PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise Drug Trials Snapshot Phase II Developer:Theracos, Inc.
Conditions
Phases
Recruitment
Interventions
Sponsor/Collaborators
Diabetes Mellitus Type 2
Phase 2
Completed
Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)
Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).
The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)
To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.
Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)
To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)
A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)
To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)
To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.
Preparation of (2-chloro-5-iodophenyl)methan-1-ol
A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.
Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex
Example 1A
Preparation of 2-cyclopropoxyethanol (1)
To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)
To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)
A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)
To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)
To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.
To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7
[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8
A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References: 1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates, whichmaybeacommonchallengetootherSGLT2inhibitors.31 Anotherpatent disclosedbyPiramal Enterprises suggesteda similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34 Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2 whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane, whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was reported in 92% yield. Details on stereoselectivity of this approachwerenotdisclosed. Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol 3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether 3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.
(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447− 453. (29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79. (30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive review of small-molecule drugs for the treatment of type 2 diabetes mellitus: Synthetic approaches and clinical applications. Eur. J. Med. Chem. 2024, 267, No. 116185. (31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F. Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A. Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev. 2018, 22, 467−488. (32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.; Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′ position as potent and selective renal sodium-dependent glucose co transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes. Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470. (33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the preparation of benzyl-benzene C-glycosides via coupling reaction as potential SGLT2 inhibitors. US 20130267694, 2013. (34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal, D. Aprocess for the preparation of SGLT2 inhibitor and intermediates thereof. WO 2018207113, 2018. (35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu, B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside derivatives as antidiabetic agents. WO 2009026537, 2009.
Bexagliflozin (Brenzavvy) On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108]. The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring. BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol. Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453. [105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng, B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293. [106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M. P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin. Pharmacother. 22 (2021) 2095–2103. [107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am. Soc. Nephrol. 18 (2023) 279–289. [108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr. Drug Saf. 13 (2018) 84–91. [109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd, J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives as Antidiabetic Agents, 2009. WO2009026537A1.
.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Fruquintinib, also known as HMPL-013, is an orally available, small molecule inhibitor of vascular endothelial growth factor receptors (VEGFRs), with potential anti-angiogenic and antineoplastic activities.
HMPL-013, a novel small molecule compound that selectively inhibits vascular endothelial growth factor receptor (VEGFR), is in phase III clinical studies at Hutchison MediPharma for the treatment of locally advanced or metastatic colorectal cancer. Phase II clinical trials are also ongoing for the treatment of non-squamous non-small cell lung cancer.
Early clinical development is under way at the company for the treatment of gastric cancer in combination with paclitaxel.
Fruquintinib’s mechanism of action is the inhibition of all three forms of VEGF receptors (VEGFR-1, 2, 3). Competitive advantages over currently marketed therapies are the compound’s unique kinase profile, a highly potent efficacy and excellent kinase selectivity, large safety margin, a broad spectrum antitumor activity and a low cost of goods.
Upon oral administration, fruquintinib inhibits VEGF-induced phosphorylation of VEGFRs 1, 2, and 3 which may result in the inhibition of migration, proliferation and survival of endothelial cells, microvessel formation, the inhibition of tumor cell proliferation, and tumor cell death. Expression of VEGFRs may be upregulated in a variety of tumor cell types.
In 2013, the company entered into a licensing, co-development, and commercialization agreement in China with Eli Lilly.
Angiogenesis is a physiological process of growing new blood vessels from pre-existing vessels. It takes place in a healthy subject to heal wounds, i.e., restoring blood flow to tissues after injury or insult.
Excessive angiogenesis may be triggered by certain pathological conditions such as cancer, age-related macular degeneration, and chronic inflammatory disease. As a result, new blood vessels feed diseased tissues and destroy normal tissues. In cancer, new blood vessels also allow tumor cells to escape into the circulation and lodge in other organs.
Vascular endothelial growth factor (VEGF), a homodimeric glycoprotein, and its receptors, e.g., kinase insert domain receptor (KDR), constitute an important angiogenic pathway. Studies have shown that inhibition of KDR resulted in endothelial cell apoptosis and, thus, suppression of angiogenesis. See Rubin M. Tuder, Chest, 2000; 117: 281. KDR inhibitors are therefore potential candidates for treating an angiogenesis-related disorder.
Written by Richard Daverman, PhD, Executive Editor, Greg B. Scott.
Hutchison MediPharma, a division of Chi-Med reported that fruquintinib met its primary endpoint in a second proof-of-concept China trial, this time as a treatment for advanced non-squamous non-small cell lung cancer. The company said fruquintinib “clearly” met its primary endpoint of progression-free survival, though specific data are being held for a scientific meeting. In 2013, Hutchison out-licensed China rights for the drug to Lilly. In May, the first proof-of-concept trial triggered two payments from Lilly to HMP totaling $18 million. More details…. http://www.chinabiotoday.com/articles/20150904
EXAMPLE 1 Synthesis of 6-(6,7-dimethoxyquinazolin-4-yloxy)-N,2-dimethylbenzofuran-3-carboxamide:
To a solution of 4-chloro-6,7-dimethoxyquinazoline (1 equiv.) in 2 ml CH3CN were added 6-hydroxy-N,2-dimethylbenzofuran-3-carboxamide (1 equiv.) and K2CO3 (1.5 equiv.). The mixture was refluxed under stirring for 10 hr. After the solvent was evaporated, the residue was washed with water, dried over MgSO4, filtered, concentrated, and purified by column chromatography to give the title compound in a yield of 85%.
Example a: 6- (6,7-dimethoxy-quinazolin-4-oxo) -N, 2- dimethyl-benzofuran-3-carboxamide
[0048]
[0049] 4-Chloro-6,7-dimethoxy-quinazoline (1 mmol) was dissolved in 2 ml of acetonitrile, followed by addition of 6-hydroxy -N, 2- dimethyl-benzofuran-3- amide (1 mmol) and potassium carbonate (1.5 mmol).The reaction mixture was heated at reflux for 10 hours, concentrated to dryness, washed with water, and purified to give the desired product, yield 85%.
Method for the simplified production of (3-chloro-4-fluoro-phenyl)- 7-(3-morpholino-4-yl-propoxy)-6-nitro-quinazoline-4-yl]-amine or (3-chloro-4-fluoro-phenyl)- 7-(3-morpholino-4-yl-propoxy)-6-amino-quinazoline-4-yl]-amine
Mr To has been a Director since 2000 and an Executive Director and Chairman since 2006. He is also Chairman of the Remuneration Committee and a member of the Technical Committee of the Company. He is managing director of Hutchison Whampoa (China) Limited (“Hutchison China”) and has been with Hutchison China for over thirty years, building its business from a small trading company to a billion dollar investment group. He has negotiated major transactions with multinationals such as Procter & Gamble, Lockheed, Pirelli, Beiersdorf, United Airlines and British Airways.
Mr To’s career in China spans more than thirty years and he is well known to many of the top Government leaders in China. Mr To is the original founder of Hutchison Whampoa Limited’s healthcare business and has been instrumental in the acquisitions made to date. He received a First Class Honours Bachelor’s Degree in Mechanical Engineering from Imperial College, London and an MBA from Stanford University’s Graduate School of Business.
Christian Hogg, M.B.A. Chief Executive Officer, Hutchison China MediTech Limited and Director, Hutchison MediPharma Holdings Limited
Mr Hogg has been an Executive Director and Chief Executive Officer since 2006. He is also a member of the Technical Committee of the Company. He joined Hutchison Whampoa (China) Limited in 2000 and has since led all aspects of the creation, implementation and management of the Company’s strategy, business and listing. This includes the creation of the Company’s start-up businesses and the acquisition and operational integration of assets that led to the formation of the Company’s China joint ventures.
Prior to joining Hutchison China, Mr Hogg spent ten years with Procter & Gamble starting in the US in Finance and then Brand Management in the Laundry and Cleaning Products Division. Mr Hogg then moved to China to manage P&G’s detergent business followed by a move to Brussels to run P&G’s global bleach business. Mr Hogg received a Bachelor’s degree in Civil Engineering from the University of Edinburgh and an MBA from the University of Tennessee.
Weiguo Su, Ph.D. Executive Vice President and Chief Scientific Officer
Dr. Su has headed all drug discovery and research since he joined, including creating our R&D strategy, the formation and growth of research platform, and the research and discovery of each and every small molecule drug candidate in the Company’s portfolio.
Prior to joining in 2005, Dr. Su spent 15 years with Pfizer’s US R&D organization. Dr. Su delivered several high quality new drug candidates during his time with Pfizer, most recently as a director in the Medicinal Chemistry Department.
He received his Ph.D. and post-doctoral fellowship in Chemistry from Harvard University under the guidance of Nobel Laureate Professor E. J. Corey, and his Bachelor’s degree in Chemistry from Fudan University in Shanghai, China.
USAN (AB-55) BOCOCIZUMAB
PRONUNCIATION boe” koe siz’ ue mab
THERAPEUTIC CLAIM Treatment of dyslipidemia
CHEMICAL NAME
1. Immunoglobulin G2, anti-(human neural apoptosis-regulated proteinase
1)(human-Mus musculus monoclonal PF-04950615 heavy chain), disulfide
with human-Mus musculus monoclonal PF-04950615 light chain, dimer
2. Immunoglobulin G2-kappa, anti-[human proprotein convertase subtilisin/hexin type 9 (neural apoptosis-regulated convertase 1, PC9)], humanized mouse monoclonal antibody; gamma 2 heavy chain (1-444) [humanized VH (Homo sapiens IGHV1-46-1*03 (90.8%) -(IGHD)-IGHJ6*01) [8.8.11] (1-118)-Homo sapiens IGHG2*01 CH2A100>S(327),CH2P101>S(328) (119-444)] (132-214′)-
disulfide with kappa light chain (1′-214′) [humanized V-KAPPA (Homo sapiensIGKV1-39*01 (88.2%)-IGKJ2*01 [6.3.9] (1′-107′)-IGKC*01 (108′-214′)]; dimer
(220-220”:221-221”:224-224”:227-227”)-tetrakisdisulfide
MOLECULAR FORMULA C6414H9918N1722O2012S54
MOLECULAR WEIGHT 145.1 kDa
TRADEMARK None as yet
SPONSOR Pfizer, Inc.
CODE DESIGNATIONS RN316, PF-04950615
CAS REGISTRY NUMBER 1407495-02-6
WHO NUMBER 9840
A phase 2b study of statin patients was presented at the 2014 American College of Cardiology. Monthly or bimonthly injections resulted in significantly reduced LDL-C at week 12.
The Phase 3 SPIRE trials plan to enroll 17,000 patients to measure cardiovascular risk. High risk and statin intolerant subjects will be included.
24 Mar 2015 Shionogi plans a phase III trial in Thrombocytopenia (in patients with chronic liver disease) in USA (NCT02389621)
31 Dec 2014 Preregistration for Thrombocytopenia in Japan (PO)
08 Nov 2013 Phase II development is ongoing in the US and the Europe
Process for preparing intermediates of an optically active 1,3-thiazole containing thrombopoietin receptor agonist Also claims crystalline forms of lusutrombopag intermediates and a process for preparing lusutrombopag. Shionogi is developing lusutrombopag, a small-molecule thrombopoietin mimetic, as an oral tablet formulation for treating thrombocytopenia.
In December 2014, an NDA was submitted in Japan. In May 2015, the drug was listed as being in phase III development for thrombocytopenia in the US and Europe.
The lusutrombopag, a low molecular-human thrombopoietin receptor agonist, its chemical formula, “(E) -3- [2,6-Dichloro-4- [4- [3 – [(S) -1-hexyloxyethyl] – 2-methoxyphenyl] -thiazol- 2-ylcarbamoyl] -phenyl] is a -2-methylacrylic acid “. lusutrombopag is represented by the following chemical structural formula.
Eltrombopag is represented by the following chemical structural formula.
Avatrombopag is represented by the following chemical structural formula.
Totrombopag choline is represented by the following chemical structural formula.
Synthesis of (R)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (C-3A) (not included in the present invention) and (S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (C-3B)
According to the same method as in Example 1, an optically active compound (C-3A) and an opticallly active compound (C-3B) were synthesized from (RS)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (B-3) obtained in Reference Example 3.Optically active compound (C-3A)Melting point: 139-141°C UNDESIRED
Second step: Synthesis of (S)-1-bromo-3-(1-hexyloxyethyl)-2-methyloxybenzene (18)
Using the same method as that of the second step of Example 3, the compound (18) was obtained from the compound (17) at a yield of 96%.
Optical rotation: -29.8 ± 0.6 degrees (CHCl3, c = 1.055, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.42 (3H, d, J = 6.5 Hz), 1.54 (2H, m), 3.29 (2H, m), 3.85 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 7.02 (1H, t, J = 7.9 Hz), 7.39 (1H, dd, J = 7.8 Hz, J = 1.7 Hz), 7.45 (1H, dd, J = 7.9 Hz, J = 1.7 Hz)
Third step and fourth step: Synthesis of (S)-4-(3-(1-hexyloxyethyl)-2-methyloxyphenyl)thiazole-2-amine (20)
Using the same method as that of the fourth step of Example 3, the compound (19) was obtained from the compound (18), subsequently according to the same method as that of the fourth step, the compound (20) was obtained.
Methods respectively for producing optically active compound having agonistic activity on thrombopoietin receptors and intermediate of said compound
(Step 1) Synthesis of compound (VII ‘) under a nitrogen atmosphere, it was dissolved compound 1 (2.00kg) in 1,2-dimethoxyethane (28.0kg). 25% LDA tetrahydrofuran – heptane – ethyl benzene solution (13.20kg) was added dropwise over 1 hour at -55 ℃, and stirred for 30 minutes. It was added dropwise over 40 minutes to 1,2-dimethoxyethane (3.0kg) solution of N- formyl morpholine (3.74kg) at -55 ℃, and stirred for 1 hour. 1,2-dimethoxyethane (3.0kg) solution of 2-phosphono-propanoic acid triethyl (3.74kg) was added dropwise over 45 minutes at 0 ℃, and stirred for 2 hours. 35% aqueous solution of sulfuric acid (15.8kg) was added dropwise over 40 minutes to the reaction solution. Water (16.0kg) was added and extracted. The resulting organic layer was washed with water (8.0kg), and the solvent was evaporated under reduced pressure. Acetonitrile (16.0kg) was added, and the mixture was stirred for 1 hour at 25 ℃, and the mixture was stirred and cooled to 0 ℃ 5 hours and 30 minutes. The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (16.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Over 1 hour and then cooled to 30 ℃, and the mixture was stirred for 45 minutes. Over 40 minutes and then cooled to 5 ℃, and the mixture was stirred for 3 hours.The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (13.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Furthermore, up to 30 ℃ over 1 hour and then cooled and stirred for 70 minutes. Over 30 minutes and then cooled to 5 ℃, and the mixture was stirred for 4 hours. I precipitated crystals were collected by filtration. Washed with 5 ℃ acetonitrile (3.2kg), and dried to give the compound (VII ‘) (1.63kg, 51.2% yield). NMR (CDCl 3 ) delta ppm: 8.07 (s, 2H), 7.47 (s, 1H), 4.32 (Q, 2H, J = 7.0 Hz), 1.79 (s, 3H), 1.38 (t, 3H, J = 7.0 Hz) Results of powder X-ray diffraction and I shown in Figure 1 and Table 3. [Table 3] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 8.1 ± 0.2 °, 16.3 ± 0.2 °, 19.2 ± 0.2 °, 20.0 ± 0. 2 °, the peak was observed at 24.8 ± 0.2 °, and 39.0 ± 0.2 ° degrees.
(Synthesis of Compound (XI ‘))
(Step 2) Synthesis of Compound 4 under a nitrogen atmosphere over Compound 3 (3.00kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (11.40kg) 1 hour at 25 ℃ in The dropped, and stirred for 2 hours. 1mol / L isopropylmagnesium chloride in tetrahydrofuran solution (0.56kg) was added at 25 ℃, and stirred for 2 hours. To the reaction mixture N- methoxymethyl -N- methylacetamide the (1.45kg) was added dropwise over at 25 ℃ 40 minutes, and stirred for 80 minutes. 7% hydrochloric acid (9.7kg) was added to the reaction mixture, and the mixture was extracted with toluene (11.0kg). The resulting organic layer twice with water (each 7.5kg) washed, the solvent was evaporated under reduced pressure to give Compound 4 (2.63kg). NMR (CDCl 3 ) delta ppm: 7.69 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.55 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.05 (t, 1H, J = 7.7 Hz), 3.88 (s, 3H), 2.64 (s, 3H) ppm:
(Step 3) Synthesis of Compound 5 Under a nitrogen atmosphere, chloro [(1S Compound 4 (2.63kg), 2S) -N- ( p- toluenesulfonyl) -1,2-diphenyl-ethane diamine] (p- cymene) ruthenium (II) (28.6g), it was added to tetrahydrofuran (1.3kg) and triethylamine (880.0g). Formic acid (570.0g) was added dropwise over 6 hours at 40 ℃, and stirred for 1 hour. In addition 3.5% hydrochloric acid (14.4kg) to the reaction mixture, and the mixture was extracted with toluene (13.0kg).The organic layer was washed with 3.5% hydrochloric acid (14.4kg) and water (7.5kg), the solvent was concentrated under reduced pressure to obtain a toluene solution of Compound 5 (4.44kg).
(Step 4) Synthesis of Compound 6 under a nitrogen atmosphere, it was a potassium hydroxide (6.03kg) was dissolved in water (6.0kg). To the solution, it added tetrabutylammonium bromide (182.0g) and toluene solution of Compound 5 (4.44kg). 1-bromo-hexane (2.79kg) was added dropwise over 1 hour at 60 ℃, and the mixture was stirred for 4 hours. And extracted by adding water (4.4kg) to the reaction solution. The resulting organic layer was filtered through powdered cellulose and extracted with toluene (3.0kg) and water (7.6kg) to the filtrate. The solvent it was evaporated under reduced pressure from the organic layer. Toluene operation of evaporated under reduced pressure and the solvent by the addition of a (7.8kg) was repeated five times to obtain a toluene solution of Compound 6 (10.0kg).
(Step 5) Synthesis of Compound 7 under a nitrogen atmosphere, magnesium powder (301.0g), in tetrahydrofuran (1.3kg), the compound in toluene (6.4kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (432.0g) 6 In addition of the toluene solution (0.50kg) at 30 ℃, and the mixture was stirred for 2 hours. Toluene solution of Compound 6 (9.50kg) was added dropwise over 3 hours at 50 ℃, and stirred for 2 hours. 1-bromo-hexane (746.0g) was added at 50 ℃, and the mixture was stirred for 1 hour. It was added dropwise over 1 hour at 5 ℃ toluene (5.3kg) solution of 2-chloro -N- methoxy -N- methyl-acetamide (1.78kg), and stirred for 1 hour. 3.7% hydrochloric acid (16.7kg) was added to the reaction mixture, and the mixture was extracted. The obtained organic layer was washed with water (15.0kg), and concentrated under reduced pressure to give a toluene solution of Compound 7 (8.25kg).
