
Home » Posts tagged 'fda' (Page 2)
Tag Archives: fda
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves new diagnostic imaging agent FLUCICLOVINE F-18 to detect recurrent prostate cancer

FLUCICLOVINE F-18
Cyclobutanecarboxylic acid, 1-amino-3-(fluoro-18F)-, trans- [
- Molecular FormulaC5H818FNO2
- Average mass132.124 Da
May 27, 2016
Release
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
Prostate cancer is the second leading cause of death from cancer in U.S. men. In patients with suspected cancer recurrence after primary treatment, accurate staging is an important objective in improving management and outcomes.
“Imaging tests are not able to determine the location of the recurrent prostate cancer when the PSA is at very low levels,” said Libero Marzella, M.D., Ph.D., director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research. “Axumin is shown to provide another accurate imaging approach for these patients.”
Two studies evaluated the safety and efficacy of Axumin for imaging prostate cancer in patients with recurrent disease. The first compared 105 Axumin scans in men with suspected recurrence of prostate cancer to the histopathology (the study of tissue changes caused by disease) obtained by prostate biopsy and by biopsies of suspicious imaged lesions. Radiologists onsite read the scans initially; subsequently, three independent radiologists read the same scans in a blinded study.
The second study evaluated the agreement between 96 Axumin and C11 choline (an approved PET scan imaging test) scans in patients with median PSA values of 1.44 ng/mL. Radiologists on-site read the scans, and the same three independent radiologists who read the scans in the first study read the Axumin scans in this second blinded study. The results of the independent scan readings were generally consistent with one another, and confirmed the results of the onsite scan readings. Both studies supported the safety and efficacy of Axumin for imaging prostate cancer in men with elevated PSA levels following prior treatment.
Axumin is a radioactive drug and should be handled with appropriate safety measures to minimize radiation exposure to patients and healthcare providers during administration. Image interpretation errors can occur with Axumin PET imaging. A negative image does not rule out the presence of recurrent prostate cancer and a positive image does not confirm the presence of recurrent prostate cancer. Clinical correlation, which may include histopathological evaluation of the suspected recurrence site, is recommended.
The most commonly reported adverse reactions in patients are injection site pain, redness, and a metallic taste in the mouth.
Axumin is marketed by Blue Earth Diagnostics, Ltd., Oxford, United Kingdom
Patent
http://www.google.com/patents/WO2014023775A1?cl=en
The non-natural amino acid [ F]-l-amino-3-fluorocyclobutane-l-carboxylic acid
([18F]-FACBC, also known as [18F]-Fluciclovine) is taken up specifically by amino acid transporters and has shown promise for tumour imaging with positron emission tomography (PET).
A known synthesis of [18F]-FACBC begins with the provision of the protected precursor compound 1 -(N-(t-butoxycarbonyl)amino)-3 –
[((trifluoromethyl)sulfonyl)oxy]-cyclobutane-l-carboxylic acid ethyl ester. This precursor compound is first labelled with [18F]-fluoride:

II before removal of the two protecting groups:

IT III
EP2017258 (Al) teaches removal of the ethyl protecting group by trapping the [18F]- labelled precursor compound (II) onto a solid phase extraction (SPE) cartridge and incubating with 0.8 mL of a 4 mol/L solution of sodium hydroxide (NaOH). After 3 minutes incubation the NaOH solution was collected in a vial and a further 0.8 mL 4 mol/L NaOH added to the SPE cartridge to repeat the procedure. Thereafter the SPE cartridge was washed with 3 mL water and the wash solution combined with the collected NaOH solution. Then 2.2 mL of 6 mol/L HCl was then added with heating to 60°C for 5 minutes to remove the Boc protecting group. The resulting solution was purified by passing through (i) an ion retardation column to remove Na+ from excess NaOH and Cl~ from extra HCl needed to neutralise excess of NaOH to get a highly acidic solution before the acidic hydrolysis step, (ii) an alumina column, and (iii) a reverse-phase column. There is scope for the deprotection step(s) and/or the
purification step in the production of [18F]-FACBC to be simplified.
Example 1: Synthesis of f FIFACBC
No-carrier- added [18F]fluoride was produced via the 180(p,n)18F nuclear reaction on a GE PETtrace 6 cyclotron (Norwegian Cyclotron Centre, Oslo). Irradiations were performed using a dual-beam, 30μΑ current on two equal Ag targets with HAVAR foils using 16.5 MeV protons. Each target contained 1.6 ml of > 96% [180]water (Marshall Isotopes). Subsequent to irradiation and delivery to a hotcell, each target was washed with 1.6 ml of [160]water (Merck, water for GR analysis), giving approximately 2-5 Gbq in 3.2 ml of [160]water. All radiochemistry was performed on a commercially available GE FASTlab™ with single-use cassettes. Each cassette is built around a one-piece-moulded manifold with 25 three-way stopcocks, all made of polypropylene. Briefly, the cassette includes a 5 ml reactor (cyclic olefin copolymer), one 1 ml syringe and two 5 ml syringes, spikes for connection with five prefilled vials, one water bag (100 ml) as well as various SPE cartridges and filters. Fluid paths are controlled with nitrogen purging, vacuum and the three syringes. The fully automated system is designed for single-step fluorinations with cyclotron-produced [18F]fluoride. The FASTlab was programmed by the software package in a step-by-step time-dependent sequence of events such as moving the syringes, nitrogen purging, vacuum, and temperature regulation. Synthesis of
[18F]FACBC followed the three general steps: (a) [18F]fluorination, (b) hydrolysis of protection groups and (c) SPE purification.
Vial A contained K222 (58.8 mg, 156 μπιοΐ), K2C03 (8.1 mg, 60.8 μπιοΐ) in 79.5% (v/v)
MeCN(aq) (1105 μΐ). Vial B contained 4M HC1 (2.0 ml). Vial C contained MeCN
(4.1ml). Vial D contained the precursor (48.4 mg, 123.5 μιηοΐ) in its dry form (stored at -20 °C until cassette assembly). Vial E contained 2 M NaOH (4.1 ml). The 30 ml product collection glass vial was filled with 200 mM trisodium citrate (10 ml). Aqueous
[18F]fluoride (1-1.5 ml, 100-200 Mbq) was passed through the QMA and into the 180-
H20 recovery vial. The QMA was then flushed with MeCN and sent to waste. The trapped [18F]fluoride was eluted into the reactor using eluent from vial A (730 μΐ) and then concentrated to dryness by azeotropic distillation with acetonitrile (80 μΐ, vial C). Approximately 1.7 ml of MeCN was mixed with precursor in vial D from which 1.0 ml of the dissolved precursor (corresponds to 28.5 mg, 72.7 mmol precursor) was added to the reactor and heated for 3 min at 85°C. The reaction mixture was diluted with water and sent through the tC18 cartridge. Reactor was washed with water and sent through the tC18 cartridge. The labelled intermediate, fixed on the tC18 cartridge was washed with water, and then incubated with 2M NaOH (2.0 ml) for 5 min after which the 2M NaOH was sent to waste. The labelled intermediate (without the ester group) was then eluted off the tC18 cartridge into the reactor using water. The BOC group was hydrolysed by adding 4M HC1 (1.4 ml) and heating the reactor for 5 min at 60 °C. The reactor content with the crude [18F]FACBC was sent through the HLB and Alumina cartridges and into the 30 ml product vial. The HLB and Alumina cartridges were washed with water (9.1 ml total) and collected in the product vial. Finally, 2M NaOH (0.9 ml) and water (2.1 ml) was added to the product vial, giving a purified formulation of [18F]FACBC with a total volume of 26 ml. Radiochemical purity was measured by radio-TLC using a mixture of MeCN:MeOH:H20:CH3COOH (20:5:5: 1) as the mobile phase. The radiochemical yield (RCY) was expressed as the amount of radioactivity in the [18F]FACBC fraction divided by the total used [18F]fluoride activity (decay corrected). Total synthesis time was 43 min.
The RCY of [18F]FACBC was 62.5% ± 1.93 (SD), n=4.
/////FDA, diagnostic imaging agent, recurrent prostate cancer, fda 2016, Axumin, marketed, Blue Earth Diagnostics, Ltd., Oxford, United Kingdom, fluciclovine F 18
C1[C@@](C[C@H]1[18F])(N)C(=O)O
UPDATE
![]()
SEE EMA
| Axumin : EPAR – Summary for the public | EN = English | 06/07/2017 |
The active substance fluciclovine (18F) is prepared from the precursor AH113487 by nucleophilic substitution
of a triflate group by 18F-fluoride, followed by two deprotection steps. Due to the short half-life of the 18Ffluorine
radioisotope, each batch is prepared on the day of clinical use.
The active substance is prepared in a proprietary automated synthesiser unit. The synthesiser module is
computer-controlled. A fluid path for synthesis is provided in the form of a single use cassette (FASTlab). The
cassette contains 3 reagent vials and 3 solid phase cartridges. Two other reagent vials are supplied
separately as they have a recommended storage temperature of 2-8°C. These 2 vials are inserted into the
cassette on the day of production.
Assessment report
EMA/237809/2017 Page 13/90
Fluciclovine (18F) is produced in a continuous operation from the precursor AH113487. Due to the radioactive
nature of the process, and the short half-life of [18F] fluorine, intermediates are not isolated and there is no
opportunity for operator intervention or in-process testing. Control of the synthesis of fluciclovine (18F) from
the precursor is achieved through the automated synthesis platform, which is pre-programmed with
synthesis parameters optimised for the process. On-board detectors record transfers of radioactivity through
the fluid path at critical points and monitor temperature and pressure as appropriate so that the operator
may track the progress of the synthesis.
The active substance fluciclovine (18F) progressses immediately to purification, formulation and dispensing as
the finished product within a single, continuous operation. Validation of the manufacturing process for
fluciclovine (18F) is therefore described as part of finished product validation.
The characterisation of the active substance is in accordance with the EU guideline on chemistry of new
active substances.
As mentioned, the manufacture of the active substance and finished product takes place in a single,
continuous process. The active substance is not isolated at any point. Therefore, relevant information about
impurities is given only for the finished product.
For the same reason, information for the container closure system is provided only for the finished product.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004197/WC500230836.pdf
FDA approved a switchover from batch to the new technology for production of HIV drug Prezista, Darunavir on a line at its plant in Gurabo, Puerto Rico

Above is an Illustration example,
FDA urges companies to get on board with continuous manufacturing
The FDA gave Johnson & Johnson’s ($JNJ) Janssen drug unit the thumbs up last week for the continuous manufacturing process that it has been working on for 5 years. The agency approved a switchover from batch to the new technology for production of HIV drug Prezista on a line at its plant in Gurabo, Puerto Rico……http://www.fiercepharma.com/manufacturing/fda-urges-companies-to-get-on-board-continuous-manufacturing
 |
|
 |
SEE……http://www.en-cphi.cn/news/show-29367.html
Just after opening a refurbished manufacturing facility in Cape Town, South Africa earlier this year, pharma giant Johnson & Johnson ($JNJ) recently opened the doors to its Global Public Health Africa Operations office there.
The company has invested $21 million (300 million rand) in the facilities. The global public health facility will focus on HIV, tuberculosis and maternal, newborn and child health, South Africa – The Good News reported.
“This (investment) tells us that South Africa has the capability to provide a facility for world-class manufacturing,” Rob Davies, minister of the Department of Trade and Industry told the publication.
Johnson & Johnson, which has operated in South Africa for more than 86 years, planned to close the Cape Town manufacturing plant by the end of 2008 but was persuaded to keep the facility open for local manufacturing to serve sub-Saharan business. By 2015, the plant was cited by J&J as the most-improved in cost competitiveness from 30 company plants worldwide.
Earlier this month, the FDA gave J&J’s Janssen drug unit the go-ahead to proceed with the continuous manufacturing process it’s been working on for 5 years. The agency approved a switchover from batch to the new technology for production of HIV drug Prezista, Darunavir on a line at its plant in Gurabo, Puerto Rico.

