GS-4997, GS-4977, Selonsertib
Selonsertib; 1448428-04-3; GS-4997; UNII-NS3988A2TC; NS3988A2TC; 5-(4-cyclopropyl-1H-imidazol-1-yl)-2-fluoro-N-(6-(4-isopropyl-4H-1,2,4-triazol-3-yl)pyridin-2-yl)-4-methylbenzamide
5-(4-cyclopropylimidazol-1-yl)-2-fluoro-4-methyl-N-[6-(4-propan-2-yl-1,2,4-triazol-3-yl)pyridin-2-yl]benzamide
- 5-(4-Cyclopropyl-1H-imidazol-1-yl)-2-fluoro-4-methyl-N-[6-[4-(1-methylethyl)-4H-1,2,4-triazol-3-yl]-2-pyridinyl]benzamide
- 5-(4-Cyclopropyl-1H-imidazol-1-yl)-2-fluoro-4-methyl-N-{6-[4-(propan-2-yl)-4H-1,2,4-triazol-3-yl]pyridin-2-yl}benzamide
| Molecular Formula: |
C24H24FN7O |
| Molecular Weight: |
445.502 g/mol |
-
-
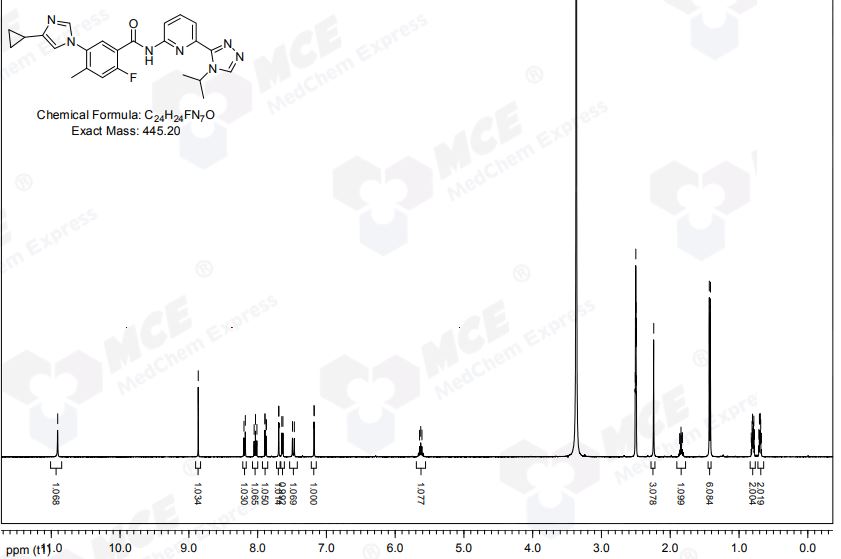
NMR https://file.medchemexpress.com/batch_PDF/HY-18938/Selonsertib-HNMR-25028-MedChemExpress.pdf

Selonsertib is an orally bioavailable inhibitor of apoptosis signal-regulating kinase 1 (ASK1; IC50 = 3.2 nM), which is involved in a variety of conditions, including fibrosis, oxidative stress, and inflammation, among others.1 A formulation containing selonsertib showed antifibrotic activity in a Phase II clinical trial. Clinical trials are ongoing for other conditions, including severe alcoholic hepatitis and nonalcoholic steatohepatitis.
- Originator Gilead Sciences
- Class Benzamides; Cardiovascular therapies; Imidazoles; Pyridines; Triazoles
- Mechanism of Action MAP kinase kinase kinase 5 inhibitors
Highest Development Phases
- Phase III Non-alcoholic steatohepatitis
- Phase II Alcoholic hepatitis; Diabetic nephropathies; Non-alcoholic fatty liver disease; Pulmonary arterial hypertension
Most Recent Events
- 13 Apr 2018 Efficacy data from a phase II trial in Non-alcoholic fatty liver disease presented at the The International Liver Congress™ 2018 of the European Association for the Study of the Liver (EASL-2018)
- 13 Apr 2018 Gilead completes enrolment in the STELLAR 3 phase III trial for Non-alcoholic steatohepatitis in US, Argentina, Australia, Austria, Belgium, Brazil, Canada, France, Germany, Hong Kong, India, Israel, Italy, Japan, South Korea, Malaysia, Mexico, Netherlands, New Zealand, Poland, Portugal, Puerto Rico, Singapore, Spain, Switzerland, Taiwan, Turkey, and United Kingdom (NCT03053050)
- 13 Apr 2018 Gilead completes enrolment in the STELLAR 4 phase III trial for Non-alcoholic steatohepatitis in the US, Australia, Austria, Belgium, Canada, France, Germany, Hong Kong, India, Israel, Italy, Japan, South Korea, Mexico, New Zealand, Poland, Puerto Rico, Singapore, Spain, Switzerland, Taiwan, and United Kingdom ( NCT03053063)
Apoptosis signal -regulating kinase 1 (ASK1) is a member of the mitogen-activated protein kinase kinase kinase (“MAP3K”) family that activates the c-Jun N-terminal protein kinase (“JNK”) and p38 MAP kinase (Ichijo, H., Nishida, E., e, K., Dijke, P. T., Saitoh, M., Moriguchi, T., Matsumoto, K., Miyazono, K., and Gotoh, Y. (1997) Science, 275, 90-94).
ASK1 is activated by a variety of stimuli including oxidative stress, reactive oxygen species (ROS), LPS, TNF-a, FasL, ER stress, and increased intracellular calcium concentrations (Hattori, K., Naguro, I., Runchel, C, and Ichijo, H. (2009) Cell Comm. Signal. 7: 1-10; Takeda, K., Noguchi, T., Naguro, I., and Ichijo, H. (2007) Annu. Rev. Pharmacol. Toxicol. 48: 1-8.27; Nagai, H., Noguchi, T., Takeda, K., and Ichijo, I. (2007) J. Biochem. Mol. Biol. 40: 1-6).
Phosphorylation of ASK1 protein can lead to apoptosis or other cellular responses depending on the cell type. ASK1 activation and signaling have been reported to play an important role in a broad range of diseases including neurodegenerative, cardiovascular, inflammatory,
autoimmune, and metabolic disorders. In addition, ASK1 has been implicated in mediating organ damage following ischemia and reperfasion of the heart, brain, and kidney (Watanabe et al. (2005) BBRC 333, 562-567; Zhang et al, (2003) Life Sci 74-37-43; Terada et al. (2007) BBRC 364: 1043-49).
ROS are reported be associated with increases of inflammatory cytokine production, fibrosis, apoptosis, and necrosis in the kidney. (Singh DK, Winocour P, Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol 201 1 Mar;7(3): 176- 184; Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001 Dec 13; 414(6865):813-820; Mimura I, Nangaku M. The suffocating kidney:
tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol 2010 Nov; 6(1 1):667- 678).
Moreover, oxidative stress facilitates the formation of advanced glycation end-products (AGEs) that cause further renal injury and production of ROS. (Hung KY, et al. N- acetylcysteine-mediated antioxidation prevents hyperglycemia-induced apoptosis and collagen synthesis in rat mesangial cells. Am J Nephrol 2009;29(3): 192-202).
Tubulointerstitial fibrosis in the kidney is a strong predictor of progression to renal failure in patients with chronic kidney diseases (Schainuck LI, et al. Structural-functional correlations in renal disease. Part II: The correlations. Hum Pathol 1970; 1 : 631-641.).
Unilateral ureteral obstruction (UUO) in rats is a widely used model of tubulointerstitial fibrosis. UUO causes tubulointerstital inflammation, increased expression of transforming growth factor beta (TGF-β), and accumulation of myofibroblasts, which secrete matrix proteins such as collagen and fibronectin. The UUO model can be used to test for a drug’s potential to treat chronic kidney disease by inhibiting renal fibrosis (Chevalier et al., Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy, Kidney International (2009) 75, 1 145-1152.
Thus, therapeutic agents that function as inhibitors of ASK1 signaling have the potential to remedy or improve the lives of patients in need of treatment for diseases or conditions such as neurodegenerative, cardiovascular, inflammatory, autoimmune, and metabolic disorders. In particular, ASK1 inhibitors have the potential to treat cardio-renal diseases, including kidney disease, diabetic kidney disease, chronic kidney disease, fibrotic diseases (including lung and kidney fibrosis), respiratory diseases (including chronic obstructive pulmonary disease (COPD) and acute lung injury), acute and chronic liver diseases.
U.S. Publication No. 2007/0276050 describes methods for identifying AS 1 inhibitors useful for preventing and/or treating cardiovascular disease and methods for preventing and/or treating cardiovascular disease in an animal.
WO2009027283 discloses triazolopyridine compounds, methods for preparation thereof and methods for treating autoimmune disorders, inflammatory diseases, cardiovascular diseases and neurodegenerative diseases.
U.S. Patent Publication No. 2001/00095410A1, published January 13, 201 1, discloses compounds useful as ASK-1 inhibitors. U.S. Patent Publication No. 2001/00095410A1 relates to compounds of Formula (I):
SYN
WO 2016106384
PRODUCT PATENT
WO 2013112741
https://patents.google.com/patent/WO2013112741A1/en
InventorGregory Notte Original AssigneeGilead Sciences, Inc. Priority date 2012-01-27
SCHEME 1

