
Home » Posts tagged 'serine/threonine kinase inhibitor'
Tag Archives: serine/threonine kinase inhibitor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ocadusertib
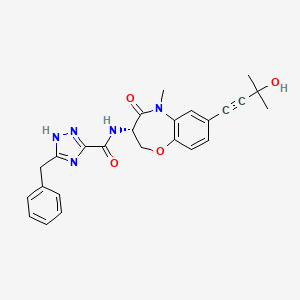
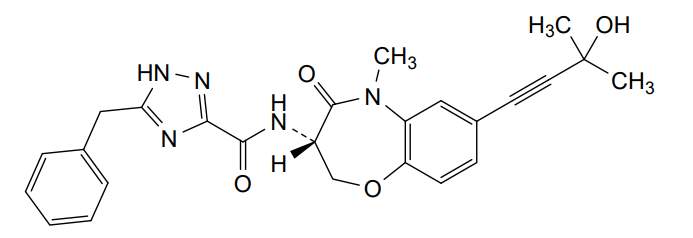
Ocadusertib
CAS 2382811-41-6
MF C25H25N5O4 MW 459.5 g/mol
5-benzyl-N-[(3S)-7-(3-hydroxy-3-methylbut-1-ynyl)-5-methyl-4-oxo-2,3-dihydro-1,5-benzoxazepin-3-yl]-1H-1,2,4-triazole-3-carboxamide
5-benzyl-N-[(3S)-7-(3-hydroxy-3-methylbut-1-yn-1-yl)-5-methyl-4-oxo-2,3,4,5-tetrahydro-1,5-benzoxazepin-3-yl]-1H-1,2,4-triazole-3-carboxamide
serine/threonine kinase inhibitor, LY3871801, R552, LY 3871801, R 552, S53J4A7ME4
- OriginatorRigel Pharmaceuticals
- DeveloperEli Lilly and Company; Rigel Pharmaceuticals
- ClassAnti-inflammatories; Antirheumatics; Small molecules
- Mechanism of ActionRIPK1 protein inhibitors
- Phase IIRheumatoid arthritis
- No development reportedUnspecified
- 28 Mar 2025No recent reports of development identified for phase-I development in Unspecified(In volunteers) in Singapore (PO, Suspension)
- 14 Nov 2024Pharmacodynamics data from preclinical trials in Rheumatoid arthritis presented at the ACR Convergence 2024 (ACR-2024)
- 14 Nov 2024Safety and pharmacokinetics data from a phase I trial in Rheumatoid arthritis presented at the ACR Convergence 2024 (ACR-2024)
Ocadusertib (LY3871801/R552) is an oral, potent, and selective small-molecule RIPK1 inhibitor developed by Rigel Pharmaceuticals and Eli Lilly for autoimmune and inflammatory diseases. It is currently in Phase 2 clinical trials for treating moderate-to-severe rheumatoid arthritis. 
Key Aspects of Ocadusertib:
- Mechanism of Action: It inhibits receptor-interacting serine/threonine-protein kinase 1 (RIPK1), which blocks necroptotic (cell death) responses and, consequently, reduces inflammation.
- Target Indications: Primarily focused on rheumatoid arthritis, it has also been investigated for psoriasis and general inflammatory joint conditions.
- Development Status: As of late 2025, it is in Phase 2 clinical trials (NCT05848258), with previous trials evaluating its safety, tolerability, and pharmacokinetics in healthy volunteers.
- Characteristics: It is designed to be a selective inhibitor, showing no significant inhibition in a broad panel of other kinases.
ACR Meeting Abstracts +3
Ocadusertib is a small molecule drug. The usage of the INN stem ‘-sertib’ in the name indicates that Ocadusertib is a serine/threonine kinase inhibitor. Ocadusertib is under investigation in clinical trial NCT05848258 (An Adaptive Phase 2a/2b Study of LY3871801 in Adult Participants With Rheumatoid Arthritis). Ocadusertib has a monoisotopic molecular weight of 459.19 Da.
