
Home » 2013 (Page 3)
Yearly Archives: 2013
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FALDAPREVIR

FALDAPREVIR
801283-95-4
(1R,2S)-1-{[(2S,4R)-4-[{8-bromo-7-methoxy-2-[2-(2-methylpropanamido)-1,3-thiazol-4-yl]quinolin-4-yl}oxy]-1-[(2S)-2-{[(cyclopentyloxy)carbonyl]amino}-3,3-dimethylbutanoyl]pyrrolidine-2-carboxamido]-2-ethenylcyclopropane-1-carboxylic acid
Boehringer Ingelheim (Originator)
BI-201335 is an HCV NS3 protease inhibitor awaiting approval in the E.U. by Boehringer Ingelheim for the treatment of chronic hepatitis C, in combination with pegylated Interferon and ribavirin.
Faldaprevir (formerly BI 201335) is an experimental drug for the treatment of hepatitis C. It is being developed by Boehringer-Ingelheim and is currently in Phase III trials.[1]
Faldaprevir is a hepatitis C virus protease inhibitor.
Faldaprevir is being tested in combination regimens with pegylated interferon and ribavirin, and in interferon-free regimens with other direct-acting antiviral agents including BI 207127.
Data from the SOUND-C2 study, presented at the 2012 AASLD Liver Meeting, showed that a triple combination of faldaprevir, BI 207127, and ribavirin performed well in HCV genotype 1b patients.[2] Efficacy fell below 50%, however, for dual regimens without ribavirin and for genotype 1a patients.
- Efficacy and Safety of BI 201335 (Faldaprevir) in Combination With Pegylated Interferon-alpha and Ribavirin in Treatment-naïve Genotype 1 Hepatitis C Infected Patients (STARTverso 1). Cliicaltrials.gov. March 6, 2013.
- Interferon-free hepatitis C treatment with faldaprevir proves safe and effective in people with cirrhosis. Alcorn, K. Aidsmap.com. 20 November 2012.
Phase II clinical trials are also ongoing for the treatment of patients with chronic genotype-1a hepatitis C virus (HCV) infection, in combination with PPI-668 and BI-207127.
In 2007, fast track designation was assigned to the compound in the U.S. for the treatment of chronic genotype-1 hepatitis C (HCV).
Protease inhibitors that are active against NS3/4a are a fertile area of research, not least because of the early promise shown by the two already-approved agents
 Faldaprevir
FaldaprevirProtease inhibitors that are active against NS3/4a are a fertile area of research. Boehringer Ingelheim’s compound faldaprevir is currently in Phase III trials.1 In one 24-week trial in 429 treatment-naïve patients with genotype-1 hepatitis C infection, subjects were given standard peg-interferon and ritonavir therapy plus placebo, or standard therapy plus either 120mg or 240mg of faldaprevir either with or without a three day lead-in of standard therapy alone, or standard therapy plus the higher dose of faldaprevir.
ADDN LIT
Discovery of a potent and selective noncovalent linear inhibitor of the hepatitis C virus NS3 protease (BI 201335)
J Med Chem 2010, 53(17): 6466
WO 2010033444
WO 2004103996
| US6323180 | Aug 5, 1999 | Nov 27, 2001 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| US7514557 * | May 23, 2005 | Apr 7, 2009 | Boehringer Ingelheim International Gmbh | Process for preparing acyclic HCV protease inhibitors |
| US7585845 * | May 20, 2004 | Sep 8, 2009 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US20050020503 * | May 20, 2004 | Jan 27, 2005 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US20120059033 | Mar 9, 2011 | Mar 8, 2012 | Boehringer Ingelheim International Gmbh | Crystalline Salts of a Potent HCV Inhibitor |
| USRE40525 | Sep 30, 2005 | Sep 30, 2008 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C inhibitor tri-peptides |
| WO2000009543A2 | Aug 9, 1999 | Feb 24, 2000 | Boehringer Ingelheim Ca Ltd | Hepatitis c inhibitor tri-peptides |
| WO2004087741A1 | Mar 25, 2004 | Oct 14, 2004 | Boehringer Ingelheim Int | Crystalline phases of a potent hcv inhibitor |
| WO2004103996A1 | May 19, 2004 | Dec 2, 2004 | Boehringer Ingelheim Int | Hepatitis c inhibitor compounds |
| WO2011112761A1 | Mar 10, 2011 | Sep 15, 2011 | Boehringer Ingelheim International Gmbh | Crystalline salts of a potent hcv inhibitor |
………………………………………….

EXAMPLES Example 1 Preparation of Quinoline Starting Material Compound 11

Step 1
The dianion of amide 1 (prepared exactly as described above, from 1.00 g amide 1) was cooled to −78° C., then 2.19 mL perfluorooctyl bromide (8.46 mmol, 1.75 eq.) was added dropwise via syringe over 5 minutes. The dark-colored reaction mixture was then placed in a −10° C. bath. After two hours, 10 mL 1N HCl was cautiously added, and the mixture extracted with EtOAc (2×25 mL), dried (MgSO4), and the solvents removed in vacuo. The residue was then chromatographed on silica gel eluting with 4:1 Hexane:EtOAc to give 1.13 g bromoamide 5 (81%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.12 (br s, 1H), 8.04 (dd, J=1.3, 8.4 Hz, 1H), 7.24 (t, J=8.3 Hz, 1H), 6.63 (dd, J=1.3, 8.3 Hz, 1H), 3.87 (s, 3H), 1.33 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 176.57 (s), 155.74 (s), 136.98 (s), 128.34 (d), 113.63 (d), 106.86 (d), 103.07 (s), 56.26 (q), 40.20 (s), 27.45 (q).
Step 2
0.25 g bromoamide 5 (0.87 mmol, 1 eq.), 2.0 mL con. HCl (24 mmol, 28 eq.), and 1.0 mL diglyme were heated at 100° C. for 24 hours. The mixture was then cooled and filtered (product). The filtrate was evaporated in vacuo using H2O to azeotropically remove all solvents. The residue was triturated with EtOAc to cause precipitation of additional product, which was also filtered. The combined solids were dried to give 0.16 g (77%) of bromoaniline 6.HCl as a light brown solid. 1H NMR (400 MHz, CDCl3) δ: 7.09 (t, J=8.1 Hz, 1H), 6.61 (d, J=8.0 Hz, 1H), 6.47 (d, J=8.1 Hz, 1H), 3.84 (br s, 2H), 3.77 (s, 3H).
Step 3
Bromoanisidine.HCl (5.73 g, 24.0 mmol), Aluminumtrichloride (3.52 g) and chlorobenzene (15.0 mL) are charged into an oven dried 100 mL three necked flask at rt (temperature rise to 30° C.). The resulting mixture is then stirred for 10 min then cooled to 0-5° C. followed by slow addition of acetonitrile (1.89 mL, 36.0 mmol) followed by addition of BCl3 (2.82 g), transferred as gas (or liquid) into the reaction mixture, keeping the temperature below 5° C. The resulting mixture is then stirred at rt for 20 min then heated to 85-100° C. for 16 h. HPLC indicate completion of the reaction (SM<0.5% at 220 nm). The mixture is cooled down to 50° C. then Toluene (15 mL) was added followed by slow addition of IPA (11.1 mL) then slow addition of water (32 mL) at 50° C. The resulting mixture stirred for additional 2 h at this temperature then 3 g Celite was added and the stirred mixture cooled to rt. Filtration then wash of the organic fraction with water 1×15 mL, 2×15 m: 5% NaHCO3, 1×15 mL water then concentration under reduced pressure provided 3.92-4.4 g of the desired product in 68-72% isolated yield. 1H NMR (400 MHz, CDCl3) δ: 7.72 (d, J=9.0 Hz, 1H), 7.1 (br s, 2H), 6.28 (d, J=9.1 Hz, 1H), 3.94 (s, 3H), 2.55 (s, 3H).
Step 4
Oxalyl chloride (8.15 mL) is added dropwise to the cold mixture (10±5° C.) of Thiazole acid 8 (20.18 g) is dissolved in THF (300 mL) and DMF (300 μL) over a period of ˜5 min keeping the internal temperature at 10±5° C. The reaction mixture becomes yellow and homogenous. The cooling bath is removed and the mixture is allowed to reach ambient temperature over a period of ˜30 min. Gas evolution is observed. The mixture is stirred at ambient temperature for 30 min to 1 hour. A solution of aniline 7 (19.8 g), DMAP (140 mg) and THF (35 mL) was added at 10±5° C. Et3N (13.2 mL) was added in portions at 10±5° C. over a period of 10 min. The ice bath was removed and mixture was heated to 65±2° C. and stirred overnight (18 h). The mixture was allowed to reach ambient temperature, diluted with EtOAc (150 mL) and washed with water (150 mL). NaHCO3(5%, 225 mL) was added to the organic portion and the mixture was stirred at ambient temperature for 30 min. The organic portion was concentrated under reduced pressure at approx. 40° C. EtOAc (150 mL) was added to the resulting material and the residual water was removed and the mixture was concentrated under reduced pressure at approx. 40° C. (to azeotrope water). EtOAc (94 mL) was added and the resulting slurry was stirred for 2-6 h and filtered. The solid was washed with EtOAc (30 mL) followed by heptane (30 mL) and air dried for 1 h to give the desired product in 70% yield.
1H NMR (400 MHz, CDCl3) δ: 1.32 (d, 6H, J=7.8 Hz), 2.58 (s, 3H), 2.65-2.72 (m, 1H), 3.98 (s, 3H), 6.83 (d, 1H, J=8.7 Hz), 7.70 (d, 1H, J=8.7 Hz), 7.86 (s, 1H), 8.98 (bs, 1H), 10.13 (bs, 1H).
Step 5
In a 2 L flask was placed potassium t-butoxide (112 g). Dry DME was added at room temperature (exothermic: temperature went up to 35° C.). The resultant solution was heated to ca. 80° C., and amide (88 g) was added in 10 portions slowly so temperature was kept between 80-85° C. Upon completion, reaction mixture was stirred at 85° C. for 2 hours. Solid precipitated during the reaction. HPLC analysis indicated that the reaction was completed at this point (conversion: 100%). The reaction mixture was cooled to room temperature and then to 10° C. with a cool bath. Aqueous 2N HCl solution (ca. 500 ml) was added slowly so temperature was kept under 25° C. to quench the reaction mixture. pH was adjusted to 4-5. About 100 ml of water was added (Note: amount of water may need adjustment to facilitate filtration), and the resulting suspension was stirred at room temperature for 5-10 hours. Product was isolated by filtration, washing with THF and drying under vacuum. Yield: 81 g, 96% yield.
1H-NMR (400 M Hz, DMSO-d6): 1.14 (6H, d, J=6.8 Hz, i-Pr), 2.48 (1H, hept., J=6.8 Hz, i-Pr), 3.99 (3H, s, MeO), 6.75 (1H, s, H-3), 7.24 (1H, d, J=8.5 Hz, H-6), 8.10 (1H, d, J=8.5 Hz, H5), 8.22 (1H, s, H-5′), 9.87 (1H, s, OH), 12.40 (1H, s, amide NH).
Step 6
In a 100 ml flask was placed starting material quinoline (4.22 g) and dioxane (40 ml). POCl3 (4.6 g) was added, and the mixture was heated to 75° C. After 2 hours, HPLC showed the reaction finished (99.7% conversion). Reaction mixture was cooled to room temperature, and then poured to 100 ml saturated NaHCO3 solution and 20 ml EtOAc. The resulting suspension was stirred for 3 hours. Product was isolated by filtration, washing with EtOAc and drying under vacuum. Yield: 4.0 g, 90.9%.
1H-NMR (400 M Hz, CDCl3): 1.14 (6H, d, J=6.8 Hz, i-Pr), 2.76 (1H, hept., J=6.8 Hz, i-Pr), 4.05 (3H, s, MeO), 7.68 (1H, d, J=8.5 Hz, H-6), 8.07 (1H, s, H-3), 8.13 (1H, s, H-5′), 8.20 (1H, d, J=8.5 Hz, H5), 12.30 (1H, s, amide NH).
Example 2 Preparation of Dipeptide Acid Compound 13 Starting Material

A 250 mL 3-neck flask with a thermocouple, nitrogen inlet, and magnetic stir bar was charged with N-cyclopentyloxy carbonyl-tert-L-leucine (20.0 g, 82.2 mmol, 1.0 eq.), 1-hydroxy-benzotriazole (12.73 g, 90.42 mmol, 1.1 eq), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (17.33 g, 90.42 mmol, 1.1 eq.) The flask was purged with nitrogen, and the stiffing started. Anhydrous DMF (62 mL) was added to the flask and the mixture was stirred for 20 minutes at room temperature (about 24° C.). The reaction was mildly exothermic, the internal temperature rose to 29° C. Solid trans-4-hydroxyproline methyl ester HCl (14.93 g, 82.2 mmol, 1.0 eq) was added to the reaction in one portion. Using a syringe, diisopropyl ethyl amine (14.36 mL, 82.2 mmol, 1.0 eq) was added to the reaction dropwise over 25 min. The internal temperature rose to 34.5° C. from 29° C. The reaction was stirred for 1.75 h, forming 12. The reaction was then quenched with 0.1 M HCl (100 mL), the internal temperature rose to 34° C. The reaction was extracted three times with 75 mL of ethyl acetate, and the organic layers were combined. The organic layer was washed with 75 mL H2O, and 2×75 mL of sat. NaHCO3. The organic layer (about 235 mL) was transferred to a 500 mL flask fitted with a mechanical stirrer, shortpath distillation head, internal and external thermocouples, and distilled to minimal stirrable volume under house vacuum (˜110 mm Hg) below 35° C. internal temperature with an oil bath temperature of 40° C. To this crude mixture of 12 was then added tetrahydrofuran (150 mL) and it was distilled to minimum stirrable volume. Tetrahydrofuran (100 mL) was added to the flask, and it was again distilled to minimum stirrable volume. The distillation head was replaced with an addition funnel. Tetrahydrofuran, (100 mL) and methanol (50 mL) were added to the flask, and the solution stirred for about 15 minutes. A 3.2 M solution of LiOH (77 mL, 246.6 mmol, 3 eq.) was charged to the addition funnel, and added over 45 minutes. The temperature rose from 22° C. to 29° C., and the reaction mixture became slightly cloudy. The mixture was cooled in a cold water bath, then the reaction was quenched by slow (45 min.) addition of 4 M HCl (58-65 mL) to adjust the pH to 3.5, causing a slight increase in temperature to 27° C. The flask was fitted with a distillation head, and the methanol and tetrahydrofuran were removed by distillation at reduced pressure, with a bath temperature of 40° C., internal temperature below 30° C. The mixture was extracted twice with 150 mL of MTBE. The MTBE solution was concentrated at reduced pressure, (350 mmHg) to minimum stirrable volume. 50 mL of MTBE was added, it was removed by distillation, internal temp below 35° C. The reaction was a clear viscous liquid, 20 mL of MTBE was added, the mixture was heated to 50° C., solution was clear, the oil bath was turned off, and the solution cooled to rt, ˜24° C. over 1.5 h. To the resultant slurry was then added 60 mL MTBE, stirred 2 h, then the slurry was filtered, using ˜20 mL MTBE to transfer the mixture. The solid was then dried under vacuum at 35° C. to constant weight, 16.4 g (52%), to give the ⅓ MTBE solvate compound 13 as a colorless solid, m.p. 117-124° C.; αD=−58.6 (c 2.17, MeOH); 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 6.76 (d, J=9.3 Hz, 1H), 5.15 (s, 1H), 4.92 (m, 1H), 4.31 (br s, 1H), 4.26 (t, J=8.3 Hz, 1H), 4.19 (d, J=9.3 Hz, 1H), 3.63 (m, 2H), 3.06 (s, 1H, (MTBE)), 2.08 (m, 1H), 1.87-1.48 (m, 9H), 1.09 (s, 3H, (MTBE)), 0.92 (s, 9H).
Example 3 Preparation of Tripeptide Acid Compound 16 Starting Material

In a 25 ml flask 14 was dissolved in 3 ml DMF. HOBt (149 mg, 1.1 mmol), EDC (211 mg, 1.1 mmol), 13 (290 mg, 1.0 mmol) and i-Pr2NEt (129 mg, 1.0 mmol) were added in the given order at room temperature. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was poured into 15 ml aqueous NaHCO3 and extracted with ethyl acetate (20 ml). The organic layer was washed with HCl (0.5 N, 2×10 ml) and saturated aqueous NaHCO3 (10 ml). After removal of solvent by rotary evaporation, 15 was obtained as a white solid. 0.46 g (95% yield). 1H-NMR (400 M Hz, CDCl3): 0.96 (s, 9H), 1.35 (1H, dd, J=3.0, 4.5 Hz), 1.45-1.90 (m, 9H), 1.77 (1H, dd, J=3.0, 4.0 Hz), 2.00-2.09 (1H, m), 2.45-2.52 (1H, m), 3.02 (1H, br), 3.50 (1H, dd, J=11.0, 3.0 Hz), 3.58 (3H, s), 3.99 (1H, d, J=11.0 Hz), 4.18 (1H, d, J=9.0 Hz), 4.43 (1H, br), Hz), 4.63 (1H, t, J=8.0 Hz), 4.93-5.00 (1H, m), 5.04 (1H, dd, J=10.5, 2.0 Hz), 5.20 (1H, d, J=18.0 Hz), 5.20-5.25 (1H, m), 5.65-5.77 (1H, ddd, J=18.0, 10.5, 2.0 Hz), 7.78 (1H, br) ppm.
320 mg ester 15 (0.667 mmol, 1 eq.) was dissolved in 6.7 mL THF+3.4 mL MeOH at ambient temperature under N2. To this solution was then added 3.34 mL 1.6 M LiOH (5.34 mmol, 8 eq.) dropwise over 5 minutes. After 1.5 hours, the solvents were removed in vacuo, and the residue diluted with 15 mL EtOAc+10 mL sat’d NaCl, then 1N HCl was added until pH 3.45 was reached. The phases were separated and the aqueous phase reextracted with 15 mL EtOAc. The combined EtOAc layers were washed with H2O (1×50 mL), dried (MgSO4), and the solvents removed in vacuo to give an oil. The oil was azeotroped with MTBE (1×15 mL), and the residue dried under high vacuum to give 320 mg of 16 (100%) as a colorless foam. Exact mass calc’d for C23H35N3O7: 465.25. Found (ES−): 464.29; 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 12.40 (br s, 1H), 8.49 (s, 1H), 6.77 (d, J=8.2 Hz, 1H), 5.71 (m, 1H), 5.22-4.85 (m, 4H), 4.36-4.10 (m, 3H), 3.80-3.21 (m, 4H), 2.00-1.42 (m, 11H), 0.92 (s, 9H).
Example 4 Dipeptide SNAr Approach to Amorphous Compound (1)

