
Home » Posts tagged 'solid tumors'
Tag Archives: solid tumors
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pepinemab, VX 15
(Heavy chain)
QVQLVQSGAE VKKPGSSVKV SCKASGYSFS DYYMHWVRQA PGQGLEWMGQ INPTTGGASY
NQKFKGKATI TVDKSTSTAY MELSSLRSED TAVYYCARYY YGRHFDVWGQ GTTVTVSSAS
TKGPSVFPLA PCSRSTSEST AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
YSLSSVVTVP SSSLGTKTYT CNVDHKPSNT KVDKRVESKY GPPCPPCPAP EFLGGPSVFL
FPPKPKDTLM ISRTPEVTCV VVDVSQEDPE VQFNWYVDGV EVHNAKTKPR EEQFNSTYRV
VSVLTVLHQD WLNGKEYKCK VSNKGLPSSI EKTISKAKGQ PREPQVYTLP PSQEEMTKNQ
VSLTCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG SFFLYSRLTV DKSRWQEGNV
FSCSVMHEAL HNHYTQKSLS LSLGK
(Light chain)
DIVMTQSPDS LAVSLGERAT INCKASQSVD YDGDSYMNWY QQKPGQPPKL LIYAASNLES
GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSNEDPY TFGQGTKLEI KRTVAAPSVF
IFPPSDEQLK SGTASVVCLL NNFYPREAKV QWKVDNALQS GNSQESVTEQ DSKDSTYSLS
STLTLSKADY EKHKVYACEV THQGLSSPVT KSFNRGEC
(Disulfide bridge: H22-H96, H132-L218, H145-H201, H224-H’224, H227-H’227, H259-H319, H365-H423, H’22-H’96, H’132-L’218, H’145-H’201, H’259-H’319, H’365-H’423, L23-L92, L138-L198, L’23-L’92, L’138-L’198)
Pepinemab
VX15/2503
Antineoplastic, Anti-human semaphorin 4D antibody
Monoclonal antibody
Treatment of solid tumors, multiple sclerosis and Huntington’s disease
| Formula | C6442H9910N1702O2052S48 |
|---|---|
| MOL WGT | 145481.0022 |
- Moab VX15/2503
- Pepinemab
- UNII-BPZ4A29SYE
- VX-15
- VX15
- VX15/2503
| Product name | Pepinemab Biosimilar – Anti-SEMA4D mAb – Research Grade |
|---|---|
| Source | CAS 2097151-87-4 |
| Species | Chimeric,Humanized |
| Expression system | Mammalian cells |
- OriginatorVaccinex
- DeveloperBristol-Myers Squibb; Children’s Oncology Group; Emory University; Merck KGaA; National Cancer Institute (USA); Teva Pharmaceutical Industries; UCLAs Jonsson Comprehensive Cancer Center; Vaccinex
- ClassAntibodies; Antidementias; Antineoplastics; Immunotherapies; Monoclonal antibodies
- Mechanism of ActionCD100 antigen inhibitors
- Orphan Drug StatusYes – Huntington’s disease
- New Molecular EntityYes
- Phase IIHuntington’s disease
- Phase I/IIAlzheimer’s disease; Non-small cell lung cancer; Osteosarcoma; Solid tumours; Squamous cell cancer
- Phase IColorectal cancer; Malignant melanoma; Pancreatic cancer
- No development reportedMultiple sclerosis
- 22 May 2021Pepinemab is still in phase I trials for Colorectal cancer and Pancreatic cancer in USA (NCT03373188)
- 17 May 2021Phase-I/II clinical trials in Squamous cell cancer (Combination therapy, Late-stage disease, Metastatic disease, Recurrent, Second-line therapy or greater) in USA (IV) (NCT04815720)
- 17 May 2021Vaccinex plans a phase I/II trial for Alzheimer’s disease (In volunteers), in H2 2021
Semaphorin 4D (SEMA4D) plays a role in multiple cellular processes that contribute to the pathophysiology of neuroinflammatory/neurodegenerative diseases. SEMA4D is, therefore, a uniquely promising target for therapeutic development.
Pepinemab is a novel monoclonal antibody that blocks the activity of SEMA4D, and preclinical testing has demonstrated the beneficial effects of anti-SEMA4D treatment in a variety of neurodegenerative disease models. Vaccinex is committed to the development of this potentially important antibody that has the potential to help people with different neurodegenerative disorders that share common mechanisms of pathology.
Note: Pepinemab (VX15/2503) is an investigational drug currently in clinical studies. It has not been demonstrated to be safe and effective for any disease indication. There is no guarantee that pepinemab (VX15/2503) will be approved for the treatment of any disease by the U.S. Food and Drug Administration or by any other health authority worldwide.
////////////////////Pepinemab, VX15/2503, vx 15, Antineoplastic, Anti-human semaphorin 4D antibody, Monoclonal antibody, solid tumors, multiple sclerosis, Huntington’s disease, PEPTIDES
NEW DRUG APPROVALS
ONE TIME
$10.00
CC-90010
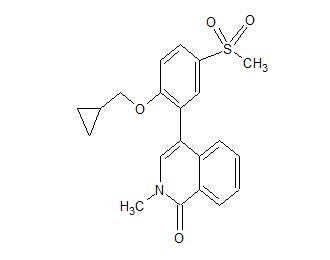
CC-90010
C21 H21 N O4 S, 383.46
CAS 1706738-98-8
1(2H)-Isoquinolinone, 4-[2-(cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]-2-methyl-
- 4-[2-(Cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]-2-methyl-1(2H)-isoquinolinone
- 4-[2-(Cyclopropylmethoxy)-5-(methanesulfonyl)phenyl]-2-methylisoquinolin-1(2H)-one
- 4-[2-(Cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one
Quanticel Pharmaceuticals Inc, Michael John BennettJuan Manuel BetancortAmogh BoloorStephen W. KaldorJeffrey Alan StaffordJames Marvin Veal
![]()
Celgene (now a wholly owned subsidiary of Bristol-Myers Squibb ) , following its acquisition of Quanticel , is developing CC-90010, an oral inhibitor of BET (bromodomain and extraterminal) proteins, for the potential treatment of solid tumors and non-Hodgkin’s lymphoma. In August 2019, a phase I trial for diffuse astrocytoma, grade III anaplastic astrocytoma and recurrent glioblastoma was planned
PATENT
WO2018075796 claiming solid composition comprising a bromodomain inhibitor, preferably 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one in crystalline form A.
PATENT
WO2015058160 (compound 89, page 103).

