
Home » Posts tagged 'organic chemistry' (Page 6)
Tag Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Temsirolimus

TEMSIROLIMUS
Proline CCI-779
Torisel, NCGC00167518-01
LAUNCHED 2007
PFIZER
- CCI 779
- CCI-779
- HSDB 7931
- Temsirolimus
- Torisel
- UNII-624KN6GM2T
- WAY-CCI 779
Inhibits mTOR protein
For the treatment of renal cell carcinoma (RCC). Also investigated for use/treatment in breast cancer, lymphoma (unspecified), rheumatoid arthritis, and multiple myeloma.
An ester analog of rapamycin. Temsirolimus binds to and inhibits the mammalian target of rapamycin (mTOR), resulting in decreased expression of mRNAs necessary for cell cycle progression and arresting cells in the G1 phase of the cell cycle. mTOR is a serine/threonine kinase which plays a role in the PI3K/AKT pathway that is upregulated in some tumors
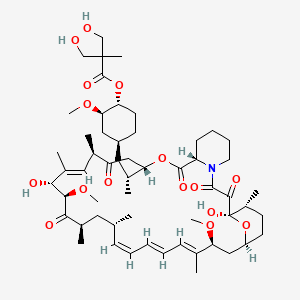
(1R,2R,4S)-4-{(2R)-2-[(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-1,5,11,28,29-pentaoxo-1,4,5,6,9,10,11,12,13,14,21,22,23,24,25,26,27,28,29,31,32,33,34,34a-tetracosahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontin-3-yl]propyl}-2-methoxycyclohexyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate
cas 162635-04-3
Temsirolimus is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by Wyeth Pharmaceuticals and approved by the FDA in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
Molecular Formula: C56H87NO16
Molecular Weight: 1030.28708
Temsirolimus (CCI-779) is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by WyethPharmaceuticals and approved by the U.S. Food and Drug Administration (FDA) in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
Temsirolimus is a specific inhibitor of mTOR and interferes with the synthesis of proteins that regulate proliferation, growth, and survival of tumor cells. Treatment with temsirolimus leads to cell cycle arrest in the G1 phase, and also inhibits tumor angiogenesis by reducing synthesis of VEGF.
The product had been under development by Wyeth Pharmaceutical for the treatment of pancreas cancer and metastatic breast cancer, multiple sclerosis (MS) and rheumatoid arthritis (RA); however, no recent development for these indications has been reported. Pfizer had been developing the compound for the treatment of sarcoma.
Temsirolimus holds orphan drug designation in both the U.S. and the E.U. for the treatment of renal cell carcinoma. Orphan drug designation was received in the U.S. in 2006 for the treatment of mantle-cell lymphoma.
mTOR (mammalian target of rapamycin) is a kinase enzyme inside the cell that collects and interprets the numerous and varied growth and survival signals received by tumor cells. When the kinase activity of mTOR is activated, its downstream effectors, the synthesis of cell cycle proteins such as cyclin D and hypoxia-inducible factor-1a (HIF-1a) are increased. HIF-1a then stimulates VEGF. Whether or not mTOR kinase is activated, determines whether the tumor cell produces key proteins needed for proliferation, growth, survival, and angiogenesis.
mTOR is activated in tumor cells by various mechanisms including growth factor surface receptor tyrosine kinases, oncogenes, and loss of tumor suppressor genes. These activating factors are known to be important for malignant transformation and progression.mTOR is particularly important in the biology of renal cancer (RCC) owing to its function in regulating HIF-1a levels. Mutation or loss of the von Hippel Lindau tumor-suppressor gene is common in RCC and is manifested by reduced degradation of HIF-1a. In RCC tumors, activated mTOR further exacerbates accumulation of HIF-1a by increasing synthesis of this transcription factor and its angiogenic target gene products.
Rapamycin 42-ester with 3-hydroxy-2-(hydroxymethyl)-2-methylpropionic acid (CCl-779) is an ester of rapamycin which has demonstrated significant inhibitory effects on tumor growth in both in vitro and in vivo models.
CCl-779 may delay the time to progression of tumors or time to tumor recurrence which is more typical of cytostatic rather than cytotoxic agents. CCl-779 is considered to have a mechanism of action that is similar to that of sirolimus. CCl-779 binds to and forms a complex with the cytoplasmic protein FKBP, which inhibits an enzyme, mTOR (mammalian target of rapamycin, also known as FKBP12-rapamycin associated protein [FRAP]). Inhibition of mTOR’s kinase activity inhibits a variety of signal transduction pathways, including cytokine-stimulated cell proliferation, translation of mRNAs for several key proteins that regulate the G1 phase of the cell cycle, and IL-2-induced transcription, leading to inhibition of progression of the cell cycle from G1 to S. The mechanism of action of CCl-779 that results in the G1-S phase block is novel for an anticancer drug.
The preparation and use of hydroxyesters of rapamycin, including CCl-779, are disclosed in U.S. Pat. No. 5,362,718. A regiospecific synthesis of CCl-779 is described in U.S. Pat. No. 6,277,983.
CCl-779 can be synthesized by the non-regioselective acylation of rapamycin, as described in U.S. Pat. No. 5,362,718. The synthesis, however, is complicated by mixtures of the desired 42-ester, with 31-esterified rapamycin, as well as 31, 42-diesterified rapamycin and unreacted rapamycin.
CCl-779 can also be prepared by the acylation of the 31-silyl ether of rapamycin with a ketal of bis-(hydroxymethyl)propionic acid, followed by removal of the 31-silyl ether and ketal protecting group from the bis-(hydroxymethyl) propionic acid, as described in U.S. Pat. No. 6,277,983. However, the crude 42-monoester produced from this regioselective synthesis requires further purification by column chromatography to remove residual amounts of diester by-products and unreacted rapamycin starting material.
Temsirolimus (CCI-779), an mTOR kinase Inhibitor of formula (I) is an antineoplastic agent indicated for the treatment of advanced renal cell carcinoma.Temsirolimus is a Rapamycin 42 ester with [3-hydroxy-2-(hydroxymethyl)-2-methylpropanoic acid and was first disclosed by Skotnicki et al in US Patent No. 5,362,718.

Several processes for the preparation of Temsirolimus have been reported in the literature such as those described in US 5,362,718; US 6,277,983 and US 7, 153,957.
US Patent No 5,362,718 discloses a process for the preparation of different rapamycin 42 esters including Temsirolimus as per the scheme given below (Scheme-I).

Scheme-I: Synthesis of Temsirolimus as disclosed in US Patent No. 5,362,718
The process is non-regioselective and hence results in 31-estehfied rapamycin, 31 , 42 diesterified rapamycin and unreacted rapamycin along with the desired rapamycin-42 ester.
US Patent No. 6,277,983 reports a process for the preparation of Temsirolimus by using 31 , 42 bis silyl intermediates as per the scheme shown below (Scheme-ll).

Scheme-ll: Synthesis of Temsirolimus as disclosed in US Patent No. 6,277,983 US Patent No. 7, 153,957 reports a process for the preparation of Temsirolimusby using boronate intermediate as per the scheme shown below (Scheme-Ill).

Scheme-Ill: Synthesis of Temsirolimus as disclosed in US Patent No. 7, 153,957
 Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS “Amano” (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS “Amano” (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
………………………………………………..
SYNTHESIS
https://www.google.co.in/patents/EP0763039A1
Example 11
Rapamycin 42-ester with 2.2-bis-(hydroxymethyl)propionic acid
A solution of the product of Example 10 (2.8 g, 2.65 mmol) in 50 mL THF and
25 mL IN HCl was stirred at room temperature for 4 h. The mixture was diluted with water and extracted three times with EtOAc. The combined organic phases were washed with saturated NaHCO3 solution, saturated NaCl solution, dried over MgSO4, filtered and evaporated to a yellow oily solid. Purification by flash chromatography (3X with EtOAc) afforded the title compound (1.6 g, 59 %).
(-)FAB-MS mlz 1029.6 (M-), 590.4 (southern fragment), 437.3 (northern fragment). !H NMR (400 MHz, d-6 DMSO) δ 4.5 (m, 1 H, C(42)H), 3.45 (s, 4 H), 1.04 (s, 3 H).
*3C NMR (100.6 MHz, d-6 DMSO) δ 174.2, 63.7, 63.6, 49.9, 16.8.
Example 10 Rapamycin 42-ester with 2.2.5-trimethyl.1.3_dioxane-5-carboxyric acid
To a solution of the 2,2-bis(hydroxymethyl)propionic acid isopropylidene ketal (1.041 g, 5.98 mmol) (prepared according to the procedure of Bruice, J. Am. Chem. Soc. 89: 3568 (1967)) and triethylamine (0.83 mL, 5.98 mmol) in 20 mL anhydrous THF at 0 °C under nitrogen was added 2, 4, 6-trichlorobenzoyl chloride (0.93 mL, 5.98 mmol) and the resultant white suspension was stirred 5 h at room temperature. The precipitate was removed by vacuum filtration, rinsing the flask and filter cake with an additional 10 mL dry THF. The filtrate was concentrated by rotary evaporation to a white solid. The residue was dissolved in 20 mL dry benzene, then rapamycin (5.47 g, 5.98 mmol) and DMAP (0.731 g, 5.98 mmol) were added. After stirring overnight at room temperature, the mixture was diluted with EtOAc, washed with H2O and saturated NaCl (aq), dried over MgSO4, filtered and evaporated to a yellow oil. Flash chromatography (5X with 60% EtOAc-hexane) afforded the title compound (2.2 g, 34 %) as a white solid.
(-)FAB-MS mlz 1069.5 (M-), 590.3 (southern fragment), 477.2 (northern fragment). –■H NMR (400 MHz, d-6 DMSO) δ 4.57 (m, 1 H, C(42)H), 4.02 (d, 2 H), 3.60 (d, 2 H), 1.34 (s, 3 H), 1.24 (s, 3 H), 1.06 (s, 3 H). 1 C NMR (100.6 MHz, d-6 DMSO) δ 173.2, 99.0, 65.0, 22.2, 18.1.
…………………………………………..
SYNTHESIS
https://www.google.co.in/patents/US7153957
This scheme



Preparation of 5-Methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [A]
To a suspension of 2,2-bis(hydroxymethyl)propionic acid (131 g, 0.98 mole) in tetrahydrofuran (500 ml) was added a solution of phenylboronic acid (122 g, 1.0 mole) in tetrahydrofuran (500 ml). The mixture was stirred for 3 h and toluene (1.0 L) was added. Water was removed by azeotropic distillation with toluene. Heptanes (500 ml) was added to the precipitated product, heated to reflux and cooled. The mixture was filtered and washed with heptanes (2×300 ml). The solids were dried under vacuum at 70–75° C. until constant weight to give 94% yield. 1H NMR: δ (DMSO-d6) 7.65 (d, 2H, Ar), 7.40 (m, 3H, Ar), 4.35 (d, 2H, CH2), 3.92 (d, 2H, CH2), 1.17 (s, 3H, CH3)
Preparation of Rapamycin 42-ester with 5-methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [B]
As described in U.S. Pat. No. 6,277,983 (2001) a 3 L flask was charged with rapamycin (100 g, 0.104 mole) and dissolved in ethyl acetate (1.50 L). The solution was cooled to 5–10° C. Imidazole (30 g, 0.44 moles, 4.23 eq.) was added and dissolved. Under nitrogen protection, trimethylsilyl chloride (44 g, 0.405 mole, 4.0 eq.) was added over 30–40 min while maintaining the temperature at 0–5° C. during the addition. The mixture was held for a minimum of 0.5 h. The reaction was monitored by TLC (30:70 acetone:heptane eluent). The reaction was complete when all of the rapamycin was consumed.
Two to three drops of the reaction mixture were removed and retained as a 31,42-bis(trimethylsilyl) rapamycin reference standard. 0.5 N Sulfuric acid (300 mL) was added to the 3 L flask over 0.5 h maintaining the temperature 0–5° C. The mixture was stirred vigorously and held for 5 h. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone:heptane eluent). The reaction was complete when essentially no 31,42-bis-(trimethylsilyl) rapamycin was present. The layers were separated and the lower aqueous layer was back extracted with ethyl acetate (500 mL). The combined organic layers were washed with saturated brine (500 mL) and saturated sodium bicarbonate (2×200 mL) until pH 8 was obtained. The organic layer was washed with water (2×500 mL) and brine (500 ml) until pH 6 to 7 was obtained. The solution was dried over magnesium sulfate (100 g) for 30 min, filtered into a 2 L flask and concentrated to a volume of 135 ml. Ethyl acetate (500 ml) was added and concentrated to a volume of 135 ml. The water chase was repeated once more with ethyl acetate (500 ml). Methylene chloride (300 ml) was added and the solution held until needed in the next step.
A 3 L flask equipped with mechanical stirrer was charged with compound [A] (75 g, 0.341 mole) in methylene chloride (400 mL). Diisopropylethylamine (66.1 g, 0.51 mole) was added dropwise over 20 mins and rinsed with methylene chloride (25 mL). 2,4,6-Trichlorobenzoyl chloride (80 g, 0.328 mole) was added and rinsed with methylene chloride (25 mL). The mixture was held at 0–5° C. for 4 h, and cooled to −10±5° C.
The solution of 31-trimethylsilyl rapamycin was added to the 3 L flask containing the mixed anhydride, and rinsed with methylene chloride (25 mL). A solution of dimethylamino pyridine (48.5 g, 0.397 mole) in methylene chloride (150 mL) was prepared, added over 1.5 h, maintaining the temperature <−8° C., and rinsed with methylene chloride (25 mL). The mixture was held for 12 h at −11 to −5° C. The reaction mixture was quenched with 1 N sulfuric acid (600 ml) keeping the temperature <10° C. The mixture was stirred and held for 30 mins. The pH of the upper aqueous layer was ≦2. The layers were separated, and the lower organic layers washed with brine (450 ml), saturated sodium bicarbonate (500 mL) until pH ≧8. The organic layer was washed with water (450 ml) until pH 6–7 was obtained. The solution was concentrated, acetone (250 ml) added and concentrated. This was repeated with another portion of acetone (250 ml) and concentrated.
The solution was diluted with acetone. 0.5 N Sulfuric acid (500 ml) was added dropwise over 30 mins keeping the pot temperature 0–5° C. The mixture was held for a minimum of 5 h, during which time, the product precipitated out of solution. Aqueous sodium bicarbonate (30 g in 375 ml water) was added dropwise over 30 minutes keeping the pot temperature 0 to 5° C.; the mixture was held for a minimum of 30 minutes. Acetic acid (25 ml) was added until pH was 5–6 keeping the pot temperature <10° C. The mixture was warmed to room temperature and held for 16 h. The solid product was filtered and washed with water (2×100 ml) followed by 1:1 acetone:water (2×100 ml). The cake was purified in acetone (375 ml) to give 65 g (58% overall from rapamycin) of product [B]. LC/MS: using an electrospray interface in the positive ion mode afforded the molecular ion [M+Na]=1138.5 atomic mass units (amu).
Preparation of Rapamycin 42-ester with 2,2-bis(hydroxymethyl)-propionic acid, [C]
Compound [B] (200 g, 0.179 mole), was dissolved in tetrahydrofuran (600 ml), 2-methyl-2,4-pentanediol (42.3 g, 0.358 mole, 2.0 eq.) was added and the mixture stirred for a minimum of 3 h. The reaction mixture was concentrated to a foam. Diethyl ether (1.0 L) was added and the mixture stirred for 2 h. Heptanes (1.0 L) was added dropwise over 1 h and the mixture stirred for 2 h. The mixture was filtered and the solid product washed with heptanes (500 ml). The solids were re-dissolved in acetone (400 ml), re-treated with 2-methyl-2,4-pentanediol (21.1 g, 0.179 mole, 1 eq.) in acetone (200 ml), clarified through a 0.2 micron cartridge filter, and rinsed with acetone (200 ml). The solution was concentrated to a foam, diethyl ether (1.0 L), pre-filtered through a 0.2 micron cartridge filter, was added and the mixture stirred for 2 h. The mixture was co-precipitated by adding pre-filtered heptanes (1.0 L). The precipitated solids were filtered and washed with ether:heptane (2×500 ml). The solids were dried (55 to 60° C., 10 mm Hg, minimum 24 h) to give 159 g (86%) of product [C]. LC/MS: using APCl in the positive ion mode afforded the molecular ion [M+NH4]=1047.0 amu. The 1H NMR of the product (CCl-779) was identical to the product described in example 11 of U.S. Pat. No. 5,362,718 (1994).
…………………………………
Synthesis
http://www.google.com/patents/WO2005100366A1
Example 1 – Synthesis of Proline CCI-779

