stirred at room temperature for 2 hours. The reaction was quenched with sat. aqueous NH 4Cl solution and extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by flash column chromatography on silica gel (petroleum ether to petroleum ether/ethyl acetate = 2/1) to afford 2-3.
[0185]
Step 4: To a solution of 2-3 (746 mg, 2 mmol) and DIEA (387 mg, 3 mmol) in MeCN (20 mL) was added POCl 3(367 mg, 2.4 mmol) dropwise at room temperature. The resulting mixture was stirred at 80℃ for 45 minutes, followed by addition of DIEA (3.87 g, 30 mmol) and a solution of 1-5 (1.58 g, 4 mmol) in MeCN (10 mL) dropwise at -10℃. After stirring at room temperature for 1 hour, the reaction was quenched with ice-water and the mixture was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by flash column chromatography on silica gel (dichloromethane to dichloromethane/methanol = 10/1) to afford 2-4.
[0186]
Step 5: A mixture of 2-4 (8 mg, 0.15 mmol) , 3-fluoro-2- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) aniline (42 mg, 0.18 mmol) , Pd (dppf) Cl 2(13 mg, 0.018 mmol) and KOAc (40 mg, 0.41 mmol) in dioxane (3 mL) /H 2O (1 drop) was stirred at 80℃ for 2 hours under nitrogen atmosphere. The mixture was diluted with water and extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na 2SO 4and concentrated. The residue was purified by a Prep-HPLC (acetonitrile with 0.05%of TFA in water (30%to 65%) to afford 2. LCMS (ESI, m/z) : [M+H] += 599.1; HNMR (400 MHz, methanol-d 4, ppm) : δ 8.73 (s, 1H) , 8.26-8.22 (m, 1H) , 7.15-7.09 (m, 1H) , 6.84-6.74 (m, 1H) , 6.53 (d, J = 8.4 Hz, 1H) , 6.42-6.38 (m, 1H) , 6.30-6.24 (m, 1H) , 5.83-5.78 (m, 1H) , 5.01 (brs, 1H) , 4.91-4.83 (m, 1H) , 4.53-4.29 (m, 2H) , 3.96-3.89 (m, 1.5H) , 3.54-3.50 (m, 0.5H) , 1.82-1.75 (m, 1H) , 1.73-1.66 (m, 1H) , 1.47 (d, J = 6.8 Hz, 3H) , 1.37-1.27 (m, 3H) , 1.16-1.05 (m, 4H) , 1.03-0.97 (m, 2H) , 0.88-0.83 (m, 2H) . FNMR (376 MHz, methanol-d 4, ppm) : δ -114.9 (1F) , -125.6 (1F) .
[0187]
Compound 2 prepared via the above procedure was slurried in EtOAc, and filtered to provide Compound 2 in a crystalline form A. About 1.1%of residual EtOAc was detected by 1H-NMR, corresponding to weight loss at 120 –290 ℃ in TGA (FIG. 5B) . Two overlapped endothermic peaks were observed by DSC (FIG. 5B) . Compound 2 in Form A was heated to 250 ℃ and DSC profile of the residual solid was unchanged, suggesting the overlapped peak was due to melting with crystal form transformation. Thus, the starting material was an anhydrate.
[0188]
Form A was very soluble in DCM (> 92 mg/mL) and soluble (20 –33 mg/mL) in MeOH, butanone, THF, ACN and acetone. In other solvents, Form A was practically insoluble
SYN
CN112585129
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN321747237&_cid=P11-MEJTN6-36089-1
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Garsorasib (D-1553), marketed as Anfangning, is an orally bioavailable KRAS G12C inhibitor jointly developed by InventisBio and Chia Tai Tianqing Pharmaceutical Group [40]. This compound is specifically engineered to target the KRAS G12C mutation, a prevalent oncogenic driver in multiple cancers, including NSCLC. In 2024, the NMPA granted conditional approval for Garsorasib to treat adult patients with advanced NSCLC harboring the KRAS G12C mutation, who have undergone at least one prior systemic therapy regimen [41]. Garsorasib exerts its pharmacological effects through selective and irreversible binding to the KRAS G12C mutant protein, thereby immobilizing it in an inactive GDP-bound conformation. This binding event effectively disrupts the activation of downstream signaling path
ways, including mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K), resulting in diminished tumor cell proliferation and survival. The clinical efficacy of Garsorasib has been
confirmed in a Phase II clinical trial (NCT05383898) involving patients with advanced NSCLC harboring the KRAS G12C mutation. The trial reported an ORR of 52.0 % and a DCR of 88.6 %. Additionally, the
median PFS was observed to be 9.1 months, while the median overall survival (OS) reached 14.1 months, both indicative of significant antitumor activity within this patient cohort. In terms of safety, Garsorasib
exhibited a generally favorable tolerability profile [42]. The most common treatment-related adverse events included diarrhea, nausea, and elevated liver enzymes, which were predominantly of grade 1 or 2
severity.The synthesis of Garsorasib, depicted in Scheme 10, initiates with Suzuki-Miyaura coupling of Gars-001 and cyclopropylboronic acid, affording Gars-002 [43]. Gars-003 undergoes nucleophilic acylation with acryloyl chloride to yield Gars-004. TFA-mediated Boc deprotection of Gars-004 affords Gars-005. In parallel, Gars-006 is sequentially acylated with oxalyl chloride and aminated with ammonia to form Gars-007. DCE-mediated acylation of Gars-007, followed by concentration and coupling with Gars-002 in MeCN, produces Gars-008.KHMDS-catalyzed intramolecular cyclization of Gars-008 generates Gars-009. DIEA-catalyzed intermediate generation enables nucleophilic coupling with Gars-005 to assemble Gars-010. Final Suzuki-Miyaura coupling of Gars-010 with 3-fluoro-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline delivers Garsorasib.
[40] W. Luo, J. Zhu, W. Zhang, A. Yu, W. Zhou, K. Xu, Efficacy and toxicity of drugs
targeting KRAS(G12C) mutation in non-small cell lung cancer: a meta-analysis,
Expert Rev. Anticancer Ther. 23 (2023) 1295–1303.
[41] Z. Li, X. Dang, D. Huang, S. Jin, W. Li, J. Shi, X. Wang, Y. Zhang, Z. Song, J. Zhang,
W. Zhuang, X. Liu, L. Jiang, X. Meng, M. Zhao, J. Zhou, L. Zhang, P. Wang, H. Luo,
J. Yang, S. Cang, X. Wang, L. Zhang, S. Lu, Garsorasib in patients with KRAS
(G12C)-mutated non-small-cell lung cancer in China: an open-label, multicentre,
single-arm, phase 2 trial, Lancet Respir. Med. 12 (2024) 589–598.
