
Home » Antiulcerative
Category Archives: Antiulcerative
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Fexuprazan, Abeprazan
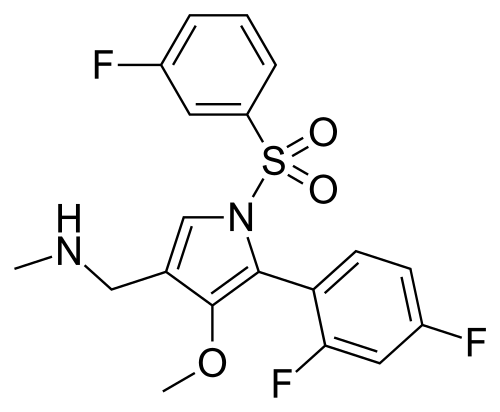
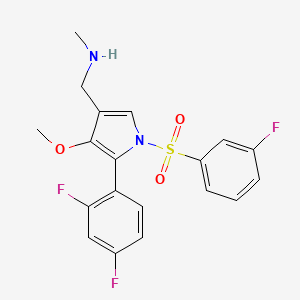
Fexuprazan, Abeprazan; DWP14012; DWP-14012
- CAS 1902954-60-2
- BE52S2C1QT
1-[5-(2,4-difluorophenyl)-1-(3-fluorophenyl)sulfonyl-4-methoxypyrrol-3-yl]-N-methylmethanamine
WeightAverage: 410.41
Monoisotopic: 410.091198078
Chemical FormulaC19H17F3N2O3S
Fexuprazan (trade name Fexuclue) is a drug for the treatment of gastroesophageal reflux disease (GERD).[1] It is a potassium-competitive acid blocker,[2] which is a class of drugs suppressing gastric acids.[3][4]
Fexuprazan is approved for clinical use in South Korea,[4][5] Mexico,[6] Philippines,[7] Chile,[8] and Ecuador.[9]
Abeprazan is under investigation in clinical trial NCT04341454 (Study to Evaluate the Efficacy and Safety of DWP14012 in Patients With Acute or Chronic Gastritis).
Proton pump inhibitors (PPIs) typified by omeprazole, which inhibit gastric acid secretion, are widely used in clinical settings. However, existing PPIs are accompanied by problems in terms of effectiveness and side effects. Specifically, since existing PPIs are unstable under acidic conditions, they are often formulated as enteric agents. in need. In addition, the existing PPI exhibits variation in therapeutic effect due to metabolic enzyme polymorphism and drug interaction with drugs such as diazepam, so improvement is desired.
In addition, since PPI is a prodrug activated by gastric acid and acts only on the active proton pump, the maximum drug expression time is delayed, the effect of suppressing acid secretion at night is poor, and it has disadvantages such as having to take it before meals. exist. In addition, PPI is mainly metabolized through the CYP2C19 enzyme, and there is a large difference in efficacy between individuals due to the genetic polymorphism of the CYP2C19 enzyme.
In order to improve the disadvantages of PPI as described above, a potassium-competitive gastric acid secretion inhibitor (Potassium-Competitive Acid Blocker, P-CAB) is attracting attention. Potassium competitive gastric acid secretion inhibitor strongly and rapidly inhibits gastric acid secretion by reversibly and competitively binding with K + ions to proton pump (H + /K + -ATPase), an enzyme involved in the final stage of gastric acid secretion in parietal cells. These P-CAB formulations show strong inhibition of the normal acidity (pH 1-3) in the stomach compared to the PPI formulations. However, pharmacological activity, which decreases as the pH increases, is required for gastric P-CAB preparations, and some P-CAB preparations show pharmacological activity that maintains pharmacological activity even when the pH increases, and some related side effects have been reported. In addition, since P-CAB preparations are mainly metabolized through the CYP3A4 enzyme, the difference in efficacy between individuals is relatively small, and concerns about interactions with drugs metabolized by the CYP2C19 enzyme are relatively low.
International Patent Publication No. WO2019/013310 A1 discloses bonoprazan as a potassium-competitive acid secretion inhibitor.
However, it was confirmed that vonoprazan induces severe hypergastrinemia compared to the existing PPI drug lansoprazole. Such hypergastrinemia can include enterochromaffin-like (ECL)-cell hyperplasia; parietal cell hyperplasia; fundic gland polyp; It can cause problems such as bone loss, damaged bone quality, and fractures. In fact, it has been reported that vonoprazan is associated with the development of gastric neuroendocrine tumors in carcinogenicity studies in mice and rats. However, discontinuation of administration of P-CAB or PPI-based drugs such as vonoprazan restores excess gastric acid and causes indigestion, so despite the above problems, drug administration cannot be easily stopped.
On the other hand, PPI is used for the prevention of gastric and duodenal ulcers by administration of nonsteroidal anti-inflammatory drugs (NSAIDs). However, it has been reported that bonoprazan aggravates the damage to the small intestine caused by various types of NSAIDs. For example, NSAID-induced gastrointestinal damage includes edema, erythema, submucosal hemorrhage, erosion, and ulceration. From this point of view, clinically, in the case of vonoprazan, there may be significant limitations in combination with NSAID drugs.
There are two major mechanisms by which drugs such as NSAIDs or alcohol cause damage to the gastrointestinal mucosa: a local irritant effect and a systemic effect. The local irritant effect occurs due to ion-trap and mitochondrial damage, and systemically due to the decrease in prostaglandin and NO (nitric oxide). In addition to mitochondrial damage caused by oxidative stress, damage to vascular endothelial cells causes microcirculation disorders, making the gastrointestinal mucosa very vulnerable to damage and interfering with the mucosal damage recovery mechanism. Due to the combined action of these mechanisms, damage to the mucous membrane of the gastrointestinal tract, ie, gastric ulcer, enteropathy, etc. may occur or be severe.
Accordingly, even considering the effect of bonoprazan in terms of suppressing gastric acid secretion, its use is bound to be very limited due to the above potential problems.
Separately, Helicobacter pylori ( H. pylori ) is known as one of the main causes of gastrointestinal diseases such as chronic gastritis and peptic ulcer and gastric cancer. Although the prevalence of Helicobacter pylori in our country is gradually decreasing, a prevalence of more than 50% is still being reported. In particular, Helicobacter pylori is related to digestive diseases, and the importance of antibacterial treatment agents is increasing day by day. In particular, as reported in several studies, antibacterial treatment of Helicobacter pylori reduces the occurrence of bleeding in peptic ulcer. For this antibacterial therapy, in general, patients take clarithromycin and amoxicillin along with gastric acid inhibitors such as PPI as the first-line treatment. For multi-drug use of PPIs and antibiotics, the risk of drug-drug interactions must be low, and the risk of such interactions can be predicted through in vitro CYP inhibition, CYP/UGT phenotyping, and CYP induction tests.
However, additional or repeated administration of various antibiotics is required up to the second and third treatment, and side effects and resistance have been reported. Therefore, by reducing gastric acidity, the antibacterial effect of antibiotics on Helicobacter pylori (H. pylori ) is enhanced, and long-term dose reduction of gastric acidity, for example, proton-potassium pump inhibitory ability, etc. The need to develop a visible drug is emerging.
In addition, in the case of an oral drug, the bioavailability, which is the rate at which the administered drug enters the systemic circulation and is used in the body, is measured. High bioavailability is one of the essential elements of oral drugs because the higher the bioavailability, the higher the rate and extent to which the active ingredient or part of the drug is absorbed and utilized at the site of action. In general, such bioavailability increases as absorption through the gastrointestinal tract is higher and the degree of first-pass effect is lower. , is affected by the size and shape of the particles, and the surface area of the particles.
It is also important that the concentration of the drug in the target organ, in this case the gastric tissue, is maintained as well as the bioavailability in the circulatory system. Therefore, drug distribution and maintenance to the target organ, gastric tissue, is judged to be an important pharmacokinetic characteristic in P-CAB drug development.
On the other hand, somatostatin, also known as growth hormone-inhibiting hormone (GHIH), is a cyclic peptide expressed in the gastrointestinal tract, pancreas, hypothalamus and central nervous system. It is secreted by D cells of the stomach and pancreas and acts as a paracrine regulator of gastric acid secretion, and suppresses gastric acid secretion by inhibiting gastric G cell gastrin secretion and parietal cell acid secretion. Activation of somatostatin receptors by somatostatin analogs and somatostatin receptor agonists inhibit gastrin secretion, thereby regulating histamine release from ECL cells and inhibiting acid secretion. In actual animal models and hypergastrinemia patients, it has been reported that the somatostatin analogue decreased the total gastric acid secretion by decreasing gastrin secretion and gastric acid response.
