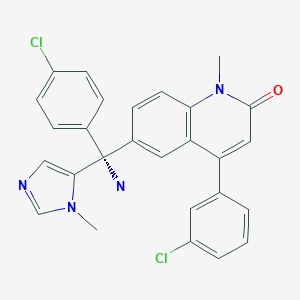
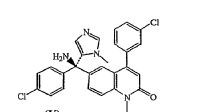
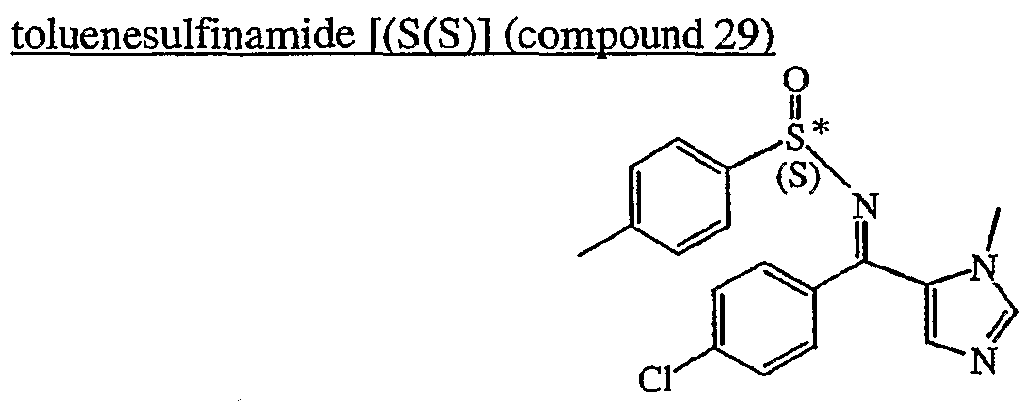
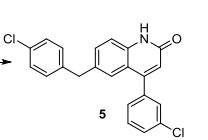
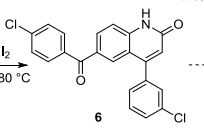
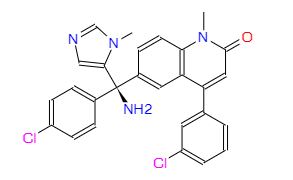
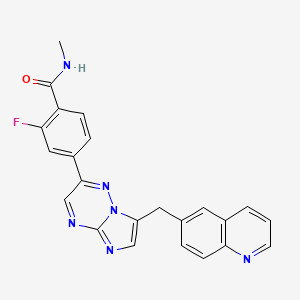
2-fluoro-N-methyl-4-{7-[(quinolin-6-yl)methyl]imidazo[1,2-b][1,2,4]triazin-2-yl}benzamide
Capmatinib has been used in trials studying the treatment of Melanoma, Gliosarcoma, Solid Tumors, Colorectal Cancer, and Hepatic Impairment, among others.
Capmatinib has been used in trials studying the treatment of Melanoma, Gliosarcoma, Solid Tumors, Colorectal Cancer, and Hepatic Impairment, among others
Example 8 4-Bromo-3-fluoro-N-methoxy-iV-methylbenzamide (3)
To a suspension of 4-bromo-3-fluorobenzoic acid (1, 967.9 g, 4.4 mol) in dichloromethane (5.9 L) and DMF (21 mL) was slowly added a solution of oxalyl chloride ((COCl)2, 560 mL, 6.4 mol, 1.45 equiv) in dichloromethane (520 mL) at room temperature. The resulting reaction mixture was stirred at room temperature for 20 h and then cooled to 0 0C by ice-water bath. iV,0-dimethyl hydroxylamine hydrochloride (826 g, 8.4 mol, 1.9 equiv) was added followed by slow addition of triethylamine (TEA, 2.5 L, 17.7 mol, 4.0 equiv) at 0 0C. The reaction mixture was then gradually warmed to room temperature and stirred at room temperature overnight. Once the coupling reaction was complete, the reaction mixture was washed with saturated aqueous sodium bicarbonate solution (NaHCO3, 2 L). The aqueous phase was back extracted with dichloromethane (1 L). The combined organic phases were washed with water (1 L), brine (1 L), and concentrated under reduced pressure. The resulting solid residue was dissolved into methyl tert-butyl ether (MTBE, 5 L), washed sequentially with water (5 x 1 L), brine (1 L), and dried over anhydrous sodium sulfate (Na2SO4). The filtrated solution was concentrated under reduced pressure and the resulting solid was dried in a vacuum oven at 45 0C to afford 4-bromo-3-fluoro-Λr-methoxy-Λr-methylbenzamide (3, 1106 g, 1153 g theoretical, 95.9% yield) which was used for the subsequent reaction without further purification. For 3: 1H NMR (400 MHz, DMSO-J6) δ ppm 7.78 (t, IH, J= 7.47 Hz), 7.56 (dd, IH, J= 9.3, 1.6 Hz), 7.18 (d, IH, J= 8.1 Hz), 3.53 (s, 3H), 3.25(s, 3H); C9H9BrFNO2 (MW 262.08), LCMS (EI) mle 262.0/ 264.0 (M+ + H).
Scheme 1 (Examples 8-14)

C7H4BrFO2 C7H3BrCIFO C9H9BrFNO2 MoI. Wt: 219.01 MoI. Wt: 237.45 MoI. Wt: 26208

C8H6BrFO C8H4BrFO2 Ci2Hi4BrFO3 MoI. Wt: 217.04 MoI. Wt: 231.02 MoI. Wt: 305.14

13
C22H13FN3 MoI. Wt: 380.38
Example 9 l-(4-Bromo-3-fluorophenyl)ethanone (4)
To a solution of crude 4-bromo-3-fluoro-N-methoxy-iV-methylbenzamide (3, 1106 g, 4.2 mol) in anhydrous tetrahydrofuran (THF, 11 L) was slowly added a 3.0 M solution of methylmagnesium chloride (MeMgCl, 2.5 L, 7.5 mol, 1.7 equiv) in THF at 0 0C. The resulting reaction mixture was stirred at 0 0C for 2 h and then quenched very carefully with saturated aqueous ammonium chloride (NH4Cl, 1.5 L). The resulting solution was concentrated under reduced pressure to remove most of THF. The residue was then diluted with ethyl acetate (EtOAc, 5 L) and the resulting solution was washed with water (2 L). The aqueous phase was extracted with ethyl acetate (EtOAc, 2 x 2 L). The combined organic phases were washed with water (2 L), brine (2 L) and dried over anhydrous sodium sulfate (Na2SO4). The filtered solution was concentrated under reduced pressure and the resulting solid was dried in a vacuum oven at 45 0C to afford l-(4-bromo-3-fluorophenyl)ethanone (4, 890.8 g, 911.6 g theoretical, 97.7% yield) as a solid which was used in the subsequent reaction without further purification. For 4: 1H NMR (400 MHz, OMSO-d6) δ ppm 7.89-7.84 (m, 2H), 7.71 (dd, IH, J= 8.30, 1.87 Hz), 2.57 (s, 3H).
Example 10 2-(4-Bromo-3-fluorophenyl)-2-oxoacetaldehyde (5)
To a solution of l-(4-bromo-3-fluorophenyl)ethanone (4, 890.8 g, 4.1 mol) in DMSO (4 L) was slowly added a solution of 48% aqueous hydrogen bromide (HBr, 1420 mL, 12.5 mol, 3.0 equiv). The reaction temperature was gradually increased from 2O0C to 50 0C during the course of the addition. The reaction mixture was subsequently heated to 60 0C and stirred at 60 0C overnight. The resulting dimethyl sulfide was removed by distillation and the residue was poured into ice water (28 L). The resulting yellow precipitate was collected by filtration (save the filtrate) and washed with water (5 L). The yellow solid was dissolved in ethyl acetate (EtOAc, 5 L), washed with brine (1 L) and dried over anhydrous sodium sulfate (Na2SO4). The solution was then concentrated under the reduced pressure and the resulting solid was dried in a vacuum oven at 45 0C to give the desired product, 2-(4-bromo-3-fluorophenyl)-2-oxoacetaldehyde, as its hydrate (hydrate of 5, 730.6 g, 1020.9 g theoretical, 71.6% yield). The aqueous phase (filtrate) was extracted with ethyl acetate (3 x 5 L) and the combined organic phase was washed with water (2 x 2 L), brine (2 L) and dried over anhydrous sodium sulfate (Na2SO4). The solution was concentrated under reduced pressure and the resulting solid was dried in a vacuum oven at 45 0C to give the second crop of 2-(4-bromo-3-fluorophenyl)-2-oxoacetaldehyde hydrate (hydrate of 5, 289.4 g, 1020.9 g theoretical, 28.3% yield; total 1020 g, 1020.9 g theoretical, 99.9% yield) which was used in the subsequent reaction without further purification. For hydrate of 5: H NMR (400 MHz, DMSO-</6) δ ppm 8.00-7.70 (m, 3H), 6.69 (br s, 2H), 5.59 (s, IH).
