
Home » Posts tagged 'MERCK' (Page 3)
Tag Archives: MERCK
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Suvorexant- FDA panel backs Merck & Co sleep drug but at low doses

[(7R)-4-(5-chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone
Suvorexant
may23,2013
A panel of experts at the US Food and Drug Administration has recommended Merck & Co’s insomnia drug suvorexant when given in lower dosages but rejected the higher dose that the company was seeking.———read more at
Suvorexant (MK-4305) is a dual orexin receptor antagonist in development by Merck & Co.[1][2][3] Suvorexant works by turning off wakefulness rather than by inducing sleep.[4] It is not currently approved for commercial use, but it has completed three Phase III trials.[5]The recent FDA review showed that the drug is associated with increased somnolence the next day and users of higher doses had an increased rate of suicidal ideation. [6] It is one of two such compounds currently in development, the other being GlaxoSmithKline‘s SB-649,868.
- Cox, Christopher D.; Breslin, Michael J.; Whitman, David B.; Schreier, John D.; McGaughey, Georgia B.; Bogusky, Michael J.; Roecker, Anthony J.; Mercer, Swati P. et al. (2010). “Discovery of the Dual Orexin Receptor Antagonist [(7R)-4-(5-Chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone (MK-4305) for the Treatment of Insomnia”. Journal of Medicinal Chemistry 53 (14): 5320–32. doi:10.1021/jm100541c. PMID 20565075. edit
- Baxter, Carl A.; Cleator, Ed; Brands, Karel M. J.; Edwards, John S.; Reamer, Robert A.; Sheen, Faye J.; Stewart, Gavin W.; Strotman, Neil A. et al. (2011). “The First Large-Scale Synthesis of MK-4305: A Dual Orexin Receptor Antagonist for the Treatment of Sleep Disorder”. Organic Process Research & Development 15 (2): 367–75.doi:10.1021/op1002853. edit
- Winrow, Christopher J.; Gotter, Anthony L.; Cox, Christopher D.; Doran, Scott M.; Tannenbaum, Pamela L.; Breslin, Michael J.; Garson, Susan L.; Fox, Steven V. et al. (2011). “Promotion of Sleep by Suvorexant—A Novel Dual Orexin Receptor Antagonist”.Journal of Neurogenetics 25 (1–2): 52–61. doi:10.3109/01677063.2011.566953.PMID 21473737. edit
- Kahn, Howie (June 1, 2012). “Sleep Better”. In Koerth-Baker, Maggie. 32 Innovations That Will Change Your Tomorrow. New York Times. Retrieved November 29, 2012.
- Three completed trials:
- ClinicalTrials.gov NCT01097629 Safety and Efficacy Study in Primary Insomnia Patients-Study B (4305-029)
- ClinicalTrials.gov NCT01021813 A Long Term Safety Study of MK4305 in Patients With Primary Insomnia (4305-009 AM3)
- ClinicalTrials.gov NCT01097616 Safety and Efficacy Study in Primary Insomnia Patients- Study A (4305-028)
- http://www.usatoday.com/story/news/nation/2013/05/20/fda-merck-insomnia-drug/2326921/
Enantioselective Synthesis of a Dual Orexin Receptor Antagonist.
Org. Lett. 2012; 14: 3458-3461

Orexins A and B are excitatory neuropeptides that stimulate wakefulness. Suvorexant is a dual orexin receptor antagonist that is in phase III clinical trials for the treatment of insomnia. The key step in the asymmetric synthesis depicted is a tandem enzymatic transamination–annulation sequence (F → G → H).
A previous synthesis of suvorexant (N. A. Strotman et al. J. Am. Chem. Soc. 2011, 133, 8362) involved an asymmetric Ru-catalyzed reductive amination in the construction of the diazepane ring. The present route benefits from the circumvention of transition-metal catalysis and dichloromethane as solvent.
Drug Spotlight: Zioptan, Tafluprost, Merck,