This example describes a method for the synthesis of the proline analog of CCI- 779, which is illustrated in the scheme provided above.
A.
Preparation of 31, 42-Bis (trimethylsilyl) proline rapamycin (Compound B)
A 3 -neck 50 mL flask was charged with proline rapamycin (compound A in the scheme) (1.47 g, 1.63 mmol), imidazole (0.45 g, 6.6 mmol, 4 eq.) and ethyl acetate (22.5 mL). The magnetically stirred mixture became cloudy. The mixture was cooled to 0-5°C. Under nitrogen protection, trimethylsilyl chloride (0.62 g, 5.7 mmol, 3.5 eq.) was added over 0.5 h via syringe while maintaining the temperature at 0-5°C during the addition. The syringe was rinsed with 2.5 ml ethyl acetate and the mixture held for 0.75 hours (0.75 h), whereupon a white precipitate was formed. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone :heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The mixture was shaken and allowed to settle. The upper organic layer was spotted against the starting material (proline rapamycin). The reaction was complete when no more starting material was present.
B.
Preparation of 31 -trimethylsilyl proline rapamycin, Compound E
When the above reaction was complete, 2-3 drops of the reaction mixture was removed and retained for the following step as the 31,42-bis(trimethylsilyl) proline rapamycin reference standard. To the 50 ml flask was added 0.5 N sulfuric acid (4.5 mL) over 0.5 h maintaining the temperature at 0-5 °C. The mixture became less cloudy. The mixture was held for 2.5 h and was monitored by thin layer chromatography (TLC, 30:70 acetone:heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The reaction aliquot was shaken and allowed to settle. The upper organic layer was spotted against the 31 ,42-bis(trimethylsilyl) proline rapamycin reference. The reaction was complete when essentially no 31,42-bis(trimethylsilyl) proline rapamycin was present. Ethyl acetate (5 mL) was added and the layers separated. The lower aqueous layer is extracted with ethyl acetate (7.5 mL). The combined organic layers were washed with brine (7.5 mL), by washing with saturated sodium bicarbonate (6 mL) followed by washing water (3 x 7.5 mL), in that order. The pH of the last water wash was 6-7. The organic layer was washed again with brine (7.5 mL) and dried over sodium sulfate (4 g) for 20 min. The mixture was filtered into a 250 mL flask and concentrated to dryness.
The solid was dried at room temperature under high vacuum (10 mmHg or less) for 20 h.
Weight = 1.51 g of an off-white foam.
C.
Preparation of Intermediate, Compound F:
A 3 -neck 100 mL flask equipped with mechanical stirrer was charged with
2,2,5-trimethyl[l,3-dioxane]-5-carboxylic acid, Compound C (0.63 g, 3.6 mmol) in methylene chloride (7.5 mL). Dusopropylethylamine (0.77 g, 5.9 mmol) was added, followed by a rinse with methylene chloride (1 mL). 2,4,6-Trichlorobenzoyl chloride (0.85 g, 3.5 mmol) was added, followed by a rinse with methylene chloride (1.5 mL).
The mixture was held at room temperature for 4.5 h. The solution was cooled to -12 ±
2°C. 31 -Trimethylsilyl proline rapamycin, compound E, (1.51 g) in methylene chloride (8 mL) was dissolved and added to the 100 mL flask. Methylene chloride (2 mL) was added as a rinse. A solution of dimethylamino pyridine (DMAP) (0.77 g, 6.8 mmol) in methylene chloride (3 mL) was prepared and added to the 100 mL flask over
2.5 h maintaining the temperature -12 ± 2 °C. Methylene chloride (1 mL) was added as a rinse. The mixture was held for 16 h and was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL water and 0.2 mL ethyl acetate. The HPLC sample was prepared by withdrawing 2 drops of the upper organic layer, blowdrying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04
Wavelength : λ = 280 nm Flow rate : 1 mL / min
Time : 60 min
Retention times : Compound E ~14.0-14.5 min Compound F -33.4-33.8 min
The reaction was complete when < 0.5% of starting material was present. The reaction mixture was quenched with water (6 mL). Methylene chloride (10 mL) was added and the layers separated. The aqueous layer was extracted with methylene chloride (10 mL). The combined organic layers were washed with 0.5 N sulfuric acid (12 mL), brine (10 mL), saturated sodium bicarbonate (6 mL), and water (3 x 10 mL) in that order. The pH of the last water wash was 6-7. The clear yellow solution was concentrated to a foam. The solid was dried at room temperature under high vacuum (10 mmHg or less) for 24 h. Weight = 1.88 g of a yellow foam.
D.
Preparation of crude proline CCI-779
A 1-neck 50 mL flask equipped with mechanical stirrer was charged with Compound F in THF (18.8 mL, 10 vols) and then cooled to 0 – 5 °C (or about -2.5°C). 2 N sulfuric acid (9.4 mL, 5 vols) was added over 2.5 h. After complete addition, the mixture was warmed to 2.5 °C and then held for 45 h. The reaction was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 0.25 mL ethyl acetate. The HPLC sample was prepared by withdrawing 5 drops of the upper organic layer, blow drying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04 Wavelength : λ= 280 nm Flow rate : 1 mL / min Time : 60 min Retention times Compound F ~33.4-33.8 min Desilylated Compound F ~10.5-11.5 min (intermediate) Proline CCI-779 -5.0-5.5 min The desilylated intermediate of compound F was formed first. The reaction was complete when < 0.5% of the silylated analog remained. Ethyl acetate (27 mL) and brine (7.5 mL) was added and the layers separated. The aqueous layer was extracted with ethyl acetate (10 mL). The combined organic layers were washed with brine (10 mL), saturated sodium bicarbonate (7.5 mL), and water (3 x 7.5 mL) in that order. The pH of the last water wash was 6-7. The mixture was dried over sodium sulfate (5 g) for 30 min, filtered into a 250 L flask and concentrated to dryness. Weight = 1.58 g of a yellow foam.
E.
Chromatographic purification of crude proline CCI-779
A silica gel column (31.6 g, 60 A, 200-400 mesh) (22 cm length x 2.5 cm diameter) was prepared and conditioned with 15:85 acetone:HPLC grade hexane (1 L). The yellow crude proline CCI-779 (1.58 g) in acetone (1.58 mL) was prepared and chromatographed. The column was eluted with the remaining 15:85 acetone :hexane mixture followed by 25:75 acetone:hexane (4 L). The positive fractions were combined and concentrated to dryness. The resulting foam was dried at 35 °C, high vacuum (i.e., 10 mmHg or less) for 24 h. Weight = 1.12 g of a light yellow foam.
F.
Ether treatment of proline CCI-779
A 1 -neck 50 mL flask was charged with proline CCI-779 ( 1.12 g) and dissolved in ether (1.5 mL). The mixture was held for 2 h. The ether was stripped to give a foam. The foam was dried at 35 °C, under high vacuum (10 mmHg or less) for 12 h then at room temperature overnight (12 h). Weight = 1.09 g.
*H NMR (500 and 600 MHz, DMSO-d6) δ 5.45 (H-l), 6.12 (H-2), 6.27 (H-3), 6.41 (H-4), 6.20 (H-5), 3.66 (H-7), 1.14 and 1.86 (H-8), 4.02 (H-9), 1.19 and 1.81 (H-10), 1.52 (H-11), 2.03 (H-12), 3.23 and 3.54 (H-18), 1.76 (H-19), 2.20 and 1.89 (H-21), 4.22 (H-22), 4.87 (H-25), 2.28 and 2.70 (H-26), 3.22 (H-28), 5.11 (H-29), 4.04 (H-31), 4.17 (H-32), 2.25 (H-34), 0.985 and 1.38 (H-35), 2.22 (H-36), 1.76 (H-37), 0.961 and 1.11 (H-38), 1.31 (H-39), 0.726 and 1.90 (H- 40), 3.14 (H-41), 4.46 (H-42), 1.22 and 1.81 (H-43), 0.888 and 1.60 (H-44), 1.60 (H-45), 3.05 (H-46, OCH3), 0.697 (H-47), 6.48 (H-48), 0.821 (H-49), 1.76 (H-50), approx. 5.1- 5.3 (H-51), 3.17 (H-52, OCH3), 0.755 (H-53), 0.966 (H-54), 0.805 (H-55), 3.29 (H-56, OCH3), 3.46 (H-59), 1.01 (H-60), approx. 4.3-4.7 (0-61)
13C NMR (75 MHz, DMSO- d6) δ 139.12 (C-1), 130.53 (C-2), 132.49 (C-3), 127.08 (C-4), 127.21 (C-5), 137.12 (C-6), 81.93 (C-7), 40.40 (C-8), 65.83 (C-9), 29.45 (C-10), 25.87 (C-l l), 34.21 (C-12), 99.25 (C-13), 198.17 (C-15), 165.55 (C-16), 47.01 (C-18), 24.04 (C-19), 28.93 (C-21), 58.50 (C-22), 170.44 (C-23), 73.24 (C-25), 39.96 (C-26), 207.67 (C-27), 44.51 (C-28), 123.92 (C-29), 136.56 (C-30), 75.84 (C-31), 84.86 (C-32), 209.49 (C-33), 40.76 (C-34), 39.20 (C-35), 35.05 (C-36), 32.73 (C-37), 38.42 (C-38), 32.06 (C-39), 36.01 (C-40), 80.12 (C- 41), 75.92 (C-42), 29.25 (C-43), 30.24 (C-44), 10.27 (C-45), 55.48 (C-46, OCH3), 15.46 (C-47), 15.59 (C-49), 14.41 (C-50), 56.56 (C-52, OCH3), 12.67 (C-53), 21.50 (C-54), 14.89 (C-55), 57.27 (C-56, OCH3), 174.22 (C-57), 49.90 (C-58), 63.59 and 63.98 (C-59), 16.82 (C-60). MS [M+NH ] 1033.5, [ESI(+), M+Na+] 1038.7.
Example 3 – Synthesis of CCI-779:

A. Synthesis of CCI-779 via intermediate A Method 1 : A mixture of rapamycin (6 g), vinyl ester I (2 g), lipase PS-C “Amano” II (6 g) in anhydrous TBME (36 mL) was heated at 45 °C under Ar2 for 2 days. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, the oily residue was added to heptane while stirring. The batch was then cooled to -15 °C for 2 h, collect the solid on the Buchner funnel and washed with cold heptane, A was obtained as off-white solid, crude yield : 98%.MS (El): 1070 Above crude A (6g), dissolved in n-PrOH (24 mL) cooled to 0 °C with an ice-water bath, to this solution was added aqueous H2S04 (12 mL, 1.2N). The mixture was stirred for 24 h at 0°C and was then added to cold phosphate buffer (300 ml, pH=7.8), collect the solid on a Buchner funnel and washed with DI water and dry under vacuum, silica gel column purification eluting with hexane-acetone furnished CCI-779 as a white solid (5.2 g, 90%). MS (El): 1030 Method 2: A mixture of rapamycin (30.0 g, 32.8 mmol), vinyl ester I (10.0 g, 50 mmol), lipase PS-C “Amano” II (30 g) and molecular sieves (5 A) (10.0 g) in anhydrous TBME (150 mL) was heated at 42-43 °C under Ar2 for 48 hours. THF (100 mL) was added to dissolve the precipitation and the mixture was cooled to room temperature. Enzyme was removed by filtration and washed with THF (200 mL), the filtrate was concentrated to about 60 mL and diluted with THF (320 mL). The solution was then cooled to 0-5 °C, H2S04 (180 mL, 2N) was added dropwise over lh. The mixture was stirred for 48 h at 0-5 °C or until the disappearance of A as monitored by TLC. The mixture was diluted with brine (300 mL) and extracted with EtOAc (three times). The combined organic layer was washed with H20, 5% NaHC03, then brine and dried
(MgS04). Evaporation of solvent gave a light yellowish semi solid which was purified by flash chromatography (hexane/acetone, 2:1) to give CCI-779 as a white solid (30.77 g, 91% for two steps). B. Synthesis of CCI-779 via intermediate B: A mixture of rapamycin (3 g), vinyl ester II (1.2 g), lipase PS-C “Amano” II (5 g) in anhydrous TBME (45 mL) was heated at 45 °C under Ar2 for 60 h. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, MeOH (20 mL) was added to the residue and concentrated to dryness. Silica gel column purification of crude eluting with hexane-acetone furnished CCI-779 as a white solid (2.3 g), and recovered rapamycin (0.81 g). The yield is 93% based on the recovered rapamycin.
proline analog of CCI-779 (proline-rapamycin42-ester with 2,2-bis(hydroxymethyl)propionic acid or proline-CCI-779) and methods of synthesizing same. Proline-CCI-779 is an active drug substance useful in oncology and other associated indications (immunosuppression, anti-inflammatory, anti-proliferation and anti-tumor). In one aspect, the synthesis of proline-CCI-779 is accomplished through bis- silylation of proline rapamycin, mono-de-protecting 31 ,42-bis-trimethylsilyl proline rapamycin, and acylating the mono-silyl proline rapamycin followed by hydrolysis. In another aspect, the invention provides a two-step enzymatic process involving a regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to give CCI-779.
Example 4 – Synthesis of Proline-CCI-779 The enzymatic procedure of the invention can also be applied to the synthesis of proline CCI-779 from proline-rapamycin under essentially the same conditions as described in Example 2, procedure A for the synthesis of CCI-779 from rapamycin.

proline-rapamycin proline-CCI-779
………………….
more info added for readers
synthesis of CCI-779 or Proline CCI-779 (Temsirolimus) which is useful as an antineoplastic agent having the structure

It is stated to be effective in multiple applications, including inhibition of tumor growth, the treatment for multiple sclerosis and rheumatoid arthritis.
2. The Prior Arts

U.S. Pat. No. 7,202,256 disclosed methods for the synthesis of CCI-779 (Temsirolimus), providing two-step enzymatic process involving regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to obtain the CCI-779 (as shown in scheme 1). A number of drawbacks of the synthesis route depicted in scheme 1 are high-priced PdCl2 and poisonous trimethylboroxine.


A selective synthesis of 42-monoacylated product was previously conducted by reacting rapamycin 31,42-bis-silyl ether, and then the 42-sily ether protection group is selectively removed to provide rapamycin-OH-31-sily ether (U.S. Pat. No. 5,563,145). In addition, a regioselective process for the preparation of CCI-779 is also described in U.S. Pat. No. 6,277,983 (Scheme2). First, rapamycin (compound 4b) is treated with excess chlorotrimethylsilane to form rapamycin31,42-bis-trimethylsilyl ether (compound 5), and then 42-trimethylsilyl ether protection group is selectively removed in mild acid to provide rapamycin 42-OH-31-trimethylsilyl ether (compound 6). This free 42-OH was then acylated with 2,4,6-trichlorobenzyl mixed anhydride of 2,2,5-trimethyl[1,3-dioxane]-5-carboxylic acid (compound 7) at −15° C. for 16 h to give rapamycin 31-trimethylsilyl ether 42-ester (compound 8). Following treatment with mild acid for a certain period, CCI-779 can be isolated. 2,4,6-trichlorobenzyl chloride is irritant, moisture sensitive and costly.

Further, as below-depicted in Scheme 3, U.S. Pat. No. 7,153,957 disclose another method for the CCI-779. It can be prepared by the acylation of 31-silyl ether of rapamycin with the anhydride derived from the 2-phenylboronate acid to give rapamycin 31-silyl ether, 42-boronate. Thereafter, it is hydrolyzed under mild acid condition to form rapamycin 42-ester boronate. After being treated with a suitable diol, CCI-779 was obtained (Scheme 3). Mixed anhydride is not satisfactory for commercial scale synthesis because it can be kept stable only for 48 hr at −5˜0° C., not durable for longer time.
synthesis ofTemsirolimus in a more economic way.

…………..
PAPERS
CCI-779
Drugs Fut 2002, 27(1): 7
Drugs Fut 2002, 27(1): 7
Organic Letters, 2005 , vol. 7, 18 pg. 3945 – 3948 seenmr
PATENTS
| United States | 5362718 | APPROVED 1994-04-18 | EXPIRY 2014-04-18 |
| Canada | 2429020 | 2009-05-26 | 2021-11-13 |
| Canada | 2187024 | 2004-08-10 | 2015-04-14 |
|
6-13-2012
|
N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF
|
|
|
11-18-2011
|
N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF
|
|
|
8-17-2011
|
N-Hydroxyamide Derivatives and Use Thereof
|
|
|
7-6-2011
|
Sulfonyl Amino Cyclic Derivatives and Use Thereof
|
|
|
11-24-2010
|
Benzothiazole Formulations and Use Thereof
|
|
|
11-19-2010
|
Indazole Compounds for Treating Inflammatory Disorders, Demyelinating Disorders and Cancers
|
|
|
9-31-2010
|
Process for preparation of temsirolimus
|
|
|
4-23-2010
|
COMBINATION OF BENZIMIDAZOLE ANTI-CANCER AGENT AND A SECOND ANTI-CANCER AGENT
|
|
|
10-21-2009
|
Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants
|
|
|
6-12-2009
|
Administration of an Inhibitor of HDAC and an mTOR Inhibitor
|
|
6-8-2007
|
Methods for preparing crystalline rapamycin and for measuring crystallinity of rapamycin compounds using differential scanning calorimetry
|
|
|
4-11-2007
|
Proline CCI-779, production of and uses therefor, and two-step enzymatic synthesis of proline CCI-779 and CCI-779
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
7-12-2006
|
CCI-779 Isomer C
|
| US5362718 | 18 Apr 1994 | 8 Nov 1994 | American Home Products Corporation | Rapamycin hydroxyesters |
| US6197967 | 13 Dec 1999 | 6 Mar 2001 | Clariant Gmbh | Process for the preparation of paraoxadiazolyphenylboronic acids |
| US6277983 | 27 Sep 2000 | 21 Aug 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO1995028406A1 | 14 Apr 1995 | 26 Oct 1995 | American Home Prod | Rapamycin hydroxyesters, process for their preparation and pharmaceutical compositions containing them |
| US7553843 | 6 Dec 2006 | 30 Jun 2009 | Wyeth | Process for the preparation of purified crystalline CCI-779 |
| US7605258 | 16 Oct 2007 | 20 Oct 2009 | Wyeth | Processes for the synthesis of individual isomers of mono-peg CCI-779 |
| US7622578 | 6 Dec 2006 | 24 Nov 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US7625726 | 29 Sep 2008 | 1 Dec 2009 | Wyeth | Process for preparing rapamycin 42-esters and FK-506 32-esters with dicarboxylic acid, precursors for rapamycin conjugates and antibodies |
| US7875612 | 24 Apr 2002 | 25 Jan 2011 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US7910594 | 13 May 2003 | 22 Mar 2011 | Endocyte, Inc. | Vitamin-mitomycin conjugates |
| US8026276 | 25 Jul 2003 | 27 Sep 2011 | Wyeth Llc | Parenteral CCI-779 formulations containing cosolvents, an antioxidant, and a surfactant |
| US8044200 | 14 Mar 2006 | 25 Oct 2011 | Endocyte, Inc. | Synthesis and purification of pteroic acid and conjugates thereof |
| US8105568 | 10 Jul 2009 | 31 Jan 2012 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| US8288557 | 22 Jul 2005 | 16 Oct 2012 | Endocyte, Inc. | Bivalent linkers and conjugates thereof |
| US8299116 | 10 Aug 2011 | 30 Oct 2012 | Wyeth Llc | CCI-779 concentrate formulations |
| US8455539 | 15 Oct 2012 | 4 Jun 2013 | Wyeth Llc | CCI-779 concentrate formulations |
| US8465724 | 18 Aug 2006 | 18 Jun 2013 | Endocyte, Inc. | Multi-drug ligand conjugates |
| US8470822 | 7 May 2010 | 25 Jun 2013 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US8524893 | 28 Jan 2011 | 3 Sep 2013 | Fresenius Kabi Oncology Limited | Process for the preparation of temsirolimus and its intermediates |
| WO2011092564A2 | 20 Jan 2011 | 4 Aug 2011 | Fresenius Kabi Oncology Ltd | Process for the preparation of temsirolimus and its intermediates |
Orphan Drug Designation Granted for Epidiolex in Dravet syndrome by the FDA

Cannabidiol Seven Expanded Access INDs granted by FDA to U.S. physicians to treat with Epidiolex 125 children suffering from intractable epilepsy syndromes -
LONDON, Nov. 15, 2013
GW Pharmaceuticals plc (AIM: GWP, Nasdaq: GWPH, “GW”) announced today that the U.S. Food and Drug Administration (FDA) has granted orphan drug designation for Epidiolex(R), our product candidate that contains plant-derived Cannabidiol (CBD) as its active ingredient, for use in treating children with Dravet syndrome, a rare and severe form of infantile-onset, genetic, drug-resistant epilepsy syndrome. Epidiolex is an oral liquid formulation of a highly purified extract of CBD, a non-psychoactive molecule from the cannabis plant. Following receipt of this orphan designation, GW anticipates holding a pre-IND meeting with the FDA in the near future to discuss a development plan for Epidiolex in Dravet syndrome.
Dravet syndrome is a rare pediatric epilepsy syndrome with a distinctive but complex electroclinical presentation. Onset of Dravet syndrome occurs during the first year of life with clonic and tonic-clonic seizures in previously healthy and developmentally normal infants. Prognosis is poor and patients typically develop intellectual disability and life-long ongoing seizures. There are approximately 5,440 patients with Dravet in the United States and an estimated 6,710 Dravet patients in Europe. These figures may be an underestimate as this syndrome is reportedly underdiagnosed.
In addition to GW’s clinical development program for Epidiolex in Dravet syndrome, which is expected to commence in 2014, GW has also made arrangements to enable independent U.S. pediatric epilepsy specialists to treat high need pediatric epilepsy cases with Epidiolex immediately. To date in 2013, a total of seven “expanded access” INDs have been granted by the FDA to U.S. clinicians to allow treatment with Epidiolex of approximately 125 children with epilepsy. These children suffer from Dravet syndrome, Lennox-Gastaut syndrome, and other pediatric epilepsy syndromes. GW is aware of further interest from additional U.S. and ex-U.S. physicians to host similar INDs for Epidiolex. GW expects data generated under these INDs to provide useful observational data during 2014 on the effect of Epidiolex in the treatment of a range of pediatric epilepsy syndromes.
“I, together with many colleagues in the U.S. who specialize in the treatment of childhood epilepsy, very much welcome the opportunity to investigate Epidiolex in the treatment of Dravet syndrome. The FDA’s timely approval of the orphan drug designation for Epidiolex in Dravet syndrome is a key milestone that comes after many years of reported clinical cases that suggest encouraging evidence of efficacy for CBD in this intractable condition,” stated Dr. Orrin Devinsky, Professor of Neurology, Neurosurgery and Psychiatry in New York City. “With GW now making plans to advance Epidiolex through an FDA development program, we have the prospect for the first time of fully understanding the science of CBD in epilepsy with a view to making an appropriately tested and approved prescription medicine available in the future for children who suffer from this debilitating disease.”
“GW is proud to be at the forefront of this important new program to treat children with Dravet Syndrome and potentially other forms of intractable childhood epilepsy. For families in these circumstances, their lives are significantly impacted by constant and often times very severe seizures in children where all options to control these seizures have been exhausted,” stated Dr. Stephen Wright, GW’s R&D Director. “GW intends to advance a full clinical development program for Epidiolex in Dravet syndrome as quickly as possible, whilst at the same time helping families in the short term through supporting physician-led INDs to treat intractable cases. Through its efforts, GW aims to provide the necessary evidence to confirm the promise of CBD in epilepsy and ultimately enabling children to have access to an FDA-approved prescription CBD medicine.”
“This orphan program for Epidiolex in childhood epilepsy is an important corporate strategic priority for GW. Following receipt of today’s orphan designation, GW now intends to commence discussions with the FDA regarding the U.S. regulatory pathway for Epidiolex,” stated Justin Gover, GW’s Chief Executive Officer. “GW intends to pursue this development in-house and retains full commercial rights to Epidiolex.”
About Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition — generally a disease or condition that affects fewer than 200,000 individuals in the U.S. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication.
About GW Pharmaceuticals plc
Founded in 1998, GW is a biopharmaceutical company focused on discovering, developing and commercializing novel therapeutics from its proprietary cannabinoid product platform in a broad range of disease areas. GW commercialized the world’s first plant-derived cannabinoid prescription drug, Sativex(R), which is approved for the treatment of spasticity due to multiple sclerosis in 22 countries. Sativex is also in Phase 3 clinical development as a potential treatment of pain in people with advanced cancer. This Phase 3 program is intended to support the submission of a New Drug Application for Sativex in cancer pain with the U.S. Food and Drug Administration and in other markets around the world. GW has established a world leading position in the development of plant-derived cannabinoid therapeutics and has a deep pipeline of additional clinical-stage cannabinoid product candidates targeting epilepsy (including an orphan pediatric epilepsy program), Type 2 diabetes, ulcerative colitis, glioma and schizophrenia. For further information, please visit http://www.gwpharm.com.
Cannabidiol (CBD) is one of at least 85 cannabinoids found in cannabis.It is a major constituent of the plant, second to tetrahydrocannabinol (THC), and represents up to 40% in its extracts. Compared with THC, cannabidiol is not psychoactive in healthy individuals, and is considered to have a wider scope of medical applications than THC, including to epilepsy, multiple sclerosis spasms, anxiety disorders, bipolar disorder,schizophrenia,nausea, convulsion and inflammation, as well as inhibiting cancer cell growth. There is some preclinical evidence from studies in animals that suggests CBD may modestly reduce the clearance of THC from the body by interfering with its metabolism.Cannabidiol has displayed sedative effects in animal tests. Other research indicates that CBD increases alertness. CBD has been shown to reduce growth of aggressive human breast cancer cells in vitro, and to reduce their invasiveness.

