
Home » Posts tagged 'organic chemistry' (Page 14)
Tag Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Orexigen files obesity drug Contrave for approval in Europe

Orexigen files obesity drug Contrave for approval in Europe
Orexigen Therapeutics has submitted the Marketing Authorization Application (MAA) for Contrave, an investigational weight-loss drug to the European Medicines Agency (EMA).
 The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
read all at
http://www.pharmatopics.com/2013/10/orexigen-files-obesity-drug-contrave-approval-europe/
Tetraphase to Present New Data at IDWeek on Eravacycline’s Potential Activity in Treating Serious Respiratory Infections

eravacycline
http://www.ama-assn.org/resources/doc/usan/eravacycline.pdf
1-Pyrrolidineacetamide, N-[(5aR,6aS,7S,10aS)-9-(aminocarbonyl)-7-(dimethylamino)-
4-fluoro-5,5a,6,6a,7,10,10a,12-octahydro-1,8,10a,11-tetrahydroxy-10,12-dioxo-2-
naphthacenyl]-
(4S,4aS,5aR,12aS)-4-(dimethylamino)-7-fluoro-3,10,12,12a-tetrahydroxy-1,11-dioxo-9-
[(pyrrolidin-1-ylacetyl)amino]-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide
MOLECULAR FORMULA C27H31FN4O8
MOLECULAR WEIGHT 558.6
SPONSOR Tetraphase Pharmaceuticals, Inc.
CODE DESIGNATION TP-434
CAS REGISTRY NUMBER 1207283-85-9
WHO NUMBER 9702
Tetraphase Pharmaceuticals Inc. (NASDAQ:TTPH) today announced that it will present two posters at IDWeek 2013 that examine the potential of its lead antibiotic candidate eravacycline to treat serious multi-drug resistant (MDR) infections. The first will highlight positive results of a Phase 1 study assessing the bronchopulmonary disposition safety and tolerability of eravacycline in healthy men and women; this study represents the first clinical assessment of eravacycline for potential use in treating pneumonia. The second poster will provide the results of a study that examined the activity of eravacycline in vitro against multiple Gram-negative and Gram-positive pathogens to set quality-control limits for monitoring eravacycline activity in future testing programs.
http://www.pharmiweb.com/pressreleases/pressrel.asp?ROW_ID=79335#.Uk6MOoanonU
More: http://www.pharmiweb.com/pressreleases/pressrel.asp?ROW_ID=79335#.Uk6MOoanonU#ixzz2gkEMTtQm
Eravacycline (TP-434) is a synthetic fluorocycline antibiotic in development.
Eravacycline (TP-434 or 7-fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline) is a novel fluorocycline that was evaluated for antimicrobial activity against panels of recent aerobic and anaerobic Gram-negative and Gram-positive bacteria. Eravacycline showed potent broad spectrum activity against 90% of the isolates (MIC90) in each panel at concentrations ranging from ≤0.008 to 2 μg/mL for all species panels except Pseudomonas aeruginosa and Burkholderia cenocepacia (MIC90 values of 32 μg/mL for both organisms). The antibacterial activity of eravacycline was minimally affected by expression of tetracycline-specific efflux and ribosomal protection mechanisms in clinical isolates. Further, eravacycline was active against multidrug-resistant bacteria, including those expressing extended spectrum β-lactamases and mechanisms conferring resistance to other classes of antibiotics, including carbapenem resistance. Eravacycline has the potential to be a promising new IV/oral antibiotic for the empiric treatment of complicated hospital/healthcare infections and moderate-to-severe community-acquired infections.
Tetraphase’s lead product candidate, eravacycline, has also received an award from Biomedical Advanced Research and Development Authority (BARDA) that provides for funding to develop eravacycline as a potential counter-measure to certain biothreat pathogens. It is worth up to USD 67 million.
Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development
Eravacycline (TP-434)
We are developing our lead product candidate, eravacycline, as a broad-spectrum intravenous and oral antibiotic for use as a first-line empiric monotherapy for the treatment of multi-drug resistant (MDR) infections, including MDR Gram-negative bacteria. We developed eravacycline using our proprietary chemistry technology. We completed a successful Phase 2 clinical trial of eravacycline with intravenous administration for the treatment of patients with complicated intra-abdominal infections (cIAI) and have initiated the Phase 3 clinical program.
Eravacycline is a novel, fully synthetic tetracycline antibiotic. We selected eravacycline for development from tetracycline derivatives that we generated using our proprietary chemistry technology on the basis of the following characteristics of the compound that we observed in in vitro studies of the compound:
- potent antibacterial activity against a broad spectrum of susceptible and multi-drug resistant bacteria, including Gram-negative, Gram-positive, atypical and anaerobic bacteria;
- potential to treat the majority of patients as a first-line empiric monotherapy with convenient dosing; and
- potential for intravenous-to-oral step-down therapy.
In in vitro studies, eravacycline has been highly active against emerging multi-drug resistant pathogens like Acinetobacter baumannii as well as clinically important species ofEnterobacteriaceae, including those isolates that produce ESBLs or are resistant to the carbapenem class of antibiotics, and anaerobes.
Based on in vitro studies we have completed, eravacycline shares a similar potency profile with carbapenems except that it more broadly covers Gram-positive pathogens like MRSA and enterococci, is active against carbapenem-resistant Gram-negative bacteria and unlike carbapenems like Primaxin and Merrem is not active against Pseudomanas aeruginosa. Eravacycline has demonstrated strong activity in vitro against Gram-positive pathogens, including both nosocomial and community-acquired methicillin susceptible or resistantStaphylococcus aureus strains, vancomycin susceptible or resistant Enterococcus faecium andEnterococcus faecalis, and penicillin susceptible or resistant strains of Streptococcus pneumoniae. In in vitro studies for cIAI, eravacycline consistently exhibited strong activity against enterococci and streptococci. One of the most frequently isolated anaerobic pathogens in cIAI, either as the sole pathogen or often in conjunction with another Gram-negative bacterium, is Bacteroides fragilis. In these studies eravacycline demonstrated activity against Bacteroides fragilis and a wide range of Gram-positive and Gram-negative anaerobes.
Key Differentiating Attributes of Eravacycline
The following key attributes of eravacycline, observed in clinical trials and preclinical studies of eravacycline, differentiate eravacycline from other antibiotics targeting multi-drug resistant infections, including multi-drug resistant Gram-negative infections. These attributes will make eravacycline a safe and effective treatment for cIAI, cUTI and other serious and life-threatening infections for which we may develop eravacycline, such as ABSSSI and acute bacterial pneumonias.
- Broad-spectrum activity against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria. In our recently completed Phase 2 clinical trial of the intravenous formulation of eravacycline, eravacycline demonstrated a high cure rate against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria. In addition, in in vitro studies eravacycline demonstrated potent antibacterial activity against Gram-negative bacteria, including E. coli; ESBL-producing Klebsiella pneumoniae; Acinetobacter baumannii; Gram-positive bacteria, including MSRA and vancomycin-resistant enterococcus, or VRE; and anaerobic pathogens. As a result of this broad-spectrum coverage, eravacycline has the potential to be used as a first-line empiric monotherapy for the treatment of cIAI, cUTI, ABSSSI, acute bacterial pneumonias and other serious and life-threatening infections.
- Favorable safety and tolerability profile. Eravacycline has been evaluated in more than 250 subjects in the Phase 1 and Phase 2 clinical trials that we have conducted. In these trials, eravacycline demonstrated a favorable safety and tolerability profile. In our recent Phase 2 clinical trial of eravacycline, no patients suffered any serious adverse events, and safety and tolerability were comparable to ertapenem, the control therapy in the trial. In addition, in the Phase 2 clinical trial, the rate at which gastrointestinal adverse events such as nausea and vomiting that occurred in the eravacycline arms was comparable to the rate of such events in the ertapenem arm of the trial.
- Convenient dosing regimen. In our recently completed Phase 2 clinical trial we dosed eravacycline once or twice a day as a monotherapy. Eravacycline will be able to be administered as a first-line empiric monotherapy with once- or twice-daily dosing, avoiding the need for complicated dosing regimens typical of multi-drug cocktails and the increased risk of negative drug-drug interactions inherent to multi-drug cocktails.
- Potential for convenient intravenous-to-oral step-down. In addition to the intravenous formulation of eravacycline, we are also developing an oral formulation of eravacycline. If successful, this oral formulation would enable patients who begin intravenous treatment with eravacycline in the hospital setting to transition to oral dosing of eravacycline either in hospital or upon patient discharge for convenient home-based care. The availability of both intravenous and oral administration and the oral step-down will reduce the length of a patient’s hospital stay and the overall cost of care.
Additionally, in February 2012, Tetraphase announced a contract award from the Biomedical Advanced Research and Development Authority (BARDA) worth up to $67 million for the development of eravacycline, from which Tetraphase may receive up to approximately $40 million in funding. The contract includes pre-clinical efficacy and toxicology studies; clinical studies; manufacturing activities; and associated regulatory activities to position the broad-spectrum antibiotic eravacycline as a potential empiric countermeasure for the treatment of inhalational disease caused by Bacillus anthracis, Francisella tularensis and Yersinia pestis.\
……………………………………………………………………………………….