SNAr Protocol 1: A 100 mL 3-neck round bottom flask was charged with 1.93 g 13 (5.00 mmol, 1 eq.), then evacuated/Ar filled (3×), then 17.0 mL DMSO was added via syringe to give a clear, colorless solution. The flask was again evacuated/Ar filled (3×), then 2.53 g t-BuOK (22.5 mmol, 4.5 eq.) was added neat, at once. An exotherm to a maximum of 31.5° C. was observed. The flask was evacuated/Ar filled (3×), then stirred under house vacuum (˜60 mm) for one hour, and some foaming (-t-BuOH) was observed. The vacuum was relieved to Ar, then 2.20 g 11 (5.00 mmol, 1 eq.) was added neat, at once. An exotherm to 28.6° C. was observed. The flask was evacuated/Ar filled (3×), then stirred under house vacuum protected from light at ambient temperature. After 6.5 h the vacuum was relieved to Ar and a sample removed for HPLC, which showed <2% unreacted 11. The flask was then cooled in a cold water bath to 18° C., and 1.72 mL glacial HOAc (30 mmol, 6 eq.) was then added via syringe over ˜10 minutes. An exotherm to 20.5° C. was observed. The mixture was stirred for 10 minutes, then added dropwise over 15 minutes into a second flask containing a well-stirred solution of 30 mL pH 3.5H2O (˜0.001M HCl) at 18° C., causing a precipitate to form immediately, and giving an exotherm to 21.0° C. 2.0 mL DMSO was used to wash the residue into the aqueous mixture, followed by a wash of 5.0 mL ˜0.001M HCl. The resulting suspension was stirred for 15 minutes, then 30 mL of a 1:1 mixture of EtOAc:MTBE was added, and the mixture agitated vigorously for 15 minutes. Agitation was stopped and the phases were allowed to separate. Rapid phase separation and formation of 2 clear phases with no rag layer was seen. The lower aqueous phase was then reextracted with 30 mL of 1:1 EtOAc:MTBE (same fast separation), and the organic extracts were combined and saved. The aqueous phase was discarded as waste.
The organic solution was then washed with H2O (3×30 mL), again all extractions gave rapid separation of phases and no rag layer, then the EtOAc was distilled to minimal stirrable volume. The residue was then azeotroped with 30 mL THF (2×), again distilling to minimal stirrable volume. The resultant slurry of crude 18 was used immediately in the peptide coupling. Exact mass calc’d for C34H42BrN5O8S: 759.19. Found (MS−): 757.92.
SNAr Protocol 2: 1.00 g 13 (2.59 mmol, 1 eq.) and 1.35 g 11 (2.59 mmol, 1 eq.) were charged to a dry flask. The flask was then evacuated/Ar filled (3×), then 10 mL dry DMSO was added via syringe. The flask was again evacuated/Ar filled (3×), then cooled to 19° C. with a cold water bath. To this mixture was then added a 2M solution of KDMO/heptane (5.71 mL, 11.7 mmol, 4.5 eq.) dropwise over 30 minutes. After six hours, HPLC showed the reaction as complete. The reaction was quenched with 0.89 mL HOAc (6 eq.), and added slowly to 25 mL stirring H2O, causing a precipitate to form. The mixture was then extracted with IPAc (2×25 mL). The combined IPAc phases were washed with H2O (1×25 mL), dried (MgSO4), and the solvents removed in vacuo to give a solid, which was azeotroped with MeCN (1×25 mL), and then diluted with heptane to give a slurry. The slurry was filtered and dried to give 1.80 g 18 (91%).
Peptide Coupling Protocol 1: To the THF slurry of crude 18 from SNAr Protocol 1 (taken as 5.00 mmol, 1 eq.) under Ar at ambient temperature in a flask protected from light was added 1.72 g 14 (5.5 mmol, 1.1 eq.) and 25 mL THF. The solution was then cooled to 5° C. under Ar, then 0.958 mL DIEA (5.50 mmol, 1.1 eq.) was added dropwise via syringe over 5 minutes. 5 minutes after the DIEA addition was completed, 0.85 g HOBT hydrate (6.00 mmol, 1.2 eq.), and 1.05 g EDC (5.50 mmol, 1.1 eq.) was then added neat, at once. The flask was then removed from the cold bath and the resultant mixture was then stirred at ambient temperature under Ar for 4 hours. A sample was withdrawn for HPLC which showed <2% unreacted 18 remained. The mixture was cooled to 5° C., then 40 mL 0.1N HCl was added dropwise via addition funnel over 5 minutes, followed by 40 mL EtOAc. The mixture was well agitated for 15 minutes, then agitation was stopped and the phases were allowed to separate. The lower aqueous phase was then reextracted with 40 mL EtOAc and the organic phases were combined and saved. The aqueous phase was discarded as waste. The organic solution was then washed with H2O (1×40 mL), sat’d NaHCO3 (2×40 mL), and again H2O (1×40 mL), then distilled to minimal stirrable volume. The residue was then azeotroped with MTBE (2×40 mL), and again distilled to minimal stirrable volume. The residue was dried under high vacuum to give 4.70 g of crude 19 as an orange solid, with HPLC purity of 78.3%. This material was then chromatographed on silica gel eluting with 2:1 EtOAc:Hexane to give 3.01 g (68% over 2 steps) pure 19 as a yellow powder. Exact mass calc’d for C41H51BrN6O9S: 882.26, MS+: 883.30. 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 12.32 (s, 1H), 8.69 (s, 1H), 8.14 (d, J=9.2 Hz, 1H), 8.03 (s, 1H), 7.45 (s, 1H), 7.33 (d, J=9.4 Hz, 1H), 6.97 (d, J=8.6 Hz, 1H), 5.65 (m, 1H), 5.40 (s, 1H), 5.20 (dd, J=1.5, 17 Hz, 1H), 5.06 (dd, J=1.6, 10.2 Hz, 1H), 5.56 (s, 1H), 4.46 (m, 1H), 4.37 (d, J=9 Hz, 1H), 4.08 (m, 1H), 3.99 (s, 3H), 3.90 (m, 1H), 3.56 (s, 3H), 2.81 (m, 1H), 2.51 (m, 1H), 2.25 (m, 1H), 2.07 (m, 1H), 1.70-1.32 (m, 7H), 1.30 (m, 3H), 1.15 (d, J=8.1 Hz, 6H), 0.95 (s, 9H).
Peptide Coupling Protocol 2: A 5 L 4-neck RBF fitted with mech. stirrer, addition funnel, and thermocouple was charged with 69.57 g 14 (222 mmol, 1.3 eq.), then evacuated/Ar filled (3×). To this was then added a 200 mL THF solution of 18 (contains 129.85 g 171 mmol, 1 eq.), then 523 mL THF was charged to bring the final THF volume to 1 L. The mixture was then cooled to 4.0° C. under Ar. 38.67 mL DIEA (222 mmol, 1.3 eq.) was then added dropwise via addition funnel over 10 minutes, as the internal temperature fell to 2.4° C. The mixture was aged 5 minutes, then 29.98 g HOBT H2O (222 mmol, 1.3 eq.) was added, followed by 42.57 g EDC (222 mmol, 1.3 eq.). The internal temperature was then 3.6° C. The bath was then removed. The internal temperature rose to 20.5° C. over 90 minutes. 4 h after the EDC addition was completed, HPLC showed the reaction was complete. The mixture was cooled to 4.0° C., then 750 mL 0.1N HCl was added over 30 minutes via addition funnel, giving an exotherm to 9.5° C. To this mixture was then added 250 mL sat’d NaCl, followed by 1 L IPAc. After 5 min. vigorous stirring, the mixture was added to a separatory funnel, and the phases were separated. The lower aq. phase was then reextracted with 500 mL IPAc, and the IPAc phases combined. These were then washed successively with H2O (1×1 L), sat’d NaHCO3 (1×1 L), and then H2O (1×1 L). The mixture was then mech. stirred for 12 h to precipitate quinoline 7. The mixture was then filtered through a medium-fritted funnel, and the filtrate distilled until minimal stirrable volume was reached. The residue was then azeotroped with MTBE (2×400 mL), and again distilled to minimal stirrable volume. The residue was dried under high vacuum to give 128 g of 19 as a yellow solid, with HPLC purity of 89%.
140 mg 19 (0.158 mmol, 1 eq.) was dissolved in 1.6 mL THF+0.80 mL MeOH at ambient temperature under N2. To this solution was then added 0.79 mL 1.6 M LiOH (1.27 mmol, 8 eq.) dropwise over 5 minutes. After 1.5 h, the organic solvents were removed in vacuo, and the residue diluted with 10 mL EtOAc+10 mL sat’d NaCl. The pH was then adjusted to 5.75 with 1N HCl. The mixture was agitated vigorously for one hour, then the phases were separated. The aqueous phase was reextracted with 10 mL EtOAc. The combined EtOAc phases were then washed with H2O (2×25 mL), dried (MgSO4, and the solvents removed in vacuo to give 125 mg of Compound (1) (91%) as an amorphous yellow powder.
Example 5 Tripeptide SNAr Approach to Amorphous Compound (1)

233 mg tripeptide acid 16 (0.50 mmol) was charged to a flask, then the flask was evacuated/Ar filled (3×). 1.7 mL DMSO was then added, and the mixture evacuated/Ar filled (3×). The mixture was then cooled in a cold water bath, then 317 mg t-BuOK (2.82 mmol, 5.63 eq.) were added. The flask was again evacuated/Ar filled (3×), then stirred under 60 mm vacuum for one hour. 220 mg quinoline 11 (0.50 mmol, 1 eq.) was then added, and the flask evacuated/Ar filled (3×), then stirred under 60 mm vacuum in the dark at ambient temperature for 3 hours. 0.30 mL HOAc was then added, then the resulting solution was added to 25 mL 0.001 M HCl, causing a precipitate to form. The slurry was filtered, washing the solids with 25 mL H2O. The solid was dried under N2 for 2 hours, then chromatographed on silica gel eluting with EtOAc to give 226 mg (52%) of Compound (1) as an amorphous yellow solid.
Additional methods for preparing amorphous Compound (1) can be found in U.S. Pat. Nos. 6,323,180, 7,514,557 and 7,585,845, which are herein incorporated by reference.
Example 6 Preparation of Type A of Compound (1)
Amorphous Compound (1) (Batch 7, 13.80 g) was added to a 1000 ml three neck flask. Absolute ethanol (248.9 g) was added to the flask. While stirring, the contents of the flask were heated at 60 degrees C./hr to ˜74 degrees C. (Solids do not dissolve at 74 degrees C.). Water (257.4 g) was then added linearly over 4 hr to the resulting slurry while stirring and maintaining the temperature at 74 degrees C. After the water addition was complete, the temperature was reduced linearly to ambient temperature at 8 degrees C./hr and then held at ambient temperature for 6 hrs while stiffing. The resulting solids were collected by filtration and washed with 50 ml of 1/1 (w/w) EtOH/Water. The wet solids were dried on the funnel for 30 minutes by sucking N2 through the cake. (XRPD analysis on this sample indicates that the pattern is similar to the EtOH solvate). The solids were then dried at 65-70 degrees C. under vacuum (P=25 in Hg) and a nitrogen bleed for 1.5 hr. The resulting solids (12.6 g, 95.5% corrected yield) were confirmed by XRPD as being Type A Compound (1).
The unique XRPD pattern and DSC curve of Type A Compound (1) is shown in FIGS. 1 and 2.
Example 7 Preparation of the Sodium Salt of Compound (1)—Method 1
2.1 g of amorphous sodium salt of Compound (1) and 8.90 g of acetone was added to a vial and stirred at ambient temperature for 3 hr. The slurry was filtered off mother liquors and the resulting solids were dried for 20 minutes under nitrogen flow for 20 minutes. 1.51 g of crystalline sodium salt of Compound (1) as solids was collected.
Example 8 Preparation of the Sodium Salt of Compound (1)—Method 2
15.6 g of Type A of Compound (1), 175 ml of acetone and 3.6 ml of water was added to a 250 ml reactor and heated to 53 degrees C. to dissolve the solids. 900 ul of 10.0 N NaOH was added to reactor and the solution was seeded with Type A. The seeded solution was stirred at 53 degrees C. for 10 minutes. A second 900 ul portion of 10.0 N NaOH was added and the system was stirred at 53 degrees C. for 30 minutes over which a slurry developed. The slurry was cooled to 19 degrees C. at a cooling rate of 15 degrees C. per hour and held overnight at 19 degrees C. The final resulting slurry was filtered and the wet solids were washed with 15 ml of acetone. Dried solids for 1 hr at 52 degrees C. under vacuum with a nitrogen flow and then exposed the solids to lab air for one hour. Collected 12.1 g of Compound (1) crystalline sodium salt solids.
Example 11 Preparation of the Sodium Salt of Compound (1)—Method 5
At room temperature a solution of sodium ethoxide in ethanol (21 weight %; 306 ml) was added to a solution of Compound (1) (745 g) in THF (2000 ml) and water (76.5 ml) while stiffing. After stiffing for 30 minutes, the mixture was filtered and the filter was washed with THF (85 ml). The resulting solution was warmed to 65° C. and treated with filtered butyl acetate (6640 ml, optionally pre-warmed to 65° C.) within 30 minutes. Seeding crystals (0.50 g) were added, and the mixture was stirred at 65° C. for 2 hours, while crystallization starts after about 30 minutes. The suspension was cooled to 50° C. within 1 hour and stirred at this temperature for an additional hour. The title compound was isolated by filtration, washed with filtered butyl acetate (765 ml, optionally pre-warmed to 50° C.) and dried at 65° C. for about 16 h giving Compound (1) crystalline sodium salt (˜725 g).
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8399484 | Sep 16, 2009 | Mar 19, 2013 | Boehringer Ingelheim International Gmbh | Combination therapy for treating HCV infection |
| US8530497 | Mar 9, 2011 | Sep 10, 2013 | Boehringer Ingelheim International Gmbh | Crystalline salts of a potent HCV inhibitor |
| WO2013144193A1 | Mar 27, 2013 | Oct 3, 2013 | Boehringer Ingelheim International Gmbh | Combination therapy for treating hcv infection in specific patient subgenotype sub-population |
Biota Reports That Laninamivir Octanoate is Approved for the Prevention of Influenza in Japan

Laninamivir
(4S,5R,6R)-5-acetamido-4-carbamimidamido-6-[(1R,2R)-3-hydroxy-2-methoxypropyl]-5,6-dihydro-4H-pyran-2-carboxylic acid
| Formula | C13H22N4O7 |
|---|---|
| Mol. mass | 346.33638 g/mol |
cas 203120-17-6,
Laninamivir (L174000) prodrug; a novel long-acting neuraminidase inhibitor.
laninamivir octanoate
 472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
Daiichi Sankyo (Originator)
R-118958 is a potent, long-acting neuraminidase inhibitor (LANI) approved and launched in 2010 in Japan as an inhalable formulation for the treatment of influenza A and influenza B in adults and pediatric patients. In 2013 the product was approved in Japan for the prevention of influenza A and influenza B.
| 5-(Acetylamino)-4-[(aminoiminomethyl)amino]-2,6-anhydro-3,4,5-trideoxy-7-O-methyl-D-glycero-D-galacto-non-2-enonic Acid 9-Octanoate |
| (2R,3R,4S)-3-Acetamido-4-guanidino-2-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-3,4-dihydro-2H-pyran-6-carboxylic Acid |
| (4S,5R,6R)-5-Acetamido-4-guanidino-6-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-5,6-dihydro-4H-pyran-2-carboxylic Acid |
| CS 8958 |
ATLANTA, Dec. 20, 2013 (GLOBE NEWSWIRE) — Biota Pharmaceuticals, Inc.
(Nasdaq:BOTA) (“Biota” or the “Company”) today reported that Daiichi Sankyo Company, Limited (“Daiichi Sankyo”) has been granted regulatory approval in Japan to manufacture and market Inavir(R) Dry Powder Inhaler 20mg (generic name laninamivir octanoate) for the prevention of influenza A and B. Inavir(R) was successfully developed and launched by Daiichi Sankyo in Japan for treatment of influenza A and B viruses in October, 2010. Biota is developing laninamivir octanoate outside of Japan for the treatment of influenza, and is currently conducting a large, multi-national Phase 2 trial of laninamivir octanoate in adults infected with influenza. In 2003, the Company and Daiichi Sankyo entered into a collaboration and license agreement to develop long-acting neuraminidase inhibitors, including laninamivir octanoate, and in March 2009, the parties entered into a commercialization agreement, pursuant to which Daiichi Sankyo obtained exclusive marketing rights to laninamivir octanoate in Japan.http://www.pharmalive.com/biota-flu-drug-okd-in-japan

Laninamivir (CS-8958) is a neuraminidase inhibitor which is being researched for the treatment and prophylaxis of Influenzavirus A and Influenzavirus B.[1] It is currently in Phase III clinical trials. [2]
Laninamivir was approved for influenza treatment in Japan in 2010 and is currently marketed under the name “Inavir” by Daiichi Sankyo. Biota Pharmaceuticals [3] and Daiichi Sankyo co-own Laninamivir. On 1st April 2011, BARDA awarded up to an estimated U$231m to Biota Pharmaceuticals (Formerly Biota Holdings Ltd) wholly owned subsidiary, Biota Scientific Management Pty Ltd, for the advanced development of Laninamivir in the US. [4]
patent
|
8-13-2010
|
DRUG FOR TREATMENT OF INFLUENZA
|
The recent flu scares – first H5N1 bird flu and then H1N1 swine flu – transformed Roche’s neuraminidase inhibitor Tamiflu (oseltamivir) into a household name, along with GSK’s Relenza (zanamivir). Both of these require twice-daily dosing, and the orally available oseltamivir is the first choice, but resistance is starting to appear.
A new neuraminidase inhibitor, laninamivir, is being developed by Daiichi Sankyo.5 When administered as the octanoate prodrug form, it appears that a single dose might be sufficient to treat influenza, weekly doses could be preventative, and it is active against extremely pathogenic H5N1 strains.
Laninamivir octanoateIn a double blind, randomised, placebo-controlled Phase I study in 76 healthy male volunteers, subjects were given inhaled single doses of 5, 10, 20, 40, 80 or 120mg of the prodrug, or twice-daily doses of 20 or 40mg for three days.6 No adverse events were observed, and while the prodrug disappeared from the plasma with a half-life of about two hours, the laninamivir itself was much more slowly eliminated, with a half-life of the order of three days, suggesting the potential for giving long-lasting activity against influenza.
In another Phase I trial, a total of 20 healthy subjects with renal function ranging from normal to severely impaired were given single inhaled 20mg doses of the prodrug.7 The degree of renal impairment did not affect the maximum concentration or the time to achieve it, but the half-life increased as renal function reduced. This indicates that the rate-limiting step for elimination is drug release rate to plasma from tissues rather than renal excretion. It was well tolerated, but systemic exposure increased with increasing renal impairment.
It has also been compared with oseltamivir in patients with influenza. A total of 186 children under 10 who had had febrile influenza symptoms for no longer than 36 hours were randomised to receive 20 or 40mg of laninamivir octanoate as a single inhalation or 2mg/kg oseltamivir orally twice a day for five days.8
The new drug gave a significant reduction, of 61 hours for the 40mg group and 66 for the 20mg group, in median time to illness alleviation compared with oseltamivir in those with oseltamivir-resistant H1N1 influenza A. However, there was no significant difference in the time to alleviation of illness with H3N2 influenza A, or influenza B.
The most common side-effects were gastrointestinal problems.
In a Phase III trial, a total of 1,003 adult patients with febrile influenza symptoms for no more than 36 hours were given similar doses to those in the trial in children.9 Median time to alleviation of illness was 73h for 40mg, 86h for 20mg, and 74h for oseltamivir, and the proportion of patients shedding virus at day 3 was significantly lower in the 40mg group than for those given oseltamivir.
- Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nasu H, Kubo S (January 2009).“CS-8958, a prodrug of the new neuraminidase inhibitor R-125489, shows long-acting anti-influenza virus activity”. Antimicrobial Agents and Chemotherapy53 (1): 186–92.doi:10.1128/AAC.00333-08. PMC2612152. PMID18955520.
- Hayden F (January 2009). “Developing new antiviral agents for influenza treatment: what does the future hold?”. Clinical Infectious Diseases. 48. Suppl 1 (S1): S3–13.doi:10.1086/591851. PMID19067613.
- http://www.biotapharma.com
- http://www.biotapharma.com/?page=1021001&subpage=1021019
5. T. Honda et al. Synthesis and in vivo influenza virus-inhibitory effect of ester prodrug of 4-guanidino-7-O-methyl-Neu5Ac2en, Bioorg Med Chem Lett 2009, 19(11): 2938
6. H. Ishizuka et al. J. Clin. Pharmacol. 2010, 50, 1319
7. H. Ishizuka et al. J. Clin. Pharmacol. 2010, epub ahead of print, doi 10.1177/0091270010361914
8. N. Sugaya and Y. Ohashi, Antimicrob. Ag. Chemother. 2010, 54, 2575
9 A. Watanabe et al. Clin. Inf. Dis. 2010, 51, 1167
A new route toward 2-acetamido-4-O-methyl-2-deoxy-D-mannopyranose from a Ferrier derivative of tri-O-acetyl-D-glucal, which contributes to aldolase-catalyzed synthesis of laninamivir (CS-8958)
Tetrahedron 2013, 39(37): 7931

