
Home » Posts tagged 'GENERIC DRUG' (Page 10)
Tag Archives: GENERIC DRUG
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Type 2 diabetic patients treated with DPP-4, Linagliptin experience reductions in blood glucose levels
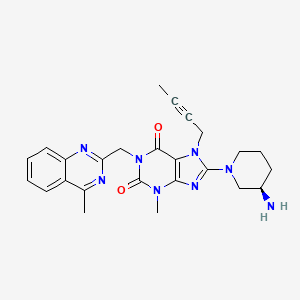
linagliptin
C25H28N8O2
CAS : 668270-12-0
Molecular Weight: 472.54
Purity: > 98%
(R)-8-(3-aminopiperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-1H-purine-2,6(3H,7H)-dione
8-(3R)-3-aminopiperidinyl)-7-butyn-2-yl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione
Solubility: Up to 25 mM in DMSO
Synonyms: BI-1356, BI1356, Linagliptin, Tradjenta, Trajenta
BI-1356 (Linagliptin) is a highly potent and selective dipeptidyl peptidase 4 (DPP-4) inhibitor (IC50 = 1 nM) for treatment of type II diabetes. [1] BI-1356 can increase incretin levels (GLP-1 and GIP), which increases insulin secretion and inhibits glucagon release, decreases gastric emptying, and decreases blood glucose levels. BI-1356 shows 10,000-fold more selectivity for DPP-4 against other protease/peptidases, including DPP-8, DPP-9, trypsin, plasmin, and thrombin, It is a DPP-4 inhibitor developed by Boehringer Ingelheim for the treatment of type II diabetes.
Linagliptin is a highly potent, selective DPP-4 inhibitor with IC50 of 1 nM.
“This study provides much-needed data on glucose-lowering treatment of elderly people with Type 2 Diabetes, inadequately controlled with common anti-hyperglycaemic agents”

Data published in The Lancet showed that elderly people with Type 2 Diabetes (T2D) treated for 24 weeks with the dipeptidyl peptidase-4 (DPP-4) inhibitor linagliptin, marketed by Boehringer Ingelheim and Eli Lilly and Company, experienced significant reductions in blood glucose levels (HbA1c) compared with those receiving placebo. In addition, the overall safety and tolerability profile of linagliptin was similar to placebo, with no significant difference in hypoglycaemia
INTRODUCTION
Linagliptin (BI-1356, trade names Tradjenta and Trajenta) is a DPP-4 inhibitor developed by Boehringer Ingelheim for treatment of type II diabetes.
Linagliptin (once-daily) was approved by the US FDA on 2 May 2011 for treatment of type II diabetes.[1] It is being marketed by Boehringer Ingelheim and Lilly.
-
Linagliptin, namely 8-(3R)-3-aminopiperidinyl)-7-butyn-2-yl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione, of formula (A), is a long acting inhibitor of dipeptidylpeptidase-IV (DPP-IV) activity, at present under development for the treatment of type II diabetes mellitus.

-
The synthesis of Linagliptin is reported in US 7,407,955 , according to the scheme below, where 8-bromo xanthine of formula (B) is condensed with 3-(R)-Boc-aminopiperidine of formula (C) to obtain a compound of formula (D), which is converted to Linagliptin (A) by deprotection of the amine function

-
Optically active 3-aminopiperidine protected as the tert-butylcarbamate (Boc), compound (C), although commercially available, is very expensive and difficult to prepare; moreover in this process impurities are very difficult to remove, particularly on an industrial scale, in particular because of the Boc protective group. For this reason,US 2009/0192314 discloses a novel process for the preparation of Linagliptin (A) which makes use of a 3-(R)-aminopiperidine protected as a phthalimide of formula (E).

-
Accordingly, a compound of formula (E) can be prepared starting from 3-aminopyridine by hydrogenation, reaction with phthalic anhydride, resolution through diastereoisomeric salts using expensive D-tartaric acid, and then cleavage of the tartrate salt.
-
This intermediate is, however, still expensive and its use in the substitution reaction of the bromine derivative of formula (B) is still poorly efficient, as it takes place under drastic reaction conditions.
-
As it can be noted, these processes make use of drastic reaction conditions, or expensive, difficult to prepare starting materials, thus negatively affecting costs. There is therefore the need for an alternative synthetic route to provide Linagliptin or a salt thereof with high enantiomeric and chemical purity, from low cost starting materials.



US ‘955 is schematically represented in scheme

U.S. Patent No. 7,820,815 (“US ‘815) discloses a process for preparation of Linagliptin wherein it is prepared by deprotecting 1 -[(4-methyl-quinazolin-2-yl)methyl]-3- methyl-7-(2-butyn-1 -yl)-8-(3-(R)-phthalimidopiperidin-1 -yl)-xanthine of formula Ilia in presence of ethanolamine. The 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2- butyn-1 -yl)-8-(3-(R)phthalimidopiperidin-1 -yl)-xanthine is prepared by condensing 1 -[(4- l methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromo xanthine of formula III with (R)-3-phthalimidopiperidine of formula I la. The process disclosed in US ‘815 is schematically represented in scheme-ll.

Scherre
PCT Publications WO 2004/018468 and WO 2006/048427 describe synthesis of Linagliptin. Crystalline forms of Linagliptin, Forms A, B, C, D, and E are described in the PCT Publication No. WO 2007/128721. According to WO 2007/128721, Linagliptin prepared according to Publication No.
WO 2004/018468 is present in ambient temperature as a mixture of two enantiotropic polymorphs. The temperature at which the two polymorphs transform into one another is 25±15° C. The pure high temperature form (polymorph A), can be obtained by heating the mixture to temperatures>40° C. The low temperature form (polymorph B) is obtained by cooling to temperatures<10° C.”.
According to WO 2007/128721, the transition point between forms A and B is at room temperature, such that they exist as a polymorphic mixture. In addition, WO 2007/128721 teaches that form D “is obtained if polymorph C is heated to a temperature of 30-100° C. or dried at this temperature”. Since the procedure to obtain form C according to this application includes drying at 70° C., the dried form C is expected to be obtained in admixture with form D.
WO 2007/128721 teaches that Form E is obtained only at high temperatures (after melting of form D at 150±3° C.), and therefore is not relevant industrially.
PATENT