Example 89: 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
Step 1 : 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one
[00344] A suspension of 4-bromo-2-methylisoquinolin-l-one (100 mg, 0.42 mmol), bis(pinacolato)diboron (214 mg, 0.84 mmol), Pd(dppf)Cl2 (31 mg, 0.04 mmol) and potassium acetate (104 mg, 1.05 mmol) in dioxane (2 mL) under nitrogen was warmed up to 90 °C for 135 minutes. It was then cooled down to room temperature and diluted with ethyl acetate (8 mL). The mixture was washed with aqueous saturated solution of NaHC03 (8 mL) and brine (8 mL). The organic phase was separated, dried over Na2S04, filtered and concentrated under reduced pressure. The residue was purifed by normal phase column chromatography (10-90% EtOAc/Hexanes) to give the title compound (44 mg, 37%). 1H NMR (CDC13, 400 MHz) δ 8.43 (d, J = 7.9 Hz, 1 H), 8.40 (dd, J = 8.2 Hz, 0.9 Hz, 1 H), 7.68 (s, 1 H), 7.65 (ddd, J = 8.2, 8.2, 1.1 Hz, 1 H), 7.46 (t, J = 7.5 Hz, 1 H), 3.63 (s, 3H), 1.38 (s, 12H). LCMS (M+H)+ 286. Step 2: 4-[2-(cyclopropylmethox -5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00345] The title compound was prepared in a manner similar to Example 18, step 3, substituting 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene for 4-bromo-2-methylisoquinolin-l(2H)-one and 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one for N-benzyl-2-methoxy-5-(tetramethyl-l,3,2-dioxaborolan-2-yl)benzamide. 1H NMR (DMSO-d6, 400 MHz) δ 0.09 (m, 2 H), 0.29 (m, 1H), 0.35 (m, 1H),
0.94 (m, 1H), 3.22 (s, 3H), 3.57 (s, 3H), 3.95 (m, 2H), 7.16 (d, J = 7.9 Hz, 1H), 7.37 (d, J =
8.8 Hz, 1H), 7.53 (m, 2H), 7.65 (t, J = 7.6 Hz, 1H), 7.81 (d, J = 2.4 Hz, 1H), 7.97 (dd, J = 8.8,
2.4 Hz, 1H), 8.30 (d, J = 8.1 Hz, 1H). LCMS (M+H)+ 384.
[00346] Alternatively, 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one can be prepared as described below.
Step 1 : 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one
[00347] A mixture of 4-bromo-2-methylisoquinolin-l-one (8.0 g, 33.6 mmol),
bis(pinacolato)diboron (17.1 g, 67.2 mmol), KOAc (6.6 g, 67.2 mmol), Pd2(dba)3 (3.1 g, 3.36 mmol) and X-Phos (1.6 g, 3.36 mmol) in anhydrous dioxane (200 mL) was stirred at 60 °C for 12 h. The reaction mixture was concentrated and the residue was purified by column chromatography on silica gel (PE : EA = 15 : 1) to give the title compound (6.0 g, 62 %) as a solid.
Step 2: 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00348] The title compound from Step 1 (5.0 g, 17.5 mmol), 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene (6.4 g, 21 mmol), K3PO4 (9.3 g, 43.9 mmol) and Pd(dppf)Cl2 (1.4 g, 1.75 mmol) in a dioxane/water (100 mL / 10 mL) mixture were stirred at 60 °C for 12 hrs. The reaction mixture was concentrated under reduced pressure and the residue was purified by column chromatography on silica gel (EA : DCM = 1 : 4).
Appropriate fractions were combined and concentrated under reduce pressure. The resultant solid was recrystallized from DCM / MTBE (1 : 1, 50 mL) to give the title compound (4.0 g, 60 %) as a white solid. 1H NMR: (CDC13, 400 MHz) δ 8.51 (dd, Ji = 8.0 Hz, J2 = 0.8 Hz, 1 H), 7.98 (dd, Ji = 8.4 Hz, J2 = 2.4 Hz, 1 H), 7.86 (d, J = 2.4 Hz, 1 H), 7.53 (m, 2 H), 7.16 (d, J = 7.6 Hz, 1 H), 7.10 (m, 2 H), 3.88 (m, 2 H), 3.66 (s, 3 H), 3.09 (s, 3 H), 1.02-0.98 (m, 1 H), 0.44-0.38 (m, 2 H), 0.11-0.09 (m, 2 H). LCMS: 384.1 (M+H)+
Patent
WO-2020023438
A process for preparing bromodomain inhibitor, particularly 4-[2(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one (having HPLC purity of 99%; compound 1; (hereafter referred to as C-90010)) and its hydrates, solvates, prodrugs and salts comprising the reaction of a substituted 4-(methylsulfonyl)phenol compound with a quinoline derivative, followed by purification is claimed. Also claimed are novel intermediates of CC-90010 and their processes for preparation. Further claimed are novel crystalline form of CC-90010. CC-90010 is known and disclosed to be a bromodomain containing protein inhibitor, useful for treating cancer.
Scheme 10: Synthesis of Compound 1
[0090] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compound 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223g, l.OSmol). Agitation
was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum purged three times with nitrogen. The batch was heated to about 65 to about 75 °C (or any temperature in between and including these two values) and contents stirred for at least about 16 hours until reaction was complete by HPLC analysis. The batch was cooled to about 60 to about 70 °C (or any temperature in between and including these two values), agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at about 60 to about 70°C (or any temperature in between and including these two values). Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at about 60 to about 70°C (or any temperature in between and including these two values). After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at about 60 to about 70°C (or any temperature in between and including these two values). The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to about 60 to about 65°C (or any temperature in between and including these two values) and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at about 60 to about 65°C (or any temperature in between and including these two values). The slurry was cooled to about 15 to about 25°C (or any temperature in between and including these two values) over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at about 40°C with nitrogen purge. Yield: 139g of 1.
[0091] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox et al., Chem. Soc. Rev., 43: 412 (2014). Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used.
Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used.
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cb (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 fCC-900101 bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCh(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 (CC-90010! bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
33 -a
and Form 5. Supporting data (XRPD, DSC, photomicroscopy) for all three forms is provided in the examples below.
[0093] The stmcture of Compound 1 (CC-90010) is shown below:
Example 1: Synthesis of Compound 1
[0217] Synthesis of compound 1 was accomplished according to Scheme 1 below. Referring to Scheme 1, synthesis commenced with bromination of starting material 4-(methylsulfonyl)phenol 4, to produce compound 5. Compound 5 was O-alkylated with (bromomethyl)cyclopropane to produce compound 6. Boronate Compound 2 was then formed by borylation of Compound 6 with Pd catalyst and bis(pinacolato)diboron to produce transient Compound 7, which was subsequenctly treated with diethanolamine (DBA) to afford cross-coupling partner Compound 2. Cross-coupling partner Compound 3 was formed in one pot starting from commercially available Compound 8. Compound 8 was N-methylated and brominated to afford Compound 3. Compounds 2 and 3 were cross-coupled (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)) to afford the target compound 1.
Scheme 1: Synthesis of compound 1
1.1: Bromination of 4
[0218] The bromination of Compound 4 to produce Compound 5 itself is simple, however stopping at the mono-brominated Compound 5 was challenging. The bis-brominated Compound 5-a (see Scheme 2 below) is a particularly pernicious impurity as it couples downstream to form a di ffi cult-to-purge impurity.
Scheme 2: Bromination of Compound 4
[0219] The key to high purity with reasonable yield was to exploit the solubility differences of the starting material Compound 4 (46 mg/ml at 20 °C) and the product Compound 5 (8 mg/ml) in CH2CI2. These solubility differences are summarized in Table 3 below.
[0220] This solubility difference is exploited by performing the reaction at a high
concentration to drive Compound 5 out of solution once formed, thereby minimizing its ability to react further with the brominating reagent to form Compound 5-a diBr. The reaction is seeded with Compound 5 to initiate its crystallization.
[0221] In Fig. 22 (Conversion of Compound 4 to Compound 5: Effect of Sulfuric Acid) it can be seen that in the absence of acid the initial reaction to Compound 5 is rapid, however the conversion plateaus at about 30% Compound 5. The main side product was found to be the impurity Compound 5-a diBr (see Fig. 23: Conversion of Compound 5 and Compound 5-a diBr: No H2SO4). Addition of increasing amounts of sulfuric acid leads to a higher conversion to desired Compound 5.
[0222] Fig. 24 (Compound 4 to Compound 5 Reaction Profile: Portion-wise Addition of NBS, Seeding) depicts further reaction control. The portion-wise addition ofNBS after addition of catalytic sulfuric acid minimizes the temperature rise, and the addition of Compound 5 after an initial NBS charge promotes the reactive crystallization of Compound 5. After about 6 to 7 hours of reaction it can be seen that the major product is Compound 5, with only a small (<5%) of the di-brominated impurity formed. In contrast, in a reaction where Compound 4 and all of the NBS were charged followed by the addition of 4 volumes of methylene chloride, a rapid exotherm resulted and undesired Compound 5-a diBr was found to be the major product.
[0223] Thus, the reaction was run under a high concentration in CH2CI2 with a portion-wise solid addition of NBS (to control both availability of the electrophile and the exotherm). An end of reaction slurry sample typically showed not more than 5% of the starting material Compound 4 remaining. After filtration the crude cake was washed with cold CH2CI2 and the OkCk-washed filter cake contained not more than 0.5% by weight dibrominated Compound 5-a. It also contained a large amount of HPLC-silent succinimide.
[0224] The following procedure was carried out: Compound 4 (25g, 145mmol) followed by CH2CI2 (lOOmL) were added to a reaction vessel and agitated. The batch was adjusted to 17 °C to 23 °C. Sulfuric acid was charged (2.7mL, Slmmol) to the batch maintaining 17 °C to 23°C. The batch was stirred at 17 °C to 23 °C for 10 minutes to 20 minutes. The first portion of A-bromosuccimide (NBS) was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The second portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The batch was seeded with
Compound 5 (0.02wt) and stirred for ca. 30 min at 17 °C to 23 °C to induce crystallization.