AN EXAMPLE NOT RELATED TO DARUNAVIR
References
International Symposium on Continuous Manufacturing of Pharmaceuticals
Implementation, Technology & Regulatory
![]()
![]()
May 20-21, 2014 (Link to 2016 Meeting Website)
Continuous Bioprocessing
https://iscmp.mit.edu/white-papers/white-paper-4
READ
Achieving Continuous Manufacturing: Technologies and Approaches for Synthesis, Work-Up and Isolation of Drug Substance
https://iscmp.mit.edu/white-papers/white-paper-1
//////
//////FDA, HIV drug, Prezista, Darunavir, Gurabo, Puerto Rico
FDA issues rule for data collection of antimicrobial sales and distribution by animal species

May 10, 2016
Release
The U.S. Food and Drug Administration finalized a rule today that revises its annual reporting requirements for drug sponsors of all antimicrobials sold or distributed for use in animals intended for human consumption or food-producing animals. Companies are now required to provide estimates of sales broken down by major food-producing species (cattle, swine, chickens and turkeys) in addition to the overall estimates they already submit on the amount of antimicrobial drugs they sell or distribute for use in food-producing animals.
The new sales data will improve the agency’s understanding of how antimicrobials are sold and distributed for use in major food-producing species and help further target efforts to ensure judicious use of medically important antimicrobials.
Section 105 of the Animal Drug User Fee Amendments of 2008 (ADUFA 105) requires antimicrobial drug sponsors to annually report to the FDA the amount of all antimicrobial drugs they sell and distribute for use in food-producing animals, including those antibiotics that are not used in human medicine. ADUFA 105 also requires the FDA to prepare summary reports of sales and distribution information received from drug sponsors each year, by antimicrobial class for classes with three or more distinct sponsors, and to provide those summaries to the public. Prior to finalizing this rule, animal drug sponsors were not required to submit sales or distribution data by particular species.
Adding the requirement for sponsors to report species-specific sales estimates will also complement the data collection plan the FDA is developing, as part of the National Strategy for Combating Antibiotic-Resistant Bacteria (CARB), with the U.S. Department of Agriculture and the Centers for Disease Control and Prevention, to obtain additional on-farm use and resistance data. The collection of data from multiple sources, including enhanced sales data from antimicrobial animal drug sponsors, is important for providing a comprehensive and science-based picture of antimicrobial drug use and resistance in animal agriculture.
“This information will further enhance FDA’s ongoing activities related to slowing the development of antimicrobial resistance to help ensure that safe and effective antimicrobial new animal drugs will remain available for use in human and animal medicine,” said Dr. William T. Flynn, D.V.M., M.S., deputy director for science policy in the FDA’s Center for Veterinary Medicine.
The final rule also includes a provision to improve the timeliness of annual reports by requiring the FDA to publish its summary report of the antimicrobial sales and distribution information it collects for each calendar year by Dec. 31 of the following year.
The rule was proposed in May 2015, and takes into consideration hundreds of public comments from the veterinary community, animal feed manufacturing and livestock production associations, drug manufacturers, consumer groups and other stakeholders. Drug sponsors are required to comply with the reporting requirements in the final rule when submitting their reports covering the period of calendar year 2016.
///////FDA , data collection, antimicrobial sales, distribution, animal species
FDA approves new drug Venclexta (venetoclax) for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality

April 11, 2016
Release
The U.S. Food and Drug Administration today approved Venclexta (venetoclax) for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a chromosomal abnormality called 17p deletion and who have been treated with at least one prior therapy. Venclexta is the first FDA-approved treatment that targets the B-cell lymphoma 2 (BCL-2) protein, which supports cancer cell growth and is overexpressed in many patients with CLL.
According to the National Cancer Institute, CLL is one of the most common types of leukemia in adults, with approximately 15,000 new cases diagnosed each year. CLL is characterized by the progressive accumulation of abnormal lymphocytes, a type of white blood cell. Patients with CLL who have a 17p deletion lack a portion of the chromosome that acts to suppress cancer growth. This chromosomal abnormality occurs in approximately 10 percent of patients with untreated CLL and in approximately 20 percent of patients with relapsed CLL.
“These patients now have a new, targeted therapy that inhibits a protein involved in keeping tumor cells alive,” said Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option for their specific condition.”
The efficacy of Venclexta was tested in a single-arm clinical trial of 106 patients with CLL who have a 17p deletion and who had received at least one prior therapy. Trial participants took Venclexta orally every day, beginning with 20 mg and increasing over a five-week period to 400 mg. Results showed that 80 percent of trial participants experienced a complete or partial remission of their cancer.
Venclexta is indicated for daily use after detection of 17p deletion is confirmed through the use of the FDA-approved companion diagnostic Vysis CLL FISH probe kit.
The most common side effects of Venclexta include low white blood cell count (neutropenia), diarrhea, nausea, anemia, upper respiratory tract infection, low platelet count (thrombocytopenia) and fatigue. Serious complications can include pneumonia, neutropenia with fever, fever, autoimmune hemolytic anemia, anemia and metabolic abnormalities known as tumor lysis syndrome. Live attenuated vaccines should not be given to patients taking Venclexta.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. These are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions. Venclexta also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Venclexta is manufactured by AbbVie Inc. of North Chicago, Illinois, and marketed by AbbVie and Genentech USA Inc. of South San Francisco, California. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular of Des Plaines, Illinois.
GALETERONE
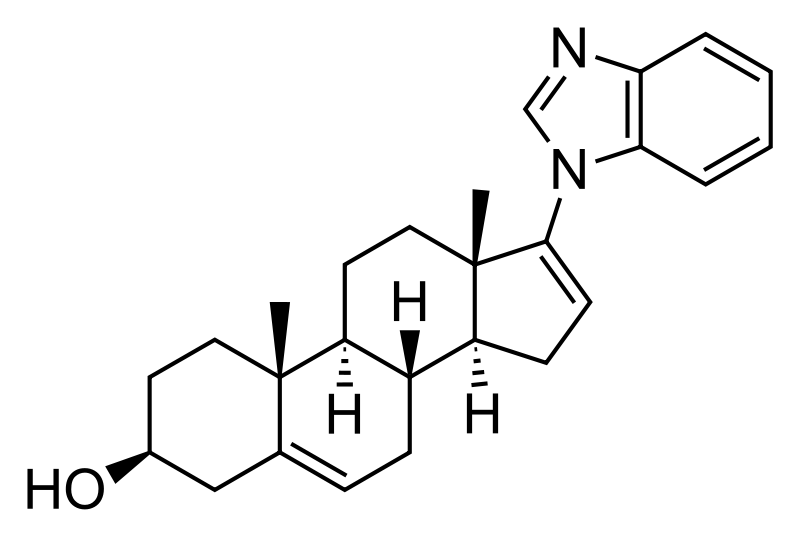
Galeterone
SYNTHESIS SEE BELOW
A SARM potentially for the treatment of prostate cancer.
Research Code, TOK-001; VN; 124; 124-1; 1241
TOK-001; Galeterone; 851983-85-2; VN/124; UNII-WA33E149SW; VN/124-1;
CAS No. 851983-85-2(Galeterone)
(3S,8R,9S,10R,13S,14S)-17-(benzimidazol-1-yl)-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15-decahydro-1H-cyclopenta[a]phenanthren-3-ol
Fast track 2012 f
| Molecular Formula: | C26H32N2O |
|---|---|
| Molecular Weight: | 388.54508 g/mol |