Infection of poultry with H5N1 avian influenza virus has been expanding since 2003 in wide areas including Asia, Europe and Africa. Humans infected with this virus have been found not only in Asia but also in Middle East and Africa. If a new type of H5N1 influenza virus has appeared and its infection has started, it is believed that the infection will rapidly expand to cause a worldwide spread (i.e., influenza pandemic) because most people do not possess immunity against that virus and influenza viruses spread via droplet infection and airborne infection. More than half of human patients infected with H5N1 influenza virus have died so far. Thus, the virus is highly pathogenic. It is known that three influenza pandemics, the Spanish Flu, the Asian Flu and the Hong Kong Flu, occurred in the 20th century. In the Spanish Flu which caused the largest number of patients, it is estimated that about 20-40 million people died in the world and about 0.5 million people in Japan.
According to a report from Japanese Ministry of Health, Labour and Welfare made in November, 2005, if a new type influenza virus has spread, the number of patients who will consult medical doctors in Japan as a result of infection with that virus is estimated about 18-25 million. Further, when the pathogenicity of that new type influenza virus is severe, the number of inpatients is estimated about 0.2 million while the number of dead is estimated about 0.64 million. Therefore, not only health hazard but also significant influences upon social activities are feared.
Thus, a new type influenza can cause a highly severe disease. Early development of effective therapeutics is demanded.
Although it is reported that zanamivir (in particular, zanamivir hydrate) and oseltamivir (in particular, oseltamivir phosphate or oseltamivir carboxylate) which are influenza therapeutics with neuraminidase inhibitory activity show an inhibitory activity against H5N1 influenza virus, compounds with more excellent activity are desired (Non-Patent Document 1 or 2). Further, H5N1 influenza virus strains against which oseltamivir does not show any inhibitory activity (i.e., oseltamivir resistant virus strains) have been reported. Compounds which possess an inhibitory activity against these oseltamivir resistant H5N1 influenza virus strains are desired (Non-Patent Document 1 or 2).
Compounds represented by formula (I) are known to be useful as influenza therapeutics with neuraminidase inhibitory activity (Patent Documents 1 to 3). However, it has not been reported that these compounds have an inhibitory activity against H5N1 influenza virus. Further, the structures of the compounds represented by formula (I) resemble the structure of zanamivir but are completely different from the structure of oseltamivir.
Non-Patent Document 1: Nature, 2005, vol. 437, p. 1108
Non-Patent Document 2: N. Engl. J. Med., 2005, vol. 353, (25):2667-72
Patent Document 1: U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946)
Patent Document 2: U.S. Pat. No. 6,451,766 (Japanese Patent Publication No. Hei 10-109981)
Patent Document 3: U.S. Pat. No. 6,844,363 (Japanese Patent Publication No. 2002-012590)

………………………
Preparation Example 1 5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid

(1) Diphenylmethyl 5-acetamido-4-(N,N-bis-t-butyloxycarbonyl)guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoate (3.46 g, 4.1 mmol) disclosed in Example 35 (i) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was dissolved in methylene chloride (27 ml) and trifluoroacetic acid (14 ml). The resultant solution was stirred at room temperature overnight. The reaction solution was concentrated to dryness under reduced pressure, followed by three cycles of azeotropic distillation to dryness with toluene (5 ml). The resultant oily material was dissolved in ethyl acetate (10 ml). The solution was poured into a saturated aqueous solution of sodium hydrogencarbonate (50 ml). The pH of the resultant solution was adjusted to 8.5 by addition of 20% aqueous solution of sodium carbonate. Then, the solution was stirred at room temperature for 3 hr and its pH was adjusted to 5.0 with hydrochloric acid (3 ml), followed by stirring at room temperature for another 1 hr. The solution was further stirred for 1 hr while ice-cooling. Subsequently, precipitating crystals were suction filtered and vacuum dried for 10 hr at an external temperature of 50° C. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (0.97 g; yield 51%).
Infrared Absorption Spectrum (KBr) ν max cm−1: 3412, 2929, 2856, 1676, 1401, 1320, 1285, 1205, 1137, 722.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.88 (1H, d, J=2.5 Hz), 4.45 (3H, m), 4.27 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.15 (1H, m), 3.47 (21-1, m), 3.42 (3H, s), 2.37 (2H, t, J=7.4 Hz), 2.10 (3H, s), 1.31 (2H, m), 1.20-1.40 (8H, m), 0.85-0.95 (3H, m).
13C Nuclear Magnetic Resonance Spectrum (100 MHz, CD3OD) δ ppm: 176.5, 173.7, 164.7, 158.9, 146.7, 108.7, 80.1, 78.0, 69.3, 66.8, 61.4, 52.4, 35.1, 32.8, 30.2, 30.1, 26.0, 23.7, 22.8, 14.4.
(2) The subject compound was also obtained by the method described below.
5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 35 (ii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was subjected to reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque), 100 g] and eluted with methanol/water (0/1-1/1-2/1). Fractions containing the compound of interest were vacuum concentrated. The precipitating crystals were suction filtered and vacuum dried. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (1.2 g; yield 49%). The property data of the resultant compound were consistent with those of the compound obtained in (1) above.
Preparation Example 2 5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid

5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 28 (viii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was purified in an ion-exchange resin column [Dowex-50X; (i) water and (ii) 5% aqueous ammonium solution] and further purified by reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque); water]. Fractions containing the compound of interest were vacuum concentrated. The resultant solid was washed with methanol, filtered and dried to thereby yield the subject compound (1.43 g) as a colorless solid.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.64 (1H, d, J=2.0 Hz), 4.43 (2H, m), 4.22 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.00 (1H, m), 3.85 (1H, dd, J=10.0 Hz, 2.4 Hz), 3.68 (1H, dd, J=10.0 Hz, 5.5 Hz), 3.58 (1H, m), 3.43 (3H, s).
………………………….


…………………………..
Process W is known as a method for manufacturing a compound represented by the formula (Ia), which is embraced in a compound represented by the formula (I) or a pharmacologically acceptable salt thereof, (hereinafter also referred to as “compound (Ia)”; the same shall be applied with respect to other (Patent Document 1). In Process W, n-Hep represents a 1-heptyl group.


Process X is known as a method for manufacturing compound (Ib), which is embraced in compound (I) or a pharmacologically acceptable salt thereof (Patent Document 2). Compound (IVk) is a synthetic intermediate in Process W. In Process X, n-Hep represents a 1-heptyl group.

Process Y is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 1). The procedures from compound (IVc) to compound (IVe) and from compound (IVf) to compound (IVh) in Process Y are the same as in Process W.


Process Z is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 2). In Process Z, the procedure from compound (IVf) to compound (IVh) is the same as in Process W, and the procedure from compound (IVh) to compound (IIIa) is the same as in Process Y.

From the viewpoint of industrial production, the aforementioned Process W, Process Y, or Process Z could be improved in points such as the following:
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
update…………

:ACIE 10.1002/anie.201408138
Scheme zanamivir and Lanimiwei is based on N- acetylneuraminic acid as a starting material, the price is more expensive (ca.13000RMB / kg). Ma recently from Shanghai Institute of Organic Chemistry greatly researcher on ACIE published zanamivir, Lanimiwei and CS-8958 is more simple synthetic route. References: ACIE 10.1002 / anie.201408138
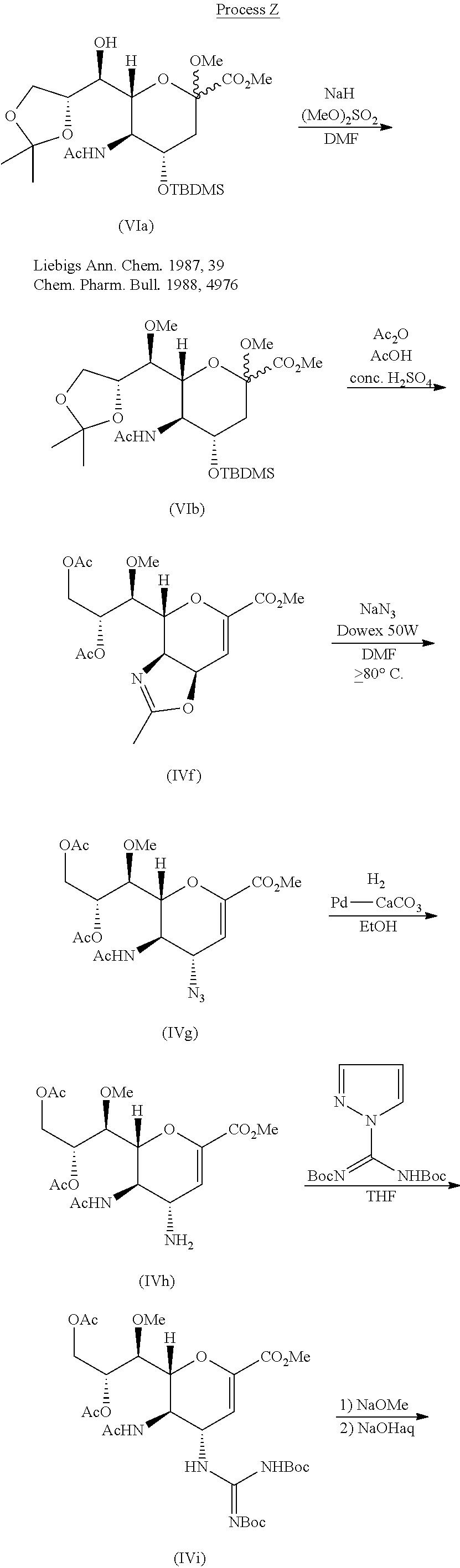
LEDIPASVIR , 来迪派韦 , Ледипасвир , ليديباسفير


Carbamic acid, N-((1S)-1-(((6S)-6-(5-(9,9-difluoro-7-(2-((1R,3S,4S)-2-((2S)-2-((methoxycarbonyl)amino)-3-methyl-1-oxobutyl)-2-azabicyclo(2.2.1)hept-3-yl)-1H-benzimidazol-6-yl)-9H-fluoren-2-yl)-1H-imidazol-2-yl)-5-azaspiro(2.4)hept-5-yl)carbonyl)-2-me
Chemical Formula:C52H60F2N8O7
Molecular Weight:947.08


The structure of ledipasvir was unambiguously confirmed by 1 H, 13C and 19F NMR spectroscopy, UV spectroscopy, IR spectroscopy, high resolution mass spectrometry, elemental analysis and X-ray crystallography. LDV-AS is a white to tinted (off-white, tan, yellow, orange, or pink), slightly hygroscopic crystalline solid. It shows pH dependent solubility in aqueous media: it is slightly soluble in pH 2.3 buffer but practically insoluble in pH 4-7.5 buffers. It is freely soluble in ethanol and DMSO and slightly soluble in acetone. Ledipasvir is chiral and possesses 6 stereogenic centres and enantiomeric purity is controlled in starting material specifications. Three crystalline forms are known and ledipasvir acetone solvate is the designated commercial form. The first step for finished product manufacture involves the dissolution of ledipasvir in ethanol followed by spray-drying and thus precise control of morphology and particle size is not considered important. Ledipasvir is a chemical substance not previously authorised as a medicinal product in the European Union. Furthermore, it is not a salt, complex, derivative or isomer, (nor mixture of isomers), of a previously authorised substance. Whilst it contains some structural features in common with daclastavir, it is metabolically stable and the applicant presented data indicating that there are no common active metabolites. Therefore, the therapeutic moieties are not the same. Ledipasvir thus meets the definition of a New Active Substance according to the Notice to Applicants (NtA), Vol 2A, Chapter 1, Annex 3.
The mode of action of ledipasvir has not been directly established but indirect evidence is consistent with the compound targeting the NS5A molecule. In vitro resistance selection and cross-resistance studies, and the lack of HCV enzyme or kinase inhibition was taken to support the conclusion that ledipasvir targets NS5A as its mode of action. Ledipasvir has shown antiviral activity against HCV genotypes 1a and 1b with mean EC50 values of 0.031 and 0.004 nM, respectively. Antiviral activity determined as EC50 against genotypes 2 to 6 ranged from 0.15 to 530 nM. Ledipasvir showed no relevant antiviral activity at the highest concentration tested, or the highest concentration without cytotoxicity, against other virus such as bovine viral diarrhea virus (BVDV), RSV, HBV, HIV-1, HRV, influenza A and B, and a panel of flaviviruses (including West Nile virus, yellow fever virus, dengue virus, and banzai virus). Cytotoxicity of ledipasvir was characterised by CC50 of 4029 to >50000 nM using different cell lines (1b-Rluc-2, Huh-luc, 1a-HRlucp, Hep G2, SL3, Huh7, Hep-2, AD-38 and MT4 cells). Ledipasvir at 10 µM showed significant binding to 3 ion channels and 1 receptor in a radioligand binding assay screen against a panel of 68 mammalian ion channels and receptors. The IC50s of ledipasvir were 0.210 and 3.47 μM against sodium channel site 2 and calcium channel L-type (dihydropyridine), respectively. A 50% inhibition of androgen receptor was noted at 10 μM. Ledipasvir activity against 442 kinases was assessed using a quantitative polymerase chain reaction (qPCR)-based competition assay. Results showed weak competition for binding of 2 kinases, Bruton’s tyrosine kinase (BTK) and homeodomain-interacting protein kinase 1 (HIPK1) at 0.1 and 1 μM, respectively. Taking into account the high protein binding, >99.5%, of ledipasvir the large margin between unbound maximum clinical plasma levels (0.8 nM) and potential ion channel/receptor inhibition indicates limited clinical relevance.
Ledipasvir (formerly GS-5885) is a drug for the treatment of hepatitis C that was developed by Gilead Sciences.[1] After completingPhase III clinical trials, on February 10, 2014 Gilead filed for U.S. approval of a ledipasvir/sofosbuvirfixed-dose combination tablet for genotype 1 hepatitis C.[2][3] The ledipasvir/sofosbuvir combination is a direct-acting antiviral agent that interferes with HCV replication and can be used to treat patients with genotypes 1a or 1b without PEG-interferon or ribavirin.
Ledipasvir is an inhibitor of the hepatitis C virusNS5A protein.
Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of the nucleotide analog inhibitor sofosbuvir, ledipasvir, and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[4][5] The sofosbuvir/ledipasvir coformulation is being tested with and without ribavirin. In February 2014 Gilead has filed for United StatesFood and Drug Administration (FDA) approval of ledipasvir/sofosbuvir oral treatment, without interferon and ribavirin.[6]
On October 10, 2014 the FDA approved the combination product ledipasvir 90 mg/sofosbuvir 400 mg called Harvoni.[7]


https://www.google.co.in/patents/WO2013184698A1
CLIP
SYN


https://www.google.co.in/patents/WO2013184702A1









PATENT
https://www.google.co.in/patents/US8088368
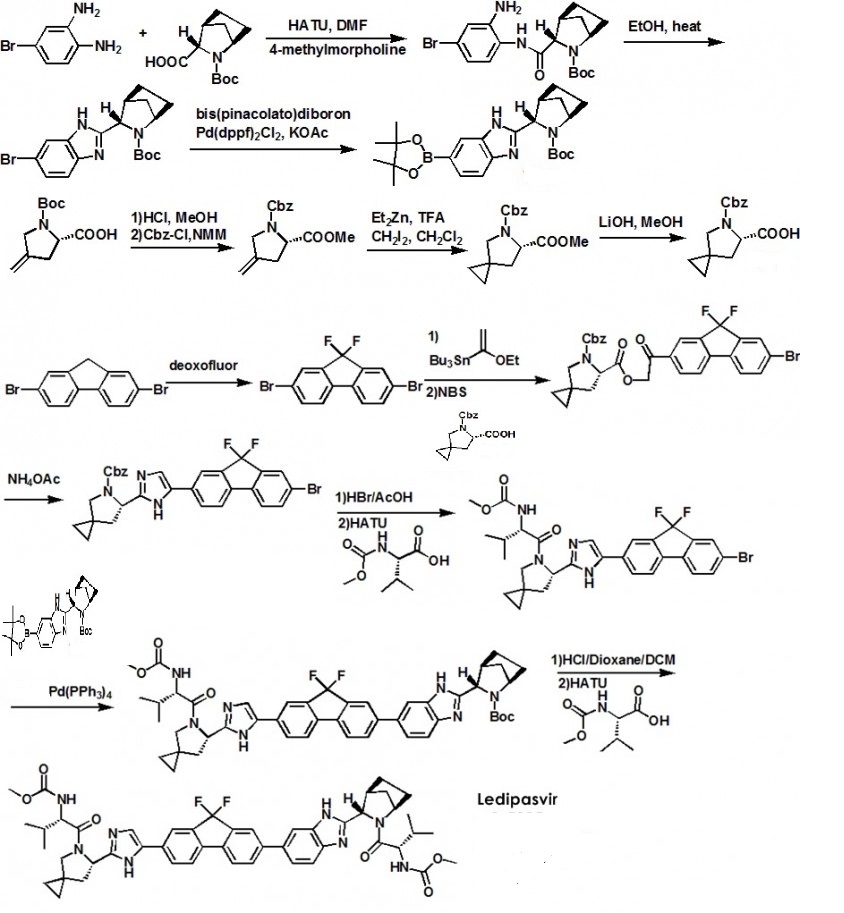
Example ED Preparation of Intermediate 5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-methyl ester

4-Methylene-pyrrolidine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester
Example ED′


2,7-Dibromo-9,9-difluoro-9H-fluorene
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-[2-(7-bromo-9,9-difluoro-9H-fluoren-2-yl)-2-oxo-ethyl]ester
6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carboxylic acid benzyl ester
(1-{6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester
(1-{3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
https://www.google.co.in/patents/US8088368

2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester: A mixture of 2-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carboxylic acid tert-butyl ester (324 mg, 0.627 mmol), 3-[6-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (1.1 eq., 304 mg), [1,1′ bis(diphenylphosphino)ferrocene]dichloropalladium(II)(3%, 15 mg), tetrakis(triphenylphosphine)palladium (3%, 22 mg) and potassium carbonate (3.3 eq., 285 mg) in 10 mL DME and 3 mL water was heated to 90° C. under Argon for 3 hours. The reaction mixture was cooled and diluted with ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was dried (MgSO4), concentrated and purified by flash column chromatography (silica gel, 20 to 100% ethyl acetate/hexane) to give 2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester (361 mg, yield 77%). LCMS-ESI−: calc’d for C43H46F2N6O4: 748.86. Found: 749.2 (M+H+).
PATENTS
SEE
WO 2010132601
WO 2013040492
WO 2013059630
WO 2013059638
CLIP
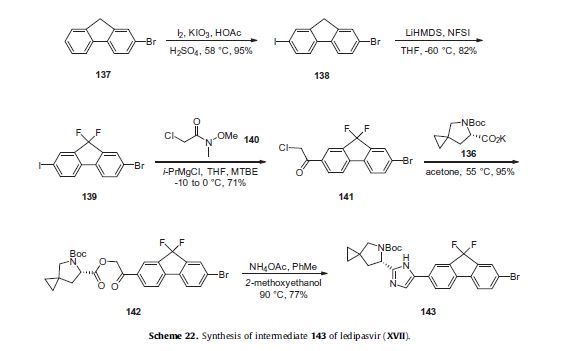
Ledipasvir (Harvoni) Ledipasvir is a potent NS5A inhibitor that is approved for use in combination with sofosbuvir, a nucleotide inhibitor of viral polymerase, for the treatment of chronic hepatitis C virus genotype 1 infection.14,130,131 This combination was discovered and developed at Gilead Sciences and is marketed as the fixed combination with brand name of Harvoni. The synthesis of ledipasvir has been reported in the literature132 and the routes shown in Schemes 22–24 below represent the most efficient and largest scale sequence reported in the patent literature.133,134 The synthesis of the spirocyclopropane proline intermediate 136 is described in Scheme 21. Bis-iodination of cyclopropane-1,1-diyldimethanol (131) in the presence of triphenylphosphine gave diiodide 132 in 70% yield. N-Boc-glycine ethyl ester (133) was then treated with sodium hydride followed by diiodide 132 to give the protected proline analog 134 in 61% yield. Saponification of the ester followed by a classical resolution with (1S,2R)-amino-indanol gave enantomerically pure salt 135. Liberation of the free acid with 1 M HCl followed by treatment with potassium tert-butoxide provided enantiopure potassium salt 136 in high yield. The synthesis of the difluoro-fluorene Suzuki coupling intermediate 143 is described in Scheme 22. Iodination of 2-bromofluorene (137) produced aryl iodide 138 in 95% yield, which was then treated with lithium hexamethyldisilazide and N-fluorobenzenesulfonimide (NFSI) to give the difluoro intermediate 139 in 82% yield. Formation of the Grignard reagent of 139 through reaction with isopropylmagnesium chloride followed by condensation with Weinreb amide 140 gave chloroketone 141 in 71% yield. The potassium salt of the cyclopropyl proline intermediate 136 (described in Scheme 21) was coupled with 141 to give keto ester 142 in high yield. Heating 142 with ammonium acetate resulted in formation of the imidazole ring in intermediate 143 in 77% yield. The completion of the synthesis of ledipasvir is described in Scheme 23. Commercially available (1R,3S,4S)-N-Boc-2-azabicyclo [2.2.1]heptane-3-carboxylic acid (144) was coupled to 4-bromo- 1,2-benzenediamine (145) using EDC/HOBt to give a mixture ofamides 146a/146b in 72% yield. Heating mixture 146a/146b with acetic acid affected cyclization to benzimidazole 147 in 94% yield. Palladium mediated coupling of bromide 147 to bis(pinacolato)diboron gave intermediate148 which was then coupled in the same reaction vessel to bromide 143 generated in Scheme 22. This was followed by formation of the oxalate salt to give the protected central core of ledipasvir (149) in good overall yield. Removal of the amine protecting groups gave diamine 150 which was coupled to two equivalents of Moc-valine (151) via EDC/HOBt to give ledipasvir XVII in 73% yield. 19. Lobeglitazone sulfate



