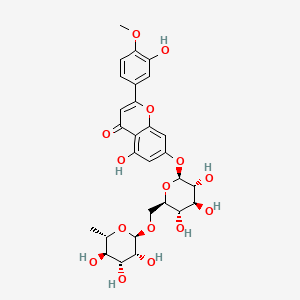
DIOSMIN
- Molecular FormulaC28H32O15
- Average mass608.545 Da
5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-7-{[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-({[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxy}methyl)oxan-2-yl]oxy}-4H-chromen-4-one
3′,5,7-Trihydroxy-4′-methoxyflavone-7-rutinoside
4H-1-Benzopyran-4-one, 7-((6-O-(6-deoxy-α-L-mannopyranosyl)-β- D-glucopyranosyl)oxy)-5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-
4H-1-Benzopyran-4-one, 7-((6-O-(6-deoxy-α-L-mannopyranosyl)-β-D-glucopyranosyl)oxy)-5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-
4H-1-Benzopyran-4-one, 7-[[6-O-(6-deoxy-α-L-mannopyranosyl)-β-D-glucopyranosyl]oxy]-5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-
5-Hydroxy-2-(3-hydroxy-4-methoxyphenyl)-4-oxo-4H-chromen-7-yl 6-O-(6-deoxy-α-L-mannopyranosyl)-β-D-glucopyranoside
Barosmin / Dalfon (Servier) / Detralex / Diosven / Dioven / Diovenor / Flebosmil / Flebosten / Hemerven / Insuven / Litosmil / Varinon / Ven-detrex / Venosmine, Barosmin, Diosmine, Venosmine, Diosmil,
Diosmin is a bioflavonoid that strengthens vascular walls.
Diosmin is a semisynthetic drug indicated for the treatment of venous disease. Diosmin is a flavone that can be found in the plant Teucrium gnaphalodes. Diosmin is available as a prescription medicine in several European countries, and is available as a nutritional supplement in the United States and the rest of Europe. It should be noted that clinical studies have been inconclusive and no articles have been published pertaining to its use in the treatment of vascular disease. When used in rats, diosmin has been effective at mitigating hyperglycaemia, and may also have antineurodegenerative properties.
Diosmin is a flavone, a member of the flavonoid family. Diosmin aglycone is diosmetin. It can be found in Teucrium gnaphalodes, a plant endemic to the Iberian Peninsula.[1]


IR KBR





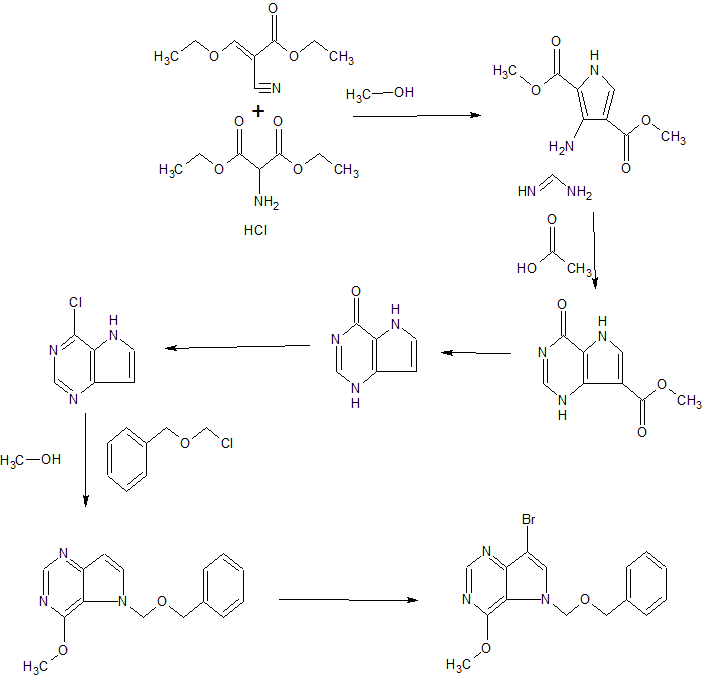
SYNTHESIS

Drug
Diosmin is a semisynthetic flavonoid molecule derived from citrus d (modified hesperidin). It is an oral phlebotropic drug used in the treatment of venous disease, i.e., chronic venous insufficiency (CVI) including spider and varicose veins, leg swelling (edema), stasis dermatitis and venous ulcers. It is also used as a stand-alone or surgical adjunctive therapy in hemorrhoidal disease (HD).
There are extensive clinical trials that show diosmin improves all stages of venous disease including venous ulcers and improves quality of life.[2] There are no prospective studies in arterial disease.
