
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ranbaxy to introduce malarial treatment Synriam in African nations

Ranbaxy to introduce malarial treatment Synriam in African nations
Ranbaxy Laboratories has obtained regulatory approval to introduce India’s first new chemical entity (NCE) Synriam (arterolane maleate 150mg and piperaquine phosphate 750mg drug) in seven African countries.
read at
Synriam is a new age therapy recommended to treat uncomplicated Plasmodium falciparum malaria in adults. It was launched in India in April 2012.
The product was also launched in Uganda and is set to be introduced in Nigeria, Senegal, Cameroon, Guinea, Kenya and Ivory Coast by the end of January 2015.

Arterolane

Arterolane, also known as OZ277 or RBx 11160,is a substance being tested for antimalarial activity[1] by Ranbaxy Laboratories.[2] It was discovered by US and European scientists who were coordinated by the Medicines for Malaria Venture (MMV).[3] Its molecular structure is uncommon for pharmacological compounds in that it has both an ozonide group and an adamantane substituent.[4]
Phase III clinical trials of arterolane, in combination with piperaquine, began in India in 2009.[5] When clinical trial results were disappointing, the MMV withdrew support[2] and Ranbaxy continued developing the drug combination on its own.

Ranbaxy launched India’s first new drug, SynriamTM, treating Plasmodium falciparummalaria in adults. The drug provides quick relief from most malaria-related symptoms, including fever, and has a high cure rate of over 95 %.
Just one tablet per day is required, for three days, instead of two to four tablets, twice daily, for three or more days with other medicines. The drug is independent of dietary restrictions for fatty foods or milk.
Ranbaxy developed Synriam as a fixed-dose combination of arterolane maleate and piperaquine phosphate, where arterolane is the new chemical entity (NCE) that was developed as an alternative to artemisinin. It is the first recently developed antimalarial not based on artemisinin, one of the most effective treatments for malaria, which has shown problems with resistance in recent years. Arterolane was discovered by a collaborative drug discovery project funded by the Medicines for Malaria Venture. Since SynriamTM has a synthetic source, unlike artemisinin-based drugs, production can be scaled up whenever required and a consistent supply can be maintained at a low cost.

The new drug, has been approved by the Drug Controller General of India (DCGI) for marketing in India and conforms to the recommendations of the World Health Organization (WHO) for using combination therapy in malaria. Ranbaxy is also working to make it available in African, Asian and South American markets where Malaria is rampant. SynriamTM trials are ongoing for Plasmodium vivax malaria and a paediatric formulation.
Derek Lowe of the famous In the Pipeline blog had written about arterolane in 2009. At the time it was in Phase III trial, which I assumed were the trials that Ranbaxy was conducting. But it turned out that arterolane was developed by a collaboration between researchers in the US, the UK, Switzerland and Australia who were funded by the World Health Organization and Medicines for Malaria Venture (a Swiss non-profit).
They published this work in Nature in 2004 and further SAR (Structure Activity Relationship) studies in J Med Chem in 2010. So Ranbaxy did not develop the drug from scratch? But the press release quotes Arun Sawhney, CEO and Managing Director of Ranbaxy which misleads people to think so: “It is indeed gratifying to see that Ranbaxy’s scientists have been able to gift our great nation its first new drug, to treat malaria, a disease endemic to our part of the world.
This is a historic day for science and technology in India as well as for the pharmaceutical industry in the country. Today, India joins the elite and exclusive club of nations of the world that have demonstrated the capability of developing a new drug”. So Ranbaxy mixes a known active compound (piperaquine) with a new compound that someone else found to be active (arterolane) and claims that they developed a new drug?
In an interview in LiveMint, Sawhney says, “Ranbaxy spent around $30 million on Synriam and the contribution from DST [India’s Department of Science & Technology] was Rs.5 crore.
The drug went through several phases of development since the project began in 2003. We did not look at this as a commercial development. Instead, this is a CSR [Corporate Social Responsibility] venture for us.” That’s a give away because developing a new drug from scratch has to cost more than $30 million + Rs.50 million.
- Ranbaxy Laboratories Limited (Ranbaxy), India
![]() now taken over by sun
now taken over by sun
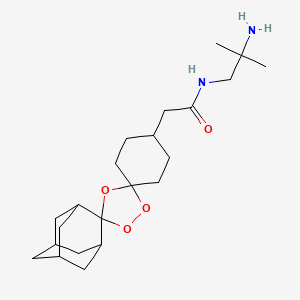
SynriamTM
|
|
Description SynriamTM is a fixed dose combination of two antimalarial active ingredients arterolane maleate and piperaquine phosphate.
Arterolane maleate is a synthetic trioxolane compound. The chemical name of arterolane maleate is cis-adamantane-2-spiro-3’-8’-[[[(2’-amino-2’ methylpropyl) amino] carbonyl] methyl] 1’,2’,4’-trioxaspiro [4.5] decane hydrogen maleate. The molecular formula is C26H40N2O8 and molecular weight is 508.61. The structural formula is as follows:

| MALARIA |
| Malaria is one of the most prevalent and deadly parasitic diseases in the world. Up to 289 million cases of malaria may have occurred in 2010, causing between 660,000 and 1.25 million deaths, mainly in Africa and mostly of children younger than 5 years. |
| (WHO: http://www.who.int/malaria/publications/world_malaria_report_2012/en/index.html; Fidock, D. A. Eliminating Malaria. Science 2013, 340, 1531-1533.) |
|


Malaria, the most common parasitic disease of humans, remains a major health and economic burden in most tropical countries. Large areas of Central and South America, Hispaniola (Haiti and the Dominican Republic), Africa, the Middle East, the Indian subcontinent, Southeast Asia, and Oceania are considered as malaria-risk areas. It leads to a heavy toll of illness and death, especially amongst children and pregnant women.
According to the World Health Organization, it is estimated that the disease infects about 400 million people each year, and around two to three million people die from malaria every year. There are four kinds of malaria parasites that infect human: Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale and Plasmodium malariae.
Malaria spreads from one person to another by the bite of mosquito, Anopheles gambiae, which serves as vector. When a mosquito sucks the blood of human, sporozoites are transfused into the human body together with saliva of the mosquito. The sporozoites enter into the hepatocytes, reproduce asexually and finally enter into the blood stream. The parasites continue to multiply inside the red blood cells, until they burst and release large number of merozoites.
This process continues, destroying a significant number of blood cells and causing the characteristic paroxysm (“chills and fever”) associated with the disease. In the red blood cells, some of the merozoites become male or female gametocytes. These gametocytes are ingested by the mosquito when it feeds on blood. The gametocytes fuse in the vector’s gut; sporozoites are produced and are migrated to the vector’s salivary glands.
The clinical symptoms of malaria are generally associated with the bursting of red blood cells causing an intense fever associated with chills that can leave the infected individual exhausted and bedridden. More severe symptoms associated with repeat infections and/or infection by Plasmodium falciparum include anaemia, severe headaches, convulsions, delirium and, in some instances, death.
Quinine, an antimalarial compound that is extracted from the bark of cinchona tree, is one of the oldest and most effective drugs in existence. Chloroquine and mefloquine are the synthetic analogs of quinine developed in 1940’s, which due to their effectiveness, ease of manufacture, and general lack of side effects, became the drugs of choice. The downside to quinine and its derivatives is that they are short-acting and have bitter taste.
Further, they fail to prevent disease relapses and are also associated with side effects commonly known as “Chinchonism syndrome” characterized by nausea, vomiting, dizziness, vertigo and deafness. However, in recent years, with the emergence of drug- resistant strains of parasite and insecticide-resistant strains of vector, the treatment and/or control of malaria is becoming difficult with these conventional drugs.
Malarial treatment further progressed with the discovery of Artemisinin
(qinghaosu), a naturally occurring endoperoxide sesquiterpene lactone isolated from the plant Artemisia annua (Meshnick et al., Microbiol. Rev. 1996, 60, p. 301-315; Vroman et al., Curr. Pharm. Design, 1999, 5, p. 101-138; Dhingra et al., 2000, 66, p. 279-300), and a number of its precursors, metabolites and semi-synthetic derivatives which have shown to possess antimalarial properties. The antimalarial action of artemisinin is due to its reaction with iron in free heme molecules of the malaria parasite, with the generation of free radicals leading to cellular destruction. This initiated a substantial effort to elucidate its molecular mechanism of action (Jefford, dv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297) and to identify novel antimalarial peroxides (Dong and Vennerstrom, Expert Opin. Ther. Patents 2001, 1 1, p. 1753-1760).
Although the clinically useful artemisinin derivatives are rapid acting and potent antimalarial drugs, they have several disadvantages including recrudescence,
neurotoxicity, (Wesche et al., Antimicrob. Agents. Chemother. 1994, 38, p. 1813-1819) and metabolic instability (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43). A fair number of these compounds are quite active in vitro, but most suffer from low oral activity (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43 and van Agtmael et al., Trends Pharmacol. Sci., 1999, 20, p. 199-205). Further all these artemisinin derivatives are conventionally obtained from plant source and are therefore expensive.
As the cultivation of the plant material is dependent on many factors including the weather conditions, the supply source thus becomes finite and there are chances of varying yield and potency. This leads to quality inconsistencies and supply constraints. As malaria is more prevalent in developing countries, a switch to cheaper and effective medicine is highly desirable.
Thus there exists a need in the art to identify new peroxide antimalarial agents, especially those which are not dependent on plant source and can be easily synthesized, are devoid of neurotoxicity, and which possess improved solubility, stability and pharmacokinetic properties.
Following that, many synthetic antimalarial 1 ,2,4-trioxanes (Jefford, Adv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297), 1,2,4,5-tetraoxanes (Vennerstrom et al., J. Med. Chem., 2000, 43, p. 2753-2758), and other endoperoxides have been prepared. Various patents/applications disclose means and method for treating malaria using Spiro or dispiro 1,2,4-trioxolanes for example, U.S.
Patent Application No. 2004/0186168 and U.S. Patent Nos. 6,486, 199 and 6,825,230. The present invention relates to solid dosage forms of the various spiro or dispiro 1 ,2,4- trioxolanes antimalarial compounds disclosed in these patents/applications and are incorporated herein by reference.
Active compounds representing various Spiro and dispiro 1 ,2,4-trioxolane derivatives possess excellent potency, efficacy against Plasmodium parasites, and a lower degree of neurotoxicity, in addition to their structural simplicity and ease of synthesis. Furthermore, these compounds have half-lives which are believed to permit short-term treatment regimens comparing favorably to other artemisinin-like drugs. In general, the therapeutic dose of trioxolane derivative may range between about 0.1-1000 mg/kg/day, in particular between about 1-100 mg/kg/day. The foregoing dose may be administered as a single dose or may be divided into multiple doses. For malaria prevention, a typical dosing schedule could be, for example, 2.0-1000 mg/kg weekly beginning 1-2 weeks prior to malaria exposure, continued up to 1-2 weeks post-exposure.
Monotherapy with artemisinin (natural or synthetic) class of drugs might cure the patients within 3 days, however perceiving the potential threat of the malarial parasite developing resistance towards otherwise very potent artemisinin class of drugs, WHO had strictly called for an immediate halt to the provision of single-drug artemisinin malaria pills. Combination therapy in case of malaria retards the development of resistance, improve efficacy by lowering recrudescence rate, provides synergistic effect, and increase exposure of the parasite to the drugs.
Artemsinin based combinations are available in the market for a long time.
Artemether-lumafentrine (Co-artem®) was the first fixed dose antimalarial combination containing an artemisinin derivative and has been known since 1999. This combination has passed extensive safety and efficacy trials and has been approved by more than 70 regulatory agencies. Co-artem® is recommended by WHO as the first line treatment for uncomplicated malaria.
Other artemisinin based combinations include artesunate and amodiaquine (Coarsucam®), and dihydroartemisin and piperaquine (Eurartesim®). Unfortunately, all the available artemisinin based combinations have complicated dosage regimens making it difficult and inconvenient for a patient to comply completely with the total prescribed duration. For example, the dosage regimen of Co-artem®for an adult having body weight of more than 35 kg includes 6 doses over three days.
The first dose comprises four tablets initially, the second dose comprises four tablets after eight hours, the third to sixth doses comprise four tablets twice for another two days; making it a total of 24 tablets. The dosage regimen of Coarsucam® for an adult having body weight of more than 36 kg or age above 14 years includes three doses over three days; each dose comprises two tablets; making it a total of six tablets. The dosage regimen of Eurartesim® for an adult having body weight between 36 kg – 75 kg includes 3 doses over three days, each dose comprises of three tablets, making it a total of nine tablets.
It is evident that the available artemisinin-based combinations have a high pill burden on patients as they need to consume too many tablets. As noted above, this may increase the possibility of missing a few doses, and, consequently, could result in reduced efficacy due to non-compliance and may even lead to development of resistance for the drug. Therefore, there is an urgent and unmet need for anti-malarial combinations with a simplified daily dosing regimen that reduces the pill burden and would increase patient compliance.
Apart from simplifying the regimen, there are certain limitations for formulators developing formulations with trioxolones, the first being their susceptibility to degradation in presence of moisture that results in reduced shelf lives. Another is their bitter taste, which can result in poor compliance of the regimen or selection of another, possibly less effective, therapeutic agent.
……………………..
PATENT
http://www.google.st/patents/US6906205

……………………
PATENT
http://www.google.st/patents/WO2013008218A1?cl=en
structural Formula II.

Formula II
Active compound includes one or more of the various spiro and dispiro trioxolane derivatives disclosed in U.S. Application No. 2004/0186168 and U.S. Patent Nos.
6,486,199 and 6,825,230, which are incorporated herein by reference. These trioxolanes are relatively sterically hindered on at least one side of the trioxolane heterocycle which provides better in vivo activity, especially with respect to oral administration. Particularly, spiro and dispiro 1,2,4-trioxolanes derivatives possess excellent potency and efficacy against Plasmodium parasites, and a lower degree of neurotoxicity.
The term “Active compound I” herein means cis-adamantane-2-spiro-3′-8′-[[[(2′- amino-2′-methylpropyl)amino]carbonyl]-methyl]- 1 ‘,2′,4′-trioxaspiro[4.5]decane hydrogen maleate. The Active compound I may be present in an amount of from about 5% to about 25%, w/w based on the total dosage form.

………………
PATENT
http://www.google.st/patents/WO2007138435A2?cl=en
A synthetic procedure for preparing compounds of Formula I, salts of the free base c«-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]- 1 ‘, 2′, 4′-trioxaspiro [4.5] decane has been disclosed in U.S. 6,906,205.

The process for the preparation of compounds of Formula I wherein a compound of Formula II (wherein R is lower alkyl) is reacted with a compound of Formula III (wherein R is lower alkyl) to obtain compound of Formula IV;


Formula Formula IV
followed by hydrolysis of the compounds of Formula IV to give a compound of Formula V;

Formula V followed by the reaction of the compound of Formula V with an activating agent, for example, methyl chloroformate, ethyl chloroformate, propyl chloro formate, n-butyl chloro formate, isobutyl chloroformate or pivaloyl chloride leads to the formation of mixed anhydride, which is reacted in situ reaction with 1 ,2-diamino-2-methyl propane to give a compound of Formula VI; and

Formula Vl reacting the compound of Formula VI with an acid of Formula HX (wherein X can be the same as defined earlier) to give compounds of Formula I.
Example 1 : Preparation of O-methyl-2-adamantanone oxime
To a solution of 2-adamantanone (50 g, 0.3328 mol, 1 equiv.) in methanol (0.25 lit), sodium hydroxide solution (15 g, 0.3761mol, 1.13 equiv, in 50 mL water) was added followed by methoxylamine hydrochloride (37.5 g x 81.59% Purity= 30.596 g, 0.366 mol, 1.1 equiv) at room temperature under stirring. The reaction mixture was stirred at room temperature for 1 to 2 h. The reaction was monitored by HPLC. The reaction mixture was concentrated at 40- 45°C under vacuum to get a thick residue. Water (250 mL) was added at room temperature and the reaction mixture was stirred for half an hour. The white solid was filtered, washed with water (50 mL), and dried at 40 to 45°C under reduced pressure. O-methyl 2- adamantanone oxime (57 g, 95 % yield) was obtained as a white solid.
(M++l) 180, 1HNMR (400 MHz, CDCl3 ): δ 1.98 – 1.79 (m, 12H), 2.53 (s, IH), 3.46 ( s, IH), 3.81 (s, 3H).
Example 2: Preparation of 4-(methoxycarbonvmethvPcvclohexanone A high pressure autoclave was charged with a mixture of methyl (4- hydroxyphenyl)acetate (50 g, 0.30 mol), palladium ( 5g) (10 %) on carbon (50 % wet) and O- xylene (250 mL). The reaction mixture was stirred under 110 to 115 psi of hydrogen pressure for 7 to 8 h at 1400C. The reaction was monitored by HPLC. The reaction mixture was then cooled to room temperature, and the catalyst was filtered off. Filtrate was concentrated under reduced pressure to get 4-(methoxycarbonylmethyl)cyclohexanone as light yellow to colorless oily liquid (48.7 g, 97.4 %).
(M++!) 171, ‘ HNMR (400 MHz, CDCl 3): δ 1.48 – 1.51 ( m, 2H), 2.1 1-2.07 (m, 2H), 2.4- 2.23 (m, 7H), 3.7 (s, 3H).
Example 3: Preparation of methyl (Is, 4s)-dispiro [cyclohexane-l, 3′-f 1,2,4] trioxolane-5′, 2″-tricvclor3.3.1.13–71decan1-4-ylacetate
A solution of O-methyl-2-adamantanone oxime (example 1) (11.06 g, 61.7 mmol, 1.5 equiv.) and 4-(methoxycarbonymethyl)cyclohexanone (example 2) (7.0 g, 41.1 mmol, 1 equiv.) in cyclohexane ( 200ml) and dichloromethane (40 mL) was treated with ozone (ozone was produced with an OREC ozone generator [0.6 L/min. O2, 60 V] passed through an empty gas washing bottle that was cooled to -780C). The solvent was removed after the reaction was complete. After removal of solvents, the crude product was purified by crystallization from 80% aqueous ethanol (200 mL) to afford the title compound as a colorless solid. Yield: 10.83 g, 78%, mp: 96-980C; 1HNMR (500 Hz3CDCl3): δ 1.20-1.33 (m, 2H), 1.61-2.09 (m, 5 21H), 2.22 (d, J = 6.8Hz, 2H), 3.67(s,3H).
Example 4: Preparation of (Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″- tricvclo [3.3.1.13‘7] decanl-4-ylacetic acid
Sodium hydroxide (3.86 g, 96.57 mmol, 3 equiv.) in water (80 mL) was added to a solution of methyl (\s, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo
10 [3.3.1.I3‘7] decan]-4-ylacetate (example 3) (10.83 g, 32.19 mmol, 1 equiv.) in 95% ethanol (150 mL). The mixture was stirred at 500C for about 4 h, cooled to O0C, and treated with IM hydrochloric acid (129ml, 4 equiv). The precipitate was collected by filtration, washed with 50 % aqueous ethanol (150 mL) and dried in vacuum at 40 0C to give the title compound as colorless solid. Yield: 9.952 g, 96%, mp: 146-1480C ( 95% ethanol), 1HNMR (500 Hz,
15 CDCl3): δ 1.19-1.41 (m,2H), 1.60-2.05 (m,21H), 2.27 (d, J=6.8 Hz,2H).
Example 5: Preparation of c?s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, T , 4′-trioxaspiro [4.5] decane
Method A:
(Is, 4s)-dispiro[cyclohexane- 1 ,3 ‘-[ 1 ,2,4]trioxolane-5 ‘,2 ‘ ‘-tricyclo[3.3.1.13‘7]decan]-4-
.0 ylacetic acid (example 4) (5 g ,15.5mmol, 1 equiv) was mixed with triethylamine (2.5 g , 24.8 mmol, 1.6 equiv) in 100ml of dichloromethane. The reaction mixture was cooled to – 1O0C to 00C. Ethyl chloro formate (1.68 g, 17 mmol, 1.0 equiv) in 15 mL dichloromethane was charged to the above reaction mixture at – 100C to 00C. The reaction mixture was stirred at the same temperature for 10 to 30 minutes. The resulting mixed anhydride reaction mixture
15 was added dropwise to a previously prepared solution of l,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv), in 100 mL dichloromethane at -100C to O0C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the same temperature till the reaction was complete. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete
>0 within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (50 mL) was charged, organic layer was separated and washed with 10% sodium bicarbonate solution (50 mL) and water (50 mL) at room temperature. The organic layer was dried over sodium sulphate and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (50ml) was added to obtain residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. The solid was dried under reduced pressure at room 5 temperature.
Yield: 5.2 g (85.4 %), (M++l) 393, 1HNMR (400 MHz, DMSO-J6 ): δ 0.929 ( s, 6H), 1.105 – 1.079 (m, 2H), 1.887-1.641 (m, 21H), 2.030-2.017 (d, 2H), 2.928 (d, 2H).
Method B:
(Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo [3.3.1.I3‘7]
10 decan]-4-ylacetic acid (example 4) (10 g, 31mmol, 1 equiv) was treated with isobutyl chloroformate (4.5 g, 33mmol, 1.1 equiv) in presence of organic base like triethyl amine (5 g, 49.6mmol, 1.6 equiv) at 00C to 7°C in 250ml of dichloromethane. The solution was stirred at O0C to 7°C for aboutlO to 30 minutes. To the above reaction mixture, previously prepared solution of l,2-diamino-2-methylpropane (3.27 g, 37 mmol, 1.2 equiv), in 50 mL of
15 dichloromethane was added at O0C to 7°C in one lot. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. Reaction was complete within 2 h. The reaction nitrogen atmosphere was maintained throughout the reaction. Water (250 mL) was charged, organic
20 layer was separated and washed with 10% sodium bicarbonate solution (200 mL) and water (100 mL) at room temperature and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (100ml) was added to the residue, under stirring, at room temperature. The mixture was filtered and washed with chilled hexane (10 mL). The resultant solid was dried under reduced pressure at room temperature. Yield: 10.63 g (87%), (M++l) 393, 1HNMR
>5 (400 MHz, DMSO-J6 ) :δ 0.928 ( s, 6H), 1.102 – 1.074 (m, 2H), 1.859-1.616 (m, 21H), 2.031- 2.013 (d, 2H), 2.94-2.925 (d, 2H). Method C:
(\s, 4s)-dispiro[cyclohexane-l,3′-[l,2,4]trioxolane-5′,2″-tricyclo[3.3.1.13>7]decan]-4- ylacetic acid (example 4) (5 g, 15.5mmol, 1 equiv) was treated with pivaloyl chloride (1.87 g, 15.5 mmol, 1 equiv) and triethylamine (2.5gm, 24.8mmol, 1.6 equiv) at -15°C to -100C in dichloromethane (125 mL). The solution was stirred at -150C to -100C for aboutlO to 30 minutes. It resulted in the formation of mixed anydride. To the above reaction mixture, previously prepared solution of 1 ,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv) in 25 mL dichloromethane was added at -15°C to -100C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (125 mL) was charged, organic layer was separated and washed with 50 mL of 10% sodium bicarbonate solution and 125 mL of water, respectively at room temperature. Finally solvent was removed at 25 to 4O0C under reduced pressure. 50 mL of 5% Ethyl acetate – hexane solvent mixture was added to the residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. Solid was dried under reduced pressure at room temperature. Yield: 5.03 g (83 %), (M++l) 393, 1JINMR (400 MHz, OMSO-d6 ):δ 0.93 ( s, 6H), 1.113 – 1.069 (m, 2H), 1.861-1.644 (m, 21H), 2.033-2.015 (d, 2H), 2.948-2.933 (d, 2H).
Example 6: Preparation of c/s-adamantane-2-spiro-3′ -8 ‘-πT(2′-amino-2′ -methyl propyl) amino! carbonyl] methyli-l ‘, 2\ 4′-U-JoXaSpJrQ [4.51 decane maleate To a solution of c/s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, 2′, 4′-trioxaspiro [4.5] decane (example 5) (60 g, 0.153 moles) in ethanol (150 mL) was added a solution of maleic acid (17.3 g, 0.15 moles, 0.98 equiv. in ethanol 90 mL) and the reaction mixture was stirred for about 1 h. To this clear solution, n- heptane (720 mL) was added at room temperature in 1 h and the reaction mixture was stirred for 3 h. It was then cooled to 0 to 100C and filtered. The cake was washed with n-heptane (60 mL) and dried under vacuum at 40-450C.
Yield: 67 g, 77.4%, mp: 1490C (decomp), (M++l) 393.5, 1HNMR (300 MHz, DMSO-^ ): δ 1.05-1.11 (2H,m), 1.18 (6H,s), 1.64-1.89 (21H,m), 2.07(2H,d), 3.21 (2H,d), 6.06 (2H,d), 7.797 (2H, bs), 8.07 (IH, t).