[42] Z. Li, Z. Song, Y. Zhao, P. Wang, L. Jiang, Y. Gong, J. Zhou, H. Jian, X. Dong,
W. Zhuang, S. Cang, N. Yang, J. Fang, J. Shi, J. Lu, R. Ma, P. Wu, Y. Zhang,
M. Song, C.W. Xu, Z. Shi, L. Zhang, Y. Wang, X. Wang, Y. Zhang, S. Lu, D-1553
(garsorasib), a potent and selective inhibitor of KRAS(G12C) in patients with
NSCLC: phase 1 study results, J. Thorac. Oncol. 18 (2023) 940–951.
[43] X. Dai, Y. Wang, Y. Jiang, Y. Liu, Z. Shi, Z. Wang, L. Tao, Z. Han, H. Niu, J. Weng,
Heterocyclic Compounds, Preparation Methods and Uses Thereof in the Treatment
of Cancers, 2020 CN112585129A.
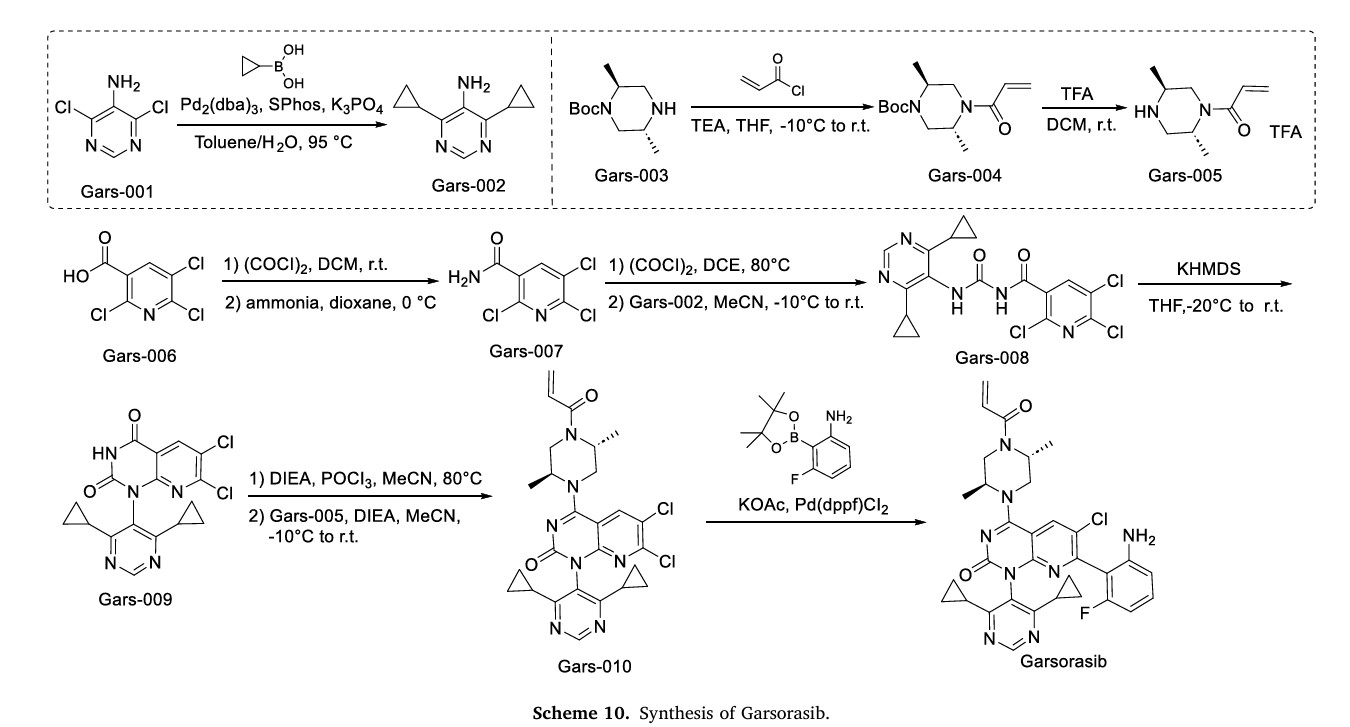
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- D‐1553: A novel <scp>KRASG12C</scp> inhibitor with potent and selective cellular and in vivo antitumor activityPublication Name: Cancer SciencePublication Date: 2023-05-09PMCID: PMC10323112PMID: 37158138DOI: 10.1111/cas.15829
Methods of treating a ras protein-related disease or disorder
Publication Number: US-2025049810-A1
- Heterocyclic compounds, preparation methods and uses thereofPublication Number: US-2022226328-A1Priority Date: 2019-12-18
- Heterocyclic compounds, preparation methods and uses thereofPublication Number: WO-2020233592-A1Priority Date: 2019-05-21
- Heterocyclic compounds, preparation and methods and uses thereofPublication Number: US-11091481-B2Priority Date: 2019-05-21Grant Date: 2021-08-17
- Heterocyclic compounds, preparation and methods and uses thereofPublication Number: US-2021198255-A1Priority Date: 2019-05-21
- Heterocyclic compounds, preparation methods and uses thereofPublication Number: US-2021355125-A1Priority Date: 2019-05-21
- Medicament for treatment and/or prevention of cancerPublication Number: WO-2023008462-A1Priority Date: 2021-07-27
- Heterocyclic compounds, preparation methods and uses thereofPublication Number: US-2021260067-A1Priority Date: 2019-12-18
- Heterocyclic compounds, preparation methods and uses thereofPublication Number: WO-2021120045-A1Priority Date: 2019-12-18
- Heterocyclic compounds, preparation methods and uses thereofPublication Number: WO-2021121330-A1Priority Date: 2019-12-18
- Heterocyclic compounds, preparation methods and uses thereofPublication Number: US-11241437-B2Priority Date: 2019-12-18Grant Date: 2022-02-08
- Medicament for treatment and/or prevention of cancerPublication Number: AU-2022320304-A1Priority Date: 2021-07-27
- Medicament for treatment and/or prevention of cancerPublication Number: CA-3227706-A1Priority Date: 2021-07-27
- Medicines for the treatment and/or prevention of cancerPublication Number: CN-117677398-APriority Date: 2021-07-27
- Medicament for treatment and/or prevention of cancerPublication Number: EP-4378478-A1Priority Date: 2021-07-27
- Medicines for the treatment and/or prevention of cancerPublication Number: KR-20240041917-APriority Date: 2021-07-27
- Composition for preventing or treating c-myc-associated diseases through inhibition of expression of ubiquitin-specific protease 47 (usp47), and use thereofPublication Number: WO-2024085603-A1Priority Date: 2022-10-18
- Chimeric small molecules for labeling proteins with immunogenic moieties and methods of use thereofPublication Number: WO-2024081675-A2Priority Date: 2022-10-10
- Macrocyclic heterocycles and uses thereofPublication Number: US-11912708-B2Priority Date: 2022-04-20Grant Date: 2024-02-27
- Macrocyclic heterocycles and uses thereofPublication Number: US-2023339952-A1Priority Date: 2022-04-20
- Macrocyclic heterocycles and uses thereofPublication Number: WO-2023205701-A1Priority Date: 2022-04-20
- Composition for cancer prevention or treatment containing met inhibitor and k-ras inhibitorPublication Number: WO-2024191132-A1Priority Date: 2023-03-10
- Combination therapy and related methodsPublication Number: TW-202436340-APriority Date: 2023-02-06
- Combination therapy and related methodsPublication Number: US-2024343813-A1Priority Date: 2023-02-06
- Combination therapy and related methodsPublication Number: WO-2024167880-A1Priority Date: 2023-02-06