- An Adaptive Phase 2a/2b Study of LY3871801 in Adult Participants With Rheumatoid ArthritisCTID: NCT05848258Phase: Phase 2Status: RecruitingDate: 2025-12-09
- A Study of LY3871801 in Healthy Asian and Non-Asian ParticipantsCTID: NCT05960851Phase: Phase 1Status: CompletedDate: 2024-01-10
- A Drug Interaction Study of LY3871801 in Healthy ParticipantsCTID: NCT05602675Phase: Phase 1Status: CompletedDate: 2023-04-18
- A Study of LY3871801 in Healthy ParticipantsCTID: NCT05222399Phase: Phase 1Status: CompletedDate: 2022-03-18
SYN
PAT
PAT
WO 2014/125444
PAT
- [WO2021046407]
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021046407&_cid=P20-MM02H8-46041-1
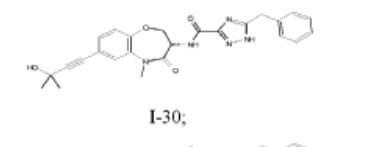
I-30: (S)-5-benzyl-N-(7-(3-hydroxy-3-methylbut-1-yn-1-yl)-5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][1,4]oxazepin-3-yl)-1H-1,2,4-triazole-3-carboxamide;
(S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][1,4]oxazepin-3-yl)-4H-1,2,4-triazole-3-carboxamide (WO 2014/125444), having a structure as illustrated below, was used as a comparative compound and was examined using a similar protocol as described by WO 2014/125444. This comparison
compound exhibited 93% inhibition at a dose of 30 mg/kg according to WO 2014/125444; however, in the inventors hands, the compound inhibited only 70% at 30 mg/kg. In comparison, compound I-30 of the present disclosure achieved greater than 85% inhibition at a dose of just 5 mg/kg using the similar assay protocol described above.