[0225] The third portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23 °C and stirred for at least 30 min. NBS (6.5g, 36.5 mmol) was charged to the batch at 17 °C to 23 °C and stirred for at least 30 min. Additional CH2CI2 was charged (50mL) to the batch while maintaining 17 °C to 23 °C to aid in agitation and transfer for filtration. The batch was stirred at 17 °C to 23 °C until complete by HPLC analysis (~20 – 40 h). The product was collected by suction filtration. The filter cake was slurry washed with CH2CI2 (3 x 50mL) at 17 °C to 23 °C (target 20 °C). The filter cake was slurry washed with purified water (3.0vol) at 65 °C to 75 °C for 2 to 3 hours. Then, the filter cake was slurry washed with purified water (3 x 1.0 vol, 3 x 1.0 wt) at 17 °C to 23°C. The wet cake was dried under vacuum with nitrogen bleed at 60 °C. Yield: 27g 5 (74% molar) >97% by weight. ¾ NMR (500 MHz, de-DMSO) 8.01 (1H, d, 4J = 2.1 Hz, RO-Ar meta- H ), 7.76 (1H, dd, J = 8.6 and 4J = 2.1 Hz, RO-Ar meta-H ), 7.14 (1H, d, J = 8.6 Hz, RO-Ar ortho- H), 3.38 (1H, br s, OH), 3.20 (3H, s,
CHJ); MS (ES-) calc. 249/251; found 249/251. Melting point (MP): (DSC) 188 °C.
[0226] The above procedure allowed for the following modifications. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as acetonitrile, tetrahydrofuran, or 2- methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from about 2X vol to about 20 X vol (with respect to Compound 4). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 1.9 equiv. Bromination reagent addition: The brominating reagent can be added all at once, portion wise in about 2 to about 20 portions, or continuously. The addition times can vary from about 0 to about 72 hours. Temperature: Reaction temperatures from about 0 °C to about 40 °C could be used. Acids: Different acids can be envisioned, including benzenesulfonic acid, para-toluenesulfonic acid, triflic acid, hydrobromic acid, and trifluoroacetic acid. Isolation: Instead of directly filtering the product and washing with methylene chloride and water, at the end of reaction an organic solvent capable of dissolving Compound 5 could be charged, followed by an aqueous workup to remove succinimide, and addition of an antisolvent or solvent exchange to an appropriate solvent to crystallize Compound 4. Drying: A temperature range of about 10 to about 60 °C could be used for drying.
[0227] An alternative process to Compound 5 has also been developed. This process is advantageous in that it does not use a chlorinated solvent, and provides additional controls over the formation of the Compound 5-a dibromo impurity. See Oberhauser, T. J Org. Chem 1997, 62, 4504-4506. The process is as follows. Compound 4 (10 g, 58 mmol) and acetonitrile (100 ml) were charged to the reactor and agitated. The batch was cooled to -20 °C. Triflic acid (CF3SO3H or TfOH, 5.5 mL, 62 mmol) was charged while maintaining a batch temperature of -10 to -25 °C. N-bromosuccinimide was charged (NBS, 11.4 g, 64 mmol), stirred at -10 to -25 °C for 30 minutes, then warmed to 20 °C over 3 to 4 hours. Agitation was continued at 15 °C to 25 °C until reaction completion. If the reaction conversion plateaued before completion, the reaction was cooled to -5 to -15 °C, and additional NBS was added, the amount based off of unreacted starting material, followed by warming to 15 °C to 25 °C and reacting until complete.
[0228] After reaction completion, the batch was warmed to 40 °C to 50 °C and concentrated under reduced pressure to 40 mL. The batch was cooled to -5 °C to -15 °C and the resulting product solids were filtered off. The solids were slurry washed three times, each with 20 mL water, for at least 15 minutes. The final cake was dried at 50 °C to 60 °C under reduced pressure to furnish 10 g of 5 containing less than 0.1% MeCN, 0.07% water, and 0.1% triflic acid (TfOH) by weight.
[0229] Alternatives to the above procedure employing MeCN and TfOH are as follows. Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination Reagent Stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 2 equiv. Drying: A temperature range of about 10 °C to about 60 °C could be used for drying.
[0230] The impurity 5-a is was prepared and characterized as follows. 10 g of Compound 4 and sulfuric acid (35 mol%) were dissolved in MeOH (10 vol). The mixture was set to stir at 20 °C to 25 °C for 5-10 min and 2.0 equivalents of NBS were charged in one portion. The resulting yellow mixture was stirred for three days at 20-25 °C. The batch was concentrated under reduced pressure and the resulting solid was slurried in water at 95-100 °C for 3 hours. After a second overnight slurry in CH2CI2 at room temperature, the batch was filtered and dried to give a white solid 5-a (15.0 g, 78%). ¾ NMR (500 MHz, de-DMSO), 8.05 (2H, s, ArH), 3.40 (1H, br s, HO-Ar), 3.28 (3H, s, CH3); MS (ES‘) calc. 327/329/331; found
327/329/331; MP (DSC): 226 °C (onset 221 °C, 102 J/g); lit. 224-226 °C.
1.2: O-alkylation of 5 to produce 6
[0231] Compound 6 was prepared according to Scheme 7 below.
Scheme 7: O-alkylation of 5 to produce 6
[0232] Compound 5 (100 g, 398 mmol) and methyl ethyl ketone (MEK, 700 mL) were charged to the reaction vessel and agitated. Potassium carbonate (K2CO3, 325 mesh 82.56 g, 597 mmol) was then charged to the stirred reaction vessel at 15 °C to 25 °C.
Bromomethylcyclopropane (64.4 mL, 664 mmol) was charged to the reaction vessel over at least 1 hour, maintaining the temperature between 15 °C to 25 °C. MEK (200 mL) was added into the reactor and the reactor heated to 65 to 75 °C. The contents of the reaction vessel were stirred at 65 to 75°C for approximately 10 hours until reaction was complete by HPLC analysis. Water (3.0 vol, 3.0wt) was charged to the vessel maintaining the temperature at 65 to 75 °C. The batch was stirred at 65 to 75 °C. The phases were allowed to separate at 65°C to 75 °C and the lower aqueous phase was removed. Water (300 mL) was charged to the vessel maintaining the temperature at 65 °C to 75 °C. The batch was agitated for at least 10 minutes at 65 to 75 °C. The phases were allowed to separate at 65 °C to 75 °C and the lower aqueous phase was removed. The water wash was repeated once. The temperature was adjusted to 40 to 50°C. The mixture was concentrated to car. 500 mL under reduced pressure. The mixture was distilled under reduced pressure at up to 50 °C with MEK until the water content was <1.0% w/w. n-heptane (500mL) was charged to the vessel maintaining the temperature at 40 to 50 °C. The mixture was continuously distilled under vacuum with n-heptane (300mL), maintaining a 1L volume in the reaction vessel. Compound 6 seeds (0.0 lwt) were added at 40 to 50 °C. The mixture was continuously distilled under reduced pressure at up to 50 °C with n-heptane (300mL) while maintaining 1L volume in the reactor. The batch was cooled to 15 to 25 °C and aged for 2 hours. The product was collected by suction filtration. The filter cake was washed with a solution of 10% MEK in n-heptane (5vol) at 15 to 25°C. The filter cake was dried under reduced pressure at up to 40 °C under vacuum with nitrogen flow to afford 95g of 6. 1H NMR (500 MHz, de-DMSO) 8.07 (1H, d, 4J = 2.2 Hz, ArH), 7.86 (1H, d, J = 8.7 Hz, meta-ArH), 7.29 (1H, d, J = 8.8 Hz, ortho-AiK),
4.04 (2H, d, J = 6.9 Hz, OCH2CH), 3.21 (3H, s, CH3), 1.31-1.24 (1H, m, OCH), 0.62- 0.58 (2H, m, 2 x CHCHaHb), 0.40-0.37 (2H, m, 2 x CHC¾Hb); MS (ES+) calc. 305/307; found 305/307; MP: (DSC) 93 °C.
[0233] The following modifications of the above reaction, synthesis of 6 from 5, may be employed as well. Solvent: Different solvents could be used, for example acetone, methyl isobutyl ketone, ethyl acetate, isopropyl acetate, acetonitrile, or 2-methyl tetrahydrofuran. Reaction volume: Reaction volumes of 3 to 30 volumes with respect to 3 could be used. Base: Different inorganic bases, such as cesium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Also, organic bases, such as trimethylamine or diisopropyldiimide could be used. Base particle size: Different particle sizes of potassium carbonate from 325 mesh could be used. Reaction temperature: A lower temperature, such
as 50 °C could be used. A higher temperature, such as about 100 °C could be used. Any temperature above the boiling point of the solvent could be run in a pressure vessel.
Isolation: Different solvent ratios of MEK to n-heptane could be used. Different amounts of residual water can be left. Different amounts of seeds, from 0 to 50% could be used.
Seeding could take place later in the process and/or at a lower temperature. An un-seeded crystallization can be employed. A different isolation temperature, from 0 °C to 50 °C could be used. A different wash could be used, for example a different ratio of MEK to n-heptane. A different antisolvent from n-heptane could be used, such as hexane, pentane, or methyl tert-butyl ether. Alternatively, the batch could be solvent exchanged into a solvent where Compound 3 has a solubility of less than 100 mg/ml and isolated from this system. Drying: A temperature range of 10 to 60 °C could be used for drying.
[0234] Compound 10, shown below may also be formed as a result of O-alkylation of unreacted 4 present in product 5, or alternatively from or via a palladium mediated proteodesbromination or proteodesborylation in subsequent chemistry discussed in Example 1.3 below.
[0235] Preparation of methylsulfonylphenyl(cyclopropylmethyl) ether 10: Compound 4 (0.86 g, 5.0 mmol) and K2CO3 (1.04 g, 7.5 mmol) were slurried in acetone (17 mL, 20 vols). Cyclopropylmethyl bromide (0.73 mL, 7.5 mmol) was added in several small portions over ~1 minute and the reaction mixture heated to 50 °C for 48 hours, then cooled to 25 °C. Water (5.0 mL) was added with stirring and the acetone was evaporated on a rotary evaporator from which a fine white solid formed which was filtered off and returned to a vessel as a damp paste. A 1 : 1 mixture of MeOH/ water (8 mL) was added and heated to 40 °C with stirring. After 1 hour, the white solid was filtered off. Some residual solid was washed out with fresh water that was also rinsed through the cake, which was then isolated and left to air dry over the two days to give a dense white solid 10 (1.00 g, 88%). ¾ NMR (500 MHz, CDCb) 7.85
(2H, d, J = 8.8 Hz, RO-Ar ortho-H), 7.00 (2H, d, J = 8.8 Hz, RO-Ar meta- H), 3.87 (2H, d, J = 7.0 Hz, OCH2CH), 3.02 (3H, s, CHs), 1.34-1.23 (1H, m, OCH2CH), 0.72-0.60 (2H, m, 2 x CHCHflHb), 0.42-0.31 (2H, m, 2 x CHCH^.
1.3: Synthesis and Isolation Coupling Partner Boronic Ester 2