- A composition for preventing or treating c-Myc-related diseases through suppression of expression of USP47, and use thereofPublication Number: KR-20240055632-APriority Date: 2022-10-18
- Ectonucleotide pyrophosphatase/phosphodiesterase 1 (enpp1) inhibitor combinations and uses thereofPublication Number: WO-2025007955-A1Priority Date: 2023-07-06
- Methionine adenosyltransferase 2a (mat2a) inhibitor combinations and uses thereofPublication Number: WO-2024217502-A1Priority Date: 2023-04-19
- Combinations of kras inhibitor and atr inhibitor for the treatment of cancerPublication Number: WO-2024213699-A1Priority Date: 2023-04-14
- Anti-lipocalin-2 antibodies and uses thereofPublication Number: WO-2024191972-A2Priority Date: 2023-03-13
- Composition for Cancer Prevention or Treatment Including MET inhibitors and K-RAS inhibitorsPublication Number: KR-20240138493-APriority Date: 2023-03-10
- A kind of preparation method of 2,5,6-trichloronicotinic acidPublication Number: CN-117263854-APriority Date: 2023-09-15
- Protein tyrosine phosphatase degraders and uses thereofPublication Number: WO-2025030006-A2Priority Date: 2023-08-02
- Protein tyrosine phosphatase inhibitor combinationsPublication Number: WO-2025030008-A1Priority Date: 2023-08-02
- Methods of treating a ras related disease or disorderPublication Number: US-2025041295-A1Priority Date: 2023-07-14
- Methods of treating a ras related disease or disorderPublication Number: WO-2025019318-A2Priority Date: 2023-07-14
///////Garsorasib, D 1553, Chia Tai Tianqing, CHINA 2024, APPROVALS 2024, Anfangning, 2559761-14-5, P491NE9G6Z
Fulzerasib
Fulzerasib
GFH925
CAS No. : 2641747-54-6
| Molecular Weight | 617.07 |
|---|---|
| Formula | C32H30ClFN6O4 |
(7R)-16-chloro-15-(2-fluoro-6-hydroxyphenyl)-9-methyl-12-(4-methyl-2-propan-2-ylpyridin-3-yl)-5-prop-2-enoyl-2,5,9,12,14-pentazatetracyclo[8.8.0.02,7.013,18]octadeca-1(10),13,15,17-tetraene-8,11-dione
CHINA 2024, APPROVALS 2024, Innovent Biologics, DUPERT
Fulzerasib (Dupert®; Innovent Biologics/GenFleet Therapeutics) is an orally active small molecule inhibitor of the KRAS G12C mutant protein being developed for the treatment of solid tumors harboring the KRAS G12C oncogenic driver mutation, including non-small cell lung cancer (NSCLC) and colorectal cancer. Fulzerasib received its first approval on 21 August 2024 in China, for the treatment of adults with KRAS G12C-mutated advanced NSCLC who have received at least one line of systemic therapy. This conditional approval was based on the positive results of a single-arm, phase II study. This article summarizes the milestones in the development of fulzerasib leading to this first approval for KRAS G12C-mutated advanced NSCLC.
Fulzerasib (GFH925) is an irreversible KRAS G12C inhibitor, has a synergistic anti-cancer effect with cetuximab (HY-P9905)..
PAPER
https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c03183
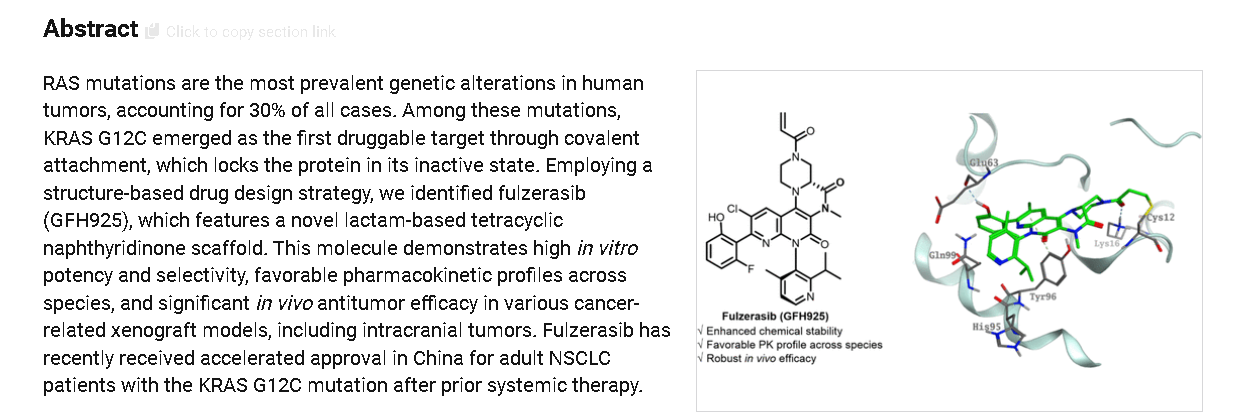
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021083167&_cid=P20-MEJIF1-91906-1
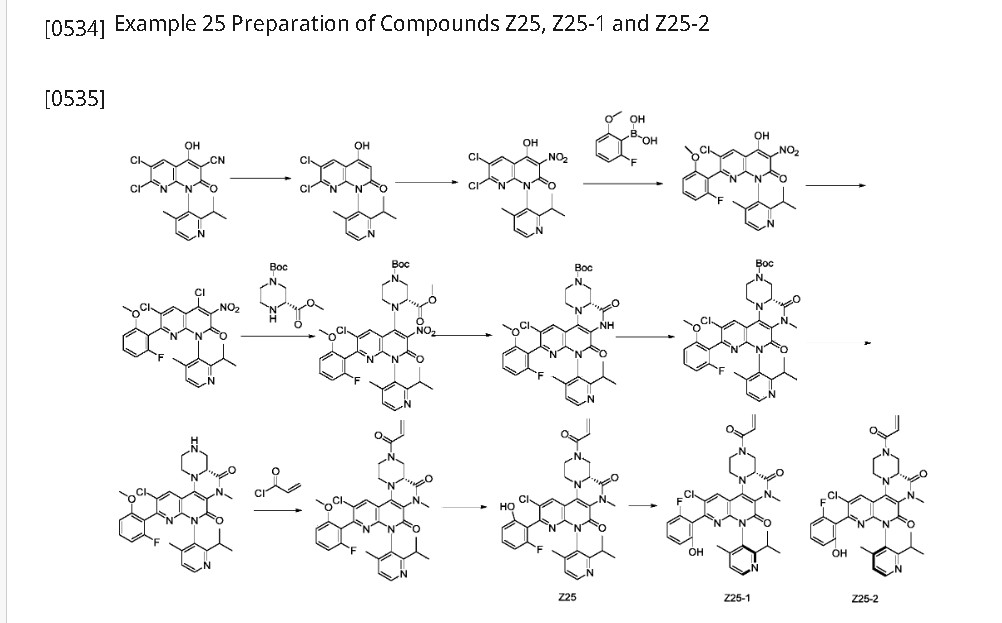
Step 1: Suspend 6,7-dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carbonitrile (30.0 g, 77.319 mmol) in a mixture of 1,4-dioxane (120 mL) and water (120 mL). Slowly add concentrated sulfuric acid (120 mL). Stir at 120°C for 36 hours. Pour the cooled reaction mixture into 200 mL of ice water, adjust the pH to 2-3 with sodium carbonate, and extract with ethyl acetate (1000 mL x 2). Combine the ethyl acetate phases, dry over anhydrous sodium sulfate, filter, and vacuum-dry the filtrate to obtain 6,7-dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-1,8-naphthyridine-2(1H)-one (24 g, Y: 85.7%) as a light brown solid. ES-API: [M+H]
[0537]Step 2: 6,7-Dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-1,8-naphthyridin-2(1H)-one (3.16 g, 8.705 mmol) was dissolved in acetic acid (15 mL). Sodium nitrite (100 mg, 1.58 mmol) and concentrated nitric acid (5.0 mL, 74.52 mmol) were added sequentially. The reaction was stirred at room temperature for 30 minutes. The reaction solution was slowly poured into 100 mL of ice water. The precipitated solid was filtered, and the filter cake was washed with 20 mL of ice water and dried under vacuum to obtain 6,7-dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (3.5 g, Y: 92%) as a yellow solid. ES-API: [M+H]
