
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
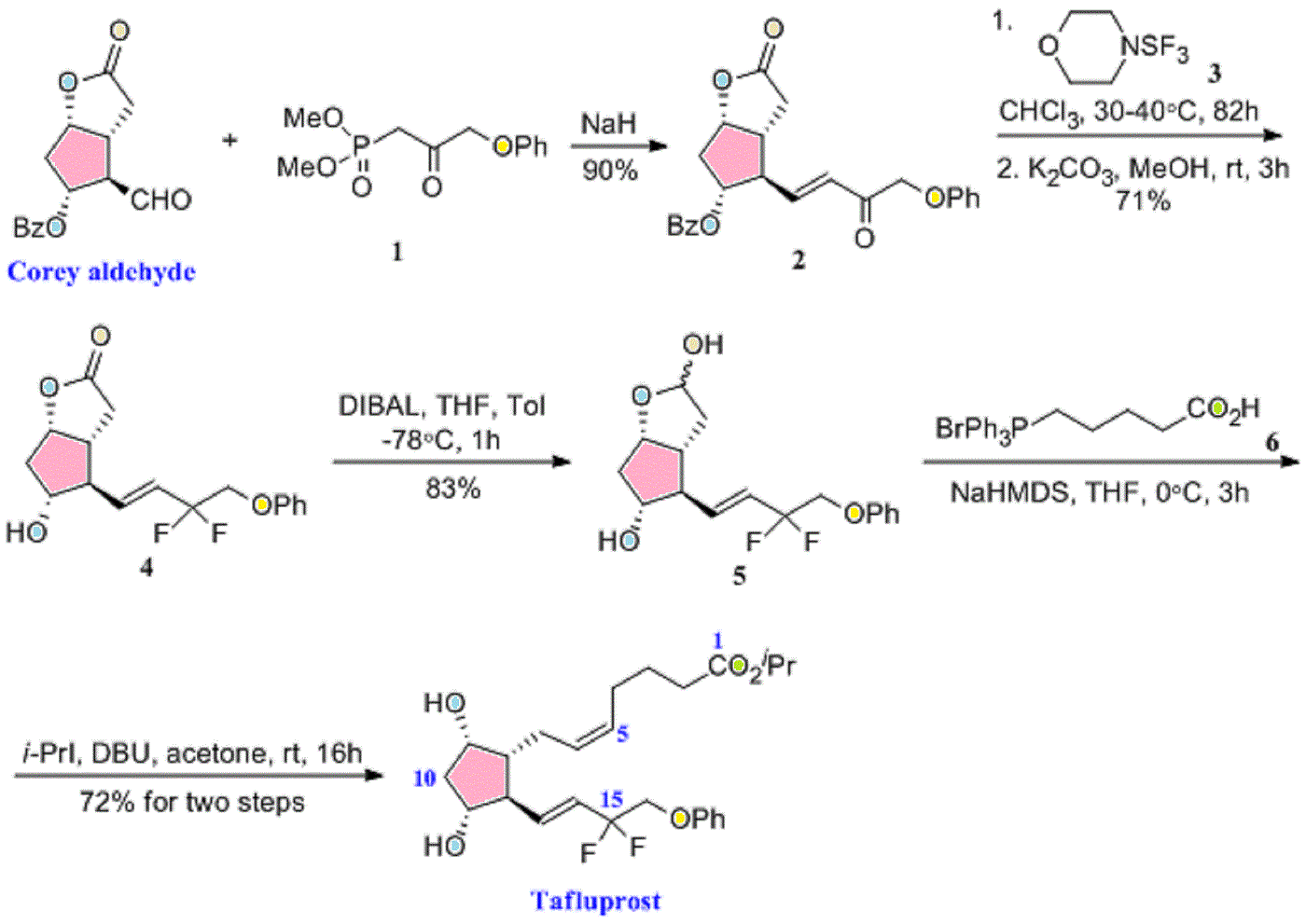
Drug Spotlight: Zioptan, Tafluprost, Merck,

 |
|---|
isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate,
Drug: Zioptan
Generic molecule: tafluprost
Company: Merck
Approval date: Feb. 10, 2012
The scoop: Merck says this is the first (get ready for a mouthful) preservative-free prostaglandin analog ophthalmic solution and is for treating elevated eye pressure in some patients with the most common form of glaucoma. Merck sells the ointment in the U.S. and most of Europe, while it licensed it to Japanese drugmaker Santen in Japan, Germany and northern Europe.
Tafluprost (trade names Taflotan, marketed by Santen Pharmaceutical Co. and Zioptan, by Merck (U.S.)) is a prostaglandin analogue used topically (as eye drops) to control the progression of glaucoma and in the management of ocular hypertension. It reduces http://en.wikipedia.org/wiki/Intraocular_pressure”; rel=”nofollow”>intraocular pressure by increasing the outflow of aqueous fluid from the eyes.[1][2]
Taflotan contains 15 µg/ml Tafluprost. Taflotan sine is a preservative-free, single-dose formulation containing 0.3 ml per dose.[3]

|
|
| Systematic (IUPAC) name | |
|---|---|
| isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate | |
| Clinical data | |
| Trade names | Saflutan, Taflotan, Tapros, Zioptan |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Identifiers | |
| CAS number | 209860-87-7 |
| ATC code | S01EE05 |
| PubChem | CID 6433101 |
| ChemSpider | 8044182 |
| UNII | 1O6WQ6T7G3 |
| ChEBI | CHEBI:66899 |
| ChEMBL | CHEMBL1963683 |
| Chemical data | |
| Formula | C25H34F2O5 |
| Mol. mass | 452.531266 g/mol |


- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Santen Home Page
- Gelbe Liste (in German)

European Patent No. 8509621 discloses a process for the preparation of tafluprost. In the first step, (3afl,4fl,5fl,6aS)-4-formyl-2-oxohexahydro-2 — cyclopenta[b]furan-5-ylbenzoate (CTAF 1 (i)) is condensed with dimethyl (2-oxo-3- phenoxypropyl)-phosphonate in the presence of lithium chloride and triethylamine, to provide (3aft,4F?,5F?,6aS)-2-oxo-4-((£)-3-oxo-4-phenoxybut-1 -en-1 -yl)hexahydro-2H- cyclopenta[b]-furan-5-ylbenzoate (CTAF1 ). In the second step, CTAF 1 is reacted with morpholinosulfurtrifluoride to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-2-oxohexahydro-2 –cyclopenta-[b]furan-5-yl benzoate (CTAF2). CTAF 2 is debenzoylated by potassium carbonate in methanol, to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-5-hydroxyhexahydro-2H- cyclopenta[b]furan-2-one(CTAF 3), which is further reduced by diisobutyl aluminum hydride (DIBALH) to provide (3af?,4f?,5f?,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 – yl) hexahydro-2H-cyclopenta[b]furan-2,5-diol (CTAF 4). CTAF 4 is then treated with (4- carboxybutyl)triphenylphosphonium bromide, in the presence of potassium bis(trimethylsilyl)amide in THF, to provide (Z)-7-((1 f?,2f?,3f?,5S)-2-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-3,5-dihydroxycyclopentyl)hept-5-enoic acid (“tafluprost free acid,” CTAF5), which is reacted with isopropyl iodide in the presence of DBU to provide (Z)- isopropyl 7-((1 F?,2F?,3F?,5S)-2-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-3,5-dihydroxy- cyclopentyl)hept-5-enoate (“tafluprost,” CTAF 6). The reaction sequence is summarized in Scheme 1 .

CTAF 1(i)
CTAF 1 CTAF 2

U.S. Patent Application Publication No. 2010/0105775A1 discloses amino acid salts of prostaglandins. The application also discloses a process for the preparation of prostaglandins, comprising forming an amino acid salt of a prostaglandin and converting the amino acid salt to the prostaglandin.
EXAMPLE 1 : Preparation of CTAF 1

CTAF1(i)
CTAF1
To a stirred suspension of sodium hydride (60% dispersion in mineral oil, 0.217 g, 5.429 mmol) in THF (5 ml_) was added a solution of dimethyl (2-oxo-3- phenoxypropyl)phosphonate(1 .21 g, 4.705 mmol) in THF (2 ml_), over 15 minutes at 0- 5°C under a nitrogen atmosphere. The mixture was warmed to 25-35 , 0.5 M zinc chloride solution in THF (9.4 ml_, 4.705 mmol) was added over 10 minutes, and then the mixture was stirred for 15 minutes at 25-35<€. CTAF1 (i) (3af?,4F?,5F?,6aS)-4-formyl-2- oxohexahydro-2 –cyclopenta[b]furan-5-yl benzoate (1 g) in dichloromethane (10 ml_) was added over 5 minutes at 25-35 °C. The temperature was raised to 35-40 °C and the mixture was stirred for 2hours under a nitrogen atmosphere. The mixture was cooled to 15°C and the reaction was quenched by adding acetic acid (0.2 mL), followed by adding saturated ammonium chloride solution (10 mL), and further stirring for 15 minutes. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (5 mL). The combined organic layers were evaporated under reduced pressure below 50°C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (0.9 g, 61 %yield).
EXAMPLE 2: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (5 g, 0.0123 mol) in dichloromethane (100 mL) was added diethylaminosulfurtrifluoride (13 mL, 0.09841 mol) at 0-5 °C under a nitrogen atmosphere. The temperature was raised to 25-35 °C and maintained for 24 hours under a nitrogen atmosphere at the same temperature. The mass was slowly added into a saturated sodium bicarbonate solution (75 mL) at 0-5 °C. Temperature was raised to 25- 35 °C, the layers were separated, and the aqueous layer was extracted with dichloromethane (2×25 mL). The combined organic layer was washed with water (25mL) and dried over sodium sulfate (5 g). The organic layer was evaporated to dryness under reduced pressure below 40 °C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (4.2 g, 79% yield). EXAMPLE 3: Preparation of CTAF 4

CTAF 2 CTAF 4
CTAF 2 (2.30 g, 5.37 mmol) was dissolved in toluene (25 mL) and the solution was cooled to -65 °C under nitrogen. Diisobutyl aluminum hydride (1 .5 M in toluene, 1 1 .8 mL, 17.7mmol) was added over 15 minutes at -61 to -65 . The mixture was stirred for 3hours and then the reaction was quenched by adding methanol (1 .5 mL). Sulfuric acid (1 M, 25 mL) was added and the temperature rose to -20°C during the addition. Methyl t-butyl ether (MTBE) (10 mL) was added and the mixture was allowed to warm to room temperature. The organic phase was separated and the aqueous phase was extracted with MTBE (2x 10 mL). The combined organic phase was washed with water (10 mL), saturated aqueous sodium bicarbonate (10 mL), and then brine (10 mL). The washes were back-extracted with MTBE (10 mL). The combined organic phases were dried with magnesium sulfate, filtered, and evaporated to give a colourless oil (2.20 g). The crude product was chromatographed on silica (60 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), and then with ethyl acetate, to give CTAF 4 as a colourless oil (1 .71 g, 97% yield).
EXAMPLE 4: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (20 g, 0.0492 mol) in dichloromethane(400 mL) was added diethylaminosulfurtrifluoride (52 mL, 0.393 mol) at 0-10°C under a nitrogen atmosphere. The temperature was raised to 25-35 and maintained for 96hours under a nitrogen atmosphere at that temperature. The mass was slowly added to a saturated NaHCOs solution (600 mL) at 0-10°C. The mixture was heated to 25-35 <€ and filtered through aCelite bed. The layers were separated and the aqueous layer was extracted with DCM (2×100 mL). The combined organic layer was washed with 10% NaCI solution (100 mL) and evaporated to dryness under reduced pressure below 40°C. The residue was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane.
Column purified material was dissolved in MTBE (80 mL) at 40°C and stirred for 30 minutes at that temperature. Diisopropyl ether (160 mL) was added at 35-40 and stirring continued for 30 minutes at 35-40 . Cooled the mass to 5-15°C and stirred for 30 minutes at that temperature. The solid was filtered, washed with a mixture of MTBE and diisopropyl ether (DIPE) (1 :2 by volume, 60 mL), and dried at 40°C under vacuum, to afford pure CTAF2 (12.0 g, 57% yield).
EXAMPLE 5: Preparation of CTAF 5