SCHEME 2

COUPLING

GIVES

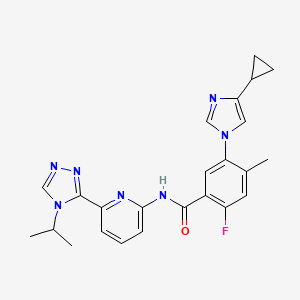
The name of the compound of the present invention as generated using ChemBioDraw Ultra 11.
is 5-(4-cyclopropyl- 1 H-imidazol- 1 -yl)-N-(6-(4-isopropyl-4H- 1 ,2,4-triazol-3 -yl)pyridin-2-yl)-2- fluoro-4-methylbenzamide also known as 5-((4-cyclopropyl-lH-imdazol-l-yl)-2-fluoro-N-(6-(4- isopropyl-4H- 1 ,2,4-triazole-3 -yl)pyridine-2-yl)-4-methylbenzamide.
One method of preparing compounds of formula (I) is shown in Reaction Schemes 1 and 2 below.
Scheme 1
Preparation of Compound A
To a solution of methyl 6-aminopicolinate (432 g, 2.84 mol) in MeOH (5 L) was added NH2NH2.H2O (284 g, 5.68 mol, 2.0 eq.). The reaction mixture was heated under reflux for 3 hr and then cooled to room temperature. The precipitate formed in the mixture was collected by filtration, washed with EA (2 L><2) and then dried in vacuo to give compound A (405 g, 94% yield) as white solid.
Preparation of compound B
A mixture of compound A (405 g, 2.66 mol) in dimethylformamide-dimethylacetal (DMF-DMA) (3.54 L) was heated under reflux for 18 hr, cooled to room temperature and then concentrated under reduced pressure. The residue was taken up in EA (700 mL) and heated at 50°C for 20 min. After being cooled to room temperature, the solid was collected by filtration and dried in vacuo to give compound B (572 g, 82% yield) as white solid.
Preparation of C
To a solution of compound B (572 g, 2.18 mol) in a mixture of CH3CN-AcOH (3.6 L, 4:1) was added propan-2-amine (646 g, 5.0 eq.). The resulting mixture was heated under reflux for 24 hr and then cooled to room temperature, and the solvent was removed under reduced pressure. The residue was dissolved in water (2.8 L) and 1 N aqueous NaOH was added to a pH of 8.0 H. The precipitate was collected by filtration and the filtrate was extracted with EA (500 mLx3). The combined organic layers were dried over anhydrous Na2S04, and then concentrated to a volume of 150 mL. To this mixture at 0°C was slowly added PE (400 mL) and the resulting suspension was filtered. The combined solid was re-crystallized from EA-PE to give compound C (253 g, 57% yield) as off-white solid.
1H- MR (400 MHz, CDC13): δ 8.24 (s, 1 H), 7.52 (m, 2 H), 6.51 (dd, J = 1.6, 7.2 Hz, 1 H), 5.55 (m, 1 H), 4.46 (bs, 2 H), 1.45 (d, J = 6.8 Hz, 6 H). MS (ESI+) m/z: 204 (M+l)+.
Compound C is a key intermediate for the synthesis of the compound of formula (I). Thus, an object of the present invention is also the provision of the intermediate compound C,
its salts or protected forms thereof, for the preparation of the compound of formula (I). An example of a salt of the compound C is the HC1 addition salt. An example of a protected form of compound C is the carbamate compound such as obtained with Cbz-Cl. Protective groups, their preparation and uses are taught in Peter G.M. Wuts and Theodora W. Greene, Protective Groups in Organic Chemistry, 2nd edition, 1991, Wiley and Sons, Publishers. Scheme 2
Preparation of the Compound of formula (I) continued:
Formula (I)
Compound 6 is a key intermediate for the synthesis of the compound of formula (I). Thus an object of the present invention is also the provision of intermediate compound 6,
6
salts or protected forms thereof, for the preparation of the compound of formula (I). An example of a salt of the compound 6 is the HC1 addition salt. An example of a protected form of the compound 6 is an ester (e.g. methyl, ethyl or benzyl esters) or the carbamate compound such as obtained with Cbz-Cl. Protective groups, their preparations and uses are taught in Peter G.M. Wuts and Theodora W. Greene, Protective Groups in Organic Chemistry, 2nd edition, 1991, Wiley and Sons, Publishers. Step 1 – Preparation of 5-amino-2-fluoro-4-methylbenzonitrile – Compound (2)
The starting 5-bromo-4-fluoro-2-methylaniline (1) (20g, 98 mmol) was dissolved in anhydrous 1-methylpyrrolidinone (100 mL), and copper (I) cyanide (17.6g, 196 mmol) was added. The reaction was heated to 180°C for 3 hours, cooled to room temperature, and water (300 mL) and concentrated ammonium hydroxide (300 mL) added. The mixture was stirred for 30 minutes and extracted with EA (3 x 200 mL). The combined extracts were dried over magnesium sulfate, and the solvent was removed under reduced pressure. The oily residue was washed with hexanes (2 x 100 mL), and the solid dissolved in dichloromethane and loaded onto a silica gel column. Eluting with 0 to 25% EA in hexanes gradient provided 5-amino-2-fluoro- 4-methylbenzonitrile (10.06g, 67.1 mmol). LC/MS (m/z:151 M+1).
Step 2 – Preparation of 5-(2-cvclopropyl-2-oxoethylamino)-2-fluoro-4-methylbenzonitrile – Compound (3)
5-Amino-2-fluoro-4-methylbenzonitrile (12g, 80mmol) was dissolved in anhydrous N,N- dimethylformamide (160 mL) under nitrogen, and potassium carbonate (13.27g, 96 mmol) and potassium iodide (14.61g , 88mmol) were added as solids with stirring. The reaction was stirred for 5 minutes at room temperature and then bromomethyl cyclopropylketone (20.24 mL, 180 mmol) was added. The reaction mixture was heated to 60°C for 3 hours, and then the solvents removed under reduced pressure. The residue was dissolved in EA (400 mL) and washed with 400 mL of water. The organic layer was dried over magnesium sulfate, and solvent was removed under reduced pressure. The residue was re-dissolved in a minimum amount of EA, and hexanes were added to bring the solution to 3: 1 hexanes: EA by volume. The product precipitated out of solution and was collected by filtration to provide 5-(2-cyclopropyl-2- oxoethylamino)-2-fluoro-4-methylbenzonitrile (14.19g, 61.2 mmol). LC/MS (m/z : 233, M+1)
Step 3 – Preparation of 5-(4-cvclopropyl-2-mercapto-lH-imidazol-l -yl)-2-fluoro-4- methylbenzonitrile – Compound (4)
5-(2-Cyclopropyl-2-oxoethylamino)-2-fluoro-4-methylbenzonitrile (14.19g, 61.2mmol) was dissolved in glacial acetic acid (300 mL). Potassium thiocyanate (11.9g, 122.4mmol) was added as a solid with stirring. The reaction mixture was heated to 110°C for 4 hours at which time the solvent was removed under reduced pressure. The residue was taken up in dichloromethane (200 mL) and washed with 200 mL water. The aqueous extract was extracted with (2 x 200 mL) additional dichloromethane, the organic extracts combined and dried over magnesium sulfate. The solvent was removed under reduced pressure and the oily residue was re-dissolved in EA (50 mL) and 150 mL hexanes was added. A dark layer formed and a stir bar was added to the flask. Vigorous stirring caused the product to precipitate as a peach colored solid. The product was collected by filtration, to yield 5-(4-cyclopropyl-2-mercapto-lH- imidazol-l-yl)-2-fluoro-4-methylbenzonitrile, (14.26g, 52.23 mmol). Anal. LC/MS (m/z : 274, M+1)
Step 4 – Preparation of 5-(4-cyclopropyl-lH-imidazol -yl)-2-fluoro-4-methylbenzonitrile – Compound (5)
In a 500 mL three neck round bottom flask was placed acetic acid (96 mL), water (19 mL) and hydrogen peroxide (30%, 7.47 mL, 65.88 mmol). The mixture was heated to 45°C with stirring under nitrogen while monitoring the internal temperature. 5-(4-Cyclopropyl-2- mercapto-lH-imidazol-l-yl)-2-fluoro-4-methylbenzonitrile (6.00g, 21.96 mmol) was then added as a solid in small portions over 30 minutes while maintaining an internal temperature below 55°C. When addition of the thioimidazole was complete the reaction was stirred for 30 minutes at a temperature of 45 C, and then cooled to room temperature, and a solution of 20% wt/wt sodium sulfite in water (6 mL) was slowly added. The mixture was stirred for 30 minutes and solvents were removed under reduced pressure. The residue was suspended in 250 mL of water and 4N aqueous ammonium hydroxide was added to bring the pH to ~10. The mixture was extracted with “dichloromethane (3 x 200ml), the organics combined, dried over magnesium sulfate, and the solvent was removed under reduced pressure. The residue was dissolved in 20 mL EA, and 80 mL of hexanes were added with stirring. The solvents were decanted off and an oily residue was left behind. This process was repeated and the product, 5-(4-cyclopropyl-lH- imidazol-l-yl)-2-fluoro-4-methylbenzonitrile was obtained as a viscous oil (5.14 g, 21.33 mmol) Anal. LC/MS (m/z: 242, M+1)
Step 5 – Preparation of 5-(4-cvclopropyl-lH-imidazol-l-yl)-2-fluoro-4-methylbenzoic acid hydrochloride (6)
5-(4-Cyclopropyl-lH-imidazol-l-yl)-2-fluoro-4-methylbenzonitrile (1 1.21g, 46.50mmol) was placed in a round bottom flask fitted with a reflux condenser, and suspended in 38% hydrochloric acid (200 mL). The mixture was heated to 100°C for 4.5 hours, and then cooled to room temperature. Solvent was removed under reduced pressure to give a pink solid, to which was added 100ml of EA. The solid product was collected by filtration and washed with 3 xlOO mL EA. To the solid product was added 100 mL 10% methanol in dichloromethane, the mixture stirred, and the filtrate collected. This was repeated with 2 more 100ml portions of 10% methanol in dichloromethane. The filtrates were combined and solvent was removed under reduced pressure, to provide crude 5-(4-cyclopropyl-lH-imidazol-l -yl)-2-fluoro-4- methylbenzoic acid hydrochloride. No further purification was carried out (1 1.13g, 37.54mmol). Anal. LC/MS (m/z: 261 , M+1)
Step 6 – Preparation of 5-(4-cvclopropyl- 1 H-imidazol- 1 -yl)-2-fluoro-N-(6-(4-isopropyl-4H- l,2,4-triazol-3-yl)pyridin-2-yl)-4-methylbenzamide – formula (I)
5-(4-Cyclopropyl- 1 H-imidazol- 1 -yl)-2-fluoro-4-methylbenzoic acid hydrochloride (1.5g,
5.07mmol) was suspended in anhydrous 1 ,2-dichlorom ethane (25 mL) at room temperature. Oxalyl chloride (0.575ml, 6.59mmol) was added with stirring under nitrogen, followed by N,N- dimethylformamide (0.044ml, 0.507mmol). The ; mixture was stirred for 4 hr at room temperature, and then the solvent was removed under reduced pressure. The residue was dissolved in 25 mL anhydrous dichloromethane. 6-(4-isopropyl-4H-l ,2,4-triazol-3-yl)pyridin-2- amine (1.13g, 5.58mmol) (compound C) and 4-dimethylaminopyridine (0.62g, 5.07 mmol) were rapidly added with stirring under nitrogen. The reaction was stirred for 2 hours at room temperature and aqueous saturated NaHC03 (15 mL) was added. The mixture was stirred for 10 minutes, and the layers were separated, and the aqueous layer was washed 1 x 20 mL dichloromethane. The combined organics were dried (MgS04), filtered and concentrated. The residue was dissolved in a minimum amount of CH3CN and water was slowly added until solids precipitated from the mixture. The solid was collected by filtration and dried to give 5-(4- cyclopropyl-lH-imidazol-l -yl)-2-fluoro-N-(6-(4-isopropyl-4H-l ,2,4-triazol-3-yl)pyridin-2-yl)- 4-methylbenzamide in -96% purity (1.28g, 2.88 mmol). Anal. LC/MS (m/z: 446, M+1). The material was further purified by RP-HPLC (reverse phase HPLC) to obtain an analytically pure sample as the HC1 salt.

