
Home » Posts tagged 'FDA 2016' (Page 2)
Tag Archives: FDA 2016
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves new medication for dry eye disease, Xiidra (lifitegrast ophthalmic solution)

lifitegrast
Xiidra (lifitegrast ophthalmic solution)

FDA approves new medication for dry eye disease
July 12, 2016
Release
The U.S. Food and Drug Administration approved Xiidra (lifitegrast ophthalmic solution) for the treatment of signs and symptoms of dry eye disease, on Monday, July 11, 2016. Xiidra is the first medication in a new class of drugs, called lymphocyte function-associated antigen 1 (LFA-1) agonist, approved by the FDA for dry eye disease.
“Normal tear production is needed for clear vision and eye health,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “This approval will provide a new treatment option for patients with dry eye disease.”
Dry eye disease includes a group of conditions in which the eye does not produce an adequate volume of tears or when the tears are not of the correct consistency. The chance of experiencing dry eye increases with age, affecting approximately five percent of the adult population age 30-40 and 10 to 15 percent of adults over age 65, and is more common among women. When severe and left untreated, this condition can lead to pain, ulcers or scars on the part of the eye called the cornea. Dry eye can make it more difficult to perform some activities, such as using a computer or reading for an extended period of time, and it can decrease tolerance for dry environments, such as the air inside an airplane.
The safety and efficacy of Xiidra was assessed in over a thousand patients, in four separate, randomized, controlled studies. These studies included patients 19–97 years of age, of which the majority were female (76 percent). Patients were randomized equally to receive either Xiidra eyedrops or placebo eyedrops, which were used twice a day for twelve weeks. The studies found that groups treated with Xiidra demonstrated more improvement in both the signs and the symptoms of eye dryness than the groups treated with placebo.
The most common side effects of Xiidra include eye irritation, discomfort or blurred vision and an unusual taste sensation (dysgeusia).
Dry eye disease does not routinely occur in children. Safety and efficacy in pediatric patients below the age of 17 years has not been studied.
Xiidra is manufactured by Shire US Inc., of Lexington, Massachusetts.
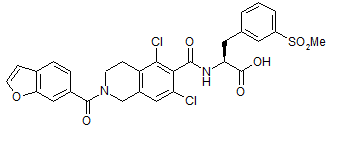
Common name: Lifitegrast, SAR 1118
Molecular Formula: C29H24Cl2N2O7S
CAS Registry Number: 1025967-78-5
Molecular Weight: 615.49
Activity: SAR 1118 is a potent novel small molecule lymphocyte function-associated antigen-1 (LFA-1/ICAM-1) antagonist for a broad range of ocular inflammatory conditions including dry eye and diabetic macular edema.
Intermediates:

FDA approves new diagnostic imaging agent FLUCICLOVINE F-18 to detect recurrent prostate cancer

FLUCICLOVINE F-18
Cyclobutanecarboxylic acid, 1-amino-3-(fluoro-18F)-, trans- [
- Molecular FormulaC5H818FNO2
- Average mass132.124 Da
May 27, 2016
Release
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
Prostate cancer is the second leading cause of death from cancer in U.S. men. In patients with suspected cancer recurrence after primary treatment, accurate staging is an important objective in improving management and outcomes.
“Imaging tests are not able to determine the location of the recurrent prostate cancer when the PSA is at very low levels,” said Libero Marzella, M.D., Ph.D., director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research. “Axumin is shown to provide another accurate imaging approach for these patients.”
Two studies evaluated the safety and efficacy of Axumin for imaging prostate cancer in patients with recurrent disease. The first compared 105 Axumin scans in men with suspected recurrence of prostate cancer to the histopathology (the study of tissue changes caused by disease) obtained by prostate biopsy and by biopsies of suspicious imaged lesions. Radiologists onsite read the scans initially; subsequently, three independent radiologists read the same scans in a blinded study.
The second study evaluated the agreement between 96 Axumin and C11 choline (an approved PET scan imaging test) scans in patients with median PSA values of 1.44 ng/mL. Radiologists on-site read the scans, and the same three independent radiologists who read the scans in the first study read the Axumin scans in this second blinded study. The results of the independent scan readings were generally consistent with one another, and confirmed the results of the onsite scan readings. Both studies supported the safety and efficacy of Axumin for imaging prostate cancer in men with elevated PSA levels following prior treatment.
Axumin is a radioactive drug and should be handled with appropriate safety measures to minimize radiation exposure to patients and healthcare providers during administration. Image interpretation errors can occur with Axumin PET imaging. A negative image does not rule out the presence of recurrent prostate cancer and a positive image does not confirm the presence of recurrent prostate cancer. Clinical correlation, which may include histopathological evaluation of the suspected recurrence site, is recommended.
The most commonly reported adverse reactions in patients are injection site pain, redness, and a metallic taste in the mouth.
Axumin is marketed by Blue Earth Diagnostics, Ltd., Oxford, United Kingdom
Patent
http://www.google.com/patents/WO2014023775A1?cl=en
The non-natural amino acid [ F]-l-amino-3-fluorocyclobutane-l-carboxylic acid
([18F]-FACBC, also known as [18F]-Fluciclovine) is taken up specifically by amino acid transporters and has shown promise for tumour imaging with positron emission tomography (PET).
A known synthesis of [18F]-FACBC begins with the provision of the protected precursor compound 1 -(N-(t-butoxycarbonyl)amino)-3 –
[((trifluoromethyl)sulfonyl)oxy]-cyclobutane-l-carboxylic acid ethyl ester. This precursor compound is first labelled with [18F]-fluoride:

II before removal of the two protecting groups:

IT III
EP2017258 (Al) teaches removal of the ethyl protecting group by trapping the [18F]- labelled precursor compound (II) onto a solid phase extraction (SPE) cartridge and incubating with 0.8 mL of a 4 mol/L solution of sodium hydroxide (NaOH). After 3 minutes incubation the NaOH solution was collected in a vial and a further 0.8 mL 4 mol/L NaOH added to the SPE cartridge to repeat the procedure. Thereafter the SPE cartridge was washed with 3 mL water and the wash solution combined with the collected NaOH solution. Then 2.2 mL of 6 mol/L HCl was then added with heating to 60°C for 5 minutes to remove the Boc protecting group. The resulting solution was purified by passing through (i) an ion retardation column to remove Na+ from excess NaOH and Cl~ from extra HCl needed to neutralise excess of NaOH to get a highly acidic solution before the acidic hydrolysis step, (ii) an alumina column, and (iii) a reverse-phase column. There is scope for the deprotection step(s) and/or the
purification step in the production of [18F]-FACBC to be simplified.
Example 1: Synthesis of f FIFACBC
No-carrier- added [18F]fluoride was produced via the 180(p,n)18F nuclear reaction on a GE PETtrace 6 cyclotron (Norwegian Cyclotron Centre, Oslo). Irradiations were performed using a dual-beam, 30μΑ current on two equal Ag targets with HAVAR foils using 16.5 MeV protons. Each target contained 1.6 ml of > 96% [180]water (Marshall Isotopes). Subsequent to irradiation and delivery to a hotcell, each target was washed with 1.6 ml of [160]water (Merck, water for GR analysis), giving approximately 2-5 Gbq in 3.2 ml of [160]water. All radiochemistry was performed on a commercially available GE FASTlab™ with single-use cassettes. Each cassette is built around a one-piece-moulded manifold with 25 three-way stopcocks, all made of polypropylene. Briefly, the cassette includes a 5 ml reactor (cyclic olefin copolymer), one 1 ml syringe and two 5 ml syringes, spikes for connection with five prefilled vials, one water bag (100 ml) as well as various SPE cartridges and filters. Fluid paths are controlled with nitrogen purging, vacuum and the three syringes. The fully automated system is designed for single-step fluorinations with cyclotron-produced [18F]fluoride. The FASTlab was programmed by the software package in a step-by-step time-dependent sequence of events such as moving the syringes, nitrogen purging, vacuum, and temperature regulation. Synthesis of
[18F]FACBC followed the three general steps: (a) [18F]fluorination, (b) hydrolysis of protection groups and (c) SPE purification.
Vial A contained K222 (58.8 mg, 156 μπιοΐ), K2C03 (8.1 mg, 60.8 μπιοΐ) in 79.5% (v/v)
MeCN(aq) (1105 μΐ). Vial B contained 4M HC1 (2.0 ml). Vial C contained MeCN
(4.1ml). Vial D contained the precursor (48.4 mg, 123.5 μιηοΐ) in its dry form (stored at -20 °C until cassette assembly). Vial E contained 2 M NaOH (4.1 ml). The 30 ml product collection glass vial was filled with 200 mM trisodium citrate (10 ml). Aqueous
[18F]fluoride (1-1.5 ml, 100-200 Mbq) was passed through the QMA and into the 180-
H20 recovery vial. The QMA was then flushed with MeCN and sent to waste. The trapped [18F]fluoride was eluted into the reactor using eluent from vial A (730 μΐ) and then concentrated to dryness by azeotropic distillation with acetonitrile (80 μΐ, vial C). Approximately 1.7 ml of MeCN was mixed with precursor in vial D from which 1.0 ml of the dissolved precursor (corresponds to 28.5 mg, 72.7 mmol precursor) was added to the reactor and heated for 3 min at 85°C. The reaction mixture was diluted with water and sent through the tC18 cartridge. Reactor was washed with water and sent through the tC18 cartridge. The labelled intermediate, fixed on the tC18 cartridge was washed with water, and then incubated with 2M NaOH (2.0 ml) for 5 min after which the 2M NaOH was sent to waste. The labelled intermediate (without the ester group) was then eluted off the tC18 cartridge into the reactor using water. The BOC group was hydrolysed by adding 4M HC1 (1.4 ml) and heating the reactor for 5 min at 60 °C. The reactor content with the crude [18F]FACBC was sent through the HLB and Alumina cartridges and into the 30 ml product vial. The HLB and Alumina cartridges were washed with water (9.1 ml total) and collected in the product vial. Finally, 2M NaOH (0.9 ml) and water (2.1 ml) was added to the product vial, giving a purified formulation of [18F]FACBC with a total volume of 26 ml. Radiochemical purity was measured by radio-TLC using a mixture of MeCN:MeOH:H20:CH3COOH (20:5:5: 1) as the mobile phase. The radiochemical yield (RCY) was expressed as the amount of radioactivity in the [18F]FACBC fraction divided by the total used [18F]fluoride activity (decay corrected). Total synthesis time was 43 min.
The RCY of [18F]FACBC was 62.5% ± 1.93 (SD), n=4.
/////FDA, diagnostic imaging agent, recurrent prostate cancer, fda 2016, Axumin, marketed, Blue Earth Diagnostics, Ltd., Oxford, United Kingdom, fluciclovine F 18
C1[C@@](C[C@H]1[18F])(N)C(=O)O
UPDATE
![]()
SEE EMA
| Axumin : EPAR – Summary for the public | EN = English | 06/07/2017 |
The active substance fluciclovine (18F) is prepared from the precursor AH113487 by nucleophilic substitution
of a triflate group by 18F-fluoride, followed by two deprotection steps. Due to the short half-life of the 18Ffluorine
radioisotope, each batch is prepared on the day of clinical use.
The active substance is prepared in a proprietary automated synthesiser unit. The synthesiser module is
computer-controlled. A fluid path for synthesis is provided in the form of a single use cassette (FASTlab). The
cassette contains 3 reagent vials and 3 solid phase cartridges. Two other reagent vials are supplied
separately as they have a recommended storage temperature of 2-8°C. These 2 vials are inserted into the
cassette on the day of production.
Assessment report
EMA/237809/2017 Page 13/90
Fluciclovine (18F) is produced in a continuous operation from the precursor AH113487. Due to the radioactive
nature of the process, and the short half-life of [18F] fluorine, intermediates are not isolated and there is no
opportunity for operator intervention or in-process testing. Control of the synthesis of fluciclovine (18F) from
the precursor is achieved through the automated synthesis platform, which is pre-programmed with
synthesis parameters optimised for the process. On-board detectors record transfers of radioactivity through
the fluid path at critical points and monitor temperature and pressure as appropriate so that the operator
may track the progress of the synthesis.
The active substance fluciclovine (18F) progressses immediately to purification, formulation and dispensing as
the finished product within a single, continuous operation. Validation of the manufacturing process for
fluciclovine (18F) is therefore described as part of finished product validation.
The characterisation of the active substance is in accordance with the EU guideline on chemistry of new
active substances.
As mentioned, the manufacture of the active substance and finished product takes place in a single,
continuous process. The active substance is not isolated at any point. Therefore, relevant information about
impurities is given only for the finished product.
For the same reason, information for the container closure system is provided only for the finished product.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004197/WC500230836.pdf
Albutrepenonacog alfa
Albutrepenonacog alfa
recombinant factor IX
(Idelvion®)Approved, 2016-03-04 USFDA
A recombinant albumin-human coagulation factor IX (FIX) fusion protein indicated for the treatment and prevention of bleeding in patients with hemophilia B.
![]()
Research Code CSL-654
| Type | Recombinant coagulation factor |  |
| Source | Human | |
| Molecular Formula | C5077H7846N1367O1588S67 | |
| Molecular Weight | ~125000 |
Other Names
- Albutrepenonacog alfa
Protein Sequence
Sequence Length: 1018modified (modifications unspecified)
- Originator CSL Behring
- Class Albumins; Antihaemorrhagics; Blood coagulation factors; Recombinant fusion proteins
- Mechanism of Action Blood coagulation factor replacements; Factor X stimulants
- Orphan Drug Status Yes – Haemophilia B
- Marketed Haemophilia B
Most Recent Events
- 21 Mar 2016 Launched for Haemophilia B (In adolescents, In children, In adults) in USA (IV) – First global launch
- 07 Mar 2016 Preregistration for Haemophilia B in Australia (IV) before March 2016
- 04 Mar 2016 Registered for Haemophilia B (In children, In adolescents, In adults) in USA (IV)
| Latest Stage of Development | Approved |
| Standard Indication | Hemophilia |
| Indication Details | Treat and prevent bleeding episodes in hemophilia B patients; Treat hemophilia B |
| Regulatory Designation | U.S. – Orphan Drug (Treat and prevent bleeding episodes in hemophilia B patients); EU – Orphan Drug (Treat and prevent bleeding episodes in hemophilia B patients); Switzerland – Orphan Drug (Treat and prevent bleeding episodes in hemophilia B patients) |
-
BNF Category:Antifibrinolytic drugs and haemostatics (02.11)
Pharmacology: Albutrepenonacog alfa is a recombinant factor IX (rIX-FP) albumin fusion protein, designed to exhibit an extended half-life. Factor IX has a short half-life which necessitates multiple injections. Epidemiology: Haemophilia B is a genetic disorder caused by missing or defective factor IX, a clotting protein. It has a prevalence of around 1 in 50,000 live births in the UK and is more common in males. In 2012-13, there were 476 hospital admissions in England due to haemophilia B, accounting for 508 finished consultant episodes and 125 bed days. Indication: Haemophilia B