References
- Dong, Yuxiang; Wittlin, Sergio; Sriraghavan, Kamaraj; Chollet, Jacques; Charman, Susan A.; Charman, William N.; Scheurer, Christian; Urwyler, Heinrich et al. (2010). “The Structure−Activity Relationship of the Antimalarial Ozonide Arterolane (OZ277)”. Journal of Medicinal Chemistry 53 (1): 481–91. doi:10.1021/jm901473s. PMID 19924861.
- Blow to Ranbaxy drug research plans at LiveMint.com, Sep 21 2007
- Vennerstrom, Jonathan L.; Arbe-Barnes, Sarah; Brun, Reto; Charman, Susan A.; Chiu, Francis C. K.; Chollet, Jacques; Dong, Yuxiang; Dorn, Arnulf et al. (2004). “Identification of an antimalarial synthetic trioxolane drug development candidate”. Nature 430 (7002): 900–4.doi:10.1038/nature02779. PMID 15318224.
- In the Pipeline: “Ozonides As Drugs: What Will They Think Of Next?”, by Derek Lowe, November 23, 2009, at Corante.com
- Indian company starts Phase III trials of synthetic artemisinin, May 4 2009, at the WorldWide Antimalarial Resistance Network
- http://www.nature.com/nature/journal/v430/n7002/full/nature02779.html
|
5-27-2011
|
PROCESS FOR THE PREPARATION OF DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS (OZ277)
|
|
|
2-13-2009
|
STABLE DOSAGE FORMS OF SPIRO AND DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS
|
|
|
6-15-2005
|
Spiro and dispiro 1,2,4-trioxolane antimalarials
|
|
|
11-31-2004
|
Spiro and dispiro 1,2,4-trixolane antimalarials
|
ANTIMALARIALS




http://www.rsc.org/chemistryworld/2013/03/new-antimalarial-drug-class-resistance-elq-300-quinolone

http://www.nature.com/nrd/journal/v9/n11/full/nrd3301.html
Structure of NITD609; the 1R,3Sconfiguration is fundamental for its antimalarial activity
FDA gives green light to Novartis acromegaly drug Pasireotide

Pasireotide, Signifor; SOM 320; HY-16381; 396091-73-9
Cyclo[4(R)-[N-(2-aminoethyl)carbamoyloxy]-L-prolyl-L-phenyl-glycyl-D-tryptophyl-L-lysyl-(4-O-benzyl)-L-tyrosyl-L-phenylalanyl]bis(L-aspartic acid)
Regulators in the USA has approved a long-acting release of Novartis’ Signifor as a treatment for acromegaly.
Read more at: http://www.pharmatimes.com/Article/14-12-16/FDA_gives_green_light_to_Novartis_acromegaly_drug.aspx#ixzz3M8Ibn14Q
clinical…..https://clinicaltrials.gov/search/intervention=Pasireotide+OR+SOM-230
Pasireotide (SOM230, trade name Signifor[1]) is an orphan drug approved in the U.S. and Europe for the treatment of Cushing’s disease in patients who fail or are ineligible for surgical therapy.[2][3] It was developed by Novartis. Pasireotide is a somatostatinanalog which has a 40-fold increased affinity to somatostatin receptor 5 than other somatostatin analogs.
The drug showed therapeutical potential in a recent study (PASPORT-CUSHINGS B2305) where 162 patients were treated with either 2x 600 µg or 2x 900 µg pasireotide s.c. daily.[4] The effectiveness of the treatment was checked by the UFC-value (urinary free cortisol) after six months of treatment. The mean reduction of UFC after six months was 47.9%, which also lead to amelioration of clinical symptoms such as blood pressure, cholesterol value, and weight loss.[5]
Pasireotide was approved by the EMEA in October 2009[6] and by the FDA in December 2012.[7]
At present, it is in phase III clinical trials at Novartis for the treatment of carcinoid tumors and symptoms that are not adequately controlled by somatostatin analogues (Sandostatin). Phase II clinical development is also under way at the company for the treatment of gastric dumping syndrome, metastatic carcinoid tumors, meningioma and pituitary adenoma and for the treatment of hepatocellular carcinoma in combination with everolimus. Early clinical trials are also ongoing for the treatment of patients with metastatic melanoma or Merkel cell carcinoma. A phase I clinical trial for the treatment of alcoholic cirrhosis has been completed. The company intends to file for approval in 2007 for these indications. Novartis and Thomas Jefferson University are conducting phase II clinical trials for the treatment of prostate cancer, alone or in combination with everolimus. The Mayo Clinic is conducting phase II clinical trials for the treatment of polycystic liver disease. Phase III clinical trials had been ongoing for the reduction of post-pancreatectomy fistula, leak, and abscess; however, in 2010 these trials were suspended. In 2004, orphan drug designation was assigned in the E.U. for the treatment of functional gastroenteropancreatic endocrine tumors. In 2009, orphan drug designation was received in the U.S. and the E.U. for the treatment of Cushing’s disease and acromegaly. The designation for the treatment of Cushing’s disease was assigned in Australia in 2011 and in Japan in 2012. In 2013, orphan drug designation was assigned in Australia for the treatment of acromegaly.

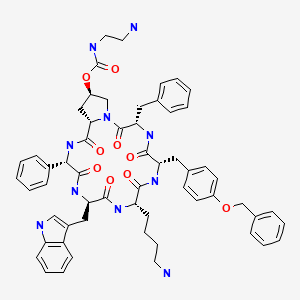
SIGNIFOR (pasireotide diaspartate) injection is prepared as a sterile solution of pasireotide diaspartate in a tartaric acid buffer for administration by subcutaneous injection. SIGNIFOR is a somatostatin analog. Pasireotide diaspartate, chemically known as (2-Aminoethyl) carbamic acid (2R,5S,8S,11S,14R,17S,19aS)-11-(4-aminobutyl)-5-benzyl-8-(4-benzyloxybenzyl)-14-(1H-indol-3ylmethyl)-4,7,10,13,16,19-hexaoxo-17-phenyloctadecahydro-3a,6,9,12,15,18hexaazacyclopentacyclooctadecen-2-yl ester, di[(S)-2-aminosuccinic acid] salt, is a cyclohexapeptide with pharmacologic properties mimicking those of the natural hormone somatostatin.
The molecular formula of pasireotide diaspartate is C58H66N10O9 • 2C4H7NO4 and the molecular weight is 1313.41. The structural formula is:
 SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
Each glass ampule contains:
| 0.3 MG | 0.6 MG | 0.9 MG | |
| Pasireotide diaspartate | 0.3762* | 0.7524* | 1.1286* |
| Mannitol | 49.5 | 49.5 | 49.5 |
| Tartaric acid | 1.501 | 1.501 | 1.501 |
| Sodium hydroxide | ad pH 4.2 | ad pH 4.2 | ad pH 4.2 |
| Water for injection | ad 1ml | ad 1ml | ad 1ml |
| * corresponds to 0.3/0.6/0.9 mg pasireotide base Note: Each ampule contains an overfill of 0.1ml to allow accurate administration of 1 ml from the ampule. |
|||
 |
|
| Systematic (IUPAC) name | |
|---|---|
| [(3S,6S,9S,12R,15S,18S,20R)-9-(4-aminobutyl)-3-benzyl-12-(1H-indol-3-ylmethyl)-2,5,8,11,14,17-hexaoxo-15-phenyl-6-[(4-phenylmethoxyphenyl)methyl]-1,4,7,10,13,16-hexazabicyclo[16.3.0]henicosan-20-yl] N-(2-aminoethyl)carbamate | |
| Clinical data | |
| Trade names | Signifor |
| Licence data | EMA:Link |
| Legal status |
|
| Routes | Subcutaneous |
| Identifiers | |
| CAS number | 396091-73-9 |
| ATC code | H01CB05 |
| PubChem | CID 9941444 |
| UNII | 98H1T17066 |
| Synonyms | SOM230 |
| Chemical data | |
| Formula | C58H66N10O9 |
| Mol. mass | 1107.26 g/mol |
Pasireotide is a multiligand somatostatin analogue with high binding affinity to somatostatin receptors sst1, sst2, sst3 and sst5. Novartis Oncology, a division of Novartis, filed for approval in the E.U. for the treatment of Cushing’s syndrome in 2010. A positive opinion was granted in 2011 and final approval was obtained in 2012. The E.U.’s first launch took place in Germany in June 2012. Also in 2011, Novartis filed an NDA in the U.S. seeking approval of the compound for the treatment of Cushing’s syndrome; however, the application was withdrawn the same year due to an issue related to chemistry, manufacturing and controls. In November 2012, the product was recommended for approval in the U.S. for Cushing’s syndrome. In December 2012, final FDA approval was granted. Phase III clinical trials are ongoing in Japan for this indication. In 2014, the product was approved in the E.U and the U.S. for the treatment of adult patients with acromegaly for whom surgery is not an option or has not been curative and who are inadequately controlled on treatment with a first-generation somatostatin analogue (SSA).
- http://www.google.com/patents/EP2310042B1?cl=en
-
The present invention relates to a new use of Somatostatin (SRIF) peptidomimetics (also referred to as Somatostatin- or SRIF-analogs).
-
Somatostatin is a tetradecapeptide having the structure

-
The somatostatin class is a known class of small peptides comprising the naturally occurring somatostatin-14 and analogues having somatostatin related activity, e.g. as disclosed by A.S. Dutta in Small Peptides, Vol.19, Elsevier (1993). By “somatostatin analog” as used herein is meant any straight-chain or cyclic polypeptide having a structure based on that of the naturally occurring somatostatin-14 wherein one or more amino acid units have been omitted and/or replaced by one or more other amino radical(s) and/or wherein one or more functional groups have been replaced by one or more other functional groups and/or one or more groups have been replaced by one or several other isosteric groups. In general, the term covers all modified derivatives of the native somatostatin-14 which exhibit a somatostatin related activity, e.g. they bind to at least one of the five somatostatin receptor (SSTR), preferably in the nMolar range.
-
Natural somatostatin binds and activates all 5 somatostatin receptors (SSTR1-5) with nmol efficacy and thus causes its multiple physiological effects.
-
Synthetically available somatostatin analogs differ in their binding affinity to the different somatostatin receptor subtypes and often bind selectively to one or few subtypes with significantly higher affinity.
-
Somatostatin analogs of particular interest according to the present invention have a high binding affinity to human SSTR1,2,3,5 and have been described e.g. in WO 97/01579 , the contents of which being incorporated herein by reference. Said somatostatin analogs comprise the amino acid sequence of formula I-(D/L)Trp-Lys-X1 -X2 - Iwherein X1 is a radical of formula (a) or (b)

wherein R1 is optionally substituted phenyl, wherein the substituent may be halogen, methyl, ethyl, methoxy or ethoxy,
R2 is -Z1-CH2-R1, -CH2-CO-O-CH2-R1,
wherein Z1 is O or S, and
X2 is an α-amino acid having an aromatic residue on the Cα side chain, or an amino acid unit selected from Dab, Dpr, Dpm, His,(Bzl)HyPro, thienyl-Ala, cyclohexyl-Ala and t-butyl-Ala, the residue Lys of said sequence corresponding to the residue Lys9 of the native somatostatin-14. -
Somatostatin analogs of particular interest which have a high binding affinity to human SSTR1,2,3,5 have also been described e.g. inWO02/10192. Said somatostatin analogs comprise the compound of formula

also called cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] or pasireotide, as well as diastereoisomers and mixtures thereof, in free form, in salt or complex form or in protected form. Phg means -HN-CH(C6H5)-CO- and Bzl means benzyl.




…………………
http://www.google.com/patents/WO2002010192A2?cl=en
Example 1 : Cyclo[{4-(NH2-C2H4-NH-CO-O-
a) Synthesis of Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
L-hydroxyproline methylester hydrochloride is reacted with Fmoc-OSu in aqueous 1.0 N sodium carbonate/THF at room temperature. After completion of the reaction, Fmoc-Pro(4- OH)-OMe is isolated by precipitation. Fmoc-Pro(4-OH)-OMe is then added dropwise into a solution of trisphosgene (0.6 eq.) in THF to give a chlorocarbonate intermediate. After 1 h dimethylaminopyridine (1.0 eq.) and N-Boc-diaminoethane (6.0 eq.) are added and the reaction is stirred at room temperature. After completion of the reaction, the solvent is removed in vacuo and the resulting Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMe is extracted from a two phase system of ethyl acetate/0.1 M HCI to give crude product (MH+ = 554) which is purified by crystallization from ethyl acetate. The methyl ester is then cleaved to the free acid by treatment with 1 N NaOH in dioxane/water and the product Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH is purified on silica gel, [(M+Na)]+= 562).
b) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH Commercially available Fmoc-Tyr(Bzl)-O-CH2-Ph(3-OCH3)-O-CH2-Polystyrene resin (SASRIN-resin, 2.4 mM) is used as starting material and carried through a standard protocol consisting of repetitive cycles of Nα-deprotection (Piperidine/DMF, 2:8), repeated washings with DMF and coupling (DIPCI: 4.8 mM/HOBT: 6mM, DMF). The following amino acid- derivatives are sequentially coupled: Fmoc-Lys(Boc)-OH, Fmoc-DTrp(Boc)-OH, Fmoc-Phg- OH, Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH, Fmoc-Phe-OH. Couplings (2 eq. amino acids) are continued or repeated until completion, i.e. until complete disappearance of residual amino groups which is monitored by a negative ‘Kaiser* Ninhydrin test. Before cleavage of the completely assembled protected linear peptide from its resin support the Nα-Fmoc protection from the last residue is removed.
c) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH After washings with CH2CI2) the peptide-resin is transferred into a column or a stirred suction filter and the peptide fragment is cleaved and eluted with a short treatment with 2% TFA in CH2CI2 within 1 h. The eluate is immediately neutralized with a saturated NaHCO3 solution. The organic solution is separated and evaporated and the side chain protected precursor (MH+ = 1366) is cyclized without further purification.
d) cyclo[-Pro(4-OCO-NH-CH2-CH2-NH2)-Phg-DT -Lys-Tyr(Bzl)-Phe-], trifluoroacetate The above linear fragment is dissolved in DMF (4 mM), cooled to minus 5°C and treated with 2 eq. DIPEA then 1.5 eq. of DPPA and stirred until completion (ca. 20h) at 0-4°C. The solvent was almost completely removed in vacuo; the concentrate is diluted with ethyl acetate, washed with NaHCO3, water, dried and evaporated in vacuo.
For deprotection the residue is dissolved at 0°C in TFA H2O 95:5 (ca.50 mM) and stirred in the cold for 30 min. The product is then precipitated with ether containing ca. 10 eq. HCI, filtered, washed with ether and dried. In order to completely decompose remaining Indole-N carbaminic acid the product is dissolved in 5% AcOH and lyophilized after 15 h at ca. 5°C. Preparative RP-HPLC is carried out on a C-18 10 μm STAGROMA column (5-25 cm) using a gradient of 0.5% TFA to 0.5% TFA in 70% acetonitrile. Fractions containing the pure title compound are combined, diluted with water and lyophilized. The lyophilisate is dissolved in water followed by precipitation with 10% Na2CO3 in water. The solid free base is filtered of, washed with water and dried in vacuum at room temperature. The resulting white powder is directly used for the different salts.
Example 2: Cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] in salt form a. Acetate
Conversion to the acetate salt form is carried out using an ion-exchange resin (e.g. AG 3- X4). MS (ESI): m/z 524.5 [M+2H]2+ [α]D 20= -42°, c=0.26 in AcOH 95%
b. Aspartate
Conversion to the mono- or di-aspartate is obtained by reacting 1 equivalent of the compound of Example 1 with 1 or 2 equivalent of aspartic acid in a mixture of acetonitrile/water 1 :3. The resulting mixture is frozen and lyophilized. The di-aspartate may also be obtained by dissolving the compound of Example 1 in water/acetonitrile 4:1, filtering, loading on a an ion-exchange resin, e.g. BioRad AG4X4 column, and eluting with water/acetonitrile 4:1. The eluate is concentrated, frozen and lyophilized. [ ]D 20= -47.5°, c= 2.5mg/ml in methanol
……………..
WO2013/174978 A1
http://www.google.im/patents/WO2013174978A1?cl=ru
………………………..
WO2013/131879 A1,
………………………..
WO2005/53732 A1,
http://www.google.com/patents/WO2005053732A1?cl=en
……………………………
Journal of Medicinal Chemistry, 2003 , vol. 46, 12 pg. 2334 – 2344
http://pubs.acs.org/doi/abs/10.1021/jm021093t

A rational drug design approach, capitalizing on structure−activity relationships and involving transposition of functional groups from somatotropin release inhibitory factor (SRIF) into a reduced size cyclohexapeptide template, has led to the discovery of SOM230 (25), a novel, stable cyclohexapeptide somatostatin mimic that exhibits unique high-affinity binding to human somatostatin receptors (subtypes sst1−sst5). SOM230 has potent, long-lasting inhibitory effects on growth hormone and insulin-like growth factor-1 release and is a promising development candidate currently under evaluation in phase I clinical trials.

