
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Motesanib (AMG-706)

Motesanib (AMG-706)
Amgen Inc.

Motesanib (AMG 706) is an experimental drug candidate originally developed by Amgen[1] but is now being investigated by theTakeda Pharmaceutical Company. It is an orally administered small molecule belonging to angiokinase inhibitor class which acts as an antagonist of VEGF receptors, platelet-derived growth factor receptors, and stem cell factor receptors.[2] It is used as thephosphatesalt motesanib diphosphate.
Motesanib, also known as AMG-706, is an orally administered multikinase inhibitor that selectively targets VEGF receptors, platelet-derived growth factor receptors, and Kit receptors.
Clinical trials
Motesanib was originally investigated for effectiveness against advanced nonsquamous non-small-cell lung cancer (NSCLC), withPhase II trials indicating an effectiveness comparable to bevacizumab when they were both used in combination withpaclitaxel/carboplatin.[3] However a later and more detailed Phase III trial failed to show any benefit for the treatment of NSCLC.[2][4]A second Phase III trial was started in 2012,[5] which focused on patients from Asian backgrounds (performed on the bases ofsubgroup analysis)[6] however this also failed to meet its primary endpoint.[7]
The drug has undergone a Phase II evaluation as first-line therapy for breast cancer[2] however this study found no evidence to support further investigation.[8] Phase II testing against persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas was also unsuccessful.[9]
There have also been 2 separate Phase II clinical trials for thyroid cancer which have both shown promising results.[10][11][12]
Developed at Amgen, the compound is also being evaluated as both monotherapy and in combination with other agents in the treatment of breast, colorectal, lung, thyroid and ovarian cancers. Clinical trials for the treatment of bladder cancer have been terminated.
The National Cancer Institute had been evaluating the potential of the drug in patients with low-grade neuroendocrine tumors; however, no recent development has been reported for this research. The FDA awarded fast track status to motesanib in 2004. In 2008, the compound was licensed to Takeda in Japan.

AMG-706 is synthesized as follows: 1-Acetyl-3,3-dimethyl-6-nitroindoline (I) is reduced by catalytic hydrogenation over Pd/C, giving the aminoindoline (II), which is then coupled with 2-chloronicotinoyl chloride (III) in the presence of DIEA to yield the corresponding nicotinamide (IV). Subsequent condensation of (IV) with neat 4-(aminomethyl)pyridine (V) at 120 °C affords the 2-aminonicotinamide derivative (VI). The N-acetyl group of (VI) is finally removed by acidic hydrolysis to furnish the title compound (1,2).
,………………………………………
US 2003125339
http://www.google.com/patents/US20030125339
………………………………………………….
US 2003225106
https://www.google.com/patents/US20030225106
EXAMPLE 133
[2295]

N-(3,3-Dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
Step A—Preparation of 1-acetyl-6-amino-3,3-dimethylindoline
1-Acetyl-3,3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL), the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc:CH2Cl2 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O—204.27.
Step B—Preparation of N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from 1-acetyl-6-amino-3,3-dimethylindoline (Step A) by the method described in Example 82.
Step C—Preparation of N-(3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N5O—373.45.
…………………….
http://www.google.com/patents/WO2012063085A3?cl=en
Example 133

N- (3, 3-Dimethy1indolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) }carboxamide Step A – Preparation of l-acetyl-6-amino-3 , 3- dimethylindoline l-Acetyl-3 , 3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL) , the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc :CH2C12 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O-204.27.
Step B – Preparation of N-(l-acetyl- 3 , 3-dimethylindolin-6- yl) (2-[ (4-pyridylmethyl) amino] (3-pyridyl) } carboxamide The titled compound was prepared from l-acetyl-6- amino-3 , 3-dimethylindoline (Step A) by the method described in Example 82.
Step C – Preparation of N- (3 , 3-dimethylindolin-6-yl) {2- [ (4- pyridylmethyl) amino] (3-pyridyl) }carboxamide
The titled compound was prepared from N-(l-acetyl- 3 , 3-dimethylindolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) } carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N50-373.45.
References
- Stafford, edited by Rongshi Li, Jeffrey A. (2009). “Chapter 5. Discovery of Motesanib”. Kinase inhibitor drugs. Hoboken, N.J.: Wiley. pp. 113–130. ISBN 978-0-470-27829-1.
- “Amgen and Takeda’s NSCLC Drug Fails in Phase III Study”. 30 Mar 2011.
- Blumenschein Jr, G. R.; Kabbinavar, F.; Menon, H.; Mok, T. S. K.; Stephenson, J.; Beck, J. T.; Lakshmaiah, K.; Reckamp, K.; Hei, Y.- J.; Kracht, K.; Sun, Y.- N.; Sikorski, R.; Schwartzberg, L. (14 February 2011). “A phase II, multicenter, open-label randomized study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced nonsquamous non-small-cell lung cancer”. Annals of Oncology 22 (9): 2057–2067. doi:10.1093/annonc/mdq731.
- Jump up^ Scagliotti, G. V.; Vynnychenko, I.; Park, K.; Ichinose, Y.; Kubota, K.; Blackhall, F.; Pirker, R.; Galiulin, R.; Ciuleanu, T.-E.; Sydorenko, O.; Dediu, M.; Papai-Szekely, Z.; Banaclocha, N. M.; McCoy, S.; Yao, B.; Hei, Y.-j.; Galimi, F.; Spigel, D. R. (2 July 2012). “International, Randomized, Placebo-Controlled, Double-Blind Phase III Study of Motesanib Plus Carboplatin/Paclitaxel in Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer: MONET1”. Journal of Clinical Oncology 30 (23): 2829–2836. doi:10.1200/JCO.2011.41.4987. PMID 22753922.
- “Takeda Initiates Phase 3 Trial of Motesanib in Japan and Additional Asian Countries”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J. H.; Shinkai, T.; Takeda, K.; Kim, S.- W.; Hsia, T.- C.; Li, R. K.; Tiangco, B. J.; Yau, S.; Lim, W.- T.; Yao, B.; Hei, Y.- J.; Park, K. (13 January 2014). “Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis”.Annals of Oncology 25 (2): 529–536. doi:10.1093/annonc/mdt552.
- Jump up^ “Takeda Announces Phase 3 MONET-A Study Evaluating Motesanib (AMG 706) in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer Does Not Meet Primary Endpoint”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Martin, Miguel; Roche, Henri; Pinter, Tamas; Crown, John; Kennedy, M John; Provencher, Louise; Priou, Frank; Eiermann, Wolfgang; Adrover, Encarna; Lang, Istvan; Ramos, Manuel; Latreille, Jean; Jagiełło-Gruszfeld, Agnieszka; Pienkowski, Tadeusz; Alba, Emilio; Snyder, Raymond; Almel, Sachin; Rolski, Janusz; Munoz, Montserrat; Moroose, Rebecca; Hurvitz, Sara; Baños, Ana; Adewoye, Henry; Hei, Yong-Jiang; Lindsay, Mary-Ann; Rupin, Matthieu; Cabaribere, David; Lemmerick, Yasmin; Mackey, John R (April 2011). “Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study”. The Lancet Oncology 12 (4): 369–376. doi:10.1016/S1470-2045(11)70037-7. PMID 21429799.
- Schilder, R.J.; Sill, M.W.; Lankes, H.A.; Gold, M.A.; Mannel, R.S.; Modesitt, S.C.; Hanjani, P.; Bonebrake, A.J.; Sood, A.K.; Godwin, A.K.; Hu, W.; Alpaugh, R.K. (April 2013). “A phase II evaluation of motesanib (AMG 706) in the treatment of persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas: A Gynecologic Oncology Group study”. Gynecologic Oncology 129 (1): 86–91. doi:10.1016/j.ygyno.2013.01.006. PMID 23321064.
- Motesanib Diphosphate Provides Anticancer Activity Among Patients with Progressive Thyroid Cancer, CancerConnect.com
- Jump up^ Schlumberger, M. J.; Elisei, R.; Bastholt, L.; Wirth, L. J.; Martins, R. G.; Locati, L. D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.-P.; Eschenberg, M. J.; Sun, Y.-N.; Juan, T.; Stepan, D. E.; Sherman, S. I. (29 June 2009). “Phase II Study of Safety and Efficacy of Motesanib in Patients With Progressive or Symptomatic, Advanced or Metastatic Medullary Thyroid Cancer”.Journal of Clinical Oncology 27 (23): 3794–3801. doi:10.1200/JCO.2008.18.7815. PMID 19564535.
- Sherman, Steven I.; Wirth, Lori J.; Droz, Jean-Pierre; Hofmann, Michael; Bastholt, Lars; Martins, Renato G.; Licitra, Lisa; Eschenberg, Michael J.; Sun, Yu-Nien; Juan, Todd; Stepan, Daniel E.; Schlumberger, Martin J. (3 July 2008). “Motesanib Diphosphate in Progressive Differentiated Thyroid Cancer”. New England Journal of Medicine 359 (1): 31–42.doi:10.1056/NEJMoa075853. PMID 18596272.
External links

