
Home » Posts tagged 'organic synthesis' (Page 17)
Tag Archives: organic synthesis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Idenix Pharmaceuticals Announces Samatasvir (IDX719) Poster Presentations at the Asian Pacific Association for the Study of the Liver (APASL) Conference

New Hepititis C Virus Therapy
Idenix Pharmaceuticals Announces Samatasvir (IDX719) Poster Presentations at …
Wall Street Journal JUNE 6, 2013
… Inc. (Nasdaq:IDIX), a biopharmaceutical company engaged in the discovery and development of drugs for the treatment of human viral diseases, today announced three poster presentations featuring clinical and preclinical data for samatasvir (IDX719), …
READ ALL AT
http://online.wsj.com/article/PR-CO-20130606-905746.html
Idenix Pharmaceuticals, the Cambridge-based biotechnology company, announced that their drug, IDX719, was granted a Fast Track designation by the FDA. IDX719, an NS5A inhibitor, is designed to treat chronic hepatitis C virus (HCV) infection in patients. The Fast Track designation will enable Idenix Pharmaceuticals to shave precious time off their predicted timeline for a new drug application (NDA), and even increase interaction with the FDA to guarantee a quicker review and a shorter time to market.
Biocon Seeks Partner to Sell Rival Drug to J J’s Stelara

Managing Director Kiran Mazumdar-Shaw, BIOCON
Photographer-Namas Bhojani/Bloomberg
Rapid commercialization of Alzumab “will be transformational for us,” said Kiran Mazumdar-Shaw, chairman and managing director of Biocon Ltd.
Managing Director Kiran Mazumdar-Shaw, is seeking a partner to help with expertise and funding for the tests needed for approval in the U.S., she said in an interview. Biocon plans to file for permission in the year ending March to sell in the North American nation, and aims to start marketing Alzumab in that country two to three years later, she said.
Alzumab would provide a novel therapy for a plaque-causing form of the immune disorder that would compete with best-selling products from Johnson & Johnson, AbbVie Inc. and Pfizer Inc. (PFE) The biologic psoriasis treatment, made from living cells, will help Biocon enter a market that it estimates will be valued at $8 billion by 2016.
READ ALL AT
http://www.ukmi.nhs.uk/applications/ndo/record_view_open.asp?newDrugID=5630
Itolizumab (Alzumab) is a ‘first in class’ humanized IgG1 monoclonal antibody developed by Biocon. It selectively targets CD6, a pan T cell marker involved in co-stimulation, adhesion and maturation of T cells. Itolizumab, by binding to CD6, down regulates T cell activation, causes reduction in synthesis of pro-inflammatory cytokines and possibly plays an important role by reducing T cell infiltration at sites of inflammation.[1] A double blind, placebo controlled, phase III treat –Plaq study of Itolizumab, successfully met the pre-specified primary end-point of significant improvement in PASI-75 (Psoriasis Area and Severity Index) score after 12 weeks of treatment in patients with moderate to severe psoriasis compared to placebo.[2] Biocon has received marketing authorization for the drug from the Drugs Controller General of India (DCGI).[3]
- http://www.biocon.com/docs/PR_080113.pdf?subLink=news
- http://www.thehindubusinessline.com/companies/article2789996.ece
- http://www.pharmabiz.com/NewsDetails.aspx?aid=73075&sid=2

Cadila banks on diabetes drug, Lipaglyn, Saroglitazar

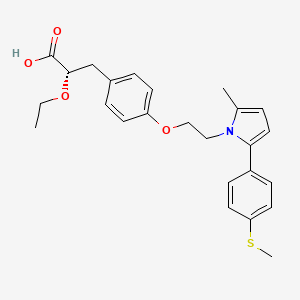
(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid magnesium salt
- saroglitazar
- ZYH1 compound
-
- E0YMX3S4JD
- cas no 495399-09-2
Saroglitazar, Lipaglyn
| Molecular Weight | 439.56706 g/mol |
|---|---|
| Molecular Formula | C25H29NO4S |


 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Cadila banks on diabetes drug
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet …http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp

Mumbai, June 5: Cadila Healthcare will launch a homegrown drug against diabetes by the third quarter of this year.
The Drug Controller General of India has approved its drug — Lipaglyn — to treat “diabetic dyslipidemia”.
Diabetic dyslipidemia is a condition where a person is diabetic and has elevated levels of total cholesterol. Over 80 per cent of diabetic patients are dyslipidemic.
http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp
Zydus Cadila said it is looking for partnership to market its new chemical entity (NCE) Lipaglyn, to be used for treating a type of diabetes in developed and developing markets. “Lipaglyn is the first glitazar to be approved in the world and the first NCE discovered and developed indigenously by an Indian pharma company.
The new drug is expected to be launched in Q3 of this fiscal in the country,” Zydus Cadila Chairman and Manging Director Pankaj Patel told reporters.
The company has spent USD 250 million in developing Lipaglyn and aims to spend another USD 150-200 million to launch the drug in overseas markets in next 3-5 years period, Patel said, adding that the company is looking for marketing partnerships.
“We expect this to be a blockbuster drug, which means over USD 1 billion sales a year, when the drug is sold globally, he said. The market for this drug is estimated at Rs 100 crore in the local market over the next three years and having market potential size of over USD 30 billion in the world market, he said.
Zydus Cadila took about eight years to develop the molecule and conducted clinical trials on more than 1,000 patients in India, Patel said, adding that the company is yet to finalise the price, but believes that it will be reasonably priced in the local market.
The company said that the Indian drug regulator Drug Controller General of India (DCGI) has approved Lipaglyn to be used for treating ‘diabetic dyslipidemia’.
| Saroglitazar, is a drug for the treatment of diabetic dyslipidemia and hypertriglyceridemia with Type 2 diabetes mellitus not controlled by statin therapy. Its trade name is Lipaglyn. It is also a 1,2-Diarylpyrroles derivative, which can be used in the preparation of Nonsteroidal anti-inflammatory drugs (NSAIDs). |
| References: Khanna, I. K., et al.: J. Med. Chem., 40, 1619 (1997) |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid | |
| Clinical data | |
| Trade names | Lipaglyn |
| Pregnancy cat. |
|
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 495399-09-2 |
| ATC code | None |
| PubChem | CID 60151560 |
| ChemSpider | 32079086 |
| Chemical data | |
| Formula | C25H29NO4S |
| Mol. mass | 439.56 g/mol |
MORE DETAILS
Saroglitazar (INN, trade name Lipaglyn) is a drug for the treatment of type 2 diabetes mellitus and dyslipidemia. It is approved for use in India by the Drug Controller General of India.[1] Saroglitazar is indicated for the treatment of diabetic dyslipidemia andhypertriglyceridemia with type 2 diabetes mellitus not controlled by statin therapy. In clinical studies, saroglitazar has demonstrated reduction of triglycerides (TG), LDL cholesterol, VLDL cholesterol, non-HDL cholesterol and an increase in HDL cholesterol a characteristic hallmark of atherogenic diabetic dyslipidemia (ADD). It has also shown favorable glycemic control by reducing the fasting plasma glucose and HBA1c in diabetes patients.
Zydus-Cadila has developed and launched saroglitazar (ZYH-1; Lipaglyn; structure shown), a lipid metabolism modulator, a potent PPAR-alpha agonist with relatively weak PPAR-gamma activity, an insulin sensitizer (glucose-lowering agent), for the once-daily oral treatment of metabolic disorders, including diabetic dyslipidemia and hypertriglyceridemia
In June 2013, the Drug Controller General of India (DCGI) approved the drug for launch in India ; in September 2013, the drug was launched . The company is also developing the drug for the potential treatment of lipodystrophy. In May 2014, a phase III trial was initiated . In June 2012, the company was seeking to outlicense the drug for regional/global partnerships
By June 2012, an NDA filing had been made for dyslipidemia. In June 2013, the DCGI approved the drug for launch in India . By September 2013, the drug was launched for dyslipidemia and hypertriglyceridemia .
Mechanism of action
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of theperoxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action onPPARγ improves insulin resistance and consequently lowers blood sugar.[2]
_MoA.png)
Clinical trials
The approval for saroglitazar was based on the results obtained from clinical studies, which were conducted for more than eight years.
The studies evaluated the efficacy, safety, pharmacokinetics and pharmacodynamics of the drug. Phase I clinical trials on saroglitazar were conducted in 2005. The highest dose of saroglitazar evaluated in a Phase I trial was 128 mg, several times the estimated therapeutic doses (1–4 mg). The pharmacokinetics of saroglitazar support a once daily dosage schedule. No serious adverse events were reported.[3] Phase II studies were completed in 2006.
The Phase III clinical trials were conducted between 2008 and 2011. The first Phase III clinical trials on saroglitazar compared saroglitazar 4 mg dose with pioglitazone 45 mg. The results of the study demonstrated that patients who were administered with saroglitazar 4 mg dose showed reduction in LDL cholesterol and triglycerides, and increase in HDL cholesterol. The study also showed that saroglitazar administered patients showed a reduction in fasting plasma glucose and glycosylated hemoglobin.
Saroglitazar 2 mg and 4 mg significantly reduced (P < 0.001) plasma triglycerides from baseline by 26.4% (absolute change ± SD: −78.2 ± 81.98 mg/dL) and 45% (absolute change ± SD −115.4 ± 68.11 mg/dL), respectively, as compared to pioglitazone -15.5% (absolute change ± SD: −33.3 ± 162.41 mg/dL) at week 24. Saroglitazar 4 mg treatment also demonstrated marked decrease in low-density lipoprotein (5%), very-low-density lipoprotein (45.5%), total cholesterol (7.7%), and apolipoprotein-B (10.9%).[4]
The second Phase III clinical trials on saroglitazar were conducted to evaluate the diabetic dyslipidemic patients insufficiently controlled with statin therapy. The second Phase III study results showed that patients treated with saroglitazar showed pronounced beneficial effect on both the lipid and glycaemic parameters.
At Week 12, saroglitazar 2-mg and 4-mg tablets significantly reduced mean plasma triglyceride levels by -45.5±3.03% and -46.7±3.02% (mean±SE), respectively, and the difference was significant (P<0.001) compared with placebo. Saroglitazar 2 mg demonstrated significant decrease in levels of non-HDL-C, very LDL-C, total cholesterol, and fasting plasma glucose. Additionally, saroglitazar 4 mg also significantly reduced LDL-C and apolipoprotein B levels. Saroglitazar was found to be safe and well tolerated by patients.[5]
Safety
Saroglitazar was found to be safe and well tolerated during the clinical program. In Phase III trials, There was no edema or weight gain reported in any of the study arms. During this study, subjects were monitored for cardiac events, ECG abnormalities, and cardiac function by 2-D ECHO at the start of the study, at the end of 12 weeks, and at 24 weeks after the last dose of the study drug. There were no adverse events reported as far as cardiac safety is concerned.
After 12 weeks of treatment, there were a no significant changes in hemoglobin, liver enzymes (alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, andγ-glutamyl transferase), renal function (creatinine, enhanced glomerular filtration rate, and blood urea nitrogen), CPK, and high-sensitivity C-reactive protein in the saroglitazar and placebo arms.[6][7]
In Phase I clinical trials saroglitazar was used up to 128 mg and found well tolerated. No serious adverse events were reported. Adverse events were generally mild and moderate in nature and did not show any clinically relevant findings in clinical laboratory investigations, physical examinations, vital signs and electrocardiograms.[8]
PAPER
A new enantioselective synthesis of (S)-2-ethoxy-3-(4-hydroxyphenyl)propanoic acid esters (EEHP and IEHP), useful pharmaceutical intermediates of PPAR agonists
Tetrahedron Lett 2014, 55(21): 3223
http://www.sciencedirect.com/science/article/pii/S0040403914006200