Diosmin is currently a prescription medication in some European countries (under the Dio-PP, Venotec, Daflon etc. tradenames), and is sold as a nutritional supplement in the United States.
Diosmin has been found to be effective in mitigating hyperglycemia in diabetic rats.[3] It is also speculated that diosmin might have potential in the treatment of neurodegenerative diseases,[4] such as Alzheimer’s disease.
Mechanisms
Diosmin improves lymphatic drainage by increasing the frequency and intensity of lymphatic contractions, and by increasing the total number of functional lymphatic capillaries. Furthermore, diosmin with hesperidine decreases the diameter of lymphatic capillaries and the intralymphatic pressure.Diosmin prolongs the vasoconstrictor effect of norepinephrine on the vein wall, increasing venous tone, and therefore reducing venous capacitance, distensibility, and stasis. This increases the venous return and reduces venous hyperpressure present in patients suffering from CVI.
At the microcirculation level, diosmin reduces capillary hyperpermeability and increases capillary resistance by protecting the microcirculation from damaging processes.
Diosmin reduces the expression of endothelial adhesion molecules (ICAM1, VCAM1), and inhibits the adhesion, migration, and activation of leukocytes at the capillary level. This leads to a reduction in the release of inflammatory mediators, principally oxygen free radicals and prostaglandins (PGE2, PGF2a).
Society and culture
Diosmin is distributed in the U.S. as a dietary supplement.[5][6]
Diosmin was first reported by O. A. Osterle and G. Wander in HeIv. Chim. Acta. 8, 519 – 536, 1925 and is a naturally occurring flavonoid glycoside that can be isolated from various plant sources, i.e from the peel of the citrus fruit or hesperidin. Diosmin is a protecting agent and is used for the treatment of chronic venous insufficiency, lymphedema, hemorrhoids and varicose veins. It has been also used for other therapeutic purposes such as cancer, premenstrual syndrome, colitis, and diabetes.
The several references are reported in the prior art for conversion of hesperidin to diosmin.
Zemplen and Bogner, in Ber. 76, 452, 1943 reported monobromination of acetylated flavanones by liquid bromine in chloroform solution in presence of ultraviolet radiation to obtain flavone derivative by following loss of hydrogen bromide and deacetylation with alcoholic alkali. The conversion of hesperidin to diosmin reported is 37%.
In the journal reference, J. Org. Chem., 16, 930 – 933, 1951, by N. B. Lorette et. al. N-bromosuccinimide was used for the bromination of acetylated hesperidin in chloroform and benzoyl peroxide was used as a catalyst. Diosmin yield was 44%.
Studies in Organic Chemistry (Amsterdam) (1982), Volume Date 1981, 11, 115-119 describes conversion of Hesperidin, neohesperidin and naringin to diosmin, neodiosmin, and rhoifolin respectively by dehydrogenation with iodine in pyridine. Tianran Chanwu Yabjiu Yu Kaif (2006), 18(6), 896-899 describes separation and purification of diosmin by macroporous resins, and reported 95% pure diosmin.
ES459076 describes the preparation of diosmin by bromination and debromination of hesperidin acetate in tetrahydrofuran with 2-carboxy ethyl triphenyl phosphonium bromide followed by saponification with potassium tertiary butoxide.
ES465156 describes diosmin preparation by reaction of hesperidin with aqueous sodium hydroxide, iodine and pyridine with 66% yield.
DE2740950 describes iodination-dehydroiodination of hesperidin in the presence of pyridine and iodine resulting 89% of diosmin.
EP52086 claims a process for the preparation of diosmin comprising of total acetylation of hesperidin or related flavone by heating it in acetic anhydride and pyridine followed by selective dehydrogenation or oxidation by means of SeO2 in isoamyl alcohol and then deprotection by means of alkaline hydrolysis with inorganic bases under hot condition. The isolated diosmin is purified by base acid treatment with overall reported yield is of 60%.
US4078137 describes a process for diosmin comprising of acetylation of hesperidin, thereby brominating it and the brominated product is hydrolysed to isolate diosmin with bromine content less then 0.1% with over all 65% yield.
In BE 904614, diosmin was prepared by iodination of hesperidin followed by elimination of HI. In the process, iodine in dimethylformamide and pyridine were successively added to hesperidin and the resulting mixture was heated at 100°C to give 96% pure diosmin.
EP 860443 describes the process that involves reaction of hesperidin with iodine in presence of pyridine at reflux temperature for 5 hours. The reaction mixture is cooled to 5°C and the isolated diosmin is purified using base acid treatment to get the quality of diosmin above 90% with 75 % yield.
FR2760015 provides industrial dehydrogenation of hesperidin with potassium iodide in DMSO in presence of cone. H2SO4 resulted in diosmin with 73% yield and pharmacopoeial quality.