(Step 6) Synthesis of Compound (II ‘) under a nitrogen atmosphere, thiourea (1.03kg), in ethanol (1.2kg) and 65 ℃ toluene solution of compound 7 (8.25kg) in toluene (6.3kg) over 3 hours was added dropwise and stirred for 2 hours. The reaction solution was extracted by adding 0.7% hydrochloric acid (30.6kg), and washed twice with water (30.0kg). Ethanol in the organic layer (9.5kg), and extracted by addition of heptane (10.0kg) and 3.5% hydrochloric acid (5.9kg). The resulting aqueous layer with 4% hydrochloric acid (1.5kg) and ethanol (3.5kg) merged the aqueous layer was extracted from the organic layer, the ethanol was washed with heptane (10.0kg) (3.1kg) It was added. 8% aqueous sodium hydroxide (6.0kg) was added dropwise over at 5 ℃ 30 minutes, and stirred for 20 minutes. 8% aqueous sodium hydroxide (5.8kg) was added dropwise over a period at 5 ℃ 15 minutes.The precipitated crystals were collected by filtration, washed with 45% aqueous ethanol (10.9kg) and water (15.0kg) (crude crystals of Compound (II ‘)). The resulting crude crystals were dissolved in 50 ℃ in ethanol (8.1kg), over a period of 1 hour and then cooled to 10 ℃, and the mixture was stirred for 30 minutes. Water (10.0kg) over 2 hours was added dropwise and stirred for 30 minutes. The precipitated crystals were collected by filtration, washed with 50% aqueous ethanol (7.5kg) and water (10.0kg) (crystals of the compound after recrystallization from ethanol / water system (II ‘)). The resulting crystals were dissolved at 55 ℃ in toluene (1.6kg) and heptane (1.3kg), over 1 hour and cooled to 20 ℃, and stirred for 30 minutes. Heptane (6.3kg) over a period of 30 minutes was added dropwise and stirred for 15 minutes. The obtained crystals precipitated were collected by filtration, washed with a mixed solvent of toluene (0.3kg) and heptane (2.3kg), and dried to give compound (II ‘) (1.67kg, 44.5% yield) a (crystalline compound after recrystallization from toluene / heptane system (II ‘)).
NMR (CDCl 3 ) delta ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, M), 1.35 (3H, D, J = 6.5 Hz), 1.48 (2H, M), 3.25 ( 2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) it is shown in Figure 2 and Table 4 the results of powder X-ray diffraction. [Table 4] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 12.5 ± 0.2 °, 13.0 ± 0.2 °, 13.6 ± 0.2 °, 16.4 ± 0. 2 °, 23.0 ± 0.2 °, a peak was observed at 24.3 ± 0.2 ° degrees. Above, each of the compounds (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) crystallographic purity of the results of the , Fig. 3, I 4 and 5 as well as Table 5. [Table 5](HPLC was measured by the above method A.) As shown in the results of the above table, as compared to recrystallization from ethanol / water, recrystallized with toluene / heptane system, compounds having a high optical purity it is possible to manufacture a crystal of (II ‘). Next, the above-mentioned compound (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) results of crystals of HPLC of the respectively, Fig. 6, I 7 and 8 and Table 6. [Table 6] (units, .N.D shows the peak area of the (%). is, .HPLC to indicate not detected was measured by the above method B.) As shown in the results of Table, with ethanol / water system Compared to recrystallization, recrystallization from toluene / heptane system is found to be efficiently remove organic impurities A and organic impurities B.
(Step 7) Compound ‘Synthesis of DMSO adduct of (VIII) Under a nitrogen atmosphere, the compound (II ‘) (1.50kg) and compound (VII’) (1.43kg) in ethyl acetate (17.6kg) and triethylamine (1.09kg) were sequentially added, was dissolved.Diphenyl phosphorochloridate the (1.46kg) was added dropwise over 1 hour at 50 ℃, and the mixture was stirred for 3 hours. The reaction mixture was cooled to 25 ℃, after the addition of 2.6% hydrochloric acid (8.1kg), and extracted. The resulting organic layer to 6.3% aqueous solution of sodium hydroxide (3.2kg) and 14% aqueous sodium carbonate (5.2kg) was added and stirred for 20 minutes. Adjusted to pH7.5 with 8.3% hydrochloric acid and extracted. The organic layer it was washed with 4.8% sodium chloride aqueous solution (11.0kg). DMSO and (16.5kg) was added, and the mixture was concentrated under reduced pressure.DMSO and (5.8kg) was added, over a period at 40 ℃ 30 minutes was added dropwise water (0.9kg), and stirred for 1 hour. Over a period of 30 minutes, cooled to 25 ℃, and the mixture was stirred for 30 minutes. Over at 25 ℃ 30 minutes was added dropwise water (1.4kg), and the precipitated crystals were collected by filtration. After washing with 90% DMSO solution (10.0kg) and water (27.0kg), to obtain crystals of DMSO adduct and dried to Compound (VIII ‘) (2.98kg, 95.2% yield).
In the powder X-ray diffraction spectrum, diffraction angle (2θ): 5.2 ° ± 0.2 °, 7.0 ° ± 0.2 °, 8.7 ° ± 0.2 °, 10.5 ° ± 0.2 °, 12.3 ° ± 0.2 °, 14.0 ° ± 0.2 °, 15.8 ° ± 0.2 °, 19.3 ° ± 0.2 °, 22.5 ° peak was observed to ± 0.2 ° and 24.1 ° ± 0.2 °. TG / DTA analysis result it is shown in Figure 10. Then, each result of HPLC of concentrated dry solid and the above DMSO adduct crystals described in the following Reference Examples 1, 11 and 12, 13 and 14, and I are shown in Table 8. [Table 8] (unit, .HPLC showing peak areas of (%) was measured by the above methods C.) As shown in the results of the above Table, when compared with the extract, DMSO adduct of the compound (VIII ‘) The in the crystal, less residual organic impurities D, and it found to be about 56% removal.
(Step 8) under nitrogen atmosphere, DMSO adduct of the compound (VIII ‘) and (2.50kg) it was dissolved in ethanol (15.8kg). 24% sodium hydroxide aqueous solution (1.97kg) was added dropwise over a period at 45 ℃ 30 minutes to the solution and stirred for 3 hours. The reaction mixture was cooled to 25 ℃, water was added (20.0kg) and ethanol (7.8kg). 18% hydrochloric acid (2.61kg) was added dropwise over at 25 ℃ 30 minutes, followed by addition of seed crystals prepared according to the method described in Patent Document 23. After stirring for 3 hours and allowed to stand overnight. Thereafter, the precipitated crystals were collected by filtration, to give after washing with 50% aqueous ethanol solution (14.2kg), and dried to a compound (XI ‘) (1.99kg, 93.9% yield).
Patent Document 1: JP-A-10-72492 JP
Patent Document 2: WO 96/40750 pamphlet
Patent Document 3: JP-A-11-1477 JP
Patent Document 4: Japanese Unexamined Patent Publication No. 11-152276
Patent Document 5: International Publication No. 00/35446 pamphlet
Patent Document 6: JP-A-10-287634 JP
Patent Document 7: WO 01/07423 pamphlet
Patent Document 8: International Publication WO 01/53267 pamphlet
Patent Document 9: International Publication No. 02 / 059 099 pamphlet
Patent Document 10: International Publication No. 02/059100 pamphlet
Patent Document 11: International Publication No. 02/059100 pamphlet
Patent Document 12: International Publication No. 02/062775 pamphlet
Patent Document 13: International Publication No. 2003/062233 pamphlet
Patent Document 14: International Publication No. 2004/029049 pamphlet
Patent Document 15: International Publication No. 2005/007651 pamphlet
Patent Document 16: International Publication No. 2005/014561 pamphlet
Patent Document 17: JP 2005-47905 Japanese
patent Document 18: Japanese Patent Publication No. 2006-219480
Patent Document 19: Japanese Patent Publication No. 2006-219481
Patent Document 20: International Publication No. 2007/004038 pamphlet
Patent Document 21: International Publication No. 2007/036709 pamphlet
Patent Document 22: International Publication No. 2007/054783 pamphlet
Patent Document 23: International Publication No. 2009/017098 pamphlet
Non-Patent Document 1: Proceedings of the National Akademyi of Science of the United State of America (…. Proc Natl Acad Sci USA) 1992, Vol. 89, p 5640-5644.
Non-Patent Document 2: Journal of Organic (.. J. Org Chem) Chemistry 1984, Vol. 49, p 3856-3857.
Non-Patent Document 3: (.. J. Org Chem). Journal of Organic Chemistry, 1992, Vol. 57, p 6667-6669
Non-Patent Document 4:. Shinretto (Synlett) 2004 year Vol. 6, p 1092-1094
101 – Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia
Masami Takayama1, masami.takayama@shionogi.co.jp, Hajime Yamada3, Hiroshi Takemoto2, Takeshi Shiota2, Yoshikazu Tanaka2, Noriko Yamane2, Kouji Takahashi2, Naoki Oyabu3, Kenji Kuwabara3, Itsuki Oshima2, Kenzo Koizumi3, Hiroshi Yoshida3, Ayumu Nogami3, Tomomi Yamada3, Yutaka Yoshida3, Takami Murashi3, Shinichiro Hara2. (1) Department of Strategic Research Planning Offices, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan, (2) Department of Innovative Drug Discovery Research Laboratories, Shionogi & CO.,LTD, Toyonaka, Osaka 561-0825, Japan, (3) Department of Medicinal Research Laboratories, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan
As a drug candidate of thrombocytopenia, Lusutrombopag (S-888711) is in Phase III clinical trial stage right now. It is been proven that Lusutrombopag (S-888711) is excellent property in safety and efficacy by clinical trials. In this meeting, we will present in detail about the history of drug discovery of Lusutrombopag.Because Lusutrombopag (S-888711) acts specifically to human TPO receptor, we prepared TPOR-Ki/Shi mice expressing a mouse-human chimeric TPOR for evaluating the efficacy. This TPOR-Ki/Shi mice worked very well as an evaluation model of drug efficacy, so we were able to select Lusutrombopag from many candidate compounds. In this meeting, we will present the results of the efficacy in TPOR-Ki/Shi mice of Lusutrombopag and the similar drug (Eltrombopag). Sunday, March 16, 2014 07:00 PM General Poster Session (07:00 PM – 10:00 PM) Location: Dallas Convention Center Room: Hall E Monday, March 17, 2014 08:00 PM Sci-Mix (08:00 PM – 10:00 PM) Location: Dallas Convention Center Room: Hall F
On July 31, 2018, the Food and Drug Administration approved lusutrombopag (Mulpleta, Shionogi Inc.) for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure.
Approval was based on two randomized, double-blind, placebo-controlled trials (L-PLUS 1 and L-PLUS 2, NCT02389621) involving 312 patients with chronic liver disease and severe thrombocytopenia who were undergoing an invasive procedure and had a platelet count less than 50 x 109/L. Patients were randomized 1:1 to receive 3 mg of lusutrombopag or placebo once daily for up to 7 days.
In L-PLUS 1, 78% of patients (38/49) receiving lusutrombopag required no platelet transfusion prior to the primary invasive procedure, compared with 13% (6/48) who received placebo (95% CI for treatment difference: 49%, 79%; p<0.0001). In L-PLUS 2, 65% (70/108) of patients who received lusutrombopag required no platelet transfusion prior to the primary invasive procedure or rescue therapy for bleeding from randomization through 7 days after the procedure, compared with 29% (31/107) receiving placebo (95% CI for treatment difference: 25%, 49%; p<0.0001).
The most common adverse reaction in ≥ 3% of patients was headache.
The recommended lusutrombopag dosage is 3 mg orally once daily with or without food for 7 days.
Healthcare professionals should report all serious adverse events suspected to be associated with the use of any medicine and device to FDA’s MedWatch Reporting System or by calling 1-800-FDA-1088.
Follow the Oncology Center of Excellence on Twitter @FDAOncology.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
Filgotinib is an orally available inhibitor of JAK1/JAK2 and TYK2 in phase III clinical development at Galapagos and Gilead for the treatment of rheumatoid arthritis, moderate or severe Crohn’s disease and ulcerative colitis
In 2017, orphan drug designation was assigned to the compound in the U.S. for the treatment of pediatric Crohn’s disease and pediatric ulcerative colitis.
GlaxoSmithKline had been developing filgotinib preclinically for the treatment of rheumatoid arthritis pursuant to a license; however, in 2010, the compound was re-acquired by Galapagos. In 2012, the product was licensed to Abbott for development and marketing. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. The license agreement between Galapagos and Abbott was terminated in September 2015, Galapagos regaining all rights to the product. The same year, Galapagos and Gilead entered into a global partnership and Gilead obtained the global rights of codevelopment and commercialization for the treatment of inflammatory diseases
Filgotinib (GLPG0634) is an orally-available, selective inhibitor of JAK1 (Janus kinase 1) for the treatment of rheumatoid arthritis and potentially other inflammatory diseases. Filgotinib (GLPG0634) dose-dependently inhibited Th1 and Th2 differentiation and to a lesser extent the differentiation of Th17 cells in vitro. GLPG0634 was well exposed in rodents upon oral dosing, and exposure levels correlated with repression of Mx2 expression in leukocytes. The JAK1 selective inhibitor GLPG0634 (Filgotinib) is a promising novel therapeutic with potential for oral treatment of rheumatoid arthritis and possibly other immune-inflammatory diseases. Filgotinib (GLPG0634) is currently in a Phase 2 study in Crohn’s disease.
3D
Mechanism of action
Filgotinib is a Janus kinase inhibitor with selectivity for subtype JAK1 of this enzyme. It is considered a promising agent as it inhibits JAK1 selectively. Less selective JAK inhibitors (e.g. tofacitinib) are already being marketed. They show long-term efficacy in the treatment of various inflammatory diseases. However, their lack of selectivity leads to dose-limiting side effects.[1] It is thought that inhibition of all JAK isoenzymes is beneficial in rheumatoid arthritis. However, pan-JAK inhibition might also lead to unwanted side effects that might not outweigh its benefits. This is the rationale for the development of newer and more selective inhibitors like filgotinib.
The signal transmission of large numbers of proinflammatorycytokines is dependent on JAK1. Inhibition of JAK2 may also contribute to the efficacy against RA. Nonetheless it is thought that JAK2 inhibition might lead to anemia and thrombopenia by interference witherythropoietin and thrombopoietin and granulocyte-macrophage colony-stimulating factor. Therefore one might prefer to choose a more selective JAK1 inhibitor as a primary therapeutic option. Filgotinib exerts a 30-fold selectivity for JAK1 compared to JAK2.[2] It is however still to be seen to what extent JAK2 inhibition should be avoided.
Novel crystalline forms of filgotinib salts, particularly hydrochloride salt, useful for treating JAK-mediated diseases eg inflammatory diseases, autoimmune diseases, proliferative diseases, allergy and transplant rejection. Galapagos and licensee AbbVie are developing filgotinib, a selective JAK-1 inhibitor, for treating rheumatoid arthritis (RA) and Crohn’s disease (CD). In August 2015, the drug was reported to be in phase 2 clinical development for treating RA and CD. The drug is also being investigated for the treatment of colitis and was discovered as part of the company’s arthritis alliance with GSK; however in August 2010 Galapagos reacquired the full rights. See WO2013189771, claiming use of filgotinib analog for treating inflammatory diseases. Also see WO2010010190 (co-assigned with GSK and Abbott) and WO2010149769 (assigned to Galapagos) claiming filgotinib, generically and specifically, respectively.
Clinical trials and approval
The efficacy of filgotinib is currently studied in a phase2b program (DARWIN trial 1, 2) with involvement of 886 rheumatoid arthritis patients and 180 Crohn’s disease patients.
Phase 1 study
It was shown in phase 1 studies that the pharmacokinetics of filgotinib metabolism is independent of hepatic CYP450 enzymatic degradation. The drug metabolism is however mediated by carboxylesterases. There is no interference reported with the metabolism of methotrexate nor with any of the investigated transport proteins.[3]
Phase 2 study: Proof of concept (2011)
In november 2011 Galápagos released the results of their phase 2 study (identification: NCT01384422, Eudract: 2010-022953-40) in which 36 patients were treated who showed a suboptimal clinical response to methotrexate treatment. Three groups of twelve patients were treated either with 200 mg filgotinib in a single dose, 200 mg divided in two doses or placebo. The primary end-point was the ACR20 score, which monitors improvements in the symptomatology of the patient. After the scheduled 4 weeks of treatment, 83% of the respondents showed an improved ACR20-score. Half of the treated patients showed a complete (or near complete) remission of the disease. There were no reports ofanemia nor changes in lipidemia. The company stated in their press release that filgotinib is the first selective JAK1 inhibitor that shows clinical efficacy. As a result of this study, the company stated that “GLPG0634 shows one of the highest initial response rates ever reported for rheumatoid arthritis treatments”.[4]
DARWIN 1 trial
The DARWIN 1 trial is a 24 week double blindplacebo-controlled trial with 599 rheumatoid arthritis patients enrolled. All participants have moderate to severe RA and showed an insufficient response to standard methotrexate treatment. The trial compares three dosages of filgotinib as a once or twice per day regimen. During the trial all participants remain on their methotrexate treatment. According to the company, the results of this trial are expected in July 2015.[5]
DARWIN 2 trial
The DARWIN 2 trial is a double blind placebo-controlled trial with 280 rheumatoid arthritis patients enrolled who show an insufficient response to standard methotrexate treatment. This trial, in contrast to the previous DARWIN 1 trial, methotrexate is discontinued. Therefore, this trial investigates filgotinib as a monotherapy.[6] The recruitment of DARWIN trial 2b ended in november 2014.[7] Preliminary results are expected in the second quarter of 2015 and a full completion of the study is expected in the third quarter of 2015.
DARWIN 3 trial
Patients who complete DARWIN 1 and 2 will be eligible for DARWIN 3.
COSY PREDICT
Time line
june 2011: results of first phase 2 trial
november 2014: initiation of DARWIN 1 and 2 trials
april 2015: expected date of DARWIN 1 trial results
june 2015: expected date of DARWIN 2 trial results
GALAPAGOS NV [BE/BE]; Generaal De Wittelaan L11/A3 B-2800 Mechelen (BE) (For All Designated States Except US). MENET, Christel Jeanne Marie [FR/BE]; (BE) (For US Only). SMITS, Koen Kurt [BE/BE]; (BE) (For US Only)
Condensation of 2-amino-6-bromopyridine (I) with ethoxycarbonyl isothiocyanate (II) in CH2Cl2 gives 1-(6-bromopyridin-2-yl)-3-carboethoxythiourea (III), which upon cyclization with hydroxylamine hydrochloride (IV) in the presence of DIEA in EtOH/MeOH yields 2-amino-5-bromo[1,2,4]triazolo[1,5-a]pyridine (V). N-Acylation of amine (V) with cyclopropanecarbonyl chloride (VI) using Et3N in acetonitrile, and subsequent treatment with methanolic ammonia furnishes the carboxamide (VII) (1-3), which upon Suzuki coupling with 4-(hydroxymethyl)phenylboronic acid (VIII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C, followed by bromination with PBr3 in CHCl3 affords intermediate (IX). Condensation of benzyl bromide derivative (IX) with thiomorpholine-1,1-dioxide (X) using DIEA in CH2Cl2/MeOH yields filgotinib (1,2). Alternatively, condensation of (4-bromomethylphenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (XI) with thiomorpholine 1,1-dioxide (X) in the presence of DIEA in CH2Cl2/MeOH gives intermediate (XII), which undergoes Suzuki coupling with aryl bromide (VII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C to afford the target filgotinib
The present invention is based on the discovery that the compound of the invention is able to act as an inhibitor of JAK and that it is useful for the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6. In a specific aspect the compound is an inhibitor of JAKl and JAK2. The present invention also provides methods for the production of this compound, a pharmaceutical composition comprising this compound and methods for treating inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 by administering the compound of the invention.
Accordingly, in a first aspect of the invention, a compound of the invention is provided having a formula (I):
