PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Vadadustat, also known as AKB-6548 and PG-1016548, is a potent Hypoxia-inducible factor-proline dioxygenase inhibitor. AKB-6548 works by inhibiting hypoxia inducible factor-prolyl hydroxylase (HIP-PH), leading to stabilization and increased levels of HIFα. In turn HIFα improves production of hemoglobin and red blood cells (RBCs), while maintaining normal levels of erythropoietin (EPO) in patients. We believe this differentiated mechanism of action has the potential to be safer than that of injectable recombinant erythropoietin stimulating agents (rESAs), avoiding supra-physiological levels of EPO and saturation of EPO receptors for prolonged periods of time.
Akebia Therapeutics, under license from Procter & Gamble, and sublicensee Mitsubishi Tanabe Pharma are developing vadadustat, an orally active small-molecule hypoxia-inducible factor prolyl hydroxylase (HIF-PH) inhibitor that stabilize HIF2-α, as a once-daily formulation, for treating anemia. Also the company is investigating AKB-6899, an oral HIF-PH inhibitor, for treating cancer and ocular diseases. In March 2016, the IND application was opened. Aerpio Therapeutics, a spinoff of Akebia, is investigating AKB-4924, a HIF2-α stabilizer, which inhibits HIF prolyl hydroxylase-2, for treating inflammatory bowel disease and wound healing
Hypoxia-inducible factor (HIF) is a transcription factor that is a key regulator of responses to hypoxia. In response to hypoxic conditions, i.e., reduced oxygen levels in the cellular environment, HIF upregulates transcription of several target genes, including those encoding erythropoietin. HIF is a heteroduplex comprising an alpha and beta subunit. While the beta subunit is normally present in excess and is not dependent on oxygen tension, the HIF-alpha subunit is only detectable in cells under hypoxic conditions. In this regard, the accumulation of HIF-alpha is regulated primarily by hydroxylation at two proline residues by a family of prolyl hydroxylases known as HIF prolyl hydroxylases, wherein hydroxylation of one or both of the proline residues leads to the rapid degradation of HIF-alpha. Accordingly, inhibition of HIF prolyl hydroxylase results in stabilization and accumulation of HIF-alpha {i.e., the degradation of HIF-alpha is reduced), thereby leading to an increase in the amount of HIF-alpha available for formation of the HIF heterodimer and upregulation of target genes, such as the Erythropoietin gene. Conversely, activation of HIF prolyl hydroxylase results in destabilization of HIF-alpha {i.e., the degradation of HIF-alpha is increased), thereby leading to a decrease in the amount of HIF-alpha available for formation of the HIF heterodimer and downregulation of target genes, such as VEGF.
The family of hypoxia inducible factors includes HIF- 1 -alpha, HIF-2-alpha, and HIF-3 -alpha.
A new class of prolyl hydroxylase inhibitors and their use to treat or prevent diseases ameliorated by modulation of hypoxia-inducible factor (HIF) prolyl hydroxylase are described in U.S. Patent No. 7,811,595, which is incorporated herein by reference in its entirety. The synthesis of such prolyl hydroxylase inhibitors is described in U.S. Patent Publication No.2012/0309977, which is incorporated herein by reference in its entirety. Such compounds inhibit HIF prolyl hydroxylase, thereby stabilizing HIF-alpha. As a consequence of stabilizing HIF-alpha, endogenous erythropoietin (EPO) production is increased. As with all drugs, proper doses and dosing regimens for treating patients having diseases such as anemia are essential for achieving a desired or optimal therapeutic effect without adverse effects or unwanted side-effects. Indeed, many active compounds fail in clinical trials because an effective and safe dosing regimen cannot be found.
Vadadustat (also known as AKB-6548) in anemia secondary to chronic kidney disease (CKD)
We are developing our lead product candidate, vadadustat, to be the potential best-in-class hypoxia inducible factor–prolyl hydroxylase inhibitor for the treatment of anemia secondary to CKD.
HIF inhibitor Vadadustat (Code AKB-6548) The chemical name N- [5- (3- chlorophenyl) -3-hydroxypyridine-2-carbonyl] glycine,
Vadadustat is a treatment for anemia associated with chronic kidney disease oral HIF inhibitor, is an American biopharmaceutical company Akebia Therapeutics invention in the research of new drugs, has completed Phase II pivotal clinical trial treatment studies, successfully met the researchers set given the level of hemoglobin in vivo target and good security, a significant effect, and phase III clinical trials.
U.S. Patent Publication US20120309977 synthetic route for preparing a Vadadustat: A 3-chlorophenyl boronic acid and 3,5_-dichloro-2-cyanopyridine as starting materials, by-catalyzed coupling methoxy substituted, cyano hydrolysis and condensation and ester hydrolysis reaction Vadadustat, process route is as follows:
Since the entire synthetic route 12 steps long, complicated operation, high cost.U.S. Patent No. 1 2 ^ ¥ disclosed 20070299086 & (^ (Scheme 3 1118 seven seven to 3,5-dichloro-2-cyanopyridine starting material, first-dichloro substituted with benzyloxy, then cyano hydrolysis, condensation, hydrogenation and deprotection trifluorosulfonyl, to give N- [5- trifluoromethanesulfonyloxy-3-hydroxypyridine-2-carbonyl) glycine methyl ester, 3-chlorophenyl and then boronic acid catalyzed coupling reactions, the final ester hydrolysis reaction Vadadustat, process route is as follows:
The synthesis steps long, intermediate products and final products contain more impurities and byproducts, thus purified requires the use of large amounts of solvents, complicated operation, low yield, and because the hydrogenation reaction is a security risk on the production, not conducive to the promotion of industrial production, it is necessary to explore a short process, simple operation, low cost synthetic method whereby industrial production Vadadus tat fit.
Example 1
A) Preparation of N- (3,5_-dichloro-2-carbonyl) glycine methyl ester:
3,5-dichloro-2-pyridinecarboxylic acid (19.2g, 0.10mol) and N, N’_ carbonyldiimidazole (24.3g, 0.15mol) was dissolved in N, N- dimethylformamide (100 mL ), was added glycine methyl ester hydrochloride (15.18,0.12111〇1), 11 was added dropwise diisopropylethylamine (51.7g, 0.40mol), the reaction mixture was stirred 35 ° C for 8 hours, TLC determined the completion of reaction gussets The reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was adjusted to neutral by adding ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give N- (3,5- dichloro-pyridin-2 – carbonyl) glycine methyl ester, an off-white solid (21.6g), a yield of 82.0%, this reaction step is as follows:
1234567 B) Preparation of N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester: 2
1 (3,5-dichloro-2-carbonyl) glycine methyl ester (20 (^, 〇1 76111111), 3-chlorophenyl boronic acid (13.18, 3 83.7mmol), [l, l’- bis (diphenylphosphino) ferrocene] dichloropalladium (2.8g, 3.8mmol), potassium carbonate (14.2g, 4 0. lmo 1) and N, N- dimethylformamide (75mL) was added The reaction flask, the reaction mixture was heated to 60 ° C for 20 hours the reaction was stirred for 5:00, point TLC plates to determine completion of the reaction, the reaction solution was cooled to room temperature, was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, sulfuric acid 6 magnesium dried and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane was recrystallized to give N- [5- (3- chlorophenyl) -3-7-chloro-2-carbonyl] glycine methyl ester, white solid (19.7g), yield 76.4%, this reaction step is as follows:
C) Preparation of N_ [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine:
N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester (19 (^, 56111 111〇1) and sodium methoxide (7.6g, 0.14mol) was dissolved in methanol (150 mL), the reaction mixture was heated to 65 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution was cooled to room temperature, water (300mL) was stirred for 3h, cooled to 0 ° C, stirred for 2h, precipitated solid was filtered, the filter cake was dried to give N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine, off-white solid (17.4 g of), a yield of 96.5%, of the reaction steps are as follows:
D) Preparation Vadadustat:
N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine (16.68,51.7111111〇1) and 48% hydrobromic acid solution (52mL, 0.46mol) added to the reaction bottle, the reaction mixture was heated to 100 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution cooled square ~ 5 ° C, was slowly added 50% sodium hydroxide solution was adjusted to pH 2 at 0 -5 ° C under crystallization 3h, the filter cake washed with ethyl acetate and n-hexane mixed solvent of recrystallization, in finished Vadadustat, off-white solid (15.6g), a yield of 98.0%, this reaction step is as follow
Lanthier et al. (U.S. Patent Application 2012/0309977) described a procedure for synthesizing a compound of Formula (II) starting from 3-chloroboronic acid and 3,5-dichloropicolinonitrile, as shown in the scheme below:
which has an X-ray powder diffraction pattern as shown in FIG. 1. In certain embodiments, Form A of Compound (I) has an X-ray powder diffraction pattern comprising one, two, three, four, or five peaks at approximately 18.1 , 20.3, 22.9, 24.0, and 26.3 °2Θ; and wherein the crystalline Compound (I) is substantially free of any other crystalline form of Compound (I).
Compound (I) as prepared according to e.g., U.S. 7,811,595 and/or U.S. Patent Application No. 13/488,554 and then subjecting the resulting Compound (I)
(I),
to a procedure comprising
a) preparing a solution of Compound (I) in 2-methyltetrahydrofuran;
b) adding n-heptane;
c) heating the suspension {e.g., to about 40-50 °C);
d) cooling the suspension {e.g., to about 0-10 °C); and
c) isolating the crystals.
SYNTHESIS
US 2015361043
Synthesis of vadadustat and its intermediates is described. The process involves Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile with (3-chlorophenyl)boronic acid, selective chloride displacement, simultaneous hydrolysis of nitrile and methyl ether, activation with CDI, condensation with methyl glycinate hydrochloride and finally ester hydrolysis. The process is simple and provides high product yield with high quality. Vadadustat is expected to be useful for the treatment of renal failure anemia (1). Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile (I) with (3-chlorophenyl)boronic acid (II) in the presence of PdCl2(dppf) and K2CO3 in DMF yields 3-chloro-5-(3-chlorophenyl)pyridine-2-carbonitrile (III), which upon selective chloride displacement with NaOMe in refluxing MeOH affords methyl ether (IV). Hydrolysis of nitrile and methyl ether in intermediate (IV) with HBr or HCl at 100 °C furnishes 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (V). After activation of carboxylic acid (V) with CDI or pivaloyl chloride and DIEA in DMSO, condensation with methyl glycinate hydrochloride (VI) in the presence of DIEA provides vadadustat methyl ester (VII). Finally, hydrolysis of ester (VII) with NaOH in H2O/THF produces the target vadadustat (1).
FIG. 1 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitors.
FIG. 2 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor ester prodrugs.
FIG. 3 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor amide prodrugs.
Example 1 describes a non-limiting example of the disclosed process for the preparation of a prolyl hydroxylase ester pro-drug
EXAMPLE 1Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): To a 100 mL round bottom flask adapted for magnetic stirring and equipped with a nitrogen inlet was charged (3-chlorophenyl)boronic acid (5 g, 32 mmol), 3,5-dichloro-2-cyanopyridine (5.8 g, 34 mmol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (0.1 g, 0.13 mmol), dimethylformamide (50 mL) and water (5 mL). The reaction solution was agitated and heated to 45° C. and held at that temperature for 18 hours after which the reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to room temperature and the contents partitioned between ethyl acetate (250 mL) and saturated aqueous NaCl (100 mL). The organic phase was isolated and washed a second time with saturated aqueous NaCl (100 mL). The organic phase was dried for 4 hours over MgSO4, the MgSO4 removed by filtration and the solvent removed under reduced pressure. The residue that remained was then slurried in methanol (50 mL) at room temperature for 20 hours. The resulting solid was collected by filtration and washed with cold methanol (50 mL) then hexanes (60 mL) and dried to afford 5.8 g (73% yield) of an admixture containing a 96:4 ratio of the desired regioisomer. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H)
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): To a 500 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (10 g, 40 mmol), sodium methoxide (13.8 mL, 60 mmol) and methanol (200 mL). With stirring, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to room temperature and combined with water (500 mL). A solid began to form. The mixture was cooled to 0° C. to 5° C. and stirred for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 9.4 g (96% yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (1 g, 4 mmol) and a 48% aqueous solution of HBr (10 mL). While being stirred, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction contents was then cooled to 0° C. to 5° C. with stirring and the pH was adjusted to approximately 2 by the slow addition of 50% aqueous NaOH. Stirring was then continued at 0° C. to 5° C. for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 1.03 g (quantitative yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a nitrogen inlet tube was charged 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (1 gm, 4 mmol), N,N′-carbonyldiimidazole (CDI) (0.97 g, 6 mmol) and dimethyl sulfoxide (5 mL). The reaction mixture was stirred at 45° C. for about 1 hour then cooled to room temperature. Glycine methyl ester hydrochloride (1.15 g, 12 mmol) is added followed by the dropwise addition of diisopropylethylamine (3.2 mL, 19 mmol). The mixture was then stirred for 2.5 hours at room temperature after which water (70 mL) was added. The contents of the reaction flask was cooled to 0° C. to 5° C. and 1N HCl was added until the solution pH is approximately 2. The solution was extracted with dichloromethane (100 mL) and the organic layer was dried over MgSO4 for 16 hours. Silica gel (3 g) is added and the solution slurried for 2 hours after which the solids are removed by filtration. The filtrate is concentrated to dryness under reduced pressure and the resulting residue was slurried in methanol (10 mL) for two hours. The resulting solid was collected by filtration and washed with cold methanol (20 mL) then hexane and the resulting cake is dried to afford 0.85 g of the desired product as an off-white solid. The filtrate was treated to afford 0.026 g of the desired product as a second crop. The combined crops afford 0.88 g (68% yield) of the desired product. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use
EXAMPLE 2Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): A 20 L reactor equipped with a mechanical stirrer, dip tube, thermometer and nitrogen inlet was charged with (3-chlorophenyl)boronic acid (550 g, 3.52 mol), 3,5-dichloro-2-cyanopyridine (639 g, 3.69 mol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (11.5 g, 140 mmol), and dimethylformamide (3894 g, 4.125 L). The reaction solution was agitated and purged with nitrogen through the dip-tube for 30 minutes. Degassed water (413 g) was then charged to the reaction mixture while maintaining a temperature of less than 50° C. 25 hours. The reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to 5° C. and charged with heptane (940 g, 1.375 L) and agitated for 30 minutes. Water (5.5 L) was charged and the mixture was further agitated for 1 hour as the temperature was allowed to rise to 15° C. The solid product was isolated by filtration and washed with water (5.5 L) followed by heptane (18881 g, 2750 ML). The resulting cake was air dried under vacuum for 18 hours and then triturated with a mixture of 2-propanol (6908 g, 8800 mL0 and heptane (1 g, 2200 mL0 at 50° C. for 4 hours, cooled to ambient temperature and then agitated at ambient temperature for 1 hour. The product was then isolated by filtration and washed with cold 2-propanol (3450 g, 4395 mL) followed by heptane (3010 g, 4400 mL). The resulting solid was dried under high vacuum at 40° C. for 64 hours to afford 565.9 g (65% yield) of the desired product as a beige solid. Purity by HPLC was 98.3. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H).