(4-Carboxybutyl)triphenylphosphonium bromide (10.32 g, 23.3 mmol, 4 eq) was suspended in THF (20 mL) under a nitrogen atmosphere and cooled to 5°C. NaHMDS solution (1 M in THF, 46.6 mL, 46.6 mmol, 8 eq) was added over 10 minutes. The red/orange mixture was stirred for 30 minutes. A solution of CTAF 4 (1 .90 g, 5.82 mmol) in THF (10 mL) was added over 30 minutes at 0-3 . The mixture was stirred for 1 .5hours and then the reaction was quenched by adding water (30 mL) and the masswas warmed to room temperature. The aqueous phase was separated and the organic phase was washed with water (20 mL). The combined aqueous phases were washed with MTBE (30 mL). The organic phases up to this point were discarded. The aqueous phase was acidified with 2M hydrochloric acid (14 mL, to pH 3-4) and extracted with ethyl acetate (2×30 mL). The combined ethyl acetate layers were washed with brine (20 mL), dried with magnesium sulfate, filtered, and evaporated under reduced pressure to give CTAF 5 asa yellow oil (8.60 g).
A 2.96 g sample was removed and the remainder (5.64 g) was chromatographed on silica (30 g) eluting with ethyl acetate to give purified CTAF 5 (1 .41 g) asa yellow oil. NMR analysis showed approximately 90% purity, remainder triphenyl phosphine oxide.
EXAMPLE 6: Preparation of CTAF 5 DCHA salt

CTAF 5 CTAF 5 DCHA sa t
CTAF5 (1 1 .72 g, 90% purity, 25.7 mmol, containing 1 .4% trans isomer) was dissolved in acetone (60 mL). Dicyclohexylamine (4.66 g, 25.7 mmol) was added and the mixture was stirred at room temperature overnight. The solid was filtered and washed with acetone (6 mL), then dried to give the DCHA salt (12.93 g, 85% yield, 0.29% trans-isomer).
A sample (7.03 g) was further purified by recrystallisation. It was dissolved in hot acetone (30 mL) and cooled to room temperature with stirring. The mixture was stirred for 3 hours, filtered and the solid was washed with acetone (3 mL) and dried to give a white solid (6.41 g, 91 % recovery, 0.1 1 % trans-isomer).
A PXRD pattern of the product is shown as Fig. 1 , obtained using copper Ka radiation. In the drawing, the y-axis is intensity units and the x-axis is the 2-theta angle, in degrees. EXAMPLE 7: Pre aration of CTAF 6

CTAF 5 DCH A sa l ^ I AI- O
CTAF 5 DCHA salt (5.80 g, 9.80 mmol) was suspended in ethyl acetate (20 mL). Sulfuric acid (1 M, 20 mL) was added and the mixture was stirred until a clear solution was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (2×20 mL). The combined organic layers were washed with water (15 mL) and brine (15 mL), dried with magnesium sulfate, filtered, and evaporated. The residue was dissolved in acetone (40 mL) and charged into a jacketed vessel at 30°C. 1 ,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (8.95 g, 58.8 mmol) was added, then 2- iodopropane (10.0 g, 58.8 mmol) was added, and the mixture was stirred for 20hours. The mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (30 mL) and aqueous potassium dihydrogen orthophosphate (8 g) in water (50 mL). The organic phase was separated and the aqueous was extracted with ethyl acetate (30 mL). The combined organic phases were washed with brine (20 mL), dried with magnesium sulfate, filtered and evaporated to give a yellow oil (4.83 g). The crude product was chromatographed on silica (130 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), to give CTAF 6 (3.98 g, 90% yield) as a colorless oil.
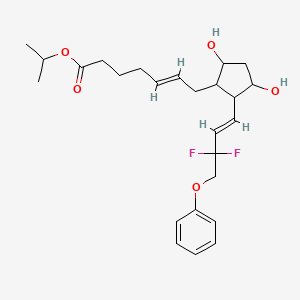
DRUG SPOTLIGHT-Afinitor (everolimus) , Novartis:

Afinitor (everolimus)
40-O-(2-hydroxyethyl)-rapamycin
Company: Novartis
Approval Status: Approved July 2012
Treatment Area: hormone receptor-positive, HER2-negative breast cancer
Everolimus is a derivative of Rapamycin (sirolimus), and works similarly to Rapamycin as an mTOR (mammalian target of rapamycin) inhibitor. It is currently used as an immunosuppressant to prevent rejection of organ transplants. In a similar fashion to other mTOR inhibitors Everolimus’ effect is solely on the mTORC1 protein and not on the mTORC2 protein.
159351-69-6 CAS NO
BRANDS
| Afinitor | Novartis |
| Certican | Novartis |
| VOTUBIA | Novartis |
| Zortress | Novartis |
Afinitor (everolimus), an inhibitor of mTOR (mammalian target of rapamycin), is an antineoplastic agent.
Afinitor is specifically approved for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane, after failure of treatment with letrozole or anastrozole.
Afinitor is supplied as a tablet for oral administration. The recommended dose of Afinitor for breast cancer is 10 mg, to be taken once daily, at the same time every day, either consistently with food or consistently without food.
FDA Approval
The FDA approval of Afinitor for the treatment of advanced hormone receptor-positive, HER2-negative breast cancer was based on a randomized, double-blind, multicenter study in 724 postmenopausal women with estrogen receptor-positive, HER 2/neu-negative advanced breast cancer with recurrence or progression following prior therapy with letrozole or anastrozole.
Everolimus is indicated for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane, after failure of treatment with letrozole or anastrozole. Indicated for the treatment of adult patients with progressive neuroendocrine tumors of pancreatic origin (PNET) with unresectable, locally advanced or metastatic disease. Indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) after failure of treatment with sunitinib or sorafenib. Indicated for the treatment of adult patients with renal angiomyolipoma and tuberous sclerosis complex (TSC), not requiring immediate surgery. Indicated in pediatric and adult patients with tuberous sclerosis complex (TSC) for the treatment of subependymal giant cell astrocytoma (SEGA) that requires therapeutic intervention but cannot be curatively resected.
Everolimus (RAD-001) is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors for use in a number of cancers.
It is marketed by Novartis under the tradenames Zortress (USA) and Certican (Europe and other countries) in transplantation medicine, and Afinitor in oncology.
AFINITOR (everolimus), an inhibitor of mTOR, is an antineoplastic agent.
The chemical name of everolimus is (1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18- dihydroxy-12-{(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl}-19,30-dimethoxy15,17,21,23,29,35-hexamethyl-11,36-dioxa-4-aza-tricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20pentaone.
The molecular formula is C53H83NO14 and the molecular weight is 958.2. The structural formula is:
 |
AFINITOR Tablets are supplied for oral administration and contain 2.5 mg, 5 mg, 7.5 mg, or 10 mg of everolimus. The tablets also contain anhydrous lactose, butylated hydroxytoluene, crospovidone, hypromellose, lactose monohydrate, and magnesium stearate as inactive ingredients.
AFINITOR DISPERZ (everolimus tablets for oral suspension) is supplied for oral administration and contains 2 mg, 3 mg, or 5 mg of everolimus. The tablets for oral suspension also contain butylated hydroxytoluene, colloidal silicon dioxide, crospovidone, hypromellose, lactose monohydrate, magnesium stearate, mannitol, and microcrystalline cellulose as inactive ingredients.
- R.N Formica Jra, K.M Lorberb, A.L Friedmanb, M.J Biaa, F Lakkisa, J.D Smitha, M.I Lorber (March 2004). “The evolving experience using everolimus in clinical transplantation”. Elsevier 36 (2): S495–S499.
- “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 2009-03-30. Retrieved April 6, 2009.
- “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 2010-04-22. Retrieved April 26, 2010.
- “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 Nov 2010.
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- “US FDA approves Novartis drug Afinitor for breast cancer”. 20 Jul 2012.
PATENTS
|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 6440990 | 1993-09-24 | 2013-09-24 |
| Canada | 2145383 | 2004-11-16 | 2013-09-24 |
| Canada | 2225960 | 2004-05-11 | 2016-07-12 |
| United States | 7297703 | 1999-12-06 | 2019-12-06 |
|
10-28-2011
|
METHODS OF TREATMENT
|
|
|
1-21-2011
|
ANTI-IGF1R
|
|
|
1-14-2011
|
HISTONE H2AX (HH2AX) BIOMARKER FOR FTI SENSITIVITY
|
|
|
3-24-2010
|
Thermal treatment of a drug eluting implantable medical device
|
|
|
1-13-2010
|
Therapeutic phosphonate compounds
|
|
|
10-21-2009
|
Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants
|
|
|
10-16-2009
|
Heparin Prodrugs and Drug Delivery Stents Formed Therefrom
|
|
|
9-11-2009
|
PHOSPHONATE COMPOUNDS HAVING IMMUNO-MODULATORY ACTIVITY
|
|
|
12-31-2008
|
Phosphonate compounds having immuno-modulatory activity
|
|
|
10-8-2008
|
Anti-inflammatory phosphonate compounds
|
|
6-27-2008
|
Genes Involved in Neurodegenerative Conditions
|
|
|
10-24-2007
|
Fluid treatment of a polymeric coating on an implantable medical device
|
|
|
7-11-2007
|
Oxepane isomer of 42-O-(2-hydroxy)ethyl-rapamycin
|
|
|
2-9-2007
|
40-O-(2-hydroxy)ethyl-rapamycin coated stent
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
9-8-2006
|
Anti-inflammatory phosphonate compounds
|
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO2007135397A1 * | May 18, 2007 | Nov 29, 2007 | Christoph Beckmann | 36 -des (3 -methoxy-4 -hydroxycyclohexyl) 36 – (3 -hydroxycycloheptyl) derivatives of rapamycin for the treatment of cancer and other disorders |
| EP0663916A1 | Sep 24, 1993 | Jul 26, 1995 | Novartis AG | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5665772 | Sep 24, 1993 | Sep 9, 1997 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US20030125800 | Apr 24, 2002 | Jul 3, 2003 | Shulze John E. | Drug-delivery endovascular stent and method for treating restenosis |
…………………………………….
Rapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:

See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.

(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis

Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):

According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.

(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).