Example 1: Preparation of a compound of formula (II) with X=OEt
-
The bromoxanthine of formula (B) prepared according to US 7,407, 955 (28.2 g, NMR title 90%, 56.0 mmols) and L-(+)-tartrate salt of (R)-ethylnipecotate (22.4 g, 72.8 mmols) are suspended in 50 mL of 1-methyl-2-pyrrolidone. The suspension is heated at 100° under stirring and, maintaining such temperature, diisopropylethylamine (38.3 ml, 224 mmols) is slowly dropwise added. The suspension is moderately refluxed for 2 hours. The mixture is cooled to 30°C and 400 mL of are dropwise added under vigorous stirring. The suspension is stirred for 30 minutes, then filtered off and the solid is washed with 100 mL of water. 27 g of solid product are obtained after drying with a 90% yield.
-
1H-NMR (300 MHz, CDCl3), δ 8.02 (d, 1H), 7.87 (d, 1H), 7.76 (t, 1H), 7.51 (t, 1H), 5.55 (s, 2H), 4.90 (s, 2H), 4.25 – 4.10 (m, 2H), 3.82 (dd, 1H), 3.65 – 3.51 (m, 4H), 3.33 (dd, 1H), 3.15 (m, 1H), 2.88 – 2.72 (m, 4H), 2.08 (m, 1H), 1.92 – 1.73 (m, 6H), 1.27 (t, 3H).
Example 2: Preparation of a compound of formula (II) with X=OH
-
The compound of formula (II) having X = OEt, prepared according to Example 1 (27 g, 51 mmols), is suspended in 270 mL of MeOH and 4.1 g of NaOH scales and 13.7 mL of water are added under stirring. The reaction mixture is maintained under stirring for 2 hours at reflux temperature and then cooled to 40°C and diluted with 400 ml of water.
-
[0080]The mixture is then acidified by adding 6.6 mL of acetic acid and the solid is filtered off and washed with water and dried under vacuum at 50°C, obtaining 21 g of product, with a yield of 82%.
-
1H-NMR (300 MHz, DMSO-d6), δ 8.11 (d, 1H), 7.85 (t, 1H), 7.80 (d, 1H), 7.62 (t, 1H), 5.30 (s, 2H), 4.87 (s, 2H), 3.79 (dd, 1H), 3.57 (m, 1H), 3.38 (s, 3H), 3.33 (dd, 1H), 3.10 (m, 1H), 2.85 (s, 3H), 2.62 (m, 1H), 1.95 (m, 1H), 1.78 – 1.60 (m, 6H).
Example 3: Preparation of a compound of formula (IV) with R = OCH(CH3)2
-
The compound of formula (II) with X=OH prepared according to Example 2 (0.5 g; 1 mmol), 5 ml of isopropanol and trietylamine (0.17 ml, 1.2 mmols) are mixed under stirring. 0.3 g of diphenylphosphorylazide (DPPA) are added in a sole portion. The mixture is heated at reflux temperature for 2 hours under stirring. The mixture is then cooled to room temperature and the solid is filtered off and washed with 2 ml of isopropyl alcohol. The solid is dried under vacuum at 50°C obtaining 0.4 g of product with a yield of 72%.
-
1H-NMR (300 MHz, DMSO-d6), δ 8.12 (d, 1H), 7.85 (t, 1H), 7.80 (d, 1H), 7.63 (t, 1H), 5.28 (s, 2H), 4.85 (s, 2H), 4.75 (ep, 1H), 4.27 (d, 1H), 3.78-3.55 (m, 4H), 3.35 (s, 3H), 2.85 (s, 3H), 1.85 – 1.60 (m, 6H). 1.42 (m, 1H), 1.02 (d, 6H).
Example 4: Preparation of Linagliptin
-
The carbamate of formula (IV), prepared according to Example 3 (400 mg, 0.72 mmols), is dissolved in 5 ml of 32% HCl in water. The reaction mixture is maintained under stirring at 65-70°C for 7 hours and then cooled to room temperature. The pH of the solution is brought to about 8-9 by treatment with 30% NaOH in water and the obtained suspension is stirred for 10 minutes and then filtered off. The solid is dissolved in 10 ml of AcOEt, the solution is filtered and the filtrate is evaporated under reduced pressure. 250 mg of Linagliptin are obtained with a yield of 73%.
Example 5: Preparation of a compound of formula (IV) with R = S(CH2)11CH3
-
The compound of formula (II) with X =OH, prepared according to Example 2 (3.0 g, 6 mmols), 30 ml of acetonitrile and triethylamine (1.09 ml, 7.8 mmols) are mixed together. Subsequently, 1.55 ml (7.2 mmols) of diphenylphosphorylazide (DPPA) are added. The reaction mixture is heated at reflux temperature for 1 hour under stirring and then cooled to 60°C and treated with dodecanethiol (1.87 ml, 7.8 mmols). The mixture is maintained under stirring at the same temperature for 30 minutes and then cooled to 25°C. The formed solid is filtered off and washed with 10 ml of acetonitrile. The solid is dried under vacuum at 60°C obtaining 3.5 g of product with a yield of 85%.
-
1H-NMR (300 MHz, DMSO-d6), δ 8.21 (d, 1H), 7.88 (t, 1H), 7.83 (d, 1H), 7.64 (t, 1H), 5.30 (s, 2H), 4.86 (s, 2H), 3.85 (m, 1H), 3.70 (d, 1H), 3.56 (d, 1H), 3.38 (s, 3H), 3.10-2.87 (m, 3H), 2.85 (s, 3H), 2.74 (t, 2H), 1.90-1.60 (m, 3H), 1.74 (s, 3H), 1.60-1.40 (m, 2H), 1.38-1.10 (m, 18H), 0.82 (t, 3H).
Example 6: Preparation of Linagliptin
-
The thiocarbamate of formula (IV) (10 g, 14,3 mmols), prepared according to Example 5, is dissolved in 100 mL of N-methylpyrrolidone (NMP) and treated with a 30% NaOH solution (7.6 g, 57.0 mmols). The reaction mixture is stirred for 3 hours and then diluted with water and acidified by adding concentrated H2SO4. The mixture is extracted with hexane and brought to pH 9.5 by adding 30% NaOH and repeatedly extracted with dichloromethane. The dichloromethane phases are collected and washed with water and then dried over Na2SO4, filtered and concentrated under reduced pressure. The so obtained oily residue is then dissolved in methyl tert-butyl ether (MTBE) and the mixture is maintained under stirring for 2 hours, then cooled to 0-5°C and the so obtained solid is filtered off, washed with MTBE and dried under vacuum at 50°C till constant weight. 4.2 g of Linagliptin with a yield of 63% are obtained.
Example 7: Preparation of a compound of formula (IV) with R=C7H5N2S (2-mercaptobenzoimidazole)
-
The compound of formula (II) with X =OH, prepared according to Example 2 (2.0 g, 4 mmols), 20 ml of acetonitrile and triethylamine (0.8 ml, 5.6 mmols) are mixed together. Subsequently, 1.43 g (5.2 mmols) of diphenylphosphorylazide (DPPA) are added. The reaction mixture is heated at reflux temperature for 1 hour under stirring and then cooled to 60°C and treated with 2-marcaptobenzimidazole (0.8 g, 5.2 mmols). The mixture is maintained under stirring at the same temperature for 30 minutes, then cooled to 25°C and evaporated under reduced pressure with Rotavapor®. The residue is treated with 50 ml of dichloromethane (CH2Cl2) and washed with 2X20 ml of 5% NaOH. The organic phase is dried over Na2SO4, filtered and concentrated under reduced pressure and the residue is triturated with 30 ml of MTBE. The so obtained solid is filtered off, dried under vacuum at 60°C till constant weight obtaining 2.5 g of light brown powder.
Example 8: Preparation of Linagliptin
-
Starting from the compound of formula (IV) as obtained in example 7 and following the procedure of example 6, product Linagliptin is obtained.
PAPER
DOI: 10.1039/C5OB01111F
http://pubs.rsc.org/en/content/articlelanding/2015/ob/c5ob01111f#!divAbstract
By employing a rhodium–Duanphos complex as the catalyst, β-alkyl (Z)-N-acetyldehydroamino esters were smoothly hydrogenated in a highly efficient and enantioselective way. Excellent enantioselectivities together with excellent yields were achieved for a series of substrates. An efficient approach for the synthesis of the intermediate of the orally administered anti-diabetic drugs Alogliptin and Linagliptin in the DPP-4 inhibitor class was also developed.