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): A 20 L reactor equipped with a mechanical stirred, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (558 g, 2.24 mol) and sodium methoxide (25% solution in methanol, 726.0 g, 3.36 mol). With agitation, the reaction solution was heated to reflux for 24 hours, resulting in a beige-colored suspension. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to 5° C. and then charged with water (5580 mL). The resulting slurry was agitated for 3 hours at 5° C. The solid product was isolated by filtration and washed with water (5580 mL) until the filtrate had a pH of 7. The filter cake was air dried under vacuum for 16 hours. The filter cake was then charged back to the reactor and triturated in MeOH (2210 g, 2794 mL) for 1 hour at ambient temperature. The solid was collected by filtration and washed with MeOH (882 g, 1116 mL, 5° C.) followed by heptane (205 mL, 300 mL), and dried under high vacuum at 45° C. for 72 hours to afford 448 g (82% yield) of the desired product as an off-white solid. Purity by HPLC was 97.9%. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer, nitrogen inlet and 25% aqueous NaOH trap was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (440.6 g, 1.8 mol) and 37% aqueous solution of HCl (5302 g). While being agitated, the reaction solution was heated to 102° C. for 24 hours. Additional 37% aqueous HCl (2653 g) was added followed by agitation for 18 hours at 104° C. The reaction contents was then cooled to 5° C., charged with water (4410 g) and then agitated at 0° C. for 16 hours. The resulting precipitated product was isolated by filtration and washed with water until the filtrate had a pH of 6 (about 8,000 L of water). The filter cake was pulled dry under reduced pressure for 2 hours. The cake was then transferred back into the reactor and triturated in THF (1958 g, 2201 mL) at ambient temperature for 2 hours. The solid product was then isolated by filtration and washed with THF (778 g, 875 mL) and dried under reduced pressure at 5° C. for 48 hours to afford 385 g (89% yield) of the desired product as an off-white solid. HPLC purity was 96.2%. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (380 g, 1.52 mol) and diisopropylethylamine (DIPEA) (295 g, 2.28 mol). With agitation, the solution was cooled to 3° C. and charged with trimethylacetyl chloride (275.7 g, 2.29 mol) while maintaining a temperature of less than 11° C., The mixture was then agitated at ambient temperature for 2 hours. The mixture was then cooled to 10° C. and charged with a slurry of glycine methyl ester HCl (573.3 g, 4. 57 mol) and THF (1689 g, 1900 mL), then charged with DIPEA (590.2 g, 4.57 mol) and agitated at ambient temperature for 16 hours. The mixture was then charged with EtOH (1500 g, 1900 mL) and concentrated under reduced pressure to a reaction volume of about 5.8 L. The EtOH addition and concentration was repeated twice more. Water (3800 g) was then added and the mixture was agitated for 16 hours at ambient temperature. The resulting solid product was isolated by filtration and washed with a mixture of EtOH (300 g, 380 mL) and water (380 g), followed by water (3800 g), dried under reduced pressure for 18 hours at 50° C. to afforded 443 g (91% yield) of the desired product as an off-white solid. Purity by HPLC was 98.9%. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
Scheme II herein below outlines and Example 2 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase inhibitor from an ester prodrug.
EXAMPLE 3{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3 -chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 50 mL flask is charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (0.45 g, 1.4 mmol), tetrahydrofuran (4.5 mL) and 1 M NaOH (4.5 mL, 4.5 mmol). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was adjusted to pH 1 with concentrated HCl and the solution was heated at 35° C. under vacuum until all of the tetrahydrofuran had been removed. A slurry forms as the solution is concentrated. With efficient stirring the pH is adjusted to ˜2 with the slow addition of 1 M NaOH. The solid which forms was collected by filtration, washed with water, followed by hexane, then dried under vacuum to afford 0.38 g (88% yield) of the desired product as a white solid. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use.
EXAMPLE 4{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (440 g, 1.42 mol), tetrahydrofuran (3912 g, 4400 mL) and 1 M NaOH (4400 mL). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was acidified to a pH of 2 with slow addition of 2M HCl (2359 g). The resulting mixture was concentrated under reduced pressure to a volume of about 7.5 L. Ware (2210 g) was added and the solution cooled to ambient temperature and agitated for 18 hours. The solid product was isolated by filtration and washed with water (6 L). the crude product was transferred back into the reactor and triturated with 2215 g o deionized water at 70° C. for 16 hours. The mixture was cooled to ambient temperature, The solid product was isolated by filtration and washed with water (500 mL) and dried under reduced pressure at 70° C. for 20 hours to afford 368 g (87% yield) of the desired product as an off-white solid. Purity by HPLC was 99.3%. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
Scheme III herein below outlines and Example 3 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase amide prodrug.
EXAMPLE 55-(3-Chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide
Preparation of 5-(3-chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide (6): To a solution of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (749 mg, 3 mmol) in DMF (20 mL) at room temperature under N2 is added 1-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide (EDCI) (0.925 g, 5.97 mmol) and 1-hydroxybenzo-triazole (HOBt) (0.806 g, 5.97 mmol). The resulting solution is stirred for 15 minutes then 2-aminoacetamide hydrochloride (0.66 g, 5.97 mmol) and diisopropylethylamine (1.56 ml, 8.96 mmol) are added. The reaction is monitored by TLC and when the reaction is complete the reaction mixture is concentrated under reduced pressure and H2O added. The product can be isolated by normal work-up: The following data have been reported for compound (6). 1H NMR (250 MHz, DMSO-d6) δ ppm 12.46 (1H, s), 9.17 (1H, t, J=5.9 Hz), 8.55 (1H, d, J=2.0 Hz), 7.93 (1H, d, J=0.9 Hz), 7.75-7.84 (2H, m), 7.49-7.60 (3H, m), 7.18 (1H, s), 3.91 (2H, d, J=5.9 Hz). HPLC-MS: m/z 306 [M+H]+.
Scheme IV herein below depicts a non-limiting example the hydrolysis of an amide pro-drug to a prolyl hydroxylase inhibitor after removal of a R10 protecting group
Beuck S, Schänzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test Anal. 2012 Nov;4(11):830-45. doi: 10.1002/dta.390. Epub 2012 Feb 24. Review. PubMed PMID: 22362605.
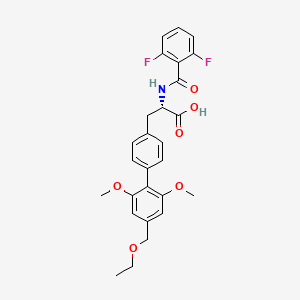
Solid forms of 2-(5-(3-fluorophenyl)-3-hydroxypicolinamido)acetic acid, compositions, and uses thereof
clip
Oct 6, 2015
Akebia Reaches Agreement with FDA and EMA on Vadadustat Global Phase 3 Program
Plans to Initiate Phase 3 PRO2TECT™ Clinical Program by Year-End
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Akebia Therapeutics, Inc. (NASDAQ: AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced the successful completion of the End-of-Phase 2 Meeting process with the United States Food and Drug Administration (FDA) and the Scientific Advice Process with the European Medicines Agency (EMA) for its lead product, vadadustat (formerly AKB-6548), for patients with anemia related to non-dialysis dependent chronic kidney disease (NDD-CKD). The company has reached agreement with both the FDA and EMA regarding key elements of the Phase 3 program, known as the PRO2TECT™ program, and expects to launch the program later this year.
The PRO2TECT™program includes two separate studies and will collectively enroll approximately 3,100 NDD-CKD patients across 500 sites globally. The correction study will address anemia patients not currently being treated with recombinant erythropoiesis stimulating agents (rESAs). The conversion study includes patients currently receiving rESA who will be converted to either vadadustat or the active control with the goal of maintaining their baseline hemoglobin levels. Both studies will include a 1:1 randomization and an open label, active-control, non-inferiority design. Primary endpoints include an efficacy assessment of the hemoglobin response and an assessment of cardiovascular safety measured by major adverse cardiovascular events.
“Akebia’s Phase 3 program is designed to provide the medical community and regulators with a clear understanding of vadadustat’s potential benefit and safety advantages over rESAs, the current standard of care worldwide and, with a positive outcome, to establish vadadustat as the best-in-class treatment option for patients with renal anemia,” stated John P. Butler, President and Chief Executive Officer of Akebia. “We are pleased that the regulators are in agreement regarding the importance of an active-control trial as this design is the most clinically relevant and commercially valuable, and will allow us the quickest path to full enrollment. We are now moving rapidly to launch these studies and advance our goal of bringing forward new treatment options for patients suffering from renal anemia.”
“This Phase 3 program builds on the positive data from our Phase 2 program in NDD-CKD patients which demonstrated that once-daily vadadustat can control and maintain hemoglobin levels in a clinically relevant range while minimizing fluctuations in hemoglobin levels that are associated with increased cardiovascular safety risks,” stated Brad Maroni, M.D., Chief Medical Officer at Akebia. “These two Phase 3 event-driven studies are designed to establish the safety and efficacy of vadadustat in the setting of contemporary clinical practice patterns, and support regulatory approvals globally.”
In addition, Akebia discussed with the FDA and EMA a parallel Phase 3 program, known as the INNO2VATE™ program, for vadadustat in patients with anemia related to chronic kidney disease who are undergoing dialysis (DD-CKD). Akebia expects to formalize its Phase 3 program in DD-CKD patients after presenting the results from its recently completed Phase 2 study to both regulatory agencies.
About Vadadustat (Formerly AKB-6548)
Vadadustat is an oral therapy currently in development for the treatment of anemia related to chronic kidney disease (CKD). Vadadustat is designed to stabilize HIF, a transcription factor that regulates the expression of genes involved with red blood cell (RBC) production in response to changes in oxygen levels, by inhibiting the hypoxia-inducible factor prolyl hydroxylase (HIF-PH) enzyme. Vadadustat exploits the same mechanism of action used by the body to naturally adapt to lower oxygen availability associated with a moderate increase in altitude. At higher altitudes, the body responds to lower oxygen availability with increased production of HIF, which coordinates the interdependent processes of iron mobilization and erythropoietin (EPO) production to increase RBC production and, ultimately, improve oxygen delivery.
As a HIF stabilizer with best-in-class potential, vadadustat raises hemoglobin levels predictably and sustainably, with a dosing regimen that allows for a gradual and controlled titration. Vadadustat has been shown to improve iron mobilization, potentially eliminating the need for intravenous iron administration and reducing the overall need for iron supplementation.
About Anemia Related to CKD
Approximately 30 million people in the United States have CKD, with an estimated 1.8 million of these patients suffering from anemia. Anemia results from the body’s inability to coordinate RBC production in response to lower oxygen levels due to the progressive loss of kidney function, which occurs in patients with CKD. Left untreated, anemia significantly accelerates patients’ overall deterioration of health with increased morbidity and mortality. Renal anemia is currently treated with injectable rESAs, which are associated with inconsistent hemoglobin responses and well-documented safety risks.
About Akebia Therapeutics
Akebia Therapeutics, Inc. is a biopharmaceutical company headquartered in Cambridge, Massachusetts, focused on delivering innovative therapies to patients with kidney disease through HIF biology. The company has completed Phase 2 development of its lead product candidate, vadadustat, an oral therapy for the treatment of anemia related to CKD in both non-dialysis and dialysis patients.
clip
Akebia Announces Positive Top-Line Results from its Phase 2 Study of Vadadustat in Dialysis Patients with Anemia Related to Chronic Kidney Disease
-Treatment with Vadadustat Successfully Maintained Mean Hemoglobin Levels Following Conversion from rESA Therapy-
-Vadadustat Demonstrated a Favorable Safety Profile with Once Daily and Three Times per Week Dosing-
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Akebia Therapeutics, Inc. (NASDAQ:AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced positive top-line results from its Phase 2 study of vadadustat (formerly AKB-6548) in dialysis patients with anemia related to chronic kidney disease (CKD). The study achieved its primary objective, indicating that vadadustat maintained stable hemoglobin (HGB) levels throughout the 16-week treatment period following conversion from recombinant erythropoiesis-stimulating agent (rESA) therapy. Vadadustat demonstrated a favorable safety profile with no drug-related serious adverse events and no deaths. The results highlight the potential of vadadustat, dosed either once daily or three times per week, to safely and predictably manage and sustain HGB levels in CKD patients undergoing dialysis.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy”
The open-label, multi-center, 94 patient study was designed to evaluate the ability of vadadustat to maintain hemoglobin levels in patients undergoing hemodialysis who were previously being treated with rESAs. Patients were assigned to one of three dose cohorts: once daily vadadustat at a starting dose of 300mg, once daily vadadustat at a starting dose of 450mg, or vadadustat three times per week in conjunction with the patient’s hemodialysis schedule at a starting dose of 450mg. The study achieved its primary endpoints of maintaining stable hemoglobin levels over 16 weeks of treatment in all three cohorts of patients converting from rESAs to vadadustat.