C24H24FN7O-HCI. 446.2 (M+1). 1H-NMR (DMSO): δ 1 1.12 (s, 1H), 9.41 (s, 1H), 9.32 (s, 1H), 8.20 (d, J = 8.4 Hz, 1H), 8.07 (t, J = 8.4 Hz, 1 H), 7.95 (d, J = 6.4 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.79 (s, 1H), 7.59 (d, J = 10.4 Hz, 1H), 5.72 (sept, J = 6.8 Hz, 1H), 2.29 (s, 3H), 2.00-2.05 (m, 1H), 1.44 (d, J = 6.8 Hz, 6H), 1.01-1.06 (m, 2H), 0.85-0.89 (m, 2H).
PATENT
US 9067933
US 20150342943
WO 2016187393
WO 2016025474
WO 2016112305
WO 2017205684
WO 2017210526
WO 2018013936
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(3), 400-404
https://www.sciencedirect.com/science/article/pii/S0960894X17311861?via%3Dihub
https://ars.els-cdn.com/content/image/1-s2.0-S0960894X17311861-mmc1.pdf

PAPER
ACS Medicinal Chemistry Letters (2017), 8(3), 316-320
https://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00481
https://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.6b00481/suppl_file/ml6b00481_si_001.pdf
Apoptosis signal-regulating kinase 1 (ASK1/MAP3K) is a mitogen-activated protein kinase family member shown to contribute to acute ischemia/reperfusion injury. Using structure-based drug design, deconstruction, and reoptimization of a known ASK1 inhibitor, a lead compound was identified. This compound displayed robust MAP3K pathway inhibition and reduction of infarct size in an isolated perfused heart model of cardiac injury.
PATENT
FORM I TO IX POLYMORPHS
WO 2016105453
https://patents.google.com/patent/WO2016105453A1/zh-CN
Compound I is known to exhibit ASK1 inhibitory activity and is described in, for example, U.S. Patent No. 8,742,126, which is hereby incorporated by reference in its entirety. Compound I has the formula:

Compound I
Compound I can be synthesized according to the methods described in U.S. Patent No. 8,742,126 or U.S. Provisional Application No. 62/096,391, U.S. Provisional Application No. 62/269,064 and PCT Application PCT/US2015/067511 (filed on even date herewith and titled “Processes for Preparing ASK1 Inhibitors”), all of which are incorporated by reference in their entirety.
The present disclosure provides forms of Compound I and salts, co-crystals, hydrates, and solvates thereof. Also described herein are processes for making the forms of Compound I, pharmaceutical compositions comprising crystalline forms of Compound I and methods for using such forms and pharmaceutical compositions in the treatment of diseases mediated by ASK1 disregulation.
Thus, one embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I Form I) characterized by an X-ray powder diffractogram comprising the following peaks: 16.7, 21.3, and 22.8 °2Θ ± 0.2 °2Θ, as determined on a diffractometer using Cu-Kct radiation at a wavelength of 1.5406 A.
Another embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I Form II) characterized by an X-ray powder diffractogram comprising the following peaks: 11.2, 16.6, and 17.4 °2Θ ± 0.2 °2Θ, as determined on a diffractometer using Cu-Κα radiation at a wavelength of 1.5406 A.
Another embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I Form III) characterized by an X-ray powder diffractogram comprising the following peaks: 5.1, 10.2, and 25.3 °2Θ ± 0.2 °2Θ, as determined on a diffractometer using Cu-Κ radiation at a wavelength of 1.5406 A.
Another embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I FormIV) characterized by an X-ray powder diffractogram comprising the following peaks: 7.2, 12.6, and 19.3 °2Θ ± 0.2 °2Θ, as determined on a diffractometer using Cu-Κα radiation at a wavelength of 1.5406 A.
Another embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I FormV) characterized by an X-ray powder diffractogram comprising the following peaks: 9.7, 13.3, and 16.4 °2Θ ± 0.2 °2Θ, as determined on a diffractometer using Cu-Κα radiation at a wavelength of 1.5406 A.
Another embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I FormVI) characterized by an X-ray powder diffractogram comprising the following peaks: 8.8, 23.2, and 23.5 °2Θ ± 0.2 °2Θ, as determined on a diffractometer using Cu-Κα radiation at a wavelength of 1.5406 A.
Another embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I FormVII) characterized by an X-ray powder diffractogram comprising the following peaks: 8.2, 14.2, and 22.9 °2Θ ± 0.2 °2Θ as determined on a diffractometer using Cu-Κα radiation at a wavelength of 1.5406 A.
Another embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I FormVIII) characterized by an X-ray powder diffractogram comprising the following peaks: 8.4, 19.3, and 24.3 °2Θ ± 0.2 °2Θ as determined on a diffractometer using Cu-Κα radiation at a wavelength of 1.5406 A.
Another embodiment is crystalline 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyI-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (Compound I FormIX) characterized by an X-ray powder diffractogram comprising the following peaks: 6.9, 14.3, 23.7, and 24.8 °2Θ ± 0.2 °2Θ as determined on a diffractometer using Cu-Κα radiation at a wavelength of 1.5406 A.
Another embodiment is amorphous 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide.
Some embodiments provided herein relate to crystalline forms of salts or co-crystals of Compound I.
The compound, 5-(4-cyclopropyl-lH-imidazol-l-yl)-N-(6-(4-isopropyl-4H-l,2,4-triazol-3-yl)pyridin-2-yl)-2-fluoro-4-methylbenzamide (also known as 5-((4-cyclopropyl-lH-imidazol-l-yl)-2-fluoro-N-(6-(4-isopropyl-4H-l,2,4-triazole-3-yl)pyridine-2-yl)-4-methylbenzamide)) designated herein as Compound I, has the formula:

Compound I exhibits an EC50 value of about 2 nanomolar in an ASK1 293 cell-based assay. The experimental protocol for this assay is known in the art and is described in U.S. Patent No. 8,742,126, which is hereby incorporated by reference in its entirety.
The present disclosure relates to various crystalline forms of Compound I, and processes for making the crystalline forms. Compound I also provides forms further described herein as “Compound I Form I,” “Compound I Form II,” “Compound I Form III,” “Compound I Form TV,” “Compound I Form V,” “Compound I Form VI,” “Compound I Form VII,” “Compound I Form VIII,” “Compound I Form IX,” and “amorphous Compound I.” In some embodiments, such forms of Compound I may be a solvate or a hydrate.
Additional crystalline forms of Compound I are also further described herein. In some embodiments, crystalline forms of Compound I may include salts or co-crystals of Compound I. Salts or co-crystals of Compound I may have the following formula:

• X
PATENT
WO 2016106384
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016106384&recNum=31&docAn=US2015067511&queryString=EN_ALL:nmr%20AND%20PA:(gilead%20sciences)&maxRec=1065
As described generally above, the disclosure provides in some embodiments processes for making a compound of formula (A).
Scheme 1 represents an exemplary synthesis of a compound of formula (A) and can be carried out according to the embodiments described herein. It is contemplated that the exemplary synthesis shown in Scheme 1 may be particularly advantageous. For example, the synthesis employs less toxic starting materials (i.e., using Compound (H) in place of its corresponding analog having bromide at the tosylate position), avoids toxic reagents (i.e., CuCN), and employs less toxic solvents (i.e., using dichloromethane instead of dichloroethane), including at the final step of the synthesis. The synthesis also can utilize milder reaction conditions (i.e., avoids high temperatures needed for cyanation, etc.), can avoid the use of heavy metals, and can require less purification steps (e.g. avoid column chromatography). The particular reaction conditions and reagents employed in Scheme 1 are discussed below.
Scheme 1

Compound (B)

Scheme 2

Compound (A)
Scheme 3

Compound (E) Compound (A)
EXAMPLES
The compounds of the disclosure may be prepared using methods disclosed herein and routine modifications thereof which will be apparent given the disclosure herein and methods well known in the art. Conventional and well-known synthetic methods may be used in addition to the teachings herein. The synthesis of compounds described herein, may be accomplished as described in the following examples. If available, reagents may be purchased commercially, e.g. from Sigma Aldrich or other chemical suppliers. Unless otherwise noted, the starting materials for the following reactions may be obtained from commercial sources.
Example 1: Synthesis of Compound (A)

Compound (C)

MeCN Toluene, /‘Pr2EtN
Compound (J) Compound (H)

ompound F
(COCI)2, DMF 
Compound (D-a)

Compound (B) J Compound (A) Hydroxytosylation of Compound (J) to form Compound (H)