Motesanib Diphosphate (AMG-706)
857876-30-3 diphosphate
453562-69-1 (free base)
N-(2,3-Dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide diphosphate
3-Pyridinecarboxamide, N-(2,3-dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-, phosphate (1:2)
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide diphosphate
| 569.4 | |
| Formula | C22H23N5O.2H3PO4 |
|---|
 |
|
| Names | |
|---|---|
| IUPAC name
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-[(pyridin-4-ylmethyl)amino]pyridine-3-carboxamide
|
|
| Other names
AMG 706
|
|
| Identifiers | |
| 453562-69-1 |
|
| ChEMBL | ChEMBL572881 |
| ChemSpider | 9842625 |
| Jmol-3D images | Image |
| PubChem | 11667893 |
| Properties | |
| C22H23N5O | |
| Molar mass | 373.45 g·mol−1 |
Stats today
6.8 lakh views on this blog
…………………..
TAKEDA, JAPAN
![]()

TOKYO HO

Takeda Pharmaceutical CEO Yasuchika Hasegawa
 Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.
Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.

Christophe Weber (L), the new president of Takeda Pharmaceutical Co., and CEO Yasuchika Hasegawa pose

 Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
 OSAKA
OSAKA

Dotonbori, Osaka, Japan
 OSAKA
OSAKA

AZD 3264 an IKK2 Inhibitor from Astra Zeneca

AZD 3264
MW 441.50
CAS 1609281-86-8
Inhibition of IkB-kinase IKK2 has been identified as one of the novel pathways to treat inflammatory conditions such as asthma, chronic pulmonary obstructive disorder (COPD) and rheumatoid arthritis
……………………..
PATENT
WO 2003010158
https://www.google.com/patents/WO2003010158A1?cl=en

The synthesis began with the aromatic nucleophilic substitution reaction of 2-fluorobromobenzene (2) with (S)-N-Boc-3-pyrrolidinol 3 to give the bromo intermediate 4, which was borylated via halogen metal exchange using n-hexLi in THF followed by treatment with triisopropyl borate and acidic work-up to give the boronic acid intermediate 5. Suzuki coupling of the boronic acid 5 with bromothiophene 6(2)afforded the intermediate 7. Intermediate 7 was subjected to regioselective bromination using bromine in acetic acid. This reaction was nonregioselective and yielded 17% of the required isomer 8. The bromo compound 8 was coupled with isoxazole boronate ester 9 by another Suzuki reaction to get the title compound. The overall yield of the synthesis was <6%.


………………………..
PAPER
http://pubs.acs.org/doi/full/10.1021/op500105n

An efficient and scalable synthesis of AZD3264 is described in which the differential reactivities of various halogen atoms have been employed. The process involves five linear chemical steps with three isolated stages starting from commercially available fragments.
Elemental impurities – A database to facilitate the risk assessment of active ingredients and excipients
DRUG REGULATORY AFFAIRS INTERNATIONAL

One of the main demands of the Guideline ICH Q3D is to carry out risk assessments on metallic impurities. A database with analytical data provides a valuable support. Learn more about the data sharing using the new elemental impurities database.
Released in December 2014, the ICH Q3D Guideline on Elemental Impurities contains extensive specifications for the control of a total of 24 elements (21 metals, 3 metalloids) that can be present as impurities in pharmaceutical products. Main sources can be
- Active ingredients
- Excipients (including water)
- Processing auxiliaries and catalysts
- Production equipment
- Container and closure systems
The Guideline ICH Q3D calls for a risk assessment with regard to the presence of metallic impurities in various dosage forms, taking into account the respective limit values. The main factors of influence are to be included (see fishbone diagram on p. 6 of the Guideline). The risks identified in a comprehensive analysis…
View original post 343 more words
ECA and PQG publish Chapter 6 of the interpretation of the ECA and PQG publish Chapter 6 of the interpretation of the EU GDP Guideline
DRUG REGULATORY AFFAIRS INTERNATIONAL

The ECA Foundation and the Pharmaceutical Quality Group (PQG) have been working on the interpretation of different chapters of the EU GDP Guideline. Now the group has finalized the work on chapter 6 – Complaints, Returns, Suspected Falsified Medicinal Products & Medicinal Product Recalls. Read more about the GDP Guidance Chapter 6.
The ECA Foundation and the Pharmaceutical Quality Group (PQG) have been working on the interpretation of different chapters of the EU GDP Guideline. The interpretation of five chapters have been published already. The following 5 Guidance chapters on the EU GDP Guideline are available:
Chapter 1: Quality Management
Chapter 9: Transportation (also contains a template for a Technical Agreement)
Chapter 7: Outsourced Activities
Chapter 2: Personnel
Chapter 5: Operations
Now the group has finalized the work on chapter 6 – Complaints, Returns, Suspected Falsified Medicinal Products & Medicinal Product Recalls. Chapter 6 of the EU GDP…
View original post 124 more words
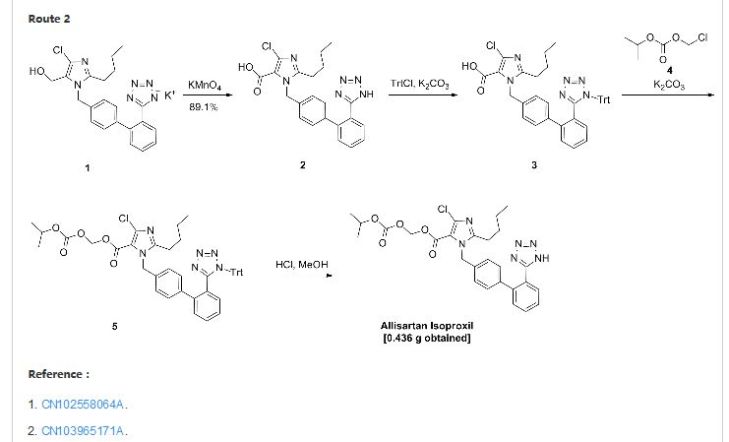
Allisartan isoproxil

Allisartan isoproxil
CAS: 947331-05-7
553.01, C27 H29 Cl N6 O5
An angiotensin II receptor antagonist used to treat mild to moderate essential hypertension.
Approved china, cfda July 1 2012
![]()
Shanghai Allist Pharmaceutical, Inc.
Allist Shanghai Pharmaceutical Co., Ltd.