Mechanism of action
Linagliptin is an inhibitor of DPP-4, an enzyme that degrades the incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). Both GLP-1 and GIP increase insulin biosynthesis and secretion from pancreatic beta cells in the presence of normal and elevated blood glucose levels. GLP-1 also reduces glucagon secretion from pancreatic alpha cells, resulting in a reduction in hepatic glucose output. Thus, linagliptin stimulates the release of insulin in a glucose-dependent manner and decreases the levels of glucagon in the circulation.
PAPER

PATENT
http://www.google.com/patents/WO2013098775A1?cl=en
In one aspect, the application provides a process for preparation of Linagliptin comprising reacting (R)-piperidine-3-amine of formula II or an acid addition salt thereof with 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine of formula III in the presence of a suitable base in an inert organic solvent.

In another aspect, the application provides Linagliptin or a pharmaceutically acceptable salt thereof, having less than about 0.15 area % of potential process related impurities viz., regio-impurity of the formula la, bromo-impurity of the formula lb and S- isomer as measured by HPLC.

L nag pt n S- somer
Example 1 : Preparation of Linagliptin
a) Preparation of 3-methyl-7-(2-butyn-l-yl)-8-bromo-xanthine (compound of formula IV)
3-Methyl-8-bromo-xanthine (30 gm) and N,N-dimethylformamide (170 ml_) were charged into a 1000 ml_ round bottomed flask equipped with a mechanical stirrer. Diisopropylethylamine (DIPEA, 1 5.9 gm) and 1 -bromo-2-butyne (16.2 gm) were added at 30°C. The reaction mixture was heated to 85 °C and maintained the temperature for 4 hours. The reaction mixture was cooled to 30°C and pre cooled water (300 ml_) was added. The solid formed was collected by filtration and washed with pre cooled water (150 ml_) and diethyl ether (30 ml_). The solid was dried in oven under vacuum at 50°C to get 30.9 gm of the title compound.
(b) Preparation of 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8- bromoxanthine (compound of formula III) 3-Methyl-7-(2-butyn-l-yl)-8-bromo-xanthine (10 gm) and Ν,Ν-dimethylacetamide (150 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (9.3 gm) and 2-(chloromethyl)-4- methylquinazoline (6.8 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 90 °C and maintained the temperature for 8 hours. The reaction mixture was cooled to 30°C and water (450 mL) was added and the mixture was stirred for 1 hour at 30°C. The solid formed was collected by filtration and washed with water (150 mL). The wet cake was charged into 500 mL round bottomed flask and toluene (220 mL) was added and the mixture was heated to reflux temperature and maintained for 1 hour. The mixture was cooled to 10°C and maintained for 2 hours. The solid was collected by filtration and washed with toluene (50 mL). The solid was dried in oven under vacuum at 80°C to get 10.8 gm of the title compound. Purity by HPLC: 99.59%
(c) Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (5 gm) and Ν,Ν-dimethylformamide (DMF, 50 mL) were charged into a 500 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (4.57 gm) and (R)-piperidine-3-amine dihydrochloride (2.86 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 80 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to room temperature and DMF was evaporated under vacuum, then dichloromethane (DCM, 50 mL) was added, and stirred for 15 minutes. The reaction mixture was filtered to separate out the non- dissolved material and the non-dissolved material was washed with 15 mL of dichloromethane. The dichloromethane was evaporated under vacuum to give 4 gm of crude Linagliptin.
Example 2: One pot process for preparation of Linagliptin
3-Methyl-8-bromo-xanthine (5 gm) and Ν,Ν-dimethylformamide (DMF, 28.5 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Diisopropylethylamine (DIPEA, 2.6 gm) and 1 -bromo-2-butyne (2.7 gm) were added at 30 °C. The reaction mixture was heated to 85 °C and maintained at this temperature for 4 hours. The reaction mixture is cooled to 30°C and Ν,Ν-dimethylformamide (DMF, 100 ml_) was added. Potassium carbonate (4.4 gm) and 2-(chloromethyl)-4- methylquinazoline (4.2 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 85 °C and maintained at this temperature for 4 hours. The reaction mixture was cooled to 30°C and Ν,Ν-dimethylformamide (DMF, 90 ml_) was added. Potassium carbonate (8.3 gm) and (R)-piperidine-3-amine dihydrochloride (5.2 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 80 °C and maintained at this temperature for 8 hours. The reaction mixture was cooled to 30 °C and DMF was evaporated under vacuum. Dichloromethane (DCM, 30 ml_) was added and stirred for 15 minutes. The reaction mixture was filtered to separate out the undissolved material and the undissolved material was washed with dichloromethane (30 ml_). The dichloromethane was evaporated under vacuum and 10% acetic acid (100 ml_) was added. The resulted solution was stirred for 30 minutes and washed with dichloromethane (25 ml_x3). The pH of the aqueous layer was adjusted to 8.5 with 10% aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (25 ml_x2) and the dichloromethane was evaporated under vacuum to get 1 .2 gm of Linagliptin.
Example 3: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 ml_) were charged into a 1000 ml_ round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine dihydrochloride (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95°C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 ml_). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at room temperature. The aqueous layer was separated and washed with 60 ml_ of dichloromethane. The aqueous layer was charged into another flask and 200 ml_ of dichloromethane and 100 ml_ of aqueous sodium hydroxide solution was added drop-wise at 30 °C. The mixture was stirred for one hour at 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45°C. Isopropyl alcohol (100 mL) was added to the residue and stirred for 3 hours at room temperature. Filtered the compound and washed with isopropyl alcohol (20 mL) and dried the compound at below 60 °C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 99.0%
Example 4: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 95 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to room temperature and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at room temperature. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at room temperature. The mixture was stirred for one hour at room temperature and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Hexane (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with Hexane (40 mL) and dried the compound at below 60°C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 98.92%
Example 5: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95°C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at 30 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at 30°C. The mixture was stirred for one hour at 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Toluene (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with Toluene (40 mL) and dried the compound at below 60 °C under vacuum to give 16.8 gm of Linagliptin. Purity: 98.91 %, PXRD pattern: Fig. 2.
Example 6: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at 30 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at room temperature (pH is > 10). The mixture was stirred for one hour 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Ethyl acetate (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with ethyl acetate (40 mL) and dried the compound at below 60 °C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 98.72%
Example 7: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (4 gm) and methyl isobutyl ketone (MIBK 100 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (3.7 gm) and (R)-piperidine-3-amine dibenzoyl-D-tartrate (6.1 gm) were added to the reaction mixture at 26°C. The reaction mixture was heated to 100°C and maintained at that temperature for 6 hours. The reaction mixture was cooled to 30 °C and filtered, and the salt was washed with MIBK (8 mL). The filtrate was charged into another flask and added slowly 10% aqueous acetic acid solution (40 mL) and stirred for one hour at 26°C. The aqueous layer was separated and washed with 12 mL of dichloromethane. The aqueous layer was charged into another flask and 40 mL of dichloromethane and 20 mL of 16 % aqueous sodium hydroxide solution was added drop-wise at 26°C. The mixture was stirred for one hour at 26 °C and the organic layer was separated and the aqueous layer was extracted with 20 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Isopropyl alcohol (8 mL) was added to the residue and evaporated under vacuum at below 45 °C. Isopropyl alcohol (16 mL) was added to the residue and stirred for 2 hours at 2Q°C. Filtered the compound and washed with isopropyl alcohol (4 mL) and dried the compound at 60 °C under vacuum to give 3.2 gm of Linagliptin. PXRD pattern: Fig. 2, Chemical Purity: 98.68%, Chiral Purity: 99.82%, S-isomer content: 0.12%, Regio impurity: 0.57%, Bromo impurity: 0.28%