Gastric acid suppression by taking drugs such as PPI suppresses somatostatin secretion by D cells and promotes gastrin secretion by G cells by a feedback mechanism to induce hypergastrinemia. Gastrin promotes epithelial cell growth to induce oxyntic cell hyperplasia in the gastric body and increase parietal cell mass. This leads to proliferation of adenoma cells and hyperplasia of ECL cells, which may increase the risk of neuroendocrine tumors. In addition, the frequency of neuroendocrine tumors among tumors occurring in the duodenum is relatively high, and it is known that gastrin secretion is the most common form in neuroendocrine tumors occurring in the duodenum, accounting for approximately 65% of the total. It has been confirmed that the group taking bonoprazan tends to have a higher blood gastrin level than the group taking the existing PPI formulation due to the feedback mechanism of excessive gastric acid suppression. Because hypergastrinemia stimulates intestinal endocrine cells and may increase the risk of neuroendocrine tumors, studies are ongoing regarding the safety of long-term use.
Inhibition of gastrin secretion through somatostatin receptor activation has been reported to inhibit ECL cell hyperproliferation. In fact, synthetic peptide analogues of somatostatin with indications for endocrine diseases such as acromegaly, neuroendocrine tumors (NETs), and digestive system diseases such as upper gastrointestinal bleeding Sandostatin® (octreotide acetate) and Somatuline® Depot (lanreotide) are gastric neuroendocrine It has been reported to inhibit the overgrowth of ECL cells by inhibiting gastrin secretion in tumors.
In addition, there have been reports of anti-inflammatory responses through somatostatin receptor activation. Somatostatin is a type of neuropeptide that suppresses neurogenic inflammation and regulates the secretion of hormones and neurotransmitters. It is known to inhibit neurogenic inflammation and to be involved in nociception. Somatostatin is known to control the secretion of hormones and neurotransmitters to suppress neuronal inflammation and to be involved in nociception. Inflammatory somatostatin inhibits the proliferation of T lymphocytes and granulocytes in addition to controlling the neuroendocrine system. Somatostatin analogs are known to increase the expression of the anti-inflammatory factor IL-10 and inhibit the expression of the pro-inflammatory factors IFN-γ and TNF-α. As a result, the anti-inflammatory role of somatostatin has been mainly reported in studies related to inflammatory bowel disease (IBD). It is known that the level of intestinal somatostatin is reduced in patients with IBD, and it is known that the higher the level of inflammation in the intestine, the lower the level of somatostatin. In fact, it has been reported that the somatostatin analogue octreotide improved the symptoms of IBD in patients and animal models.
REF
- The efficacy and safety of fexuprazan in treating erosive esophagitis: a phase III, randomized, double‐blind, multicenter studyPublication Name: Journal of Gastroenterology and HepatologyPublication Date: 2024-01-22PMID: 38251791DOI: 10.1111/jgh.16471
- Review of the clinical development of fexuprazan for gastroesophageal reflux–related diseasePublication Name: European Journal of Clinical PharmacologyPublication Date: 2023-06-22PMID: 37344679DOI: 10.1007/s00228-023-03521-4
- Efficacy and Safety of Fexuprazan in Patients with Acute or Chronic GastritisPublication Name: Gut and LiverPublication Date: 2023-02-15PMCID: PMC10651377PMID: 36789577DOI: 10.5009/gnl220457
- Randomized controlled trial to evaluate the efficacy and safety of fexuprazan compared with esomeprazole in erosive esophagitisPublication Name: World Journal of GastroenterologyPublication Date: 2022-11-28PMCID: PMC9730436PMID: 36504556DOI: 10.3748/wjg.v28.i44.6294
- Editorial: acid suppression with potassium‐competitive acid blockers—dismissing genotype concerns. Authors’ replyPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2020-12-17PMID: 33333601DOI: 10.1111/apt.16158
- Editorial: acid suppression with potassium‐competitive acid blockers dismissing genotype concernsPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2020-12-17PMID: 33333607DOI: 10.1111/apt.16139
- Pharmacodynamics and pharmacokinetics of DWP14012 (fexuprazan) in healthy subjects with different ethnicitiesPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2020-10-27PMID: 33111337DOI: 10.1111/apt.16131
- Safety, tolerability, pharmacodynamics and pharmacokinetics of <scp>DWP</scp>14012, a novel potassium‐competitive acid blocker, in healthy male subjectsPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2018-06-04PMID: 29863280DOI: 10.1111/apt.14818
PATENTS
SearchSubmit searchSort byPublication Number – A to ZPublication Number – Z to APriority Date – OldestPriority Date – Most RecentGrant Date – OldestGrant Date – Most Recent