 |
|---|
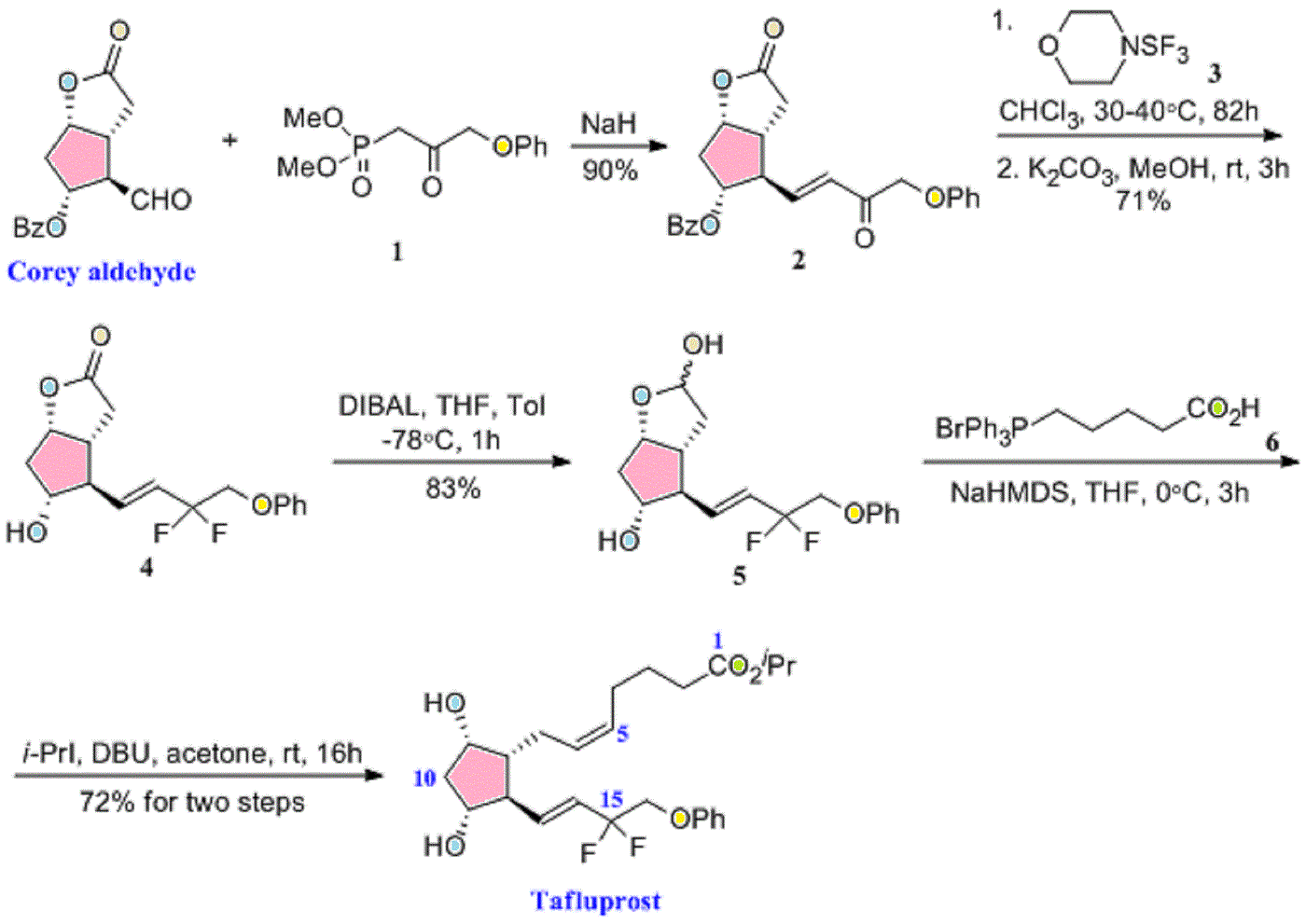
isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate,
Drug: Zioptan
Generic molecule: tafluprost
Company: Merck
Approval date: Feb. 10, 2012
The scoop: Merck says this is the first (get ready for a mouthful) preservative-free prostaglandin analog ophthalmic solution and is for treating elevated eye pressure in some patients with the most common form of glaucoma. Merck sells the ointment in the U.S. and most of Europe, while it licensed it to Japanese drugmaker Santen in Japan, Germany and northern Europe.
Tafluprost (trade names Taflotan, marketed by Santen Pharmaceutical Co. and Zioptan, by Merck (U.S.)) is a prostaglandin analogue used topically (as eye drops) to control the progression of glaucoma and in the management of ocular hypertension. It reduces http://en.wikipedia.org/wiki/Intraocular_pressure”; rel=”nofollow”>intraocular pressure by increasing the outflow of aqueous fluid from the eyes.[1][2]
Taflotan contains 15 µg/ml Tafluprost. Taflotan sine is a preservative-free, single-dose formulation containing 0.3 ml per dose.[3]

|
|
| Systematic (IUPAC) name | |
|---|---|
| isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate | |
| Clinical data | |
| Trade names | Saflutan, Taflotan, Tapros, Zioptan |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Identifiers | |
| CAS number | 209860-87-7 |
| ATC code | S01EE05 |
| PubChem | CID 6433101 |
| ChemSpider | 8044182 |
| UNII | 1O6WQ6T7G3 |
| ChEBI | CHEBI:66899 |
| ChEMBL | CHEMBL1963683 |
| Chemical data | |
| Formula | C25H34F2O5 |
| Mol. mass | 452.531266 g/mol |


- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Santen Home Page
- Gelbe Liste (in German)