WO2000011009 describes reaction of hesperidin with iodine in presence of pyridine and anhydrous alkaline earth metal base. The process involves purifying the reaction mass using morpholine followed by base acid treatment which resulted in diosmin with 80% yield and purity of diosmin meets with pharmacopoeial norms.
EP 1086953 discloses the process for purification of diosmin by reacting with pulverized zinc in aqueous solution followed by filtration and acidification.
Diosmin which is produced by many of the prior art processes is often found to contain impurities and is contaminated with various byproducts, for instance hesperidin, Isorhoifin, acetyl lisovanilone, 6-Iododiosmin, linarin, diosmetin and other organic volatile impurities. Some of the major impurities are resulted from hesperidin during extraction. The impurities of hesperidin have a major effect on the final assay of diosmin. The impurities vary depending upon the source of hesperidin. It is worthy to note that direct crystallization of crude diosmin with aqueous base acid solution does not necessarily improve the assay / purity of diosmin.
The process described in Studies in Organic Chemistry (Amsterdam) (1982), Volume Date 1981, 11, 115-119 is different from the present inventors process.
Although ES459076 teaches the preparation of diosmin by bromination and debromination of hesperidin acetate in tetrahydrofuran with 2-carboxy ethyl triphenyl phosphonium bromide, it does not teach about the final purity of diosmin with pharmaceutical quality as required.
ES465156 and DE2740950 although disclose method of preparation, does not teach a process that gives yields as are taught by present invention.
EP52086 and US4078137 uses acetic anhydride for acetylation of hesperidine with yields around 60%, which are phenomenally less as compared with yields of process described by present invention.
Sequence of addition of reactants in the process as taught by BE 904614 is different than the teachings of the present invention.
FR2760015 teaches use of different reactants under conditions that are different from the teachings of the present invention.
Invention disclosed in present application does not use morpholine as disclosed in WO2000011009.
Though there are reported several processes for preparation of diosmin in the prior art, present invention describes a novel systematic process for the preparation of diosmin by converting hesperidin to diosmin at optimum level i.e. % conversion, and keeping the impurities at minimum level which results in consistently pure diosmin with good yield and the desired quality. The process allows recovery and recycle of major contributing chemicals and solvents such as methanol, pyridine and iodine, without impact on quality, purity or yield of the process making the process more economical and ecofriendly. It is surprisingly found that quality diosmin output obtained is independent of hesperidin quality used

Title: Diosmin
CAS Registry Number: 520-27-4
CAS Name: 7-[[6-O-(6-Deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-4H-1-benzopyran-4-one
Additional Names: 3¢,5,7-trihydroxy-4¢-methoxyflavone-7-rutinoside; 5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-7-(O6-a-L-rhamnopyranosyl-b-D-glucopyranosyloxy)chromen-4-one; 5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-7-b-rutinosyloxy-4H-chromen-4-one; diosmetin 7-b-rutinoside; barosmin; buchu resin
Trademarks: Diosmil (Bellon); Diosven (CT); Diovenor (Innotha); Flebosmil (Bouchara); Flebosten (Bonomelli); Hemerven (Interdelta); Insuven (Berenguer); Litosmil (Evans); Tovene (Kali-Chemie); Varinon (Exa); Ven-Detrex (Zyma); Venosmine (Geymonat)
Molecular Formula: C28H32O15
Molecular Weight: 608.54
Percent Composition: C 55.26%, H 5.30%, O 39.44%
Literature References: Naturally occurring flavonic glycoside; rhamnoglycoside of diosmetin, q.v. Isolation from various plant sources: O. A. Oesterle, G. Wander, Helv. Chim. Acta 8, 519 (1925). Elucidation of structure: G. Zemplén, R. Bognár, Ber. 76, 452 (1943). Prepn from hesperidin, q.v.: eidem, ibid.; N. B. Lorette et al., J. Org. Chem. 16, 930 (1951). Isoln from lemon peel (Citrus limon Linn. Rutaceae): R. M. Horowitz, J. Org. Chem. 21, 1184 (1956); from Zanthoxylum avicennae, Rutaceae: H. R. Arthur et al.,J. Chem. Soc. 1956, 632; H. R. Arthur et al., ibid. 1959, 4007; from flowers of Sophora microphylla Ait. Leguminosae: L. H. Briggs et al., ibid. 1960, 1955. Toxicology studies: H. Heusser, W. Osswald, Arch. Farmacol. Toxicol. 3, 33 (1977). NMR spectrum: J. L. Nieto, A. M. Gutierrez, Spectrosc. Lett. 19, 427 (1986). Mechanism of action: C. Boudet, L. Peyrin, Arch. Int. Pharmacodyn. 283,312 (1986). Pharmacology: J. R. Caseley-Smith, J. R. Caseley-Smith, Agents Actions 17, 1 (1985); M. Damon et al., Arzneim.-Forsch. 37, 1149 (1987). HPLC determn in biological fluids: D. Baylocq et al., Ann. Pharm. Fr. 41, 115 (1983). Clinical study in post-phlebitic ulcers: M. C. Nguyen, K. Morere, Gaz. Med. 92, 71 (1985); in acute hemorrhoids: A. Tajana et al., Minerva Med. 79,387 (1988). Clinical trial in chronic venous insufficiency: R. Laurent et al., Int. Angiol. 7, Suppl. 2, 39 (1988).