- Novel 4-methoxypyrrole derivative or salt thereof and pharmaceutical composition containing the samePublication Number: JP-6244498-B1Priority Date: 2015-04-27Grant Date: 2017-12-06
- Novel 4-methoxy pyrrole derivatives or salts thereof and pharmaceutical composition comprising the samePublication Number: KR-20160127646-APriority Date: 2015-04-27
- Novel 4-methexipirols derivatives or salts thereof and pharmaceutical compositions thereofPublication Number: RU-2663895-C1Priority Date: 2015-04-27Grant Date: 2018-08-13
- 4-methoxy pyrrole derivatives or salts thereof and pharmaceutical composition comprising the samePublication Number: US-10100010-B1Priority Date: 2015-04-27Grant Date: 2018-10-16
- Novel 4-methoxy pyrrole derivatives or salts thereof and pharmaceutical composition comprising the samePublication Number: WO-2016175555-A2Priority Date: 2015-04-27
SYN
https://pubs.acs.org/doi/10.1021/acsomega.4c04507
PAT
https://patents.google.com/patent/WO2023211843A1/en
Patent Citations (4)
Publication numberPriority datePublication dateAssigneeTitle
WO2016175555A2 *2015-04-272016-11-03Daewoong Pharmaceutical Co., Ltd.Novel 4-methoxy pyrrole derivatives or salts thereof and pharmaceutical composition comprising the same
US20190031609A1 *2016-03-252019-01-31Daewoong Pharmaceutical Co., Ltd.Novel acid addition salt of 1-(5-(2,4-difluorophenyl)-1-((3-fluorophenyl)sulfonyl)-4-methoxy-1h-pyrrol-3-yl)-n-methylmethanamine
US20200146974A1 *2017-07-072020-05-14Cj Healthcare CorporationComposition for injection
WO2021256861A1 *2020-06-172021-12-23일동제약(주)Novel acid secretion inhibitor and use thereof
PAT
https://patents.google.com/patent/WO2016175555A2/en
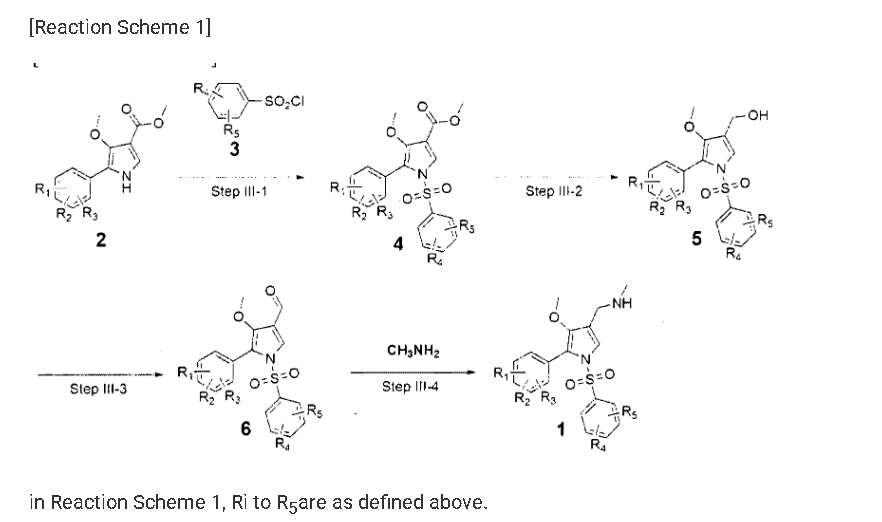
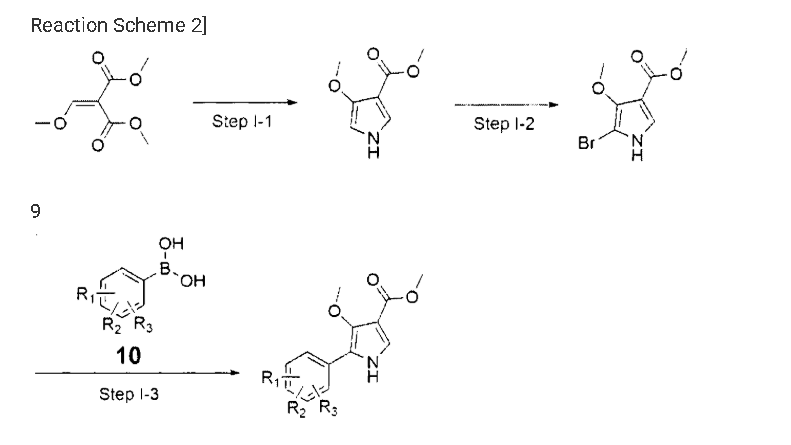
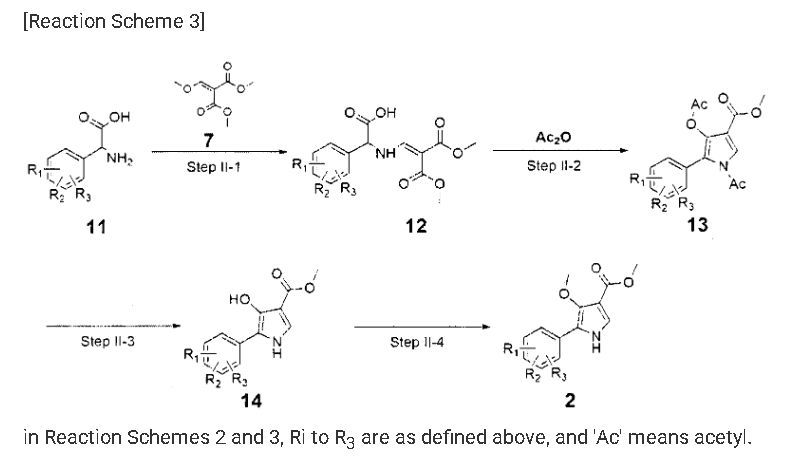
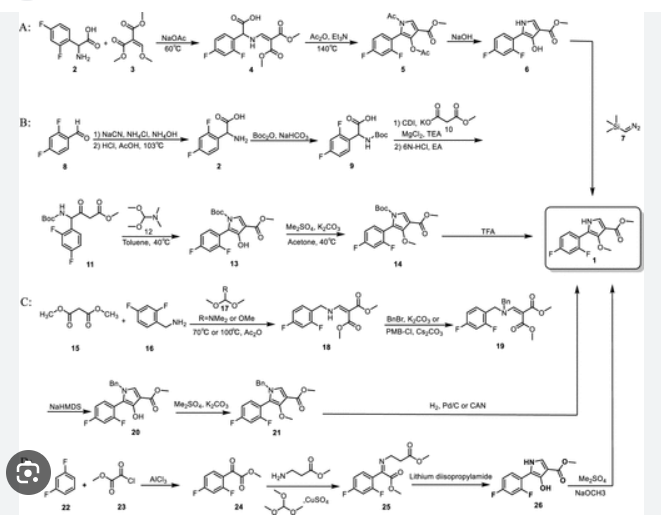
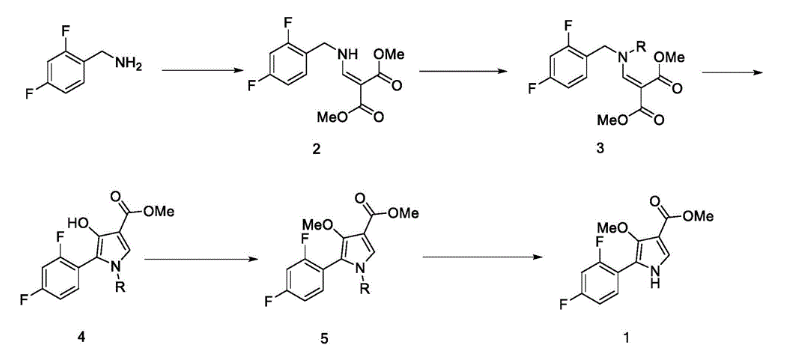
Example 8: Preparation of l-(5-(2,4-difluorophenyI)-l-((3-fluorophenyl)sulfonyI)-
4- metho\ -lH-pyrrol-3-yl)-N-methylmethanamine hydrochloride

(Step 8-1) Preparation of 2-(2,4-difluorophenyl)-2-((3-methoxy-2- (methoxycarbonyl)-3-oxoprop-l-en-l-yl)amino)acetic acid
2,4- Di fluorophenyl glycine (150.0 g, 801.5 mmol), dimethyl 2- (methoxymethylene)malonate (126.9 g, 728.6 mmol), and sodium acetate (65.8 g, 801 .5 mmol) were added to methanol (800.0 ml), and then refJuxed at 60°C for 4 hours. The reaction mixture was cooled to room temperature, and concentrated under reduced pressure to remove about 70% of methanol, and then filtered. The resulting solid was dried reduced pressure to give 190.0 g of the title compound. (Yield: 79.2%) Ή-NMR (500 MHz, CDC13): 8.02-7.99 (m, 1H), 7.45-7.40 (m, lH), 7.00-6.95 (m, 2H), 5.16 (s, lH), 3.74 (s, 3H), 3.76 (s, 3H)
PAT
https://patents.google.com/patent/WO2021256861A1/en
PAT
https://patents.google.com/patent/CN112094219A/en

Synthesis of Compound 1
In a 500ml reaction flask were charged 10 g of compound 5B, 100 ml of acetonitrile, 50 ml of water, 56 g of ceric ammonium nitrate, and reacted at room temperature for 12 hours. 100 ml of water and 100 ml of ethyl acetate are added. The mixture was allowed to stand for separation, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with water and saturated brine in this order, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a crude product of Compound 1. The crude product was crystallized from ethyl acetate and n-heptane to give 6.1 g of compound 1 in 85.6% yield as a pale yellow solid.
Syn
https://pubs.acs.org/doi/10.1021/acs.oprd.5c00255



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Ramani A, Merchant A, Cash BD (August 2023). “Review of the clinical development of fexuprazan for gastroesophageal reflux-related disease”. European Journal of Clinical Pharmacology. 79 (8): 1023–1029. doi:10.1007/s00228-023-03521-4. PMID 37344679. S2CID 259222741.
- Kim GH, Choi MG, Kim JI, Lee ST, Chun HJ, Lee KL, et al. (November 2023). “Efficacy and Safety of Fexuprazan in Patients with Acute or Chronic Gastritis”. Gut and Liver. 17 (6): 884–893. doi:10.5009/gnl220457. PMC 10651377. PMID 36789577.
- Jeong YS, Kim MS, Lee N, Lee A, Chae YJ, Chung SJ, et al. (May 2021). “Development of Physiologically Based Pharmacokinetic Model for Orally Administered Fexuprazan in Humans”. Pharmaceutics. 13 (6): 813. doi:10.3390/pharmaceutics13060813. PMC 8229463. PMID 34072547.
- Kim MS, Lee N, Lee A, Chae YJ, Chung SJ, Lee KR (June 2022). “Model-Based Prediction of Acid Suppression and Proposal of a New Dosing Regimen of Fexuprazan in Humans”. Pharmaceuticals. 15 (6): 709. doi:10.3390/ph15060709. PMC 9230547. PMID 35745628.
- “펙수클루정40밀리그램(펙수프라잔염산염)” [Fexuclue tablets 40 mg (fexuprazan hydrochloride)]. nedrug.mfds.go.kr (in Korean).
- “Daewoong Pharma’s GER drug gets product OK from Mexico”. Korea Economic Daily. 19 October 2023.
- Park IH. “Daewoong launches GERD treatment Fexuclu in Philippines”. KED Global. Retrieved 4 April 2025.
- Lee JH (14 March 2023). “Daewoong wins approval for GERD treatment Fexuclu in Chile”. KED Global. Retrieved 4 April 2025.