PATENT
WO 2003009841
http://www.google.co.in/patents/WO2003009841A1?cl=en
PATENT
US 20030236254
http://www.google.com/patents/US20030236254
PATENT
US 20140099333
http://www.google.com/patents/US20140099333
PATENT
http://patentscope.wipo.int/search/en/WO2014174524
 (I)
(I)
The compound as claimed in claim 1 wherein R is -SMe and M+ is Mg+2.
The compound of claim 1 is Saroglitazar.
wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. r .
PATENT
3-Aryl-2-hydroxy propanoic acid derivatives serve as a key intermediate for the synthesis of many pharmaceutically important compounds especially, peroxime proliferator activated receptor (PPAR) agonist.
Optically active 3-aryl-2-alkoxy propanoic acid and its esters, particularly, ethyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (EEHP) and isopropyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (IEHP) are versatile chiral pharmacophores present in many pharmaceutically important compounds, especially in peroxisome proliferator activated receptor (PPAR) agonists that have beneficial effects in treating Type 2 diabetes.
Several PPAR agonists, in particular PPAR α/γ dual agonists, commonly termed as glitazars (Ragaglitazar, Tesaglitazar, Navaglitazar etc.), as shown in the figure below were developed by many pharmaceutical companies that have a potential application in the treatment of Type 2 diabetes and dyslipidemia.
However, many of these drugs were discontinued due to their undesirable side effects, but some of them still have great potential [For example, Saraglitazar (LipaglynTM) developed by Zydus Cadila got approval in India for the treatment of diabetic dyslipidemia or hypertriglyceridemia]. Several PPAR α/γ agonists possessing chiral (S)-l moieties are shown below.

Tesaglitazar Naveglitazar
In addition, these derivatives find an application in photosensitive materials, sweetening agents, treatment of certain eating disorders etc. Therefore, these compounds have attracted a great deal of attention of synthetic chemists and different methods of preparation of the compound of formula (S)-l have been extensively studied.
Generally, the reported protocols for the synthesis involve chiral pool approaches starting from L-tyrosine and its derivatives (Refer WO 02/24625, US 6559335B2, WO 2003/027084), asymmetric synthesis (Org. Lett. 2005, 7, 1947, US 2007/0149804) and resolution processes using chiral amines or enzymes (WO 2000/026200, WO 2001/11073, Org. Process Res. Dev. 2003, 7, 82, Org. Process Res. Dev. 2004, 8, 838, Tetrahedron Asymmetry 2009, 20, 2594).
Some of these methods have disadvantages such as expensive chiral starting materials and catalysts, low enantioselectivity and overall yields, problems associated with the O-alkylation step which often leads to the loss of optical purity, and many others.
The processes described in WO20026200 (Rao et. al.) uses benzyl bromide for benzylation, which is highly lachrymatory. Again, in the processes described, the debenzylation of the final intermediate was done by using Pd/C under pressure, which escalates the process economics.
WO2003024915 describes a process for the preparation 3-aryl-2-hydroxy propanoic acid derivatives from 3-(4-hydroxyphenyl)-2-oxopropanoic acid.
WO 2003008362 describes 3-Aryl-2-hydroxy propanoic acid derivatives of formula I and the preparation thereof.