Albutrepenonacog alfa was approved by the U.S. Food and Drug Administration (FDA) on March 4, 2016. It was developed and marketed as Idelvion® by CSL Behring.
Albutrepenonacog alfa is a recombinant albumin-human coagulation factor IX (FIX) fusion protein, which replaces the missing FIX needed for effective hemostasis. It is indicated for the treatment and prevention of bleeding in children and adults with hemophilia B.
Idelvion® is available as injection (lyophilized powder) for intravenous use, containing 250 IU, 500 IU, 1000 IU or 2000 IU of albutrepenonacog alfa in single-use vials. In control and prevention of bleeding episodes and perioperative management, the required dosage is determined using the following formulas: Required Dose (IU) = Body Weight (kg) x Desired Factor IX rise (% of normal or IU/dL) x (reciprocal of recovery (IU/kg per IU/dL)). In routine prophylaxis, the recommended dose is 25-40 IU/kg (for patients ≥12 years of age) or 40-55 IU/kg (for patients <12 years of age) every 7 days.
On 25 February 2016, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product IDELVION, intended for treatment and prophylaxis of bleeding in patients with Haemophilia B. IDELVION was designated as an orphan medicinal producton 04 February 2010. The applicant for this medicinal product is CSL Behring GmbH.
IDELVION will be available as 250 IU, 500 IU, 1000 IU and 2000 IU Powder and solvent for solution for injection. The active substance of IDELVION is albutrepenonacog alfa, an antihaemorrhagic, blood coagulation factor IX, (ATC code: B02BD04). It works as replacement therapy and temporarily increases plasma levels of factor IX, helping to prevent and control bleeding.
The benefits with IDELVION are its ability to stop the bleeding when given on demand and prevent bleeding when used as routine prophylaxis or for surgical procedures. The most common side effects are injection site reaction and headache.
The full indication is: “the treatment and prophylaxis of bleeding in patients with Haemophilia B (congenital factor IX deficiency)”. Idelvion can be used in all age groups. It is proposed that IDELVION be prescribed by physicians experienced in the treatment of haemophilia B.
Detailed recommendations for the use of this product will be described in the summary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission.
| Name | Idelvion |
|---|---|
| INN or common name | albutrepenonacog alfa |
| Therapeutic area | Hemophilia B |
| Active substance | albutrepenonacog alfa |
| Date opinion adopted | 25/02/2016 |
| Company name | CSL Behring GmbH |
| Status | Positive |
| Application type | Initial authorisation |
//////Albutrepenonacog alfa, CSL-654, Idelvion; Recombinant factor IX – CSL Behring, Recombinant factor IX fusion protein linked with human albumin, rFIX-FP – CSL Behring; rIX-FP, Orphan Drug Status, Haemophilia B, recombinant factor IX , FDA 2016
update
| Human medicines European public assessment report (EPAR): Idelvion, albutrepenonacog alfa, Hemophilia B, 11/05/2016, |
Reslizumab