- Kim JE. “Daewoong receives approval for its GERD drug Fexuclue in Ecuador”. KED Global. Retrieved 4 April 2025.
| Clinical data | |
|---|---|
| Trade names | Fexuclue |
| Other names | Abeprazan; DWP14012; DWP-14012 |
| ATC code | A02BC10 (WHO) |
| Legal status | |
| Legal status | Rx in South Korea, Mexico |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1902954-60-2 |
| PubChem CID | 122662112 |
| DrugBank | DB16078 |
| ChemSpider | 68006985 |
| UNII | BE52S2C1QT |
| KEGG | D13012 |
| ChEMBL | ChEMBL4594445 |
| Chemical and physical data | |
| Formula | C19H17F3N2O3S |
| Molar mass | 410.41 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Fexuprazan, Abeprazan, DWP14012, DWP-14012
LANSOPRAZOLE
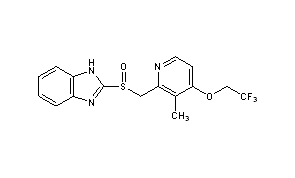

Lansoprazole
- Molecular FormulaC16H14F3N3O2S
- Average mass369.362 Da
Lansoprazole, AG-1749, ABT-006, CG-4801, A-65006, Ogast, Lanzor, Lanzo, Agopton, Opiren, Bamalite, Takepron, Lansox, Lansox, Ogastro, Monolitum, Prevacid, Zoton103577-45-3[RN]
1H-Benzimidazole, 2-[[[3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridinyl]methyl]sulfinyl]-
лансопразол [Russian] [INN], لانسوبرازول [Arabic] [INN], 兰索拉唑 [Chinese] [INN]
CAS Registry Number: 103577-45-3
CAS Name: 2-[[[3-Methyl-4-(2,2,2-trifluoro-ethoxy)-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazole
Additional Names: 2-(2-benzimidazolylsulfinylmethyl)-3-methyl-4-(2,2,2-trifluoroethoxy)pyridine
Manufacturers’ Codes: A-65006; AG-1749
Trademarks: Agopton (Takeda); Lansox (Takeda); Lanzor (Aventis); Limpidex (Sigma-Tau); Ogast (Takeda); Prevacid (TAP); Takepron (Takeda); Zoton (Wyeth)
Molecular Formula: C16H14F3N3O2S, Molecular Weight: 369.36
Percent Composition: C 52.03%, H 3.82%, F 15.43%, N 11.38%, O 8.66%, S 8.68%
Literature References: Gastric proton-pump inhibitor. Prepn: A. Nohara, Y. Maki, EP174726; eidem,US4628098 (both 1986 to Takeda).HPLC determn in plasma: T. Uno et al., J. Chromatogr. B816, 309 (2005). Pharmacology: H. Satoh et al.,J. Pharmacol. Exp. Ther.248, 806 (1989). Mechanism of action study: H. Nagaya et al.,ibid.252, 1289 (1990). Clinical pharmacology and effect on human gastric acid secretion: P. Müller et al.,Aliment. Pharmacol. Ther.3, 193 (1989). Review of pharmacology and clinical experience: H. D. Langtry, M. I. Wilde, Drugs54, 473-500 (1997). Comparative clinical trial with esomeprazole in erosive esophagitis: C. W. Howden et al., Clin. Drug Invest.22, 99 (2002).
Properties: mp 178-182° (dec).
Melting point: mp 178-182° (dec)
Therap-Cat: Antiulcerative., Keywords: Antiulcerative; Gastric Proton Pump Inhibitor.
Lansoprazole, sold under the brand name Selanz SR among others, is a medication which reduces stomach acid.[2] It is used to treat peptic ulcer disease, gastroesophageal reflux disease, and Zollinger–Ellison syndrome.[3] Effectiveness is similar to other proton pump inhibitors (PPIs).[4] It is taken by mouth.[2] Onset is over a few hours and effects last up to a couple of days.[2]
Common side effects include constipation, abdominal pain, and nausea.[2][5] Serious side effects may include osteoporosis, low blood magnesium, Clostridium difficile infection, and pneumonia.[2][5] Use in pregnancy and breastfeeding is of unclear safety.[1] It works by blocking H+/K+-ATPase in the parietal cells of the stomach.[2]
Lansoprazole was patented in 1984 and came into medical use in 1992.[6] It is available as a generic medication.[3] In 2017, it was the 188th most commonly prescribed medication in the United States, with more than three million prescriptions.[7][8]
Medical uses
Lansoprazole is used for treatment of:[5]
- Ulcers of the stomach and duodenum, and NSAID-induced ulcers
- Helicobacter pylori infection, alongside antibiotics (adjunctive treatment), treatment to kill H. pylori causing ulcers or other problems involves using two other drugs besides lansoprazole known as “triple therapy“, and involves taking twice daily for 10 or 14 days lansoprazole, amoxicillin, and clarithromycin
- Gastroesophageal reflux disease
- Zollinger-Ellison syndrome[9]
There is no good evidence that it works better than other PPIs.[4]
Side effects
Side effects of PPIs in general[10] and lansoprazole in particular[11] may include:[5]
- Common: diarrhea, abdominal pain[12]
- Infrequent: dry mouth, insomnia, drowsiness, blurred vision, rash, pruritus
- Rarely and very rarely: taste disturbance, liver dysfunction, peripheral oedema, hypersensitivity reactions (including bronchospasm, urinary, angioedema, anaphylaxis), photosensitivity, fever, sweating, depression, interstitial nephritis, blood disorders (including leukopenia, leukocytosis, pancytopenia, thrombocytopenia), arthralgia, myalgia, skin reactions[13] including (erythroderma[14] Stevens–Johnson syndrome, toxic epidermal necrolysis, bullous eruption)
PPIs may be associated with a greater risk of hip fractures and Clostridium difficile-associated diarrhea.[5]: 22
Interactions
Lansoprazole interacts with several other drugs, either due to its own nature or as a PPI.[15]
- PPIs reduce absorption of antifungals (itraconazole and ketoconazole) [16] and possibly increase digoxin in plasma
- Increases plasma concentrations of cilostazol (risk of toxicity)
Lansoprazole possibly interacts with, among other drugs:
- sucralfate
- ampicillin
- bisacodyl
- clopidogrel
- delavirdine
- fluvoxamine
- iron salts
- voriconazole
- aminophylline and theophylline
- astemizole
Chemistry
It is a racemic 1:1 mixture of the enantiomers dexlansoprazole and levolansoprazole.[17] Dexlansoprazole is an enantiomerically pure active ingredient of a commercial drug as a result of the enantiomeric shift. Lansoprazole’s plasma elimination half-life (1.5 h) is not proportional to the duration of the drug’s effects to the person (i.e. gastric acid suppression).[18]
History
Main article: Discovery and development of proton pump inhibitors
Lansoprazole , available in the name of Selanz SR, was originally synthesized at Takeda and was given the development name AG 1749.[19] Takeda patented it in 1984 and the drug launched in 1991.[20] In the United States, it was approved for medical use in 1995.[21]
Society and culture

Prevacid 30 mg
Patents
The lansoprazole molecule is off-patent and so generic drugs are available under many brand names in many countries;[22] there are patents covering some formulations in effect as of 2015.[23] Patent protection expired on 10 November 2009.[24][25]
Availability
Since 2009, lansoprazole has been available over the counter (OTC) in the U.S. as Prevacid 24HR[26][27] and as Lansoprazole 24HR.[28] In Australia, it is marketed by Pfizer as Zoton.[citation needed]
Research
In vitro experiments have shown that lansoprazole binds to the pathogenic form of tau protein.[29] As of 2015 laboratory studies were underway on analogs of lansoprazole to explore their use as potential PET imaging agents for diagnosing tauopathies including Alzheimer’s disease.[29]
SYN
English: doi: 10.1248/cpb.38.2853
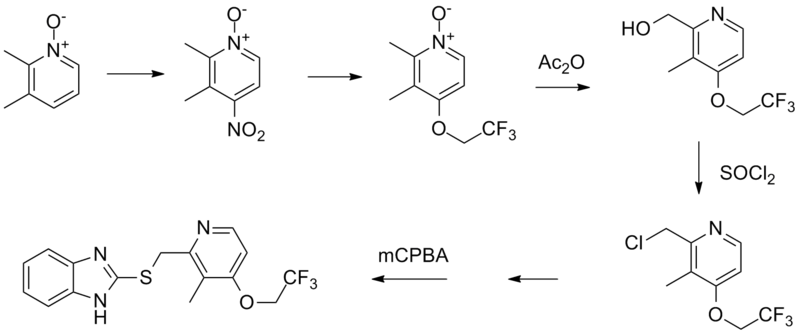
SYN
Method of synthesis
i. 2,3-dimethyl-4-nitropyridine-1-oxide is reacted with 2,2,2-trifluoroethanol in presence of potassium carbonate to give 2,3-dimethyl-4-(2,2,2-trifluoro-ethoxy)pyridine-1-oxide.
ii. The compound so formed is treated with acetic anhydride in acidic conditions followed by nutrilizing with sodium hydroxide solution to get 2-hydroxymethyl-3-methyl-4-(2,2,2-trifluoroethoxy)-pyridine
iii. Last is treated with thionyl chloride followed by reaction with 2-mercaptobenzimidazole to get 2-[3-methyl-4-(2,2,2-trifluoroethoxy)pyrid-2-ylmethylthio]benzimidazole.