European Patent No. 8509621 discloses a process for the preparation of tafluprost. In the first step, (3afl,4fl,5fl,6aS)-4-formyl-2-oxohexahydro-2 — cyclopenta[b]furan-5-ylbenzoate (CTAF 1 (i)) is condensed with dimethyl (2-oxo-3- phenoxypropyl)-phosphonate in the presence of lithium chloride and triethylamine, to provide (3aft,4F?,5F?,6aS)-2-oxo-4-((£)-3-oxo-4-phenoxybut-1 -en-1 -yl)hexahydro-2H- cyclopenta[b]-furan-5-ylbenzoate (CTAF1 ). In the second step, CTAF 1 is reacted with morpholinosulfurtrifluoride to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-2-oxohexahydro-2 –cyclopenta-[b]furan-5-yl benzoate (CTAF2). CTAF 2 is debenzoylated by potassium carbonate in methanol, to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-5-hydroxyhexahydro-2H- cyclopenta[b]furan-2-one(CTAF 3), which is further reduced by diisobutyl aluminum hydride (DIBALH) to provide (3af?,4f?,5f?,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 – yl) hexahydro-2H-cyclopenta[b]furan-2,5-diol (CTAF 4). CTAF 4 is then treated with (4- carboxybutyl)triphenylphosphonium bromide, in the presence of potassium bis(trimethylsilyl)amide in THF, to provide (Z)-7-((1 f?,2f?,3f?,5S)-2-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-3,5-dihydroxycyclopentyl)hept-5-enoic acid (“tafluprost free acid,” CTAF5), which is reacted with isopropyl iodide in the presence of DBU to provide (Z)- isopropyl 7-((1 F?,2F?,3F?,5S)-2-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-3,5-dihydroxy- cyclopentyl)hept-5-enoate (“tafluprost,” CTAF 6). The reaction sequence is summarized in Scheme 1 .

CTAF 1(i)
CTAF 1 CTAF 2

U.S. Patent Application Publication No. 2010/0105775A1 discloses amino acid salts of prostaglandins. The application also discloses a process for the preparation of prostaglandins, comprising forming an amino acid salt of a prostaglandin and converting the amino acid salt to the prostaglandin.
EXAMPLE 1 : Preparation of CTAF 1

CTAF1(i)
CTAF1
To a stirred suspension of sodium hydride (60% dispersion in mineral oil, 0.217 g, 5.429 mmol) in THF (5 ml_) was added a solution of dimethyl (2-oxo-3- phenoxypropyl)phosphonate(1 .21 g, 4.705 mmol) in THF (2 ml_), over 15 minutes at 0- 5°C under a nitrogen atmosphere. The mixture was warmed to 25-35 , 0.5 M zinc chloride solution in THF (9.4 ml_, 4.705 mmol) was added over 10 minutes, and then the mixture was stirred for 15 minutes at 25-35<€. CTAF1 (i) (3af?,4F?,5F?,6aS)-4-formyl-2- oxohexahydro-2 –cyclopenta[b]furan-5-yl benzoate (1 g) in dichloromethane (10 ml_) was added over 5 minutes at 25-35 °C. The temperature was raised to 35-40 °C and the mixture was stirred for 2hours under a nitrogen atmosphere. The mixture was cooled to 15°C and the reaction was quenched by adding acetic acid (0.2 mL), followed by adding saturated ammonium chloride solution (10 mL), and further stirring for 15 minutes. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (5 mL). The combined organic layers were evaporated under reduced pressure below 50°C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (0.9 g, 61 %yield).
EXAMPLE 2: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (5 g, 0.0123 mol) in dichloromethane (100 mL) was added diethylaminosulfurtrifluoride (13 mL, 0.09841 mol) at 0-5 °C under a nitrogen atmosphere. The temperature was raised to 25-35 °C and maintained for 24 hours under a nitrogen atmosphere at the same temperature. The mass was slowly added into a saturated sodium bicarbonate solution (75 mL) at 0-5 °C. Temperature was raised to 25- 35 °C, the layers were separated, and the aqueous layer was extracted with dichloromethane (2×25 mL). The combined organic layer was washed with water (25mL) and dried over sodium sulfate (5 g). The organic layer was evaporated to dryness under reduced pressure below 40 °C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (4.2 g, 79% yield). EXAMPLE 3: Preparation of CTAF 4

CTAF 2 CTAF 4
CTAF 2 (2.30 g, 5.37 mmol) was dissolved in toluene (25 mL) and the solution was cooled to -65 °C under nitrogen. Diisobutyl aluminum hydride (1 .5 M in toluene, 1 1 .8 mL, 17.7mmol) was added over 15 minutes at -61 to -65 . The mixture was stirred for 3hours and then the reaction was quenched by adding methanol (1 .5 mL). Sulfuric acid (1 M, 25 mL) was added and the temperature rose to -20°C during the addition. Methyl t-butyl ether (MTBE) (10 mL) was added and the mixture was allowed to warm to room temperature. The organic phase was separated and the aqueous phase was extracted with MTBE (2x 10 mL). The combined organic phase was washed with water (10 mL), saturated aqueous sodium bicarbonate (10 mL), and then brine (10 mL). The washes were back-extracted with MTBE (10 mL). The combined organic phases were dried with magnesium sulfate, filtered, and evaporated to give a colourless oil (2.20 g). The crude product was chromatographed on silica (60 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), and then with ethyl acetate, to give CTAF 4 as a colourless oil (1 .71 g, 97% yield).
EXAMPLE 4: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (20 g, 0.0492 mol) in dichloromethane(400 mL) was added diethylaminosulfurtrifluoride (52 mL, 0.393 mol) at 0-10°C under a nitrogen atmosphere. The temperature was raised to 25-35 and maintained for 96hours under a nitrogen atmosphere at that temperature. The mass was slowly added to a saturated NaHCOs solution (600 mL) at 0-10°C. The mixture was heated to 25-35 <€ and filtered through aCelite bed. The layers were separated and the aqueous layer was extracted with DCM (2×100 mL). The combined organic layer was washed with 10% NaCI solution (100 mL) and evaporated to dryness under reduced pressure below 40°C. The residue was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane.
Column purified material was dissolved in MTBE (80 mL) at 40°C and stirred for 30 minutes at that temperature. Diisopropyl ether (160 mL) was added at 35-40 and stirring continued for 30 minutes at 35-40 . Cooled the mass to 5-15°C and stirred for 30 minutes at that temperature. The solid was filtered, washed with a mixture of MTBE and diisopropyl ether (DIPE) (1 :2 by volume, 60 mL), and dried at 40°C under vacuum, to afford pure CTAF2 (12.0 g, 57% yield).
EXAMPLE 5: Preparation of CTAF 5