Example 11 l-(4-Bromo-3-fluorophenyl)-2,2-diethoxyethanone (6)
A 22 L flask was charged with the hydrate of (4-bromo-3-fluorophenyl)-2-oxoacetaldehyde (5, 1020 g, 4.41 mol), toluene (7.5 L), triethyl orthoformate (1633 g, 1.8 L, 11.04 mol, 2.5 equiv), para-toluene sulfonic acid (33.5 g, 0.176 mol, 0.4 equiv) at room temperature, and the resulting reaction mixture was heated to 110 0C and stirred at 1 10 0C for 6 h. When HPLC showed that the reaction was complete, the reaction mixture was cooled down to room temperature before being poured into a 50 L separation funnel along with ethyl acetate (7.5 L) and the saturated aqueous sodium bicarbonate solution (NaHCO3, 3 L). The mixture was stirred and the layers were separated. The aqueous layer was extracted with ethyl acetate (2 L). The combined organic layers were washed with brine (4 L), dried with sodium sulfate (Na2SO4), and concentrated under the reduced pressure to afford crude l-(4-bromo-3-fluorophenyl)-2,2-diethoxyethanone (6, 1240 g, 1345.7 g theoretical, 92.1% yield) which was used in the subsequent reaction without further purification. For 6: 1H NMR (400 MHz, DMSO-J6) δ ppm 7.94-7.94 (m, 2H), 7.78 (dd, IH, J= 8.51, 2.08 Hz), 5.40 (s, IH), 3.77-3.60 (m, 4H), 1.16-1.14 (m, 6H).
Example 12 6-(4-Bromo-3-fluorophenyl)-l,2,4-triazin-3-amine (7)
A 22 L flask was charged with l-(4-bromo-3-fluorophenyl)-2,2-diethoxyethanone (6, 1240 g, 4.07 mol), ethanol (11 L), water (1.4 L), potassium hydroxide (KOH, 910 g, 16.3 mol, 4.0 equiv), and aminoguanidine bicarbonate (1105 g, 8.13 mol, 2.0 equiv) at room temperature. The resulting reaction mixture was then heated to 75 0C for 14 h. When HPLC showed the condensation reaction was deemed complete, the reaction mixture was cooled down to room temperature before being filtered. The filtrate was then concentrated under the reduced pressure to remove the most of the solvents. The residual aqueous solution was extracted with ethyl acetate (EtOAc, 3 x 6 L). The organic layers were combined and concentrated under the reduced pressure to give a dark brown solid. This solid was dissolved in ethanol (4 L) and the resulting solution was treated with a solution of 0.2 M aqueous hydrochloric acid solution (4 L). The resulting slurry was subsequently heated to 50 0C for 6 h before being allowed to cool down to room temperature. A solution of saturated aqueous sodium bicarbonate solution (NaHCO3, 2 L) was slowly added to the slurry and the resulting mixture was then concentrated under the reduced pressure to remove most of the solvents. The aqueous residue was then treated with ethyl acetate (20 L) to dissolve the solids. The two layers were separated and the aqueous layer was extracted with ethyl acetate (2 x 2 L). The combined organic layers were concentrated under the reduced pressure. The dark brown solids were treated with methyl ter/-butyl ether (MTBE, 4 L) and the resulting slurry was heated to 30 0C and stirred at 30 0C for 30 min. The mixture was filtered and the solids (green to orange in color) were collected (save the filtrate) and washed with methyl tert-buty\ ether (MTBE, 2 L) to give the first crop of the crude desired product (7). The filtrate was evaporated under the reduced pressure, and the resulting dark brown solids were treated with methyl tert-butyl ether (MTBE, 2 L). The resulting slurry was heated to 30 0C and stirred at 30 0C for 30 min. The mixture was filtered to give the second crop of the crude desired product (7) which was washed with MTBE (1 L). The combined solids were dried in vacuum at 40 – 45 0C to afford 6-(4-bromo-3-fluorophenyl)-l,2,4-triazin-3-amine (7, 585 g, 1095.1 g theoretical, 53.4 % yield) which was used in the subsequent reaction without further purification. For 7: 1H NMR (400 MHz, DMSO-J6) δ ppm 8.86 (s, IH), 7.97 (d, IH, J= 10.79 Hz), 7.81 (m, 2H), 7.52 (br s, 2H); C9H6BrFN4 (MW 269.07), LCMS (EI) mle 269.0/271.1 (M+ + H).
Example 13 6-((2-(4-Bromo-3-fluorophenyl)imidazo[l,2-6][l,2,4]triazin-7-yl)methyl)quinoline (12) l-(2-Chloro-l-hydroxy-3-(quinolin-6-yl)propyl)pyrrolidine-2,5-dione (11, 228 g, 0.74 mol, 1.1 equiv) and 6-(4-bromo-3-fluorophenyl)-l,2,4-triazin-3-amine (7, 181 g, 0.673 mol) were suspended in 1-butanol (1800 mL) and the resulting suspension was heated to 110 0C and stirred at 110 0C for 18 h (the reaction mixture becomes homogeneous at this point). The reaction mixture was then gradually cooled down to room temperature before being further cooled down to 10 0C in an ice bath. The resulting yellow solid was collected by filtration (save the 1 -butanol filtrates), washed with cold 1-butanol (3 x 100 mL) and dried by suction. This solid was then suspended in the saturated aqueous sodium bicarbonate solution (NaHCO3, 500 mL) and the resulting suspension was stirred at room temperature for 1 h to neutralize the corresponding hydrochloride salt. The free base was then filtered, washed with water (500 mL) and dried in a vacuum oven at 45 0C for 18 h to afford the first crop of the crude 6-((2-(4-bromo-3-fluorophenyl)imidazo[l,2-6][l,2,4]triazin-7-yl)methyl)quinoline (12, 125.1 g, 292.3 g theoretical, 42.8% yield). The 1-butanol filtrates were then concentrated under the reduced pressure and the resulting solids were dissolved in dichloromethane (CH2Cl2, 2 L). The solution was wash with the saturated aqueous sodium bicarbonate solution (NaHCO3, 1 L), dried over sodium sulfates (Na2SO4), and concentrated under the reduced pressure. The residue was then purified by flash column chromatography (SiO2, O – 10% MeOH-CH2Cl2 gradient elution) to afford the second crop of 6-((2-(4-bromo-3-fluorophenyl)imidazo[l,2-&][l ,2,4]triazin-7-yl)methyl)-quinoline (12, 19.7 g, 292.3 g theoretical, 6.7% yield; total 144.8 g, 292.3 g theoretical, 49.5% yield) as yellow solids. For 12: 1H NMR (400 MHz, DMSO-^6) δ ppm 9.23 (s, IH), 9.11 (dd, IH, J= 4.98, 1.55 Hz), 8.85 (d, IH, J= 8.09 Hz), 8.25 – 8.18 (m, 2H), 8.12 -8.00 (m, 3H), 7.93 – 7.86 (m, 3H), 4.70 (s, 2H); C21H13BrFN5 (MW 434.26), LCMS (EI) mle 434.00/435.95 (M+ + H).
Example 14 2-Fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-6][l,2,4]triazin-2-yl)benzonitrile (13)
6-((2-(4-Bromo-3-fluorophenyl)imidazo[ 1 ,2-b] [ 1 ,2,4]triazin-7-yl)methyl)quinoline (12, 200 g, 0.461 mol), zinc cyanide (ZnCN2, 32.7 g, 0.277 mol, 0.6 equiv), zinc powder (Zn, 6.0 g, 0.093 mol, 0.2 equiv) and Pd(dppf)2Cl2 (22.6 g 0.028 mol, 0.06 eqiv) were suspended in premixed solution of ΛyV-dimethyl acetamide (DMAC, 2000 mL) and water (H2O, 40 mL). The resulting suspension was then degassed with a stream of nitrogen for 20 min before being heated to 110 0C and stirred at 110 0C for 1 – 2 h (homogeneous solution was observed). When LC/MS indicated the reaction was deemed complete, the reaction mixture was cooled first to room temperature and then in an ice bath to 5 0C. The cooled reaction mixture was diluted with a mixture of the saturated aqueous ammonium chloride solution (aq. NH4Cl), the concentrated ammonium hydroxide aqueous solution (aq. NH4OH), and water (4:1 :4 by volumn, 8.1 L) and the resulting mixture was stirred at room temperature for 30 min. The resulting solids were collected by filtration and dried in a vacuum oven overnight at 45 0C to afford the crude desired product (13). This crude material was then purified by flash chromatography (SiO2, gradient elution with 1% triethylamine in dichloromethane, 2.5 % acetone and 1% triethylamine in dichloromethane, 5.0 % acetone and 1% triethylamine in dichloromethane, and 10.0 % acetone and 1% triethylamine in dichloromethane sequentially) to afford the pure 2-fluoro-4-(7-(quinolin-6-ylmethyl)-imidazo[l,2-έ][l,2,4]triazin-2-yl)benzonitrile (13, 127.4 g, 175.4 g theoretical, 72.6% yield) as yellow solids. For 13: 1H NMR (400 MHz, DMSO-^6) δ ppm 9.24 (s, IH), 8.81 (dd, IH, J= 4.15, 1.66 Hz), 8.26 – 8.12 (m, 4H), 8.02 (s, IH), 7.95 – 7.93 (m, 2H), 7.76 (dd, IH, J= 8.71, 2.08 Hz), 7.47 (dd, IH, J= 8.70, 4.15 Hz), 4.62 (s, 2H); C22HnFN6 (MW 380.38), LCMS (EI) mle 381.0 (M+ + H).