wherein Rland R2 may be same or different and represent hydrogen or (CI- C6) alkyl.
The process is depicted in Scheme 1 below.
Scheme 1

In another process variant as in Scheme 2, WO’362 discloses a process for the preparation of novel 3-aryl-2 -hydroxy propanol and their derivatives of the formula (I)

wherein OR and OR together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms, which comprises: i) reducing the compound of formula (III) where R represents hydrogen or alkyl group, R3 represents benzyl to a compound of formula (IV) where R3 represents benzyl, ii) cyclizing the compound of formula (IV) to a compound of formula (V) where ORl and OR2 together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms and R3 represents benzyl and iii) debenzylating the compound of formula (V) in the presence of metal catalysts to yield pure compound of formula (I).
Scheme 2

Both the processes described in WO’362 result in poor overall yield and further fail to describe the preparation of compound of formula V using different alkylating agents. This document exemplifies the compound of formula V with similar ether groups as it fails to teach selective alkylation of formula IV.
WO2005019152 discloses an improved process for the preparation of compound of the general formula (la) and (lb).

Wherein, Rl represent H or (C1-C6) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like. R2 represents (Ci-Ce) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t- butyl and the like. R3 represents H, protecting groups such as benzyl, substituted benzyl, (C1-C3) alkyl and like.
The compound of general formula (la) is prepared according to the following schemes 3 and 4.
Scheme 3

Both the processes start with selective O-alkylation or O-aralkylation of L-Tyrosine of formula (2a) using a base, a chelating agent, an alkyl or aralkyl halide in the presence of solvents to obtain the compound of formula (3a), which is diazotized to obtain formula (4a) which upon dialkylation using an excess of alkylating agent and excess base, in presence of suitable solvent to obtain optically pure compound of formula (la). Alternatively, compound of formula (4a) may be selectively esterified to obtain compound of formula (5a), which is subsequently O-alkylated to obtain compound of formula (la) (Scheme 2).
However, the above processes have many disadvantages such as multistep synthesis including protection & deprotection and low overall yield. Further, low temperature diazotization on industrial scale is not viable. Moreover, the starting material is very expensive and hence escalates the process.
In the light of the foregoing, development of a new, alternate enantio-selective synthetic route to these important chiral intermediates, which are simple and can preserve the optical purity at the C-2 carbon of 3-Aryl-2-hydroxy propanoic acid derivatives, is highly desirable. There is a need for an efficient process for synthesis of 3-Aryl-2-hydroxy propanoic acid derivatives of formula (S)-l in high enantiopurity and good overall yield from commercially available starting material.
PATENT
saroglitazar magnesium alongwith its intermediates may be prepared by the reaction scheme- 1, scheme-2 and scheme-3 as shown below, which is also the scope of the present invention.

Scheme-1

EXAMPLES
Example-l:
Preparation of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, sodium methoxide (165 g) and toluene (1000.0 ml) were added under nitrogen environment and cooled to 8°C to 12°C. Methyl acetoacetate (331.55 g) was added dropwise and stirred for 1 hour. 2-bromo-l-(4-methyl sulfonyl phenyl) ethanone (500.0 g) compound (El) in toluene (1500.0 ml) and sodium sulfate
(75.0 g) mixture was stirred for 10 min and filtered at 25° to 35°C. The filtrate as obtained was added dropwise into the previous reaction mixture and stirred at 30°C to 35°C for 30 min. The organic layer was collected and washed with 10% sodium bicarbonate solution. The separated organic layer was collected and washed with water. 2-[2-(4-Methyl sulfanyl-phenyl)-2-oxo-ethyl]-3-oxo-butynic acid methyl ester as obtained in toluene layer is diluted with methanol (2500 ml) and sodium hydroxide solution (89.75 g) in water (2500 ml) was added and heated to 50° to 55°C for 1 hour. The layers were separated and the toluene layer was collected and heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (57.3 g) and ethanol amine (143.9 g) were added and heated to 105° to 1 15°C for removing water azeotropically. The toluene layer was separated and triethyl amine (271.85 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (282.5 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum to obtain the residue. The residue was dissolved in toluene (1500 mL) and used for further process.
ExampIe-2:
Preparation of methanesulfonic acid 2-f2-methyl-5-(4-methylsulfanyl-pheny0-pyrrol- 1-viyethyl ester (Al)