Process research and development of the first fully synthetic broad spectrum 7-fluorotetracycline in clinical development is described. The process utilizes two key intermediates in a convergent approach. The key transformation is a Michael–Dieckmann reaction between a suitable substituted aromatic moiety and a key cyclohexenone derivative. Subsequent deprotection and acylation provide the desired active pharmaceutical ingredient in good overall yield.
Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development
FDA Grants QIDP Designation to Eravacycline, Tetraphase’s Lead Antibiotic Product Candidate
– Eravacycline designated as a QIDP for complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI) indications –
July 15, 2013 08:30 AM Eastern Daylight Time
WATERTOWN, Mass.–(BUSINESS WIRE)–Tetraphase Pharmaceuticals, Inc. (NASDAQ: TTPH) today announced that the U.S. Food and Drug Administration (FDA) has designated the company’s lead antibiotic product candidate, eravacycline, as a Qualified Infectious Disease Product (QIDP). The QIDP designation, granted for complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI) indications, will make eravacycline eligible to benefit from certain incentives for the development of new antibiotics provided under the Generating Antibiotic Incentives Now Act (GAIN Act). These incentives include priority review and eligibility for fast-track status. Further, if ultimately approved by the FDA, eravacycline is eligible for an additional five-year extension of Hatch-Waxman exclusivity.
Aerial Biopharma announce positive Phase II results for narcolepsy drug
ADX-N05, ARL-N05, SKL-N05
Aerial Biopharma announce positive Phase II results for narcolepsy drug
A new drug to treat excessive daytime sleepiness associated with narcolepsy has shown positive results from a phase 2b clinical trial, US-based Aerial Biopharma has announced this week.
read all at
Secukinumab
Secukinumab is an anti-IL17A drug being investigated for a number of inflammatory conditions. For plaque psoriasis, Novartis is planning to evaluate a dose of 150 mg subcutaneously compared with placebo.
The primary outcome measure of the planned Phase III trial named ERASURE is to evaluate the efficacy in patients with moderate to severe chronic plaque-type psoriasis. Novartis is also planning to evaluate secukinumab dosed at either 150 or 300 mg versus Enbrel (enterecept) 50 mg in a Phase III trial entitled FIXTURE.
Final data collection for the primary outcome measures in both ERASURE and FIXTURE are anticipated in March 2013.
Secukinumab is a human monoclonal antibody designed for the treatments of uveitis,rheumatoid arthritis, and psoriasis. It targets member A from the cytokine family ofinterleukin 17.[1][2]
Secukinumab was developed by Novartis Pharma AG and has completed Phase II clinical trials for plaque psoriasis in 2011.[3]
CAS registry numbers
- 875356-43-7 (heavy chain)
- 875356-44-8 (light chain)
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Secukinumab”. American Medical Association.
- ^ Hueber, W.; Patel, D. D.; Dryja, T.; Wright, A. M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M. H.; Psoriasis Study, P.; Durez, P. P.; Tak, J. J.; Gomez-Reino, C. S.; Rheumatoid Arthritis Study, R. Y.; Foster, C. M.; Kim, N. S.; Samson, D. S.; Falk, D.; Chu, Q. D.; Callanan, K.; Nguyen, A.; Uveitis Study, F.; Rose, K.; Haider, A.; Di Padova, F. (2010). “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis”. Science Translational Medicine 2 (52): 52ra72.doi:10.1126/scitranslmed.3001107. PMID 20926833. edit
- ^ Papp K.A. et al. ‘Secukinumab efficacy and safety preliminary results from a phase II subcutaneous dose-ranging study in the treatment of moderate-to-severe plaque psoriasis.’ Presented at: 20th Congress of the European Academy of Dermatology and Venereology; 20-24 October, 2011; Lisbon, Portugal.

Dimeric Thymosin beta-4……..accelerates the rate of wound healing

Structure of a Longitudinal Actin Dimer Assembled by Tandem W Domains
Thymosin beta 4 (Tβ4) is a peptide with 43 amino acids that is critical for repair and remodeling tissues on the skin, eye, heart, and neural system following injury
Thymosin beta-4 is a protein that in humans is encoded by the TMSB4X gene.
The protein consists (in humans) of 43 amino acids (msdkpdmaei ekfdksklkk tetqeknplp sketieqekq ages) molWt 4921

NMR structure of a β-thymosin. Both thymosin α1 and β-thymosins areintrinsically unstructured proteins, i.e. they lack a stable fold when free in aqueous solution. This structure, mostly alpha helix, was artificially stabilised by an organic solvent. The thymosin illustrated, originally named β9 is the cow orthologue of human β10
It has been studied in a number of clinical trials.
The thymosin beta-4 peptide, if used after a heart attack, might reactivate cardiacprogenitor cells to repair damaged heart tissue.
Doping in Sports
Thymosin beta-4 was allegedly used by some players in various Australian football codes and is under investigation by the Australian Sports Anti-Doping Authority for anti-doping violations (Feb/Mar 2013):
https://theconversation.edu.au/cronulla-sharks-and-thymosin-beta-4-is-it-doping-12694
FDA, EMA Accept Omeros Ophthalmology Product NDA
OMS302
US and European Regulators Accept for Review OMS302 Marketing Applications
— OMS302 Remains on Track for Planned 2014 Commercial Launch —
SEATTLE, Oct. 2, 2013 /PRNewswire/ — Omeros Corporation (NASDAQ: OMER) announced today that the New Drug Application (NDA) for its ophthalmology product, OMS302, has been confirmed for filing by the U.S. Food and Drug Administration (FDA), which means that the application, submitted in July of this year, is sufficiently complete to permit a substantive review. The company also announced that its Marketing Authorization Application (MAA) for OMS302, submitted last month, has been validated by the European Medicines Agency (EMA). Validation of the MAA confirms that the submission package is administratively complete and is ready for formal review by Europe’s Committee for Medicinal Products for Human Use (CHMP).
read all at
http://www.pharmalive.com/fda-ema-accept-omeros-opthamology-product-nda
Isavuconazole – Basilea reports positive results from study

This post is updated in sept 2015……..
Isavuconazole (BAL4815; trade name Cresemba) is a triazole antifungal drug. Its prodrug, isavuconazonium sulfate (BAL8557), was granted approval by the U.S. Food and Drug Administration (FDA) on March 6, 2015[1]
During its Phase III drug trials, Astellas partnered with Basilea Pharmaceutica, the developer of the drug, for rights to co-development and marketing of isavuconazole. [2]
On May 28, 2013, Basilea Pharmaceutica announced it had been granted orphan drug status by the FDA for treatment of aspergillosis.[3] Since then, it has also been granted orphan drug status for the treatment of invasive candidiasis.[4]

Isavuconazonium sulfate (BAL8557)—a prodrug of isavuconazole.
CLINICAL TRIALS…LINK
PATENTS
|
6-27-2012
|
Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole
|
|
|
11-18-2011
|
Antifungal Composition
|
|
|
9-29-2010
|
PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS
|
|
|
12-3-2008
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
3-14-2007
|
N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
11-3-2004
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
10-10-2001
|
Azoles for treatment of fungal infections
|
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.

Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:



……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
http://www.sciencedirect.com/science/article/pii/S0960894X02008922
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.

-

-
Scheme 1.
Chemistry
Scheme 1.We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.

-
Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%).
-
Figure options

-
Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).

read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
Clinical trials
There have been three phase III clinical trials of isavuconazole, ACTIVE, VITAL and SECURE. As of June 2015, SECURE and VITAL have been presented in abstract form and results from ACTIVE have not been released.[9]
The SECURE trial compared voriconazole and isavuconazole in invasive fungal infections due to aspergillus. Isuvaconazole was found to be non-inferior to voriconazole, anothertriazole antifungal, with all cause mortality at 18.6%, compared to 20.2% in the voriconazole group. It additionally demonstrated a similar side effect profile.[10]
Data from the VITAL study showed that isavuconazole could be used in treatment of invasive mucormycosis, but did not evaluate its clinical efficacy for this indication.[11]
The ACTIVE trial is a comparison of isuvaconazole and caspofungin for invasive candida infections and results are anticipated in the second half of 2015.[12][13]
References
- [1]
- Saboo, Alok. “Basilea Announces Global Partnership With Astellas for Its Antifungal Isavuconazole.” FierceBiotech. N.p., 24 Feb. 2010. Web.
- “Basilea reports isavuconazole orphan drug designation by U.S. FDA.” Market Wired. 28 May 2013.
- “FDA Grants Orphan Drug Designation to Astellas for Isavuconazole for the Treatment of Invasive Candidiasis.” News Releases. Astellas. 3 Nov 2014.
- Cresemba (isovuconazole sulfate) [prescribing information]. Astella Pharma US, Inc. Revised March 2015.
- Jump up^ “Aspergillosis.” Centers for Disease Control and Prevention. Centers for Disease Control and Prevention, 08 Sept. 2014.
- Jump up^ “Astellas Receives FDA Approval for CRESEMBA® (isavuconazonium Sulfate) for the Treatment of Invasive Aspergillosis and Invasive Mucormycosis.” PR Newswire. N.p., 6 Mar. 2015.
- Jump up^ “Isavuconazonium.” Micromedex Solutions. Truven Health Analytics, n.d. Web. <www.micromedexsolutions.com>.
- Jump up^ Pettit, Natasha N.; Carver, Peggy L. (2015-07-01). “Isavuconazole A New Option for the Management of Invasive Fungal Infections”. Annals of Pharmacotherapy 49 (7): 825–842.doi:10.1177/1060028015581679. ISSN 1060-0280. PMID 25940222.
- Mujais, A. “2014: M-1756. A Phase 3 Randomized, Double-Blind, Non-Inferiority Trial Evaluating Isavuconazole (ISA) vs. Voriconazole (VRC) for the Primary Treatment of Invasive Fungal Disease (IFD) Caused by Aspergillus spp. or other Filamentous Fungi (SECURE): Outcomes by Malignancy Status”. http://www.icaaconline.com. Retrieved 2015-06-19.
- “Abstract: An Open-Label Phase 3 Study of Isavuconazole (VITAL): Focus on Mucormycosis (IDWeek 2014)”. idsa.confex.com. Retrieved 2015-06-19.
- Ltd., Basilea. “Basilea Pharmaceutica – Portfolio – Isavuconazole”. http://www.basilea.com. Retrieved 2015-06-19.
- “Isavuconazole (BAL8557) in the Treatment of Candidemia and Other Invasive Candida Infections – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2015-06-19.
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
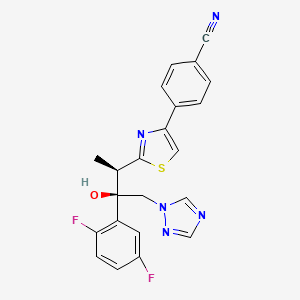
4-{2-[(1R,2R)-(2,5-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
|
|
| Clinical data | |
| Trade names | Cresemba (prodrug form) |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, intravenous |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 6918485 |
| ChemSpider | 5293682 |
| UNII | 60UTO373KE |
| ChEBI | CHEBI:85979 |
| ChEMBL | CHEMBL409153 |
| NIAID ChemDB | 416566 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Molecular mass | 437.47 g/mol |


/////
Vortioxetine ボルチオキセチン 臭化水素酸塩 – FDA Approves Brintellix to Treat Major Depressive Disorder

Vortioxetine
ボルチオキセチン 臭化水素酸塩
1-[2-(2,4-dimethylphenyl)sulfanylphenyl]piperazine
Lu AA21004
| VORTIOXETINE; CAS 508233-74-7;
1-(2-((2,4-Dimethylphenyl)thio)phenyl)piperazine; Lu AA21004; UNII-3O2K1S3WQV; C18H22N2S; |
|
| Molecular Formula: | C18H22N2S |
|---|---|
| Molecular Weight: | 298.44568 g/mol |
Vortioxetine Hydrobromide

C18H22N2S.HBr : 379.36
[960203-27-4] HYDROBROMIDE
Vortioxetine is an atypical antipsychotic and antidepressant indicated for the treatment of major depressive disorder (MDD). It is classified as a serotonin modulator and simulator (SMS) as it has a multimodal mechanism of action towards the serotonin neurotransmitter system whereby it simultaneously modulates one or more serotonin receptors and inhibits the reuptake of serotonin. More specifically, vortioxetine acts via the following biological mechanisms: as a serotonin reuptake inhibitor (SRI) through inhibition of the serotonintransporter, as a partial agonist of the 5-HT1B receptor, an agonist of 5-HT1A, and an antagonist of the 5-HT3, 5-HT1D, and 5-HT7 receptors. SMSs were developed because there are many different subtypes of serotonin receptors, however, not all of these receptors appear to be involved in the antidepressant effects of SRIs. Some serotonin receptors seem to play a relatively neutral or insignificant role in the regulation of mood, but others, such as 5-HT1A autoreceptors and 5-HT7 receptors, appear to play an oppositional role in the efficacy of SRIs in treating depression.
Sept. 30, 2013 — The U.S. Food and Drug Administration today approved Brintellix (vortioxetine) to treat adults with major depressive disorder.
Major depressive disorder (MDD),
 Commonly referred to as depression, is a mental disorder characterized by mood changes and other symptoms that interfere with a person’s ability to work, sleep, study, eat and enjoy once-pleasurable activities. Episodes of depression often recur throughout a person’s lifetime, although some may experience a single occurrence.
Commonly referred to as depression, is a mental disorder characterized by mood changes and other symptoms that interfere with a person’s ability to work, sleep, study, eat and enjoy once-pleasurable activities. Episodes of depression often recur throughout a person’s lifetime, although some may experience a single occurrence.
READ ALL AT
http://www.drugs.com/newdrugs/fda-approves-brintellix-major-depressive-disorder-3918.html
The disease: Major depression
The developers: Lundbeck, Takeda
Vortioxetine (vor-tye-OX-e-teen, code name Lu AA21004) is an experimental drug currently under development by Lundbeck and Takeda for the treatment of major depressive disorder (MDD) and generalized anxiety disorder (GAD).Commercial names chosen are Brintellix and Rexulti.
Regulatory approval for the treatment of MDD for the European market has been filed in September 2012, for the United States in October 2012, and filing for Canada should follow. Filing for the Japanese market is expected in 2013.