Compound (J) Compound (H)
Koser’s reagent, PhI(OH)OTs, (1.0 eq.) and acetonitrile (5 vols) are charged to a flask. Cyclopropylmethyl ketone (Compound (J), 1.2 eq.) is charged and the mixture is heated to about 70 °C to about 75 °C. Once the reaction is complete, the contents are cooled and concentrated. The residue is diluted in dichloromethane (about 2.5 vols) and washed with water (2 x about 1 to 2 volumes). The organic phase is concentrated to approximately 1.5 vols and the product is triturated with hexanes (about 1.5 to 2 vols) and concentrated to remove dichloromethane and the distilled volume is replaced with hexanes. The slurry is agitated for about two hours, filtered and washed with hexanes. The solids are dried under vacuum at about 40 °C to afford Compound (H). 1H MR (400 MHz, DMSO-d6): δ 7.82 (d, 2H, J= 8.0 Hz), 7.49 (d, 2H, J= 8.0 Hz), 4.98 (s, 2H), 2.42 (s, 3H), 2.02-2.08 (m, 1H), 0.95-0.91 (m, 2H), 0.89-0.82 (m, 2H). 13C MR (100 MHz, DMSO-de): 202.39, 145.60, 132.76, 130.57, 128.12, 72.98, 21.52, 17.41, 11.39.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in lieu of Koser’s reagent, alternative reagents may include, but are not limited to, (diacetoxyiodo)benzene organosulfonic acid, (diacetoxyiodo)benzene and p-toluenesulfonic acid, iodosylbenzene/p-toluenesulfonic acid, m-chloroperbenzoic acid/p-toluenesulfonic acid, poly(4-hydroxy tosyloxyiodo)styrenes, N-methyl-O-tosylhydroxylamine, Dess-Martin periodinane/p-toluenesulfonic acid, HlCVp-toluenesulfonic acid, and o-iodoxybenzoic acid/p-toluenesulfonic acid. Various solvents, such as toluene, benzene, tetrahydrofuran, 2-methyltetrahydrofuran, dichloromethane, and chloroform, may be employed. The reaction may take place at temperatures that range from about 20 °C to about 100 °C.
Alkylation of Compound (H) with Compound (I) to form Compound (G)
Co

To a mixture of Compound (I) (1.0 equiv) and Compound (H) (1.1 equiv) in toluene (5 vols) is charged iPr2 Et (2.1 equiv). The mixture is heated to about 90 to about 100 °C and aged for about less than 10 hours. Upon completion, the mixture is cooled and diluted with water (about 5 to about 6 vols). The biphasic mixture is separated and the organic solution is washed sequentially with aq. H4C1 (about 27 wt%, about 2 to about 3 vols), aq. NaHC03 (about 9 wt%, about 2 to about 3 vols), and aq. NaCl (about 15 wt%, about 1 vols). The organic solution is dried over Na2S04, filtered, and washed with toluene (about 2 to about 3 vols). The solution is concentrated under vacuum at about 45 °C and the residue is crystallized by the addition of hexane at about 20 °C to about 25 °C and at about 10 °C to about 15 °C. The slurry is filtered, washed with cooled isopropanol (about 1 vol) and dried under vacuum at about 37 °C to about 43 °C to afford Compound (G). 1H NMR(400 MHz, DMSO-d6): δ 7.05 (d, 1H, J= 12.0 Hz), 6.51 (d, lH, J= 8.0 Hz), 5.27 (t, 1H, J= 4.0 Hz), 4.17 (d, 2H, J= 4.0 Hz), 2.21-2.14 (m, 1H), 2.10 (s, 3H), 0.96-0.86 (m, 4H). 13C NMR (100 MHz, DMSO-d6): 208.17, 151.63, 149.32, 143.99, 143.97, 123.81, 123.74, 118.13, 117.90, 112.87, 105.09, 104.88, 53.72, 18.33, 17.43, 17.42, 10.85.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative bases, including but not limited to organic bases (e.g., DBU and DMAP), alkali metal bases (e.g., NaH), hexamethyldisilazane bases (e.g, sodium, potassium and lithium hexamethyldisilazide), carbonate bases (e.g., Cs2C03, Na2C03), and potassium tert-butoxide. Various solvents, such as THF, MTBE, 2-MeTHF, acetonitrile, dioxane, benzene, DMF, DMAc, NMP, may be employed. The reaction may take place at temperatures that range from about -78 °C to about 100 °C.
Formylation of Compound (G) to form Compound (F)

Acetic anhydride (4 equiv) is added to aqueous formic acid (about 3 to about 4 vols) at about 0 °C to about 5 °C and the mixture is agitated. Compound (G) (1.0 equiv) in DCM (about 3 vols) is charged. The reaction is aged at about 0 to about 5 °C until it is deemed complete. Upon reaction completion, water (about 4 vols) is charged and the mixture is adjusted to about pH 8-9 by the addition of 40-50% aqueous NaOH with the content temperature maintained between about 0 °C to about 15 °C. The biphasic mixture is separated and the aqueous solution is extracted with dichloromethane (about 6 vols). The organic solution is washed with saturated aqueous NaCl (about 4 vols), dried over Na2S04, and filtered. Compound (F) is carried forward to the next step as a solution in dichloromethane without further purification. 1H MR (400 MHz, DMSO-de): δ (mixture of amide rotamers) 8.17 (s, 1H), 8.14 (s, 1H), 7.61 (d, 1H, J= 8.0 Hz), 7.45 (d, 1H, J= 8.0 Hz), 7.42 (d, 1H, J= 12.0 Hz), 7.33 (d, 1H, J= 12.0 Hz), 4.87 (s, 2H), 4.68 (s, 2H), 2.25 (s, 3H), 2.16 (s, 3H), 2.12-2.03 (m, 1H), 0.98-0.85 (m, 4H). 13C MR (100 MHz, DMSO-de): 206.68 (204.85), 163.71 (163.22), 158.95 (158.69), 156.51 (156.35), 139.09 (139.02), 138.61 (138.53), 137.58 (137.55), 133.35 (133.34), 132.45, 119.02 (118.79), 118.58 (118.36), 105.35 (105.03), 104.77 (104.55), 58.68, 55.40, 17.84 (17.77).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in lieu of acetic anhydride and formic acid, acetic acid monoanhydride with carbonic acid or trifluoroacetic anhydride with formic acid may be used. Various solvents, such as chloroform, acetonitrile, isopropyl acetate, or THF, may be employed. The reaction may take place at temperatures that range from about -10 °C to about 40 °C.
Imidazole Cyclization to Form Compound (E)

To a solution of Compound (F) (1.0 equiv) in DCM is charged acetic acid (about 5 vols). The solution is concentrated under vacuum at about 35 °C to remove the bulk of DCM and ammonium acetate (3.9 equiv) is added. The mixture is heated to about 110 °C to about 115 °C and agitated until the reaction is deemed complete. The reaction is cooled, diluted with water (about 10 vols) and iPrOAc (about 6 vols). The mixture is adjusted to about pH 8-9 by the addition of 40-50% aqueous NaOH. The biphasic mixture is separated. Sodium chloride (about 0.3 wt equiv wrt Compound (F)) is charged to the aqueous layer and the aqueous layer is extracted with iPrOAc (about 2 vols). The organic solution is washed with water (about 5 vols) and aq. NaCl (about 10 wt%, about 4 to about 5 vols). The solution is concentrated under vacuum and solvent exchanged to about 2-3 vols Ν,Ν-di methyl acetamide (DMAc). Water (about 5 to about 6 vols) is charged to afford Compound (E) as a slurry. The slurry is filtered and washed sequentially with DMAc/water, water, and hexanes. The resulting solids are dried under vacuum at about 55 °C to afford Compound (E). 1H NMR (400 MHz, DMSO-d6): δ 7.68 (d, 1H, J= 4.0 Hz), 7.64 (d, 1H, J= 1.0 Hz), 7.46 (d, 1H, J= 12.0 Hz), 7.12 (d, 1H, J= 1.0 Hz), 2.12 (s, 3H), 1.85-1.79 (m, 1H), 0.81-0.76 (m, 2H), 0.70-0.66 (2H). 13C NMR (100 MHz, DMSO-d6): 159.11, 156.67, 156.67, 143.94, 137.36, 136.19, 136.11, 134.44, 134.41, 131.21, 131.20, 119.05, 118.82, 116.21, 105.56, 105.34, 17.72, 17.71, 9.26, 7.44.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in lieu of ammonium acetate, alternative sources of ammonia may be used, including but not limited to ammonium formate and ammonium hydroxide. Various solvents, such as toluene, benzene, and isopropanol, may be employed. The reaction may take place at temperatures that range from about 80 °C to about 120 °C.
Carboxylation o Compound (E) to form Compound (D)

Compound (E) then 15 10 25 c Compound (D)
A mixture of Compound (E) (1.0 equiv) in THF (about 15 vols) was cooled to about -10 to about 0 °C and a solution of iPrMgCl (2.0 M in THF, 1.2 equiv) was charged slowly to maintain the internal temperature below about 5 °C. The mixture was stirred for about 1 hour at about -5 to about 5 °C after which C02 was bubbled slowly into the mixture (exothermic). The addition is continued until the exotherm subsides and the internal temperature typically increases to about 15 to about 25 °C after the addition. Upon reaction completion, the mixture is concentrated under vacuum to approximately 3 vols and water (about 6 to about 7 vols) is added, followed by about 1 vol 6M HC1. MTBE (about 10 vols) is added and the biphasic mixture is separated. A solution of 6 M HC1 is added slowly to the aqueous layer to adjust the pH (initially at > 10) to approximately 4.8. The mixture is seeded with Compound (D) (if necessary), which was formed according to the procedure outlined above, and the resultant slurry is cooled slowly to about 0 °C to about 5 °C and aged. The slurry is filtered, washed with water (about 4 vols), isopropanol (about 4 vols), followed by n-heptane (about 6 vols). The solids are dried under vacuum at about 40 °C to afford Compound (D). 1H NMR (400 MHz, DMSO-d6): δ 7.69 (d, 1H, J= 2.0 Hz), 7.67 (d, 1H, J= 8.0 Hz), 7.40 (d, 1H, J= 8.0 Hz), 7.15 (d, 1H, J= 2.0 Hz), 2.20 (s, 3H), 1.87-1.80 (m, 1H), 0.81-0.77 (m, 2H), 0.71-0.67 (m, 2H). 13C NMR (100 MHz, DMSO-d6): 164.52, 164.48, 161.68, 159.12, 143.95, 141.63, 141.53, 137.34, 133.21, 133.18, 129.70, 119.85, 119.61, 118.08, 117.97, 116.25, 18.02, 9.21, 7.48.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative bases, including but not limited to organolithium bases (e.g., MeLi, «-BuLi, t-BuLi, and sec- uLi) and Grignard bases (e.g., MeMgCl, «-BuMgCl, and PhMgCl). Various solvents, such as 2-MeTHF, dioxane, MTBE, and Et20, may be employed. The reaction may initially take place at temperatures that range from about -20 °C to about 40 °C and then continue at temperature that range from about -10 °C to about 50 °C.
Conversion o Compound (D) to form Compound (D-a)