Example 15 6-(3,3-Diethoxyprop-l-ynyl)quinoline (22)
A mixture of 6-bromoquinoline (8, 2.63 g, 12.6 mmol), propargylaldehyde diethyl acetal (3.73 niL, 25.2 mmol, 2.0 equiv), triethylamine (TEA, 12.7 mL, 90.8 mmol, 7.2 equiv), copper(I) iodide (CuI, 24.0 mg, 0.126 mmol, 0.01 equiv), and triphenylphosphine (PPh3, 0.39716 g, 1.5142 mmol, 0.12 equiv) in JV^V-dimethylformamide (DMF, 15.6 mL, 202 mmol) was degassed with nitrogen bubbling for 5 min. Palladium acetate (Pd(OAc)2, 0.08499 g, 0.3786 mmol, 0.03 equiv) was added and the mixture was degassed with nitrogen bubbling for 5 min. The reaction mixture was heated to 90 0C under nitrogen with stirring. After 3 h and 10 min, HPLC indicated that the reaction was complete. The reaction mixture was diluted with ethyl acetate (EtOAc, 100 mL) and washed with water (H2O, 2 x 100 mL). The aqueous layer was extracted with ethyl acetate (EtOAc, 20 mL). The combined organic extracts were then concentrated under the reduced pressure to give the crude product as a black oil. The crude product was purified by flash column chromatography (SiO2, 0 – 40% EtOAc in hexane gradient elution) to afford 6-(3,3-diethoxyprop-l-ynyl)quinoline (22, 3.2 g, 3.22 g theoretical, 99% yield) as a colorless oil. For 22: 1H NMR (400 MHz, OMSO-d6) δ ppm 8.92 (dd, IH, J= 4.35 Hz, 1.86 Hz), 8.36 (d, IH, J= 8.40 Hz, 1.66 Hz), 8.20 (d, IH, J= 1.78 Hz), 7.99 (d, IH, J= 8.71 Hz), 7.76 (dd, IH, J= 8.71 Hz, 1.87 Hz), 7.57 (dd, IH, J= 8.09 Hz, 4.05 Hz), 5.58 (s, IH), 3.75 – 3.55 (m, 4H), 1.17 (t, 6H, J= 7.16 Hz); Ci6H17NO2 (MW 255.31), LCMS (EI) m/e 256.0 (M+ + H).
Scheme 2 (Examples 15-18)

Method C



Example 16 6-(3,3-Diethoxypropyl)quinoline (23)
Method A. 3,3-Diethoxy-l-propene (548 g, 4.2 mol, 1.75 equiv) was added to a 22 L flask charged with 0.5 M solution of 9-borabicyclo[3.3.1] nonane in tetrahydrofuran (9-BBN solution in THF, 8.4 L, 4.2 mol, 1.75 equiv) at room temperature (the internal temperature raised to 40 0C) over 1 h.. The resulting reaction mixture was stirred at room temperature for overnight. At which time 1H NMR of an aliquot of the reaction mixture indicated that all the 3,3-diethoxy-1-propene had been consumed. 6-Bromoquinoline (8, 500 g, 2.4 mol, 1.0 equiv), potassium carbonate (K2CO3, 662 g, 4.8 mol, 2.0 equiv), tricyclohexylphosphine (67.4 g, 0.24 mol, 0.1 equiv), palladium acetate (Pd(OAc)2, 27 g, 0.12 mol, 0.05 equiv) and water (90 mL) were added to the reaction mixture in that order followed by degassing with nitrogen for 0.5 h. The reaction mixture was then heated to reflux for 4 h. Once TLC and LC/MS showed that the starting material had been consumed, the reaction mixture was cooled to room temperature with stirring before being quenched with water (7.5 L) and ethyl acetate (EtOAc, 7.5 L). The layers were separated and the aqueous layer was extracted with ethyl acetate (EtOAc, 4 L). The combined organic layers were washed with a saturated brine solution (NaCl, 4 L), dried over magnesium sulfate (MgSO4) and concentrated under the reduced pressure. The residue was purified by column chromatography (SiO2, 10 – 60% of ethyl acetate in heptane gradient elution) to afford 6-(3,3-diethoxypropyl)quinoline (23, 520 g, 622.4 g theoretical, 83.5% yield) as a colorless oil. For 23: 1HNMR (DMSO-</6, 300MHz) δ ppm 8.81 (dd, IH, J= 4.23 Hz, 1.73 Hz), 8.28 (d, IH, J= 8.07 Hz), 7.91 (d, IH, J= 8.62 Hz ), 7.75 (s, IH), 7.61 (dd, lH, J= 8.63 Hz, 1.92 Hz), 7.46 (dd, IH, J= 8.25 Hz, 4.22 Hz), 4.46 (t, IH, J= 5.60 Hz), 3.61 – 3.38 (m, 4H), 2.79 (t, 2H, J= 8.53 Hz), 1.95 -1.85 (m, 2H), 1.11 (t, 6H, J= 6.84 Hz); Ci6H21NO2 (MW 259.34), LCMS (EI) m/e 260.2 (M+ + H).
Method A-Alternative. 9-BBN was generated in situ and used to prepare compound 23 as discribed as follows: under a nitrogen atmosphere anhydrous 1 ,2-dimethoxyethane (DME, 47.0 mL) was charged into a 500 mL 3-neck flask equipped with a distillation apparatus. Borane-dimethyl sulfide complex (12.1 g, 151 mmol, 2 equiv) was added and the solution temperature increased from 20 to 22 0C. To this solution, 1 ,5-cyclooctadiene (16.3 g, 151 mmol, 2 equiv) was added dropwise over a period of 30 min to maintain a reaction temperature of 50 – 60 0C, during which time a small amount of dimethyl sulfide was collected by the distillation apparatus. The reaction mixture was then distilled under nitrogen until the distillate temperature reach 84 0C. The distillates collected had a volume of ~ 21 mL. The oil bath was removed and anhydrous THF (49 mL) was added. A small sample of the reaction mixture was taken for 1H NMR analysis and the result indicated the olefin was consumed. This 9-BBN solution was used directly for the next step.
To the above 9-BBN solution, 3,3-diethoxy-l-propene (19.3 g, 142 mmol, 1.89 equiv) was added dropwise while maintaining the temperature below 30 0C. The reaction is slightly exothermal and white precipitate slowly dissolved. The reaction mixture was then stirred at room temperature for 18 h.
To the solution prepared above, 6-bromoquinoline (8, 15.7 g, 75.4 mmol, 1 equiv), tricyclohexylphosphine (1.27 g, 4.52 mmol, 0.06 equiv), potassium carbonate (20.8 g, 151 mmol, 2 equiv), and water (0.421 mL, 23.4 mmol) were added. The mixture was degassed with nitrogen bubbling for 10 – 15 min. Palladium acetate (Pd(OAc)2, 0.508 g, 2.26 mmol, 0.03 equiv) was added and the nitrogen bubbling was continued for an additional 10 min. The reaction mixture was heated to 75 0C and maintained at 75 – 78 0C for 2 – 3 h. When HPLC showed the completion of the reaction, the heating was discontinued and the reaction mixture was cooled to room temperature. Ethyl acetate (EtOAc, 162 mL) and water (H2O, 162 mL) were added and the organic layer was separated. The aqueous layer was extracted with ethyl acetate (EtOAc, 2 x 60 mL) and the combined organic extracts were dried over sodium sulfate (Na2SO4) and concentrated under the reduced pressure. The residue was purified by flash column chromatography (silica gel, 0 – 40% EtOAc in hexane gradient elution) to afford 6-(3,3-diethoxypropyl)quinoline (23, 17.6 g, 19.6 g theoretical, 90% yield) as a clear oil, which was found to be identical to the meterial made from Method A in every comparable aspect.
Method B. A mixture of 6-(3,3-diethoxyprop-l-yn-l-yl)quinoline (22, 56 mg, 0.22 mmol) and 10% palladium on carbon (5 mg) in THF (5 mL) was hydrogenated under H2 at 1 atm for 6 h. The reaction mixture was filtered through a celite bed and the celite bed was washed with THF (2 x 2 mL). The combined filtrates were concentrated under the reduced pressure to afford 6-(3,3-diethoxypropyl)quinoline (23, 56 mg, 57 mg theoretical, 98% yield) as a clear oil, which was found to be sufficiently pure to be used in the subsequent reaction without further purification and was identical to the meterial made from Method A in every comparable aspect.