Example 8: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine dihydrochloride (8.4 gm) were added to the reaction mixture at 26°C. The reaction mixture was heated to ‘\ 00 °C and maintained at that temperature for 4 hours. The reaction mixture was cooled to 30 °C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 200 mL of 10% aqueous acetic acid solution and stirred for 30 minutes at 28 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) were added drop- wise at 28°C (pH is > 10). The mixture was stirred for one hour at 28°C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and divided into 5 equal parts.
Part 1 : The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (16 mL) was added to the residue stirred for 30 minutes at 28 °C and 48 mL of MTBE was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 8 mL of MTBE and dried the compound at 65 °C under vacuum to give 3.0 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.46%, Regio impurity: 0.37%, Bromo impurity: 0.03%
Part 2: The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (24 mL) was added to the residue stirred for 30 minutes at 28 °C and the resulted solution was cooled to 5°C and stirred for 1 hour. Filtered the compound and washed with 5 mL of chilled methanol and dried the compound at 65°C under vacuum to give 3.0 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.41 %, Regio impurity: 0.38%, Bromo impurity: 0.03%
Part 3: The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (20 mL) was added to the residue stirred for 30 minutes at 28 °C and 20 mL of MTBE was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 8 mL of MTBE and dried the compound at 65 °C under vacuum to give 2.8 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.47%, Regio impurity: 0.36%, Bromo impurity: 0.03%.
Part 4: The organic layer was distilled off completely under vacuum at 45 °C. Isopropyl alcohol (8 mL) was added to the residue and distilled off completely under vacuum at 45 °C. Methanol (16 mL) was added to the residue stirred for 30 minutes at 28 °C and 16 mL of isopropyl alcohol was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 4 mL of isopropyl alcohol and dried the compound at 65 °C under vacuum to give 2.9 gm of Linagliptin. PXRD pattern: Fig. 1 .
Chemical Purity: 99.44%, Regio impurity: 0.38%, Bromo impurity: 0.02%.
Part 5: The organic layer was distilled off completely under vacuum at 45 °C. Ethyl acetate (8 mL) was added to the residue and distilled off completely under vacuum at 45 °C. Ethyl acetate (16 mL) was added to the residue stirred for 30 minutes at 28°C and 16 mL of methanol was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 4 mL of ethyl acetate and dried the compound at 65 °C under vacuum to give 0.7 gm of Linagliptin. PXRD pattern: Fig. 2.
Chemical Purity: 99.57%, Regio impurity: 0.29%, Bromo impurity: 0.02%
Example 9: Purification of Linagliptin
Linagliptin (3.5 gm) was dissolved in 10% aqueous acetic acid and stirred for 15 minutes. Dichloromethane (50 mL) was added to the solution and stirred for 30 minutes. The aqueous layer was separated and the pH of this layer was adjusted to 8.5 using 10% aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (50 mLx2). The dichloromethane was evaporated under vacuum to give 3 gm of Linagliptin.
Example 10: Purification of Linagliptin
Linagliptin (31 gm) and methanol (124 mL) were charged into 500 mL round bottomed flask and the solution was heated to 40 °C and stirred for 60 minutes. Charcoal (3 gm) was added to the clear solution and stirred for 30 minutes. The solution was filtered through Hy-flow and the Hy-flow bed was washed with methanol (30 mL). Filtrate was charged into 1000 mL round bottomed flask and methyl tertiary butyl ether was added drop-wise to the solution and stirred for 2 hours at 30 °C. The precipitate so formed was filtered and the wet cake was washed with methyl tertiary butyl ether (30 mL) to get 25.6 gm of pure Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.57%, Chiral purity: 99.73%, Regio impurity: 0.10%, Bromo impurity: 0.1 %
Example 1 1 : Purification of Linagliptin
Linagliptin (4 gm) and methanol (24 mL) were charged into 100 mL round bottomed flask and the solution is heated to 50 °C and stirred for 60 minutes. Methyl tertiary butyl ether (MTBE, 80mL) was charged into 500 mL round bottomed flask and the methanol solution containing linagliptin was added drop-wise at 27 °C and stirred for 2 hours at same temperature. The precipitate formed was filtered and the wet cake was washed with methyl tertiary butyl ether (8 mL) to get 2.6 gm of pure Linagliptin. PXRD pattern: Fig. 2, Bromo impurity content: 0.04%.
Example 12: Purification of Linagliptin
a) Preparation of linagliptin-(D)-tartrate
Linagliptin (10 gm) and methanol (300 mL) were charged into 1000 mL round bottomed flask and (D)-tartaric acid solution (3.3 gm of (D)-tartaric acid in 100 mL of methanol) was added at 26 °C. The solution was heated to 65 °C and stirred for 60 minutes. The solution was cooled to 28 °C and stirred for 2 hours at 27 °C. The precipitate formed was filtered and the wet cake was washed with methanol (20 mL) and the solid was dried under vacuum at 55°C to get 8.3 gm of Linagliptin-(D)-tartrate. PXRD pattern: Fig. 4. Chemical Purity: 99.72%, Chiral purity: 99.89%, Regio impurity: 0.08%, Bromo impurity: 0.05%, S-isomer: 0.1 1%.
b) Isolation of pure Linagliptin
Linagliptin-(D)-tartrate (8 gm) and water (100 mL) were charged into 1000 mL round bottomed flask and stirred for 30 minutes at 26 °C. Dichloromethane (80 mL) was added to the solution and cooled to 5°C. Aqueous sodium hydroxide solution (0.6 gm of NaOH is added to 20 mL of water) was added to the mixture at 5°C and maintained for 1 hour. Layers were separated and aqueous layer was extracted with dichloromethane (20 mL). Combined both organic layers and dried over sodium sulphate and distilled off the organic layer under vacuum at 45 °C. Hexane (20 mL) was added to the crude and stirred for 1 hour at 26°C. The precipitate was filtered and washed with 4 mL of hexane and dried the compound at 60°C under vacuum to give 6 gm of pure Linagliptin. PXRD pattern: Fig. 2, Chemical Purity: 99.67%, Chiral purity: 99.85%, (S)-isomer content: 0.1 5%, Regio impurity: 0.09%, Bromo impurity: 0.07%.
PATENT
http://www.google.com/patents/US20130123282
-
[0181]8-Bromo-3-methylxanthine was reacted with 1-bromo-2-butyne in the presence of base in a mixture of N-methyl pyrrolidone and toluene mixture. The reaction mixture was heated overnight. The reaction completion was determined, and the mixture was then cooled to ambient temperature. A solid precipitate formed on cooling precipitation. The product, 3-Methyl-7-(2-butyne-1-yl)-8-bromoxanthine, having greater than 95% purity was isolated by filtration and washed with toluene.
- Example 34Preparation of (R)-8-(3-amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (Form-XXII): A. 3-Methyl-7-(2-butyne-1-yl)-8-bromoxanthine
Example 35Preparation of 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione
-
[0182]3-Methyl-7-(2-butine-1-yl)-8-bromoxanthine was reacted with 2-(chloromethyl)-4-methylquinazoline in the presence of base under phase transfer catalyst using a N-methyl pyrrolidone/toluene mixture as the reaction solvent. The reaction mixture was heated overnight. When the reaction was complete, the reaction mixture was cooled to ambient temperature. A solid precipitate formed and was separated by filtration and washed with toluene and then with water to provide the product, 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione having more than 97% purity.
Example 36Preparation of (R)-8-(3-Amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (Form-XXII)
-
[0183](R)-3-N-tert-Butoxycarbonylaminopiperidine was reacted with 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione in the presence of base. The reaction mixture was heated overnight. When the reaction was complete, the reaction mixture was cooled to ambient temperature. The cooled reaction mixture was washed several times with water and separated. The resulting 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine organic solution was greater than 95%. Purified by HPLC. An excess of aqueous HCl solution was added to the obtained 1-[(4-methylquinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine organic solution. The resulting mixture was stirred under heating until complete conversion was observed. Aqueous base was added to the reaction. The resulting mixture was stirred and separated. The organic phase was washed with aqueous base and separated. A non-polar or moderately polar solvent was added to the resulting organic phase. The mixture was partially concentrated to achieve precipitation, and the concentrated mixture was cooled and filtered to provide the wet crude product. The crude product was re-crystallized from alcohol, filtered and dried in vacuum oven with heating to afford dry solid Form-XXII of (R)-8-(3-amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione having more than 98% purity.
Clinical trials
Results in 2010 from a Phase III clinical trial of linagliptin showed that the drug can effectively reduce blood sugar.[2]