[0017] The compound of the invention is a novel inhibitor of JAK that appears to exhibit a dramatically improved in vivo potency as compared to structurally similar compounds. In a particular embodiment the compound of the invention is an inhibitor of JAKl and JAK2. In particular it appears to exhibit this increase in potency at lower in vivo exposure levels compared to structurally similar compounds. The use of a compound with these improvements is expected to result in a lower dosage requirement (and therefore an improved dosing schedule).
General Synthetic Method Scheme 1
1. RCOCI, Et3N 2. NH3 / MeOH CH3CN, 20 0C 2O 0C
wherein Ar represents phenyl-Ll-heterocycloalkyl, where Ll is a bond, -CH2– or -CO- and the heterocycloalkyl group is optionally substituted.
[00117] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5 0C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20 0C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
[00118] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH
(1 :1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 0C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition Of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-t/β) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (IH, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 :1, M+H+, 100%).
7.7.3 General procedure for mono-acylation to afford intermediate (4):
[00119] To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN
(150 mL) at 5 0C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2x50mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A
Preparation of compounds of the invention via Suzuki coupling (5):
[00120] An appropriate boronic acid (2eq.) is added to a solution of bromo intermediate (4) in
1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters
XBridge Prep Cl 8 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using
MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
[00121] 2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)- [l,2,4]triazolo[l,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide formation (General Method)
[00122] An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 00C. The acid chloride (Bl, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method C
Wherein R3a or R3b together with the nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reductive alkylation (general method)
[00123] An appropriate amine (2 eq.), cyclopropanecarboxylic acid (for example cyclopropanecarboxylic acid [5-(4-formyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridine-2-yl]-amide) prepared by method A (1 eq.) and Ti(OPr)4 are mixed and stirred at room temperature for 3 hrs. The mixture is diluted in ethanol and Na(CN)BH3 (leq.) is added. The resulting solution is stirred at room temperature for 16 hrs. The mixture is diluted in water and filtered. The filtrate is washed with ethanol. The combined solvent phases are evaporated under vacuum. The final compound is isolated by preparative HPLC.
Method D
wherein R1 and R2 together with the Nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reaction ofalkylation
[00124] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and Et3N (2 eq) (or AgCO3) are dissolved in DCM/MeOH (4:1 v:v) under N2 and an amine (2 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSθ4. Organic layers are filtered and evaporated. The final compound is isolated by flash chromatography.
Suzuki coupling
[00125] The obtained boronic acid (2eq.) is added to a solution of cyclopropanecarboxylic acid
(5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide (4) in 1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (This reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Synthesis of the compound of the invention and comparative examples
Compound l(the compound of the invention)
Step 1:
[00126] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and DIPEA
(2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgS O4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
(l.leq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00128] Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 500C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative route to Compound l(the compound of the invention):
Step 1:
[00129] 4-(Hydroxymethyl)phenylboronic acid (l.leq.) was added to a s o luti o n o f cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:
[00130] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)- [l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSθ4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:
[00131] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin- 2-yl]-amide (leq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO^ Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
JAK inhibitor N-(5-(4-(1,1-dioxothiomorpholinyl)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide, and methods for preparing the four crystal forms, wherein the four crystal forms respectively are a crystal form H1, a crystal form H2, a crystal form H3 and a crystal form H4,
The present invention relates to cryst. filgotinib free base (I), a method of its prepn. and a pharmaceutical compn. comprising the same. Thus, reacting the compd. II with thiomorpholine dioxide afforded 92% I (purity of 98.6%) which was then converted into its hydrochloride salt. Pharmaceutical compn. comprising compd. I was disclosed.
POLYMORPH
CN 105669669
The present invention provides a crystal form A, B, D, G and M of N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide hydrochloride.
PAPER
Future Medicinal Chemistry (2015), 7(2), 203-235. | Language: English, Database: CAPLUSA review. The discovery of the JAK-STAT pathway was a landmark in cell biol. The identification of these pathways has changed the landscape of treatment of rheumatoid arthritis and other autoimmune diseases. The two first (unselective) JAK inhibitors have recently been approved by the US FDA for the treatment of myelofibrosis and rheumatoid arthritis and many other JAK inhibitors are currently in clin. development or at the discovery stage. Research groups have demonstrated the different roles of JAK member and the therapeutic potential of targeting them selectively. ………..
Synthetic Preparation of the Compound of the Invention and Comparative Examples
The compound of the invention and the comparative examples can be produced according to the following scheme.
wherein Ar represents phenyl-L1-heterocycloalkyl, where L1 is a bond, —CH2— or —CO— and the heterocycloalkyl group is optionally substituted.
General 1.1.1 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)
To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5° C. is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20° C.) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (1H, br s, NH), 8.81 (1H, d, J=7.8 Hz, H-3), 8.15 (1H, br s, NH), 7.60 (1H, t, J=8.0 Hz, H-4), 7.32 (1H, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1:1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20° C.) for 1 h. 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1:1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-d6) δ 7.43-7.34 (2H, m, 2×aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1:1, M+H+, 100%).
1.1.3 General Procedure for Mono-Acylation to Afford Intermediate (4)
To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN (150 mL) at 5° C. is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp. (for 1-16 h) to hydrolyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2×50 mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A Preparation of Compounds of the Invention Via Suzuki Coupling (5):
An appropriate boronic acid (2 eq.) is added to a solution of bromo intermediate (4) in 1,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140° C. for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 90° C. for 16 h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSO4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide Formation (General Method)
An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 0° C. The acid chloride (B1, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
…
Synthesis of the Compound of the Invention and Comparative Examples Compound 1 (the Compound of the Invention) Step 1:
2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
STEP 2: Suzuki coupling
4-[4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-1,1-dioxide (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 50° C., the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative Route to Compound 1 (the Compound of the Invention): Step 1:
4-(Hydroxymethyl)phenylboronic acid (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:
To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSO4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:
Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders
Also claims a method for preparing filgotinib hydrochloride trihydrate. The present filing forms a pair with this week’s filing, WO2015117980, claiming a tablet composition comprising filgotinib hydrochloride.
The compound cyclopropanecarboxylic acid {5-[4-(l,l-dioxo-thiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5-a]pyridin-2-yl -amide (Compound 1), which has the chemical structure:
is disclosed in our earlier application WO 2010/149769 (Menet C. J., 2010) as being an inhibitor of JAK and as being useful in the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, allergy, transplant rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 or interferons. Hereafter this compound is named Compound 1. The data presented in WO 2010/149769 demonstrate that despite similar in vitro activities, Compound 1 has unexpectedly high in vivo potency compared with structurally similar compounds.
[00205] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) is added portionwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated without further purification.
[00206] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5°C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction
mixture is then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3 x 600 niL) and air-dried to afford the desired product. The thiourea may be used as such for the next step without any purification. lH (400 MHz, CDC13) δ 12.03 (1H, br s), 8.81 (1H, d), 8.15 (1H, br s), 7.60 (1H, t), 7.32 (1H, dd), 4.31 (2H, q), 1.35 (3H, t).
[00207] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) is added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids are washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
[00208] To a solution of the 2-amino-triazolopyridine obtained in the previous step (7.10 g, 33.3 mmol) in dry MeCN (150 mL) at 5°C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et20 (50 mL). The solids are collected by filtration, washed with H20 (2x50mL), acetone (50 mL) and Et20 (50 mL), then dried in vacuo to give the desired compound.
1.1.3. Compound 1
[00209] 4-[4-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)-benzyl] hiomoφholine , l -dioxide (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo.
[00210] The final compound is obtained after purification by flash chromatography.
[00211] Alternatively, after completion of the reaction, a palladium scavenger such as 1 ,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cool down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HC1 is added, and after stirring at room temperature, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at room temperature, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H20, treated with a palladium scavenger (e.g. SMOPEX 234) at 50°C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the desired compound as a free base.
[00212] 4-(Hydroxymethyl)phenylboronic acid (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water
(4:1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo. The resulting mixture is used without further purification.
1.2.2. Step 2: Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5- a Jpyridin-2-ylJ -amide
[00213] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl] -amide (1.0 eq) in chloroform is slowly added phosphorus tribromide (1.0 eq.). The reaction mixture is stirred at room temperature for 20 h, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer is dried over anhydrous MgSO i, filtered and concentrated to dryness. The resulting white residue is triturated in dichloromethane/diethyl ether 2:1 to afford the desired product.
1.2.3. Step 3:
[00214] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (l eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5: 1 v:v) under N2 and thiomorpho line 1,1-dioxide (1.1 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is dissolved in DCM, washed with water and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated by column chromatography using EtOAc to afford the desired product.
1.1.1. Synthesis of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (Intermediate 3)
led to 5 °C was added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture was then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gave a solid which was collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea was used as such in the next step without any purification.
[00158] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) was added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture was stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) was then added and the mixture slowly heated to reflux (Note: bleach scrubber was required to quench H2S evolved). After 3 h at reflux, the mixture was allowed to cool and filtered to collect the precipitated solid. Further product was collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids were washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound was used as such in the next step without any purification.
[00160] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portion wise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
1.1.3. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- a ridin-2-ylamine (Formula I)
[00161] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide (l .leq.) was added to a solution of 5-bromo-[l,2,4]triazolo[l,5-a]pyrid in-2-ylamine (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90°C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhydrous MgSC>4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00164] The compound according to Formula II may be synthesized according to the procedure described in WO 2010/149769.
1.2.2. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- aJpyridin-2-ylamine (Formula I)
[00165] The compound according to Formula I can also be produced by hydrolysis of the compound accor ing to Formula II:
[00166] Hydrochloric acid 30% aq (12.06 kg; 3.9 rel. volumes) was added to a slurry of the compound according to Formula II (3.45 kg; 1.0 equiv.) in demineralized water (10.0 kg; 3.0 rel. volumes). Subsequently, a line rinse was performed with demineralized water (3.4 kg; 1.0 rel. volumes). The reaction mixture was heated to 80±5°C for 14.5 h. After completion of the reaction (conversion > 99%>), the reaction mixture was cooled to 20±5°C. The reaction mixture was diluted with demineralized water (6.8 kg; 2.0 rel. volumes) and sodium hydroxide 33%> aq (9.52 kg; 3.7 rel volumes) was dosed at such a rate that the temperature of the reactor contents remained below 35°C. An additional amount of sodium hydroxide 33%> aq (2.55 kg; 1.0 rel. volumes) was needed to get the pH > 10. The product was filtered off, washed twice with demineralized water (1.5 rel. volumes) and dried under vacuum for 1 h, thus yielding the crude compound according to Formula I.
[00167] The crude compound according to Formula I (5.70 kg) was re-slurried in demineralized water (23.0 kg; 8.5 rel. volumes). Hydrochloric acid 30%> aq (1.65 kg; 0.7 rel. volumes) and demineralized water (4.3 kg; 1.6 rel. volumes) were added and the reaction mixture was stirred at 20±5°C for 45 min. As the compound according to Formula I was not dissolved completely, the reaction mixture was stirred at 45±5°C for 1 h. The reaction mixture was filtered and the residue was washed with demineralized water (2.0 kg 0.75 rel. volumes). Sodium hydroxide 33%> aq (1.12 kg; 0.6 rel volumes) was added to the filtrate. An additional amount of sodium hydroxide 33%> aq (1.01 kg) was needed to get the pH > 10. The resulting reaction mixture was stirred at 20±5°C for about 3 h. The product was filtered off, washed twice with demineralized water (4.1 kg; 1.5 rel. volumes), and twice with methyl tert-butyl ether (MTBE; 3.0 kg; 1.5 rel. volumes) and dried under vacuum for 15.5 h on the filter. The product was further dried in a vacuum oven at 40±5°C for 202 h, thus affording the desired compound according to Formula I.
Namour, Florence; Diderichsen, Paul Matthias; Cox, Eugène; Vayssière, Béatrice; Van der Aa, Annegret; Tasset, Chantal; Van’t Klooster, Gerben (2015-02-14). “Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection”. Clin Pharmacokinet. Epub ahead of print.doi:10.1007/s40262-015-0240-z.
Van Rompaey, L; Galien, R; Van der Aar, E; Clement-Lacroix, P; Van der Aar, E; Nelles, L; Smets, B; Lepescheux, L; Cristophe, T; Conrath, K; Vandeghinste, N; Vayssiere, B; De Vos, S; Fletcher, S; Brys, R; Van’t Klooster, G; Feyen, J; Menet, C (2013-10-01). “Preclinical characterization of GLPG0634, a selective inhibitor of JAK1 for the treatment of inflammatory diseases”. J Immunol. 191(7). doi:10.4049/jimmunol.1201348.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
(3RS,11Brs)-9,10-di((2H3)methoxy)-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydro-2H-benzo(a)quinolizin-2-one
Treatment of Chorea Associated with Huntington Disease
SD-809 was granted Orphan Drug Designation for the treatment of HD by the FDA in November 2014 and became part of Teva’s CNS portfolio with the acquisition of Auspex Pharmaceuticals in May 2015.
Teva announced that the New Drug Application (NDA) for SD-809 (deutetrabenazine) has been accepted by the U.S. Food and Drug Administration (FDA) for the treatment of chorea associated with Huntington disease (HD), a rare and fatal neurodegenerative disorder caused by the progressive breakdown of nerve cells in the brain that affects about five to seven people per 100,000 in western countries, according to the World Health Organization.
Chemically tetrabenazine is cis rac -1, 3, 4, 6, 7, 1 lb-hexahydro-9, 10-dimethoxy-3-(2- methylpropyl)-2Hbenzo[a]quinolizin-2-one and it is represented by compound of structural formula I.
Formula 1
The proprietary name of tetrabenazine is Xenazine and is marketed by Biovail Americas. Xenazine is indicated for the treatment of chorea associated with Huntington’s disease. U.S. patent no. 2,830,993 discloses a process for the preparation of tetrabenazine compound of structural formula I wherein 1 -carbethoxymethyl-6, 7-dimethoxy-l , 2, 3, 4- tetrahydroisoquinoline compound of structural formula IV is being reacted with mono- isobutylmalonic acid dimethyl ester compound of structural formula V and paraformaldehyde in methanol solvent to get l-carbethoxymethyl-2 (2, 2-dicarbomethoxy-4-methyl-n-pentyl)-6, 7- dimethoxy-1, 2, 3, 4-tetrahydroisoquinoline compound of structural formula VI. The 1- carbethoxymethyl-2(2,2-dicarbomethoxy-4-methyl-n-pentyl)-6,7-dimethoxy-l ,2,3,4- tetrahydroisoquinoline compound of structural formula VI is subjected to Dieckmann cyclization , hydrolysis and decarboxylation to get tetrabenazine compound of structural formula I, which is recrystallized from di-isopropyl ether solvent.
Formula I
Scheme I
U. S. patent no. 4,678,792 discloses a process for the preparation of 6, 7-dimethoxy-3, 4- dihydroisoquinoline compound of structural formula VII wherein 2-(3, 4-dimethoxyphenyl)- ethylamine compound of structural formula II is being reacted with chloral hydrate at 120°C to get N-formyl-2-(3, 4-dimethoxyphenyl)-ethylamine compound of structural formula III. The N- formyl-2-(3, 4-dimethoxyphenyl)-ethylamine compound of structural formula III is further reacted with polyphosphoric acid to get 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII. The 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII is being used as an intermediate for the preparation of tetrabenazine compound of structural formula I.
Formula III
Formula II
Polyphosphoric acid
Formula VII
Scheme II
Bull. Korean Chem. Soc. 2002 Volume (23). No. l , page no. 149 discloses N-formylation of various amines and alcohols with formic acid in toluene.
U.S. patent publication no. 2010/0130480 discloses a process for the preparation of 6, 7- dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII by reacting 2-(3, 4- dimethoxyphenyl)-ethylamine compound of structural formula II with hexamethylenetetramine in presence of acetic acid or trifluoroacetic acid.
Hexamethylenetetramine