Example 17 3-(Quinolin-6-yl)propanal (9)
Method 1. A 22 L flask was charged with tris(dibenzylideneacetone)dipalladium(0) (70.0 g, 0.076 mol, 0.015 equiv), tri-tert-butylphosphonium tetrafluoroborate (44 g, 0.152 mol, 0.03 equiv), and dioxane (12 L) at room temperature. The resulting solution was then degassed with a steady stream of nitrogen for 20 min before 6-bromoquinoline (8, 1055 g, 5.07 mol, 1.0 equiv), allyl alcohol (588 g, 10.1 mol, 2.0 equiv), and 7V-methyl-iV-cyclohexylcyclohexylamine (1186 g, 6.08 mol, 1.2 equiv) were added at room temperature. The resulting reaction mixture was stirred at 50 – to 55 °C for 8 – 12 h. When TLC and LC/MS showed that the reaction was deemed complete, the reaction mixture was cooled to room temperature before methyl fert-butyl ether (MTBE, 10 L) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 10 min before being filtered through a plug of celite. The filtrate was concentrated under the reduced pressure and the residue was purified by flash column chromatography (SiO2, 20 – 80 % ethyl acetate in heptane gradient elution) to afford 3-(quinolin-6-yl)propanal (9, 495 g, 939.1 g theoretical, 52.7%) as a yellow oil, which solidified partially upon standing at 0 – 5 0C. For 9: 1H NMR (400 MHz, DMSO-J6) δ ppm 9.75 (t, IH, J= 1.24 Hz), 8.83 (dd, IH, J= 4.15 Hz, 1.66 Hz), 8.25 (dd, IH, J= 8.3, 1.03 Hz), 7.93 (d, IH, J= 8.71 Hz), 7.76 (d, IH, J= 1.45 Hz), 7.64 (dd, IH, J= 8.72 Hz, 2.08 Hz), 7.48 (dd, IH, J= 8.30 Hz, 4.36 Hz), 3.05 (t, 2H, J= 7.26 Hz), 2.89 (t, 2H, J= 7.26 Hz); Ci2HnNO (MW 185.22), LCMS (EI) We 186 (M+ + H).
Method 2. A solution of 6-(3,3-diethoxypropyl)quinoline (23, Method A of Example 16, 520 g , 2.08 mol, 1.0 equiv) in ethyl acetate (EtOAc, 2.2 L) was cooled to 0 0C before a 2 N aqueous hydrochloric acid (HCl) solution (2.2 L) was added over 1 h while keeping the reaction temperature below 5 0C. The resulting reaction mixture was stirred for an additional 2 h at 0 – 5 0C. When TLC and HPLC/MS indicated the reaction was complete, the reaction was quenched with an ice cold 3 N aqueous sodium hydroxide (NaOH) solution at 0 °C until the pH was between 8 to 9. The layers were separated and the aqueous layer was extracted with ethyl acetate (EtOAc, 2 L). The combined organic layers were washed with brine (2 L), dried with sodium sulfate (Na2SO4), and concentrated under the reduced pressure to afford crude 3-(quinolin-6-yl)propanal (9, 385.3 g, 385.3 g theoretical, 100%) as a yellow oil, which was found to be identical to the material obtained from Method 1 in every comparable aspect. Since this crude material was found to be sufficiently pure, it was used directly in subsequent reaction without further purification.
Method 5. A 22 L flask charged with 0.5 M solution of 9-borabicyclo[3.3.1] nonane in tetrahydrofuran (9-BBN, 5.75 L, 2.89 mol, 2.0 equiv) and tetrahydrofuran (THF, 6 L) was treated with 3,3-diethoxy-l-propene (393 g, 3.02 mol, 2.10 equiv) at 0 – 5 0C and the resulting reaction mixture was subsequently warmed to room temperature and stirred at room temperature for 14 h. 6-Bromoquinoline (8, 300 g, 1.44 mol, 1.0 equiv), palladium acetate (Pd(OAc)2, 16.1 g, 0.072 mol, 0.05 equiv), potassium carbonate (K2CO3, 398 g, 2.89 mol, 2.0 equiv), tricyclohexylphosphine (22.3 g, 0.079 mol, 0.055 equiv), and water (52 g, 2.8 mol) were added to the reaction mixture at room temperature before being degassed with nitrogen for 1 h. The resulting reaction mixture was heated to 75 0C for 1 h. When TLC and LC/MS showed the reaction was deemed complete, the reaction mixture was cooled to room temperature and water (2 L) was added to dissolve the salts. The resulting mixture was then concentrated under the reduced pressure to a volume of approximately 4 L before being filtered through a plug of Celite. The Celite plug was washed with ethyl acetate (EtOAc, 2 L). The filtrate was concentrated under the reduced pressure to a volume of approximately 2 L and this residual solution was then added slowly over 5 min to a flask containing a 2.0 M aqueous hydrochloric acid (HCl) solution (2 L) at 0 – 5 °C. The resulting solution was stirred at 0 – 5 °C for 14 h before being quenched with saturated aqueous sodium bicarbonate (NaHCO3) solution at 0 0C until the pH was between 8 to 9. The layers were separated and the aqueous layer was extracted with ethyl acetate (EtOAc, 2 L). The combined organic layers were washed with brine (1 L), dried with sodium sulfate (Na2SO4), and concentrated under the reduced pressure. The residue, which contains the crude 3-(quinolin-6-yl)propanal (9) was purified by flash column chromatography (SiO2, 20 – 80 % ethyl acetate in heptane gradient elution) to afford 3-(quinolin-6-yl)propanal (9, 139 g, 266.7 g theoretical, 52.1%) as a yellow oil, which was found to be identical to the material obtained from Methods 1 and 2.
Example 18 l-(2-Chloro-l-hydroxy-3-(quinolin-6-yI)propyl)pyrrolidine-2,5-dione (11)
Method I. A solution of 3-(quinolin-6-yl)propanal (9, 407 g, 2.2 mol, 1.0 equiv) in chloroform (CHCl3, 1700 mL) was cooled to 0 0C before proline (52 g, 0.44 mol, 0.2 equiv) and iV-chlorosuccinimide (NCS, 303 g, 2.31 mol, 1.05 equiv) were added. The resulting reaction mixture was allowed to slowly warm to room temperature (becomes homogeneous) and stirred at room temperature for overnight. The reaction was exothermal to around 40 0C when it reaches room temperature and a precipitate had formed at this point. Once TLC and LC/MS showed that the reaction was deemed complete, the reaction mixture was diluted with ethyl acetate (EtOAc, 1700 mL) and the resulting mixture was cooled to 0 0C. The solid was collected by filtration and the collected wet solid cake was placed in a flask and triturated with water (750 mL). The resulting suspension was stirred at room temperature for 30 min before the solids were collected by filtration. The collected solids were washed with water (250 mL) and methyl tert-bntyl ether (MTBE, 500 mL) and dried in a vacuum oven at 45 0C to constant weight to afford l-(2-chloro-l-hydroxy-3-(quinolin-6-yl)propyl)pyrrolidine-2,5-dione (11, 378.7 g, 701.3 g theoretical, 54 % yield) as off-white powder. For 11: 1HNMR (DMSO-J6, 400MHz) δ ppm 8.86 (dd, IH, J= 4.15 Hz, 1.66 Hz), 8.33 (dd, IH, J= 8.51 Hz, 1.04 Hz), 7.98 (d, IH, J= 8.72 Hz), 7.85 (d, IH, J= 1.66 Hz), 7.68 (dd, IH, J= 8.51 Hz, 1.87 Hz), 7.51 (dd, IH, J= 8.29 Hz, 4.15 Hz), 7.36 (d, IH, J = 7.05 Hz), 5.28 (dd, IH, J= 9.54 Hz, 6.85 Hz), 5.07 (dt, IH, J= 9.75 Hz, 2.70 Hz), 3.65 (dd, IH, J= 14.52 Hz, 2.49 Hz), 3.09 (dd, IH, J= 14.52 Hz, 9.75 Hz), 2.64 (s, 4H); C16H15ClN2O3 (MW 318.75), LCMS (EI) m/e 319.2 (M+ + H).
Method II. A solution of 3-quinolin-6-ylpropanal (9, 74.8 g, 0.404 mol) in acetonitrile (202 mL, 3.87 mol) was cooled to 0 0C before L-proline (4.70 g, 0.0404 mol, 0.10 equiv), benzoic acid (4.96 g, 0.0404 mol, 0.10 equiv), and iV-chlorosuccinimide (NCS, 57.8 g, 0.424 mol, 1.05 equiv) were added at 0 0C. The reaction mixture was stirred at 0 °C for 3 h and the resulting clear solution was allowed to warm to room temperature and stirred at room temperature for 18 h. The reaction mixture became a thick suspension and LCMS showed the completion of the reaction. Ethyl acetate (EtOAc, 202 mL) was added to the reaction mixture and the resulting mixture was stirred at room temperature for 1 h. The solids were collected by filtration, washed with ethyl acetate (EtOAc, 100 mL) and dried under vacuum at 40 – 45 0C to constant weight to afford l-(2-chloro-l-hydroxy-3-(quinolin-6-yl)propyl)pyrrolidine-2,5-dione (11, 88.8 g, 128.8 g theoretical, 69 % yield) as an off-white powder, which was found to be identical to the material made from method I in every comparable aspect.