Compound (D) Compound (D-a)
To a mixture of Compound (D) (1.0 equiv) in methanol (about 4 vols) at about 15 °C to about 25 °C is charged concentrated HC1 (1.1 equiv relative to Compound (D)). The mixture is aged until most of the Compound (D) is dissolved, seeded with Compound (D-a) (0.005 equiv), which was formed according to the procedure outlined above, and MTBE (about 3 vols relative to the amount of seed) is charged slowly. The slurry is aged, filtered, and rinsed with MTBE (5 vols) and the solids are dried under vacuum at about 40 °C to afford Compound (D-a). 1H MR (400 MHz, DMSO-de): δ 9.34 (s, 1H), 8.00 (d, 1H, J= 8.0 Hz), 7.76 (d, 1H, J= 2.0 Hz), 7.54 (d, 1H, J= 12.0 Hz), 2.25 (s, 3H), 2.08-2.01 (m, 1H), 1.05-1.00 (m, 2H), 0.92-0.88 (m, 2H). 13C MR QOO MHz, DMSO-d6): 164.08, 164.05, 162.73, 160.14, 142.11, 142.01, 137.11, 135.91, 131.14, 131.11, 130.73, 120.19, 119.96, 118.78, 118.39, 118.27, 17.71, 8.24, 6.13.
Carboxylation o Compound (E) to form Compound (D) Hydrate

Compound (E) then 15 10 25 °c Compound (D) Hydrate
A mixture of Compound (E) (1.0 equiv) in THF (about 15 vols) was cooled to about -10 to about 0 °C and a solution of iPrMgCl (2.0 M in THF, 1.2 equiv) was charged slowly to maintain the internal temperature below about 5 °C. The mixture was stirred for about 1 hour at about -5 to about 5 °C after which C02 was bubbled slowly into the mixture (exothermic). The addition is continued until the exotherm subsides and the internal temperature typically increases to about 15 to about 25 °C after the addition. Upon reaction completion, the mixture is concentrated under vacuum to approximately 3 vols and water (about 6 to about 7 vols) is added, followed by about 1 vol 6 M HC1. MTBE (about 10 vols) is added and the biphasic mixture is separated. A solution of 6 M HC1 is added slowly to the aqueous layer to adjust the pH (initially at > 10) to approximately 4.8. The mixture is seeded with Compound (D) (if necessary), which was formed according to the procedure outlined above, and the resultant slurry is cooled slowly to about 0 °C to about 5 °C and aged. The slurry is filtered and washed with water (about 4 vols). The solids are dried under vacuum at about 40 °C to afford Compound (D) hydrate. 1H NMR (400 MHz, DMSO-d6): δ 7.69 (d, 1H, J= 2.0 Hz), 7.67 (d, 1H, J= 8.0 Hz), 7.40 (d, 1H, J = 8.0 Hz), 7.15 (d, 1H, J= 2.0 Hz), 2.20 (s, 3H), 1.87-1.80 (m, 1H), 0.81-0.77 (m, 2H), 0.71-0.67 (m, 2H). 13C NMR (100 MHz, DMSO-d6): 164.52, 164.48, 161.68, 159.12, 143.95, 141.63, 141.53, 137.34, 133.21, 133.18, 129.70, 119.85, 119.61, 118.08, 117.97, 116.25, 18.02, 9.21, 7.48.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative bases, including but not limited to organolithium bases (e.g., MeLi, «-BuLi, t-BuLi, and sec- uLi) and Grignard bases (e.g., MeMgCl, «-BuMgCl, and PhMgCl). Various solvents, such as 2-MeTHF, dioxane, MTBE, and Et20, may be employed. The reaction may initially take place at temperatures that range from about -20 °C to about 40 °C and then continue at temperature that range from about -10 °C to about 50 °C.
Acid Chloride Formation Using Compound (D-a) to Form Compound (B)

Compound (B)
To a mixture of Compound (D-a) (1.0 equiv), DCM (about 10 vols) and DMF (0.1 equiv), a solution of oxalyl chloride (about 1.7 equiv) was slowly charged to maintain the internal temperature below about 30 °C. The mixture was stirred for about 1 hour at about 20 °C after which time the mixture is distilled to about about 4 vols total volume. DCM (about 5 vols) is repeatedly charged and the mixture distilled to about 4 vols total volume. DCM is then charged to bring the total volume to about 12 vols of Compound (B). The solution is carried forward to the next step without further purification.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in lieu of Compound (D-a), compound (D) may be used. Additionally, in lieu of oxalyl chloride and DMF, thionyl chloride, PC15, and PCI3 may be used. Various
solvents, such as MeCN, THF, and MTBE, may be employed. In some embodiments, additives may be used, including but not limited to trimhetylsilyl chloride, water, HC1, or tetrabutyl ammonium chloride. The reaction may take place at temperatures that range from about -20 °C to about 40 °C.
Acid Chloride Formation Using Compound (D) Hydrate to Form Compound (B)

To a mixture of Compound (D) hydrate (1.0 equiv), DCM (about 10 vols) and DMF (0.1 equiv), a solution of oxalyl chloride (1.2 equiv) was slowly charged to maintain the internal temperature below about 30 °C. The mixture was stirred for about 1 hour at about 20 °C after which time the mixture is distilled to about about 4 vols total volume. DCM (about 5 vols) is repeatedly charged and the mixture distilled to about 4 vols total volume. DCM is then charged to bring the total volume to about 12 vols of Compound (B). The solution is carried forward to the next step without further purification.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in lieu of Compound (D) hydrate, compound (D) may be used.
Additionally, in lieu of oxalyl chloride and DMF, thionyl chloride, PC15, and PCI3 may be used. Various solvents, such as MeCN, THF, and MTBE, may be employed. In some embodiments, additives may be used, including but not limited to trimhetylsilyl chloride, water, HC1, or tetrabutyl ammonium chloride. The reaction may take place at temperatures that range from about -20 °C to about 40 °C.
mide Bond Formation to form Compound (A)

Compound (C) 15 to 25 °C Compound (A)
Compound (C) was synthesized as described in U.S. Patent No. 8,742, 126, which is hereby incorporated by reference in its entirety.
To a solution of Compound (B) (about 1 equiv in about 12 vols DCM) was charged diisopropylethyl amine (1.0 equiv) followed by Compound (C) (1.05 equiv). Upon reaction completion, 5% aqueous sodium hydroxide (about 5 vols) is added and the layers of the biphasic mixture are separated. A solution of 10% aqueous citric acid (about 2 vols) is charged to the organic layer and the layers of the biphasic mixture are separated. Water (about 5 vols) is charged to the organic layer and the layers of the biphasic mixture are separated. The organic solution is filtered, and the solution is solvent swapped to about 15% DCM in EtOH under vacumm at about 45 °C. The mixture is seeded with about 0.001 equiv of Compound (A), which was synthesized as described by U.S. Patent No. 8,742,126, and the resultant slurry is aged at about 45 °C. An additional 2-3 vols solvent is distilled in vacuo and then heptane (about 10 vols) is charged slowly and the slurry is aged, cooled to about 20 °C, filtered and washed with 1 :2 EtOH:heptane (about 3 vols). The solids are dried under vacuum at about 40 °C to afford Compound (A). Characterization data for Compound (A) matches that disclosed in U.S. Patent No. 8,742,126.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative bases may be used, including but not limited to Et3N, pyridine, and DMAP. Various solvents, such as 2-MeTHF, toluene, MTBE, and chloroform, may be employed. The reaction may take place at temperatures that range from about 0 °C to about 40 °C.
In lieu of Compound (B), Compound (D) or activated esters thereof may be employed.
Coupling reagents may also be employed; non-limiting examples of such reagents include
propane phosphonic acid anhydride (T3P®), Ι, -carbonyldiimidazole, EDC/HOBt or other imide coupling reagents, isobutylchloroformate (to generate an isobutyl ester), and pivoyl chloride (to generate a pivalate ester).
Example 2: Alternative Synthesis of Compound (D)
C 
ompound (K) Compound (L)
Compound (D)
Coupling of Compound (K) and Compound (L-a) to provide Compound (D)

Compound (K) Compound (L-a) Compound (D)

Compound 2-1 Compound 2-2
Compound (L-a) (1.0 eq), Compound (K) (1.5 eq), potassium phosphate (5.0 eq), copper
(I) oxide (0.05 eq), and 8-hydroxyquinoline, Compound 2-2 (0.2 eq) were combined with degassed DMSO (about 6 vols). The reaction mixture was heated to about 95 °C to about 105 °C and stirred for about 22 h. Upon reaction completion, the mixture was cooled to ambient temperature and diluted with water (about 6 vols) and isopropyl acetate (about 5 vols). The aqueous layer was washed with isopropyl acetate (about 5 vols), and the pH was adjusted to about 6 by the addition of 8 M HC1. The solution was seeded with about about 0.003 equiv of Compound (D) seed, which was synthesized as described in U.S. Patent No. 8,742, 126, and the pH was further adjusted to pH about 4.8. The resultant slurry was cooled to about 0 °C for about 2 h, filtered, and washed with cold dilute HC1 (pH about 4.8, about 2 vols) and cold isopropyl alcohol (about 2 vols) to provide Compound (D). 1H NMR (400 MHz, DMSO-d6): δ 7.69 (d,
1H, J= 2.0 Hz), 7.67 (d, 1H, J= 8.0 Hz), 7.40 (d, 1H, J= 8.0 Hz), 7.15 (d, 1H, J= 2.0 Hz), 2.20 (s, 3H), 1.87-1.80 (m, 1H), 0.81-0.77 (m, 2H), 0.71-0.67 (m, 2H). 13C MR (100 MHz, DMSO-d6): 164.52, 164.48, 161.68, 159.12, 143.95, 141.63, 141.53, 137.34, 133.21, 133.18, 129.70, 119.85, 119.61, 118.08, 117.97, 116.25, 18.02, 9.21, 7.48.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative bases may be used, including but not limited to carbonate bases (such as CS2CO3, K2C03, and Na2C03). In lieu of Cu20, alternative catalysts may be used, such as CuOAc, Cul, CuBr, and [(CuOTf)2-benzene complex]. Non-limiting examples of alternative ligands include phenanthroline ligands (such as 4,7-dimethoxy-l, 10-phenanthroline (Compound 2-1) and 1,10-phenanthroline), aminoarenethiols (such as 2-((dimethylamino)methyl)benzenethiol), oxime-phospine oxides, phosphoramidites, 2-aminopyrimidine diols (such as 2-aminopyrimidine-4,6-diol), and oxime-phosphine oxides (such as 2-hydroxybenzaldehyde oxime). In some embodiments, additives may be used, including but not limited to polyethyleneglycol and/or water, Et4NHC03, and cetryltrimethylammonium bromide.
In lieu of Compound (L-a), alternative starting material can be used, including but not limited to 5-bromo-2-fluoro-4-methylbenzoic acid, 2-fluoro-4-methyl-5-(((trifluoromethyl)sulfonyl)oxy)benzoic acid, and 2-fluoro-4-methyl-5-(tosyloxy)benzoic acid. Additionally, in lieu of the free base of Compound (K), various salts of Compound (K) may be used, such as the besylate salt.
Various solvents may be used, including but not limited to DMF, DMAc, DMSO, butyronitrile, xylenes, EtCN, dioxane, and toluene. The reaction may take place at temperatures that range from about 80 °C to about 150 °C.
Coupling of Compound (L-b) with Compound (K) to provide Compound (D)