[0236] The final bond forming step to Compound 1 is a Suzuki-Miyaura coupling between Compounds 2 and 3, as shown in Scheme 3 below (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)). Early studies demonstrated that the boronic ester of the isoquinolinone Compound 3-a had poor physical attributes and solid phase stability (Kaila, N. et al., J. Med Chem., 57: 1299-1322 (2014)). The pinacolatoboronate of the O-alkyl phenol, Compound 7, had acceptable solid phase stability and could be isolated via crystallization.
Scheme 3: Suzuki-Miyaura coupling between 2 and 3
[0237] Process robustness studies for the isolation of Compound 7, however, indicated that Compound 7 has poor solution stability, decomposing primarily to the proteodeborylated compound 10, as shown in Scheme 4 below. This was particularly problematic as the isolation process involved a solvent exchange from 2-MeTHF (2-methyl tetrahydrofuran) to iPrOAc (isopropyl acetate), which is not a fast unit operation on scale.
Scheme 4: Modification of 7
[0238] A search for a more stable boronic ester was undertaken. Early attempts targeted making N-methyliminodiacetic acid (MID A) boronate Compound 2-a (E. Gilis and M. Burke,“Multi step Synthesis of Complex Boronic Acids from Simple MIDA Boronates,” J Am. Chem. Soc., 750(43): 14084-14085 (2008)), however, all attempts resulted in product decomposition. Applicant then turned to a relatively obscure boronate formed by the addition of diethanolamine to Compound 7 (Bonin et al., Tetrahedron Lett., 52: 1132-1135 (2011)). Addition of diethanolamine to a solution of Compound 7 led to rapid ester formation and concomitant crystallization of Compound 2.
[0239] The discovery of boronic ester Compound 2 allowed for a simple, fast, high-yielding, high-purity process comprising the following procedure. Tetrahydrofuran (THF, 1500mL) was charged to a flask containing Compound 6 (100g, 328 mmol), bis(pinacolato)diboron (90.7g, 357 mmol) and cesium acetate (CsOAc, 158g, 822 mmol). The system was vacuum purged three times with nitrogen. Pd(PPh3)2Cl2 (13.8g, 20 mmol) was charged to the reaction and the system was vacuum purged three times with nitrogen. The reaction was then heated to 55 to 65°C.
[0240] The batch was stirred for approximately 8 hours until reaction was complete by HPLC analysis. The batch was cooled to 15 to 25 °C (target 20 °C ) and charged with silica gel (20g) and Ecosorb C-941 (20g). After lh, the mixture was filtered to remove solid. The residual solids were washed twice, each with THF (300mL). The filtrate and washes were combined. In a separate vessel, diethanolamine (34.5mL, 360 mmol) was dissolved in THF (250 mL). The diethanolamine solution in THF (25mL) was then charged to the batch. After 10 minutes, the batch was seeded with 2 (1 g) and aged for 1 to 2 hours. The remaining of the diethanolamine solution in THF was charged to the batch over at least 2 hours and the slurry was stirred for at least 2 hours. The product 2 was collected by suction filtration. The wet cake was washed thrice with THF (200mL). The material was dried under vacuum at 40 °C with nitrogen purge yielding 94.6g of 2.
[0241] The reaction to synthesize Compound 2 from Compound 6 described above may be modified as follows. Solvent: Different solvents from THF could be used, such as 1,4 dioxane or 2-methyltetrahydrofuran. Reaction volume: The reaction volume can be varied from 4 to 50 volumes with respect to compound 2. Catalyst and base: Different palladium catalyst and bases can be used for the borylation. Examples can be found in Chow et al., RSC Adv., 3 : 12518-12539 (2013). Borylation reaction temperature: Reaction temperatures from room temperature (20 °C) to solvent reflux can be used. Carbon/ Silica treatment:
The treatment can be performed without silica gel. The process can be performed without a carbon treatment. Different carbon sources from Ecosorb C-941 can be used. Different amounts of silica, from 0.01X to IX weight equivalents, can be used. Different amounts of Ecosorb C-941, from 0.01X to IX weight equivalents, can be used. Crystallization: A different addition rate of diethanolamine can be used. Different amounts of diethanolamine, from 1.0 to 3.0 molar equivalents can be used. A different cake wash with more or less THF can be used. Different amount of seeds from 0.0001X wt to 50X wt can be used.
Alternatively, the process can be unseeded. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
[0242] The subsequent Suzuki-Miyaura coupling between Compounds 2 and 3 also proceeded well, providing over 20 kg of crude compound 1 with an average molar yield of 80% and LCAP of 99.7%.
1.4: Synthesis of Coupling Partner 3
[0243] Cross-coupling partner 3 was prepared by two different processes corresponding to Schemes 8 and 9 shown below.
Scheme 8: Process A for preparation of 3
[0244] According to Process A, Compound 9 (100g, 628 mmol) was dissolved in acetonitrile (450 mL) at room temperature. In a separate vessel, N-bromosuccinimide (NBS, 112g, 628 mmol) was suspended in acetonitrile (1 L). Compound 9 in acetonitrile was charged to the NBS slurry over at least 45 minutes. The contents of the reaction vessel were warmed to 45 °C to 55 °C and the batch stirred until the reaction was complete by HPLC analysis. The batch was cooled to 35 °C to 45 °C and ensured dissolution. Norit SX plus carbon (lOg) was charged to the mixture and the reaction mixture adjusted to 55 °C to 60 °C. The mixture was stirred at 55 °C to 60 °C for about lh and the mixture filtered at 55 °C to 60 °C to remove solids. The solids were washed with acetonitrile (500mL) at 55 °C to 60 °C. The volume of the combined filtrate was reduced to 900 mL by distilling off acetonitrile under reduced pressure. The batch with Compound 3 (lg) and stirred at 35 °C to 45 °C for at least 60 minutes. The contents of the reaction vessel were cooled to 15 °C to 25 °C over at least 1 hour. Water (2000 mL) was charged to the reaction vessel over at least 90 minutes and the slurry aged for at least 60 minutes. The product was collected by suction filtration. The cake was washed with a premixed 5% solution of acetonitrile in water (300mL). The wet cake was dried under vacuum at 40 °C with nitrogen purge. Yield: 120g of 3.
[0245] The above procedure, Process A for this synthesis of 3, may be practiced with alternative reagents and conditions as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 9). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent Stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
Scheme 9: Process B for preparation of 3
[0246] According to Process B, Compound 3 can be formed starting from 8 via non-isolated compound 9 as follows. Compound 8 (80 g, 55 mmol), cesium carbonate (CS2CO3, 215 g, 66 mmol), and acetonitrile (800 mL) were charged to the reactor. The temperature was adjusted from 15 to 25 °C and iodomethane charged to the reactor (Mel, 86 g, 0.61 mol) while maintaining a batch temperature below 25 °C. The batch was heated to 40 °C and agitated for 10 hours to form Compound 9. The batch was cooled to 25 °C, filtered into a fresh reactor to remove solids, and the solids washed twice with acetonitrile. The combined organic layers were concentrated via atmospheric distillation to about 320 mL.
[0247] In a separate reactor N-bromosuccinimide (NBS, 98.1 g, 0.55 mol) was charged to acetonitrile (800 mL) and agitated. The batch containing Compound 9 was transferred to the NBS solution while maintaining a batch temperature of 15 to 25 °C. The batch was heated to 45 to 55 °C and agitated for at least 4 hours to allow for reaction completion to Compound 3. Upon reaction completion, Norit SX Plus activated carbon (8 g) was charged, and agitated at 45 to 55 °C for one hour. The batch was filtered into a fresh vessel, the Norit SX plus cake was washed with 400 ml of 45 to 55 °C acetonitrile. The acetonitrile layers were combined, cooled to 35 to 45 °C, and distilled under reduced pressure to 720 mL. The batch was adjusted to a temperature of 40 °C, charged with Compound 3 seeds (0.8 g), agitated for one hour, cooled to 15 to 25 °C over at least on hour, then charged with water (1600 mL) over at least two hours. The mixture was agitated for an additional one to two hours, filtered, the cake washed with a premixed 5% solution of acetonitrile in water (240 mL). The wet cake was dried under vacuum at 40°C with nitrogen purge. Yield: 52 g of 3.
[0248] Process B to synthesize Compound 3, described above, may be modified as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 8).
Alkylating reagent: Alternative methylating reagents to methyl iodide can be used such as dimethylsulfate. Alkylating reagent stoichiometry: 1 to 10 molar equivalents of methyl iodide may be used. Base: Different inorganic bases, such as potassium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Brominating agents:
Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. Seeding levels from 0.0001% to 50% can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying.
1.5: Cross-coupling of 2 and 3 to Produce Target Compound 1
[0249] 1 is synthesized by Suzuki cross-coupling of 3 and 2 according to Scheme 10 and as described below.
Scheme 10: Synthesis of 1
[0250] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compovmd 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223 g, l.OSmol). Agitation was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum
purged three times with nitrogen. The batch was heated to 65 to 75°C and contents stirred for at least 16 hours until reaction was complete by HPLC analysis. The batch was cooled to 60 to 70°C, agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at 60 to 70°C. Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at 60 to 70°C. After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at 60 to 70°C. The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to 60 to 65°C and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at 60 to 65°C. The slurry was cooled to 15 to 25°C over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at 40°C with nitrogen purge. Yield: 139g of 1.
[0251] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox, Alister, J.J., Lloyd-Jones, Guy C. Chem. Soc. Rev., 2014, 43, 412. Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1);
cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5); Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compound 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compoxmd 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
[0253] Formic acid is one ICH Class 3 solvent in which Compound 1 is highly soluble, having a solubility greater than 250 mg/ml at 20 °C. The solubility curve of Compound 1 in formic acid-Water is quite steep (see Figure 7), which enables a volumetrically efficient process.