Formula II Formula VII
U.S. patent publication no. 2008/0167337 discloses a process for the preparation of tetrabenazine compound of structural formula I wherein 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII is reacted with 3-dimethylaminomethyl-5-methyl-hexan-2-one methiodide compound of structural formula VIII to get crude tetrabenazine compound. The crude tetrabenazine compound was purified by employing flash column chromatography technique and
Formula VIII Formula I
The prior-art processes for preparing N-formyl-2-(3, 4-dimethoxyphenyl)-ethylamine compound of structural formula III produces below mentioned compound of structural formula XVII, XVIII, XIX, XX, XXI and XXII as a by-product of the reaction due to the demethylation and formylation of resulting hydroxy compounds.
Formula XX Formula XXI Formula XXII
The compounds of structural formula XVII, XVIII, XIX, XX, XXI and XXII are being carry- forwarded into the further steps of reactions of preparing tetrabenazine compound of structural formula I and therefore there is a need in the art to develop an improved process of preparing 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII, which obviates the prior-art problems. Accordingly there is provided a process of preparing tetrabenazine compound of structural formula I wherein 6, 7-dimethoxy-3, 4-dihydroisoquinoline compound of structural formula VII is being formed without the formation of above mentioned compounds of structural formula XVII, XVIII, XIX, XX, XXI and XXII.
EXAMPLE: PROCESS FOR THE PREPARATION OF SUBSTANTIAL PURE CRYSTALLINE FORM A OF TETRABENAZINE
Stage A: Process for the preparation of 6, 7-dimethoxy-3, 4-dihydroisoquinoIine
Step 1 : Process for the preparation of N-formyl-2-(3, 4-dimethoxyphenyl)-ethylamine
A solution of 2-(3, 4-dimethoxyphenyl)-ethylamine (500gm) in toluene (2000ml) was added formic acid (150gm) at 25°C, the resulting reaction mixture was diluted with toluene (500ml) and heated up to 45°C. The reaction mixture was maintained at 40-45°C for 5 hours and then the resulting reaction mixture was concentrated under reduced pressure at 50°C to get the title compound
Yield: 570gm
Purity: 99.98% (By HPLC)
Step 2: Process for the preparation of 6, 7-dimethoxy-3, 4-dihydroisoquinoline
A solution of N-formyl-2-(3, 4-dimethoxyphenyl)-ethylamine (250gm) obtained from step 1 in toluene (500ml) and polyphosphoric acid (50gm) was heated at 110°C for 5 hours. The resulting reaction mixture was cooled to 50°C, quenched with water (500ml) and pH of the resulting solution was adjusted to about 8.3 with aqueous solution of sodium hydroxide [sodium hydroxide (690gm) + water (690ml)]. The resulting reaction mass was extracted by ethyl acetate (2 1250ml), dried over anhydrous sodium sulfate (50gm) and concentrated under reduced pressure to get 6, 7-dimethoxy-3, 4-dihydroisoquinoline (190gm).
Yield: 215gm
Purity: 99.67% (By HPLC)
Stage B: Process for the preparation of 3-((dimethylamino) methyi)-5-methylhexan-2-one methiodide
Step 1 : Process for the preparation of 3-((dimethylamino) methyl)-5-methylhexan-2-one Dimethylamine hydrochloride (180gm) and paraformaldehyde (lOOgm) were added to a solution of 5-methylhexan-2-one (900ml) in methanol (1600ml). The resulting reaction mass was heated at reflux for 12 hours, and then the pH was adjusted to about 8.75 with aqueous solution of sodium hydroxide [sodium hydroxide(90gm) + water (900ml)] at 25 °C. The resulting reaction solution was extracted by toluene (2x1234ml). The organic layer was dried over anhydrous sodium sulfate (50gm) and concentrated under reduced pressure to get title compound.
Yield: 900gm
Purity: 99.80% (By HPLC)
Step 2: Process for the preparation of 3-((dimethylamino) methyl)-5-methylhexan-2-one methiodide
Methyl iodide (323gm) was added dropwise to a solution of 3-((dimethylamino) methyl)-5- methylhexan-2-one (195gm) obtained from step 1 , in ethyl acetate (1650ml) at 25-30°C in 30 minutes. The resulting reaction mixture was stirred at 25 °C for 12 hours and then the resulting solids were filtered, washed with water (200ml) and suck-dried to get wet compound (400gm). The wet compound was slurried with water (1000ml) at 25°C for 1 hour and then it was again filtered, washed with water (200ml) and dried at 45-50°C to get title compound
Yield: 300gm
Purity: 99.86% (By HPLC)
Stage C: Preparation of substantial pure crystalline form A of Tetrabenazine
3-((Dimethylamino) methyl)-5-methylhexan-2-one methiodide (80gm) was added to the solution of 6, 7-dimethoxy-3, 4-dihydroisoquinoline (40gm) in isopropanol (288ml) at 25°C and the resulting reaction mass was heated at 40-45°C for 15 hours. The resulting insoluble material was filtered, washed with isopropanol (80ml) and filtrate was concentrated under reduced pressure up to the 150ml reaction volume. The reaction solution was diluted with methylene dichloride (1200ml) and water (1000ml) and pH was adjusted to 8.5 with sodium hydroxide solution [10%, 100ml]. The organic layer was separated, washed with water (3 x 1000ml) and concentrated under reduced pressure to obtain residue. The residue was dissolved in methanol (300ml) at 50°C, and resulting solution was treated with an activated carbon (20gm) at 50-60°C for 30minutes and then it was filtered and filtrate was further stirred at 20-25°C for 2 hours. The resulting solids were filtered, washed with methanol (150ml), dried at 50-55°C for 8 hours. The resulting solids were milled, sifted through 40 mesh sieve and micronized.
A concise synthesis of tetrabenazine and dihydrotetrabenazine is described. The key feature of this synthesis is the intramolecular aza-Prins-type cyclization of an amino allylsilane via oxidative C–H activation.
The TBZ (4) for these reactions was prepared by reacting 3,4-dihydro-6,7-dimethoxyisoquinoline (3) and the Mannich base (2) as shown in Scheme 1.14 The α,β-unsaturated TBZ (5), which was the original substrate, was obtained by further treatment with chloranil in refluxing benzene.
Tetrabenazine (4a)
To a solution of 3,4-dihydro-6,7-dimethoxyisoquinoline hydrochloride (3, 3.5 g, 15.4 mmol) in cold H2O (20 mL) in an ice water bath, was added 3-(dimethylaminomethyl)-5-methyl-2-hexanone (2, 3.15 g, 18.3 mmol) as the free base with stirring. Precipitate formed within 3 h, and stirring was continued until the solid-gummy precipitate prevented stirring. The mixture was allowed to stand at RT (room temperature) for 3 days. The solid–gum mixture was filtered, and the yellow solid–gum mixture was dissolved in hot MeOH. The solution was chilled at −10°C for 18 h. The pale yellow solid was filtered to give 2.1 g (43%) of TBZ (4a).
Example 10 Removal The Boc Protecting Group From First Intermediate 12 And Amino Cyclization Provide (+)-Tetrabenazine XVII
[0063] First intermediate 12 (1.0 eq) was dissolved in 10% Me2S- dichloromethane to provide an 82 mM solution. The solution was cooled to 0 0C and triisopropylsilane (1.1 eq.) followed by TFA (precooled to 0 0C) was added to the reaction mixture to provide a final concentration of 41 mM. The reaction mixture was permitted to stir at 0 0C for 1 h. Following the allotted time the reaction mixture was quenched at 0 0C by the addition of saturated aqueous potassium carbonate solution and concentrated under reduced pressure to remove the majority of the dimethylsulfide. The mixture was extracted with five portions of dichloromethane, and the combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure to provide the crude product as a yellow solid. The crude product was recrystallized from 3.5% dimethoxyethane in hexanes. The resulting colorless crystals were washed with hexanes to provide pure (+)- tetrabenazine (XVII) 46%: mp 126.0 0C (3.5% DME-hexanes) (a crystal polymorph was observed at 116 0C); [α]26D +37.2 (c 0.41, CH2Cl2); 1H NMR (CD2Cl2) δ 0.89 (apparent t, J = 7.2 Hz, 6H), 0.98 (ddd, J = 12, 6.0, 4.0 Hz, IH), 1.59-1.68 (m, IH), 1.74 (ddd, J = 12, 5.9, 5.7 Hz, IH), 2.32 (apparent t, J = 11.7 Hz, IH), 2.46 (apparent t, J = 12.3 Hz, IH), 2.55 (ddd, J = 12, 10.0, 3.8 Hz, IH), 2.65-2.73 (m, 2H), 2.83 (dd, J = 5.5, 2.8Hz, IH), 2.97-3.07 (m, IH), 3.07-3.14 (m, IH), 3.25 (dd, J =9.7, 6.3 Hz, IH), 3.47 (apparent d, J = 12Hz, IH), 3.75 (s, 3H), 3.77 (s, 3H), 6.55 (s, IH), 6.60 (s, IH) 13C NMR (CD2Cl2) δ 21.98, 23.02, 25.51, 29.46, 35.16, 47.47, 47.63, 50.47, 55.87, 56.01, 61.47, 62.46, 108.46, 111.72, 126.37, 128.96, 147.65, 147.98, 209.72; HRMS-(ESI+) calcd for (C19H27NO3 + H) ([M+H]+ 318.2069, found 318.2082.
NBI-98854 (CAS # 1025504-59-9), (S)-(2R,3R,l lbR)-3-isobutyl-9,10-dimethoxy-2,3,4,6,7,1 lb-hexahydro-lH-pyrido[2,l-a]isoquinolin-2-yl 2-amino-3-methylbutanoate, is a VMAT2 inhibitor. NBI-98854 is currently under investigation for the treatment of movement disorders including tardive dyskinesia. WO 2008058261; WO 2011153157; and US 8,039,627. NBI-98854, a valine ester of (+)-a-dihydrotetrabenazine, in humans is slowly hydrolyzed to (+)-a-dihydrotetrabenazine which is an active metabolite of tetrabenazine.
[0193] Jgrt-butyl 3,4-dihydroxyphenethylcarbamate : A solution of dopamine
hydrochloride (209 g, 1.11 mol, 1.00 equiv), sodium carbonate (231 g, 2.75 mol, 2.50 equiv) and di-tert-butyl dicarbonate (263 g, 1.21 mol, 1.10) in 2.4 L tetrahydrofuran / water (5: 1) was stirred at 20°C for 2.5 h. After the starting material was consumed completedly, the reaction was diluted with ethyl acetate (2 L) and washed with water (2×600 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under reduced pressure until two volumes of solvent was left. The precipitated solid was isolated by filtration and dried under vacuum to give 254 g (91%) of ieri-butyl 3,4-dihydroxyphenethylcarbamate as white solid. Ή-ΝΜΪ (300 MHz, CDC13) 8.72 (s, 1H), 8.62 (s, 1H), 6.79 (m, 1H), 6.62 (m, 1H), 6.51 (m, 1H), 6.40 (m, 1H), 3.03 (m, 2H), 2.50 (m, 2H), 1.37 (s, 1H). LC-MS: m /z = 254 (MH) +.
Step 2
[0194] D6-fert-butyl 3,4-dimethoxyphenethylcarbamate: A solution of ieri-butyl 3,4-dihydroxyphenethylcarbamate (127 g, 397 mmol, 1.00 equiv), potassium carbonate (359.3 g, 2.604 mmol, 3.00 equiv) and 18-crown-6 (1,4,7,10,13,16-hexaoxacyclooctadecane ) (68.64 g, 0.26 mmol, 0.03 equiv) in acetone (800 mL) was stirred at 38°C. After 30 min., CD3I (362 g, 2.604 mmol, 3.00 equiv) was added to the reaction, and the mixture was stirred at 38°C for 12 h. Then an additional CD3I (120 g, 0.868 mmol, 1.00 equiv) was added to the solution and the solution was stirred for 5 h. Then the mixture was cooled to room temperature and the solid was filtered. The filtrate was concentrated under vacuum. The resultant solid was dissolved in H2O (300 mL) and extracted with EA (3×300 mL), the organic layers was combined and concentrated under vacuum to give 114 g (79%) of de-tert-butyl 3,4-dimethoxyphenethylcarbamate as white
[0195] D6-2-(3,4-dimethoxyphenyl)ethanamine: A solution of de-tert-butyl 3,4-dimethoxyphenethylcarbamate (128 g, 455.26 mmol, 1.00 equiv) in ethyl acetate (1.5 L) was stirred at room temperature. Then HC1 gas was introduced into the reaction mixture for 2h. The precipitated solid was isolated by filtration. The solid was dissolved in 300 mL of water. The pH value of the solution was adjusted to 12 with sodium hydroxide (solid). The resulting solution was stirred for 1 h at 5-10°C. The resulting solution was extracted with 6×800 mL of ethyl acetate and the organic layers combined, dried over sodium sulfate, and concentrated under vacuum to give 64 g (78%) of d6-2-(3,4-dimethoxyphenyl)ethanamine as yellow oil.
[0196] D6-N-r2-(3,4-dimethoxy-phenyl)ethyllformamide: A solution of d6-2-(3,4-dimethoxyphenyl)ethanamine (69 g, 368 mmol, 1.00 equiv) in ethyl formate(250 mL) was heated under reflux overnight. The solution was concentrated under vacuum to give 71 g (91%) of d6-N-[2-(3,4-dimethoxy-phenyl)ethyl]formamide as yellow solid. The crude solid was used in next step without purification. ^-NMR (300 MHz, CDCb) £8.17 (s, 1H), 6.81 (m, 3H), 5.53 (br, 1H).3.59 (m, 2H), 2.81 (t, 2H, / = 6.9 Hz). LC-MS: m /z = 216 (MH) +.
Step 5
[0197] D6-6,7-dimethoxy-3,4-dihvdroisoguinoline: A solution of d6-N-[2-(3,4-dimethoxy-phenyl)ethyl]formamide (71 g, 329 mmol, 1.00 equiv) in phosphorus oxychloride (100 mL) was stirred at 105°C for 1 h. Then the solution was concentrated under vacuum to remove
phosphorus oxychloride. The residual oil was dissolved in ice / water. The solution was made basic with potassium carbonate with cooling. The basic aqueous solution was extracted with dichloromethane. The collected organic phase was dried using sodium sulfate and then filtered. The dichloromethane was removed by concentration under vacuum to give an orange oil.
Purification by silica gel (ethyl acetate:petroleum ether = 1: 1 ~ ethyl acetate) to give 43 g (66%) of d6-6,7-dimethoxy-3,4-dihydroisoquinoline as orange solid (yield 66%). Ή-ΝΜΡ (300 MHz, CDC13) 8.24 (s, 1H), 6.82 (s, 1H), 6.68 (s, 1H), 3.74 (m, 2H), 2.69 (t, 2H, J = 12 Hz). LC-MS: m /z = 198 (MH) +.
Step 6
[0198] Trimethyl(5-methylhex-2-en-2-yloxy)silane: To a cold (-78°C), stirred solution of j-PrMgBr (500 mL of 2 M solution in tetrahydrofuran, 1 mol, 1.00 equiv) in anhydrous tetrahydrofuran (1 L) was added Cul (19.02 g, 0.1 mol, 0.10 equiv) and the resultant mixture was stirred for 15 min at -78°C. Anhydrous hexamethylphosphorous triamide (358.4 g, 2 mmol, 2 equiv) was added and after 20 min, a solution of methyl vinyl ketone (70 g, 0.1 mol, 1.00 equiv), trimethylsilyl chloride (217 g, 0.2 mol, 2.00 equiv), in tetrahydrofuran (200 mL) was added dropwise over 30 min. After the reaction mixture was stirred at -78 °C for lh, triethylamine (20.2g, 200 mmol, 2.00 equiv) was added and the resulting mixture stirred for 10 min at 0 °C. To this was added ie/ -butyl methyl ether (2 L), and the solution was washed with 5% ammonia solution (6×300 mL). Then the organic phase was dried over sodium sulfate and concentrated under vacuum at 25°C to give 155 g crude product as yellow liquid. The liquid was purified by distilling (64-68°C/40 mmHg) to provide 118 g (63.3%) of trimethyl(5-methylhex-2-en-2-
[0199] 3-r(Dimethylamino)methyl1-5-methylhexan-2-one: To a stirred solution of trimethyl(5-methylhex-2-en-2-yloxy)silane (118 g, 633 mmol, 1.00 equiv) in anhydrous acetonitrile (800 mL) was added N-methyl-N-methylenemethanaminium iodide (128.8 g, 696.3 mmol, 1.10 equiv) in several batches and the resultant mixture was stirred at 20°C overnight. Then the solution was concentrated under vacuum to remove the solvent. The residue was dissolved in 400 mL 1 N HC1 (aq.) and extracted with ieri-butyl methyl ether. Then the water phase was basiced with 2 N aq. NaOH and extracted with ie/ -butyl methyl ether. The organic phase was dried and concentrated under vacuum. The liquid was purified by distilling (80°C/0.5 mmHg) to provide 50 g (46%) of 3-[(dimethylamino)methyl]-5-methylhexan-2-one as a colorless oil. XH-NMR (300 MHz, J6-DMSO) £0.92 (d, 3H), 0.98 (d, 3H), 1.11-1.23 (m, 1H), 1.23-1.38 (m, 1H), 1.54-1.70 (m, 1H), 2.30 (s, 3H), 3.01 (s, 9H), 3.10-3.32 (m, 2H), 3.81-3.88 (m, 1H).
Step 8
[0200] 2-Acetyl-N,N V,4-tetramethylpentan-l-aminium iodide: A solution of 3-[(dimethylamino)methyl]-5-methylhexan-2-one (50 g, 15.00 mmol, 1.00 equiv) and methyl iodide (4.26 g, 30.00 mmol, 2.00 equiv) in 50 mL diethyl ether was stirred overnight at room temperature. The precipitated solid was isolated by filtration and dried under vacuum to give 79 g (86%) of 2-acetyl-N,N,N,4-tetramethylpentan-l-aminium iodide as white solid. XH-NMR (300 MHz, Je-DMSO) 0.89-0.98 (m, 6H), 1.11-1.20 (m, 1H), 1.40 (m, 1H), 1.66 (m, 1H), 2.30 (s, 3H), 3.01(s, 9H), 3.21 (m, 2H), 3.85 (m, 1H).
Step 9
[0201] Ρό- (±) -tetrabenazine : A solution of d6-6,7-dimethoxy-3,4-dihydroisoquinoline (33.4 g, 169 mmol, 1.10 equiv) and 2-acetyl-N,N,N,4-tetramethylpentan-l-aminium iodide (48 g, 153 mmol, 1.00 equiv) in 300ml of methanol was heated under reflux for 48 h. Then 150 mL water was added. The solution was cooled to room temperature. The precipitated solid was isolated by filtration and dried under vacuum to give 38 g of crude d6-tetrabenazine as yellow solid. The crude tetrabenazine was dissolved in ieri-butyl methyl ether (15 volumes), the mixture was heated until the solid was almost dissolved. The yellow solid which was unsolvable was filtered. The filtrate was concentrated under vacuum until 2 volumes ieri-butyl methyl ether was left. The solid was filtered and collected. The above solid was dissolved in ethanol (4 volumes), then the mixture was heated until the solid was dissolved. The solution was stirred and cooled to room temperature at the rate of 20°C/h. Then the mixture was stirred at 0°C for lh. The precipitated solid was isolated by filtration and dried under vacuum to give 25 g (50.4%) of tetrabenazine-<d6 as white solid.
SYNTHESIS OF N-FORMYL-3,4-DI-t-BUTOXYCARBONYLOXY-6-(TRIMETHYLSTANNYL)-L-PHENYLALANINE ETHYL ESTER AND ITS REGIOSELECTIVE RADIOFLUORODESTANNYLATION TO 6-[18F]FLUORO-L-DOPA
DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; NARASIMHAN, N. S. ET AL: “Unusual products in Bischler-Napieralski reaction“, XP002659743, retrieved from STN Database accession no. 1981:46871
DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; WANG, CHENG-XUE ET AL: “Synthesis of rutaecarpine and quinazolone compounds“, XP002659737, retrieved from STN Database accession no. 2009:92700
A novel process for the synthesis of tetrabenazine (1) and deutetrabenazine (2), two well-known drugs used for the treatment of chorea associated with Huntington’s disease, has been developed. All of the reaction parameters were optimized through a series of reactions and by using Design of Experiment techniques. The newly developed methods are industrially scalable and employ cheap, commercially available raw materials and hence are highly efficient. The added advantage is that the developed processes evade the use of genotoxic alkylating agents and therefore could be considered as safe and viable alternatives to the existing methods.
Tetrabenazine (1) was invented by Hoffmann-La-Roche (Nutley, NJ, USA). It was also known as Ro 1-9569, Nitoman, and Xenazine. It is a benzoquinolizine derivative with the chemical name 1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-3-(2-methylpropyl)-2H-benzo[α]quinolizin-2-one . Initially it was developed as an antipsychotic agent.Despite 50 years of medicinal background, the U.S. Food and Drug Administration (FDA) approved 1 on August 15, 2008, for the treatment of chorea associated with Huntington’s disease.
Chemical structures of 1 and 2.
Deutetrabenazine (2) (trade name Austedo) is a stable, nonradioactive deuterium analogue of the approved drug tetrabenazine in which the six hydrogen atoms of the 9- and 10-methoxy (−OCH3) substituents have been replaced by deuterium atoms . Deutetrabenazine was found to be more effective for the treatment of chorea associated with Huntington’s disease because of improved pharmacokinetic properties compared with the nondeuterated drug tetrabenazine. Deutetrabenazine was originally developed by Auspex Pharmaceuticals (La Jolla, CA, USA). In 2015, Teva acquired Auspex Pharmaceuticals and submitted a new drug application (NDA) in the United States for the treatment of Huntington’s disease. On April 3, 2017, Teva Pharmaceutical received approval from the FDA to market deutetrabenazine as the first deuterated drug for the treatment of chorea associated with Huntington’s disease.(4) Both 1 and 2 are racemic mixtures .
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये। DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on google plus Googleplus
Join me on Researchgate
amcrasto@gmail.com
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Merck Alzheimer’s drugs Verubecestat (MK-8931) is an oral β- amyloid precursor protein cleaving enzyme (BACE1 or β-secretase enzyme) inhibitor, is currently in Phase III clinical trials
C17 H17 F2 N5 O3 S, 409.41 Mechanism: Oral β- amyloid precursor protein cleavage enzyme (BACE) inhibitors Indications: Alzheimer’s disease Development progress: phase III clinical Companies: Merck
Verubecestat (MK-8931) is a small-molecule inhibitor of beta-secretase cleaving enzyme (BACE) 1 and BACE2 in development by Merck for the treatment of Alzheimer’s Disease.
MK-8931 is a beta-secretase 1 (BACE1) inhibitor in phase III development for the treatment of amnestic mild cognitive impairment (aMCI) due to Alzheimer’s disease at Merck & Co. The company is also conducting phase II/III trials for the treatment of Alzheimer’s type dementia.