(4-Carboxybutyl)triphenylphosphonium bromide (10.32 g, 23.3 mmol, 4 eq) was suspended in THF (20 mL) under a nitrogen atmosphere and cooled to 5°C. NaHMDS solution (1 M in THF, 46.6 mL, 46.6 mmol, 8 eq) was added over 10 minutes. The red/orange mixture was stirred for 30 minutes. A solution of CTAF 4 (1 .90 g, 5.82 mmol) in THF (10 mL) was added over 30 minutes at 0-3 . The mixture was stirred for 1 .5hours and then the reaction was quenched by adding water (30 mL) and the masswas warmed to room temperature. The aqueous phase was separated and the organic phase was washed with water (20 mL). The combined aqueous phases were washed with MTBE (30 mL). The organic phases up to this point were discarded. The aqueous phase was acidified with 2M hydrochloric acid (14 mL, to pH 3-4) and extracted with ethyl acetate (2×30 mL). The combined ethyl acetate layers were washed with brine (20 mL), dried with magnesium sulfate, filtered, and evaporated under reduced pressure to give CTAF 5 asa yellow oil (8.60 g).
A 2.96 g sample was removed and the remainder (5.64 g) was chromatographed on silica (30 g) eluting with ethyl acetate to give purified CTAF 5 (1 .41 g) asa yellow oil. NMR analysis showed approximately 90% purity, remainder triphenyl phosphine oxide.
EXAMPLE 6: Preparation of CTAF 5 DCHA salt

CTAF 5 CTAF 5 DCHA sa t
CTAF5 (1 1 .72 g, 90% purity, 25.7 mmol, containing 1 .4% trans isomer) was dissolved in acetone (60 mL). Dicyclohexylamine (4.66 g, 25.7 mmol) was added and the mixture was stirred at room temperature overnight. The solid was filtered and washed with acetone (6 mL), then dried to give the DCHA salt (12.93 g, 85% yield, 0.29% trans-isomer).
A sample (7.03 g) was further purified by recrystallisation. It was dissolved in hot acetone (30 mL) and cooled to room temperature with stirring. The mixture was stirred for 3 hours, filtered and the solid was washed with acetone (3 mL) and dried to give a white solid (6.41 g, 91 % recovery, 0.1 1 % trans-isomer).
A PXRD pattern of the product is shown as Fig. 1 , obtained using copper Ka radiation. In the drawing, the y-axis is intensity units and the x-axis is the 2-theta angle, in degrees. EXAMPLE 7: Pre aration of CTAF 6

CTAF 5 DCH A sa l ^ I AI- O
CTAF 5 DCHA salt (5.80 g, 9.80 mmol) was suspended in ethyl acetate (20 mL). Sulfuric acid (1 M, 20 mL) was added and the mixture was stirred until a clear solution was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (2×20 mL). The combined organic layers were washed with water (15 mL) and brine (15 mL), dried with magnesium sulfate, filtered, and evaporated. The residue was dissolved in acetone (40 mL) and charged into a jacketed vessel at 30°C. 1 ,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (8.95 g, 58.8 mmol) was added, then 2- iodopropane (10.0 g, 58.8 mmol) was added, and the mixture was stirred for 20hours. The mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (30 mL) and aqueous potassium dihydrogen orthophosphate (8 g) in water (50 mL). The organic phase was separated and the aqueous was extracted with ethyl acetate (30 mL). The combined organic phases were washed with brine (20 mL), dried with magnesium sulfate, filtered and evaporated to give a yellow oil (4.83 g). The crude product was chromatographed on silica (130 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), to give CTAF 6 (3.98 g, 90% yield) as a colorless oil.
FDA accepts Merck BLA for investigational allergy immunotherapy tablet