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 4-(methylthio)benzaldehyde (10 g), methyl vinyl ketone (3.63 g), triethylamine (9.95 g) and 3-methyl-5-(2-hydroxyethyl)-4-methyI thiazolium iodide (stetter
catalyst) (2.8 g) were heated to 70°C to 80°C and maintained overnight. The reaction mixture was cooled to room temperature and ethanol (100 mL) was added. The reaction mixture was stirred for 30 min and filtered. The product was washed with ethanol and dried to obtain 1 ,4-diketo compound (CI).
1 ,4-diketo compound (CI) obtained above and toluene (50 mL) were heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (5.7 g) and ethanol amine (14.4 g) were added and heated to 105° to 1 15°C and cooled to 25°C. Triethyl amine (27.2 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (28.3 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum, methanol (2500 ml) was added and heated to 55° to 65 °C and charcoalized for 30 min. The reaction mixture was filtered and washed with methanol. The reaction mixture was cooled to 25° to 35°C and stirred for 30 min. Reaction mass was further cooled to -5° to 5°C and filtered. The wet-cake was washed with methanol and dried to obtain compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.2).
Example-3:
Purification of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 70 g methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l -yl]-ethyl ester (Al) and 420 mL ethyl acetate were added at 25°C. The reaction mixture was stirred for 30 min to obtain clear solution. 3.5 g charcoal was added and stirred for 30 min. The reaction mixture was filtered and washed with ethyl acetate. The filtrate was concentrated and 315 mL methanol was added. The reaction mixture was stirred for 2 hours at 25°C and cooled to 0°C. The product precipitated was filtered and washed with methanol to obtain crystalline
compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.3).
Example-4:
Preparation of saroglitazar magnesium (T)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) (100.0 g) and toluene (1300.0 ml) were charged and reaction mixture was heated to 45° to 55°C. Potassium carbonate (58.0 g) was added and stirred for 30 min. Toluene solution of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol- 1 -yl]-ethyl ester (Al) (150.24 g) obtained in example- 1, 18-Crown-6 (5.0 g) and THF (200.0 ml) were added and heated to 75°C to 85°C for 36 hour, The reaction mixture was cooled to 25° to 35°C and water (1000.0 ml) was added and stirred for 15 min. The separated aqueous layer was treated with toluene (200.0 ml) and stirred for 15 min. The organic, layers were combined and washed with caustic solution (600.0 ml). The separated organic layer was washed with water (600.0 ml) and characoalized with HP-120 (5.0 g) charcoal and stirred for 30 min and filtered. The filtrate was added sodium hydroxide 20.14 g solution in water (200.0 ml) and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with n-butyl acetate. The separated aqueous layer was added magnesium acetate tetrahydrate solution (90.0 g) in water (100.0 ml) and stirred for 1 hour. The aqueous layer was extracted with methylene dichloride (2000 ml). The separated organic layer was washed with sodium chloride solution and charcoalized. The charcoalized solution was filtered and filtrate was distilled to remove toluene completely. The residue was diluted with toluene (1000 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
The reaction of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al) and 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) may also be performed in similar manner as above in absence of phase transfer catalyst 18-Crown-6.
ExampIe-5:
Preparation of saroglitazar (S)-(-)-phenyl ethylamine salt:

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, residue-A obtained in example- 1 and ethanol (400 mL) were stirred for 15 min. Sodium hydroxide 20.14 g solution in water (200.0 ml) was added and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with isopropyl acetate (400 mL). The separated aqueous layer was diluted with isopropyl acetate (500 mL) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with (S)-(-)-phenyl ethylamine (55.94 g) and stirred for 2 hours at 25°C and 30 min at 45°C. The reaction mixture was cooled to 0°C and stirred for 2 hours, filtered and washed with isopropyl acetate. The wet-cake was dried to obtain saroglitazar phenyl ethylamine salt.
ExampIe-6:
Preparation of saroglitazar magnesium from saroglitazar (SH-)-phenyl ethylamine salt:
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, saroglitazar phenyl ethylamine wet-cake obtained in example-7 and isopropyl acetate (800 mL) were added at 25°C. The reaction mixture was diluted with water (400.0 ml) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with sodium hydroxide solution (20.14 g) in water (200 mL) and stirred for 30 min. The separated aqueous layer was treated with magnesium acetate tetrahydrate (2.29 g) in water (5 mL) solution and stirred for 60 min. The reaction mixture was extracted with methylene dichloride (800 mL). The methylene dichloride was complete removed by distillation under vacuum below 40°C to obtain the residue. The residue was diluted with methylene dichloride (50 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain substantially amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
References
- “Zydus Group launches new diabetic drug”. The Times of India. Jun 6, 2013.
- “Lipaglyn (Saroglitazar) for Treating Hypertriglycerdemia in Type II Diabetes, India”. Drug Development and Technology.
- “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
- “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
-
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
- 7 “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- 8 “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
by WORLD DRUG TRACKER
DR ANTHONY
do not miss out on updates
see my update at https://newdrugapprovals.org/2015/03/09/saroglitazar-magnesium-new-patent-wo-2015029066-cadila-healthcare-ltd/ 9 may 2015
////////////
CCO[C@@H](Cc1ccc(cc1)OCCn2c(ccc2c3ccc(cc3)SC)C)C(=O)O
OXYTOCIN , THE LOVE MOLECULE