[0538]Step 3: To a 100 mL three-necked round-bottom flask, add 6,7-dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (3.5 g, 8.570 mmol), (2-fluoro-6-methoxyphenyl)boronic acid (5.8 g, 34.10 mmol), tetrakistriphenylphosphine palladium (1.15 g, 0.9956 mmol), sodium carbonate (3.5 g, 33.02 mmol), 10 mL of water, and 40 mL of dioxane. Under nitrogen, stir at 100°C for 2-3 hours. After completion, cool the reaction mixture to room temperature, add 80 mL of water and 100 mL of methyl tert-butyl ether, and extract once. The aqueous phase was adjusted to pH 3-5 with 1M hydrochloric acid solution and extracted with ethyl acetate (200 mL x 2). The ethyl acetate phases were combined, dried over anhydrous sodium sulfate, filtered, and the filtrate was dried under vacuum to afford 6-chloro-7-(2-fluoro-6-methoxyphenyl)-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (4.5 g, crude) as a pale yellow solid. ES-API: [M+H]
[0539]Step 4: 6-Chloro-7-(2-fluoro-6-methoxyphenyl)-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (4.6 g, 8.57 mmol) was dissolved in acetonitrile (30 mL). Phosphorus oxychloride (7.5 g, 48.92 mmol) and N,N-diisopropylethylamine (10.5 g, 81.24 mmol) were added sequentially. The reaction mixture was gradually heated to 80°C and stirred for 30 minutes. The reaction solution was concentrated, 30 mL of cold acetonitrile was added, and the mixture was added dropwise to 150 mL of saturated sodium bicarbonate solution under an ice-water bath. The mixture was extracted with ethyl acetate (200 mL x 2). The ethyl acetate phases were combined and washed once with 200 mL of saturated brine. The reaction mixture was dried over anhydrous sodium sulfate and filtered. The organic phase was dried and concentrated, and the crude product was purified by flash silica gel column chromatography (EtOAc/PE: 0-50%) to afford 4,6-dichloro-7-(2-fluoro-6-methoxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (3.05 g, Y: 76%) as a yellow solid. ES-API: [M+H]
[0540]Step 5: 4,6-Dichloro-7-(2-fluoro-6-methoxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (2.5 g, 4.843 mmol) was dissolved in N,N-dimethylacetamide (25 mL). 1-(tert-butyl)-3-methyl(R)-piperazine-1,3-dicarboxylate (3.5 g, 14.34 mmol) and N,N-diisopropylethylamine (2.0 g, 15.47 mmol) were added sequentially. The reaction mixture was stirred at 120°C for 2 hours. 80 mL of ethyl acetate was added to the reaction mixture, and the mixture was washed three times with 80 mL of saturated brine. The ethyl acetate phase was dried and concentrated, and the crude product was purified on a flash silica gel column (EtOAc/PE: 0-80%) to afford 1-(tert-butyl)-3-methyl (3R)-4-(6-chloro-7-(2-fluoro-6-methoxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-2-oxo-1,2-dihydro-1,8-naphthyridin-4-yl)piperazine-1,3-dicarboxylate (2.7 g, Y: 77%) as a yellow solid. ES-API: [M+H]
[0541]Step 6: 1-(tert-Butyl)3-methyl(3R)-4-(6-chloro-7-(2-fluoro-6-methoxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-2-oxo-1,2-dihydro-1,8-naphthyridin-4-yl)piperazine-1,3-dicarboxylate (2.7 g, 3.728 mmol) was dissolved in acetic acid (30 mL), iron powder (835 mg, 14.91 mmol) was added, and the reaction was stirred at 80 °C for 30 minutes. The reaction mixture was concentrated, and 200 mL of ethyl acetate and 100 mL of saturated sodium bicarbonate were added sequentially. The suspension was filtered through celite, and the filter cake was washed with ethyl acetate. The organic phase was separated and washed sequentially with 100 mL of saturated sodium bicarbonate and 150 mL of saturated brine. The mixture was dried and concentrated to give (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylic acid tert-butyl ester (2.70 g, crude) as a yellow solid. ES-API: [M+H]+ = 663.2.