Alzheimer’s disease (AD) is a devastating, progressive neurodegenerative disease that is associated with up to 80% of the estimated 47 million cases of dementia worldwide and is a leading cause of death in the United States.(1, 2) As the elderly population increases, the worldwide incidence of dementia is expected to nearly triple to approximately 132 million by 2050, creating an unsustainable socioeconomic burden.(1) Currently available therapies, which include acetylcholinesterase inhibitors and the N-methyl-d-aspartate receptor antagonist memantine, produce modest and transient improvement in cognitive function but do not alter the progression of AD.(3) Treatments that delay or halt disease progression by targeting the underlying causes of AD would have lasting impacts on patient function and quality of life and would address an urgent unmet medical need.
Two histopathological hallmarks are invariably observed in the brains of AD patients, namely, extracellular amyloid plaques composed primarily of β-amyloid (Aβ) peptides and intraneuronal neurofibrillary tangles composed primarily of aggregates of abnormally phosphorylated tau protein. Aβ peptides are formed by two sequential cleavages of the amyloid precursor protein (APP), first by β-site amyloid precursor protein cleaving enzyme 1 (BACE1) followed by cleavage of the resulting C-terminal fragment C99 by γ-secretase. This cleavage sequence results in production of a family of Aβ peptides, of which Aβ40 is the most abundant isoform and Aβ42 is more highly prone to aggregate into neurotoxic, oligomeric species.(4) According to the amyloid hypothesis, aberrant production and/or accumulation of Aβ peptides, principally Aβ42, over a period of decades is causative of the underlying disease pathogenesis that ultimately leads to neuronal cell death.(4, 5) In addition to the invariant presence of amyloid plaques in the brains of AD patients, the amyloid hypothesis is underpinned by several other lines of evidence. First, many distinct neurodegenerative diseases are associated with the invariant presence of abnormal protein aggregates analogous to amyloid plaques. Second, low levels of Aβ42 in the CSF are a reasonably good diagnostic/prognostic biomarker for AD. Finally, and most significantly, early onset autosomal dominant familial AD is associated with mutations in APP and the presenilin proteins (which are components of the γ-secretase enzyme), and all of these mutations share the common phenotype of increasing total Aβ levels or the relative proportion of Aβ42.(6) Given this multifaceted evidence supporting the role of Aβ peptides in AD progression, substantial efforts have been invested in the development of amyloid-lowering therapies as a disease-modifying approach to AD treatment.(7) Prominent among these has been inhibition of BACE1 to reduce or prevent production of the Aβ peptides. This approach has been further supported by the recent finding that a rare mutation (A673T) near the BACE1 cleavage site in APP reduces Aβ peptide production and is associated with reduced risk of developing AD and improved cognitive function in the elderly.(8)
BACE1 is a membrane-bound aspartyl protease expressed primarily in the central nervous system (CNS), is the sole enzymatic activity responsible for the initial β-site APP cleavage, and is required for Aβ peptide production in vivo.(9) In the brain, BACE1 is expressed mainly in neurons and cleaves APP predominantly in the endosomal compartments where the acidic pH is near the optimum for its enzymatic activity (pH 5).(10) Since the characterization of BACE1 more than 15 years ago,(11) there have been intensive efforts to overcome the challenges of identifying small molecule inhibitors that can penetrate the CNS and inhibit the formation of centrally derived Aβ peptides.(12) These efforts have been driven by evidence that BACE1 inhibition, in comparison with γ-secretase inhibition and antiamyloid immunotherapy, may be an inherently safer amyloid-lowering approach,(7) a notion that has been informed by an evolving understanding of BACE1 biology. In this regard, Bace1 knockout mice have been reported to have a number of subtle phenotypes, including reduction of central and peripheral nerve myelination, and several putative BACE1 substrates other than APP have recently been proposed.(10, 13, 14) However, many of these phenotypes and substrates remain to be independently confirmed, have little if any functional consequence, are not recapitulated by pharmacological inhibition of BACE, or may be mitigated through partial BACE1 inhibition.(8, 10, 13-15)
.
Smiles: C [C @] 1 (CS (= O) (= O) N (C (= N1) N) C) c2cc (ccc2F) NC (= O) c3ccc (cn3) F
The amine A (Scheme 3a, step 4) (13.7 g) in n-butanol (150 mL) was added a slurry solution of cyanogen bromide (5M, in MeCN).The resulting mixture was heated to reflux for 4 hours.The mixture was concentrated to 1/3 of original volume.To this mixture was added Et20 (200 mL).The resulting solid was removed by filtration, and the solid was washed with Et20 (2x).The solid was partitioned between EtOAc and saturated Na2CO3 (aq).The aqueous layer was extracted with EtOAc (3x).The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated to give 10.6 g
Scheme 10:
The nitro compound (Scheme 3b) (2. 50 g, 6. 0 mmol) of Et0H (150 mL) was degassed (To this solution was bubbled with nitrogen time 3 min).To this solution was added Pd / C (10% w / w, 50% water, 698 mg).The mixture was placed in a nitrogen atmosphere.Exhaust, and backfilled with H2 (3x).The obtained mixture at room temperature, followed by stirring under H2 balloon for 2 hours.Bubbling nitrogen gas, and the mixture was purged, filtered through Celite, and concentrated.Small plug filtered through a silica gel column, eluting with EtOAc, and the product was purified to give the aniline (2. 2g, 97%).
These steps were performed using similar procedures to those described in steps 1-4 of Scheme 1a.
Step 5:
To a solution of the amine from step 4 (10.5 g, 36 mmol) in CH2Cl2 (200 mL) was added benzoylisothiocyanate (4.3 mL, 1.1 eq.). The resulting solution was stirred at RT for 2.5 days. Additional benzoylisothiocyanate (0.86 mL, 0.2 eq.) was added and the solution was stirred at RT for an additional 2 hours. The solution was then concentrated in vacuo.
A portion of this material (6.5 g, ˜14 mmol) was dissolved in MeOH (200 mL). To this solution was added Na2CO3 (s) (1.52 g, 14 mmol). The resultant mixture was stirred at RT for 45 min. After that time, a slight excess of HOAc was added to the solution. The mixture was then concentrated. The residue was partitioned between CH2Cl2 and ½ sat. NaHCO3 (aq.). The aqueous layer was extracted with CH2Cl2 (3×). The combined organic layers were dried over Na2SO4, filtered and concentrated. The thiourea (˜4.9 g) was carried onto the next reaction without further purification.
Step 6:
Example 15 was prepared using a method similar to that described in Scheme 1a step 6.
To a shiny of amine A (Scheme 3a step 4) (13.7 grams) in n-butanol (150 mL) was added a solution of cyanogen bromide (5M in MeCN). The resultant mixture was heated to reflux for 4 hours. The mixture was concentrated to ⅓ of the original volume. To the mixture was added Et2O (200 mL). The resultant solid was removed via filtration and the solid was washed with Et2O (2×). The solid was partitioned between EtOAc and sat. Na2CO3 (aq.). The aqueous layer was extracted with EtOAc (3×). The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated to afford 10.6 grams of Ex. 15. This material was converted to the t-butyl carbamate using a procedure similar to that described in Scheme 3.
Step 7:
A mixture of the bromide (3.00 g, 6.92 mmol), benzophenone imine (1.39 mL, 8.30 mmol), Pd2(dba)3 (0.634 g, 0.692 mmol), John-Phos (0.413 g, 1.38 mmol), sodium tert-butoxide (2.13 g, 22.1 mmol), and toluene (51 mL) was degassed (vacuum/N2). The mixture was then stirred at 65° C. under nitrogen for 3 h. After this time, the reaction mixture was cooled to room temperature and filtered through a pad of Celite and rinsed with ethyl acetate (100 mL). The filtrate was concentrated under reduced pressure. The residue was then dissolved in methanol (76 mL) and the resulting solution was charged with hydroxyl amine hydrochloride (2.16 g, 31.1 mmol) and sodium acetate (2.55 g, 31.1 mmol). The reaction mixture was stirred at room temperature for 40 min. After this time, the reaction mixture was concentrated under reduced pressure. The resulting residue was dissolved in ethyl acetate (200 mL) and washed with saturated aqueous sodium bicarbonate (100 mL), water (100 mL), and brine (100 mL). The organic layer was then dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (silica, 0-100% ethyl acetate/heptane) to afford the amino pyridine (0.880 g, 34%).
To a flame-dried flask was added a pyridyl bromide (Table IIb, Entry 15, 1.5 g, 3.3 mmol), Pd2(dba)3 (305 mg, 0.3 mmol), (2-biphenyl)di-tert-butylphosphine (200 mg, 0.7 mmol), sodium tert-butoxide (1.02 g, 0.011 mmol), benzophenone imine (670 ul, 4 mmol), and toluene (21 mL). The mixture was evacuated under vacuum and back-filled with N2 (3×). The mixture was stirred at 60° C. for 1 h. After filtration through celite, the filtrate was concentrated. The crude residue was dissolved in 36 mL of methanol, and hydroxyl amine hydrochloride (458 mg, 6.6 mmol) and sodium acetate (541 mg, 6.6 mmol) were added. The reaction was stirred for 35 min and then quenched with saturated aqueous sodium bicarbonate. The mixture was extracted with ethyl acetate, and the combined organic portions were dried over magnesium sulfate and concentrated. The crude residue was purified by a flash silica column (50% ethyl acetate/hexane) to get an aminopyridine product (730 mg, 68%).
A solution of the nitro compound (Scheme 3b) (2.50 g, 6.0 mmol) in EtOH (150 mL) was degassed by bubbling N2 through the solution for 3 min. To this solution was added Pd/C (10% w/w, 50% H2O, 698 mg.). The mixture was placed under an atmosphere of N2. The atmosphere was evacuated and back-filled with H2 (3×). The resulting mixture was stirred at RT under a H2 balloon for 2 h. The mixture was purged by bubbling N2 through it, filtered through Celite and concentrated. The product was purified by filtering through a small plug of silica gel column eluting with EtOAc to afford the aniline (2.2 g, 97%).
ENTRY 25
MH+: 410.0, HPLC1.79 min, LCMSMETHOD D
Method D:
Column: Agilent Zorbax SB-C18 (3.0×50 mm) 1.8 uM
Mobile phase: A: 0.05% Trifluoroacetic acid in water
B: 0.05% Trifluoroacetic acid in acetonitrile
Gradient: 90:10 (A:B) for 0.3 min, 90:10 to 5:95 (A:B) over 1.2 min, 5:95 (A:B) for 1.2 min.
Flow rate: 1.0 mL/min
UV detection: 254 and 220 nm
Mass spectrometer: Agilent 6140 quadrupole
update…………..
Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-Dioxide Derivative Verubecestat (MK-8931)–A β-Site Amyloid Precursor Protein Cleaving Enzyme 1 Inhibitor for the Treatment of Alzheimer’s Disease
Departments of †Discovery Chemistry, ‡Neuroscience, §Pharmacokinetics, Pharmacodynamics, and Drug Metabolism, ΔTranslational Medicine, #Structural Chemistry, ⊥Molecular and Materials Characterization, ∥Pharmaceutical Sciences and Clinical Supply, and ¶Toxicological Sciences, MRL, Merck & Co. Inc., 2015 Galloping Hill Road, Kenilworth, New Jersey 07033, United States
∇Albany Molecular Research Inc., 26 Corporate Circle, Albany, New York 12203, United States
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
Abstract
Verubecestat 3 (MK-8931), a diaryl amide-substituted 3-imino-1,2,4-thiadiazinane 1,1-dioxide derivative, is a high-affinity β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitor currently undergoing Phase 3 clinical evaluation for the treatment of mild to moderate and prodromal Alzheimer’s disease. Although not selective over the closely related aspartyl protease BACE2, verubecestat has high selectivity for BACE1 over other key aspartyl proteases, notably cathepsin D, and profoundly lowers CSF and brain Aβ levels in rats and nonhuman primates and CSF Aβ levels in humans. In this annotation, we describe the discovery of 3, including design, validation, and selected SAR around the novel iminothiadiazinane dioxide core as well as aspects of its preclinical and Phase 1 clinical characterization.
(a) Wortmann, M.Dementia: a global health priority – highlights from an ADI and World Health Organization reportAlzheimer’s Res. Ther.2012, 4, 40– 43, DOI: 10.1186/alzrt143
Haass, C.; Selkoe, D. J.Soluble protein oligomers in neurodegeneration: Lesson from the Alzheimer’s amyloid β-peptideNat. Rev. Mol. Cell Biol.2007, 8, 101– 112, DOI: 10.1038/nrm2101
(a) Hardy, J.; Selkoe, D. J.The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeuticsScience2002, 297, 353– 356, DOI: 10.1126/science.1072994
(b) Karran, E.; Mercken, M.; De Strooper, B.The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeuticsNat. Rev. Drug Discovery2011, 10, 698– 712, DOI: 10.1038/nrd3505
Roberds, S. L.; Anderson, J.; Basi, G.; Bienkowski, M. J.; Branstetter, D. G.; Chen, K. S.; Freedman, S. B.; Frigon, N. L.; Games, D.; Hu, K.; Johnson-Wood, K.; Kappenman, K. E.; Kawabe, T. T.; Kola, I.; Kuehn,R.; Lee, M.; Liu, W.; Motter, R.; Nichols, N. F.; Power, M.; Robertson, D. W.; Schenk, D.; Schoor, M.; Shopp, G. M.; Shuck, M. E.; Sinha, S.; Svensson, K. A.; Tatsuno, G.; Tintrup, H.; Wijsman, J.; Wright, S.; McConlogue, L.BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeuticsHum. Mol. Genet.2001, 10, 1317– 1324
Vassar, R.; Kuhn, P.-H.; Haass, C.; Kennedy, M. E.; Rajendran, L.; Wong, P. C.; Lichtenthaler, S. F.Function, therapeutic potential and cell biology of BACE proteases: current status and future prospectsJ. Neurochem.2014, 130, 4– 28, DOI: 10.1111/jnc.12715
(a) Vassar, R.; Bennett, B. D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E. A.; Denis, P.; Teplow, D. B.; Ross, S.; Amarante, P.; Loeloff, R.; Luo, Y.; Fisher, S.; Fuller, J.; Edenson, S.; Lile, J.; Jarosinski, M. A.; Biere, A. L.; Curran, E.; Burgess, T.; Louis, J.-C.; Collins, F.; Treanor, J.; Rogers, G.; Citron, M.β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACEScience1999,286, 735– 741
(b) Sinha, S.; Anderson, J. P.; Barbour, R.; Basi, G. S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H. F.; Frigon, N.; Hong, J.; Jacobson-Croak, K.; Jewett, N.; Keim, P.; Knops, J.; Lieberburg, I.; Power, M.; Tan, H.; Tatsuno, G.; Tung, J.; Schenk, D.; Seubert, P.; Suomensaari, S. M.; Wang, S.; Walker, D.; Zhao, J.; McConlogue, L.; John, V.Purification and cloning of amyloid precursor protein β-secretase from human brainNature1999, 402, 537– 540, DOI: 10.1038/990114
(c) Hussain, I.; Powell, D.; Howlett, D. R.; Tew, D. G.; Meek,T. D.; Chapman, C.; Gloger, I. S.; Murphy, K. E.; Southan, C. D.; Ryan, D. M.; Smith, T. S.; Simmons, D. L.; Walsh, F. S.; Dingwall, C.; Christie, G.Identification of a novel aspartic protease (Asp 2) as β-SecretaseMol. Cell. Neurosci.1999, 14, 419– 427, DOI: 10.1006/mcne.1999.0811
(d) Yan, R.; Bienkowski, M. J.; Shuck, M. E.; Miao, H.; Tory, M. C.; Pauley, A. M.; Brashler, J. R.; Stratman, N. C.; Mathews, W. R.; Buhl, A. E.; Carter, D. B.; Tomasselli, A. G.; Parodi, L. A.; Heinrikson, R. L.; Gurney,M. E.Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activityNature1999,402, 533– 537, DOI: 10.1038/990107
(e) Lin, X.; Koelsch, G.; Wu, S.; Downs,D.; Dashti, A.; Tang, J.Human aspartic protease memapsin 2 cleaves the β-secretase site of β-amyloid precursor proteinProc. Natl. Acad. Sci. U. S. A.2000, 97, 1456– 1460, DOI: 10.1073/pnas.97.4.1456
(a) Oehlrich, D.; Prokopcova, H.; Gijsen, H. J. M.The evolution of amidine-based brain penetrant BACE1 inhibitorsBioorg. Med. Chem. Lett.2014, 24, 2033– 2045, DOI: 10.1016/j.bmcl.2014.03.025
(b) Yuan, J.; Venkatraman, S.; Zheng, Y.; McKeever, B. M.; Dillard, L. W.; Singh, S. B.Structure-based design of β-site APP cleaving enzyme 1 (BACE) inhibitors for the treatment of Alzheimer’s disease. diseaseJ. Med. Chem.2013, 56, 4156– 4180, DOI: 10.1021/jm301659n
(c) Probst, G.; Xu, Y. z.Small-molecule BACE1 inhibitors: a patent literature review (2006 – 2011)Expert Opin. Ther. Pat.2012, 22, 511– 540, DOI: 10.1517/13543776.2012.681302
(a) Willem, M.; Garratt, A. N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C.Control of peripheral nerve myelination by the β-Secretase BACE1Science2006, 314, 664– 666, DOI: 10.1126/science.1132341
(b) Hu, X.; Hicks, C. W.; He, W.; Wong, P.; Macklin, W. B.; Trapp, B. D.; Yan, R.Bace1 modulates myelination in the central and peripheral nervous systemNat. Neurosci.2006, 9, 1520– 1525, DOI: 10.1038/nn1797
Cheret, C.; Willem, M.; Fricker, F. R.; Wende, H.; Wulf-Goldenberg, A.; Tahirovic, S.; Nave, K.-A.; Saftig,P.; Haass, C.; Garratt, A. N.; Bennett, D. L.; Birchmeier, C.Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindlesEMBO J.2013, 32, 2015– 2028, DOI: 10.1038/emboj.2013.146
McConlogue, L.; Buttini, M.; Anderson, J. P.; Brigham, E. F.; Chen, K. S.; Freedman, S. B.; Games, D.; Johnson-Wood, K.; Lee, M.; Zeller, M.; Liu, W.; Motter, R.; Sinha, S.Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic miceJ. Biol. Chem.2007,282, 26326– 26334, DOI: 10.1074/jbc.M611687200
Wang, Y.-S.; Strickland, C.; Voigt, J. H.; Kennedy, M. E.; Beyer, B. M.; Senior, M. M.; Smith, E. M.; Nechuta, T. L.; Madison, V. S.; Czarniecki, M.; McKittrick, B. A.; Stamford, A. W.; Parker, E. M.; Hunter, J. C.; Greenlee, W. J.; Wyss, D. F.Application of fragment-based NMR screening, X-ray crystallography, structure-based design, and focused chemical library design to identify novel μM leads for the development of nM BACE-1 (β-site APP cleaving enzyme 1) inhibitorsJ. Med. Chem.2010, 53, 942– 950, DOI: 10.1021/jm901472u
The CAS name represents the 3-aminothiadiazine tautomer, and the structures depicted in the manuscript represent the 3-iminothiadiazinane tautomer.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये। DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Toremifene is a first generation selective estrogen receptor modulator (SERM). Like TAMOXIFEN, it is an estrogen agonist for bone tissue and cholesterol metabolism but is antagonistic on mammary and uterine tissue.
The company GTx is conducting phase III clinical trials for the prevention of prostate cancer in men who have been diagnosed with high grade prostatic intraepithelial neoplasia (PIN).
Toremifene citrate is an oral selective estrogen receptor modulator (SERM) which helps oppose the actions of estrogen in the body. Licensed in the United States under the brand name Fareston, toremifene citrate is FDA-approved for use in advanced (metastatic)breast cancer. It is also being evaluated for prevention of prostate cancer under the brand name Acapodene.[1]
In 2007 the pharmaceutical company GTx, Inc was conducting two different phase 3 clinical trials; First, a pivotal Phase clinical trial for the treatment of serious side effects of androgen deprivation therapy (ADT) (especially vertebral/spine fractures and hot flashes, lipid profile, and gynecomastia) for advanced prostate cancer, and second, a pivotal Phase III clinical trial for the prevention of prostate cancer in high risk men with high grade prostatic intraepithelial neoplasia, or PIN. Results of these trials are expected by first quarter of 2008[2]
An NDA for the first application (relief of prostate cancer ADT side effects) was submitted in Feb 2009,[3] and in Oct 2009 the FDA said they would need more clinical data, e.g. another phase III trial.[4]
Originally developed at Orion, toremifene was subsequently licensed to Nippon Kayaku in Japan and to Asta Medica (now, part of Meda) in Germany.
Toremifene (Toremifene), chemical name (Z) -4- chloro-1,2-diphenyl–1- [4- (2- (N, N- dimethylamino) ethoxy yl) phenyl] -1-butene, having the structure I.Toremifene to tamoxifen (Tamoxifen) analogues with anti-estrogenic activity, can be used in the treatment of hormone-dependent breast cancer, and its E-isomer has the presence of estrogenic activity, E isomers toremifene may counteract anti-estrogenic activity, and therefore isomeric purity is essential toremifene.Toremifene was developed in 1983 by the Finnish Famos company, listed in 1996 by the Orion company in the EU, the trade name Fareston, 2002 to enter the country, the trade name of toremifene.
RJ Toivola et al., European Patent EP95875, disclosed in U.S. Patent US4696949A synthetic route toremifene, that following a synthetic route, the synthetic route to phenol as a raw material, by acylation, rearrangement, alkyl group and an addition reaction to give 1,2-diphenyl -1- [4- [2- (N, N- dimethylamino) ethoxyphenyl]] – 1,4-diol (Compound 5) as the key intermediate, further HCl in ethanol or hydrochloric acid elimination reaction occurs, then get toremifene thionyl chloride reaction.The main problem with this approach is that the elimination reaction of the compound 5 in ethanol occurs when hydrochloric acid or concentrated hydrochloric acid, the resulting triaryl alcohol butyrate (Compound 6) having a Z / E configuration, both the ratio of 1: 2 ~ 2: 1, stereo selectivity is not high, and there are 5% of the cyclization by-product; on the Z / E configuration Miyoshi butyric fractional crystallization of alcohol, you can get pure Z-type Miyoshi butyric alcohol , but the yield is only 41%; then, Z-type Miyoshi butyric alcohol chlorination reaction occurs in the action of thionyl chloride, the purified product toremifene.