2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carboxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester,
2-Butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)biphenyl-4-ylmethyl]-1H-imidazole-5-carboxylic acid isopropoxycarbonyloxymethyl ester
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester
Allisartan is an orally-available angiotensin AT1 antagonist in phase II clinical trials at Shanghai Allist Pharmaceutical for the treatment of mild to moderate essential hypertension.
Shanghai Allist Pharmaceutical PHASE 2 for Hypertension

CN200710094021.4 and CN201110289695.6 disclose the preparation of Alicante medoxomil, the inventor repeated, the proceeds of crystal and Chinese patent CN200710094131.0 consistent disclosed.

Allisartan isoproxil
Angiotensin II AT-1 receptor antagonist
Essential hypertension
Amorphous form of allisartan isoproxil is claimed in WO 2015062498. Useful for treating hypertension. Shenzhen Salubris Pharmaceuticals, in collaboration with Allist, has developed and launched allisartan isoproxil. In October 2012, Shenzhen Salubris signed a strategic cooperation framework agreement with Allist Pharmaceutical for the production and marketing of allisartan isoproxil. Family members of the product case of allisartanWO2007095789, expire in the EU and in the US in 2026. For a prior filing see WO2009049495 (assigned to Allist Pharmaceuticals), claiming the crystalline form of allisartan and its method of preparation.
The compound of formula (I) is an Ang II receptor antagonist. Its chemical name is 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carb-oxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester. Chinese Patent CN101024643A describes the structure, and its use as antihypertensive drugs.

As regards to the solid physical properties of the compound of formula (I), the patent document of CN101024643A discloses that it is a white solid, and its melting point is 134.5-136° C. However, CN101024643A dose not disclose the crystalline structure of the compound of formula (I).

 CHINA
CHINA
NEW PATENT
WO-2015062498
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015062498
……………………..
PATENT
http://www.google.com/patents/CN103965171A?cl=en
Hypertension is a major disease threat to human health, looking for efficiency, low toxicity anti-hypertensive drugs can help relieve social pressures and family responsibilities, with good social and economic benefits.
Angiotensin II (Ang II) is the renin – angiotensin – aldosterone system (RAAS) main vasoconstrictor hormone, which plays an important role in the pathobiology of many chronic diseases, particularly its the role of blood pressure regulation is particularly prominent, and therefore Ang II receptor is believed to be a good target for the development of anti-hypertensive drugs.
EP0253310 discloses a series of imidazole derivatives, DuPont declared and obtained by the study of losartan potassium-listed in 1994, was the first non-peptide Ang II receptor antagonist anti-hypertensive drugs. Thereafter, he listed a series of losartan antihypertensive drugs: candesartan cilexetil, valsartan, irbesartan, telmisartan and olmesartan medoxomil, etc. (EP0253310, W02005049587, GB2419592, EP1719766, US5196444) .
The losartan potassium in the body, the active metabolite EXP3174 has a stronger antihypertensive effect than losartan potassium, but EXP3174 polar molecular structure, is difficult to form passive absorption by diffusion through the cell membrane. US5298915 discloses five carboxyl ester group transformation EXP3174 is a series of derivatives, focusing on the compound HN-65021, and discloses hypotensive test results HN-65021 administered by the oral route, its hypotensive activity with chlorine Similar losartan potassium (BritishJouurnal ofClinical Pharmacology, 40,1995,591).
CN200680000397.8 _5_ discloses a class of imidazole carboxylic acid derivatives, namely Alicante medoxomil compound 8 has a good blood pressure lowering effect, the structure of formula I, the preparation method disclosed in this patent document follows the route A, losartan potassium by oxidation, the protecting group into an ester, deprotected to give a compound of formula I, the route step oxidation process of hydroxyl to carboxyl groups, will be reduced to very fine granular potassium permanganate, manganese dioxide, filtration This manganese mud time-consuming, inefficient, polluting; the second step conversion was about 70%, and post-processing cumbersome; byproducts and produced the first two steps more. This makes the high cost of the entire route, not suitable for the production of amplification.

CN200710094021.4 discloses another method for preparing the compounds of formula I, the following route B, the starting material by nucleophilic substitution, oxidation, an ester, a tetrazole ring to obtain a compound of formula I, the first step of the method nucleophilic substitution easy to generate an imidazole ring -3 para isomer impurities difficult to remove; the last step into the ring to use sodium azide, operating dangerous.

CN201210020174.5 disclosed a series of anti-hypertensive compound and preparation method, the following line C, the temperature control in the first step of its preparation O ~ 5 ° C, a mixed solution of acetone and water, with a 5% aqueous solution of sodium hypochlorite oxidation, yield 70%, the second step use of potassium permanganate, manganese dioxide will produce the same, and a yield of only 40%, the first two steps total yield of 28%, is very low, and the post-treatment methods are by column separation, the first two steps are used are organic and inorganic mixed solvent is not conducive to recovery, not suitable for scale-up.






Example 8 2-Butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil crude)

To a 20L reactor 9800ml of methanol, stirring was started, the rotational speed is added at 200r / min 1225.3g solid compound of formula II, and heated to reflux. The reaction 8-10h evacuation HPLC detection, the formula II compound residue <1.0% seen as a response endpoint. After reaching the end of the reaction the heating was stopped, continued stirring speed of 180r / min. About 3_4h fell 20_25 ° C, colorless transparent crystalline solid precipitated. The reaction mixture was cooled to continue to 15-20 ° C, to maintain 15-20 ° C with stirring 3h, the reaction mixture was filtered to give a pale yellow clear filtrate. The filtrate was concentrated under reduced pressure to move 20L flask, vacuum degree of 0.075MPa, 40_45 ° C methanol distilled off under until no distillate. 800ml of absolute ethanol was added, a vacuum degree of 0.075MPa, 40-45 ° C under distillation until no distillate.
900ml of absolute ethanol was added, heated to reflux. N-heptane was added slowly 1100ml, reflux 15min, to -10 ° c / h speed cooled to 15 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration lh, was Allie medoxomil crude (800.lg, yield 93.8%).Purification was used directly in the next step without drying.
Example 9 2-butyl-4-chloro-_1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole-5-carboxylic acid, 1 – [(isopropylamino oxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

850ml of absolute ethanol was added to the 3L reaction vessel was charged with crude Alicante medoxomil (800.lg, 1.45mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1300ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration, the purified Alicante medoxomil (780.9g, 97.6% yield).
Example 10 2-butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

950ml of absolute ethanol was added to the 5L reaction vessel was charged with crude Alicante medoxomil (549.9g, 1.72mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1200ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = cake was washed with a mixture of 1/3, and dried under reduced pressure after filtration to obtain a purified Alicante medoxomil (540.0g, 98.2% yield).
Example 122-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester (compound 8)