Depression
In May 22 2011, Lundbeck presented the results of four phase III trials on vortioxetine at the 2011 Annual Meeting of the American Psychiatric Association. A statistically significant effect was shown in two of the studies (one for active treatment using the Hamilton Depression Rating Scale (HAM-D), the second as a maintenance treatment), vortioxetine failed to prove superiority over placebo in a third (again using the HAM-D) and the fourth was nullified by an exceptionally high placebo response (according to the Montgomery-Åsberg Depression Rating Scale (MADRS)).
In July 2011, Lundbeck published the results of a double-blind, randomized, placebo-controlled clinical trial with venlafaxine as an active reference. It was found to be superior to placebo in treating MDD while having fewer side effects than venlafaxine. Similarly, in May 2012, Lundbeck published the results of a double-blind, randomized, placebo-controlled clinical trial with duloxetine evaluating vortioxetine in elderly depressed patients, and it was found superior to placebo, with fewer side effects than duloxetine.
In May 2012, Lundbeck disclosed the results of three phase III clinical trials, showing vortioxetine’s superiority over placebo according to the MADRS.
In August 2012, a randomized, double-blind trial confirms the superiority of vortioxetine over placebo according to all measures, excepted the Sheehan Disability scale.
In September 2012, a randomised, double-blind trial reveals that a dose of 5mg shows superiority over placebo only in patients that suffer from comorbid anxiety.This is consistent with results from another trial published in December 2012, demonstrating that 2.5 mg and 5 mg doses are ineffective.
Anxiety
August 2012, contradictory results of two randomized, double-blind trial were published. While the first demonstrated vortioxetine’s superiority over the placebo, the second showed that the drug had no efficacy, leading the authors to question the designs of the different trials.

United States Patent Number: 7,144,884 , 8,476,279
related to Chinese patent: CN1319958 C , CN1561336 A; CN1319958C, CN1561336A
patent validity: January 9, 2023 (U.S. Patent Number: 7,144,884), October 2, 2022 (U.S. Patent No.: 8,476,279)
peak annual sales (estimated): $ 2 billion
drug companies: Lundbeck (Lundbeck), Takeda (Takeda)
Wal antidepressant drug Paxil (Brintellix, Vortioxetine) for – 1 – Preparation – [2 – (2,4 methyl) phenyl] piperazine process
Method II:
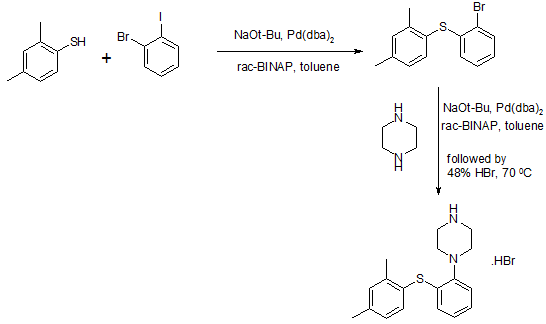
815g of the NaOBut (8,48 mo1), 844 g of piperazine Qin (9,8 mol), 6,6 g of Pd (dba) 2 (11,48 mmol) and 13,6 g of rac-BINAP (21, 84 mmol) was stirred for 50 minutes with 4L ofbenzene. Then, 840 g 2 – bromo – iodobenzene (2,97 mol) and 1.5L of Yue added to the mixture with benzene, and the stirring was continued for 30 minutes. Finally, 390.8g of 2,4 -thiophenol (2,83 mol) was added together with 1.5L toluene. The resulting suspension was heated to reflux and reflux was continued for 5 hours. The reaction mixture was cooled overnight. 2L of water was added and stirred for l hour and then filtered through a filter aid, the resulting mixture. Then, the filtrate was washed with brine 3xlL. Subsequently, the combined aqueous phase extracted with 600ml of benzene. Then, Yue The combined benzene phase was heated to 70 ° C, then adding 329.2ml 48-wt. / HBr (aq.) and 164.6ml water o’s. The mixture was cooled to room temperature overnight. Final product was collected by filtration (l-[2 – (2,4 – di曱group – phenylsulfanyl) – phenyl] – piperazine hydrobromide Qin), and dried under vacuum (60 0 C), to give 895g of product (84% yield).
Method III:
The benzene is placed 500ml three-necked 1L round bottom flask equipped with a mechanical stirrer and add 809mg Pd2dba3 (0.88mmol; 0.5 mol%) and 952 mg DPEPhos (1.77 mmol; 0.5mol-%). The deep red solution was purged with nitrogen for 5 minutes, then add 100g2-bromo-iodobenzene (353 mmol) and 48.9 g 2,4 – bis thiophenol (353 mmol). Add 43.6g KOBut (389 mmol) caused an exothermic reaction, so that the temperature rise of 20 ° C 42 ° C, while forming a non-uniform mixture, and the color changed from deep red to orange / brown. The force of the suspension under nitrogen was heated to port 100 ° C. After only 20 minutes, HPLC showed complete conversion to have l-(2 – bromo – phenylsulfanyl) -2,4 – Yue group – benzene. The mixture was cooled to 40 ° C, was added to 600ml 15-wt% NaCl, and stirred for 5 minutes. The organic phase was separated, and the aqueous phase was washed 2xl00mwith benzene. The combined organic phase was washed with HCl (aq) NaCl and washed with 100ml 2M 100ml 15-wt%, and then Na 2 S04 dried by activated charcoal (10 g) at reflux for 15 minutes, filtered twice and evaporated to 107.3 g of orange-red oil (103%), the oil was found by HPLC purity of 98%.
To 90 g of the orange-red oil (307 mmol) in 500ml of anhydrous toluene was added 57 g boc-piperazine Qin (307 mmol), degassed with nitrogen for 5 minutes, was added 1.4g Pd2dba3 (1.53 mmol- %; 0.5 mol%) and 2.9g mc-BINAP (4.6 mmol; 1.5 mol-%), degassed and then another 2 minutes, then add 35.4 g of NaOtBu (368 mmol), and heated to 80 ° C for 18 hours. HPLC showed complete conversion to have the reaction mixture was cooled to RT, filtered, and the filter cake was washed with 2 x 100ml of曱benzene. % NaCl, washed twice in Na2S04 dried, added charcoal, refluxed for 30 minutes, filtered twice and evaporated to 140.7 g of a brown oil (4 – – The combined filtrates with 2 x 150ml 15 [2 – (2, 4 – di曱group – phenylsulfanyl) -. phenyl]-BOC-piperazine Qin). The resulting crude oil was dissolved in 300ml MeOH and 200ml 6MHCl (aq.) and refluxed for l hour, after which HPLC showed complete deprotection. After cooling to RT, the vacuum on a rotary evaporator to remove曱alcohol was added 20ml of concentrated NaOH (pH was measured to 13-14), after which the mixture with 1000ml EtOAc – 15 minutes from stirring. The organic phase was collected and dried 300ml 15wtQ /. Saline extraction in Na2S04 dried, and added 46.3 g of fumaric acid in 300mlMeOH (399 mmol) was added. The mixture was heated to reflux, cooled to room temperature and then placed in the tank (-18. C) overnight. The precipitate was collected, washed with 100ml and 100ml of acetone with EtOAc, and dried in vacuo (50 ° C), to give 103.2g of l-[2 – (2,4 – di group – phenylsulfanyl) – phenyl] – piperazine. Qin fumarate (249mmo1), as a white powder, overall yield 81%, determined by LC-MS and the purity was 99% fumarate. Use EtOAc/H20 / concentrated NaOH to the fumarate salt into the free base (l-[2 – (2,4 – dimethyl – phenylsulfanyl) – phenyl] – piperazine Qin), The organic phase was washed with brine, dried over Na 2 S04 sulfate, filtered and to the filtrate was added 34ml48-wto / o of HBr (aq.), to form a white solid precipitated. The solid was collected, and the solid was washed with 1000ml H20 boiling process, the resultant was cooled to room temperature and purified by forming a slurry. The final product was collected by filtration (l-[2 – (2,4 – digroup – phenylsulfanyl) – phenyl] – piperazine hydrobromide Qin Kr), and dried in vacuo (50 ° C), to produce 83g of white powder (total yield 71%).
Source:
1) Bang-Andersen B, Ruhland T, Jørgensen M, Smith G, Frederiksen K, Jensen KG, Zhong H, Nielsen SM, Hogg S, Mørk A, Stensbøl TB “Discovery of 1 -. [2 – (2,4 – dimethylphenylsulfanyl) phenyl] PIPERAZINE (Lu AA21004): a novel multimodal Major Compound for the treatment of depressive disorder. ” Journal of Medicinal Chemistry 54 (9): 3206-21.
2) Thomas Ruhland, Garrick Paul Smith, Benny Bang-Andersen, Ask Puschl, Ejner Knud Moltzen, Kim Andersen,; Phenyl-piperazine derivatives as serotonin reuptake inhibitors; US patent number 7144884 ; also published as CA2462110A1, CA2462110C , CN1319958C, CN1561336A, DE60225162D1, DE60225162T2, DE60233608D1, EP1436271A1, EP1436271B1, EP1749818A2, EP1749818A3, EP1749818B1, US7138407, US7148238, US7683053, US8110567, US8476279, US20050014740, US20060084662, US20060089368, US20070060574, US20110009423, US20120302553, WO2003029232A1; H. Lundbeck A / S;
T · Rouland, G · P · Smith, B · Bang – Anderson, A · Pi Shier, E · K · Moore Cen, K · Anderson; as serotonin reuptake inhibitors phenyl piperazine derivatives matter; CN 1319958 C
T · Rouland, G · P · Smith, B · Bang – Anderson, A · Pi Shier, E · K · Moore Cen, K · Anderson; as serotonin reuptake inhibitors phenyl piperazine derivatives; CN 1561336 A
3) Benny Bang-Andersen; Phenyl-piperazine derivatives as serotonin reuptake inhibitors; US patent number 8476279 B2 ; Also published as CA2462110A1, CA2462110C, CN1319958C, CN1561336A, DE60225162D1, DE60225162T2, DE60233608D1, EP1436271A1, EP1436271B1, EP1749818A2, EP1749818A3, EP1749818B1, US7138407, US7144884, US7148238, US7683053, US8110567, US20050014740, US20060084662, US20060089368, US20070060574, US20110009423, US20120302553, WO2003029232A1; H. Lundbeck A / S;
4) Kim Lasse Christensen; Process for the manufacture of 1 – [2 – (2,4-dimethyl-phenylsulfanyl)-phenyl]-piperazine; PCT application, WO2013102573 A1
5) Benny Bang-Andersen, Joergen Brodersen, Andre Faldt, Rene Holm, Morten Joergensen, De Diego Heidi Lopez, Michael J Mealy, Arne Moerk, Nicholas Moore, Lone Munch Ringgaard, Michael Harold Rock, Tine Bryan Stensboel; 1 – [2 – (2, 4-dimethylphenylsulfanyl)-phenyl] piperazine as a compound with combined serotonin reuptake, 5-ht3 and 5-ht1a activity for the treatment of cognitive impairment; WO2007144005 A1
Updated oct 2015…………….