| residue | group | δ 1H [ppm] | δ 13C [ppm] | residue | group | δ 1H [ppm] | δ 13C [ppm] |
| 1 | l-phenylglycine |
| 1 | NH | 9.73 | 1 | α-CH | 6.47 | 59.3 | |||
| 1 | 2/6-CH | 8.02 | 127.3 | 1 | CO | 169.6 | |||
| 1 | 3/5-CH | 7.41 | 129.1 | 1 | 1-C | 141.0 |
| 1 | 4-CH | 7.21 | 128.0 | |
| 2 | d-tryptophane |
| 2 | 1‘-NH | 12.20 | 2 | α-CH | 5.28 | 55.6 | |||
| 2 | NH | 10.34 | 2 | β-CH2 | 3.72 | 3.30 | 28.5 | ||
| 2 | 7-CH | 7.65 | 112.0 | 2 | CO | 173.9 | |||
| 2 | 4-CH | 7.43 | 119.2 | 2 | 8-C | 137.5 | |||
| 2 | 2-CH | 7.28 | 124.7 | 2 | 9-C | 128.3 | |||
| 2 | 6-CH | 7.23 | 121.6 | 2 | 3-C | 110.3 |
| 2 | 5-CH | 6.96 | 119.2 | |
| 3 | l-lysine |
| 3 | NH | 10.10 | 3 | δ-CH2 | 1.41 | 1.32 | 31.5 | ||
| 3 | α-CH | 4.62 | 55.2 | 3 | γ-CH2 | 0.89 | 23.5 | ||
| 3 | ε-CH2 | 2.80 | 41.0 | 3 | CO | 171.9 | |||
| 3 | β-CH2 | 1.87 | 1.32 | 31.6 | 3 | NH3+ | a |
| 4 | (4-O-benzyl)-l-tyrosine | |||
| 4 | NH | 7.99 | 4 | 7-CH2 | 4.92 | 69.9 | |||
| 4 | 2‘/6‘-CH | 7.46 | 128.0 | 4 | β-CH2 | 3.46 | 3.10 | 39.7 | |
| 4 | 3‘/5‘-CH | 7.37 | 128.9 | 4 | CO | 171.8 | |||
| 4 | 4‘-CH | 7.30 | 128.2 | 4 | 4-C | 157.9 | |||
| 4 | 2/6-CH | 7.21 | 131.5 | 4 | 1‘C | 137.9 | |||
| 4 | 3/5-CH | 6.85 | 114.7 | 4 | 1-C | 129.8 |
| 4 | α-CH | 5.23 | 53.1 | |
| 5 | l-phenylalanine |
| 5 | NH | 9.82 | 5 | α-CH | 4.42 | 53.9 | |||
| 5 | 2/6-CH | 7.38 | 130.0 | 5 | β-CH2 | 3.23 | 3.06 | 37.8 | |
| 5 | 3/5-CH | 7.27 | 129.3 | 5 | CO | 171.2 | |||
| 5 | 4-CH | 7.16 | 127.6 | 5 | 1-C | 136.3 |
| 6 | (γ-O-diaminoethylcarbamate)-l-hydroxyproline | ||||||
| 6 | 2-NH | 8.04 | 6 | 4-CH2 | 2.95 | 42.4 | |||
| 6 | γ-CH | 5.23 | 70.9 | 6 | β-CH2 | 2.63 | 1.25 | 37.0 | |
| 6 | α-CH | 4.22 | 60.6 | 6 | CO | 170.7 | |||
| 6 | δ-CH2 | 4.12 | 51.4 | 6 | 1-CO | 156.7 | |||
| 6 | 3-CH2 | 3.42 | 44.5 | 6 | 4-NH3+ | a |
| A | acetate |
| A | CH3 | 2.20 | 22.1 | A | CO | 174.3 |
a The NH3+ protons are part of the water peak at 5.82 ppm.
References
- Signifor® (pasireotide) Official Website for healthcare professionals outside the US http://www.signifor.com/
- “Novartis drug Signifor® approved in the EU as the first medication to treat patients with Cushing’s disease”. Retrieved 2012-07-08.
- Mancini et al. Therapeutics and Clinical Risk Management 2010;6:505-516
- Colao et al. Pasireotide (SOM230) provides clinical benefit in patients with Cushing’s disease: results from a large, 12-month, randomized-dose, double-blind, Phase III study, Abstract OC1.7. European Neuroendocrine Association (ENEA) 14th Congress, 2010:62-63
- U.S. National Library of Medicine: Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. http://www.ncbi.nlm.nih.gov/pubmed/18957506?dopt=Abstract
- EMEA Approval for Pasireotide
- “FDA Approves Pasireotide for Cushing’s Disease”.
| WO2005117830A1 | 6 Jun 2005 | 15 Dec 2005 | Camurus Ab | Liquid depot formulations |
| WO2006075124A1 * | 9 Dec 2005 | 20 Jul 2006 | Camurus Ab | Somatostatin analogue formulations |
| WO2006131730A1 | 6 Jun 2006 | 14 Dec 2006 | Camurus Ab | Glp-1 analogue formulations |
| WO2007096055A1 * | 7 Feb 2007 | 30 Aug 2007 | Novartis Ag | Combination of somatostatin-analogs with different selectivity for human somatostatin receptor subtypes |
| WO2010003939A1 * | 7 Jul 2009 | 14 Jan 2010 | Novartis Ag | Use of pasireotide for the treatment of endogenous hyperinsulinemic hypoglycemia |
| US20090155193 * | 9 Dec 2005 | 18 Jun 2009 | Fredrik Joabsson | Topical Bioadhesive Formulations |
LEXIPAFANT

GR-167089
ISV-611
………………………………
Cadila banks on diabetes drug, Lipaglyn, Saroglitazar
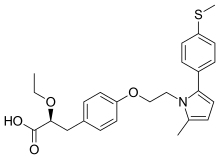
(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid magnesium salt
- saroglitazar
- ZYH1 compound
- E0YMX3S4JD
- cas no 495399-09-2
Saroglitazar, Lipaglyn
| Molecular Weight | 439.56706 g/mol |
|---|---|
| Molecular Formula | C25H29NO4S |
 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Cadila banks on diabetes drug
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet …http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp

Mumbai, June 5: Cadila Healthcare will launch a homegrown drug against diabetes by the third quarter of this year.
The Drug Controller General of India has approved its drug — Lipaglyn —…
View original post 2,491 more words
FDA approves Gardasil 9 for prevention of certain cancers caused by five additional types of HPV

GARDASIL also helps protect girls and young women ages 9 to 26 against approximately 70% of vaginal cancer cases and up to 50% of vulvar cancer cases.
GARDASIL may not fully protect everyone, nor will it protect against diseases caused by other HPV types or against diseases not caused by HPV. GARDASIL does not prevent all types of cervical cancer, so it’s important for women to continue routine cervical cancer screenings. GARDASIL does not treat cancer or genital warts. GARDASIL is given as 3 injections over 6 months.
尼达尼布 ニンテダニブ NINTEDANIB For Idiopathic pulmonary fibrosis

NINTEDANIB, BBIF 1120, Intedanib
Boehringer Ingelheim Corp
Launched 2014 USA….Idiopathic pulmonary fibrosis
| chinese, japanese | 尼达尼布 ニンテダニブ |
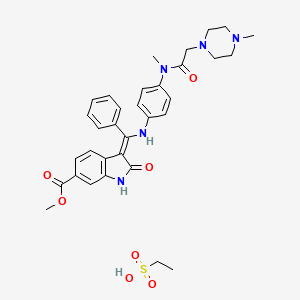
Ethanesulfonic acid – methyl (3Z)-3-{[(4-{methyl[(4-methyl-1-piperazinyl)acetyl]amino}phenyl)amino](phenyl)methylene}-2-oxo-6-indolinecarboxylate (1:1)
Nintedanib esylate
Cas 656247-18-6 [RN]
Methyl (3Z)-3-[({4-[N-methyl-2-(4-methylpiperazin-1-yl)acetamido]phenyl}amino)(phenyl)methylidene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylate ethanesulfonate
Nintedanib esylate [USAN]
(3Z)-2,3-Dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-1H-indole-6-carboxylic acid methyl ester ethanesulfonate
1H-Indole-6-carboxylic acid, 2,3dihydro-3-[[[4-[methyl[(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-,methyl ester, (3Z)-, ethanesulfonate (1:1)
ニンテダニブエタンスルホン酸塩
Highly crystalline (mp = 305 °C) and exhibits a log P of 3.0 and good aqueous solubility (>20 mg/mL in water)…..J. Med. Chem., 2015, 58 (3), pp 1053–1063

Trade Name:Ofev® / Vargatef®
MOA:Tyrosine kinase inhibitor
Indication:Idiopathic pulmonary fibrosis (IPF); Non small cell lung cancer (NSCLC)
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
Nintedanib (formerly BIBF 1120) is a small molecule tyrosine-kinase inhibitor, targeting vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet derived growth factor receptor (PDGFR) being developed by Boehringer Ingelheim as an anti-angiogenesis anti-cancer agent under the trade name Vargatef, and recently approved for treatment of idiopathic pulmonary fibrosis as Ofev.
Mechanism of action
Nintedanib is an indolinone-derived drug that inhibits the process of blood vessel formation (angiogenesis). Angiogenesis inhibitors stop the formation and reshaping of blood vessels in and around tumours, which reduces the tumour’s blood supply, starving tumour cells of oxygen and nutrients leading to cell death and tumour shrinkage. Unlike conventional anti-cancer chemotherapy which has a direct cell killing effect on cancer cells, angiogenesis inhibitors starve the tumour cells of oxygen and nutrients which results in tumour cell death. One of the advantages of this method of anti-cancer therapy is that it is more specific than conventional chemotherapy agents, therefore results in fewer and less severe side effects than conventional chemotherapy.
The process of new blood vessel formation (angiogenesis) is essential for the growth and spread of cancers. It is mediated by signaling molecules (growth factors) released from cancer cells in response to low oxygen levels. The growth factors cause the cells of the tumour’s blood vessel to divide and reorganize resulting in the sprouting of new vessels in and around the tumour, improving its blood supply.
Angiogenesis is a process that is essential for the growth and spread of all solid tumours, blocking it prevents the tumour from growing and may result in tumour shrinkage as well as a reduction in the spread of the cancer to other parts of the body. Nintedanib exerts its anti-cancer effect by binding to and blocking the activation of cell receptors involved in blood vessel formation and reshaping (i.e. VEGFR 1-3, FGFR 1-3 AND PDGFRα and β). Inhibition of these receptors in the cells that make up blood vessels (endothelial cells, smooth muscle cells and pericytes) by Nintedanib leads to programmed cell death, destruction of tumor blood vessels and a reduction in blood flow to the tumour. Reduced tumour blood flow inhibits tumor cell proliferation and migration hence slowing the growth and spread of the cancer.[1]
Adverse effects
Preclinical studies have shown that nintedanib binds in a highly selective manner to the ATP binding pocked of its three target receptor families, without binding to similarly shaped ATP domains in other proteins, which reduces the potential for undesirable side effects.[2]
The most common side effects observed with nintedanib were reversible elevation in liver enzymes (10-28% of patients) and gastrointestinal disturbance (up to 50%). Side effects observed with nintedanib were worse with the higher 250 mg dose, for this reason subsequent trials have used the equally clinically effective 200 mg dose.[1][2][3][4][5][6][7][8][9]
Nintedanib inhibits the growth and reshaping of blood vessels which is also an essential process in normal wound healing and tissue repair. Therefore a theoretical side effect of nintedanib is reduced wound healing however, unlike other anti-angiogenic agents, this side effect has not been observed in patients receiving nintedanib.
Studies
Preclinical studies have demonstrated that nintedanib selectively binds to and blocks the VEGF, FGF and PDGF receptors, inhibiting the growth of cells that constitute the walls of blood vessels (endothelial and smooth muscle cells and pericytes) in vitro. Nintedanib reduces the number and density of blood vessels in tumours in vivo, resulting in tumour shrinkage.[1][2] Nintedanib also inhibits the growth of cells that are resistant to existing chemotherapy agents in vitro, which suggests a potential role for the agent in patients with solid tumours that are unresponsive to or relapse following current first line therapy.[10]
Early clinical trials of nintedanib have been carried out in patients with non-small cell lung, colorectal, uterine, endometrial, ovarian and cervical cancer and multiple myeloma.[4][5][7][8][9] These studies reported that the drug is active in patients, safe to administer and is stable in the bloodstream. They identified that the maximum tolerated dose of nintedanib is 20 0 mg when taken once a day.
Clinical studies
In the first human trials, nintedanib halted the growth of tumours in up to 50% of patients with non-small cell lung cancer and 76% of patients with advanced colorectal cancer and other solid tumours.[4][8] A complete response was observed in 1/26 patients with non-small cell lung and 1/7 patients with ovarian cancer treated with nintedanib. A further 2 patients with ovarian cancer had partial responses to nintedanib.[8][9]
Two phase II trials have been carried out assessing the efficacy, dosing and side effects of nintedanib in non-small cell lung and ovarian cancer. These trials found that nintedanib delayed relapse in patients with ovarian cancer by two months[6] and that overall survival of patients with non-small cell lung who received nintedanib was similar to that observed with the FDA approved VEGFR inhibitor sorafenib. These trials also concluded that increasing the dose of the nintedanib has no effect on survival.[3]
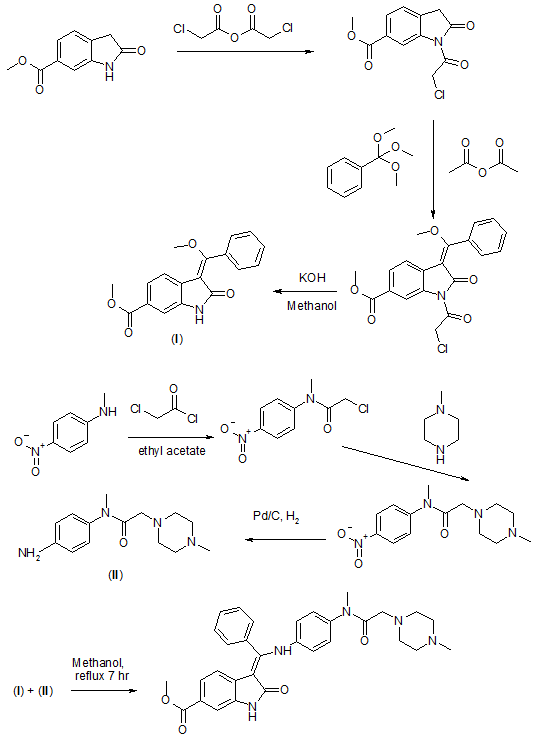
SYNTHESIS
WO2009071523A1
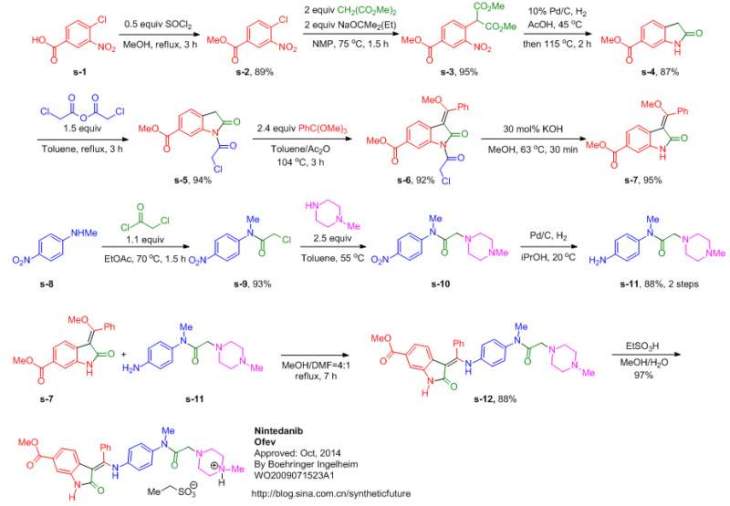
MORE SYNTHESIS

Reference:1. WO0127081A1.
2. US6762180B1.
3. J. Med. Chem. 2009, 52, 4466-4480.

Reference:1. WO2009071523A1 / US8304541B2.

Reference:1. CN104262232A.

Reference:1. CN104844499A.
Current clinical trials
Nintedanib is being tested in several phase I to III clinical trials for cancer. Angiogenesis inhibitors such as nintedanib may be effective in a range of solid tumour types including; lung, ovarian, metastatic bowel, liver and brain cancer. Patients are also being recruited for three phase III clinical trials that will evaluate the potential benefit of nintedanib when added to existing 1st line treatments in patients with ovarian.[11] and 2nd line treatment in non-small cell lung cancer [12][13] The phase III trials of nintedanib in lung cancer have been named LUME-Lung 1 and LUME-Lung 2.
Current phase II trials are investigating the effect of nintedanib in patients with metastatic bowel cancer, liver cancer and the brain tumour: glioblastoma multiforme.[14]
Phase III trials are investigating the use of nintedanib in combination with the existing chemotherapy agents permexetred and docetaxel in patients with non-small cell lung cancer,[15] and in combination with carboplatin and paclitaxel as a first line treatment for patients with ovarian cancer.[16]
A phase III clinical trial was underway examining the safety and efficacy of nintedanib on patients with the non-cancerous lung condition idiopathic pulmonary fibrosis.[17] Nintedanib, under the brand name Ofev, was approved by the FDA for treatment of idiopathic pulmonary fibrosis on 15 Oct 2014. [18]
In terms of clinical development, additional phase III clinical trials are ongoing for the treatment of epithelial ovarian cancer, fallopian tube or primary peritoneal cancer, in combination with chemotherapy, and for the treatment of refractory metastatic colorectal cancer. Phase II clinical trials are also ongoing at the company for the treatment of glioblastoma multiforme, previously untreated patients with renal cell cancer, and for the treatment of patients with unresectable malignant pleural mesothelioma. The National Cancer Center of Korea (NCC) is evaluating the compound in phase II studies as second line treatment for small cell lung cancer (SCLC). The Centre Oscar Lambret is also conducting phase II clinical trials for the treatment of breast cancer in combination with docetaxel. Phase II trials are under way at EORTC as second line therapy for patients with either differentiated or medullary thyroid cancer progressing after first line therapy. The compound is also in early clinical development for the treatment of cancer of the peritoneal cavity, hepatocellular carcinoma, acute myeloid leukemia and ovarian cancer. Clinical trials have been completed for the treatment of prostate cancer and for the treatment of colorectal cancer. Boehringer Ingelheim is also conducting phase I/II clinical trials for the treatment of NSCLC and acute myeloid leukemia in addition to low-dose cytarabine. Phase I clinical studies are ongoing at the company for the treatment of epithelial ovary cancer and for the treatment of patients with mild and moderate hepatic impairment. The company had been evaluating the compound in early clinical trials for the treatment of prostate cancer in combination with docetaxel, but recent progress reports for this indication are not available at present.
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
PAPER
http://pubs.acs.org/doi/full/10.1021/jm501562a
Nintedanib: From Discovery to the Clinic

Nintedanib (BIBF1120) is a potent, oral, small-molecule tyrosine kinase inhibitor, also known as a triple angiokinase inhibitor, inhibiting three major signaling pathways involved in angiogenesis. Nintedanib targets proangiogenic and pro-fibrotic pathways mediated by the VEGFR family, the fibroblast growth factor receptor (FGFR) family, the platelet-derived growth factor receptor (PDGFR) family, as well as Src and Flt-3 kinases. The compound was identified during a lead optimization program for small-molecule inhibitors of angiogenesis and has since undergone extensive clinical investigation for the treatment of various solid tumors, and in patients with the debilitating lung disease idiopathic pulmonary fibrosis (IPF). Recent clinical evidence from phase III studies has shown that nintedanib has significant efficacy in the treatment of NSCLC, ovarian cancer, and IPF. This review article provides a comprehensive summary of the preclinical and clinical research and development of nintedanib from the initial drug discovery process to the latest available clinical trial data.
-
Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt,U.; Walter, R.; Hilberg, F.Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120) J. Med. Chem.2009, 52, 4466– 4480
-
2.Roth, G. J.; Sieger, P.; Linz, G.; Rall, W.; Hilberg, F.; Bock, T. 3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone monoethanesulphonate and the use thereof as a pharmaceutical composition. WO2004/013099. 2004.
-
3.Merten, J.; Linz, G.; Schnaubelt, J.; Schmid, R.; Rall, W.; Renner, S.; Reichel, C.; Schiffers, R. Process for the manufacture of an indolinone derivative. WO2009/071523. 2009
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm900431g

Inhibition of tumor angiogenesis through blockade of the vascular endothelial growth factor (VEGF) signaling pathway is a new treatment modality in oncology. Preclinical findings suggest that blockade of additional pro-angiogenic kinases, such as fibroblast and platelet-derived growth factor receptors (FGFR and PDGFR), may improve the efficacy of pharmacological cancer treatment. Indolinones substituted in position 6 were identified as selective inhibitors of VEGF-, PDGF-, and FGF-receptor kinases. In particular, 6-methoxycarbonyl-substituted indolinones showed a highly favorable selectivity profile. Optimization identified potent inhibitors of VEGF-related endothelial cell proliferation with additional efficacy on pericyctes and smooth muscle cells. In contrast, no direct inhibition of tumor cell proliferation was observed. Compounds 2 (BIBF 1000) and 3 (BIBF 1120) are orally available and display encouraging efficacy in in vivo tumor models while being well tolerated. The triple angiokinase inhibitor 3 is currently in phase III clinical trials for the treatment of nonsmall cell lung cancer.
PATENT
The present invention relates to a beneficial treatment of tumours in patients suffering from NSCLC, and to a clinical marker useful as predictive variable of the responsiveness of tumours in patients suffering from NSCLC. The present invention further relates to a method for selecting patients likely to respond to a given therapy, wherein said method optionally comprises the use of a specific clinical marker. The present invention further relates to a method for delaying disease progression and/or prolonging patient survival of NSCLC patients, wherein said method comprises the use of a specific clinical marker.
The monoethanesulphonate salt form of this compound presents properties which makes this salt form especially suitable for development as medicament. The chemical structure of 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)- 1 -phenyl-methylene] -6-methoxycarbonyl-2-indolinone-monoethanesulphonate (ΓΝΝ name nintedanib esylate) is depicted below as Formula Al .
Formula Al

This compound is thus for example suitable for the treatment of diseases in which angiogenesis or the proliferation of cells is involved. The use of this compound for the treatment of immunologic diseases or pathological conditions involving an
immunologic component is being described in WO 2004/017948, the use for the treatment of, amongst others, oncological diseases, alone or in combination, is being described in WO 2004/096224 and WO 2009/147218, and the use for the treatment of fibrotic diseases is being described in WO 2006/067165.
A method using biomarkers for monitoring the treatment of an individual with the compound 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-l -phenyl-methylene] -6-methoxycarbonyl-2-indolinone or a pharmaceutically acceptable salt thereof, wherein it is determined if a sample from said individual comprises a biomarker in an amount that is indicative for said treatment, is disclosed in WO 2010/103058.
PATENT
http://www.google.com/patents/US20110201812
The present invention relates to a process for the manufacture of a specific indolinone derivative and a pharmaceutically acceptable salt thereof, namely 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate, to new manufacturing steps and to new intermediates of this process.
The indolinone derivative 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate are known from the following patent applications: WO 01/027081, WO 04/013099, WO 04/017948, WO 04/096224 and WO 06/067165. These patent applications disclose the compound, a process for its manufacture, a specific salt form of this compound and the use of the compound or its salt in a pharmaceutical composition to treat oncological or non-oncological diseases via inhibition of the proliferation of target cells, alone or in combination with further therapeutic agents. The mechanism of action by which the proliferation of the target cells occurs is essentially a mechanism of inhibition of several tyrosine kinase receptors, and especially an inhibition of the vascular endothelial growth factor receptor (VEGFR).