28 MAR 2013
The US FDA has accepted Merck’s biologics license application (BLA) for an investigational allergy immunotherapy tablet (AIT), Timothy grass pollen (Phleum pratense).
The application includes safety and efficacy data of the investigational sublingual dissolvable tablet from Phase III trials including a long-term, multi-season trial.
Merck Research Laboratories senior vice president, global scientific strategy, franchise head, infectious diseases and interim franchise head, respiratory & immunology Jeffrey Chodakewitz said, “We are pleased to have achieved this important milestone in the development of our investigational grass pollen AIT, which, if approved, would represent a potential new option for allergy specialists to offer appropriate allergic rhinitis patients.”
The grass pollen (Phleum pratense) AIT is designed to generate an immune response targeting the root cause of allergic rhinitis.
The company has collaborated with ALK-Abello for grass pollen (Phleum pratense) AIT development in North America.
Phase III trial of Merck’s Vytorin passes vital safety test
mar 13 2013
Merck & Co’s stock enjoyed a boost yesterday after it revealed it has been given permission to continue a late-stage trial of its cholesterol buster Vytorin.
The Whitehouse Station, New Jersey-based firm must have a breathed a sigh of relief when the Data Safety Monitoring Board issued a green light for the Phase III IMPROVE for a second time, having found no significant safety concerns raised by the data.
After an earlier planned review of data last year, the Board, rather unusually, said it would undertake a second interim analysis at a later date, which had led to some concerns that there may be issues that could lead to the trial being halted, according to media reports.
However, it seems these fears are unfounded at this point, as the18,000-plus patient study – which is designed to determine whether Vytorin is more effective at reducing the risk of heart attack, stroke and death in patients with heart disease than simvastatin alone – has been cleared to conclude.
The drugmaker said the trial should finish in September next year, and it will no doubt be hoping for a positive outcome to prove the benefits of Vytorin – a combination of the generic simvastatin and the still-patented Zetia (ezetimibe) – and breathe a little new life into its heart franchise.
Citi Investment Research analyst Andrew Baum, however, expressed doubt in a research note the final analysis will show Merck’s drug is more effective than generic competition, according to the Associated Press.
 |
|
|---|---|
 |
|
| Combination of | |
| Ezetimibe | via Niemann-Pick C1-Like 1 protein |
| Simvastatin | Statin HMG-CoA reductase inhibitor |
Phase 1- MERCK , Study of MK-8109 (Vintafolide) Given With Chemotherapy in Participants With Advanced Cancers

vintafolide
cas no 742092-03-1
http://www.ama-assn.org/resources/doc/usan/vintafolide.pdf
N-(4-{[(2-amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoyl)-L-γ-glutamyl-L-α- aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine disulfide with methyl (5S,7R,9S)- 5-ethyl-9-[(3aR,4R,5S,5aR,10bR,13aR)-3a-ethyl-4,5-dihydroxy-8-methoxy-6-methyl-5- ({2-[(2-sulfanylethoxy)carbonyl]hydrazinyl}carbonyl)-3a,4,5,5a,6,11,12,13a-octahydro- 1H-indolizino[8,1-cd]carbazol-9-yl]-5-hydroxy-1,4,5,6,7,8,9,10-octahydro-2H-3,7- methanoazacycloundecino[5,4-b]indol-9-carboxylate
Vincaleukoblastin-23-oic acid, O4-deacetyl-, 2-[(2-mercaptoethoxy)carbonyl]hydrazide, disulfide with N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-γ- glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine
Vintafolide is a water-soluble, folate-receptor-targeted conjugate of folate and the vinca alkaloid desacetylvinblastine monohydrazide (DAVLBH) with potential antineoplastic activity. The folate moiety of folate-vinca alkaloid conjugate EC145 binds to folic acid receptors on the tumor cell surface and the agent is internalized via folate receptor-mediated endocytosis, delivering the tubulin-binding DAVLBH moiety directly into the tumor cell; DAVLBH binding to tubulin results in the disruption of microtubule assembly-disassembly dynamics, cell cycle arrest, and tumor cell apoptosis. Folic acid receptors are frequently upregulated on the surfaces of many tumor cell types. DAVLBH is a derivative of the natural product vinblastine.
http://clinicaltrials.gov/show/NCT01688791
ClinicalTrials.gov Identifier:
Vintafolide is a derivative of the anti-mitotic chemotherapy drug vinblastine.[1] chemically linked to folic acid. The vintafolide molecule was designed to specifically target the toxic vinblastine group to cancer cellsthat overexpress the folic acid receptor.[2] Vintafolide is being studied for treatment of late-stage ovarian cancer and mid-stage non-small cell lung cancer.
Merck & Co. acquired the development and marketing rights to this experimental cancer drug from Endocyte in April 2012. Endocyte had planned to file for marketing approval for vintafolide in the third quarter of 2012. The drug received an orphan drug status in Europe in March 2012.[3] Endocyte remains responsible for the development and commercialization of etarfolatide, a non-invasive companion diagnostic imaging agent used to identify folate receptor positive tumor cells that may be susceptible to vintafolide.[4]
- Statement on a nonproprietary name adopted by the USAN Council, United States Adopted Names (USAN) Council, 6 April 2012
- Dosio F, Milla P, Cattel L. EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers. Curr Opin Investig Drugs. 2010 Dec;11(12):1424-33. Review. PubMed PMID: 21154124.
- Endocyte soars on cancer drug deal with Merck, Reuters, US Edition, Mon Apr 16, 2012.
- Merck, Endocyte in Development Deal Wed, Drug Discovery and Development. 04/25/2012

Chemical structure of EC-145
(source: THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, 2011, 336(2):336–343)
Phase 3 , breast cancer, Ridaforolimus (MK-8669; AP23573; formerly Deforolimus) Merck, license,Ariad Pharmaceuticals