[0254] Initial attempts to recrystallize crude Compound 1 involved dissolving in formic acid, polish filtering, and charging polish filtered water to about 20% supersaturation, followed by seeding with the thermodynamically most stable form (Form 1), followed by slow addition of water to the final solvent ratio, filtration, washing, and drying. Applicant observed that during the initial water charge, if the batch self-seeded it formed a thick slurry. X-ray diffraction (XRD), differential scanning calorimetry (DSC), and photomicroscopy demonstrated that a metastable form was produced. Once seeded with Form 1, the batch converted to the desired form (Form 1) prior to the addition of the remaining water. This process worked well during multiple lab runs, consistently delivering the desired form and purity with about 85% yield.
[0255] Unfortunately, upon scale-up, the batch did not convert to Form 1 after seeding. Additional water was charged and the batch began to convert to the desired form (mix of Form 1 and the metastable form by X-ray powder diffraction (XRPD)). When additional water was charged, the XRPD indicated only the metastable form. After a few hours with no change, Applicant continued the water charge to the final solvent ratio, during which time the batch eventually converted to Form 1. This process is summarized in Figure 8.
[0256] It was subsequently found by closer analysis of the plant and laboratory retains that a new metastable form was formed during scale up, with a similar, but different XRPD pattern. This form (metastable B) could be reproduced in the laboratory, but only when the batch has a high formic acid:water ratio and is seeded with Form 1. Without Form 1 seeds, metastable A is the kinetic form. Both metastable forms converted to Form 1 with additional water and/or upon drying, leading Applicant to believe that the metastable forms are formic acid solvates. These findings are summarized in Figure 9.
[0257] While there is little risk in not being able to control the final form, there is a risk of forming a difficult-to-stir slurry which can lead to processing issues. The crystallization procedure was therefore modified to keep a constant formic acid-water ratio. This was performed by charging 2.4X wt. formic acid and 1.75X wt. water (final solvent composition)
to the crystallizer with 0.03X wt. Form 1 seeds, and performing a simultaneous addition of Compound 1 in 6. IX wt. formic acid and 4.4X wt. water. The batch filtered easily and was washed with formic acid/water, then water, and dried under reduced pressure to yield 8.9 kg of Compound 1 (92% yield) with 99.85% LCAP and N.D. formic acid.
Example 2: Exemplary high throughput experimentation reaction
[0258] The following procedure is an exemplary high throughput experimentation reaction.
[0259] An overview of the reaction is shown below in Scheme 5:
Scheme 5: Reaction conditions tested for cross-coupling reaction of 2 and 3
[0260] Pd catalysts were dosed into the 24-well reactor vial as solutions (100 pL of 0.01 M solution in tetrahydrofuran (THF) or dichloroethane (DCE) depending upon the solubility of the ligand). Plates of these ligands are typically dosed in advance of the reaction, the solvent is removed by evacuation in an evaporative centrifuge and plates are stored in the glovebox. The catalysts screened in the coupling are the following: XPhos, SPhos, CataCXium A, APhos, P(Cy)3, PEPPSI-IPent. For the first five ligands, these were initially screened as the Buchwald Pd G2/G3 precatalysts.
[0261] To the plates was then added a stock solution of Compound 3 (10 pmol) and Compound 2 (12 pmol) dissolved in the following solvents: dimethylformamide (DMF),
tetrahydrofuran (THF), butanol (/r-BuOH), and toluene. The base was then added as a stock solution (30 mmol) in 20 mL of water.
[0262] A heatmap summarizing catalyst performance is shown in Figs. 10A and 10B. High performance liquid chromatography (HPLC) yields for this screening span from <5% up to -85%. Larger circles indicate higher yield. Lighter circles indicate higher cleanliness.
[0263] A similarly designed screening of base and solvent also indicate that a range of alcoholic solvents (methanol, ethanol, propanol, 2-butanol, 2-propanol, and /-amyl alcohol) are also all viable in this coupling chemistry. Bases such as potassium phosphate, potassium carbonate, potassium acetate, and potassium hydroxide were all successful in achieving the coupling. Fig. 10B shows a heatmap with HPLC yields ranging from -50 – 95%. Larger, darker circles indicate higher yield.
[0264] This chemistry from microvial screening has been scaled to a laboratory process. To a 3 -necked jacketed 250 mL flask equipped with overhead stirring, nitrogen inlet, and thermocouple was added Compound 3 (1.0 eq, 4.00 grams), Compound 2 (1.2 eq, 1.71 x wt), potassium carbonate (3.0 eq, 1.74 x wt). The reactor was inerted three times and then degassed 2-propanol (24 x vol.) followed by degassed water (6 x vol) was then added.
Stirring was then initiated at 300 rpms. The reactor was then stirred and blanketed with nitrogen for 1 hour. The catalyst was then added (0.01 eq, 0.028 x wt) and stirring continued (300 rpms) and the reactor was heated into the Tj = 65 °C.
[0265] After 2 hours, with full conversion confirmed analytically, trioctylphosphine (0.1 eq, 0.16 x wt) dosed, and reaction mixture allowed to cool slowly to room temperature hours.
The reaction mixture was then filtered, washed with 2-propanol (4 x vol), 2-propanol: water (4: 1, 4 x vol), and then with water (4 x vol). Note: If 2 is dimer present in cake, an additional ethyl acetate (EtOAc) wash (4 x vol) can be added for purging. The cake was then transferred to a vacuum oven to dry overnight at 40 °C, -40 cm Hg, under nitrogen flow. After transfer to a bottle, 6.03 grams of 1 were isolated, 98.6% assay, 91% overall yield.
Scheme 6: Alternative reagents and solvents for cross-coupling
[0266] Based on the previously delineated results, it was expected that a variety of monodentate (PPI13 [triphenylphosphine], PBu3 [tributylphosphine], etc) and bidentate phosphines (dppf [1,1 ‘-bis(diphenylphosphino)ferrocene], BINAP [2,2 -bis(diphenylphosphino)- 1 , 1 -binaphthyl], Xantphos [4,5-bis(diphenylphosphino)-9,9-dimethylxanthene], dppe [l,2-bis(diphenylphosphino)ethane], etc) ligated to any number of Pd sources (Pd halides, Pd(H) precatalyts, Pd(0) sources) could reasonably be employed to arrive at the Compound 1 crude material. A range of organic solvents ranging from non-polar (heptane, benzene), protic (alcohols), polar aprotic (dimethylsulfoxide, dimethylformamide, dimethylacetamide, acetonitrile) as well as a variety of esters and ketones (acetone, 2-butanone, ethylacetate) should also serve as effective solvents for this reactivity. Finally, inorganic bases of varying strength (phosphates, carbonates, acetates, etc) along with organic variants such as triethylamine, l,8-diazabicyclo(5.4.0)undec-7-ene, and others in a wide pKa range are viable as stoichiometric basic additives.
Example 3: Exemplary Compound 5 process
[0267] The purpose of this example was to describe an exemplary process for making Compound 5.
[0268] Charge 4 (lOg, 58mmol) and acetonitrile (lOOmL) to a reaction vessel and start the stirrer. Adjust the batch to -18 °C to -22 °C (target -20 °C). Charge triflic acid (5.5mL, 62mmol) to the batch maintaining -10 °C to -25 °C (target -20 °C). Stir the batch at -10 °C to -25 °C (target -20 °C) for 10 to 20 minutes. Charge NBS (11.38g, 64mmol) to the batch at -10 °C to -25 °C (target -20 °C) and stir for ca. 30 min at -10 °C to -25 °C (target -20 °C). Warm the batch to 20 °C over 3-4 hours (reaction will occur when internal temp is between 5 °C and 15 °C). Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and sample for reaction completion.
[0269] If Compound 4 relative to Compound 5 is more than 5%:
[0270] Cool the bath to -5 °C to -15 °C (target -10 °C) (cooling below 0 °C to ensure selectivity). Charge NBS to the batch according to the follow formula: Mass of NBS = (% Compound 4 x lOg). Warm the batch to 20 °C over 1-2 hours. Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and check reaction for completion. Proceed to next line.
[0271] If Compound 4 relative to Compound 5 is less than 5%:
[0272] Warm the batch to 40 °C to 50 °C (target 48 °C). Concentrate the batch under reduced pressure to a final volume of ~40mL. Cool the batch to -15 °C to -5 °C (target -10 °C) and stir for ca. lh. Filter the batch by suction filtration. Slurry wash the filter cake with purified water (3 x 20mL) at 15 °C to 25 °C (target 20 °C) for 10 to 15 minutes each wash. Remove a sample of the filter cake for analysis by ¾ NMR. Continue washing cake until the residual succimide is below 1.0%mol% relative to 5. Dry the filter cake at up to 60°C under vacuum and nitrogen purge. Analyse the 5 by HPLC analysis (97%w/w to 99%w/w). Expected yield: 60-85% theory (90-110% w/w).
Example 4: Purification of Compound 1 (CC-90010) by crystallization from formic acid and water.
[0273] This example describes a method for the purification of Compound 1 by
crystallization from formic acid and water. Also detailed are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1.
[0274] Figure 11 shows XH NMR of Compound 1 (CC-90010). Solvent: d6DMSO; and Figure 12 shows microscopy of Compound 1 (CC-90010) Form I. Figure 13 shows XRPD of Compound 1 (CC-90010) Form I, with peak information detailed in Table 6:
PATENT
US 20190008852
WO 2018081475
US 20180042914
WO 2016172618
WO 2015058160
/////////CC-90010, solid tumors , non-Hodgkin’s lymphoma, PHASE 1, CANCER, QUANTICEL
CS(=O)(=O)c4cc(C1=CN(C)C(=O)c2ccccc12)c(OCC3CC3)cc4
Elacridar
Elacridar
C34H33N3O5, 563.6 g/mol
依克立达;gw0918
UNII-N488540F94
143851-98-3 (monoHCl)
N-[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]phenyl]-5-methoxy-9-oxo-10H-acridine-4-carboxamide
GF120918
Elacridar (GF120918)
GF-120918
GG-918
GW-120918
GW-918
GF-120918A (HCl)
GlaxoSmithKline (previously Glaxo Wellcome ) was developing elacridar, an inhibitor of the multidrug resistance transporter BCRP (breast cancer resistant protein), as an oral bioenhancer for the treatment of solid tumors.
Elacridar is an oral bioenhancer which had been in early clinical trials at GlaxoSmithKline for the treatment of cancer, however, no recent development has been reported. It is a very potent inhibitor of P-glycoprotein, an ABC-transporter protein that has been implicated in conferring multidrug resistance to tumor cells.
SYN