iv. Above formed compound is reacted with m-chloro-perbenzoic acid to get lansoprazole.[2]
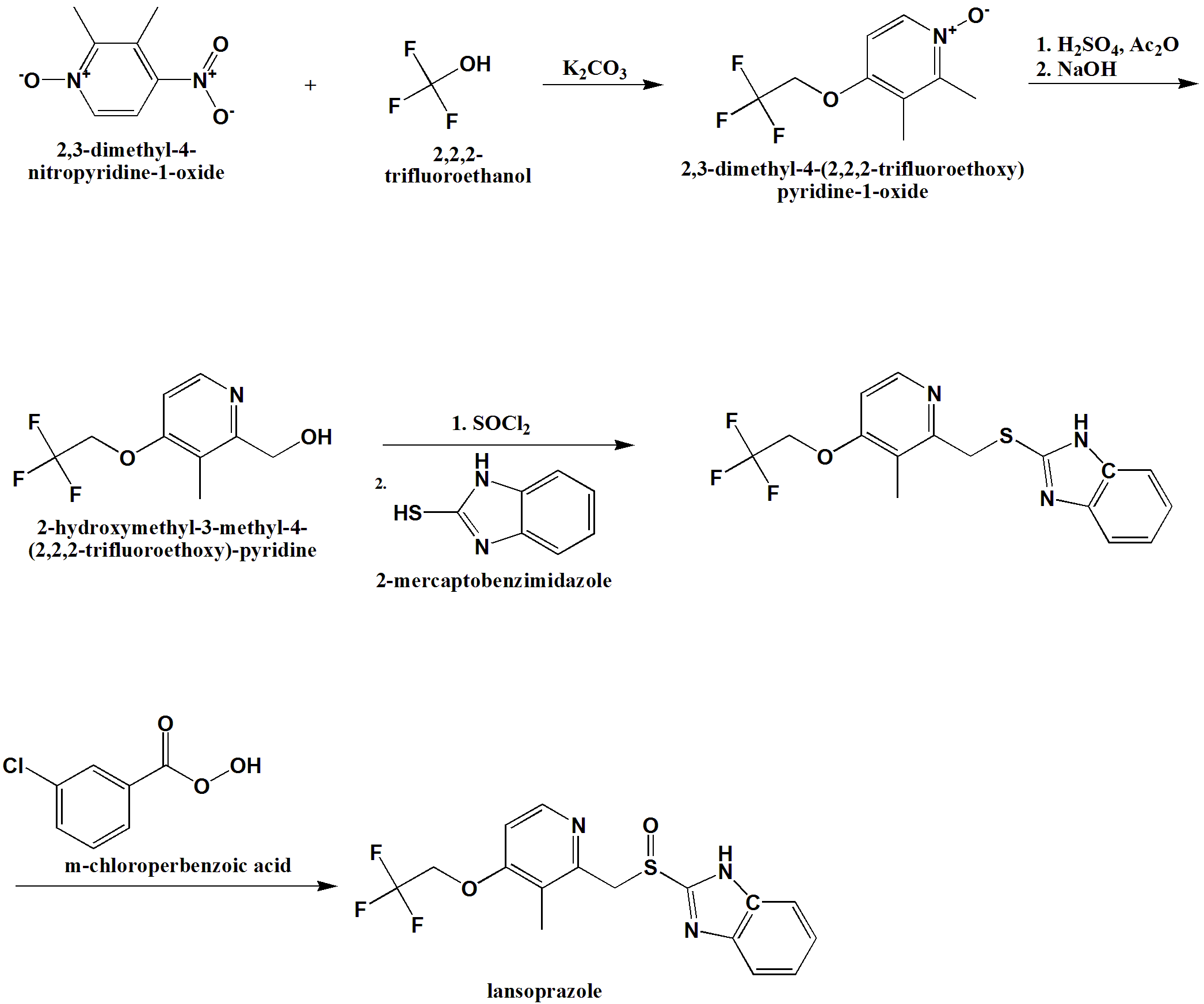
SYN
Proton Pump Inhibitors
Ruben Vardanyan, Victor Hruby, in Synthesis of Best-Seller Drugs, 2016
Lansoprazole–Prevacid
Lansoprazole (37.3) is the second approved gastric acid pump inhibitor. The common approach for the synthesis of lansoprazole involves coupling of mercapto-benzimidazole (37.24) with a new 2-chloromethylpyridine derivative (37.32) followed by oxidation of the prochiral sulfide group with m-chloroperbenzoic acid or hydrogen peroxide was first disclosed by Nohara and Maki [73], with followed improvements in patents [74-78] and briefly summed up in papers [79-80].
Lansoprazole synthesis is represented on the Scheme 37.4.

In principle it repeats the synthesis Scheme of omeprazole, differing in details and characteristics, for example, in place of 2,3,5-collidine (37.15) as a starting material, 2,3-lutidine (37.27) was selected, and the methoxy group in the fourth position of pyridine ring was replaced by the 2,2,2-trifluoroethoxy group.
Another interesting approach has been demonstrated [81]. In this case, 2-chloromethyl-3-methyl-4-(2,2,2-trifluoroethoxy)pyridine (37.32) was prepared starting with 3-picoline (37.34), which was oxidized using peracids (i.e., m-chloroperoxybenzoic acid) to produce 3-methylpyridineN-oxide (37.35). The obtained product was nitrated with fuming nitric acid to produce 3-methyl-4-nitropyridine N-oxide (37.36). The prepared N-oxide was treated with dimethylsulfate at 65 to 70°C to form N-methoxypyridinium salt (37.37), the aqueous solution of which on cooling was treated with sodium cyanide to produce an after formation of intermediate (37.38) and elimination of methanol 2-cyano-3-methyl-4-nitropyridine (37.39). This method for the synthesis of 2-cyanopyridines via addition of cyanide ion to N-alkoxy-quaternary salts of pyridines, supplements the plethora of Reissert-Kaufmann reactions in the quinoline and isoquinoline series previously described [82]. The nitro group in (37.39) was replaced by the 2,2,2-trifluoroethoxy group by a direct reaction with sodium trifluoroethoxide in trifluoroethanol that produced ether (37.40). The next step—transformation of nitrile group in prepared 2-cyanopyridine (37.40) to 2-carboxypyridine (37.41)—was carried out in a one-pot procedure by heating the 2-cyano compound in the presence of concentrated sulfuric acid followed by reaction of the intermediate amide with sodium nitrite under aqueous acidic conditions [83,84]. The obtained acid was esterified in methanol with a catalytic amount of sulfuric acid to produce ester (37.42). The ester (37.42) was reduced by NaBH4, producing the above-described 2-hydroxymethyl- pyridine derivative (37.31) followed by a reaction with thionyl chloride in dioxane that produced the required 2-chloromethylpyridine compound (37.32). Direct reaction of the last with 2-mercaptobenzimidazole (37.34) in methanol, even without use of any base, produced a sulfide (37.33) in high yield. The oxidation of the last to lansoprazole (37.3) has been carried out by various oxidants and catalysts, which, together with the desired sulfoxide, produced a certain amount of overoxidized product. Oxidizing sulfide (37.33) with a new oxidation method made up of the use of the composite metal oxide catalyst, LiNbMoO6, in methanol and 35% H2O2 as an oxidant sulfide (37.33) was successfully oxidized to desired lansoprazole (37.3) (Scheme 37.5.).

Lansoprazole is the second inhibitor of the gastric H+/K+-ATPase to be marketed for the treatment of peptic ulcer disease and reflux esophagitis, erosive esophagitis, and Zollinger-Ellison syndrome. It is an inhibitor of gastric acid secretion and also exhibits antibacterial activity against H. pylori in vitro. More common side effects of lansoprazole are diarrhea and skin rash or itching. Less-common side effects are abdominal pain, joint pain, nausea, vomiting, and increased or decreased appetite [85-91].
SYN
| AU 8545895; EP 0174726; ES 8607288; JP 1986050978; US 4628098; US 4689333 |
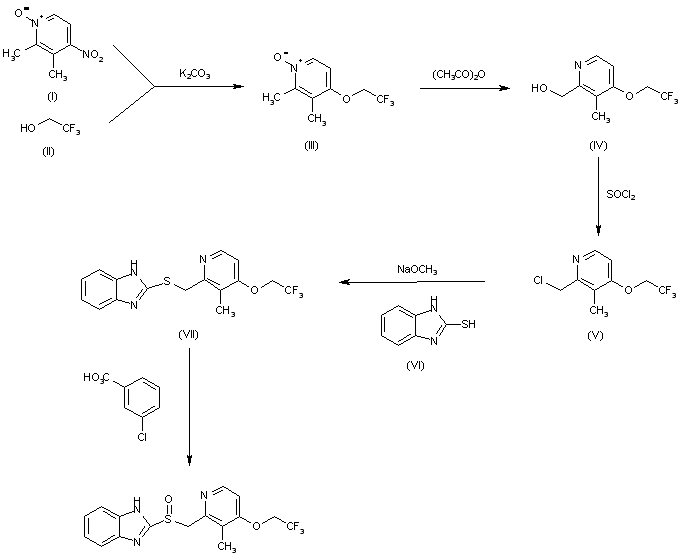
The condensation of 2,3-dimethyl-4-nitropyridine N-oxide (I) with 2,2,2-trifluoroethanol (II) by means of K2CO3 in hot HMPT gives 2,3-dimethyl-4-(2,2,2-trifluoroethoxy)pyridine N-oxide (III), which by isomerization in acetic anhydride at 100 C is converted to 2-(hydroxymethyl)-3-methyl-4-(2,2,2-trifluoroethoxy)pyridine (IV). The reaction of (IV) with SOCl2 in refluxing CHCl3 affords the corresponding chloromethyl derivative (V), which is condensed with 2-mercaptobenzimidazole (VI) by means of sodium methoxide in refluxing methanol to yield 2-(2-benzimidazolylthiomethyl)-3-methyl-4-(2,2,2-trifluoroethoxy)pyridin (VII). Finally, this compound is oxidized with m-chloroperbenzoic acid in CHCl3.