Reslizumab
(Cinqair®) Approved Active, FDA 2016-03-23
An interleukin-5 (IL-5) antagonist used to treat severe asthma.
CAS 241473-69-8
![]()
Research Code CDP-835; CEP-38072; CTx-55700; SCH-5570; SCH-55700; TRFK-5,

Anti-interleukin-5 monoclonal antibody – Celltech/Schering-Plough
Reslizumab was approved by the U.S. Food and Drug Administration (FDA) on March 23, 2016. It was developed and marketed as Cinqair® by Teva.
Reslizumab is an interleukin-5 antagonist, which binds to human IL-5 and prevents it from binding to the IL-5 receptor, thereby reducing eosinophilic inflammation. It is indicated for the maintenance treatment of patients with severe asthma in patients aged 18 years and older.
Cinqair® is available as injection for intravenous infusion, containing 100 mg of reslizumab in 10 mL solution in single-use vials. The recommended dose is 3 mg/kg once every four weeks.
- Originator Celltech R&D; Schering-Plough
- Developer Celltech R&D; Teva Pharmaceutical Industries
- Class Antiasthmatics; Monoclonal antibodies
- Mechanism of Action Interleukin 5 receptor antagonists
- Orphan Drug Status Yes – Oesophagitis
- 23 Mar 2016 Registered for Asthma in USA (IV) – First global approval
- 04 Mar 2016 Pooled efficacy data from two phase III trials in Asthma presented at the 2016 Annual Meeting of the American Academy of Allergy, Asthma and Immunology (AAAAI-2016)
- 10 Dec 2015 Preregistration for Asthma in Canada (IV)
Reslizumab (trade name Cinqair) is a humanized monoclonal antibody intended for the treatment of eosinophil-meditated inflammations of the airways, skin and gastrointestinal tract.[1] The FDA approved reslizumab for use with other asthma medicines for the maintenance treatment of severe asthma in patients aged 18 years and older on March 23, 2016. Cinqair is approved for patients who have a history of severe asthma attacks (exacerbations) despite receiving their current asthma medicines.[2]