Compound (L-b) Compound (K) Compound (D)
Compound (L-b) (1 equiv), Compound (K) (1.2 equiv), and Cu(OAc)2 (1 equiv) was added methanol (about 20 vols) followed by pyridine (2.2 equiv). The mixture was then stirred at about 23 °C for about 16 h, then at about 45 °C for about 4 h.The reaction mixture was diluted with methanol (about 60 vols), filtered though a pad of celite and concentrated in vacuo to afford Compound (D) . 1H MR (400 MHz, DMSO-d6): δ 7.69 (d, 1H, J= 2.0 Hz), 7.67 (d, 1H, J= 8.0 Hz), 7.40 (d, 1H, J= 8.0 Hz), 7.15 (d, 1H, J= 2.0 Hz), 2.20 (s, 3H), 1.87-1.80 (m, 1H), 0.81-0.77 (m, 2H), 0.71-0.67 (m, 2H). 13C MR (100 MHz, DMSO-d6): 164.52, 164.48, 161.68, 159.12, 143.95, 141.63, 141.53, 137.34, 133.21, 133.18, 129.70, 119.85, 119.61, 118.08, 117.97, 116.25, 18.02, 9.21, 7.48.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in lieu of Compound (L-b), 2-fluoro-4-methyl-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzoic acid may be used. In lieu of Compound (K), the besylate salt of Compound (K) may be used.
Various copper reagents can be employed, such as Cu(OTf)2, Cu20, and CuBr.
Alternative bases include but are not limited to triethylamine and N,N-diisopropylethylamine. Various solvents, such as DCM and DMF, may be employed. The reaction may take place at temperatures that range from about 23 °C to about 100 °C and under an atmosphere of oxygen or nitrogen.
Example 3: Alternative Synthesis of Compound (C)
C

Compound (C)
Coupling of Compound (O) with Compound (N-a) to form Compound (M)

Compound (O) Compound (N-a)
Compound (M)
To a mixture of Compound (O) (1.0 equiv), Compound (N-a) (1.6 equiv), PdCl2(PPh3)2 (65 mol%), Cs2C03 (2.0 equiv), and Cul (4.7 mol%) was charged dioxane (10 mL). The mixture
was degassed and then heated to about 95 °C to about 105 °C. After a period of about 20 hours, the mixture was cooled to ambient temperature. The reaction mixture was diluted with EtOAc (about 10 vols), washed with water (about 10 vols) and the layers of the biphasic mixture were separated. The organic layer was dried over MgS04 and concentrated in vacuo. The crude residue was purified by silica gel chromatography to afford Compound (M). 1H NMR (400
MHz, DMSO-de): δ 8.95 (s, 1H), 8.16-8.04 (m, 2H), 7.67 (d, 1H, J= 8.4 Hz), 5.34 (sep, 1H, J = 6.6 Hz), 1.50 (d, 6H, 6.6 Hz). 13C NMR (100 MHz, DMSO-d6): 149.90, 149.58, 148.36, 144.11, 141.62, 125.27, 122.92, 48.91, 23.42.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative catalysts may be other Pd (II) complexes or Pd(0) complexes with trialkyl or triarylphosphine ligands, including but not limited to: Pd(PPh3)4, Pd2dba3/PPh3, Pd(OAc)2/dppf, Pd2dba3/dppp, Pd(OAc)2/PPh3, Pd(OAc)2/dppe, Pd2dba3/dppf. Various bases may be used, such as a carbonate base (e.g. K2C03 or Na2C03). Various solvents, such as DMF, DMAc, DMSO, butyronitrile, and NMP, may be employed. The reaction may take place at temperatures that range from about 80 °C to about 150 °C.
Conversion of Compound (M) to form Compound (C)

Compound (M) Compound (C)
To a mixture of Compound (M) (1.0 equiv), Pd(OAc)2 (2.0 mol%), rac-BINAP (3.0 mol%), and Cs2C03 (1.4 equiv), was charged dioxane (about 9 vols) followed by benzophenone imine (2.0 equiv). The mixture was degassed, sealed and then heated to about 75 °C to about 85 °C under nitrogen. After a period of about 20 hours, the mixture was cooled to ambient temperature, and HC1 (6 M, about 8 vols) was charged until the pH of the reaction mixture was about 1 to about 2. The solution was maintained at ambient temperature for about 15 minutes, then NaOH (30 wt.%, about 1 to about 2 vols) was charged until the pH of the reaction mixture was about 8-9. The reaction mixture was concentrated in vacuo, slurried in MeOH (about 22 vols), and filtered to remove gross solids, which were washed with MeOH (2 x about 3 vols). The resulting solution was concentrated in vacuo, adsorbed onto celite and purified by silica gel chromatography to provide compound (C). LRMS [M+H]+: 204.08.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative catalysts may be other Pd (II) complexes or Pd(0) complexes with trialkyl or triarylphosphine ligands, including but not limited to: Pd(PPh3)4, Pd2dba3/PPh3, Pd(OAc)2/dppf, Pd2dba3/dppp, Pd(OAc)2/PPh3, Pd(OAc)2/dppe, Pd2dba3/dppf,
Pd2dba3/CyJohnPhos, Pd2dba3/P(t-Bu)3. Various ammonia sources may be used such as
LiHMDS or ammonium hydroxide. Various carbonate bases (e.g. K2C03 or Na2C03) or phosphate bases such as K3P04 may be used. Various solvents, such as THF, DMAc, DMSO, and NMP, may be employed. The reaction may take place at temperatures that range from about 75 °C to about 150 °C and pressures ranging from about 15 to about 50 psig.
Example 4: Alternative Synthesis of Compound (C)
Co 
mpound (O)
Compound (C)
Coupling of Compound (O) with Compound (P-a) to form Compound (C)
C

)
To a mixture of Compound (O) (1.0 equiv), Compound (P-a) (1.0 equiv), PdCl2(PPh3)2 (10 mol%), Cs2C03 (2.0 equiv), and Cul (4.7 mol%) was charged dioxane (about 20 vols). The mixture was degassed and then heated to about 95 °C to about 105 °C. After a period of about 20 to about 40 hours, the mixture was cooled to ambient temperature. The reaction mixture was diluted with EtOAc (about 40 vols) and the organic layer was washed with water (about 40 vols) The layers of the biphasic mixture were separated and the aqueous phase was extracted with
EtOAc (about 40 vols). The combined organic phases were concentrated in vacuo. To the residue was charged IPA (about 20 vols), and the resulting suspension was stirred at about 40 °C to about 50 °C for about 1 h and then stirred at ambient temperature for about 16 h. The suspension was cooled to about 5 °C, filtered and washed with cold IPA (about 4 vols). The resulting solids were dried at about 40 °C to afford Compound (C). 1H NMR (400 MHz, DMSO-d6): δ 8.77 (s, 1H), 7.51 (t, 1H, J= 8.0 Hz), 7.18 (d, 1H, J= 4.0 Hz), 6.53 (d, 1H, J= 8.0 Hz), 6.17 (s, 1H), 5.53 (sep, 1H, J= 8.0 Hz), 1.42 (d, 6H, J= 8.0 Hz). 13C NMR (100 MHz, DMSO-d6): 159.59, 151.18, 146.25, 142.97, 138.41, 111.90, 108.88, 48.12, 23.55.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative catalysts may be other Pd (II) complexes or Pd(0) complexes with trialkyl or triarylphosphine ligands, including but not limited to: Pd(PPh3)4, Pd2dba3/PPh3, Pd(OAc)2/dppf, Pd2dba3/dppp; Pd(OAc)2/PPh3; Pd(OAc)2/dppe; Pd2dba3/dppf, Pd(OAc) 2/(m-tolyl)3P, Pd(OAc)2/JohnPhos; PdCl2dppf, Pd(OAc)2/(o-tolyl)3P; PdCl2(AmPhos)2; Pd(OAc) 2/(cyclohexanlyl)3P. Various bases may be used, such as a carbonate base (e.g. K2C03 or Na2C03). Various solvents, such as DMF, DMAc, DMSO, butyronitrile, and NMP, may be employed. The reaction may take place at temperatures that range from about 80 °C to about 150 °C.
Coupling of Compound (O) with Compound (P-b) to form Compound (C)
Co