Derivative Type: Monohydrate
Molecular Formula: C28H32O15.H2O
Molecular Weight: 626.56
Percent Composition: C 53.67%, H 5.47%, O 40.86%
Properties: mp 275-277° (dec) (Zemplén). Also reported as fine needles from aq pyridine or aq DMF, mp 283° (dec) (Briggs). uv max (ethanol): 255, 268, 345 nm (log e 4.28, 4.25, 4.30). Practically insol in water, alcohol.
Melting point: mp 275-277° (dec) (Zemplén); mp 283° (dec) (Briggs)
Absorption maximum: uv max (ethanol): 255, 268, 345 nm (log e 4.28, 4.25, 4.30)
Derivative Type: Flavonoid extract
Trademarks: Daflon (Servier); Flebopex (Profarma); Flebotropin (Bago)
Therap-Cat: Capillary protectant.
Keywords: Vasoprotectant.
PAPER
Lee, Sanghyun; Natural Product Sciences 2002, VOL 8(4), P127-128
Siciliano, Tiziana; Journal of Agricultural and Food Chemistry 2004, VOL 52(21), P6510-6515
Yin, Feng; Zhongguo Tianran Yaowu 2004, VOL 2(3), P149-151
Markovic, D.; Farm. Glasnik 1949, VOL 5(No. 7;No. 8), P135-48;153-62
Nakaoki, Tahitiro; Yakugaku Zasshi 1938, VOL 58, P639-47(in German 197-201)
Wander, G.; Pharmaceutical Journal 1925, VOL 115, P520
Morita, Naokata; Yakugaku Zasshi 1967, VOL 87(3), P319-20
“PhysProp” data were obtained from Syracuse Research Corporation of Syracuse, New York (US)
Narasimhachari, N.; Proceedings – Indian Academy of Sciences, Section A 1949, VOL 30A, P151-62
Horowitz, Robert M.; Journal of Organic Chemistry 1956, VOL 21, P1184-5
paper
Spectroscopy Letters , An International Journal for Rapid Communication , Volume 19, 1986 – Issue 5, 1H NMR Spectra at 360 MHz of Diosmin and Hesperidin in DMSO Solution
PAPER
Journal of Natural Products, 2013, vol. 76, 1, pg. 8 – 12
https://pubs.acs.org/doi/suppl/10.1021/np300460a/suppl_file/np300460a_si_001.pdf
1 H NMR, 13C NMR, HMQC and HMBC spectra of diosmin (5) ……………….S2




PAPER
Journal of Molecular Liquids, 2014, vol. 199, pg. 35 – 41

PATENT
https://patents.google.com/patent/CN102653549A/en
BRIEF DESCRIPTION
[0012] BRIEF I: iodine purification process Figure 2: Synthesis of diosmin roadmap
detailed description
[0013] Main reaction: The 80Kg Hesperidin, 40Kg soda ash, 400kg90% ethanol, 80L pyridine, 24kg iodine successively into reactor closed good pot opening, with stirring and heated to 110 ° c with a microwave, heat stirring, until the orange leather glycosides completely dissolved, about ten minutes. Hesperidin is completely dissolved, the solvent was slowly added to 80L of pyridine, combined with sodium iodide 8Kg, heated to 110 ° C, the reaction was stirred for 3-4 hr incubation, the sample is then detected by HPLC detection method, when a peak area less than hesperidin the reaction was terminated when 5% diosmin peak area, heat recovery of the solvent pyridine.
[0014] The filter press: End recovered 25Kg pyridine was added a paste of methanol, was press iodine recycling of waste, the recovery is completed, washed with purified water of 62 ° C, colorless and transparent until the washing water to the water 3 t, remove the filter cake to afford crude diosmin, 125. 4Kg.