Teva Announces FDA Acceptance of the Biologics License Application for Reslizumab
Investigational Biologic for the Treatment of Inadequately Controlled Asthma in Patients with Elevated Blood Eosinophils Accepted for Review
JERUSALEM–(BUSINESS WIRE)–Jun. 15, 2015– Teva Pharmaceutical Industries Ltd., (NYSE: TEVA) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the Biologics License Application (BLA) for reslizumab, the company’s investigational humanized monoclonal antibody (mAb) which targets interleukin-5 (IL-5), for the treatment of inadequately controlled asthma in adult and adolescent patients with elevated blood eosinophils, despite an inhaled corticosteroid (ICS)-based regimen.
“Despite currently available medicines, uncontrolled asthma remains a serious problem for patients, physicians and healthcare systems, highlighting the need for targeted new treatment options,” said Dr. Michael Hayden, President of Global R&D and Chief Scientific Officer at Teva Pharmaceutical Industries Ltd. “The reslizumab BLA filing acceptance represents a significant milestone for Teva as we work toward serving a specific asthma patient population that is defined by elevated blood eosinophil levels and inadequately controlled symptoms despite standard of care therapy. In clinical trials, patients treated with reslizumab showed significant reductions in the rate of asthma exacerbations and significant improvement in lung function. If approved, we believe reslizumab will serve as an important new targeted treatment option to achieve better asthma control for patients with eosinophil-mediated disease.”
The BLA for reslizumab includes data from Teva’s Phase III BREATH clinical trial program. The program consisted of four separate placebo-controlled Phase III trials involving more than 1,700 adult and adolescent asthma patients with elevated blood eosinophils, whose symptoms were inadequately controlled with inhaled corticosteroid-based therapies. Results from these studies demonstrated that reslizumab, in comparison to placebo, reduced asthma exacerbation rates by at least half and provided significant improvement in lung function and other secondary measures of asthma control when added to an existing ICS-based therapy. Common adverse events in the reslizumab treatment group were comparable to placebo and included worsening of asthma, nasopharyngitis, upper respiratory infections, sinusitis, influenza and headache. Two anaphylactic reactions were reported and resolved following medical treatment at the study site.
Results from the reslizumab BREATH program were recently presented at the American Thoracic Society 2015 Annual Meeting and the American Academy of Allergy, Asthma and Immunology 2015 Annual Meeting, in addition to being published in The Lancet Respiratory Medicine. The BLA for reslizumab has been accepted for filing by the FDA for standard review, with FDA Regulatory Action expected in March 2016.
About Reslizumab
Reslizumab is an investigational humanized monoclonal antibody which targets interleukin-5 (IL-5). IL-5 is a key cytokine involved in the maturation, recruitment, and activation of eosinophils, which are inflammatory white blood cells implicated in a number of diseases, such as asthma. Elevated levels of blood eosinophils are a risk factor for future asthma exacerbations. Reslizumab binds circulating IL-5 thereby preventing IL-5 from binding to its receptor.
About Asthma
Asthma is a chronic (long term) disease usually characterized by airway inflammation and narrowing of the airways, which can vary over time. Asthma may cause recurring periods of wheezing (a whistling sound when you breathe), chest tightness, shortness of breath and coughing that often occurs at night or early in the morning. Without appropriate treatment, asthma symptoms may become more severe and result in an asthma attack, which can lead to hospitalization and even death.
About Eosinophils
Eosinophils are a type of white blood cell that are present at elevated levels in the lungs and blood of many asthmatics. Evidence shows that eosinophils play an active role in the pathogenesis of the disease. IL-5 has been shown to play a crucial role in maturation, growth and activation of eosinophils. Increased levels of eosinophils in the sputum and blood have been shown to correlate with severity and frequency of asthma exacerbations.
About Teva
Teva Pharmaceutical Industries Ltd. (NYSE and TASE: TEVA) is a leading global pharmaceutical company that delivers high-quality, patient-centric healthcare solutions to millions of patients every day. Headquartered in Israel, Teva is the world’s largest generic medicines producer, leveraging its portfolio of more than 1,000 molecules to produce a wide range of generic products in nearly every therapeutic area. In specialty medicines, Teva has a world-leading position in innovative treatments for disorders of the central nervous system, including pain, as well as a strong portfolio of respiratory products. Teva integrates its generics and specialty capabilities in its global research and development division to create new ways of addressing unmet patient needs by combining drug development capabilities with devices, services and technologies. Teva’s net revenues in 2014 amounted to $20.3 billion. For more information, visit www.tevapharm.com.
The U.S. Food and Drug Administration today approved Cinqair (reslizumab) for use with other asthma medicines for the maintenance treatment of severe asthma in patients aged 18 years and older. Cinqair is approved for patients who have a history of severe asthma attacks (exacerbations) despite receiving their current asthma medicines.
Asthma is a chronic disease that causes inflammation in the airways of the lungs. During an asthma attack, airways become narrow making it hard to breathe. Severe asthma attacks can lead to asthma-related hospitalizations because these attacks can be serious and even life-threatening. According to the Centers for Disease Control and Prevention, as of 2013, more than 22 million people in the U.S. have asthma, and there are more than 400,000 asthma-related hospitalizations each year.
“Health care providers and their patients with severe asthma now have another treatment option to consider when the disease is not well controlled by their current asthma therapies,” said Badrul Chowdhury, M.D., Ph.D., director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Cinqair is administered once every four weeks via intravenous infusion by a health care professional in a clinical setting prepared to manage anaphylaxis. Cinqair is a humanized interleukin-5 antagonist monoclonal antibody produced by recombinant DNA technology in murine myeloma non-secreting 0 (NS0) cells. Cinqair reduces severe asthma attacks by reducing the levels of blood eosinophils, a type of white blood cell that contributes to the development of asthma.
The safety and efficacy of Cinqair were established in four double-blind, randomized, placebo‑controlled trials in patients with severe asthma on currently available therapies. Cinqair or a placebo was administered to patients every four weeks as an add-on asthma treatment. Compared with placebo, patients with severe asthma receiving Cinqair had fewer asthma attacks, and a longer time to the first attack. In addition, treatment with Cinqair resulted in a significant improvement in lung function, as measured by the volume of air exhaled by patients in one second.
Cinqair can cause serious side effects including allergic (hypersensitivity) reactions. These reactions can be life-threatening. The most common side effects in clinical trials for Cinqair included anaphylaxis, cancer, and muscle pain.
Cinqair is made by Teva Pharmaceuticals in Frazer, Pennsylvania.