The condensation of 2-(4-nitrophenyl)ethyl bromide with 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline by means of K2CO3 and KI in DMF at 100 C gives 6,7-dimethoxy-2-[2-(4-nitrophenyl)ethyl]-1,2,3,4-tetrahydroisoquinoline,
Which is reduced with H2 over Pd/C in ethanol to yield the corresponding amine . Finally, this compound is condensed with 5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxylic acid by means of DCC and HOBt in DMF to afford the target carboxamide.
The intermediate 5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxylic acidhas been obtained as follows: The condensation of 2-amino-3-methoxybenzoic acid with 2-bromobenzoic acid by means of K2CO3 and copper dust give the diphenylamine , which is cyclized to the target acridine Elacridar by means of POCl3 in refluxing acetonitrile.
PATENT
WO-2019183403
Deuterated analogs of elacridar as P-gp/BCRP inhibitor by preventing efflux useful for treating cancer.
Elacridar, previously referred to as GF120918, is a compound with the structure of 9,10-dihydro-5-methoxy-9-oxo-N-[4-[2-(1 ,2,3,4-tetrahydro- 6,7-dimethoxy-2-isoquinolinyl)ethyl] phenyl]-4-acridine-carboxamide or, as sometimes written, N-4-[2-(1 ,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy- 9-oxo-4-acridine carboxamide. Elacridar was originally described as a P-gp selective inhibitor but is now recognized as a dual P-gp/BCRP inhibitor. (Matsson P, Pedersen JM, Norinder U, Bergstrom CA, and Artursson P 2009 Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm Res 26:1816-1831 ).
003 Elacridar has been examined with some success both in vitro and in vivo as a P-gp and BCRP inhibitor. By way of example, in cancer patients, coadministration of elacridar with therapeutic agents such as paclitaxel (P-gp substrate) and topotecan (BCRP substrate) improved their oral absorption – presumably by preventing efflux into the intestinal lumen by P-gp/BCRP pumps located in the Gl tract. Similarly, in rodents, elacridar has been coadministered with some success with pump substrates such as morphine, amprenavir, imatinib, dasatinib, gefitinib, sorafenib, and sunitinib to increase drug levels in the brain (by blocking efflux mediated by P-gp and BCRP at the blood brain barrier). A summary of some of these studies can be found in a study report by Sane et al. (Drug Metabolism And Disposition 40:1612-1619, 2012).
004 Administration of elacridar has several limitations. By way of example, elacridar has unfavorable physicochemical properties; it is practically insoluble in water, making it difficult to formulate as, for example, either an injectable or oral dosage form. Elacridar’s poor solubility and high lipophilicity result in dissolution rate-limited absorption from the gut lumen.
005 A variety of approaches have been pursued in order to increase efficacy of elacridar. For example, United States Patent Application Publication 20140235631 discloses a nanoparticle formulation in order to increase oral bioavailability.
006 Sane et al. (Journal of Pharmaceutical Sciences, Vol. 102, 1343-1354 (2013)) report a micro-emulsion formulation of elacridar to try and overcome its dissolution-rate-limited bioavailability.
007 Sawicki et al. (Drug Development and Industrial Pharmacy, 2017 VOL. 43, NO. 4, 584-594) described an amorphous solid dispersion formulation of freeze dried elacridar hydrochloride-povidone K30-sodium dodecyl sulfate. However, when tested in healthy human volunteers, extremely high doses (e.g. 1000 mg) were required to achieve a Cmax of 326 ng/ml. (Sawicki et al. Drug Deliv. and Transl.
Res. Published online 18 Nov 2016).
008 Montesinos et al. (Mol Pharm. 2015 Nov 2; 12(11 ):3829-38) attempted several PEGylated liposome formulations of elacridar which resulted in a partial increase in half life, but without an increase in efficacy when co-administered with a therapeutic agent.
009 Because of the great unpredictability in the art and poor correlations in many cases between animal and human data, the value of such formulation attempts await clinical trial.
0010 Studies of the whole body distribution of a microdose of 11C elacridar after intravenous injection showed high level accumulation in the liver (Bauer et al. J Nucl Med. 2016;57:1265-1268). This has led some to suggest that systemic levels of elacridar are also substantially limited by clearance in the liver.
0011 A potentially attractive strategy for improving metabolic stability of some drugs is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the rate of formation of inactive metabolites by replacing one or more hydrogen atoms with deuterium atoms.
Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the absorption, distribution, metabolism, excretion and/or toxicity (‘ADMET’) properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
0012 Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, M I et al, J Pharm Sci, 1975, 64:367-91 ; Foster, A B, Adv Drug Res 1985, 14:1 -40 (“Foster”); Kushner, D J et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, M B et al, Curr Opin Drug Discov Devel, 2006, 9:101 -09 (“Fisher”)). The results have been variable and unpredictable. For some compounds, deuteration indeed caused decreased metabolic clearance in vivo. For others, no change in metabolism was observed. Still others demonstrated increased metabolic clearance. The great unpredictability and variability in deuterium effects has led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting metabolism (see Foster at p. 35 and Fisher at p. 101 ).
0013 The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem. 1991 , 34, 2871 -76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
0014 Considering elacridar’s challenging physicochemical and ADMET properties in humans, in spite of recent formulation advancements, there remains a need in the art for elacridar analogs that can achieve higher, less variable levels in the systemic circulation, at the blood-brain barrier, and elsewhere to optimize efflux inhibition.
Example 1 : Synthesis of Instant Analogs and Compositions
00179 This example demonstrates a synthetic method for making elacridar analogs, deuterium substitutions based upon the deuteration of the starting compounds. The synthesis and the analog numbers refer to Figure 4.
00180 Step 1
00181 A 12L three-neck flask was charged with compound 1 (270.5 g, 1.618 mol), compound 2 (357.8 g, 1.78 mol, 1.1 eq.), K2C03 (447 g, 3.236 mol, 2.0 eq), Cu (20.6 g, 0.324 mol, 0.2 eq.) and ethanol (2.7 L) and the resulting mixture was heated to reflux under nitrogen for 1 hour. The reaction mixture was cooled to room
temperature after the reaction progress was checked with LC-MS. Water (2.7 L) was added and the mixture was filtered through a pad of Celite. The Celite was washed with water (1.35L) and the combined filtrate was adjusted to pH~2 by addition of concentrated HCI (~410 mL) over 15 min. The resulting suspension was stirred at 10°C for 1.5 hours and the solid was filtered, washed with water (2.7 L) and dried at 45°C using a vacuum oven for 2 days to give compound 3 (465 g, ~100%) as a yellow solid.
00182 Step 2
00183 A suspension of compound 3 (498 g, 1.734 mol) in acetonitrile (4.0 L) was heated to reflux under stirring. To the suspension was added POCb (355.5 mL,
3.814 mol, 2.2 eq.) drop-wise over 2h. The mixture was heated at reflux for 2.5h and then cooled to 30 °C. To the mixture was slowly added water (3.0 L) and the resultant thick slurry was heated to reflux for 1 5h. The slurry was cooled to 10 °C and filtered. The solid was washed with water (2 X 1.0 L), acetonitrile (2 X 1.0 L) and dried using a vacuum oven overnight at 45 °C to afford compound 4 (426 g, 91.3%) as a yellow solid.
00184 Step 3:
00185 A 12L three-neck flask was charged with compound 5 (475g, 2.065 mol), compound 6 (474.8g, 2.065 mol), K2C03 (314g, 2.273 mol), Kl (68.6g, 0.413 moL) and DMF (2.5L) and the resulting mixture was heated to 70 °C and stirred for 2.5 hours. After LC-MS showed that the reaction was complete, the mixture was cooled to 50 °C and methanol (620 ml_) was added. Then the mixture was cooled to 30 °C and water (4.75 L) was added. The resulting suspension was cooled to 10 °C and for 1 hour. The solid was filtered, washed with water (2 X 2.5 L) and air dried for 2 days to afford the compound 7 (630 g, 89.1 %) as a yellow solid.
00186 Step 4
00187 To a solution of compound 7 (630 g, 1.84 mol) in THF/ethanol (8 L at 1 :1 ) was added Pd/C (10%, 50% wet, 30 g). The mixture was stirred under an
atmosphere of hydrogen (1 atm, balloon) at 15-20 °C for 4h. The reaction mixture was filtered through a pad of Celite and the pad was washed with TFIF (1.0 L). The filtrate was concentrated to 3 volumes under vacuum and hexanes (4.0 L) was added. The resulting slurry was cooled to 0 °C and stirred for 1 h. The solid was filtered and washed with hexanes (2 X 500 ml_) and air dried overnight to afford the compound 8 (522 g, 90.8%) as an off -white solid.
00188 Step 5
00189 A 5L three-neck flask was charged with compound 4 (250 g, 0.929 mol, 1 eq.), compound 8 (290 g, 0.929mol, 1 eq.) and DMF (2.5 L) and the resulting mixture was stirred at room temperature until it became a clear solution. To the solution was added TBTU (328 g, 1.021 mol, 1.1 eq.), followed by triethylamine (272 ml_, 1.95 mol, 2.1 eq.) and the resulting mixture was stirred at room temperature under nitrogen overnight. The mixture was poured slowly into water (7.5 L) with stirring and the resulting suspension was stirred for 1 hour at room temperature. The solid was filtered and washed with water (2 X 7 L). The solid thus obtained was dried using a vacuum oven at 50 °C for two days and 509.0 g (97.3%) of compound 9 was obtained as yellow solid.
00190 Step 6
00191 300.0 g (0.532 mol) of compound 9 was suspended in acetic acid (1.2 L) and heated to 70 °C. The resultant solution was hot filtered and heated to 70°C again. Preheated ethanol (70 °C, 3.6 L) was then added. To this solution was added concentrated HCI (66.0 ml_, 0.792 mol, 1.5 eq.) dropwise over 30 min. The resulting solution was stirred at 70°C until crystallization commenced (~about 20 min). The suspension was cooled to room temperature over 3h, filtered, washed with ethanol (2 X 1.8 L) and dried using a vacuum oven at 60°C over the weekend to afford compound 10 (253.0 g, 79.2%) as a brown solid.
Example 2 Manufacture of a Deuterated Elacridar analog EE60.
00192 EE60 is synthesized by the procedure shown in Figure 4 and as continued in Figure 5.
00193 The structure of EE60 is confirmed as follows: Samples of 5 pi are measured using an LC system comprising an UltiMate 3000 LC Systems (Dionex, Sunnyvale, CA) and an 2996 UV diode array detector (Waters). Samples are injected on to a 100 x 2mm (ID) 3.5 pm ZORBAX Extend-C18 column (Agilent, Santa Clara, CA). Elution is done at a flow rate of 0.4 mL/min using a 5 minute gradient from 20% to 95% B (mobile phase A was 0.1 % FICOOFI in water (v/v) and mobile phase B was methanol). 95% B is maintained for 1 min followed by re-equilibration at 20% B. Chromeleon (v6.8) is used for data acquisition and peak processing.
Example 3: Manufacture of a Deuterated Elacridar analog EE59
00194 EE59 was synthesized by the procedure shown in Figure 6.
00195 The resulting yellowish brown precipitate was removed by filtration and the filter cake was dried overnight (72 mg). Analysis of the filter cake by LCMS indicated the presence of a single peak at multiple wavelengths (215 nm, 220 nm, 254 nm,
280 nm); each peak confirmed the presence of the desired product (LC retention time, 5.3 min; m/z = 575 [(M+FI)+]).
00196 1H NMR of EE598 revealed 1H NMR (400 MHz, DMSO-d6) d 12.3 ( s , 1H), 10.6 (s, 1H), 8.51-8.46 (m, 2H), 7.80 (d, J = 8.8 Hz, 1H), 7.66 (d, J = 7.6 Hz, 2H), 7.45-7.38 (m, 2H), 7.32-7.25 (m, 3H), 6.66 (d, J = 6.8 Hz, 2H), 3.62 (s, 2H), 2.86 (t, J = 6.8 Hz, 2H), 2.66 (m, 4H).
Example 4: Demonstration of superior properties of instant analogs and compositions: in vivo ADMET.
00197 Pharmacologic studies are performed according to Ward KW et al (2001 Xenobiotica 317783-797) and Ward and Azzarano (JPET 310:703-709, 2004).
Briefly, instant analogs are administered solutions in 10% aqueous polyethylene glycol-300 (PEG-300) or 6% Cavitron with 1 % dimethyl sulfoxide, or as well triturated suspensions in 0.5% aqueous HPMC containing 1 % Tween 80. Blood samples are collected at various times up to 48 h after drug administration; plasma samples are prepared and at “70°C until analysis.
00198 Mice. Instant analogs are administered to four groups of animals by oral gavage (10 ml/kg dose volume). Three groups receive instant analogs as a suspension at 3, 30, or 300 mg/kg, and the fourth group receive instant analogs as a solution in Cavitron at 3 mg/kg. Blood sampling in mice is performed via a tail vein at 0.5, 1 , 2, 4, 8, 24, and 32 h postdose.
00199 Rats. A total of seven groups of animals receive instant analogs by oral gavage (10 ml/kg). Three groups receive instant analogs as a suspension at 3, 30, or 300 mg/kg, and a fourth and fifth group each receive instant analogs as a solution in Cavitron or PEG-300, respectively, at 3 mg/kg. A sixth and seventh group of rats with indwelling hepatic portal vein catheters receive instant analogs by oral gavage (10 ml/kg) as a suspension at 3 or 30 mg/kg, respectively. Blood sampling in rats are performed via a lateral tail vein; samples are also obtained from the hepatic portal vein catheter. Blood samples are obtained before dosing and at 5, 15, 30, and 45 min, and 1 ,1.5, 2, 3, 4, 6, 8, 10, 24, and 32 h postdose.
00200 Dogs. Dogs receive instant analogs by lavage (4 ml/kg) on three separate occasions with dosages at 3 and 30 mg/kg as a suspension and 3 mg/kg as a solution in Cavitron. Blood samples are obtained from a cephalic vein and from the hepatic portal vein catheter before dosing and at 5, 15, 30, and 45 min and 1 , 1.5, 2, 3, 4, 6, 8, 10, 24, 32, and 48 h postdose.
00201 Monkeys. Monkeys receive instant analogs by oral gavage (8 ml/kg dose volume) on three separate occasions at dosages of 3 and 30 mg/kg as a suspension and 3 mg/kg as a solution in Cavitron. Blood samples are obtained from a femoral vein via an indwelling catheter and from the hepatic portal vascular access port
before dosing and at 5, 15, and 30 min and 1 , 1.5, 2, 4, 6, 8, 10, 24, 32, and 48 h postdose.
00202 Humans. Healthy volunteers receive instant analogs orally at doses ranging from 25 mg to 1000 mg. Blood samples are obtained and analyzed for analog concentrations at 0, 15 min, 30 min, 45 min, 60 min, 90 min, 120 min, 180 min, 2 hr, 4 hr, 6hr, 8 hr, 12 hr, 24 hr, and 48 h after administration .
Analytical Methods
00203 Instant analogs are isolated from samples by precipitation with acetonitrile and quantified by LC/MS/MS coupled with an atmospheric pressure chemical ionization interface (475°C). Internal standards [in acetonitrile/10 mM ammonium formate, pH 3.0; 95:5 (v/v)] are added to 50 pi samples and vortexed and centrifuged for 30 min at 4000 rpm. The supernatants are injected onto the LC/MS/MS system using an HTS PAL autosampler (CTC Analytics, Zwingen, Switzerland) coupled to an Aria TX2 high-throughput liquid chromatographic system using turbulent flow technology (Cohesive Technologies, Franklin, MA) in focus mode. The mobile phase consists of a mixture of 0.1 % formic acid in water and 0.1 % formic acid in
acetonitrile. The turbulent flow column is a 0.5 X 50-mm Cyclone P column
(Cohesive Technologies) in series to a 2 X 20 mm, 4 pm Polar RP (Phenomenex, Torrance, CA) analytical column. Positive-ion multiple reaction monitoring is used for the detection of instant analogs and internal standard and the selected precursor and product ions are mlz 564 and 252, respectively. Using a (1/x) weighted linear regression analysis of the calibration curve, linear responses in analyte/internal standard peak area ratios are observed for instant analog concentrations ranging from 2 to 10,000 ng/ml.
00204 Alternatively, useful analytical methods to demonstrate the surprising and superior properties of the instant elacridar analogs are the methods as described by Stokvis et al, J Mass Spectr 2004: 39: 1122-1130.
PATENT
WO2014018932
claiming nano-particle composition comprising breast cancer resistance protein inhibitor (eg elacridar). Family member of the elacridar
PAPER
J Med Chem 1995, 38(13): 2418
PATENT
Product PATENT WO9212132
PATENT
US5604237
NMR includes d 2.60-2.95 (m,8H,CH2); 3.58 (s,2H,N–CH2 –Ph); 3.72 (s,6H,OMe); 4.05 (s,3H,OMe acridone); 6.78 (2s,2H,Ar.isoquinoline), 7.20-7.88 (m,8H,Ar.), 8.48 (t,2H,H1 and H8 acridone), 10.60 (s, 1H,CONH), 12.32 (s, 1H,NH acridone)
///////////Elacridar, GF-120918, GG-918 , GW-120918, GW-918, GF-120918A (HCl), solid tumors, GSK, GLAXO
[11C]-elacridar
HEC-68498
HEC-68498, CT-365
CAS 1621718-37-3
N-[5-(3-Cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxy-3-pyridinyl]-2,4-difluorobenzenesulfonamide
HEC Pharm , Calitor Sciences Llc; Sunshine Lake Pharma Co Ltd
PHASE 1, idiopathic pulmonary fibrosis and solid tumors
Phosphoinositide 3-kinase inhibitor; mTOR inhibitor