Vortioxetine (vor-tye-oks-e-teen, trade name Trintellix) is an atypical antidepressant (a serotonin modulator and stimulator) made by Lundbeck and Takeda.[1]
Vortioxetine [1-[2-(2,4-Dimethylphenyl-sulfanyl)-phenyl]-piperazine] is an orally administered small molecule developed as once-daily treatment of major depressive disorder (MDD) and generalized anxiety disorder (GAD). As a drug, Vortioxetine is a bis-aryl-sulphanyl amine compound that combines serotonin (5-HT) reuptake inhibition with other characteristics, including receptor activity modulation.
Animal and in vitro studies indicate that several neurotransmitter systems may be impacted by vortioxetine, with the drug enhancing levels of 5-HT, noradrenaline, dopamine, acetylcholine and histamine in certain areas of the brain, as well as modulating γ-aminobutyric acid and glutamate neurotransmission. Results from additional animal models suggest vortioxetine may also improve measures of cognitive function, such as memory. In healthy volunteers, single or repeated administration of vortioxetine (10 mg) did not impair cognitive function, psychomotor performance or driving ability in a placebo-controlled study.
Medical use
Vortioxetine is used as first-line treatment for major depressive disorder.[1][2][3][4][5]
Pharmacokinetics
Vortioxetine reaches peak plasma concentration (Cmax) within 7 to 11 hours post-administration (Tmax), and its mean terminal half-life (t½) is ≈ 66 hours. Steady-state plasma concentrations are typically reached within two weeks.[1] It has no active metabolites (i.e. it is not a prodrug).[1]
Research
Vortioxetine has been studied in several clinical trials as a potential treatment for general anxiety disorder but results were inconsistent.[9][10]
History
Vortioxetine was discovered by scientists at Lundbeck who reported the rationale and synthesis for the drug (then called Lu AA21004) in a 2011 paper.[7][11]
In 2007, the compound was in Phase II clinical trials, and Lundbeck and Takeda entered into a partnership in which Takeda paid Lundbeck $40 million upfront, with promises of up to $345 million in milestone payments, and Takeda agreed to pay most of the remaining cost of developing the drug. The companies agreed to co-promote the drug in the US and Japan, and that Lundbeck would receive a royalty on all such sales. The deal included another drug candidate, tedatioxetine (Lu AA24530), and could be expanded to include two other Lundbeck compounds.[12]
Vortioxetine was approved by the U.S. FDA for the treatment of major depressive disorder (MDD) in adults in September, 2013,[13] and it was approved in Europe later that year.[14]
Vortioxetine was previously trademarked as Brintellix in the United States, but on May 2, 2016, the US FDA approved a name change to Trintellix in order to avoid confusion with the blood-thinning medication ticagrelor (Brilinta).[15]
WO2015155153, SYNTHESIS OF VORTIOXETINE VIA (2,4-DIMETHYLPHENYL)(2-IODOPHENYL)SULFANE INTERMEDIATE
| LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI) | |
| Inventors:ZUPANCIC, Borut; (SI) |
Vortioxetine is disclosed as Example 1 e in WO 2003/029232 A1 and is described as being prepared analogously to Example 1 . The process used to prepare Example 1 involves the preparation of 1 -(2-((2-(trifluoromethyl)phenyl)thio)phenyl)piperazine on a solid polystyrene support, followed by decomplexation using visible light irradiation, and purification by preparative LC-MS and ion-exchange chromatography. The overall yield for the preparation of vortioxetine is described as 17%.
Several alternative palladium catalyzed processes for the preparation of vortioxetine are described in Examples 17 to 25 of WO 2007/144005 A1 . These processes describe the preparation of vortioxetine from 2,4-dimethylthiophenol and 2-bromoiodobenzene (or 1 ,2-dibromobenzene) starting materials via a 1 -(2-bromo-phenylsulfanyl)-2,4-dimethyl-benzene intermediate. Each of these processes involves the use of a palladium catalyst and a phosphine ligand.
The preparation of vortioxetine is also described by Bang-Andersen et al. in J. Med. Chem. (201 1 ), Vol. 54, 3206-3221 . Here, in a first step, te/t-butyl 4-(2-bromophenyl)piperazine-1 -carboxylate intermediate is prepared from Boc-piperazine and 2-bromoiodobenzene in a palladium catalyzed coupling reaction. te/t-Butyl 4-(2-bromophenyl)piperazine-1 -carboxylate is then reacted with 2,4-dimethylthiophenol, again in the presence of palladium catalyst and a phosphine ligand, to provide Boc-protected vortioxetine. In the final step, vortioxetine is deprotected using hydrochloric acid to give vortioxetine hydrochloride.
WO 2013/102573 A1 describes a reaction between 1 -halogen-2,4-dimethyl-phenyl, 2-halogen-thiophenol and an optionally protected piperazine in the presence of a base and a palladium catalyst consisting of a palladium source and a phosphine ligand.
Each of the above processes has disadvantages. The process described in WO 2003/029232 is low yielding and unsuitable for the large scale production of vortioxetine, whereas the processes described in WO 2007/144005 A1 , WO 2013/102573 A1 and by Bang-Andersen et al. require the use of expensive starting materials, palladium catalyst and phosphine ligand. In addition, the toxicity of palladium is well known, Liu et al. Toxicity of Palladium, Toxicology Letters, 4 (1979) 469-473, and the European Medicines Agency’ s Guideline on the Specification for Residues of Metal Catalysts sets clear limits on the permitted daily exposure to palladium arising from palladium residue within drug substances, http://www.ema.europa.eu. Thus it would be desirable to avoid the use of a palladium catalyst in the synthesis of vortioxetine and the subsequent purification steps required to remove palladium residue from the final pharmaceutical product.
The invention is described below in further detail by embodiments, without being limited thereto.
A general concept of the process of the present invention may be represented in Scheme 1 .