SYN
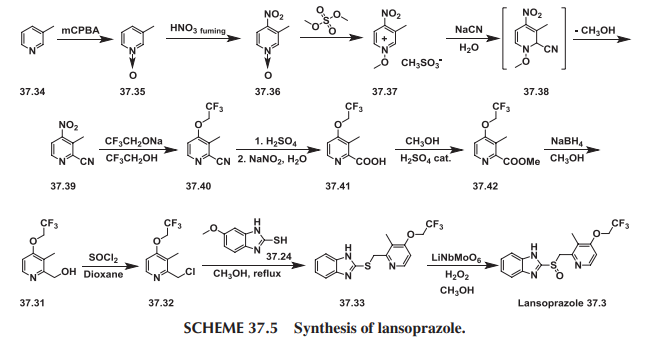
SYN
Chemical Synthesis
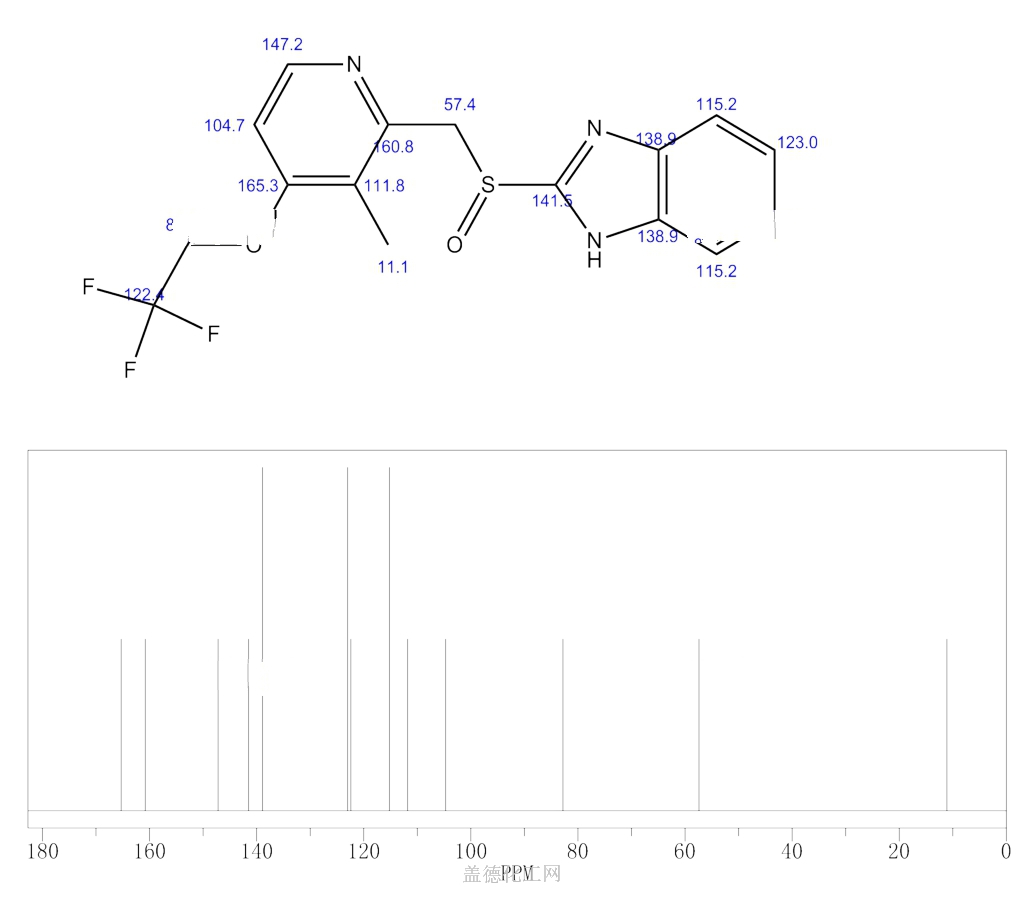
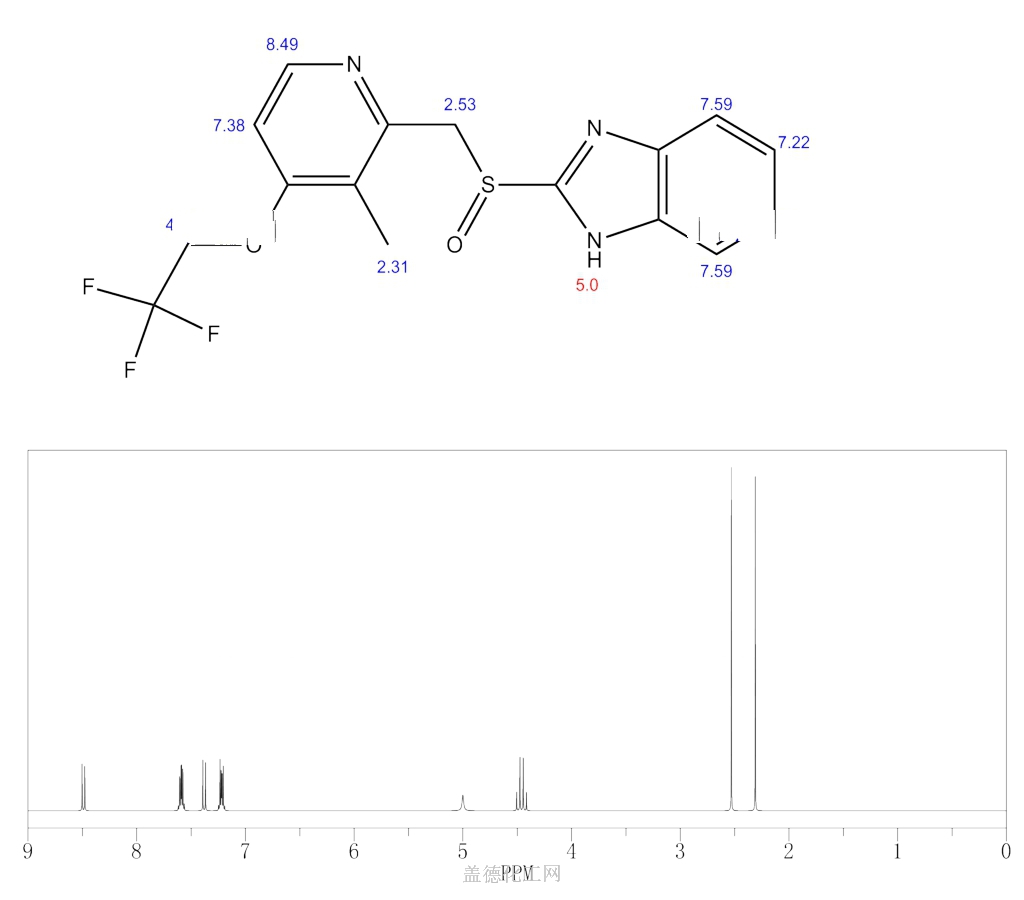
Similar to the synthesis of the chiral sulfoxide of armodafinil vide supra, the preparation of the chiral sulfoxide of lansoprazole utilized the catalytic oxidation method developed by Kagan and co-workers (the Scheme). Two routes have been reported that describe the preparation of dexlansoprazole on large scale. The first route developed by Takeda reacts commercially available thioether 29, also used to make lansoprazole, under the Kagan asymmetric oxidation conditions and the alternative route utilizes the cheaper commercial intermediate nitrosulfide 30 in the analogous asymmetric oxidation by Kagan). Thus, the catalyst complex consisting of (+)-DET, Ti(OiPr)4 and water was formed in the presence of thioether 29 in toluene at 30–40°C. The reaction mixture was then cooled to 5 °C and DIPEA and cumene hydroperoxide (CMHP) were added to give, after aqueous work-up and in situ crystallization from the organic layer, dexlansoprazole (VI) in 98% ee. No yield was given in the patent. An alternate, but similar, sequence was also described wherein the nitrosulfide intermediate 30 was subjected to similar oxidative conditions that gave intermediate nitro compound 31 in 80% yield and 98% ee. Compound 31 was treated with KOH and trifluoroethanol to provide dexlansoprazole (VI).