Only 8% yield of the corresponding compound (4B)

and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
Xolair gains final NICE approval

NICE is recommending Novartis’ asthma drug Xolair as a cost effective treatment for young children and adults with a severe form of the condition in England.
Novartis’ Xolair (omalizumab) is now recommended as an option for treating severe, persistent confirmed allergic immunoglobulin E (IgE)-mediated asthma in people aged six years and older.
read more at http://www.pharmatimes.com/Article/13-04-24/Xolair_gains_final_NICE_approval.aspx
Omalizumab (trade name Xolair, Roche/Genentech and Novartis) is a humanized antibody approved for patients 12 years and older with moderate to severe allergic asthmain the United States and with severe, persistent allergic asthma in many other countries. It has also been approved for pediatric patients 6 to 11 years old with severe, persistent allergic asthma in the European Union. Omalizumab’s cost is high, ranging from $6,000 to $24,000 a year and hence omalizumab is mainly prescribed for patients with severe, persistent asthma, which cannot be controlled even with high doses of corticosteroids.
Omalizumab is a recombinant DNA-derived humanized IgG1k monoclonal antibody that selectively binds to free human immunoglobulin E (IgE) in the blood and interstitial fluid and to membrane-bound form of IgE (mIgE) on the surface of mIgE-expressing B lymphocytes. Unlike an ordinary anti-IgE antibody, omalizumab does not bind to IgE that is already bound by the high affinity IgE receptor (FcεRI) on the surface of mast cells,basophils, and antigen-presenting dendritic cells. IgE is commonly involved in type I hypersensitivity, which manifests the most prevalent allergic diseases. It has been estimated that as high as 20 to 40% of the populations who live a western lifestyle in economically advanced countries are affected by allergy and seek medical help.[1] While allergy occurs more frequently in individuals with higher serum IgE levels, such a correlation is only statistical and not absolute. Some allergic individuals have very low serum IgE, and some people with very high IgE have no allergic problems.
Xolair received approval by the U.S. Food and Drug Administration (FDA) in 2003 for treating patients 12 years and older with moderate to severe allergic asthma. It has also received approval in many other countries for treating patients 12 years and older with severe, persistent allergic asthma. Xolair was approved by the European Union in 2009 for treating patients 6 to 12 years old with severe, persistent allergic asthma. Thus, the primary use of Xolair is for patients mostly with severe, persistent allergic asthma, uncontrollable with oral or injectable corticosteroids.[2] The efficacy is more evident among severe asthmatics than among those with moderately severe disease. The response rates among treated severe “allergic” asthma patients are 60-80% or higher, probably depending on the patient screening procedures used by the various clinical groups of different specialties. Because 30-40% of adult asthma cases are not related to allergy and unresponsive to Xolair, a reliable way to identify treatable patients has been a subject of considerable research interest. The primary benefits for the responding patients are reduced numbers of exacerbations, improved lung function, reduced numbers of emergency visits to the doctors, reduced days of hospitalization, and increased quality of life measurements. The other major benefit is that most responding patients can reduce or spare entirely the use of corticosteroids, which cause multiple serious side effects, when used at high doses for extended periods.
Due to the requirement for long-term administration and hence the high cost of a Xolair treatment regimen, and to the concern over long-term safety, Xolair treatment is not yet very common, especially in developing countries where medical funds are relatively scarce. Another barrier to Xolair’s wide use is its injectable dosage form, which requires the patient to visit a physician’s office or clinic every 2 to 4 weeks during treatment. In August 2010, the National Institute for Clinical Excellence (NICE) in the United Kingdom ruled that Xolair should not be prescribed on the National Health Service (NHS) to children under 12, causing widespread condemnation from asthma charities.[3] NICE concluded that the high costs of the compound, over £250 per vial, did not represent a sufficiently high increase in quality of life. Additionally, as IgE could be a natural defense against parasitic diseases, treatment is usually not recommended when living in environments where the presence of parasites is common.
The website clinicaltrials.gov reveals that 102 clinical trials on omalizumab on various clinical indications have been finished or are in progress as of June 23, 2012. Among those more than 70 are multi-center, placebo-controlled phase II or III trials.[4] The tested indications are in the areas of allergic asthma, perennial and seasonal allergic rhinitis, peanut allergy, latex allergy, atopic dermatitis,chronic urticaria, idiopathic anaphylaxis, mastocytosis, eosinophilic gastroenteritis, nasal polyposis, and others. The relatively recent clinical trial results, which indicate that omalizumab has excellent effects on patients with recalcitrant, antihistamine-resistant chronic idiopathic urticaria, including those cases of autoimmune cause,[5][6] have generated much excitement among dermatologists as well as patients affected by chronic urticaria. Omalizumab has also been studied in combination with allergen-based specificimmunotherapy (allergy shots) for the purpose of reducing anaphylactic reactions when receiving allergen immunizations and of accelerating immunization schedule and dosing, so as to achieve therapeutic effects in shorter treatment periods and in broader patient populations.[7][8][9]
While data from additional clinical trials are required for omalizumab to be reviewed by governmental regulatory bodies for approval for any of the new indications, the data from finished trials thus far indicate that omalizumab is efficacious and safe for treating various IgE-mediated allergic or non-allergic diseases
- http://www.aaaai.org/about-the-aaaai/newsroom/allergy-statistics.aspx
- Davydov L (January 2005). “Omalizumab (Xolair) for treatment of asthma”. Am Fam Physician 71 (2): 341–2. PMID 15686303.
- “Asthma drug ruling ‘nonsensical'”. BBC News. August 12, 2010.
- http://clinicaltrials.gov/ct2/results?term=omalizumab
- Saini S, Rosen KE, Hsieh HJ, Wong DA, Conner E, Kaplan A, Spector S, Maurer M (September 2011). “A randomized, placebo-controlled, dose-ranging study of single-dose omalizumab in patients with H1-antihistamine-refractory chronic idiopathic urticaria”. J. Allergy Clin. Immunol. 128 (3): 567–73.e1. doi:10.1016/j.jaci.2011.06.010. PMID 21762974.
- Kaplan AP, Joseph K, Maykut RJ, Geba GP, Zeldin RK (September 2008). “Treatment of chronic autoimmune urticaria with omalizumab”. J. Allergy Clin. Immunol. 122 (3): 569–73. doi:10.1016/j.jaci.2008.07.006. PMID 18774392.
- Casale TB, Busse WW, Kline JN, Ballas ZK, Moss MH, Townley RG, Mokhtarani M, Seyfert-Margolis V, Asare A, Bateman K, Deniz Y (January 2006). “Omalizumab pretreatment decreases acute reactions after rush immunotherapy for ragweed-induced seasonal allergic rhinitis”. J. Allergy Clin. Immunol. 117 (1): 134–40. doi:10.1016/j.jaci.2005.09.036. PMID 16387596.
- Kopp MV, Hamelmann E, Zielen S, Kamin W, Bergmann KC, Sieder C, Stenglein S, Seyfried S, Wahn U; DUAL study group (October 2008). “Combination of omalizumab and specific immunotherapy is superior to immunotherapy in patients with seasonal allergic rhinoconjunctivitis and co-morbid seasonal allergic asthma”. Clin Exp Allergy 39 (2): 271–9. doi:10.1111/j.1365-2222.2008.03121.x.PMID 19016798.
- Massanari M, Nelson H, Casale T, Busse W, Kianifard F, Geba GP, Zeldin RK (February 2010). “Effect of pretreatment with omalizumab on the tolerability of specific immunotherapy in allergic asthma”. J. Allergy Clin. Immunol. 125 (2): 383–9.doi:10.1016/j.jaci.2009.11.022. PMID 20159249.
Merck’s Lambrolizumab Designated As A Breakthrough Therapy By FDA
Lambrolizumab
STRUCTURAL FORMULA of Lambrolizumab
Heavy chain
QVQLVQSGVE VKKPGASVKV SCKASGYTFT NYYMYWVRQA PGQGLEWMGG 50
INPSNGGTNF NEKFKNRVTL TTDSSTTTAY MELKSLQFDD TAVYYCARRD 100
YRFDMGFDYW GQGTTVTVSS ASTKGPSVFP LAPCSRSTSE STAALGCLVK 150
DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTKT 200
YTCNVDHKPS NTKVDKRVES KYGPPCPPCP APEFLGGPSV FLFPPKPKDT 250
LMISRTPEVT CVVVDVSQED PEVQFNWYVD GVEVHNAKTK PREEQFNSTY 300
RVVSVLTVLH QDWLNGKEYK CKVSNKGLPS SIEKTISKAK GQPREPQVYT 350
LPPSQEEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS 400
DGSFFLYSRL TVDKSRWQEG NVFSCSVMHE ALHNHYTQKS LSLSLGK 447
Light chain
EIVLTQSPAT LSLSPGERAT LSCRASKGVS TSGYSYLHWY QQKPGQAPRL 50′
LIYLASYLES GVPARFSGSG SGTDFTLTIS SLEPEDFAVY YCQHSRDLPL 100′
TFGGGTKVEI KRTVAAPSVF IFPPSDEQLK SGTASVVCLL NNFYPREAKV 150′
QWKVDNALQS GNSQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV 200′
THQGLSSPVT KSFNRGEC 218′
Disulfide bridges
22-96 22”-96” 23′-92′ 23”’-92”’ 134-218′ 134”-218”’ 138′-198′ 138”’-198”’
147-203 147”-203” 226-226” 229-229” 261-321 261”-321” 367-425 367”-425”
Glycosylation sites (N)
Asn-297 Asn-297”
MOLECULAR FORMULA C6504H10004N1716O2036S46 (peptide)
MOLECULAR WEIGHT 146.3 kDa (peptide)
TRADEMARK None as yet
SPONSOR Merck Sharp & Dohme Corp.
CAS REGISTRY NUMBER 1374853-91-4
THERAPEUTIC CLAIM Antineoplastic, immunomodulatory, immunotherapy
CHEMICAL NAMES
1. Immunoglobulin G4, anti-(human protein PDCD1 (programmed cell death 1))
(human-Mus musculus monoclonal heavy chain), disulfide with human-Mus
musculus monoclonal light chain, dimer
2. Immunoglobulin G4, anti-(human programmed cell death 1); humanized
mouse monoclonal [228-L-proline(H10
-S>P)]γ4 heavy chain (134-218′)-disulfide
with humanized mouse monoclonal κ light chain dimer (226-226”:229-229”)-
bisdisulfide
04/24/2013
Merck announced that the U.S. Food and Drug Administration (FDA) has designated lambrolizumab (MK-3475) as a Breakthrough Therapy for the treatment of patients with advanced melanoma. Lambrolizumab is Merck’s investigational antibody therapy targeting the programmed death receptor (PD-1) that is currently being evaluated for the treatment of patients with advanced melanoma, and other tumor types.
Merck & Co. Inc., also known as MSD outisdeUS and Canada, a provider of health solutions through prescription medicines, vaccines, biologic therapies, animal health, and consumer care products, Wednesday announced that the US Food and Drug Administration or FDA has designated its investigational antibody therapy, Lambrolizumab or MK-3475 as a Breakthrough Therapy for the treatment of patients with advanced melanoma, and other tumor types.
The Food and Drug Administration Safety and Innovation Act includes a provision that allows sponsors to request that an investigational drug be designated as a Breakthrough Therapy.
Lambrolizumab is targeting Programmed Death receptor or PD-1 that is currently being evaluated for the treatment of patients with advanced melanoma, and other tumor types. Advanced melanoma accounts for more than 80 percent of skin cancer-related deaths and one to two percent of all cancer deaths in the United States.
Gary Gilliland, senior vice president and oncology franchise head of Merck Research Laboratories stated, “”The FDA’s decision to place lambrolizumab in a category that may enable expedited development and review is an important milestone for Merck as we advance ongoing programs in multiple cancer indications.”
FDA spokeswoman Sandy Walsh noted that “the concept behind ‘breakthrough’ is that, with increased communication, FDA will work with new drug developers to help design efficient ways to study the safety and effectiveness of their drug.” The agency indicated that it has received 45 requests for breakthrough status designation, with 11 requests approved and 18 requests rejected. Other drugs granted breakthrough status include: Johnson & Johnson and Pharmacyclics’ ibrutinib for mantle cell lymphoma and other cancers; Novartis’ LDK378 for certain patients with non-small-cell lung cancer; and Pfizer’s palbociclib for breast cancer.
The Merck drug is an antibody designed to help the body’s immune system go after cancer cells. Lambrolizumab, also known as MK-3475, specifically targets the “programmed death” 1 receptor, or PD-1, which cancer cells can exploit to escape destruction by the immune system. Bristol-Myers Squibb Co. also is developing an anti-PD-1 drug, nivolumab, that is in Phase 3 testing.