Scheme 3 (Examples 19-21)

15 21, dihydrochloride
C23H17FN6O C23H19Q2FN6O MoI Wt 412 42 MoI Wt 485 34
Example 19
2-Fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-Z>] [l,2,4]triazin-2-yl)benzoic acid (14)
A suspension of 2-fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l ,2-&][l ,2,4]triazin-2-yl)benzonitrile (13, 277.5 g, 0.73 mol, 1.0 equiv) in concentrated hydrochloric acid (2500 mL) and water (250 mL) was heated to 1000C (homogenous at this point) and stirred at around 100 0C for 18 h. When LC/MS indicated the reaction was deemed complete, the reaction mixture was cooled down to 70 – 80 0C before being diluted with water (2500 mL). The resulting diluted reaction mixture was then cooled down to room temperature (yellow solid forms at 40 – 50 0C) and subsequent to 0 – 5 0C. The solids were then collected by filtration, washed with a small amount of IN aqueous HCl (100 mL), and dried in a vacuum oven at 45 0C to constant weight to afford 2-fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-έ][l,2,4]triazin-2-yl)benzoic acid (14, 271 g, 291.5 g theoretical, 93% yield) as yellow to bright-yellow powders. For 14: 1H NMR (400 MHz, OMSO-d6) δ ppm 9.34 (s, IH), 9.23 (dd, IH, J- 5.19 Hz, 1.45 Hz), 9.08 (d, IH, J= 8.29 Hz), 8.38 (d, IH, J= 8.92 Hz), 8.30 (d, IH, J= 1.24 Hz), 8.18 (dd, IH, J= 8.72 Hz, 1.87 Hz), 8.12 (s, IH), 8.08 – 8.00 (m, 4H), 4.75 (s, 2H); C22H16Cl2FN5O2 (MW 472.30), C22H14FN5O2 (free base: MW 399.38), LCMS (EI) mle 400.0 (M+ + H).
Example 20 2-Fluoro-7V-methyl-4-(7-(quiiiolin-6-ylmethyl)imidazo[l,2-^][l,2,4]triazin-2-yl)benzainide
(15).
A suspension of 2-fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l ,2-b][l ,2,4]triazin-2-yl)benzoic acid (14, 431.4 g, 0.914 mol, 1.0 equiv) and (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP, 570 g, 1.1 mol, 1.2 equiv) in ΛyV-dimethylformamide (DMF, 3700 mL) was treated with a solution of 2 M methylamine in THF (1830 mL, 3.656 mol, 4.0 equiv) over 15 min at room temperature. The reaction temperature increased to 30 0C during the addition of methylamine and the reaction mixture became homogeneous once the addition of methylamine was complete. Triethylamine (TEA, 382 mL, 2.742 mol, 3.0 equiv) was then added to the reaction mixture and the resulting reaction mixture was stirred at room temperature for 2 – 4 h. When LC/MS showed the coupling reaction was deemed complete, the reaction mixture was treated with water (950 mL). The resulting suspension was cooled down to 0 – 5 0C in an ice-bath and stirred at 0 – 5 0C for 30 min. The solids were collected by filtration and washed with water (200 mL). The wet solid cake was then suspended in a mixture of water and acetonitrile (1/1 by volume, 2000 mL) and the resulted suspension was stirred at room temperature for Ih. The solids were collected by filtration, washed with water and acetonitrile, and dried in a vacuum oven at 40 – 45 0C to constant weight to afford 2-fluoro-Λ/-methyl-4-(7-(quinolin-6-ylmethyl)imidazo[l ,2-έ>][l ,2,4]triazin-2-yl)benzamide (15, 322 g, 377 g theoretical, 85.4% yield) as yellow to bright-yellow powders. For 15: 1H NMR (400 MHz, DMSO-J6) δ ppm 9.20 (s, IH), 8.82 (dd, IH, J= 4.05, 1.56 Hz), 8.38 (br m, IH), 8.27 (dd, IH, J= 8.50 Hz, 1.25 Hz), 8.06 – 7.93 (m, 5H), 7.81 – 7.74 (m, 2H), 7.49 (dd, IH, J= 8.40 Hz, 4.35 Hz), 4.62 (s, 2H), 2.78 (d, 3H, J= 4.36 Hz); C23H17FN6O (MW 412.42), LCMS (EI) mle 413.1 (M+ + H).
Example 21
2-Fluoro-Λr-methyl-4-(7-(quinoIin-6-ylmethyl)imidazo[l,2-6][l,2,4]triazm-2-yl)benzamide dihydrochloride (21, dihydrochloride)
A suspension of 2-fluoro-iV-methyl-4-[7-(quinolin-6-ylmethyl)imidazolo[l ,2-6][l,2,4]triazin-2-yl]benzamide (15, 421.2 g, 1.021 mol) in methanol (MeOH, 6600 mL) was heated to 55 0C before a premixed solution of aqueous concentrated hydrochloric acid (cone.
HCl, 37 wt.%, 12 M, 420 mL, 5.10 mol, 5.0 equiv) in isopropyl alcohol (IPA, 1510 mL) was added dropwise at 55 0C. The resulting clear solution was stirred at 55 0C for 30 min before methyl tert-butyl ether (MTBE, 6750 mL) was added via an additional runnel over 30 min. The solids were slowly precipitated out after addition of methyl tert-butyl ether. The resulting mixture was stirred at 55 0C for an additional 1 h before being gradually cooled down to room temperature. The mixture was stirred at room temperature for overnight. The solids were collected by filtration, washed with methyl tert-butyl ether (MTBE, 3 x 500 mL), and dried in vacuum oven at 45 – 55 0C to constant weight. The desired 2-fluoro-Λr-methyl-4-[7-(quinolin-6-ylmethyl)imidazolo[l,2-£][l,2,4]triazin-2-yl]benzamide dihydrochloride (21, dihydrochloride, 470.7 g, 495.5 g theoretical, 95% yield) was obtained as off-white to light yellow crystalline solids. For 21 (dihydrochloride): mp (decom.) 222 0C; 1H NMR (400 MHz, DMSO-J6) δ ppm 9.46 (s, IH), 9.25 (dd, IH, J= 5.4 Hz, 1.4 Hz), 9.12 (d, IH, J= 8.3 Hz), 8.51 (m, IH), 8.47 (d, IH, J= 0.9 Hz), 8.34 (d, IH, J= 1.3 Hz), 8.23 (s, IH), 8.21 (dd, IH, J= 9.0 Hz, 1.8 Hz), 8.09-8.02 (m, 3H), 7.79 (dd, IH, J= 7.5 Hz, 8.3 Hz), 4.77 (s, 2H), 2.78 (s, 3H, J= 4.5 Hz); 13C NMR (100 MHz, DMSO-^6) δ ppm 163.4, 159.4 (d, J= 249.9 Hz), 145.8, 145.4, 144.5, 143.8, 140.4, 138.8, 136.8, 135.9, 135.7 (J= 8.6 Hz), 131.2 ( J= 3.1 Hz), 130.7, 128.7, 128.2, 126.2 (J- 14.9 Hz), 126.0, 123.1 (J= 3 Hz), 122.5, 121.0, 114.9 (J= 5.6 Hz), 28.4, 26.3; 19F NMR (376.3 MHz, DMSO-^6) δ ppm -113.2; C23H17FN6O (free base, MW 412.42), LCMS (EI) mle 413.1 (M+ + H) and 435.0 (M+ + Na).
Scheme 4 (Examples 22-25)

C7H3BrFN C13H15BFN2O2 MoI. Wt: 200.01 MoI. Wt: 247.07 Example 22 l,2,4-Triazin-3-amine (16)
An aqueous solution of glyoxal (57 Kg of 40 wt% aqueous solution, 393 mol, 0.73 equiv) was added to a suspension of aminoguanidine bicarbonate (73 Kg, 536.3 mol) in water (400 L) at room temperature. The evolution of carbon dioxide (CO2) began almost immediately. The reaction mixture was then stirred at room temperature for 18 h and the evolution of gas had virtually ceased after about 2 h. The reaction mixture was then filtered, and the filtrate was evaporated to dryness under the reduced pressure. The residue was then extracted with cold methanol (MeOH, 3 x 120 L), and the combined methanol solution was cooled down to 0 – 5 0C before being filtered to remove the residual solids. The filtrate was then concentrated under the reduced pressure, and the residue was recrystallized in acetonitrile to afford l,2,4-triazin-3-amine (16, 34 Kg, 37.76 Kg theoretical, 90% yield) as fine, white needles. For 16: 1H NMR (400 MHz, DMSO-J6) δ ppm 8.54 (d, IH, J- 2.33 Hz), 8.20 (d, IH, J= 2.33 Hz), 7.15 (br s, 2H).