- Originator HEC Pharm
- Developer HEC Pharm; Sunshine Lake Pharma
- Class Anti-inflammatories; Antifibrotics; Isoenzymes
- Mechanism of Action 1 Phosphatidylinositol 3 kinase inhibitors; MTOR protein inhibitors
- Phase I Idiopathic pulmonary fibrosis
- 22 May 2018 Phase-I clinical trials in Idiopathic pulmonary fibrosis in USA (PO) (NCT03502902)
- 24 Apr 2018 Sunshine Lake Pharma in collaboration with Covance plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in China , (NCT03502902)
- 19 Apr 2018 Preclinical trials in Idiopathic pulmonary fibrosis in China (PO)
- US 20140234254
- CN 103965199
CN 103965199

CN 103965199

Sunshine Lake Pharma , a subsidiary of HEC Pharm is developing an oral capsule formulation of HEC-68498 (phase 1, in July 2019) sodium salt, a dual inhibitor of phosphoinositide-3 kinase and the mTOR pathway, for the treatment of idiopathic pulmonary fibrosis and solid tumors
HEC 68498 is an oral inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin in clinical development at HEC Pharm for the treatment of idiopathic pulmonary fibrosis. A phase I trial is under way in healthy volunteers.
The phosphoinositide 3-kinases (PI3 kinases or PI3Ks), a family of lipid kinases, have been found to play key regulatory roles in many cellular processes including cell survival, proliferation and differentiation. The PI3K enzymes consist of three classes with variable primary structure, function and substrate specificity. Class I PI3Ks consist of heterodimers of regulatory and catalytic subunits, and are subdivided into 1A and 1B based on their mode of activation. Class 1A PI3Ks are activated by various cell surface tyrosine kinases, and consist of the catalytic pl lO and regulatory p85 subunits. The three known isoforms of Class 1A pl lO are pl lOot, rΐ ΐqb, and rΐ ΐqd, which all contain an amino terminal regulatory interacting region (which interfaces with p85), a Ras binding domain, and a carboxy terminal catalytic domain. Class IB PI3Ks consist of the catalytic (pl lOy) and regulatory (p 101 ) subunits and are activated by G-protein coupled receptors. (“Small-molecule inhibitors of the PI3K signaling network” Future Med. Chem ., 2011, 3, 5, 549-565).
[0004] As major effectors downstream of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), PI3Ks transduce signals from various growth factors and cytokines into intracellular massages by generating phospholipids, which activate the serine-threonine protein kinase ART (also known as protein kinase B (PKB)) and other downstream effector pathways. The tumor suppressor or PTEN (phosphatase and tensin
homologue) is the most important negative regulator of the PI3K signaling pathway. (“Status of PBK/Akt/mTOR Pathway Inhibitors in Lymphoma.” Clin Lymphoma, Myeloma Leuk , 2014, 14(5), 335-342.)
[0005] The signaling network defined by phosphoinositide 3-kinases (PI3Ks), AKT and mammalian target of rapamycin (mTOR) controls most hallmarks of cancer, including cell cycle, survival, metabolism, motility and genomic instability. The pathway also contributes to cancer promoting aspects of the tumor environment, such as angiogenesis and inflammatory cell recruitment. The lipid second messenger produced by PI3K enzymes, phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3; also known as PIP3), is constitutively elevated in most cancer cells and recruits cytoplasmic proteins to membrane-localized‘onco’ signal osomes.
[0006] Cancer genetic studies suggest that the PI3K pathway is the most frequently altered pathway in human tumors: the PIK3CA gene (encoding the PI3K catalytic isoform pl lOa) is the second most frequently mutated oncogene, and PTEN (encoding phosphatase and tensin homolog, the major PtdIns(3,4,5)P3 phosphatase) is among the most frequently mutated tumor suppressor genes. In accord, a recent genomic study of head and neck cancer found the PI3K pathway to be the most frequently mutated. Indeed, even in cancer cells expressing normal PI3K and PTEN genes, other lesions are present that activate the PI3K signaling network (that is, activated tyrosine kinases, RAS and AKT, etc ). As a net result of these anomalies, the PI3K pathway is activated, mutated or amplified in many malignancies, including in ovarian cancer (Campbell et al., Cancer Res., 2004, 64, 7678-7681; Levine et al., Clin. Cancer Res., 2005, 11, 2875-2878; Wang et al., Hum. Mutat., 2005, 25, 322; Lee et al., Gynecol. Oncol. ,2005, 97, 26-34), cervical cancer, breast cancer (Bachman et al.,· Cancer Biol., Ther, 2004, 3, 772-775; Levine et al., supra; Li et al., Breast Cancer Res. Treat., 2006, 96, 91-95; Saal et al., Cancer Res., 2005, 65, 2554-2559; Samuels and Velculescu, Cell Cycle, 2004, 3, 1221-1224), colorectal cancer (Samuels et al., Science, 2004, 304, 554; Velho et al., Eur. J. Cancer, 2005, 41, 1649-1654), endometrial cancer (Oda et al ., Cancer Res., 2005, 65, 10669-10673), gastric carcinomas (Byun et al., M. J. Cancer, 2003 , 104, 318-327; Li et al., supra; Velho et al., supra; Lee et al., Oncogene, 2005 , 24, 1477-1480), hepatocellular carcinoma (Lee et al., id), small and non-small cell lung cancer (Tang et al., Lung Cancer 2006, 11, 181-191; Massion et al , Am. J. Respir. Crit. Care Med., 2004, 170, 1088-1094), thyroid carcinoma (Wu et al., J. Clin. Endocrinol. Metab., 2005, 90, 4688-4693),
acute myelogenous leukemia (AML) (Sujobert et al., Blood, 1997, 106, 1063-1066), chronic myelogenous leukemia (CML) (Hickey et al., J. Biol. Chem ., 2006, 281, 2441-2450), glioblastomas (Hartmann et al. Jlcta Neuropathol (Bert ), 2005, 109, 639-642; Samuels et al., supra), Hodgkin and non-Hodgkin lymphomas (“PI3K and cancer: lessons, challenges and opportunities”, Nature Reviews Drug Discovery., 2014, 13, 140).
[0007] The PI3K pathway is hyperactivated in most cancers, yet the capacity of PI3K inhibitors to induce tumor cell death is limited. The efficacy of PI3K inhibition can also derive from interference with the cancer cells’ ability to respond to stromal signals, as illustrated by the approved PI3K5 inhibitor idelalisib in B-cell malignancies. Inhibition of the leukocyte-enriched PI3K5 or RI3Kg may unleash antitumor T-cell responses by inhibiting regulatory T cells and immune-suppressive myeloid cells. Moreover, tumor angiogenesis may be targeted by PI3K inhibitors to enhance cancer therapy. (“Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy”, Cancer Discov., 2016, 6(10), 1090-1105.)
[0008] mTOR is a highly conserved serine-threonine kinase with lipid kinase activity and participitates as an effector in the PI3K/AKT pathway. mTOR exists in two distinct complexes, mTORCl and mTORC2, and plays an important role in cell proliferation by monitoring nutrient avaliability and cellular energy levels. The downstream targets of mTORCl are ribosomal protein S6 kinase 1 and eukaryotic translation initiation factor 4E-binding protein 1, both of which are crucial to the regulation of protein synthesis. (“Present and future of PI3K pathway inhibition in cancer: perspectives and limitations”, Current Med. Chem., 2011, 18, 2647-2685).
[0009] Knowledge about consequences of dysregulated mTOR signaling for tumorigenesis comes mostly from studies of pharmacologically disruption of mTOR by repamycin and its analogues such as temsirolimus (CCI-779) and everolimus (RADOOl).Rapamycin was found to inhibit mTOR and thereby induce G1 arrest and apoptosis. The mechanism of rapamycin growth inhibition was found to be related to formation of complexes of rapamycin with FK-binding protein 12 (FKBP-12). These complexes then bound with high affinity to mTOR, preventing activation and resulting in inhibition of protein translation and cell growth. Cellular effects of mTOR inhibition are even more pronounced in cells that have concomitant inactivation of PTEN. Antitumor activity of rapamycin was subsequently identified, and a number of rapamycin analogues such as temsirolimus and everolimus have been approved by the US Food and Drug
Administration for the treatment certain types of cancer.
[0010] Fibrosis is the formation of excess fibrous connective tissue in an organ or tissue in a reparative or reactive process. Examples of fibrosis include, but are not limited to pulmonary fibrosis, liver fibrosis, dermal fibrosis, and renal fibrosis. Pulmonary fibrosis, also called idiopathic pulmonary fibrosis (IPF), interstitial diffuse pulmonary fibrosis, inflammatory pulmonary fibrosis, or fibrosing alveolitis, is a lung disorder and a heterogeneous group of conditions characterized by abnormal formation of fibrous tissue between alveoli caused by alveolitis comprising cellular infiltration into the alveolar septae with resulting fibrosis. The effects of IPF are chronic, progressive, and often fatal.
[0011] The clinical course of IPF is variable and largely unpredictable. IPF is ultimately fatal, with historical data suggesting a median survival time of 2-3 years from diagnosis. A decline in forced vital capacity (FVC) is indicative of disease progression in patients with IPF and change in FVC is the most commonly used endpoint in clinical trials. A decline in FVC of 5% or 10% of the predicted value over 6-12 months has been associated with increased mortality in patients with IPF.
[0012] Our understanding of the pathogenesis of IPF has evolved from that of a predominantly inflammatory disease to one driven by a complex interplay of repeated epithelial cell damage and aberrant wound healing, involving fibroblast recruitment, proliferation and differentiation, and culminating in excess deposition of extracellular matrix. This shift in knowledge prompted a change in the type of compounds being investigated as potential therapies, with those targeted at specific pathways in the development and progression of fibrosis becoming the focus.
[0013] In patients with IPF, the mechanisms whereby PI3K/mTOR inhibitors act may involve inhibition of kinases such as PI3Ks and mTOR. This results in inactivation of cellular receptors for mediators involved in the development of pulmonary fibrosis. As a result, fibroblast proliferation is inhibited and extracellular matrix deposition is reduced. (“Update on diagnosis and treatment of idiopathic pulmonary fibrosis”, J Bras Pneumol. 2015, 41(5), 454-466.)
[0014] Accordingly, small-molecule compounds that specially inhibit, regulate and/or modulate the signal transduction of kinases, particularly including PI3K and mTOR as described above, are desirable as a means to prevent, manage, or treat proliferative disorders and fibrosis, particular idiopathic pulmonary fibrosis in a patient. One such small-molecule is A-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfon-amide, which has the chemical structure as shown in the following:
[0015] WO 2014130375A1 described the synthesis of N-(5 -(‘3 -cyanopyrazol o [l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (Example 3) and also disclosed the therapeutic activity of this molecule in inhibiting, regulating and modulating the signal transduction of protein kinases.
[0016] Different salts and solid state forms of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, improving the dissolution profile, stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also provide improvements to the final dosage form, for example, if they serve to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
Different salts and solid state forms of /V-(5-(3-cyanopyrazolo[l,5- ]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide are described herein.
PATENT
WO2014130375 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014130375
claiming new pyrazolo[1,5-a]pyridine derivatives are PI3K and mTOR inhibitors, useful for treating proliferative diseases
Example 3 N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
Step 1) 5-bromopyrazolo[1,5-a]pyridine
[196] A solution of ethyl 5-bromopyrazolo[1,5-a]pyridine-3-carboxylate (240
mmol) in 40% H2SO4 (12 mL) was stirred at 100 °C for 4 hours, then cooled to rt, and neutralized to pH=7 with aq. NaOH (6 M) in ice bath. The resulted mixture was extracted with DCM (25 mL x 2). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (175 mg, 99.5%).
MS (ESI, pos. ion) m/z: 196.9 [M+H]+.
Step 2) 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde
[197] To a solution of 5-bromopyrazolo[1,5-a]pyridine (175 mg, 0.89 mmol) in DCM (6 mL) was added (chloromethylene)dimethyliminium chloride (632 mg, 3.56 mmol). The reaction was stirred at 44 °C overnight, and concentrated in vacuo. The residue was dissolved in saturated NaHCO3 aqueous solution (25 mL) and the resulted mixture was then extracted with EtOAc (25 mL x 3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (225 mg, 100%).
MS (ESI, pos. ion) m/z: 225.0 [M+H]+.
Step 3) (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime
[198] To a suspension of 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde (225 mg, 1 mmol) in EtOH (10 mL) and H2O (5 mL) was added hydroxylamine hydrochloride (104 mg, 1.5 mmol). The reaction was stirred at 85 °C for 2 hours, then cooled to rt, and concentrated in vacuo. The residue was adjusted to pH=7 with saturated NaHCO3 aqueous solution. The resulted mixture was then filtered and the filter cake was dried in vacuo to give title compound as a yellow solid (240 mg, 99%).
MS (ESI, pos. ion) m/z: 240.0 [M+H]+.
Step 4) 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile
[199] A solution of (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime (240 mg,
1 mmol) in Ac2O (6 mL) was stirred at 140 °C for 18 hours, then cooled to rt, and concentrated in vacuo. The residue was washed with Et2O (1 mL) to give the title compound as a yellow solid (44 mg, 22.5%).
MS (ESI, pos. ion) m/z: 222.0 [M+H]+.
Step 5) N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
[200] 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (612 mg, 1.5 mmol), 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile (222 mg, 1 mmol), Pd(dppf)Cl2·CH2Cl2 (16 mg, 0.02 mmol) and Na2CO3 (85 mg, 0.8 mmol) were placed into a two-neck flask, then degassed with N2 for 3 times, and followed by adding 1,4-dioxane (5 mL) and water (1 mL). The resulted mixture was degassed with N2 for 3 times, then heated to 90 °C and stirred further for 5 hours. The mixture was cooled to rt and filtered. The filtrate was concentrated in vacuo and the residue was purified by a silica gel column chromatography (PE/EtOAc (v/v) = 1/2) to give the title compound as a light yellow solid (400 mg, 81.6%).
MS (ESI, pos. ion) m/z: 442.0 [M+H]+;
1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.37 (s, 1H), 9.02 (d, J = 7.2 Hz, 1H), 8.67 (s, 1H), 8.60 (d, J = 2.2 Hz, 1H), 8.26-8.16 (m, 2H), 7.82-7.72 (m, 1H), 7.57 (dd, J = 13.2, 5.8 Hz, 2H), 7.21 (t, J= 8.5 Hz, 1H), 3.67 (s, 3H).
PATENT
WO-2019125967
The invention relates to salts of pyrazolo[l,5-a]pyridine derivatives and use thereof, specifically relates to salt of /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl) -2,4-difluorobenzenesulfonamide (compound of formula (I)) and use thereof, further relates to composition containing said salts above. The salts or the composition can be used to inhibit/modulate protein kinases, further prevent, manage or treat proliferative disorders or pulmonary fibrosis in a patient.
Amorphous form of mono-sodium salt of HEC-68498 , useful for treating a proliferative disorder or pulmonary fibrosis.
The invention is further illustrated by the following examples, which are not be construed as limiting the invention in scope.
[00108] /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluoroben zenesulfonamide can be prepared according to the synthetic method of example 3 disclosed in WO2014130375 Al.
//////////////HEC-68498, HEC 68498, HEC68498, HEC Pharm , Calitor Sciences, Sunshine Lake Pharma, PHASE 1, proliferative disorder, pulmonary fibrosis, idiopathic pulmonary fibrosis, solid tumors, CT-365 , CT 365 , CT365
Fc1ccc(c(F)c1)S(=O)(=O)Nc2cc(cnc2OC)c3ccn4ncc(C#N)c4c3
CRD 1152, CURADEV PHARMA PRIVATE LTD
Several candidates….one is…….CRD1152
ONE OF THEM IS CRD 1152
Kynurenine pathway regulators (solid tumors)