)
A solution of Compound (O) (1.0 equiv) in THF (about 20 vols) was degassed with nitrogen. The solution was cooled to about -55 °C to about -70 °C and a solution of n-BuLi (1.6 M solution in hexane, 1.0 equiv) was added over about 15 to about 20 minutes. The suspension was stirred for about 15 to about 25 minutes at about -55 °C to about -60 °C, followed by the slow addition of ZnCl2 (0.5 M solution in THF, 1 equiv). The suspension was stirred for about 30 minutes and warmed to ambient temperature. To a separate flask was charged Compound (P-b) (1.0 equiv) and Pd(PPh3)4 (231 mg, 4.4 mol%) in dioxane (about 20 vols). The mixture was degassed and transferred to the flask containing the organozinc intermediate. The mixture was sealed and heated to about 115 °C to about 125 °C for about 15 hours then cooled to ambient temperatureThe reaction mixture was concentrated in vacuo at ambient temperature and triturated with MTBE (about 10 mL) to afford Compound (C). 1H NMR (400 MHz, DMSO-d6): δ 8.77 (s, 1H), 7.51 (t, 1H, J= 8.0 Hz), 7.18 (d, 1H, J= 4.0 Hz), 6.53 (d, 1H, J= 8.0 Hz), 6.17 (s, 1H), 5.53 (sep, 1H, 7= 8.0 Hz), 1.42 (d, 6H, 7= 8.0 Hz). 13C NMR (100 MHz, DMSO-d6): 159.59, 151.18, 146.25, 142.97, 138.41, 111.90, 108.88, 48.12, 23.55.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, for the metallation, in lieu of n-BuLi, other organolithium reagents (such as t-BuLi, MeLi, and s-BuLi) or Grignard reagents (such as iPrMgCl and PhMgCl) may be used. In lieu of 1 equivalent of ZnCl2, 0.5 equivalent of ZnCl2 or ZnCl2 with LiCl, ZnBr2, or Znl2 can be used. Alternative solvents to THF can include 2-MeTHF, MTBE, or Et20, and this reaction may take place at temperatures that range from about -78 °C to about -40 °C.
Additionally, during the coupling reaction, alternative catalysts may be other Pd (II) complexes or Pd(0) complexes with trialkyl or triarylphosphine ligands, such as Pd(PPh3)4.
Various solvents, such as NMP, THF, butyronitrile, and toluene, may be employed. The reaction may take place at temperatures that range from about 80 °C to about 140 °C.
Example 5: Alternative Synthesis for Compound (D) 
Compound (E) Compound (Q) Compound (D)
Carboalkoxylation to form Compound (Q)
CO (1 atm)

Compound (E)
Compound (Q)
To a reaction flask was added 1-butanol (7 volumes). Compound (E) (1 equiv) was added followed by K2C03 (1.5 equiv) and Pd(dppf)Cl2 (0.02 equiv) and the reaction was placed under a CO atmostphere. The reaction mixture was heated at about 90 °C until reaction completion. The reaction contents were cooled to ambient temperature, the reaction mixture was filtered through a pad of Celite to remove solids, and then rinsed forward with EtOAc. The mother liquor was washed with water and brine, and dried over Na2S04, filtered, and concentrated to afford Compound (Q). Purification by flash chromatography afforded Compound (Q): 1H MR (400 MHz, CDC13) δ 7.77 (d, J = 6.7 Hz, 1H), 7.39 (s, 1H), 7.08 (d, J= 10.8 Hz, 1H), 6.74 (s, 1H), 4.31 (t, J= 6.6 Hz, 2H), 2.20 (s, 3H), 1.87 (m, 1H), 1.73 (tt, J= 6.7, 6.6 Hz, 3H), 1.43 (tq, J= 7.3, 7.4 Hz), 0.94 (t, J= 7.4 Hz, 3H), 0.88 (m, 2H), 0.79 (m, 2H); Exact mass for Ci8H22N202F [M+H], 317.2. Found [M+H], 317.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative catalysts may be used. Non-limiting examples include other Pd (II) complexes or Pd(0) complexes with trialkyl or triarylphosphine ligands, such as
PdCl2(dppf) or Pd(OAc)2 with PPh3, xantphos, tBu3P-HBF4, dppe, dppb, dpcb, tBu-dppf, and (Ad)2P(nBu). Alternative bases can be used, such as other carbonate bases (such as Cs2C03, and Na2C03), NaOAc, KOAc, or organic bases such as TMEDA, Et3N, and iPr2NEt. Various solvents may be employed, such as 1-butanol with other co-solvents (e.g. DMF). The reaction may take place at temperatures that range from about 70 °C to about 115 °C and at CO pressures of about 5 to about 50 psig.
Hydrolysis of Compound (Q) to Compound (D)

Compound (Q) Compound (D)
To a reaction flask was added Compound (Q) (1.0 equiv) and MeOH (7 volumes). A 25% NaOH solution (5 equiv) was then added dropwise. Consumption of Compound (D) was observed after about 1.5 hours at which point the pH of the solution was carefully adjusted to about 1 by the addition of 6 N HC1. Methanol was removed under vacuum to afford a solid which was isolated by filtration. The crude product was first triturated in THF and then filtered. This solid was then triturated in CH2Cl2/MeOH (9: 1) and filtered. Concentration of the mother liquor afforded Compound (D). 1H MR (400 MHz, CD3OD) δ 8.87 (s, 1H), 7.94 (d, J = 6.6 Hz, 1H), 7.43 (s, 1H), 7.31 (d, J= 1 1.5 Hz, lH), 2.21 (s, 3H), 1.96 (m, 1H), 1.04 (m, 2H), 0.81 (m, 2H); LRMS: Calculated mass for C14H14N2O2F [M+H], 261.1. Found [M+H], 261.
Alternative reagents and reaction conditions to those disclosed above may also be employed.
For example, an alternative hydroxide base, including but not limited to KOH, LiOH, and CsOH, may be used in lieu of NaOH. Various solvents may be employed, such as THF, EtOH, and 2-propanol. The reaction may take place at temperatures that range from about 0 °C to about 50 °C.
Example 6: Alternative Synthesis of Compound (A)

Com ound C

(A)
Compound (E) (1 equiv.), Compound (C) (1 equiv.), DMF (about 16 vols), Et3N (1.5 equiv.), Pd(OAc)2 (0.02 equiv.), and Ad2P(«-Bu) (0.04 equiv.) were combined and the contents were purged with N2 followed by CO and then pressurized with CO (20 psi). The reaction mixture was heated to about 95 °C to about 105 °C. After about 24 hours, the reaction was allowed to cool to about 20 °C to about 30 °C to afford Compound (A).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative catalysts may be used. Non-limiting examples include other Pd (II) complexes or Pd(0) complexes with trialkyl or triarylphosphine ligands, such as
PdCl2(PPh3)2, PdCl2(A-Phos)2 or Pd(OAc)2 with PPh3. Alternative bases can be used, including but not limited to other organic bases (such as iPr2NEt and TMEDA) and inorganic bases (such as NaOAc, KOAc, Na2C03, and Cs2C03). Various solvents, NMP, dioxane, and toluene, may be employed. The reaction may take place at temperatures that range from about 90 °C to about 120 °C and at CO pressures of about 20 psig to about 60 psig.
Example 7: Alternative Synthesis of Compound (A)

Compound (A)
Compound (D) (1.0 equiv), Compound (C) (1.05 equiv), 4-(dimethylamino)pyridine (1.0 equiv), ethyl acetate (about 4 V) and diisopropylethylamine (1.2 equiv) were combined and the resulting slurry was charged T3P® as a 50 wt% solution in ethyl acetate (2.0 equiv) over about 3 min at about 20 °C. During the addition, a small exotherm was observed. The mixture was stirred at about 20 °C for about 24 h. After reaction completion, 0.5 M aqueous hydrochloric acid (about 5 vols was added, and the mixture was stirred for about 15 min. Stirring was then stopped, and the phases were allowed to separate. Then, the aqueous phase was reintroduced to the reactor. The pH of the aqueous solution was then adjusted to about 7 with a 5 wt% solution of aqueous sodium hydroxide (about 12 vols). The resulting slurry was stirred for about 12 h at about 20 °C and then filtered, and the reactor was rinsed forward with water (about 3 vols). The filter cake was washed with isopropanol (2 vols), and the resulting solids were dried under vacuum at about 45 °C to provide Compound (A).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in lieu of T3P®, other coupling reagents may be used, including but not limited to Ι, Γ-carbonyldiimidazole, isobutyl chloroformate, pivoyl chloride, EDC-HCl/HOBt, thionyl chloride, and 4-(4,6-dimethoxy-l,3,5-triazin-2-yl)-4-methylmorpholinium chloride. Alternative bases may be used, including but not limited organic amines (such as trialkyl amine bases (for example, triethylamine), N-methyl morpholine, and the like) and carbonates (such as lithium carbonates, sodium carbonates, cesium carbonates, and the like). Various solvents, such as DCM, THF, DMF, ethyl acetate, MTBE, toluene, MP, DMAc, acetonitrile, dichloroethane,
2-MeTHF, and cyclopentyl methyl ether, may be employed. The reaction may take place at temperatures that range from about -10 °C to about 60 °C or from about 0 °C to about 30 °C.
Example 8: Alternative Synthesis of Compound (C)

Compound (8-b)
The mixture of Compound (8-a) and Compound (8-b) is dissolved in about 10 volumes of process water. The solution is heated to about 80 °C, and the solution is allowed to age for about 6 hours. Upon reaction completion, the solution is cooled to about 60 °C. The reaction mixture is seeded with 0.001 equiv of Compound (C), which was obtained by suitable means, and cooled to about 0 °C. Compound (C) is filtered from the cold aqueous solution to yield the product.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, instead of the mixture of Compuond (8-a) and (8-b), the reaction may be carried out with Compound (8-a) or Compound (8-b). Additionally, other organic acids may be used, including but not limited to acetic acid and trifluoroacetic acid. Various solvents, such as toluene, dimethylacetamide, MP, and 2-MeTHF, may be employed. The reaction may take place at temperatures that range from about 80 °C to about 110 °C or about 100 °C.
rnative Synthesis of Compound (C)

Compound (9-c)
Compound (C) may be synthesized as described in U.S. Patent No. 8,742, 126, which is hereby incorporated by reference in its entirety. Additionally, when starting with Compound (9-a), it was found that Compound (C) may be formed through two additional intermediates, Compound (9-b) and Compound (9-c). LRMS for Compound (9-b): Calculated mass, C14H14N2O2F [M+H], 235.1; Found [M+H], 235.9. LRMS for Compound (9-c): Calculated mass, C14H14N2O2F [M+H], 207.1; Found [M+H], 208.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in lieu of acetic acid, other organic acids may be used, including but not limited to trifluoroacetic acid. Various solvents, such as toluene, dimethylacetamide, NMP, 2-MeTHF, acetic acid, and water, may be employed. The reaction may take place at
temperatures that range from about 80 °C to about 110 °C or about 100 °C.
Example 10: Alternative Synthesis of Compou