PAT
- RIP1 inhibitory compounds and methods of making and using the samePublication Number: CN-112368278-BPriority Date: 2018-05-03Grant Date: 2025-06-17
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021002237-A1Priority Date: 2018-05-03
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: AU-2019262144-A1Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021002236-A1Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021040053-A1Priority Date: 2018-05-03
- RIP1-inhibiting compounds and methods of making and using the samePublication Number: CN-112368278-APriority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2023113841-A1Priority Date: 2018-05-03
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-11370765-B2Priority Date: 2018-05-03Grant Date: 2022-06-28
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-11377428-B2Priority Date: 2018-05-03Grant Date: 2022-07-05
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021238153-A2Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: EP-4248975-A2Priority Date: 2018-05-03
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-12116354-B2Priority Date: 2018-05-03Grant Date: 2024-10-15
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-11332451-B2Priority Date: 2018-05-03Grant Date: 2022-05-17
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-11370764-B2Priority Date: 2018-05-03Grant Date: 2022-06-28
- RIP1 inhibitory compounds and methods of obtaining and using themPublication Number: MD-3788044-T2Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2019337907-A1Priority Date: 2018-05-03
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-10815206-B2Priority Date: 2018-05-03Grant Date: 2020-10-27
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021009537-A1Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2020407332-A1Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: EP-3788044-A1Priority Date: 2018-05-03



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

///////ocadusertib, serine/threonine kinase inhibitor, LY3871801, R552, LY 3871801, R 552, S53J4A7ME4
Nacresertib
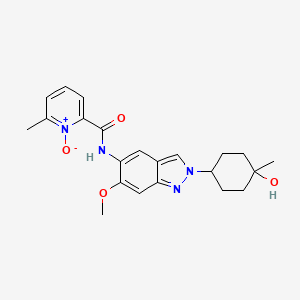
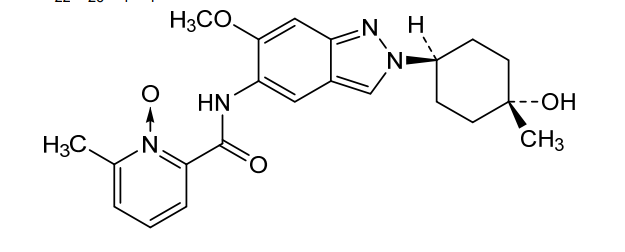
Nacresertib
CAS 2629977-59-7
MF C22H26N4O4, 410.5 g/mol
N-[2-(4-hydroxy-4-methylcyclohexyl)-6-methoxyindazol-5-yl]-6-methyl-1-oxidopyridin-1-ium-2-carboxamide
2-({2-[(1r,4r)-4-hydroxy-4-methylcyclohexyl]-6-methoxy-2H-indazol-5-yl}carbamoyl)-6-methylpyridine
1-oxide
serine/threonine kinase inhibitor, MB3QBD4BE7,
SYN
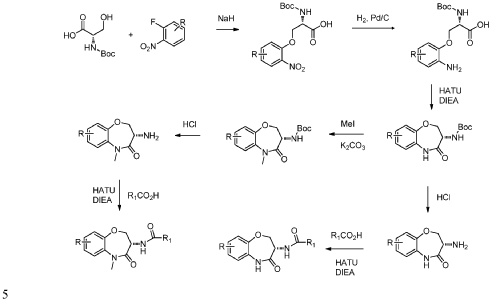
Example 1: Synthesis of Compound 001
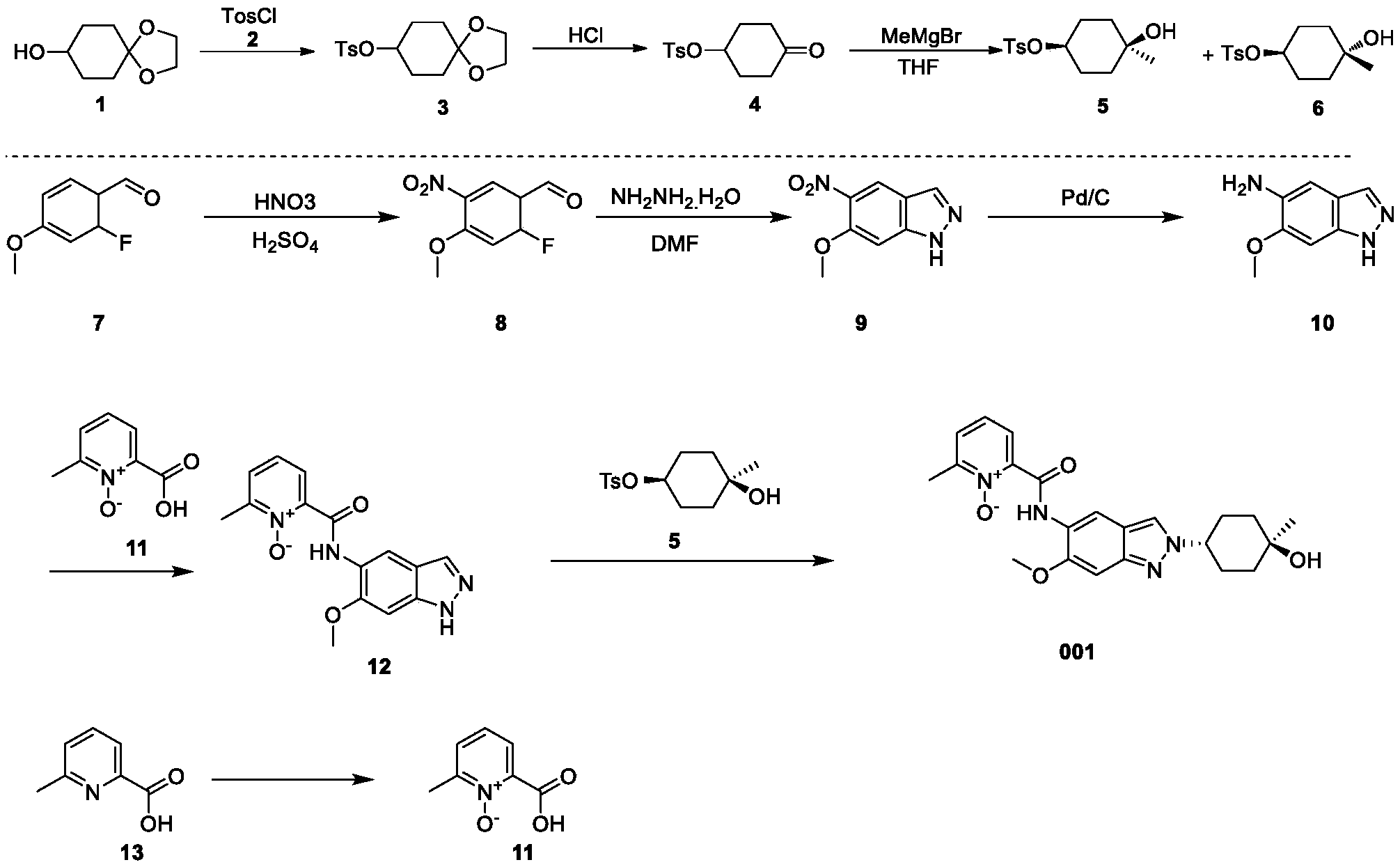
ynthesis of compound 001, namely 2-((2-(trans-4-hydroxy-cis-4-methylcyclohexyl)-6-methoxy-2H-indazol-5-yl)carbamoyl)-6-methylpyridine 1-oxide [0133]