Scheme 1 : General representation of the basic synthetic concept of the present invention.
Scheme 2.

X = NH2: lb
Scheme 2: Representation of a particular synthetic embodiment of the present invention.
Compound III can also be prepared from 2,4-dimethylbenzenethiol (II) and 1 -fluoro-2-nitrobenzene (l”‘a) or 1 -chloro-2-nitrobenzene (l'”b). In the first step (2,4-dimethylphenyl)(2- nitrophenyl)sulfane (III’) is formed and in the second reaction step nitro group is reduced to ami

Z = F: l”‘a
Z = CI: l”‘b
Scheme 3: Representation of a particular synthetic embodiment of the present invention.
Example 7: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-iodophenyl)sulfane V (0.34 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and 2-phenylphenol (68 mg, 0.4 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120°C for 20 h. Water (10 mL) is then added and product is extracted to EtOAc (3 x 10 mL). Combined organic layers were washed with water (3 x 10 mL) and brine (2 x 10 mL) and dried over Na2S04. After evaporation of the solvent crude product is purified by chromatography to afford title compound: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-
3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J= 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Example 8: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine (vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-iodophenyl)sulfane V (0.34 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and N,N-diethyl-2-hydroxybenzamide (39 mg, 0.2 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120 ^ for 20 h. Water (10 mL) is then added and product is extracted to EtOAc (3 x 10 mL). Combined organic layers were washed with water (3 x 10 mL) and brine (2 x 10 mL) and dried over Na2S04. After evaporation of the solvent crude product is purified by chromatography to afford title compound: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J= 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Example 9: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine hydrobromide
(vortioxetine HBr, VII.HBr)

To a solution of vortioxetine VII (1 .80 g, 6.03 mmol) in iPrOAc (20 mL) at room temperature 48% HBr (0.68 mL, 6.03 mmol) was slowly added. Obtained mixture was stirred at room temperature for 1 h, white precipitate was then filtered off, washed with acetone (2 x 20 mL), and dried to afford title compound VII.HBr as a white powder (2.15 g, 94% yield): H NMR (DMSO-d6, 500 MHz) δ 2.23 (s, 3H), 2.32 (s, 3H), 3.15-3.27 (m, 8H), 6.40 (m, 1 H), 6.96 (m, 1 H), 7.08-7.17 (m, 3H), 7.24 (m, 1 H), 7.32 (d, J= 7.8 Hz, 1 H), 8.85 (br, 2H).
Reference Example 1 : Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of piperazine (1 .0 g, 1 1 .6 mmol), NaOtBu (1 .37 g, 13.8 mmol), Pddba2 (40 mg, 0.07 mmol), and 1 ,3-bis(2,6-di-i-propylphenyl)imidazolium chloride (24 mg, 0,07 mmol) in dry and degassed toluene (10 mL) is stirred at room temperature for 1 h. (2,4-Dimethylphenyl)(2-iodophenyl)sulfane V (1 .32 g, 3.86 mmol) is then added, reaction mixture is heated to l OO’C and stirred for 24 h. After cooling to room temperature to the reaction mixture water (5 mL) and Celite (0.4 g) is added. After stirring for 20 min salts are filtered off, organic layer is separated, washed with brine (2 x 10 mL), dried over Na2S04 and solvent is evaporated to afford crude product, which is then purified by chromatography to afford title compound as yellowish crystals: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H),
Reference Example 2: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of piperazine (1 .29 g, 15.0 mmol), NaOtBu (1 .77 g, 17.8 mmol), Pddba2 (52 mg, 0.09 mmol), and rac-BINAP (93 mg, 0,15 mmol) in dry and degassed toluene (10 mL) was stirred at room temperature for 1 h. (2,4-Dimethylphenyl)(2-iodophenyl)sulfane V (1 .70 g, 5.0 mmol) was then added, reaction mixture was heated to 100°C and stirred for 24 h. After cooled to room temperature to the reaction mixture water (5 mL) and Celite (0.4 g) were added. After stirring for 20 min salts were filtered off, organic layer was separated, washed with brine (2 x 10 mL), dried over Na2S04 and solvent was evaporated to afford product as an orange oil (1 .41 g, 95% yield): H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J = 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Comparative Example 1 : Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-bromohenyl)sulfane V” (0.29 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and 2-phenylphenol (68 mg, 0.4 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120°C for 20 h. Vortioxetine VII was not formed.
Comparative Example 2: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-bromophenyl)sulfane V (0.29 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and N,N-diethyl-2-hydroxybenzamide (39 mg, 0.2 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120 ^ for 20 h. Vortioxetine VII was not formed.
WO2007144005A1: Industrial process

Discovery of 1-[2-(2,4-Dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): A Novel Multimodal Compound for the Treatment of Major Depressive Disorder