130. Gentile, I.; Buonomo, A. R.; Borgia, F.; Castaldo, G.; Borgia, G. Expert Opin.Invest. Drugs 2014, 23, 561.
131. Smith, M. A.; Chan, J.; Mohammad, R. A. Ann. Pharmacother. 2015, 49, 343.132. Link, J. O.; Taylor, J. G.; Xu, L.; Mitchell, M.; Guo, H.; Liu, H.; Kato, D.;Kirschberg, T.; Sun, J.; Squires, N.; Parrish, J.; Keller, T.; Yang, Z. Y.; Yang, C.;Matles, M.; Wang, Y.; Wang, K.; Cheng, G.; Tian, Y.; Mogalian, E.; Mondou, E.;Cornpropst, M.; Perry, J.; Desai, M. C. J. Med. Chem. 2014, 57, 2033.
133. Guo, H.; Kato, D.; Kirschberg, T. A.; Liu, H.; Link, J. O.; Mitchell, M. L.; Parrish, J.P.; Squires, N.; Sun, J.; Taylor, J.; Bacon, E. M.; Canales, E.; Cho, A.; Cottell, J. J.;Desai, M. C.; Halcomb, R. L.; Krygowski, E. S.; Lazerwith, S. E.; Liu, Q.;Mackman, R.; Pyun, H. J.; Saugier, J. H.; Trenkle, J. D.; Tse, W. C.; Vivian, R. W.;Schroeder, S. D.; Watkins, W. J.; Xu, L.; Yang, Z. Y.; Kellar, T.; Sheng, X.; Clarke,M. O. N. H.; Chou, C. H.; Graupe, M.; Jin, H.; McFadden, R.; Mish, M. R.;Metobo, S. E.; Phillips, B. W.; Venkataramani, C. WO Patent 2010132601A1,2010.
134. Scott, R. W.; Vitale, J. P.; Matthews, K. S.; Teresk, M. G.; Formella, A.; Evans, J.W. US Patent 2013324740A1, 2013.
135. Jin, S. M.; Park, C. Y.; Cho, Y. M.; Ku, B. J.; Ahn, C. W.; Cha, B.-S.; Min, K. W.;Sung, Y. A.; Baik, S. H.; Lee, K. W.; Yoon, K.-H.; Lee, M.-K.; Park, S. W. Diab.Obes. Metab. 2015, 17, 599.
136. Lee, H. W.; Ahn, J. B.; Kang, S. K.; Ahn, S. K.; Ha, D.-C. Org. Process Res. Dev.2007, 11, 190.
137. Lee, H. W.; Kim, B. Y.; Ahn, J. B.; Kang, S. K.; Lee, J. H.; Shin, J. S.; Ahn, S. K.; Lee,S. J.; Yoon, S. S. Eur. J. Med. Chem. 2005,
PAPER
The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection
http://pubs.acs.org/doi/abs/10.1021/jm401499g?prevSearch=LEDIPASVIR&searchHistoryKey=
http://pubs.acs.org/doi/pdf/10.1021/jm401499g
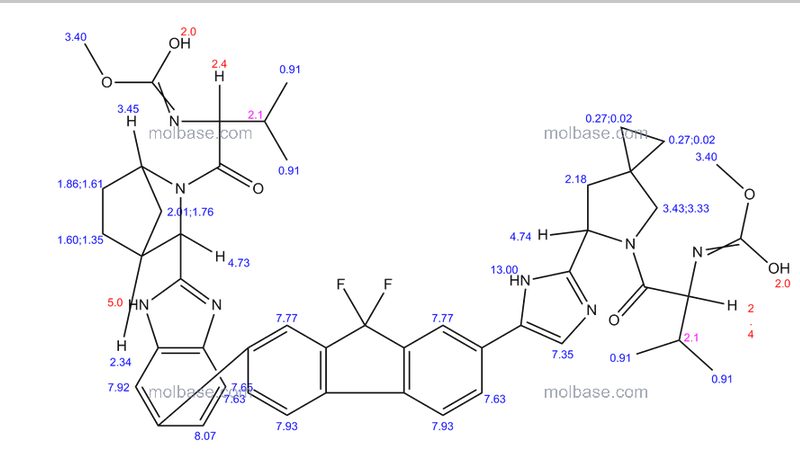
1H-NMR: 300 MHz, (dmso-d6) δ: 8.20-7.99 (m, 8H), 7.73 (s, 2H), 7.37 – 7.27
(m, 2H), 5.25 (dd, J = 7.2 Hz, 1H), 4.78 (s, 1H) 4.54 (s, 1H), 4.16 (m, 1H), 4.02 (m,
1H), 3.87 (m,1H), 3.74 (m, 1H), 3.55 (s, 3H), 3.53 (s, 3H), 2.75 (m, 1H), 2.25 (m,
2H), 2.09 – 2.04 (m, 2H), 1.88 – 1.79 (m, 2H), 1.54 (m, 1H), 0.94 – 0.77 (m, 15H)
0.63 (m, 4H) ppm.
19F-NMR: 282 MHz, (dmso-d6) δ: -109.1 ppm [-74.8 ppm TFA].
HRMS (ESI-TOF) m/z: [M + H]+
calc’d for C49H55F2N8O6: 889.4207; Found: 889.4214.
methyl [(2S)-1-{(6S)-6-[5-(9,9-difluoro-7-{2-[(1R,3S,4S)-2-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-azabicyclo[2.2.1]hept-3-yl]-1H-benzimidazol-6-yl}-9H-fluoren-2-yl)-1H-imidazol-2-
yl]-5-azaspiro[2.4]hept-5-yl}-3-methyl-1-oxobutan-2-yl]carbamate (39 NOS IS LEDISPAVIR
PATENT
Synthesis of 25
25
B. Synthesis of 26 and 27
25 26 27
[0186] To a flask was charged 25 (20.00 g, 0.083 mol), 4-bromo-l,2-benzenediamine (16.74 g, 0.089 mol, 1.08 equiv.), hydroxybenzotriazole (HOBt) (13.96 g, 0.091 mol, 1.1 equiv.), and l-ethyl-3-(3-dimethylaminopropyl) carbodiimide HC1 (EDC.HC1) (17.48 g, 0.091 mol, 1.1 equiv.). The flask was cooled in an ice bath, and was charged with N,N- dimethylacetamide (DMAc, 80 mL). The reaction was allowed to cool to ca. 10 °C with stirring. N-methylmorpholine (NMM) (27.34 mL, 0.249 mol, 3 equiv.) was added over 5 minutes keeping the internal temperature below 20 °C. The reaction was stirred at rt for 20 h. Upon reaction completion, the reaction mixture was added to MTBE (200 mL) and water (600 mL) in a separatory funnel and was gently shaken. The layers were allowed to separate, and the aqueous layer was removed. The aqueous layer was extracted twice with MTBE (50 mL), and the organic extracts were combined. The combined organic extracts were then extracted with water (500 mL), forming a mixture that did not separate well. The mixture was filtered over an appropriate solid support and the layers were separated. The organic phase was concentrated under vacuum, and the resulting residue was dissolved in diisopropyl ether (100 mL). The solution was cooled to ca. 5 °C with stirring. Acetic acid (5.22 mL, 0.091 mol, 1.1 equiv.) was added slowly keeping the internal temperature below 10 °C, and the resulting suspension was stirred 2 h at 5 °C. The thick suspension was then filtered, and the solid was rinsed with diisopropyl ether (100 mL), followed by heptane (100 mL). The cake was dried under vacuum to give the product as a light-beige solid as a mixture of regioisomers 26 and 27 (28.19 g, 72%, >99% AN). 1H NMR (400 MHz, DMSO) mixture of 26 & 27 (data is for the two rotamers of the major regioisomer): δ 9.25 (s, 0.5H), 9.13 (s, 0.5H), 7.08 (d, J= 8.3 Hz, 0.5H); 7.06 (d, J= 8.2 Hz, 0.5H), 6.92 (d, J= 2.2 Hz, 0.5H), 6.89 (d, J= 2.1 Hz, 0.5H), 6.71 (dd, J= 8.4, 2.2, 0.5H), 6.66 (dd, J= 8.4, 2.2, 0.5H), 5.10 (br s, 1H), 5.05 (br s, 1H), 4.15 (br s, 0.5H), 4.10 (br s, 0.5H), 3.76 (s, 1H), 2.64 (br s, 1H), 1.96- 1.88 (m, 1H), 1.77-1.67 (m, 1H), 1.67-1.19 (m, 4H), 1.41 (s, 4.5H), 1.33 (s, 4.5H). MS-ESI+: [M + H]+ calcd for Ci8H25Br03N3, 410.1, 412.1; found, 410.0, 412.0
[0187] The disclosure provides in some embodiments the use of other coupling reagents. These include but are not limited to N,N”-dicyclohexylcarbodiimide (DCC), NJV- diisopropylcarbodiimide (DIC), 6-chloro-2,4-dimethoxy-s-triazine (CDMT), O- benzotriazole-N^N^A^-tetramethyl-uronium-hexafluoro-phosphate (HBTU), and 2-(7-Aza- 1H- benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU).
[0188] The amine base also can be varied or omitted completely. For instance the amine is selected from tertiary amines (R3N), 2,6-lutidine, pyridine, dicyclohexylmethylamme, and N- methylmorpholine (NMM).
[0189] Suitable solvent alternatives are selected from DMF, NMP, dialkyl and cyclic ethers R20, THF, 2-MeTHF, DCM, DCE, toluene, EtOAc, IP Ac, acetone, MIBK, and MEK.
[0190] Suitable temperatures for the reaction range from about -20 °C to 80 °C.
NMR PREDICT

1H/13C NMR PREDICT


COSY
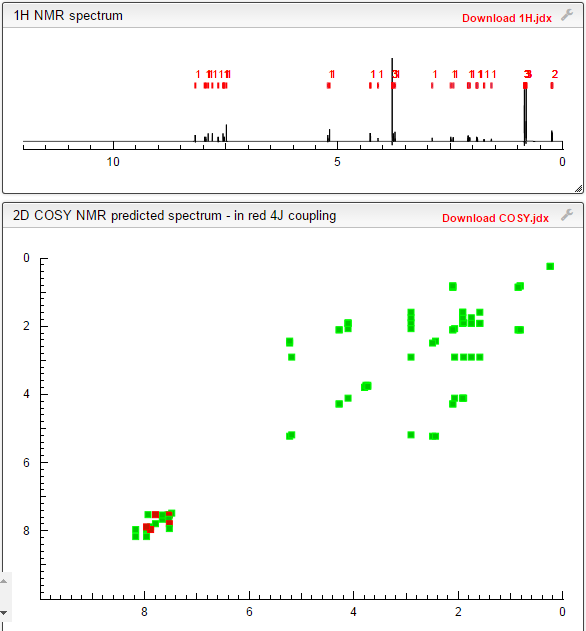
Links
1)Link, John O.et al; The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection; Journal of Medicinal Chemistry (2013), Ahead of Print.DOI:10.1021/jm401499g
2)Ray, Adrian S. et al; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., WO2013040492
3) Delaney, William E. et al ; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., wo2012087596
4) Delaney, William E., IV et al; Preparation of quinoline derivatives and analogs for use in the treatment of hepatitis C virus infection in combination with ribavirin; PCT Int. Appl., wo2011156757
5) Guo, Hongyan et al; Preparation of biaryls, arylheteroaryls, heteroaryls, biarylacetylenes and related compounds end-capped with amino acid or peptide derivatives as antiviral agents; PCT Int. Appl., WO2010132601
6)Phase III (Sofosbuvir + Ledipasvir) ION-1 study: (Clinical Trial number: NCT01701401):
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection
7) Phase III (Sofosbuvir + Ledipasvir) ION-2 study: (Clinical Trial number: NCT01768286)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/GS-5885 Fixed-Dose Combination ± Ribavirin for 12 and 24 Weeks in Treatment-Experienced Subjects With Chronic Genotype 1 HCV Infection
8) Phase III (Sofosbuvir + Ledipasvir) ION-3 study: (Clinical trial number: NCT01851330)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection

References
- “Ledipasvir” (PDF). United States Adopted Name.
- “Ledipasvir-submitted-to-FDA”.
- “GS-5885”. Gilead Sciences.
- ELECTRON: 100% Suppression of Viral Load through 4 Weeks’ Post-treatment for Sofosbuvir + Ledipasvir (GS-5885) + Ribavirin for 12 Weeks in Treatment-naïve and -experienced Hepatitis C Virus GT 1 Patients. Gane, Edward et al. 20th Conference on Retroviruses and Opportunistic Infections. March 3–6, 2013. Abstract 41LB.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, Liz. HIVandHepatitis.com. 4 March 2013.
- “Gilead Files for U.S. Approval of Ledipasvir/Sofosbuvir Fixed-Dose Combination Tablet for Genotype 1 Hepatitis C”. Gilead Sciences. 10 February 2014.
- “U.S. Food and Drug Administration Approves Gilead’s Harvoni® (Ledipasvir/Sofosbuvir), the First Once-Daily Single Tablet Regimen for the Treatment of Genotype 1 Chronic Hepatitis C”. 10 October 2014. Retrieved 10 October 2014.
- Afdhal, N; Zeuzem, S; Kwo, P; Chojkier, M; Gitlin, N; Puoti, M; Romero-Gomez, M; Zarski, J. P.; Agarwal, K; Buggisch, P; Foster, G. R.; Bräu, N; Buti, M; Jacobson, I. M.; Subramanian, G. M.; Ding, X; Mo, H; Yang, J. C.; Pang, P. S.; Symonds, W. T.; McHutchison, J. G.; Muir, A. J.; Mangia, A; Marcellin, P; Ion-1, Investigators (2014). “Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection”. New England Journal of Medicine 370 (20): 1889–98. doi:10.1056/NEJMoa1402454. PMID 24725239.
- http://www.gilead.com/~/media/Files/pdfs/medicines/liver-disease/harvoni/harvoni_pi.pdf
- http://www.hepatitisc.uw.edu/page/treatment/drugs/ledipasvir-sofosbuvir
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Methyl N-[(2S)-1-[(6S)-6-[5-[9,9-Difluoro-7-[2-[(1S,2S,4R)-3-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]-3-azabicyclo[2.2.1]heptan-2-yl]-3H-benzimidazol-5-yl]fluoren-2-yl]-1H-imidazol-2-yl]-5-azaspiro[2.4]heptan-5-yl]-3-methyl-1-oxobutan-2-yl]carbamate
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 76% |
| Protein binding | >99% |
| Metabolism | No cytochromemetabolism |
| Biological half-life | 47 hrs |
| Identifiers | |
| CAS Registry Number | 1256388-51-8 |
| ATC code | None |
| ChemSpider | 29271894 |
| ChEBI | CHEBI:85089 |
| Chemical data | |
| Formula | C49H54F2N8O6 |
| Molecular mass | 889.00 g/mol |
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT
////////////////GS-5885, LEDIPASVIR
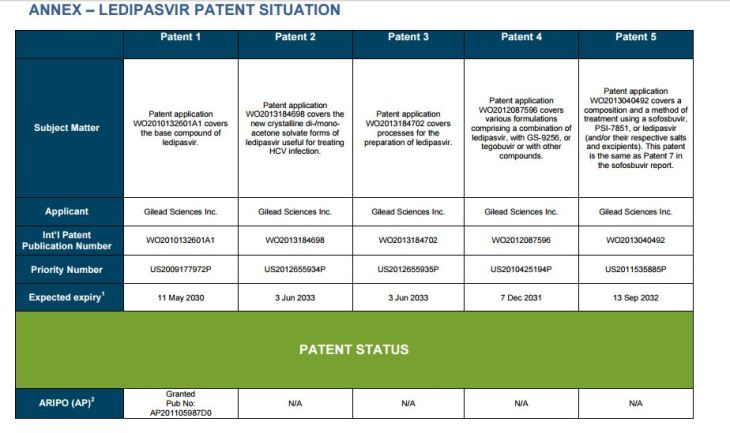
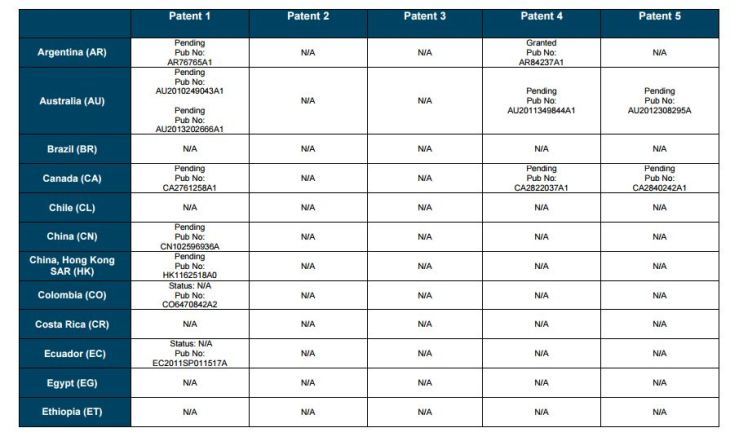
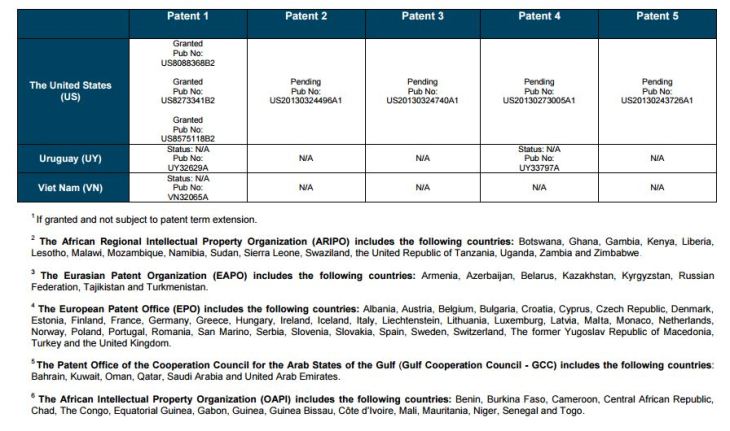
PATENT 1
Patent application WO2010132601A1 (primary patent) discloses the base compound of ledipasvir. The application claims a general structural formula (Markush) of new amide compounds useful for treating disorders associated with HCV. This patent, if granted, serves as a blocking patent preventing competitors from making the product. The claims are very broad, using a Markush structure of antiviral agents. As per the WIPO ISR, claims 1-19 are novel and inventive. However, according to the ISR, all remaining claims (claims 20 to 173), covering a large number of compounds, lack both novelty and inventive step, due to lack of support from the patent specification and in the light of prior art. Prosecution at the USPTO Three patents have been granted in the United States: US8088368B2, claiming the base compound by general structural formula; US8273341B2 (a division of US8088368B2), claiming a method of inhibiting HCV; and US8575118B2 (a continuation of US8273341B2 and a division of US8088368B2), claiming specific amide compounds not covered in the other two related patents. The examination report of US8088368B2 reveals that the application was allowed after the applicant cancelled and amended claims on Markush substuents. The examination report of US8273341B2 reveals that the application was allowed after the applicant amended a claim ‘A method of treating HCV’ to ‘A method of inhibiting HCV´. The examination report of US8575118B2 reveals that the application was allowed after the applicant cancelled claims already covered by the related patents, and limited claims to four specific compounds. Patent 1 has been filed in various jurisdictions: The patent has been granted by the ARIPO, in South Africa, and the United States. The patent (or a related patent) is pending in Argentina, Australia, Canada, China, as well as China, Hong Kong SAR, the EAPO, the EPO, Israel, India, Japan, New Zealand, Singapore, and Ukraine. Legal status is not available for Colombia, Ecuador, Mexico, Peru, Uruguay, and Viet Nam. 13 Litigation / Opposition on Patent 1 In December 2013, Gilead Sciences filed apatent infringement lawsuit against Abbott Laboratories and AbbVie Inc., in the United States District Court for the District of Delaware (case Number: 1:13cv02034). The case involves Gilead Sciences patents US8088368B2, US8273341B2, and US8575118B2.
PATENT 2 Patent application WO2013184698A1 is a product and process patent, claiming new crystalline solvate forms of ledipasvir useful for treating a subject suffering from HCV infection. The application also claims processes of manufacture of such amorphous and crystalline forms with specific X-ray diffraction peaks, and compositions and combinations comprising them. The application has just recently been published and no written opinion on patentability is available at this stage. As per the available information (details available in the Annex): The patent is pending at the EPO and the United States. There are no litigation or opposition procedures reported.
PATENT 3 Patent application WO2013184702A1 is a process patent, claiming processes for the preparation of ledipasvir. The disclosure also provides compounds that are synthetic intermediates to compounds of ledipasvir. The claims are moderately narrow covering crystalline and amorphous forms of ledipasvir with specific X-ray diffraction peaks. The application has just recently been published and no written opinion on patentability is available at this stage. As per the available information (details available in the Annex): The patent is pending at the EPO and the United States. There are no litigation or opposition procedures reported.
PATENT 4 Patent application WO2012087596A1 is a formulation patent, claiming various formulations comprising a combination of ledipasvir with GS-9256, or tegobuvir or with other compounds. The application also claims methods of treatment with the said combinations for reducing viral load in a person infected with HCV. 14 As per the WIPO ISR, the application is novel but not inventive in comparison to the closest prior art retrieved during the search. The combinations claimed in the instant application are not disclosed in the prior art, thus the combinations are novel. However, the prior art discloses various combinations, therefore, the problem to be solved through the invention should be new combinations with fewer side effects. Further, no experimental data of synergism has been provided to support double, triple, or quadruple combinations. Thus, according to the ISR, the instant invention cannot be regarded as inventive. As per the available information (details available in the Annex): The patent has been granted in Argentina. The patent is pending in Australia, Canada, the EPO, and the United States. Legal status is not available for Japan and Uruguay. There are no litigation or opposition procedures reported.
PATENT 5 Patent application WO2013040492A2 is a formulation and method of use patent, claiming compositions and a method of using the combination for the treatment of HCV. Drug combinations are used, and the compositions include sofosbuvir, PSI-7851 and ledipasvir. Since the application claims a group of compounds of Markush structure, it gives the claims a broad scope. As per the WIPO ISR the application is novel but lacks the inventive step in light of prior art. The invention lacks an inventive step as it would be obvious to a person skilled in the art to combine the diastereoisomer of the present invention, disclosed in the prior art, with other antiviral agents to provide an alternative HCV therapy. As per the available information (details available in the Annex): The patent is pending in Australia, Canada, the EPO, and the United States. There are no litigation or opposition procedures reported. This patent is listed in the sofosbuvir report as Patent No. 7
http://www.who.int/phi/implementation/ip_trade/ledipasvir_report_2014-09-02.pdf