Galeterone (TOK-001 or VN/124-1) is a novel steroidal antiandrogen under development by Tokai Pharmaceuticals for the treatment of prostate cancer. It possesses a unique dual mechanism of action, acting as both an androgen receptor antagonist and an inhibitor of CYP17A1, an enzyme required for the biosynthesis of the androgens.[1] It shows selectivity for 17,20-lyase over 17-hydroxylase.[2]
As of 2016, galeterone is being compared to enzalutamide in a phase III clinical trial (ARMOR3-SV) for AR-V7-expressing metastatic castration-resistant prostate cancer.[3][4]
Specific Androgen Receptor Modulator CYP17 Inhibitor TOK-001 is an orally bioavailable small-molecule androgen receptor modulator and CYP17 lyase inhibitor with potential antiandrogen activity. Galeterone exhibits three distinct mechanisms of action: 1) as an androgen receptor antagonist, 2) as a CYP17 lyase inhibitor and 3) by decreasing overall androgen receptor levels in prostate cancer tumors, all of which may result in a decrease in androgen-dependent growth signaling. Localized to the endoplasmic reticulum (ER), the cytochrome P450 enzyme CYP17 (P450C17 or CYP17A1) exhibits both 17alpha-hydroxylase and 17,20-lyase activities, and plays a key role in the steroidogenic pathway that produces progestins, mineralocorticoids, glucocorticoids, androgens, and estrogens.
About Galeterone
Tokai’s lead product candidate is galeterone, a highly-selective, oral small molecule with the potential to transform the treatment of prostate cancer. We are focusing our late-stage development of galeterone on the treatment of men with metastatic, castration-resistant prostate cancer, or CRPC, whose prostate tumor cells express the AR-V7 splice variant.
We are conducting ARMOR3-SV, a Phase 3 clinical trial of galeterone evaluating whether administration of galeterone results in a statistically significant increase in radiographic progression-free survival as compared to Xtandi® (enzalutamide), an oral therapy currently approved for the treatment of CRPC, in AR-V7 positive metastatic CRPC patients. ARMOR3-SV is the first pivotal trial in prostate cancer to employ a precision medicine approach for patient selection. For more information regarding ARMOR3-SV, click here.
Galeterone has been studied in over 250 subjects in Phase 1 and Phase 2 clinical trials, including in CRPC patients with and without the AR-V7 splice variant. In these trials, galeterone demonstrated good tolerability and showed clinically meaningful reductions in levels of prostate specific antigen, or PSA, a biochemincal marker used to evaluate prostate cancer patients for signs of response to therapy.
We are currently focusing our late-stage development of galeterone on AR-V7 positive metastatic CRPC patients because it represents an unmet need in prostate cancer and our precision medicine approach provides an efficient development path. Based on the data we and our collaborators have produced to date, we also believe there is rationale for the broader clinical exploration of galeterone in the future.
Galeterone acts by disrupting the androgen receptor signaling pathway. This pathway is activated by the binding of male hormones (also known as androgens), such as testosterone and dihydrotestosterone (DHT) to androgen receptors in prostate cancer cells.
Galeterone disrupts the activation of the androgen receptor pathway in three ways:
- Androgen receptor degradation, which reduces the amount of androgen receptor protein in tumor cells. There are no currently marketed drugs whose mechanism of action entails degradation of the androgen receptor. Therefore, galeterone represents a potential first-in-class therapeutic opportunity.
- CYP17 enzyme inhibition, which blocks the synthesis of testosterone. This mechanism has been validated clinically by Zytiga (abiraterone). Zytiga must be co-administered with the steroid prednisone in order to minimize the risk of a potentially fatal side effect called mineralocorticoid excess. Unlike Zytiga, galeterone has not been shown in clinical trials to cause mineralocorticoid excess and, as a result, does not require co-administration of steroids. As a result, we believe that galeterone may be easier to administer, provide convenience for patients and enhance patient compliance.
- Androgen receptor inhibition, which blocks the binding of testosterone or DHT with the androgen receptor. This mechanism has been validated clinically by Xtandi® (enzalutamide), which is also currently approved for the treatment of CRPC. Xtandi™ has shown a risk of grand mal seizures in clinical trials. We have not had any reports of seizures in clinical trials of galeterone and, therefore, galeterone may have certain safety advantages over Xtandi.
Tokai retains global rights to galeterone. We intend to commercialize galeterone in the United States on our own, and to seek a partner to further develop and commercialize galeterone outside of the United States.
Galeterone has been granted Fast Track designation by U.S. Food and Drug Administration for the treatment of CRPC. Fast Track designation is designed to facilitate the development and expedite review of drugs intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs.
 Androgen receptor degradation, which reduces the amount of androgen receptor protein in the tumor cells.
Androgen receptor degradation, which reduces the amount of androgen receptor protein in the tumor cells.
 Androgen receptor antagonism, which blocks the binding of testosterone or DHT with the androgen receptor.
Androgen receptor antagonism, which blocks the binding of testosterone or DHT with the androgen receptor.
 Inhibition of the enzyme CYP17, which blocks the synthesis of testosterone.
Inhibition of the enzyme CYP17, which blocks the synthesis of testosterone.
Figure 3: The structures of abiraterone, orteronel and galeterone.
From CYP17 inhibitors—abiraterone, C17,20-lyase inhibitors and multi-targeting agents
- Nature Reviews Urology 11,32–42 (2014)
- doi:10.1038/nrurol.2013.274