U.S. Patent US5491173A also reported another synthetic route toremifene namely the following two synthetic routes.The route to the aryl ketone (Compound 7) with a phenyl Grignard reagent addition reaction of ketone carbonyl groups to give triaryl-butanediol (compound 5), which is the elimination of toremifene and chlorinated reaction products happen again.
Chinese Patent Publication No. CN1125716A application reported an efficient synthesis of Z-type Miyoshi butyric alcohol (compound 6) method, US4696949A compared with the US patent, the method mild conditions, reduce the acid concentration and reaction temperature, reaction time, triarylphosphine butanediol (Compound 5) in concentrated hydrochloric acid or concentrated hydrochloric isopropanol or ethanol effect of concentrated hydrochloric acid, can be 60-78% selectivity and 95% yield of type 2 Miyoshi butyric alcohol But after Publication No. 0 02,126,969 attached eight patent applications after the inventor repeated experiments show that the technique disclosed in the patent application programs can not achieve their claimed technical effect.
Publication No. CN102126969A of Chinese patent applications through the intermediate Miyoshi butyric alcohol occurs at a catalytic converter configuration of concentrated hydrochloric acid, while taking advantage of differences in solubility, so E- type Miyoshi butyric alcohol continuously into Z-type Miyoshi butyric alcohol (compound 6) precipitates, thereby undermining the balance, so that one of the E-type Miyoshi butyric alcohol continuously into Z-type Miyoshi butyric alcohol (compound 6) to give the Z-Miyoshi butyric alcohol ( Compound 6) and then get toremifene thionyl chloride after chlorination.Although to some extent, improve the yield, but increased operating procedure, is not conducive to industrial production.
Currently, the key intermediate is patent protected, and z-type Miyoshi butyric alcohol (compound 6) stereoselective low yield and isolated intermediates, to solve this problem, to overcome technical barriers to foreign pharmaceutical companies, urgent need to find a simple process, low cost, easy to separate and viable for large-scale production of synthetic routes.
To achieve the above object, according to one aspect of the present invention, there is provided a method of synthesizing toremifene, synthetic method comprising: a step S1, so that a compound having the structural formula II with a compound B having the structural formula III C occurs Mike Murray to give compound D having the structural formula IV; step S2, the Compound D and Compound E or Compound E of the hydrochloride salt of the formula V having a phenolic hydroxyl group on the occurrence of a selective alkylation reaction, to give a compound having the structural formula VI F; step S3, the compound F is reacted with thionyl chloride to give toremifene, wherein,
Formula II is:
Structural formula III as follows:
Formula IV is
Of formula V is C1CH2CH2N (CH 3) 2; formula VI is
FIG. 1 illustrates the present invention obtained in Example 1 H NMR spectrum of compound D of implementation;
FIG. 2 shows the 1 H NMR spectrum of the present invention, the compound obtained in Example F;
FIG. 3 shows the present invention is a proton nuclear magnetic resonance spectrum of toremifene obtained in Example.
Figure 1, which shows a spectrum of results for Che bandit? (400 cm take, 01 ^ 0) 3 = 9.20 (! 8,1 1), 7.37 (^ = 7.4 to take, 2 1!), 7.30- 7. 23 (m, 3H), 1.22- 7. 15 (m, 2H), 7. 15 – 7. 06 ( m, 3H), 6. 61 (dd, J = 9. 0, 2. 2Hz, 2H), 6. 49 -. 6. 32 (m, 2H), 4 48 (s, 1H), 3 30 (. m, 2H), 2 55 (t, J = 7. 5Hz, 2H);. F proton nuclear magnetic resonance spectrum of the compound attached to the
The synthetic routes above synthetic method are as follows:
Synthesis of toremifene:
To a 2L reaction flask 1. 1L of toluene, 110g (0. 28mol) obtained in the above step Z configuration compound F, mixed to obtain a sixth system, the cooling system to the sixth mixed 0~5 ° C , was slowly added dropwise 99. 93g (0. 84mol) thionyl chloride addition was complete the formation of the seventh mixed system, the mixed system was slowly warmed to a seventh ll〇 ° C, for 1 hour to obtain a third product system, stop The third product heating and cooling system to 15~25 ° C, the third product system slowly poured into 1L of water, adding NaOH solution to a pH 9~10 and get the second system, the second in and a system for liquid separation, and the resulting aqueous phase to obtain a second solution was extracted with 1L toluene extraction, the organic phase of the second extraction solution and liquid separation were combined and concentrated to give crude toremifene, the crude product was mass ratio of 1 : mixed solvent of ethyl acetate and acetone 1 crystals to give 103. 7g toremifene products.
Synthesis of toremifene:
[0062] To a 5L reaction flask 3. 3L of toluene, 110g (0. 28mol) obtained in the above step Z configuration compound F, mixed to obtain a sixth system, the cooling system to the sixth mixed 0~5 ° C , was slowly added dropwise 33. 31g (0. 28mol) thionyl chloride addition was complete the formation of the seventh mixed system, the mixed system was slowly warmed to a seventh ll〇 ° C, after the reaction for 6 hours to obtain a third product system, stop The third product heating and cooling system to 15~25 ° C, the third product system slowly poured into 1L of water, potassium carbonate solution to a pH 9~10 and get the second system, the second and system for liquid separation, and the resulting aqueous phase to obtain a second solution was extracted with 1L ethyl acetate, the organic phase after the second extraction solution and liquid separation were combined and concentrated to give crude toremifene, the crude product was quality ratio was crystallized from acetone to give 92. 2g toremifene products.
Purity of toremifene following method:
[0107] to take the product, add the mobile phase dissolved and diluted into 1ml of 1. Omg solution containing, according to HPLC octadecylsilane bonded silica as a filler to square 1% trifluoroacetic acetic acid aqueous solution (A) and acetonitrile (B) as the mobile phase gradient elution (T = Omin 10% B; T = lOmin 95% B; T = 12min 100% B; T = 15min 10% B), detection wavelength 210nm; area normalization method to calculate the Z configuration purity compound F, where F Z configuration compound retention time of 6. 76min.The purity of the above calculation or Z configuration detection obtained compound D, compound D Z configuration and E configuration of the weight ratio, toremifene yield and purity are reported in Table 1 below.
c) 4-chloro-1,2-diphenyl-1-[4-[2-(N ,N-dimethylamino)ethoxy]phenyl]-1-butene (Z and E)
(Z)-isomer: The reaction is performed under dry conditions. 42.4 g of (Z)-1,2-diphenyl-1-[4-[2-(N,N-dimethylamino )ethoxy]phenyl]-1-buten-4-ol are dissolved in 250 ml of chloroform. Then 23.8 g of thionyl chloride areadded dropwise. The mixture is refluxed 3 h. The solvent is evaporated, after which the product is recrystallized from ethyl acetate. The yield ofthe hydrochloride salt is 36.7 g (83%), m.p. 194°-6° C. The base can be liberated from the Salt with 1M sodium carbonate solution, after which the base is extracted in toluene. The toluene solution is dried and the solvent is evaporated. The free base has m.p. 108°-10° C. (from acetone).
The citric acid salt can be prepared as follows: 40.6 g of the (Z)-isomer as a free base are dissolved in 175 ml of warm acetone and 24.3 g of citric acid are dissolved in 100 ml of warm acetone. The solutions are combined and the mixture is allowed to cool. The citrate, m.p. 160°-162° C., is collected by filtration.
(E)-isomer: The compound is prepared from (E)-1,2-diphenyl-1-[4-[2-(N ,N-dimethylamino)ethoxy]phenyl]-1-buten-4-ol in the same manner as the corresponding (Z)-isomer. The hydrochloride salt is crystallized from toluene. The yield is 35.8 g (81%) of a product having m.p. 183°-5° C. The base can be liberated from the salt in the same manner as the corresponding (Z)-isomer. It has m.p. 69°-71° C. (from hexane).
4-chloro-1,2-diphenyl-1-[4-[2-(N ,N-diethylamino)ethoxy]phenyl ]-1-butene (Z and E)
43.3 g of 1,2-diphenyl-1-[4-[2-(N,N-diethylamino)ethoxy]phenyl]butane-1,4-diol (pureenantiomer pairs or their mixture: m.p. of (RR,SS)-pair is 107°-9° C.)is suspended in 250 ml of toluene, after which 25ml toluene is distilled off to dry the solution. The mixture is cooled to 0° C. with stirring. While stirring and keeping the temperature at 0° C. or a little below, 47.6 g of thionyl chloride of good qualityare added. The mixture is stirred for 1 h at 0° C. and the temperature is then allowed to rise to 22° C. The mixture is stirred at 80° C. until the reaction is completed (about 3 h). After that, water is added to decompose the excess of thionyl chloride followed by 20% sodium hydroxide solution to liberate the product from itshydrochloride salt. The aqueous layer is discarded and the toluene layer iswashed with water. Then the solvent is evaporated to leave a mixture of (Z)- and (E)isomers (Z:E 7:3) as an oil in quantitative yield.
(Z)-isomer: The (Z)-isomer is isolated from the isomer mixture above as thehydrochloride salt because of the low melting point of the free base. The m.p. of the hydrochloride salt is 178°-80° C. The (Z)-isomermay be freed from its salt by any normal method.
Price N, Sartor O, Hutson T, Mariani S. Role of 5a-reductase inhibitors and selective estrogen receptor modulators as potential chemopreventive agents for prostate cancer. Clin Prostate Cancer 2005;3:211-4. PMID 15882476
Androgen Decline in the Aging Male (Andropause) in phase 2
Fispemifene is the Z-isomer of the compound of formula (I)
WO 01/36360 describes a group of SERMs, which are tissue-specific estrogens and which can be used in women in the treatment of climacteric symptoms, osteoporosis, Alzheimer’s disease and/or cardiovascular diseases without the carcinogenic risk. Certain compounds can be given to men to protect them against osteoporosis, cardiovascular diseases and Alzheimer’s disease without estrogenic adverse events (gynecomastia, decreased libido etc.). Of the compounds described in said patent publication, the compound (Z)-2-{2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethoxy}ethanol (also known under the generic name fispemifene) has shown a very interesting hormonal profile suggesting that it will be especially valuable for treating disorders in men. WO 2004/108645 and WO 2006/024689 suggest the use of fispemifene for treatment or prevention of age-related symptoms in men, such as lower urinary tract symptoms and diseases or disorders related to androgen deficiency in men.
Quatrx had been conducting phase II clinical development for the treatment of androgen decline in the aging male. Unlike testosterone replacement therapies that are typically topical or injection therapies, fispemifene is an oral treatment and is not a formulation of testosterone. Fispemifene utilizes the body’s normal feedback mechanism to increase testosterone levels. Originally developed at Hormos, QuatRx gained rights to the drug candidate following a merger of the companies pursuant to which Hormos became a wholly-owned subsidiary of QuatRx.
Known methods for the syntheses of compounds like ospemifene and fispemifene include rather many steps. WO 02/090305 describes a method for the preparation of fispemifene, where, in a first step, a triphenylbutane compound with a dihydroxysubstituted butane chain is obtained. This compound is in a second step converted to a triphenylbutene where the chain is 4-chlorosubstituted. Then the desired Z-isomer is crystallized. Finally, the protecting group is removed to release the ethanol-ethoxy chain of the molecule.
Fispemifene is a selective estrogen receptor modulator (SERM) studied in phase II clinical trials at Forendo Pharma for the treatment low testosterone in men. The compound is also in phase II clinical studies at Apricus for the treatment of men with secondary hypogonadism.
In 2013, Forendo Pharma acquired the drug from Hormos Medical for the treatment of male low testosterone.
In 2014, Apricus Biosciences acquired U.S. rights for development and commercialization
EXAMPLE 2 2-{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethoxy}-ethanol (Compound I)
{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethoxy}-acetic acid ethyl ester was dissolved in tetrahydrofuran at room temperature under nitrogen atmosphere. Lithium aluminium hydride was added to the solution in small portions until the reduction reaction was complete. The reaction was quenched with saturated aqueous ammonium chloride solution. The product was extracted into toluene, which was dried and evaporated in vacuo. The residue was purified with flash chromatography with toluene/triethyl amine (9.5:0.5) as eluent. Yield 68%.
is prepared from benzyl bromide and 2-(2-chloroethoxy)ethanol by the method described in literature (Bessodes, 1996).
b) {4-[2-(2-Benzyloxyethoxy)ethoxy]phenyl}phenylmethanone
The mixture of 4-hydroxybenzophenone (16.7 g, 84.7 mmol) and 48 % aqueous sodium hydroxide solution (170 ml) is heated to 80 °C. Tetrabutylammonium bromide (TBABr) (1.6 g, 5.1 mmol) is added and the mixture is heated to 90 °C. [2-(2-Chloroethoxy)ethoxymethyl]benzene (18. g, 84.7 mmol) is added to the mixture during 15 min and the stirring is continued for additional 3.5 h at 115-120 °C. Then the mixture is cooled to 70 °C and 170 ml of water and 170 ml of toluene are added to the reaction mixture and stirring is continued for 5 min. The layers are separated and the aqueous phase is extracted twice with 50 ml of toluene. The organic phases are combined and washed with water, dried with sodium sulphate and evaporated to dryness. Yield 31.2 g.
Another method to prepare {4-[2-(2-benzyloxyethoxy)ethoxy]phenyl}phenyl- methanone is the reaction of 2-(2-benzyloxyethoxy)ethyl mesylate with 4- hydroxybenzophenone in PTC-conditions.
c) 1- {4-[2-(2-Benzyloxyethoxy)ethoxy]phenyl} – 1 ,2-diphenyl -butane- 1 ,4-diol
R = BENZYL
Lithium aluminum hydride (1.08 g, 28.6 mmol) is added into dry tetrahydrofuran (60 ml) under nitrogen atmosphere. Cinnamaldehyde (6.65 g, 50 mmol) in dry tetrahydrofuran (16 ml) is added at 24-28 °C. The reaction mixture is stirred at ambient temperature for 1 h. {4-[2-(2- Benzyloxyethoxy)ethoxy]phenyl}-phenyl-methanone (14.0 g, 37 mmol) in dry tetrahydrofuran (16 ml) is added at 50-55 °C. The reaction mixture is stirred at 60 °C for 3 h. Most of tetrahydrofuran is evaporated. Toluene (70 ml) and 2 M aqueous hydrogen chloride (50 ml) are added. The mixture is stirred for 5 min and the aqueous layer is separated and extracted with toluene (30 ml). The toluene layers are combined and washed with 2M HC1 and water, dried and evaporated. The product is crystallized from isopropanol as a mixture of stereoisomers (8.8 g, 50 %).
Η NMR (CDCI3 ): 1.75-2.10 (m, 2H), 3.20-4.16 (m, 1 OH), 4.52 and 4.55 (2s, together 2H), 6.61 and 6.88 (2d, together 2H), 6.95-7.39 (m, 15H), 7.49 and 7.57 (2d, together 2H).
1 – {4- [2-(2-Benzyloxy-ethoxy)ethoxy]phenyl} – 1 ,2-diphenyl -butane- 1 ,4-diol (10.0 g, 19.5 mmol) is dissolved in toluene (50 ml). Triethylamine (2.17 g, 21.4 mmol) is added to the solution and the mixture is cooled to -10 °C. Thionyl chloride (6.9 g, 58.5 mmol) is added to the mixture at -10 – ±0 °C. The mixture is stirred for 1 hour at 0-5 °C, warmed up to 70 °C and stirred at this temperature for 4 hours. Solvent is evaporated, the residue is dissolved to toluene, washed three times with 1M HC1 solution and twice with water. The Z-isomer of the product is crystallized from isopropanol-ethyl acetate. Yield 3.0 g. The filtrate is purified by flash chromatography to give E-isomer.
Z- 1 – {4-[2-(2-Benzyloxy-ethoxy)ethoxy]phenyl} -4-chloro- 1 ,2-diphenyl -but- 1-ene (3.8 g, 7.4 mmol) is dissolved in ethyl acetate under nitrogen atmosphere , Zn powder (0.12 g, 1.85 mmol) and acetyl chloride (1.27 g, 16.3 mmol) are added and the mixture is stirred at 50 °C for 3 h (Bhar, 1995). The reaction mixture is cooled to room temperature, water (10 ml) is added and stirring is continued for additional 10 min. The aqueous layer is separated and the organic phase is washed with 1 M aqueous hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in methanol (16 ml) and water (4 ml). The acetate ester of the product is hydrolysed by making the mixture alkaline with sodium hydroxide (1 g) and stirring the mixture at room temperature for 1 h. Methanol is evaporated, water is added and the residue is extracted in ethyl acetate and washed with 1 M hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in toluene (25 ml), silica gel (0.25 g) is added and mixture is stirred for 15 min. Toluene is filtered and evaporated to dryness. The residue is crystallised from heptane-ethyl acetate (2:1). The yield is 71 %.
E-2- {2- [4-(4-Chloro- 1 ,2-diphenyl-but- 1 -enyl)phenoxy]ethoxy} ethanol is prepared analogously starting from E-l-{4-[2-(2-benzyloxy- ethoxy)ethoxy]phenyl} -4-chloro- 1,2-diphenyl-but-l-ene. The product is purified by flash chromatography with toluene-methanol (10:0.5) as eluent.
Debenzylation of 1 – {4-[2-(2-benzyloxy-ethoxy)ethoxy]phenyl} -4-chloro- 1 ,2- diphenyl-but- 1-ene is also carried out by hydrogenation with Pd on carbon as a catalyst in ethyl acetate-ethanol solution at room temperature.
Use of a selective estrogen receptor modulator for the manufacture of a pharmaceutical preparation for use in a method for the treatment or prevention of androgen deficiency
Use of a selective estrogen receptor modulator for the manufacture of a pharmaceutical preparation for use in a method for the treatment or prevention of androgen deficiency
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Gemigliptin was initially developed solely by LG Life Sciences. In 2010, Double-Crane Pharmaceutical Co. (DCPC) joined with LGLS to co-develop the final compound and collaborate on the marketing of the drug in China. LGLS also announced on Nov., 2010 that NOBEL Ilac has been granted rights to develop and commercialize gemigliptin in Turkey.
Gemigliptin, a dipeptidyl peptidase IV (CD26; DPP-IV; DP-IV) inhibitor, is currently undergoing phase III clinical trials at LG Life Sciences as an oral treatment for type II diabetes. The company is also testing the compound in phase II/III clinical studies for the treatment of patients with cisplatin-induced acute kidney injury.
DPP IV inhibitors have glucose-lowering effects mediated by GLP-1 incretin hormone which is inactivated by DPP IV. In 2010, gemigliptin was licensed to Beijing Double-Crane Pharmaceutical by LG Life Sciences for distribution and supply in China for the treatment of type 2 diabetes.
A New Drug Application (NDA) for gemigliptin in the treatment of type 2 diabetes was submitted to the Korea Food & Drug Administration (KFDA) in July 2011. Then on June 27, 2012, the KFDA has approved the manufacture and distribution of LG Life Sciences’ diabetes treatment, Zemiglo, the main substance of which is gemigliptin. Clinical trials for evaluating the safety and efficacy of gemigliptin in combination with metformin have been completed.
…………
Efficient synthesis of gemigliptin, a potent and selective DPP-4 inhibitor for the treatment of type 2 diabetes mellitus, has been developed. Gemigliptin were prepared from two key API starting materials, DP18 and DP57, in 75~80% yield and >99% purity over three steps under the GMP control: coupling, deprotection of N-Boc group, and final crystallization with L-tartaric acid. All steps were conducted in the same solvent system and the intermediates were isolated by simple filtration without distillation of solvent. The established process was validated obviously through the three consecutive batches for a commercial production.
The NDA for gemigliptin was submitted to KFDA in July, 2011 and it was approved on June 27, 2012. By the end of 2012, gemigliptin will be marketed in Korea as Zemiglo which is the fifth new DPP-4 inhibitor diabetes treatment in the world.
Mechanism of action