OXYTOCIN
1-({(4R,7S,10S,13S,16S,19R)-19-amino-7-(2-amino-2-oxoethyl)-10-(3-amino-3-oxopropyl)-16-(4-hydroxybenzoyl)-13-[(1S)-1-methylpropyl]-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentaazacycloicosan-4-yl}carbonyl)-L-prolyl-L-leucylglycinamide
Oxytocin is a mammalian neurohypophysial hormone that acts primarily as a neuromodulator in the brain.
Oxytocin plays roles in sexual reproduction, in particular during and after childbirth. It is released in large amounts after distension of the cervix and uterus during labor, facilitating birth, maternal bonding, and, after stimulation of the nipples, breastfeeding. Both childbirth and milk ejection result from positive feedback mechanisms.
Recent studies have begun to investigate oxytocin’s role in various behaviors, including orgasm, social recognition, pair bonding, anxiety, and maternal behaviors. For this reason, it is sometimes referred to as the “love hormone”. There is some evidence that oxytocin promotes ethnocentric behavior, incorporating the trust and empathy of in-groups with their suspicion and rejection of outsiders. Furthermore, genetic differences in the oxytocin receptor gene (OXTR) have been associated with maladaptive social traits such as aggressive behaviour.
Oxytocin , sometimes known as the “love molecule” or the “trust molecule” plays an important role in many processes. These include uterine contractions during childbirth, sexual arousal, lactation, puberty, orgasm, facial recognition, trust, memory formation and pair bonding.
Oxytocin is a cyclic peptide hormone with just nine amino acids in sequence (CYIQNCPLG) that also acts as a neurotransmitter in the brain where it is produced in the hypothalamus. It was the first ever polypeptide hormone to be sequenced and synthesized biochemically, work for which the American biochemist Vincent du Vigneaud was awarded the 1955 Nobel Prize in Chemistry.

Together with the neuropeptide argipressin (arginine vasopressin), it is believed to influence social cognition and behaviour. First shown in mice, recent studies have shown that also in humans simply sniffing a spray containing oxytocin increases a person’s level of trust in others.

References
- Lee, H.J., Macbeth, A.H., Pagani, J.H. and Young, W.S. (2009) Oxytocin: the great facilitator of life. Prog. Neurobiol. (Amsterdam, Neth.) 88, 127–151.
- du Vigneaud, V., Ressler, C., Swan, J.M., Roberts, C.W., Katsoyannis, P.G. and Gordon, S. (1953) The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J. Am. Chem. Soc. 75, 4879–4880..
- Kosfeld, M., Heinrichs, M., Zak, P.J., Fischbacher, U. and Fehr, E. (2005) Oxytocin increases trust in humans. Nature 435, 673–676.

Oxytocin (ball-and-stick) bound to its carrier protein neurophysin (ribbons) based on: “Crystal structure of the neurophysin-oxytocin complex” Rose, J.P., Wu, C.K., Hsiao, C.D., Breslow, E., Wang, B.C. (1996) Nat.Struct.Biol. 3: 163-169

Avanir said the FDA agreed to a faster development process for its experimental drug AVP-786 and will allow the company to use some data from studies of Nuedexta in its applications for AVP-786

6/may/2013
Avanir Pharmaceuticals Inc. announced that the Food and Drug Administration will allow it to speed research on a newer version of its drug Nuedexta.
Avanir said the FDA agreed to a faster development process for its experimental drug AVP-786 and will allow the company to use some data from studies of Nuedexta in its applications for AVP-786.
Avanir plans to start human clinical trials of the drug after it completes some limited preclinical testing. The company said the FDA’s decision could reduce the cost of developing the drug and allow it to win marketing approval sooner.
Nuedexta is a treatment for pseudobulbar affect, a condition that involves involuntary emotional outbursts like laughing or crying. It is associated with brain disease or injury. Net revenue from the drug more than doubled to $31.4 million over the first six months of the company’s current fiscal year. That was almost all of the Aliso Viejo, Calif., company’s revenue.
Nuedexta was approved in February 2011. The drug combines two ingredients: dextromethorphan, a common ingredient in cough and cold medicines that can suppress coughing, and quinidine, which is used to treat abnormal heart rhythms.
Avanir is also studying Nuedexta as a treatment for diabetic nerve pain, agitation in patients with Alzheimer’s disease, central nerve pain in multiple sclerosis, and levodopa-induced dyskinesia in Parkinson’s disease. Dyskinesias are involuntary movements tied to most treatments used to manage Parkinson’s.
Canaccord Genuity analyst Ritu Baral said Avanir considers AVP-786 to be a safer version of Nuedexta because it contains less quinidine. The drug also has stronger patent protection and is patent protected until 2030, a few years longer than Nuedexta.
In a telephone interview, Baral said the FDA’s decision could speed approval of AVP-786 by two to four years. She said Avanir may start late-stage testing of the drug in the second half of 2014 depending on the results of current studies of Nuedexta.
OPRD PAPER-An Improved Manufacturing Process for the Antimalaria Drug artemether