To a 100 ml of one-necked flask, 0.523 g of material, 0.124 g of potassium carbonate, 5 ml of N,N-dimethylacetamide were added in turn. The solution was stirred at room temperature for 20 minutes. Then 0.562 g of 1-chloromethyl isopropyl carbonate was added and the mixture was reacted at 45-50° C. for 16 hours. After the reaction was completed, the mixture solution was filtered, and 30 ml of water was added into the filtrate. The resulting mixture was extracted with 30 ml of ethyl acetate twice. The organic phase was dried and concentrated to give 1.724 g of oil, which was directly used in the next reaction without purification.
10 ml of dioxane and 5 ml of 4 mol/L HCl were added, and the resulting mixture was reacted at room temperature for 16 hours. The reaction was stopped and the solution was adjusted to pH 6-7 using aqueous sodium bicarbonate solution. The solution went turbid, and was extracted with ethyl acetate. The organic phase was washed with saturated brine, dried, concentrated to give 0.436 g of 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester.
In addition, the following reaction condition can be used to deprotect the protecting group. To 1.7 g of oily product, 5 ml absolute methanol was added and the mixture was heated slowly to reflux and stirred for 8 hours. When the insoluble solid disappeared totally, the mixture was discontinued to heating and cooled to 5° C. The white solid precipitated, and was separated by filtration, and the filter cake was washed with a small quantity of methanol. The combined filtrate was concentrated to dryness to give 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester with the yield of 70%.
1H-NMR (CDCl3) δ H (ppm): 0.89 (t, 3H, J=14.6), 1.24 (d, 6H, J=6.3), 0.37 (m, 2H, J=22.1), 1.69 (m, 2H, J=30.5), 2.64 (t, 2H, J=15.5), 4.81 (m, 1H, J=12.4), 5.54 (s, 2H), 5.86 (s, 2H), 6.95-7.64 (8H), 8.08 (d, 1H, J=7.42)
ESI(+) m/z: 552.7
Mp: 134.5-136° C.




| WO2005011646A2 * | 20 Jul 2004 | 10 Feb 2005 | Nicoletta Almirante | Nitrooxy derivatives of losartan, valsatan, candesartan, telmisartan, eprosartan and olmesartan as angiotensin-ii receptor blockers for the treatment of cardiovascular diseases |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8455526 * | 6 Jun 2008 | 4 Jun 2013 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| US20100168193 * | 6 Jun 2008 | 1 Jul 2010 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| USRE44873 | 31 Jul 2006 | 29 Apr 2014 | Salubris Asset Management Co., Ltd. | Imidazole-5-carboxylic acid derivatives, the preparation method therefor and the uses thereof |
| CN101024643A | 20 Feb 2006 | 29 Aug 2007 | 上海艾力斯医药科技有限公司 | Imidazo-5-carboxylic-acid derivatives, its preparing method and use |
| US5298519 * | 24 Sep 1992 | 29 Mar 1994 | Chemish Pharmazeutische Forschungsgesellschaft M.B.H. | Acylals of imidazole-5-carboxylic acid derivatives, and their use as angiotensin (II) inhibitors |
……………….
update……………..
Example 1
Weigh 25g 2- butyl-4-chloro-1- [2 ‘- (1-trityl–1H- tetrazol-5-yl) -1,1’-biphenyl – methyl] – imidazole 5-carboxylic acid, 1 – [(isopropoxy) – carbonyloxy] -, methyl ester, was added to a 500ml three-necked flask, methanol was added 200ml, refluxed for 9h, methanol was distilled off under reduced pressure to give crude Alicante medoxomil .
To the residue (i.e., medoxomil crude Alicante) were added 33ml of isopropanol and 66ml of n-heptane, heated to 76 ℃ stirred for 2h. After cooling to 60 ℃ stirring for 1h, and then the system was slowly cooled to 0 ℃, stirring was continued for 3h. Filtered, the filter cake was washed with n-heptane. At 40 ℃ 8 hours and dried in vacuo to give 15.3g Alicante medoxomil (purity 99.3%) as a XRD spectrum as shown in Figure, the main peak of the diffraction peaks as shown in the following table, the DSC spectrum shown in figure II . Compared with the published crystal, the crystal obtained by the absence of significant electrostatic phenomena.
Shanghai , CHINA







RG-1577, EVT 302, Sembragiline, RO-4602522
RG-1577, EVT 302, Sembragiline, RO-4602522
CAS 676479-06-4, MW 342.36
- C19 H19 F N2 O3
- Acetamide, N-[(3S)-1-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-3-pyrrolidinyl]-
UNII-K3W9672PNJ
RG-1577, a selective and reversible monoamine oxidase B inhibitor, for treating AD (phase 2 clinical, as of May 2015).
Family members of the product case for RG-1577 (WO2004026825) hold protection in EU until 2023 and expire in US in 2024 with US154 extension. Follows on from WO2006097197, claiming a process for preparing RG-1577.
Alzheimer‘s Disease is a brain disease that slowly destroys memory and thinking skills, up to loss of the ability to carry out the simplest tasks. It is the most common cause of dementia among older people. Mild Alzheimer‘s Disease manifests itself in memory loss and small changes in other cognitive abilities, e.g getting lost, trouble handling money and managing daily tasks, having some mood and personality changes, etc.
In the stage of Moderate Alzheimer‘s Disease, the control of language, reasoning, sensory processing, and conscious thought are impacted. Memory loss and con usion grow worse, e.g patients have problems recognizing family and friends and become unable to learn new things, etc. hallucinations, delusions, and paranoia may occur. .Severe Alzheimer‘s Disease is the final stage. Patients cannot communicate anymore and are completely dependent.
N-[(3S)-l-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide has previously been described in the art. 1 WO 2006/097197 2 and WO 2006/0972703 relate to methods for preparing enantiomerically pure 4-pyrrolidinophenylbenzyl ether derivatives.
![]()
The processes of the prior art hamper from several drawbacks (e.g. long reaction sequence, low overall yield also due to loss of half of the product in the classical resolution step, the need for a chromatographic purification to remove by-products formed in the Mitsunobu reaction) and are therefore less suitable for the preparation of N-[(3S)-l-[4-[(3-fluorophenyl) methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide on large scale.
Most Recent Events
- 01 Aug 2014Roche completes a phase I trial in volunteers in USA (NCT02104648)
- 14 May 2014Roche completes enrolment in the MAyflOwer RoAD trial for Alzheimer’s disease (combination therapy, adjunctive treatment) in Australia, Canada, Czech Republic, France, Germany, Italy, Poland, South Korea, Spain, Sweden the United Kingdom and the USA (NCT01677754)
- 01 Apr 2014Roche initiates enrolment in a phase I trial in healthy volunteers in USA (NCT02104648)
http://www.evotec.com/uploads/media_library/10/2012-09_Evotec_Company_presentation_September_e.pdf

……………………..
WO2004026825
http://www.google.com/patents/WO2004026825A1?cl=en
………………….
WO2006097197
http://www.google.com/patents/WO2006097197A1?cl=en
……………………………………………..
PATENT
WO 2015063001
Novel, crystalline polymorphic forms A and B of a pyrrolidone derivative ie RG-1577, useful for treating Alzheimer’s disease (AD). Roche and its Japanese subsidiary Chugai, under license from Evotec, which previously licensed the drug from Roche, are developing RG 1577
formula 1 via the following routes