SYNTHESIS
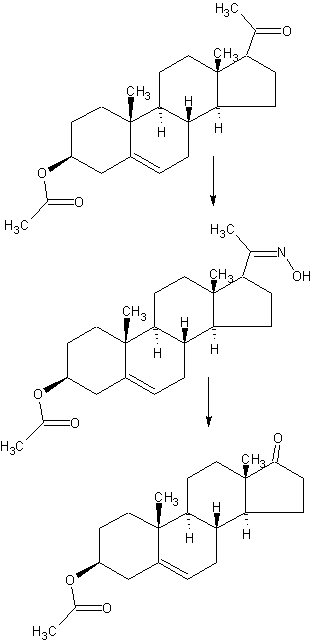
DETAILED DESCRIPTION
1J loss reaction.
(1) raw material specifications to match.
acetate pregnancy dehydropregnenolone: toluene + ethanol: Batch steep: hydrochloric acid amine light = 1: 3: 0 4: 0.213, which pregnenolone acetate pregnancy 160kg, toluene + ethanol 320kg + 160kg, approved Steep 64kg, hydrochloric acid amine light 34kg.
(2) process operation.
In the first input 1000L tank oximation with hydroxylamine hydrochloride in pyridine, and then pumped into a mixed solvent of toluene and ethanol, the reaction solution was stirred and heated to complete dissolution, pregnancy-dehydropregnenolone acetate was added and heated under reflux for 3 hours, cooling and crystallization, The Department conducted into the centrifuge centrifugal drying, apply a recovery from the mother liquor, rinse with warm water mixture to no foam, centrifugal drying, drying to a moisture at 0.2% or less, that acetic acid in pregnancy dehydropregnenolone oxime (oxime compounds) 163kg, content of 99%, a melting point of 202-204 ° C, a yield of about 102% (for pregnenolone acetate pregnancy weight ratio).
2, heavy drain hydrolysis reaction.
(1) raw material specifications to match.
acetate pregnancy dehydropregnenolone waning: Benzene: Batch steep: phosphorus oxychloride and toluene: HCl + water = 1: 6 5: 0 4: 1: 3.5, which acetate pregnancy alcohol one hand 163kg, benzene 1060kg, batch steep 64kg, phosphorus oxychloride and toluene 80kg + 80kg, hydrochloric acid + water 245kg + 325kg.
(2) process operation.
The first drying 2000L rearrangement reaction tank, then pumped to the reaction tank benzene, alcohol into acetate pregnancy oxime, pulls out into benzene, stirring heated to reflux until the reaction mixture is completely dissolved, cooling to 1 (TC When, pyridine, of the reaction liquid at temperatures down to 6 ° C, start dropping a mixed solution of previously prepared phosphorus oxychloride and toluene (1: 1 mass ratio), slowly dropping, dropping control, first After slow fast reaction when dropping liquid temperature control in 4-8 ° C, the addition was complete, the reaction solution at 9-12 ° C for 3 hours the first time under.
After incubation, a solution has been a mixed solution of hydrochloric acid and water, good preparation, while dropping the reaction liquid temperature is controlled at 15-25 ° C, the addition was complete, the reaction solution at 15-25 ° C under a second Insulation 1. 5-2 hours. After incubation, stand 40 minutes, then points to lower acidic water layer, the remaining upper layer was added 0.3 times the amount of 30-35 ° C in the brine and let stand 20 minutes, a second watershed, sub lower aqueous layer was then allowed to stand for 30 minutes, a third water diversion, to give the final weight of the upper layer reaction solution was drained.
3, the red Dingding steam distillate process.
The rearrangement reaction liquid was pumped to punch distillate tank, conduct atmospheric distillate punch, has been rushed to the reaction mixture was distilled benzene mixed solvent only, at the start of the steam valve not to open too much, so as not to rush material, distillation after cooling discharge, centrifugal drying, washing with tap water to neutral, and then into the oven dried to a moisture in the square. 5% acetic acid in dehydroepiandrosterone (rearrangement thereof) The crude product is about 142kg, content of about 97.5%, a melting point of 160 ° C _165 ° C or so, yield about 88% (for acetate pregnancy dehydropregnenolone weight ratio).
4, refining processes.
The drying in acetic acid Dehydroepiandrosterone crude into refined tin, adding 8 times the weight of the crude methanol and 0.10 times the weight of activated carbon, heat, stirring to dissolve, reflux billion. 5 hours, filtered , concentrated, cooled to about 5 ° C, the discharge




| Patent ID | Date | Patent Title |
|---|---|---|
| US2011034428 | 2011-02-10 | Treatment of Prostate Cancer |
| US7875599 | 2011-01-25 | C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens, in vitro biological activities, pharmacokinetics and antitumor activity |
| US2010137269 | 2010-06-03 | Novel C-17-Heteroaryl Steroidal Cyp17 Inhibitors/Antiandrogens: Synehesis, In Vitro Biological Activities, Pharmacokinetics and Antitumor Activity |
| US2010048914 | 2010-02-25 | Novel C-17-Heteroaryl Steroidal Cyp17 Inhibitors/Antiandrogens, In Vitro Biological Activities, Pharmacokinetics and Antitumor Activity |
| US2010048913 | 2010-02-25 | Novel C-17-Heteroaryl Steroidal CYP17 Inhibitors/Antiandrogens Synthesis In Vitro Biological Activities, Pharmacokinetics and Antitumor Activity |
| US2010048912 | 2010-02-25 | Novel C-17-Heteroaryl Steroidal CYP17 Inhibitors/Antiandrogens, In Vitro Biological Activities, Pharmacokinetics and Antitumor Activity |
| US2010048524 | 2010-02-25 | Novel C-17-Heteroaryl Steroidal CYP17 Inhibitors/Antiandrogens Synthesis In Vitro Biological Activities, Pharmacokinetics and Antitumor Activity |
| US2010047338 | 2010-02-25 | Novel C-17-Heteroaryl Steroidal CYP17 Inhibitors/Antiandrogens, In Vitro Biological Activities, Pharmacokinetics and Antitumor Activity |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2013336962 | 2013-12-19 | AZIRIDINE BISPHENOL ETHERS AND RELATED COMPOUNDS AND METHODS FOR THEIR USE |
| US8569393 | 2013-10-29 | UV-LED curable compositions and inks |
| US2013203615 | 2013-08-08 | ANTIANDROGEN THERAPY MONITORING METHODS AND COMPOSITIONS |
| US2012309861 | 2012-12-06 | PHOTOINITIATORS FOR UV-LED CURABLE COMPOSITIONS AND INKS |
| US2012237502 | 2012-09-20 | METHOD FOR TREATING BREAST CANCER AND OVARIAN CANCER |
| US2011319369 | 2011-12-29 | COMBINATION OF A 17 ALPHA-HYDROXYLASE/C17, 20-LYASE INHIBITOR WITH AN ADDITIONAL THERAPEUTIC AGENT |
| US2011312924 | 2011-12-22 | NOVEL STEROIDAL CYP17 INHIBITORS/ANTIANDROGENS |
| US2011312916 | 2011-12-22 | NOVEL PRODRUGS OF STEROIDAL CYP17 INHIBITORS/ANTIANDROGENS |
| US2011118219 | 2011-05-19 | NOVEL PRODRUGS OF C-17-HETEROARYL STEROIDAL CYP17 INHIBITORS/ANTIANDROGENS: SYNTHESIS, IN VITRO BIOLOGICAL ACTIVITIES, PHARMACOKINETICS AND ANTITUMOR ACTIVITY |
| US2011105445 | 2011-05-05 | ANDROGEN RECEPTOR INACTIVATION CONTRIBUTES TO ANTITUMOR EFFICACY OF CYP17 INHIBITORS IN PROSTATE CANCER |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015051179 | 2015-02-19 | NOVEL STEROIDAL CYP17 INHIBITORS/ANTIANDROGENS |
| US2015005265 | 2015-01-01 | METHODS AND COMPOSITIONS FOR COMBINATION THERAPY USING P13K/MTOR INHIBITORES |
| US2014371261 | 2014-12-18 | INDOMETHACIN ANALOGS FOR THE TREATMENT OF CASTRATE-RESISTANT PROSTATE CANCER |
| US2014371181 | 2014-12-18 | NOVEL PRODRUGS OF STEROIDAL CYP17 INHIBITORS/ANTIANDROGENS |
| US2014343024 | 2014-11-20 | TREATMENT OF PROSTATE CANCER |
| US2014288037 | 2014-09-25 | NOVEL COMPOSITIONS AND METHODS FOR TREATING PROSTATE CANCER |
| US2014288036 | 2014-09-25 | NOVEL C-17-HETEROARYL STEROIDAL CYP17 INHIBITORS/ANTIANDROGENS, IN VITRO BIOLOGICAL ACTIVITIES, PHARMACOKINETICS AND ANTITUMOR ACTIVITY |
| US2014274983 | 2014-09-18 | NOVEL PRODRUGS OF C-17-HETEROARYL STEROIDAL CYP17 INHIBITORS/ANTIANDROGENS: SYNTHESIS, IN VITRO BIOLOGICAL ACTIVITIES, PHARMACOKINETICS AND ANTITUMOR ACTIVITY |
| US2014107085 | 2014-04-17 | Bifunctional AKR1C3 Inhibitors/Androgen Receptor Modulators and Methods of Use Thereof |
| US2013336962 | 2013-12-19 | AZIRIDINE BISPHENOL ETHERS AND RELATED COMPOUNDS AND METHODS FOR THEIR USE |