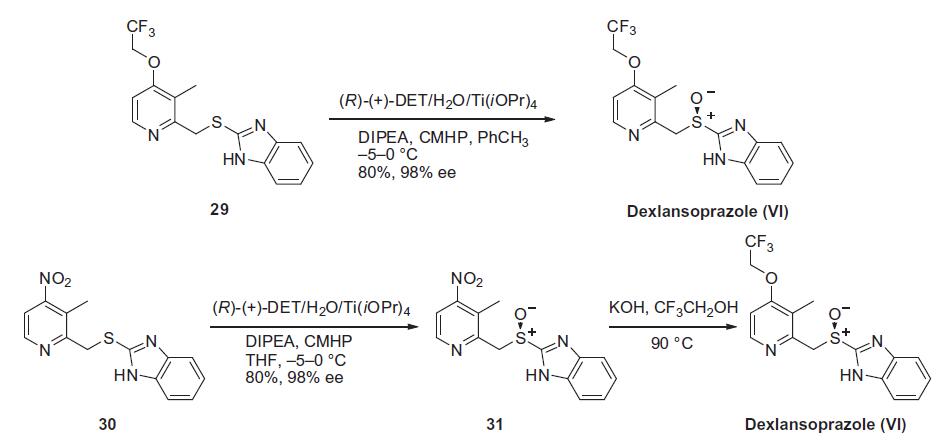
R-(+)-Lansoprazole Preparation Products And Raw materials
PATENT
https://patents.google.com/patent/WO2008087665A2/enA number of substituted 2-(2-pyridylmethyl) sulfinyl-lH-benzimidazole derivatives are reported as gastric proton pump inhibitors. These benzimidazole derivatives include lansoprazole, omeprazole, pantoprazole, and rabeprazole. The Lansoprazole is generally represented by the following chemical formula I

US 4,628,098 & 4,689,333 describes lansoprazole having its chemical name (2-[[[3-methyl-4-(2, 2, 2-trifluoro-ethoxy)-2-pyridinyl] methyl] sulfinyl]-lH-benzimidazole. As a characteristic shared with other benzimidazole derivatives (e.g., omeprazole and pantoprazole), lansoprazole can inhibit gastric acid secretion, and thus commonly used as an antiulcer agent. Several methods for preparing Lansoprazole are known. The majority of these methods involve the use of a lansoprazole precursor that contains a thioether group. The thioether group is oxidized in the last step of preparation to form the lansoprazole. These patents (‘098 and ‘333) further describes the oxidation of the thioether group using m-chloroperbenzoic acid, per acid, sodium bromite, sodium hypochlorite, or hydrogen peroxide as the oxidizing agent and the reaction solvent is halogenated hydrocarbon, ether, amide, alcohol, or water.US 6,002,011 describe the crystallization of Lansoprazole from the same ethanol: water system, containing traces of ammonia. This patent discloses a reslurry method in water, which permits to obtain more stable “solvent free” Lansoprazole. This patent fails to disclose the level of purity for Lansoprazole. In addition, the ethanol and water are difficult to eliminate. Even after intensive drying, Lansoprazole still contains solvent and is unstable under storage. US 6,180,652 describe the presence of sulfone derivative. Formation of sulfone derivative brings about the drawback of low yield of the desired sulfoxide. Although attempts have been made to separate the sulfone derivative from Lansoprazole, it is not a simple task, given their very similar structures and physicochemical properties. This patent also describes a method for separation of Lansprazole from its sulfone derivative, by converting to an acetone complex of the Lansoprazole salt & hence is purified in this method. Lansoprazole and other 2-(2- pyridylmethyl) sulfinylbenzimidazole derivatives tend to lose stability and undergo decomposition when contaminated with traces of a solvent, particularly water, in their crystal structure. It is desirable that the benzimidazole crystals be solvent free (i.e., residual solvent should be reduced to a minimum).US 6,909,004 describes the method of purifying Lansoprazole, comprising the steps of: a) providing a solution of lansoprazole in a solvent selected from an organic solvent or a mixture of organic solvent and water in the presence of an amine compound; b) combining the provided solution with an acid, and c)isolating the purified Lansoprazole. The amine compound is present in 1:1, mole: mole, ratio relative to the lansoprazole. Solution is in an organic solvent selected from the group consisting of alcohols, acetone, 2-butanone, dimethylformamide and tetrahydrofuran. The alcohol consisting of ethanol, methanol, n-propanol, & iso-propanol.US 7022859 & US 7060837 provides a method for preparing a substantially pure Lansoprazole containing less than about 0.2% (wt/wt) impurities including sulfone/sulfide derivatives. The present invention also provides a process for recrystallizing Lansoprazole to obtain a Lansoprazole containing less than about 0.1% (wt/wt) water.US 2004/010151 disclose a method of preparing crystalline Lansoprazole form A, comprising the steps of: a) preparing a solution of Lansoprazole in a solvent selected from the group consisting of methanol, n-butanol, acetone, methylethylketone, ethyl acetate, dimethyl sulfoxide, dimethylforniamide and their mixtures optionally with water; and b) isolating crystalline Lansoprazole form A.US 2005/020638 describe the process of preparing a stable Lansoprazole, comprising the steps of: a) crystallizing a Lansoprazole from an organic solvent or a mixture of organic solvent and water in the presence of a weak base; and b) isolating a stable Lansoprazole. An amorphous form of Lansoprazole prepared by spray drying method has been described (Farm. Vest. vol. 50, p. 347 (1999)). Curin et al. describe an ethanole solvate form and an ethanole-hydrate form of Lansoprazole (Farm. Vest. vol. 48, pp. 290-291 (1997). Kotar et al. describe two lansoprazole polymorphs, designated as crystalline Lansoprazole forms A and B, (Eur. J. Pharm. Sci. vol. 4, p. 182 (1996 Supp). According to Kotar, each of the crystalline Lansoprazole forms A and B exhibits a different DSC curve. In fact, crystalline Lansoprazole form B is unstable and can undergo a solid-solid transition to form crystalline Lansoprazole form A. No XRD data for crystalline Lansoprazole forms A and B, and fails to disclose processes for preparing these crystalline forms. No indication was found in the literature regarding the existence of other crystalline Lansoprazole forms other than the known forms A, B, ethanolate and ethanolate- hydrate.WO 00/78729 is discloses a phenomenon of polymorphism in Lansoprazole. The crystalline forms , I and II. The form I find application as an active ingredient of pharmaceutical compositions.WO 03/082857 disclose a method of preparing crystalline Lansoprazole form A, comprising the steps of: a) preparing a solution of Lansoprazole in a solvent selected from the group consisting of methanol, n-butanol, acetone, methylethylketone, ethyl acetate, dimethyl sulfoxide, dimethylformamide and their mixtures optionally with water; and b) isolating crystalline Lansoprazole form A.WO 2004/046135 describe the process for preparing a stable Lansoprazole compound, comprising the steps of: a) crystallizing a Lansoprazole from an organic solvent or a mixture of organic solvent and water in the presence of an amine; and b) isolating a stable Lansoprazole compound, wherein the stable Lansoprazole compound comprises greater than 500 ppm and not more than about 3,000 ppm water.Since proton pump inhibitors of the benzimidazole-type are very susceptible to degradation under acidic or neutral conditions, the reaction mixture is usually worked-up under basic conditions. These basic conditions may decompose any unwanted oxidizing agent still present in the reaction mixture and may also neutralize any acid formed when the oxidizing agent is consumed in the oxidation reaction. The main problem with the oxidation reaction to convert the sulfide intermediates of formula (II) into the sulfoxide compounds of formula (I) is over- oxidation, i.e. oxidation from sulfoxides of formula (I) to sulfones of formula (III) ; N-oxide of formula (IV) & chlorinating impurities ( V).The formation of sulfones of formula (III) due to over-oxidation is almost impossible to avoid and can be kept to a minimum by performing the oxidation reaction at a low temperature and restricting the amount of oxidizing agent. Typically the amount of oxidizing agent is less than 1 molar equivalent of the starting material, i.e. sulfide intermediates of formula (II), which inevitably results in a less than 100% conversion of starting material. Usually the amount of oxidizing agent is a compromise between maximum conversion of starting material, maximum formation of sulfoxides of formula (I) and minimum formation of unwanted sulfones of formula (III). Chlorinating impurities (V) are observed when chlorinating oxidizing agent such as sodium hypochlorite is used for oxidation reaction. Furthermore removal of the sulfones of formula (III) & chlorinating impurities (V) has often proved to be difficult, time-consuming and costly, in particular when high performance chromatography on an industrial scale is needed. Another problem with the benzimidazole-type is very susceptible to degradation when exposed to high temperatures for removal of solvents during distillation.Thus, there is continuing need to obtain 2-(2-pyridylmethyl) sulfϊnyl-lH-benzirnidazoles (e.g., Lansoprazole) that are free of contaminants including sulfone and sulfide derivatives. There has also -been a long-felt need for a method to prepare Lansoprazole having reduced water content (<0.1% wt/wt water).SCHEME : ]