Compound (10-a) Compound (C)
Compound (10-a) (1 equiv), toluene (about 20 vols), N-isopropylformamide (3.00 equiv), isopropylamine (3.00 equiv) and trifluoroacetic acid (2.50 equiv) were sequentially
combined. The vial was sealed and heated to about 100 °C. After about 22 h, the vial was cooled to room temperature and the contents were analyzed by HPLC. Compound (C) was observed by HPLC.
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, other organic acids may be used, including but not limited to acetic acid. Various solvents, such as dimethylacetamide, MP, and acetic acid, may be employed. The reaction may take place at temperatures that range from about 80 °C to about 110 °C or about 100 °C.
Example 11: Alternative Synthesis of Compound (C)

Compound (10-a) Compound (11 -b) Compound (C)
Compound (10-a) (1.0 equiv), toluene (about 12 volumes), 79 wt% 
dimethylformimidamide (3.0 equiv), isopropylamine (3.0 equiv) and trifluoroacetic acid 2.5 equiv) were combined and heated to about 100 °C. After about 22 h, the reaction mixture was cooled to room temperature. The mixture was seeded with Compound (C), which was obtained by suitable means, and cooled to about 0 °C. After about 30 min, the heterogeneous mixture was filtered and the vial was rinsed forward with toluene (about 25 vols). The solid was collected and dried under vacuum at about 40 °C to provide Compound (C).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, organic acids may be used, including but not limited to acetic acid. Various solvents, such as acetic acid, dimethylacetamide, and NMP, may be employed.
Alternative organic amines may also be added. The reaction may take place at temperatures that range from about 80 °C to about 110 °C or about 90 °C to about 100 °C.
Example 12: Alternative Synthesis of Compound (C)

Compound (10-a) Compound (C)
A suitable reactor fitted with a reflux condenser was charged with acyl hydrazide (1 equiv), toluene (6 volumes), isopropylamine (7.20 equiv) andN.N-dimethylformamide dipropyl acetal (2.70 equiv). To the resulting slurry was charged acetic acid (1.50 equiv) over about 2 min at about 20 °C. During the addition, an exotherm was observed. The mixture was heated to about 95 °C for about 20 h. After reaction completion, the mixture was concentrated under vacuum at about 80 °C. The mixture was diluted with water (10 volumes), and the resulting biphasic solution was concentrated under vacuum at about 80 °C. Water was added (3 volumes), and the solution is heated to about 85 °C. The resulting solution was cooled to about 60 °C and seeded with Compound (C), which was obtained by suitable means. The resulting slurry was aged for about 30 min and then cooled to about 20 °C over about 1 h and aged for about 15 h. The resulting slurry was cooled to about 5 °C and aged for about 3 h. The cold slurry is filtered and the reactor is rinsed forward with cold water (15 mL). The resulting solids were dried under vacuum at about 40 °C to give Compound (C).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative formamide reagents may be used, such as dimethyl formamide diethyl acetal, dimethyl formamide diisopropyl acetal, dimethyl formamide disec-butyl acetal, dimethyl formamide diisobutyl acetal, and the like. Other organic acids may be used, including but not limited to trifluoroacetic acid, chloroacetic acid, and methanesulfonic acid. Various solvents, such as acetic acid, dimethylacetamide, 2-MeTHF, NMP, isobutyl acetate, isobu
Phase 2 Data for Selonsertib in Nonalcoholic Steatohepatitis (NASH) Presented at The Liver Meeting® 2016
— Results Demonstrate Improvement in Fibrosis Stage among NASH Patients with Moderate to Severe Fibrosis —
BOSTON–(BUSINESS WIRE)–Nov. 14, 2016– Gilead Sciences (Nasdaq:GILD) today announced detailed results from an open-label Phase 2 trial evaluating the investigational apoptosis signal-regulating kinase 1 (ASK1) inhibitor selonsertib (formerly GS-4997) alone or in combination with the monoclonal antibody simtuzumab (SIM) in patients with nonalcoholic steatohepatitis (NASH) and moderate to severe liver fibrosis (fibrosis stages F2 or F3). The data demonstrate regression in fibrosis that was, in parallel, associated with reductions in other measures of liver injury in patients treated with selonsertib for 24 weeks. These data were presented in a late-breaking abstract session at The Liver Meeting® 2016 in Boston (#LB-3).
Patients receiving selonsertib demonstrated improvements in several measures of liver disease severity, including fibrosis stage, progression to cirrhosis, liver stiffness (measured by magnetic resonance elastography, MRE) and liver fat content (measured by magnetic resonance imaging (MRI)-proton density fat fraction, PDFF). Data for these efficacy endpoints are summarized in the table below. As no differences were observed between combination and monotherapy, results are presented for selonsertib (18 mg and 6 mg) with/without SIM and for SIM alone. Additionally, patients with fibrosis improvement demonstrated reductions in hepatic collagen content, liver biochemistry (e.g., serum ALT) and the apoptosis marker, cytokeratin-18, supporting the biological activity of selonsertib.
| Endpoint (Week 24) |
|
|
Selonsertib
18 mg ± SIM
|
|
|
Selonsertib
6 mg ± SIM
|
|
|
SIM |
| Fibrosis Improvement ≥1 Stage from Baseline* |
|
|
43% (n=13/30) |
|
|
30% (n=8/27) |
|
|
20% (n=2/10) |
| Progression to Cirrhosis |
|
|
3% (n=1/30) |
|
|
7% (n=2/27) |
|
|
20% (n=2/10) |
| ≥15% Reduction in Liver Stiffness by MRE |
|
|
20% (n=5/25) |
|
|
32% (n=7/22) |
|
|
0% (n=0/7) |
| ≥30% Reduction in Liver Fat by MRI-PDFF |
|
|
26% (n=8/31) |
|
|
13% (n=3/24) |
|
|
10% (n=1/10) |
|
|
*Fibrosis staged according to the NASH Clinical Research Network (CRN) classification by a central pathologist blinded to treatment group.
|
|
Selonsertib demonstrated no dose-related increases in treatment-emergent adverse events or serious adverse events. Headache, nausea and sinusitis were the most common adverse events in patients receiving selonsertib.
“Currently, no approved treatments exists for NASH, and patients with advanced fibrosis would potentially benefit from new options to halt and/or reverse the progression of their disease,” said Rohit Loomba, MD, MHSc, lead study author and Director, NAFLD Research Center, Director of Hepatology, Professor of Medicine, Vice Chief, Division of Gastroenterology, University of California San Diego School of Medicine. “After only 24 weeks of therapy, selonsertib exhibited promising anti-fibrotic activity in this study, which was the first known multi-center NASH clinical trial to use centrally-assessed MRE, MRI-PDFF, in addition to liver biopsy as endpoints. Based on these data, selonsertib represents an important investigational drug candidate for further clinical trials in patients with NASH and significant fibrosis.”
Other Gilead NASH data being presented at The Liver Meeting include results from Phase 1 studies evaluating the investigational selective, non-steroidal Farnesoid X receptor (FXR) agonist GS-9674. Data from a Phase 1 study demonstrated the biological activity and safety profile of GS-9674 in healthy volunteers and support the evaluation of this compound in patients with NASH and cholestatic liver disorders (#1077 and #1140). Phase 2 studies with GS-9674 are ongoing in patients with NASH, primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC).
Additionally, preclinical data for the combination of selonsertib and GS-9674 in a rodent model of advanced fibrosis suggested that the combination of selonsertib and GS-9674 resulted in greater anti-fibrotic activity than either agent alone (#1588). These preclinical data support clinical evaluation of combination approaches with selonsertib and GS-9674 in patients with NASH and advanced fibrosis.
Selonsertib, GS-9674 and simtuzumab have not been determined to be safe or efficacious.
About Selonsertib and the Study
Selonsertib is an investigational small molecule inhibitor of ASK1, a protein that promotes inflammation, apoptosis (cell death) and fibrosis in settings of oxidative stress. Oxidative stress can be increased in many pathological conditions including liver diseases such as NASH.
This Phase 2, randomized, open-label trial evaluated the safety, tolerability and efficacy of selonsertib alone or in combination with SIM in 72 patients with NASH and fibrosis stages F2 (n=25) or F3 (n=47). Eligible patients were randomized (2:2:1:1:1) to receive selonsertib 6 mg (n=20), selonsertib 18 mg (n=22), selonsertib 6 mg plus SIM 125 mg (n=10), selonsertib 18 mg plus SIM 125 mg (n=10) or SIM 125 mg alone (n=10) for 24 weeks. Selonsertib was administered orally once daily and SIM was administered via weekly subcutaneous injection.
About Gilead’s Clinical Programs in NASH
Gilead is advancing a pipeline of novel investigational therapies for the treatment of NASH with advanced fibrosis. Gilead is currently planning or conducting Phase 2 and Phase 3 clinical trials evaluating single-agent and combination therapy approaches against multiple core pathways associated with NASH – metabolic dysfunction, inflammation and fibrosis. Compounds in development include the ASK1 inhibitor, selonsertib; the FXR agonist, GS-9674; and an inhibitor of acetyl-coA carboxylase (ACC), GS-0976, currently being evaluated in a Phase 2 study in patients with NASH.
About Gilead Sciences
Gilead Sciences is a biopharmaceutical company that discovers, develops and commercializes innovative therapeutics in areas of unmet medical need. The company’s mission is to advance the care of patients suffering from life-threatening diseases. Gilead has operations in more than 30 countries worldwide, with headquarters in Foster City, California.
/////////Selonsertib, GS-4997, PHASE 3, GILEAD, GS-4997, GS-4977
CC1=C(C=C(C(=C1)F)C(=O)NC2=CC=CC(=N2)C3=NN=CN3C(C)C)N4C=C(N=C4)C5CC5

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....





















































 [75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.

 [85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.



























 approved by the FDA")