| CN101691392A * | Sep 17, 2009 | Apr 7, 2010 | 扬州市天平化工厂有限公司 | Method for preparing 3beta-acetoxyl group-5androstene-17ketone |
| CN102212099A * | Apr 2, 2011 | Oct 12, 2011 | 邵阳市科瑞化学品有限公司 | Synthesis method for dehydroepiandrosterone |
| CN102603839A * | Jan 13, 2012 | Jul 25, 2012 | 宜城市共同药业有限公司 | Preparation method of dehydroepiandrosterone |
| CN102746356A * | Jul 17, 2012 | Oct 24, 2012 | 湖北芳通药业股份有限公司 | Process for producing dehydroepiandrosterone acetate through homogeneous phase method |
| Reference | ||||
|---|---|---|---|---|
| 1 | * | 石诚等: “5-雄甾烯-3β-醇-17-酮-3-醋酸酯的工艺研究“, 《山东化工》, vol. 41, no. 1, 31 December 2012 (2012-12-31) | ||
| 2 | * | 石诚等: “醋酸妊娠双烯醇酮肟的工艺研究“, 《广州化工》, vol. 39, no. 23, 31 December 2011 (2011-12-31), pages 78 – 79 | ||
References
- 1 Brawer MK (2008). “New treatments for castration-resistant prostate cancer: highlights from the 44th annual meeting of the american society of clinical oncology, may 30-june 3, 2008, chicago, IL”. Rev Urol 10 (4): 294–6. PMC 2615106. PMID 19145273.
- 2
- Yin L, Hu Q (2014). “CYP17 inhibitors–abiraterone, C17,20-lyase inhibitors and multi-targeting agents”. Nat Rev Urol 11 (1): 32–42. doi:10.1038/nrurol.2013.274. PMID 24276076.
- 3
- “A Study of Galeterone Compared to Enzalutamide In Men Expressing Androgen Receptor Splice Variant-7 mRNA (AR-V7) Metastatic CRPC – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2016-02-27.
- 4
Silberstein, John L.; Taylor, Maritza N.; Antonarakis, Emmanuel S. (2016-04-01). “Novel Insights into Molecular Indicators of Response and Resistance to Modern Androgen-Axis Therapies in Prostate Cancer”. Current Urology Reports 17 (4): 29. doi:10.1007/s11934-016-0584-4. ISSN 1534-6285. PMID 26902623.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
17-(1H-benzimidazol-1-yl)androsta-5,16-dien-3β-ol
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 851983-85-2 |
| PubChem | CID 11188409 |
| ChemSpider | 9363493 |
| KEGG | D10125 |
| Chemical data | |
| Formula | C26H32N2O |
| Molar mass | 388.25 |
///////
C[C@]12CC[C@@H](CC1=CC[C@@H]3[C@@H]2CC[C@]4([C@H]3CC=C4N5C=NC6=CC=CC=C65)C)O
CC12CCC(CC1=CCC3C2CCC4(C3CC=C4N5C=NC6=CC=CC=C65)C)O
FDA´s Emerging Technology Applications Program – Draft Guidance
![]()
FDA´s Emerging Technology Applications Program – Draft Guidance
The FDA recently published a draft guidance for industry on the “Advancement of Emerging Technology Applications”. The draft guidance provides recommendations to pharmaceutical companies interested in participating in a program involving the submission of CMC information containing emerging manufacturing (including testing, packaging and labeling, and quality control) technology to FDA. Find out more about the draft guidance for industry “Advancement of Emerging Technology Applications to Modernize the Pharmaceutical Manufacturing Base“..
On December 23, 2015, the FDA published a draft guidance for industry “Advancement of Emerging Technology Applications to Modernize the Pharmaceutical Manufacturing Base“. Comments and suggestions regarding this draft document should be submitted within 60 days of publication.
The draft guidance provides recommendations to pharmaceutical companies interested in participating in a program involving the submission of CMC (chemistry, manufacturing, and controls) information containing emerging manufacturing (including testing, packaging and labeling operations, and quality control) technology to FDA.
The program is open for new drug applications (INDs), original or supplemental new drug application (NDA), abbreviated new drug application (ANDA), or biologic license application (BLA). It only affects the quality section of a submission (CMC and facility-related information).
The development of emerging manufacturing technology, like, for example, aseptic manufacturing facilities with highly automated systems and isolators, may lead to improved manufacturing, and therefore improved product quality and availability throughout a product´s lifecycle.
Pharmaceutical companies can submit questions and proposals about the use of these technologies to a group within CDER (Emerging Technology Team – ETT).
The draft guidance is a follow-on to the FDA guidance for industry “PAT – A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance” which describes the concept that quality cannot be tested into products. It should be built-in or should be present by design. Through the ETT, FDA intends to encourage the adoption of innovative approaches by leveraging existing resources of FDA to facilitate regulatory reviews of submissions.
Examples of emerging technology elements include an innovative or novel:
- Product manufacturing technology, such as the dosage form;
- Manufacturing process (e.g., design, scale-up, and/or commercial scale);
- Testing technology.
Interested parties should submit a written meeting request to participate in the ETT program at least three months prior to the planned application (IND, ANDA, BLA, NDA) submission date. In addition to the items outlined in the FDA guidance “Formal Meetings Between the FDA and Sponsors or Applicants” the request should also include the following items:
- A brief description of the proposed testing, process, and/or proposed technology;
- A brief explanation why the proposed testing, process, and/or technology are substantially novel and unique;
- A description of how the proposed testing and/or technology could modernize pharmaceutical manufacturing and thus improve product safety, identity, strength, quality, or purity;
- A summary of the development plan and any perceived roadblocks to technical or regulatory implementation;
- A timeline for submission.
The request should generally not exceed five pages and FDA expects to notify companies of its decision regarding acceptance into the program within 60 days of receipt of the request. Once accepted into the program, the participant can engage with ETT and CMC in accordance with existing meeting procedures and guidances (e.g. above mentioned FDA guidance on Formal Meetings).
For further information, please find all the details in the draft guidance “Advancement of Emerging Technology Applications to Modernize the Pharmaceutical Manufacturing Base“.
Statistics and Process Validation: current Findings of the FDA