Scheme:
. J. Med Chem 2009, 52, 6433..
J. Med Chem 2007, 50, 6450…

References
- H. Spreitzer (September 1, 2008). “Neue Wirkstoffe – BI-1356”. Österreichische Apothekerzeitung (in German) (18/2008): 918.
- Wang, Y, Serradell, N, Rosa, E, Castaner, R (2008). “BI-1356”. Drugs of the Future 33 (6): 473–477. doi:10.1358/dof.2008.033.06.1215244.
- ^ “FDA Approves Type 2 Diabetes Drug from Boehringer Ingelheim and Lilly”. 3 May 2011.
- “Four Phase III Trials Confirm Benefits of BI’s Oral, Once-Daily Type 2 Diabetes Therapy”. Genetic Engineering & Biotechnology News. 28 June 2010.
| CN101735218A * | Dec 17, 2009 | Jun 16, 2010 | 廖国超 | Piperidine carbamic acid ester derivative and application thereof |
| US7407955 | Aug 12, 2003 | Aug 5, 2008 | Boehringer Ingelheim Pharma Gmbh & Co., Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US20040097510 * | Aug 12, 2003 | May 20, 2004 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US20090192314 | Mar 30, 2009 | Jul 30, 2009 | Boehringer Ingelheim International Gmbh | Process for the preparation of chiral 8-(3-aminopiperidin-1yl)-xanthines |
| WO2005085246A1 * | Feb 12, 2005 | Sep 15, 2005 | Boehringer Ingelheim Int | 8-[3-amino-piperidin-1-yl]-xanthine, the production thereof and the use in the form of a dpp inhibitor |
| Reference | ||
|---|---|---|
| 1 | CHIRALITY vol. 7, 1995, pages 90 – 95 | |
| 2 | * | JEAN L ET AL: “A convenient route to 1-benzyl 3-aminopyrrolidine and 3-aminopiperidine“, TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 42, no. 33, 13 August 2001 (2001-08-13), pages 5645-5649, XP004295831, ISSN: 0040-4039, DOI: DOI:10.1016/S0040-4039(01)00985-6 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014033746A2 * | Aug 6, 2013 | Mar 6, 2014 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Process for the preparation of dipeptidylpeptidase inhibitors |
| WO2014059938A1 * | Oct 17, 2013 | Apr 24, 2014 | 2Y-Chem, Ltd. | Method for preparing important intermediate of linagliptin |
| WO2014097314A1 * | Dec 16, 2013 | Jun 26, 2014 | Mylan Laboratories Ltd | An improved process for the preparation of linagliptin |
| WO2010072776A1 * | Dec 22, 2009 | Jul 1, 2010 | Boehringer Ingelheim International Gmbh | Salt forms of organic compound |
| CN101784270A * | Aug 15, 2008 | Jul 21, 2010 | 贝林格尔.英格海姆国际有限公司 | Pharmaceutical composition comprising a glucopyranosyl-substituted benzene derivative |
| CN102127080A * | Nov 2, 2005 | Jul 20, 2011 | 贝林格尔.英格海姆国际有限公司 | Method for producing chiral 8-(3-amino-piperidin-1-yl)-xanthines |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015067539A1 * | Oct 31, 2014 | May 14, 2015 | Chemelectiva S.R.L. | Process and intermediates for the preparation of linagliptin |
| WO2015087240A1 | Dec 9, 2014 | Jun 18, 2015 | Ranbaxy Laboratories Limited | Process for the preparation of linagliptin and an intermediate thereof |
| WO2015107533A1 * | Sep 1, 2014 | Jul 23, 2015 | Harman Finochem Limited | A process for preparation of 1h-purine-2,6-dione, 8-[(3r)-3-amino-1-piperidinyl]-7 (2-butyn-1-yl)-3,7-dihydro-3-methyl-1-[(4-methyl-2quinazolinyl) methyl] and its pharmaceutically acceptable salts |
| Eckhardt M, et al. 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J Med Chem. 2007; 50(26):6450-3. Pubmed ID: 18052023 | |
| 2. | Thomas L, et al. (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J Pharmacol Exp Ther. 2008; 325(1):175-82. Pubmed ID: 18223196 |
//////////BI-1356, BI1356, Linagliptin, Tradjenta, Trajenta, DPP-IV, DPP-4 inhibitor
Yusuf Hamied – Cipla
Name: Yusuf Hamied
Title: Chairman and Managing Director, Cipla
http://www.fiercebiotech.com/special-reports/yusuf-hamied-cipla
Yusuf Hamied made his name a decade ago when he faced down Big Pharma on patents for HIV/AIDS drugs and Cipla started selling them at a cost of about $1 a day. His disdain for what he considers Big Pharma’s “obscene prices” born out of monopolies is well documented. Hamied has the industry’s rapt attention again with his new attack on cancer meds and his avowal that Cipla will soon take on biologics.
Read more: Yusuf Hamied – Cipla – FierceBiotech http://www.fiercebiotech.com/special-reports/yusuf-hamied-cipla#ixzz2b9oexHpH
Subscribe at FierceBiotech
Yusuf Khwaja Hamied is a leading Indian scientist and chairman of Cipla, a socially conscious generic pharmaceuticals company founded by his father Khwaja Abdul Hamiedin 1935.[2]
Born in Vilnius, Lithuania, Yusuf Hamied was raised in Bombay (now known as Mumbai). His Indian Muslim father and Russophone Jewish mother met in Berlin, where they both were graduate students. He holds a Ph.D. in chemistry from Christ’s College, Cambridge. He still uses his chemistry notebooks from Cambridge when he develops new syntheses of drugs.[3]