In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route A

1
In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route B

In a certain embodiment, present invention relates to a crystalline polymorph of a compound of formula 1.

synthesize a compound of formula 1 from a compound of formula 7

compound of formula 6 to a compound of formula 7

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 6 via the intermediate 6a to a compound of formula 7

further comprising reacting a compound of formula 3 with a compound of formula 5 to a compound of formula 6

comprising reacting a compound of formula 2 to a compound of formula 3

2 3
In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 10 to a compound of formula 6

eacting a compound of formula 9 with a compound of formula 5 to a compound of formula 10

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 8 to a compound of formula 9

(lS’)-N-[l-[4-(3-fluoro-benzyloxy)-phenyl]-5-oxo-pyrrolidin-3-yl-]acetamide (1)
To a suspension of chloride (7) (37.9 g, 100 mmol) in 2-methyltetrahydrofurane (600 ml) was added under vigorous stirring at 0°C 1.65 M potassium ie/t-butoxide in THF (75.5 ml, 125 mmol, ACROS) over 2.5 h. After additional stirring at 0°C for 1 h, the cold suspension was hydrolyzed with 0.1 M HCl (600 ml) and the reaction mixture was stirred at 30°C for 0.5 h. The organic layer was washed with water (300 ml), dried (Na2S04) and filtered. Removal of the solvent by rotary evaporation (50°C/>10 mbar) afforded 32.1 g crystalline residue, which was dissolved in 2-butanone (400 ml) at ca. 95°C and hot filtered. Crystallization, which was induced by seeding and cooling to room temperature and 0°C (4 h) afforded 25.4 g (74.2%) of the titled compound (1) as an off-white, crystalline powder,
Mp. 162-164°C (polymorph B).
Ee >99.8%, [cc]D20 = – 17.8 (DMF; c = 1).
1H NMR (400 MHz, DMSO- 6) δ ppm 1.82 (s, 3H), 2.34 (dd, J1=n. l, J2=3.9, 1H), 2.84 (dd, J/=17.1, J2=8.2, 1H), 3.55 (dd, J/=10.2, J2=3.2, 1H), 4.07 (dd, J/=10.2, J2=6.7, 1H), 4.32-4.41 (m, 1H), 5.13 (s, 2H), 7.02 & 7.55 (d, J=9.1, each 1H), 7.11-7.19 (m, 1H), 7.24-7.31 (m, 1H), 7.40-7.47 (m, 1H), 8.40 (d, J=6.4, 1H).
ESI-MS (m/z) 343 [M+H]+, 365 [M+Na]\. Anal.Calcd for Ci9H19FN203 (342.37): Calcd. C, 66.66; H, 5.59; N, 8.18; F, 5.02; O, 14.02. Found C, 66.76; H, 5.48; N, 8.13; F, 5.03; O, 13.99.
Crystallized (1) form previous step (9.5 g, 0.028 mol) was dissolved in 2-butanone (290 mL) upon heating. The hot solution was filtered over charcoal. The solution was concentrated by removal of 2-butanone (200 mL) by distillation prior to seeded cooling crystallization. Filtration, washing with chilled 2-butanone and drying at 50°C/25 mbar/16h afforded 9.18 g (93.9% corrected yield) of the title compound (1) as a crystalline powder of polymorphic form B with an assay of 100.4 %(w/w) and a purity of 99.97 %(area) (by HPLC).
Alternatively, to a stirred suspension of hydroxyamide (6) (30.0 g, 0.083 mol) in toluene (500 ml) was added at 50°C within 45 minutes thionyl chloride (10.40 g, 0.087 mol) and the resulting mixture was stirred for 3h at 50°C. The mixture was then heated up to 92°C and subsequently stirred at this temperature for 15 h. The Suspension was then cooled to 50°C and toluene was removed by distillation under reduced pressure. The distillation residue was cooled to ambient temperature and treated with N-methylpyrrolidone (210 ml) to obtain an almost clear solution. This solution was then cooled to -10°C and subsequently treated at this temperature within 2h with a solution of potassium iert-butoxide (12.40 g, 0.111 mol) in THF (60 g). The resulting mixture was stirred for another 60 minutes at -10°C, then warmed up to room temperature within 60 minutes and subsequently stirred at room temperature for 6 h. The reaction mixture was quenched with water (150 g) and the pH was adjusted with acetic acid (approx. 1.8 g) to pH 7-8. The mixture was then heated to 30-45°C and THF and toluene were distilled off under reduced pressure (<200 mbar) to obtain a clear NMP/water mixture (400 ml). This mixture was heated to 45°C and 260 mg of seed crystals were added. Water (320 ml) was then added within 3 h whereby the product crystallized. The resulting suspension was cooled to room temperature within 3 h and subsequently stirred at this temperature for 2 h. Filtration and washing of the filter cake with a mixture of water (100 ml) and N-methylpyrrolidone (20 ml) and subsequently only with water (150 ml) afforded after drying (70°C/10 mbar/20 h) 26.2 g (92%) of the title compound (1) as a crystalline powder with an assay of 99.6 %(w/w) and a purity of 99.7 %(area) (by HPLC).

HPLC
Purity (HPLC): Column: XSelect Phenyl Hexyl x2, 150 x 4.6mm, 3.5um. Starting
Pressure: 226 bar; temp.: 50°C. Inj. vol.: 2.0 μΐ^ + wash. Flow: 1.0 ml/min. Det: 204 nm. A: Water + 5% ACN, 77-2% in 7 min., hold for 1 min.; B: 0.1% HCOOH, 18% isocratic; C: MeOH, 5-80% in 7 min., hold for 1 min. Sample prep.: 2 mg/ml ACN. Retention times: β-acid 5.93 min., diacid 6.18 min., cc-acid 6.89 min., diester 6.96 min.
ee determination(HPLC): Column: Chiralpak IA-3 100 x 4.6mm, 3um; 91 bar, 2ml/min; temp.: 30°C. Inj. vol.: 10.0 μL· Det.: 206 nm. A: n-heptane, 80%; B: EtOH, 20%. Sample prep.: 4 mg/ml EtOH. Retention times: D-enantiomer 2.21 min., L-enantiomer 2.71 min
………………….
US 20050065204
EXAMPLE 11
Preparation of (S)-1-(4-Hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic Acid
8.00 g Polyethyleneglycol 6000 was dissolved in 150 mL (100 mM) magnesium acetate buffer pH 6.0 under stirring, and the solution added to a stirred suspension of 10.00 g (42.51 mmol) (RS)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid methyl ester (99.7%) in 40 mL methylcyclohexane. The mixture was heated to 28° C. and the pH readjusted to 6.0 with 2 M NaOH. The reaction was started by adding 33.2 mg Candida cylindraceae cholesterase (16.88 kU/g), and the pH was maintained at 6.0 by the controlled addition of 1.0 M NaOH solution under stirring. After a total consumption of 20.35 mL (20.35 mmol) 1.0 M sodium hydroxide solution (after 17.1 h; 47.9% conversion) the reaction mixture was passed through a sintered glass filter. The filtrate spontaneously separated into an aqueous and an organic phase.The aqueous phase was washed with 2×200 mL ethyl acetate to remove uncleaved ester. The aqueous phase was set to pH 4.0 with 25% sulfuric acid and concentrated in vacuo to a volume of ca. 80 mL (bath 60° C.). The solution was cooled to 1° C. (formation of white precipitate/crystals) and the pH set to 1.5 with 25% sulfuric acid. The precipitate/crystals were stirred overnight at 1° C., filtered off on a sintered glass filter (washed with a minimum amount of water) and dried overnight on high vacuum (RT, 6×10−2 mbar) to give 4.32 g (19.53 mmol; 45.9%) (S)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid. Analysis: HPLC (area A226nm): 99.3%, 0.7% ester. 98.9%ee. The product contains 5.3% water (according to Karl Fischer determination) and 2.1% (w/w) PEG (according to NMR).
| Company | Evotec AG |
| Description | Small molecule monoamine oxidase B (MAO-B) inhibitor |
| Molecular Target | Monoamine oxidase B (MAO-B) |
| Mechanism of Action | Monoamine oxidase B (MAO-B) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Alzheimer’s disease (AD) |
| Indication Details | Treat Alzheimer’s disease (AD) |
| Regulatory Designation | |
| Partner |
//////////
Chūō, japan