Example 23 6-Bromo-l,2,4-triazin-3-amine (17)
A solution of 1 ,2,4-triazin-3-amine (16, 33 Kg, 343.4 mol) in water (500 L) and acetonitrile (300 L) was treated with jV-bromosuccinimide (NBS, 66 Kg, 370 mol, 1.08 equiv) at 5 – 15 0C, and the resulting reaction mixture was stirred at 10 – 15 0C for 1 – 4 h. When TLC and LC/MS showed that the bromination reaction was deemed complete, the reaction mixture was treated with an aqueous solution of saturated sodium carbonate (Na2CO3). The resulting solution was then extracted with ethyl acetate (EtOAc, 3 x 500 L). The combined organic extracts were washed with water (2 x 100 L), dried over magnesium sulfate (MgSO4), and concentrated under the reduced pressure to afford 6-bromo-l,2,4-triazin-3-amine (17, 10.3 Kg, 60 Kg theoretical, 17.2% yield) as yellow to brown powders. For 17: 1H NMR (400 MHz, DMSO-J6) δ ppm 8.39 (s, IH), 7.47 (br, 2H); C3H3BrN4 (MW 174.99), LCMS (EI) mle 175.0/176.9 (M+ + H).
Example 24 2-Fluoro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzonitrile (19) Step 1. A solution of 2-fluro-4-bromobenzonitrile (18, 12.5 Kg, 62.5 mol) in anhydrous tetrahydrofuran (THF, 30 L) was treated with a solution of isopropylmagnesium chloride generated from magnesium (Mg, 1.8 Kg, 150 mol, 1,2 equiv) an 2-chloropropane (7.2 Kg, 92 mol, 1.47 equiv) in THF (20 L) and 2-(2-(dimethylamino)ethoxy)-τV/vr-dimethylethanamine (11 Kg, 69 mol, 1.1 equiv) at room temperature. The resulting mixture was then stirred at 12 – 20 0C for an additional 2 h before being treated with trimethylborate (9 Kg, 86.7 mol, 1.4 equiv) at 10 -15 0C. The reaction mixture was stirred at 7 – 16 0C for 40 min. When TLC and LC/MS showed that the reaction was deemed complete, the reaction mixture was quenched with 1 N aqueous hydrochloric acid (HCl, 35 Kg) at room temperature. The quenched aqueous reaction mixture was then extracted with ethyl acetate (EtOAc, 4 x 35 L). The combined organic extracts were washed with water (50 L), dried over magnesium sulfate (MgSO4), and concentrated under the reduced pressure. The residual solids were then recrystallized from acetonitrile (20 L) and hexanes (45 L) to afford the corresponding crude 3-fluoro-4-cyanophenyl boronic acid (5.0 Kg, 48% yield).
Step 2. A suspension of the crude 3-fluoro-4-cyanophenyl boronic acid (9.2 Kg, 55.8 mol) in cyclohexane (150 L) was treated with pinacol (13.2 Kg, 111.6 mol, 2.0 equiv) at room temperature, and the resulting reaction mixture was warmed to 40 0C for 4 h. When TLC and LC/MS showed that the reaction was deemed complete, the reaction mixture was cooled down to room temperature before being washed with water (2 x 75 L). The organic layer was then dried over magnesium sulfate (MgSO4) and concentrated under the reduced pressure to afford 2-fluoro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzonitrile (19, 11.8 Kg, 13.8 Kg theoretical, 85.6% yield) as a light yellow solid. For 19: 1H NMR (300 MHz, DMSO-J6) δ ppm 7.92 (t, IH, J- 7.00 Hz), 7.62 (m, 2H), 1.29 (s, 12 H).
Example 25 4-(3-Amino-l,2,4-triazin-6-yI)-2-fluorobenzonitrile (20).
A mixture of 6-bromo- 1,2, 4-triazin-3 -amine (17, 100.0 g, 571.47 mmol) and 2-fluoro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzonitrile (19, 145.43 g, 588.61 mmol, 1.03 equiv) in 1,4-dioxane (1200 mL) was stirred at room temperature for 10 min before potassium carbonate (K2CO3, 355.4 g, 2572 mmol) in water (600 mL) was added to give a deep red solution. The mixture was degassed by bubbling with nitrogen for 10 min before 1,1′-bis(diphenyl phosphino)ferrocene dichloropalladium(II) complex with dichloromethane (1 :1) (Pd(dppf)2Cl2, 14.14 g, 17.14 mmol, 0.03 equiv) was added at room temperature. The resulting reaction mixture was degassed by bubbling with nitrogen for 10 min and then heated at 86 0C under nitrogen. After 2 h, HPLC showed that the reaction was deemed complete, and the reaction mixture was cooled to room temperature and then to 0 – 5 0C with an ice-water bath. 1 ,4-Dioxane (400 mL) was added to the cooled reaction mixture before a solution of 3.3 M aqueous hydrochloric acid solution (HCl, 1900 mL) was added dropwise with stirring to adjust pH to 0.40- 0.93. The mixture was stirred at room temperature for 30 min and filtered. The solid collected was stirred with 1,4-dioxane (260 mL) and then added IN HCl (400 mL). The mixture was stirred at room temperature for 10 min and filtered. The filtrate was combined with the filtrate obtained earlier and washed with ethyl acetate (EtOAc, 2 x 2 L). The combined ethyl acetate extracts was extracted with 1 N aqueous hydrochloric acid solution (HCl, 3 x 200 mL). The combined aqueous solution was then treated with activated charcoal (20 g) and stirred at room temperature for 30 min. The mixture was filtered through a celite bed and the filtrate was cooled to 0 – 5 0C with an ice- water bath. A solution of 50% of sodium hydroxide in water (NaOH, 240 mL, 4500 mmol) was added drowise at 5-12 0C to adjust pH tolθ.6 – 11.6. The mixture was stirred at 0 – 5 0C for 30 min and then filtered. The solids collected were washed with aqueous ammonium hydroxide (1 to 3 of 28% concentrated NH4OH to water, 1900 mL) and dried under vacuum at 40 – 45 0C to constant weight to afford 4-(3-amino-l,2,4-triazin-6-yl)-2-fluorobenzonitrile (20, 101.2 g, 122.9 g theoretical, 82.3% yield) as a off-white powder. For 20: 1H NMR (400 MHz, DMSO-J6) δ ppm 8.94 (s, IH), 8.12 (d, IH, J= 11.41 Hz), 8.08 – 8.00 (m, 2 H), 7.71 (br s, 2 H); Ci0H6FN5 (MW 215.19), LCMS (EI) mle 215.9 (M+ + H).
Scheme 5 (Example 26)

20 13
C10H6FN5 C16H15ON2O3 C22H13FN6 MoI. Wt: 215.19 MoI. Wt 318.75 MoI. Wt: 380.38 Example 26 2-Fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-Z>][l,2,4]triazin-2-yl)benzonitrile (13).
Step 1. A 22 L reactor equipped with a overhead stirring, a thermocouple, a distillation apparatus, and a nitrogen inlet was purged with nitrogen before 4-(3-amino-l,2,4-triazin-6-yl)-2-fluorobenzonitrile (20, 300 g, 1.39 mol), l-(2-chloro-l-hydroxy-3-(quinolin-6-yl)propyl)pyrrolidine-2,5-dione (11, 635 g, 1.99 mol, 1.43 equiv), and ethylene glycol (3.0 L) were charged to the reactor at room temperature. The resulting reaction mixture was heated to 130-140 °C with nitrogen bubbled through continuously. The distillate was collected with the distillation apparatus. After 3 – 4 h, HPLC indicated the reaction was deemed complete (presence of < 1.5% of starting material 20). The reaction mixture was gradually cooled to room temperature. A 2.5% aqueous sodium carbonate solution (Na2CO3, 14.1 L) was added with stirring to the reactor over 60 min and the mixture was stirred at room temperature for 1 – 2 h. The mixture was then filtered, and the solid was washed with water (9.6 L) and dried under vacuum to afford the desired crude product (13, 980.4 g), which was combined with several other batches for purification as described below.
Step 2. A solution of crude product (13, 2754 g) in methylene chloride (CH2Cl2, 37.8 L) and methanol (0.54 L) was treated with silica gel (SiO2, 2700 g) at room temperature, and the resulting mixture was stirred at room temperature for 90 min. The mixture was filtered and the filter cake was washed with a mixture OfCH2Cl2(18 L) and methanol (0.26 L). The combined filtrates were treated with silica gel (SiO2J 800 g) and the resulting mixture was stirred at room temperature for 90 min and then filtered. The filter cake was washed with a mixture of CH2Cl2 (18 L) and methanol (0.26 L). The combined filtrates were concentrated under the reduced pressure at 20 – 60 0C to about 8 – 12 L. The residue was treated with a mixture of isopropanol (IPA) and water (1 : 1 , 9 L) in portions and the distillation was continued at 1 atm pressure until the temperature reached 68 – 75 0C. The mixture was cooled to room temperature and the solids were collected by filtration. The solids collected were washed with isopropanol (IPA, 3.6 L) and dried under vacuum at 40 – 45 0C to constant weight to afford pure 2-fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-ό][l,2,4]triazin-2-yl)benzonitrile (13, 940.27g) as a bright yellow powder.