The “new” FDA’s process validation guideline has been effective since January 2011. One considerable change was made to the original validation guideline from 1987 to put a significantly greater emphasis on statistics in the context of process validation. So far, relatively few inspection deficiencies had been observed by the FDA with regard to statistics. At a conference in September 2015 co-sponsored by the FDA, Grace McNally – Senior FDA official – reported about current “findings” in the 483s deficiency reports and in Establishment Inspection Reports (EIR). Now, deficiencies regarding statistical problematics can also be found here.
For example, it has been criticised that a (statistical) sampling plan had be misinterpreted. Wrong AQL values with regard to the number of samples have been noted based on MIL-STD-105D. Moreover, it has been criticised that the company didn’t know the operation characteristics of its sampling plan.
Another criticised “finding” was that PPQ batches had been considered as “accepted” when all in-process controls and release specifications were met. It has also been criticised that no intra-batch variabilities have been examined. In addition, it has been noticed that there was no information available in the validation plan concerning the assessment of the process itself. There was also no indication about the objective of the determination of inter-batch variabilities.
Although OOS results had been found in 2 out of 4 PPQ batches, reduced IPC tests have been recommended in the PPQ report giving the justification that this was a standard procedure. Regarding this point, the FDA criticises the lack of scientific rationales for reduced sampling and monitoring. Interestingly, Grace McNally mentions possibilities for rationales of IPC sampling plans and the adaptation to a reduced size. In this context, she refers to the ANSI/ASQ Z1.4 norm and ISO 2859 whereby it is expressly pointed out that the ANSI norm recommends the production of at least 10 successful batches before reducing testing. According to the ISO norm even 15 successful batches are necessary.
The FDA notified a tablet process, criticising the fact that no rationales for warning and action limits were available. Furthermore, it has been criticised that no analyses on variabilities were available although they had been required internally and no capacity indices had been determined. There have been no analyses on the distribution of data, neither planned nor performed. The FDA also remarked that the calculation of variabilities is necessary to be able to make statements about process capacities.
Conclusion: Reinforcing the emphasis on statistics in the US FDA Process Validation Guideline from 2011 hasn’t been really often addressed in the official deficiencies reports. This seems to be changing.


////////////Statistics, Process Validation, current Findings, FDA
FDA approves new treatment for HIV