DPP-4 is a serine protease located on the cell surfaces throughout the body. In plasma, DPP-4 enzyme rapidly inactivates incretins including GLP-1 and GIP which are produced in the intestine depending on the blood glucose level and contribute to the physiological regulation of glucose homeostatis. Active GLP-1 and GIP increase the production and release of insulin by pancreatinc beta cells. GLP-1 also reduces the scretion of glucacon by pancreatic alpha cells, thereby resulting in a decreased hepatic glucose production. However these incretins are rapidly cleaved by DPP-4 and their effects last only for a few minutes. DPP-4 inhibitors block the cleavage of the gliptins and thus lead to an increasee insulin level and a reduced glucagon level in a glucose-dependent way. This results in a decrease of fasting and postprandial glycemia, as well as HbA1c levels.[2]
Preclinical studies
Gemigliptin is a competitive, reversible DPP-4 inhibitor (IC50 = 16 nM) with excellent selectivity over other critical human proteases such as DPP-2, DPP-8, DPP-9, elastase,trypsin, urokinase and cathepsin G. Gemigliptin was rapidly absorbed after single oral dosing and the compound was eliminated with a half-life of 3.6 h, 5.2 h, and 5.4 h in the rat, dog, and monkey, respectively.
The bioavailability of gemigliptin in the rat, dog, and monkey was species-dependent with the values of 94%, 73%, and 26%, respectively. Following the oral administration of gemigliptin in the rat, dog and monkey, about 80% inhibition of plasma DPP-4 activity were observed at the plasma levels of 18 nM, 14 nM and 4 nM, respectively.
In the diet-induced obese (DIO) mice, gemigliptin reduced glucose excursion during OGTT in a dose dependent manner with the minimum effective dose of 0.3 mg/kg and enhanced glucose-stimulated plasma GLP-1 increase in a dose dependent manner reaching the maximum effect at the dose of 1 mg/kg.
Following 4 week oral repeat dosing in the DIO mice, gemigliptin reduced significantly HbA1c with the minimum effective dose of 3 mg/kg. In the beagle dog, gemigliptin significantly enhanced active GLP-1, decreased glucagon, and reduced glucose excursion during OGTT following a single dosing.
Studies on animals suggest its positive effect on hepatic and renal fibrosis .[3][4] Data on human patients are still inconclusive .[5]
Clinical studies
The dose-range finding phase 2 study was performed and 145 patients (91men and 54 women) with type 2 diabetes mellitus were enrolled. All three doses (50,100 and 200 mg groups) of gemigliptin significantly reduced the HbA1c from baseline compared to the placebo group without a significant difference between the doses.
Subjects with a higher baseline HbA1c (≥8.5%) had a greater reduction in HbA1c. Insulin secretory function, as assessed using homeostasis model assessment-beta cell, C-peptide and the insulinogenic index, improved significantly with gemigliptin treatment. Insulin sensitivity, as assessed using homeostasis model assessment-insulin resistance, also improved significantly after 12 weeks of treatment.
The 50 and 200 mg groups had significantly reduced total cholesterol and low-density lipoprotein cholesterol levels at 12 weeks compared to the placebo group.
The incidences of adverse events were similar in all study subjects. Gemigliptin monotherapy (50 mg for 12 weeks) improved the HbA1c, FPG level, oral glucose tolerance testresults, β-cell function and insulin sensitivity measures, and was well tolerated in subjects with type 2 diabetes.
Results of Phase 3 clinical trials which have been finished recently will be updated near future.
-difluoro-2-oxpiperidin-l-yl)methyl]-3-oxpropyl}carbamate obtained in
PREPARATION 143 was used. [1962] 1K NMR (CD3OD) δ 5.05-4.92 (2H, m), 3.98-3.91 (2H, m), 3.85-3.79 (2H, m),
3.70-3.59 (2H, m), 3.54-3.48 (IH, m), 3.36-3.33 (2H, m), 3.24 (IH, bra), 3.14 (IH, bra), 2.83-2.76 (IH, m), 2.72-2.53 (3H, m), 2.43-2.34 (2H, m) [1963] Mass (m/e) 490 (M+l)
[1966] 14 mg of the title compound was obtained in a yield of 17% at the same manner as in PREPARATION 45, except that 43.7 mg (0.138 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-[2(S)-2-methyl-5-oxomoφholin-4-yl]-butanoic acid obtained in PREPARATION 55 and 42.5 mg (0.138 mmol) of 2,4-bis(trifluoromethyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine hydrochloric acid salt (product of PREPARATION 127) were used.
[1967] 1K NMR (CDCl3) δ 5.85-5.83 (IH, m), 5.09-4.92 (IH, m), 4.95-4.78 (IH, m),
4.23-4.08 (3H, m), 4.04-3.76 (3H, m), 3.73-3.66 (IH, m), 3.46-3.38 (IH, m), 3.36-3.21 (2H, m), 3.18-3.10 (2H, m), 2.96-2.81 (IH, m), 2.61-2.50 (IH, m), 1.43-1.41 (9H, m), 1.28-1.24 (3H, m)
PREPARATION 1: Synthesis of diethyl 2,2-difluoropentanedioate
To a solution of ethyl bromodifluoroacetate (33.2 g) in tetrahydrofuran (94.0 g) was added ethyl acrylate (8.2 g) and copper powder (10.9 g). After heating to 50℃, TMEDA (9.5 g) was added dropwise and the reaction mixture was then stirred for 3 hours at the same temperature. Upon disappearance of ethyl acrylate as the starting material, to the reaction solution was added methyl t-butyl ether (MTBE, 73.7 g) followed by addition of 10% aqueous ammonium chloride solution (49.8 g) dropwise, and the mixture was then stirred for 30 minutes. The remaining copper residue was removed by filtration through a celite, and methyl t-butyl ether (MTBE, 66.3 g) was added to separate the layers. The separated organic layer was washed successively with 10% aqueous NH4Cl solution (66.3 g) and 3 N aqueous hydrochloric acid solution (99.6 g) in order and then distilled under reduced pressure to obtain 55.0 g of the desired title compound.
PREPARATION 2: Synthesis of ethyl 4,4-difluoro-5-hydroxypentanoate
14.8 g of the compound obtained from the above Preparation 1 was diluted with ethanol (20.4 g) and tetrahydrofuran (69.1 g) and then cooled to 0℃. To this solution was slowly added sodium borohydride (NaBH4, 3.5 g) stepwise while keeping the internal temperature below 30℃. After confirming completion of the reaction by 1H NMR, the reaction solution was cooled to the temperature of 10℃ and 10% aqueous ammonium chloride solution (77.7 g) was slowly added. The remaining boron compound was filtered through celite, and the filtrate was distilled under reduced pressure to remove tetrahydrofuran. Then, ethyl acetate (105.2 g) was added to separate the layers, and the organic layer was distilled under reduced pressure to obtain 10.8 g of the title compound.
EXAMPLE 1: Synthesis of ethyl 4,4-difluoro-5-{[(trifluoromethyl)sulfonyl]oxy}- pentanoate
To the solution of 10.8 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (100.2 g) was added pyridine (7.0 g), and then the mixture was cooled to -5.0℃. After completion of cooling, trifluoromethane sulfonic acid anhydride (20.1 g) was slowly added dropwise while keeping the reaction temperature below 6.3℃. After stirring the reaction solution for 30 minutes, 1.5 N hydrochloric acid solution was added dropwise at 0℃ to separate the layers. The aqueous layer as separated was back-extracted twice with dichloromethane (33.4 g), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 19.7 g of the title compound as a yellow oil.
EXAMPLE 2-1: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate
To the solution of 100.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (300.0 ml) was added pyridine (65.7 g), and the mixture was then cooled to -10.0℃. After completion of cooling, nonafluorobutanesulfonic anhydride (477.4 g) was slowly added dropwise. After stirring the reaction solution for 3 hours, 1.0 N hydrochloric acid solution (300.0 ml) was added dropwise to separate the layers. The aqueous layer as separated was back extracted once with dichloromethane (500.0 ml), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 177.5 g of the title compound.
EXAMPLE 2-2: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate
To the solution of 500.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (1000.0 ml) was added triethylamine (389.0 g), and the mixture was then cooled to 0℃. After completion of cooling, perfluorobutanesulfonyl chloride (948.80 g) was slowly added dropwise. The reaction solution was stirred for 3 hours at room temperature, distilled under reduced pressure, dissolved in methyl t-butyl ether (MTBE, 3000.0 ml) and then washed three times with water. The organic layer thus obtained was dehydrated with magnesium sulfate, filtered through a celite and then distilled under reduced pressure to obtain 960.0 g of the title compound.
EXAMPLE 3: Synthesis of methyl (2S)-2-[(tert-butoxycarbonyl)amino]-4-oxo- pentanoate
To 25.0 g of the starting material, (3S)-3-[(t-butoxycarbonyl)amino]-4-oxo- pentanoic acid, was added t-butanol (96.9 g) followed by the addition of Boc2O (25.4 g) and dimethylaminopyridine (DMAP, 62.0 g, 0.5 mol%) at room temperature, and the reaction mixture was then stirred for 23 hours at 40℃. Upon completion of the reaction, ethylene dichloride (62.3 g) in t-butanol was added, and the mixture was then distilled under reduced pressure to obtain 30.7 g of the title compound.
EXAMPLE 4: Synthesis of tert-butyl (3S)-3-[(tert-butoxycarbonyl)amino]-4-hydroxy- butanoate
30.7 g of the compound obtained from the above Example 3 was dissolved in ethanol (112.3 g) and, after lowering the internal temperature to 10.5℃ sodium borohydride (NaBH4, 5.7 g) was slowly added dropwise. This reaction solution was stirred while maintaining the temperature below 22℃. After confirming completion of the reaction by 1H NMR and TLC, to the reaction solution was slowly added 3.0 N hydrochloric acid solution (30.7 g) dropwise at the internal temperature of 10℃ followed by addition of diluted 0.2% hydrochloric acid solution (100.0 g). The reaction solution was adjusted to pH 3~4 with addition of 9.0% aqueous hydrochloric acid solution, and then back-extracted twice with ethyl acetate (100.0 g) and toluene (44.0 g). The organic layer thus obtained was distilled under reduced pressure to obtain 25.1 g of the title compound.
EXAMPLE 5: tert-butyl (3S)-[(tert-butoxycarbonyl)amino]-4-[(methylsulfonyl)oxy]- butanoate
To 25.1 g of the compound obtained from the above Example 4 was added dichloromethane (133.0 g) and triethylamine (148.0 g), and the mixture was then cooled to 0℃. To this reaction solution was slowly added methanesulfonyl chloride (11.8 g) diluted with dichloromethane (39.9 g) dropwise for 50 minutes while maintaining the internal temperature below 12℃. After completion of the reaction, the reaction solution was washed with 0.5 N aqueous hydrochloric acid solution (120.0 g) and water (100.4 g), and then distilled under reduced pressure to obtain 31.5 g of the title compound.
EXAMPLE 6: Synthesis of tert-butyl (3S)-4-azido-3-[(tert-butoxycarbonyl)amino]- butanoate
Sodium azide (NaN3, 11.6 g) was diluted with dimethylacetamide (DMAc, 260.0 g). After elevating the internal temperature to 80℃, a solution of 31.5 g of the compound, as obtained from the above Example 5, diluted with dimethylacetamide (DMAc, 45.0 g) was added thereto. The reaction proceeded at 80℃ for 2 hours. To the reaction solution were added toluene (251.0 g) and water (320.0 g) to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 24.0 g of the title compound.
EXAMPLE 7: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate
To 21.0 g of the compound obtained from the above Example 6 was added tetrahydrofuran (93.3 g) followed by the addition of triphenylphosphine (PPh3, 21.0 g) at 40℃, the mixture was stirred for 2 hours at the same temperature, and water (3.8 g) was then added thereto. The reaction solution was distilled under reduced pressure, and the resulting triphenylphosphine oxide solid was diluted with toluene (26.0 g) and n-hexane (41.0 g), and then filtered off. The filtrate was adjusted to pH 2~3 with 1.0 N aqueous hydrochloric acid solution (110.0 g) and then subjected to separation of the layers. To remove any residual triphenylphosphine oxide solid, the aqueous layer obtained above was washed with dichloromethane (100.0 g) and then adjusted to pH 8~9 with 28% aqueous ammonia solution (7.6 g). The aqueous solution thus obtained was extracted with dichloromethane (100.0 g) and distilled under reduced pressure to obtain 8.5 g of the title compound as a white solid.
EXAMPLE 8: Synthesis of N,N-dibenzyl-L-N(Boc)-aspartamide 4-tert-butyl ester
N-Boc-L-aspartic acid 4-t-butyl ester (29.0 g, 0.10 mol) was added to THF (200 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (13.0 ml, 0.10 mol) followed by addition of N-methyl morpholine (12.0 ml, 0.10 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise dibenzylamine (21.1 ml, 0.11 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (300.0 mL) and 1 N hydrochloric acid to separate the layers, and distilled under reduced pressure to precipitate a solid. The solid was filtered and washed with ethyl acetate (100 ml), and then the washings were concentrated by distillation again under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (41.7 g, 0.89 mol).
Mass (ESI, m/z): 491 (M+Na), 469 (M+H), 413 (M-55).
EXAMPLE 9: Synthesis of N, N-diallyl-L-N(Boc)-aspartamide 4-tert-butyl ester
L-N(Boc)-aspartic acid 4-t-butyl ester (5.00 g, 17.3 mol) was added to THF (50 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (2.26 ml, 17.3 mol) followed by addition of N-methyl morpholine (1.90 ml, 17.3 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise diallylamine (2.35 ml, 19.0 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (60 ml) and 1 N hydrochloric acid and, after separating the layers, concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (6.0 g, 16.3 mol).
Mass (ESI, m/z): 391 (M+Na), 369 (M+H), 313 (M-55).
EXAMPLE 10: Synthesis of N,N-dibenzyl-4-amino-3(S)-N(Boc)-aminobutanoic acid 4-tert-butyl ester
10.0 g of the compound obtained from the above Example 8, Ru3(CO)12 (136 mg, 1mol%), and diphenylsilane (19.7 ml, 106.7 mmol) were added to tetrahydrofuran (50 ml), and the reaction solution was stirred under reflux for over 40 hours. The reaction solution was extracted with ethyl acetate (200 ml) and concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (4.7 g, 10.5 mmol).
EXAMPLE 11: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- 4-oxobutanoate
360.0 g of the starting material, N-Boc-Asp(O-t-Bu)OH, together with Boc2O (353.0 g) and ammonium bicarbonate (NH4HCO3, 123.9 g) was added to dimethylformamide (1174.6 g), and pyridine (61.0 g) was added dropwise thereto at room temperature, and the reaction mixture was then stirred for about 3 hours. Upon completion of the reaction, water (1440 ml) and toluene (1800 ml) were added to the reaction solution and stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to remove t-butanol and toluene to obtain the title compound, which was directly used in the next reaction.
EXAMPLE 12: Synthesis of (S)-tert-butyl 3-(tert-butoxycarbonylamino)-3-cyanopropanoate
To the compound obtained from Example 11 was added dimethylformamide (1019.5 g) followed by addition of cyanuric chloride (112.0 g) dropwise for 1.5 hours at temperature below 25℃. The reaction solution was stirred for one hour at room temperature, and then 0.1 N aqueous sodium hydroxide solution (1850.0 g) and toluene (1860 ml) were added thereto to separate the layers. The organic layer thus obtained was washed once again with water (700 ml) and then distilled under reduced pressure to obtain 318.3 g of the title compound.
EXAMPLE 13: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate
To 212.1 g of the compound obtained from the above Example 12 was added acetic acid (4000 ml) followed by addition of 20 wt% Pd(OH)2 (1.1 g) at 40℃. The mixture was stirred for 8 hours while keeping the internal temperature below 45℃ and 3 atmospheric pressure of hydrogen. Upon completion of the reaction, the reaction solution was distilled under reduced pressure to remove acetic acid, diluted with toluene (640 L) and then filtered through a celite. To the filtrate was added 0.25 N aqueous hydrochloric acid solution (1060 ml) to separate the layers. The aqueous layer thus obtained was basified with aqueous ammonia solution (543.1 g) and then extracted with methyl t-butyl ether (MTBE, 1000 ml). The organic layer thus obtained was distilled under reduced pressure to obtain 185.0 g of the title compound.
EXAMPLE 14: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid t-butyl ester
Triethylamine (13.2 g) was added to 16.0 g of the compound obtained from the above Example 1 or 2-1 or 2-2, and 14.1 g of the compound obtained from the above Example 7 or 13, and the mixture was then stirred for 21 hours at 40℃. Then, dichloromethane (154.8 g) and acetic acid (18.3 g) were added, and the mixture was stirred for 5 hours at room temperature. To the resulting reaction solution was added 0.5 N aqueous hydrochloric acid solution (116.8 g) and then, the mixture was stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 23.6 g of the title compound.
EXAMPLE 15: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid
23.6 g of the compound obtained from the above Example 14 was added to dichloromethane (20.0 g) followed by addition of H3PO4 (30.0 g), and the mixture was stirred for 16 hours at room temperature. After confirming the detachment of all of t-butyl group and t-butyloxycarbonyl group, the reaction solution was adjusted to pH 7.0~8.0 with 10 N aqueous hydrogen peroxide, and Boc2O (16.0 g) was added thereto. After completion of the addition, 10 N aqueous hydrogen peroxide was used to maintain the pH of the reaction solution at 8.0~9.0. After stirring for 3 hours, the resulting sodium phosphate was filtered off, and the filtrate was then adjusted to pH 2.0~3.0 with 3.0 N aqueous hydrochloric acid solution. The resulting solid was filtered and dried under nitrogen to obtain 14.5 g of the title compound.
For the title compound resulting from the above, its enantiomeric isomers―i.e. S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess of the enantiomeric isomers (S vs. R form) (enantiomeric excess; ee) was then calculated as being ee > 99%. On the other hand, in case of the Comparative Example prepared according to the prior method based on WO 06/104356, as described below, the excess (ee) of enantiomeric isomers (S vs. R form) was 80%. From this, it can be identified that the compound of formula (2) having an optically high purity could be obtained according to the method of the present invention.
COMPARATIVE EXAMPLE 1: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
COMPARATIVE EXAMPLE 1-1: Synthesis of methyl 5-amino-4,4-difluoro- pentanoate HCl
To 10.0 g of the compound obtained from Example 1 was added 40 ml of anhydrous ammonia solution (7 M solution in methanol), and the mixture was stirred for 3 hours. The reaction solution was distilled and 30 ml of hydrochloric acid solution saturated with methanol was added dropwise thereto. The reaction mixture was stirred at room temperature and then distilled to obtain 7.2 g of the title compound as a white solid.
COMPARATIVE EXAMPLE 1-2: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
To the solution of the compound (1.93 g), as obtained from the above Example 4, dissolved in dichloromethane (20.0 g) and H2O (4.0 g) were added NaBr (0.8 g) and TEMPO (11 mg, 1 mol%). To this reaction solution was slowly added a solution of 5% NaOCl (11.5 g) and NaHCO3 (1.7 g) dissolved in H2O (12.0 g) dropwise for about 2 hours while maintaining the temperature below 5℃. Upon completion of dropwise addition, the reaction solution was stirred for 30 minutes to separate the layers. To the organic layer thus obtained was added the compound (1.6 g) obtained from the above Comparative Example 1-1. After stirring for 15 minutes at room temperature, NaBH(OAc)3 (2.23 g) was added to the reaction solution. After stirring for about 19 hours, 10% aqueous NaHCO3 solution (20.0 g) and 0.5 N aqueous hydrochloric acid solution (20.0 g) were added dropwise to the reaction solution to separate the layers. The organic layer thus obtained was dehydrated under anhydrous MgSO4 to obtain 2.0 g (yield 73%) of the same title compound as Example 14, as a yellow solid. For the title compound resulting from the above, its enantiomeric isomers―i.e., S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess (ee) of the enantiomeric isomers (S vs. R form) was then calculated as being ee = 80%.
Pyrrolidinone anilines as progesterone receptor modulators
Footnotes
Lim KS, Kim JR, Choi YJ, Shin KH, Kim KP, Hong JH, Cho JY, Shin HS, Yu KS, Shin SG, Kwon OH, Hwang DM, Kim JA, Jang IJ (October 2008). “Pharmacokinetics, pharmacodynamics, and tolerability of the dipeptidyl peptidase IV inhibitor LC15-0444 in healthy Korean men: a dose-block-randomized, double-blind, placebo-controlled, ascending single-dose, Phase I study”. Clin Ther30 (10): 1817–30. doi:10.1016/j.clinthera.2008.10.013. PMID19014837.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....