Vadadustat was well tolerated among patients in all three dose cohorts. Treatment-emergent adverse events (TEAEs) with vadadustat were balanced across the cohorts. Serious adverse events (SAEs) were reported in 13 subjects (13.8%), well within the expected range for this patient population. There were no drug-related SAEs and no deaths reported in the study.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy,” said Brad Maroni, M.D., Chief Medical Officer at Akebia. “We are impressed with the consistency in hemoglobin levels across the duration of the study, which highlights the ability of vadadustat to control and maintain hemoglobin levels in this patient population. Furthermore, the results indicate that daily and three times per week dosing regimens are both viable options for patients on dialysis.”
John P. Butler, President and Chief Executive Officer of Akebia, stated, “These results further confirm vadadustat as a potential best-in-class anemia treatment for CKD patients, and reinforce our confidence in this product candidate as we advance toward our Phase 3 program. Adding these results to the 12 other clinical studies we have completed, we are confident in the potential for vadadustat to treat anemia in a broad array of patients with CKD. We are pleased to have successfully completed this stage of our drug development and look forward to initiating Phase 3 studies.”
Complete efficacy and safety data from this Phase 2 study will be presented at an upcoming medical meeting.
About the Phase 2 Study Design of Vadadustat in Dialysis Patients with Anemia Related to CKD
The Phase 2 multi-center, open-label study evaluated 94 patients over 16 weeks of treatment, at 20 dialysis centers in the United States, including an assessment of HGB response to the starting dose of vadadustat during the first 8 weeks, followed by an assessment of HGB response to algorithm-guided dose adjustments of vadadustat during the subsequent 8 weeks of treatment. The study enrolled three cohorts, each consisting of approximately 30 CKD patients with anemia undergoing dialysis who were switched from injectable rESA therapy to vadadustat. Patients in the first two cohorts received once daily doses of vadadustat, while patients in the third cohort received vadadustat three times per week in conjunction with their hemodialysis schedule.
Jump up^Gupta N, Wish JB. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am J Kidney Dis. 2017 Jun;69(6):815-826. . doi:10.1053/j.ajkd.2016.12.011. PMID28242135. Missing or empty |title= (help)
Jump up^Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM. Clinical Trial of Vadadustat in Patients with Anemia Secondary to Stage 3 or 4 Chronic Kidney Disease. Am J Nephrol. 2017;45(5):380-388. . doi:10.1159/000464476. PMID28343225. Missing or empty |title= (help)
Vadadustat (Vafseo). Vadadustat (28) is a hypoxia inducible factor prolyl hydroxylase (HIF-PH) inhibitor developed by Akebia Therapeutics. It was approved by the European Commission in April 2023, and recently also by the USFDA, for the treatment of symptomatic anemia associated with chronic kidney disease in adults receiving chronic maintenance dialysis. Vadadustat acts by inhibiting HIFPH, 214 which results in increases of endogenous erythropoietin production, red blood cell synthesis, and iron mobilization. 215 While a number of syntheses of vadadustat (28) have been published in previous patents 216−228 and a journal article, 229 Akebia Therapeutics has published two patents regarding the large-scale preparation of vadadustat (Scheme 52). 218,226 The key intermediate nitrile 28.3 could be accessed in two steps: the neat SNAr reaction between commercially available 2,3,5trichloropyridine (28.1) and 4-DMAP to generate pyridinium salt 28.2, followed by a second SNAr reaction of 28.2 with NaCN. The Suzuki coupling between 28.3 and 3-chlorophenyl boronic acid (28.4) gave the biaryl 28.5, and the subsequent SNAr reaction of 28.5 with NaOMe replaced the 3-chloro substitution on the pyridine ring with a methoxy group, generating intermediate 28.6. Global acidic hydrolysis of both methyl ether and nitrile group in 28.6 gave the 3 hydroxypicolinic acid 28.7. Treatment of 28.7 with DIPEA and excess pivaloyl chloride (PivCl) resulted in the formation of mixed anhydride 28.8 with concomitant acylation of the 3 hydroxy group. Without isolation of 28.8, glycine methyl ester hydrochloride (28.9) was then charged with additional DIPEA to generate the corresponding amide 28.10. The residual amount (∼0.5%) of 28.7 in 28.10 was hard to remove, but this impurity could be effectively rejected with an extra amount of DIPEA during workup and solvent switch. Finally, the Opivaloyl group and methyl ester were both removed via basic hydrolysis, giving vadadustat (28) in about 90% yield from 28.7.
REF
(215) Pergola, P. E.; Spinowitz, B. S.; Hartman, C. S.; Maroni, B. J.; Haase, V. H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016, 90, 1115−1122. (216) Lanthier, C. M.; Gorin, B.; Oudenes, J.; Dixon, C. E.; Lu, A. Q.; Copp, J. D.; Janusz, J. M. Preparation of [(3-hydroxypyridine-2carbonyl)amino]alkanoic acids, esters and amides as prolyl hydroxylase inhibitors. US 20120309977, 2012. (217) Li, X.; Chen, J. Process for the preparation of vadadustat. CN105837502, 2016. (218) Gorin, B. I.; Lanthier, C. M.; Luong, A. B. C.; Copp, J. D.; Gonzalez, J. Process for preparing 2-[[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino]acetic acid. WO 2019217550, 2019. (219) Kou, J.; Li, Y.; Xiao, Q.; Lin, B.; Sun, J.; Wang, Z.; Luo, Z.;Huang, F. Preparation method of vadadustat. CN 110903238, 2020. (220) Machida, K.; Yasukouchi, H.; Nishiyama, A. Method for producing vadadustat intermediate. WO 2020217733, 2020. (221) Xiao, Q.; Lin, B.; Kou, J.; Sun, J.; Qiu, X.; Wang, Z.; Luo, Z.;Huang, F. Preparation of vadadustat intermediate. CN 111848505,2020
(222) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Li, Y.; Sun, J.; Jin, L.; Luo, Z.; Huang, F. Preparation of vadadustat and intermediate thereof. CN 111205222, 2020. (223) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Luo, Z.; Huang, F.; Li, Y. Preparation of vadadustat and intermediate thereof. CN 111423367, 2020. (224) Xiao, Q.; Qiu, X.; Lin, B.; Kou, J.; Li, Y.; Sun, J.; Wang, Z.; Luo, Z.; Huang, F. Preparation of vadadustat. CN 111320577, 2020. (225) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Qiu, X.; Cai, X.; Li, Y.; Luo, Z.; Huang, F. Method for preparing vadadustat and intermediate thereof. WO 2021179540, 2021. (226) Jurkauskas, V.; Jung, Y. C.; Kwon, T.; Kannan, A.; Gondi, V. B. Manufacturing process for 3,5-dichloropicolinonitrile for synthesis of vadadustat. WO 2022006427, 2022. (227) Chen, Z.; Zheng, Y.; Zhang, L.; Yu, C.; Liu, L.; He, B. Preparation of a pyridine compound used for the preparation of vadadustat. CN 117843565, 2024. (228) Patel, K. R.; Thakrar, V. H.; Mehta, T. B.; Wagh, A. G.; Patel, J. A.; Patil, R. R.; Solanki, Y. U.; Ladumor, C. B. A process for the preparation of Vadadustat or salts thereof. WO 2024079708, 2024. (229) Lin, B. Y.; Kou, J. P.; Wu, S. M.; Cai, X. R.; Xiao, Q. B.; Li, Y. L.; Hu, J.; Li, J. B.; Wang, Z. Q. Development of a robust and scalable process for the large-scale preparation of Vadadustat. Org. Process Res. Dev. 2021, 25, 960−968.
.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Mitsubishi reported that tenatoprazole was still in Phase I clinical trials in 2007[4]:27 and again in 2012.[3]:17
Tenatoprazole has an imidazopyridine ring in place of the benzimidazole moiety found in other proton pump inhibitors, and has a half-life about seven times longer than other PPIs.[5]
Tenatoprazole is a novel imidazopyridine derivative and has an imidazopyridine ring in place of the benzimidazole moiety found in other proton pump inhibitors. It is activated more slowly than other proton pump inhibitor, but its inhibition is resistant to reversal.Tenatoprazole has an extended plasma half-life in comparison with those of all other proton pump inhibitors; this makes it more potent in the treatment of nocturnal acid breakthrough than esomeprazole, one of the most popular proton pump inhibitors.
Tenatoprazole belongs to the class of covalent proton pump inhibitors (PPIs), which is converted to the active sulfenamide or sulfenic acid by acid in the secretory canaliculus of the stimulated parietal cell of the stomach.This active species binds to luminally accessible cysteines of the gastric H+,K+-ATPase, resulting in disulfide formation and acid secretion inhibition.Tenatoprazole binds at the catalytic subunit of the gastric acid pump with a stoichiometry of 2.6 nmol mg−1 of the enzyme in vitro. In vivo, maximum binding of tenatoprazole was 2.9 nmol mg−1of the enzyme at 2 h after intravenous (IV) administration.
Tenatoprazole, or (+)-5-methoxy-2-{[(4-methoxy-3,5-dimethyl-2-pyridyl) methyl] sulfinyl} imidazo-[4,5-b] pyridine, is described in Patent No. EP 254,588. It belongs to the group of drugs considered as proton pump inhibitors, which inhibit the secretion of gastric acid and are useful in the treatment of gastric and duodenal ulcers. It can also be used to treat gastro-oesophageal reflux, digestive bleeding and dyspepsia, because of its relatively long elimination half-life, as described in the application for French patent No. FR 02. 13113.
The first known derivative of this series of proton pump inhibitors was omeprazole, described in Patent No. EP 001,529, which is endowed with properties which inhibit the secretion of gastric acid and is widely employed as an anti-ulcerative in human therapeutics.
In addition to omeprazole, other proton pump inhibitors are well known, and particular mention can be made of rabeprazole, pantoprazole and lansoprazole, which all exhibit structural analogy and lansoprazole, which all exhibit structural analogy and belong to the group of pyridinyl methyl sulfinyl benzimidazoles. These compounds are sulfoxides presenting with asymmetry at the level of the sulphur atom, and therefore generally take the form of a racemic mixture of two enantiomers.
Like omeprazole and other sulfoxide with an analogue structure, tenatoprazole has an asymmetric structure and may therefore be present in the form of a racemic mixture or of its enantiomers. Thus it may exist in the form of its two enantiomers with R and S configurations, or (+) or (−), respectively.
Recent studies have shown that, unlike all the other proton pump inhibitors such as, for example, omeprazole or lansoprazole, and unexpectedly, tenatoprazole is endowed with a markedly prolonged duration of action, resulting from a plasma half-life which is about seven times longer. Thus the clinical data collected have shown that tenatoprazole enables a degree of symptom relief and healing of gastric lesions which is superior to that achieved by other drugs belonging to the same therapeutic category of proton pump inhibitors, which thus allows its effective use in the treatment of atypical and oesophageal symptoms of gastro-oesophageal reflux, digestive bleeding and dyspepsia, as indicated above.
Studies performed by the application have made it possible to show that the two enantiomers contribute differently to the properties of tenatoprazole, and that the two enantiomers, (+) and (−) exhibit significantly different pharmacokinetic properties. Thus it is possible to prepare medicinal products with specific activity by isolating the enantiomers, and these enantiomers themselves exhibit a different pharmacokinetic profile from that of the known racemic mixture. It then becomes possible to use each of these enantiomers more effectively in precise indications for the treatment of perfectly identified pathologies.
Anti-ulcer drug tenatoprazole (tenatoprazole) is a new proton pump inhibitor, by the Japanese company Tokyo Tanabe, Japan’s Mitsubishi Corporation and Japan’s Hokuriku pharmaceutical companies jointly developed, has passed Phase II clinical trials. It acts on gastric parietal cells, reducing treatment of gastric ulcer, duodenal ulcer, reflux wall cell H + / K + -ATP activity, inhibition of gastric acid secretion, and H. pylori antibacterial activity, mainly for esophagitis and Zhuo – Ellison syndrome and gastric acid secretion disorders related diseases. Compared with the same types of drugs, Tenatoprazole suppress H + / K + -ATP enzyme activity is stronger, more stable, its efficacy than similar products currently widely used in clinical omeprazole strong 7 times. It has not been in the domestic market, nor ratified the production, with broad market prospects and development potential.
Proton pump inhibitors (proton pump inhibitors) for the treatment of acid-related diseases, the past ten years a wide range of clinical applications, better effect of the drug. It can quickly pass through the stomach wall membrane, gathered in a strongly acidic secretory tubules, and H + / K + -ATP enzyme (proton pump) thiol groups covalently bonded to form a disulfide bond, proton pump inactivation, inhibition of the enzyme H + / K + transport, so as to achieve the effect of acid suppression. Proton pump inhibitors and conventional clinical application of gastric acid suppression drugs H2 receptor antagonists compared with different sites of action and have different characteristics, namely acid-suppressing effect at night is good, rapid onset of acid inhibition strong and long time, easy to take these drugs can quickly and efficiently inhibit gastric acid secretion and clearance of Helicobacter pylori, it is widely used gastric ulcer, duodenal ulcer, reflux esophagitis and Zhuo – Ellison syndrome and other diseases treatment. Currently, proton pump inhibitors has been listed on the main omeprazole, lansoprazole, pantoprazole, rabeprazole and esomeprazole.Physical and
chemical properties ofwhite or white crystalline powder, melting point 174 ~ 175 ℃. Soluble in chloroform, insoluble in alcohol and water.