SUMMARY The search revealed patents filed with respect to ledipasvir by the Sponsor as well as a nonSponsor. The ledipasvir Sponsor patent collection comprises 5 different patents (patent families) with 47 family members published in 23 jurisdictions. The majority of these patent applications are still pending in the respective patent offices (see Patents 1 to 5 in the Annex). Patent 1 is the primary patent, claiming the base compound through a Markush claim, along with various substituents. Where granted, this patent can prevent competitors from making ledipasvir. Patents 2 and 3 claim processes to make ledipasvir and thus if granted will require competitors to design around these patents and use other production processes. The chemical product itself is not protected. Patents 4 and 5 claim combinations of different HCV drugs with ledipasvir, and their formulations. There is competition in the field by AbbVie, Inc., which filed formulation patents. Note: The search also revealed two patents that are relevant for all seven reports. Patent applications WO2013059630A1 and WO2013059638A1 inter alia claim the use of combinations of unnamed direct-acting antiviral agents for treating HCV, where the treatment does not include administration of interferon or ribavirin, and the treatment lasts between 8-12 weeks. The description and the dataset for these two patents can be found in the Working Paper on ombitasvir (Patents No 3 and 4). These patents are in litigation. Detailed information can be found in the Working Paper on sofosbuvir under Patent No 2.
World Drug Tracker: LEDIPASVIR
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
LEDIPASVIR
Biological Activity of Ledipasvir
Ledipasvir(GS5885) is an inhibitor of the hepatitis C virus NS5A protein. Ledipasvir is an experimental drug for the treatment of hepatitis C.
IC50 Value: 141 nM (EC50, JFH1/3a-NS5A hybrid replicon) [1]
Target: HCV NS5A
in vitro: Against JFH1/3a-NS5A, DCV was more potent (EC(50) = 0.52 nM) than GS-5885 (EC(50) = 141 nM). DCV sensitivity was increased against JFH1/3a-NS5A-M28V (EC50 = 0.006 nM), A30V (EC(50) = 0.012 nM), and E92A (EC(50) = 0.004 nM) while the NS5A-A30K and -Y93H variants exhibited reduced sensitivity to DCV (EC50 values of 23 nM and 1120 nM, respectively) and to GS-5885 (EC50 values of 1770 nM and 4300 nM, respectively) [1].
in vivo: GS-5885 was well tolerated and resulted in median maximal reductions in HCV RNA ranging from 2.3 log(10) IU/ml (1 mg QD) to 3.3 log(10) IU/ml (10 mg QD in genotype 1b and 30 mg QD). E(max) modeling indicated GS-5885 30 mg was associated with>95% of maximal antiviral response to HCV genotype 1a. HCV RNA reductions were generally more sustained among patients with genotype 1b vs. 1a. Three of 60 patients had a reduced response and harbored NS5A-resistant virus at baseline. NS5A sequencing identified residues 30 and 31 in genotype 1a, and 93 in genotype 1b as the predominant sites of mutation following GS-5885 dosing. Plasma pharmacokinetics was consistent with QD dosing [2].
Toxicity:
Clinical trial: Combination Therapy for Chronic Hepatitis C Infection. Phase 2
Clinical Information of Ledipasvir
| Product Name | Sponsor Only | Condition | Start Date | End Date | Phase | Last Change Date |
|---|---|---|---|---|---|---|
| Ledipasvir | Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-12 | 31-DEC-14 | Phase 3 | 12-SEP-13 |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-13 | 31-JAN-15 | Phase 3b | 11-NOV-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-MAY-13 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-DEC-10 | 30-APR-14 | Phase 2b | 28-AUG-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-JUN-13 | Phase 2 | 22-AUG-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-APR-13 | Phase 2b | 03-OCT-12 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-13 | 31-JAN-15 | Phase 3 | 11-NOV-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-MAY-13 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-12 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-APR-13 | Phase 2 | 03-OCT-12 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-JUN-13 | Phase 2b | 22-AUG-13 |
update………..
![]()

WO 2016145990, Ledipasvir, New patent, SHANGHAI FOREFRONT PHARMACEUTICAL CO., LTD
(WO2016145990) METHOD OF PREPARATION FOR LEDIPASVIR AND DERIVATIVE THEREOF, AND INTERMEDIATE COMPOUND FOR PREPARATION OF LEDIPASVIR
SHANGHAI FOREFRONT PHARMCEUTICAL CO., LTD [CN/CN]; Room 1306, No.781 Cailun Road China (Shanghai) Pilot Free Trade Zone, Pudong New Area Shanghai 201203 (CN)
HUANG, Chengjun; (CN).
FU, Gang; (CN).
FU, Shaojun; (CN).
WEI, Zhewen; (CN).
LI, Wei; (CN).
ZHANG, Xixuan; (CN)
chinese machine translation please bear………..
SMILES COC(=O)N[C@@H](C(C)C)C(=O)N1CC2(CC2)C[C@H]1c3ncc([nH]3)c4ccc5c6ccc(cc6C(F)(F)c5c4)c7ccc8nc([nH]c8c7)[C@@H]9[C@H]%10CC[C@H](C%10)N9C(=O)[C@@H](NC(=O)OC)C(C)C
DARUNAVIR


DARUNAVIR
206361-99-1 CAS NO
[(1S,2R)-3-[[(4-Aminophenyl)sulfonyl] (2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester
M. P.:- 72-74 °C (dec)
MW: 547.66
Darunavir and processes for its preparation are disclosed in EP0715618, W09967417, EP1725566 and Bioorganic & Medicinal Chemistry Letters (2004), 14(4), 959-963.
J Med Chem. 2013 May 23;56(10):4017-27. doi: 10.1021/jm400231v
US20050250845 discloses various pseudopolymorphs of darunavir and processes for their preparation. According to this application, “pseudopolymorph” is defined as a crystalline form of a compound in which solvent molecules are incorporated in the lattice structure. The Form B disclosed in the patent application is a pseudopolymorph wherein water is used as solvent. The thermogravimetric experiments of the Form B shows weight loss of 3.4% in the temperature range 25-78°C (water), 5.1% in the temperature range 25-1 10°C (ethanol and water) and further 1.1% weight loss (ethanol) in temperature range 110-200° C. Further at the drying step the Form B showed about 5.6% weight loss. The obtained dried product was hygroscopic and it adsorbed up to 6.8% water at high relative humidity. Amorphous form of darunavir is disclosed in US20050250845 and the publication in J.Org. Chem. 2004, 69, 7822 – 7829.
US 7700645 patent disclosed amorphous Darunavir, various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. Journal of Organic Chemistry 2004, 69, 7822-7829 disclosed amorphous Darunavir is obtained by purification with column chromatography in 2% methanol in chloroform as eluent. PCT publication WO2010086844A1 disclosed crystalline dimethylsulfoxide solvate and crystalline tetrahydrofuran solvate of darunavir. The publication also disclosed the amorphous darunavir having the IR spectrum with characteristic peaks at about 1454 and 1365 cm“1
PCT publication WO201 1083287A2 disclosed crystalline darunavir hydrate substantially free of any non aqueous solvent.
Drug information:- Darunavir is an Anti-microbial drug further classified as anti-viral agent of the class protease inhibitor. It is used either single or in combination with other drugs for the treatment of human immunodeficiency virus.
Darunavir (brand name Prezista, formerly known as TMC114) is a drug used to treat HIV infection. It is in the protease inhibitor class. Prezista is an OARAC recommended treatment option for treatment-naïve and treatment-experienced adults and adolescents.Developed by pharmaceutical company Tibotec, darunavir is named after Arun K. Ghosh, the chemist who discovered the molecule at the University of Illinois at Chicago. It was approved by the Food and Drug Administration (FDA) on June 23, 2006.[2]
Darunavir is a second-generation protease inhibitor (PIs), designed specifically to overcome problems with the older agents in this class, such as indinavir. Early PIs often have severe side effects and drug toxicities, require a high therapeutic dose, are costly to manufacture, and show a disturbing susceptibility to drug resistant mutations. Such mutations can develop in as little as a year of use, and effectively render the drugs useless.
Darunavir was designed to form robust interactions with the protease enzyme from many strains of HIV, including strains from treatment-experienced patients with multiple resistance mutations to PIs.
Darunavir received much attention at the time of its release, as it represents an important treatment option for patients with drug-resistant HIV. Patient advocacy groups pressured developer Tibotec not to follow the previous trend of releasing new drugs at prices higher than existing drugs in the same class. Darunavir was priced to match other common PIs already in use, such as the fixed-dose combination drug lopinavir/ritonavir.
PREZISTA (darunavir) is an inhibitor of the human immunodeficiency virus (HIV-1) protease.
PREZISTA (darunavir), in the form of darunavir ethanolate, has the following chemical name: [(1S,2R)-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]-carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester monoethanolate. Its molecular formula is C27H37N3O7S • C2H5OH and its molecular weight is 593.73. Darunavir ethanolate has the following structural formula:
 |
Darunavir ethanolate is a white to off-white powder with a solubility of approximately 0.15 mg/mL in water at 20°C.
|
|
4-11-2012
|
METHODS FOR THE PREPARATION OF HEXAHYDROFURO[2,3-b]FURAN-3-OL
|
|
|
12-28-2011
|
Substituted Aminophenylsulfonamide Compounds as Hiv Protease Inhibitor
|
|
|
12-23-2011
|
POLYMORPHS OF DARUNAVIR
|
|
|
12-14-2011
|
METHODS FOR THE PREPARATION OF N-ISOBUTYL-N-(2-HYDROXY-3-AMINO-4-PHENYLBUTYL)-P-NITROBENZENESULFONYLAMIDE DERIVATIVES
|
|
|
11-30-2011
|
Protease inhibitor precursor synthesis
|
|
|
6-31-2011
|
PROCESS FOR THE PREPARATION OF (3R,3AS,6AR)-HEXAHYDROFURO [2,3-B] FURAN-3-YL (1S,2R)-3-[[(4-AMINOPHENYL) SULFONYL] (ISOBUTYL) AMINO]-1-BENZYL-2-HYDROXYPROPYLCARBAMATE
|
|
|
9-29-2010
|
Aminophenylsulfonamide Derivatives as Hiv Protease Inhibitor
|
|
|
8-11-2010
|
Process for the preparation of (3R,3aS,6aR)-hexahydrofuro [2,3-b] furan-3-yl (1S,2R)-3[[(4-aminophenyl) sulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate
|
|
|
7-30-2010
|
RELATING TO ANTI-HIV TABLET FORMULATIONS
|
|
|
7-30-2010
|
COMBINATION FORMULATIONS
|
|
7-2-2010
|
METHODS AND INTERMEDIATES USEFUL IN THE SYNTHESIS OF HEXAHYDROFURO [2,3-B]FURAN-3-OL
|
|
|
5-7-2010
|
METHODS AND COMPOSITIONS FOR TREATING HIV INFECTIONS
|
|
|
4-21-2010
|
Pseudopolymorphic forms of a hiv protease inhibitor
|
|
|
9-21-2007
|
Immunoassays, Haptens, Immunogens and Antibodies for Anti-HIV Therapeutics
|
|
|
6-23-2006
|
Method for treating HIV infection through co-administration of tipranavir and darunavir
|
|
|
6-3-2005
|
Combination of cytochome p450 dependent protease inhibitors
|
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010086844A1 | Dec 8, 2009 | Aug 5, 2010 | Mapi Pharma Hk Limited | Polymorphs of darunavir |
| WO2011048604A2 * | Sep 16, 2010 | Apr 28, 2011 | Matrix Laboratories Limited | An improved process for the preparation of darunavir |
| WO2011083287A2 | Oct 6, 2010 | Jul 14, 2011 | Cipla Limited | Darunavir polymorph and process for preparation thereof |
| CN102584844A * | Jan 11, 2011 | Jul 18, 2012 | 浙江九洲药业股份有限公司 | Darunavir crystal form and method for preparing same |
| US6248775 | Apr 8, 1999 | Jun 19, 2001 | G. D. Searle & Co. | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| US7700645 | May 16, 2003 | Apr 20, 2010 | Tibotec Pharmaceuticals Ltd. | Pseudopolymorphic forms of a HIV protease inhibitor |
| Reference | ||
|---|---|---|
| 1 | JOURNAL OF ORGANIC CHEMISTRY vol. 69, 2004, pages 7822 – 7829 | |
| 2 | * | VAN GYSEGHEM E ET AL: “Solid state characterization of the anti-HIV drug TMC114: Interconversion of amorphous TMC114, TMC114 ethanolate and hydrate“, EUROPEAN JOURNAL OF PHARMACEUTICAL SCIENCES, ELSEVIER, AMSTERDAM, NL, vol. 38, no. 5, 8 December 2009 (2009-12-08), pages 489-497, XP026764329, ISSN: 0928-0987, DOI: 10.1016/J.EJPS.2009.09.013 [retrieved on 2009-09-24] |
Virus-encoded proteases, which are essential for viral replication, are required for the processing of viral protein precursors. Interference with the processing of protein precursors inhibits the formation of infectious virions. Accordingly, inhibitors of viral proteases may be used to prevent or treat chronic and acute viral infections. Darunavir has HIV protease inhibitory activity and is particularly well suited for inhibiting HIV-I and HIV -2 viruses. Darunavir, chemically (1 S^R.S’R.S’aS.e’aRJ-fS’he ahydrofuro^.S-b ]furanyl-[3-( 4-aminobenzenesulfonyl)isobutylamino [- 1-benzyl-zhydroxypropyl]carbamate. Darunavir is represented by the following structure:
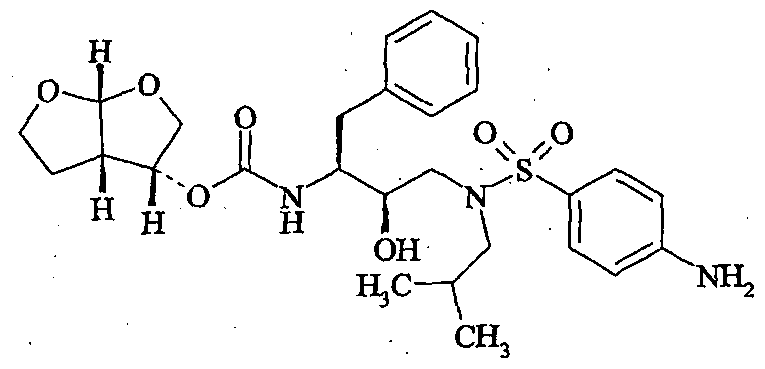
Darunavir and its pharmaceutically acceptable salts were disclosed in US 6248775 patent, wherein Darunavir is prepared by condensing 2R-hydroxy-3-[[(4-aminophenyl)sulfonyl](2- methylpropyl)amino]-1S(phenylmethyl)propylamine with hexahydro-furo[2,3-b]furan-3-ol in anhydrous acetonitrile in the presence of anhydrous pyridine and Ν,Ν’-disuccinimidyl carbonate at ambient temperature.
US 7700645 patent disclosed amorphous Darunavir, various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. Journal of Organic Chemistry 2004, 69, 7822-7829 disclosed amorphous Darunavir is obtained by purification with column chromatography in 2% methanol in chloroform as eluent. PCT publication WO2010086844A1 disclosed crystalline dimethylsulfoxide solvate and crystalline tetrahydrofuran solvate of darunavir. The publication also disclosed the amorphous darunavir having the IR spectrum with characteristic peaks at about 1454 and 1365 cm“1
PCT publication WO201 1083287A2 disclosed crystalline darunavir hydrate substantially free of any non aqueous solvent.
Darunavir Ethanolate, has the chemical name: [(1 S, 2R)-3-[[(4-aminophenyl) sulfonyl](2- methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3/?, 3aS, 6a/?)- hexahydrofuro[2,3-i>]furan-3-yl ester monoethanolate and has the following structural formula:

Darunavir and its process are first disclosed in US 6248775, wherein 2 ?-hydroxy-3-[[(4- aminophenyl)sulfonyl](2-methylpropyl)amino]-1 S(phenylmethyl) propylamine (4) is reacted with (3R, 3aS, 6aR)-hexahydrofuro[2,3- >]furan-3-ol in anhydrous acetonitrile in the presence of N, W-disuccinimidyl carbonate, anhydrous pyridine at ambient temperature followed by workup to get Darunavir (Scheme A).
Scheme A

Darunavir
US 20050250845 disclosed the various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. The same application disclosed the amorphous Darunavir by Raman spectra without process details.
WO 2005063770 discloses process for the preparation of Darunavir ethanolate, wherein 2R-hydroxy-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-1 S-(phenylmethyl)propyl amine (4) is reacted with (3R, 3aS, 6a ?)-hexahydrofuro[2,3-b]furan-3-ol in the presence of N, /V-disuccinimidyl carbonate, triethylamine, 41% methylamine in ethanol in a mixture of ethyl acetate and acetonitrile followed by workup and crystallization from ethanol to get Darunavir ethanolate (Scheme B).
Scheme B

In the prior art process, compound of formula 4 condensed with (3/?, 3aS, 6aR)- hexahydrofuro[2,3-6]furan-3-ol in large excess of solvent or solvent mixture containing large excess of base or mixture of bases to get Darunavir. Further, the obtained products by the processes described in the prior art are not satisfactory, from purity point of view. We have repeated the Darunavir synthetic procedures as described in the prior art and found that relatively large amounts of impurities were obtained along with Darunavir (Table-1) which need repeated crystallizations in different solvents to get desired quality of the final product resulting in poor yields. Among other impurities, the carbonic acid [(1/?,2S)-1-{((4-amino-benzenesulfonyl)-isobutyl-amino)-methyl}-2-((3R,3aSI6aR)- hexahydro-furot2,3-/3]furan-3-yloxycarbonylamino)-3-phenyl-propylester (3R,3aS,6aR)- hexahydro-furo[2,3-ft]furan-3-yl ester (difuranyl impurity of formula 1) is identified.