EXAMPLE 1Synthesis of the 6-methoxycarbonyl-2-oxindole in accordance with the process shown in synthesis scheme CSynthesis of benzoic acid, 4-chloro-3-nitro-, methylester
-
- 20 kg of 4-chloro-3-nitro-benzoic acid (99.22 mol) is suspended in 76 L methanol. 5.9 kg thionylchloride (49.62 mol) is added within 15 minutes and refluxed for about 3 hours. After cooling to about 5° C., the product is isolated by centrifugation and drying at 45° C.
- Yield: 19.0 kg (88.8% of theoretical amount)
- Purity (HPLC): 99.8%
Synthesis of propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester
-
- 12.87 kg of malonic acid, dimethylester (97.41 mol) is added to a hot solution (75° C.) of 10.73 kg sodium-tert.amylate (97.41 mol) in 35 L 1-methyl-2-pyrrolidinone (NMP). A solution of 10 kg benzoic acid, 4-chloro-3-nitro-, methylester (46.38 mol) in 25 L 1-methyl-2-pyrrolidinone is added at 75° C. After stirring for 1.5 hours at about 75° C. and cooling to 20° C., the mixture is acidified with 100 L diluted hydrochloric acid to pH 1. After stirring for 1.5 hours at about 5° C., the product is isolated by centrifugation and drying at 40° C.
- Yield: 13.78 kg (95.4% of theoretical amount)
- Purity (HPLC): 99.9%
- Alternatively, propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester can be synthesized as follows:
- 33.1 kg of malonic acid, dimethylester (250.6 mol) and 27.0 kg benzoic acid, 4-chloro-3-nitro-, methylester (125.3 mol) are subsequently added to a solution of 45.1 kg sodium-methylate (250.6 mol) in 172 kg 1-methyl-2-pyrrolidinone (NMP) at 20° C. After stirring for 1.5 hours at about 45° C. and cooling to 30° C., the mixture is acidified with 249 L diluted hydrochloric acid. At the same temperature, the mixture is seeded, then cooled to 0° C. and stirred for an additional hour. The resulting crystals are isolated by centrifugation, washed and dryed at 40° C.
- Yield: 37.5 kg (86% of theoretical amount)
- Purity (HPLC): 99.7%
Synthesis of 6-methoxycarbonyl-2-oxindole
A solution of 13 kg propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester (41.77 mol) in 88 L acetic acid is hydrogenated at 45° C. and under 40-50 psi in the presence of 1.3 kg Pd/C 10%. After standstill of the hydrogenation, the reaction is heated up to 115° C. for 2 hours. The catalyst is filtered off and 180 L water is added at about 50° C. The product is isolated after cooling to 5° C., centrifugation and drying at 50° C.
-
- Yield: 6.96 kg (87.2% of theoretical amount)
- Purity (HPLC): 99.8%
EXAMPLE 2Synthesis of the “chlorimide” (methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate)

Method 1
6-methoxycarbonyl-2-oxindole (400 g; 2.071 mol) is suspended in toluene (1200 ml) at room temperature. Chloroacetic anhydride (540 g; 3.095 mol) is added to this suspension. The mixture is heated to reflux for 3 h, then cooled to 80° C. and methyl cyclohexane (600 ml) is added within 30 min. The resulting suspension is further cooled down to room temperature within 60 min. The mother liquor is separated and the solid is washed with ice cold methanol (400 ml). The crystals are dried to afford 515.5 g (93.5%) of the “chlorimide” compound as a white solid. 1H-NMR (500 MHz, DMSO-d6) δ: 8.66 (s, 1H, 6-H); 7.86 (d, J=8.3 Hz, 1H, 8-H); 7.52 (d, J=8.3 Hz, 1H, 9-H); 4.98 (s, 2H, 15-H2); 3.95 (s, 3H, 18-H3); 3.88 (s, 2H, 3-H2). 13C-NMR (126 MHz, DMSO-d6) δ: 174.7 (C-2); 36.0 (C-3); 131.0 (C-4); 140.8 (C-5); 115.7 (C-6); 128.9 (C-7); 126.1 (C-8); 124.6 (C-9); 166.6 (C-10); 165.8 (C-13); 46.1 (C-15); 52.3 (C-18). MS: m/z 268 (M+H)+. Anal. calcd. for C12H10ClNO4: C, 53.85; H, 3.77; Cl, 13.25; N, 5.23. Found: C, 52.18; H, 3.64; Cl, 12.89; N, 5.00.
Method 2
6-Methoxycarbonyl-2-oxindole (10 g; 0.052 mol) is suspended in n-butyl acetate (25 ml) at room temperature. To this suspension a solution of chloroacetic anhydride (12.8 g; 0.037 mol) in n-butyl acetate (25 ml) is added within 3 min. The mixture is heated to reflux for 2 h, then cooled to 85° C. and methyl cyclohexane (20 ml) is added. The resulting suspension is further cooled down to room temperature and stirred for 2 h. The mother liquor is separated and the solid is washed with methanol (400 ml) at ambient temperature. The crystals are dried to afford 12.7 g (91.5%) of the “chlorimide” compound as a slightly yellow solid.
EXAMPLE 3Synthesis of the “chlorenol” (methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)

Method 1
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to not less than 104° C. and trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 60 min. During the addition period and subsequent stirring at the same temperature for 3 h, volatile parts of the reaction mixture are distilled off. The concentration of the reaction mixture is kept constant by replacement of the distilled part by toluene (40 ml). The mixture is cooled down to 5° C., stirred for 1 h and filtrated. The solid is subsequently washed with toluene (14 ml) and with a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying, 16.3 g (91.7%) of the “chlorenol” compound are isolated as slightly yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.73 (d, J=1.5 Hz, 1H, 6-H); 8.09 (d, J=8.0 Hz, 1H, 9-H); 7.90 (dd, J=8.1; 1.5 Hz, 1H, 8-H); 7.61-7.48 (m, 5H, 21-H, 22-H, 23-H, 24-H, 25-H); 4.85 (s, 2H, 18-H2); 3.89 (s, 3H, 27-H3); 3.78 (s, 3H, 15-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 165.9 (C-2+C16); 103.9 (C-3); 127.4; 128.6; 130.0; 135.4 (C-4+C-5+C-7+C-20); 115.1 (C-6); 126.1 (C-8); 122.5 (C-9); 166.7 (C-10); 173.4 (C-13); 58.4 (C-15); 46.4 (C-18); 128.6 (C-21+C-22+C-24+C-25); 130.5 (C-23); 52.2 (C-27). MS: m/z 386 (M+H)+. Anal. calcd. for C20H16ClNO5: C, 62.27; H, 4.18; Cl, 9.19; N, 3.63. Found: C, 62.21; H, 4.03; Cl, 8.99; N, 3.52.
Method 2
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in xylene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 4 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with xylene (14 ml) and a mixture of xylene (8 ml) and ethyl acetate (8 ml). After drying 14.3 g (81.0%) of the “chlorenol” compound are isolated as yellow crystals.
Method 3
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 3 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with toluene (14 ml) and a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying 15.3 g (87.3%) of the “chlorenol” compound are isolated as fawn crystals.
EXAMPLE 4Synthesis of the “enolindole” (methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)

Method 1
A solution of potassium hydroxide (0.41 g, 0.006 mol) in methanol (4 ml) is added at 63° C. to a suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.020 mol) in methanol (32 ml). The mixture is then stirred for 30 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 6.0 g (94.6%) of the “enolindole” compound as yellow crystals. 1H-NMR (500 MHz, CDCl3) δ: 8.08 (s, 1H, 1-H); 7.88 (d, J=7.8 Hz, 1H, 9-H); 7.75 (m, 1H, 8-H); 7.52-7.56 (m, 3H, 18-H, 19-H, 20-H); 7.40-7.45 (m, 3H, 6-H, 17-H, 21-H); 3.92 (s, 3H, 23-H3); 3.74 (s, 3H, 13-H3). 13C-NMR (126 MHz, CDCl3) δ: 168.8 (C-2); 107.4 (C-3); 130.8 (C-4); 138.2 (C-5); 109.4 (C-6); 128.2 and 128.3 (C-7, C-16); 123.5 (C-8); 123.1 (C-9); 170.1 (C-11); 57.6 (C-13); 167.2 (C-14); 128.7 and 128.9 (C-17, C-18, C-20, C-21); 130.5 (C-19); 52.1 (C-23). MS (m/z): 310 (M+H)+. Anal. calcd. for C18H15NO4: C, 69.89; H, 4.89; N, 4.53. Found: C, 69.34; H, 4.92; N, 4.56.
Method 2
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (7.0 g; 0.018 mol) in methanol (28 ml) is heated to reflux. Within 3 min, a solution of sodium methoxide in methanol (0.24 g, 30 (w/w), 0.001 mol) is added to this suspension. The mixture is then stirred for 30 min, cooled to 5° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (9 ml) and dried to afford 5.4 g (89.7%) of the “enolindole” compound as yellow crystals.
Method 3
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.021 mol) in methanol (32 ml) is heated to reflux. A solution of sodium methoxide in methanol (0.74 g, 30% (w/w), 0.004 mol), further diluted with methanol (4 ml), is added dropwise to this suspension. The mixture is then stirred for 90 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 5.9 g (91.2%) of the “enolindole” compound as yellow crystals.
EXAMPLE 5Synthesis of the “chloroacetyl” (N-(4-nitroanilino)-N-methyl-2-chloro-acetamide)

Method 1
A suspension of N-methyl-4-nitroaniline (140 g; 0.920 mol) in ethyl acetate (400 ml) is heated to 70° C. Within 90 min, chloro acetylchloride (114 g; 1.009 mol) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 60° C. and methyl cyclohexane (245 ml) is added. The suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (285 ml) and the precipitate is dried to afford 210.4 g (92.7%) of the “chloroacetyl” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.29 (d, J=8.5 Hz, 2H, 1-H+3-H); 7.69 (d, J=8.5 Hz, 2H, 4-H+6-H); 4.35 (s, 2H, 9-H2); 3.33 (s, 3H, 12-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 124.6 (C-1+C-3); 145.6 (C-2); 127.4 (C-4+C-6); 148.6 (C-5); 165.6 (C-8); 42.7 (C-9); 37.2 (C-12). MS (m/z): 229 (M+H)+. Anal. calcd. for C9H9ClN2O3: C, 47.28; H, 3.97; N, 12.25. Found: C, 47.26; H, 3.99; Cl, 15.73; N, 12.29.
Method 2
A suspension of N-methyl-4-nitroaniline (20.0 g; 0.131 mol) in ethyl acetate (20 ml) is heated to 60° C. Within 15 min, a solution of chloro acetic anhydride (26.0 g; 0.151 mol) in ethyl acetate (60 ml) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 75° C. ° C. and methyl cyclohexane (80 ml) is added. After seeding at 60° C., the suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (40 ml) and the precipitate is dried to afford 25.9 g (83.3%) of the “chloroacetyl” compound as grey crystals.
EXAMPLE 6Synthesis of the “nitroaniline” (N-(4-nitrophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide) and of the “aniline” (N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide)

Method 1
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (20.0 g; 0.087 mol) in toluene (110 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (21.9 g; 0.216 mol) is added dropwise. After purging of the dropping funnel with toluene (5 ml) the reaction mixture is stirred for 2 h at 55° C., cooled to ambient temperature and washed with water (15 ml). The organic layer is diluted with isopropanol (100 ml) and Pd/C (10%; 1.0 g) is added. After subsequent hydrogenation (H2, 4 bar) at 20° C. the catalyst is removed. Approximately ⅘ of the volume of the resulting solution is evaporated at 50° C. The remaining residue is dissolved in ethyl acetate (20 ml) and toluene (147 ml) heated to 80° C., then cooled to 55° C. and seeded. The reaction mixture is further cooled to 0° C. and stirred for 3 h at the same temperature. After filtration, the solid is washed with ice cold toluene (40 ml) and dried to afford 20.2 g (88.0%) of the “aniline” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 6.90 (d, J=8.5 Hz, 2H, 4-H+6-H); 6.65 (d, J=8.5 Hz, 2H, 1-H+3-H); 5.22 (2H, 19-H2); 3.04 (s, 3H, 9-H3); 2.79 (s, 2H, 11-H2); 2.32 (m, 4H, 13-H2+17-H2); 2.23 (m, 4H, 14-H2+16-H2); 2.10 (s, 3H, 18-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 114.0 (C-1+C-3); 148.0 (C-2); 127.6 (C-4+C-6); 131.5 (C-5); 168.9 (C-8); 36.9 (C-9); 58.5 (C-11); 52.4 (C-13+C-17); 54.6 (C-14+C-16); 45.7 (C-18). MS (m/z): 263 (M+H)+. Anal. calcd. for C14H22N4O: C, 64.09; H, 8.45; N, 21.36. Found: C, 64.05; H, 8.43; N, 21.39.
Method 2
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (14.5 g; 0.063 mol) in ethyl acetate (65 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (15.8 g; 0.156 mol) is added dropwise. After purging of the dropping funnel with ethyl acetate (7 ml) the reaction mixture is stirred at 50° C. for 90 min, cooled to ambient temperature and washed with water (7 ml). The organic layer is diluted with isopropanol (75 ml) and dried over sodium sulphate. After separation of the solid, Pd/C (10%; 2.0 g) is added and the solution is hydrogenated (H2, 5 bar) at ambient temperature without cooling. Subsequently the catalyst is removed by filtration and the solvent is evaporated at 60° C. The remaining residue is dissolved in ethyl acetate (250 ml) and recrystallized. After filtration and drying 10.4 g (60.4%) of the “aniline” compound are isolated as white crystals.
EXAMPLE 7Synthesis of the “anilino” (3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone)

Method 1
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (10.0 g; 0.032 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (8.6 g; 0.032 mol) in a mixture of methanol (72 ml) and N,N-dimethylformamide (18 ml) is heated to reflux. After 7 h of refluxing the suspension is cooled down to 0° C. and stirring is maintained for additional 2 h. The solid is filtered, washed with methanol (40 ml) and dried to afford 15.4 g (88.1%) of the “anilino” compound as yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 11.00 (s, 1H, 23-H); 12.23 (s, 19-H); 7.61 (t; J=7.1 Hz, 1H, 33-H); 7.57 (t, J=7.5 Hz, 2H, 32-H+34-H); 7.50 (d, J=7.7 Hz, 2H, 31-H+35-H); 7.43 (d, J=1.6 Hz, 1H, 29-H); 7.20 (dd, J=8.3; 1.6 Hz, 1H, 27-H); 7.13 (d, J=8.3 Hz, 2H, 14-H+18-H); 6.89 (d, 8.3 Hz, 2H, 15-H+17-H); 5.84 (d, J=8.3 Hz, 1H, 26-H); 3.77 (s, 3H, 40-H3); 3.06 (m, 3H, 12-H3); 2.70 (m, 2 H, 8-H2); 2.19 (m, 8H, 2-H2, 3-H2, 5-H2, 6-H2); 2.11 (s, 3H, 7-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 54.5 (C-2+C-6); 52.2 (C-3+C-5); 45.6 (C-7); 59.1 (C-8); 168.5 (C-9); 36.6 (C-12); 140.1 (C-13); 127.6 (C-14+C-18); 123.8 (C-17+C-15); 137.0 (C-16); 158.3 (C-20); 97.5 (C-21); 170.1 (C-22); 136.2 (C-24); 128.9 (C-25); 117.2 (C-26); 121.4 (C-27); 124.0 (C-28); 109.4 (C-29); 131.9 (C-30); 128.4 (C-31+C-35); 129.4 (C-32+C-34); 130.4 (C-33); 166.3 (C-37); 51.7 (C-40). MS (m/z): 540 (M+H)+. Anal. calcd. for C31H33N5O4: C, 69.00; H, 6.16; N, 12.98. Found: C, 68.05; H, 6.21; N, 12.81.
Method 2
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (20.0 g; 0.064 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (17.1 g; 0.065 mol) in methanol (180 ml) is heated to reflux for 7.5 h. The resulting suspension is cooled down to 10° C. within 1 h and stirring is maintained for 1 h. After filtration, the solid is washed with ice cold methanol (80 ml) and dried to afford 31.0 g (89.0%) of the “anilino” compound as yellow crystals.
EXAMPLE 8Synthesis of the 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone, monoethanesulfonate

A suspension of 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone (30.0 g; 0.055 mol) in methanol (200 ml) and water (2.4 ml) is heated to 60° C. Aqueous ethanesulfonic acid (70% (w/w); 8.75 g; 0.056 mol) is added to the reaction mixture. The resulting solution is cooled to 50° C., seeded and then diluted with isopropanol (200 ml). The mixture is further cooled to 0° C. and stirred for 2 h at this temperature. The precipitate is isolated, washed with isopropanol (120 ml) and dried to furnish 35.1 g (97.3%) of the monoethanesulfonate salt of the compound as yellow crystals. 1H-NMR (400 MHz, DMSO-d6) δ: 12.26 (s, 11-H); 10.79 (s, 1H, 1-H); 9.44 (s, 1H, 24-H); 7.64 (m, 1H, 32-H); 7.59 (m, 2H, 31-H+33-H); 7.52 (m, 2H, 30-H+34-H); 7.45 (d, J=1.6 Hz, 1H, 7-H); 7.20 (dd, J=8.2; 1.6 Hz, 1H, 5-H); 7.16 (m, 2H, 14-H+16-H); 6.90 (m, 2H, 13-H+17-H); 5.85 (d, J=8.2 Hz, 1H, 4-H); 3.78 (s, 3H, 37-H3); 3.45-2.80 (broad m, 4H, 23-H2+25-H2); 3.08 (s, 3H, 28-H3); 2.88 (s, 2H, 20-H2); 2.85-2.30 (broad m, 4H, 22-H2+26-H2); 2.75 (s, 3H, 27-H3); 2.44 (q, J=7.4 Hz, 2H, 39-H2); 1.09 (t, J=7.4 Hz, 3H, 38-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 9.8 (C-38); 36.6 (C-28); 42.3 (C-27); 45.1 (C-39); 51.7 (C-37); 48.9 (C-22+C-26); 52.6 (C-23+C-25); 57.5 (C-20); 97.7 (C-3); 109.5 (C-7); 117.3 (C-4); 121.4 (C-5); 123.8 (C-13+C-17); 124.1 (C-6); 127.7 (C-14+C-16); 128.4 (C-30+C-34); 128.8 (C-9); 129.5 (C-31+C-33); 130.5 (C-32); 132.0 (C-29); 168.5 (C-9); 136.3 (C-8); 137.3 (C-12); 139.5 (C-15); 158.1 (C-10); 166.3 (C-35); 168.0 (C-19); 170.1 (C-2). MS (m/z): 540 (M(base)+H)+. Anal. calcd. for C33H39N5O7S: C, 60.17; H, 6.12; N, 10.63; S, 4.87. Found: C, 60.40; H, 6.15; N, 10.70; S, 4.84.
CLIPS

CLIPS
see
http://www.yaopha.com/2014/07/09/synthesis-of-vargatef-nintedanib-boehringer-ingelheim-idiopathic-pulmonary-fibrosis-drug/

CLICK ON PIC
Updates………..
PATENT
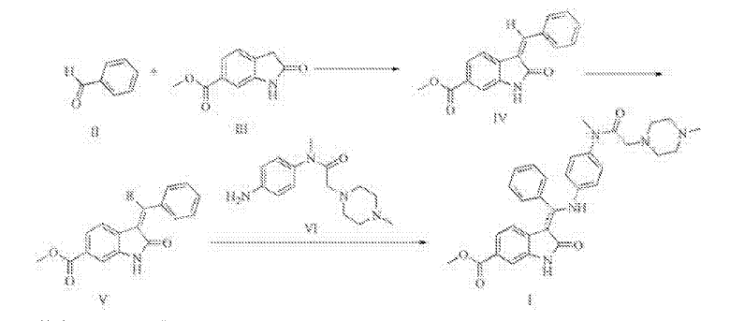
MACHINE TRANSLATED FROM CHINESE
Synthesis of Trinidad Neeb (I),
A 500ml reaction flask was charged 30g of compound V, 22.5g compound of the VI, ethanol 300ml, sodium bicarbonate and 15g, the reaction was heated to reflux for 2 hours, the reaction mixture was added to 600ml of water, there are large amount of solid precipitated, was filtered, the cake washed with 100ml washed once with methanol, a yellow solid 41.9g refined Trinidad Neeb (I). Yield 92.7%.
4 bandit R (400MHz, dmso) δ11 · 97 (s, 1H), 8.38 (s, 1H), 7.97 (dd, J = 11.9, 5.0Hz, 2H), 7.67 (d, J = 8.1Hz, 1H), 7.16 (ddd, J = 26.9, 22.1, 7.0Hz, 5H), 6.85 (d, J = 8.6Hz, 2H), 6.63 (d, J = 8.7Hz, 2H), 3.90 (s, 3H), 2.99 (s, 3H), 2.69 (s, 2H), 2.51-2.24 (m, 8H), 2.20 (s, 3H) MS:. m / z540 (m + 1) + 2 Example: Preparation of compound IV 250ml reaction flask was added 28.7g of 2- oxindole-6-carboxylate, 130ml ethanol, stirred open, then added 30.3ml (31.8g) benzaldehyde, 2.97 mL piperidine was heated to 70 ° C-80 after ° C for 2 hours, allowed to cool to 20 ° C- 30 ° C, the precipitate was filtered, the filter cake was washed with absolute ethanol, 50 ° C 5 hours and dried in vacuo give a yellow solid 38.7g (IV of), yield: 92.4% Preparation of compound V square in 500ml reaction flask was added 30g compound IV, dichloromethane 360ml, cooled with ice water to 0-5 ° C, 71/92 bromine 3.lml (9.7g), drop finished warmed to 20- 30 ° C, 3 hours after the reaction, the reaction solution was washed once with 150ml dichloromethane layer was concentrated oil was done by adding 200ml ethanol crystallization, filtration, 60 ° C and dried under vacuum 36.lg white solid (V ), yield: 93 · 8%.
After Trinidad Technip (I) are synthesized in the reaction flask was added 500ml of 30g compound V, 33.0g compound of the VI, ethanol 300ml, sodium bicarbonate, 15g, was heated to reflux for 2 hours, the reaction mixture was added to 600ml water, there are large amount of solid precipitated, was filtered, the filter cake washed once with 100ml methanol obtained 42.3g of yellow solid was purified by Technip Trinidad (I). Yield 93.6%.
ΧΗNMR (400MHz, dmso) δ11.94 (s, 1Η), 8.36 (s, 1H), 7.96 (dd, J = 11.9, 5.0Hz, 2H), 7.67 (d, J = 8.1Hz, 1H) , 7.16 (ddd, J = 26.9, 22.1, 7.0Hz, 5H), 6.85 (d, J = 8.6Hz, 2H), 6.61 (d, J = 8.7Hz, 2H), 3.90 (s, 3H), 2.99 ( s, 3H), 2.65 (s, 2H), 2.50-2.30 (m, 8H), 2.20 (s, 3H) MS:. m / z540 (m + 1) + square
PATENT
(I) 2.30g, yield 85.3%. Melting point 241 ~ 243 ℃, Mass spectrum (the EI): m / Z 540 (the M + the H), 1 the H NMR (of DMSO D . 6 ): 2.27 (S, 3H), 2.43 (m, 8H), 2.78 (S, 2H) , 3.15 (s, 3H), 3.82 (s, 3H), 5.97 (d, J = 8.3Hz, 1H), 6.77 (d, J = 8.7Hz, 1H), 6.96 (d, J = 8.6Hz, 2H) , 7.32-7.62 (m, 8H), 8.15 (s, 1H), 12.15 (s, 1H).
CLIPS
http://pubs.rsc.org/en/content/articlelanding/2015/ay/c5ay01207d#!divAbstract