Amgen: Phase III Melanoma treatment, Talimogene Laherparepvec Improves Survival
 |
|
|---|---|
| Transmission electron micrograph of an unmodified herpes simplex virus |
Talimogene Laherparepvec
Amgen Presents Interim Overall Survival Data From Phase 3 Study Of Talimogene Laherparepvec In Patients With Metastatic Melanoma
T-VEC was engineered from herpes simplex 1 (HSV-1), a relatively innocuous virus that normally causes cold sores. A number of genetic modifications were made to the virus in order to:
- Attenuate the virus (so it can no longer cause herpes)
- Increase selectivity for cancer cells (so it destroys cancer cells while leaving healthy cells unharmed)
- Secrete the cytokine GM-CSF (a protein naturally secreted in the body to initiate an immune response)
T-VEC has a dual mechanism of action, destroying cancer both by directly attacking cancer cells and also by helping the immune system to recognize and destroy cancer cells. T-VEC is injected directly into a number of a patient’s tumors. The virus invades both cancerous and healthy cells, but it is unable to replicate in healthy cells and thus they remain unharmed. Inside a cancer cell, the virus is able to replicate, secreting GM-CSF in the process. Eventually overwhelmed, the cancer cell lyses (ruptures), destroying the cell and releasing new viruses, GM-CSF, and an array of tumor-specific antigens (pieces of the cancer cell that are small enough to be recognized by the immune system).
The GM-CSF attracts dendritic cells to the site. Dendritic cells are immune cells that process and present antigens to the immune system so that the immune system can then identify and destroy whatever produced the antigen. The dendritic cells pick up the tumor antigens, process them, and then present them on their surface to cytotoxic (killer) T cells. Now the T cells are essentially “programmed” to recognize the cancer as a threat. These T cells lead an immune response that seeks and destroys cancer cells throughout the body (eg, tumors and cancer cells that were not directly injected with T-VEC).

In this way, T-VEC has both a direct effect on injected tumors and a systemic effect throughout the entire body. Because the adaptive immune system “remembers” a target once it has been identified, there is high likelihood that the effect of an oncolytic virus like T-VEC will be durable (eg, prevent relapse). And it is for this reason that T-VEC does not need to be injected into every tumor, just a few in order to start the immune process.
Clinical efficacy in unresectable melanoma has been demonstrated in Phase II and Phase III clinical trials.
The Phase II clinical trial was published in the Journal of Clinical Oncology in 2009. 50 patients with advanced melanoma (most of whom had failed previous treatment) were treated with T-VEC. The overall response rate (patients with a complete or partial response per RECIST criteria) was 26% (16% complete responses, 10% partial responses). Another 4% of patients had a surgical complete response, and another 20% had stable disease for at least 3 months. On an extension protocol, 3 more patients achieved complete responses, and overall survival was 54% at 1 year and 52% at 2 years—demonstrating that responses to T-VEC are quite durable.
Consistent with other immunotherapies, some patients exhibited initial disease progression before responding to therapy because of the time it takes to generate the full immune response. Responses were seen in both injected and uninjected tumors (including those in visceral organs), demonstrating the systemic immunotherapeutic effect of T-VEC. Treatment was extremely well tolerated, with only Grade 1 or 2 drug-related side effects, the most common being mild flu-like symptoms.

|

|
Amgen announced the initial results of the Phase III OPTiM trial on Mar. 19, 2013. This global, randomized, open-label trial compared T-VEC with subcutaneously administered GM-CSF (2:1 randomization) in 430 patients with unresectable stage IIIB, IIIC or IV melanoma. The primary endpoint was durable response rate (DRR), defined as a complete or partial tumor response lasting at least 6 months and starting within 12 months of treatment.

T-VEC was proven to offer superior benefits in metastatic melanoma. DRR was achieved in 16% of patients receiving T-VEC compared with only 2% in the GM-CSF control group (P<.0001). The greatest benefit was seen in patient with stage IIIB or IIIC melanoma, with a 33% DRR vs 0% with GM-CSF. The objective response rate (any response) with T-VEC was 26%, with an impressive 11% of patients experiencing a complete response (complete disappearance of melanoma throughout the body). This demonstrated once again that T-VEC has a systemic immune effect that destroys distant, uninjected tumors. According to Financial Times one of the investigators involved questioned the ethics of the trial design, as the control arm received subcutaneous GM-CSF instead of standard care

A trend toward improved survival with T-VEC was observed in a pre-specified interim analysis of this endpoint, with the final survival data (event-driven) expected in late 2013. At the interim analysis, T-VEC was associated with a 21% reduced risk of death. The most common side effects with T-VEC were fatigue, chills, and fever. No serious side effect occurred in more than 3% of patients in either arm of the study.
The investigators concluded that “T-VEC represents a novel potential [treatment] option for melanoma with regional or distant metastases.” The success of T-VEC in the OPTiM trial represents the first Phase III proof of efficacy for a virus-based oncolytic immunotherapy.
Ziprasidone


Ziprasidone
Ziprasidone
CAS 146939-27-7
CAS Name: 5-[2-[4-(1,2-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one
Additional Names: 5-(2-(4-(1,2-benzisothiazol-3-yl)piperazinyl)ethyl)-6-chlorooxindole
Manufacturers’ Codes: CP-88059
Molecular Formula: C21H21ClN4OS
Molecular Weight: 412.94
Percent Composition: C 61.08%, H 5.13%, Cl 8.59%, N 13.57%, O 3.87%, S 7.77%
Hydrochloride monohydrate
CAS Registry Number: 138982-67-9; 122883-93-6 (anhydrous)
Manufacturers’ Codes: CP-88059-1
Trademarks: Geodon (Pfizer); Zeldox (Pfizer)
Molecular Formula: C21H21ClN4OS.HCl.H2O
Molecular Weight: 467.41
Percent Composition: C 53.96%, H 5.18%, Cl 15.17%, N 11.99%, O 6.85%, S 6.86%
Literature References: Prepn: D. J. M. Allen et al., EP 586191; eidem, US 5312925 (1993, 1994 both to Pfizer).
Properties: White to slightly pink powder. Also prepd as the hemihydrate, mp >300°.
Melting point: mp >300°
Therap-Cat: Antipsychotic.
Ziprasidone (marketed as Geodon, Zeldox by Pfizer) was the fifth atypical antipsychotic to gain approval (February 2001) in the United States. It is approved by the U.S. Food and Drug Administration (FDA) for the treatment of schizophrenia, and acute mania and mixed states associated withbipolar disorder. Its intramuscular injection form is approved for acute agitation in schizophrenic patients for whom treatment with just ziprasidone is appropriate.
Ziprasidone is also used off-label for depression, bipolar maintenance, mood disorders, anxiety, aggression, dementia, attention deficit hyperactivity disorder, obsessive compulsive disorder, autism, and post-traumatic stress disorder.

Ziprasidone synthesis: John A. Lowe, Arthur A. Nagel. Pfizer Inc. U.S. Patent 4,831,031 (1989).
The oral form of ziprasidone is the hydrochloride salt, ziprasidone hydrochloride. The intramuscular form, on the other hand, is the mesylate salt, ziprasidone mesylate trihydrate, and is provided as a lyophilized powder.
Ziprasidone, chemically named (5-[2-{4-(l,2-benzisothiazol-3-yl)piperizin- 0 1 -yl } ethyl] -6-chlorooxindole)hydrochloride hydrate, is a substituted benzisothiazolylpiperazine. The free base of ziprasidone has the following structure:

Ziprasidone and some of its uses are described by U.S. Patent Nos. 4,831,031 and 0 5,312,925.
Like clozapine and risperidone, ziprasidone is a highly potent and selective 5-HT2receptor and dopamine D2 receptor antagonist. Seeger, T.F. et al, J. Pharmacol. Exp. Ther.. 275(1): 101-1 13 (1995). Ziprasidone is characterized as an antipsychotic, but may also have anxiolytic and antidepressant effects due to its ability to inhibit serotonin and 5 noradrenaline reuptake. Davis, R. and Markham, A., CNS Drugs, 8(2):154-159 (1997). The therapeutic potential of ziprasidone may also be enhanced by its high affinity for the 5-HT1A, 5-HT1D, 5-HT2Creceptor subtypes. Seeger, T.F. et al, J. Pharmacol. Exp. Ther.. 275(1):101-113 (1995).
The metabolism of ziprasidone is complex. When administered orally to
30 healthy humans, the drug is extensively metabolized by at least four major pathways: 1) N- dealkylation of the ethyl side chain attached to the piperazinyl nitrogen; 2) oxidation at sulfur resulting in the formation of sulfoxide or sulfone; 3) reductive cleavage of the bensisothiazole moiety; and 4) hydration of the C=N bond and subsequent sulfur oxidation or N-dearylation of the benzisothiazole moiety. Prakash, C. et al, Drug Metab. Dispos.,
35 25(7):863-872 (1997). At least 12 human metabolites have been identified: ziprasidone sulfoxide (ZIP-SO); ziprasidone sulfone (ZIP-SO2); 3-(piperazine-l -yl)-l,2-benzisothiazole (BITP); BITP sulfoxide; BITP sulfone; 6-chloro-5-(2-piperazinJ-yl-ethyl)JJ-dihydro- indol-2-one; 6-chloro-5-(2- {4-[imino-(2-mercapto-phenyl)methyl]-piperazin- 1 -yl} ethyl)- 1 ,3-dihydro-indol-2-one; 6-chloro-5-(2- {4-[imino-(2-methylsulfanyl-phenyl)methyI]- piperazin-1-yl} ethyl)- l,3-dihydro-indol-2-one; S-methyl-dihydro-ziprasidone; S-methyl- dihydro-ziprasidone sulfoxide; dihydro-ziprasidone sulfoxide; and (6-chloro-2-oxo-2,3- dihydro-lH-indol-5-yl)acetic acid. Two metabolites, ZIP-SO and ZIP-SO2, both of which are formed by oxidation of the ziprasidone sulfur atom are discussed herein. These metabolites have the following structures:

Ziprasidone Sulfoxide (ZIP-SO)

Ziprasidone Sulfone (ZIP-SO2)
Both ZIP-SO and ZIP-SO2 are minor metabolites, and account for less than about 10% and less than about 3% of ziprasidone metabolites found in human urine, respectively. Prakash, C. et al, Drug Metab. Dispos.. 25(7):863-872 (1997). It has been reported that neither metabolite likely contributes to the antipsychotic activity of ziprasidone. Prakash, C. et al, Drug Metab. Dispos., 25(7):863-872 (1997). Indeed, it has been reported that ziprasidone metabolites in general are not active at the D2 and 5-HT2A receptor sites. Ereshefsky, L., JL Clin. Psvch.. 57(suppl. l l):12-25 (1996).
Ziprasidone offers a number of benefits, but unfortunately many adverse effects are associated with its administration. Examples of adverse affects of ziprasidone include, but are not limited to, nausea, somnolence, asthenia, dizziness, extra-pyramidal symptoms, akathisia, cardiovascular disturbances, male sexual dysfunction, and elevated serum liver enzyme levels. Davis, R. and Markham, A., CNS Drugs, 8(2): 154-159 (1997). These adverse effects can significantly limit the dose level, frequency, and duration of drug therapy. It is thus desirable to find a compound which possesses advantages of ziprasidone but fewer of its disadvantages.
3. SUMMARY OF THE INVENTION
This invention relates to novel methods using, and compositions comprising, ziprasidone metabolites, preferably, ziprasidone sulfoxide and ziprasidone sulfone. These metabolites, prior to the present invention, have been reported to have little or no in vivo activity. The present invention encompasses the in vivo use of these metabolites, and their incorporation into pharmaceutical compositions and single unit dosage forms useful in the treatment and prevention of disorders that are ameliorated by the inhibition of serotonin reuptake at 5-HT2receptors and/or the inhibition of dopamine reuptake at dopamine D2 receptors. Such disorders include psychotic and neuroleptic disorders. In a preferred embodiment, ziprasidone metabolites are used in the treatment or prevention of neuroleptic and related disorders in mammals, including humans.

-
Ziprasidone (5-(2-(4-(1,2-benzisothiazol-3-yl-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2-(1H)-indol-2-one) is a potent antipsychotic agent and is useful for treating various disorders including schizophrenia, anxiety and migraine pain. Ziprasidone has been approved by the FDA for treatment of schizophrenia and goes by the brand name Geodon™ in the United States. Ziprasidone has also been indicated as useful for treating Tourette’s Syndrome (United States Patent 6,127,373), glaucoma and ischemic retinopathy (EP 985414 A2), and psychiatric conditions including dementia of the Alzheimer’s type, bipolar disorders, mood disorders, panic disorders, agoraphobia, social phobia, panic disorder, post-traumatic stress disorder, acute stress disorder, substance-induced anxiety disorder, anxiety disorders not otherwise specified, dyskinesias and behavioral manifestations of mental retardation, conduct disorder, and autistic disorder (United States Patent 6,245,766).
-
United States Patent 4,831,031 describes a genus of compounds encompassing ziprasidone and the synthesis of such compounds. Another method for synthesizing ziprasidone is described in United States Patent 5,206,366. A method for specifically synthesizingziprasidone hydrochloride monohydrate is described in United States Patent 5,312,925. A method for synthesizing ziprasidone mesylate dihydrate is described in United States Patent 6,245,765; and a method for synthesizing ziprasidone mesylate trihydrate is described in United States Patent 6,110,918. United States Patents 5,338,846; 5,359,068; and 6,111,105 also describe methods for synthesizing ziprasidoneand/or intermediates therefore.
-
The structure of ziprasidone can be depicted as:

(H. Howard, et al., “Ziprasidone Hydrochloride”, Drugs of the Future1994, 19(6): 560-563. As can be seen from the structure above, the compound ziprasidone comprises a chlorine atom.
-
Methods of introducing halogens into organic compounds are summarized in many organic text books. For example, J. March,Advanced Organic Chemistry, 4th Edition, pp. 587-591, and references cited therein, has a discussion of halogenation chemistry. More specifically, formation of chloro-aromatic compounds are frequently formed by a variety of methods also well known to those skilled in the art, and again summarized in J. March, Advanced Organic Chemistry, 4th Edition, Chapter 11, “Aromatic Electrophilic Substitution”. The chemistry to add a halogen, or more specifically a chlorine, to an aromatic group is thus well known to those skilled in the art. It is also known that such chemistry usually results in some mixtures of molecules, one of which is commonly the unreacted starting material not containing the chlorine atom. Further, over-chlorination is a problem well known to those skilled in the art; it is common to form some dichloro-compound impurities when the mono-chloro is desired and some trichloro-compound impurities when the dichloro- is desired. Over-chlorination is typically controlled by limiting the amount of the chlorinating reagent used. Unfortunately, control of over-chlorinated analogs in the drug substance by limiting the amount of chlorinating reagent utilized in the introduction of the aromatic chlorine substituent would be expected to result in more of a des-chloro impurity (unreacted starting material not containing the chlorine atom).
-
-
6-chlorooxindole (6-chloro-1,3-dihydro-2H-indol-2-one).
-
Although there are many known routes to 6-chlorooxindole, starting materials therefore are typically a substituted 4-chlorotoluene or 1,4-dichloro-nitrobenzene (see, G. J. Quallich and P. M. Morrissey,Synthesis, 1993, 51-53; and references cited therein; and F. R. Busch and R. J. Shine, “Development of an Efficient Process to 6-Chlorooxindole”, presented at the 208th ACS National Meeting in Washington D.C. in the Symposium on Technical Achievements in Organic Chemistry, 1994, (talk #126).). However, the concept of controlling chlorinated isomers, over-chlorination, or des-chloro impurities for the synthesis of 6-chlorooxindole is not described in the prior art. Other methods of synthesizing 6-chlorooxindole can be determined by a person of ordinary skill in the art, and such methods are included in the step of obtaining a batch of 6-chlorooxindole for the above-described method of this invention. Furthermore, a batch of 6-chlorooxindole can be obtained by purchase from manufacturers of organic chemicals, for example Plaistow, Ltd., Little Island, County Cork, Ireland or Finorga, Route de Givors, 38670 Chasse-Sur-Rhone, France.
-
Ziprasidone has two major fragments, benzisothiazol and substituted oxindole. In from 2 – mercapto acid methyl ester ( 1 ), the alkaline conditions with hydroxylamine-O-sulfonic acid reaction ring closure under alkaline conditions to obtain 5 . 5 3 can also be prepared from the disulfide, disulfides 3 by three methods (anthranilic acid by diazotization pass sulfur dioxide gas, o-fluorinated thiol acid and two xenon reaction, or dibromoethoxychlorophosphonazo acid and sulfur in copper iodide reaction), 3 and chlorinated sulfoxide and sulfone chlorination reaction of 4 , 4and ammonia reaction again 5 . 5 by chlorination with phosphorus oxychloride, the reaction of piperazine 7 . 7 may be made of the compound 8 ( 8 can be from 2 – cyano bromobenzene After the i-PrMgCl, ZnBr 2 , S 2 Cl 2 prepared in one-pot reaction) was prepared in DMSO and directly in the hot reaction piperazine.
Oxindole fragment from 6 – chloro-indol-2 – one ( 10 ) starts, the FC acylation later reduction with triethylsilane 12 , 12 and 7 occurs in alkaline aqueous solution S N 2 reaction with hydrochloric acid salt to obtain ziprasidone hydrochloride.
United States Patent 5,206,366,