image credit http://anewmerckreviewed.wordpress.com/
DRUG SPOTLIGHT –CUBICIN, DAPTOMYCIN
Daptomycin

N-decanoyl-L-tryptophyl-L-asparaginyl-L-aspartyl-L-threonylglycyl-
L-ornithyl-L-aspartyl-D-alanyl-L-aspartylglycyl-D-seryl-threo-3-methyl-L-glutamyl-3-anthraniloyl-L-alanine[egr]1-lactone
Daptomycin is a lipopeptide antibiotic used in the treatment of systemic and life-threatening infections caused by Gram-positive organisms. It is a naturally occurring compound found in the soil saprotroph Streptomyces roseosporus. Its distinct mechanism of action makes it useful in treating infections caused by multi-resistant bacteria. It is marketed in the United States under the trade name Cubicin by Cubist Pharmaceuticals.

The compound LY 146032 was discovered by researchers at Eli Lilly and Company in the late 1980s.LY 146032 showed promise in Phase I/II clinical trials for treatment of infection caused by Gram-positive organisms. Lilly ceased development because high-dose therapy was associated with adverse effects on skeletal muscle, including myalgia and potential myositis.
The rights to LY 146032 were acquired by Cubist Pharmaceuticals in 1997, which following U.S. Food and Drug Administration (FDA) approval in September 2003 for use in people older than 18 years began marketing the drug under the trade name CUBICIN. Cubicin is marketed in the EU and in several other countries by Novartis following its purchase of Chiron Corporation, previous licensee.

Daptomycin has a distinct mechanism of action, disrupting multiple aspects of bacterial cell membrane function. It appears to bind to the membrane and cause rapid depolarization, resulting in a loss of membrane potential leading to inhibition of protein, DNA and RNA synthesis, which results in bacterial cell death
US HERBS- FIGHT CANCER WITH BROCCOLI

An example of the superfoods is the broccoli. Broccoli belongs to the cruciferous group of vegetables. Naturally, broccoli is an an excellent source of vitamins, minerals and fiber. These are elements from Nature that have been extensively studied.
Epidemiological studies provideevidence that the consumption of this vegetable protects against cancer. The protection against cancer is mainly derived from altering estrogen metabolism and antioxidant properties, enhancing detoxification, decreasing carcinogen coumpound activation, slowing tumor growth and inducing cancer cell apoptosis (death). Such attributes qualify broccoli as a superfood.
American Institute of Cancer Research estimates that a daily intake of three servings would potentially reduce cancer rates by 20%.
Broccoli contains certain chemicals that may reduce the risk of colorectal or other cancers, although it is not clear which individual compounds may be responsible for the protective effects. While research in this area continues, the best advice at this time to reduce cancer risk is to eat a wide variety of vegetables. It is reasonable to include broccoli as part of a balanced diet.
roccoli has been around for more than 2,000 years but has only been commercially grown in the United States since the 1920s. Today, more than 90% of the broccoli harvested in the United States comes from California, although it is also grown in other parts of the country.
About 2 decades ago, researchers first suggested a possible link between diets high in cruciferous vegetables (a group of plants including cauliflower, cabbage, broccoli, and Brussels sprouts)) and a lower risk of cancer. However, it was not until the 1990s that certain chemicals found in broccoli were identified as possible cancer-preventing compounds. In 1997, a study was published that noted broccoli sprouts had higher levels of one of these compounds than mature broccoli.
Broccoli is a plant in the cabbage family, whose large flower head is used as a vegetable. The word broccoli, from the Italian plural of broccolo, refers to “the flowering top of a cabbage”. Broccoli is usually boiled or steamed but may be eaten raw and has become popular as a raw vegetable in hors d’œuvre trays. The leaves may also be eaten.
Broccoli is classified in the Italica cultivar group of the species Brassica oleracea. Broccoli has large flower heads, usually green in color, arranged in a tree-like structure on branchessprouting from a thick, edible stalk. The mass of flower heads is surrounded by leaves. Broccoli most closely resembles cauliflower, which is a different cultivar group of the same species.
Broccoli was derived from cultivated leafy cole crops in the Northern Mediterranean in about the 6th century BCE. Since the Roman Empire, broccoli has been considered a uniquely valuable food among Italians. Broccoli was brought to England from Antwerp in the mid-18th century by Peter Scheemakers. Broccoli was first introduced to the United States by Italian immigrants but did not become widely known there until the 1920s
Broccoli is high in vitamin C and dietary fiber; it also contains multiple nutrients with potent anti-cancer properties, such as diindolylmethane and small amounts of selenium. A single serving provides more than 30 mg of vitamin C and a half-cup provides 52 mg of vitamin C. The 3,3′-Diindolylmethane found in broccoli is a potent modulator of theinnate immune response system with anti-viral, anti-bacterial and anti-cancer activity. Broccoli also contains the compound glucoraphanin, which can be processed into an anti-cancer compound sulforaphane, though the benefits of broccoli are greatly reduced if the vegetable is boiled. Broccoli is also an excellent source of indole-3-carbinol, a chemical which boosts DNA repair in cells and appears to block the growth of cancer cells.
Boiling broccoli reduces the levels of suspected anti-carcinogenic compounds, such as sulforaphane, with losses of 20–30% after five minutes, 40–50% after ten minutes, and 77% after thirty minutes. However, other preparation methods such as steaming,microwaving, and stir frying had no significant effect on the compounds.
Broccoli has the highest levels of carotenoids in the brassica family.[17] It is particularly rich in lutein and also provides a modest amount of beta-carotene.
A high intake of broccoli has been found to reduce the risk of aggressive prostate cancer. Broccoli consumption may also help prevent heart disease.
Broccoli sprouts are often suggested for their health benefits
| Nutritional value per 100 g (3.5 oz) | |
|---|---|
| Energy | 141 kJ (34 kcal) |
| Carbohydrates | 6.64 g |
| – Sugars | 1.7 g |
| – Dietary fiber | 2.6 g |
| Fat | 0.37 g |
| Protein | 2.82 g |
| Water | 89.3 g |
| Vitamin A equiv. | 31 μg (4%) |
| – beta-carotene | 361 μg (3%) |
| – lutein and zeaxanthin | 1403 μg |
| Thiamine (vit. B1) | 0.071 mg (6%) |
| Riboflavin (vit. B2) | 0.117 mg (10%) |
| Niacin (vit. B3) | 0.639 mg (4%) |
| Pantothenic acid (B5) | 0.573 mg (11%) |
| Vitamin B6 | 0.175 mg (13%) |
| Folate (vit. B9) | 63 μg (16%) |
| Vitamin C | 89.2 mg (107%) |
| Vitamin E | 0.78 mg (5%) |
| Vitamin K | 101.6 μg (97%) |
| Calcium | 47 mg (5%) |
| Iron | 0.73 mg (6%) |
| Magnesium | 21 mg (6%) |
| Manganese | 0.21 mg (10%) |
| Phosphorus | 66 mg (9%) |
| Potassium | 316 mg (7%) |
| Zinc | 0.41 mg (4%) |
| Link to USDA Database entry Percentages are relative to US recommendations for adults. Source: USDA Nutrient Database
Broccoli is considered a good source of nutrients because it is rich in vitamin C, carotenoids (vitamin A-like substances), fiber, calcium, and folate. Broccoli is also a source of many substances called phytochemicals, or plant chemicals, that may have anticancer properties. For example, broccoli contains several compounds called isothiocyanates, including sulforaphane and indole-3-carbinol (I3C), which have been touted as possible anti-cancer agents in recent years. Early studies have shown these substances may act as anti-oxidants and may boost detoxifying enzymes in the body. Some studies have also suggested they may alter the levels of estrogen in the body, which might affect breast cancer risk.The chemical composition of broccoli and other cruciferous vegetables is complex, which makes it hard to determine which compound or combination of compounds may provide protection against cancer. Eating a wide variety of plant-based foods may be the best way to get the necessary components.Some researchers suggest that small amounts of broccoli sprouts may protect against the risk of cancer as effectively as much larger amounts of the mature vegetable. We are not aware of any clinical studies that have been done in humans to verify this claim.Another substance in broccoli, indole-3-carbinol (I3C), seems to alter estrogen levels and may also raise levels of protective enzymes in the body. Several studies of cancer cells growing in laboratory dishes or flasks have shown it may slow or stop the growth of breast, prostate, and other cancer cells. Some early studies in animals have shown similar results. Small studies in humans have found it may prevent the development of pre-cancerous growths in the cervix, as well as growths called papillomas in the throat. Again, larger studies are needed to find out what benefits I3C may have against cancer. THE MOLECULES The active molecules are Indole-3-carbinol (1H-indol-3-ylmethanol IUPAC name) OR I3C and isothiocyanates (mostly sulforaphane: 1-Isothiocyanato-4-methylsulfinylbutane). Indole-3-carbinol has a indole with a hydroxymethyl group that represents the hydrophilic group. Sulfurofane is a isothiocyanate, it means that it has a –N=C=S chemical group, formed by substituting sulfur for oxygen in the isocyanate group, bounded to a big alkyl chain containing a sulfinyl S=O group. Both indole-3-carbinol and sulphoraphane derive from glucosinolates. Glucosinolates are a class of organic compounds that contain sulfur and nitrogen and are derived from glucose and an amino acid. They occur as secondary metabolites of almost all plants of the order Brassicales. GENERAL EFFECTS ON HEALTH I3C has been shown to have a chemopreventive action on several human cancers. The first and greatest effects concern breast and cervical estrogen-dependant cancer. Later, many researches managed to relate I3C with the prevention from colon, lung and prostate cancer too. The micronutrient indole-3-carbinol: implications for disease and chemoprevention, 2000 Sulforaphane, like I3C, is useful against many types of cancer. Moreover, it has antimicrobial properties, as it appears to help eradicate Helicobacter Pylori from the stomach. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer |
|