Nintedanib
|
|
| Systematic (IUPAC) name | |
|---|---|
| Methyl (3Z)-3-{[(4-{methyl[(4-methylpiperazin-1-yl)acetyl]amino}phenyl)amino](phenyl)methylidene}-2-oxo-2,3-dihydro-1H-indole-6-carboxylate | |
| Clinical data | |
| Trade names | Vargatef, Ofev |
| AHFS/Drugs.com | Consumer Drug Information |
| Pregnancy cat. |
|
| Legal status | |
| Routes | Oral and intravenous |
| Identifiers | |
| CAS number | 656247-17-5 |
| ATC code | None |
| Chemical data | |
| Formula | C31H33N5O4 |
| Mol. mass | 539.6248 g/mol |
References
- Hilberg, F.; G. J. Roth, M. Krssak, S. Kautschitsch, W. Sommergruber, U. Tontsch-Grunt, P. Garin-Chesa, G. Bader, A. Zoephel, J. Quant, A. Heckel, W. J. Rettig (2008). “BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy”. Cancer Res 68 (12): 4774–82. doi:10.1158/0008-5472.CAN-07-6307. ISSN 1538-7445. PMID 18559524.
- Hilberg, F.; U. Tontsch-Grunt, F. Colbatzky, A. Heckel, R. Lotz, J.C.A. van Meel, G.J. Roth (2004). “BIBF1120 a novel, small molecule triple angiokinase inhibitor: profiling as a clinical candidate for cancer therapy”. European Journal of Cancer Supplements 2 (50).
- Reck, M.; R. Kaiser; C. Eschbach; M. Stefanic; J. Love; U. Gatzemeier; P. Stopfer; J. von Pawel (2011). “A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer”. Ann Oncol. ISSN 1569-8041.
- Okamoto, I.; H. Kaneda, T. Satoh, W. Okamoto, M. Miyazaki, R. Morinaga, S. Ueda, M. Terashima, A. Tsuya, A. Sarashina, K. Konishi, T. Arao, K. Nishio, R. Kaiser, K. Nakagawa (2010). “Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors”. Mol Cancer Ther 9 (10): 2825–33. doi:10.1158/1535-7163.MCT-10-0379. ISSN 1538-8514. PMID 20688946.
- Mross, K.; M. Stefanic, D. Gmehling, A. Frost, F. Baas, C. Unger, R. Strecker, J. Henning, B. Gaschler-Markefski, P. Stopfer, L. de Rossi, R. Kaiser (2010). “Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors”. Clin Cancer Res 16 (1): 311–9. doi:10.1158/1078-0432.CCR-09-0694. ISSN 1078-0432. PMID 20028771.
- Ledermann, J.A. (2009). “A randomised phase II placebo-controlled trial using maintenance therapy to evaluate the vascular targeting agent BIBF 1120 following treatment of relapsed ovarian cancer (OC)”. J Clin Oncol 27 (15s): (suppl; abstr 5501).
- Kropff, M.; J. Kienast; G. Bisping; W. E. Berdel; B. Gaschler-Markefski; P. Stopfer; M. Stefanic; G. Munzert (2009). “An open-label dose-escalation study of BIBF 1120 in patients with relapsed or refractory multiple myeloma”. Anticancer Res 29 (10): 4233–8. ISSN 1791-7530. PMID 19846979.
- Ellis, P. M.; R. Kaiser; Y. Zhao; P. Stopfer; S. Gyorffy; N. Hanna (2010). “Phase I open-label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non-small cell lung cancer patients”. Clin Cancer Res 16 (10): 2881–9. doi:10.1158/1078-0432.CCR-09-2944. ISSN 1078-0432. PMID 20460487.
- du Bois, A.; J. Huober; P. Stopfer; J. Pfisterer; P. Wimberger; S. Loibl; V. L. Reichardt; P. Harter (2010). “A phase I open-label dose-escalation study of oral BIBF 1120 combined with standard paclitaxel and carboplatin in patients with advanced gynecological malignancies”. Ann Oncol 21 (2): 370–5. doi:10.1093/annonc/mdp506. ISSN 1569-8041. PMID 19889612.
- Xiang, Q. F.; F. Wang; X. D. Su; Y. J. Liang; L. S. Zheng; Y. J. Mi; W. Q. Chen; L. W. Fu (2011). “Effect of BIBF 1120 on reversal of ABCB1-mediated multidrug resistance”. Cell Oncol (Dordr) 34 (1): 33–44. doi:10.1007/s13402-010-0003-7. ISSN 2211-3436.
- “Boehringer Ingelheim – AGO-OVAR 12 / LUME-Ovar 1 Trial Information”. 2011.
- “Boehringer Ingelheim – LUME-Lung 2 Trial Information”. 2011.
- “Boehringer Ingelheim – LUME-Lung 1 Trial Information”. 2011.
- http://clinicaltrials.gov/ct2/results?term=++%09+BIBF+1120&phase=1
- http://clinicaltrials.gov/ct2/show/NCT00805194 Phase III LUME-Lung 1: BIBF 1120 Plus Docetaxel as Compared to Placebo Plus Docetaxel in 2nd Line Non Small Cell Lung Cancer
- http://clinicaltrials.gov/ct2/show/NCT01015118 Phase III BIBF 1120 or Placebo in Combination With Paclitaxel and Carboplatin in First Line Treatment of Ovarian Cancer
- http://clinicaltrials.gov/ct2/show/NCT01335477 Safety and Efficacy of BIBF 1120 at High Dose in Idiopathic Pulmonary Fibrosis Patients II
- “FDA approves Ofev to treat idiopathic pulmonary fibrosis”. 2014.
-
F. Hilberg et al. Cancer Res. 2008, 68, 4774
2. M. Reck et al. Ann. Oncol. 2011, 22, 1374
3. M. Reck et al. J. Clin. Oncol. 2013 (suppl.), Abst LBA8011
4. N. H. Hanna et al. J. Clin. Oncol. 2013, 2013 (suppl.), Abst 8034
5. J.A. Ledermann et al. J. Clin Oncol. 2011, 29, 3798
6. Glioblastoma: A. Muhac et al. J. Neurooncol. 2013, 111, 205
7. O. Bouche et al. Anticancer Res. 2011, 31, 2271
8. T. Eisen et al. J. Clin. Oncol. 2013 (suppl.), Abst. 4506
MORE…………….
Reference:
[6]. Japan PMDA.
[7]. Drug@FDA, NDA205832 Pharmacology Review(s).
[8]. Med. Chem. 2015, 58, 1053-1063.
[9]. Drug@EMA, EMEA/H/C/002569 Vargatef: EPAR-Assessment Report.
[10]. Drug Des. Devel. Ther. 2015, 9, 6407-6419.
[11]. Cancer Res. 2008, 68, 4774-4782.
[12]. J. Med. Chem. 2009, 52, 4466-4480.
Merten, J.; et. al. Process for the manufacture of an indolinone derivative. US20110201812A1
2. Roth, G. J.; et. al. 3-z-[1-(4-(n-((4-methyl-piperazin-1-yl)-methylcarbonyl)-n-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition. WO2004013099A1
3. Roth, G. J.; et. al. Design, Synthesis, and Evaluation of Indolinones as Triple Angiokinase Inhibitors and the Discovery of a Highly Specific 6-Methoxycarbonyl-Substituted Indolinone (BIBF 1120). J Med Chem, 2009, 52(14), 4466-4480. -

ニンテダニブエタンスルホン酸塩
Nintedanib Ethanesulfonate
C31H33N5O4.C2H6O3S : 649.76
[656247-18-6]US7119093 * Jul 21, 2003 Oct 10, 2006 Boehringer Ingelheim Pharma Gmbh & Co. Kg 3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition ///////////////
ICOTINIB
ICOTINIB
4-((3-ethynylphenyl)amino)-6,7-benzo-12-crown-4-quinazoline
N-(3-Ethynylphenyl)-7,8,10,11,13,14-hexahydro[1,4,7,10]tetraoxacyclododecino[2,3-g]quinazolin-4-amine
[1,4,7,10]Tetraoxacyclododecino[2,3-g]quinazolin-4-amine, N-(3-ethynylphenyl)-7,8,10,11,13,14-hexahydro-
BPI 2009H, UNII-JTD32I0J83
610798-31-7 CAS BASE

Icotinib Hydrochloride, 1204313-51-8, CS-0918, HY-15164, Conmana Zhejiang Beta Pharma Ltd.
CLINICALS………http://clinicaltrials.gov/search/intervention=Icotinib
Icotinib Hydrochloride (BPI-2009H), or Icotinib, is a highly selective, first generation epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI). EGFR is an oncogenic driver and patients with somatic mutations, particularly an exon 19 deletion or exon 21 L858R mutation, within the tyrosine kinase domain have activating mutations that lead to unchecked cell proliferation. Overexpression of EGFR causes inappropriate activation of the anti-apoptotic Ras signaling pathway, found in many different types of cancer. Icotinib is a quinazoline derivative that binds reversibly to the ATP binding site of the EGFR protein, preventing completion of the signal transduction cascade.[1]
Clinical Evaluation
Icotinib is indicated for the treatment for EGFR mutation-positive, advanced or metastatic non-small cell lung cancer (NSCLC) as a second-line or third-line treatment, for patients who have failed at least one prior treatment with platinum-based chemotherapy. The ICOGEN trial was a double-blind, head-to-head phase III study comparing icotinib with gefitinib in all-comers. From 27 centers in China, 399 patients were randomized between the two treatments testing for a primary objective of progression-free survival and secondary objectives of overall survival, time to progression, quality of life, percentage of patients who achieved an objective response, and toxic effects. The ICOGEN results showed icotinib to have a median PFS of 4.6 months (95% CI 3.5 – 6.3) as compared to gefitinib which has a PFS of 3.4 months (95% CI 2.3 – 3.8). After the study was completed, post-hoc analysis revealed that in the icotinib treatment group, patients with activating EGFR mutations showed improved PFS as compared to patients with wild-type EGFR. Icotinib also was associated with fewer adverse events than gefitinib when considering all grades of reactions together (61% versus 70% respectively, p = 0.046).[2] The phase IV ISAFE trial evaluated 5,549 patients and showed icotinib to have an overall response rate of 30% and a low adverse event rate of 31.5%.[3]
Regulatory Approvals
Icotinib was approved in China by the SFDA in June, 2011.[4] Since approval, Icotinib has treated over 40,000 patients in China successfully and is now undergoing global development.
January 2014, Beta Pharma, Inc. was given a “May Proceed” from the US FDA to conduct a Phase I study for the evaluation of icotinib as a treatment of EGFR+ Non-Small Cell Lung Cancer (NSCLC).
Icotinib is a potent and specific EGFR inhibitor with IC50 of 5 nM, including the EGFR, EGFR(L858R), EGFR(L861Q), EGFR(T790M) and EGFR(T790M, L858R). Phase 4.Icotinib hydrochloride is the epidermal growth factor receptor kinase targeting a new generation of targeted anti-cancer drugs, completely independent from the original tumor clinical practitioners and experts of science, through eight years of the development, its first adaptation disease is advanced non-small cell lung cancer. Icotinib is an orally available quinazoline-based inhibitor of epidermal growth factor receptor (EGFR), with potential antineoplastic activity. Icotinib selectively inhibits the wild-type and several mutated forms of EGFR tyrosine kinase. This may lead to an inhibition of EGFR-mediated signal transduction and may inhibit cancer cell proliferation. EGFR, a receptor tyrosine kinase, is upregulated in a variety of cancer cell types. Icotinib was approved in China in 2011
Icotinib has been found to be noninferior to gefitinib in patients with non-small-cell lung cancer (NSCLC), according to reports from the phase III Chinese double-blind ICOGEN study.
“[I]cotinib is a valid therapeutic option for patients with non-small-cell lung cancer as a second-line or third-line treatment, although patients might find taking icotinib three times a day an inconvenience,” write Yan Sun (Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China) and colleagues.
Icotinib is an oral epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) that has exhibited good antitumor activity in phase II studies. However, it has a shorter half-life than gefitinib, another TKI, which means that it needs to be taken more often.
…
-
Tyrosine kinase receptors are trans-membrane proteins that, in response to an extracellular stimulus, propagate a signaling cascade to control cell proliferation, angiogenesis, apoptosis and other important features of cell growth. One class of such receptors, epidermal growth factor receptor (EGFR) tyrosine kinases, are over-expressed in many human cancers, including brain, lung, liver, bladder, breast, head and neck, esophagus, gastrointestinal, breast, ovary, cervix or thyroid cancer.
-
EGFR is expressed in many types of tumor cells. Binding of cognate ligands (including EGF, TGFα (i.e., Transforming Growth Factor-α) and neuregulins) to the extracellular domain causes homo- or heterodimerization between family members; the juxtaposition of cytoplasmic tyrosine kinase domains results in transphosphorylation of specific tyrosine, serine and threonine residues within each cytoplasmic domain. The formed phosphotyrosines act as docking sites for various adaptor molecules and subsequent activation of signal transduction cascades (Ras/mitogen-activated, PI3K/Akt and Jak/STAT) that trigger proliferative cellular responses.
-
Various molecular and cellular biology and clinical studies have demonstrated that EGFR tyrosine kinase inhibitors can block cancer cell proliferation, metastasis and other EGFR-related signal transduction responses to achieve clinical anti-tumor therapeutic effects. Two oral EGFR kinase inhibitors with similar chemical structures are Gefitinib (Iressa; AstraZeneca), approved by the U.S. FDA for advanced non-small cell lung cancer in 2003 (and later withdrawn), and Erlotinib Hydrochloride (Tarceva; Roche and OSI), approved by the U.S. FDA for advanced non-small cell lung cancer and pancreatic cancer treatment in 2004.
-
Chinese Patent Publication No. CN1305860C discloses the structure of 4-[(3-ethynyl-phenyl)amino]-6,7-benzo-12-crown-quinoline (free base) on page 29, Example 15, Compound 23.
Icotinib was launched in China in August 2011, after approval by the State Food and Drug Administration. It is a targeted EGFR tyrosine kinase inhibitor that, like erlotinib (Tarceva) and gefitinib (Iressa), shows benefit in patients with EGFR m+ NSCLC.
……………………………………..
http://www.google.com/patents/EP2392576A1
-
Formula I (Icotinib hydrochloride):

Method 1:

Method 2:

Method 3:
-

-
BPI-02 is obtained by recrystallization.

http://www.google.com/patents/EP2392576A1 Example 1Step 1
-

-
Preparation: 16 kg (400 mol) of sodium hydroxide was dissolved in 80 L of water in a 400 L reactor, and then 18.8 L (140 mol) of triethylene glycol, 32 L of THF were added into the reactor. After cooling below 5 °C, a solution of 47.84 kg (260 mol) of tosyl chloride and 50 L of THF was added dropwise. Following the addition, the reaction mixture was kept at this temperature for 2 hours, and it was then poured into 240 L of ice water. The precipitate was formed and filtered, washed with a small amount of water, and dried. 58.64 kg of BPI-01 as a white crystalline powder was yielded at 91.4%. mp: 77-80 °C, HPLC: 97%. TLC (petroleum ether: ethyl acetate = 1:1) Rf = 0.87.
-
NMR data: 1H-NMR (CDCl3): δ ppm: 7.78 (d, 4H, J = 10.4 Hz, benzene protons by sulfonyl group); 7.34 (d, 4H, J = 11.6 Hz, benzene protons by methyl group); 4.129 (dd, 4H, J = 5.6 Hz, ethylene protons by the sulfonyl group); 3.64 (dd, 4H, J = 5.6 Hz, ethylene protons away from the sulfonyl group); 3.517 (s, 4H, ethylene protons in the middle); 2.438 (s, 6H, methyl protons on the benzene).

Step 2
-

-
Preparation: A solution containing 3.64 kg (20 mol) of ethyl 3,4-dihydroxybenzoate and 12.4 kg (89.6 mol) of potassium carbonate in 300 L of N,N-dimethylformamide was stirred and heated to 85-90 °C for about 30 minutes. A solution of 9.17 kg (20 mol) of BPI-01 in 40 L of N,N-dimethylformamide was added dropwise over 1.5-2 hours. After the addition, the reaction was kept for 30 minutes; the reaction completion was confirmed by TLC (developing solvent: petroleum ether:ethyl acetate = 1:1, Rf = 0.58). The reaction mixture was removed from the reactor and filtered. Then, the filtrate was evaporated to remove N,N-dimethylformamide; 240 L of ethyl acetate was added to dissolve the residue. After filtration and vacuum evaporation, the residual solution was extracted with 300 L of petroleum ether. After evaporation of the petroleum ether, the residual solids were re-crystallized with isopropanol in a ratio of 1:2.5 (W/V); 1.68 kg of BPI-02 as a white powder was obtained in a yield of 28%. mp: 73-76 °C, HPLC: 96.4%. NMR data: 1H-NMR (CDCl3): δ ppm: 7.701 (d, 1H, J = 2.4 Hz, benzene proton at position 6); 7.68 (s, 1 H, benzene proton at position 2); 6.966 (d, 1H, J = 10.8 Hz, benzene proton at position 5); 4.374-3.81 (q, 2H, J = 9.6 Hz, methylene protons of the ethyl); 3.78-4.23 (dd, 12H, J = 4.8 Hz, crown ether protons); 1.394 (t, 3H, J = 9.6 Hz, methyl protons of the ethyl). MS: m/z 296.

Step 3
-

-
Preparation: A solution of 592 g (2 mol) of BPI-02 and 600 mL of acetic acid in a 5 L reaction flask was cooled to 0°C; 1640 mL (25.4 mol) of concentrated nitric acid was slowly added. The internal temperature should not exceed 10 °C. While cooled below 0°C, 1 L of concentrated sulfuric acid was added dropwise. The internal temperature should not be higher than 5°C. After the addition, the reaction was kept at 0-5 °C for 1-2 hours. After completion of the reaction, the reaction solution was poured into 15 L of ice water in a plastic bucket. After mixing, filtration, and re-crystallization in ethanol, 449 g of BPI-03 as a light yellow to yellow crystalline powder was obtained in 65.7% yield. mp: 92-95 °C, HPLC: 98.2%. TLC (petroleum ether: ethyl acetate =1:1) Rf = 0.52. NMR data: 1H-NMR (CDCl3): δ ppm: 7.56 (s, 1H, benzene proton at position 5); 7.20 (s, 1H, benzene proton at position 2); 4.402 (q, 2H, J = 9.2 Hz, methylene protons of the ethyl); 4.294 (dd, 12H, J = 4.8 Hz, crown ether protons); 1.368 (t, 3H, J = 9.2 Hz, methyl protons of the ethyl).

Step 4
-

-
Preparation: In a 3 L hydrogenation reactor, 2 L of methanol and 195 g (0.57 mol) of BPI-03 were added, and then 63 mL of acetyl chloride was slowly added. After a short stir, 33 g of Pd/C containing 40% water was added. The reaction was conducted under 4 ATM hydrogen until hydrogen absorption stopped, and then the reaction was kept for 1-2 hours. After completion of the reaction, the reaction mixture was transferred into a 5 L reactor. After filtration, crystallization, and filtration, the product was obtained. The mother liquor was concentrated under vacuum, and more product was obtained. The combined crops were 168 g of BPI-04 as a white to pink crystalline powder in a yield of 85%. mp: 198-201 °C, HPLC: 99.1 %. TLC (petroleum ether: ethyl acetate = 1:1) Rf = 0.33. NMR data: 1H-NMR (DMSO-d6): δ ppm: 8-9 (br., 3H, 2 protons of the amino group and a proton of the hydrochloric acid); 7.37 (s, 1H, benzene proton at position 5); 6.55 (s, 1H , benzene proton at position 2); 4.25 (q, 2H, J = 7.06 Hz, methylene protons of the ethyl); 4.05 (dd, 12H, J = 4.04 Hz, crown ether protons); 1.31 (t, 3H, J = 7.06 Hz, methyl protons of the ethyl).