Ridaforolimus
572924-54-0
(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28Z,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl dimethylphosphinate
Dimethyl-phosphinic Acid C-43 Rapamycin Ester
42-(dimethylphosphinate) Rapamycin
- AP 23573
- AP23573
- Deforolimus
- MK 8669
- MK-8669
- MK8669
- Ridaforolimus
- Taltorvic
- UNII-48Z35KB15K
An mTOR inhibitor for the treatment of cancer.
Ridaforolimus (MK-8669; AP23573; formerly Deforolimus)
Merck, under exclusive worldwide license agreement with Ariad Pharmaceuticals
Method of Action: Oral inhibitor of mammalian target of rapamycin inhibitor (mTOR)
Indications/Phase of Trial: Maintenance therapy for metastatic soft-tissue sarcoma and bone sarcomas after at least four chemotherapy cycles (under review after receiving Complete Response letter from FDA in June; NME)
Ridaforolimus is an investigational small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN, known to be important to malignancy.
TARGET- mTOR
Ridaforolimus (also known as AP23573 and MK-8669; formerly known as Deforolimus[1]) is an investigational targeted and small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN known to be important to malignancy. Blocking mTOR creates a starvation-like effect in cancer cells by interfering with cell growth, division, metabolism, and angiogenesis.
It has had promising results in a clinical trial for advanced soft tissue and bone sarcoma.
RIDAFOROLIMUS
NMR….http://file.selleckchem.com/downloads/nmr/S102201-Deforolimus-HNMR-Selleck.pdf
HPLC . http://file.selleckchem.com/downloads/hplc/S102201-Deforolimus-HPLC-Selleck.pdf
MSDS..http://www.selleckchem.com/msds/Deforolimus-MSDS.html
| Commercial arrangements |
Ridaforolimus is being co-developed by Merck and ARIAD Pharmaceuticals. On May 5, 2010, Ariad Pharmaceuticals and Merck & Company announced a clinical development and marketing agreement. With this agreement, Ariad received $125 million in upfront payments from Merck and $53 million in milestone payments. Future payments are triggered upon acceptance of the NDA by the FDA with another payment when the drug receives marketing approval. There are similar milestones for acceptance and approval in both Europe and Japan. Other milestone payments are tied to revenue goals for the drug.[2] ARIAD has opted to co-promote ridaforolimus in the U.S. Merck plans to submit a New Drug Application (NDA) for ridaforolimus to the U.S. Food and Drug Administration (FDA) and a marketing application in the European Union in 2011.[3]
Clinical trials
Phase III SUCCEED
On June 6, 2011, Ariad and Merck announced detailed results from the largest randomized study ever in the soft tissue and bone sarcoma population, the Phase III SUCCEED clinical trial. SUCCEED evaluated oral ridaforolimus, in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. In this patient population, ridaforolimus improved progression-free survival (PFS) compared to placebo, the primary endpoint of the study. The complete study results were presented by Sant P. Chawla, M.D., director, Sarcoma Oncology Center, Santa Monica, CA, during the 2011 American Society of Clinical Oncology (ASCO) annual meeting.
The SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) trial was a randomized (1:1), placebo-controlled, double-blind study of oral ridaforolimus administered at 40 mg/day (five of seven days per week) in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. Oral ridaforolimus was granted a Special Protocol Assessment (SPA) by the FDA for the SUCCEED trial.
Based on 552 progression-free survival (PFS) events in 711 patients, (ridaforolimus (N=347), placebo (N=364) determined by an independent radiological review committee, the study achieved its primary endpoint of improvement in PFS, with a statistically significant (p=0.0001) 28 percent reduction in the risk of progression or death observed in those treated with ridaforolimus compared to placebo (hazard ratio=0.72).
Median PFS was 17.7 weeks for those treated with ridaforolimus compared to 14.6 weeks in the placebo group. Furthermore, based on the full analysis of PFS determined by investigator assessment, there was a statistically significant (p<0.0001) 31 percent reduction by ridaforolimus in the risk of progression or death compared to placebo (hazard ratio=0.69). In the investigator assessment analysis, median PFS was 22.4 weeks for those treated with ridaforolimus compared to 14.7 weeks in the placebo group [4
EU WITHDRAWAL IN NOV 2012
Merck, known as MSD outside the U.S. and Canada, announced today that it has formally notified the European Medicines Agency (EMA) of Merck’s decision to withdraw the Marketing Authorisation Application (MAA) for ridaforolimus.
The application for Marketing Authorisation for ridaforolimus was accepted by the EMA in August 2011. At the time of the withdrawal it was under review by the Agency’s Committee for Medicinal Products for Human Use (CHMP). In its letter to the EMA, Merck said that the withdrawal of ridaforolimus was based on the provisional view of the CHMP that the data available to date and provided in the Marketing Authorisation Application were not sufficient to permit licensure of ridaforolimus in the European Union for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor.
Although the application for these uses was withdrawn, Merck is studying ridaforolimus in combination with other drugs in other tumor types. The withdrawal of the European application of ridaforolimus for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor does not change Merck’s commitment to the ongoing clinical trials with ridaforolimus.
Description
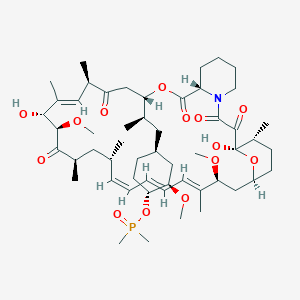
42-(dimethylphosphinate) Rapamycin (Ridaforolimus) represented by the following formula I:

2. Description of RelatedArt
The mammalian target of Rapamycin (mTOR) is known as a mechanistic target of Rapamycin (H), which is found in the studies of Rapamycin. On the other hand, 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I) is a derivative of Rapamycin (II), which is also a kind of mTOR inhibitor. Ridaforolimus (I) can inhibit cell division and possibly lead to tumor cell death. Hence, there are many studies related to solid tumor treatments and blood cancer treatments using Ridaforolimus (I). In addition, in 2011, Merck also applied a certification of this compound against soft tissue and bone cancer.
U.S. Pat. No. 7,091,213 discloses a process for preparing 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I), and the process thereof is shown in the following Scheme I.

In this process, a solution of Rapamycin (II) in dichloromethane (DCM) was respectively added with 2,6-di-tert-butyl-4-methylpyridine or 3,5-lutidine as a base, and followed by the addition of a solution of dimethylphosphinic chloride (DMP-Cl) to perform a phosphorylation reaction at 0° C., under a stream of N2(g). The crude product was purified by flash chromatography (eluted with MeOH/DCM/EtOAc/hexane=1:10:3:3) to provide 42-(dimethyl- phosphinate) Rapamycin (Ridaforolimus) (I), which is a phosphorylated compound at 42-hydroxyl position of Rapamycin (II). In addition, this patent also disclosed a side product of 31,42-bis(dimethyl phosphinate) Rapamycin (III), which is a phosphorylated compound at both 31- hydroxyl position and 42- hydroxyl position of Rapamycin (II).
…………………..
SYNTHESIS
Some additional transformations of potential interest to the practitioner are shown below, including the preparation of reagents for generating the described C-43 phosphorus-containing rapalogs:
Preparation of Diakyl/diaryl Chlorophoshates

Preparation of Alkyl Halide Phosphonates

Illustrative routes for using the foregoing sorts of reagents to prepare certain rapalogs of this invention are shown below.

The synthesis of compounds of this invention often involves preparation of an activated form of the desired moiety “J”, such as a phosphoryl chloride as shown above (e.g. (R)(RO)P—Cl or RR′P(═O)—Cl, etc), and reaction of that reagent with rapamycin (or the appropriate rapalog) under conditions yielding the desired product, which may then be recovered from residual reactants and any undesired side products. Protecting groups may be chosen, added and removed as appropriate using conventional methods and materials.
Purification of Compounds of the Invention
A variety of materials and methods for purifying rapamycin and various rapalogs have been reported in the scientific and patent literatures and may be adapted to purification of the rapalogs disclosed herein. Flash chromatography using a BIOTAGE prepacked cartridge system has been particularly effective. A typical protocol is disclosed in the Examples which follow.
Physicochemical Characterization of Compounds of the Invention
The identity, purity and chemical/physical properties of the rapalogs may be determined or confirmed using known methods and materials, including HPLC, mass spectral analysis, X ray crystallography and NMR spectroscopy. High resolution 1D 1H and 31P NMR spectra acquired using a typical relaxation delay of 3 seconds have proved useful, as has reverse phase HPLC analysis (analytical column, 3 micron particle size, 120 ansgstrom pore size, thermostatted to 50° C. with a mobile phase of 50% acetonitrile, 5% methanol and 45% water (all % s by volume), for example, in an isocratic elution system, with elution of product and impurity peaks followed by UV detection at 280 nanometers). Normal phase HPLC may also be used, especially to evaluate the level of residual rapamycin or rapalog by-products. The presence of residual solvent, heavy metals, moisture and bioburden may be assessed using conventional methods.
Example 9
Dimethyl-phosphinic Acid C-43 Rapamycin Ester