[0542]Step 7: To a 150 mL sealed tube was added tert-butyl (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylate (2.7 g, 3.728 mmol), 30 mL of acetone, anhydrous potassium carbonate (2.2 g, 15.94 mmol), and iodomethane (5.4 g, 38.03 mmol). The tube was sealed and the reaction was stirred at 55°C for 18 hours. The reaction mixture was added with 150 mL of ethyl acetate, washed three times with 100 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column (EtOAc/PE: 0-80%) to obtain (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylic acid tert-butyl ester (2.2 g, Y: 87%) as a yellow solid. ES-API: [M+H]
[0543]Step 8: Tert-butyl (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylate (517 mg, 0.7549 mmol) was dissolved in dichloromethane (8 mL) and trifluoroacetic acid (2 mL) was added. After stirring at room temperature for 2 hours, the reaction mixture was concentrated to afford (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (530 mg, crude), which was used directly in the next reaction. ES-API: [M+H]
[0544]Step 9: (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (530 mg, 0.7549 mmol) was dissolved in dichloromethane (15 mL) and triethylamine (3.0 mL, 21.62 mmol) was added. The reaction mixture was cooled to 0°C and acryloyl chloride (100 mg, 1.1048 mmol) was added dropwise. The reaction was stirred at 0°C for 15 minutes. 80 mL of dichloromethane was added to the reaction solution, and the mixture was washed with 100 mL of saturated aqueous NaHCO₃
and 80 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column (EtOAc/PE: 0-60%) to obtain (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (280 mg, Y: 59%) as a yellow solid. ES-API: [M+H]
[0545]Step 10: In an ice-water bath, (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (280 mg, 0.444 mmol) was added to dry dichloromethane (6.0 mL), and then boron tribromide (5.0 mL, 5.0 mmol) was added. The mixture was warmed to room temperature and reacted overnight. Under ice-water bath conditions, the reaction solution was added dropwise to a saturated sodium bicarbonate solution, extracted twice with dichloromethane (80 mL), dried, and concentrated. The crude product was purified by flash silica gel column chromatography (EtOAc/PE: 0-60%) to give (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-hydroxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (Z25, 233 mg, Y: 85%).
[0546]Step 11: Compound Z25 was separated by preparative chiral HPLC (column type: IA: 10 μm, 30*250 mm, mobile phase: hexane:EtOH = 60:40, flow rate: 25 ml/min, column temperature) to obtain: an atropisomer compound Z25-1 (76.8 mg, peak 1, retention time 2.531 min, Y: 34%).
1 H NMR (500 MHz, DMSO-d
6 )δ10.03(d,J=18.4Hz,1H),8.52(d,J=7.3Hz,1H),8.43(d,J=4.7Hz,1H),7.23(d,J=9.6Hz,2H),7.08(dd,J=16 .6,10.5Hz,1H),6.74–6.62(m,2H),6.15(d,J=16.8Hz,1H),5.75(d,J=10.7Hz,1H),4.73(d,J=14.2Hz,1H),4.4 6 (d, J = 12.9 Hz, 1H), 4.00 (s, 1H), 3.61 (d, J = 10.0 Hz, 1H), 3.51 (s, 1H), 3.34 (s, 3H), 3.22 (s, 1H), 2.64 (t, J = 11.5 Hz, 1H), 2.48–2.42 (m, 1H), 1.98 (d, J = 5.1 Hz, 3H), 1.03 (t, J = 6.9 Hz, 3H), 0.86 (t, J = 7.9 Hz, 3H). ES-API: [M+H]
+ = 617.2. And another atropisomer compound Z25-2 (70 mg, peak 2, retention time 3.683 min, Y: 31%).
1 H NMR (500MHz, CDCl
3 )δ8.64–8.59(m,1H),8.35(s,1H),8.07(s,1H),7.27–7.20(m,2H),7.14–7.02(m,1H),6.75–6.63(m,2H),6.39(dd,J=17.0,2.0Hz,1H),5.88 –5.77(m,1H),4.91(d,J=14.0Hz,1H),4.83(d,J=13.0Hz,1H),3.72–3.58(m,2H),3.50(s,3H),3.43(d,J=12.0Hz,1H),3.1 6(t,J=13.0Hz,1H),2.91(t,J=12.0Hz,1H),2.82-2.73(m,1H),1.93(s,3H),1.24(d,J=7.0Hz,3H),1.12(d,J=7.0Hz,3H). ES-API: [M+H]
+ =617.2. The isomers were detected by analytical chiral HPLC (column type: IA: 5 μm, 4.6*150 mm, mobile phase: hexane:EtOH=60:40, flow rate: 1 ml/min, column temperature=30°C).