[0134]Cesium carbonate (985 mg) was added to 5 mL of a DMF solution containing compound 12 (300 mg) and compound 5 (344 mg) at 25°C. The reaction mixture was stirred at 90°C for 16 hours. The reaction mixture was then added to 30 mL of water and extracted with ethyl acetate (10 mL * 3). The organic phase was concentrated under reduced pressure, and the residue was purified by preparative high-performance liquid chromatography (HPLC) using a column (CH3CN :
H2O ( 0.1 % NH4HCO3 ) = 15-45%, UV: 214 nm, flow rate: 15 mL/min) to obtain compound 001 (70 mg, yield 17%).[0135]
1 H NMR (400MHz, DMSO-d6): δ14.16(s,1H),8.78(s,1H),8.34(s,1H),8.32-8.30(m,1H),7.77(d,J=7.6Hz,1H),7.58(t,J=8.0Hz,1H),7.13(s,1 H),4.45(s,1H),4.43-4.40(m,1H),3.95(s,3H),2.53(s,3H),2.09-2.00(m,4H),1.68-1.58(m,4H),1.22(s,3H).LCMS: Rt=3.646min,[M+H] + =411.1.
SYN
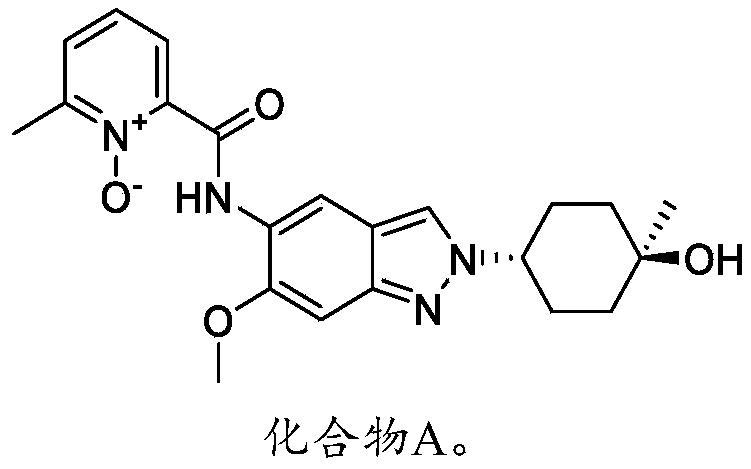
2-((2-(trans-4-hydroxy-cis-4-methylcyclohexyl)-6-methoxy-2H-indazol-5-yl)carbamoyl)-6-methylpyridine 1-oxide as shown in the following formula:
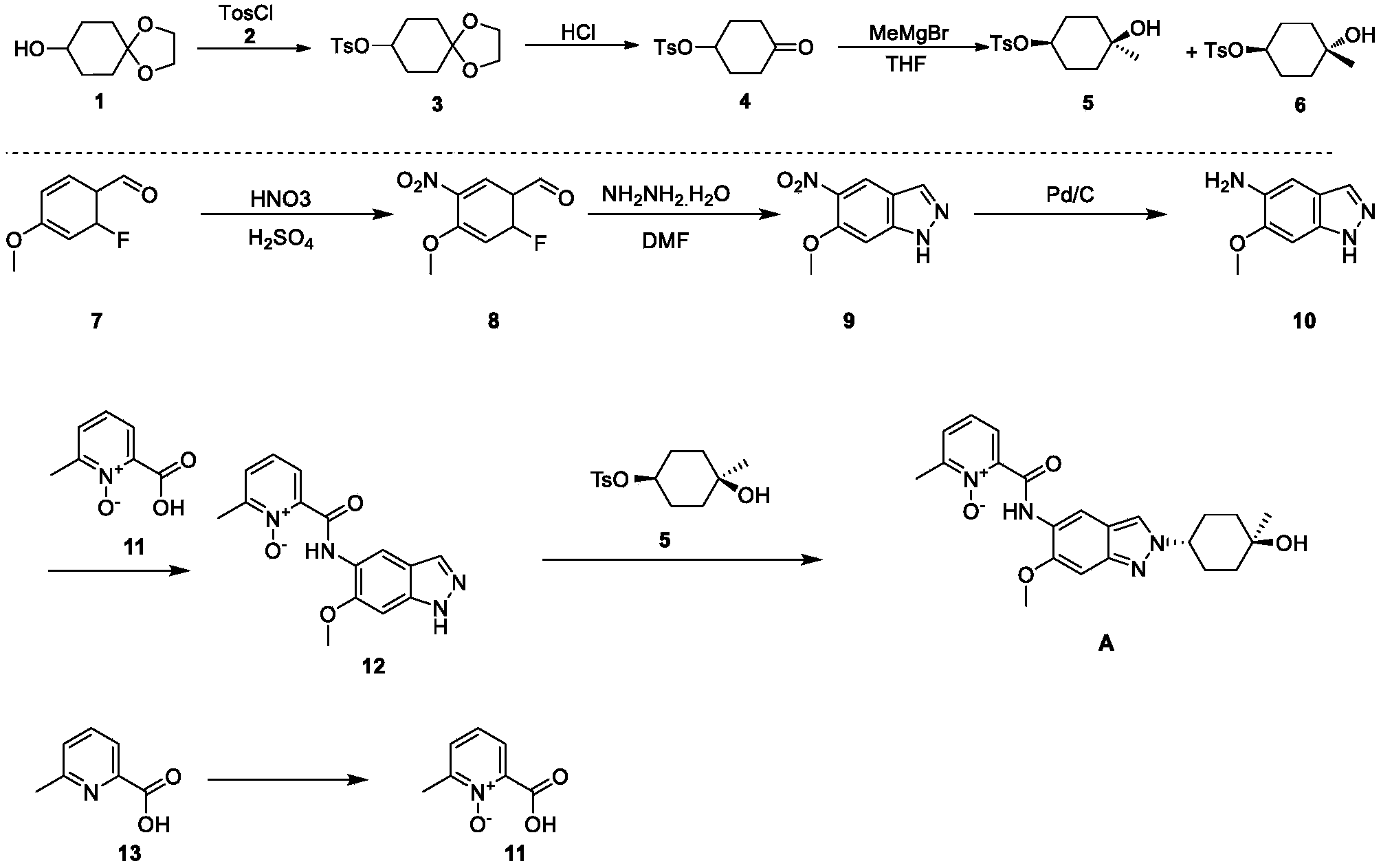
[0257](1) Synthesis of compound 3
[0258]DMAP (42.5 g), compound 2 (63.4 g), and triethylamine (63.9 g) were added sequentially to a 500 mL solution of compound 1 (50 g) in dichloromethane at 15°C, and the mixture was stirred at 25°C for 18 hours. Dichloromethane (200 mL) was added to the reaction mixture, followed by washing with water (300 mL × 2), then with 1 M dilute hydrochloric acid (300 mL × 3). The organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain 98 g of a yellow solid, yield: 99% (i.e., compound 3).