Step 5
-

-
Preparation: 1105 g (3.175 mol)of BPI-04, 4810 g (106.9 mol) of formamide, and 540 g (8.55 mol) of ammonium formate were added to a 10 L 3-neck bottle. The reaction mixture was heated to 165 °C under reflux for 4 hours. After cooling to room temperature, 3 L of water was added, and then the mixture was stirred for 10 minutes. After filtration, washing, and drying, 742 g of BPI-05 as a white crystalline powder was obtained in a yield of 80%. mp: 248-251 °C, HPLC: 99.78%. TLC (chloroform: methanol = 8:1) Rf = 0.55. NMR data: 1H-NMR (DMSO-d6): δ ppm: 12.06 (s, 1H, NH of the quinazoline); 8.0 (d, 1H, J = 3.28 Hz, proton of the quinazoline position 3); 7.62 (s, 1H, proton of the quinazoline position 6); 7.22 (s, 1H, proton of the quinazoline position 9); 4.25 (dd, 12H, J = 4.08 Hz, crown ether protons).

Step 6
-

-
Preparation: 337 g (1.13 mol) of BPI-05, 7.1 L of chloroform, 1.83 L (19.58mol) of POCI3 and 132 ml of N,N-dimethylformamide were added to a 10 L 3-neck bottle. The reaction mixture was stirred at reflux temperature. After dissolution, reaction completion was checked by TLC (developing solvent: chloroform: methanol = 15:1, Rf = 0.56); the reaction took approximately 8 hours to complete. Then, the reaction solution was cooled and evaporated under vacuum to dryness. The residue was dissolved in 4 L of chloroform; 4 kg of crushed ice was poured into the solution and the mixture was stirred for 0.5 hours. After separation, the aqueous phase was extracted twice with 2 L of chloroform. The organic phases were combined, 4 L of ice water was added and the pH was adjusted with 6 N NaOH to pH 8-9 while the temperature was maintained below 30 °C. After separation, the organic phase was washed with saturated NaCl, dried over anhydrous sodium sulfate and the solvents removed by vacuum evaporation. The residual solids were washed with acetone and filtered; 268 g of BPI-06 as a white crystalline powder was obtained in a yield of 77% with mp: 164-167°C and HPLC purity of 99%. NMR data: 1H-NMR (CDCl3): δ ppm: 8.89 (s, 1H, proton of the quinazoline position 2); 7.68 (s, 1H, proton of the quinazoline position 9); 7.42 (s, 1H, proton of the quinazoline position 6); 4.38-3.81 (dd, 12H, J = 3.88 Hz, crown ether protons).

Step 7
-
-
Preparation of the compound of the present invention: To a suspension of 20.8 g of BPI-06 in 500 mL of ethanol was added 25 mL of N,N-dimethylformamide and a solution of 8.98 g m-acetylene aniline in 200 mL of isopropanol. The reaction mixture was stirred at room temperature for 5 minutes until dissolved completely, and then the reaction solution was heated at reflux for 3 hours. After concentration and drying, the residual solids were dissolved in ethyl acetate, washed with water, and dried over anhydrous sodium sulfate. Thus, 27.1 g of the compound of Formula I was obtained as a white crystalline powder. NMR data: 1H-NMR (Bruker APX-400, solvent: DMSO-d6, TMS as internal standard): δ ppm: 3.58 (dd, 2H, two protons of the crown position 12); 3.60 (dd, 2H, two protons of the crown position 13); 3.73 (dd, 2H, two protons of the crown position 10); 3.80 (dd, 2H, two protons of the crown position 15); 4.30 (s, 1H, proton of the alkynyl); 4.34 (dd, 2H, two protons of the crown position 16); 4.40 (dd, 2H, two protons of the crown position 9); 7.39 (d, 1H, benzene proton at position 25); 7.46 (dd, 1H, benzene proton at position 26); 7.49 (s, 1H, proton of the quinazoline position 6); 7.82 (d, 1H, benzene proton at position 27); 7.94 (t due dd, 1H, proton of the quinazoline position 19); 8.85 (s, 1H, benzene proton at the position 23); 8.87 (s, 1H, proton of the quinazoline position 2); 11.70 (s, 1H, proton of the aromatic amine as salt); 14-16 (bs, 1H, hydrochloride), see Figure 5. NMR data: 13C-NMR (DMSO-d6), see Figure 6. Mass spectrometry (MS): Instrument: ZAB-HS, testing conditions: EI, 200°C, 700ev, MS measured molecular weight: m/z 427.

…………………………………..

Synthesis of compound 1 A
1 Synthesis of Compound 2

2
79.5g 3,4 – dihydroxybenzene nitrile, 272g of potassium carbonate, acetonitrile (6L) was added to a 10L three-necked reaction flask, and dissolved with stirring, heated to reflux and reflux was added dropwise an acetonitrile solution of the compound 1 (compound 1, 200 g; acetonitrile , 2L), and completion of the dropping, the HPLC monitoring of the completion of the reaction, the mixture was cooled to room temperature, filtered, and the solvent was removed, and the resulting solid was washed with ethyl acetate was dissolved, filtered, and the filtrate was concentrated, the resulting residue was dissolved in petroleum ether by rotary evaporation, the resulting solid was purified to give 18.9g of the compound 2.
1 LAI MR (CDC1 3-Sppm): 7.30 ~ 7.33 (m, 1H); 7.25 (s, 1H); 6.97-6.99 (d, 1H); 4.19 – 4.23 (m, 4H); 3.83 ~ 3.91 (m, 4H); 3.77 (s, 4H). MS: (M + H) +250 2 Synthesis of compound A

2 A
41.6g of compound 2 was dissolved in 580ml of acetic acid, dropwise addition of 83ml of fuming nitric acid at 30 ° C under completion of the dropping, the dropwise addition of 42ml of concentrated sulfuric acid at 30 ° C under the reaction at room temperature overnight, TLC monitoring completion of the reaction, the reaction solution was poured into ice water 4L , the precipitated solid was filtered, washed with cold water (500 mL X 2), vacuum 35 ° C and dried crude A compound 46g, isopropanol recrystallization was purified to give 33g of compound A.
1 LAI MR (CDC1 3-Sppm): 7.90 (s, 1H); 7.36 (s, 1H); 4.33 ~ 4.36 (m, 4H); 3.87 ~ 3.89 (m, 4H); 3.737 (s, 4H). Embodiment of Example 2 Synthesis of Compound B

AB
32g of compound A, 30.5g of iron powder, 5% acetic acid solution in methanol 1070ml 2L reaction flask was heated to reflux
TLC monitoring of the end of the reaction cooled and concentrated, dissolved in ethyl acetate, filtered, dried over anhydrous NaS0 4 23g of compound B. The solvent was removed.
1HNMR (d 6-DMSO-Sppm): 7.07 (s, 1H); 6.36 (s, 1H); 5.73 (s, 2H); 3.95 ~ 4.22 (m, 4H); 3.77-3.78 (m, 2H); 3.34 3.62 (m, 6H).Embodiment of Example 3 Synthesis of Compound CI

B CI
500mL three-necked flask, the Add 5g compound B, 5g v, v-dimethyl formamide dimethyl acetal and 160ml of dioxane was heated to reflux the TLC monitoring progress of the reaction, the reaction time is about 12 hours, after the end of the reaction The reaction solution was cooled to room temperature, spin-dry to give 5.8g of compound Cl.
1 LAI MR (CDCl 3-Sppm): 7.56 (s, 1H); 7.15 (s, 1H); 6.51 (s, 1H); 4.12-4.18 (m, 4H); 3.89-3.91 (m, 2H); 3.78 -3.80 (m, 6H); 3.07 (s, 6H); Example 4 Icotinib Synthesis

5 g of the compound Cl, 2.2 g inter-aminophenyl acetylene, 230ml of acetic acid was added to a 500 ml reaction flask was heated to 100 ° c,
TLC monitoring of the reaction. The end of the reaction, the reaction system spin dry methanol was added, and shock dispersion, filtration, wash with methanol, 5g Icotinib.
^ M (d 6-DMSO-5ppm): 11.98 (s, IH); 9.50 (s, IH); 8.53 (s 1H); 8.14 (s, IH); 8.04-8.05 (m, IH); 7.90-7.92 (m, IH); 7.38-7.42 (m, IH); 7.31 (s IH); 7.20-7.22 (m, IH); 4.29-4.30 (m, 4H); 4.21 (s, IH); 3.74-3.81 ( m, 4H); 3.64 (s, 4H); 1.91 (s, 3H); Synthesis Example 5 Exe hydrochloride erlotinib

Exeter for Nick for; s
700mg Icotinib Add to a 100 ml reaction flask, add 40 ml of methanol, stirred pass into the hydrogen chloride gas or concentrated hydrochloric acid, and filtered to give crude hydrochloric acid Icotinib after, and purified by recrystallization from isopropanol to give 760mg hydrochloride Icotinib.
1HNMR (d 6-DMSO-Sppm): 11.37 (s, IH); 8.87 (s, IH); 8.63 (s, IH); 7.90 (s, IH); 7.78-7.80 (d, IH); 7.48-7.52 (m, IH); 7.40-7.41 (m, 2H); 4.36-4.38 (d, 4H); 4.30 (s, IH); 3.75-3.81 (d, 4H); 3.61 (s, 4H); Example 6 Synthesis of Compound B

AB
25g of compound A, 25 g of iron powder, 3% acetic acid in methanol solution 900ml with Example 2 are the same, to give 16.6g of compound B.
Embodiment of Example 7 Synthesis of Compound B

AB
40 g of compound A, 40 g of iron powder and 7% acetic acid in methanol solution was 1200ml, in Example 2, to give 28.4g of compound B.
Example 8 Compound B Synthesis

AB
25 g of compound A, 5 g of Pd / C in 3% acetic acid in methanol solution 900ml Add 2L reaction flask, of the hydrogen, TLC monitoring of the end of the reaction, filtered, and the solvent was removed to give 17g of compound B.
Example 9 Compound B Synthesis

AB
40g of compound A, 17 g of magnesium and 5% acetic acid in methanol solution 1200ml, in Example 2, to give 25.2g of compound B. Example 10 Compound B Synthesis

AB
25 g of compound A, 32.5g of zinc powder and 5% acetic acid in methanol solution 900ml with Example 2 are the same, to give 17.1g of compound B.
Example Synthesis of compound 11 B

AB
25g of compound A, 28 g of iron powder, 5% trifluoroacetic acid in methanol solution 700ml, in Example 2, 16g of compound B.
Embodiment Example 12 Synthesis of Compound C1

3g compound B, 3G v, v-dimethyl formamide dimethyl acetal and 140ml of dioxane, reflux the reaction time is 10-11 hours, the other in the same manner as in Example 3 to give 3.2g of the compound Cl.
Example 13 Synthesis of Compound C1
8g compound B, 8G N, v-dimethyl formamide dimethyl acetal and 180ml of dioxane under reflux for a reaction time of approximately 12-13 hours, with the same manner as in Example 3 to give 8.7g of compound C. Embodiment Example 14 Synthesis of Compound CI

3g compound B, 3 g of N, N-dimethyl formamide dimethyl acetal and 140ml of toluene, the reaction time is 13-15 hours under reflux, with the same manner as in Example 3 to give 2.9g of the compound Cl.
Example 15 Synthesis of Compound C1

The same as in Example 14, except that reaction time is 10 hours, to obtain 2.6g compound Cl t
Embodiment Example 16 Synthesis of Compound C1

500mL three-necked flask, add 3 g of compound B, 3.7 g v, v-dimethylformamide, diethyl acetal and 140ml of dioxane was heated to reflux, TLC monitoring the progress of the reaction, the reaction time of approximately 11-12 hours, After completion of the reaction, the mixture was cooled to room temperature, spin-dry the reaction solution to give 2.5g of the compound Cl.
Example 17 Synthesis of Compound C1

G of compound B, 5.1 g of the N, N-dimethyl formamide di-t-butyl acetal was dissolved in 140ml dioxane was heated to reflux the TLC monitoring progress of the reaction, the reaction time of approximately 11-12 hours after the completion of the reaction, was cooled to room temperature, the reaction solution was spin-dry to give 2.6g of the compound Cl.
Embodiment Example 18 Synthesis of Compound CI

3g compound B, 4.4g N, N-dimethyl formamide diisopropyl acetal was dissolved in 140ml dioxane was heated to reflux, tlc monitoring the progress of the reaction, the reaction time of approximately 11-12 hours after the completion of the reaction, was cooled to room temperature, the reaction solution was spin-dry to give 2.4g of the compound Cl.
The implementation of the synthesis of Example 19 Icotinib

3g compound Cl, 1.3 g inter-aminophenyl acetylene, 130 ml of acetic acid was added 250 ml reaction flask and heated to 70-80
V, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.8g Icotinib. Implementation of Example 20 Icotinib synthesis

C1 Icotinib
. Example 25 Icotinib Hydrochloride synthesis

Icotinib Hydrochloride
The 500mg Icotinib Add to a 100 ml reaction flask, add 30ml of ethanol was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 515mg hydrochlorideIcotinib. Example 26 Icotinib Hydrochloride Synthesis
500mg Icotinib Add 100 ml reaction flask, add 40 ml of tetrahydrofuran was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochlorideIcotinib. EXAMPLE 27 Icotinib Hydrochloride Synthesis

500mg Icotinib Add 100 ml reaction flask, add 50 ml of isopropanol and stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochloride Icotinib.
………………………………………………………………….
http://www.google.com/patents/EP2392576A1 NMR data: 1H-NMR (Bruker APX-400, solvent: DMSO-d6, TMS as internal standard): δ ppm: 3.58 (dd, 2H, two protons of the crown position 12); 3.60 (dd, 2H, two protons of the crown position 13); 3.73 (dd, 2H, two protons of the crown position 10); 3.80 (dd, 2H, two protons of the crown position 15); 4.30 (s, 1H, proton of the alkynyl); 4.34 (dd, 2H, two protons of the crown position 16); 4.40 (dd, 2H, two protons of the crown position 9); 7.39 (d, 1H, benzene proton at position 25); 7.46 (dd, 1H, benzene proton at position 26); 7.49 (s, 1H, proton of the quinazoline position 6); 7.82 (d, 1H, benzene proton at position 27); 7.94 (t due dd, 1H, proton of the quinazoline position 19); 8.85 (s, 1H, benzene proton at the position 23); 8.87 (s, 1H, proton of the quinazoline position 2); 11.70 (s, 1H, proton of the aromatic amine as salt); 14-16 (bs, 1H, hydrochloride), see Figure 5. NMR data: 13C-NMR (DMSO-d6), see Figure 6. Mass spectrometry (MS): Instrument: ZAB-HS, testing conditions: EI, 200°C, 700ev, MS measured molecular weight: m/z 427.
………………………..
NEW PATENT
Zhejiang Beta Pharma Incorporation, 浙江贝达药业有限公司
http://www.google.co.in/patents/WO2013064128A1?cl=en
General synthetic route
Compound A, the present invention is provided for availability, but are not limited to, the following synthetic route to achieve:

The present invention is to provide beta available but are not limited to, the following synthetic route is now:

A BETA
The present invention is to provide a compound C, can be used, but are not limited to, the following synthetic route to achieve:

Wherein
And are independently selected from the group consisting of methyl, ethyl, propyl or isopropyl, or
, And they are connected in common to the N atom form a 3-7 membered ring. R 3 and R4 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, iso-butyl or benzyl group, or,
R 3 and R4 to form a 3-7 membered ring.
The present C can be used for the direct preparation of Icotinib:

Wherein
And are independently selected from the group consisting of methyl, ethyl, propyl or isopropyl, or
, And they are connected in common to the N atom form a 3-7 membered ring.

Icotinib
Icotinib Hydrochloride
Example Synthesis of compound 1 A
1 Synthesis of Compound 2

2
79.5g 3,4 – dihydroxybenzene nitrile, 272g of potassium carbonate, acetonitrile (6L) was added to a 10L three-necked reaction flask, and dissolved with stirring, heated to reflux and reflux was added dropwise an acetonitrile solution of the compound 1 (compound 1, 200 g; acetonitrile , 2L), and completion of the dropping, the HPLC monitoring of the completion of the reaction, the mixture was cooled to room temperature, filtered, and the solvent was removed, and the resulting solid was washed with ethyl acetate was dissolved, filtered, and the filtrate was concentrated, the resulting residue was dissolved in petroleum ether by rotary evaporation, the resulting solid was purified to give 18.9g of the compound 2.
1 LAI MR (CDC1 3-Sppm): 7.30 ~ 7.33 (m, 1H); 7.25 (s, 1H); 6.97-6.99 (d, 1H); 4.19 – 4.23 (m, 4H); 3.83 ~ 3.91 (m, 4H); 3.77 (s, 4H). MS: (M + H) +250 2 Synthesis of compound A

2 A
41.6g of compound 2 was dissolved in 580ml of acetic acid, dropwise addition of 83ml of fuming nitric acid at 30 ° C under completion of the dropping, the dropwise addition of 42ml of concentrated sulfuric acid at 30 ° C under the reaction at room temperature overnight, TLC monitoring completion of the reaction, the reaction solution was poured into ice water 4L , the precipitated solid was filtered, washed with cold water (500 mL X 2), vacuum 35 ° C and dried crude A compound 46g, isopropanol recrystallization was purified to give 33g of compound A.
1 LAI MR (CDC1 3-Sppm): 7.90 (s, 1H); 7.36 (s, 1H); 4.33 ~ 4.36 (m, 4H); 3.87 ~ 3.89 (m, 4H); 3.737 (s, 4H). Embodiment of Example 2 Synthesis of Compound B

AB
32g of compound A, 30.5g of iron powder, 5% acetic acid solution in methanol 1070ml 2L reaction flask was heated to reflux
TLC monitoring of the end of the reaction cooled and concentrated, dissolved in ethyl acetate, filtered, dried over anhydrous NaS0 4 23g of compound B. The solvent was removed.
1HNMR (d 6-DMSO-Sppm): 7.07 (s, 1H); 6.36 (s, 1H); 5.73 (s, 2H); 3.95 ~ 4.22 (m, 4H); 3.77-3.78 (m, 2H); 3.34 3.62 (m, 6H). Embodiment of Example 3 Synthesis of Compound CI

B CI
500mL three-necked flask, the Add 5g compound B, 5g v, v-dimethyl formamide dimethyl acetal and 160ml of dioxane was heated to reflux the TLC monitoring progress of the reaction, the reaction time is about 12 hours, after the end of the reaction The reaction solution was cooled to room temperature, spin-dry to give 5.8g of compound Cl.
1 LAI MR (CDCl 3-Sppm): 7.56 (s, 1H); 7.15 (s, 1H); 6.51 (s, 1H); 4.12-4.18 (m, 4H); 3.89-3.91 (m, 2H); 3.78 -3.80 (m, 6H); 3.07 (s, 6H); Example 4 Icotinib Synthesis

5 g of the compound Cl, 2.2 g inter-aminophenyl acetylene, 230ml of acetic acid was added to a 500 ml reaction flask was heated to 100 ° c,
TLC monitoring of the reaction. The end of the reaction, the reaction system spin dry methanol was added, and shock dispersion, filtration, wash with methanol, 5g Icotinib.
^ M (d 6-DMSO-5ppm): 11.98 (s, IH); 9.50 (s, IH); 8.53 (s 1H); 8.14 (s, IH); 8.04-8.05 (m, IH); 7.90-7.92 (m, IH); 7.38-7.42 (m, IH); 7.31 (s IH); 7.20-7.22 (m, IH); 4.29-4.30 (m, 4H); 4.21 (s, IH); 3.74-3.81 ( m, 4H); 3.64 (s, 4H); 1.91 (s, 3H);
Synthesis Example 5 Exe hydrochloride erlotinib

Exeter for Nick for; s
700mg Icotinib Add to a 100 ml reaction flask, add 40 ml of methanol, stirred pass into the hydrogen chloride gas or concentrated hydrochloric acid, and filtered to give crude hydrochloric acid Icotinib after, and purified by recrystallization from isopropanol to give 760mg hydrochloride Icotinib.
1HNMR (d 6-DMSO-Sppm): 11.37 (s, IH); 8.87 (s, IH); 8.63 (s, IH); 7.90 (s, IH); 7.78-7.80 (d, IH); 7.48-7.52 (m, IH); 7.40-7.41 (m, 2H); 4.36-4.38 (d, 4H); 4.30 (s, IH); 3.75-3.81 (d, 4H); 3.61 (s, 4H);
Example 18 Synthesis of Compound CI

3g compound B, 4.4g N, N-dimethyl formamide diisopropyl acetal was dissolved in 140ml dioxane was heated to reflux, tlc monitoring the progress of the reaction, the reaction time of approximately 11-12 hours after the completion of the reaction, was cooled to room temperature, the reaction solution was spin-dry to give 2.4g of the compound Cl.
The implementation of the synthesis of Example 19 Icotinib

3g compound Cl, 1.3 g inter-aminophenyl acetylene, 130 ml of acetic acid was added 250 ml reaction flask and heated to 70-80
V, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.8g Icotinib. Implementation of Example 20 Icotinib synthesis

C1 Icotinib
8g compound Cl, 3.5g inter-aminophenyl acetylene, dissolved in 380ml of acetic acid, heated to 100-120 ° C, TLC monitoring of the reaction. Spin dry the reaction system, by adding ethanol shock dispersion, filter, the ethanol wash 7.2g Icotinib. Implementation of Example 21 Icotinib Synthesis

The C1 Exeter erlotinib reaction temperature of 120-15CTC Example 4 was 2.2 g Icotinib.
Example 22 Icotinib Synthesis
3g compound Cl, 1.8 g inter-aminophenyl acetylene and 130 ml of acetic acid was added 250 ml reaction flask and heated to 90-100C, TLC monitoring of the reaction. Spin dry the reaction system, isopropanol shock dispersion, filtration, isopropyl alcohol wash was 2.9g Icotinib.
The implementation of the synthesis of Example 23 Icotinib

3G compound CI and 1.3 g of m-aminophenyl acetylene dissolved in 130ml of formic acid was heated to 80-90 ° C, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.7g Icotinib.
Example 24 Icotinib synthesis