DURECT Announces Submission of New Drug Application for POSIDUR™ (SABER®-Bupivacaine)

Bupivacaine
DURECT Announces Submission of New Drug Application for POSIDUR™ (SABER®-Bupivacaine)
“We’re extremely pleased to submit this NDA for POSIDUR to the FDA. If approved by the FDA, POSIDUR will provide a non-opioid alternative treatment option for post-surgical pain,” James E. Brown, D.V.M., President and CEO of DURECT, stated, “Treating post-surgical pain with a true long-acting local anesthetic has the potential benefit of reducing the need for opioids and their associated systemic side effects that can prolong the time to recovery and result in extended hospital stays.”
We expect that the FDA will notify us whether our NDA submission has been accepted for filing within 74 days of submission, which the FDA bases on their initial 60-day review of the completeness of our application. If accepted for filing, the FDA would be expected to assign a PDUFA date of 10 months after the submission.
About POSIDUR
POSIDUR is a post-operative pain relief depot that utilizes DURECT’s patented SABER®technology to deliver bupivacaine to provide up to three days of pain relief after surgery. We are in discussions with potential partners regarding licensing development and commercialization rights to POSIDUR, for which we hold worldwide rights.
About DURECT Corporation
DURECT is a specialty pharmaceutical company developing innovative drugs for pain and other chronic diseases, with late-stage development programs including REMOXY®, POSIDUR™, ELADUR®, and TRANSDUR®-Sufentanil. DURECT’s proprietary oral, transdermal and injectable depot delivery technologies enable new indications and superior clinical/commercial attributes such as abuse deterrence, improved convenience, compliance, efficacy and safety for small molecule and biologic drugs. For more information, please visit www.durect.com.
DRUG SPOTLIGHT- Pemetrexed

Pemetrexed
(2S)-2-{[4-[2-(2-amino-4-oxo-1,7-dihydro
pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino}
pentanedioic acid
N-[4-[2-(2-amino-4,7-dihydro-4-oxo-3H-pyrrolo[2,3-d]-pyrimidin-5-yl)ethyl] benzoyl]-L-glutamic acid or N-[4-[2-(2-amino-4,7-dihydro-4-oxo-1 H-pyrrolo [2,3-d]-pyrimidin-5-yl)ethyl] benzoyl]-L-glutamic acid
GENERIC LICENSING NEWSLETTER TODAY 23 APRIL 2013 REPORTED, SEE LINK BELOW
http://www.leadformix.com/ef1/preview_campaign.php?lf1=775434470d357512625317e6516156
 |
PEMETREXED
Pemetrexed is a chemotherapy drug used in the treatment of pleural mesothelioma as well as non-small cell lung cancer.Used in combination with cisplatin for the treatment of malignant pleural mesothelioma in adults whose disease is unresectable or who otherwise are not candidates for potentially curative surgery. Also used as a monotherapy for the treatment of locally advanced or metastatic non-small cell lung cancer (NSCLC) after prior chemotherapy.Click here to contact Logenex about this product.
|
Pemetrexed (brand name Alimta) is a chemotherapy drug manufactured and marketed by Eli Lilly and Company. Its indications are the treatment of pleural mesothelioma andnon-small cell lung cancer.
The molecular structure of pemetrexed was developed by Edward C. Taylor at Princeton University and clinically developed by Indianapolis based drug maker, Eli Lilly and Company in 2004.
 PEMETREXED
PEMETREXED
Pemetrexed is chemically similar to folic acid and is in the class of chemotherapy drugs called folate antimetabolites. It works by inhibiting three enzymes used in purine andpyrimidine synthesis—thymidylate synthase (TS), dihydrofolate reductase (DHFR), andglycinamide ribonucleotide formyltransferase[1][2] (GARFT). By inhibiting the formation of precursor purine and pyrimidine nucleotides, pemetrexed prevents the formation of DNAand RNA, which are required for the growth and survival of both normal cells and cancer cells.
Pemetrexed disodium is chemically described as L-Glutamic acid, N-[4-[2- (2-amino-4,7-dihydro-4-oxo-1 H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]- disodium salt heptahydrate, represented by the chemical structure of Formula (I).

Formula I
Pemetrexed is an anti-folate anti-neoplastic agent that exerts its action by disrupting folate-dependent metabolic processes essential for cell replication. It is believed to work by inhibiting three enzymes that are required in purine and pyrimidine biosynthesis — thymidylate synthase (TS), dihydrofolate reductase (DHFR), and glycinamide ribonucleotide formyl transferase (GARFT). Pemetrexed is available in the market under the brand name ALIMTA®.
Taylor et al., in describe pemetrexed, its related compounds and pharmaceutically acceptable cation. Chelius et al., in WO 01/14379 A2 disclose pemetrexed disodium crystalline hydrate Form I and process for preparation thereof.
Chelius et al., in WO 01/62760 disclose pemetrexed disodium heptahydrate crystalline Form Il and process for the preparation thereof.
Journal of Organic Process Research & Development, Volume 3, 1999, page 184 describes a process for the preparation of pemetrexed diacid. Busolli et al., in WO200802141 1 disclose process for preparation of pharmaceutically acceptable salt of pemetrexed diacid.
Busolli et al., in WO2008021405A1 disclose seven crystalline forms of pemetrexed diacid designated as Form A, B, C, D, E, F, & G and processes for preparation thereof.
In February 2004, the Food and Drug Administration approved pemetrexed for treatment of malignant Pleural Mesothelioma, a type of tumor of the lining of the lung, in combination with cisplatin[3] for patients whose disease is either unresectable or who are not otherwise candidates for curative surgery.[4] In September 2008, the FDA granted approval as a first-line treatment, in combination with cisplatin, against locally-advanced and metastatic non-small cell lung cancer (NSCLC) in patients with non-squamous histology. A Phase III study showed benefits of maintenance use of pemetrexed for non-squamous NSCLC.Activity has been shown in malignant peritoneal mesothelioma.Trials are currently testing it against esophagus and other cancers.
MECHANISM

Pemetrexed is also recommended in combination with carboplatin for the first-line treatment of advanced non-small cell lung cancer.However, the relative efficacy or toxicity of pemetrexed-cisplatin versus pemetrexed-carboplatin has not been established beyond what is generally thought about cisplatin or carboplatin doublet drug therapy

In addition to the brand name Alimta, this drug is also marketed in India by Abbott Healthcare as Pleumet and by Cadila Healthcare asPemecad.
-
Pemetrexed disodium is a multitargeted antifolate agent approved as a single agent for the treatment of non-small cell lung cancer, and in combination with cisplatin for the treatment of patient with malignant pleural mesothelioma, under the trade name Alimta®.
Pemetrexed disodium is available in a number of crystalline forms. -
Barnett et al, Organic Process Research & Development, 1999, 3, 184-188 discloses synthesis and crystallization of pemetrexed disodium from water-ethanol. The product obtained by the process disclosed herein is the 2.5 hydrate of pemetrexed disodium.
-
United States patent number 7,138,521 discloses a crystalline heptahydrate form of pemetrexed disodium, which has enhanced stability when compared to the known 2.5 hydrate.
-
To date workers have concentrated on producing stable crystalline forms of pemetrexed disodium and there has been no disclosure of any non-crystalline form of this active.
-
We have now found a new form of pemetrexed disodium, which is an amorphous form, as characterized by powder X-ray diffraction. Surprisingly, we have found that it is possible to prepare an amorphous form of pemetrexed disodium and that this form is stable. The amorphous form of the invention is stable contrary to expectations. The amorphous form of pemetrexed disodium of the present invention is stable as it retains it’s amorphous character under a variety of storage conditions. The amorphous form of the present invention is particularly advantageously characterized by a bulk density in the range of 0.15 to 0.35 gm/ml.
N-[4-[2-(2-amino-4,7-dihydro-4-oxo-3H-pyrrolo[2,3-d]-pyrimidin-5-yl)ethyl] benzoyl]-L-glutamic acid or N-[4-[2-(2-amino-4,7-dihydro-4-oxo-1 H-pyrrolo [2,3-d]-pyrimidin-5-yl)ethyl] benzoyl]-L-glutamic acid (also known as
“Pemetrexed”)

R = H: Pemetrexed; I
R = Na: Pemetrexed Disodium; II is a known compound. Pemetrexed Disodium is an known anticancer agent. It is clinically active in several solid tumors and approved for treatment of malignant pleural mesothelioma (MPM) and metastatic non-small cell lung cancer (NSCLC). Pemetrexed Disodium is supplied as a sterile lyophilized powder for intravenous administration.
The compound of formula I including pharmaceutically salts thereof as well as a process for its preparation is at first and specifically disclosed in EP patent no. 0432677 B1. The preparation and isolation of Pemetrexed (compound of formula I) as its Disodium salt (compound of formula II) was described for the first time in WO patent no. 9916742 A1 and in Drugs of the future 1998, 23(5), 498-507 as well as by Charles J. Barnett et al. in Organic Process Research & Development, 1999, 3, 184-188 and by Peter Norman in Current Opinion in Investigational Drugs 2001 , 2(11 ), 1611-1622.
Detailed information about the crystalline form of Pemetrexed Disodium prepared according to the process as described above were not provided but it is reported by Charles J. Barnett et al. in Organic Process Research & Development, 1999, 3, 184-188 that the disodium salt II was obtained as a hygroscopic solid.
The first crystalline form of Pemetrexed Disodium has been described in WO patent no. 0114379 designated Disodium MTA Hydrate Form I (MTA = multi- targeted antifolate, disodium N-[4-[2-(2-amino-4,7-dihydro-4-oxo-3H- pyrrolo[2,3-d]-pyrimidin-5-yl)ethyl]benzoyl]-L-glutamic acid salt). The Disodium MTA Hydrate Form I obtained according to the Examples 2, 3 and 4 contained different amounts of water (Example 2: water = 9.1%; Example 3: water = 17.7%; Example 4: water = 11.7%). The Disodium MTA Hydrate Form I has a typical XRD pattern as shown in Figure 4 (the corresponding 2theta values have been calculated from the provided d-spacing values).
An improved crystalline form of Pemetrexed Disodium has been disclosed in WO patent no. 0162760. It is teached that Pemetrexed Disodium can exist in the form of a heptahydrate (Form II; theoretical amount of water: approx 21%) which is much more stable than the previously known 2.5 hydrate (Form I; theoretical amount of water: 8.7%). The Pemetrexed Disodium Heptahydrate Form (Form II) has a typical XRD pattern as shown in Figure 5 (the corresponding 2theta values have been calculated from the provided d- spacing values).
The Chinese patent no. 1778802 describes a hydrate or trihydrate form of Pemetrexed Disodium. The preparation of Pemetrexed Disodium hydrate or trihydrate includes crystallization from water and water soluble solvent. An overview of the X ray powder diffraction data for Pemetrexed Disodium Hydrate provided in Chinese patent no. 1778802 is shown in Figure 6.
The WO patent no. 2008124485 disclose besides crystalline Forms of the diacid Pemetrexed also amorphous Pemetrexed Disodium as well as a crystalline Form III thereof including a composition containing a major amount of amorphous Form and a minor amount of crystalline Form III of Pemetrexed Disodium. An overview of the X ray powder diffraction data for Pemetrexed Disodium crystalline Form 3 is shown in Figure 7.
EP patent application no. 2072518 disclose a stable amorphous form of Pemetrexed Disodium.
-
According to the more recent US 5,416,211 , which is incorporated herein by reference, pemetrexed can be synthesized from 4-[2-(2-amino-4,7-dihydro-4-oxo-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoic acid of formula 1, obtained from simple precursors, in accordance with the following Scheme 1:

-
This second method seems to be used also for the industrial preparation of the active ingredient. In fact, the same type of synthesis scheme is also described in C. J. Barnett, T. W. Wilson and M. E. Kobierski, Org. Proc. Res. & Develop., 1999, 3, 184-188, in which the experimental examples refer to a scale of the order of tens of kgs.