This product and other proton pump inhibitors as compared to chemically stable. China had 34 omeprazole preparations from Portugal, Brazil, India, China and other 13 countries, the stability of the measurements were made. The results showed that only six products (18%) during the trial showing good physical and chemical stability of. 27 products (79%) less (including Chinese product), the active ingredient a significant chemical decomposition, color and physical properties such as dissolution, are also a corresponding change. The results of a stability test designed to compare the various proton pump inhibitors show investigated eight days at 60 ℃, relative humidity of 75%, after omeprazole decomposition only 3% of the active ingredient, the tenatoprazole 77% of the data, said Alpha pantoprazole stability far superior to omeprazole, is already developed similar products in the most promising products.
Synthesis
Matsuishi, N.; Takeda, H.; Iizumi, K.; Murakami, K.; Hisamitsu, A. US Patent 4,808,596, 1989
Synthesis of Tenatoprazole 1 commences with the coupling of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 in the presence of potassium hydroxide affords 4 with 73% yield in ethanol and chloroform. The oxidation of the penultimate sulfide intermediate4 with m-CPBA in chloroform (100 vol) afforded 1
synthesis of 1 begins with the solvent-free condensation of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 to deliver the sulfide intermediate4 with 98% yield.
The final step of the synthesis is the oxidation of the sulfide intermediate with m-CPBA to form tenatoprazole 1. The sulfide intermediate 4 on treatment with 0.9 equiv of m-chloroperbenzoic acid (m-CPBA) at −10 to −15 °C afforded the crude tenatoprazole which was isolated as its sodium salt. The sodium salt of tenatoprazole 5 was purified by recrystallsation using dimethyl formamide and ethyl acetate (2:1 ratio) to yield the pure crystalline tenatoprazole sodium 5. Treatment of tenatoprazole sodium 5 with dil. HCl in the presence of acetone and water afforded the pure tenatoprazole 1
Tenatoprazole is a new type of gastric H + / K + -ATP enzyme inhibitors (proton pump inhibitor PPI), the chemical name 5-methoxy-2- (4-methoxy-3, 5-dimethyl-2-methylsulfinyl) imidazole and W, 5-b] pyridine, useful in the treatment of gastric ulcer, duodenal ulcer, reflux esophagitis and Zhuo – Ai syndrome and gastric acid secretion disorders related diseases.The drug was developed by Japan’s Tokyo Tanabe, Japan’s Mitsubishi Corporation and Japan’s Hokuriku pharmaceutical companies.Compared with other varieties of the same type, which inhibit H + / K + -ATP enzyme activity is stronger, ulcers of various tests are effective, and significantly improve the stability compared with other proton pump inhibitors.
US patent US4808596 “hidazo [4,5_b] pyridine compounds and pharmaceutical compositions containing same)) synthesis process disclosed Tenatoprazole the below formula:
By The route of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride with 2-mercapto-5-methoxy-imidazole, 5-b] pyridine under basic conditions condensation of Intermediate 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine, and then oxidizing the Thai duly omeprazole.This route for the synthesis of pull azole classic line, many pull azoles such as omeprazole can be synthesized by a similar route, this route mild condition, simple operation.But the route condensation, oxidation treatment after use of large amounts of toxic solvent chloroform, is not conducive to industrial scale; lower oxidation yields, the resulting Tenatoprazole containing unreacted starting materials 2- [2_ (3,5 – dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine, further comprising a sulfone by-product, N- oxide, N- oxide sulfone, These by-products may interfere with purification of tenatoprazole.
Japanese Patent invention Wo 丨 J JP05222038 “5_methoxy-2- [[(4_methoxy-3, 5-dimethyl-2-pyridyl) methyl] thio] imidazo [4,5 ~ b] pyridine and intermediates)) male
Synthesis open Tenatoprazole the below formula:
4-chloro-2-chloromethyl-3,5-dimethylpyridine -N- oxide 2_ mercapto _5_ methoxy-imidazo – [4, 5-b] pyridine in alkaline under condensation of Intermediate 5-Methoxy-2- (4-chloro-3,5-dimethyl-2-methylthio Bi) imidazo W, 5-b] pyridine-oxide -N- ( yield 82%), then refluxed in a solution of sodium methoxide in methanol to give 5-methoxy-2- (4-oxo-3,5-dimethyl-2-methyl sulfide) imidazo W , 5-b] pyridine -N- oxide (income ¥ 71%), and then at room temperature in methylene chloride, phosphorus trichloride treated with deoxy (yield 95%), and finally oxidation in Tenatoprazole (income Rate not reported).The novel synthetic route, mild reaction conditions, simple operation, the yield of each step is higher, but the route is too long resulting in a total yield is not high, prolonged and rising production costs.
To a reaction flask was added 2-mercapto-5-methoxy-imidazole, 5-b] pyridine 18. lg, 12g of sodium hydroxide and water 144. 8g, stirred and dissolved at 25 ° C, was added dropwise within Ih 20g of the 2-chloromethyl-dimethyl-4-methoxy _3,5- pyridine hydrochloride and 60g of water were mixed solution dropwise at 25 ° C the reaction 2h, the reaction is completed, filtered, washed with water 144. 8g, 36. 2mL ethanol and washed to obtain a wet powder; wet powder was dried at 50 ° C in vacuo to constant weight to give 2- [2_ (3,5-dimethyl) -4-methoxy-pyridylmethyl sulfide -5 – methoxy] imidazo [4,5-b] pyridine 32. Og;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine 30g, dichloromethane 300g, methanol 15g, and dissolved with stirring; cooled to -10 ° C, was added dropwise the 15g and 485g m-chloroperbenzoic acid in methylene chloride mixed solution, dropwise addition the reaction temperature was controlled at -10 ° C, the dropping time of the pool; the dropwise addition, the temperature control at -10 ° C, the reaction 30min; completion of the reaction, at 10 ° C by the dropwise addition of lithium hydroxide and 135g water 15g mixed solution, drip complete, insulation stirred Ih; filtered cake was washed with acetone 60mL, get wet powder; wet powder was dried at 35 ° C under vacuum to constant weight to give Tenatoprazole lithium salt ^ g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, acetone 63mL, water IOOmL, cooling M0 ° C, dropping lmol within lh / L hydrochloric pH7 0, drops. Albert, stirring 30min; the filter cake washed with water 50mL, washed with acetone and 50mL, wet powder was dried at 35 ° C under vacuum to constant weight to give Tenatoprazole 19. Sg.
Example 2:
a) 2- [2- (3,5-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5_b] pyridine (4) Preparation: To the reaction flask was added 2-mercapto-5-methoxy-imidazo 44,5-b] pyridine 18. lg, 11. 2g of potassium hydroxide and water 217mL, stirred and dissolved at! 35 ° C, was added dropwise within 2h by the 33. 3g of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride and 133. 2mL water mixed solution, dropwise at 35 ° C the reaction 4h, the reaction is completed, filtration, water 217mL, 72. 4mL ethanol and washed to obtain a wet powder; wet powder was dried at 60 ° C in vacuo to constant weight to give 2- [2- (3,5-dimethyl) -4-methoxy-pyridylmethyl sulfide -5-methoxy-yl] imidazo W, 5-b] pyridine 33. Ig;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 400mL, methanol 50mL, stirring to dissolve; cooled to _15 ° C, was added drop by the m-chloroperoxybenzoic acid 16g of mixed solution of dichloromethane and 400mL , the process reactor temperature control was added dropwise at -20 ° C, the dropping time 2. 5h; the dropwise addition, the temperature control _15 ° C, the reaction 35min; completion of the reaction, at 15 ° C by the dropwise addition of 20g of hydrogen Lithium oxide and 200mL water mixed solution, drip completed, insulation mixing 1. 5h; filtration, the filter cake washed with acetone 90mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 6g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, ethanol 75mL, water 150mL, cooled to 10 ° C, dropping 6mol / L hydrochloric pH8 0 within 2h,. drops Albert, stirring 40min; the filter cake washed with water 100mL, washed with acetone IOOmL, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 5g.
Example 3:
a) 2- [2- (3,5-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5_b] pyridine (4) Preparation: To the reaction flask was added 2-mercapto-5-methoxy-imidazo 44,5-b] pyridine 18. lg, 8.4g of lithium hydroxide and water 180ml, stirred and dissolved at 30 ° C, was added dropwise within 1. 5h by the Guang .6g 2-chloro-3,5-dimethyl-4-methoxy-pyridine hydrochloride and 90mL water mixed solution, drop end at 30 ° C reaction 3h, the reaction is complete, filtration, water 217mL , washed with 85mL ethanol to obtain a wet powder; wet powder was dried at 55 ° C in vacuo to constant weight to give 2- [2- (3,5-dimethyl) -4-methoxy-5-pyridylmethyl sulfide oxy] imidazo [4,5-b] pyridine 32. 4g;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 600mL, methanol 60mL, stirring to dissolve; cooled to -20 ° C, was added drop by the m-chloroperoxybenzoic acid 18g of mixed solution of dichloromethane and 600mL , dropwise addition the reaction temperature is controlled at _20 ° C, the dropping time of the pool; the dropwise addition, the temperature control at _20 ° C, the reaction 40min; completion of the reaction, at 20 ° C by the dropwise addition of lithium hydroxide and 300mL 30g water mixed solution, drip complete insulation mixing tank; filter, the filter cake washed with acetone and 120mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 7g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, methanol 75mL, water 120mL, cooled to 5 ° C, dropping dilute hydrochloric acid within 1 5h tune pH7 5,.. drops Albert, stirring 35min; the filter cake washed with water 75mL, 75mL acetone washed, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 6g.
Example 4:
a) 2- [2- (3,5-dimethyl) -4-methoxy-picolyl thioether _5_ methoxy] imidazo [4,5_b] pyridine ⑷ Preparation of: To a solution The reaction flask was added 2-mercapto-5-methoxy imidazole -½, 5-b] pyridine 18. lg, IOg sodium hydroxide and water 150ml, stirred and dissolved at 30 ° C, the 1. 5h dropwise added from 21 . 5g of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride and 90mL water mixed solution, dropwise at 30 ° C the reaction 3h, completion of the reaction, was filtered, washed with water 217mL, The wet powder was washed with ethanol to give 85mL; wet powder was dried at 55 ° C in vacuo to constant weight to give 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide ] imidazo [4,5-b] pyridine 32. 3g;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 500mL, methanol 60mL, stirring to dissolve; cooled to -20 ° C, was added drop by the m-chloroperoxybenzoic acid 18g of mixed solution of dichloromethane and 500mL , the process reactor temperature control was added dropwise at -20 ° C, the dropping time pool; the dropwise addition, the temperature control in -20 ° C, the reaction 40min; completion of the reaction, at 20 ° C by the dropwise addition of lithium hydroxide 30g and 300mL water mixed solution, drip complete insulation mixing tank; filter, the filter cake washed with acetone and 120mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 6g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, isopropanol 75mL, water 120mL, cooled to 5 ° C, dropping 3mol / L hydrochloric within 1 5h. . pH7 5, drops Albert, stirring 35min; the filter cake washed with water 75mL, 75mL acetone washed, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 7g.
PAPER
An Improved Synthesis of Antiulcerative Drug: Tenatoprazole
Department of Research and Development, Srini Pharmaceuticals Ltd., Plot No. 10, Type-C, Road No. 8, Film Nagar, Jubilee Hills, Hyderabad-500033, Andhra Pradesh, India, Department of Chemistry, Osmania University, Tarnaka, Hyderabad-500007, Andhra Pradesh, India and Research and Development, Integrated Product Development Organization, Innovation Plaza, Dr. Reddy’s Laboratories Ltd., Bachupally, Qutubullapur, R. R. Dist. 500 072, Andhra Pradesh, India
* To whom correspondence should be addressed. Telephone: +91 9490783736. E-mail: drkvr_ou@yahoo.com;kvgr1951@rediffmail.com., †Srini Pharmaceuticals Ltd.
, ‡Osmania University.
, §Dr. Reddy’s Laboratory Ltd.
An efficient, cost-effective and multikilogram-scale process for the synthesis of tenatoprazole 1, an antiulcerative drug, is described. The key steps in this synthesis involve the coupling of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 to yield 4 and its subsequent oxidation with m-CPBA to produce sulfoxide 1. The process has been scaled up for the multikilogram-scale of compound 1 with an overall yield of 72%. The new process requires no purification process and affords the target compound 1 with 99.8% purity by HPLC.
the (+) enantiomer of tenatoplazole can be obtained by using chloroform, an industrially acceptable solvent, in accordance with the method proposed by Umemura et al. (J. Org. Chem. 1993, 58, 4592) as follows:
Example 1 (−)-5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridineThe conditions for preparative chromatography, shown as an example, are as follows:
Column: 265×110 mm ChiralPak®
Chiral Stationary Phase selector of the Amylose tris type [(S)-a methylbenzylcarbamate]
Flow rate: 570 ml/min
Detection: UV 240 nm
Temperature: Ambient temperature
These conditions are implemented on a liquid preparative chromatography apparatus.
Introduce approximately 2 g of the racemic mixture if tenatoprazole exhibiting purity higher than 99.5%. The (−) enantiomer is identified by measuring the angle of optical rotation, which must be laevogyre. This measurement can be performed directly on the column, the product being dissolved in the solvent (acetonitrile).