References
- 1Walsh, GM (2009). “Reslizumab, a humanized anti-IL-5 mAb for the treatment of eosinophil-mediated inflammatory conditions”. Current opinion in molecular therapeutics 11 (3): 329–36. PMID 19479666.
- 2http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491980.htm
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491980.htm
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from rat) |
| Target | IL-5 |
| Clinical data | |
| Trade names | Cinquil |
| Identifiers | |
| ATC code | R03DX08 (WHO) |
| ChemSpider | none |
/////////CDP-835, CEP-38072, CTx-55700, SCH-5570, SCH-55700, TRFK-5, Reslizumab, Cinqair®, teva, interleukin-5 (IL-5) antagonist, severe asthma, FDA 2016, Orphan Drug StatuS
FDA approves new drug Venclexta (venetoclax) for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality

April 11, 2016
Release
The U.S. Food and Drug Administration today approved Venclexta (venetoclax) for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a chromosomal abnormality called 17p deletion and who have been treated with at least one prior therapy. Venclexta is the first FDA-approved treatment that targets the B-cell lymphoma 2 (BCL-2) protein, which supports cancer cell growth and is overexpressed in many patients with CLL.
According to the National Cancer Institute, CLL is one of the most common types of leukemia in adults, with approximately 15,000 new cases diagnosed each year. CLL is characterized by the progressive accumulation of abnormal lymphocytes, a type of white blood cell. Patients with CLL who have a 17p deletion lack a portion of the chromosome that acts to suppress cancer growth. This chromosomal abnormality occurs in approximately 10 percent of patients with untreated CLL and in approximately 20 percent of patients with relapsed CLL.
“These patients now have a new, targeted therapy that inhibits a protein involved in keeping tumor cells alive,” said Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option for their specific condition.”
The efficacy of Venclexta was tested in a single-arm clinical trial of 106 patients with CLL who have a 17p deletion and who had received at least one prior therapy. Trial participants took Venclexta orally every day, beginning with 20 mg and increasing over a five-week period to 400 mg. Results showed that 80 percent of trial participants experienced a complete or partial remission of their cancer.
Venclexta is indicated for daily use after detection of 17p deletion is confirmed through the use of the FDA-approved companion diagnostic Vysis CLL FISH probe kit.
The most common side effects of Venclexta include low white blood cell count (neutropenia), diarrhea, nausea, anemia, upper respiratory tract infection, low platelet count (thrombocytopenia) and fatigue. Serious complications can include pneumonia, neutropenia with fever, fever, autoimmune hemolytic anemia, anemia and metabolic abnormalities known as tumor lysis syndrome. Live attenuated vaccines should not be given to patients taking Venclexta.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. These are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions. Venclexta also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Venclexta is manufactured by AbbVie Inc. of North Chicago, Illinois, and marketed by AbbVie and Genentech USA Inc. of South San Francisco, California. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular of Des Plaines, Illinois.
Defibrotide


Defibrotide sodium is an oligonucleotide mixture with profibrinolytic properties. The chemical name of defibrotide sodium is polydeoxyribonucleotide, sodium salt. Defibrotide sodium is a polydisperse mixture of predominantly single-stranded (ss) polydeoxyribonucleotide sodium salts derived from porcine intestinal tissue having a mean weighted molecular weight of 13-20 kDa, and a potency of 27-39 and 28-38 biological units per mg as determined by two separate assays measuring the release of a product formed by contact between defibrotide sodium, plasmin and a plasmin substrate. The primary structure of defibrotide sodium is shown below.
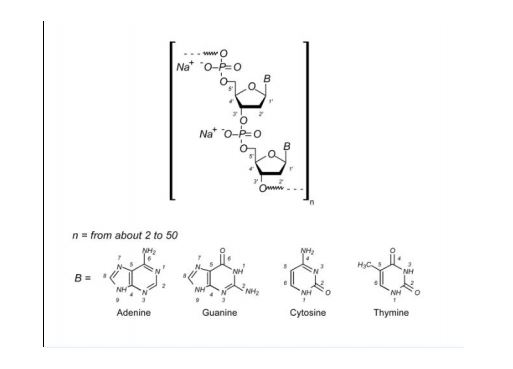
DEFITELIO (defibrotide sodium) injection is a clear, light yellow to brown, sterile, preservative-free solution in a single-patient-use vial for intravenous use. Each milliliter of the injection contains 80 mg of defibrotide sodium and 10 mg of Sodium Citrate, USP, in Water for Injection, USP. Hydrochloric Acid, NF, and/or Sodium Hydroxide, NF, may have been used to adjust pH to 6.8-7.8.
Defibrotide is the sodium salt of a mixture of single-stranded oligodeoxyribonucleotides derived from porcine mucosal DNA. It has been shown to have antithrombotic, anti-inflammatory and anti-ischemic properties (but without associated significant systemic anticoagulant effects). It is marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries, but is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide is used to treat or prevent a failure of normal blood flow (occlusive venous disease, OVD) in the liver of patients who have had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others.
In 2012, an IND was filed in Japan seeking approval of the compound for the treatment of veno-occlusive disease.
Approved 3/30/3016 US FDA, defibrotide sodium, (NDA) 208114