MORE INFO UPDATED
Ziprasidone is an antipsychotic agent with the following chemical name: 5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one of formula (I)
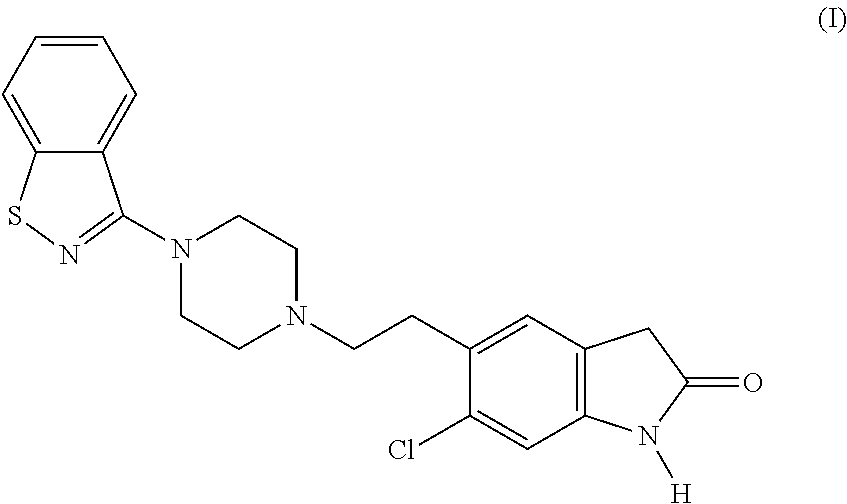
Ziprasidone is disclosed in U.S. Pat. Nos. 4,831,031 and 5,312,925 (assigned to Pfizer). Ziprasidone inhibits synaptic reuptake of serotonin and norepinephrine. No appreciable affinity was exhibited for other receptor/binding sites tested, including the cholinergic muscarinic receptor. The mechanism of action of ziprasidone, as with other drugs having efficacy in schizophrenia, is unknown. However, it has been proposed that this drug’s efficacy in schizophrenia is mediated through a combination of dopamine type 2 (D 2) and serotonin type 2 (5HT 2) antagonism.Ziprasidone’s antagonism of histamine H receptors may explain the somnolence observed with this drug.
U.S. Pat. No. 5,312,925 (Pfizer Inc.) describes a process for the synthesis of monohydrate of 5-(2-(4-(1,2-benzisothiazol-3-yl)piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride and its characterization based on IR, XRD and moisture content. The ‘925 patent also discloses that the hemihydrate may be obtained by the process described in Example 16 of U.S. Pat. No. 4,831,031 and its characterization by IR, XRD and moisture content. It also discloses the IR, XRD and moisture content of anhydrous Ziprasidone hydrochloride. According to the invention in the ‘925 patent, Ziprasidone of water content of 3.97, 2.55 and 0.37% were used for the IR and XRD study of Ziprasidone hydrochloride monohydrate, hemihydrate and anhydrous. In this invention, the monohydrate ofZiprasidone hydrochloride was prepared by reacting anhydrous 5-(2-(4-(1,2-benzisothiazol-3-yl)piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one with aqueous hydrochloric acid. The temperature range of the reaction was maintained between 60 to 65° C. and aqueous hydrochloride used for salt formation was around 0.7 M. Depending on the reaction temperature and other conditions, the reaction times were set around 3 to 24 hours. The final product thus obtained was dried carefully in monitored conditions to make certain that water content was from about 3.8% to about 4.5% to obtain the stable monohydrate.
U.S. Pat. No. 6,150,366, discloses a manufacturing process of ziprasidonehydrochloride monohydrate, comprises: 1) dissolving, ziprasidone free base in a solvent comprising THF and water, in a volume ratio of about 22-35 unit volumes of THF to about 1.5-8 volumes of water; 2) heating the solution resulting from step (1); 3) adding HCl to the solution resulting from step (2); and 4) cooling the solution resulting from step (3) and crystals collected by filtration and drying.
U.S. Pat. No. 5,206,366 and U.S. Pat. No. 5,338,846 describe a process for preparing ziprasidone by reacting 1-(1,2-benzisothiazol-3-yl) piperazine with 5-(2-chloroethyl)-6-chloro-oxindole in water with a neutralizing agent such as sodium carbonate under reflux.
J. Med. Chem. 1996, 39, 143-148 discloses preparation of ziprasidone by reacting 1-(1,2-benzisothiazol-3-yl)piperazine with 5-(2-bromoethyl)-6-chloro-oxindole in isoamyl alcohol solvent in the presence of sodium carbonate.
Some salts of ziprasidone, and in particular, its hydrochloride salt is a potent commercial antipsychotic agent useful in the treatment of various disorders, including schizophrenia and anxiety diseases. Ziprasidone hydrochloride is currently marketed under the proprietary name of Geodon. Other salts ofziprasidone are also reported to be effective for the treatment of the same type of diseases.
Some of the processes described in the aforementioned patents necessitate the use of ion-exchange catalyst (i.e. sodium iodide) and/or phase transfer catalysts (for example tetra butyl ammonium bromide or tetra butyl phosphoriium bromide) in order for the coupling reaction producing ziprasidone to take place. For example, U.S. Pat. No. 4,831,031 indicates that arylpiperazinyl-ethyl (or butyl)-heterocydic compounds may be prepared by reacting piperazines of the formula II with compounds of the formula III as follows in [Scheme 1]:

Wherein Hal is fluoro, chloro, bromo or iodo; and Ar, n, X and Y are as defined therein with reference to formula I. According to the ‘031 patent the coupling reaction is generally conducted in a polar solvent, such as a lower alcohol, dimethylformamide or methylisobutylketone, and in the presence of a weak base and that, preferably, the reaction is carried out in the presence of a catalytic amount of sodium iodide, hydrogen chloride and neutralizing agent such as sodium carbonate.
In some instances, the ziprasidone obtained was purified by column chromatography, thus making the process impractical for large-scale preparations. Another process uses potentially explosive gases such as hydrogen in the presence of catalysts, for example zinc, palladium on carbon, followed by acid treatment to carry out a reduction and cyclization of an intermediate, in order to obtain ziprasidone.
Despite various processes disclosed in the prior art for the preparation of ziprasidone and salts thereof, still there is a need for a good process for producing ziprasidone and pharmaceutically acceptable acid addition salts of ziprasidone thereof, in high purity. One of the major problems faced in the prior art is formation of sticky material and difficult stirrability of the reaction mass. This problem is especially acute in large scale manufacturing.
picked up from polish site…translation is machine, please bear for errors
The first stages are two simple reactions: reduction of Wolf-Kiżnera and Friedel-Crafts

The next step is to reduce the use of triethylsilane and trifluoroacetic acid [2], and then the coupling with a compound 5 to obtain the final product.

These reactions przyspożyły organic chemist problems, while the synthesis of compound5 was a challenge to Pfizer entourage coped in two ways:
a) method 1

a) method 1

b) method 2

In the second method, we have marked with an interesting transition.As you zoom scale but found that method 2 is the only possible one. Just destroy the product hydrochloride and get a clean API ready for tableting. [1] Bhugra D. The global prevalence of schizophrenia .. “PLoS Medicine”. 5 (2), pp. E151, 175 [2] Tetrahedron Letters “Selectivities in Ionic Reductions of Alcohols and Ketones with Triethyisilane / Trifluoroacetic Acid” 38, (6), 1997, pp. 1013-1016
1H NMR PREDICT

13C NMR PREDICT

http://www.google.com/patents/EP1476162A1?cl=en
Scheme 1
Stepl

MW = 167.59 MW = 244.08 Step 2

MW = 244.08 MW = 230.09 Step 3

MW = 230.09 MW = 255.76

MW = 412.94 Step 4

ziprasidone hydrochloride
MW = 412.94 monohydrate MW = 467.42
Scheme 2

Example 1 : Synthesis of Ziprasidone
Step 1 : Friedel-Crafts Acylation of 6-chloro-1 ,3-dihydro-2H-indol-2-one
Methylene chloride (310 L) and aluminum chloride (172.3 kg) were combined. Chloroacetyl chloride (66.7 kg) was added, and the resulting mixture was stirred for
45 minutes. 6-Chloro-1 ,3-dihydro-2/-/-indol-2-one (61.8 kg) was added. The reaction mixture was stirred at 28 to 32° C for 19.5 hours and then cooled to 15 to 20 C. Water (805 L) was cooled to 5 to 10 C. The reaction was quenched by the slow addition of the reaction mixture to the cold water. After the quench was complete, the mixture was heated to reflux, and the methylene chloride was removed by atmospheric distillation at 43 to 57° C. The resulting mixture was cooled to 15 to 20° C and stirred for 1 hour. The solids were isolated by filtration and washed with water (114 L) followed by methanol (114 L).- The solids were dried in a suitable dryer.
6-Chloro-5-(chloroacetyl)-1,3-dihydro-2/-/-indol-2-one, yield: 91.3 kg (101.4%). Note: A weight yield in excess of 100% resulted due to small amounts of residual salts which were removed in the following step.
The resulting 6-chloro-5-(chloroacetyl)-1,3-dihydro-2H-indol-2-one was carried through the following step in portions, one of which is detailed below.
Step 2: Trifluoroacetic Acid/Silane Reduction of 6-Chloro-5-(chloroacetyl)-1 ,3- dihydro-2H-indol-2-one
Trifluoroacetic acid (278 kg) and (74.2 kg) were combined and stirred slowly at 24 to 28° C. Triethylsilane (77.9 kg) was charged to the stirring mixture. The reaction temperature was allowed to exotherm slightly during this addition and was maintained between 50 to 62° C during the reaction period. , The reaction mixture was stirred for 8 hours, cooled to 38 C, and sampled for reaction completion. The reaction mixture was stirred at 50 to 54° C for an additional 3 hours. After the reaction was determined to be complete, the reaction mixture was cooled to 18° C, and quenched with water (594 L). The resulting slurry was stirred for 30 minutes at 10 to 15° C, and the solids were isolated by filtration. The product was rinsed from the tank and the product cake was washed with water (83 L) followed by methanol (76 L).
In each of two batches of equal size, tetrahydrofuran (742 L), Darco KB-B (1.9 kg), and the wet product cake were combined and heated to reflux. The resulting mixture was stirred at reflux for 30 minutes and filtered through a sparkler filter (pre- coated with filteraid) at 50 to 60° C to remove the carbon. The tank and sparkler were rinsed with hot tetrahydrofuran (38 L). Following the filtration the two batches were combined. The solution was concentrated in vacuo and stirred at 4 to 5° C for 1 hour. The solids were isolated by filtration and washed with cold tetrahydrofuran (38
L). The solids were dried in vacuo at 45 to 73° C until a loss on drying of 0.45% was achieved, giving 6-Chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one, yield: 60.1 kg (85.9%).
The resulting 6-chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one was combined with material of comparable quality and carried through the following step.
Step 3: Coupling of 6-Chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one and 3-(1-Piperazinyl)-1 ,2-benzisothiazole Monohydrochloride
Water (780 L) and sodium carbonate (126.0 kg) were combined and the mixture was stirred to dissolve. 3-(1-Piperazinyl)-1 ,2-benzisothiazole monohydrochloride (155.0 kg) and 6-chloro-5-(2-chloroethyl)-1 ,3-dihydro-2W-indol-2- one (150.4 kg) were added, and the reaction mixture was heated to reflux (-100° C). After 24 and 28 hours, the reaction slurry was sampled for reaction completion assay. The reaction was determined to be complete after the assay of the second sample. Water (1251 L) was added and the slurry was cooled to temperatures between 18 to
22° C. The solids were isolated by filtration and washed with water (302 L). The water wet solids were combined with isopropanol (940 L) and the resulting mixture was stirred for approximately 2 hours at ambient temperature. The solids were isolated by filtration, washed with isopropanol (89 L), and dried in vacuo at less than 43° C, giving 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3- dihydro-2H-indol-2-one, yield: 202.8 kg (80.8%).
The resulting 5-[2-[4-(2,3-benzisothiazol-3-yl)-1 -piperazinyl]ethyl]-6-chloro- 1 ,3-dihydro-2/- -indol-2-one was divided into two portions. These batches were carried separately through the following additional purification and resulted in material of comparable quality. The processing of one of these batches is detailed below.
Step 3R: Purification of 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]- 6-chloro-1 ,3-dihydro-2H-indol-2-one 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H- indol-2-one (51 kg), filteraid (4 kg) and tetrahydrofuran (2678 L) were combined. The mixture was heated to reflux (-65° C) for -1 hour, filtered while maintaining the temperature above 55° C, and rinsed with tetrahydrofuran (570 L). The product rich filtrate was partially concentrated in vacuo. 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1- piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H-indol-2-one (51 kg), filteraid (4 kg) and tetrahydrofuran (2675 L) were combined. The mixture was heated to reflux (-65° C) for -1 hour, filtered while maintaining the temperature above 55° C, and rinsed with tetrahydrofuran (560 L). The product rich filtrate was combined with the partially concentrated mixture above and concentrated in vacuo. The resulting mixture was cooled to 0 to 5° C. The solids were isolated by filtration, washed with filtered tetrahydrofuran (113 L), and dried in vacuo at less than 41° C, giving ziprasidone, free base, yield: 79.3 kg (77.7 %).
A portion of the batch was combined with material of comparable quality which had been recrystallized separately and the batch was carried through the following step.
Example 2: Crystallization Salt Formation of Ziprasidone Hydrochloride Monohydrate Tetrahydrofuran (2715 L), water (307 L), and 5-[2-[4-(2,3-benzisothiazol-3-yl)-
1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2 – -indol-2-one (100.0 kg) were combined, heated to reflux (- 64° C), and stirred for -30 minutes. The solution was filtered and rinsed with tetrahydrofuran (358 L).
Water (203 L) and concentrated hydrochloric acid (29 L) were combined and stirred at ambient temperature. The resulting aqueous hydrochloric acid solution was charged to the 5-[2-[4-(2,3-benzisothiazpl-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3- dihydro-2 – -indol-2-one solution over a period of 27 minutes. The reaction mixture was cooled .to temperatures between 1 and 5° C over a period of -2 hours. The mixture was stirred between 1 and 5 C for -10 hours. The solids were isolated by filtration, washed with cold tetrahydrofuran (358 L), and dried until a water content of
4.1% was obtained.
Ziprasidone Hydrochloride Monohydrate, yield: 108.6 kg (96.0 % weight yiplrl)
The solids were milled on a Bauermeister mill. Example 3: Purification of 6-Chloro-5-(2-chloroethyl)-1,3-dihydro-2H-indol-2- one To Remove 5-(2-Chloroethyl)-1,3-dihydro-2H-indol-2-one
A 100 mL round bottom flask equipped with a magnet stirrer and reflux condenser was charged with 4.0 g (17.4 mmoles) of 6-chloro-5-(2-chloroethyl)-1 ,3- dihydro-2H-indol-2-one (Compound 3) and 36 mL of acetonitriie and 4.0 mL of water were added. The slurry was gently heated and stirred overnight (-18 hrs at -78° C). The heating was then removed and the slurry cooled to 0 to 5° C, and stirred for an additional hour. The product was collected by filtration, washed with a small portion of acetonitriie and the product dried under vacuum at 50° C, to give 3.77 g (94.3% yield) of 6-chloro-5-(2-chloroethyl)-1,3-dihydro-2H-ιndol-2-one. The level of the des- chloro impurity had been reduced from 1280 ppm to 230 ppm.
Example 4: Experimental Determination of Purge Factor for Compound 6 (1,3-Dihydro-2/Y-indol-2one)
A batch of 6-chloro-1 ,3-dihydro-2W-indol-2-one which contained a very high content of 1 ,3-dihydro-2λ7-indol-2-one was selected. This was intentionally selected so that higher levels of the impurity would be easier to measure, and to determine the purge factor for this impurity. An additional reason for this strategy of starting with material which was very high in the impurity for purposes of determining the purge factor of the impurity was to avoid having the material purge to less than the limit of analytical detection during the synthesis; thus resulting in a zero value in the final product. Since the purge factor is a ratio, it is not meaningful to divide by a zero result. (The material with the high level of impurity was used for this experiment but was NOT subsequently used in any studies with human subjects.) A batch of 6- chloro-1 ,3-dihydro-2H-indol-2-one which contained 4000 ppm of jl ,3-dihydro-2 – – ιndol-2-one was processed through the standard synthetic process according to Examples 1 and 2 above.
Following the first two steps of the synthesis, the level of the corresponding des-chloro impurity was measured, using the method described. It was found that
1700 ppm of 5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one (Compound 8 of Scheme 2, above) was present in 6-chloro-5-(2-chloroethyl)-1,3-dihydro-2 – -indol-2-one (Compound 3 of Scheme 1 , above). The processing was continued to 5-[2-[4-(1 ,2)- benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H-indol-2-one hydrochloride monohydrate, where it was determined that 600 ppm of 5-[2-[4-(1,2)- benzisothiazol-3-yl)-1-piperazinyl]ethyl]-1 ,3-dihydro-2H-indol-2-one (Compound 9 of Scheme 2, above) was present.
Thus the purge factor through the entire synthesis for the des-chloro analogs was from 4000 ppm to 600 ppm, or approximately a 6-fold decrease. Minor run to run variations in processing can lead to small differences in the yield and quality of the materials produced. A 20% error in the reproducibility of the impurity formation, that is if 500 ppm in one run expecting between 400 and 600 ppm in other experiments, is then allowed for. In the case of the synthesis described in Examples 1 and 2, with 5 processing steps, the additive experimental error could result in as much as a 2-fold difference in the level of the impurity. Thus, for the purpose of setting the upper limit, where the drug is going to be used by human subjects a conservative 3-fold purge factor was utilized. Therefore, to insure that the product produced would not contain over 100 ppm of 5-[2-[4-(1 ,2)-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-1,3-dihydro-2H- indol-2-one (Compound 9), a limit of 300 ppm of 1 ,3-dihydro-2H-indol-2-one
(Compound 6) in 6-chloro-1 ,3-dihydro-2H-indol-2-one (Compound 1) was determined.
…………………………………………..

The condensation of 1-(1,2-benzoisothiazol-3-yl)piperazine (I) with 6-chloro-5-(2-chloroethyl)-2-indolinone (II) in refluxing water or refluxing methyl isobutyl ketone gives the target indolinone derivative.
AU 8812537; EP 0281309; JP 1988301861

Wolff-Kishner reduction of 6-chloroisatin (I) gives 6-chlorooxindole (II), which is treated with chloroacetyl chloride under Friedel-Crafts conditions to yield 5-chloroacetyl-6-chlorooxindole (III). The ketone (III) is reduced using triethylsilane in trifluoroacetic acid to produce 6-chloro-5-(2-chloroethyl)oxindole (IV). 1,2-Benzisothiazolin-3-one (V) is converted to 3-chloro-1,2-benzisothiazole (VI) using phosphorus oxychloride and is then condensed with piperazine to provide 1-(1,2-benzisothiazol-3-yl)piperazine (VII). Finally, intermediate (VII) is alkylated by compound (IV) in the presence of sodium carbonate in water and is converted to the salt with aqueous hydrochloric acid.
US 4831031

WO 9500510
The nitration of 2,5-dichlorotoluene (I) with HNO3 in H2SO4/AcOH gives 2,5-dichloro-4-methylnitrobenzene (II), which is treated with t-butoxybis(dimethylamino)methane (III) in refluxing THF to yield 2,5-dichloro-4-[2-(dimethylamino)vinyl]nitrobenzene (IV). The condensation of (IV) with 1-(1,2-benzoisothiazol-3-yl)piperazine (V) in AcOH affords the disubstituted piperazine (VI), whose double bond is reduced by means of NaBH(OAc)3 in dichloroethane/AcOH to provide the saturated compound (VII). The condensation of (VII) with dimethyl malonate (VIII) by means of KOH in NMP gives the alkylated malonic ester (IX), which is hydrolyzed and monodecarboxylated with refluxing 3N HCl to yield the phenylacetic acid (X). The esterification of (X) with SOCl2 and methanol affords the methyl ester (XI), which is finally cyclized to the target indolone by reduction of its nitro group with sodium hydrosulfite in refluxing THF/ethanol. Alternatively, compound (VII) can be condensed with methyl cyanacetate (XII) by means of KOH in NMP to give the alkylated cyanacetic ester (XIII), which is hydrolyzed with refluxing 3N HCl to afford the already reported phenylacetic acid (X).
……………………
J Label Compd Radiopharm 1994,34(2),117
The Friedel Crafts condensation of 6-chloroindolin-2-one (I) with 14C labeled 2-chloroacetyl chloride (II) by means of AlCl3 in CS2 gives 6-chloro-5-(2-chloroacetyl)indolin-2-one (III), which is reduced with trimethylsilane in TFA to yield the labeled chloroethyl derivative (IV). Finally, this compound is condensed with 3-(1-piperazinyl)-1,2-benzoisothiazole (V) by means of Na2CO3 in refluxing water to provide the target radiolabeled compound.