Example 2 (+)-5-methoxy-2-{(4-methoxy-3, 5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridine(R)-(+)-binaphthol 85 g (0.311 mol, 0.2 equivalence), ortho titanic acid isopropyl 42 g (0.148 mol, 0.1 equivalence), water 55 g (3.06 mol) and chloroform 7.5 L were stirred for 1 hour at room temperature. To the resultant, 5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]thio}imidazo[4,5-b]pyridine (MPI), 0.5 kg, was added and stirred for 0.5 hours at room temperature. The thus-prepared mixture was cooled to 5° C. and then 70% aqueous solution of tert-butylhydroperoxide, 0.4 L (approx. 3.0 mol, 2.0 equivalence) was added and stirred for 72 hours at the same temperature as above. After the reaction endpoint was confirmed by HPLC, an aqueous solution of sodium hydroxide was added thereto to separate the aqueous layer, thus removing foreign matter. Then, the resultant was concentrated. Ethyl acetate was added to concentrated residues, which were then heated and suspended. The thus-prepared crude crystalline substances were dissolved in water and neutralized to pH 6.8 with a diluted sulfuric acid solution which was chilled with ice. Deposited crystals were filtered, dried and recrystallized by addition of ethanol to obtain (+)-5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridine {(+)-TU-199}
When measurements were carried out, for a solubility of (+), (−) forms and a racemic form (±) of tenatoprazole in relation to water, it was found that the (+) form dissolved almost 3 times greater than the racemic body and (−) form dissolved over 2 times greater than the racemic form, exhibiting favorable physical properties in preparing drugs (refer to Table 2 below).
TABLE 2
(+) form
(−) form
(±)racemic form
Solubility (water) μg/mL
93.0
74.4
34.6
CLIPS
Tenatoprazole is a pyridinylmethylsulfinyl imidazopyridine compound, which is a weak base. This compound has three pKas. One is the pyridine pKa of pyridinylmethyl moiety and the others are the imidazole pKa and the pyridine pKa of the imidazopyridine moiety. The pyridine pKa1 enables tenatoprazole accumulation in the acidic canaliculus of the parietal cell. Protonation of the imidazopyridine ring enhances electron deficiency at the C-2 position, allowing intramolecular rearrangement to the active form. The active form is the sulfenic acid and/or cyclic sulfonamide, and reacts with luminal cysteine thiols of the enzyme to inhibit the enzyme activity
Synthesis route from 2-mercapto-5-methoxy-imidazo [4,5-b] pyridine (2) and 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride ( 3) by nucleophilic substitution synthesis of 2- (4-methoxy-3,5-dimethyl-2-methylthio) -5-methoxy-imidazo [4,5-b] pyridine (4) the oxidation of 4 1. Figure 1 is a synthesis route of tenatoprazole
《Organic Process Research & Development》 20081112 Somaiah Sripathi et al. An Improved Synthesis of Antiulcerative Drug:Tenatoprazole 第804-806页 1-6 第13卷,
2
*
《Synthetic Communication》 20080101 Liyan Dai et al. Improved Synthetic Approach to Tenatoprazole 第576-582页 1-6 第38卷,
Teneligliptin (INN; trade name Tenelia) is a pharmaceutical drug for the treatment of type 2 diabetes mellitus. It is approved for use in Japan.[1] It belongs to the class of anti-diabetic drugs known as dipeptidyl peptidase-4 inhibitors or “gliptins”.[2] {(2S,4S)-4-[4-(3-Methyl-1-phenyl-1H-pyrazol-5-yl)-1-piperazinyl]-2-pyrrolidinyl}(1,3-thiazolidin-3-yl)methanone
Teneligliptin was launched in Japan in 2012 by Mitsubishi Pharma and Daiichi Sankyo for the treatment of type 2 diabetes mellitus. In 2013, the indication was partially changed to include it as a combination therapy with existing oral hypoglycemic agents, such as biganides, alpha-glucosidaseinhibitors, rapid-acting insulin secretagogues, and insulin preparations, as well as sulfonylureas and thiazolidines that had been approved for the combination.
In 2014, the product was registered in KR for the treatment of type 2 diabetes mellitus. In 2013, Mitsubishi Tanabe Pharma filed for approval in Japan for use of the compound as combination therapy for the treatment of diabetes type 2.
3-{(2S,4S)-4-[4-(3-methyl-l -phenyl- 1 H- pyrazol-5-yl)- l-piperazinyl]-2-pyrrolidinylcarbonyl}-l , 3-thiazolidine is represented structurally by a compound of formula (I):
Teneligliptin (CAS 760937-92-6) is a novel, potent and long-lasting dipeptidyl peptidase-4 inhibitor in treatment of type 2 diabetes. Dipeptidyl-peptidase-4 (DPP- 4) inhibitor has been demonstrated to improve glycemic control, in particular postparandial hyperglycemic control.
Despite of their common mechanism of action, DPP-4 inhibitors show marked structural heterogeneity. DPP-4 inhibitors may be classified into peptidomimetic (i.e. sitagliptin, vildagliptin, saxagliptin, and anagliptin) and non-peptidomimetic (i.e. alogliptin and linagliptin) subtypes.
Teneligliptin, is chemically known as a 3- {((2S,4S)-4-(4-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-yl 25 carbonyl}thiazolidine hemipentahydrobromide hydrate and is peptidomimetic with the molecular formula of C22H30N6OS.2½HBr.xH2O and molecular weight of 642.88 g/mol for hemipentahydrobromide. The hydrate can be from mono to dihydrate.
U.S. Patent No. 7,074,794 B2 (the US ‘794) discloses teneligliptin as L-proline derivative and its pharmaceutically acceptable salts which exhibits a Dipeptidyl 5 peptidase IV (DPP-IV) inhibitory activity, which is useful for the treatment or prophylaxis of diabetes, obesity, HIV infection, cancer metastasis, dermopathy, prostatic hyperplasia, periodontitis, autoimmune diseases and the like.
The example-222 of the US ‘794 discloses the process for the preparation of teneligliptin as trihydrochloride salt U.S. Patent No. 8,003,790 B2 (the US ‘790) discloses salts of proline derivative, solvate thereof and production method thereof. In particular, the US ‘790 discloses 2.0 hydrochloride or 2.5 hydrochloride; 2.0 hydrobromide or 2.5 hydrobromide, and hydrates thereof teneligliptin.
The US ‘790 B2 further discloses different salts 15 of teneligliptin which are incorporated herein as reference in their entirety U.S. PG-Pub. No. 2011/0282058 A1 discloses salts of 3-{((2S,4S)-4-(4-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-ylcarbonyl}thiazolidine with mono-, di- and tri-basic acids or a solvate thereof. 20 International (PCT) publication No. WO 2012/165547 A1 discloses a process for preparation of teneligliptin and pharmaceutically acceptable salts thereof.
International (PCT) publication No. WO 2007/127635 A2 (the WO ‘635 A2) discloses a process for the preparation of diketo-piperazine and piperidine 25 derivatives. In particular, the WO ‘635 A2 discloses the process for preparation of 4-oxo-2-(thiazolidine-3-carbonyl)-pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (III)] by reacting piperazine with aryl halide.
International (PCT) publication No. WO 2012/099915 A1 (the WO ‘915 A1) 5 discloses the process for the preparation of deuterated thiazolidine derivatives. The WO ‘915 A1 also discloses the process for the preparation of 1-(3-methyl-1- phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by condensation of 5- chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by deprotection of Boc-protected 1-(3-methyl-1-phenyl-1Hpyrazol-5-yl)piperazine with triflouroacetic acid.
U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine by condensation of 5-chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine in presence of n-BuLi in tetrahydrofuran. Bioorganic & Medicinal Chemistry, 14(11), 3662-3671 (2006),
Bioorganic & Medicinal Chemistry, 20(16), 5033-5041 (2012) and U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate by reacting (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid with 25 thiazolidine in presence of HOBT and EDC.HCl in dimethylformamide solvent.
Bioorganic & Medicinal Chemistry, 15(2), 641-655 (2007) discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3- carbonyl)pyrrolidine-1-carboxylate by treating (2S,4S)-tert-butyl 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate with tetrabutylammonium fluoride in tetrahydrofuran.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the 5 process for the preparation of herein compound (II) after by reacting 1-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) with (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate in presence of sodium triacetoxyborohydride. There is provided different alternative processes for the preparation of teneligliptin and intermediates thereof.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) also discloses the process for the preparation of 4-[4-(5-methyl-2-phenyl-2H-pyrazol-3-yl)-piperazin- 1-yl]-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (II)] after by reacting 1-(3-methyl-1-phenyl-1H-pyrazol-5- 15 yl)piperazine [herein compound (V)] with (2S,4S)-tert-butyl 4-[[(1,1- dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate in presence of trifluoromethylsulfonic anhydride and diisopropylethylamine. 3 – [[(2S, 4S) -4- [4- (3- methyl-1-phenyl–1H- pyrazol-5-yl) -1-piperazinyl ] -2-pyrrolidinyl] carbamoyl] thiazolidine, having the formula below, is a very novel DPP-4 inhibitor potential.
World Patent Application No. W02012099915 for Ge Lieting discloses a process for the preparation route is as follows:
Journal B10rganic & Medicinal Chemistry, 2012, 20, 5705-5719 also discloses a preparation method for Ge Lieting, the route is as follows:
[0009] 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine, was prepared for the Ge Lieting key intermediate. Journals B10rganic & Medicinal Chemistry, 2012,20,5705-5719 reported the preparation of the intermediates prepared route is as follows:
[0011] The preparative route after the N-Boc-N- acetoacetyl piperazine phenylhydrazine and methanesulfonic acid in an ethanol solution of the reaction at room temperature 14h, concentrated under reduced pressure after addition of pyridine.Was added phosphorus oxychloride in pyridine, 20h post treatment reaction at room temperature the reaction system. The compound obtained above was then added trifluoroacetic acid was dissolved in methylene chloride after, after treatment at room temperature for 1.5h to give 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine.
The reaction process requires mesylate mesylate flammable, easy-absorbent deliquescence, and has a strong corrosive and irritating, easy to cause the body burns; phosphorus oxychloride, a highly toxic substance, water violent hair in the air smoke, hydrolyzed into phosphoric acid and hydrogen chloride, is very unstable, to operate a lot of trouble; trifluoroacetic acid is highly corrosive and irritant, can cause the body burns; low yield of the reaction (10%). Seeking a simple operation, high reaction yield, low cost and suitable for industrial production production process 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine has a very important role in the field of medicine.
…………………………………….
since the capture is staggered, compd 165 is not clear in above pic see below
Description: LR as Lawesson reagent (Lawesson Reagent), is a sulfur oxygen exchange reagent. The present invention provides a method for preparing key intermediates Ge Lieting method, comprising the steps of: (I) N-Boc-N- acetoacetyl piperazine Lawesson’s reagent in the presence of an organic solvent, with a phenylhydrazine of the formula occurs ⑴ reaction shown:
(2) the step (1) The product was dissolved in an organic solvent, the following formula (II) in concentrated hydrochloric acid to deprotected shown:
格列汀 refers to 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine
Example 5: Preparation of {(2^,.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -vHpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
Activated carbon (10 g) was added to a solution of the residue (obtained in Example 4) in isopropyl alcohol (1000 mL) at 30°C to 35°C. The reaction mixture was filtered through a Hyflo® bed. The filtrate was heated to a temperature of 70°C to 75°C. Hydrobromic acid (48%; 168 g) was slowly added to the filtrate at 70°C to 75°C over a period of 10 minutes to 15 minutes. The reaction mixture was stirred for 2.5 hours at 70°C to 77°C. The progress of the reaction was monitored by HPLC. After completion of the reaction, the reaction mixture was cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes. The reaction mixture was filtered to obtain a solid. The solid obtained was washed with isopropyl alcohol (2 x 200 mL), and dried at 50°C under reduced pressure for 15 hours to obtain crude {(25*,45)-4-[4-(3-methyl-l-phenyl-lH- pyrazol-5 -yl)piperazin- 1 -yl]pyrrolidin-2-yl} ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate.
Yield: 90%
Example 6: Purification of {(2^’.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -yllpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
hemipentahydrobromide hydrate (100 g; prepared according to the process of Example 5) in ethanol (700 mL) was heated at 70°C to 75°C to obtain a solution. The solution was filtered at the same temperature. The filtrate was allowed to cool to a temperature of 65 °C to 68°C, and deionized water (10 mL) was added at the same temperature. The solution was cooled to a temperature of 55°C to 60°C, and stirred at the same temperature for 2 hours. The solution was further cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes to obtain a solid. The solid was filtered, washed with ethanol (100 mL), and dried at 45°C to 50°C under reduced pressure for 18 hours to 20 hours to obtain pure {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin-l- yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone hemipentahydrobromide hydrate .
Process for the preparation of n-protected (5s)-5-(1,3-thiazolidin-3-ylcarbonyl)pyrrolidin-3-one
Patent
Submitted
Granted
Proline derivatives and use thereof as drugs [US7060722]
2005-11-03
2006-06-13
Proline derivatives and the use thereof as drugs [US7074794]
2004-06-03
2006-07-11
Proline derivatives and use thereof as drugs [US2006173056]
2006-08-03
SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US8003790]
2009-08-27
2011-08-23
METHOD OF TREATING ABNORMAL LIPID METABOLISM [US2010305139]
2010-12-02
COMBINED USE OF DIPEPTIDYL PEPTIDASE 4 INHIBITOR AND SWEETENER [US2010113382]
2010-05-06
CONCOMITANT PHARMACEUTICAL AGENTS AND USE THEREOF [US2009082256]
2009-03-26
PROPHYLACTIC/THERAPEUTIC AGENT FOR ABNORMALITIES OF SUGAR/LIPID METABOLISM [US2009088442]
2009-04-02
SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US2011282058]
2011-11-17
Joanne Bronson, Amelia Black, T. G. Murali Dhar, Bruce A. Ellsworth, and J. Robert Merritt. “Teneligliptin (Antidiabetic)”. Annual Reports in Medicinal Chemistry48: 523–524. doi:10.1016/b978-0-12-417150-3.00028-4.