[0259](2) Synthesis of compound 4
[0260]1M dilute hydrochloric acid (300 mL) was added to a tetrahydrofuran solution (50 g) of compound 3 at 15°C, and the mixture was stirred at 25°C for 20 hours. The mixture was cooled to 0°C. The pH was adjusted to 9 with 1M sodium hydroxide solution. Extraction was performed with ethyl acetate (200 mL × 3). The extract was washed with saturated sodium chloride solution (300 mL). The solution was dried over anhydrous sodium sulfate. The mixture was filtered. The solution was concentrated under reduced pressure, and the residue was slurried with petroleum ether (150 mL) to give 39 g of a white solid, 91% yield (i.e., compound 4).
[0261](3) Synthesis of compounds 5 & 6
[0262]At -40°C, a tetrahydrofuran solution (200 mL) of compound 4 (34.5 g) was added dropwise to a tetrahydrofuran solution (500 mL) of methyl magnesium bromide (85.8 mL). The mixture was stirred at -40°C for 4 hours. The reaction was quenched with a saturated ammonium chloride solution (100 mL). The mixture was extracted with ethyl acetate (500 mL × 3). The extract was washed with saturated brine (300 mL), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (petroleum ether: ethyl acetate = 5:1) to give a colorless oily compound 5 (4.3 g, 10%), a colorless oily compound 6 (7.0 g, 17%), and a mixture of 12 g.
[0264]
1H NMR (400MHz,CDCl3 ) : δ7.79(d,J=8.0Hz,2H), 7.32(d,J=8.4Hz,2H), 4.52-4.41(m,1H), 2.44(s,3H), 1.95-1.80(m,2H), 1.77-1.61(m,4H), 1.46-1.35(m,2H), 1.19(s,3H).
[0266]
1H NMR (400MHz,CDCl3 ) : δ7.79(d,J=8.4Hz,2H), 7.33(d,J=8.0Hz,2H), 4.74-4.64(m,1H), 2.44(s,3H), 1.92-1.79(m,2H), 1.77-1.62(m,4H), 1.49-1.38(m,2H), 1.23(s,3H).
[0267](4) Synthesis of compound 8
[0268]A mixture of concentrated sulfuric acid (1.6 mL, 98%) and nitric acid (1.6 mL, 70%) was added dropwise to a solution of compound 7 (2.0 g) in concentrated sulfuric acid (12 mL, 98%) at -15°C. After the addition was complete, the mixture was stirred at -15°C for 2 hours. The reaction solution was then slowly poured into ice water and stirred for 5 minutes. The mixture was filtered, washed with water, and the solid was collected and dried under reduced pressure to give 2.5 g of a yellow solid, yield: 97% (i.e., compound 8).
[0269](5) Synthesis of compound 9
[0270]Hydrazine hydrate (2.4 mL, 98%) was added to a DMF (20 mL) solution of compound 8 (2.0 g) at room temperature. After the addition was complete, the mixture was heated to 120 °C and stirred for 16 hours. After cooling to room temperature, the mixture was slowly poured into ice water and stirred. The mixture was filtered, the solid was washed with water, and the solid was collected and concentrated under reduced pressure to obtain 1.3 g of yellow solid. Yield: 67% (i.e., compound 9).
[0271](6) Synthesis of compound 10
[0272]Compound 9 (12.4 g) and palladium on carbon (7 g, 10%) were added sequentially to 400 mL of ethyl acetate at 15°C. After the addition was complete, the mixture was stirred for 18 hours under hydrogen protection at 15°C. The palladium on carbon was filtered off from the reaction solution, and the filtrate was concentrated and evaporated to dryness to obtain 10.4 g of white solid product, with a yield of 99% (i.e., compound 10).
[0273](7) Synthesis of compound 12
[0274]EDCI.HCl (2.6 g) was added to a Py (15 mL) solution of compound 10 (1.5 g) and compound 11 (1.4 g) at 25°C. The reaction mixture was stirred at 25°C for 16 hours. The reaction mixture was concentrated and evaporated to dryness. The residue was slurried by passing MeOH/H₂O ( 20 mL/20 mL) to obtain 1.3 g of a yellow solid product, with a yield of 48% (i.e., compound 12).