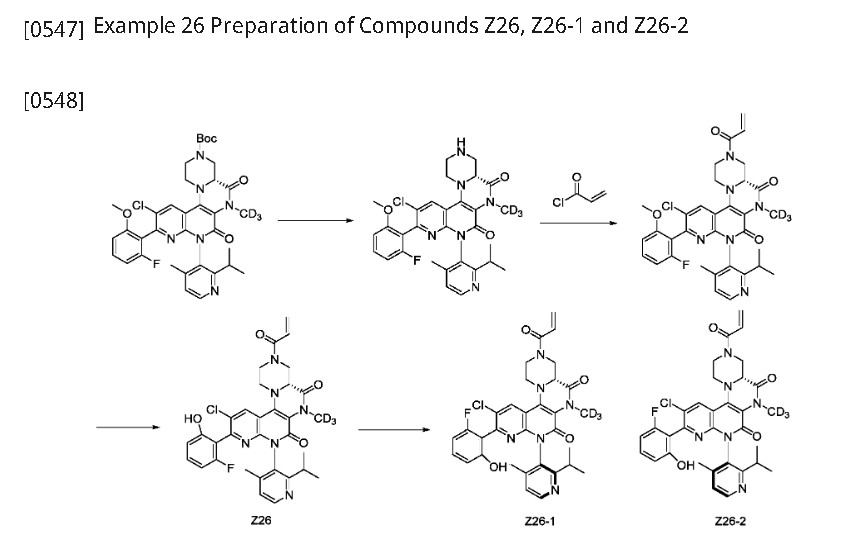
Step 1: (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylic acid tert-butyl ester (511 mg, 0.7549 mmol) was dissolved in dichloromethane (8 mL) and trifluoroacetic acid (2 mL) was added. After stirring at room temperature for 2 hours, the reaction mixture was concentrated to give (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (520 mg, crude), which was used directly in the next reaction. ES-API: [M+H]
[0550]Step 2: (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (520 mg, 0.7549 mmol) was dissolved in dichloromethane (10 mL) and triethylamine (3.0 mL, 21.62 mmol) was added. The reaction mixture was cooled to 0°C and acryloyl chloride (100 mg, 1.1048 mmol) was added dropwise. The reaction was stirred at 0°C for 15 minutes. 80 mL of dichloromethane was added to the reaction solution, and the mixture was washed with 100 mL of saturated aqueous NaHCO₃
and 80 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column (EtOAc/PE: 0-60%) to obtain (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d₃)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (232 mg, Y: 48%) as a yellow solid. ES-API: [M+H]
[0551]Step 3: Under ice-water bath conditions, (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (240 mg, 0.3791 mmol) was added to dry dichloromethane (6.0 mL), and boron tribromide (5.0 mL, 5.0 mmol) was added. The temperature was warmed to room temperature and the reaction was allowed to react overnight. Under ice-water bath conditions, the reaction solution was added dropwise to a saturated sodium bicarbonate solution, extracted twice with dichloromethane (80 mL), dried, and concentrated. The crude product was purified on a flash silica gel column (EtOAc/PE: 0-60%) to give (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-hydroxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (Z26, 187 mg, Y: 79%). [M+H]
[0552]Step 4: Compound Z26 (187 mg, 0.302 mmol) was separated by preparative chiral HPLC (column type: IA: 10 μm, 30*250 mm, mobile phase: hexane:EtOH = 60:40, flow rate: 25 ml/min, column temperature) to obtain: an atropisomer compound, arbitrarily designated as Z26-1 (68.8 mg, peak 1, retention time 2.525 min, Y: 36.7%).
1 H NMR (500 MHz, DMSO-d
6 )δ10.03(d,J=17.9Hz,1H),8.51(d,J=7.4Hz,1H),8.43(d,J=4.7Hz,1H),7.29–7.18(m,2H),7.08(dd,J =17.0,10.6Hz,1H),6.74–6.61(m,2H),6.15(d,J=16.6Hz,1H),5.75(d,J=11.5Hz,1H),4.73(d,J=13.5 Hz, 1H), 4.46 (d, J = 12.3 Hz, 1H), 4.00 (s, 1H), 3.61 (d, J = 10.5 Hz, 1H), 3.50 (s, 1H), 3.22 (s, 1H), 2.65 (t, J = 12.5 Hz, 1H), 2.49–2.42 (m, 1H), 1.98 (d, J = 5.0 Hz, 3H), 1.02 (d, J = 7.0 Hz, 3H), 0.86 (t, J = 7.9 Hz, 3H). ES-API: [M+H]
+ = 620.3. Another atropisomer, arbitrarily designated Z26-2 (63.2 mg, peak 2, retention time 3.683 min, Y: 33.79%), was obtained.
1 H NMR (400 MHz, CDCl
3 )δ8.62(d,J=4.8Hz,1H),8.35(s,1H),8.07(s,1H),7.24–7.20(m,2H),7.16–7.01(m,1H),6.74–6.6 3(m,2H),6.39(dd,J=16.8,2.0Hz,1H),5.82(dd,J=10.4,2.0Hz,1H),4.91(d,J=13.6Hz,1H),4.83(d δ (d, J = 13.6 Hz, 1H), 3.71–3.57 (m, 2H), 3.42 (d, J = 12.0 Hz, 1H), 3.16 (t, J = 12.8 Hz, 1H), 2.91 (t, J = 12.0 Hz, 1H), 2.81–2.70 (m, 1H), 1.92 (s, 3H), 1.22 (d, J = 6.8 Hz, 3H), 1.10 (d, J = 6.8 Hz, 3H). ES-API: [M+H]
+ = 620.3. The isomeric compounds were detected by analytical chiral HPLC (column type: IA: 5 μm, 4.6*150 mm, mobile phase: hexane:EtOH = 60:40, flow rate: 1 ml/min, column temperature = 30°C).
PAT
- US12054497, Compound Z25-1
- US12054497, Compound Z25-2
- US12054497, Compound Z26-1
- US12054497, Compound Z26-2
PAT
CN112390818
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN319676055&_cid=P20-MEJIKL-96783-1
| Example 25 Preparation of Z25 |
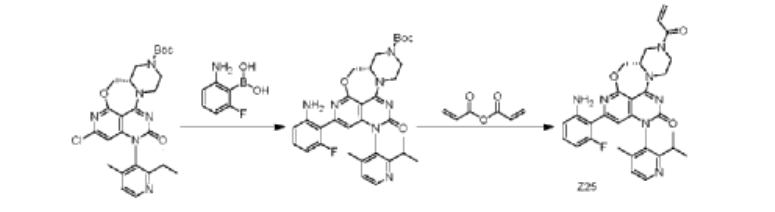
| Step 1: To a 100 mL three-necked round-bottom flask was added (S)-2-chloro-12-(2-isopropyl-4-methylpyridin-3-yl)-11-oxo-5a,6,8,9,11,12-hexahydro-4-oxo-3,7,9a,10,12-pentaazabenzo[4,5]cycloheptyl[1,2,3-de]naphthalene-7(5H)-carboxylic acid tert-butyl ester (1.4 g, 2.66 mmol), (2-amino-6-fluorophenyl)boronic acid (0.6 g, 3.87 mmol), Sphos-Pd-G2 (0.2 g, 0.21 mmol), Sphos (120 mg, 0.29 mmol), potassium phosphate (1.2 g, 5.66 mmol), 10 mL of dioxane, and 2 mL of water. The system was purged with nitrogen three times and then protected with nitrogen. The reaction was continued at 120°C for 2 h. 30 mL of ethyl acetate was added to the reaction solution, which was washed three times with 30 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column to give the target product, (S)-2-(2-amino-6-fluorophenyl)-12-(2-isopropyl-4-methylpyridin-3-yl)-11-oxo-5a,6,8,9,11,12-hexahydro-4-oxa-3,7,9a,10,12-pentaazabenzo[4,5]cyclohepta[1,2,3-de]naphthalene-7(5H)-carboxylic acid tert-butyl ester (845 mg, yield: 41%). ES-API: [M+H]+ = 602.2. |
| Step 2: Dissolve (S)-tert-butyl 2-(2-amino-6-fluorophenyl)-12-(2-isopropyl-4-methylpyridin-3-yl)-11-oxo-5a,6,8,9,11,12-hexahydro-4-oxa-3,7,9a,10,12-pentaazabenzo[4,5]cyclohepta[1,2,3-de]naphthalene 7(5H)-carboxylate (800 mg, 1.33 mmol) in dichloromethane (8 mL), and add trifluoroacetic acid (2 mL). Stir at room temperature for 2 hours. The reaction mixture is concentrated to obtain the target intermediate, which is dissolved in dichloromethane (15 mL) and triethylamine (800 mg, 87.1 mmol) is added. Cool the reaction mixture to 0°C, and add acrylic anhydride (160 mg, 1.27 mmol) dropwise. Stir the reaction mixture at 0°C for 15 minutes. The reaction mixture was added with 40 mL of dichloromethane, washed with 50 mL of saturated aqueous NaHCO₃ and 40 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column to obtain the target product, Z25(S)-7-acryloyl-2-(2-amino-6-fluorophenyl)-12-(2-isopropyl-4-methylpyridin-3-yl)-5,5a,6,7,8,9-hexahydro-4-oxa-3,7,9a,10,12-pentaazabenzo[4,5]cyclohepta[1,2,3-de]naphthalen-11(12H)-one (250 mg, yield: 34%). ES-API: [M+H] ⁺ = 556.2. 1 H NMR (500MHz, DMSO) δ8.55 (d, J=4.9Hz, 1H), 7.32 (d, J=4.9Hz, 1H), 7.04 (dd, J= 14.8,8.0Hz,1H),6.95-6.80(m,1H),6.52(d,J=8.3Hz,1H),6.36-6.13(m,4H) ,6.06-5.95(m,1H),5.78(d,J=10.3Hz,1H),4.82-4.04(m,7H),3.56(s,1H),3 .25-3.18(m,1H),2.84-2.70(m,1H),1.98(d,J=5.2Hz,3H),1.15-0.95(m,6H). |