To treat adults and children who develop hepatic veno-occlusive disease with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation
Polydeoxyribonucleotides from bovine lung or other mamalian organs with molecular weight between 15,000 and 30,000 Da
CAS 83712-60-1
Defibrotide is a polydisperse mixture of oligonucleotides produced by random, chemical cleavage (depolymerisation) of porcine DNA. It is predominantly single stranded, of varying base sequence, lengths and conformations; unfolded, folded or combined. The mean oligonucleotide length is 50 bases with a mean molecular weight of 17 ± 4 kDa. No individually defined component is at more than femtomolar concentration. The only meaningful scientific information that can be obtained about the biochemical nature of defibrotide (aside from determination of percentage of each nucleobase) is a measurement of its average length and its average percentage double stranded character. Therefore, it can be established that this active substance is of highly heterogenic nature.

Defibrotide (Defitelio, Gentium)[1] is a deoxyribonucleic acid derivative (single-stranded) derived from cow lung or porcine mucosa. It is an anticoagulant with a multiple mode of action (see below).
It has been used with antithrombin III.[2]
Jazz Pharmaceuticals plc announced that the FDA has accepted for filing with Priority Review its recently submitted New Drug Application (NDA) for defibrotide. AS ON OCT 2015
Defibrotide is an investigational agent proposed for the treatment of patients with hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), with evidence of multi-organ dysfunction (MOD) following hematopoietic stem-cell transplantation (HSCT).
Priority Review status is designated for drugs that may offer major advances in treatment or provide a treatment where no adequate therapy exists. Based on timelines established by the Prescription Drug User Fee Act (PDUFA), FDA review of the NDA is expected to be completed by March 31, 2016.
“The FDA’s acceptance for filing and Priority Review status of the NDA for defibrotide is an important milestone for Jazz and reflects our commitment to bringing meaningful medicines to patients who have significant unmet needs,” said Karen Smith, M.D., Ph.D., Global Head of Research and Development and Chief Medical Officer of Jazz Pharmaceuticals. “We look forward to continuing to work closely with the FDA to obtain approval for defibrotide for patients with hepatic VOD with evidence of MOD in the U.S. as quickly as possible, as there are no other approved therapies for treating this rare, often fatal complication of HSCT.”
The NDA includes safety and efficacy data from three clinical studies of defibrotide for the treatment of hepatic VOD with MOD following HSCT, as well as a retrospective review of registry data from the Center for International Blood and Marrow Transplant Research. The safety database includes over 900 patients exposed to defibrotide in the clinical development program for the treatment of hepatic VOD.
The compound was originally developed under a collaboration between Sanofi and Gentium. In December 2001, Gentium entered into a license and supply agreement with Sigma-Tau Pharmaceuticals, pursuant to which the latter gained exclusive rights to distribute, market and sell the product for the treatment of VOD in the U.S. This agreement was expanded in 2005 to include all of North America, Central America and South America.
Defibrotide was granted orphan drug designations from the FDA in July 1985, May 2003 and January 2007 for the treatment of thrombotic thrombocytopenic purpura (TTP), for the treatment of VOD and for the prevention of VOD, respectively. Orphan drug was also received in the E.U. for the prevention and treatment of hepatic veno-occlusive disease (VOD) in 2004 and for the prevention of graft versus host disease (GvHD) in 2013.
Pharmacokinetics
Defibrotide is available as an oral, intravenous, and intramuscular formulation. Its oral bioavailability is in the range of 58-70% of theparenteral forms. T1/2 alpha is in the range of minutes while T1/2 beta is in the range of hours in studies with oral radiolabelleddefibrotide. These data suggest that defibrotide, in spite of its macromolecular nature, is absorbed well after oral administration. Due to the drug’s short half-life, it is necessary to give the daily dose divided in 2 to 4 doses (see below).
In 2014, Jazz Pharmaceuticals (parent of Gentium) acquired the rights of the product in U.S. and in the Americas
Mode of action
The drug appears to prevent the formation of blood clots and to help dissolve blood clots by increasing levels of prostaglandin I2, E2, and prostacyclin, altering platelet activity, increasing tissue plasminogen activator (tPA-)function, and decreasing activity of tissue plasminogen activator inhibitor. Prostaglandin I2 relaxes the smooth muscle of blood vessels and prevents platelets from adhering to each other. Prostaglandin E2 at certain concentrations also inhibits platelet aggregation. Moreover, the drug provides additional beneficial anti-inflammatory and antiischemic activities as recent studies have shown. It is yet unclear, if the latter effects can be utilized clinically (e.g., treatment of ischemic stroke).
Unlike heparin and warfarin, defibrotide appears to have a relatively mild anticoagulant activity, which may be beneficial in the treatment of patients at high risk of bleeding complications. Nevertheless, patients with known bleeding disorders (e.g., hemophilia A) or recent abnormal bleedings should be treated cautiously and under close medical supervision.