[0015] Purification: 16Kg sodium hydroxide into dissolving tank, add purified water 500Kg, dissolved under stirring, and after dissolution the crude into the tank, and the water plus t I stir crystals were filtered into a stainless steel frame filter kettle, adding 42Kg hydrochloric acid, adjusted to PH 6.7, 25Kg of methanol was added, after stirring for 30min, the precipitate was allowed to stand, the I h.
[0016] Washing: The crystalline material tank into the filter press, ere washed with purified water, the washing water to colorless far, four tons of water, remove the filter cake to afford fine diosmin, 118. 5Kg. [0017] The dried, pulverized, mixed: semi-finished products into the oven dried 11.2 hours, 82 ° C temperature conditions, the dried material was crushed with a grinder, then put double cone blender and mixed overall speed 15r / min, each of the positive and negative inversion 20min, diosmin have finished 72Kg, a yield of 90.0%.
[0018] Packaging: for medical packaging with double polyethylene bags, into the drum after passing inspection, into finished products.
[0019] The recovery of iodine: iodine-containing filtrate generated pressure filtration step was slowly added sulfuric acid to adjust the PH 4, left for 5 hours, vacuum distillation, collecting high-boiling fraction, 20Kg hydrogen peroxide was slowly added, stand for 2 hours, filtered, the recovered iodine cloth, can be re-purified to obtain purified iodine! .
[0020] Processing pyridine in water: pyridine pyridine recovered after 400Kg containing moisture added to the kettle, 35Kg of potassium hydroxide was added, heated to 105 ± 5 ° C, collecting it pyridine (105 ° C before the liquid front , is defective, back again into the reaction vessel, then 105 ° C out is a good product), Hugh moisture meter by Karl Fischer detected, less than 2%.
PATENT
https://patents.google.com/patent/CN102875621A/en
diosmin chemical name is 3 ‘, 5,7-trihydroxy-4’ – methoxy flavone, i.e. (7 – {[6-0- (6-deoxy-mannose -a -L- sugar) _β -D- glucopyranosyl] oxy} -5_ hydroxy-2- (3-hydroxy-_4_ methoxyphenoxy) -4H-L–benzopyran-4-one), the following structure Figure:
[0003]
[0004] Diosmin has a comprehensive effect on vascular transfusion system to the venous system, micro-circulatory system and the lymphatic system has a powerful effect. Diosmin can be significantly reduced in addition to the adhesion of leukocytes to vascular endothelial cells, migration, inhibition of leukocyte disintegration and release of inflammatory mediators such as histamine, bradykinin, complement, leukotrienes, prostaglandins, free radical scavenging and the like, It may also reduce blood viscosity, to enhance flow of red blood cells, thus reducing the microcirculation stasis, mainly used in clinical treatment of chronic venous insufficiency.
[0005] diosmin content in natural plant is very low, direct extraction of high cost, so it is through the oxygen
Hesperidin is prepared by chemical synthesis; hesperidin formula below:
[0006]
[0007] diosmin synthesis process generally as follows:
[0008] hesperidin and an oxidant, and a solvent after mixing an alkaline reagent can be synthesized by heating the reaction Diosmin; wherein said oxidizing agent is iodine, mainly basic agent mainly inorganic bases, typically hydrogen sodium hydroxide, potassium hydroxide, sodium carbonate or potassium carbonate, an alkaline substance, etc., the solvent is pyridine or dimethylformamide.
[0009] Preparation of diosmin conventional synthesis method will inevitably pyridine or dimethylformamide as the reaction solvent, in particular in the main pyridine; as pyridine, dimethylformamide as a reaction solvent after the treatment process is not easy divisible, thus resulting in higher residual solvent in the product; the same time, since the two types of the organic solvent is pyridine, a large irritating odor on the human body have a greater toxic effects, and therefore in the production process and on the environment endanger personnel more apparent.