3g of compound C1 and 1.3g aminophenyl acetylene dissolved in 130ml of trifluoroacetic acid was heated to 70-80 ° C, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.7g Icotinib. Example 25 Icotinib Hydrochloride synthesis

Icotinib Hydrochloride
The 500mg Icotinib Add to a 100 ml reaction flask, add 30ml of ethanol was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 515mg hydrochloride Icotinib. Example 26 Icotinib Hydrochloride Synthesis
500mg Icotinib Add 100 ml reaction flask, add 40 ml of tetrahydrofuran was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochloride Icotinib. EXAMPLE 27 Icotinib Hydrochloride Synthesis

Exeter erlotinib erlotinib hydrochloride Exeter
500mg Icotinib Add 100 ml reaction flask, add 50 ml of isopropanol and stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochloride Icotinib. Example 28 Icotinib Hydrochloride synthesis

Icotinib
Icotinib Hydrochloride
 |
|
| Clinical data | |
|---|---|
| Trade names | Conmana, Icotinib |
| Legal status |
?
|
| Routes | Oral tablets |
| Pharmacokinetic data | |
| Bioavailability | 52% |
| Metabolism | Hepatic (mainly CYP3A4, lessCYP1A2) |
| Half-life | 5.5 hrs (median) |
| Excretion | >98% as metabolites, of which >90% via faeces, 9% via urine |
| Identifiers | |
| CAS number | 1204313-51-8 |
| ATC code | ? |
| PubChem | CID 22024915 |
| DrugBank | DB00530 |
| ChemSpider | 10762174 |
| UNII | 9G6U5L461Q |
| Chemical data | |
| Formula | C22H21N3O4 |
| Mol. mass | 391.420 g/mol |
References
- Sordella, R. (20 August 2004). “Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways”. Science 305(5687): 1163–1167. doi:10.1126/science.1101637. PMID 15284455.
- Shi, Yuankai; Zhang, Li; Liu, Xiaoqing; Zhou, Caicun; Zhang, Li; Zhang, Shucai; Wang, Dong; Li, Qiang; Qin, Shukui; Hu, Chunhong; Zhang, Yiping; Chen, Jianhua; Cheng, Ying; Feng, Jifeng; Zhang, Helong; Song, Yong; Wu, Yi-Long; Xu, Nong; Zhou, Jianying; Luo, Rongcheng; Bai, Chunxue; Jin, Yening; Liu, Wenchao; Wei, Zhaohui; Tan, Fenlai; Wang, Yinxiang; Ding, Lieming; Dai, Hong; Jiao, Shunchang; Wang, Jie; Liang, Li; Zhang, Weimin; Sun, Yan. “Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial”. The Lancet Oncology 14 (10): 953–961. doi:10.1016/s1470-2045(13)70355-3.
- Tan, Fenlai; Gu, Aiqin; Zhang, Yiping; Jiao, Shun Chang; Wang, Chang-li; He, Jintao; Jia, Xueke; Zhang, Li; Peng, Jiewen; Wu, Meina; Ying, Kejing; Wang, Junye; Ma, Kewei; Zhang, Shucai; You, Changxuan; Ding, Lieming; Wang, Yinxiang; Shen, Haijiao; Wan, Jiang; Sun, Yan (2013). “Safety and efficacy results of a phase IV, open-label, multicenter, safety-monitoring study of icotinib in treating advanced non-small cell lung cancer (NSCLC): ISAFE study”. ASCO 2013 Meeting: e19161.
- Chen, Xiaofeng; Zhu, Quan; Liu, Yiqian; Liu, Ping; Yin, Yongmei; Guo, Renhua; Lu, Kaihua; Gu, Yanhong; Liu, Lianke; Wang, Jinghua; Wang, Zhaoxia; Røe, Oluf Dimitri; Shu, Yongqian; Zhu, Lingjun; Chellappan, Srikumar P. (16 May 2014). “Icotinib Is an Active Treatment of Non-Small-Cell Lung Cancer: A Retrospective Study”. PLoS ONE 9 (5): e95897.doi:10.1371/journal.pone.0095897.
| WO2007138613A2 * | 12 Mar 2007 | 6 Dec 2007 | Venkateshappa Chandregowda | A process for synthesis of [6,7-bis-(2-methoxyethoxy)-quinazolin-4-yl]-(3-ethynylphenyl)amine hydrochloride |
| WO2010003313A1 | 7 Jul 2009 | 14 Jan 2010 | Zhejiang Beta Pharma Inc. | Icotinib hydrochloride, synthesis, crystallographic form, medical combination, and uses thereof |
| CN1305468C | 29 May 2003 | 21 Mar 2007 | 中国人民解放军第三○二医院 | Bolengsu compound and its preparation, medicine composition and use |
| US7078409 | 26 Mar 2003 | 18 Jul 2006 | Beta Pharma, Inc. | Fused quinazoline derivatives useful as tyrosine kinase inhibitors |
| Patent | Submitted | Granted |
|---|---|---|
| Icotinib Hydrochloride, Synthesis, Crystalline Forms, Pharmaceutical Compositions, and Uses Thereof [US2011182882] | 2011-07-28 | |
| Fused quinazoline derivatives useful as tyrosine kinase inhibitors [US7078409] | 2004-03-11 | 2006-07-18 |
FOSTEMSAVIR ,фостемсавир , فوستيمسافير , 磷坦姆沙韦 ,ホステムサビル;

Fostemsavir
GSK3684934
CAS 864953-29-7
- Molecular FormulaC25H26N7O8P
- Average mass583.490 Da
- ホステムサビル;
[3-[2-(4-benzoylpiperazin-1-yl)-2-oxoacetyl]-4-methoxy-7-(3-methyl-1,2,4-triazol-1-yl)pyrrolo[2,3-c]pyridin-1-yl]methyl dihydrogen phosphate
- BMS 663068
- BMS663068
- Fostemsavir tromethamine
- UNII-2X513P36U0
Fostemsavir tromethamine [USAN],
CAS 864953-39-9,
MW 704.6303

Fostemsavir (GSK3684934/BMS-663068) is an experimental HIV entry inhibitor and a prodrug of temsavir (BMS-626529). It is under development by [ViiV Healthcare / GlaxoSmithKline]] for use in the treatment of HIV infection. By blocking the gp120 receptor of the virus, it prevents initial viral attachment to the host CD4+ T cell and entry into the host immune cell; its method of action is a first for HIV drugs.[1] Because it targets a different step of the viral lifecycle, it offers promise for individuals with virus that has become highly resistant to other HIV drugs.[2] Since gp120 is a highly conserved area of the virus, the drug is unlikely to promote resistance to itself via generation of CD4-independent virus.[3]



Example 6Preparation of Compound I from Compound D′ (Example 5)

N-Benzoylpiperazine HCl, Compound Db, (11.73 g, 51.74 mmol) was added to a mixture of Compound D′ (14.83 g, 47.03 mmol) (prepared in Example 5) in dry THF (265 mL) and dry DMF (29.5 mL). NaOt-Bu, 30% w/w (52.3 mL, 147 mmol) was added dropwise (30 min.) keeping the temperature at 17-21° C. The resulting yellow slurry was stirred at 17-20° for 1 h more, then cooled to about 5° C. The mixture was slowly poured into cold water (90 mL) and the flask rinsed with additional water (10 mL). The pH of the resulting yellow solution was adjusted to 6-7 with slow addition (˜20 min., 5-12° C.) of 1 N HCl (105 mL). The resulting slurry was warmed and stirred at room temperature for 1.5 h. The slurry was filtered and the cake washed with water (2×60 mL) then dried in vacuo at 65-70° C. for 5 h giving 18.4 g Compound I as a white solid (82.6%), HPLC AP 99.4. 1H NMR (400 MHz, d6-DMSO): δ 2.48 (s, 3H), 3.43 (b, 4H), 3.67 (b, 4H), 3.99 (s, 3H), 7.45 (s, 5H), 7.88 (s, 1H), 8.24 (s, 1H), 9.22 (s, 1H), 12.39 (s, 1H). 13C NMR (100 MHz, d6-DMSO): 13.85, 40.65, 45.22, 56.85, 114.19, 121.02, 122.78, 123.65, 127.06, 128.42, 129.61, 129.70, 135.51, 138.59, 142.18, 149.23, 161.38, 166.25, 169.30, 185.51.
If necessary, the product could be further purified by recrystallization from acetic acid-water-ethanol, ethanol-water, or acetone-water. For example: A mixture of Compound I (25.0 g), glacial acetic acid (260 mL) and DI water (13.8 mL) was heated to 80° C. and held with stirring (overhead) until a solution was obtained (40 min.). The batch was cooled to 70° C. and seeded (0.5 g). With slow agitation (100 rpm), EtOH (300 mL) was added slowly (1 h), keeping the temperature at 70° C. The resulting slurry was kept at 70° C. for 1 h more with very slow stirring. The slurry was cooled to 20° C. over 2 hours and held at 20° C. for over 4 hours. The slurry was filtered, the wet cake washed with EtOH (125 mL), and the solid dried in vacuo at 70° C. (≧16 h), giving 22.6 g Compound I as a white solid (88.4%).

The development of a short and efficient synthesis of a complex 6-azaindole, BMS-663068, is described. Construction of the 6-azaindole core is quickly accomplished starting from a simple pyrrole, via a regioselective Friedel–Crafts acylation, Pictet–Spengler cyclization, and a radical-mediated aromatization. The synthesis leverages an unusual heterocyclic N-oxide α-bromination to functionalize a critical C–H bond, enabling a highly regioselective copper-mediated Ullmann–Goldberg–Buchwald coupling to install a challenging triazole substituent. This strategy resulted in an efficient 11 step linear synthesis of this complex clinical candidate

Attachment inhibitor BMS-663068 is currently in clinical development for the treatment of HIV infection. Key steps in the synthesis depicted are (1) a radical-mediated redox-aromatization to generate the 6-azaindole (B → C) and (2) the regioselective bromination of an N-oxide using PyBroP (D → E).
High regioselectivity was observed in the copper(I)-mediated Ullmann–Goldberg–Buchwald coupling (H → K) using the diamine ligand J (N1/N2 = 22:1), whereas a thermal SNAr reaction gave N1/N2 = 1:1. Alternative conditions for the bromination of the N-oxide D led mainly to deoxygenation.
………………………………….
http://www.google.com/patents/US20050209246
Preparation of Compound IVc

Procedure: To a solution of the acid 6-81 (3.01 g, 10 mmol) and benzoylpiperazine hydrochloride (3.39 g, 15 mmol) in DMF (50 mL) was added triethylamine (10.1 g, 100 mmol, 10 eq.), followed by 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC; 5.75 g, 30 mmol) under N2 and the mixture stirred at room temperature for 22 h after sonication and at 40° C. for 2 h. The mixture was concentrated in vacuo to remove DMF and TEA, and to the residual solution was added water (200 mL) under stirring and sonication. The precipitates formed were collected, washed with water and dried in vacuo to obtain 2.8 g (5.9 mmol, Y. 59%) of the title compound IVc as off-white solid. The filtrate was extracted with CH2Cl2 (x2). The CH2Cl2 extracts were dried (Na2SO4), filtered and concentrated to gum which was triturated with Et2O to obtain a solid. This solid was suspended and triturated with MeOH to obtain 400 mg of the title compound IVc as off-white solid. Total yield: 3.2 g (6.8 mmol, Y. 68%): MS m/z 474 (MH); HRMS (ESI) m/z calcd for C24H24N7O4 (M+H) 474.1890, found 474.1884 (Δ-1.2 ppm); 1H NMR (DMSO-d6) δ ppm 2.50 (3H, s, overlapped with DMSO peaks), 3.43 (4H, br, CH2N), 3.68 (4H, br, CH2N), 3.99 (3H, s, CH3O), 7.46 (5H, br. s, Ar—Hs), 7.88 (1H, s, indole-H-5), 8.25 (1H, s, indole-H-2), 9.25 (1H, s, triazole-H-5), 12.40 (1H, s, NH); 13C-NMR (DMSO-d6) δ ppm 13.78 ,40.58, 45.11, 56.78, 114.11, 120.95, 122.71, 123.60, 126.98, 128.34, 129.6, 135.43, 138.52, 142.10, 149.15, 161.29, 166.17, 169.22, 185.42; UV (MeOH) λ max 233.6 nm (ε 3.43×104), 314.9 nm (ε 1.73×104); Anal: Calc for C24H24N7O4.1/5H2O; C, 60.42; H, 4.94; N, 20.55, Found; C 60.42, H 5.03, N 20.65; KF (H2O) 0.75%.
This reaction can also be performed by use of HATU and DMAP to provide more consistent yield of the title compound: To a suspension of the acid 6-81 (15.6 mmol) and HATU [O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophos phonate] (8.90 g, 23.4 mmol; 1.5 eq.) in DMF (60 mL) and CH2Cl2 (60 mL) was added a mixture of DMAP (5.72 g, 46.8 mmol, 3 eq.) and benzoylpiperazine hydrochloride (5.30 g, 23.4 mmol; 1.5 eq.) in DMF (60 mL) at room temperature and the mixture was stirred under nitrogen atmosphere for 4 hrs. The mixture was concentrated in vacuo to remove CH2Cl2 and most of DMF, and to the residual solution was added water under stirring and sonication. The precipitates formed were collected, washed with water and dried in vacuo to obtain 5.38 g (11.4 mmol, Y. 72.8%) of the title compound IVc as off-white solid: HPLC >95% (AP, uv at 254 nm)
EXAMPLE 5Preparation of Ica, (Disodium Salt)

General Procedure: A suspension of IVc (0.24 g, 0.5 mmol) in anhydrous THF (4 mL) under nitrogen atmosphere was treated with sodium hydride (60% oil dispersion, 0.08 g, 2.0 mmol), and stirred until gas evolution ceased (approximately 5 minutes). The reaction mixture was treated with iodine (0.13 g, 0.5 mmol) and stirred for 2-3 minutes followed by addition of di-tert-butyl chloromethyl phosphate (1.6 g, 6.0 mmol, crude). A stream of nitrogen was allowed to pass over the reaction to facilitate the removal of much or all of the THF. The reaction mixture was stirred overnight. HPLC analysis of crude indicated starting IVc (ca. 56%) and desired adduct (ca. 32%).
Several crude reaction mixtures (a total of 6.7 mmol based on starting material IVc) were re-dissolved in dichloromethane, combined, concentrated in vacuo to remove any remaining THF. The residue was suspended in dichloromethane and TFA (1:1, approximately 40 mL total volume). The mixture was stirred for 1.5-2 hours and then solvent was removed in vacuo. The residue was suspended in dichloromethane and extracted into water (approximately 60 mL) made weakly basic with solid or aqueous sodium bicarbonate. The aqueous layer was reduced in volume by rotary evaporator if required and the solution was loaded onto a C-18 reverse phase column (approximately 80 g of C-18, YMC ODS-Aq, 50 micron) and eluted with water, followed by water containing 2.5% acetonitrile. Fractions containing pure product were pooled and organic solvent was removed by rotary evaporator. Purified product was recovered after lyophilization to give 1.00 g (1.30 mmol, 19% over 2 steps) of the title compound Ica (disodium salt) as an off-white powder: HPLC purity>99% AP at 254 nm (gradient 0-100% B/A; A 10% CH3CN-90% H2O-0.1% TFA, B 90% CH3CN-10% H2O-0.1 % TFA, gradient time 4 min, column YMC ODS-Aq 4.6×50 mm 3 micron); MS-ESI— m/z 482 (M−H minus 2Na)−; HRMS (ESI) m/z calcd for C25H27N7O8P (M+H minus 2Na)+584.1659, found 584.1651 (Δ-1.3 ppm); 1H NMR (D2O, 500 MHz) δ ppm 2.53, 2.54 (3H, 2s), 3.56 (2H, s, CH2N), 3.72 (2H, br.s, CH2N), 3.78, 3.83 (2H, 2br.s, CH2N), 3.94, 3.96 (2H, 2br.s, CH2N), 4.14 (3H, s, CH3O), 5.38, 5.40 (2H, 2d, J=11 Hz), 7.45-7.59 (5H, m, Ar—Hs), 8.07, 8.09 (1H, 2s, indole-H-5), 8.64, 8.67 (1H, 2s, indole-H-2), 8.87, 8.89 (1H, 2s, triazole-H-5); 13C NMR (125.7 MHz, D2O) δ ppm 15.43 (N-Me), 44.03, 44.47, 44.66, 45.05, 48.20, 48.82, 49.60, 50.23, 59.78 (OMe), 75.81 (NCH2O), 115.6, 126.0, 127.2, 129.6, 131.0, 131.7, 132.1, 133.5, 136.8, 147.6, 150.1, 154.2, 164.8, 170.4, 175.8, 189.2; UV (H2O) λmax 220 nm (ε 3.91×104), 249 nm (ε 2.00×104), 303 nm (ε 1.60×104); Anal: Calc for C25H24N7O8PNa2. 8H2O. 0.2NaHCO3; C, 38.39; H, 5.14; N, 12.44, P, 3.93, Na, 6.42 Found; C, 38.16; H, 4.81; N, 12.43, P, 3.72, Na, 6.05; KF (H2O) 17.3%. A less pure fractions were collected to obtain 0.22 g (0.29 mmol, Y. 4%) of the title compound Ica (disodium salt): HPLC purity>95% (AP at 254 nm).
EXAMPLE 7Preparation of Crystalline Ic (Free Acid Mono-Hydrate)

To a mixture of IVc (600 mg, 1.27 mmol) in anhydrous THF (10 ml) in an oven-dried round bottle flask under nitrogen at r.t. was added NaH (153 mg, 6.38 mmol, dry powder, 95%), and the white suspension stirred until no gas evolution was observed. The mixture was then added I2 (375 mg, 1.48 mmol), and stirred at r.t. for 3 h. To the reaction mixture was added NaH (153 mg, 6.38 mmol, dry powder, 95%), and the mixture stirred for about 5 to 10 min. The crude chloromethyl di-tert-butylphosphate (2.0 g, about 1.6 ml, 7.79 mmol) was added to the mixture, which was then stirred at r.t. for 15 h. LCMS analysis of the reaction showed a >97% conversion of the starting material. After evaporation of the volatiles, the residue was added CH2Cl2 (10 ml), cooled in an ice-water bath, slowly added TFA (10 ml) and stirred at r.t. for 3 h. The reaction mixture was then evaporated, and the residue partitioned between CH2Cl2 (50 ml) and H2O (50 ml). The CH2Cl2 layer was poured into the reaction flask that contained some undissolved brownish solid, and this mixture was extracted with a dilute aqueous NaHCO3 solution (50 ml). The aqueous mixture was purified by reverse phase preparative HPLC (solvent A: 10% MeOH-90% H2O-0.1% TFA; solvent B: 90% MeOH-10% H2O-0.1% TFA; start % B=0, final % B=100; gradient time=6 min; flow rate=45 ml/min; column: phenomenex-Luna 30×50 mm, S5; fraction collected: 3.65 to 4.05 min). The fractions collected were evaporated to dryness, and the residue dried under high vacuum to obtain the acid Ic as a pale yellow solid (356.6 mg); 1H NMR: (500 MHz, CD3OD) δ 9.05 (s, 1H), 8.46 (s, 1H), 8.04 (s, 1H), 7.47 (b s, 5H), 5.93 (d, J=12, 2H), 4.10 (s, 3H), 4.00-3.40 (b s, 8H), 2.53 (s, 3H); 19F NMR analysis showed that the material contained residual TFA, (the percentage was not quantified); Analytical HPLC method: Start % B=0, Final % B=100, Gradient time=2 min, Flow Rate=5 mL/min, Column: Xterra MS C18 7u 3.0×50 mm, LC/MS: (ES+) m/z (M+H)+=584, HPLC Rt=0.983.
172.2 mg of the purified acid Ic was dissolved in 1 ml of H2O and then about 0.3 ml of absolute EtOH (200 proof) was added. The mixture was left standing in a refrigerator (temperature about 3° C.) overnight, after which time, crystalline material was observed. The mixture was then warmed to ambient temperature, diluted with H2O to a volumn of 3 mL, and then 20 mL of MeCN was added slowly. Following the completion of addition, the mixture was stirred at r.t. for 2 h and then filtered. The solid collected (90 mg) was dried in vacuo, and then under high vacuum. This material was shown by powder x-ray studies to be crystalline; Elemental Analysis calculated for C25H26N7O8P.H2O: C 49.92; H 4.69; N 16.30; observed: C 49.66; H 4.62; N 15.99; mp=205° C. (measured by differential scanning calorimetry). The 1H NMR pattern for crystalline material was compared with that from the purified acid and both were consistent with the structure.
EXAMPLE 10Preparation of Icb (mono tromethamine salt): [3-[(4-benzoylpiperazin-1-yl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2, 3-c]pyridin-1-yl]methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)propane-1,3-diol salt (1:1). The sequence of reactions is described in Scheme for Example 10.
Scheme for Example 10


Preparation of di-tert-butyl chloromethyl phosphate

A mixture of tetrabutylammonium di-tert-butyl phosphate (57 g, 0.126 mol, Digital Specialty Chemicals) and chloroiodomethane (221 g, 1.26 mol) was stirred at room temperature for four hours before the volatiles were removed under vacuum. 500 ml of ethyl ether was added to the residue and insoluble solid was filtered away. Concentration of the filtrate in vacuo and removal of remaining volatiles using a vacuum pump provided di-tert-butyl chloromethyl phosphate as a light brown or yellow oil, which was utilized in the next step without further purification.
Preparation of IIc: (3-(2-(4-benzoylpiperazin-1-yl)-2-oxoacetyl)-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl)methyl di-tert-butyl phosphate