………………….

The bromination of 3-chloro-1,2-benzoisothiazole (I) with Br2 in AcOH using FeCl3 as catalyst gives a mixture of 3,5-dibromo-1,2-benzoisothiazole (II) and 3,7-dibromo-1,2-benzoisothiazole (III) that are separated by flash chromatography. The desired isomer (III) is condensed with piperazine (IV) in refluxing diglyme to yield 7-bromo-3-(1-piperazinyl)-1,2-benzoisothiazole (V), which is condensed with 6-chloro-5-(2-chloroethyl)indolin-2-one (VI) by means of Na2CO3 in refluxing water to afford the brominated adduct (VII). Finally, this compound is debrominated with tritium gas over a Pd/BaSO4 catalyst in THF to provide the target radiolabeled compoun
……………….
Org. Process Res. Dev., 2008, 12 (6), pp 1142–1145
DOI: 10.1021/op800105j

The current process for ziprasidone involves preparation and isolation of the key intermediate 6-chloro-5-(2-chloroethyl)oxindole. An improved process for the synthesis of this intermediate is reported here. The new process involves use of a novel Lewis acid-mediated selective deoxygenation of the precursor ketone with tetramethyldisiloxane. The new method affords the desired compound in a one-pot process obviating the need for isolation of the potentially hazardous precursor ketone. This process was successfully scaled up to multikilo scale.

A new, one-step commercial process for the preparation of 3-(1-piperazinyl)-1,2-benzisothiazole, a key intermediate in the synthesis of ziprasidone has been developed: The reaction of 2-cyanophenyl disulfide (I) with piperazine (II) by means of DMSO and isopropanol at 120-5 C.
…………………………..

J Med Chem 1996,39(1),143
A new synthesis for ziprasidone hydrochloride has been reported: The condensation of 6-chloroindolin-2-one (I) with bromoacetic acid (II) by means of polyphosphoric acid (PPA) gives 5-(bromoacetyl)-6-chloroindolin-2-one (III), which is reduced with triethylsilane and trifluoroacetic acid to the corresponding 2-bromoethyl derivative (IV). Finally, this compound is condensed with 4-(3-benzisothiazolyl)piperazine (V) by means of Na2CO3 in DMF or isobutyl methyl ketone.
……………………….
http://www.google.com/patents/WO2004050655A1?cl=en
REFERENCE EXAMPLE 1 Preparation of 5-(2-Chloro ethyl) -6-chloro oxindole
Triethylsilane (57.2 gm) was added slowly to the reaction mixture of 5-(2-
Chloro acetyl) -6-chloro oxindole (50.0 gm) and trifluoroacetic acid (175 mL) below the temperature of 45°C. The reaction was maintained at 40-45°C for 6 hours. The reaction mass was cooled to 0°C to -5°C and maintained stirring for 90 minutes. The separated solid was filtered and washed with water (50 mL). Then the wet compound was further slurred in water (250 mL) for 90 minutes. The resultant solid was filtered, washed with water (50 mL) and dried at a temperature of 70-75°C to afford 5-(2-chloroethyl) -6-chloro oxindole (43.5 gm).
REFERENCE EXAMPLE 2 Preparation of Ziprasidone base
Refluxed the reaction mixture of 5-(2-chloroethyl) -6-chloro oxindole (100 gm), 3-(l-piperazinyl)-l,2-benzisothiazole (104.7 gm), sodium carbonate (92.2 gm), sodium iodide (6.4 gm), terra butyl ammonium bromide (28 gm) and cyclohexane (1000 mL) till the reaction was completed. The reaction mass was cooled to a temperature of 30°C and the solid was filtered. To the wet compound was added water (1000 mL) and continued stirring for 45 minutes. The solid was filtered and washed with water (100 mL).
To the water wet compound was added acetone (500 mL) and there was stirring for 2 hours at room temperature. The compound was filtered and washed with acetone (200 mL) and dried at a temperature of 70-75°C to afford the Cmde Ziprasidone base (156.9 g) REFERENCE EXAMPLE 3
Preparation of Ziprasidone base
Charged 5-(2-chloroethyl) -6-chloro oxindole (50 gm), 3-(l-piperazinyl)-
1,2-benzisothiazole (47.5 gm) and cyclohexane (500 mL) into an autoclave. To this sodium carbonate (46 gm), sodium iodide (3.2 gm), terra butyl phosphonium bromide (14.8 gm) was added and the reaction was maintained at a temperature of 95-102°C and the pressure was kept at 2.5 kg/cm till the reaction was completed. The reaction mass was cooled to 30°C and water (250 mL) was added. The resulting compound was filtered and washed with water (100 mL). The wet compound was further slurred in water (500 mL), filtered and washed with water (100 mL). To the water wet compound was added acetone (500 mL) and was stirred at room temperature for 2 hours and 30 minutes. The solid was filtered, washed with acetone (100 L) and dried at a temperature of 60-65°C to afford the Ziprasidone base (65.7 gm).
EXAMPLE 1
Preparation of amorphous form of Ziprasidone hydrochloride Ziprasidone (5g) and 50 mL of acetic acid were placed into a round bottom flask and heated to 45 – 50°C. Added was 25 mL of aqueous hydrochloric acid slowly to the mixture over 20 min. Then the reaction mixture was refluxed. Water (10 mL) was added, followed by addition of 50 mL of Isopropanol. The reaction mass was cooled to 50°C and distilled off the solvent completely under vacuum. The material formed was scratched from the flask.
EXAMPLE 2
Preparation of crystalline form of Ziprasidone
Sodium carbonate (56.3 g) and 500 mL of water were placed into a round bottom flask. Added was 50 g of 3-(l-piperazinyl)-l,2-benzisothiazole hydrochloride and 50 g of 6-Chloro-5-(2-Chloroethyl) oxindole. The reaction mixture was then refluxed for
15 hours. The reaction completion was monitored by TLC. The reaction mass was cooled to room temperature. The resulting compound was filtered and washed with 50 mL of water. The wet compound and 250 mL of acetone were placed into a flask and the reaction mixture was sthred at room temperature for 2 hours. The reaction mixture was filtered to give a solid cake, which was washed with 50 mL of acetone. The wet cake and
750 mL of methanol were placed into a flask, which was heated to 50°C, and 14 mL of methane sulfonic acid was added to the solution over 20 minutes. The resulting reaction mass was cooled to room temperature and was subjected to a filtration to give a solid compound, which was washed with methanol. The wet compound and 750 mL of water were placed into a flask, and then pH of the solution was adjusted to pH 9 with caustic lye.
The reaction mixture was then stirred at room temperature for 1 hour and filtered. The filtered compound was washed with water and dried at 70°C to give 65 g of crystalline form Ziprasidone base.
……………………..
http://www.google.com/patents/US7087611
Ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride), I is a potent neuroleptic agent useful in the treatment of psychotic disorders, schizophrenia, and anxiety diseases. It is currently marketed under the proprietary name of Geodon.

Ziprasidone hydrochloride is known to exist in three crystalline forms; namely, the monohydrate, hemihydrate and anhydrous form as disclosed in U.S. Pat. Nos. 4,831,031 and 5,312,925, both of which are herein incorporated by reference. U.S. Pat. No. 5,312,925 states that ziprasidone hydrochloride monohydrate is substantially hygroscopically stable, thus alleviating potential problems due to weight changes of the active pharmaceutical ingredient during the final formulation process. Nevertheless a very low aqueous solubility is observed for this crystalline form.
U.S. Pat. No. 4,831,031 discloses that arylpiperazinyl-ethyl (or butyl)-heterocyclic compounds II may be prepared by reacting piperazines of the formula III with compounds of the formula IV as follows:

The ‘031 patent indicates that this coupling reaction is generally conducted in a polar solvent, such as a lower alcohol, dimethylformamide or methyl isobutyl ketone, and in the presence of a weak base and that, preferably, the reaction is in the further presence of a catalytic amount of sodium iodide, and a neutralizing agent for hydrochloride such as sodium carbonate. At example 16, the ‘031 patent discloses a process in which a solution of ziprasidone free base is taken up in dichloromethane and then reacted with ether saturated with HCl to afford a precipitate which is subsequently filtered, slurried with acetone and filtered again to give ziprasidone hydrochoride hemihydrate.
Additionally, the methods described in the prior art for the preparation of some of these crystalline forms, for instance ziprasidone hydrochloride anhydrate provide inconsistent reproducibility. For example, ziprasidone hydrochloride anhydrate has been prepared by prolonged drying in air at 50° C. of the corresponding monohydrate form, as disclosed in U.S. Pat. No. 5,312,925. However, repeated attempts to prepare the anhydrous form of ziprasidone hydrochloride in our laboratory by using the above mentioned conditions failed to produce the expected anhydrate and instead ziprasidone hydrochloride having variable contents of water, including that corresponding to the hemihydrate form, were obtained. Furthermore, even when more drastic conditions were used, (i.e., higher temperatures, longer drying times, under vacuum) anhydrous product was still not obtained.
EXAMPLE 3
Preparation of 5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1, 3-dihydro-2H-indol-2-one hydrochloride anhydrate.
To a 3-necked flask equipped with mechanical stirrer, thermometer and nitrogen inlet was added 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one free base (5.0 g) and 1-methyl-2-pyrrolidinone (60 mL) under nitrogen and the suspension was warmed up to 35–40° C. to dissolution. The flask was cooled to about 25° C. A 20.5% anhydrous solution of hydrogen chloride in isopropanol (6.45 g) was added and the mixture was stirred at about 25° C. for about 3 h. The product was collected by filtration on a Buchner funnel. The filter cake is rinsed twice with 10 mL of isopropanol at 20–25° C. and the damp cake transferred to a flask equipped with magnetic stirrer and nitrogen inlet. Isopropanol was added (30 mL) and the suspension stirred at 20–25° C. for about 2 h. The product was collected by filtration on a Buchner funnel. The filter cake is rinsed with 3×10 mL of isopropanol at 20–25° C. and transferred to a drying oven and dried in vacuo at 70–75° C. for 43 h. This afforded 4.52 g of anhydrous ziprasidone hydrochloride. The material contained 3.1% and 0.26% of residual NMP and IPA, respectively as determined by NMR.
………………………..
http://www.google.com/patents/WO2000059489A2?cl=en
5. EXAMPLES 5.1. EXAMPLE 1: SYNTHESIS OF ZIPRASIDONE
To a 125 mL round bottom flask equipped with an N2 inlet and condenser are added 0.73 g (3.2 mmol) 5-(2-chloroethyl)-6-chloro-oxindole, 0.70 g (3.2 mmol) N-(l,2- benzisothiazol-3-yl)piperazine, 0.68 g (6.4 mmol) sodium carbonate, 2 mg sodium iodide, and 30 mL methylisobutyl ketone. The reaction is refluxed for 40 hours, cooled, filtered, and evaporated. The residue is chromatographed on silica gel, eluting the by-products with ethyl acetate (1 L) and the product with 4 % methanol in ethyl acetate (1.5 L). The product fractions (Ry = 0.2 in 5 % methanol in ethyl acetate) are evaporated, taken up in methylene chloride, and precipitated by addition of ether saturated with HC1; the solid is filtered and washed with ether, dried, and washed with acetone. The latter is done by slurrying the solid with acetone and filtering. Ziprasidone is obtained as a high melting, non-hygroscopic solid product having an expected melting point of 288°C to 288.5°C.
……………………
Prepn: D. J. M. Allen et al., EP 586191; eidem, US 5312925 (1993, 1994 both to Pfizer).
Properties: White to slightly pink powder.
Combined serotonin (5HT2) and dopamine (D2) receptor antagonist. Prepn: J. A. Lowe III, A. A. Nagel, EP281309; eidem, US 4831031 (1988, 1989 both to Pfizer). Clinical pharmacology: C. J. Bench et al., Psychopharmacology 112, 308 (1993). HPLC determn in serum: J. S. Janiszewski et al., J. Chromatogr. B 668, 133 (1995). Receptor binding profile: A. W. Schmidt et al., Eur. J. Pharmacol. 425, 197 (2001). Review of pharmacology and clinical experience: G. L. Stimmel et al., Clin. Ther. 24, 21-37 (2002); P. D. Harvey, C. R. Bowie, Expert Opin. Pharmacother. 6, 337-346 (2005).
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4831031 * | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| US5312925 | Sep 1, 1992 | May 17, 1994 | Pfizer Inc. | Neuroleptic agents |
| US5338846 | Apr 20, 1993 | Aug 16, 1994 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds with a piperazine salt |
| US5935960 | Feb 7, 1997 | Aug 10, 1999 | Pfizer Inc. | Treatment of schizophrenia |
| US6150366 | May 27, 1999 | Nov 21, 2000 | Pfizer Inc. | Crystalline ziprasidone free base or crystalline ziprasidone hydrochloride particles with specific particle size are useful as antipsychosis agent |
| US20040152711 | Dec 4, 2003 | Aug 5, 2004 | Dr. Reddy’s Laboratories Limited | Crystal structure and amorphous form; psychological disorders |
| CA2166203A1 | Apr 6, 1994 | Jan 5, 1995 | Processes and intermediates for the preparation of 5-[2-(4-(benzoisothiazol-3-yl)-piperazin-1-yl)ethyl]- 6-chloro-1,3-dihydro-indol-2-one | |
| CA2245269A1 | Aug 7, 1998 | Feb 11, 1999 | Pfizer Prod Inc | Solid pharmaceutical dispersions with enhanced bioavailability |
| CA2252898A1 | Apr 10, 1997 | Nov 13, 1997 | Pfizer | Mesylate dihydrate salts of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one(=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO1995000510A1 | Apr 6, 1994 | Jan 5, 1995 | Pfizer | Processes and intermediates for the preparation of 5-[2-(4-(benzoisothiazol-3-yl)-piperazin-1-yl)ethyl]-6-chloro-1,3-dihydro-indol-2-one |
| WO2003070246A1 | Feb 17, 2003 | Aug 28, 2003 | Pfizer Prod Inc | Controlled synthesis of ziprasidone and compositions thereof |
| WO2004050655A1 | Dec 4, 2003 | Jun 17, 2004 | Akundi Surya Prabhakar | Polymorphic forms of ziprasidone and its hydrochloride |
| US7939662 | Dec 6, 2007 | May 10, 2011 | Apotex Pharmachem Inc. | Amorphous ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride) and processes to produce the same |
| US4831031 * | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 * | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| WO1997042190A1 * | Mar 26, 1997 | Nov 13, 1997 | Frank R Busch | Mesylate trihydrate salt of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one (=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO1997042191A1 * | Apr 10, 1997 | Nov 13, 1997 | Pfizer | Mesylate dihydrate salts of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one (=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO2003099198A2 * | May 26, 2003 | Dec 4, 2003 | Srinivasu Kilaru | A process for the preparation of oxindole derivatives |
| WO2004050655A1 * | Dec 4, 2003 | Jun 17, 2004 | Akundi Surya Prabhakar | Polymorphic forms of ziprasidone and its hydrochloride |
| US6110918 * | Mar 26, 1997 | Aug 29, 2000 | Pfizer Inc | Mesylate trihydrate salt of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihy dro-2(1H)-indol-2-one (=ziprasidone), its preparation and its use as dopamine D2 antagonist |
| US6245765 | Apr 10, 1997 | Jun 12, 2001 | Pfizer Inc | Psychological disorders |
| US7087611 | Aug 30, 2004 | Aug 8, 2006 | Apotex Pharmachem Inc. | Reacting ziprasidone with hydrochloric acid in solvent; salt formation; drying |
| US7488729 | Dec 4, 2003 | Feb 10, 2009 | Dr. Reddy’s Laboratories Limited | Polymorphic forms of ziprasidone and its hydrochloride salt and process for preparation thereof |
| US7790886 | Jan 5, 2009 | Sep 7, 2010 | Dr. Reddy’s Laboratories, Inc. | providing alcoholic solvent, desolventizing to form solid mass, isolating amorphous form; psychosis |
| US7939662 | Dec 6, 2007 | May 10, 2011 | Apotex Pharmachem Inc. | Amorphous ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride) and processes to produce the same |
| US8097610 | Aug 24, 2006 | Jan 17, 2012 | Shionogi & Co., Ltd. | Derivative having PPAR agonistic activity |
| US4831031 † | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 † | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| US5312925 † | Sep 1, 1992 | May 17, 1994 | Pfizer Inc. | Neuroleptic agents |
| US6111105 † | Oct 11, 1996 | Aug 29, 2000 | Pfizer, Inc. | Chemical intermediates for ziprasidone, a neuroleptic agent; reacting a 2-(piperidino) or (1,2-benzisoxazol-3-yl) or (2-cyanophenyl)thio),3-cyanobenzene with piperazine at a temperature from 80-170 degrees c. |
| US6245765 † | Apr 10, 1997 | Jun 12, 2001 | Pfizer Inc | Psychological disorders |
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review

Posaconazole, SCH 56592, Noxafil (Schering-Plough)
Posaconazole is a triazole antifungal drug that is used to treat invasive infections by Candida species and Aspergillus species in severely immunocompromised patients.
For prophylaxis of invasive Aspergillus and Candida infections in patients, 13 years of age and older, who are at high risk of developing these infections due to being severely immunocompromised as a result of procedures such as hematopoietic stem cell transplant (HSCT) recipients with graft-versus-host disease (GVHD), or due to hematologic malignancies with prolonged neutropenia from chemotherapy. Also for the treatment of oropharyngeal candidiasis, including oropharyngeal candidiasis refractory to itraconazole and/or fluconazole. Posaconazole is used as an alternative treatment for invasive aspergillosis, Fusarium infections, and zygomycosis in patients who are intolerant of, or whose disease is refractory to, other antifungals
Posaconazole is designated chemically as 4-[4-[4-[4-[[ (3R,5R)-5- (2,4-difluorophenyl)tetrahydro-5-(1H-1,2,4-triazol-1 -ylmethyl)-3-furanyl]methoxy]phenyl]-1 -piperazinyl]phenyl]-2-[ (1S,2S)-1 -ethyl-2- hydroxypropyl]-2,4-dihydro-3H-1,2,4-triazol-3-one with an empirical formula of C37H42F2N8O4 and a molecular weight of 700.8.
Posaconazole is used, for example, to prevent and/or treat invasive fungal infections caused by Candida species, Mucor species, Aspergillus species,Fusarium species, or Coccidioides species in immunocompromised patients and/or in patients where the disease is refractory to other antifungal agents such as amphothericin B, fluconazole, or itraconazole, and/or in patients who do not tolerate these antifungal agents.
CAS No. 171228-49-2
Posaconazole compounds have been described inU.S. Pat. Appl. No. 2003/0055067 for “Antifungal Composition with Enhanced Bioavailability,” U.S. Pat. Appl. No. 2004/0058974 for “Treating Fungal Infections,” and European Patent Publication1372394 (A1 ) for “Liquid Suspensions of Posaconazole (SCH 56592) with Enhanced Bioavailability for Treating Fungal Infections.”
| Synonyms: | Pcz;Pos;Noxafil;Sch 56592;Aids058495;Aids-058495;Posconazole;Posaconazole;Posaconazole for research;HYDROXYPROPYL]-2,4-DIHYDRO-3H-1,2,4-TRIAZOL-3-ONE |
| Molecular Formula: | C37H42F2N8O4 |
| Formula Weight: | 700.78 |
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review
Marketing Authorization Application also Filed with the European Medicines Agency
WHITEHOUSE STATION, N.J., Nov. 18, 2013–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that its New Drug Application for an investigational intravenous (IV) solution formulation of the company’s antifungal agent, NOXAFIL® (posaconazole), has been accepted for priority review by the U.S. Food and Drug Administration (FDA).http://www.pharmalive.com/mercks-noxafil-nda-gets-fda-priority-review
Posaconazole (CAS Registry Number 171228-49-2; CAS Name: 2,5-anhydro-1 ,3,4-trideoxy-2- C-(2,4-difluorophenyl)-4-[[4-[4-[4-[1-[(1S,2S)-1-ethyl-2-hydroxypropyl]-1 ,5-dihydro-5-oxo-4H- 1 ,2,4-triazol-4-yl]phenyl]-1-piperazinyl]phenoxy]methyl]-1-(1 H-1 ,2,4-triazol-1-yl)-D-threo-pentitol) which is represented by the following general formula (I)