SB683699 is an alpha4 integrin antagonist that had been studied in phase II trials at GlaxoSmithKline under a license from Mitsubishi Tanabe Pharma for the oral treatment of multiple sclerosis (MS) in Europe. GlaxoSmithKline and Tanabe Seiyaku (now Mitsubishi Tanabe Pharma) had been studying the drug candidate for the treatment of asthma, rheumatoid arthritis (RA) and Crohn’s disease
MECHANISMS/EFFECTS
HUMAN:
Similar mechanism of action to natalizumab (α4-integrin blocker), but its faster elimination could improve safety profile
In a further aspect the present invention provides for a process for the preparation of compound of formula (II) which comprises coupling the compound of formula (V)
Suitable coupling conditions for the compound of formula (V) and the compound of formula (VI) include those shown in Scheme 2. In a further aspect of the invention there is provided the compound of formula (V):
1H NMR characterisation data for the compound of formula (V) were generated on an isolated and purified batch. 1H-NMR spectra were recorded on a Bruker Avance 400 at 400MHz, using TMS as an internal reference.1H NMR (400 MHz, DMSO-D6) δ ppm 1.17 (t, J=7.09 Hz, 3 H) 2.96 (dd, J=13.82, 9.90 Hz, 1 H) 3.1 1 (dd, J=13.82, 5.26 Hz, 1 H) 4.12 (q, J=7.09 Hz, 2 H) 4.63 (ddd, J=9.78, 7.82, 5.38 Hz, 1 H) 7.15 (t, J=7.95 Hz, 2 H) 7.25 (d, J=8.31 Hz, 2 H) 7.47 – 7.55 (m, 3 H) 9.23 (d, J=7.83 Hz, 1 H).
The present invention provides a process for the preparation of the compound of formula
which process comprises the steps: a) hydrolysis of an ester of formula (I la):
Recrvstallisation of (2S)-2-{r(2,6-difluorophenyl)carbonyllamino)-3-r4′-r(ethyloxy)methyll- 2′,6′-bis(methyloxy)-4-biphenylyllpropanoic acid
(2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)- 4-biphenylyl]propanoic acid (9.38Kg) was charged into a clean reactor, followed by ethyl acetate (46.9L). The solution was heated to 50°C and filtered into the pre-warmed (35°C) crystallizing vessel. A line-wash with ethyl acetate (9.4L) was carried out. The combined ethyl acetate solutions were heated to 50°C, stirred to ensure complete dissolution. Filtered heptane (9.4L) was added maintaining the temperature at 50°C then the solution cooled to 30°C and seeded with (2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4 – [(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4-biphenylyl]propanoic acid (47g) slurried in 1 :9 ethyl acetate:heptane (0.47L). The slurry was aged for 2 hours at 30°C. Filtered heptane (75L) was added over 3 hours. The slurry was then cooled to 0°C over 1 hour. The mixture was aged at 0°C for 1 hour then the solid was filtered off, washed with isopropyl ether (29.6L and dried under vacuum at 50±3°C to give the product (8.55Kg, 91 %). Characterised by having an infrared absorption spectrum with significant absorption bands at about 754, 768, 800, 820, 849, 866, 1006, 1 100, 1 122, 1 157, 1 188, 1225, 1242, 1268, 1292, 1317, 1352, 1417, 1466, 1530, 1580, 1624, 1650, 1662, 171 1 , 1728, 2938, 3302cm“
(1) The product obtained in Example l-(4) (2.1 g) was acylated with 2 , 6-difluorobenzoyl chloride in a similar manner as described in Example 1 -(5) to give N- (2, 6-difluorobenzoyl) – 4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L-phenylalanine ethyl ester (2.75 g) . mp . 70-72 °C; IR (Nujol) 3400, 3263, 1735, 1654, 1624 cm“1; MS (APCI) m/z 500 (M+H) . (2) To a solution of the product obtained above (1.72 g) in DMSO (20 ml) were added Et3N (4.8 ml) and S03«pyridine (5.6 g) successively at room temperature. The whole mixture was stirred at room temperature for 25 minutes. The reaction mixture was poured into ice-water, and then the mixture was extracted with EtOAc. The organic layer was sequentially washed with 5% aqueous HCl, H20 and brine, dried (Na2S04) and then evaporated. The residue was purified by column chromatography (silica gel; eluent: n-hexane/EtOAc 5:1 to 1:1) to yield N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-formylphenyl) -L- phenylalanine ethyl ester (1.54 g) . mp. 114-116°C; IR (Nujol)
(3) The product obtained above (716 mg) was converted into the title compound (428 mg) in a similar manner as described in Example 1- (7) . mp . 87-89°C; IR (Neat+CHC13) 3300, 1739, 1668 cm“1; MS (APCI) m/z 528 (M+H) .
(1) The product obtained in Example 2- (4) (1.00 g) was acylated with 2 , 6-difluorobenzoyl chloride to give N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L- phenylalanine methyl ester (873 mg) in a similar manner as described in Example l-(5). IR (Nujol) 3257, 1743, 1655, 1624 cm“1; MS (APCI +Q1MS) m/z 503 (M+NH4) , 486 (M+H) . (2) The product obtained above (860 mg) was converted into the title compound (220 mg) in a similar manner as described in Example 2- (6) and (7).
The product obtained in Example 10 (200 mg) was hydrolyzed in a similar manner as described in Example 3 to give the title compound (160 mg) . The product obtained in Example 11 (220 mg) was also hydrolyzed in a similar manner as described in Example 3 to give the title compound (167 mg) . mp. 156-158°C; IR (Nujol) 1735, 1655 cm“1; MS (ESI) m/z 498 (M-H) .
wherein X1 is a halogen atom, X2 is a halogen atom, Q is a group of the formula -CH2– or -(CH2)2– and Y is a lower alkyl group, or a pharmaceutically acceptable salt thereof, which has excellent inhibitory activity against α4 integrin-mediated cell adhesion.
Thus, the present invention relates to a process for preparing a compound of the formula (I) :
wherein the symbols are the same as defined above, or a pharmaceutically acceptable salt thereof, comprising : (1) coupling a compound of the formula (VI) :
wherein Z is a leaving group, R1NH is a protected amino group and C02R is a protected carboxyl group with a compound of the formula (V) :
wherein the symbols are the same as defined above, removing the protecting group from the protected amino group, and if necessary, converting the resulting compound into a salt, to yield a compound of the formula (IV) :
wherein the symbols are the same as defined above, or a salt thereof,
(2) condensing the compound (IV) or a salt thereof with a compound of the formula (III) :
wherein the symbols are the same as defined above, a salt or a reactive derivative thereof to yield a compound of the formula (II) :
(trifluoromethanesulfonyloxy) benzene propionate are described in J. Med. Chem. , 33: 1620 (1990) and JP-A-7- 157472, respectively. 4-Bromo-3, 5-dimethoxybenzyl alcohol is described in, for example, J. Med. Chem. , 20: 299 (1977), and can also be prepared according to the following process.
Firstly, 4-bromo-3, 5-dihydroxybenzoic acid is methylated to give methyl 4-bromo-3, 5-dimethoxybenzoate, which is then reduced to yield 4-bromo-3, 5-dimethoxy benzyl alcohol. The methylation can be carried out by reacting with dimethyl sulfate in the presence of a base in a suitable solvent (e.g., ethyl acetate). The reduction can be carried out by reacting with an reducing agent (e.g., lithium alminium hydride, sodium borohydride and calcium borohydride) in a suitable solvent (e.g., tetrahydrofuran) .
EXAMPLES
The following Examples are provided to further illustrate the process of preparation according to the present invention. In the following examples, some compounds may be referred to by different compound name depending on the nomenclature, as illustrated below.
Another name 1: (2S) -2- [ (2, 6-difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) phenyl]propanoic acid
Another name 2: N- [ 2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
EXAMPLE 1 (1) Under nitrogen atmosphere, pyridine (130.3 g) and trifluoromethanesulfonic anhydride (170.4 g) were added dropwise to a solution of ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-hydroxybenzenepropionate
(170.0 g) in dichloromethane (1.7 L) at 10 ° C or below. After stirring for 1 hour at the same temperature, water
(850 ml) was added dropwise to the mixture and the mixture was stirred for 2 hours at the same temperature. The organic layer was washed with 10 % aqueous citric acid solution and aqueous saturated sodium hydrogen carbonate solution, and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy)benzenepropionate (242.5 g) as oil . MS (m/z) : 441 (M+) (2) Under nitrogen atmosphere, to a mixture of ethyl (αS)- – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzenepropionate (66.2g), 4- ethoxymethyl-2, 6-dimethoxyphenylboric acid (54.0 g) , triphenylphosphine (9.83 g) and N-methylpyrrolidone (330 ml) were added palladium acetate (1.68 g) and diisopropylamine (24.9 g ), and the mixture was heated at 90 °C. After stirring for 1 hour at the same temperature, the mixture was cooled and toluene and water were added. The organic layers were washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [[ (1, 1-dimethylethoxy) carbonyl] amino] – 4′ -ethoxymethyl-2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionate (90.1 g) as oil.
The product was dissolved in ethanol (330 ml) , and after addition of p-toluenesulfonic acid monohydrate (28.5 g) , the mixture was stirred for 2 hours at 75 °C. After cooling to room temperature, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was dissolved in ethyl acetate with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS)-α- amino-4′ -ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4- propionate p-toluenesulfonate (63.4 g) .
MS (m/z) : 387 (M+-p-toluenesulfonic acid), M.p. 127-129°C
(3) To a mixture of ethyl (αS) -α-amino-4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate p- toluenesulfonate (29.0 g) , sodium hydrogen carbonate (15. 2 g) , water (290 ml) and ethyl acetate (290 ml) was added dropwise 2, 6-difluorobenzoyl chloride (9. 6 g) at 15 °C or below and the mixture was stirred for 30 minutes at the same temperature. The ethyl acetate layer was washed with saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo. The residue was recrystallized from isopropanol-water to yield ethyl (αS) -oi- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (26.4 g) . MS (m/z) : 527 (M+) , M.p. 87-89°C (4) To a solution of sodium hydroxide (2.9 g) in water- tetrahydrofuran (317 ml-159 ml) was added ethyl (oιS)-α- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate (31.7 g) at 15°C and the mixture was stirred for 4 hours at the same temperature. After neutralizing with IN HC1, the organic solvent was removed in vacuo. The aqueous layer was cooled, the crystalline precipitates were collected by filtration and recrystallized from ethanol-water to yield (αS) -a- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionic acid (28.8 g) . MS (m/z): 499 (M+) , M.p. 154-155°C
EXAMPLE 2 (1) Under nitrogen atmosphere, a mixture of ethyl (oιS)-o:- [[ (1, 1-dimethylethoxy) carbonyl] amino] -4-bromobenzene propanoate (11.17 g) , 4-ethoxymethyl-2, 6- dimethoxyphenylboronic acid (10.80 g ), palladium acetate (0.34 g), triphenylphosphine (1.57 g) , anhydrous potassium carbonate (12.44 g) , iV-methylpyrrolidone (56 ml) and water (11 ml) was stirred for 50 minutes at 80 °C. After completion of the reaction, the mixture was cooled to room temperature and extracted with ethyl acetate and water. The organic layer was washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution, dried over magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure to yield ethyl (αS)-α- [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate (20.4 g) as oil. The product was dissolved in ethanol (100 ml) , and after addition of p-toluenesulfonic acid monohydrate (5.7 g) , the mixture was stirred for 1.5 hours at 75 °C. After cooling, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was suspended in toluene with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS) – -amino-4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate p- toluenesulfonate (13.80 g) . (2) The compound obtained in the above step (1) was treated in the same manner as described in Example 1 (2) to (4) to yield (αS) -a- [ [2 , 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionic acid. The physicochemical data were the same as that obtained in Example 1.
EXAMPLE 3
To a solution of ethyl (αS) -α- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (500 g ) in water (12.6 ml) and dioxane (50 ml) was added hydrochloric acid (12.4 g) and the mixture was stirred for 60 hours at 60 “C. The organic solvent was removed in vacuo and the aqueous layer was cooled. The crystalline precipitates were collected by filtration and recrystallized from ethanol- water to yield (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionic acid (426 mg) . The physicochemical data were the same as that obtained in Example 1.