Example 8
[0081] In addition hesperidin 1000L reaction vessel 100 g, 47 g of sodium hydroxide and 12 g of iodine, and finally adding morpholine: water (60: 40) mixed solvent O. 8 liters, stir until completely dissolved. after heating to 85-90 ° C. was stirred incubated for 9 hours. the reaction liquid becomes viscous liquid was added 3 g of sodium thiosulfate, recovered at 85-90 ° C under vacuum conditions to a 70-80% morpholinyl, after complete recovery morpholine, O. 8 liters of water was added, stirred uniformly filtered to collect the waste. washed with water to give diosmin crude product. the crude product diosmin O. 8-liter and water was added 30 g of sodium hydroxide. stirred to dissolve completely after high-speed centrifuge filtration. into the crystallizer, water was added to the filtrate I. 5 liters of sulfuric acid was added slowly acidified to PH 2-3. standing, filtered and washed with water. diosmin give crude crystals. The crude crystals add water O. 8-liter and 30 g of sodium hydroxide and stirred to dissolve completely, placed in a crystallizer tank, to force saliva I. 5 liters of sulfuric acid was added slowly acidified to PH 2-3. Standing, was filtered, washed with water the drying, grinding to give the finished Diosmin 80. I g. Product purity by HPLC 95.26%, the yield was 80.1%, iodine residual, residual solvent, associated impurities, the content of all standards
PATENT
https://patents.google.com/patent/WO2010092592A2/en
Example – 1
100 gm of hesperidin , 700 ml of pyridine, 9.8 gm of sodium hydroxide and 45.6 gm of iodine were charged in 2 liter clean glass assembly, The resulting solution was heated to 95-1050C for 9 – 10 hours. Reaction was monitored by HPLC to get hesperidin less than 1 %. The pyridine was recovered completely by distillation. Charged methanol to the resulting solid, the reaction mass was heated to reflux and filtered at room temperature. Iodine was recovered from mother liquor, solid obtained was treated with sodium thiosulfate solution and 900 ml, 5% aqueous NaOH solution. pH 2-4 was adjusted with cone, sulfuric acid. Reaction mass was filtered to obtain crude diosmin. Yield : 80 – 86 gm. Recovery of iodine from above methanol mother liquor: Distilled methanol and pyridine mixture. The obtained residue was acidified with sulfuric acid. The resulting pH was less than 1. The brown precipitate formed was filtered. The resulting filtrate was oxidized with hydrogen peroxide at 0-10°C and filtered to obtain crude iodine having assay 50 – 60 %, which was steam distilled to obtain pure iodine with assay 95 %.
Example – 2
100 gm of crude diosmin as prepared in example 1 and 1800 ml of dimethylformamide was charged in 3 liter clean glass assembly. The resulting mass was heated to 90-950C to obtain clear solution. 200 ml of water was added at 90- 950C and maintained for 30 min. The reaction mass was cooled and filtered. The wet solid was collected.
Charged wet solid obtained in 3 liter clean glass assembly and charged 900 ml of water, 900 ml of 5% aqueous NaOH solution. Distilled out approximately 900 ml of water under vacuum below 5O0C. Charged 1000 ml of water and the resulting reaction mass was treated with charcoal and filtered through hyflow. pH 1.8-2.2 was adjusted using sulfuric acid. Stirred the mass for 30 min, filtered and washed it with water, hot water. Solid was dried. Yield : 80 – 85 gm. Assay : 99.9 %.
Example – 3
100 gm of crude diosmin as prepared in example 1, 1800 ml of dimethylformamide and 1800 ml of water was charged in 5 liter clean glass assembly. The resulting solution was heated to 90-950C to obtain slurry and maintained for 30 min. Cooled the reaction mass and filtered, washed with water and hot water. The obtained solid was dried. Yield: 90 – 95 gm. Assay : 97 %. Example – 4
100 gm of crude diosmin as prepared in example 1 and 1800 ml of dimethylformamide was charged in 3 liter clean glass assembly. The resulting solution was heated to 90-950C to obtain clear solution.charged 360 ml of water at 90-950C and maintained for 30 min. The reaction mass was filtered, washed with water and with hot water. Solid obtained was dried. Yield: 90 – 95 gm. Assay : 99.5 %.
Example – 5
100 gm of crude diosmin as prepared in example 1 and 1800 ml of dimethylformamide was charged in 3 liter clean glass assembly. The resulting solution was heated to 90-950C to obtain clear solution, charged 900 ml of water at 90-950C and maintained for 30 min. The reaction mass was filtered, washed with water and with hot water. Solid obtained was dried. Solid obtained was 90 — 95 gm. Assay obtained was 98.8 %.
Example – 6
100 gm of hesperidin , 700 ml of recovered pyridine, 9.8 gm of sodium hydroxide and 45.6 gm of iodine were charged in 2 liter clean glass assembly . The resulting solution was heated to 95-1050C for 9 – 10 hrs. Reaction was monitored by HPLC. Pyridine was recovered completely by distillation. Charged methanol to the resulting solid, the reaction mass was heated to reflux and filtered at room temperature. The solid obtained was treated with sodium thiosulfate solution and 900 ml, 5% aqueous NaOH solution. pH 2-4 was adjusted with cone, sulfuric acid. Reaction mass was filtered to obtain crude diosmin. Crude diosmin obtained was 80 – 86 gm. Purity was 98.6 %.