The above reaction and purification process gave product 13 in 59 – 64% yield. The spectroscopic data of compound 13 made by this synthetic process was found to be identical to those obtained from material made by cyanation of compound 12 described previously. For 13: 1H NMR (400 MHz, OMSO-d6) δ ppm 9.24 (s, IH), 8.81 (dd, IH5 J= 4.15, 1.66 Hz), 8.26 – 8.12 (m, 4H), 8.02 (s, IH), 7.95 – 7.93 (m, 2H), 7.76 (dd, IH, J= 8.71, 2.08 Hz), 7.47 (dd, IH, J = 8.70, 4.15 Hz), 4.62 (s, 2H); C22Hi3FN6 (MW 380.38), LCMS (EI) m/e 381.0 (M+ + H).
Scheme 6 (Examples 27-29)

14 15
C22H14FN5O2 C23H17FN6O MoI. Wt: 399.38 MoI. Wt: 412.42
I I) SOd2 aq. HCl/ acetone { 2) MeNH2

15 (.{hydrochloride
C23H17FN6O C23H19CI2FN6O MoI. Wt: 412.42 MoI. Wt: 485.34
Example 27
2-Fluoro-4-(7-(quinolin-6-ylmethyl)imidazo [1,2-6] [1,2,4] triazin-2-yl)benzoic acid (14).
To a 22 L reactor equipped with a overhead stirring, a thermocouple, and a nitrogen inlet was charged compound 13 (900 g, 2.37 mol), water (0.9 L), and concentrated HCl (9.1 L) at room temperature. The resulting reaction mixture was heated at 100 0C for 12 h. When HPLC showed the reaction was complete, the reaction mixture was cooled to 90 0C and water (4.9 L) was added over 15 min while maintaining the temperature at 65 – 90 0C. The reaction mixture was further cooled to room temperature and stirred at room temperature for 3 h. The solids were collected by filtration, washed with water (1.2 L) and dried in vacuum at 40 – 45 0C to constant weight to afford 2-fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-6][l,2,4]triazin-2-yl)benzoic acid (14, 945 g, 946.5 g theoretical, 99.8% yield) as a light yellow solid, which was found to be identical to the material made by earlier method. For 14: 1H NMR (400 MHz, DMSO-J6) δ ppm 9.34 (s, IH), 9.23 (dd, IH, J= 5.19 Hz, 1.45 Hz), 9.08 (d, IH5 J= 8.29 Hz), 8.38 (d, IH, J = 8.92 Hz), 8.30 (d, IH, J= 1.24 Hz), 8.18 (dd, IH, J= 8.72 Hz, 1.87 Hz), 8.12 (s, IH), 8.08-8.00 (m, 4H), 4.75 (s, 2H); C22Hi6Cl2FN5O2 (MW 472.30), C22H14FN5O2 (free base: MW 399.38), LCMS (EI) mle 400.0 (M+ + H).
Example 28
2-Fluoro-iV-methyl-4-(7-(quinolin-6-yImethyl)iinidazo [ 1 ,2-6] [ 1 ,2,4] triazin-2-yl)benzamide
(15).
Method A. To a stirred solution of 2-fluoro-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-6][l,2,4]triazin-2-yl)benzoic acid (14, 1000 g, 2.12 mol) in acetonitrile (5 L) and CH2Cl2(10 L) were charged HOBt (358 g, 2.65 mol, 1.25 equiv), and EDC hydrochloride (508.4 g, 2.65 mol, 1.25 equiv) at room temperature. Another portion OfCH2Cl2 (10 L) was then added to the reaction mixture and the resulting reaction mixture was stirred at room temperature for 20 min. A 2.0 M solution of methylamine (MeNH2) in THF (3.44 L, 6.88 mol, 3.25 equiv) was added with stirring while maintaining the temperature at 15 – 30 0C. The reaction mixture was stirred at room temperature for 2 h before an additional portion of 2.0 M solution of methylamine (MeNH2) in THF (1.06 L, 2.12 mol, 1 equiv) was added. The reaction mixture was stirred at room temperature for 1 h and a second portion of EDC hydrochloride (406 g, 2.12 mol, 1 equiv) was added and the stirring was continued for 6 h. When HPLC showed less than 1 % of starting material (14) was remaining, the reaction mixture was concentrated under the reduced pressure at < 50 0C. During distillation acetonitile (20 L) was added and distillation was continued until the remaining volume was about 20 L. The residue was treated with an aqueous solution of 2.5% sodium carbonate (Na2CO3, 40 L) and the resulting mixture was stirred at room temperature for 30 min. The solids were collected by filtration, washed with water (3 x 4.0 L), air dried by pulling vacuum on the filter to afford the crude desired product (15). The crude solids were treated with CH2Cl2 (17.6 L) and MeOH (5.2 L) at room temperature and resulting mixture was stirred until a clear solution was obtained. The solution was filtered to remove insoluble materials. With vigorous stirring a 2.5% aqueous solution of sodium carbonate (Na2CO3, 17.6 L) was added to the filtrate and the mixture was stirred at room temperature for 60 min to give a suspension. Heptane (20 L) was added and the mixture was stirred for an additional 60 min. The mixture was filtered and the solid was washed sequentially with water (3 x 4.0 L) and heptane (4.0 L), and dried in vacuum to afford 2-fluoro-./V-methyl-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-b][l,2,4]triazin-2-yl)benzamide (15, 1095.3 g, 874.3 g theoretical) as a bright yellow solid, which was found to be not totally dry and to contain ~ 25% residual solvents. This wet solid was used directly for the subsequent dihydrochloride salt (21) formation reaction without further drying. A small sample was dried completely for spectroscopic analyses and the data were consistent with those obtained by earlier method: For 15: 1H NMR (400 MHz, DMSO-J6) δ ppm 9.20 (s, IH), 8.82 (dd, IH, J= 4.05, 1.56 Hz), 8.38 (br m, IH), 8.27 (dd, IH, J = 8.50 Hz, 1.25 Hz), 8.06 – 7.93 (m, 5H), 7.81 – 7.74 (m, 2H), 7.49 (dd, IH, J= 8.40 Hz, 4.35 Hz), 4.62 (s, 2H), 2.78 (d, 3H, J= 4.36 Hz); C23HnFN6O (MW 412.42), LCMS (EI) m/e 413.1 (M+ + H).
Method B. 2-Fluoro-4-[7-(quinolin-6-ylmethyl)imidazo[l,2-b][l,2,4]triazin-2-yl]benzoic acid dihydrochloride (14, 50.00 g, 0.1059 mol) was added toluene (300 mL) and followed by thionyl chloride (SOCl2, 77.2 mL, 1.06 mol, 10.0 equiv) at room temperature. The resulting reaction mixture was heated at 72 0C under N2 and the reaction was followed by HPLC analysis of the disappearance of the starting material benzoic acid (14). After 48 h, HPLC indicated ~4% starting material remaining and the reaction was stopped. The reaction mixture was concentrated to dryness by vacuum distillation at 40-50 0C. The residual solids were added toluene (300 mL) and the solvent was removed by vacuum distillation at 40-50 0C. THF (250 mL) was added and the mixture was cooled with an ice-water bath. A 2.0 M of methylamine (MeNH2) in THF (529 mL, 1.06 mol, 10 equiv) was added dropwise. The resulting reaction mixture was allowed to warm up to room temperature and stirred at room temperature for 17 h. Water (600 mL) was added to the reaction mixture and THF (400 – 500 mL) was removed by vacuum distillation at 40 0C. Sodium carbonate (15.60 g, 0.147 mol) was added and the mixture was stirred at room temperature for 30 min. The mixture was filtered and the solid was washed with water (3 x 30 mL) and dried. The solid was dissolved in pre-mixed methylene chloride (CH2Cl2, 1000 mL) and methanol (MeOH, 300 mL). With vigorous stirring, a solution of 0.236 M of sodium carbonate (Na2CO3) in water (1000 mL) was added dropwise. Solid was slowly precipitated out after addition of aqueous solution of sodium carbonate (Na2CO3). Hexane (1000 niL) was then added dropwise with stirring. The mixture was stirred at room temperature for 30 – 40 min and the solids were collected by filtration. The solids collected were washed with water (3 x 200 mL) and dried in vacuum at 40 – 500C to constant weight to afford 2-fluoro-Λr-methyl-4-(7-(quinolin-6-ylmethyl)imidazo[l,2-ό][l,2,4]triazin-2-yl)benzamide (15, 42.2 g, 43.67 g theoretical, 96.6% yield) as a bright yellow solid, which was found to be identical to the material made by Method A in every comparable aspect. For 15: 1H NMR (400 MHz, DMSO-J6) δ ppm 9.20 (s, IH), 8.82 (dd, IH, J= 4.05, 1.56 Hz), 8.38 (br m, IH), 8.27 (dd, IH, J= 8.50 Hz, 1.25 Hz), 8.06 – 7.93 (m, 5H), 7.81-7.74 (m, 2H), 7.49 (dd, IH, J= 8.40 Hz, 4.35 Hz), 4.62 (s, 2H), 2.78 (d, 3H, J= 4.36 Hz); C23H17FN6O (MW 412.42), LCMS (EI) mle 413.1 (M+ + H).