REFERENCE EXAMPLE 1
(1) To a mixture of 4-bromo-3, 5-dimethoxybenzylalcohol (44.5 g) , triethylammonium benzyl chloride (2.05 g) and 20% aqueous sodium hydroxide solution (288 g) was added diethyl sulfate (41.7 g) under ice-cooling, and the mixture was stirred overnight at 25-30 °C. After stirring for 1 hour at 70 °C, the mixture was cooled and extracted with toluene. The toluene layer was washed with water and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield 4-bromo-3, 5- dimethoxybenzyl ethyl ether (49.5 g) as colorless oil. MS (m/z): 276 (M++2) , 274 (M+)
(2) Under nitrogen atmosphere, to a solution of 4-bromo- 3, 5-dimethoxybenzyl ethyl ether (440.0 g) in tetrahydrofuran (4.0 L) was added dropwise n-butyl lithium (1.6 M n-hexane solution, 1.1 L) at -60°C. After stirring for 15 minutes at the same temperature, trimethyl borate (249.3 g) was added. The temperature of the mixture was gradually elevated, followed by stirring for 1 hour under ice-cooling. To the mixture was added dropwise 10% aqueous sulfuric acid solution (835 g ) . The mixture was extracted with ethyl acetate and the organic layer was washed with water and saturated aqueous NaCl solution. After drying over magnesium sulfate, the solvent was removed in vacuo. The residue was dissolved in isopropyl ether with heating and cooled. The crystalline precipitates were collected by filtration and dried to yield 4-ethyoxymethyl-2, 6- dimetoxyphenylboronic acid (312.9 g) . M.p. 59-61°C
REFERENCE EXAMPLE 2
(1) To a suspension of 4-bromo-3, 5-dihydroxybenzoic acid (95.0 kg) in ethyl acetate (950 L) were added anhydrous potassium carbonate (270.8 kg) and dimethyl sulfate (174.7 kg) . The mixture was heated at 50-80 ‘C for about 4 hours and partitioned by adding water. The organic layer was washed with water and saturated aqueous NaCl solution and concentrated under reduced pressure. The residue was suspended into methanol, stirred under heating and cooled. The crystalline precipitates were collected by filtration and dried to yield methyl 4-bromo-3, 5-dimethoxybenzoate (98.8 kg) as pale yellow crystals. MS (m/z): 277 (M++2) , 275 (M+) , M.p. 120-122°C
(2) To a solution of calcium chloride (46.5 kg) in ethanol (336 L) were added tetrahydrofuran (672 L) and methyl 4- bromo-3, 5-dimethoxybenzoate (96.0 kg) to obtain a suspension. To the suspension was added sodium borohydride
(31.7 kg) by portions at room temperature, and the mixture was stirred for about 9 hours at temperature of room temperature to 45 °C. The reaction mixture was added dropwise to aqueous HC1 solution and stirred for about 16 hours at room temperature. Organic solvent was removed in vacuo, and water (1440 L) was added to the residue and stirred for 1 hour at 50 °C. After cooling, the crystalline precipitates were collected by filtration and dried to yield 4-bromo-3, 5-dimethoxybenzyl alcohol (83.3 kg) as colorless crystals. MS (m/z): 249 (M++2), 247 (M+) , M.p. 100-102°C.
INDUSTRIAL APPLICABILITY The process for preparation of the present invention makes it possible to afford a compound of the formula (I) or a pharmaceutically acceptable salt thereof with high- purity, in a high yield and inexpensively, and, therefore, the process of the present invention is industrially very useful.
useful as calcium-sensitive receptor (CaSR) agonists for treating hyperparathyroidism. a CaSR agonist, being developed by Kyowa Hakko Kirin, under license from Mitsubishi Tanabe, for treating secondary hyperparathyroidism (phase 2 clinical, as of March 2015).
The present invention provides a novel crystal form of an arylalkylamine
compound. Specifically, a novel crystal form of
4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1- yl)phenylacetic acid has
excellent stability, and is therefore useful as an active ingredient for
a medicine. The present invention also provides an industrially
advantageous method for producing an arylalkylamine compound.
The present invention provides a novel crystal form of an arylalkylamine compound. Specifically, a novel crystal form of 4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1- yl)phenylacetic acid has excellent stability, and is therefore useful as an active ingredient for a medicine. The present invention also provides an industrially advantageous method for producing an arylalkylamine compound.
(1) To a mixed solution containing 33.5 g of 3-hydroxypiperidine and 62.7 ml of triethylamine dissolved in 250 ml of methylene chloride was added dropwise a solution of 55.7 ml of benzyloxycarbonyl chloride in 150 ml of methylene chloride, and the mixture was stirred at room temperature for 16 hours. To the reaction mixture were added a saturated aqueous citric acid and chloroform, the mixture was stirred and the liquids were separated. The organic layer was dried, the solvent was evaporated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 75.5 g of benzyl 3-hydroxypiperidine-1-carboxylate.
MS•APCI (m/z): 236 [M+H]+
(2) 800 ml of a solution of 52.4 ml of oxalyl chloride in methylene chloride was cooled to −78° C., 53.2 ml of DMSO was added dropwise to the solution, and the mixture was stirred at −78° C. for 0.5 hour. A solution of 75.5 g of benzyl 3-hydroxypiperidine-1-carboxylate dissolved in 200 ml of methylene chloride was added dropwise to the mixture, and further 293 ml of triethylamine was added dropwise to the same, and the mixture was stirred for 16 hours while a temperature thereof was gradually raised to room temperature. To the reaction mixture were added a saturated aqueous sodium bicarbonate solution and chloroform, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated to obtain 83.7 g of 1-benzyloxycarbonyl-3-piperidone. MS•APCI (m/z): 234 [M+H]+
(3) To a solution of 83.7 g of 1-benzyloxycarbonyl-3-piperidone dissolved in 1.2 liters of methylene chloride was added 55.0 g of (R)-(+)-1-(1-naphthyl)ethylamine, and after the mixture was stirred at room temperature for 2 hours, 69 ml of acetic acid and 160 g of sodium triacetoxy borohydride were added to the mixture, and the mixture was stirred at room temperature for 15 hours. To the reaction mixture was added an aqueous sodium hydroxide to make the mixture basic, and then, chloroform was added to the mixture, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 98.7 g of benzyl 3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
(4) To a solution of 40.95 g of triphosgene dissolved in 800 ml of methylene chloride was added dropwise a mixed solution containing 80.6 g of benzyl 3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate and 86.6 ml of triethylamine dissolved in 200 ml of methylene chloride at 0° C., and the mixture was stirred at room temperature for 16 hours. To the reaction mixture was added water, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was washed with 200 ml of diethyl ether, and the crystal collected by filtration was recrystallized from chloroform and diethyl ether to obtain 48.9 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate.
Further, the filtrate was purified by silica gel column chromatography (hexane:ethyl acetate=8:1→0:1) to obtain 5.82 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate and 14.5 g of benzyl (S)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate.
(5) To a solution containing 54.6 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate dissolved in 700 ml of tetrahydrofuran was added 350 ml of water, and the mixture was stirred under reflux for 15 hours. After tetrahydrofuran was evaporated, a saturated aqueous sodium bicarbonate solution and chloroform were added thereto, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 24.3 g of benzyl (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
(6) To a solution containing 24.2 g of benzyl (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate dissolved in 250 ml of methanol was added 2.5 g of palladium carbon (10% wet), and the mixture was shaked under hydrogen atmosphere at 3 atm at room temperature for 40 hours. Palladium carbon was removed, and the solvent was evaporated, the residue was washed with ethyl acetate-chloroform (10:1), and collected by filtration to obtain 15.3 g of (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine (the following Reference example Table, Reference example 3.001(a)). MS•APCI (m/z): 255 [M+H]+
(7) By using 14.5 g of benzyl (S)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate, the same treatment was carried out as in the above-mentioned (5) to obtain 4.74 g of benzyl (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
Moreover, by using 4.7 g of benzyl (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate, the same treatment was carried out as in the above-mentioned (6) to obtain 2.89 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine. MS•APCI (m/z): 255 [M+H]+
(8) To a solution of 3.46 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine dissolved in 15 ml of methanol was added dropwise 20 ml of a solution of 4M hydrochloric acid in ethyl acetate, and the mixture was stirred. The reaction mixture was concentrated under reduced pressure, diethyl ether was added to the residue, washed and dried to obtain 3.33 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine dihydrochloride
Kyowa Hakko Kirin Announces Commencement of Phase 2b Clinical Study of KHK7580 in Patients with Secondary Hyperparathyroidism in Japan
Tokyo, Japan, August 29, 2014 — Kyowa Hakko Kirin Co., Ltd. (Tokyo: 4151, President and CEO: Nobuo Hanai, “Kyowa Hakko Kirin”) today announced the initiation of a phase 2b clinical study evaluating KHK7580 for secondary hyperparathyroidism patients receiving hemodialysis in Japan.
This randomized, placebo-controlled, double-blind, parallel-group, multi-center study is designed to evaluate efficacy and safety in cohorts comprising KHK7580, its placebo and cinacalcet and initial dose of KHK7580 for secondary hyperparathyroidism patients receiving hemodialysis.
KHK7580 is a small molecular compound produced by Mitsubishi Tanabe Pharma Corporation (President & Representative Director, CEO: Masayuki Mitsuka, “Mitsubishi Tanabe Pharma”). Kyowa Hakko Kirin signed a license agreement of KHK7580 with Mitsubishi Tanabe Pharma for the rights to cooperative research, develop, market and manufacture the product in Japan and some part of Asia on March 2008.
The Kyowa Hakko Kirin Group is contributing to the health and prosperity of the world’s people by pursuing advances in life sciences and technology and creating new value.
KHK 7580 …..example 3.008 2HCl MS · APCI: 375[M + H]+ in …
The calcimimetic agent, KHK-7580, currently entering Phase III clinical trials, has now been given the INN (WHO) generic name, evocalcet. Its chemical structure has also now been published and it is, in fact, correct as proposed by Dr. Crasto (Well Done!!):
Name: Evocalcet
CAS#: 870964-67-3
Chemical Formula: C24H26N2O2
Exact Mass: 374.19943
Evocalcet is a calcium-sensing receptor agonist. The calcium-sensing receptor (CaSR) is a Class C G-protein coupled receptor which senses extracellular levels of calcium ion. The calcium-sensing receptor controls calcium homeostasis by regulating the release of parathyroid hormone (PTH). CaSR is expressed in all of the organs of the digestive system. CaSR plays a key role in gastrointestinal physiological function and in the occurrence of digestive disease. High dietary Ca2+ may stimulate CaSR activation and could both inhibit tumor development and increase the chemotherapeutic sensitivity of cancer cells in colon cancer tissues. (Last update: 12/15/2015).