Example – 7
100 gm of hesperidin , 700 ml of recovered Pyridine, 9.8 gm of sodium hydroxide and 48 gm (assay 95 %) of recovered iodine were charged in 2 liter clean glass assembly.. The resulting solution was heated to 95-1050C for 9 – 10 hours. Reaction was monitored by HPLC to get hesperidin less than 1 %. The pyridine was recovered by distillation. Charged methanol to the resulting solid, the reaction mass was heated to reflux and filtered at room temperature. The solid obtained was treated with sodium thiosulfate solution and 900 ml, 5% aqueous NaOH solution. pH 2-4 was adjusted with cone, sulfuric acid. Reaction mass was filtered to obtain crude diosmin. Yield:80 – 86 gm. Purity : 95.3 %.
PATENT
WO-2018039923
Method for preparing diosminum comprising the steps of mixing amide solvent, hesperidin, alkaline reagent and iodine, and heating the reaction to obtain diosmin . Diosmin is a naturally occurring flavonoid glycoside that can be obtained from various plant sources. It is used in therapy due to its pharmacological activity as phlebotonic and vascular protecting agent, and useful for treating chronic venous insufficiency.
1.1 Oxidation reaction: Open the vacuum pump, vacuum inhale 1500L of dimethylformamide into the reaction tank, and add sodium hydroxide to adjust the pH of the solvent between 6 and 7. Add 250.00kg of hesperidin and stir it while feeding. The material and the solvent are in full contact; 125 kg of iodine is added into the reaction tank at a constant rate for reaction; the temperature of the reaction tank is controlled between 70° C. and 100° C. for 14 hours.
1.2 Solvent recovery: After the reaction is complete, open the valve of the turnover tank and dehydration tank and close the return valve. Control the temperature of the material to start depressurizing the solvent at 90°C to 110°C. During the recovery process, attention should be paid to observe the recovery temperature and recovery conditions. When the material is dilute, stop the solvent recovery and enter the next process.
1.3 Crude product crystallization, filtration, washing: open the reaction tank vacuum valve, pump 1500L of purified water from the upper part of the reaction tank, start the mixer, stir for 10-20min, then add 1500L of purified water and stir for 1h; after the mixing is accepted, when the temperature of the crystallization liquid drops After 35°C, the crystallization liquid is pumped into the plate and frame filter press for filtration, and the filtrate is temporarily stored in the storage tank; after the filtered plate nozzle no liquid flows out, the 18000L purified water pump is pumped into the frame filter press for washing; The water wash is discharged into a waterless collection tank for sewage treatment, and the water is washed until the pH of the effluent is measured with a pH test paper of 6 to 7. The beaker sample is observed to be colorless and transparent; after the washing is completed, the water inlet valve is closed and the air pressure is turned on. The valve is air-pressed, the air pressure is controlled at 0.07~0.09 MPa, and the time is maintained for 3 hours. The filter cake is collected and put into the turnover barrel for marking.
[0051]
1.4 Secondary Dissolution and Filtration: Open the dissolving tank and stir. In the dissolving tank, add 75 kg of alkali A to 1300 L of purified water. After the solution is completely dissolved, add the filter cake in the circulating drum to the tank; after the filter cake is added and stirring is continued for 1 h, Add 1300L of purified water into the tank, stir for 30 minutes, and stand for 3 hours. Filter the filtrate with a frame filter press. The filtered solution is finely filtered by a fine filter and then pumped into a clean area crystallizer.
[0052]
1.5 secondary crystallization: open the stirrer, slowly put 160 ~ 230kg 36% hydrochloric acid into the clean area crystallization tank, the measured pH of the solution is between 5.0 ~ 6.0, after stirring 20min measured the pH of the solution should be stable and qualified , Stir 1h, make the original record of the process.
1.6 Fine filtration and washing: The crystallization liquid is vacuum-inhaled into the box type filter press for filtration; after the filtration is completed, the washed water is washed with 18,000 to 20,000 L of purified water. After the washing is completed, it is checked that the washing liquid should be colorless and transparent, and the filtrate should be filtrated. Discharge into the sewage treatment system.
1.7 Fine Drying: Open the hot air circulation oven, control the temperature at 100 °C ~ 130 °C, drying time is maintained at 10 ~ 16h; when dried 10h, timely sampling, with a quick moisture meter to determine the moisture, when the sample moisture is less than 5%, Stop drying; after passing the drying, close the oven and transport the material to the next process.
1.8 Fine-grinding: The dried product is crushed with a crusher. The crushing sieve is 80 mesh to obtain Diosmin.
The quality standards of all raw materials in Example 1 are shown in Table 1.