(I)
is known as an antifungal agent. It is available as an oral suspension (40 mg/ml) under the trademark NOXAFIL® from Schering Corporation, Kenilworth, NJ. WO95/17407 and WO 96/38443 disclose the compound having the general formula (I) and its use in treating fungal infections. Various pharmaceutical compositions comprising posaconazole and being adapted for oral, topical or parenteral use are described e.g. in WO 02/80678, U.S. Patent No. 5,972,381 , U.S. Patent No. 5,834,472, U.S. Patent No. 4,957,730 and WO 2005/117831. As was mentioned above, WO 95/17407 and WO 96/38443 disclose the compound having the general formula (I). However, during prosecution of the subsequently filed European patent application no. 98951994.7, now European patent EP 1 021 439 B1 , the applicant declared that the methods disclosed in these publications only lead to the compound of formula (I) as an amorphous solid.
Polymorphism is a phenomenon relating to the occurrence of different crystal forms for one molecule. There may be several different crystalline forms for the same molecule with distinct crystal structures and distinct and varying physical properties like melting point, XRPD pattern, IR-spectrum and solubility profile. These polymorphs are thus distinct solid forms which share the molecular formula of the compound from which the crystals are made up, however, they may have distinct advantageous physical properties which can have a direct effect on the ability to process and/or manufacture the drug product, like flowability, as well as physical properties such as solubility, stability and dissolution properties which can have a direct effect on drug product stability, solubility, dissolution, and bioavailability.
Three polymorphic forms of posaconazole designated as forms I, Il and III are described and characterized in WO 99/18097 (US-B-6,713,481 , US-B-6,958,337). Crystalline forms Il and III were found to be unstable under the conditions investigated, so that crystalline form I was considered to be useful in the development of a pharmaceutical product.
A. K. Saksena et al., WO 9517407; eidem, US 5661151 (1995, 1997 both to Schering);
eidem, Tetrahedron Lett. 37, 5657 (1996).
SCH-56592, a novel orally active broad spectrum antifungal agent35th Intersci Conf Antimicrob Agents Chemother (Sept 17-20, San Francisco) 1995,Abst F61
seeSaksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Liu, Y.-T.; Ganguly, A.K.; Bennett, F. (Schering Corp.) EP 0736030; JP 1997500658; US 5661151; US 5703079; WO 9517407
Process for the preparation of triazolonesWO 9633178
Mono N-arylation of piperazine(III): Metal-catalyzed N-arylation and its application to the novel preparations of the antifungal posaconazole and its advanced intermediateTetrahedron Lett 2002,43(18),3359
Comparative antifungal spectrum: A. Cacciapuoti et al., Antimicrob. Agents Chemother. 44, 2017 (2000).
Pharmacokinetics, safety and tolerability: R. Courtney et al., ibid. 47, 2788 (2003).
HPLC determn in serum: H. Kim et al., J. Chromatogr. B 738, 93 (2000).
Review of development: A. K. Saksena et al. inAnti-Infectives: Recent Advances in Chemistry and Structure Activity Relationships (Royal Soc. Chem., Cambridge, 1997) pp 180-199; and clinical efficacy in fungal infections: R. Herbrecht, Int. J. Clin. Pract. 58, 612-624 (2004).
synthesis 1

……………..

Synthesis of intermediate (XX): The reaction of 2-chloro-2′,4′-difluoroacetophenone (I) with sodium acetate and NaI in DMF gives 2-acetoxy-2′,4′-difluoroacetophenone (II), which by methylenation with methyltriphenylphosphonium bromide and sodium bis(trimethylsilyl)amide in THF yields 2-(2,4-difluorophenyl)-2-propen-1-ol acetate ester (III). The hydrolysis of (III) with KOH in dioxane/water affords the corresponding alcohol (IV), which is regioselectively epoxidized with titanium tetraisopropoxide and L-(+)-diethyl tartrate in dichloromethane to (S)-(-)-2-(2,4-difluorophenyl)oxirane-2-methanol (V). The reaction of (V) with 1,2,4-triazole (VI) in DMF affords (R)-2-(2,4-difluorophenyl)-3-(1,2,4-triazol-1-yl)propane-1,2-diol (VII), which is selectively mesylated with methanesulfonyl chloride and triethylamine to the monomesylate (VIII). The cyclization of (VIII) with NaH in DMF gives the oxirane (IX), which is condensed with diethyl malonate (X) by means of NaH in DMSO to yield a mixture of (5R-cis)- and (5R-trans)-5-(2,4-difluorophenyl)-2-oxo-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-carboxylic acid ethyl ester (XI). The reduction of (XI) with NaBH4 and LiCl in ethanol affords (R)-4-(2,4-difluorophenyl)-2-(hydroxymethyl)-5-(1,2,4-triazol-1-yl) pentane-1,4-diol (XII), which is selectively tosylated with tosyl chloride and triethylamine in THF to the bistosylate (XIII). The cyclization of (XIII) by means of NaH in refluxing toluene gives (5R-cis)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-methanol tosylate ester (XIV). The reaction of (XIV) with 1-(4-hydroxyphenyl)-4-(4-nitrophenyl)piperazine (XV) to obtain compound (XVI), and the following reaction sequence (XVI) to (XVII) to (XVIII) to (XIX) to (5R-cis)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin-1-yl]phenyl-3,4-dihydro-2H-1,2,4-triazol-3-one (XX) has been performed according to J Med Chem 1984, 27: 894-900.
………………….pat approved expiry
| United States | 5661151 | 1999-07-19 | 2019-07-19 |
| Canada | 2305803 | 2009-12-22 | 2018-10-05 |
| Canada | 2179396 | 2001-04-17 | 2014-12-20 |
| United States | 5703079 | 1994-08-26 | 2014-08-26 |
MORE INFO
| US Patent No | Patent expiry | |
|---|---|---|
| 5661151 | Jul 19, 2019 | |
| 5703079 | Aug 26, 2014 | |
| 6958337 | Oct 5, 2018 | |
| 8263600 | Apr 1, 2022 |
- Cornely OA, Maertens J, Winston DJ, Perfect J, Ullmann AJ, Walsh TJ, Helfgott D, Holowiecki J, Stockelberg D, Goh YT, Petrini M, Hardalo C, Suresh R, Angulo-Gonzalez D: Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med. 2007 Jan 25;356(4):348-59. Pubmed
- Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S: Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N Engl J Med. 2007 Jan 25;356(4):335-47. Pubmed
- Bhattacharya M, Rajeshwari K, Dhingra B: Posaconazole. J Postgrad Med. 2010 Apr-Jun;56(2):163-7. Pubmed
- Frampton JE, Scott LJ: Posaconazole : a review of its use in the prophylaxis of invasive fungal infections. Drugs. 2008;68(7):993-1016.Pubmed
- Schiller DS, Fung HB: Posaconazole: an extended-spectrum triazole antifungal agent. Clin Ther. 2007 Sep;29(9):1862-86. Pubmed
- Kwon DS, Mylonakis E: Posaconazole: a new broad-spectrum antifungal agent. Expert Opin Pharmacother. 2007 Jun;8(8):1167-78.Pubmed
- Groll AH, Walsh TJ: Posaconazole: clinical pharmacology and potential for management of fungal infections. Expert Rev Anti Infect Ther. 2005 Aug;3(4):467-87. Pubmed
- Rachwalski EJ, Wieczorkiewicz JT, Scheetz MH: Posaconazole: an oral triazole with an extended spectrum of activity. Ann Pharmacother. 2008 Oct;42(10):1429-38. Epub 2008 Aug 19. Pubmed
- Li Y, Theuretzbacher U, Clancy CJ, Nguyen MH, Derendorf H: Pharmacokinetic/pharmacodynamic profile of posaconazole. Clin Pharmacokinet. 2010 Jun;49(6):379-96. doi: 10.2165/11319340-000000000-00000. Pubmed
FDA panel backs Vanda body clock drug Tasimelteon for blind

Tasimelteon
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide, 609799-22-6 cas
As expected, advisors to the US Food and Drug Administration have recommended approval of Vanda Pharmaceuticals’ tasimelteon, to be sold as Hetlioz, for the treatment of non-24-hour disorder in the totally blind.http://www.pharmatimes.com/Article/13-11-14/FDA_panel_backs_Vanda_body_clock_drug_for_blind.aspx
Tasimelteon (BMS-214,778) is a drug which is under development for the treatment of insomnia and other sleep disorders.[1] It is a selective agonistfor the melatonin receptors MT1 and MT2 in the suprachiasmatic nucleus of the brain, similar to older drugs such as ramelteon.[2] It has been through Phase III trials successfully and was shown to improve both onset and maintenance of sleep, with few side effects.[3]
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
A drug being developed to treat transient insomnia in circadian rhythm sleep disorders (eg jet-lag. The drug appears to be effective in the dose range of 20 to 100mg with an advance in the melatonin rhythm of 2-3 hours with the higher dose
- ‘Time-bending drug’ for jet lag. BBC News. 2 December 2008
- Vachharajani, Nimish N., Yeleswaram, Krishnaswamy, Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences 92 (4): 760–72. doi:10.1002/jps.10348. PMID 12661062.
- Shantha MW Rajaratnam, Mihael H Polymeropoulos, Dennis M Fisher, Thomas Roth, Christin Scott, Gunther Birznieks, Elizabeth B Klerman (2009-02-07). “Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials”. The Lancet373 (9662): 482–491. doi:10.1016/S0140-6736(08)61812-7. PMID 19054552. Retrieved 2010-02-23.
- Audio interview with Joseph Hull of Harvard, spring 2011
- Vanda Pharmaceuticals seeks FDA approval
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the
suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of Cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands. This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder
(N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non- 24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping.
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary.
In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime. Tasimelteon
Tasimelteon is a circadian regulator which binds specifically to two high affinity melatonin receptors, Mella (MT1R) and Mellb (MT2R). These receptors are found in high density in the suprachiasmatic nucleus of the brain (SCN), which is responsible for synchronizing our sleep/wake cycle. Tasimelteon has been shown to improve sleep parameters in prior clinical studies, which simulated a desynchronization of the circadian clock. Tasimelteon has so far been studied in hundreds of individuals and has shown a good tolerability profile.
Tasimelteon has the chemical name: tr ns-N-[[2-(2,3-dihydrobenzofuran- 4-yl)cycloprop-lyl] methyl] propanamide, has the structure of Formula I:

Formula I
and is disclosed in US 5856529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78°C (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG- 400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MTIR. It’s affinity (¾) for MTIR is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
Metabolites of tasimelteon include, for example, those described in “Preclinical Pharmacokinetics and Metabolism of BMS-214778, a Novel
Melatonin Receptor Agonist” by Vachharajani et al., J. Pharmaceutical Sci., 92(4):760-772, which is hereby incorporated herein by reference. The active metabolites of tasimelteon can also be used in the method of this invention, as can pharmaceutically acceptable salts of tasimelteon or of its active metabolites. For example, in addition to metabolites of Formula II and III, above, metabolites of tasimelteon also include the monohydroxylated analogs M13 of Formula IV, M12 of Formula V, and M14 of Formula VI.
Formula IV

Formula V
MO

Formula VI
Thus, it is apparent that this invention contemplates entrainment of patients suffering free running circadian rhythm to a 24 hour circadian rhythm by administration of a circadian rhythm regulator (i.e., circadian rhythm modifier) capable of phase advancing and/or entraining circadian rhythms, such as a melatonin agonist like tasimelteon or an active metabolite oftasimelteon or a pharmaceutically acceptable salt thereof. Other MT1R and MT2R agonists, i.e., melatonin agonists, can have similar effects on the master body clock. So, for example, this invention further contemplates the use of melatonin agonists such as but not limited to melatonin, N-[l-(2,3-dihydrobenzofuran-4- yl)pyrrolidin-3-yl]-N-ethylurea and structurally related compounds as disclosed in US 6,211,225, LY-156735 ((R)-N-(2-(6-chloro-5-methoxy-lH-indol- 3yl) propyl) acetamide) (disclosed in U.S. Patent No. 4,997,845), agomelatine (N- [2-(7-methoxy-l-naphthyl)ethyl]acetamide) (disclosed in U.S. Patent No.
5,225,442), ramelteon ((S)-N-[2-(l,6,7,8-tetrahydro-2H-indeno- [5,4-b] furan-8- yl)ethyl]propionamide), 2-phenylmelatonin, 8-M-PDOT, 2-iodomelatonin, and 6- chloromelatonin.
Additional melatonin agonists include, without limitation, those listed in U.S. Patent Application Publication No. 20050164987, which is incorporated herein by reference, specifically: TAK-375 (see Kato, K. et al. Int. J.
Neuropsychopharmacol. 2000, 3 (Suppl. 1): Abst P.03.130; see also abstracts P.03.125 and P.03.127), CGP 52608 (l-(3-allyl-4-oxothiazolidine-2-ylidene)-4- met- hylthiosemicarbazone) (See Missbach et al., J. Biol. Chem. 1996, 271, 13515-22), GR196429 (N-[2-[2,3,7,8-tetrahydro-lH-fur-o(2,3-g)indol-l- yl] ethyl] acetamide) (see Beresford et al., J. Pharmacol. Exp. Ther. 1998, 285, 1239-1245), S20242 (N-[2-(7-methoxy napth-l-yl) ethyl] propionamide) (see Depres-Brummer et al., Eur. J. Pharmacol. 1998, 347, 57-66), S-23478 (see Neuropharmacology July 2000), S24268 (see Naunyn Schmiedebergs Arch. June 2003), S25150 (see Naunyn Schmiedebergs Arch. June 2003), GW-290569, luzindole (2-benzyl-N-acetyltryptamine) (see U.S. Patent No. 5,093,352), GR135531 (5-methoxycarbonylamino-N-acetyltrypt- amine) (see U.S. Patent Application Publication No. 20010047016), Melatonin Research Compound A, Melatonin Agonist A (see IMSWorld R&D Focus August 2002), Melatonin
Analogue B (see Pharmaprojects August 1998), Melatonin Agonist C (see Chem. Pharm. Bull. (Tokyo) January 2002), Melatonin Agonist D (see J. Pineal Research November 2000), Melatonin Agonist E (see Chem. Pharm. Bull. (Tokyo) Febrary 2002), Melatonin Agonist F (see Reprod. Nutr. Dev. May 1999), Melatonin Agonist G (see J. Med. Chem. October 1993), Melatonin Agonist H (see Famaco March 2000), Melatonin Agonist I (see J. Med. Chem. March 2000), Melatonin Analog J (see Bioorg. Med. Chem. Lett. March 2003), Melatonin Analog K (see MedAd News September 2001), Melatonin Analog L, AH-001 (2-acetamido-8- methoxytetralin) (see U.S. Patent No. 5,151,446), GG-012 (4-methoxy-2- (methylene propylamide)indan) (see Drijfhout et al., Eur. J. Pharmacol. 1999, 382, 157-66), Enol-3-IPA, ML-23 (N-2,4-dinitrophenyl-5-methoxy-tryptamine ) (see U.S. Patent No. 4,880,826), SL-18.1616, IP-100-9 (US 5580878), Sleep Inducing Peptide A, AH-017 (see U.S. Patent No. 5,151,446), AH-002 (8-methoxy- 2-propionamido-tetralin) (see U.S. Patent No. 5,151,446), and IP-101.
Metabolites, prodrugs, stereoisomers, polymorphs, hydrates, solvates, and salts of the above compounds that are directly or indirectly active can, of course, also be used in the practice of this invention.
Melatonin agonists with a MT1R and MT2R binding profile similar to that of tasimelteon, which has 2 to 4 time greater specificity for MT2R, are preferred.
Tasimelteon can be synthesized by procedures known in the art. The preparation of a 4-vinyl-2,3-dihydrobenzofuran cyclopropyl intermediate can be carried out as described in US7754902, which is incorporated herein by reference as though fully set forth.
Pro-drugs, e.g., esters, and pharmaceutically acceptable salts can be prepared by exercise of routine skill in the art.
In patients suffering a Non-24, the melatonin and Cortisol circadian rhythms and the natural day/night cycle become desynchronized. For example, in patients suffering from a free-running circadian rhythm, melatonin and Cortisol acrophases occur more than 24 hours, e.g., >24.1 hours, prior to each previous day’s melatonin and Cortisol acrophase, respectively, resulting in desynchronization for days, weeks, or even months, depending upon the length of a patient’s circadian rhythm, before the melatonin, Cortisol, and day /night cycles are again temporarily synchronized.
Chronic misalignment of Cortisol has been associated with metabolic, cardiac, cognitive, neurologic, neoplastic, and hormonal disorders. Such disorders include, e.g., obesity, depression, neurological impairments.

SAR





Figure : Melatonin receptor agonists. The applied colors indicate the mutual properties with the general melatonin receptor agonists pharmacophore.

WASHINGTON, June 5, 2013 /PRNewswire/ — Vanda Pharmaceuticals Inc. (Vanda) presented additional entrainment and patient-level clinical data at SLEEP 2013, the 27th Annual Meeting of Associated Professional Sleep Societies in Baltimore, from its SET (Safety and Efficacy of Tasimelteon) and RESET (Randomized-withdrawal study of the Efficacy and Safety of Tasimelteon to treat Non-24-Hour Disorder) Phase III studies of tasimelteon, a circadian regulator for the treatment of Non-24-Hour Disorder (Non-24) in totally blind individuals. Non-24 is a serious, rare and chronic circadian rhythm disorder that affects a majority of totally blind individuals who lack light perception and cannot entrain (synchronize) their master body clock to the 24-hour day. Currently there is no approved FDA treatment for Non-24.
In the SET study, tasimelteon achieved the primary endpoints of entrainment (synchronizing) of the melatonin (aMT6s) rhythm as compared to placebo and clinical response as measured by entrainment plus a score of greater than or equal to 3 on the Non-24 Clinical Response Scale (N24CRS). Tasimelteon also demonstrated significant improvement versus placebo across a number of sleep and wake parameters including measures of total sleep time, nap duration, and timing of sleep, as well as in the Clinical Global Impression of Change (CGI-C), an overall global functioning scale. In treated patients, daytime naps decreased by 46 minutes per day in the worst 25% of days in a cycle and nighttime sleep increased by 57 minutes per day during the worst 25% of nights in a cycle.
The RESET study demonstrated that continued treatment with 20mg of tasimelteon was required to maintain entrainment of melatonin and cortisol circadian rhythms in individuals with Non-24. Patients treated with tasimelteon maintained their clinical benefits while patients who received placebo showed significant deterioration in measures of nighttime sleep, daytime naps and timing of sleep. Furthermore, discontinuation of tasimelteon resulted in a rapid relapse of circadian entrainment and a return to misaligned circadian rhythms, reinforcing the importance of chronic therapy.
Study investigator, Steven W. Lockley, Ph.D., Associate Professor of Medicine, Division of Sleep Medicine, Brigham and Women’s Hospital, Harvard Medical School, commented, “the results clearly demonstrate that tasimelteon can entrain the circadian clock, and that continued treatment is necessary to maintain entrainment.”
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
UPDATED ON JAN 2014
TASIMELTION, an orphan drug for non24
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide
(1R-trans)-N-[[2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]pro- pananamide VEC162
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
N-(((1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl)methyl)propanamide
Bristol-Myers Squibb Company
PRODUCT PATENT
U.S. Pat. No. 5,856,529
| CAS number | 609799-22-6 |
|---|
| Formula | C15H19NO2 |
|---|---|
| Mol. mass | 245.3 g/mol |
January 31, 2014 — The U.S. Food and Drug Administration today approved Hetlioz (tasimelteon), a melatonin receptor agonist, to treat non-24- hour sleep-wake disorder (“non-24”) in totally blind individuals. Non-24 is a chronic circadian rhythm (body clock) disorder in the blind that causes problems with the timing of sleep. This is the first FDA approval of a treatment for the disorder.
Non-24 occurs in persons who are completely blind. Light does not enter their eyes and they cannot synchronize their body clock to the 24-hour light-dark cycle.
VEC-162, BMS-214778, 609799-22-6, Hetlioz, Tasimelteon (USAN/INN), Tasimelteon [USAN:INN], UNII-SHS4PU80D9,
Tasimelteon
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
SEQUENCE
Discovered by Bristol-Myers Squibb (BMS) and co-developed with Vanda Pharmaceuticals, tasimelteon is a hypnotic family benzofuran. In Phase III development, it has an orphan drug status.
JAN2014.. APPROVED FDA
In mid-November 2013 the FDA announced their recommendation for the approval of Tasimelteon for the treatment of non-24-disorder.Tasimelteon effectively resets the circadian rhythm, helping to restore normal sleep patterns.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM374388.pdf
January 2010: FDA granted orphan drug tasimelteon to disturbed sleep / wake in blind without light perception.
February 2008: Vanda has completed enrollment in its Phase III trial in chronic primary insomnia.
June 2007: Results of a Phase III trial for transient insomnia tasimelteon presented by Vanda at the 21st annual meeting of the Associated Professional Sleep Societies. These results demonstrated improvements in objective and subjective measures of sleep and its maintenance.
2004 Vanda gets a license tasimelteon (or BMS-214778 and VEC-162) from Bristol-Myers Squibb.
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
Previously, BMS-214778, identified as an agonist of melatonin receptors, has been the subject of pre-clinical studies for the treatment of sleep disorders resulting from a disturbance of circadian rhythms.The first Pharmacokinetic studies were performed in rats and monkeys.
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands.
This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non-24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping. TASIMELTION
TASIMELTION
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24 h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24 h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary. In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime.
INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
………………………..
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC

PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
…………………….
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58.
References
- ‘Time-bending drug’ for jet lag. BBC News. 2 December 2008
- Vachharajani, Nimish N., Yeleswaram, Krishnaswamy, Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences 92 (4): 760–72. doi:10.1002/jps.10348. PMID 12661062.
- Shantha MW Rajaratnam, Mihael H Polymeropoulos, Dennis M Fisher, Thomas Roth, Christin Scott, Gunther Birznieks, Elizabeth B Klerman (2009-02-07). “Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials”. The Lancet 373 (9662): 482–491. doi:10.1016/S0140-6736(08)61812-7. PMID 19054552. Retrieved 2010-02-23.
- Audio interview with Joseph Hull of Harvard, spring 2011
- Vanda Pharmaceuticals seeks FDA approval
- Recent progress in the development of agonists and antagonists for melatonin receptors.Zlotos DP.
Curr Med Chem. 2012;19(21):3532-49. Review.
7 Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist.
Vachharajani NN, Yeleswaram K, Boulton DW.J Pharm Sci. 2003 Apr;92(4):760-72.
 TASIMELTION
TASIMELTION
PATENTS
| US2010261786 | 10-15-2010 | PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE |
| US2009209638 | 8-21-2009 | TREATMENT FOR DEPRESSIVE DISORDERS |
| US6060506 | 5-10-2000 | Benzopyran derivatives as melatonergic agents |
| US5981571 | 11-10-1999 | Benzodioxa alkylene ethers as melatonergic agents |
| WO9825606 | 6-19-1998 | BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS |
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
Org. Synth. 1990, 69, 154
(−)-D-2,10-CAMPHORSULTAM
[3H-3a,6-Methano-2,1-benzisothiazole, 4,5,6,7-tetrahydro-8,8-dimethyl-2,2-dioxide, (3aS)-]
|
Submitted by Michael C. Weismiller, James C. Towson, and Franklin A. Davis1.
Checked by David I. Magee and Robert K. Boeckman, Jr..
1. Procedure
(−)-2,10-Camphorsultam. A dry, 2-L, three-necked, round-bottomed flask is equipped with a 1.5-in egg-shaped Teflon stirring bar, a 250-mL addition funnel, and a 300-mL Soxhlet extraction apparatus equipped with a mineral oil bubbler connected to an inert-gas source. The flask is charged with 600 mL of dry tetrahydrofuran (THF) (Note 1) and6.2 g (0.16 mol) of lithium aluminum hydride (Note 2). Into the 50-mL Soxhlet extraction thimble is placed 35.0 g (0.16 mol) of (−)-(camphorsulfonyl)imine (Note 3) and the reaction mixture is stirred and heated at reflux. After all of the(camphorsulfonyl)imine has been siphoned into the reaction flask (3–4 hr), the mixture is allowed to cool to room temperature. The unreacted lithium aluminum hydride is cautiously hydrolyzed by dropwise addition of 200 mL of 1 Nhydrochloric acid via the addition funnel (Note 4). After the hydrolysis is complete the contents of the flask are transferred to a 1-L separatory funnel, the lower, silver-colored aqueous layer is separated, and the upper layer placed in a 1-L Erlenmeyer flask. The aqueous phase is returned to the separatory funnel and washed with methylene chloride (3 × 100 mL). After the reaction flask is rinsed with methylene chloride (50 mL), the organic washings are combined with the THF phase and dried over anhydrous magnesium sulfate for 10–15 min. Filtration through a 300-mL sintered-glass funnel of coarse porosity into a 1-L round-bottomed flask followed by removal of the solvent on arotary evaporator gives 33.5 g (95%) of the crude (−)-2,10-camphorsultam. The crude sultam is placed in a 250-mL Erlenmeyer flask and crystallized from approximately 60 mL of absolute ethanol. The product is collected on a 150-mL sintered-glass funnel of coarse porosity and dried in a vacuum desiccator to give 31.1 g (88%) of the pure sultam. A second crop of crystals can be gained by evaporating approximately half the filtrate; the residue is crystallized as above to give 1.4 g (4%). The combined yield of white crystalline solid, mp 183–184°C, [α]D −30.7° (CHCl3, c 2.3) is92% (Note 5) and (Note 6).
2. Notes
1. Tetrahydrofuran (Aldrich Chemical Company, Inc.) was distilled from sodium benzophenone.
2. Lithium aluminum hydride was purchased from Aldrich Chemical Company, Inc.
3. (−)-(Camphorsulfonyl)imine, [(7S)-(−)-10,10-dimethyl-5-thia-4-azatricyclo[5.2.1.03,7]dec-3-ene 5,5-dioxide] was prepared by the procedure of Towson, Weismiller, Lal, Sheppard, and Davis, Org. Synth., Coll. Vol. VIII, 1993, 104.
4. The addition must be very slow at first (1 drop/5 sec) until the vigorous reaction has subsided.
5. The NMR spectrum of (−)-2,10-camphorsultam is as follows: 1H NMR (CDCl3) δ: 0.94 (s, 3 H, CH3), 1.14 (s, 3 H, CH3), 1.33 (m, 1 H), 1.47 (m,, 1 H), 1.80–2.05 (5 H), 3.09 (d, 1 H, J = 14), 3.14 (d, 1 H, J = 14), 3.43 (m, 1 H), 4.05 (br s, 1 H, NH); 13C NMR (CDCl3) δ: 20.17 (q, CH3), 26.51 (t), 31.55 (t), 35.72 (t), 44.44 (d), 47.15 (s), 50.08 (t), 54.46 (s), 62.48 (d).
6. Checkers obtained material having the same mp (183–184°C) and [α]D − 31.8° (CHCl3, c 2.3).
3. Discussion
(−)-2,10-Camphorsultam was first prepared by the catalytic hydrogenation of (−)-(camphorsulfonyl)imine overRaney nickel.2 Lithium aluminum hydride reduction was used by Oppolzer and co-workers in their synthesis of the sultam.3,4 However, because of the low solubility of the sultam in tetrahydrofuran, a large amount of solvent was required.4 In the procedure described here the amount of solvent is significantly reduced by using a Soxhlet extractor to convey the imine slowly into the reducing medium.5
Oppolzer’s chiral auxiliary,6 (−)-2,10-camphorsultam, is useful in the asymmetric Diels–Alder reaction,3,4 and for the preparation of enantiomerically pure β-substituted carboxylic acids7 and diols,8 in the stereoselective synthesis of Δ2-isoxazolines,9 and in the preparation of N-fluoro-(−)-2,10-camphorsultam, an enantioselective fluorinating reagent.10
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Shriner, R. L.; Shotton, J. A.; Sutherland, H. J. Am. Chem. Soc. 1938, 60, 2794.
- Oppolzer, W.; Chapuis, C.; Bernardinelli, G. Helv. Chim. Acta 1984, 67, 1397.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, G.; Carroll,, P. J. J. Am. Chem. Soc. 1988, 110, 8477.
- Oppolzer, W. Tetrahedron 1987, 43, 1969.
- Oppolzer, W.; Mills, R. J.; Pachinger, W.; Stevenson, T. Helv. Chim. Acta 1986, 69, 1542; Oppolzer, W.; Schneider, P. Helv. Chim. Acta 1986, 69, 1817; Oppolzer, W.; Mills, R. J.; Réglier, M. Tetrahedron Lett. 1986, 27, 183; Oppolzer, W.; Poli. G.Tetrahedron Lett. 1986, 27, 4717; Oppolzer, W.; Poli, G.; Starkemann, C.; Bernardinelli, G. Tetrahedron Lett. 1988, 29, 3559.
- Oppolzer, W.; Barras, J-P. Helv. Chim. Acta 1987, 70, 1666.
- Curran, D. P.; Kim, B. H.; Daugherty, J.; Heffner, T. A. Tetrahedron Lett. 1988, 29, 3555.
- Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087.
Org. Synth. 1990, 69, 158
(+)-(2R,8aS)-10-(CAMPHORYLSULFONYL)OXAZIRIDINE
[4H-4A,7-Methanooxazirino[3,2-i][2,1]benzisothiazole, tetrahydro-9,9-dimethyl-, 3,3-dioxide, [4aS-(4aα,7α,8aR*)]]
 |
Submitted by James C. Towson, Michael C. Weismiller, G. Sankar Lal, Aurelia C. Sheppard, Anil Kumar, and Franklin A. Davis1.
Checked by David I. Magee and Robert K. Boeckman, Jr..
1. Procedure
A. (+)-(1S)-10-Camphorsulfonamide. Into a 2-L, two-necked, round-bottomed flask, equipped with a 250-mL dropping funnel, a magnetic stirring bar, and a reflux condenser fitted with an outlet connected to a disposable pipettedipped in 2 mL of chloroform in a test tube for monitoring gas evolution, were placed 116 g (0.5 mol) ofcamphorsulfonic acid (Note 1) and 750 mL of reagent-grade chloroform. The suspension of camphorsulfonic acid was heated to reflux and 71.4 g (43.77 mL, 0.6 mol, 1.2 equiv) of freshly distilled thionyl chloride was added dropwise over a 1-hr period. Heating was continued until gas evolution (sulfur dioxide and hydrogen chloride) had ceased (approximately 9–10 hr). The resultant solution of camphorsulfonyl chloride in chloroform was converted tocamphorsulfonamide without further purification.
In a 5-L, two-necked, round-bottomed flask fitted with a 250-mL dropping funnel and a mechanical stirrer was placed a solution of 1.6 L of reagent-grade ammonium hydroxide solution and the flask was cooled to 0°C in an ice bath. The solution of the crude camphorsulfonyl chloride, prepared in the preceding section, was added dropwise to the ammonium hydroxide solution at 0–10°C over a period of 1 hr. The reaction mixture was warmed to room temperature, stirred for 4 hr, the organic layer separated, and the aqueous layer was extracted with methylene chloride (3 × 250 mL). The combined organic layers were washed with brine (250 mL) and dried over anhydrousmagnesium sulfate. Removal of the solvent on the rotary evaporator gave 104.0 g (90%) of the crudecamphorsulfonamide (Note 2) and (Note 3).
B. (−)-(Camphorsulfonyl)imine. A 1-L, round-bottomed flask is equipped with a 2-in. egg-shaped magnetic stirring bar, a Dean–Stark water separator, and a double-walled condenser containing a mineral oil bubbler connected to an inert gas source. Into the flask are placed 5 g of Amberlyst 15 ion-exchange resin (Note 4) and 41.5 g of the crude(+)-(1S)-camphorsulfonamide in 500 mL of toluene. The reaction mixture is heated at reflux for 4 hr. After the reaction flask is cooled, but while it is still warm (40–50°C), 200 mL of methylene chloride is slowly added to dissolve any(camphorsulfonyl)imine that crystallizes. The solution is filtered through a 150-mL sintered glass funnel of coarse porosity an the reaction flask and filter funnel are washed with an additional 75 mL of methylene chloride.
Isolation of the (−)-(camphorsulfonyl)imine is accomplished by removal of the toluene on the rotary evaporator. The resulting solid is recrystallized from absolute ethanol (750 mL) to give white crystals, 34.5–36.4 g (90–95%), mp225–228°C; [α]D −32.7° (CHCl3, c 1.9) (Note 5).
C. (+)-(2R, 8aS)-10-Camphorylsulfonyloxaziridine. A 5-L, three-necked, round-bottomed Morton flask is equipped with an efficient mechanical stirrer, a 125-mm Teflon stirring blade, a Safe Lab stirring bearing (Note 6), and a 500-mL addition funnel. Into the flask are placed the toluene solution of (−)-(camphorsulfonyl)imine (39.9 g, 0.187 mol)prepared in Step B and a room-temperature solution of 543 g (3.93 mol, 7 equiv based on oxone) of anhydrouspotassium carbonate dissolved in 750 mL of water. The reaction mixture is stirred vigorously and a solution of 345 g (0.56 mol, 6 equiv of KHSO5) of oxone dissolved in 1250 mL of water is added dropwise in three portions over 45 min(Note 7) and (Note 8). Completion of the oxidation is determined by TLC (Note 9) and the reaction mixture is filtered through a 150-mL sintered-glass funnel of coarse porosity to remove solids. The filtrate is transferred to a 3-L separatory funnel, the toluene phase is separated and the aqueous phase is washed with methylene chloride (3 × 100 mL). The filtered solids and any solids remaining in the Morton flask are washed with an additional 200 mL of methylene chloride. The organic extracts are combined and washed with 100 mL of saturated sodium sulfite, dried over anhydrousmagnesium sulfate for 15–20 min, filtered, and concentrated on the rotary evaporator. The resulting white solid is crystallized from approximately 500 mL of hot 2-propanol to afford, after drying under vacuum in a desiccator, 35.9 g(84%) of white needles, mp 165–167°C, [α]D +44.6° (CHCl3, c 2.2) (Note 10) and (Note 11).
(−)-(2S,8aR)-10-(camphorylsulfonyl)oxaziridine is prepared in a similar manner starting from (−)-10-camphorsulfonic acid; mp 166–167°C, [α]D +43.6° (CHCl3, c 2.2).
2. Notes
1. (1S)-(+)-10-Camphorsulfonic acid was purchased from Aldrich Chemical Company, Inc.
2. The crude sulfonamide is contaminated with 5–10% of the (camphorsulfonyl)imine, the yield of which increases on standing.
3. The 1H NMR spectrum of (+)-(1S)-10-camphorsulfonamide is as follows: (CDCl3) δ: 0.93 (s, 3 H, CH3), 1.07 (s, 3 H, CH3), 1.40–2.50 (m, 7 H), 3.14 and 3.53 (AB quartet, 2 H, CH2-SO2, J = 15.1), 5.54 (br s, 2 H, NH2).
4. Amberlyst 15 ion-exchange resin is a strongly acidic, macroreticular resin purchased from Aldrich Chemical Company, Inc.
5. The spectral properties of (−)-(camphorsulfonyl)imine are as follows: 1H NMR (CDCl3) δ: 1.03 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 1.45–2.18 (m, 6 H), 2.65 (m, 1 H), 3.10 and 3.28 (AB quartet, 2 H, CH2-SO2, J = 14.0); 13C NMR (CDCl3) δ: 19.01 (q, CH3), 19.45 (q, CH3), 26.64 (t), 28.44 (t), 35.92 (t), 44.64 (d), 48.00 (s), 49.46 (t), 64.52 (s), 195.52 (s); IR (CHCl3) cm−1: 3030, 2967, 1366. Checkers obtained material having identical melting point and [α]D−32.3° (CHCl3, c 1.8).
6. The SafeLab Teflon bearing can be purchased from Aldrich Chemical Company, Inc. A glass stirring bearing lubricated with silicone grease is unsatisfactory because the dissolved salts solidify in the shaft, causing freezing.
7. Efficient stirring is important and indicated by a milky white appearance of the solution.
8. Occasionally batches of oxone purchased from Aldrich Chemical Company, Inc., have exhibited reduced reactivity in this oxidation. Oxone exposed to moisture prior to use also gives reduced reactivity in this oxidation. If this occurs, oxone is added until oxidation is complete as determined by TLC (Note 9). Potassium carbonate is added as needed to maintain the pH at approximately 9.0. Oxone stored in the refrigerator under an inert atmosphere has shown no loss in reactivity for up to 6 months.
9. Oxidation is generally complete after addition of the oxone solution. The oxidation is monitored by TLC as follows. Remove approximately 0.5 mL of the toluene solution from the nonstirring solution, spot a 250-μm TLC silica gel plate, elute with methylene chloride, and develop with 10% molybdophosphoric acid in ethanol and heating(camphorsulfonyl)imine Rf = 0.28 and (camphorylsulfonyl)oxaziridine Rf = 0.62. If (camphorsulfonyl)imine is detected, stirring is continued at room temperature until the reaction is complete (see (Note 8)). If the reaction mixture takes on a brownish color after addition of oxone and has not gone to completion after 30 min, the reaction mixture is filtered through a 150-mL sintered-glass funnel of coarse porosity, and the solids are washed with 50 mL of methylene chloride. The aqueous/organic extracts are returned to the 5-L Morton flask and stirred vigorously and 52 g (0.08 mol, 1 equiv KHSO5) of oxone is added over 5 min and stirring continued until oxidation is complete (approximately 10–15 min).
10. The submitters employed a toluene solution of crude imine prepared in Part B and obtained somewhat higher yields (90–95%). However, the checkers obtained yields in this range on one half the scale using isolatedsulfonylimine.
11. The spectral properties of (+)-(camphorsulfonyl)oxaziridine are as follows: 1H NMR (CDCl3) δ: 1.03 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 1.45–2.18 (m, 6 H), 2.65 (d, 1 H), 3.10 and 3.28 (AB quartet, 2 H, CH2-SO2, J = 14.0); 13C NMR (CDCl3) δ: 19.45 (q, CH3), 20.42 (q, CH3), 26.55 (t), 28.39 (t), 33.64 (t), 45.78 (d), 48.16 (s), 48.32 (t), 54.07 (s), 98.76 (s). The checkers obtained material (mp 165–167°C) having [α]D +44.7° (CHCl3, c 2.2).
3. Discussion
Camphorsulfonamide, required for the preparation of the (camphorsulfonyl)imine, was previously prepared in two steps. The first step involved conversion of camphorsulfonic acid to the sulfonyl chloride with PCl5 or SOCl2. The isolated sulfonyl chloride was converted in a second step to the sulfonamide by reaction with ammonium hydroxide. This modified procedure is more efficient because it transforms camphorsulfonic acid directly to camphorsulfonamide, avoiding isolation of the camphorsulfonyl chloride.
(Camphorsulfonyl)imine has been reported as a by-product of reactions involving the camphorsulfonamide.2,3,4,5Reychler in 1898 isolated two isomeric camphorsulfonamides,2 one of which was shown to be the(camphorsulfonyl)imine by Armstrong and Lowry in 1902.3 Vandewalle, Van der Eycken, Oppolzer, and Vullioud described the preparation of (camphorsulfonyl)imine in 74% overall yield from 0.42 mol of the camphorsulfonyl chloride.6 The advantage of the procedure described here is that, by using ammonium hydroxide, the camphorsulfonyl chloride is converted to the sulfonamide in >95% yield.7 The sulfonamide is of sufficient purity that it can be used directly in the cyclization step, which, under acidic conditions, is quantitative in less than 4 hr. These modifications result in production of the (camphorsulfonyl)imine in 86% overall yield from the sulfonyl chloride.
In addition to the synthesis of enantiomerically pure (camphorylsulfonyl)oxaziridine7 and its derivatives,8 the(camphorsulfonyl)imine has been used in the preparation of (−)-2,10-camphorsultam (Oppolzers’ auxiliary),6,9 (+)-(3-oxocamphorysulfonyl) oxaziridine,10 and the N-fluoro-2,10-camphorsultam, an enantioselective fluorinating reagent.11
The N-sulfonyloxaziridines are an important class of selective, aprotic oxidizing reagents.12 13 14 Enantiomerically pure N-sulfonyloxaziridines have been used in the asymmetric oxidation of sulfides to sulfoxides (30–91% ee),15selenides to selenoxides (8–9% ee).16 disulfides to thiosulfinates (2–13% ee),5 and in the asymmetric epoxidation of alkenes (19–65% ee).17,18 Oxidation of optically active sulfonimines (R*SO2N=CHAr) affords mixtures of N-sulfonyloxaziridine diastereoisomers requiring separation by crystallization and/or chromatography.3
(+)-(Camphorylsulfonyl)oxaziridine described here is prepared in four steps from inexpensive (1S)-(+)- or (1R)-(+)-10-camphorsulfonic acid in 77% overall yield.7 Separation of the oxaziridine diastereoisomers is not required because oxidation is sterically blocked from the exo face of the C-N double bond in the (camphorsulfonyl)imine. In general, (camphorsulfonyl)oxaziridine exhibits reduced reactivity compared to other N-sulfonyloxaziridines. For example, while sulfides are asymmetrically oxidized to sulfoxides (3–77% ee), this oxaziridine does not react with amines or alkenes.7 However, this oxaziridine is the reagent of choice for the hydroxylation of lithium and Grignard reagents to give alcohols and phenols because yields are good to excellent and side reactions are minimized.19 This reagent has also been used for the stereoselective oxidation of vinyllithiums to enolates.20
The most important synthetic application of the (camphorylsulfonyl)oxaziridines is the asymmetric oxidation of enolates to optically active α-hydroxy carbonyl compounds.14,21,22,23,24 Chiral, nonracemic α-hydroxy carbonylcompounds have been used extensively in asymmetric synthesis, for example, as chiral synthons, chiral auxiliaries, and chiral ligands. This structural array is also featured in many biologically active natural products. This oxidizing reagent gives uniformly high chemical yields regardless of the counterion, and stereoselectivities are good to excellent (50–95% ee).9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 Since the configuration of the oxaziridine three-membered ring controls the stereochemistry, both α-hydroxy carbonyl optical isomers are readily available. Representative examples of the asymmetric oxidation of prochiral enolates by (+)-(2R,8aS)-camphorylsulfonyl)oxaziridine are given in Tables I and II.
This preparation is referenced from:
- Org. Syn. Coll. Vol. 8, 110
- Org. Syn. Coll. Vol. 9, 212
-
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Reychler, M. A. Bull. Soc. Chim. III 1889, 19, 120.
- Armstrong, H. E.; Lowry, T. M. J. Chem. Soc., Trans. 1902, 81, 1441.
- Dauphin, G.; Kergomard, A.; Scarset, A. Bull. Soc. Chim. Fr. 1976, 862.
- Davis, F. A.; Jenkins, Jr., R. H.; Awad, S. B.; Stringer, O. D.; Watson, W. H.; Galloy, J. J. Am. Chem. Soc. 1982, 104, 5412.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron, 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, S.; Carroll, P. J. J. Am. Chem. Soc. 1988, 110, 8477.
- Davis, F. A.; Weismiller, M. C.; Lal, G. S.; Chen, B. C.; Przeslawski, R. M. Tetrahedron Lett., 1989, 30, 1613.
- Oppolzer, W. Tetrahedron 1987, 43, 1969.
- Glahsl, G.; Herrmann, R. J. Chem. Soc., Perkin Trans. I 1988, 1753.
- Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087.
- For recent reviews on the chemistry of N-sulfonyloxaziridines, see: (a) Davis, F. A.; Jenkins, Jr., R. H. in “Asymmetric Synthesis,” Morrison, J. D., Ed.; Academic Press: Orlando, FL, 1984, Vol. 4, Chapter 4;
- Davis, F. A.; Haque, S. M. in “Advances in Oxygenated Processes,” Baumstark, A. L., Ed.; JAI Press: London, Vol. 2;
- Davis, F. A.; Sheppard, A. C. Tetrahedron 1989, 45, 5703.
- Davis, F. A.; McCauley, Jr., J. P.; Chattopadhyay, S.; Harakal, M. E.; Towson, J. C.; Watson, W. H.; Tavanaiepour, I. J. Am. Chem. Soc. 1987, 109, 3370.
- Davis, F. A.; Stringer, O. D.; McCauley, Jr., J. M. Tetrahedron 1985, 41, 4747.
- Davis, F. A.; Chattopadhyay, S. Tetrahedron Lett. 1986, 27, 5079.
- Davis, F. A.; Harakal, M. E.; Awad, S. B. J. Am. Chem. Soc. 1983, 105, 3123.
- Davis, F. A.; Wei, J.; Sheppard, A. C.; Gubernick S. Tetrahedron Lett. 1987, 28, 5115.
- Davis, F. A.; Lal, G. S.; Wei, J. Tetrahedron Lett. 1988, 29, 4269.
- Davis, F. A.; Haque, M. S.; Ulatowski, T. G.; Towson, J. C. J. Org. Chem. 1986, 51, 2402.
- Davis, F. A.; Haque, M. S. J. Org. Chem. 1986, 51, 4083; Davis, F. A.; Haque, M. S.; Przeslawski, R. M. J. Org. Chem. 1989, 54, 2021.
- Davis, F. A.; Ulatowski, T. G.; Haque, M. S. J. Org. Chem. 1987, 52, 5288.
- Davis, F. A.; Sheppard, A. C., Lal, G. S. Tetrahedron Lett. 1989, 30, 779.
- Davis, F. A.; Sheppard, A. C.; Chen, B. C.; Haque, M. S. J. Am. Chem. Soc. 1990, 112, 6679.