He is an alumnus of the Cathedral and John Connon School in Bombay. Affectionately called Yuku by his close friends, Hamied is fond of Western classical music and has been close friends with the world-famous conductor Zubin Mehta since boyhood.
Hamied is best known outside India for defying large Western pharmaceutical companies in order to provide generic AIDS drugs and treatments for other ailments primarily affecting people in poor countries. He was awarded the Padma Bhushan by the Government of India in 2005.
Hamied has led efforts to eradicate AIDS in the developing world and to give patients life-saving medicines regardless of their ability to pay,[4] and has often been characterized as a modern-day Robin Hood figure[5][6][7][8] as a result.
Former head of Johnson and Johnson Ajit Dangi says plainly “In Africa, Cipla is a temple and Dr. Hamied is God.” [9] To this Hamied has countered “I don’t want to make money off these diseases which cause the whole fabric of society to crumble”. [10]
In September 2011, in a piece about how he was trying to radically lower costs of biotech drugs for cancer, diabetes and othernoncommunicable diseases, The New York Times wrote of Hamied:
Dr. Yusuf K. Hamied, chairman of the Indian drug giant Cipla Ltd., electrified the global health community a decade ago when he said he could produce cocktails of AIDS medicines for $1 per day — a fraction of the price charged by branded pharmaceutical companies. That price has since fallen to 20 cents per day, and more than six million people in the developing world now receive treatment, up from little more than 2,000 in 2001.[11]
Yusuf Hamied has also been enormously influential in pioneering development of multi-drug combination pills (also known as fixed-dose combinations, or FDCs), notably for HIV/AIDS, tuberculosis (TB), asthma and other ailments chiefly affecting developing countries, as well as development of pediatric formulations of drugs, especially those benefiting children in poor settings.[12] These innovations have greatly expanded access to medicine and increased drug safety by ensuring proper dosages are taken. He is also highly regarded for his leading role in expanding the production of bulk drugs and “active pharmaceutical ingredients” (APIs, the active chemical components in medicines) in India.[13]

In 2009 the Yusuf Hamied Centre was opened at Christ’s College, Cambridge.[1][14]
Yusuf Hamied has been the subject of in-depth profiles in The New York Times, Time magazine, The Guardian, Le Monde, The Economist, the Financial Times, The Times (London), Corriere della Sera, Der Spiegel, Wired and numerous other leading publications, as well as on television outlets such as ABC News, the BBC, CNN and CBS’ 60 Minutes.
Yusuf Hamied was awarded the ‘Indian Of The Year’ in the category of business by CNN-IBN in 2012.[15]
- “Christ’s officially opens Yusuf Hamied Centre”. University of Cambridge News. 2009-04-20. Retrieved 2009-04-20.
- Sarah Boseley (2003-02-18). “Yusuf Hamied, generic drugs boss | World news”. London: The Guardian. Retrieved 2010-09-01.
- Selling Cheap ‘Generic’ Drugs, India’s Copycats Irk Industry, By DONALD G. McNEIL Jr, Published: December 01, 2000
- “Interview of the week: Yusuf Hamied. – United Press International | HighBeam Research – FREE trial”. Highbeam.com. 2001-02-22. Retrieved 2010-09-01.
- “[Esplora il significato del termine: Yusuf Hamied, un Robin Hood contro l’ Aids “Così sconfiggerò l’ Aids senza le multinazionali”] Yusuf Hamied, un Robin Hood contro l’ Aids “Così sconfiggerò l’ Aids senza le multinazionali””.
- Bobin, Frédéric (2010-07-06). “India fears generic drugs for poor are endangered by proposed EU trade deal”. The Guardian(London).
- “Dr. Hamied Robin Hood spoof film posters”.
- “Robin Hood and the Multinationals”.
- Hans Lofgren, The Politics of the Pharmaceutical Industry and Access to Medicine, 2012, p. 55
- Hans Lofgren, The Politics of the Pharmaceutical Industry and Access to Medicine, 2012, p. 68
- Harris, Gardiner (2011-09-18). “China and India Making Inroads in Biotech Drugs”. The New York Times.
- Hans Lofgren, The Politics of the Pharmaceutical Industry and Access to Medicine, 2012, p. 58-59
- Hans Lofgren, The Politics of the Pharmaceutical Industry and Access to Medicine, 2012, p. 63
- “The Hindu News Update Service”. Chennai, India: Hindu.com. 2009-04-22. Retrieved 2010-09-01.
- http://ibnlive.in.com/news/dr-yusuf-hamied-message-on-being-cnnibns-indian-of-the-year-2012-in-the-business-category/310908-3.html.