Table 1 Raw material quality standards
[Table 0001]
| Original accessories name |
specification |
Quality Standard |
| Hesperidin |
EC |
In line with “Hesperidin Quality Standard” |
| New solvent |
Industrial grade |
Meet the “new solvent quality standards” |
| Alkali A |
Industrial grade |
In accordance with the “Alkaline A Quality Standard” |
| iodine |
Pharmaceutical grade |
In line with “Iodine Quality Standards” |
| hydrochloric acid |
Analytical purity |
In line with the “Hydrochloric Acid Quality Standard” |
Diosmin Yield Calculation Method:
Diosmin product yield = (Diosmin Fine Quality) / (Hesperidin weight) × 100%
References
- Jump up^ Flavonoid Aglycones and Glycosides from Teucrium gnaphalodes. F. A. T. Barberán, M. I. Gil, F. Tomás, F. Ferreres and A. Arques, J. Nat. Prod., 1985, 48 (5), pages 859–860, doi:10.1021/np50041a040
- Jump up^ Jantet, G. (2002-06-01). “Chronic venous insufficiency: worldwide results of the RELIEF study. Reflux assEssment and quaLity of lIfe improvEment with micronized Flavonoids”. Angiology. 53(3): 245–256. ISSN 0003-3197. PMID 12025911.
- Jump up^ Leelavinothan Pari, Subramani Srinivasan, Antihyperglycemic effect of diosmin on hepatic key enzymes of carbohydrate metabolism in streptozotocin-nicotinamide-induced diabetic rats, Biomedicine & Pharmacotherapy, Volume 64, Issue 7, September 2010, Pages 477-481.
- Jump up^ Sirlak, Mustafa; Akar, A. Ruchan; Eryilmaz, Sadik; Cetinkanat, Elif Kuzgun; Ozcinar, Evren; Kaya, Bulent; Elhan, Atilla Halil; Ozyurda, Umit (2010-01-01). “Micronized purified flavonoid fraction in pretreating CABG patients”. Texas Heart Institute Journal. 37 (2): 172–177. ISSN 1526-6702. PMC 2851420
 . PMID 20401289.
. PMID 20401289.
- Jump up^ “Nutratech dietary supplement notification” (PDF). FDA. November 3, 2000. Archived from the original (PDF) on December 9, 2006.
- Jump up^ “Stragen Pharma dietary supplement notification” (PDF). FDA. September 6, 2005.
Publication numberPriority datePublication dateAssigneeTitle
DE2740950A1 *1977-09-101979-03-22Merck Patent GmbhA process for the production of flavones
EP0860443A1 *1997-02-211998-08-26Innokem, SARLIndustrial process for the production of diosmine starting from hesperidine
WO2000011009A2 *1998-08-192000-03-02Innokem, S.A.R.L.Method for industrial production of diosmin from hesperidin by reaction with iodine and pyridine
CN102070689A *2011-01-252011-05-25湖南圆通药业有限公司Method for producing diosmin
CN102653549A *2011-12-282012-09-05长沙富能生物技术有限公司Synthesis method of diosmin raw medicine meeting EP7 version quality standards
CN102875621A *2012-10-262013-01-16成都澜绮制药有限公司Synthesis method of diosmin
RU2481353C1 *2011-12-222013-05-10Закрытое акционерное общество “Активный Компонент”Commercial method for preparing officinal diosmin and crystalline form thereof (versions)
CN103772336A *2014-02-232014-05-07闻永举Semi-synthesis method of phenolic hydroxyl flavonoid compounds and iodine recycling method
CN103435666A *2013-07-302013-12-11李玉山Novel production technology of diosmin
CN105732744A *2016-04-292016-07-06南京正大天晴制药有限公司Method for preparing green and economic diosmin
Publication numberPriority datePublication dateAssigneeTitle
DE2740950A1 *1977-09-101979-03-22Merck Patent GmbhA process for the production of flavones
EP1086953A1 *1999-09-242001-03-28IsochemProcess for the purification of diosmine
WO2010092592A2 *2009-02-112010-08-19Elder Pharmaceuticals Ltd.Process for the preparation of diosmin
CN102653549A *2011-12-282012-09-05长沙富能生物技术有限公司Synthesis method of diosmin raw medicine meeting EP7 version quality standards
//////////DIOSMIN, диосмин , ديوسمين , 地奥司明 , Barosmin, Diosmine, Venosmine, Diosmil,
COC1=C(O)C=C(C=C1)C1=CC(=O)C2=C(O)C=C(O[C@@H]3O[C@H](CO[C@@H]4O[C@@H](C)[C@H](O)[C@@H](O)[C@H]4O)[C@@H](O)[C@H](O)[C@H]3O)C=C2O1

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






















































")