A Chūō Line (Rapid) E233 series (right) and A Chūō-Sōbu Line E231 series (June 2007)
 Chuo Dori street on a weekend afternoon
Chuo Dori street on a weekend afternoon

Recovery of Artemisinin from a Complex Reaction Mixture Using Continuous Chromatography and Crystallization


Recovery of Artemisinin from a Complex Reaction Mixture Using Continuous Chromatography and Crystallization




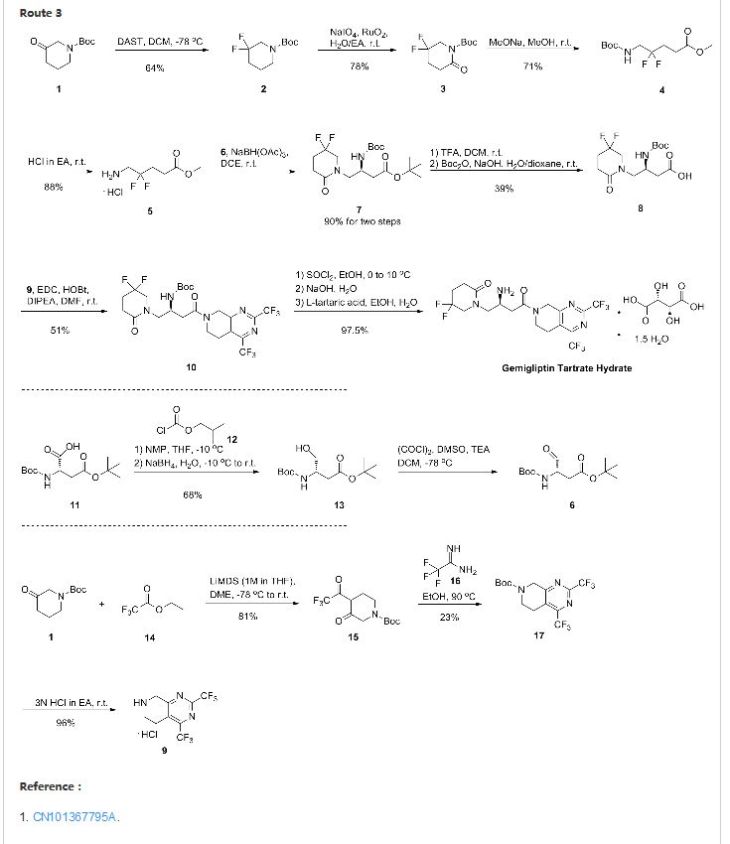
Eliglustat

ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
Eliglustat (INN, USAN;[1] trade name Cerdelga) is a treatment for Gaucher’s disease developed by Genzyme Corp that was approved by the FDA August 2014.[2] Commonly used as the tartrate salt, the compound is believed to work by inhibition ofglucosylceramide synthase.[3][4]
In March 2015, eliglustat tartrate was approved in Japan for the treatment of Gaucher disease. Eliglustat tartrate was described specifically within the US FDA’s Orange Booked listed US6916802, which is set to expire in April 2022.
In May 2015, the Orange Book also listed that eliglustat tartrate had Orphan Drug Exclusivity and New Chemical Entity exclusivity until 2019 and 2021, respectively.
it having been developed and launched as eliglustat tartrate by Genzyme (a wholly owned subsidiary of Sanofi), under license from the University of Michigan.
Eliglustat tartrate is known to act as inhibitors of glucosylceramide synthase and glycolipid, useful for the treatment of Gaucher’s disease type I and lysosome storage disease.
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
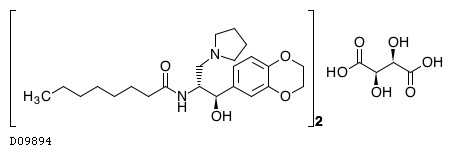
Eliglustat tartrate (USAN)
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.
Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
WO 2015059679
| Process for the preparation of eliglustat free base – comprising the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat. | |
| Dr Reddy’s Laboratories Ltd | |
| New crystalline eliglustat free base Form R1 and a process for its preparation are claimed. Also claimed is a process for the preparation of eliglustat free base which comprises the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat.Further eliglustat oxalate, its crystalline form, and a process for the preparation of crystalline eliglustat oxalate, are claimed. | |
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.

Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
Example 1 : Preparation of 5-phenyl morpholine-2-one hydrochloride
To a (S) + phenyl glycinol (100g) add N, N-diisopropylethylamine (314ml) and acetonitrile (2000ml) under nitrogen atmosphere at room temperature. It was cooled to 10- 15° C. Phenyl bromoacetate (172.4g) dissolved in acetonitrile (500ml) was added to the above solution at 15° C over a period of 30 min. The reaction mixture is allowed to room temperature and stirred for 16-20h. Progress of the reaction was monitored by thin layer chromatography. After completion of the reaction, the reaction mixture was concentrated under reduced pressure at a water bath
temperature less than 25° C to get a residue. The residue was dissolved in ethyl acetate (1000ml) and stirred for 1 h at 15-20°C to obtain a white solid. The solid material obtained was filtered and washed with ethyl acetate (200ml). The filtrate was dried over anhydrous sodium sulphate (20g) and concentrated under reduced pressure at a water bath temperature less than 25° C to give crude compound (1000g) as brown syrup. The Crude brown syrup is converted to HCI salt by using HCI in ethyl acetate to afford 5-phenyl morpholine-2-one hydrochloride (44g) as a white solid. Yield: 50%, Mass: m/z = 177.6; HPLC (% Area Method): 90.5%
Example 2: Preparation of (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4] dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one.
5-phenyl morpholine-2-one hydrochloride (100g) obtained from above stage 1 is dissolved in toluene (2500ml) under nitrogen atmosphere at 25-30°C. 1 ,4-benzodioxane-6-carboxaldehyde (185.3g) and sodium sulphate (400g) was added to the above solution and the reaction mixture was heated at 100-105°C for 72h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature less than 25° C to get a residue. The residue was cooled to 10°C, ethyl acetate (2700ml) and 50% sodium bisulphate solution (1351 ml) was added to the residue and stirred for 1 h at 10°C to obtain a white solid. The obtained white solid was filtered and washed with ethyl acetate. The separated ethyl acetate layer was washed with water (1000ml), brine (1000ml) and dried over anhydrous sodium sulphate. The organic layer was concentrated under reduced pressure at a water bath temperature of 45-50°C to get a crude material. The obtained crude material is triturated with diethyl ether (1500ml) to get a solid material which is filtered and dried under vacuum at room temperature for 2-3h to afford (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (148g) as a yellow solid. Yield: 54%, Mass: m/z = 487.7; HPLC (% Area Method): 95.4 %
Example 3: Preparation of (2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 ”^henyl-ethy^
(1 R,3S,5S,8aS)-1 !3-Bis-(2′!3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (70g) obtained from above stage 2 was dissolved in chloroform (1400ml) at room temperature. It was cooled to 0-5°C and pyrrolidone (59.5ml) was added at 0-5°C over a period of 30 minutes. The reaction mixture was allowed to room temperature and stirred for 16-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature of 40-45°C to obtain a crude. The obtained crude was dissolved in methanol (1190ml) and 1 N HCI (1 190ml) at 10-15° C, stirred for 10 minutes and heated at 80-85°C for 7h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C.The aqueous layer was extracted with ethyl acetate and the organic layer was washed with 1 N HCI (50ml). The aqueous layer was basified with saturated sodium bicarbonate solution up to pH 8-9 and extracted with ethyl acetate (3x70ml). The combined organic layers was washed with brine (100ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50-55°C to afford (2S,3R,1″S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (53g) as a yellow foamy solid. Yield: 90%, Mass: m/z = 412.7, HPLC (% Area Method): 85.1 %
Example 4: Preparation of (1 R,2R,1 “S)-1-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2”-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol.
(2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6’-yl)-3-hydroxy-2-(2”-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (2.5g) obtained from above stage 3 dissolved in Tetrahydrofuran (106ml) was added to a solution of Lithium aluminium hydride (12.2g) in tetrahydrofuran (795ml) at 0°C and the reaction mixture was heated at 60-65°C for 10h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 5- 10°C and quenched in saturated sodium sulphate solution (100ml) at 5-10°C. Ethyl acetate was added to the reaction mass and stirred for 30-45 min. The obtained solid is filtered through celite bed and washed with ethyl acetate. Filtrate was dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50°C to afford (1 R,2R, 1″S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (43.51 g) as a yellow gummy liquid. The crude is used for the next step without further purification. Yield: 85%, Mass: m/z = 398.7, HPLC (% Area Method): 77 %
Example 5: Preparation of (1 R, 2R)-2-Amino-1-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol.
(1 R,2R,1 “S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6’-yl)2-hydroxy-2-(2”-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (40g) obtained from above stage 4 was dissolved in methanol (400ml) at room temperature in a 2L hydrogenation flask. Trifluoroacetic acid (15.5ml) and 20% Pd (OH) 2 (40g) was added to the above solution under nitrogen atmosphere. The reaction mixture was hydrogenated under H2, 10Opsi for 16-18h at room temperature. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was filtered through celite bed and washed with methanol (44ml) and water (44ml). Methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C and the aqueous layer was washed with ethyl acetate. The aqueous layer was basified with 10M NaOH till the PH reaches 12-14 and then extracted with dichloromethane (2x125ml). The organic layer was dried over anhydrous sodium sulphate (3gm) and concentrated under reduced pressure at a water bath temperature of 45°C to obtain a gummy liquid. The gummy liquid was triturated with methyl tertiary butyl ether for 1 h to get a white solid, which is filtered and dried under vacuum at room temperature to afford (1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (23g) as a white solid. Yield: 82.3%, Mass (m/zj: 278.8, HPLC (% Area Method): 99.5%, Chiral HPLC (% Area Method): 97.9%
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %
……………………………..
Nmr predict
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-1h.png)
13 C NMR
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-13c.png)
CAS NO. 491833-29-5, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide
C-NMR spectral analysis
………………..
http://www.google.com/patents/WO2013059119A1?cl=en

http://www.google.com/patents/US7196205
Compound 7
(1R,2R)-Nonanoic acid[2-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide

This compound was prepared by the method described for Compound 6 using Nonanoic acid N-hydroxysuccinimide ester. Analytical HPLC showed this material to be 98.4% pure. mp 74–75° C.
1H NMR (CDCl3) δ 6.86–6.76 (m, 3H), 5.83 (d, J=7.3 Hz, 1H), 4.90 (d, J=3.3 Hz, 1H), 4.24 (s, 4H), 4.24–4.18 (m, 1H), 2.85–2.75 (m, 2H), 2.69–2.62 (m, 4H), 2.10 (t, J=7.3 Hz, 2H), 1.55–1.45 (m, 2H), 1.70–1.85 (m, 4H), 1.30–1.15 (m, 10H), 0.87 (t, J=6.9 Hz, 3H) ppm.
Intermediate 4(1R,2R)-2-Amino-1-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-3-pyrrolidin-1-yl-propan-1-ol

Intermediate 3 (5.3 g, 13.3 mmol) was dissolved in methanol (60 mL). Water (6 mL) and trifluoroacetic acid (2.05 m/L, 26.6 mmol, 2 equivalents) were added. After being placed under nitrogen, 20% Palladium hydroxide on carbon (Pearlman’s catalysis, Lancaster or Aldrich, 5.3 g) was added. The mixture was placed in a Parr Pressure Reactor Apparatus with glass insert. The apparatus was placed under nitrogen and then under hydrogen pressure 110–120 psi. The mixture was stirred for 2–3 days at room temperature under hydrogen pressure 100–120 psi. The reaction was placed under nitrogen and filtered through a pad of celite. The celite pad was washed with methanol (100 mL) and water (100 mL). The methanol was removed by rotoevaporation. The aqueous layer was washed with ethyl acetate three times (100, 50, 50 mL). A 10 M NaOH solution (10 mL) was added to the aqueous layer (pH=12–14). The product was extracted from the aqueous layer three times with methylene chloride (100, 100, 50 mL). The combined organic layers were dried with Na2SO4, filtered and rotoevaporated to a colorless oil. The foamy oil was vacuum dried for 2 h. Intermediate 4 was obtained in 90% yield (3.34 g).
Intermediate 3(1R,2R,1″S)-1-(2′,3′-Dihydro-benzo[1,4]dioxin-6′-yl)-2-(2″-hydroxy -1′-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol

To a 3-neck flask equipped with a dropping funnel and condenser was added LiAlH4 (Aldrich, 1.2 g, 31.7 mmol, 2.5 equivalents) and anhydrous THF (20 mL) under nitrogen. A solution of Intermediate 2 (5.23 g, 12.68 mmol) in anhydrous THF (75 mL) was added dropwise to the reaction over 15–30 minutes. The reaction was refluxed under nitrogen for 9 hours. The reaction was cooled in an ice bath and a 1M NaOH solution was carefully added dropwise. After stirring at room temperature for 15 minutes, water (50 mL) and ethyl acetate (75 mL) was added. The layers were separated and the aqueous layer was extracted twice with ethyl acetate (75 mL). The combined organic layers were washed with saturated sodium chloride solution (25 mL). After drying with Na2SO4 the solution was filtered and rotoevaporated to yield a colorless to yellow foamy oil. Intermediate 3 was obtained in 99% yield (5.3 g).