LANSOPRAZOLE (I) SULPHONE (III)

N-OXIDE (IV)

SULPHIDE (II)+ Chlorinated Impurities(V)General Example10 g of 2- [3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridyl] methylthio-lH-benzimidazole was suspended in 100 ml chloroform and cooled to -100C. To the above suspension 3.4 g m- chloroperbenzoic acid solution in chloroform was added over a period of 2 hrs at -10 C. After completion of reaction, reaction mass was added to sodium bicarbonate solution (500 ml) and both layers were separated. Organic layer was washed with 2 x 50 ml of hypo solution followed by washing with 3 x 200 ml sodium bicarbonate solution. Both the layers were separated. Chloroform layer was washed with sodium bicarbonate solution (0.5%; 500 ml) at room temperature. Various co-solvents mentioned in Table- 1 were added to organic layer cool slowly to -10 to 100C. Filtered and washed with chilled chloroform (10 ml) followed by sodium bicarbonate solution (0.5%, 100 ml) & dried to get pure Lansoprazole.SYNhttp://www.ijmca.com/File_Folder/116-120.pdf


join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Literatures:
Chemical and Pharmaceutical Bulletin, , vol. 38, # 10 p. 2853 – 2858
Literatures:
RECORDATI INDUSTRIA CHIMICA E FARMACEUTICA SPA Patent: WO2008/77866 A1, 2008 ; Location in patent: Page/Page column 16-17; 19 ;
Yield: ~92%
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4628098A *1984-08-161986-12-09Takeda Chemical Industries, Ltd.2-[2-pyridylmethylthio-(sulfinyl)]benzimidazolesWO2004018454A1 *2002-08-212004-03-04Teva Pharmaceutical Industries Ltd.A method for the purification of lansoprazoleUS20040049045A1 *2000-12-012004-03-11Hideo HashimotoProcess for the crystallization of (r)-or (s)-lansoprazole
Publication numberPriority datePublication dateAssigneeTitleWO2012004802A12009-07-072012-01-12Council Of Scientific & Industrial ResearchContinuous flow process for the preparation of sulphoxide compoundsCN107964005A *2017-11-102018-04-27扬子江药业集团江苏海慈生物药业有限公司A kind of preparation method of Lansoprazole
References
- ^ Jump up to:a b c “Lansoprazole Use During Pregnancy”. Drugs.com. Retrieved 3 March 2019.
- ^ Jump up to:a b c d e f “Lansoprazole Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ Jump up to:a b British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 79–80. ISBN 9780857113382.
- ^ Jump up to:a b “[99] Comparative effectiveness of proton pump inhibitors | Therapeutics Initiative”. 28 June 2016. Retrieved 14 July 2016.
- ^ Jump up to:a b c d e “Lansoprazole capsule, delayed release pellets”. DailyMed. 11 October 2016. Retrieved 31 December 2019.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 445. ISBN 9783527607495.
- ^ “The Top 300 of 2020”. ClinCalc. Retrieved 11 April 2020.
- ^ “Lansoprazole – Drug Usage Statistics”. ClinCalc. Retrieved 11 April 2020.
- ^ Hirschowitz BI, Mohnen J, Shaw S (August 1996). “Long-term treatment with lansoprazole for patients with Zollinger-Ellison syndrome”. Aliment. Pharmacol. Ther. 10 (4): 507–22. doi:10.1046/j.1365-2036.1996.10152000.x. PMID 8853754. S2CID 10668517.
- ^ British National Formulary (Free registration required) 1.3.5 Proton pump inhibitors
- ^ British National Formulary (Free registration required) Lansoprazole
- ^ “Prevacid (Lansoprazole) Drug Information: Side Effects and Drug Interactions – Prescribing Information at RxList”. RxList. Retrieved 9 February 2016.
- ^ K C Singhal & S Z Rahman, Lansoprazole Induced Adverse Effects on the Skin, Indian Medical Gazette, July 2001, Vol. CXXXV. N0. 7: 223-225
- ^ Sterry W, Assaf C (2007). “Erythroderma”. In Bolognia JL (ed.). Dermatology. St. Louis: Mosby. p. 154. ISBN 978-1-4160-2999-1..
- ^ British National Formulary (Free registration required) Lansoprazole interactions
- ^ Piscitelli, S. C.; Goss, T. F.; Wilton, J. H.; d’Andrea, D. T.; Goldstein, H; Schentag, J. J. (1991). “Effects of ranitidine and sucralfate on ketoconazole bioavailability”. Antimicrobial Agents and Chemotherapy. 35 (9): 1765–1771. doi:10.1128/aac.35.9.1765. PMC 245265. PMID 1952845.
- ^ “Pharmacy Benefit Update”. Retrieved 2 July 2014.
- ^ “Prevacid Pharmacology, Pharmacokinetics, Studies, Metabolism”. RxList.com. 2007. Archived from the original on 16 August 2000. Retrieved 14 April 2007.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 102. ISBN 9783527607495.
- ^ Chorghade, Mukund S. (2006). Drug Discovery and Development, Volume 1: Drug Discovery. John Wiley & Sons. p. 201. ISBN 9780471780090.
- ^ Mosby’s Drug Consult: Lansoprazole
- ^ drugs.com International availability of lansoprazole Page accessed 3 February 2015
- ^ drugs.com Generic lansoprazole Page accessed 3 February 2015
- ^ “Prevacid Drug Profile”. Drugpatentwatch.com. Retrieved 30 April 2020.
- ^ Teva to release Prevacid version when patent expires
- ^ “Prevacid 24 HR- lansoprazole capsule, delayed release”. DailyMed. 7 August 2019. Retrieved 31 December 2019.
- ^ “Prevacid 24 HR- lansoprazole capsule, delayed release”. DailyMed. 11 December 2019. Retrieved 31 December 2019.
- ^ “Lansoprazole 24 HR- lansoprazole capsule, delayed release”. DailyMed. 21 December 2017. Retrieved 31 December 2019.
- ^ Jump up to:a b Villemagne, VL; Fodero-Tavoletti, MT; Masters, CL; Rowe, CC (January 2015). “Tau imaging: early progress and future directions”. The Lancet. Neurology. 14 (1): 114–24. doi:10.1016/s1474-4422(14)70252-2. PMID 25496902. S2CID 10502833.
External links
- “Lansoprazole”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | /lænˈsoʊprəzoʊl/ lan-SOH-prə-zohl |
| Trade names | Prevacid, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a695020 |
| License data | EU EMA: by INNUS DailyMed: LansoprazoleUS FDA: Lansoprazole |
| Pregnancy category | AU: B3[1] |
| Routes of administration | By mouth, intravenous (IV) |
| Drug class | Proton pump inhibitor |
| ATC code | A02BC03 (WHO) |
| Legal status | |
| Legal status | AU: S2, S3, & S4UK: POM (Prescription only)US: OTC / Rx-only |
| Pharmacokinetic data | |
| Bioavailability | 80% or more |
| Protein binding | 97% |
| Metabolism | Liver (CYP3A4– and CYP2C19-mediated) |
| Elimination half-life | 1.0–1.5 hours |
| Excretion | Kidney and fecal |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 103577-45-3 |
| PubChem CID | 3883 |
| IUPHAR/BPS | 7208 |
| DrugBank | DB00448 |
| ChemSpider | 3746 |
| UNII | 0K5C5T2QPG |
| KEGG | D00355 |
| ChEBI | CHEBI:6375 |
| ChEMBL | ChEMBL480 |
| CompTox Dashboard (EPA) | DTXSID4023200 |
| ECHA InfoCard | 100.173.220 |
| Chemical and physical data | |
| Formula | C16H14F3N3O2S |
| Molar mass | 369.36 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI | |
| (verify) |
////LANSOPRAZOLE, A-65006, AG-1749, A 65006, AG 1749, лансопразол , لانسوبرازول , 兰索拉唑 , Antiulcerative, Gastric Proton Pump Inhibitor,

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
-lansoprazole-from-xtal-3D-bs-17.png){kind=link}