..
Sanofi Pasteur Initiates Phase III Study of Investigational Clostridium difficile Vaccine in the United States

Cdiffense trial to evaluate vaccine against a leading cause of life-threatening, healthcare-associated infections worldwide
SWIFTWATER, Pa., Aug. 5, 2013 /PRNewswire/ — Sanofi Pasteur, the vaccines division of Sanofi (EURONEXT: SAN and NYSE: SNY), announced today the initiation of its Phase III clinical program called Cdiffense to evaluate the safety, immunogenicity and efficacy of an investigational vaccine for the prevention of primary symptomatic Clostridium difficile infection (CDI). Clostridium difficile (C. diff) is a potentially life-threatening, spore-forming bacterium that causes intestinal disease. The risk of C. diff increases with age, antibiotic treatment and time spent in hospitals or nursing homes, where multiple cases can lead to outbreaks. The investigational vaccine is designed to help protect at-risk individuals from C. diff, which is emerging as a leading cause of life-threatening, healthcare-associated infections (HAIs)worldwide, read all at…………….
http://www.pharmalive.com/sanofi-starts-phase-iii-trial-for-clostridium-difficile-vaccine
..
…
New Findings Could Influence Development of Therapies to Treat Dengue Disease

Bristol, UK (Scicasts) – New research into the fight against Dengue, an insect-borne tropical disease that infects up to 390 million people worldwide annually, may influence the development of anti-viral therapies that are effective against all four types of the virus.
The findings, led by researchers at the University of Bristol and published in the Journal of Biological Chemistry August 2 show that there may be significant differences in specific properties of the viral proteins for the four dengue virus types.
read all at scicasts
..
…………
…
….
…
Vivimed Acquires A US FDA Approved Formulation Manufacturing Facility


FDA Grants Priority Review To New Drug Application For MNK-795
FDA Grants Priority Review To New Drug Application For MNK-795 Submitted By Depomed Licensee Mallinckrodt
Controlled Substance Analgesic Combination Product Uses Depomed’s Proprietary Acuform® Technology
NEWARK, Calif., July 29, 2013 /PRNewswire/ — Depomed, Inc. (NASDAQ:DEPO) announced today that the U. S. Food and Drug Administration (FDA) has accepted for filing a New Drug Application (NDA) from Mallinckrodt (NYSE: MNK) for MNK-795. MNK-795 is a controlled-release oral formulation of oxycodone and acetaminophen that has been studied for the management of moderate to severe acute pain where the use of an opioid analgesic is appropriate. MNK-795 is formulated with Depomed’s Acuform® drug delivery technology.
http://www.pharmalive.com/fda-grants-priority-review-to-new-drug-application-for-mnk-795
DONEPEZIL SYNTHESIS


Donepezil, marketed under the trade name Aricept by its developer Eisai and partnerPfizer, is a centrally acting reversible acetylcholinesterase inhibitor. Its main therapeutic use is in the palliative treatment of Alzheimer’s disease.Common side effects include gastrointestinal upset. It has an oral bioavailability of 100% and easily crosses the blood–brain barrier. Because it has a biological half-life of about 70 hours, it can be taken once a day.
Currently, no definitive proof shows the use of donepezil or other similar agents alters the course or progression of Alzheimer’s disease (AD). However, 6 to 12-month controlled studies have shown modest benefits in cognition and/or behavior.Pilot studies have reported donepezil therapy may potentially have effects on markers of disease progression, such as hippocampal volume. Therefore, many neurologists, psychiatrists, and primary-care physicians use donepezil in patients with Alzheimer’s disease. In 2005, the UK National Institute for Clinical Excellence (NICE) withdrew its recommendation for use of the drug for mild-to-moderate AD, on the basis of no significant improvement in functional outcome, quality of life, or behavioral symptoms. However, NICE revised its guidelines to suggest donepezil be used in moderate-stage patients for whom the evidence is strongest.

While the drug is currently indicated for mild to moderate Alzheimer’s, evidence from two clinical trials also indicates it may be effective for moderate to severe disease. An example of this is a Karolinska Institute paper published in The Lancet in early 2006, which states donepezil improves cognitive function even in patients with severe AD symptoms. In Oct. 2006 the U.S. Food and Drug Administration also approved Aricept for treatment of severe dementia.

【通用名】 Donepezil hydrochloride, BNAG, E-2020, Eranz, Memorit, Memac, Aricept
【化学名】 (?-1-Benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylmethyl)piperidine hydrochloride; (?-2-(1-Benzylpiperidin-4-ylmethyl)-5,6-dimethoxyindan-1-one hydrochloride
【CAS登记号】 120011-70-3, 123958-79-2 ([2-14C]-labeled), 142057-77-0 (deleted CAS), 120014-06-4 (free base)
【分子式】 C24-H29-N-O3.Cl-H
【分子量】 415.958
【化学活性】 Alzheimer’s Dementia, Treatment of , Analgesic and Anesthetic Drugs, Antimigraine Drugs, Attention Deficit Hyperactivity Disorder (ADHD), Treatment of, Autism, Treatment of, Cognition Disorders, Treatment of, Immunologic Neuromuscular Disorders, Treatment of, Migraine, Prophylactic Treatment of, Multiple Sclerosis, Agents for, Neurologic Drugs, Psychopharmacologic Drugs, Vascular Dementia, Treatment of, Acetylcholinesterase Inhibitors
【开发阶段】 Launched-1997
【研究机构】 Eisai (Originator), National Institute of Mental Health (Not Determined), Bracco (Licensee), Pfizer (Licensee)

Donepezil inhibiting Torpedo californicaacetylcholinesterase. See Proteopedia1eve.