The synthesis and structure−activity relationship of a novel series of compounds with combined effects on 5-HT3A and 5-HT1A receptors and on the serotonin (5-HT) transporter (SERT) are described. Compound 5m (Lu AA21004) was the lead compound, displaying high affinity for recombinant human 5-HT1A (Ki = 15 nM), 5-HT1B (Ki = 33 nM), 5-HT3A (Ki = 3.7 nM), 5-HT7 (Ki = 19 nM), and noradrenergic β1 (Ki = 46 nM) receptors, and SERT (Ki = 1.6 nM). Compound 5mdisplayed antagonistic properties at 5-HT3A and 5-HT7 receptors, partial agonist properties at 5-HT1B receptors, agonistic properties at 5-HT1A receptors, and potent inhibition of SERT. In conscious rats, 5m significantly increased extracellular 5-HT levels in the brain after acute and 3 days of treatment. Following the 3-day treatment (5 or 10 (mg/kg)/day) SERT occupancies were only 43% and 57%, respectively. These characteristics indicate that 5m is a novel multimodal serotonergic compound, and 5m is currently in clinical development for major depressive disorder.
1-[2-(2,4-Dimethylphenylsulfanyl)phenyl]piperazine Hydrochloride (5m)
ALERT HYDROCHLORIDE DATA
References
- US Label Last updated July 2014 after review in September, 2014. Versions of label are available at FDA index page Page accessed January 19, 2016
- [No authors listed] Vortioxetine. Aust Prescr. 2015 Jun;38(3):101-2. PMID 26648632Free full text
- “Relative efficacy and tolerability of vortioxetine versus selected antidepressants by indirect comparisons of similar clinical studies.”. Curr Med Res Opin 30: 2589–606. Oct 10, 2014. doi:10.1185/03007995.2014.969566. PMID 25249164.
- Köhler S, Cierpinsky K, Kronenberg G, Adli M. The serotonergic system in the neurobiology of depression: Relevance for novel antidepressants. J Psychopharmacol. 2016 Jan;30(1):13-22. PMID 26464458
- Kelliny M, Croarkin PE, Moore KM, Bobo WV. Profile of vortioxetine in the treatment of major depressive disorder: an overview of the primary and secondary literature. Ther Clin Risk Manag. 2015 Aug 12;11:1193-212. PMID 26316764 Free full text
- “Lundbeck’s “Serotonin Modulator and Stimulator” Lu AA21004: How Novel? How Good? – GLG News”.
- ^ Jump up to:a b c Bang-Andersen B, Ruhland T, Jørgensen M, et al. (May 2011). “Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder”. Journal of Medicinal Chemistry 54 (9): 3206–21. doi:10.1021/jm101459g. PMID 21486038.
- N. Moore; B. Bang-Andersen; L. Brennum; K. Fredriksen; S. Hogg; A. Mork; T. Stensbol; H. Zhong; C. Sanchez; D. Smith (August 2008). “Lu AA21004: a novel potential treatment for mood disorders”. European Neuropsychopharmacology 18 (Supplement 4): S321.doi:10.1016/S0924-977X(08)70440-1.
- Pae CU et al. Vortioxetine, a multimodal antidepressant for generalized anxiety disorder: a systematic review and meta-analysis. J Psychiatr Res. 2015 May;64:88-98. PMID 25851751
- Reinhold JA, Rickels K. Pharmacological treatment for generalized anxiety disorder in adults: an update. Expert Opin Pharmacother. 2015;16(11):1669-81. PMID 26159446
- Sanchez C, Asin KE, Artigas F Vortioxetine, a novel antidepressant with multimodal activity: review of preclinical and clinical data. Pharmacol Ther. 2015 Jan;145:43-57. PMID 25016186 Free full text
- Daniel Beaulieu for First Word Pharma. September 5th, 2007 Lundbeck, Takeda enter strategic alliance for mood disorder, anxiety drugs
- FDA approves new drug to treat major depressive disorder, U.S. Food and Drug Administration Press Announcement.
- EMA Brintellix page at EMA site Page accessed January 19, 2016
- Commissioner, Office of the. “Safety Alerts for Human Medical Products – Brintellix (vortioxetine): Drug Safety Communication – Brand Name Change to Trintellix, to Avoid Confusion With Antiplatelet Drug Brilinta (ticagrelor)”. http://www.fda.gov. Retrieved2016-05-02.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7683053 | 2010-03-23 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| US7148238 | 2006-12-12 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US7144884 | 2006-12-05 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US7138407 | 2006-11-21 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2013184291 | 2013-07-18 | THERAPEUTIC USES OF 1-[2-(2,4-DIMETHYL-PHENYLSUFLANYL)PHENYL]PIPERAZINE |
| US8476279 | 2013-07-02 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US2013115292 | 2013-05-09 | ENTERIC TABLET |
| US2012189697 | 2012-07-26 | COMPOSITIONS OF 1-[2-(2,4-DIMETHYL-PHENYLSULFANYL)-PHENYL]PIPERAZINE |
| US2012035188 | 2012-02-09 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]-PIPERAZINE |
| US8110567 | 2012-02-07 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| US2012004409 | 2012-01-05 | Purification of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine |
| US2011201617 | 2011-08-18 | Therapeutic Uses Of Compounds Having Combined SERT, 5-HT3 And 5-HT1A Activity |
| US2011009422 | 2011-01-13 | 1- [2-(2,4-DIMETHYLPHENYLSULFANYL)-PHENYL] PIPERAZINE AS A COMPOUND WITH COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF PAIN OR RESIDUAL SYMPTOMS IN DEPRESSION RELATING TO SLEEP AND COGNITION |
| US2010297240 | 2010-11-25 | 1- [2- (2,4-DIMETHYLPHENYLSULFANYL)-PHENYL] PIPERAZINE AS A COMPOUND WITH COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF COGNITIVE IMPAIRMENT |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094316 | 2015-04-02 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]PIPERAZINE |
| US8969355 | 2015-03-03 | 1-[2-(2,4 dimethylphenylsulfanyl)-phenyl]piperazine as a compound with combined serotonin reuptake, 5-HT3 and 5-HT1a activity for the treatment of cognitive impairment |
| US2015005318 | 2015-01-01 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014371453 | 2014-12-18 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014343287 | 2014-11-20 | Process for the Manufacture of 1-[2-(2,4-Dimethyl-Phenylsulfanyl)-Phenyl]-Piperazine |
| US2014315921 | 2014-10-23 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014256943 | 2014-09-11 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014248355 | 2014-09-04 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014248356 | 2014-09-04 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014163043 | 2014-06-12 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016083359 | 2016-03-24 | 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)-PHENYL]PIPERAZINE AS A COMPOUND With COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF COGNITIVE IMPAIRMENT |
| US2016060215 | 2016-03-03 | New Process For The Synthesis Of 1-(2-((2,4-Dimethylphenyl)Thio)Phenyl)Piperazine |
| US2016015706 | 2016-01-21 | CRYSTALLINE FORMS OF AN ANTIDEPRESSANT COMPOUND |
| US2016009670 | 2016-01-14 | VORTIOXETINE MANUFACTURING PROCESS |
| US9211288 | 2015-12-15 | Compositions comprising vortioxetine and donepezil |
| US2015297585 | 2015-10-22 | COMPOSITIONS COMPRISING VORTIOXETINE AND DONEPEZIL |
| US2015266841 | 2015-09-24 | Novel Crystalline Form Of Vortioxetine Hydrobromide |
| US2015150867 | 2015-06-04 | COMPOSITIONS OF 1-[2-(2,4-DIMETHYL-PHENYLSULFANYL)-PHENYL]PIPERAZINE |
| US2015110873 | 2015-04-23 | ENTERIC TABLET |
| US2015094316 | 2015-04-02 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]PIPERAZINE |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
1-[2-(2,4-Dimethyl-phenylsulfanyl)-phenyl]piperazine
|
|
| Clinical data | |
| Trade names | Trintellix, Brintellix |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 75% (peak at 7–11 hours) |
| Protein binding | 98% |
| Metabolism | extensive hepatic, primarilyCYP2D6-mediated oxidation |
| Biological half-life | 66 hours |
| Excretion | 59% in urine, 26% in feces |
| Identifiers | |
| CAS Number | 508233-74-7 |
| ATC code | N06AX26 (WHO) |
| PubChem | CID 9966051 |
| IUPHAR/BPS | 7351 |
| ChemSpider | 8141643 |
| KEGG | D10184 |
| ChEBI | CHEBI:76016 |
| Synonyms | Lu AA21004 |
| Chemical data | |
| Formula | C18H22N2S |
| Molar mass | 298.45 g/mol (379.36 as hydrobromide) |
/////////////
-
CC(C=C(C)C=C1)=C1SC2=C(N3CCNCC3)C=CC=C2
FDA Approves Perjeta for Neoadjuvant Breast Cancer Treatment