Compound 2
CAS1638121-21-7
N3-(3-Chloro-4- fluorophenyl) furo[2,3- c]pyridine-2,3- diamine

COMPD 190
CAS 1638118-99-6

COMPD248
7-Chloro-N3- (3-chloro-4- fluorophenyl) furo[2,3- c]pyridine-2,3- diamine, 166
DMSO-d6: δ 7.87 (d, J = 5.1 Hz, 1H), 7.25 (s, 2H), 7.16-7.10 (m, 2H), 6.88 (d, J = 5.1 Hz, 1H), 6.59 (dd, J′ = 6.2 Hz, J″ = 2.6 Hz, 1H), 6.48 (dt, J′ = 8.8 Hz, J″ = 6.7 Hz, J′′′ = 3.4 Hz, 1H) M + H] 312
OR
N3-(3,4- difluorophenyl)- 7-(pyridin-4- yl)furo[2,3- c]pyridine-2,3- diamine, 184
CD3CN: δ 8.72 (s, 2H), 8.26 (s, 3H), 7.07-7.03 (m, 2H), 6.47-6.40 (m, 2H), 5.74 (s, 1H), 5.55 (s, 2H) M + H] 339
OR

COMPD73
CAS 1638117-85-7
Several candidates………..CRD1152
 67
67
 66
66
| Company | Curadev Pharma Pvt. Ltd. |
| Description | Small molecule dual indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO1; IDO) inhibitor |
| Molecular Target | Indoleamine 2,3-dioxygenase (INDO) (IDO) ; Tryptophan 2,3-dioxygenase (TDO2) (TDO) |
| Mechanism of Action | Indoleamine 2,3-dioxygenase (INDO) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat cancer |
| Regulatory Designation | |
| Partner | Roche |
Hoffmann-La Roche partners with Curadev Pharma Ltd. for IDO1 and TDO inhibitors (April 20, 2015)
Curadev Pharma Pvt Ltd., founded in 2010 and headquartered in New Delhi, announced that it has entered into a research collaboration and exclusive license agreement with Roche for the development and commercialization of IDO1 and TDO inhibitors to treat cancer. The agreement covers the development of CRD1152, the lead preclinical immune tolerance inhibitor and a research collaboration with Roche’s research and early development organization to further explore the IDO and TDO pathways.
IDO1 (indoleamine-2,3-dioxygenase-1) and TDO (tryptophan-2,3-dioxygenase) are enzymes that mediate cancer-induced immune suppression. This mechanism is exploited by tumor cells as well as certain type of immune cells, limiting the anti-tumor immune response. Dual inhibition of the IDO1 and TDO pathways promises to maintain the immune response, prevent local tumor immune escape and potentially avoid resistance to other immunotherapies when used in combination, and could lead to new treatment options for cancer patients. Curadev’s preclinical lead-compound, a small-molecule that shows potent inhibition of the two rate-limiting enzymes in the tryptophan to kynurenine metabolic pathways, has the potential for mono therapy as well as combination with Roche’s broad oncology pipeline and portfolio.
Under the terms of agreement, which includes a research collaboration with Roche’s research and early development organization, Curadev will receive an upfront payment of $25 million and will be eligible to receive up to $530 million in milestone payments, as well as escalating royalties potentially reaching double digits for the first product from the collaboration developed and commercialized by Roche. Curadev is also eligible for milestones and royalties on any additional products resulting from the research collaboration.




Curadev Announces Research Collaboration and Licensing Agreement to Develop Cancer Immunotherapeutic
Curadev’s dual IDO and TDO immune tolerance inhibitor – a novel approach in cancer immunotherapy
Apr 20, 2015, 06:30 ET from Curadev
NEW DELHI, India, April 20, 2015 /PRNewswire/ —
Curadev Pharma Private Ltd. today announced that it has entered into a research collaboration and exclusive license agreement with Roche for the development and commercialization of IDO1 and TDO inhibitors. The agreement covers the development of the lead preclinical immune tolerance inhibitor and a research collaboration with Roche’s research and early development organization to further explore the IDO and TDO pathways.
IDO1 (indoleamine-2, 3-dioxygenase-1) and TDO (tryptophan-2, 3-dioxygenase) are enzymes that mediate cancer-induced immune suppression. This mechanism is exploited by tumor cells as well as certain type of immune cells, limiting the anti-tumor immune response.
Dual inhibition of the IDO1 and TDO pathways promises to maintain the immune response, prevent local tumor immune escape and potentially avoid resistance to other immunotherapies when used in combination, and could lead to new treatment options for cancer patients. Curadev’s preclinical lead-compound, a small-molecule that shows potent inhibition of the two rate-limiting enzymes in the tryptophan – to kynurenine metabolic pathways, has the potential for mono therapy as well as combination with Roche’s broad oncology pipeline and portfolio.
“We are very excited to be working with the global leader in oncology with their unrivalled expertise in clinical development,” said Arjun Surya, PhD, Chief Scientific Officer, Curadev. “The collaboration acknowledges our focused research efforts on patient-critical drug targets that have yielded a drug candidate that could make a significant difference in the development of novel treatments for patients suffering from cancer.”
Under the terms of agreement, which includes a research collaboration with Roche’s research and early development organization to further extend Curadev’s findings, Curadev will receive an upfront payment of $25 million and will be eligible to receive up to $530 million in milestone payments based on achievement of certain predetermined events and sales levels as well as escalating royalties potentially reaching double digits for the first product from the collaboration developed and commercialized by Roche. Curadev would also be eligible for milestones and royalties on any additional products resulting from the research collaboration. Roche will fund future research, development, manufacturing and commercialization costs and will also provide additional research funding to Curadev for support of the research collaboration.
About Curadev
Headquartered in New Delhi, India, Curadev Pharma Private Limited was founded in 2010 by a team of professionals from the pharmaceutical and biotech sectors with the mission to improve human health and enhance the quality of human life by accelerating the discovery and delivery of new drugs. Curadev focuses on the creation and out-licensing of pre-IND assets and IND packages for drug development.
For further information:
Curadev Partnering
Manish Tandon – VP and Chief Financial Officer, manish@curadev.in
PATENT
US20160046596) INHIBITORS OF THE KYNURENINE PATHWAY
Monali Banerjee
Sandip Middya
Ritesh Shrivastava
Sushil Raina
Arjun Surya
Dharmendra B. Yadav
Veejendra K. Yadav
Kamal Kishore Kapoor
Aranapakam Venkatesan
Roger A. Smith
Scott K. Thompson
ONE ………….Example 2
Synthesis of N3-(3-Chloro-4-fluoro-phenyl)-furo[2,3-c]pyridine-2,3-diamine (Compound 2)
Step 1: 3-Methoxymethoxy-pyridine

Step 2: 3-Methoxymethoxy-pyridine-4-carbaldehyde

Step 3: 3-Hydroxy-pyridine-4-carbaldehyde

Step 4: 4-{[3-Chloro-4-fluoro-phenylimino]-methyl}-pyridin-3-ol

Step 5: N3-(3-Chloro-4-fluoro-phenyl)-furo[2,3-c]pyridine-2,3-diamine

Monali Banerjee – Director, R&D
Ms. Banerjee has more than 10 years of research experience, during which she has held positions of increasing responsibility. Her past organizations include TCG Lifesciences (Chembiotek) and Sphaera Pharma. Ms. Banerjee is a versatile scientist with a deep understanding of the fundamental issues that underlie various aspects of drug discovery. At Curadev, she has been responsible for target selection, patent analysis, pharmacophore design, assay development, ADME/PK and in vivo and in vitro pharmacology. Ms. Banerjee holds a Masters in Biochemistry and a Bachelors in Chemistry both from Kolkata University.
writeup
|
The essential amino acid Tryptophan (Trp) is catabolized through the kynurenine (KYN) pathway. The initial rate-limiting step in the kynurenine pathway is performed by heme-containing oxidoreductase enzymes, including tryptophan 2,3-dioxygenase (TDO), indoleamine 2,3-dioxygenase-1 (IDO1), and indoleamine 2,3-dioxygenase-2 (IDO2). IDO1 and IDO2 share very limited homology with TDO at the amino acid level and, despite having different molecular structures, each enzyme has the same biochemical activity in that they each catalyze tryptophan to form N-formylkynurenine. IDO1, IDO2, and/or TDO activity alter local tryptophan concentrations, and the build-up of kynurenine pathway metabolites due to the activity of these enzymes can lead to numerous conditions associated with immune suppression.
|
|
Kynurenine pathway dysregulation and IDO1 and/or TDO activity also correlate with cardiovascular risk factors, and kynurenines and IDO1 are markers for Atherosclerosis and other cardiovascular heart diseases such as coronary artery disease (Platten et al., Science, 2005, 310(5749):850-5, Wirlietner et al. Eur J Clin Invest. 2003 July; 33(7):550-4) in addition to kidney disease. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease (Pawlak et al., Atherosclerosis, 2009, (204)1:309-314). Studies show that kynurenine pathway metabolites are associated with endothelial dysfunction markers in the patients with chronic kidney disease (Pawlak et al., Advances in Medical Sciences, 2010, 55(2):196-203).
|
///////CRD1152, CRD-1152, CRD 1152, CURADEV PHARMA PRIVATE LTD, ROCHE, IDO1 and TDO inhibitors, COLLABORATION, CANCER, indoleamine-2,3-dioxygenase-1, Hoffmann-La Roche, kynurenine pathway regulators, solid tumors