OPRD PAPER-Streamlined Process for the Conversion of Artemisinin to Artemether

Correction to A Streamlined Process for the Conversion of Artemisinin to Artemether
 The structure for β-artemether is shown above, with the correct stereochemistry shown at the anomeric (8a) position. … Assignments are correct for the α- and β-anomers of artemether and dihydroartemisinin as discussed in the text; only the structure drawings are in error. …
The structure for β-artemether is shown above, with the correct stereochemistry shown at the anomeric (8a) position. … Assignments are correct for the α- and β-anomers of artemether and dihydroartemisinin as discussed in the text; only the structure drawings are in error. …The total synthesis of artemisinin from the Isopulegol ((-)-Isopulegol) began [JACS, 1983, 624].Contrast extracted from plants, is not an economical total synthesis method, but activity was found in the total synthesis of analogues are better practical significance of a thing. In this type of terpene total synthesis of natural products stereochemical conformation analysis is also very interesting. Hu menthol with MOMCl protected hydroxy, and get a double borohydride alcohol 1. Hydroboration Addition of anti-Markovnikov rule, which is replaced by hydrogen atoms added to the side of Quito, and the boron atoms added to the less substituted side. As the front side of the double bond MOM large steric hindrance, from the double rear borane adduct, resulting product1 . Compound 1 with a benzyl group protecting the primary alcohol, HCl removal of MOM protecting, PCC oxidation of the secondary alcohol to the ketone 3 . 3 with the hydrogen generating pull enolates LDA 4 , because of steric hindrance than hydrogen methyl, the nucleophilic reaction occurs in the torus , the form compound 5 . Ketone 5 and lithium reagent 6 an addition reaction, if one equivalent of lithium reagent, the resulting product was a 1:1 8 and 9 , if the 10-fold excess of lithium reagent, the resulting product was 8:1 8 and 9 . Lithium reagent 6 as a nucleophile large volume, its addition of cyclohexanone from the equatorial position to attack (such as an intermediate state 7 as shown), so that the generated key in an upright position hydroxyl group. Equivalent of lithium reagent no stereoselectivity of the reaction, but when a large excess of lithium, when chiral ketone 5 lithium reagent of the racemic 6 kinetic resolution becomes possible. Intermediate state 7 in, R configuration of the lithium reagent to Ketones speed is faster than its enantiomer S configuration lithium reagent. So generate eight faster than 9 , and finally get 8 and 9 of the ratio of 8:1. Lithium reagent 6, TMS air resistance maximum (A-value = 2.5 kcal / mol), OMe second air resistance (A-value = 0.75 kcal / mol), so that when the attack is downward TMS, OMe and H is determined by the relative position of cyclohexanone 2,6 substituent to the size and conformation of the decision, and should also be considered in the attack Burgi-Dunitz angle, so that the stereochemistry of the product unpredictable. Compound 8after removal of the benzyl protecting the primary alcohol with excess oxidized to carboxyl groups PCC automatically generate a macrolide 10 . 10 of the vinyl silane with m -CPBA and TFA into one11 , and then generate the enol methyl desilication TBAF ethers 12 , 12 and singlet oxygen reacts13 directly after treatment with acid artemisinin.
 drugs to treat malaria")
 drugs to treat malaria")
 drugs to treat malaria")
 drugs to treat malaria")
Sanofi Updates Lantus Label in EU, ORIGIN Results on Lantus® Cardiovascular Safety Integrated Into European Union Product Label

Lantus® (insulin glargine)
June 5, 2013 –
Sanofi announced today that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has issued a positive opinion for inclusion in the Lantus® (insulin glargine) product label of safety and efficacy data from the insulin glargine cardiovascular (CV) outcomes trial ORIGIN (Outcome Reduction with Initial Glargine INtervention). The revised label is evidence of Sanofi’s ongoing commitment to further assert the well-known safety and efficacy profile of insulin glargine, the most-studied basal insulin. The indication for the use of Lantus® remains unchanged.
| Mechanism of Action |
|---|
 |
| Insulin glargine (lantus) mechanism of action. |
LANTUS (insulin glargine rdna origin injection) consists of insulin glargine dissolved in a clear aqueous fluid. Each milliliter of LANTUS (insulin glargine rdna origin injection) contains 100 IU (3.6378 mg) insulin glargine.