| Example 26 Preparation of Z26 |
| Step 1: To a solution of 7-chloro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[4,3-d]pyrimidine-2,4(1H,3H)-dione (130 mg, 0.39 mmol) in acetonitrile (3 mL) were added phosphorus oxychloride (1 mL) and N,N-diisopropylethylamine (1 mL) sequentially. The mixture was stirred at 90°C for 2 h. The reaction mixture was concentrated to afford crude 4,7-dichloro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[4,3-d]pyrimidin-2(1H)-one (130 mg). ES-API: [M+H] + = 349.3. |
| Step 2: To a solution of 4,7-dichloro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[4,3-d]pyrimidin-2(1H)-one (130 mg, 0.37 mmol) in acetonitrile (3 mL) was added N,N-diisopropylethylamine (144 mg, 1.12 mmol) and tert-butyl piperazine-1-carboxylate (70 mg, 0.37 mmol) under ice-cooling. The mixture was stirred for 30 minutes. The reaction mixture was poured into 20 mL of water and extracted with ethyl acetate (20 mL x 3). The mixture was dried over anhydrous sodium sulfate and concentrated. The mixture was then purified on a flash silica gel column (0-100% ethyl acetate/petroleum ether) to obtain tert-butyl 4-(7-chloro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[4,3-d]pyrimidin-4-yl)piperazine-1-carboxylate (140 mg) as a white solid. ES-API: [M+H] + = 499.1. |
| Step 3: Under nitrogen protection, a mixture of tert-butyl 4-(7-chloro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[4,3-d]pyrimidin-4-yl)piperazine-1-carboxylate (140 mg, 0.28 mmol), 2-fluoro-6-hydroxyphenylboronic acid (44 mg, 0.42 mmol), chloro(2-dicyclohexylphosphino-2′,6′-dimethoxy-1,1′-biphenyl)(2′-amino-1,1′-biphenyl-2-yl)palladium(II) (13 mg, 0.02 mmol), 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (10 mg, 0.02 mmol) and potassium phosphate (120 mg, 0.84 mmol) in 1,4-dioxane (4 mL) and water (1 mL) was microwaved at 120 ° C for 1 h. The reaction mixture was filtered and washed with ethyl acetate (100 mL). The filtrate was washed with saturated brine (50 mL x 3). The resulting organic phase was dried, concentrated, and purified on a flash silica gel column (0-100% ethyl acetate/petroleum ether) to afford tert-butyl 4-(7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyridinyl[4,3-d]pyrimidin-4-yl)piperazine-1-carboxylate (100 mg, yield: 62%) as a white solid. ES-API: [M+H] + = 575.2. |
| Step 4: To a solution of tert-butyl 4-(7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyridinyl[4,3-d]pyrimidin-4-yl)piperazine-1-carboxylate (100 mg, 0.17 mmol) in dichloromethane (4 mL) was added trifluoroacetic acid (1 mL) under ice-cooling. The mixture was stirred at room temperature for 2 h and concentrated to afford 7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-4-(piperazin-1-yl)pyridin[4,3-d]pyrimidin-2(1H)-one (82 mg, theoretical) as a yellow oil. ES-API: [M+H] + = 475.2. |
| Step 5: Under ice bath, add N,N-diisopropylethylamine (110 mg, 0.85 mmol) to a solution of 7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-4-(piperazin-1-yl)pyridin[4,3-d]pyrimidin-2(1H)-one (82 mg, 0.17 mmol) in dichloromethane (3 mL). After the reaction solution becomes clear, add acrylic anhydride (21 mg, 0.17 mmol) dropwise and stir for 5 minutes. The reaction solution is washed with saturated sodium bicarbonate solution (5 mL). The organic phase is dried, concentrated, and purified by preparative HPLC (ammonium bicarbonate system) to obtain a light yellow solid Z26 (12.44 mg, purity: 100%, yield: 14% ) . NMR (500MHz, DMSO) δ12.86(s,1H),9.26(s,1H),8.59(d,J=4.9Hz,1H),7.35(d,J=4.9Hz,1H),7.29(d d,J=15.0,8.2Hz,1H),6.86(dd,J=16.7,10.4Hz,1H),6.77(d,J=8.3Hz,1H),6.73-6.66(m,2H),6.21( dd,J=16.6,2.3Hz,1H),5.77(dd,J=10.4,2.3Hz,1H),4.07(d,J=5.0Hz,4H),3.88(d,J=36.8Hz,4H),2 .76(dt,J=13.6,6.8Hz,1H),1.96(s,3H),1.10(d,J=6.7Hz,3H),1.04(d,J=6.7Hz,3H).ES-API:[M+H] + =529.2. |
SYN
Fulzerasib is an orally active KRAS G12C inhibitor developed by Innovent Biologics. It selectively targets the KRAS G12C mutation in NSCLC [36,37]. In 2024, the NMPA approved Fulzerasib (brand name: Dupert) for treating adult patients with advanced NSCLC harboring the KRAS G12C mutation who have progressed after prior systemic therapy. Fulzerasib irreversibly binds to the KRAS G12C mutant protein, locking it in an inactive GDP-bound state, thereby inhibiting downstream signaling pathways such as MAPK and PI3K. This action effectively suppresses cancer cell proliferation and survival. The clinical efficacy of Fulzerasib was demonstrated in a Phase II trial (NCT05009303) involving patients with advanced NSCLC and KRAS G12C mutations [38]. In the clinical trial, Fulzerasib demonstrated an ORR of 49.1 % and a disease control rate (DCR) of 90.5 %, with a median PFS of 9.7 months, reflecting robust antitumor efficacy. The agent exhibited favorable tolerability, characterized by manageable toxicity. Treatment-related adverse events were predominantly mild to moderate in severity, with the most frequently reported being diarrhea, nausea, and elevated liver enzymes [38]. The safety profile was consistent with other KRAS G12Cinhibitors, making it a viable therapeutic option.