……………………….
Example 1 Preparation of crude pemetrexed disodium
[0023] N-[4-2-(2-Amino-4, 7-dihydro-4-oxo-
1 H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-L-glutamic Acid Diethyl Ester
4-Methylbenzenesulfonic Acid Salt and purified process water (PPW) (about 10 kg) are charged to a suitable vessel under nitrogen. The reactor is cooled to NMT 10Ό under nitrogen. Pre-cooled sodium hydroxide solution (about 1.5 kg )/PPW (about 11.4 kg) are added and the temperature is maintained at NMT 10Ό. The mixture is stirred at NMT 0 until the solid is dissolved. Pre-cooled isopropanol (about 62.8 kg) is added and the mixture temperature is maintained at NMT 5 . Pre-cooled 1 N hydrochloric acid in isopropanol is added to adjust the pH to 6.5 to 9.5, preferably between pH 7.5 to pH 8.5, at NMT 5 . The mixture is warmed to a room temperature (i.e., 15-30Ό, preferably 20-25″C) and stirred. The solids are filtered and washed with isopropanol/PPW. The wet cake is vacuum dried to provide crude pemetrexed disodium (about 2.30 kg).
Example 2 Purification of crude pemetrexed disodium to pemetrexed disodium
[0024] Crude pemetrexed disodium (about 2.1 kg) and PPW (about 23.3 kg) are charged under nitrogen to a suitable vessel at 15 to 30 . Isopropanol (about 28.3 kg) is added slowly to cloud point and stirred. Isopropanol (up to about 55 kg) is charged and stirred. The solids are filtered arid washed with isopropanol/PPW. The wet cake is vacuum dried to provide pemetrexed disodium (about 1.9 kg) (90% Yiled). 1 H NMR (D20): δ 7.51 (2H, d, J=8.0 Hz), 6.98 (2H, d, J=8.0 Hz), 6.12 (1 H, s), 4.26-4.23 (H, m), 3.60-3.54 (4H, m), 2.27-2.23 (2H, m), 2.13-2.08 (1 H, m), 2.00-1.94 (1 H, m)
HPLC
Example 7
HPLC Analysis method
-
Reagent: Water :milliQ,
Sodium perchlorate :AR Grade
Perchloric acid :AR Grade
Acetonitrile :J.T.Baker gradient
Trifluroacetic acid :AR Grade
Buffer solution: 6.1 g of sodium perchlorate into a 1000ml water. Adjust the pH to 3.0 (± 0.1) with perchloric acid.
Mobile phase A:
mixture of buffer and acetonitrile in the proportion of (90:10).
Mobile phase B:
mixture of buffer and acetonitrile in the proportion of (10 : 90).
Diluent -1 : mixture of water and acetonitrile in the ratio of 50 : 50.
Diluent -2: mixture of water and acetonitrile in the ratio of 90 : 10.
Standard Stock Solution:
Transfer accurately weighed 1.5 mg impurity-E RS and into a 200 ml volumetric flask. Dissolve in and dilute upto mark with diluent-1.
Blank solution
-
Add 10 ml diluent-2 and 50µl of 3% trifluro acetic acid to a 50 ml volumetric flask, and dilute upto mark with diluent-2.
System suitability solution
-
Transfer about 25 mg accurately weighed pemetrexed disodium sample in to a 50 ml volumetric flask. First add 10ml of diluent-2 and sonicate to dissolve the contents.Then add 50µl of 3% trifluro acetic acid (prepared in water) and add 5 ml of standard stock solution and dilute up to mark with diluent-2.
Sample preparation
-
Transfer about 25 mg accurately weighed pemetrexed disodium sample in to a 50 ml volumetric flask. First add 10ml of diluent-2 and sonicate to dissolve the contents.Then add 50µl of 3% trifluro acetic acid (prepared in water) and dilute up to mark with diluent-2 (500 µg/ml).
Chromatographic system :
-
Use a suitable high pressure liquid chromatography system equipped with Column: 250 mm x 4.6mm containing 5µ packing material (suggested column – Inertsil ODS 3V)
Detector: UV detector set to 240 nm
Cooler temp: 5°C.
Flow rate: about 1.5 ml/min. -
The system is also equipped to deliver the two phases in a programmed manner as shown in the following table :
Gradient programme :
-
[0082]
0 92 8 15 85 15 30 65 35 35 65 35 36 92 8 40 92 8
Procedure:
-
Inject 20µl of blank and system suitability solution into the chromatograph set to above conditions and record the chromatograms up to 40 min.
Calculate the resolution between pemetrexed disodium and impurity-E. The resolution should not be less than 3.0. Calculate the Number of theoretical plate and tailing factor for pemetrexed peak. Number of theoretical plate is NLT 4000 and tailing factor is NMT 2.0. -
Inject 20µl of test solution and calculate the chromatographic purity by area normalisation method.
……………………..
Synthetic Route for the Preparation of Pemetrexed Disodium
Starting from commercially available materials a novel synthetic route for the synthesis of Pemetrexed-IM8 (the dimethyl ester of Pemetrexed) was developed which was then used for the preparation of Pemetrexed Disodium (Scheme 1).


The current synthetic route for the preparation of Pemetrexed IM8 starts with an aldol-condensation reaction of Methyl-4-formylbenzoate (SM1) with 1,1-Dimethoxyacetone (SM2) to give Pemetrexed IM1a. As Pemetrexed IM1a irreversibly converts to its aldol-addition product Pemetrexed IM1b under reaction conditions the reaction mixture is directly submitted to hydrogenation (i.e. without isolation of Pemetrexed IM1a) over Pd/C to give Pemetrexed IM2. As under the hydrogenation conditions not only the double-bond of IM1a is hydrogenated but also some amount of Pemetrexed IM2 is converted to Pemetrexed IM3 (hydrogenation of the carbonyl function to the corresponding secondary alcohol) a solution of NaBH4 is added to the reaction mixture to ensure complete conversion to Pemetrexed IM3. The Pd-catalyst is removed by filtration and the reaction mixture is extracted with toluene. The combined organic layers are evaporated to give crude Pemetrexed IM3 as oil. This oil is dissolved in THF and the alcohol functionality is converted to a mesylate using MsCl and NEt3. The salts are removed by filtration, glacial acetic acid is added and THF is removed by distillation. Upon addition of water Pemetrexed IM4 crystallizes and is isolated by filtration. The dried Pemetrexed IM4 is dissolved in glacial acetic acid and gaseous HCl is added to cleave the dimethoxy acetale and liberate the aldehyde functionality of Pemetrexed IM5. Upon complete deprotection a solution of 2,6-diamino-4-hydroxypyrimidine in aq. NaOH and acetonitrile is added. Upon complete conversion the crystallized Pemetrexed IM6 is isolated by filtration. The saponification of the methyl ester of Pemetrexed IM6 to Pemetrexed IM7 is done using aqueous NaOH. Upon addition of aq. HCl first the Na-salt of Pemetrexed IM7 crystallizes from the reaction mixture. The salt is isolated by filtration, purified by slurry in a mixture of MeOH and water and then converted to Pemetrexed IM7 by pH adjustment in water using aq. HCl. Dried Pemetrexed IM7 (water content not more than 6.0%) is dissolved in DMF, activated using 1,1-carbonyldiimidazolide (CDI) and then reacted with dimethyl-L-glutamate hydrochlorid to give, upon addition of water and filtration, crude Pemetrexed IM8. This intermediate is purified by tosylate salt formation, followed by recrystallization and liberation to give pure Pemetrexed IM8. Starting with the saponification of Pemetrexed IM8 the preparation of different solid forms of Pemetrexed Disodium can be achieved.
Methods For Preparing Pemetrexed Disodium Form IV and Investigation of its Stability
An overview on the possible transformations of Pemetrexed IM8 to Pemetrexed Disodium Form IV is shown in FIG. 20.
Description of Possible Routes for the Preparation of Pemetrexed Disodium Form IV Starting from Pemetrexed IM8
All routes start with saponification of Pemetrexed IM8 in water at IT=20° C. to 30° C. using 3.25 eq of NaOH. Upon complete conversion an aqueous solution of Pemetrexed Disodium with a pH of 13.0 to 13.5 is obtained. Starting from this mixture the desired route can be accessed by addition of HCl to adjust the pH to a certain value (depending on the route, FIG. 20).