LANTUS® is a sterile solution of insulin glargine for use as an injection. Insulin glargine is a recombinant human insulin analog that is a long-acting (up to 24-hour duration of action), parenteral blood-glucose-lowering agent., LANTUS (insulin glargine rdna origin injection) is produced by recombinant DNA technology utilizing a non-pathogenic laboratory strain of Escherichia coli (K12) as the production organism. Insulin glargine differs from human insulin in that the amino acidasparagine at position A21 is replaced by glycine and two arginines are added to the C-terminus of the B-chain. Chemically, it is 21A– Gly-30Ba-L-Arg-30Bb-L-Arg-human insulin and has the empirical formula C267H404N72O78S6 and a molecular weight of 6063.
FDA Approves Revlimid (lenalidomide) for the Treatment of Patients with Relapsed or Refractory Mantle Cell Lymphoma

Lenalidomide (Revlimid)
EP 0925294; US 5635517; WO 9803502,Drugs Fut 2003, 28, 5, 425.Bioorg Med Chem Lett 1999, 9, 11, 1625
RS)-3-(4-amino-1-oxo 1,3-dihydro-2H-isoindol- 2-yl)piperidine-2,6-dione
Jun. 5, 2013– Celgene Corporation today announced the U.S. Food and Drug Administration (FDA) has approved the company’s supplemental new drug application (sNDA) for Revlimid (lenalidomide) for the treatment of patients with mantle cell lymphoma (MCL) whose disease has relapsed or progressed after two prior therapies, one of which included bortezomib.

Revlimid is used to treat a certain type of myelodysplastic syndrome (a group of conditions in which the bone marrow produces blood cells that are misshapen and does not produce enough healthy blood cells) caused by an abnormal chromosome. Revlimid is also used to treat anemia (a lack of red blood cells in the body) and along with dexamethasone for the treatment of multiple myeloma (a type of cancer of the bone marrow) who have received at least one prior therapy.
Lenalidomide (Revlimid) is a derivative of thalidomide introduced in 2004.
It was initially intended as a treatment for multiple myeloma, for which thalidomide is an accepted therapeutic treatment. Lenalidomide has also shown efficacy in the class of hematological disorders known as myelodysplastic syndromes (MDS). Lenalidomide has significantly improved overall survival in myeloma (which generally carries a poor prognosis), although toxicity remains an issue for users. It costs $163,381 per year for the average patient.
Use in USA
On June 29, 2006, lenalidomide received U.S. Food and Drug Administration (FDA) clearance for use in combination with dexamethasone in patients with multiple myeloma who have received at least one prior therapy.
As of 2011, the FDA has initiated an ongoing review of Revlimid. The review focuses on clinical trials which found that Revlimid caused an increased risk of developing new malignancies such as acute myelogenous leukemia (AML) and B-cell lymphoma.The FDA is currently advising all patients on Revlimid to continue their treatment
Use in the UK
On 23 April 2009, The National Institute for Health and Clinical Excellence (NICE) issued a Final Appraisal Determination (FAD) approving lenalidomide, in combination with dexamethasone, as an option to treat patients who suffer from multiple myeloma who have received two or more prior therapies in England and Wales.
Use in Australia -While lenalidomide is not approved for first-line treatment of multiple myeloma in Australia, in clinical trials of newly diagnosed multiple myeloma, a four-fold increase in the incidence of second primary malignancies has been observed in patients receiving lenalidomide (7.0%) compared to controls (1.8%). These included cases of acute myeloid leukaemia, myelodysplastic syndrome and solid tumours in patients receiving lenalidomide.
Management of Hepatitis C with Natural and Synthetic Medicine
Management of Hepatitis C with Natural and Synthetic Medicine by Khan Usmanghani, Asif Iqbal , Department of Basic Clinical Sciences, Faculty of Eastern Medicine , HAMDARD UNIVERSITY, Karachi, Pakistan
during the 6 th International Conference of Infection Control December 19, 2006 Liaquat National Hospital, Karachi
http://www.slideshare.net/icsp/management-of-hepatitis-c-with-natural-and-synthetic-medicine
Viral hepatitis is defined as viral infection of hepatocytes that produces necrosis and inflammation of the liver. Viral Hepatitis is a parenchymal disease of liver. This disease is caused by (a) Hepatotropic viruses A, B, C (Non-A-Non B), D,E,F,G,H (b) Other viruses like Epstein-Barr, Cytomegalovirus, and Coxsackie virus etc. Hepatitis C virus (HCV), the major causative agent of non-A and non-B hepatitis, poses a serious worldwide health problem. about 170 million people, 3% of the world’s population, are infected with HCV High prevalence rates Southeast Asian countries WHO