Dimethyl-phosphinic Acid C-43 Rapamycin Ester
To a cooled (0° C.) solution of rapamycin (0.1 g, 0.109 mmol) in 1.8 mL of dichloromethane was added 0.168 g (0.82 mmol) of 2,6-di-t-butyl-4-methyl pyridine, under a stream of N2, followed immediately by a solution of dimethylphosphinic chloride (0.062 g, 0.547 mmol) in 0.2 mL of dichloromethane. The slightly yellow reaction solution was stirred at 0° C., under an atmosphere of N2, for 3.5 h (reaction monitored by TLC). The cold (0° C.) reaction solution was diluted with ˜20 mL EtOAc then transferred to a separatory funnel containing EtOAc (150 mL) and saturated NaHCO3 (100 mL). Upon removing the aqueous layer, the organic layer was washed successively with ice cold 1N HCl (1×100 mL), saturated NaHCO3 (1×100 mL), and brine (1×100 mL), then dried over MgSO4 and concentrated. The crude product was purified by silica gel flash chromatography (eluted with 1:10:3:3 MeOH/DCM/EtOAc/hexane) to provide 0.092 g of a white solid:
1H NMR (300 MHz, CDCl3) d 4.18 (m, 1H), 4.10 (m, 1H), 3.05 (m, 1H), 1.51 (m, 6H);
31P NMR (121 MHz, CDCl3) d 53.6; 1013 m/z (M+Na).
Example 9
Alternative Synthesis
Rapamycin and dichloromethane are charged into a nitrogen-purged reaction flask. The stirred solution is cooled to approximately 0° C. (an external temperature of −5±5° C. is maintained throughout the reaction). A solution of dimethylphosphinic chloride (2.0 molar equivalents) in dichloromethane is then added over a period of approximately 8–13 minutes.
This is followed immediately by the addition of a solution of 3,5-lutidine (2.2 molar equivalents) in dichloromethane over a period of approximately 15–20 minutes. Throughout both additions, the internal temperature of the reaction sssstays below 0° C. The cooled reaction solution is stirred for 1 hour and then transferred, while still cold, to an extractor containing saturated aqueous NaHCO3 and methyl-t-butyl ether (MTBE), ethyl acetate or diethyl ether. In-process samples are removed at 30 and 60 minute time points.
Samples are prepared in a similar fashion to that described for the reaction workup. Reaction progress is monitored by TLC (1:10:3:3 MeOH/DCM/EtOAc/hexanes) and reverse-phase HPLC analyses. The isolated organic layer is successively washed with ice cold 1N HCl, saturated aqueous NaHCO3 (2×), saturated aqueous NaCl, and dried over sodium sulfate. Upon filtration and solvent removal, the residue undergoes solvent exchange with acetone followed by concentration in vacuo to provide crude product, which may be analyzed for purity by normal- and reversed-phase HPLC.
…………………….
SYNTHESIS
The process of the present invention is shown in the following Scheme II.


- “ARIAD Reports First Quarter 2009 Development Progress and Financial Results- Ridaforolimus New USAN Name to Replace Deforolimus”. ARIAD Pharmaceuticals. 2009. Retrieved 2009-05-07.
- “ARIAD – News release”. Phx.corporate-ir.net. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-03-17. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-06-06. Retrieved 2012-10-07.
|
7-11-2012
|
FULLY HUMAN ANTI-VEGF ANTIBODIES AND METHODS OF USING
|
|
|
10-28-2011
|
METHODS OF TREATMENT
|
|
|
1-21-2011
|
ANTI-IGF1R
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
4-16-2004
|
Phosphorus-containing compounds and uses thereof
|

Sanofi Pasteur has received a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) recommending market approval for Sanofi Pasteur’s 6-in-1 pediatric vaccine Hexyon/Hexacima (DTaP-IPV-Hib-HepB vaccine).
FEB22,2013
French drug major Sanofi’s vaccines subsidiary Sanofi Pasteur has received a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) recommending market approval for Sanofi Pasteur’s 6-in-1 pediatric vaccine Hexyon/Hexacima (DTaP-IPV-Hib-HepB vaccine).
Hexyon/Hexacima is the only fully liquid, ready-to-use, 6-in-1 vaccine to protect infants against diphtheria, tetanus, pertussis (whooping cough), Hepatitis B, poliomyelitis and invasive infections caused by Haemophilus influenzae type b.
The new vaccine will be commercialized under the brand name Hexyon in Western European countries by Sanofi Pasteur MSD, the joint venture between US pharma giant Merck & Co and Sanofi Pasteur, and under the brand name Hexacima in Eastern European countries by Sanofi Pasteur.
“Availability of Hexyon/Hexacima ready-to-use, 6-in-1 pediatric vaccine will raise the standard of care of vaccination for millions of children. It reduces the number of vaccination visits for infants and it is more convenient for parents to complete the recommended vaccination schedule and thus better protect their children against six major childhood diseases,” said Olivier Charmeil, president and chief executive of Sanofi Pasteur, adding: “Upon licensure, we intend to introduce Hexyon/Hexacima vaccine in countries that are looking for improved and effective solutions for public immunization programs.”
Key benefits of Hexyon/Hexacima vaccine
According to Sanofi, the key benefits of Hexyon/Hexacima include the following:
• Hexyon/Hexacima is a fully liquid, ready-to-use vaccine; no reconstitution is needed prior to administration, which improves convenience for health care professionals. It is available in vial and pre-filled syringe presentations;
• by combining six vaccines into one, the vaccine reduces the number of injections, which improves comfort and vaccination compliance for infants, and
• the use of acP (acellular pertussis) antigens and IPV (inactivated poliovirus vaccine) improves safety and reduces reactogenicity as compared to wcP (whole cell pertussis)-containing vaccines and OPV (oral polio vaccine).
Assuming licensure, Hexyon/Hexacima would be indicated for primary and booster vaccination of infants from six weeks of age in accordance with official recommendations. The CHMP positive opinion is supported by results of multi-center clinical studies involving around 5,000 infants. Phase III clinical studies comparing Hexyon/Hexacima to licensed combination vaccines demonstrated that the vaccine is safe and induces a robust immune response against all six targeted diseases.