Example 29
2-Fluoro-iV-methyl-4-(7-(quinolin-6-ylmethyl)imidazo [ 1 ,2-b] [ 1 ,2,4] triazin-2-yl)benzamide dihydrochloride (21, dihydrochloride)
2-Fluoro-vV-methyl-4-(7-(quinolin-6-ylmethyl)imidazo[l, 2-Z?][l, 2,4]triazin-2-yl)benzamide (15, 210O g, containing ~25% residual solvents) and filtered USP water (7.6 L) were charged into a 50 L reactor at room temperature. With stirring a solution of 6 M aqueous hydrochloric acid (HCl, 3 L) was added with an additional funnel. The resulting reaction mixture was stirred at room temperature for 1.5 h. Acetone (30.5 L) was added to the reactor with stirring during 1 h and the resulting mixture was stirred at room temperature for 2.5 h. The solids were collected by filtration, washed with acetone (2 x 4.3 L) and dried in vacuum to constant weight to afford 2-fluoro-iV-methyl-4-(7-(quinolin-6-ylmethyl)imidazo[l ,2-b][ 1 ,2,4]triazin-2-yl)benzamide dihydrochloride (21, dihydrochloride, 1629.2 g, 1830.6 g theoretical, 89%) as a pale yellowish crystalline powder, which was found to be identical to the material made by previous method in every comparable aspect. For 21 (dihydrochloride): 1H NMR (400 MHz, DMSO-J6) δ ppm 9.46 (s, IH), 9.25 (dd, IH, J= 5.4 Hz, 1.4 Hz), 9.12 (d, IH, J= 8.3 Hz), 8.51 (m, IH), 8.47 (d, IH, J= 0.9 Hz), 8.34 (d, IH, J= 1.3 Hz), 8.23 (s, IH), 8.21 (dd, IH, J= 9.0, 1.8 Hz), 8.09 – 8.02 (m, 3H), 7.79 (dd, IH, J= 7.5, 8.3 Hz), 4.77 (s, 2H), 2.78 (s, 3H, J= 4.5 Hz); 13C NMR (100 MHz, DMSO-J6) δ ppm 163.4, 159.4 (d, J= 249.9 Hz), 145.8, 145.4, 144.5, 143.8, 140.4, 138.8, 136.8, 135.9, 135.7 (J= 8.6 Hz), 131.2 ( J= 3.1 Hz), 130.7, 128.7, 128.2, 126.2 (J= 14.9 Hz), 126.0, 123.1 (J= 3 Hz), 122.5, 121.0, 114.9 (J= 5.6 Hz), 28.4, 26.3; 19F NMR (376.3 MHz, DMSO-Z6) δ ppm -113.2; C23H17FN6O (free base, MW 412.42), LCMS (EI) mle 413.1 (M+ + H) and 435.0 (M+ + Na).\
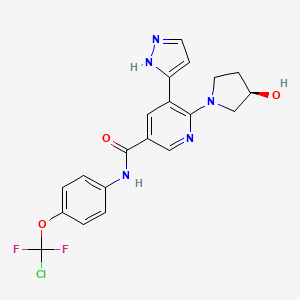
(S)-N-(54(R)-2-(2,5-difluoropheny1)-pyrrolidin-1-y1)-pyrazolo[1,5-a]pyrimidin-3-y1)-3-hydroxypyrrolidine-1-carboxamide.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

































































































































































 Mavacamten
SAR-439152; SAR 439152; SAR439152; MYK-461; MYK 461; MYK461; Mavacamten
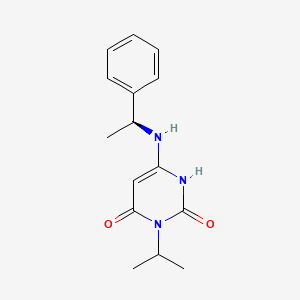
(S)-3-isopropyl-6-((1-phenylethyl)amino)pyrimidine-2,4(1H,3H)-dione
cas 1642288-47-8
Chemical Formula: C15H19N3O2
Molecular Weight: 273.336
マバカムテン;
Mavacamten
SAR-439152; SAR 439152; SAR439152; MYK-461; MYK 461; MYK461; Mavacamten
(S)-3-isopropyl-6-((1-phenylethyl)amino)pyrimidine-2,4(1H,3H)-dione
cas 1642288-47-8
Chemical Formula: C15H19N3O2
Molecular Weight: 273.336
マバカムテン;




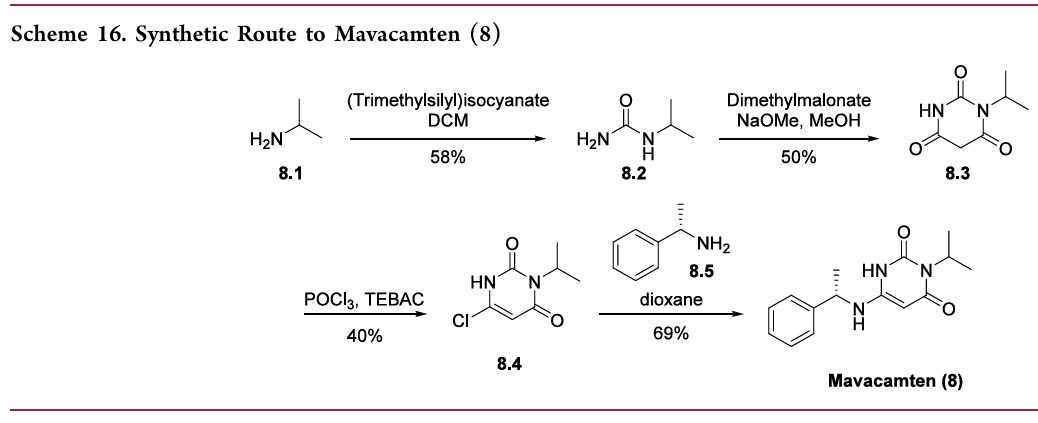
 [0072] Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0 °C was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0 °C, CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1 :20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
[0072] Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0 °C was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0 °C, CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1 :20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
 [0073] Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65 °C. After cooling to ambient temperature and then to 0 °C, the pH was carefully adjusted to 3 using aqueous concentrated HCl . The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20: 1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. . LC/MS: m/z (ES+) 171 (M+H)+.1 1H-NMR (300 MHz, de-DMSO): 5 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J = 6.0 Hz, 6H).
[0073] Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65 °C. After cooling to ambient temperature and then to 0 °C, the pH was carefully adjusted to 3 using aqueous concentrated HCl . The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20: 1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. . LC/MS: m/z (ES+) 171 (M+H)+.1 1H-NMR (300 MHz, de-DMSO): 5 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J = 6.0 Hz, 6H).
 [0074] Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50 °C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4 , and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtO Ac/petroleum ether (1 : 1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J = 6.0 Hz, 6H).
[0074] Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50 °C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4 , and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtO Ac/petroleum ether (1 : 1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J = 6.0 Hz, 6H).
 [0075] Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl) amino) pyrimidine-2,
4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3,
1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-a-methylbenzylamine (Sigma- Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80 °C for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous IN C1 (2 x 50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SC”4 and then
concentrated under reduced pressure to half the original volume to yield a precipitate.
Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, de-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J= 6.8 Hz, 1H), 4.87 (m,
1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H ), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J = 6.7 Hz, 3H), 1.36 (m, 6H).
[0075] Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl) amino) pyrimidine-2,
4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3,
1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-a-methylbenzylamine (Sigma- Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80 °C for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous IN C1 (2 x 50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SC”4 and then
concentrated under reduced pressure to half the original volume to yield a precipitate.
Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, de-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J= 6.8 Hz, 1H), 4.87 (m,
1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H ), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J = 6.7 Hz, 3H), 1.36 (m, 6H).