Conditions:-
i. Phenyl magnesium bromide, Cuprous cyanide, tetrahydrofuran, 23 °C, 1 h,
ii. t-Butyl hydroperoxide, titanium tetraisopropoxide, diethyl D-tartrate, dichloromethane, -22 °C, 24 h,
iii. Azidotrimethylsilane, titanium tetraisopropoxide, Benzene, reflux, 25 min,
iv. 2-Acetoxyisobutyryl chloride, Chloroform, 23 °C, 8 h,
v. Isobutyl amine, isopropanol, 80 °C, 12 h,
vi 4-aminobenzenesulfonyl chloride, aq. Sodium bicarbonate, dichloromethane, 23 °C, 12 h,
vii. 10% palladium on carbon, hydrogen gas (50 psi), methanol, acetic acid, tetrahydrofuran, room temperature, 2 h,
viii. [3R, 3aS,6aS]-3-hydroxyhexahydrofuro[2,3-b]-furan, disuccanamidyl carbonate, triethylamine, acetonitrile, 23 °C, 12 h
Schematic Representation for Synthesis of Darunavir
Preparation of Darunavir is described in US patent 05,158,713, and also in WO9967417 and WO9967254. Accordingly, 2-vinyloxirane 1 on reacting with phenyl magnesium bromide in presence of tetrahydrofuran solvent and cuprous cyanide catalyst give 4-phenylbut-2-ene-1-ol 2. Oxidizing 2 with t-Butyl hydroperoxide in presence of titanium tetraisopropoxide and diethyl D-tartrate using dichloromethane as solvent give [(3S)-3-benzyloxiran-2-yl]methanol 3.
Heating 3 with azidotrimethylsilane in presence of titanium tetraisopropoxide using benzene as solvent give (2S,3S)-3-azido-4-phenyl-butane-1,2-diol 4. The 1,2-dipl compound 4 underwent cyclization when treated with 2-acetoxyisobutyryl chloride in chloroform give (2S)-2-[(1S)-1-azido-2-phenyl-ethyl]oxirane 5, which was further heating with isobutylamine and isopropanol at higher temperature give (2R,3S)-3-azido-1-(isobutylamino)-4-phenyl-butan-2-ol 6. Compound 6 was reacted with 4-aminobenzenesulfonyl chloride in presence of aq. Sodium bicarbonate as base and dichloromethane as solvent resulting in to 4-amino-N-[(2R,3S)-3-azido-2-hydroxy-4-phenyl-butyl]-N-isobutyl-benzenesulfonamide 7.
Hydrogenating 7 with 10% palladium on carbon catalyst using hydrogen gas (50 psi) in methanol and tetrahydrofuran solvent in presence of small amount of acetic acid at ambient temperature resulted in to 4-amino-N-[(2R,3S)-3-amino-2-hydroxy-4-phenyl-butyl]-N-isobutyl-benzenesulfonamide 8. The final step involves reacting 8 with [3R,3aS,6aS]-3-hydroxyhexahydrofuro[2,3-b]-furan and disuccanamidyl carbonate in presence of triethylamine base and acetonitrile as solvent afford [(1S,2R)-3-[[(4-Aminophenyl)sulfonyl] (2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester also called Darunavir 9.
…………………………
http://www.google.com/patents/WO2013114382A1?cl=en
process for the preparation of amorphous Darunavir is as

Process for the preparation of intermediate 2 is as shown in below scheme.

Examples
Example -1 : Preparation of [(1S, 2S)-3-chloro-2-hydroxy-1-(phenyl methyl) propyl] carbamic acid tert-butyl ester (5).
The solution of (3S)-3-(tert-butoxycarbonyl) amino-1-chloro-4-phenyl-2-butanone (Chloromethyl ketone 6,100 g) and aluminium isopropoxide (35 g) in isoprpylalcohol was heated to mild reflux and maintained for 3 hours. After completion of reaction distilled off isopropyl alcohol up to 50 % under vacuum and the resultant mass was cooled to 25-35°C. Water was added to the distillate, pH was adjusted to 3.0-4.0 with acetic acid and maintained the stirring for 2 hours at 25-35°C. The obtained solid was filtered and washed with water. The wet cake was taken into isopropyl alcohol (400mL) and heated to reflux for 60minutes, the mass was cooled to 25-35°C again maintain the stirring for 60minutes, the obtained solid was filtered and washed with isopropyl alcohol. The wet product was dried under normal drying to get title compound 5 (yield 80 g). Example -2: Preparation of [(1 S, 2R)-3-[(2-methylpropyl) amino]-2-hydroxy-1- (phenylmethyl) propyl] carbamic acid tert-butyl ester (4).
The mixture of [(1S, 2S)-3-chloro-2-hydroxy-1-(phenylmethyl) propyl] carbamic acid tert-butyl ester (5,100 g), isobutyl amine (294 g), sodium carbonate (31.3 g) and water was heated to 60 – 65°C and maintained for 3hours. After completion of reaction water (200 mL) was added and distilled out excess isobutyl amine under vacuum at below 75°C. Water (800 mL) was added to the distillate, cooled to 25-35°C and stirred for 2 hours. The obtained solid was filtered and washed with water to get title compound 4 (yield 105 g).
Example -3: Preparation of [(1S, 2R)-3-[[(4-nitrophenyl) sulfonyl] (2-methylpropyl) amino]- 2-hydroxy-1-(phenylmethyl) propyl] carbmic acid tert-butylester (3).
[(1 S, 2R)-3-[(2-methylpropyl) amino]-2-hydroxy-1 -(phenyl methyl) propyl] carbamic acid tert-butyl ester (4, 100 gm) and triethylamine (39.04 g) was added to methylenedichloride (1200 mL) and the temperature was raised to 40°C. p-nitro benzene sulfonyl chloride solution (72.3g of p-NBSC dissolve in 300mL methylenedichloride) was added slowly at 40-45°C for 2-3 hrs. The reaction was maintained for 3hours at 40 – 45°C. After completion of the reaction, water (500 mL) was added, separated the organic layer and distilled out methylene dichloride at atmospheric pressure. Finally, strip out the methylene dichloride by using isopropyl alcohol (200 mL). Isopropyl alcohol (1000 mL) was added to the distillate and maintained the stirring for 60 minutes at 70- 80°C. Cooled the mass to 30 – 35°C, filtered and washed with Isopropyl alcohol to get title compound 3 (yield 145 g). Example – 4: Preparation of 4-Amino-N-(2R, 3S) (3-amino-2-hydroxy-4-phenylbutyl)-N- isobutyl-benzene sulfonamide (1).
(1S, 2R)-{1-benzyl-2-hydroxy-3-[isobutyl-(4-nitro-benzenesulfonyl)-amino]-propyl}-carbamic acid tert-butyl ester (3, 100g), 10% palladium carbon (10gm) and triethanolamine (2gm) were suspended in isopropyl alcohol. The reaction was heated to 40 – 45°C and maintained under 4 – 6kg/cm2 of hydrogen pressure for 3 hours. After completion of reaction, the mass was filtered and hydrochloric acid (70mL) was added to the filtered mass. The solution was heated to reflux and maintained for 2-3hours. After completion of reaction the mass was cooled to 25-35°C, the reaction mass pH was adjusted to 6.0 – 7.0 with 20% sodium hydroxide solution and distilled out isopropyl alcohol under vacuum at below 55°C. Ethanol (200mL) and water (400mL) was added to the distillate, the mass pH was adjusted to 9.0 – 10.0 with 20% sodium hydroxide solution at 25-35°C and maintained the stirring for 2 hours at 25-35°C. The mass was cooled to 0 – 5°C, filtered and wash with water. The wet product was taken into ethanol (350mL), maintained the stirring for 30minutes at reflux temperature. The mass was cooled to 2 – 4°C, stirred for 2 hours, filtered and washed with ethanol (50 mL). The wet product was dried under normal drying to get title compound 1 (Yield 60 g).
Example-5: Preparation of ethyl-2-(4,5-dihydrofuran-3-yl)-2-oxoacetate (VI).
2, 3-Dihydrofuran (250 g) was taken in toluene (2000 mL) and triethyl amine (505 g) was added to above solution. Ethyl oxalyl chloride (536.5 g) was slowly added to the above mixture by maintaining temperature at 25-30°C and maintained the stirring for 5 hours. After completion of reaction separated the organic layer, washed the organic layer with 8% sodium bicarbonate solution (2x500mL). Organic layer was distilled completely under vacuum to get title compound VI (Yield 560g).
1 H NMR : 1.38 (t, 3H), 2.93 (t, 2H), 4.34 (q, 2H), 4.63 (t, 2H), 8.02 (s, 1 H).
Example-6: Preparation of ethyl-2-(3-bromo-2-ethoxytetrahydrofuran-3-yl)-2-oxoacetate (V).
Ethyl-2-(4,5-dihydrofuran-3-yl)-2-oxoacetate (Vl, 100g) was dissolved in dichloromethane (500ml) and Ethanol (150mL) was added. The reaction mass was cooled to 5 to 10°C. N- bromosuccinimide (1 15 g) was added lot wise by maintaining temp below 10°C. Reaction mass was then stirred at 20-30°C till completion of reaction. Reaction mass was washed with sodium bicarbonate solution (2%, 3x400mL) and the organic layer was used for the next step.
Example-7: Preparation of hexahydrofuro [2, 3-b] furan-3-ol (IV).
To the solution of Ethyl-2-(3-bromo-2-ethoxy tetra hydrofuran-3-yl)-2-oxoacetate in dichloromethane (V, 500mL) as prepared in above example, sodium sulphite solution (225g was dissolved in 1700mL of water) was added at 25-35°C. Reaction mass was stirred for 5-8hours at the same temperature and separated the organic and aqueous layers. Organic layer was washed with water (340mL). Distilled out the solvent completely get ethyl-2-(2-ethoxy tetra hydrofuran-3- yl)-2-oxoacetate. Sodium borohydride (35.5g)was dissolved in ethanol (400mL) under nitrogen atmosphere, ethyl-2-(2-ethoxytetra hydrofuran-3-yl)-2-oxoacetate was dissolved in ethanol (100mL) and slowly added to above solution at 15-30°C. Reaction mass was heated to 30-45X, maintained for 5-8 hours, the reaction mass temperature was raised to 55°C and stirred for 8 hours. The reaction mass was cooled to 20-30°C, ammonium chloride solution (1 5g in 200mL water) was slowly added and stirred for 1-2hours. The reaction mass was filtered and filtrate was distilled out under vacuum to get residue. Dichloromethane (600mL) was added to residue and cooled to -10°C. Hydrochloric acid (85mL) was added slowly drop wise in 2 hours by maintaining temp -5 to 0°C, reaction mass was stirred for 60minutes at -5 to 0°C and distilled the solvent completely. The obtained residue was stripped out with isopropyl alcohol (2x200mL, 1x100mL), ethyl acetate (500mL) was added to the resultant residue, stirred for 30-60minutes and cooled to 10-15°C. The solution was filtered and filtrate was concentrated to get title compound IV (yield 56 g).
Example-8: Preparation of Hexahydrofuro [2, 3-b] furan-3-yl acetate (III).
Hexahydrofuro [2, 3-b] furan-3-ol (IV, 60g) was dissolved in dichloromethane (300mL) and cooled to 0-5°C. To the cooled solution triethylamine (58.2 g), N, N-dimethylaminopyridine (1.12g) was added, acetic anhydride (56.5g) was added for 30-60 minutes at the same temperature, the mass temperature was raised to 25-35°C and stirred for 2-4hours. After completion of reaction the mass was cooled to 10-20°C, water (120mL) was added, stirred for 30minutes, separated the organic layer, washed with 10% sodium chloride solution (120mL) and distilled out dichloromethane to get title compound (yield 72g). Further, the product was purified by fractional distillation to get pure Hexahydrofuro [2, 3-b] furan-3-yl acetate III (yield 54g).
1 H NMR : 1.9-2.09(m, 2H), 2.10(s, 3H), 3.0-3.1 (m, 1 H), 3.86-4.03(m, 2H), 3.73(dd, 1 H), 4.10(dd, 1 H), 5.19(m, 1 H), 5.72 (d, 1 H)
Example-9: Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-yl acetate (II). To the buffer solution (104.3g of sodium dihydrogen orthophosphate dissolved in 530mL of water & pH adjusted to 6.0-6.5 with saturated sodium bicarbonate solution(68g in 680 mL water) solution) hexahydrofuro [2, 3-b] furan-3-yl acetate (111,115g) and CAL-B (17.25g) was added at 25-35°C, heated to 38-45°C and stirred for 24 hours. CAL-B (17.25g) was added stirred for 16 hours, again CAL- B (11.5g) was added at 38-45°C and stirred for 16 hours (pH should maintain 6.0-6.5). The reaction mass was cooled to 20-30°C, methylenedichloride (1 150mL) was added to the mass and stirred for 30 minutes. The reaction mass was filtered through hyflowbed then separated the organic layer and washed with 10%sodiumchloride solution (575mL). Organic layer was distilled completely under vacuum to get title compound II (yield 40. Og). Example-10: Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-ol (I).
(3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-yl acetate (II, 14.0g) was dissolved in methanol (42mL). Potassium carbonate (0.34g) was added and stirred at 25-35°C for 6-8hours. Methanol was distilled out completely under vacuum, to the distillate methylenedichloride (28mL) was added, stirred the mass for 30 minutes and again distilled the solvent to get residue. Dissolved the residue in dichloromethane (56mL), the resultant solution was treated with carbon and the solvent was completely distilled out get title compound I (yield 10.5g). Example-11 : Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b]-furan-3-yl-4-nitrophenyl carbonate (2).
To the solution of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-ol (l,100g) and Bis-nitrophenyl carbonate (257.2g) in methylene dichloride (1200mL), triethylamine solution (132 g in 300 mL of methylene dichloride) was added slowly at 20-30°C for 2-3hours. Maintained the reaction at the same temperature for 8-10hours, after completion of reaction water (500mL) was added for 30- 60minut.es and settled the reaction mass then separated the organic layer. Organic layer was washed with 10% acetic acid (100mL) and 10% sodium chloride solution (500mL), distilled the organic layer and co distilled with ethyl acetate (100mL). Ethyl acetate (300mL) was added to the distillate and heated to 50-55°C for 30-45minut.es to get clear solution, the solution was cooled to 5-10°C and maintained at the same temperature for 60 minutes. The obtained solid was filtered, washed with ethanol (100mL) and dried the wet material at 40-45°C for 10-14 hours to get title compound 2 (yield 160g). Example-12: Preparation of dimethylformamide solvate of Darunavir.
To a mixture of 4-amino-N-(2r,3S)(3-amino-2-hydroxy-4-phenylbutyl)-N-lsobutyl- benzenesulfonamide (1 ,25g) and N-methyl-2-pyrrolidinone (NMPO, 50mL), a solution of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]-furan-3-yl-4-nitrophenyl carbonate (2, 8.85g) and N-methyl- 2-pyrrolidinone (75mL) was added at -5 to 0°C for 2 to 3 hours under nitrogen atmosphere. The mass temperature was slowly raised to 25 to 30°C and stirred for 6 to 8 hours. The reaction mass was quenched in to the solution of methylene chloride (125mL) and water (250mL) at 25-35°C for 30 to 45 minutes. Separated the organic layer followed by washed with 10% sodium carbonate solution (150mL), 10% sodium chloride solution (150mL) and with water (6x150mL). Organic layer was dried over sodium sulphate and distill off the solvent under vacuum at below 50°C to obtain darunavir as a residue. To the residue Ν,Ν-dimethyl formamide (50mL) was added and cooled to 0 to -5°C, water (25mL) was added to the solution and maintained for 12hours at 0 to -5 °C, the obtained solid was filtered and washed with pre-cooled mixture of N,N-dimethyl formamide & water (25mL+25mL) to get dimethylformamide solvate of darunavir.
Example-13: Preparation of non-solvated crystalline Darunavir.
To a mixture of 4-amino-N-(2r,3S)(3-amino-2-hydroxy-4-phenylbutyl)-N-lsobutyl- benzenesulfonamide (1, 25g) and N-methyl-2-pyrrolidinone (NMPO, 50mL), a solution of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]-furan-3-yl-4-nitrophenyl carbonate (2, 18.85g) and N- methyl-2-pyrrolidinone (75mL) was added at -5 – 0°C for 2 to 3 hours under nitrogen atmosphere. The mass temperature was slowly raised to 25 – 30°C and stirred for 6 to 8 hours. The reaction mass was quenched in to the solution of methylene chloride (250mL) and water (250mL) at 25- 35°C for 30 – 45 minutes. Separated the organic layer followed by washed with 10% potassium carbonate solution (5x125mL), water (5x125mL), 20% sodium chloride solution (25mL), finally washed with 20% citric acid solution (125mL). The organic layer was treated with carbon and distilled off the solvent under vacuum at below 50°C to obtain darunavir as a residue. To the residue ethylacetate (250mL) was added and cooled to 0 to -5°C, to the cooled solution hexane (225mL) was added and maintained for 12hours at 0 to -5 °C, the obtained solid was filtered, washed with pre-cooled mixture of ethylacetate and hexane (25mL+25mL) and dried the compound to get non-solvated crystalline darunavir(yield 25g).
Example -14: Preparation of Amorphous Darunavir.
Darunavir (200g) as obtained in above example was dissolved in methylene dichloride (10L) and washed with water (3×1000 mL). Organic layer was taken into agitated thin film dryer (ATFD) feed tank. Applied initial temperature about 36 – 40°C and high vacuum (580mm/Hg) to the vessel. Slowly feed the solution to the Vessel (feed rate 5L r) over 1hour finally given the methylene chloride (3L) flushing. The material is collected in the material collecter. Dried at 58 -62°C for 40 hours to get amorphous darunavir (yield 160g).
………………..
http://www.google.com/patents/WO2011048604A2?cl=en

Preparation of Durumvir ethanolate
A solution of (3R,3aS,6a ?)-hexahydrofuro[2,3-D]furan-3-yl 4-nitrophenyl carbonate (5b, 75.4 g) in A -methyl-2-pyrrolidinone (300 mL) was added to a pre-cooled (-2 ± 2°C) solution of the compound of formula 4 (100 g) in W-methyl-2-pyrrolidinone (200 mL) at -4 to 0°C over a period of 2 h. The temperature of the reaction mass was slowly raised to 25 – 30°C and maintained for 8 h. After completion of the reaction (TLC monitoring), ethyl acetate (1000 mL) and purified water (500 mL) were added to the reaction mass. The layers were separated; organic layer was washed with sodium carbonate solution (2 X 500 mL) followed by sodium chloride solution. The organic layer was concentrated; ethanol (300 mL) was added, heated to 45 – 50°C, maintained for 1 h, filtered and washed with ethanol. The wet compound was taken into a mixture of ethyl acetate- ethanol (7:93, 600 mL), heated to reflux, charcoal was added and filtered. The resultant filtrate was cooled to 0 – 5°C, filtered the separated solid and washed with ethanol. The wet compound was dried at 45°C to obtain the in 124.3 g (yield-82.5%). The obtained Darunavir ethanolate had purity of 99.79% on area by HPLC and contained 0.08% on area by HPLC of the difuranyl impurity. Preparation of Amorphous Darunavir
Example – 4
A solution of Darunavir ethanolate (200 g) in dichloromethane (10 L) was taken into ATFD Feed tank. The solvent was evaporated by fed the solution slowly to the ATFD Vessel (feed rate 5 L /h) at 36 – 40°C and high vacuum (580 mm/Hg) over 2 h and then flushed with dichloromethane (3 L). The material is collected in the material collector in 160g with the HPLC Purity of 99.60% and particle size D50 of approximately 50 micrometers and Dgo of approximately 100 to 180 micrometers. Example-5
Darunavir Ethanolate (200 gm) was dissolved in Methylene chloride (1000 ml) and solvent was evaporated by applying vacuum followed by isolation of amorphous Darunavir as a solid as such or by charging n-Heptane or Isopropyl ether. Example – 6
Darunavir Ethanolate (10 g) was dissolved in ethyl acetate (50 mL). The solution was heated to 40 – 45°C and maintained for 30 min. Ethyl acetate was distilled off under vacuum completely to get residue in the form of semisolid. n-Heptane (50 mL) was added to the residue and stirred for 30 min. at ambient temperature. The separated solid was filtered, washed the wet cake with n-heptane (5 mL) and dried at 40 – 45°C under vacuum to get 8.0 g of amorphous Darunavir.
Example – 7
Darunavir Ethanolate (10 g) was placed into a dry round bottom flask and heated to 110 – 120°C to melt and maintained under vacuum for 4 h. The reaction mass was slowly cooled to 25 – 35°C. The obtained glass type crystal was broken into powder to afford 8.5 g of amorphous Darunavir.
Example – 8
Darunavir Ethanolate (5.0 g) was suspended into glycerol (25 g), heated to 110 – 120°C under vacuum and maintained for 30min. Water (50 mL) was added to the cooled reaction mass at 25 – 35°C under stirring and the obtained suspension was stirred for 30 min at 25 – 35°C. The separated solid was filtered and dried at 40 – 45°C under vacuum to yield 3.5 g of amorphous Darunavir. Example – 9
Carbonic acid [(1 R,2S)-1-{((4-amino-benzenesulfonyl)-isobutyl-amino)-methyl}-2- ((3/?,3aS,6aR)-hexahydro-furo[2,3-ft]furan-3-yloxycarbonylamino)-3-phenyl-propylester (3R,3aS,6a ?)-hexahydro-furo[2,3- )]furan-3-yl ester (difuranyl impurity, 1).
The difuranyl impurity (1) isolated from the mother liquor by preparative HPLC using a mixture of formic acid-water (1 :99) as eluent. The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.