|
|
| Systematic (IUPAC) name | |
|---|---|
| N-[(1R,2R)-1-(2,3-Dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-(1-pyrrolidinyl)-2-propanyl]octanamide | |
| Clinical data | |
| Trade names | Cerdelga |
|
|
| Identifiers | |
| 491833-29-5 | |
| A16AX10 | |
| PubChem | CID 23652731 |
| ChemSpider | 28475348 |
| ChEBI | CHEBI:82752 |
| Chemical data | |
| Formula | C23H36N2O4 |
| 404.543 g/mol | |
- https://download.ama-assn.org/resources/doc/usan/x-pub/eligustat.pdf
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm410585.htm
- Lee, L.; Abe, A.; Shayman, J. A. (21 May 1999). “Improved Inhibitors of Glucosylceramide Synthase”. Journal of Biological Chemistry 274(21): 14662–14669. doi:10.1074/jbc.274.21.14662.
- Shayman, JA (Aug 1, 2010). “ELIGLUSTAT TARTRATE: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease.”.Drugs of the future 35 (8): 613–620. PMID 22563139.
| WO2008150486A2 * | May 30, 2008 | Dec 11, 2008 | Genzyme Corp | 2-acylaminopropoanol-type glucosylceramide synthase inhibitors |
| WO2009045503A1 * | Oct 3, 2008 | Apr 9, 2009 | Genzyme Corp | Method of treating polycystic kidney diseases with ceramide derivatives |
| WO2010014554A1 * | Jul 27, 2009 | Feb 4, 2010 | Genzyme Corporation | Glucosylceramide synthase inhibition for the treatment of collapsing glomerulopathy and other glomerular disease |
| WO2010039256A1 * | Oct 2, 2009 | Apr 8, 2010 | Genzyme Corporation | 2-acylaminopropoanol-type glucosylceramide synthase inhibitors |
………………..
SWEDEN


Alfred Nobel had the unpleasant surprise of reading his own obituary, titled The merchant of death is dead, in a French newspaper.



Nyköping (Sweden)-houses.
Fjallbacka, a colorful fishing Village along the west coast of Sweden
Knights Island, Stockholm, Sweden
 Stockholm, Sweden
Stockholm, Sweden
Sweden Stockholm
Europe Örby Änger – Sweden
 Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …

New Drug Approvals read by all Medicinal chemists across the world
As on 7 may 2015….. 6.7 lakh views
NEW DRUG APPROVALS
ALL ABOUT DRUGS, LIVE, BY DR ANTHONY MELVIN CRASTO, WORLDDRUGTRACKER, HELPING MILLIONS, 7 MILLION HITS ON GOOGLE, PUSHING BOUNDARIES, ONE LAKH PLUS CONNECTIONS WORLDWIDE, 6.7 LAKHS PLUS VIEWS ON THIS BLOG IN 206 COUNTRIES
DR ANTHONY MELVIN CRASTO



 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus 
 AMCRASTO@GMAIL.COM
AMCRASTO@GMAIL.COM

ANAGLIPTIN Spectral visit
![N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide](https://i0.wp.com/pic3.molbase.net/molpic/02/47/2473420.png)
| N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide | |
| CAS No.: | 739366-20-2 |
|---|---|
| Synonyms: |
|
| Formula: | C19H25N7O2 |
| Exact Mass: | 383.20700 |
Anagliptin chemically known as N-[2-[2-[2(S)-cyanopyrrolidin-l-yl]-2-oxoethylamino]- 2-methylpropyl]-2-methylpyrazolo[l,5-a]pyrimidine-6-carboxamide is represented by the structural formula:

Anagliptin is a dipeptidyl peptidase IV- inhibitor. United States Patent No 7345 1 80- (IJS’ 180) discloses anagliptin.
![N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide NMR spectra analysis, Chemical CAS NO. 739366-20-2 NMR spectral analysis, N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-28/002/473/2473420_1h.png)
![N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide NMR spectra analysis, Chemical CAS NO. 739366-20-2 NMR spectral analysis, N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-28/002/473/2473420_13c.png)
Example 5: Synthesis of N-[2-2[2(S)-Cyano pyrrolidin-l-yl]-2-oxoethyIamino]-2- methyIpropyl]-2-methyaIpyrazoIo [1, 5-a] pyrimidine-6-carboxamide (I, anagliptin).
1H NMR (300 MHz, CDC13): δ 1.16 (s, 6H), 2.23(m, 4H), 2.54(s, 3H), 3.25-3.51 (m, 6H), 4.78 (m, 1H), 6.53 (s, 1H), 8.05 (s, 1H), 8.93 (s, 1H), 9.22(s, 1H)
HPLC Purity: 99.71%, Chiral purity: 100%………WO2014147640A2
Kato, M.; Oka, M.; Murase, T.; Yoshida, M.; Sakairi, M.; Yamashita, S.; Yasuda, Y.; Yoshikawa, A.; Hayashi, Y.; Makino, M.; Takeda, M.; Mirensha, Y.;
Kakigami, T. Discovery and pharmacological characterization of N-[2-({2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl}amino)-2-methylpropyl]-
2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide hydrochloride (anagliptin hydrochloride salt) as a potent and selective
DPP-IV inhibitor. Bioorg. Med. Chem. 2011, 19, 7221–7227.
http://www.sciencedirect.com/science/article/pii/S0968089611007784

LATUR, MAHARASHTRA, INDIA
http://en.wikipedia.org/wiki/Latur
| Latur लातूर Lattalur, Ratnapur |
|
|---|---|
| City | |
|
Latur
Location in Maharashtra, India |
|
| Coordinates: 18.40°N 76.56°ECoordinates: 18.40°N 76.56°E | |
| Country | |
| State | Maharashtra |
| Region | Aurangabad Division |
| District | Latur |
| Settled | Possibly 7th century AD |
| Government | |
| • Body | Latur Municipal Corporation |
| • Mayor | Akhtar Shaikh |
| Area[1] | |
| • Total | 117.78 km2(45.48 sq mi) |
| Area rank | 89 |
| Elevation | 515 m (1,690 ft) |
| Population (2011) | |
| • Total | 382,754 |
| • Rank | 89th |
| • Density | 3,200/km2(8,400/sq mi) |
| Demonym | Laturkar |
| Languages | |
| • Official | Marathi |
| Time zone | IST (UTC+5:30) |
| PIN |
|
| Telephone code | 91-2382 |
| Vehicle registration | MH-24 |
| Sex ratio | 923.54 ♀/1000 ♂ |
| Literacy | 89.67 |
| Distance from Mumbai | 497 kilometres (309 mi) E (land) |
| Distance fromHyderabad | 337 kilometres (209 mi) NW (land) |
| Distance fromAurangabad, Maharashtra | 294 kilometres (183 mi) SE (land) |
| Climate | BSh (Köppen) |
| Precipitation | 666 millimetres (26.2 in) |
| Avg. summer temperature | 41 °C (106 °F) |
| Avg. winter temperature | 13 °C (55 °F) |
| http://www.citypopulation.de/world/Agglomerations.html | |





his Is The Famous ‘Ganj-Golai’ As The Central Place Of The Latur City. There Are 16 Roads Connecting To This Place And Seperate Markets i.e. Jewellers …

लातूर जिल्हयातील चित्र संग्रह
LATUR AIRPORT
 LATUR AIRPORT
LATUR AIRPORT


2012 Navratri Mahotsav in Latur

SOS Children’s Village Latur


Latur, India: Carnival Resort


Ausa Near Latur

Chakur near Latur

Vilasrao Deshmukh’s ancestral home at Babhalgaon village in Latur. Machindra Amle



UDGIR: Udgir is one of the most important towns of Latur district. Udgir has a great historical significance. It has witnessed the war between the Marathas …
 The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