November 5, 2015
Release
The U.S. Food and Drug Administration today approved Genvoya (a fixed-dose combination tablet containing elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide) as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients 12 years of age and older.
The CDC estimates that 1.2 million persons ages 13 years and older are living with HIV infection, and that more than another 150,000 persons in this age range have HIV but are unaware of their infection. Over the past decade, the number of people living with HIV has increased, while the annual number of new HIV infections has remained relatively stable.
“Today’s approval of a fixed dose combination containing a new form of tenofovir provides another effective, once daily complete regimen for patients with HIV-1 infection,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Genvoya is approved for use in HIV-infected adults and children ages 12 years and older weighing at least 35 kilograms (77 pounds) who have never taken HIV therapy (treatment-naïve) and HIV-infected adults whose HIV-1 virus is currently suppressed. While Genvoya is not recommended for patients with severe renal impairment, those with moderate renal impairment can take Genvoya.
Genvoya’s safety and efficacy in adults were evaluated in 3,171 participants enrolled in four clinical trials. Depending on the trial, participants were randomly assigned to receive Genvoya or another FDA approved HIV treatment. Results showed Genvoya was effective in reducing viral loads and comparable to the other treatment regimens.
Genvoya contains a new form of tenofovir that has not been previously approved. This new form of tenofovir provides lower levels of drug in the bloodstream, but higher levels within the cells where HIV-1 replicates. It was developed to help reduce some drug side effects. Genvoya appears to be associated with less kidney toxicity and decreases in bone density than previously approved tenofovir containing regimens based on laboratory measures. Patients receiving Genvoya experienced greater increases in serum lipids (total cholesterol and low-density lipoprotein) than patients receiving other treatment regimens in the studies.
Genvoya carries a Boxed Warning alerting patients and health care providers that the drug can cause a buildup of lactic acid in the blood and severe liver problems, both of which can be fatal. The Boxed Warning also states that Genvoya is not approved to treat chronic hepatitis B virus infection. The most common side effect associated with Genvoya is nausea. Serious side effects include new or worsening kidney problems, decreased bone mineral density, fat redistribution and changes in the immune system (immune reconstitution syndrome). Health care providers are advised to monitor patients for kidney and bone side effects. Genvoya should not be given with other antiretroviral products and may have drug interactions with a number of other commonly used medications.
Genvoya is marketed by Gilead Sciences Inc. based in Foster City, California.
/////////
FDA approves Praxbind, Idarucizumab the first reversal agent for the anticoagulant Pradaxa
October 16, 2015
Release
The U.S. Food and Drug Administration today granted accelerated approval to Praxbind (idarucizumab) for use in patients who are taking the anticoagulant Pradaxa (dabigatran) during emergency situations when there is a need to reverse Pradaxa’s blood-thinning effects.
“The anticoagulant effects of Pradaxa are important and life-saving for some patients, but there are situations where reversal of the drug’s effects is medically necessary,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval offers the medical community an important tool for managing patients taking Pradaxa in emergency or life-threatening situations when bleeding can’t be controlled.”
The FDA approved Pradaxa in 2010 to prevent stroke and systemic blood clots in patients with atrial fibrillation, as well as for the treatment and prevention of deep venous thrombosis and pulmonary embolism. Praxbind is the first reversal agent approved specifically for Pradaxa and works by binding to the drug compound to neutralize its effect. Praxbind solution is for intravenous injection.
The safety and effectiveness of Praxbind were studied in three trials involving a total of 283 healthy volunteers taking Pradaxa (i.e., people who did not require an anticoagulant). In the healthy volunteers who were given Praxbind, there was an immediate reduction in the amount of Pradaxa in participants’ blood (measured as unbound dabigatran plasma concentration) that lasted for a period of at least 24 hours. In this study, the most common side effect from use of Praxbind was headache.
Another trial included 123 patients taking Pradaxa who received Praxbind due to uncontrolled bleeding or because they required emergency surgery. In this ongoing trial, based on laboratory testing, the anticoagulant effect of Pradaxa was fully reversed in 89 percent of patients within four hours of receiving Praxbind. In this patient trial, the most common side effects were low potassium (hypokalemia), confusion, constipation, fever and pneumonia.
Reversing the effect of Pradaxa exposes patients to the risk of blood clots and stroke from their underlying disease (such as atrial fibrillation). The Praxbind labeling recommends patients resume their anticoagulant therapy as soon as medically appropriate, as determined by their health care provider.
Praxbind is approved under the FDA’s accelerated approval program, which allows the agency to approve drugs for serious conditions that fill an unmet medical need based on an effect on a surrogate or an intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. The program is designed to provide patients with earlier access to promising new drugs, but the company will be required to submit additional clinical information after approval to confirm the drug’s clinical benefit.
Praxbind and Pradaxa are both marketed by Boehringer Ingelheim of Ridgefield, Connecticut.
FDA Inspections at API Manufacturers – current Warning Letter Trends
The Warning Letters the FDA sent to active ingredient manufacturers last fiscal year, show similar patterns. Find out more about the frequent deficiencies found in the area of responsibility of quality assurance and in the handling of electronic data in production facilities for active pharmaceutical ingredients.
Taking a look at the Warning Letters the FDA issued after inspections of activesubstance manufacturers in the 2015 fiscal year, which ended on 30 September 2015, it is first of all striking that only non-American companies are among the addressees. Almost half of them are Indian companies. Overall the numbers look like this: India (3 WLs); China (2 WLs); Canada (1 WL); Thailand (1 WL); Czech Republic (1 WL).
The top issue in the Warning Letters is the non-GMP compliant handling of electronic data or missing data integrity. Each of the 8 warning letters contains the following comment in the same wording:
“Failure to prevent unauthorized access or changes to data and to provide adequate controls to prevent omission of data.”
The lack of access control on electronic (raw) data is an issue the FDA investigators have been observing for a long time, especially during inspections in pharmaceutical companies. In this as well as in the last fiscal year there were significant deficiencies in several companies – medicinal product as well as API manufacturers – as the comments in the appropriate Warning Letters show. For more information also see the GMP news Another FDA Warning Letter with Focus on “Data Integrity” and FDA Warning Letter on Data Integrity.
Ultimately these deficiencies can be traced back to a failure of the quality assurance unit which also affects other areas. In the Warning Letters, the following examples can be found for this:
- “Failure of your quality unit to ensure that materials are appropriately tested and the results are reported.”
“Failure of your quality unit to exercise its responsibility to ensure the APIs manufactured at your facility are in compliance with CGMP, and meet established specifications for quality and purity.”
Data were manipulated by laboratory staff (change of the file name), to fake results from identity tests in batches which in reality were not performed. Quality assurance was not able to uncover this manipulation.
Despite an unknown peak in the examination for residual solvents the relevant batches were released. Upon receipt of a complaint regarding this peak an examination was conducted with the result that the contamination originated in the production process itself. Preventive control measures to avoid this contamination were not established. - “Failure to adequately investigate complaints and extend the investigations to other batches that may have been affected.”
As a result of a complaint (bad smell), a cause study was initiated which was completed prior to implementation of the preventive measures again. The CAPA measures subsequently carried out were obviously not associated with the reason for the complaint. - “Failure to have appropriate controls for issuance of batch records”.
The use of document templates for batch records is out of control. These can be printed out from the production staff’s personal computers. Although there is an SOP for the control of batch records there are no appropriate training records. - “Failure to have appropriate documentation and record controls.”
Data for tracing raw materials are not available. Log entries are without date/visa and partly corrected with Tippex. There is an SOP prohibiting the use of correction fluid, however this was not trained. - “Failure to record activities at the time they are performed and destruction of original records.”
Original records of critical process data on uncontrolled memos were transferred subsequently in new report templates (after batch approvals) and then destroyed.
This selection of examples shows the lack of fundamental GMP principles which leads to a blatant misconduct of staff and ultimately to quality defects in the final product. The main responsibility usually has the quality unit, which task it actually would be to ensure a thorough training in production and quality control and to monitor compliance with the appropriate regulations. These examples of non-GMP-compliant behavior are not limited to active ingredient manufacturers; there are very similar findings in Warning Letters issued to medicinal product manufacturers. An analysis of these Warning Letters issued in the fiscal year 2015 will be part of one the coming newsletters.

/////Warning Letters, FDA, active ingredient manufacturers