1H-NMR (DMSO-cfe, 300 MHz, ppm) – δ 0.79 (d, J=6.6 Hz, 6H, 15 & 15′), 1.14-1.20 (m, 1 H, 20Ha), 1.34-1.42 (m, 1 H, 20Hb), 1.75-1.85 (m, 2H, 20’Ha & 14), 1.94-2.01(m, 1 H, 20’Hb), 2.54-2.64 (m, 2H, 8Ha & 13Ha), 2.74-2.89 (m, 3H, 8Hb, 13Hb & 19), 3.00-3.11 (m, 2H, 5Ha & 19′), 3.34-3.39 (m, 1H, 5Hb), 3.54-2.63 (m, 3H, 21 Ha & 17Ha), 3.65-3.74 (m, 3H, 21’Ha, 21 Hb &17Hb), 3.81-3.89 (m, 2H, 21’Hb & 17’Ha), 3.94-4.04 (m, 2H, 7 & 17’Hb), 4.81-4.88 (m, 1 H, 6), 4.92-4.96 (m, 1 H, 18′), 5.03-5.10 (m, 1 H, 18), 5.11 (d, J=5.4 Hz, 1 H, 22′), 5.61 (d, J=5.1 Hz, 1 H, 22), 6.03 (brs, 2H, NH2, D20 exchangeable), 6.63 (d, J=8.7 Hz, 2H, 2 & 2″), 7.15-7.28 (m, 5H, 10H, 10Ή, 11 H, 11′ & 12), 7.40 (d, J=8.7 Hz, 2H, 3 & 3′), 7.55 (d, J=9.3 Hz, 1 H, NH, D20 exchangeable).
“H-NMR (DMSO-d6, 75 MHz, ppm)- δ 19.56 & 19.81 (15C & 15’C), 25.42 (20 ), 25.47 (20C), 26.28 (14C), 35.14 (8C), 44.45(19’C), 45.01 (19C), 49.21 (5C), 53.39 (7C), 57.55 (13C), 68.70 (21 ‘C), 68.74 (21C), 69.95 (17’C), 70.20(17C), 72.65 (6C), 76.27 (18C), 79.59 (18’C), 108.70 (22’C), 108.75 (22C), 112.69 (2C), 122.56 (4C), 126.12 (12C), 128.04 (11 C & 11’C), 129.03 (10C & 10’C), 129.08 (3C), 138.03 (9C), 152.99 (1C), 153.55 (16’C), 155.32 (16C).
DIP MS: m/z (%) 1108 [M+Hf, 1131 [M+Naf
……………
http://www.google.com/patents/US20130244297

According to the present invention Darunavir having the below impurity not more than 0.1, preferably 0.05%.

………….
DARUNAVIR

CHYAVAN PRASH DABUR ; AN EVALUATION OF AYURVEDIC REMEDY IN K.P.C.A.R.C. LABORATORY TEST
CHYAVAN PRASH is an AYURVEDIC REMEDY used as RASAYANA in Ayurveda. The PRASH is also used as a Food or Food Supplement for maintaining GENERAL HEALTH CONDITION [GHC].
Above DABUR CHYAVAN PRASH container, which is tested at our Laboratory for evaluation puurposes.
Dabur branded CHYAVAN PRASH is taken randomised examination and test for evaluation of AYURVEDIC FUNDAMENTALS.The batch number of the test material container is given above.
5 gramms DABUR CHYAVAN PRASH is taken for test and examination purposes and absorbed in 100 ml solvent, used for the liquification level for laboratory test.
For Physical test and texture of the CHYAVAN PRASH, as for as prepared by me few years ago, on the similar lines , which was laid down and instructed by CHARAK SAMHITA. Although I prepared several years CHYAVAN PRASH for my patient, therefore I know well about the taste and texture of the Chyavan Prash.
A well…
View original post 330 more words
Medicinal Chemistry International: ASUNAPREVIR
Medicinal Chemistry International: ASUNAPREVIR
CLICK ABOVE
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
ASUNAPREVIR

- “A Phase 3 Study in Combination With BMS-790052 and BMS-650032 in Japanese Hepatitis C Virus (HCV) Patients”. ClinicalTrials.gov.
- C. Reviriego (2012). Drugs of the Future 37 (4): 247–254.doi:10.1358/dof.2012.37.4.1789350.
- Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- “Bristol-Myers’ Daclatasvir, Asunaprevir Cured 77%: Study”. Bloomberg. Apr 19, 2012.
- AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Bioorganic and Medicinal Chemistry Letters, 2011 , vol. 21, 7 pg. 2048 – 2054
patents
WO 2003099274, WO 2003099274, WO 2009085659
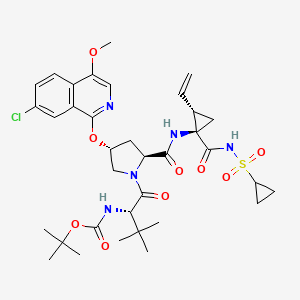
| US8202996 | 6-20-2012 | Crystalline forms of N-(tert-butoxycarbonyl)-3-methyl-L-valyl-(4R)-4-((7-chloro-4-methoxy-1-isoquinolinyl)oxy)-N- ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-L-prolinamide |
| US8163921 | 4-25-2012 | Hepatitis C Virus Inhibitors |
| US7915291 | 3-30-2011 | HEPATITIS C VIRUS INHIBITORS |
| US7449479 | 11-12-2008 | Hepatitis C virus inhibitors |
| US6995174 | 2-8-2006 | Hepatitis C virus inhibitors |










REFERENCES AND NOTES
-
- Rev. Med. Virol., 13 (2003), p. 57
-
- Hepatology, 36 (2002), p. S237
-
- Am. J. Med., 117 (2004), p. 344
-
- For a recent review on HCV anti-viral agents, see: Expert Opin. Invest. Drugs, 19 (2010), p. 63
- Curr. Opin. Pharmacol., 8 (2008), p. 522
-
- For a recent review on HCV NS3/4A protease inhibitors, see: Curr. Opin. Invest. Drugs, 10 (2009), p. 821
- Expert Rev. Anti. Infect. Ther., 7 (2009), p. 537
-
- Infect. Disord. Drug Targets, 6 (2006), p. 3
-
- Acc. Chem. Res., 41 (2008), p. 50
-
- J. Med. Chem., 47 (2004), p. 1605
-
- Antimicrob. Agents Chemother., 52 (2008), p. 4432
-
- Bioorg. Med. Chem. Lett., 18 (2008), p. 4853
-
- J. Med. Chem., 53 (2010), p. 2443
-
- Gastroenterology, 127 (2004), p. 1347
-
- J. Med. Chem., 53 (2010), p. 6466
-
- (a)Chemical and Engineering News (April 12, 2010 issue), 88, pp 30–33.
- (b)Perrone, R.K.; Wang, C.; Ying, W.; Song, A.I. WO 2009085659
-
- Sci. Transl. Med., 2 (2010), p. 30ra32


…………………………………………….
CONTD ONhttp://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
…………………………………………………………..
DR ANTHONY MELVIN CRASTO Ph.D
Zosuquidar

LY335979, RS-33295-198 (Zosuquidar)
Roche Palo Alto (Originator)
LY335979 (Zosuquidar) is a selective Pgp (P-glycoprotein) inhibitor with a Ki of 59 nM. LY335979 significantly enhanced the survival of mice implanted with Pgp-expressing murine leukemia (P388/ADR) when administered in combination with either daunorubicin, doxorubicin or etoposide.
LY335979 (Zosuquidar)
M.Wt: 636.99
Formula: C32H31F2N3O2.3HCl
Name: Zosuquidar trihydrochloride
Elemental Analysis: C, 60.34; H, 5.38; Cl, 16.70; F, 5.97; N, 6.60; O, 5.02
CAS : 167465-36-3
167354-41-8 (free base)
Roche Bioscience (Originator), Eli Lilly and Company (Licensee).
US5654304, WO1994024107A1, WO2000075121, US6570016
Drug Des Discov 1992, 9(1): 69, Bioorg Med Chem Lett 1995, 5(21): 2473, Drugs Fut 2003, 28(2): 125
Zosuquidar is currently under development. It is now in “Phase 3” of clinical tests in the United States. Its action mechanism consists of the inhibition of P-glycoproteins; other drugs with this mechanism include tariquidar and laniquidar. P-glycoproteins are proteins which convert the energy derived from the hydrolysis of ATP to structural changes in protein molecules, in order to perform coupling, thus discharging medicine from cells. If P-glycoprotein coded with the MDR1 gene manifests itself in cancer cells, it discharges much of the antineoplastic drugs from the cells, making cancer cells medicine tolerant, and rendering antineoplastic drugs ineffective. This protein also manifests itself in normal organs not affected by the cancer (such as the liver, small intestine, and skin cells in blood vessels of the brain), and participates in the transportation of medicine. The compound Zosuquidar inhibits this P-glycoprotein, causing the cancer cells to lose their medicine tolerance, and making antineoplastic drugs effective
Clinicial trials: Clinical report published in 2010 showed that zosuquidar did not improve outcome in older acute myeloid leukemia, in part, because of the presence P-gp independent mechanisms of resistance. (Blood. 2010 Nov 18;116(20):4077-85.)
Zosuquidar is a potent P-glycoprotein inhibitor, which binds with high affinity to P-glycoprotein and inhibits P-glycoprotein-mediated multidrug resistance (MDR). P-glycoprotein, encoded by the MDR-1 gene, is a member of the ATP-binding cassette superfamily of transmembrane transporters and prevents the intracellular accumulation of many natural product-derived cytotoxic agents
Zosuquidar
U.S. Patent No. 5,112,817 to Fukazawa et al. discloses certain quinoline derivatives useful as anticancer drug potentiators for the treatment of multidrug resistance. One of the initially promising active agents there-disclosed is MS-073, which has the following structure:

MS-073
U.S. Pat. Nos. 5,643,909 and 5,654,304 disclose a series of 10,11- methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. One such derivative having good activity, oral bioavailability, and stability, is zosuquidar, a compound of formula (2R)-anti-5-
3 – [4-( 10, 11 -difluoromethanodibenzosuber-5-yl)piperazin- 1 -yl]-2-hydroxypropoxy) quinoline.

Zosuquidar
Given the limitations of previous generations of MDR modulators, three preclinical critical success factors were identified and met for zosuquidar: 1) it is a potent inhibitor of P-glycoprotein; 2) it is selective for P-glycoprotein; and 3) no pharmacokinetic interaction with co-administered chemotherapy is observed.
Zosuquidar is extremely potent in vitro (Kj = 59 nM) and is among the most active modulators of P-gp-associated resistance described to date. Zosuquidar has also demonstrated good in vivo activity in preclinical animal studies. In addition, the compound does not appear to be a substrate for P-gp efflux, resulting in a relatively long duration of reversal activity in resistant cells even after the modulator has been withdrawn.
Another significant attribute of zosuquidar as an MDR modulator is the minimal pharmacokinetic (PK) interactions with several oncolytics tested in preclinical models. Such minimal PK interaction permits normal doses of oncolytics to be administered and also a more straightforward interpretation of the clinical results.
Zosuquidar is generally administered in the form of the trihydrochloride salt. Conventional zosuquidar trihydrochloride formulations include those containing zosuquidar (50 mg as free base), glycine (15 mg), and mannitol (200 mg) dissolved in enough water for injection, to yield a free base concentration of 5 mg/mL. The formulation is filled into vials and lyophilized to give a vial containing 50 mg of free base. For such formulations, a 30 mL vial size is necessary to contain 50 mg of thezosuquidar formulation. For a typical >200 mg dose of zosuquidar, multiple 50 mg vials are needed to contain the formulation, greatly increasing manufacturing costs and reducing convenience for the end user {e.g., a pharmacist). Modified Cyclodextrins
Cyclodextrins are cyclic oligomers of glucose; these compounds form inclusion complexes with any drug whose molecule can fit into the lipophile-seeking cavities of the cyclodextrin molecule. See U.S. Pat. No. 4,727,064 for a description of various cyclodextrin derivatives. Cyclodextrins of preferred embodiments can include α-, β-, and χ-cyclodextrins. The α-cyclodextrins include six glucopyranose units, the β- cyclodextrins include seven glucopyranose units, and the χ-cyclodextrins include eight glucopyranose units. The β -cyclodextrins are generally preferred as having a suitable cavity size for zosuquidar. Cyclodextrin can be in any suitable form, including amorphous and crystalline forms, with the amorphous form generally preferred. Cyclodextrins suitable for use in the formulations of preferred embodiments include the hydroxypropyl, hydroxyethyl, glucosyl, maltosyl, and maltotrosyl derivatives of β- cyclodextrin, carboxyamidomethyl-β-cyclodextrin, carboxymethyl-β-cyclodextrin, and diethylamino-β-cyclodextrin.
Pharmaceutical complexes including various cyclodextrins and cyclodextrin derivatives are disclosed in the following United States patents: U.S. Pat. No. 4,024,223; U.S. Pat. No. 4,228,160; U.S. Pat. No. 4,232,009; U.S. Pat. No. 4,351,846; U.S. Pat. No. 4,352,793; U.S. Pat. No. 4,383,992; U.S. Pat. No. 4,407,795; U.S. Pat. No. 4,424,209; U.S. Pat. No. 4,425,336; U.S. Pat. No. 4,438,106; U.S. Pat. No. 4,474,881; U.S. Pat. No. 4,478,995; U.S. Pat. No. 4,479,944; U.S. Pat. No. 4,479,966; U.S. Pat. No. 4,497,803; U.S. Pat. No. 4,499,085; U.S. Pat. No. 4,524,068; U.S. Pat. No. 4,555,504; U.S. Pat. No. 4,565,807; U.S. Pat. No. 4,575,548; U.S. Pat. No. 4,598,070; U.S. Pat. No. 4,603,123; U.S. Pat. No. 4,608,366; U.S. Pat. No. 4,659,696; U.S. Pat. No. 4,623,641; U.S. Pat No. 4,663,316; U.S. Pat. No. 4,675,395; U.S. Pat. No. 4,728,509; U.S. Pat. No. 4,728,510; and U.S. Pat. No. 4,751,095.
Chemically modified and substituted α-, β-, and χ-cyclodextrins are generally preferred over unmodified α-, β-, and χ-cyclodextrins due to improved toxicity and solubility properties. The degree of substitution of the hydroxy 1 groups of the glucopyranose units of the cyclodextrin ring can affect solubility. In general, a higher average degree of substitution of substituent groups in the cyclodextrin molecule yields a cyclodextrin of higher solubility.
Examples for Pgp inhibitors are cyclosporine A, valpodar, elacridar, tariquidar, zosuquidar, laniquidar, biricodar, S-9788, MS-209, BIBW-22 (BIBW-22-BS) , toremifene, verapamil, dexverapamil , quinine, quinidine, trans- flupentixol, chinchonine and others (J. Roberts, C. Jarry (2003) : J. Med. Chem. 46, 4805 – 4817) . The list of inhibitors of P-glycoprotein is increasing (e.g. Wang et al . (2002) : Bioorg. Med. Chem. Lett. 12, 571 – 574) .
Figure 2: Structures of BIBW-22, MS-209 and S-9788
|
7-12-2000
|
10,11-methanodibenzosuberane derivatives
|
|
|
10-17-2007
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline
|
|
|
9-2-2009
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-YL)piperazin-1-YL]-2-hydroxypropoxy}quinoline
|
……………………

U.S. Pat. Nos. 5,643,909 and 5,654,304, incorporated herein by reference, disclose a series of 10,11-methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride disclosed therein, is currently under development as a pharmaceutical agent.
U.S. pat. No. 5,654,304 (‘304), incorporated by reference herein, discloses a series of 10,11-(optionally substituted)methanodibenzosuberane derivatives useful in enhancing, the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-Difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride is disclosed in ‘304 and is currently under development as a pharmaceutical agent. WO00/75121 discloses Form I, a crystalline form of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride.
The art disclosed in U.S. Pat. No. 5,776,939, and U.S. Pat. No. 5,643,909 both incorporated herein by reference, and PCT Patent Applications (Publication numbers WO 94/24107 and 98/22112) teach the use of 1-formylpiperazine to introduce the piperazine group of the compound of formula II

Compound II is a mixture of syn isomer (III)

and anti isomer (IV)