Structures of Pemetrexed (Compound I), Pemetrexed Disodium (Compound II) and Pemetrexed Monosodium (Compound IV)
Surprisingly we found that the crucial feature of all successful transformations to Pemetrexed Disodium Form IV is the presence of Pemetrexed Monosodium during the transformation. Routes starting from pure Pemetrexed Disodium Heptahydrate, Pemetrexed Disodium 2.5 hydrate or Pemetrexed Disodium Form A in the presence of seeding crystals of Pemetrexed Disodium Form IV were not successful and resulted in isolation of Pemetrexed Disodium Form A. The same transformations, if carried out in the presence of 0.15 eq of Pemetrexed Monosodium were successful and after addition of 0.15 eq NaOH allowed the isolation of pure Pemetrexed Disodium Form IV. The use of 0.15 eq HCl instead of 0.15 eq Pemetrexed Monosodium under the same conditions resulted in isolation of Pemetrexed Disodium Form A without any Pemetrexed Disodium Form IV. Transformations via isolated Pemetrexed Monosodium gave complete transformation to Pemetrexed Disodium Form IV if either 1.0 eq NaOH were added slowly (over a period of several hours) to Pemetrexed Monosodium or if initially only 0.85 eq of NaOH (based on Pemetrexed Monosodium) were added, followed by 0.15 eq once the transformation to Pemetrexed Disodium Form IV was complete. Very fast addition (<1 min) of 1.0 eq NaOH resulted in formation of Pemetrexed Disodium Form A containing traces of Pemetrexed Disodium Heptahydrate.
Starting from Pemetrexed (compound I) the transformation to Pemetrexed Disodium Form IV would be possible if initially 1.85 eq of NaOH were added followed by 0.15 eq once the transformation was complete. Alternatively, 2.0 eq of NaOH could be added over a long period of time (i.e several hours) to achieve formation of Pemetrexed Form IV. Fast addition (<1 min) of 2.0 eq of NaOH is assumed to result in formation of Pemetrexed Disodium Form A. All these experiments show the presence of Pemetrexed Monosodium to be crucial during the transformations. This presence can be achieved by either addition of catalytic amounts of Pemetrexed Monosodium to Pemetrexed Disodium, by slow addition over several hours of NaOH to Pemetrexed Monosodium or by portionwise addition of NaOH to Pemetrexed Monosodium. Addition of catalytic amounts of HCl to Pemetrexed Disodium (in situ preparation of Pemetrexed Monosodium) failed to give Pemetrexed Disodium Form IV.
Fast addition of NaOH to Pemetrexed Monosodium results in fast formation of Pemetrexed Disodium, thereby lacking the necessary catalytic amounts of Pemetrexed Monosodium to catalyze the transformation to Pemetrexed Disodium Form IV. EtOH as solvent and water content of EtOH were found to be crucial parameters for the transformation to Pemetrexed Disodium Form IV. So far the transformation has only been observed in EtOH containing 0-4% water (v/v). A water content>4% (v/v) results in formation of Pemetrexed Disodium Heptahydrate. Under the conditions used (EtOH containing 0-4% water (v/v)) both Pemetrexed Disodium Heptahydrate and Pemetrexed Disodium 2.5 hydrate are transformed to Pemetrexed Form A. Therefore the mechanism of the transformation to Pemetrexed Disodium Form IV is assumed to proceed via Pemetrexed Disodium Form A with Pemetrexed Monosodium acting as catalyst for the transformation.
Preparation of Pemetrexed Disodium Heptahydrate
a) Preparation of Pemetrexed Disodium Heptahydrate Starting from Pemetrexed IM8
Pemetrexed Disodium Heptahydrate was prepared by adjustment of the pH of the Pemetrexed Disodium solution after saponification from pH=13 to pH=8 using HCl followed by addition of EtOH (3 times the volume of water) to achieve crystallization. Precipitated Pemetrexed Disodium Heptahydrate was isolated by filtration, washed with a mixture of EtOH and water (4:1 v/v) followed by EtOH. The wet product was dried in vacuo at 200 mbar at 20° C. to 30° C. until water content of the dried product was 20.1% to 22.1%.
b) Conversion of Pemetrexed Disodium Form A to Pemetrexed Disodium Heptahydrate
To a suspension of Pemetrexed Disodium Form A in EtOH was added water until a mixture of EtOH containing 25% water (v/v) was obtained. The resulting suspension was stirred at 20° C. to 30° C. until conversion was complete according to PXRD. Pemetrexed Disodium Heptahydrate was isolated by filtration, washed with EtOH and dried in vacuo at 200 mbar at 20° C. to 30° C. until water content of the dried product was 20.1% to 22.1%.
………………………..

- REFERENCES
- McLeod, Howard L.; James Cassidy, Robert H. Powrie, David G. Priest, Mark A. Zorbas, Timothy W. Synold, Stephen Shibata, Darcy Spicer, Donald Bissett, Yazdi K. Pithavala, Mary A. Collier, Linda J. Paradiso, John D. Roberts (Jul-2000).“Pharmacokinetic and Pharmacodynamic Evaluation of the Glycinamide Ribonucleotide Formyltransferase Inhibitor AG2034”. Clinical Cancer Research 6 (7): 2677–84.PMID 10914709. More than one of
|work=and|journal=specified (help) - Avendano, Carmen; Menendez, J. Carlos (April 2008).Medicinal Chemistry of Anticancer Drugs. Amsterdam:Elsevier. p. 37. ISBN 0-444-52824-5.
- Manegold C (August 2003). “Pemetrexed (Alimta, MTA, multitargeted antifolate, LY231514) for malignant pleural mesothelioma”. Semin. Oncol. 30 (4 Suppl 10): 32–6.doi:10.1016/S0093-7754(03)00283-5. PMID 12917819.
- National Cancer Institute: FDA Approval for Pemetrexed Disodium
| US6090168 * | Oct 6, 1999 | Jul 18, 2000 | Eli Lilly And Company | Processes and intermediates useful to make antifolates |
| US7138521 | Feb 12, 2001 | Nov 21, 2006 | Eli Lilly And Company | Crystalline of N-[4-[2-(2-Amino-4,7-dihydro-4oxo-3H-pyrrolo[2,3-D]pyrimidin-5-YL)ethyl]benzoyl]-L-glutamic acid and process therefor |
| US20030216416 * | Feb 12, 2001 | Nov 20, 2003 | Chelius Erik Christopher | Novel crystalline of n-[4-[2-(2-amino-4,7-dihydro-4oxo-3h-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid and process therefor |
| WO2001014379A2 * | Aug 15, 2000 | Mar 1, 2001 | Erik Christopher Chelius | A novel crystalline form of disodium n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo[2,3-d]-pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid salt and processes therefor |
| WO2008021405A1 * | Aug 14, 2007 | Feb 21, 2008 | Sicor Inc | Crystalline forms of pemetrexed diacid and processes for the preparation thereof |
| WO2008124485A2 * | Apr 3, 2008 | Oct 16, 2008 | Reddys Lab Ltd Dr | Solid forms of pemetrexed |
FDA Approves Amitiza [lubiprostone] for Opioid-Induced Constipation

7-[(1R,3R,6R,7R)-3-(1,1-difluoropentyl)-3-hydroxy-
8-oxo-2-oxabicyclo[4.3.0]non-7-yl]heptanoic acid
Lubiprostone
136790-76-6 cas no
- FDA Approves Supplemental New Drug Application for AMITIZA® (lubiprostone), the First Oral Treatment for Opioid-induced Constipation in Adults with Chronic Non-Cancer Pain
Apr. 23, 2013– Sucampo Pharmaceuticals, Inc. and Takeda Pharmaceuticals U.S.A., Inc. announced today that the United States (U.S.) Food and Drug Administration (FDA) has approved Sucampo’s supplemental new drug application (sNDA) for Amitiza (lubiprostone) (24 mcg twice daily) as the first and only oral medication for the treatment of opioid-induced constipation (OIC) in adult patients with chronic, non-cancer pain. The effectiveness of Amitiza in the treatment of opioid-induced constipation in patients taking diphenylheptane opioids (e.g., methadone) has not been established.

AMITIZA®
This is the third indication for Amitiza, which is also approved in the U.S. for the treatment of chronic idiopathic constipation (CIC) in adults (24 mcg twice daily) and irritable bowel syndrome with constipation (IBS-C) in adult women (8 mcg twice daily). There are more than 200 million prescriptions for opioid use in the U.S. annually, and a substantial number of these prescriptions are for non-cancer chronic pain. Scientific literature indicates that approximately 40-80% of patients taking opioids chronically for non-cancer pain report constipation.
Opioid-based medicines are widely used in the management of chronic pain, with OIC being a common adverse effect of chronic opioid use. Some patients may discontinue opioid therapy and thereby endure pain rather than suffer from the constipation the opioids cause.
Amitiza is a specific activator of ClC-2 chloride channels in the intestinal epithelium. Amitiza, via activation of apical ClC-2 channels in the intestinal epithelium, bypasses the antisecretory action of opiates.
This approval is based on results from Phase 3, well-controlled studies of 12 weeks’ duration in patients taking opioids (among them morphine, oxycodone, and fentanyl) chronically for non-cancer pain, as well as a long-term, open-label safety study, which provides additional support for use in this population. Two of the Phase 3 studies met their overall efficacy endpoint, while a third Phase 3 study did not.
OIC is a common adverse effect of chronic opioid use. Binding of opioids to peripheral opioid receptors in the gastrointestinal tract results in absorption of electrolytes, such as chloride, and subsequent reduction in small intestinal fluid. In addition, activation of enteric opioid receptors results in abnormal GI motility. Together, these processes result in OIC, which is characterized by infrequent and incomplete evacuation of stool, hard stool consistency, and straining associated with bowel movements.
|

Amitiza (lubiprostone) capsules are indicated for the treatment of chronic idiopathic constipation (CIC) in adults and opioid-induced constipation (OIC) in adults with chronic, non-cancer pain (24 mcg twice daily). The effectiveness in patients with OIC taking diphenylheptane opioids (e.g., methadone) has not been established. Amitiza is also indicated for irritable bowel syndrome with constipation (IBS-C) in women > 18 years old (8 mcg twice daily).
Lubiprostone (rINN, marketed under the trade name Amitiza) is a medicationused in the management of chronic idiopathic constipation and irritable bowel syndrome. It was approved by the U.S. Food and Drug Administration (FDA) for this purpose on 31 January 2006.
Lubiprostone is used for the treatment of chronic constipation of unknown cause and irritable bowel syndrome associated with constipation.
As of 20 July 2006, Lubiprostone had not been studied in children. There is current research underway to determine the efficacy in postoperative bowel dysfunction, and opioid-induced bowel dysfunction

- “amitiza”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- Sobrera, L. A.; Castaner, J. (2004). Drugs of the Future 29 (4): 336.
PHASE 2 -TetraLogic’s BIRINAPANT for treatment of acute myeloid leukemia, pancreatic cancer, or ovarian cancer
BIRINAPANT, Apoptosis inhibitor
(2S,2’S)-N,N’-((2S,2’S)-((3S,3’S,5R,5’R)-5,5′-((6,6′-difluoro-1H,1’H-[2,2′-biindole]-3,3′-diyl)bis(methylene))bis(3-hydroxypyrrolidine-5,1-diyl))bis(1-oxobutane-2,1-diyl))bis(2-(methylamino)propanamide)
1260251-31-7 cas no
Birinapant is an antagonist of XIAP and cIAP1 with Kd value of 45 nM and <1 nM, respectively.
US20110003877,WO 2013049350 A1
| Molecular Weight: | 806.94 |
| Birinapant Formula: | C42H56F2N8O6 |
Birinapant, also known as TL32711, is a synthetic small molecule and peptido mimetic of second mitochondrial-derived activator of caspases (SMAC) and inhibitor of IAP (Inhibitor of Apoptosis Protein) family proteins, with potential antineoplastic activity. As a SMAC mimetic and IAP antagonist, TL32711 binds to and inhibits the activity of IAPs, such as X chromosome-linked IAP (XIAP) and cellular IAPs 1 and 2. Since IAPs shield cancer cells from the apoptosis process, this agent may restore and promote the induction of apoptosis through apoptotic signaling pathways in cancer cells. IAPs are overexpressed by many cancer cell types and suppress apoptosis by binding and inhibiting active caspases-3, -7 and -9 via their baculoviral lAP repeat (BIR) domains
Birinapant is currently in Phase II clinical trials in patients with acute myeloid leukemia, pancreatic cancer, or ovarian cancer. Although these trials don’t have a control group, the emerging data are promising, TetraLogic chief executive officer John M. Gill told C&EN. (Early-stage cancer clinical trials are commonly run without placebo groups.) The birinapant trials show preliminary evidence both that the drug is having the desired effect and that this effect is associated with signs of clinical activity. Given these results, the company plans to launch randomized Phase II studies early in 2014
Birinapant, also called TL32711, is a potent antagonist for XIAP with Kd value of 45 nM and cIAP1 with Kd value <1 nM [1].
Birinapant not only binds to the isolated BIR3 domains of cIAP1, cIAP2, XIAP but the single BIR domain of ML-IAP with high affinity and degrades TRAF2-bound cIAP1 and cIAP2 rapidly accordingly inhibiting the activation of TNF-mediated NF- kB. Additionally, birinapantcan promote the formation of caspase-8: RIPK1 complex in response to TNF stimulation, which result in downstream caspasesactivation [4].
In the inorganic SUM149- and SUM190-derived cells, which with differential XIAP expression (SUM149 wtXIAP, SUM190 shXIAP) and other high cIAP1/2 but low XIAP binding affinity bivalent Smac mimetic GT13402, XIAP inhibition are needed for increasing TRAIL potency. Opposite, single agent efficacy of Birinapant is owing to pan-IAP antagonism. Rapid cIAP1 degradation was caused by birinapant, as well as NF-κB activation, PARP cleavage andcaspase activation. While combined withTNF-α, showing strong combination activity, the combination was more effective than individual. The response in spheroid models was conserved, whereas in vivo birinapant inhibited tumor growth without adding TNF-α in vitro to resistant cell lines. In a parental cell line, TNF-αcombined withbirinapantinhibited the growth of a melanoma cell line with acquired resistance to the same extent of BRAF inhibition [1, 2].
Drug treatment increased the mean [18F]ICMT-11 tumor uptake with a peak at 24 hours for CPA (40 mg/kg; AUC40-60: 8.04 ± 1.33 and 16.05 ± 3.35 %ID/mL × min at baseline and 24 hours, respectively) and 6 hours for birinapant (15 mg/kg; AUC40-60: 20.29 ± 0.82 and 31.07 ± 5.66 %ID/mL × min, at baseline and 6 hours, respectively). Voxel-based spatiotemporal analysis of tumor-intrinsic heterogeneity showed that [18F] ICMT-11 could detect the discrete pockets of caspase-3 activation. Caspase-3 activation that measured ex vivo associated with the increased tumor [18F] ICMT-11, and early radiotracer uptake predicted apoptosis, distinct from the glucose metabolism with [18F] fluorodeoxyglucose-PET, which depicted the continuous loss of cell viability [3].