back to home for more updates


DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
MK 5172 a next Generation HCV NS3/4a Protease Inhibitor

MK5172
1206524-85-7
Chemical Formula: C29H38N4O7
Exact Mass: 554.27405
Molecular Weight: 554.63462
Elemental Analysis: C, 62.80; H, 6.91; N, 10.10; O, 20.19
IUPAC/Chemical name:
(1aR,5S,8S,10R,22aR)-5-(1,1-Dimethylethyl)-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-14-methoxy-3,6-dioxo-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylic acid.
MK-5172 is a novel, competitive inhibitor of the HCV NS3/4a protease with selective, potent in vitro activity against a broad range of HCV genotypes (GTs) and known viral variants that are resistant to other protease inhibitors in development.
MK-5172 is a Next Generation HCV NS3/4a Protease Inhibitor with a Broad HCV Genotypic Activity Spectrum and Potent Activity Against Known Resistance Mutants, in Genotype 1 and 3 HCV-Infected Patients. MK-5172 exhibits excellent selectivity over other serine proteases such as elastase and trypsin (no measurable inhibition), and shows only modest inhibitory potency with chymotrypsin (IC50 = 1.5 µM; 75,000-fold selective). In the genotype 1b replicon assay, MK-5172 potently inhibits viral replication (IC50 = 2 nM) and demonstrates a modest shift in the presence of 50% NHS (EC50 = 9.5 nM). In vitro, MK-5172 inhibits the NS3/4A enzyme from genotypes 1b, 2a, 2b, and 3a with Ki values of <0.02, 0.15, 0.02, and 0.7 nM, respectively. The genotype 2a replicon is also potently inhibited by MK 5172 (EC50 = 5 nM).
Kuethe J, * Zhong Y.-L, * Yasuda N, * Beutner G, Linn K, Kim M, Marcune B, Dreher SD, Humphrey G, Pei T. Merck Research Laboratories, Rahway, USA
Development of a Practical, Asymmetric Synthesis of the Hepatitis C Virus Protease Inhibitor MK-5172.Org. Lett. 2013;
15: 4174-4177
Development of a Practical, Asymmetric Synthesis of the Hepatitis C Virus Protease Inhibitor MK-5172.Org. Lett. 2013;
15: 4174-4177

MK-5172 is a hepatitis C virus protease inhibitor. Key steps in the synthesis depicted are (1) the regioselective SNAr reaction of dichloroquinoxaline A with prolinol derivative B and (2) construction of the 18-membered macrocycle using a macrolactamization (F → G).
Comment
The medicinal chemistry route to MK-5172 is based on a ring-closing metathesis strategy (S. Harper et al.ACS Med. Chem. Lett. 2012, 3, 332). The best regioselectivity (20:1) and minimization of double substitution in the SNAr reaction of A with B was achieved using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base in polar solvents such as DMSO, NMP, or DMAc.
Pramlintide
Pramlintide (Symlin), a synthetic version of amylin, is a 37-amino acid peptide that is co-secreted with insulin by pancreatic β-cells. It was developed and approved in 2005 by the FDA for use in US patients with type I and II diabetes in conjunction with the administration of prandial insulin to improve postprandial glycemic control
 |
|---|
Pramlintide (Symlin) is a relatively new adjunct for diabetes (both type 1 and 2), developed by Amylin Pharmaceuticals (now a wholly owned subsidiary of Bristol Myers-Squibb). Pramlintide is delivered as an acetate salt.
Pramlintide is an analogue of amylin, a small peptide hormone that is released into the bloodstream by the β-cells of the pancreas along with insulin, after a meal.[1] Like insulin, amylin is completely absent in individuals with Type I diabetes.[2]
Reduction in glycated hemoglobin and weight loss have been shown in insulin-treated patients with type 2 diabetes taking pramlintide as an adjunctive therapy.[3]
By augmenting endogenous amylin, pramlintide aids in the absorption of glucose by slowing gastric emptying, promoting satiety via hypothalamic receptors (different receptors than for GLP-1), and inhibiting inappropriate secretion of glucagon, a catabolic hormone that opposes the effects of insulin and amylin. Pramlintide also has effects in raising the acute first-phase insulin response threshold following a meal.
Pramlintide has been approved by the FDA, for use by Type 1 and Type 2 Diabetics who use insulin.[4]Pramlintide allows patients to use less insulin, lowers average blood sugar levels, and substantially reduces what otherwise would be a large unhealthy rise in blood sugar that occurs in diabetics right after eating. Apart from insulin analogs, pramlintide is the only drug approved by the FDA to lower blood sugar in type 1 diabetics since insulin in the early 1920s.
Design and structure
Since native human amylin is highly amyloidogenic and potentially toxic, the strategy for designing pramlintide was to substitute residues from rat amylin, which is not amyloidogenic (but would presumably retain clinical activity). Proline residues are known to be structure-breaking residues, so these were directly grafted into the human sequence.
Amino acid sequences:
Pramlintide: KCNTATCATQRLANFLVHSSNNFGPILPPTNVGSNTY-(NH2)
Amylin: KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY-(NH2)
Rat amylin: KCNTATCATQRLANFLVRSSNNLGPVLPPTNVGSNTY-(NH2)
Pramlintide as protein is (positively charged).

- Jones MC (2007). “Therapies for diabetes: pramlintide and exenatide” (pdf). American Family Physician 75 (12): 1831–5. PMID 17619527.
- Edelman, Steve; Maier, Holly; Wilhelm, Ken (2008). “Pramlintide in the Treatment of Diabetes Mellitus”.BioDrugs 22 (6): 375–386. doi:10.2165/0063030-200822060-00004. ISSN 1173-8804.
- Hollander, Priscilla; Maggs, David G.; Ruggles, James A.; Fineman, Mark; Shen, Larry; Kolterman, Orville G.; Weyer, Christian (2004). “Effect of Pramlintide on Weight in Overweight and Obese Insulin-Treated Type 2 Diabetes Patients” (pdf). Obesity 12 (4): 661–668. doi:10.1038/oby.2004.76.ISSN 1930-7381.
- Ryan GJ, Jobe LJ, Martin R (2005). “Pramlintide in the treatment of type 1 and type 2 diabetes mellitus”. Clinical therapeutics 27 (10): 1500–12. doi:10.1016/j.clinthera.2005.10.009. PMID 16330288.

- www.symlin.com – product website
- www.amylin.com – Symlin page on the Amylin Pharmaceuticals website
| Pramlintide Acetate | |
| Pramlintide acetate is a relatively new adjunct treatment for diabetes (both type 1 and 2). |
Pramlintide Acetate, 196078-30-5,
| Synonym | Pramlintide Acetate,Pramlintide acetate hydrate |
| Molecular Formula | C171H267N51O53S2.X(C2H4O2).X(H2O) |
| Molecular Weight | 3949.39 |

FDA Approves Implanted Brain Stimulator for Epilepsy
epilepsy
THURSDAY Nov. 14, 2013 — The U.S. Food and Drug Administration on Thursday gave its approval to a new implanted device that lowers the rate of seizures among people with epilepsy
http://www.drugs.com/news/fda-approves-implanted-brain-stimulator-epilepsy-48978.html
EPROSARTAN MESYLATE

TEVETEN® (eprosartan mesylate) is a non-biphenyl non-tetrazole angiotensin II receptor (AT1) antagonist. A selective non-peptide molecule, TEVETEN® is chemically described as the monomethanesulfonate of (E)-2-butyl-1 -(p-carboxybenzyl)-α-2-thienylmethylimid-azole-5 -acrylic acid.
Its empirical formula is C23H24N2O4S•CH4O3S and molecular weight is 520.625. Its structural formula is:
 |
EPROSARTAN MESYLATE
tevetenEprosartan mesilate, SK&F-108566-J(?, SK&F-108566, Teveten SB, Navixen, Regulaten, Tevetenz, Teveten
| US 5656650 | exp Aug 12, 2014 |
CAS EPROSARTAN
144143-96-4
133040-01-4
| Chemical Name: | Eprosartan mesylate |
| Synonyms: | EPROSARTAN MESYLATE;Eprosartan Methanesulfonate;4-[[2-butyl-5-(2-carboxy-3-thiophen-2-yl-prop-1-enyl)-imidazol-1-yl]methyl]benzoic acid mesylate;4-({2-butyl-5-[(1E)-2-carboxy-2-(thiophen-2-ylMethyl)eth-1-en-1-yl]-1H-iMidazol-1-yl}Methyl)benzoic acid;(E)-α-[[2-Butyl-1-[(4-carboxyphenyl)Methyl]-1H-iMidazol-5-yl]Methylene]-2-thiophenepropanoic Acid Methanesulfonate;(αE)-α-[[2-Butyl-1-[(4-carboxyphenyl)Methyl]-1H-iMidazol-5-yl]Methylene]-2-thiophenepropanoic Acid MonoMethanesulfonate |
| CBNumber: | CB4842192 |
| Molecular Formula: | C24H28N2O7S2 |
| Formula Weight: | 520.61832 |
Eprosartan is an angiotensin II receptor antagonist used for the treatment of high blood pressure. It is marketed as Teveten byAbbott Laboratories in the United States.It is marketed as Eprozar by INTAS Pharmaceuticals in India and by Abbott Laboratorieselsewhere. It is sometimes paired with hydrochlorothiazide, marketed in the US as Teveten HCT and elsewhere as TevetenPlus.
The drug acts on the renin-angiotensin system in two ways to decrease total peripheral resistance. First, it blocks the binding ofangiotensin II to AT1 receptors in vascular smooth muscle, causing vascular dilatation. Second, it inhibits sympatheticnorepinephrine production, further reducing blood pressure.
As with other angiotensin II receptor antagonists, eprosartan is generally better tolerated than enalapril (an ACE inhibitor), especially among the elderly.[1]
- Ruilope L, Jäger B, Prichard B (2001). “Eprosartan versus enalapril in elderly patients with hypertension: a double-blind, randomized trial”. Blood Press. 10 (4): 223–9. doi:10.1080/08037050152669747. PMID 11800061.
PAT APR EXP
| Canada | 2250395 | 2005-09-06 | 2017-03-26 |
| Canada | 2115170 | 2004-05-25 | 2012-08-12 |
| United States | 5656650 | 1994-08-12 | 2014-08-12 |
| United States | 5185351 | 1993-02-09 | 2010-02-09 |
| Canada | 2115170 | 2004-05-25 | 2012-08-12 |
| United States | 5656650 | 1994-08-12 | 2014-08-12 |
| Canada | 2250395 | 2005-09-06 | 2017-03-26 |
J Med Chem1991,34,(4):1514-7
J Med Chem1993,36,(13):1880-92
Synth Commun1993,23,(22):3231-48
AU 9056901, EP 403159, JP 91115278, US 5185351.
Drugs Fut1997,22,(10):1079

Eprosartan mesylate was developed successfully by SmithKline Beecham Corporation in 1997, and marketed in Germany in 1998 under the trade-name Teveten and in the United States later in 1999. Eprosartan mesylate, as an angiotensin II receptor blocker, is an antihypertensive drug of the latest generation. Eprosartan mesylate is potent to lower systolic and diastolic pressures in mild, moderate and severe hypertensive patients, and is safe and tolerable. Eprosartan mesylate is rapidly absorbed when administrated orally, with a bioavailability of 13% and a protein binding rate of 98%. The blood peak concentration and AUC (Area Under Curve) can be elevated by about 50% in patients with liver and kidney dysfunction, or fullness after administration, and can be elevated by 2 to 3 folds in elderly patients. Eprosartan mesylate has a structure shown as follows:

U.S. Pat. No. 5,185,351 discloses a method for preparing eprosartan mesylate using Eprosartan and methanesulfonic acid in isopropanol (U.S. Pat. No. 5,185,351, Example 41 (ii)). However, it is found when following this method for preparing eprosartan mesylate in industry, an esterification reaction can occur between eprosartan and isopropanol and the following two impurities can be generated:

In addition to the above two esterification impurities, the salifying method provided by the above patent is prone to produce isopropyl mesylate. Considering currently known potential risk of gene toxicity of methylsulfonic acid ester on human as well as the stringent requirements of methylsulfonic acid ester from the Europe and the America authorities, it is important to produce eprosartan mesylate in a non-alcohol solvent during the process of producing eprosartan mesylate, since it avoids the formation of methylsulfonic acid ester and the residue thereof in the final product. Since the dosage of eprosartan mesylate is high, it is particularly important to strictly control methylsulfonic acid ester in eprosartan mesylate.
In addition, for the above salifying method, solid eprosartan is suspended in propanol at a low temperature, then methanesulfonic acid is added, about ten seconds later a great deal of eprosartan mesylate precipitate is obtained. Therefore, solid eprosartan may be embedded by the precipitated eprosartan mesylate. Since isopropyl alcohol has a high viscosity at low temperature, a heavy filtering operation burden is needed to obtain solid from isopropanol, and the obtained solid contains quite an amount of isopropanol.

Eprosartan has been obtained by several different ways: 1) The iodination of 2-butylimidazole (I) with I2 and Na2CO3 in dioxane/water gives 2-butyl-4,5-diiodoimidazole (II), which is treated with benzyl chloromethyl ether (III) and K2CO3 in DMF yielding the imidazole derivative (IV). The condensation of (IV) with N-methyl-N-(2-pyridyl)formamide (V) by means of butyllithium in THF affords 1-(benzyloxymethyl)-2-butyl-4-iodoimidazole-5-carbaldehyde (VI), which is deprotected with concentrated HCl ethanol to give 2-butyl-4-iodoimidazole-5-carbaldehyde (VII). The acylation of (VII) with methyl 4-(bromomethyl)benzoate (VIII) by means of K2CO3 in hot DMF yields 4-(2-butyl-5-formyl-4-iodoimidazol-1 ylmethyl)benzoic acid methyl ester (IX), which is deiodinated by hydrogenation with H2 over Pd/C in methanol affording compound (X). The condensation of (X) with methyl 3-(2-thienyl)propionate (XI) by means of lithium diisopropylamide (LDA) in THF gives (XII), which is acylated with acetic anhydride and dimethylaminopyridine (DMAP) in dichloromethane yielding the corresponding acetate (XIII). Elimination of acetic acid from (XIII) with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in hot toluene affords the expected propenoic ester (XIV), which is finally saponified with NaOH or KOH in ethanol/water.
…………………………………………………………………………………………………….
WO 1998035962 A1

…………………………………………………………………………………………..