[0275](8) Synthesis of compound A
[0276]
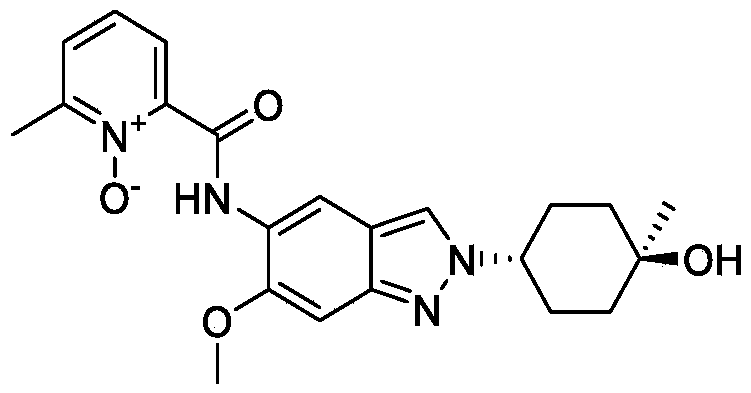
[0277]Cesium carbonate (985 mg) was added to 5 mL of a DMF solution containing compound 12 (300 mg) and compound 5 (344 mg) at 25°C. The reaction mixture was stirred at 90°C for 16 hours. The reaction mixture was then added to 30 mL of water and extracted with ethyl acetate (10 mL × 3). The organic phase was concentrated under reduced pressure, and the residue was purified by preparative high-performance liquid chromatography (HPLC) using a column (CH3CN :
H2O (
0.1 % NH4HCO3
) = 15-45%, UV: 214 nm, Flowrate: 15 mL/min) to obtain 70 mg of a yellow solid, with a yield of 17% (i.e., compound A).
[0278]
1H NMR(400MHz,DMSO-d6):δ14.16(s,1H),8.78(s,1H),8.34(s,1H),8.32-8.30(m,1H),7.77(d,J=7.6Hz,1H),7.58(t,J=8.0Hz,1H),7.13(s,1H),4.45(s,1H),4.43-4.40(m,1H),3.95(s,3H),2.53(s,3H),2.09-2.00(m,4H),1.68-1.58(m,4H),1.22(s,3H).LCMS:Rt=3.646min,[M+H] +=411.1.
PAT
- Irak inhibitor and preparation method therefor and use thereofPublication Number: EP-4015513-B1Priority Date: 2019-09-24Grant Date: 2023-11-01
- An IRAK inhibitor and its preparation method and usePublication Number: CN-114391013-BPriority Date: 2019-09-24Grant Date: 2024-01-26
- Irak inhibitor and preparation method therefor and use thereofPublication Number: US-2022298139-A1Priority Date: 2019-09-24
- A kind of IRAK inhibitor and its preparation method and usePublication Number: CN-114391013-APriority Date: 2019-09-24
- A kind of polymorphic form of compound and its preparation method and applicationPublication Number: CN-115109035-APriority Date: 2021-03-19
- Polymorphic forms of compound and preparation method therefor and application thereofPublication Number: EP-4310080-A1Priority Date: 2021-03-19
- Polymorphic forms of compound and preparation method therefor and application thereofPublication Number: US-2024182443-A1Priority Date: 2021-03-19
- Preparation method for fused pyrazole-type compoundPublication Number: US-2023250064-A1Priority Date: 2020-06-23
- An IRAK inhibitor and its preparation method and usePublication Number: CN-118146193-APriority Date: 2019-09-24
- IRAK4 inhibitor composition, and preparation method and application thereofPublication Number: CN-115252609-BPriority Date: 2022-08-01Grant Date: 2023-05-26
- Irak4 inhibitor composition, preparation method therefor and use thereofPublication Number: EP-4566607-A1Priority Date: 2022-08-01
- Composition of IRAK4 inhibitor, preparation method and application thereofPublication Number: CN-115252609-APriority Date: 2022-08-01
- Use of indazoles for treating psoriasisPublication Number: CN-114404415-APriority Date: 2022-02-25
- Use of indazole compound for treating psoriasisPublication Number: US-2025161283-A1Priority Date: 2022-02-25
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////nacresertib, serine/threonine kinase inhibitor, MB3QBD4BE7,