 Guangyao Yu, Yijun Deng, Gurpreet Singh Kapoor, Condon, Susan Rippin, Martin Graham, Angeline Mufalli, Jennifer Burns, Martin Seipel, Eric Neiman, Thomas Haimowitz, Christopher Benetatos, Yasuhiro Mitsuuchi, Srinivas Chunduru Not pictured: Divya Goel")
Birinapant was designed to reinstate cancer cells’ ability to die. Many cancers that are resistant to conventional chemotherapy drugs have defects in the cell death pathway known as apoptosis. The human body uses apoptosis every day to clear away abnormal or unwanted cells.
Apoptosis is a tightly regulated process, Condon explains, with a network of proteins that activate or block the process. TetraLogic’s target is a family of proteins called the Inhibitor of Apoptosis proteins. As their name suggests, these proteins block apoptosis. They interfere with protease enzymes that carry out cellular dismantling.
TetraLogic’s aim is to lift that blockade to restart apoptosis in tumors. Many tumors have excesses of the apoptosis inhibitor proteins relative to normal cells, so targeting this process has the potential to be less toxic to normal cells compared with conventional chemotherapy.
It turns out nature has a way of negating the inhibitor proteins’ actions—a protein known as Smac. TetraLogic’s founders demonstrated that only a tiny portion of Smac is necessary to keep the inhibitor proteins at bay—the four amino acids at the protein’s N-terminus. “Once you can get a protein down to a tetrapeptide,” about the size of a small-molecule drug, “you start getting a lot of interest from the pharma community,” Condon told C&EN.
Peptides fall apart in the body too quickly to be drugs, so Condon’s team worked with molecular mimics of the Smac tetrapeptide. Their biggest advance was realizing that combining two copies of their tetrapeptide mimics into one molecule made their compounds highly effective at reinstating apoptosis in cancer cell lines. Many proteins in the apoptosis pathway function as dimers, so using these so-called bivalent mimics against them makes sense, Condon said.
However, several of the bivalent compounds were associated with pronounced body weight loss in mice. Condon’s team eventually learned that replacing a branched side chain on their peptide mimics and adding a hydroxyl group to a proline residue improved the tolerability for the animals without impacting the antitumor effect. With that, birinapant was born.
In mice, birinapant shrank tumors. The compound has been in clinical trials since 2009, both on its own and in combination with other chemotherapy drugs such as irinotecan and gemcitabine. On the basis of other biochemical work on the apoptosis pathway, TetraLogic thinks these drugs could act in synergy with birinapant to treat cancer, Condon said.
Birinapant is currently in Phase II clinical trials in patients with acute myeloid leukemia, pancreatic cancer, or ovarian cancer. Although these trials don’t have a control group, the emerging data are promising, TetraLogic chief executive officer John M. Gill told C&EN. (Early-stage cancer clinical trials are commonly run without placebo groups.) The birinapant trials show preliminary evidence both that the drug is having the desired effect and that this effect is associated with signs of clinical activity. Given these results, the company plans to launch randomized Phase II studies early in 2014.
1H NMR











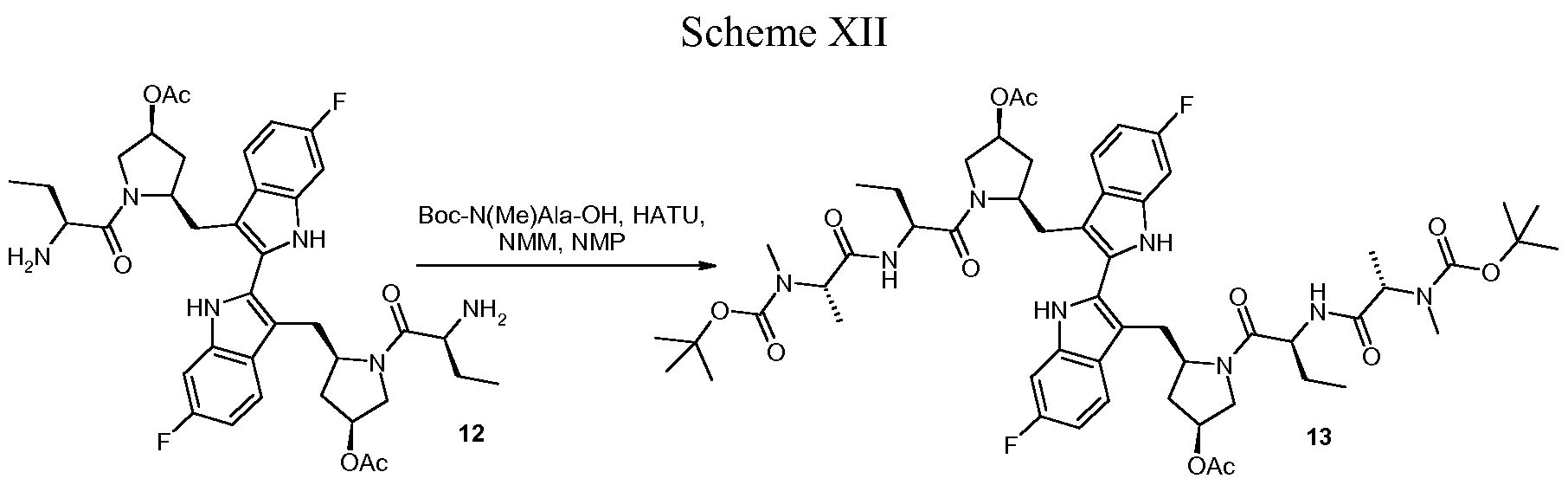


spectral data
1H NMR (300 MHz, CDC13): 511.74 (s, 2H), 8.27 (d, J= 8.7 Hz, 2H), 7.71 (dd, J= 5.4, 8.4 Hz, 2H), 7.55 (dd, J =2.4, 9.6 Hz, 2H), 6.88 (ddd, J= 2.4, 9.3, 9.3 Hz, 2H), 4.62-4.78 (m, 4H), 4.43 (dd, J= 9.3, 9.9 Hz, 2H), 4.03 (dd, J= 4.8, 11.4 Hz, 2H), 3.80 (d, J = 11.4 Hz, 2H), 3.66 (dd, J= 2.7, 14.4 Hz, 2H), 3.53 (dd, J = 11.4, 14.4 Hz, 2H), 3.11 (q, J = 6.9 Hz, 2H), 2.56 (s, 6H), 2.45 (m, 2H), 2.19 (d, J= 14.4 Hz, 2H), 1.76-2.10 (m, 6H), 1.59 (br s, 2H), 1.39 (d, J= 6.9 Hz, 6H), 1.22-1.38 (m, 2H), 1.07 (t, J = 7.2 Hz, 6H) ppm;
13C NMR (75 MHz, d6– DMSO): 5175.2, 172.8, 161.6, 158.5, 137.3, 137.2, 128.4, 128.3, 126.4, 120.8, 120.6, 109.4, 108.7, 108.4, 98.4, 98.0, 70.8, 60.2, 59.9, 56.6, 51.8, 36.4, 35.3, 28.3, 25.6, 20.0, 10.6 ppm.
Mass spectrum (ESI), m/z 807.5 [(M)+; calcd for C42H56F2N806: 806.9].
Paper
Click to access op5b00390_si_001.pdf
Click to access op5b00390_si_001.pdf
Process Development and Synthesis of Birinapant: Large Scale Preparation and Acid-Mediated Dimerization of the Key Indole Intermediate

Birinapant/TL32711 (1) is a novel bivalent antagonist of the inhibitor of apoptosis (IAP) family of proteins which is currently in clinical development for the treatment of cancer and hepatitis B virus (HBV) infection. In this report, we present a detailed description of the 1 drug substance synthesis used to support our ongoing clinical studies. Key transformations in this process included the development of a scalable, high-yielding route to acyl indole 14 as well as a two-step dimerization/oxidation of indole 19 that afforded biindole 21 in excellent yield and purity (70% yield, 2 steps; >95 area% purity by HPLC analysis). In addition, partial defluorination of 21 was observed following hydrogen-mediated benzyloxycarbonyl (Cbz) protective group removal which was obviated by the use of HBr/HOAc for this transformation. The use of commercially available amino acid derivatives afforded related impurities which proved difficult to purge in subsequent steps. Thus, defining the impurity specification for these reagents was critical to providing 1 drug substance of >99 area% chemical purity. Using this process, we have successfully prepared 1 drug substance multiple times on >500-g-scale in support of our clinical development program.
References:
1.Allensworth JL, Sauer S, Lyerly HK, et al. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-a-independent mechanism. Breast Cancer Research, 2013, 137:359-371.
2.Krepler C, Chunduru SK, Halloran MB, et al. The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clinical Cancer Research, 2013, 19 (7): 1784-1794.
3.Nguyen QD, Lavdas I, Gubbins J, et al. Temporal and Spatial Evolution of Therapy-Induced Tumor Apoptosis Detected by Caspase-3–Selective Molecular Imaging. Clinical Cancer Research, 2013, 19 (14): 3914-3924.
4.Benetatos CA, Mitsuuchi Y, Burns JM, et al. Birinapant (TL32711), a Bivalent SMAC Mimetic, Targets TRAF2-Associated cIAPs, Abrogates TNF-Induced NF-kB Activation, and Is Active in Patient-Derived Xenograft Models. 2014, 13(4):867-879.