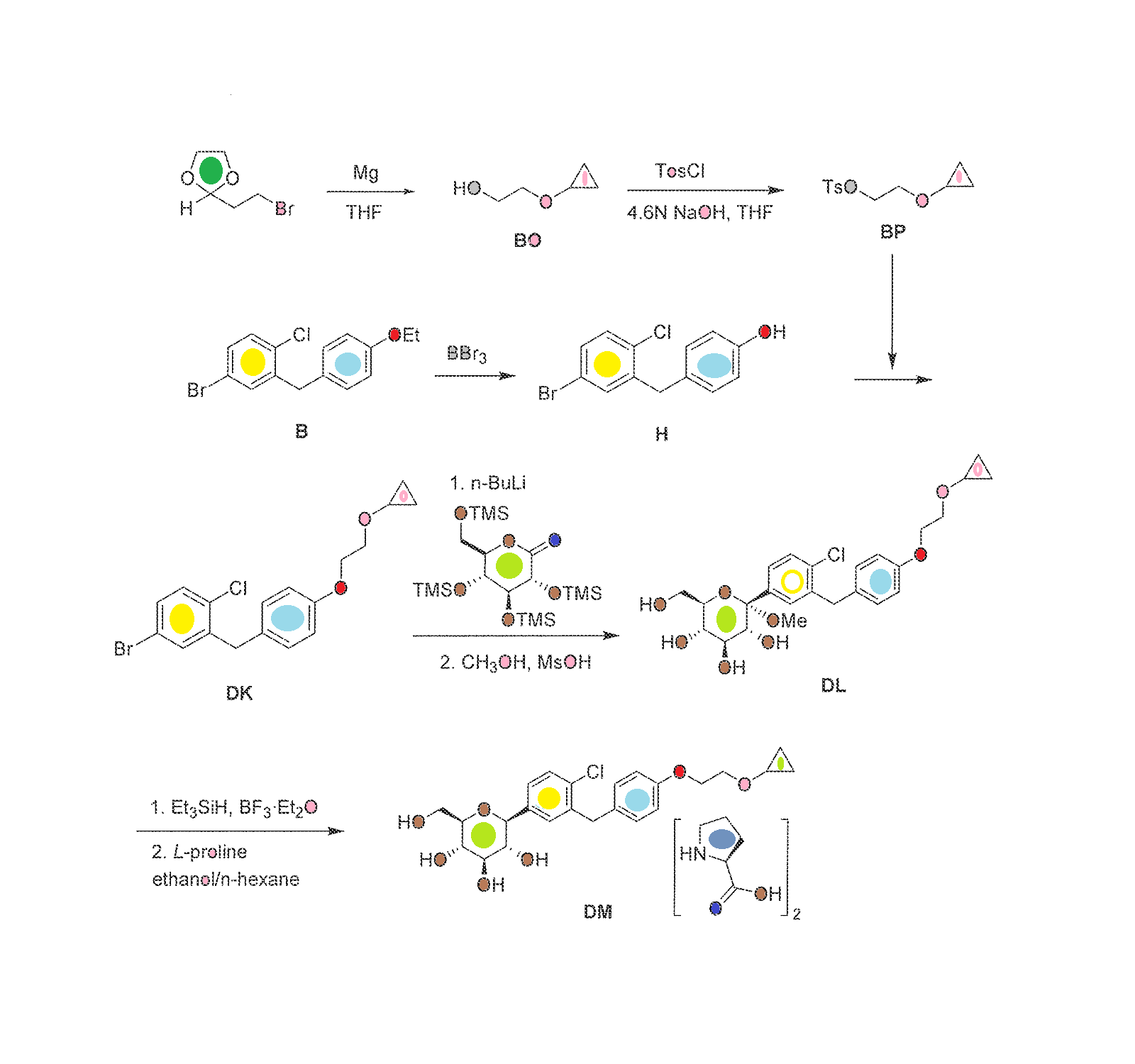











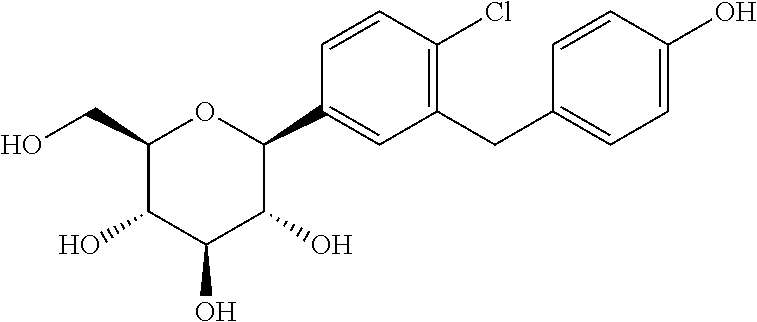



















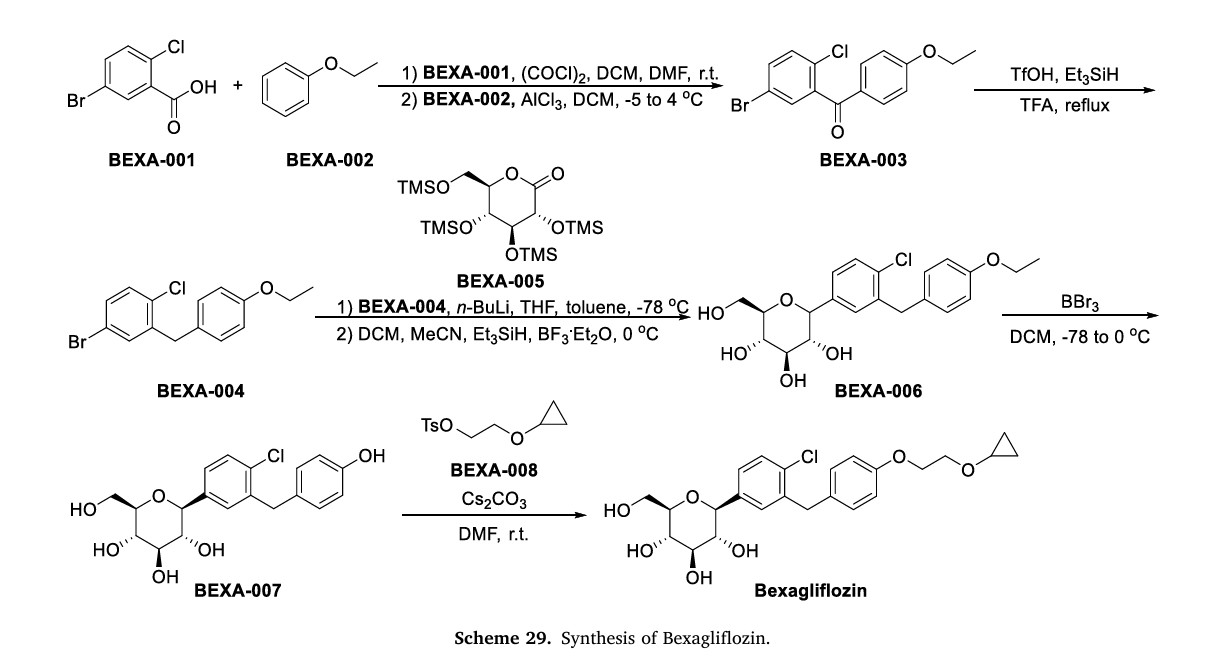

























 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



 LIONEL MY SON
LIONEL MY SON

 3D
3D
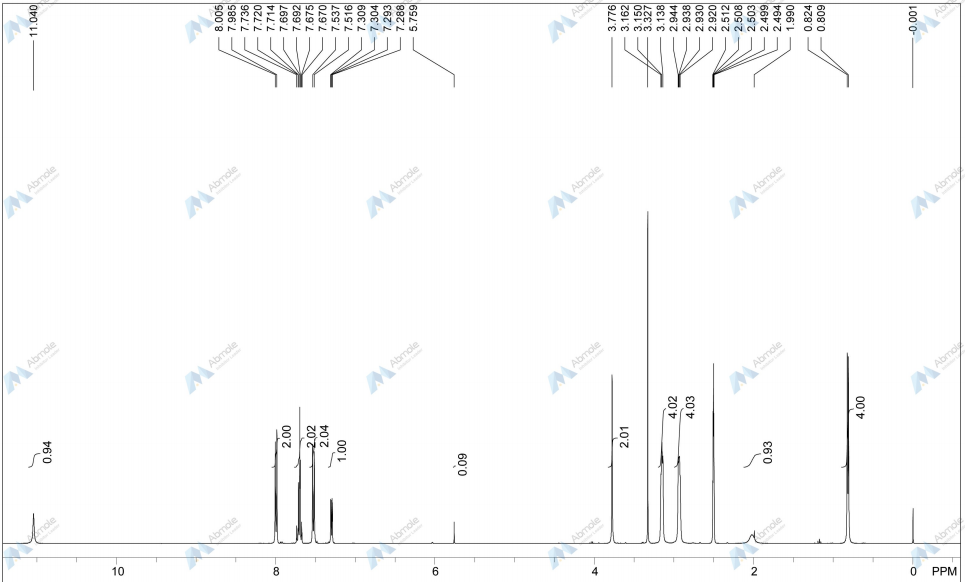



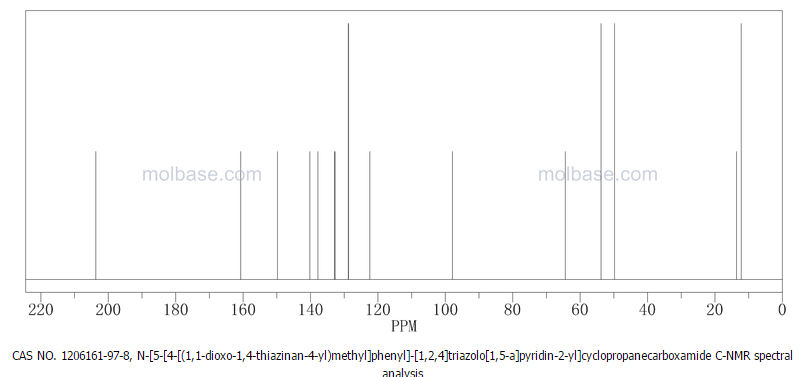

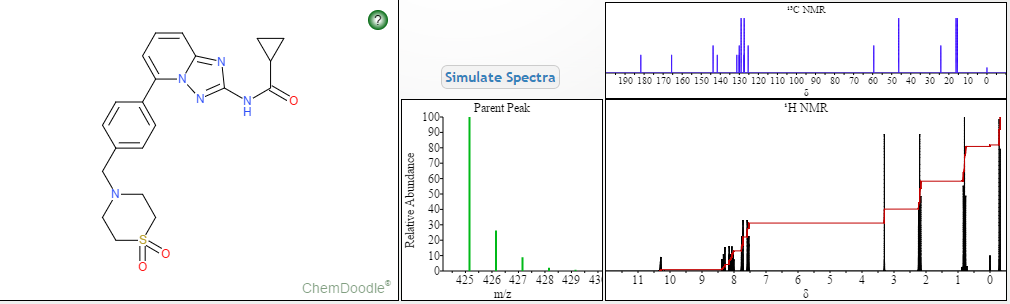
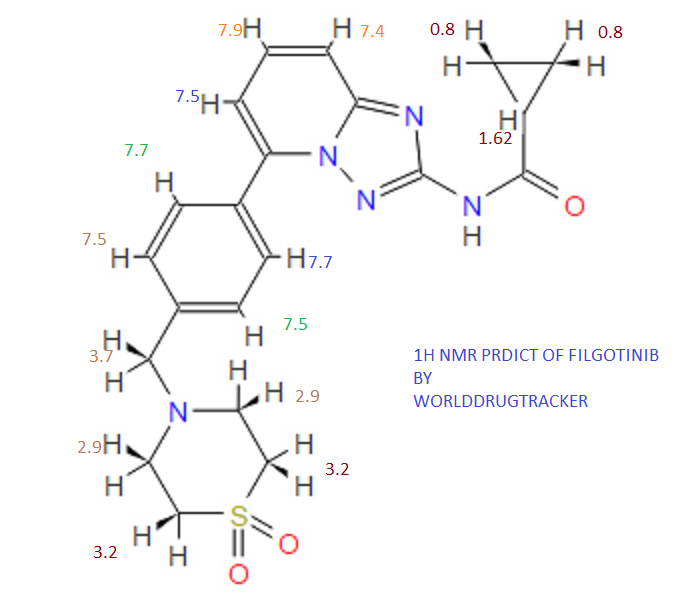
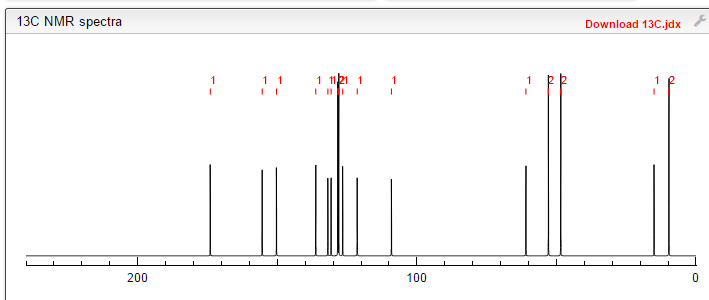
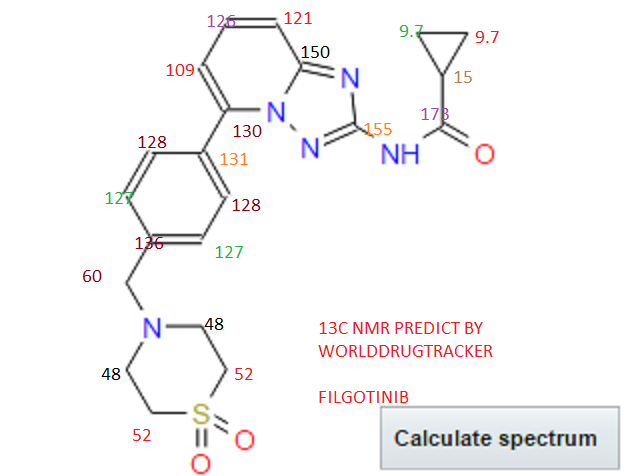
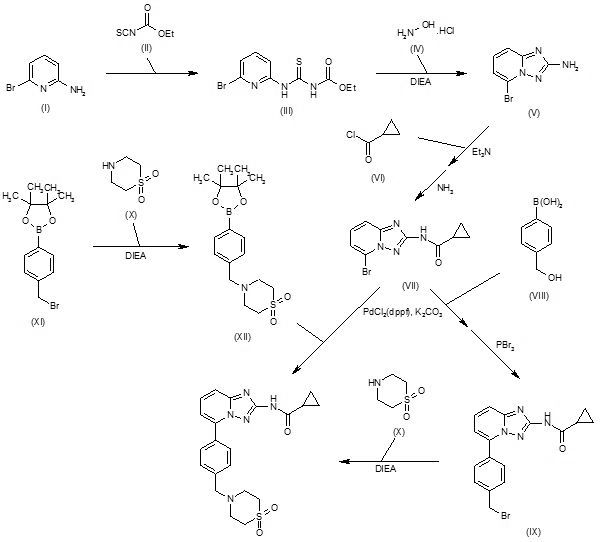














































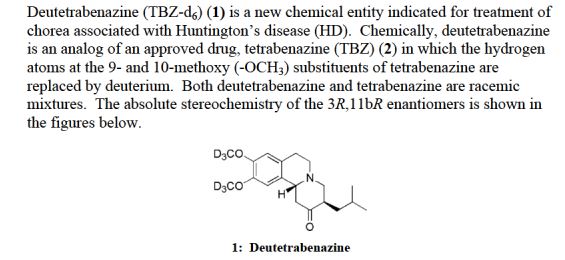

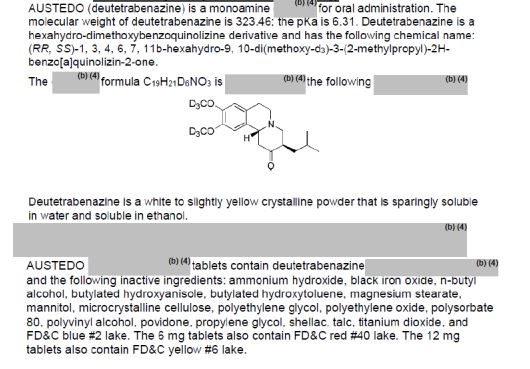


-9,10-di((2H3)methoxy)-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydro-2H-benzo(a)quinolizin-2-one")

















































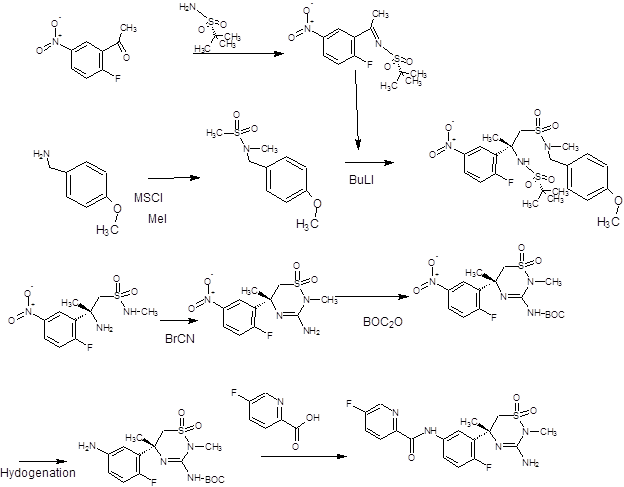

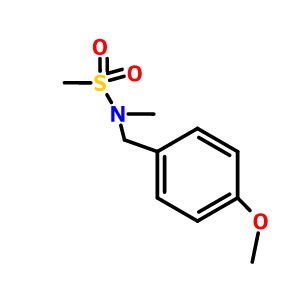
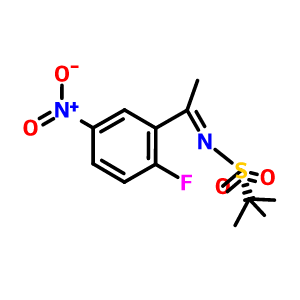

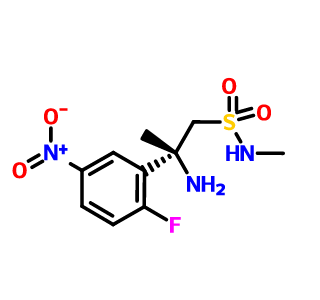
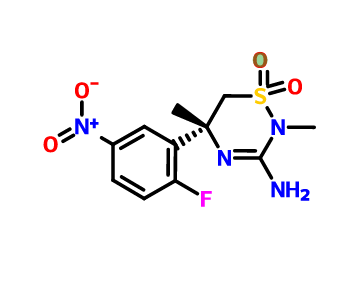
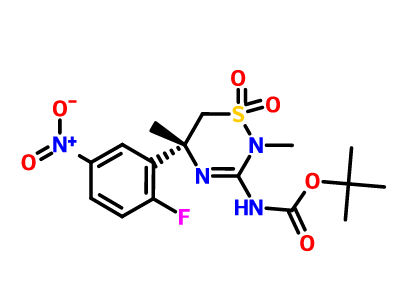
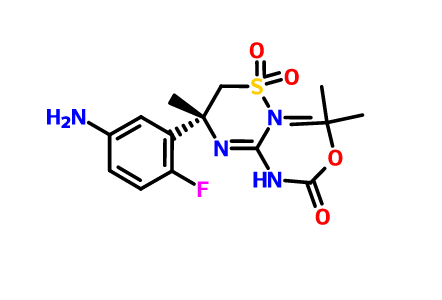



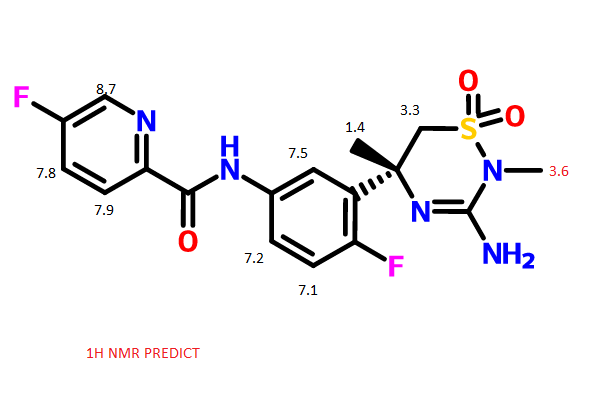

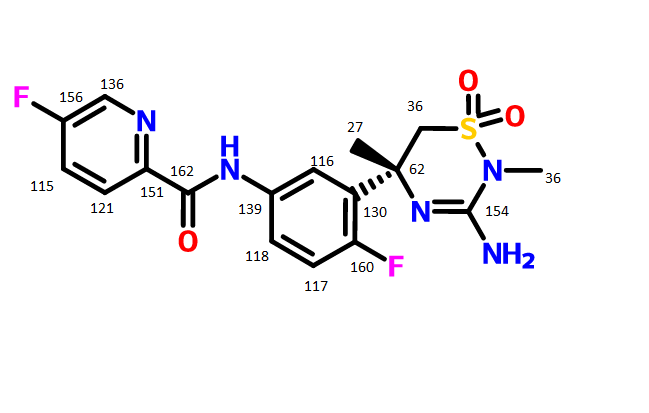


















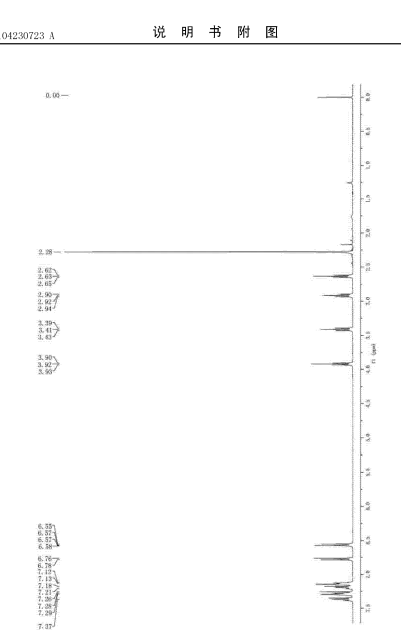










 LIONEL MY SON
LIONEL MY SON
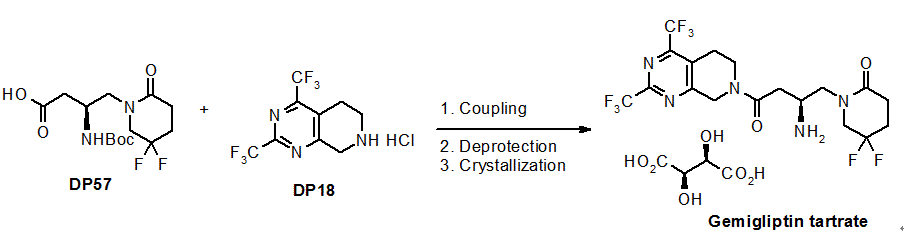





















{kind=link}