Research leading to the development of donepezil began in 1983 at Eisai, and the first Phase I clinical trial took place in 1989. In 1996, Eisai received approval from the United States Food and Drug Administration (USFDA) for donepezil under the brand Aricept, which it co-marketed with Pfizer. As of 2011, Aricept was the world’s best-selling Alzheimer’s disease treatment. The first generic donepezil became available in November 2010 with the USFDA approval of a formulation prepared by Ranbaxy Labs. In April 2011 a second generic formulation, from Wockhardt, received tentative USFDA marketing approval
| 标题: | Cyclic amine cpd., its use and pharmaceutical compsns. comprising it |
| 作者: | Sugimoto, H.; Tsuchiya, Y.; Higurashi, K.; Karibe, N.; Iimura, Y.; Sasaki, A.; Yamanashi, Y.; Ogura, H.; Araki, S.; Kosasa, T.; Kusota, A.; Kozasa, M.; Yamatsu, K. (Eisai Co., Ltd.) |
| 来源: | AU 8818216; EP 0296560; EP 0673927; EP 0742207; JP 1989079151; JP 1998067739; US 4895841; US 5100901 |
 |
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 标题: | Synthesis of 1-benzyl-4-[(5,6-dimethoxy[2-14C]-1-indanon)-2-yl]methylpiperidine hydrochloride (E-2020-14C) |
| 作者: | Sugimoto, H.; Mishima, M.; Iimura, Y. |
| 来源: | J Label Compd Radiopharm 1989,27(7),835-9 |
|
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 作者: | Casta馿r, J.; Prous, J. |
| 来源: | Drugs Fut 1991,16(1),16 |
|
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 标题: | Synthesis of 1-benzyl-4-[(5,[C-11]6-dimethoxy-1-oxoindan-2-yl)methyl]piperidine: A promising ligand for visualisation of acetylcholine esterase by PET |
| 作者: | Santens, P.; DeReuck, J.; Dierckx, R.A.; Siegers, G.; Vermeirsch, H.; De Vos, F. |
| 来源: | J Label Compd Radiopharm 2000,43(6),595 |
 |
|
| 合成路线图解说明:11C-Labeled donepezil was prepared by methylation of 2-(1-benzylpiperidin-4-ylmethyl)-6-hydroxy-5-methoxyindan-1-one (I) with 11CH3I by means of tetrabutylammonium hydroxide in DMF. | |
……………………………..
………………..
……………………….
……………………
……………………….
Donepezil hydrochloride is a useful memory enhancer introduced by the Japanese pharmaceutical company Eisai. Its preparation was described in patent no. EP 296560. In this patent Donepezil was produced by reaction of 5,6-dimethoxy-1- indanone with 1 -benzyl-4-formylpiperidine in the presence of a strong base, such as lithium diisopropylamide followed by reduction of the double bond. According to this method, Donepezil was obtained (Scheme 1). Patent application WO 99/36405 describes another process for the synthesis of Donepezil. According to this patent, 2-alkoxycarbonyl-1-indanones are reacted with (4-pyridinyl) methyl halide moiety followed by hydrolysis and decarboxylation to give the 2-(4-pyridinyl)methyl-1-indanone derivative. This is followed by reaction with benzyl halides to obtain the corresponding quaternary ammonium salt, and followed by hydrogenation of the pyridine ring to obtain Donepezil (Scheme 2).
Patent application WO 99/36405 describes another process for the synthesis of Donepezil. According to this patent, 2-alkoxycarbonyl-1-indanones are reacted with (4-pyridinyl) methyl halide moiety followed by hydrolysis and decarboxylation to give the 2-(4-pyridinyl)methyl-1-indanone derivative. This is followed by reaction with benzyl halides to obtain the corresponding quaternary ammonium salt, and followed by hydrogenation of the pyridine ring to obtain Donepezil (Scheme 2). Patent application WO 97/22584 describes the preparation of Donepezil by reaction of pyridine-4-carboxyaldehyde with malonic acid to give 3-(pyridin-4-yl)-2- propenoic acid, followed by hydrogenation of the double bond to give 3-(piperidin-4-yl)-2-propionic acid. Reaction of this intermediate with methyl chloroformate afforded 3-[N-(methyloxycarbonyl) piperidin-4-yl]propionic acid. This was followed by reaction with oxalyl chloride to give methyl 4-(2-chlorocarbonylethyl)piperidin-1-carboxylate. Reaction with 1,2-dimethoxybenzene in the presence of aluminum chloride afforded methyl 4-[3-(3,4-dimethoxyphenyl)-3-oxopropyl]piperidin-1 -carboxylate. Reaction with tetramethyldiaminomethane afforded 4-[2-(3,4-dimethoxybenzoyl)allyl] piperidin-1-carboxylate. Reaction with sulfuric acid afforded methyl 4-(5,6-dimethoxy-1-oxoindan-2-yl)methylpiperidin-1- carboxylate. This was followed by treatment with base to give 5,6-dimethoxy-2-(piperidin-4-ylmethyl) indan-1-one, then reaction with benzyl bromide afforded Donepezil (Scheme 3).
Patent application WO 97/22584 describes the preparation of Donepezil by reaction of pyridine-4-carboxyaldehyde with malonic acid to give 3-(pyridin-4-yl)-2- propenoic acid, followed by hydrogenation of the double bond to give 3-(piperidin-4-yl)-2-propionic acid. Reaction of this intermediate with methyl chloroformate afforded 3-[N-(methyloxycarbonyl) piperidin-4-yl]propionic acid. This was followed by reaction with oxalyl chloride to give methyl 4-(2-chlorocarbonylethyl)piperidin-1-carboxylate. Reaction with 1,2-dimethoxybenzene in the presence of aluminum chloride afforded methyl 4-[3-(3,4-dimethoxyphenyl)-3-oxopropyl]piperidin-1 -carboxylate. Reaction with tetramethyldiaminomethane afforded 4-[2-(3,4-dimethoxybenzoyl)allyl] piperidin-1-carboxylate. Reaction with sulfuric acid afforded methyl 4-(5,6-dimethoxy-1-oxoindan-2-yl)methylpiperidin-1- carboxylate. This was followed by treatment with base to give 5,6-dimethoxy-2-(piperidin-4-ylmethyl) indan-1-one, then reaction with benzyl bromide afforded Donepezil (Scheme 3).
 Patent application EP 711756 describes the preparation of Donepezil by reaction of 5,6-dimethoxy-1- indanone with pyridin-4-aldehyde to give 5,6-dimethoxy-2-(pyridin-4-yl)methylene indan-1-one. Reaction with benzyl bromide afforded 1-benzyl-4-(5,6-dimethoxyindan-1-on-2-ylidene)methylpyridinium bromide. Hydrogenation in the presence of platinum oxide afforded Donepezil (Scheme 4).
Patent application EP 711756 describes the preparation of Donepezil by reaction of 5,6-dimethoxy-1- indanone with pyridin-4-aldehyde to give 5,6-dimethoxy-2-(pyridin-4-yl)methylene indan-1-one. Reaction with benzyl bromide afforded 1-benzyl-4-(5,6-dimethoxyindan-1-on-2-ylidene)methylpyridinium bromide. Hydrogenation in the presence of platinum oxide afforded Donepezil (Scheme 4).

United States Patent 6844440

EP 1386607 A1
Gene Therapy for Melanoma: Progress and Perspectives

FIGURE 1.
Schematic representation of the wild type counterpart of the typically used recombinant viral vectors. (A) Gammaretroviruses and (B) lentiviruses share similar structures, but differ greatly in their genomes and their impact on cellular function. Gag, pro, pol and env genes encode structural proteins of the capsid, protease, reverse transcriptase and envelope proteins, respectively. The additional lentiviral genes perform regulatory functions as well as alter cellular function. (C) The serotype 5 adenovirus has a protein capsid (non-enveloped) and a large, complex genome that encodes critical genes for viral replication (E1a, E1b) as well as structural and functional genes that regulate both viral and cellular activities.
Introduction
Gene therapy, the therapeutic transfer of genetic information to a target cell, continues to be a promising alternative in the fight against cancer. In the case of melanoma, the use of an experimental treatment is justified since this disease is incurable in its advanced stages. Is gene therapy a viable option for the treatment of melanoma patients? In this chapter, we will attempt to answer this question by exploring the intersection between the technology of gene therapy and the biology of melanoma, a point at which opportunities for intervention are revealed.
Gene Therapy for Melanoma: Progress and Perspectives
[1] Cancer Institute of Sao Paulo, University of Sao Paulo School of Medicine, Brazil
[2] University of Sao Paulo, Biomedical Sciences Institute, Brazil

……………….