The structure of HER2 and pertuzumab
pertuzumab
Sept. 30, 2013 — The U.S. Food and Drug Administration today granted accelerated approval to Perjeta (pertuzumab) as part of a complete treatment regimen for patients with early stage breast cancer before surgery (neoadjuvant setting). Perjeta is the first FDA-approved drug for the neoadjuvant treatment of breast cancer.
Perjeta was approved in 2012 for the treatment of patients with advanced or late-stage (metastatic) HER2-positive breast cancer. HER2-positive breast cancers have increased amounts of the HER2 protein that contributes to cancer cell growth and survival
cut paste of my old article
he European Medicines Agency (EMA) has approved Roche’s PERJETA (pertuzumab) for patients with previously untreated HER2-positive metastatic breast cancer (mBC)
MARCH 5, 2013 8:59 AM / 4 COMMENTS /
The structure of HER2 and pertuzumab
march 4, 2013
Pertuzumab (also called 2C4, trade name Perjeta) is a monoclonal antibody. The first of its class in a line of agents called “HER dimerization inhibitors”. By binding to HER2, it inhibits the dimerization of HER2 with other HER receptors, which is hypothesized to result in slowed tumor growth.[1] Pertuzumab received US FDA approval for the treatment of HER2-positive metastatic breast cancer on June 8, 2012.[2] Pertuzumab was developed at Genentech and is now owned by Roche which acquired Genentech in 2009.
Clinical trials
Early clinical trials of pertuzumab in prostate, breast, and ovarian cancers have been met with limited success.[3]
The dosage of pertuzumab used in the pivotal phase III CLEOPATRA (Clinical Evaluation of Pertuzumab and Trastuzumab) trial was as follows: IV 840 mg loading dose followed by IV 420 mg every three weeks.[4]
The pharmacokinetics of intravenous pertuzumab appear to be unaffected by age and no drug-drug interaction has been reported with docetaxel. The pharmacokinetics and pharmacodynamics of pertuzumab were summarized in a Feb 2012 review by Gillian Keating.[4]
The combination of pertuzumab plus trastuzumab plus docetaxel, as compared with placebo plus trastuzumab plus docetaxel, when used as first-line treatment for HER2-positive metastatic breast cancer, significantly prolonged progression-free survival, with no increase in cardiac toxic effects in the randomized, double-blind, multinational, phase III CLEOPATRA trial.[5]
Intravenous pertuzumab is currently being evaluated in patients with breast cancer in the following trials: MARIANNE (advanced breast cancer), NEOSPHERE (early breast cancer), TRYPHAENA (HER2-positive stage II/III breast cancer) and APHINITY (HER2-positive nonmetastatic breast cancer).[4]
References
- de Bono, Johann S.; Bellmunt, J; Attard, G; Droz, JP; Miller, K; Flechon, A; Sternberg, C; Parker, C et al. (20 January 2007). “Open-Label Phase II Study Evaluating the Efficacy and Safety of Two Doses of Pertuzumab in Castrate Chemotherapy-Naive Patients With Hormone-Refractory Prostate Cancer”. Journal of Clinical Oncology 25 (3): 257–262.doi:10.1200/JCO.2006.07.0888. PMID 17235043.
- “FDA Approves Perjeta (Pertuzumab) for People With HER2-Positive Metastatic Breast Cancer” (Press release). Genentech. Retrieved 2012-06-09.
- Genentech press release – May 15, 2005
- Keating GM. Pertuzumab: in the first-line treatment of HER2-positive metastatic breast cancer. Drugs 2012 Feb 12; 72 (3): 353-60.Link text
- Baselga J, Cortés J, Kim SB, and the CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 2012 Jan 12; 366 (2): 109-19. Link text
Daclizumab
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | CD25 |
 DACLIZUMAB,
DACLIZUMAB,

Daclizumab is a humanized monoclonal antibody indicated in the United States for prophylaxis of acute organ rejection in patients receiving renal transplants.
It was marketed as Zenepax, but discontinued by Roche in 2009 due to diminishing market demand for that indication. Biogen Idec is currently conducting phase III trials for daclizumab in MS. A phase III trial started in March 2010 is being conducted to determine efficacy of preventing MS relapse.
Study dosing of daclizumab is 150 mg subcutaneously once every 4 weeks versus interferon beta-1a (Avonex) 30 mg intramuscularly given once weekly for 96 to 144 weeks.
Daclizumab (Zenapax®) (molecular wt = 144 kd.) is a humanized monoclonal antibody (IgG1) produced by recombinant DNA technology. It gained FDA approval in Dec 1997. It is known by several other names including HAT (Humanized Anti-Tac), SMART anti-Tac, anti-CD25, and humanized anti-IL2-receptor. It was developed and patented by Protein Design Laboratories (Mountain View, CA) and it is marketed by Hoffman LaRoche (Nutley, NJ ).
Daclizumab is a composite of human (90%) and murine (10%) antibody sequences. In the model below, the murine portions are shown in red and dark blue; the rest of the molecule (gray color) represents the human sequence
The study is aiming for enrollment of 1500 patients and is expected to be complete in January 2014.
more info
Daclizumab (trade name Zenapax) is a therapeutic humanized monoclonal antibody. It is used to prevent rejection in organ transplantation, especially in kidney transplants. The drug is also under investigation for the treatment of multiple sclerosis.
Daclizumab works by binding to CD25, the alpha subunit of the IL-2 receptor of T cells. The drug is marketed in the US, but not in Europe.
Uses
Prevention of organ transplants
Daclizumab is given in multiple doses, the first 1 hour before the transplant operation and 5 further doses given at two week intervals after the transplant. These saturate the receptors and prevent T cell activation and thus prevent formation of antibodiesagainst the transplant.
Like the similar drug basiliximab, daclizumab reduces the incidence and severity of acute rejection in kidney transplantation without increasing the incidence of opportunistic infections.
Daclizumab usage may also be indicated in place of a calcineurin-inhibitor (ciclosporin or tacrolimus) during the early phase after kidney transplantation, when the kidney is recovering and vulnerable to calcineurin-inhibitor toxicity. This has been shown to be beneficial in non-heart beating donor kidney transplantation.
In the United Kingdom, the National Institute for Health and Clinical Excellence (NICE) has recommended its use be considered for all kidney transplant recipients.[citation needed]
Multiple sclerosis
In 2006 it began a Phase II clinical trial that finished in 2007 as a possible multiple sclerosis (MS) treatment. Participants were nine patients with multiple sclerosis not controlled with interferon. Daclizumab was effective in reducing lesions and improving clinical scores.[1] As of June 2013, the drug is in Phase III trials for this indication.[2]
Autoimmune diseases
Daclizumab has also been used to slow the progression of autoimmune diseases, particularly that of birdshot chorioretinopathy.[3]
Common side effects with a frequency of at least 10% include sleeplessness, tremor, headache, arterial hypertension, dyspnoea, gastrointestinal side effects and oedema. In rare cases, the drug can cause severe anaphylaxis.[4]
Daclizumab must not be administered to lactating women.[4]
History
Daclizumab was developed by PDL Biopharma, building on research at the National Institutes of Health (NIH).[5] Since December 1997, it is marketed by Hoffmann-La Roche in the US.
In April 2008, Hoffmann-La Roche submitted an application to have its marketing authorisation withdrawn in the EU for commercial reasons. The drug faced diminishing market demand, according to the company. There were no safety concerns with its use. As of January 2009, its marketing authorisation has been withdrawn and the product discontinued completely.[6][7]
- Rose JW, Burns JB, Bjorklund J, Klein J, Watt HE, Carlson NG (2007). “Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results”.Neurology 69 (8): 785–789. doi:10.1212/01.wnl.0000267662.41734.1f.PMID 17709711.
- ClinicalTrials.gov NCT01462318 An Immunogenicity and Pharmacokinetics (PK) Study of DAC HYP Prefilled Syringe in Relapsing Remitting Multiple Sclerosis (RRMS) (OBSERVE)
- Sobrin L, Huang JJ, Christen W, Kafkala C, Choopong P, Foster CS (2008). “Daclizumab for treatment of birdshot chorioretinopathy”. Arch Ophthalmol. 126 (2): 186–191. doi:10.1001/archophthalmol.2007.49. PMID 18268208.
- “EPAR for Zenapax”. European Medicines Agency. 2007.
- Tsurushita, N.; Hinton, P. R.; Kumar, S. (2005). “Design of humanized antibodies: From anti-Tac to Zenapax”. Methods 36 (1): 69–83.doi:10.1016/j.ymeth.2005.01.007. PMID 15848076. edit
- British National Formulary, Edition 57
- EMEA: Withdrawal of the marketing authorisation in the European Union

{kind=link}