NaH (2.6 g, 10.3 mmol, 95% in oil, Seq.) was added slowly into a suspension of IVc (10.0 g, 21.1 mmol) in dry THF (100 ml) and the mixture was allowed to stir for 0.5 hour at room temperature. A solution of iodine (5.27 g, 20.8 mmol) dissolved in dry THF (10 ml) was added slowly into the stirring solution at a rate which prevented foaming or a violent reaction. The resultant mixture was stirred for an additional 3 hours before a second 2.6 g portion of NaH was introduced. After 15 minutes at ambient temperature di-tert-butyl chloromethyl phosphate, the entire batch of di-tert-butyl chloromethyl phosphate, obtained from step one, was added. After stirring for 16 hours, the reaction mixture was poured into iced NH4OAc (30%) (120 ml), followed by extraction with EtOAc (3×300 ml). The combined organic extracts were washed with water (100 ml) and then brine (100 ml), dried over Na2SO4, and concentrated under vacuum to afford a residue, which was purified by silica gel chromatography (elution with EtOAc/Et3N (50/1) and then EtOAc/MeOH (100/1)) to give 8.0 g (˜75% AP, ˜41% yield) of diester IIc as a light yellow solid.
1H NMR (500 MHz, CD3OD) δ8.82 (s, 1H), 8.41 (s, 1H), 8.04 (s, 1H), 7.47 (b, 5H), 6.00 (d, 2H, J=14.5 Hz), 4.10 (s, 3H), 4.00-3.40 (b, 8H), 2.49 (s, 3H), 1.28 (s, 18H); 13C NMR (125 MHz, CD3OD) δ18.6, 176.4, 172.9, 168.0, 162.6, 152.6, 147.5, 144.0, 136.5, 131.5, 130.8, 129.9, 129.1, 128.3, 126.1, 124.0, 116.2, 85.8, 75.4, 61.6, 57.7, 30.1, 22.2, 13.7; HRMS m/z: (M+H)+ calcd for C33H43N7O8P 696.29, found 696.34.
Preparation of Icb (mono L tromethamine salt): [3-[(4-benzoylpiperazin-1-yl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl]methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)propane-1,3-diol salt (1:1)

500 mg (˜p75 AP, 0.54 mmol) of diester IIc was dissolved in a mixture of water (2.5 ml) and acetone (2.5 ml). The resulting mixture was stirred at 40° C. for 16 hours to complete the solvolysis. To this reaction mixture was added 3.0M aqueous TRIS (mono tromethamine) solution to adjust pH to 3.32. Acetone (30 ml) was slowly added to the reaction mixture in 1 hour.* After complete addition of acetone, the solution was stirred overnight to complete the crystallization of Icb. The solid was collected by filtration and rinsed with 20:1 acetone-water (2×5 mL). The white crystalline solid was dried under house vacuum under nitrogen atomosphere at 50° C. for 24 h to afford 290 mg of Icb (>98.5 AP).
*After adding about 15 and 20 ml of acetone, the reaction mixture was seeded with crystalline Icb.
Icb obtained in the above operation: 1H NMR (500 MHz, CD3OD) δ8.83 (s, 1H), 8.52 (s, 1H), 8.02 (s, 1H) 7.49 (b, 5H), 5.469 (d, 2H, J=13 Hz), 4.11 (s, 3H), 4.00-3.40 (m, 8H), 3.66 (s, 6H), 2.50 (s, 3H); 13C NMR (125 MHz, CD3OD) δ185.6, 171.9, 167.4, 161.4, 151.7, 146.9, 143.8, 135.4, 130.3, 129.7, 128.8, 127.2, 124.9, 122.6, 114.3, 73.5, 61.8, 59.9, 56,5, 46.0, 41.7, 12.6. HRMS m/z: (M-trisamine+H)+ calcd for C25H27N7O8P 584.1659, found 584.1664. Anal. Calcd. C, 49.43; H, 5.29; N, 15.90; P, 4.39; found: C, 49.18; H, 5.38; N, 15.59; P, 4.26. Melting Point 203° C.
Obtained via other process (hydrolysis with TFA in methylene chloride), salt Icb is ˜1 molar mono tromethamine salt with 0.47% of water, 0.1% of acetone and 0.05% of methanol. 1H NMR (500 MHz, d6-DMSO, 30° C.) δ8.77 (s, 1H), 8.48 (s, 1H), 8.00 (s, 1H) 7.44 (b, 5H), 5.42 (d, 2H, J=15 Hz), 4.02 (s, 3H), 3.70-3.30 (m, 8H), 3.41 (s, 6H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3, 30° C.) δ184.8, 169.0, 165.8, 160.3, 150.4, 146.2, 143.2, 135.4, 129.4, 128.9, 128.2, 127.7, 126.9, 123.2, 122.2, 112.9, 72.3, 60.7, 59.0, 56.7, 13.4. MS m/z: (M-trisamine+H)+ calcd for C25H27N7O8P 584.2, found 584.0. Anal. Calcd. C, 49.11; H, 5.37; N, 15.76; P, 4.32; found: C, 48.88; H, 5.28; N, 15.71; P, 4.16. M.P. 201-205° C.
EXAMPLE 13Alternate preparation of Icb (Pro-drug of IVc)

To a 10 L reactor equipped with an overhead stirrer, thermocouple, distillation apparatus, and nitrogen inlet was charged IVc (200.00 g, 422.39 mmol), Cs2CO3 (344.06 g, 1.06 mol), KI (140.24 g, 844.81 mmol) and NMP (1.00 L, 10.38 mol). The reaction was stirred at room temperature resulting in a light brown heterogeneous suspension. Di-tert-butyl chloromethyl phosphate (273.16 g, 1.06 mol) was added via addition funnel and the reaction mixture was heated to 30° C. for 16-24 hours with stirring after which time the reaction was cooled to 5° C. To the reaction was added DCM (1.5 L) then the reaction was slowly quenched with water (3.5 L) maintaining the reaction temperature under 20° C. resulting in a biphasic mixture. The product rich bottom layer was separated, washed with water (3.5 L×3), then transferred back to the reactor. The solution was concentrated under vacuum to a volume of 1 L keeping the temperature below 25° C. IPA was added (2 L) then the reaction was concentrated under vacuum to a volume of 2 L keeping the temperature below 25° C. The reaction was then seeded with IIc (0.200 g), stirred overnight at room temperature resulting in a slurry. The slurry was filtered and the wet cake was washed with MTBE (1 L), dried in a vacuum oven at 50° C. overnight resulting in a yellow/white powder (207.1 g, 70%). 1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 8.18 (s, 1H), 7.91 (s, 1H), 7.42 (s, 5H), 5.95 (d, J=14.2 Hz, 2H), 4.06 (s, 3H), 3.97-3.36 (m, 8H), 2.50 (s, 3H), 1.27 (s, 18H); 3C NMR (100 MHz, CDCl3) δ 184.64, 170.65, 165.91, 161.60, 150.82, 145.38, 141.89, 134.96, 130.20, 129.59, 128.68, 127.58, 127.10, 124.77, 122.64, 115.22, 83.90, 83.83, 73.69, 73.63, 56.95, 46.04, 41.66, 29.61, 29.56, 13.90; ES+ MS m/z (rel. intensity) 696 (MH+,10), 640 (MH+-isobutylene, 30), 584 (MH+-2 isobutylene, 100).

To a 10 L 4 neck reactor equipped with a thermocouple, overhead stirrer, condenser and nitrogen inlet was added IIc (200.24 g, 287.82 mmol), acetone (800.00 ml, 10.88 mol) and water (800.00 ml, 44.41 mol). The reaction was heated to 40° C. and stirred for 18-24 hours. The reaction was cooled to 20° C. then tromethamine (33.62 g, 277.54 mmol) was added. The reaction was heated to 40° C. then stirred for an additional hour until all solids were dissolved. The reaction was cooled to 20° C. then filtered through a 10 micron cuno filter into a 10 L 4 neck reactor equipped with a thermocouple, overhead stirrer, and nitrogen inlet. Acetone (3 L) was added rapidly, followed by seeding with Icb (0.500 g), then additional acetone (3 L) was added. The reaction was stirred at room temperature overnight resulting in a slurry then filtered. The wet cake was washed with acetone (800 ml) then dried in a vacuum oven at 50° C. overnight resulting in a fluffy white powder (165.91 g, 82%).
Supplementary Information:
Isolation of the Free-Acid Intermediate IC:

In a 250 mL 3 neck reactor equipped with a thermocouple, overhead stirrer, condenser and nitrogen inlet was added IIc (10.0 g, 14.37 mmol), acetone (40.00 ml, 544.15 mmol) and water (40.00 ml, 2.22 mol). The reaction was heated to 40° C. and stirred for 14-24 hours. The reaction was cooled to 20° C. then stirred for three hours, resulting in a slurry. The slurry was filtered, then the wet cake washed with acetone (40.00 ml) then dried in a vacuum oven at 50° C. overnight resulting in a fluffy white powder (7.00 g, 83%). NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.47 (s, 1H), 8.06 (s, 1H), 7.45 (s, 5H), 5.81 (d, J=12.3 Hz, 2H), 4.03 (s, 3H), 3.91-3.19 (m, 8H), 2.39 (s, 3H); 13C NMR (500 MHz, DMSO-d6) δ 185.20, 169.32, 165.85, 160.75, 150.51, 146.30, 143.24, 135.53, 129.74, 129.22, 128.46, 127.34, 127.09, 123.67, 122.73, 113.94, 72.90 (d, 2JC-P=5 Hz), 57.01, 45.2 (bs), 40.8 (bs), 13.66. ES+ MS m/z (rel. intensity) 486 (MH+−H3PO4, 100).
References
- ^ HIV Attachment Inhibitor BMS-663068 Looks Good in Early Studies
- ^ HIV attachment inhibitor BMS-663068 shows good safety and efficacy in phase 2b study
- ^ Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
{3-[(4-Benzoyl-1-piperazinyl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl}methyl dihydrogen phosphate
|
|
| Other names
BMS-663068, GSK3684934
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
| KEGG | |
|
PubChem CID
|
|
| Properties | |
| C25H26N7O8P | |
| Molar mass | 583.498 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////////////////фостемсавир , فوستيمسافير , 磷坦姆沙韦 ,BMS 663068, Fostemsavir, GSK 3684934, PHASE 3, ホステムサビル;
Ripasudil hydrochloride hydrate 塩酸塩水和物 , リパスジル

Ripasudil hydrochloride hydrate
4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline;dihydrate;hydrochloride
4-Fluoro-5-[2(S)-methylperhydro-1,4-diazepin-1-ylsulfonyl]isoquinoline hydrochloride dihydrate
223645-67-8
016TTR32QF, K 115
LAUNCHED 2014Kowa

| Company | D. Western Therapeutics Institute Inc. |
| Description | Selective rho kinase inhibitor |
| Molecular Target | Rho kinase |
| Mechanism of Action | Rho kinase inhibitor |
SEE http://pdf.irpocket.com/C4576/GpH7/tLM4/sJIT.pdf
Ripasudil hydrochloride hydrate (Glanatec® ophthalmic solution 0.4 %; hereafter referred to as ripasudil) is a small-molecule, Rho-associated kinase inhibitor developed by Kowa Company, Ltd. for the treatment of glaucoma and ocular hypertension. This compound, which was originally discovered by D. Western Therapeutics Institute, Inc., reduces intraocular pressure (IOP) by directly acting on the trabecular meshwork, thereby increasing conventional outflow through the Schlemm’s canal.
As a result of this mechanism of action, ripasudil may offer additive effects in the treatment of glaucoma and ocular hypertension when used in combination with agents such as prostaglandin analogues (which increase uveoscleral outflow) and β blockers (which reduce aqueous production).
The eye drop product has been approved in Japan for the twice-daily treatment of glaucoma and ocular hypertension, when other therapeutic agents are not effective or cannot be administered. Phase II study is underway for the treatment of diabetic retinopathy.
K-115 is a Rho-kinase inhibitor as ophthalmic solution originally developed by Kowa and D Western Therapeutics Institute (DWTI). The product candidate was approved and launched in Japan for the treatment of glaucoma and ocular hypertension in 2014.
In 2002, the compound was licensed to Kowa Pharmaceutical by D Western Therapeutics Institute (DWTI) in Japan for the treatment of glaucoma. The compound is currently in phase II clinical trials at the company for the treatment of age-related macular degeneration and diabetic retinopathy.
Use of (S)-(-)-1-(4- fluoro-5-isoquinoline-sulfonyl)-2-methyl-1,4-homopiperazine (ripasudil hydrochloride, first disclosed in WO9920620), in the form of eye drops, for the treatment of retinal diseases, particularly diabetic retinopathy or age-related macular degeneration.
Follows on from WO2012105674 by claiming a combination of the same compound. Kowa, under license from D Western Therapeutics Institute, has developed the Rho kinase inhibitor ripasudil hydrochloride hydrate (presumed to be Glanatek) as an eye drop formulation for the treatment of glaucoma and ocular hypertension which was approved in Japan in September 2014..
The company is also developing the agent for the treatment of diabetic retinopathy, for which it is in phase II trial as of October 2014.
…………………….
A Practical Synthesis of (S)-tert-butyl 3-methyl-1,4-diazepane-1-carboxylate, the key intermediate of Rho-kinase inhibitor K-115
Synthesis (Stuttgart) 2012, 44(20): 3171
https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-0032-1316771
practical synthesis of (S)-tert-butyl 3-methyl-1,4-diazepane-1-carboxylate has been established for supplying this key intermediate of Rho–kinase inhibitor K-115 in a multikilogram production. The chiral 1,4-diazepane was constructed by intramolecular Fukuyama–Mitsunobu cyclization of a N-nosyl diamino alcohol starting from the commercially available (S)- or (R)-2-aminopropan-1-ol. In the same manner, an enantiomeric pair of a structural isomer were prepared for demonstration of the synthetic utility.
SEE
WO 2006137368 http://www.google.com/patents/WO2006137368A1?cl=en
WO 2012026529http://www.google.com/patents/WO2012026529A1?cl=en
The including prevention and treatment cerebral infarction, cerebral hemorrhage, subarachnoid hemorrhage, cerebrovascular disorders such as cerebral edema, the present invention relates to a salt thereof or isoquinoline derivatives useful as therapeutic agents, particularly glaucoma.
(S) – (-) -1 – (4 – fluoro-iso-5 – yl) sulfonyl – 2 – methyl -1,4 – diazepane the following formula (1):

It is a compound represented by the particular it is a crystalline water-soluble, not hygroscopic, because it is excellent in chemical stability, it is useful as a medicament has been known for its hydrochloride dihydrate ( refer to Patent Documents 1 and 2). -5 Isoquinoline of these – the sulfonamide compounds, that prophylactic and therapeutic agents for cerebral infarction, cerebral hemorrhage, subarachnoid hemorrhage, cerebrovascular disorders such as cerebral edema, is useful as a therapeutic agent for preventing and glaucoma in particular is known (1-5 see Patent Document 1).
Conventionally, for example, a method of manufacturing by the method described in Patent Document 1, as shown in the following production process has been reported preparation of said compound (Production Method 1-A).

That is, (S)-1-tert-butoxycarbonyl – 3 – by reacting the presence of triethylamine in methylene chloride-fluoro-isoquinoline (2) – methyl -1,4 – diazepane and 5 (3) – chloro-sulfonyl -4 by adding trifluoroacetic acid in methylene chloride compound (the first step), obtained following (4) to synthesize a compound (4) by deprotection to (second step) the desired compound (1) This is a method of manufacturing.
It is also an important intermediate for preparing the compound (1) (S)-1-tert-butoxycarbonyl – 3 – methyl-1 ,4 – diazepane to (3), for example, in the following manner (; see JP Production Process 1-B) that can be produced is known.

Further, on the other hand, the compound (1) (see Patent Document 1) to be manufactured manufacturing routes such as: Any (Process 2) are known.

WO 1999/20620 pamphlet WO 2006/057397 pamphlet WO 1997/028130 pamphlet JP Patent Publication No. 2006-348028 JP Patent Publication No. 2006-290827
However, it is possible to produce in the laboratory of a small amount scale, but you place the point of view for mass industrial production, environmentally harmful halogenated hydrocarbon solvent in the compound of the above-mentioned process for producing 1-A is ( problem because it is carried out coupling step (3) and 2), giving significant adverse environmental exists. Therefore, solvent of halogenated hydrocarbon other than those listed to the specification of the patent document 1, for example, I tried actually dioxane, tetrahydrofuran and the like, but the present coupling reaction will be some progress indeed, Problems reaction is not completed raw material remained even after prolonged reaction time, yield undesirably stays in at most 30% was found. Furthermore, it is hard to decompose in the environment, elimination is also difficult to dioxane is not preferred irritating to humans, and are known as compounds that potentially harmful brain, kidney and liver .
When we actually produced compound (3) by the above production method 1-B, can be obtained desired compound in good yield merged with reproducibility is difficult has further been found that. That is, in the production path, 1,4 – and is used sodium hydride with dimethyl sulfoxide in forming a diazepane ring, except that I actually doing this step, Tsu than the reproducibility of the desired compound It could not be obtained in high yield Te. Also, that this is due to the synthetic route through the unstable intermediate, that it would be converted into another compound easily found this way. limitations and potential problems of the present production process is exposed since this stability may affect the reproducibility of the reaction.
Meanwhile, an attempt to carry out mass production is actually in the Process 2, it encounters various problems. For example, it is stored as an impurity whenever I repeat step, by-products formed in each stage by tandem production process ranging from step 8 gave more complex impurity profile. Depending, it is necessary to repeat a complicated recrystallization purity obtained as a medicine until the purification, the yield in the laboratory be a good overall yield is significantly reduced in the mass production of actual example be away, it does not have industrial utility of true was found. It can be summarized as follows: Considering from the viewpoint of GMP process control required for pharmaceutical production these problems.
Requires control process and numerous complex ranging 1) to 8 step, 3 2) third step – amino-1 – in the step of reacting a propanol, a difficult to remove positional isomers are mixed, 3) The fourth step water is mixed by the minute liquid extraction operation at the time of return to the free base from oxalate require crystallization purification by oxalate in the removal of contaminants of positional isomers, in 4) fifth step, 5) sixth step The Mitsunobu by reproducibility poor require water control in the Mitsunobu reaction used in the ring closure compounds to (1) compounds in (6), 6) ring closure reaction, departing management of the reagent added or the like is generated, in 7) Seventh Step it takes a complicated purification in impurity removal after the reaction, resulting in a decrease in isolated yield. These are issues that must be solved in order to provide a stable supply of raw material for pharmaceuticals high chemical purity is required.
Thus, gentle salt thereof, or the environment isoquinoline derivative comprising a compound represented by the formula (1), the present invention provides a novel production method having good reproducibility and high purity easily and in high yield I intended.
As a result of intensive studies in view of such circumstances, the present inventors, in the manufacturing process of the final target compound shown by the following expression

(Wherein represents a fluorine, chlorine, bromine or iodine, may, R 3 and 1, R 2 R represents a C 1-4 alkyl group be the same or different from each other, and P, X 1 is a protecting group shows a, 0 to m represents an integer of 3, 0 to n is. represents an integer of 3)
Is a urea-based solvents nitrile solvents, amide solvents, sulfoxide or solvents, the solvent may be preferably used in the coupling step of the compound (III) and (II) are generally very short time With these solvents It has been found that can be converted to the desired product quantitatively. It is possible to carry out the coupling step Volume scale while maintaining a high yield by using these solvents, there is no need to use a halogenated hydrocarbon solvent to give significant adverse environment. In consideration of the process such as removal of the solvent after the reaction was further found that acetonitrile is the best among these solvents. Also, since by using hydrochloric acid with ethyl acetate solvent in step deprotection can be isolated as crystal of hydrochloride desired compound (I), without going through the manipulation of solvent evaporation complicated , it has been found that it is possible to obtain the object compound (I) is a simpler operating procedure. Since there is no need to use a halogenated hydrocarbon solvent in this deprotection step further, there is no possibility of harming the environment.
It has been found that it is possible in mass production of (II), leading to the target compound purity, in high yield with good reproducibility as compared with the conventional method compounds are important intermediates in the coupling step further. That is, was it possible to lead to the intermediate high purity and in high yield by eliminating the production of a harmful halogenated hydrocarbon solvent to the environment in this manner. 1,4 addition – in order to avoid the problems encountered in the reaction using sodium hydride in dimethyl sulfoxide in forming the diazepane ring, in order to allow the cyclization reaction at mild conditions more, as a protecting group By performing the Mitsunobu reaction using Noshiru group instead of the carbobenzyloxy group, in addition to one step shorten the manufacturing process of the whole, without deteriorating the optical purity was successfully obtained the desired compound desired.
SEE
![4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline NMR spectra analysis, Chemical CAS NO. 223645-67-8 NMR spectral analysis, 4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-28/002/466/2466109_1h.png) CAS NO. 223645-67-8, 4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline H-NMR spectral analysis |
![4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline NMR spectra analysis, Chemical CAS NO. 223645-67-8 NMR spectral analysis, 4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-28/002/466/2466109_13c.png) CAS NO. 223645-67-8, 4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline C-NMR spectral analysis |
| WO1997028130A1 | Jan 31, 1997 | Aug 7, 1997 | Hiroyoshi Hidaka | Isoquinoline derivatives and drugs |
| WO1999020620A1 | Oct 22, 1998 | Apr 29, 1999 | Hiroyoshi Hidaka | Isoquinoline derivative and drug |
| WO2006057397A1 | Nov 29, 2005 | Jun 1, 2006 | Hiroyoshi Hidaka | (s)-(-)-1-(4-fluoroisoquinolin-5-yl)sulfonyl-2-methyl-1,4homopiperazine hydrochloride dihydrate |
| JP2006290827A | Title not available | |||
| JP2006348028A | Title not available | |||
| JPH11171885A * | Title not available | |||
| JPS61227581A * | Title not available |
ORGANIC SPECTROSCOPY INTERNATIONAL……A blog to brush up spectroscopy fundamentals

ORGANIC SPECTROSCOPY INTERNATIONAL…………A blog to brush up
http://orgspectroscopyint.blogspot.in/2014/12/heavy-traffic-on-your-favorite.html
http://orgspectroscopyint.blogspot.in/


see facebook page
Organic Spectroscopy International


see/click
https://www.facebook.com/orgspectroscopyint