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











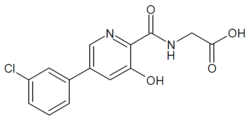
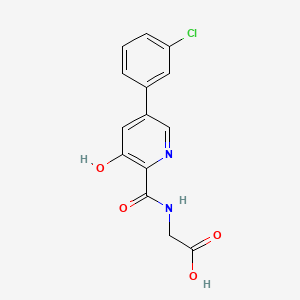



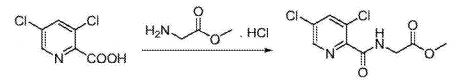
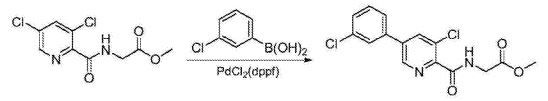
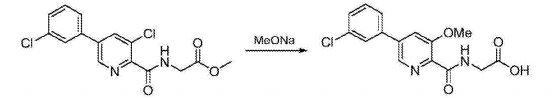
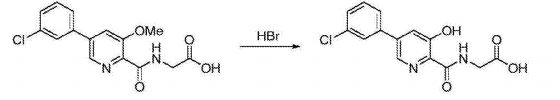


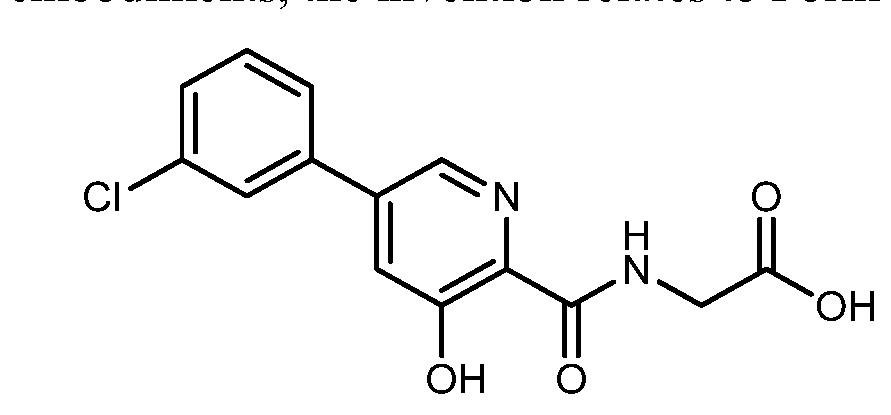
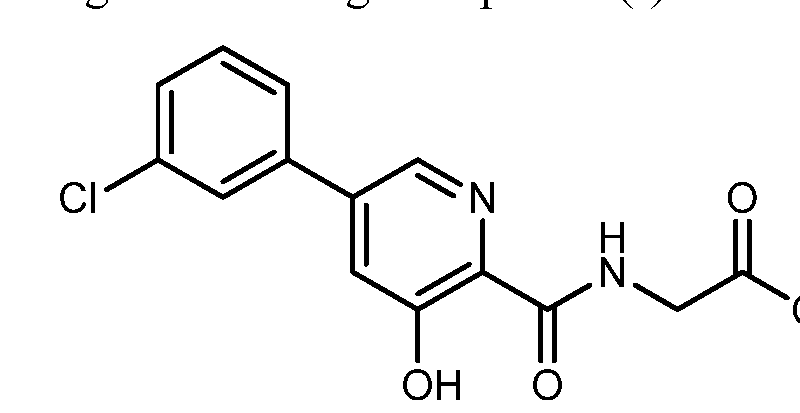







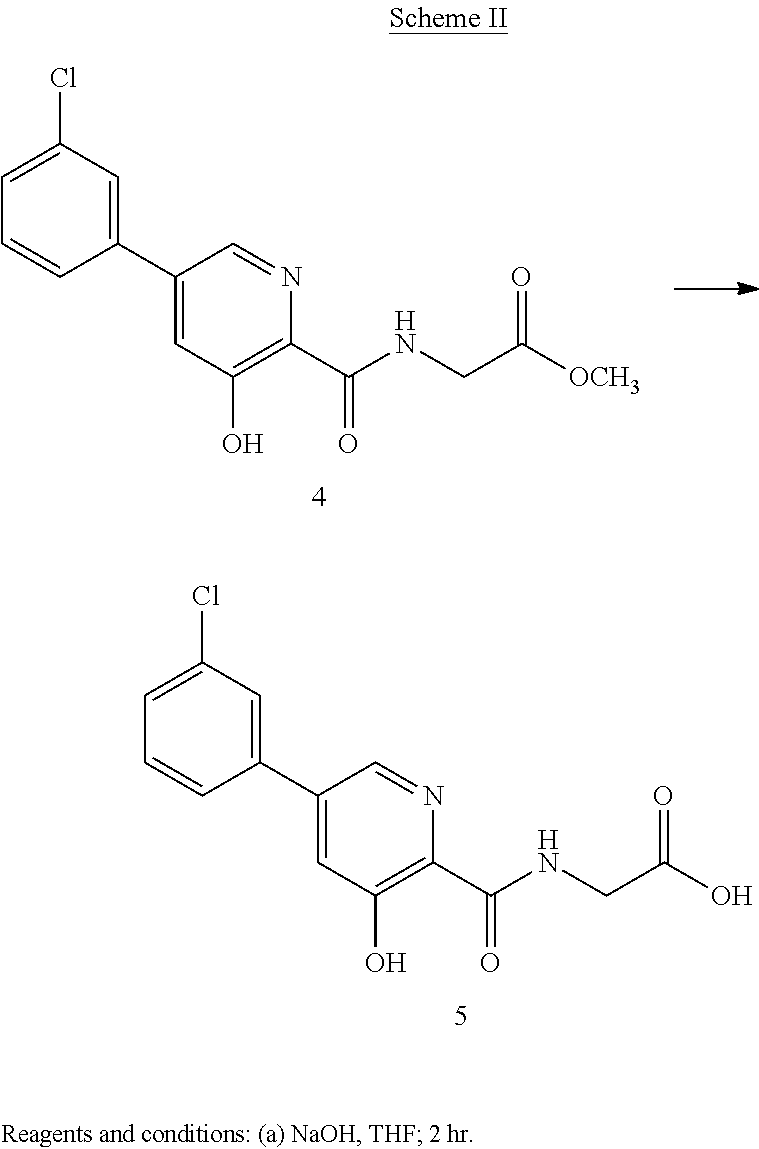
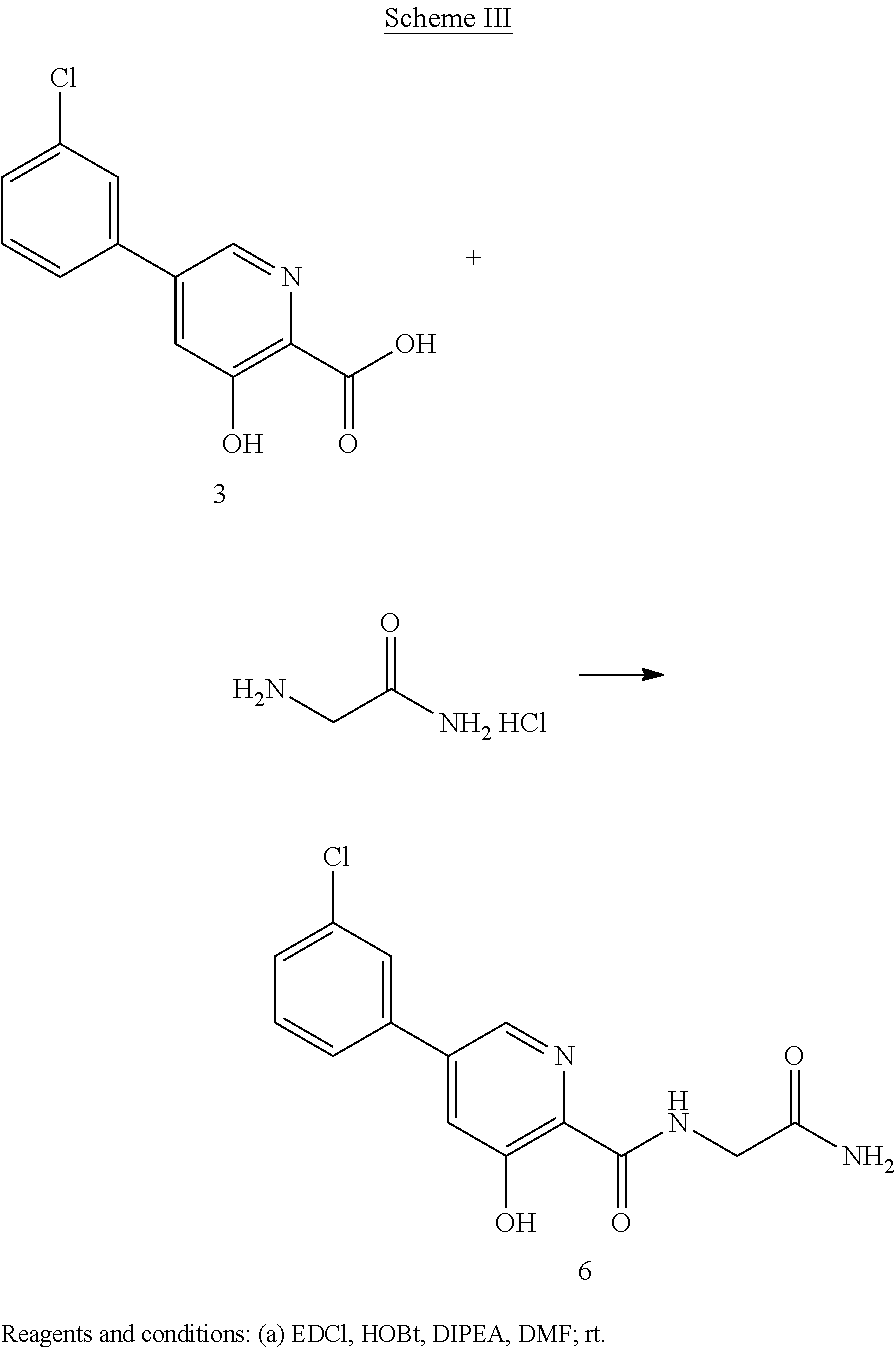




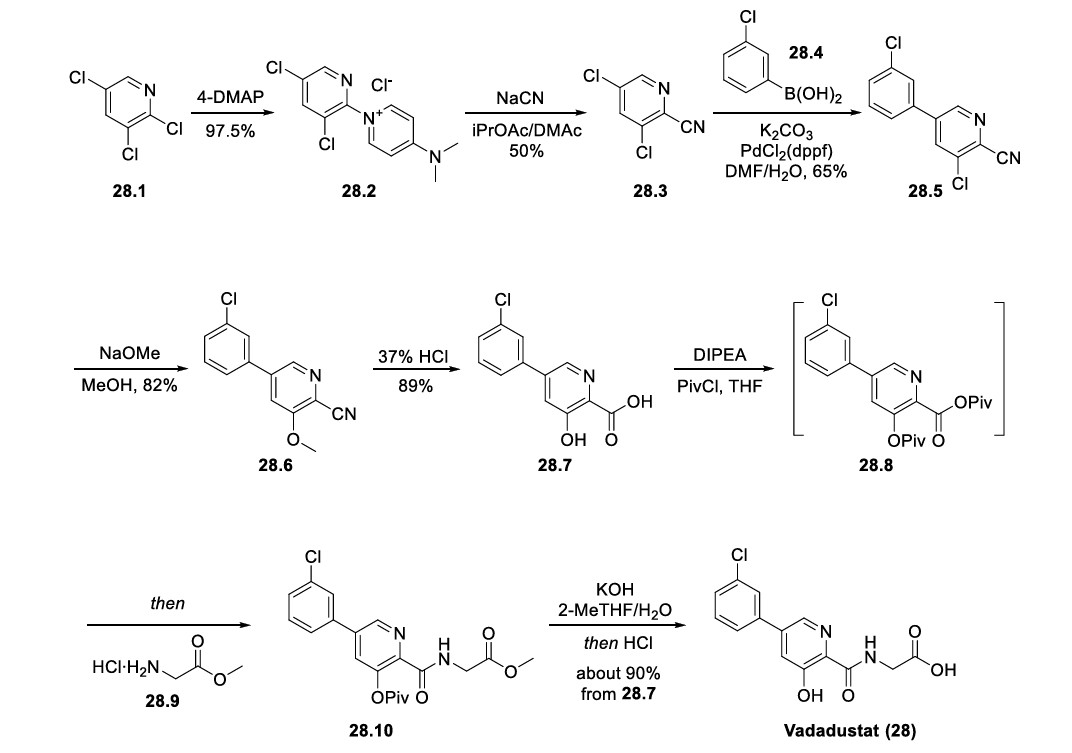






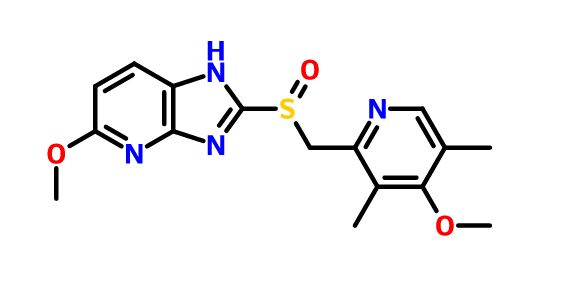


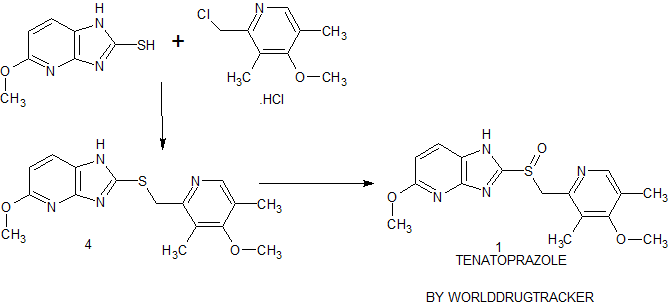
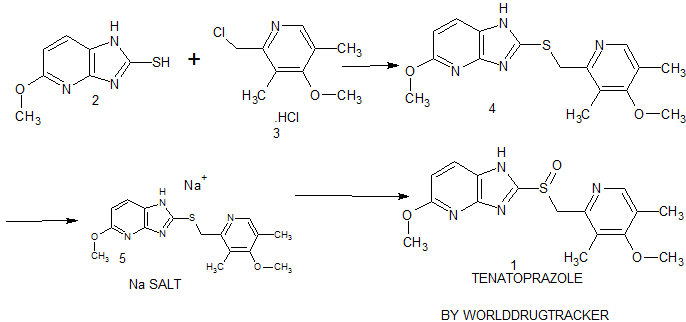










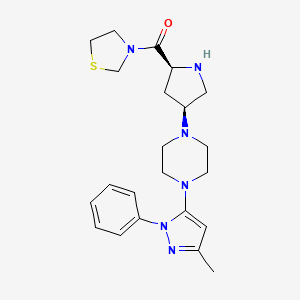







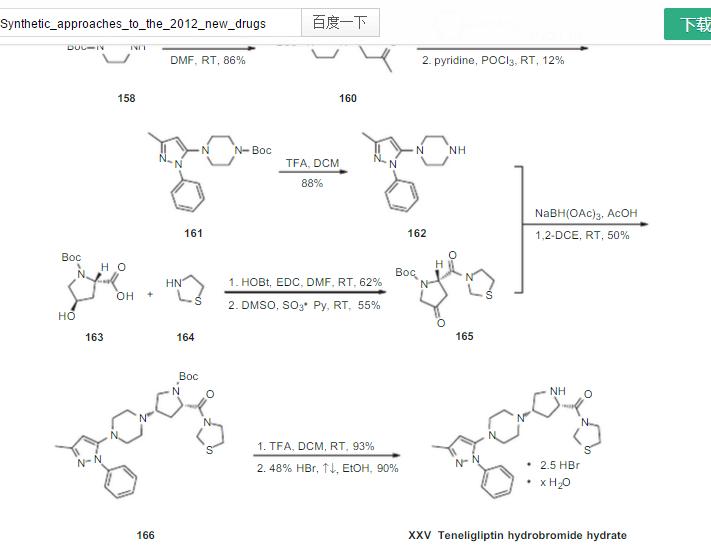
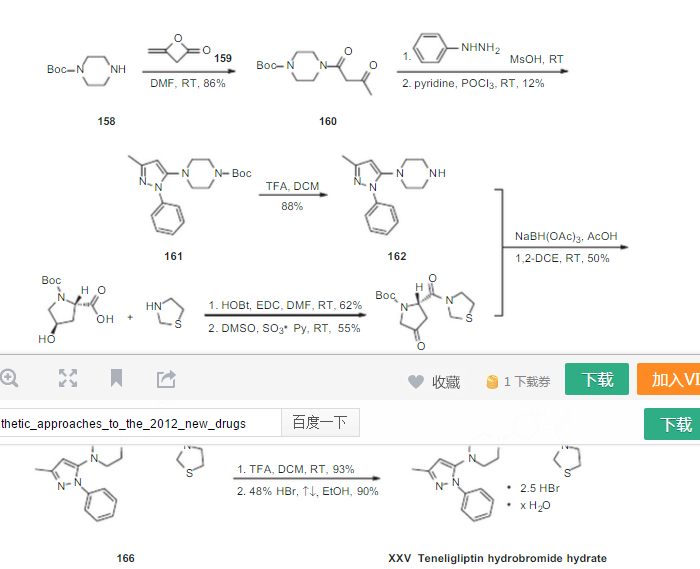






 Japan
Japan