The process as disclosed in U.S. Pat. Nos. 5,643,909 and 5,654,304 (represented by scheme A, below) involves (a) chromatographic separation(s) of the formyl piperazine compound; and (b) deformylation of the formyl piperazine compound to provide compound IV.https://www.google.co.in/patents/US6570016?cl=en
The process of the present invention uses piperazine to react with the (1aα,6α,10bα)-6-halo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cycloheptene compound or derivative, instead of formylpiperazine.
The process of the present invention is advantageous because piperazine is readily available in commercial quantities whereas 1-formylpiperazine, which was utilized in the process disclosed in U.S. Pat. No. 5,643,909 is often not readily available in commercial quantities. Additionally piperazine enjoys a significant cost advantage over 1-formylpiperazine.
The use of piperazine instead of 1-formylpiperazine is a significant advancement over the prior art because it obviates the need to deformylate or hydrolyze off the formyl group (step 6, scheme A), thereby providing fewer operational steps. U.S. Pat. No. 5,643,909 teaches the separation of the 1-formylpiperazine compounds by chromatography or repeated crystallization. The present invention obviates the need for chromatographic separations of the formylpiperazine diastereomeric addition compounds (see step 4, scheme A)
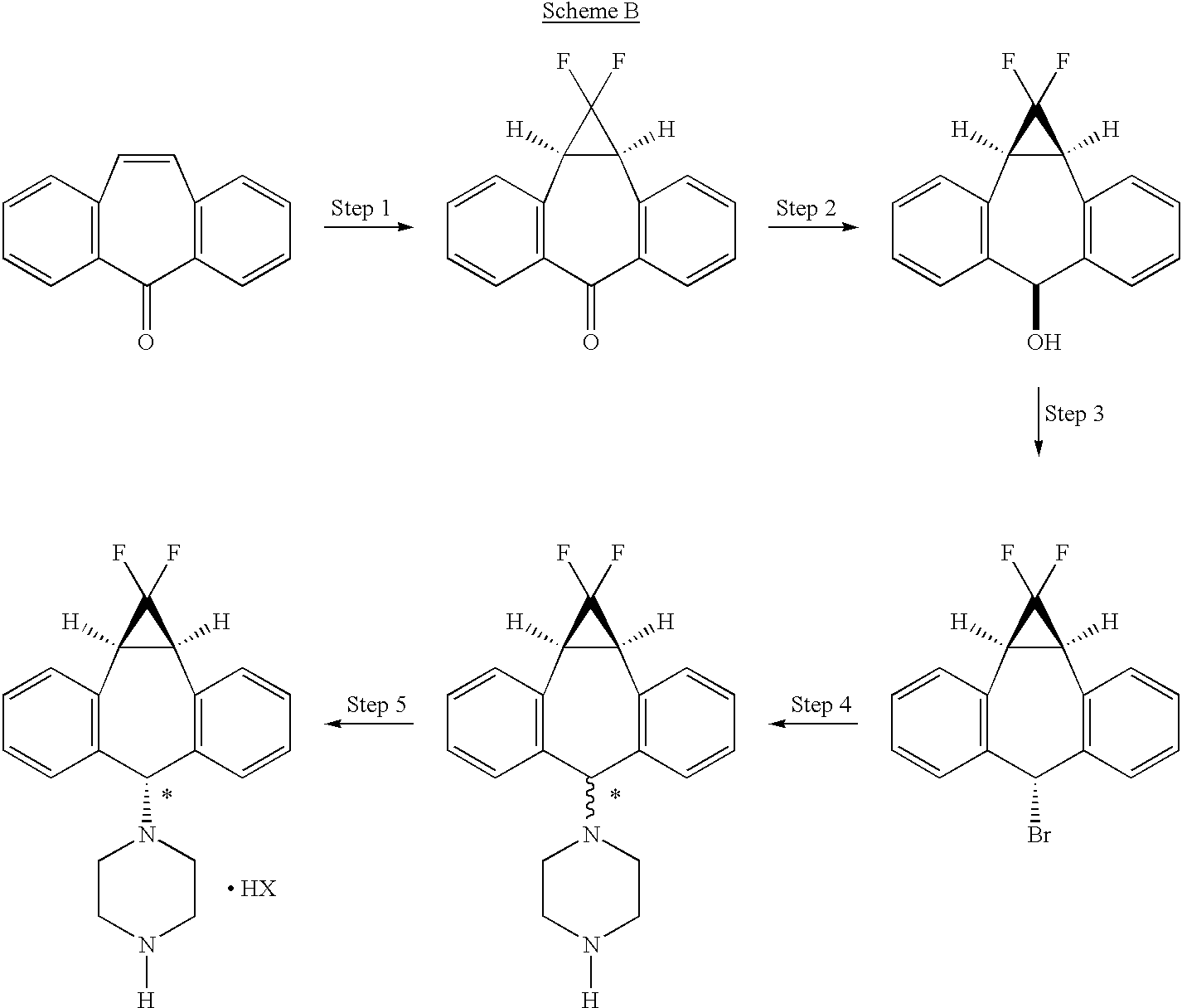
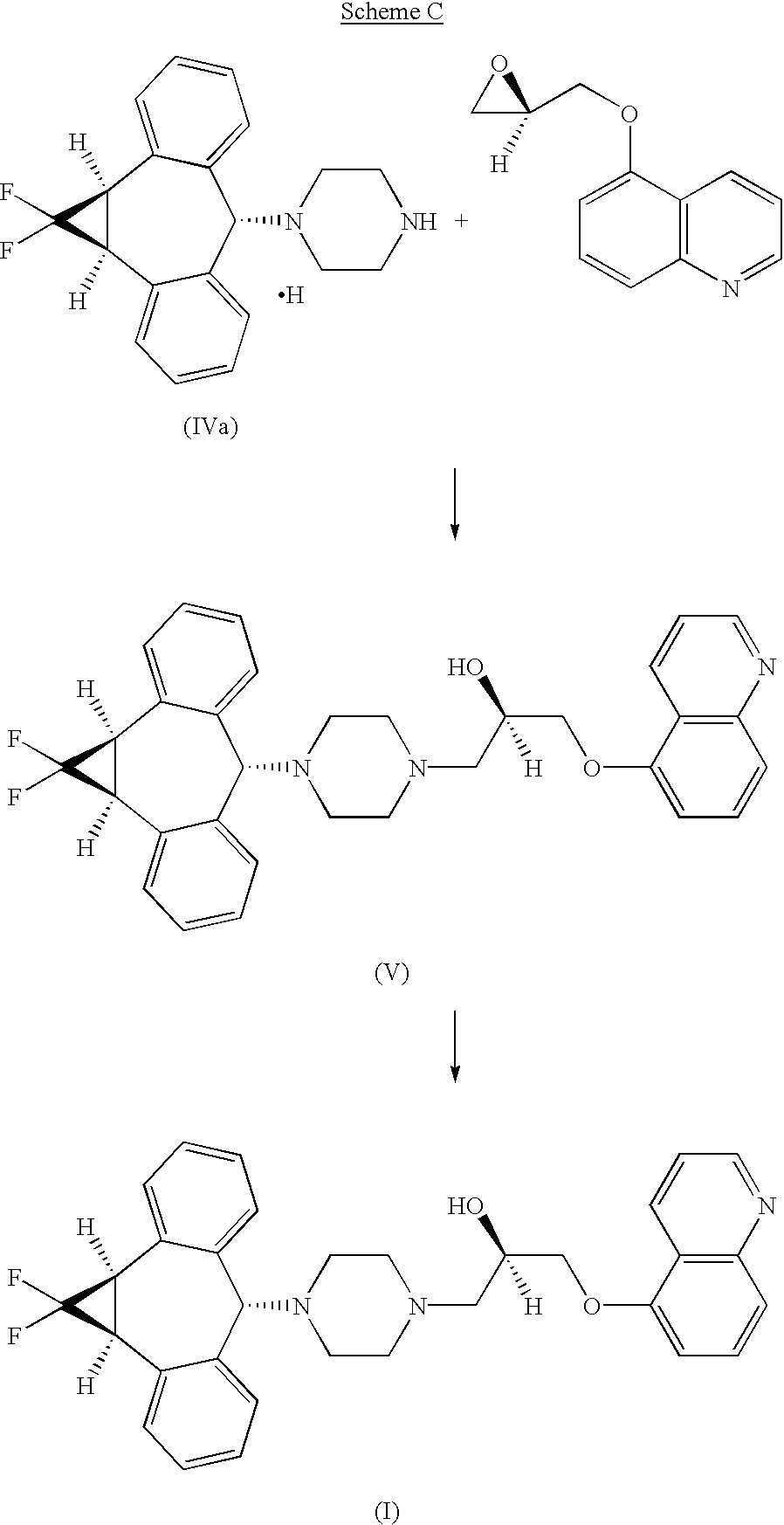
EXAMPLES
The following examples and preparations are illustrative only and are not intended to limit the scope of the invention in any way.
Preparation 1 R-1-(5-Quinolinyloxy)-2,3-epoxypropane
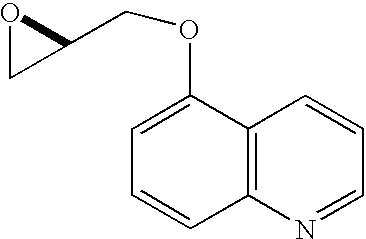
A mixture of 5-hydroxyquinoline (5.60 g, 38.6 mmol), R-glycidyl nosylate (10.0 g, 38.6 mmol), powdered potassium carbonate (11.7 g, 84.9 mmol), and N,N-dimethylformamide (100 mL) was stirred at ambient temperature until HPLC analysis (40% acetonitrile/60% of a 0.5% aqueous ammonium acetate solution, 1 mL/min, wavelength=230 nm, Zorbax RX-C8 25 cm×4.6 mm column) indicated complete disappearance of glycidyl nosylate (approximately 6 hours). The reaction mixture was filtered through paper and the filter cake was washed with 200 mL of a 3:1 mixture of MTBE and methylene chloride. The filtrate was washed with 200 mL of water and the aqueous layer was extracted four times with 100 mL of 3:1 MTBE/methylene chloride. The combined organic layers were dried over 30 grams of magnesium sulfate and the dried solution was then stirred with 50 grams of basic alumina for 30 minutes. The alumina was removed by filtration and the filter cake was washed with 200 mL of 3:1 MTBE/methylene chloride. The filtrate was concentrated to a volume of 100 mL, 300 mL of MTBE were added, and the solution was again concentrated to 80 mL. After heating to 50° C., the solution was treated with 160 mL of heptane dropwise over 15 minutes, allowed to cool to 40° C., and seeded, causing the formation of a crystalline precipitate. The mixture was stirred for two hours at ambient temperature and then at 0-5° C. for an additional 2 hours. The crystals were filtered, washed with cold heptane, and dried to provide 5.68 g (73.2%) of (2R)-1-(5-quinolinyloxy)-2,3-epoxypropane as white needles.
mp 79-81° C.;
[α]25 D−36.4° (c 2.1, EtOH);
1H NMR (500 MHz, CDCl3)δ 2.83 (dd, J=4.8, 2.7 Hz, 1H), 2.97 (m, 1H), 3.48 (m, 1H), 4.10 (dd, J=11.0, 6.0 Hz, 1H), 4.43 (dd, J=11.0, 2.7 Hz, 1H), 6.85 (d, J=7.8 Hz, 1H), 7.38 (dd, J=8.5 Hz, 4.1 Hz, 1H), 7.59 (m, 1H), 7.71 (d, J=8.5 Hz, 1H), 8.61 (m, 1H), 8.90 (m, 1H).
Example 1 (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride

Preparation of the above compound is exemplified in the following preparative steps.
Step 1 1,1-Difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6 (1H)-one

A solution of sodium chlorodifluoroacetate (350 g) in diglyme (1400 mL) was added dropwise over 4 to 8 hours, preferably over 6 hours, to a solution of 5H-dibenzo[a,d]cyclo-hepten-5-one (25 g) in diglyme (500 mL), with stirring, and under nitrogen, maintaining the reaction temperature at 160°-165° C. The cooled reaction mixture was poured into water (1.8 L) and extracted with ether (1.8 L). The organic phase was washed with water, dried over sodium sulfate (Na2SO4), and evaporated. The residue was recrystallized from ethanol, then from acetone/hexane to give 14 g of 1,1-difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6(1H)-one.
mp 149.6° C.
Flash chromatography of the combined mother liquors on silica gel, eluting with 20% acetone/hexane, gave an additional 6.5 g of the target compound.
Step 2 (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

A solution of 1,1-difluoro-1a,10b-dihydro-dibenzo[a,e]cyclopropa[c]cyclohepten-6(1H)-one (20.4 g) in tetrahydrofuran/methanol (1:2, 900 mL) was cooled in an ice bath. Sodium borohydride (12 g) was added in portions. The cooling bath was removed and the reaction mixture was stirred at ambient temperature for 2 hours, then poured into water. The product was filtered off, washed with water, and dried to give 20 g of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol (ii).
mp 230.1°-230.6° C.
Step 2A Combined Steps 1 and 2 Procedure (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

To a solution of 103.1 g (0.500 mol) of 5H-dibenzo[a,d]cyclohepten-5-one (2) in 515 mL of triethylene glycol dimethyl ether heated to between 180° C. and 210° C. was added over 7 hours, 293.3 g (2.15 mol) of chlorodifluoroacetic acid lithium salt (as a 53% by weight solution in ethylene glycol dimethyl ether). The ethylene glycol dimethyl ether was allowed to distill from the reaction as the salt addition proceeded. The GC analysis of an aliquot indicated that all of the 5H-dibenzo[a,d]cyclohepten-5-one had been consumed. The reaction was cooled to ambient temperature and then combined with 400 mL of ethyl acetate and 75 g of diatomaceous earth. The solids were removed by filtration and washed with 300 mL of ethyl acetate. The washes and filtrate were combined and the ethyl acetate was removed by concentration under vacuum leaving 635 g of dark liquid. The dark liquid was cooled to 18° C. and to this was added, over 15 minutes, 6.62 g (0.175 mol) of sodium borohydride (as a 12% by wt solution in 14 M NaOH). After stirring for 2 h the reaction was quenched by careful addition of 900 mL of a 1:3.5:4.5 solution of conc. HCl-methanol-water. The suspension was stirred for 30 min and the crude product was collected by filtration, washed with 600 mL of 1:1 methanol-water and dried to 126.4 g of dark brown solid. The crude product was slurried in 600 mL of methylene chloride, filtered, washed twice with 150 mL portions of methylene chloride, and dried to 91.6 g (71%) of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol. Gas Chromatography (GC) Conditions; Column: JW Scientific DB-1, Initial Temperature 150° C. for 5 min, 10° C./min ramp, Final temp 250° C. for 5 min. tR: intermediate, 11.5 min; reaction product (alcohol), 11.9 min; starting material, 12.3 minutes.
Step 3 Preparation of (1aα,6α,10b)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa-[c]cycloheptene

A slurry of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol (3.0 g, 11.6 mmol, 1.0 equiv) in heptane (24 mL) was treated with 48% HBr (1.58 mL, 14.0 mmol, 1.2 equiv) and the reaction was heated at reflux with vigorous stirring for 2.5 hr. Solvent was then removed by atmospheric distillation (bp 95-98° C.) until approximately 9 mL of distillate was collected. The reaction was cooled and treated with EtOAc (15 mL), Na2SO4 and activated charcoal. The mixture was stirred at RT for 15 min and filtered through hyflo. The filter cake was washed with 50:50 EtOAc:heptane and the filtrate was concentrated in vacuo to provide the title product as a crystalline solid.
mp 119° C. (3.46 g corr., 93%);
1H NMR (500 MHz CDCl3) δ 7.20-7.41 (8H, m), 5.81 (1H, s), 3.41 (2H, d, J 12.5 Hz);
13CNMR (126 MHz CDCl3) δ 141.3, 141.2, 133.5, 130.1, 129.8, 128.3, 128.2, 112.9, 110.6, 110.5, 108.3, 53.6, 30.2, 30.1, 30.0.
Anal. Calcd. For C16H11BrF2: C, 59.84; H, 3.45. Found: C, 60.13; H, 3.50.
Step 3A Preparation of (1aα,6α,10bα)-6-Bromo-1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene

To a stirred suspension of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol, (18.4 g, 71.2 mmol) in 151 mL of methylene chloride which had been cooled to 10-17° C. was added phosphorous tribromide (9.6 g, 35.6 mmol) dropwise over 15 minutes. The cooling bath was removed and the reaction was stirred for 2 hours at ambient temperature. Analysis by gas chromatography indicated complete consumption of starting material. Cold water (92 mL) and activated carbon (1.84 g) were added and the resulting mixture was stirred for 30 minutes. The activated carbon was removed by filtration through Hyflo brand filter aid and the two phases were separated. The organic phase was washed with water (184 mL×2), brine (184 ml), dried over magnesium sulfate and concentrated to dryness under vacuum, affording 21.7 g (94.8%) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene.
1H NMR (CDCl3, 300 MHz) δ 3.36 (s, 1H), 3.40 (s, 1H), 5.77 (s, 1H), 7.16-7.38 (m, 8H).
Steps 4 and 5 (1aα,6α,10bα)-1-(1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-yl)-piperazine, Hydrobromide Salt

To a solution of 237.5 g (0.739 mol) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]-cyclopropa[c]cycloheptene in 3.56 L of acetonitrile was added 207.7 g (2.41 mol) of piperazine and the mixture was heated to reflux for 2 hours, at which time analysis by gas chromatography showed complete consumption of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene (iii) and formation of a mixture of syn and anti piperazine compounds (III and IV) in an anti-syn ratio of 55:45. The reaction was cooled to about 7° C. and stirred for 30 minutes at that temperature. The reaction mixture was filtered to remove the precipitated syn-isomer (III) and the filter cake was washed with 250 mL of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 262.4 grams of a foam which was dissolved in 450 mL of acetonitrile with heating. The solution was cooled to about 12° C. in an ice bath and stirred for 1 hour at that temperature. The precipitated syn-piperazine compound of formula (III) was filtered and washed with 125 ml of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 194.1 g and dissolved in 1.19 L of ethyl acetate. The organic solution was washed sequentially with 500 mL portions of 1N sodium hydroxide, water, and saturated sodium chloride. The ethyl acetate solution was dried over sodium sulfate and concentrated to give 137.0 grams of residue which was dissolved in 1.37 L of methylene chloride and seeded with (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrobromide salt, followed by the addition of 70.8 grams of 48% aqueous hydrobromic acid. The mixture was stirred for about 45 minutes, causing the anti-isomer to crystallize as its hydrobromide salt. The crystals were filtered, washed with methylene chloride, and dried to provide purified hydrobromide salt of compound (IVa), shown by HPLC to have an anti-syn ratio of 99.3:0.7. Treatment of the isolated hydrobromide salt of compound (IVa) with aqueous sodium hydroxide, extraction into methylene chloride, separation of the aqueous layer and concentration to dryness gave 80.1 grams (33.2% yield based on starting material) of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine as the free base. Acidification of a solution of the free base in 800 mL of methylene chloride by addition of 41.2 g of 48% hydrobromic acid as described above afforded 96.4 g of pure hydrobromide salt (title compound) with an anti-syn ratio of 99.8:0.2 (HPLC), mp 282-284° C. 1H NMR (DMSO-d6) δ 2.41 (m, 4H), 3.11 (m, 4H), 3.48 (d, J=12.4 Hz, 2H), 4.13 (s, 1H), 7.2 (m, 8H), 8.65 (bs, 2H). 13C NMR (DMSO-d6) δ 28.0, 42.9, 48.0, 75.1, 108.5, 112.9, 117.3, 127.5, 128.0, 128.6, 129.6, 132.4, 141.3. IR: (KBr) 3019, 2481, 1587, 1497, 1298 cm−1. Anal. Calcd for C20H21BrF2N2: C, 58.98; H, 5.20; N, 6.88. Found: C, 58.75; H, 5.29; N, 7.05.
Step 6 Preparation of (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)propan-2-ol Trihydrochloride
A suspension of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrochloride compound of formula IVa (5.41 g, 14.9 mmol) and powdered sodium carbonate (3.16 g, 29.8 mmol) in 54 mL of 3A ethanol was stirred at ambient temperature for 1 hour. R-1-(5-quinolinyloxy)-2,3-epoxypropane (3.00 g, 14.9 mmol) was added in one portion and the reaction mixture was heated to 65° C. for 19 hours. HPLC analysis (Gradient system with solvent A (acetonitrile) and solvent B (0.02M sodium monophosphate buffer containing 0.1% triethylamine adjusted to pH 3.5 with phosphoric acid) as follows: 0-12 min, 30% solvent A/70% solvent B; 12-30 min, linear gradient from 30% to 55% solvent A/70% to 45% solvent B; 30-35 min, 55% solvent A/45% solvent B, 1 mL/min, 1=240 nm, Synchropak SCD-100 25 cm×4.6 mm column) indicated the total consumption of the piperazinyl compound of formula (IV). The mixture was allowed to cool to room temperature, filtered through a plug of silica gel, and eluted with an additional 90 mL of ethanol. The eluent was concentrated to a volume of approximately 60 mL and heated to 65° C. with stirring. A solution of HCl in ethanol (16.1 g at 0.135 g/g of solution, 59.6 mmol) was added dropwise over 10 minutes and the resultant product solution was seeded, causing the trihydrochloride salt to precipitate. The mixture was allowed to cool to ambient temperature and stirred slowly (less than 100 RPM) for 2 hours. The precipitate was filtered, washed with ethanol, and dried in vacuo at 50° C. to give the crude trihydrochloride salt which was further purified by recrystallization from methanol/ethyl acetate to provide 7.45 g (78.4%) of (2R)-anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)-propan-2-ol trihydrochloride.
Step 6a
The syn isomer compound of formula (III) isolated as described supra (combined steps 4 and 5), can be utilized to produce the corresponding syn-5-{3-[4-(10,11-difluoromethano-dibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride (XII) essentially as shown below for the free base of the anti isomer (IVa)in step 6.
https://www.google.co.in/patents/US6570016?cl=en
………………………………………
http://www.google.it/patents/WO1994024107A1?cl=en
REACTION SCHEME 1

FormuIa 1
Formula 1

Formula 2 Formula 2

Formula 3
Formula 3

Formula 4

Formula I
……………………………………….
http://www.google.com/patents/WO2000075121A3

1HNMR (500 MHz DMSO-d6) δ9.41 (2H, br. s), 7.17-7.31 (8H, m), 4.17 (1H, s), 3.52 (2H, d, J=12.4 Hz), 3.11 (4H, br. s), 2.48-2.51 (4H, m)
13CNMR (126 MHz DMSO-d6) δ142.3, 133.4, 130.5, 129.6, 129.0, 128.4, 115.9, 113.6, 111.3, 76.2, 49.0, 43.6, 29.2, 29.1, 29.0; FD MS: m/e 326 (M+).
Anal. Calcd. For C20H21ClF2N2: C, 66.20; H, 5.83; N, 7.72.
Found: C, 66.08; H, 5.90; N, 7.72.
…………………………………………..
http://www.google.com/patents/US6570016?cl=fr


(2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride
……………….
Chemical Shift Data and Peak Assignments for the Crystal Forms.

Form II has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 29.9, 50.1, 55.3, 62.0, 66.5, 72.0, 75.8, 104.8, 107.5, 108.2, 109.1, 110.2, 112.0, 118.4, 119.5, 120.1, 123.1, 128.7, 131.1, 133.0, 134.8, 136.4, 136.9, 139.9, 140.0, 142.3, 144.5, 146.6, 149.0, 144.2, 153.0 and 153.6 ppm.
Form III has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 30.3, 50.4, 59.1, 63.2, 72.8, 77.2, 109.1, 110.2, 112.2, 112.8, 118.7, 119.5, 119.9, 121.0, 122.2, 123.0, 128.9, 130.6, 132.7, 134.0, 136.4, 140.0, 141.0, 141.8, 142.5, 143.3, 146.1, 153.1, 153.8 and 154.7 ppm.