The synthetic route of Fulzerasib, shown in Scheme 9, initiates with H2SO4-mediated decyanation of Fulz-001, affording Fulz-002 [39]. Nitrosation of Fulz-002 with NaNO2 yields Fulz-003, which undergoes
Suzuki-Miyaura coupling with (2-fluoro-6-methoxyphenyl)boronic acid to construct Fulz-004. Phosphochlorination with POCl3 under DIPEA catalysis converts Fulz-004 to Fulz-005. Nucleophilic displacement with methyl (R)-1-N-Boc-piperazine-3-carboxylate assembles Fulz-006. Fe-mediated tandem Mannich cyclization/nitro reduction transforms Fulz-006 into bicyclic amine Fulz-007. Methylation with MeI generates Fulz-008, followed by TFA-mediated Boc cleavage to afford Fulz-009.
Acrylation with acryloyl chloride produces Fulz-010. Selective O-demethylation followed by chiral HPLC resolution delivers Fulzerasib
[36] Y.N. Lamb, Correction: fulzerasib: first approval, Drugs 85 (2025) 281.
[37] Y.N. Lamb, Fulzerasib: first approval, Drugs 84 (2024) 1665–1671.
[38] Q. Zhou, X. Meng, L. Sun, D. Huang, N. Yang, Y. Yu, M. Zhao, W. Zhuang, R. Guo,
Y. Hu, Y. Pan, J. Shan, M. Sun, Y. Yuan, Y. Fan, J. Huang, L. Liu, Q. Chu, X. Wang,
C. Xu, J. Lin, J. Huang, M. Huang, J. Sun, S. Zhang, H. Zhou, Y.L. Wu, Efficacy and
safety of KRASG12C inhibitor IBI351 monotherapy in patients with advanced
NSCLC: results from a phase 2 pivotal study, J. Thorac. Oncol. 19 (2024)
1630–1639.
[39] F. Zhou, T. Jiang, W. He, L. Cai, H. Yang, Z. Liu, J. Lan, Preparation of
Heteroaromatic Ring Dihydropyrimidinone Derivatives as KRAS Gene Mutation
Inhibitors Useful in the Treatment of Cancer, 2021. CN112390818A.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. Rafael Rosell, et al. KRAS G12C-mutant driven non-small cell lung cancer (NSCLC). Crit Rev Oncol Hematol. 2024 Mar:195:104228. [Content Brief][2]. Vanesa Gregorc, et al. KROCUS: A phase II study investigating the efficacy and safety of fulzerasib. Free access.June 05, 2024
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Fulzerasib: First ApprovalPublication Name: DrugsPublication Date: 2024-11-26PMID: 39587006DOI: 10.1007/s40265-024-02120-6
- Substituted heterocyclic compounds, their preparation methods and medicinal usesPublication Number: CN-117645614-APriority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: EP-4328229-A2Priority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: US-12054497-B2Priority Date: 2019-10-30Grant Date: 2024-08-06
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: US-2024158417-A1Priority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: EP-4053118-B1Priority Date: 2019-10-30Grant Date: 2024-10-09
- Substituted heterocyclic and cyclic compounds, their preparation and medical usePublication Number: CN-113853373-BPriority Date: 2019-10-30Grant Date: 2022-08-05
- Substituted heterocyclic ring compound, preparation method and medical application thereofPublication Number: CN-115057872-APriority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: EP-4053118-A1Priority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, method for preparing same, and pharmaceutical use thereofPublication Number: KR-20220106765-APriority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: US-2023084095-A1Priority Date: 2019-10-30
- Polymorph of kras inhibitor, preparation method therefor, and use thereofPublication Number: WO-2023116895-A1Priority Date: 2021-12-24
- Polymorph of kras inhibitor, preparation method therefor, and use thereofPublication Number: EP-4455147-A1Priority Date: 2021-12-24
- Polymorph of kras inhibitor, preparation method therefor, and use thereofPublication Number: US-2025051333-A1Priority Date: 2021-12-24
- Key intermediate of kras inhibitor and preparation method thereforPublication Number: WO-2022198904-A1Priority Date: 2021-03-26
- A key intermediate of a KRAS inhibitor and its preparation methodPublication Number: CN-116848111-APriority Date: 2021-03-26
- Pharmaceutical composition, and preparation method therefor and use thereofPublication Number: WO-2024061267-A1Priority Date: 2022-09-21
- Pharmaceutical composition, use thereof, and method for treating cancerPublication Number: WO-2023186075-A1Priority Date: 2022-04-01
- Pharmaceutical composition, use thereof, and method for treating cancerPublication Number: EP-4506004-A1Priority Date: 2022-04-01
- Polymorphs of KRAS inhibitors, methods of making and uses thereofPublication Number: CN-118451081-APriority Date: 2021-12-24
- Polymorph of kras inhibitor and preparation method for polymorph and use thereofPublication Number: TW-202327593-APriority Date: 2021-12-24
////////Fulzerasib, CHINA 2024, APPROVALS 2024, Innovent Biologics, DUPERT, GFH925, GFH 925, IBI351, IBI 351

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}