The drug was marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries. It is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide also received fast track designation from the FDA for the treatment of severe VOD in recipients of stem cell transplants. In 2011, the compound was licensed to Medison Pharma by Gentium in Israel and Palestine. The license covers the management of named-patient sales program and local registration, authorization, marketing, reimbursement and medical affairs for the treatment of peripheral vascular disease.
Usual indications
Defibrotide is used to treat or prevent a failure of normal blood flow (Veno-occlusive disease, VOD) in the liver of patients having had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others. Without intensive treatment, VOD is often a fatal condition, leading to multiorgan failure. It has repeatedly been reported that defibrotide was able to resolve the condition completely and was well tolerated.
Other indications are: peripheral obliterative arterial disease, thrombophlebitis, and Raynaud’s phenomenon. In very high doses, defibrotide is useful as treatment of acute myocardial infarction. The drug may also be used for the pre- and postoperative prophylaxis of deep venous thrombosis and can replace the heparin use during hemodialytic treatments.
It has been investigated for use in treatment of chronic venous insufficiency.[3]
Potential indications in the future
Other recent preclinical studies have demonstrated that defibrotide used in conjunction with Granulocyte Colony-Stimulating Factor (rhG-CSF) significantly increases the number of Peripheral Blood Progenitor Cells (Stem cells). The benefit of this increase in stem cells may be crucial for a variety of clinical indications, including graft engineering procedures and gene therapy programs. This would expand the clinical usefulness of defibrotide to a complete distinct area.
Very recently (since early 2006) combination therapy trials (phase I/II) with defibrotide plus melphalan, prednisone, and thalidomide in patients with multiple myeloma have been conducted. The addition of defibrotide is expected to decrease the myelosuppressive toxicity of melphalan. However, is too early for any definitive results at that stage.
Cautions and contraindications
- The efficacy of the drug has been reported to be poorer in patients with diabetes mellitus.
- Pregnancy: The drug should not be used during pregnancy, because adequate and well controlled human studies do not exist.
- Lactation: No human data is available. In order to avoid damage to the newborn, the nursing mother should discontinue either the drug or breastfeeding, taking into account the importance of treatment to the mother.
- Known Bleeding Disorders or Bleeding Tendencies having occurred recently: Defibrotide should be used cautiously. Before initiation of treatment, the usual coagulation values should be obtained as baseline and regularly controlled under treatment. The patient should be observed regularly regarding local or systemic bleeding events.
Side-effects
Increased bleeding and bruising tendency, irritation at the injection site, nausea, vomiting, heartburn, low blood pressure. Serious allergic reactions have not been observed so far.
Drug interactions
Use of heparin with defibrotide may increase the aPTT, reflecting reduced ability of the body to form a clot. Nothing is known about the concomitant application of other anticoagulants than heparin and dextran containing plasma-expanders, but it can be anticipated that the risk of serious bleeding will be increased considerably.
PATENT
WO 2001078761
G-CSF (CAS registry number 143011-2-7/Merck Index, 1996, page 4558) is a haematopoietic growth factor which is indispensable in the proliferation and differentiation of the progenitor cells of granulocytes; it is a 18-22 kDa glycoprotein normally produced in response to specific stimulation by a variety of cells, including monocytes, fibroblasts and endothelial cells. The term defibrotide (CAS registry number 83712-60-1) normally identifies a polydeoxyribonucleotide obtained by extraction (US 3,770,720 and US 3,899,481) from animal and/or vegetable tissue; this polydeoxyribonucleotide is normally used in the form of a salt of an alkali metal, generally sodium. Defibrotide is used principally for its anti- thrombotic activity (US 3,829,567) although it may be used in different applications, such as, for example, the treatment of acute renal insufficiency (US 4,694,134) and the treatment of acute myocardial ischaemia (US 4,693,995). United States patents US 4,985,552 and US 5,223,609, finally, describe a process for the production of defibrotide which enables a product to be obtained which has constant and well defined physico-chemical characteristics and is also free from any undesired side-effects
References
- “Jazz Pharma Acquiring Gentium for $1B”. Gen. Eng. Biotechnol. News (paper) 34 (2). January 15, 2014. p. 10.
- Haussmann U, Fischer J, Eber S, Scherer F, Seger R, Gungor T (June 2006). “Hepatic veno-occlusive disease in pediatric stem cell transplantation: impact of pre-emptive antithrombin III replacement and combined antithrombin III/defibrotide therapy”. Haematologica 91 (6): 795–800. PMID 16769582.
- Coccheri S, Andreozzi GM, D’Addato M, Gensini GF (June 2004). “Effects of defibrotide in patients with chronic deep insufficiency. The PROVEDIS study”. Int Angiol 23 (2): 100–7.PMID 15507885.
External links
- Palmer KJ, Goa KL. Defibrotide: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in vascular disorders. Drugs 1993;45:259-94.
- http://www.globalrx.com/medinfo/Defibrotide.htm
- Fisher J, Holland TK, Pescador R, Porta R, Ferro L (January 1996). “Study on pharmacokinetics of radioactive labelled defibrotide after oral or intravenous administration in rats”. Thromb. Res. 81 (1): 55–63. doi:10.1016/0049-3848(95)00213-8. PMID 8747520.
- http://www.gentium.it/Defibrotide.aspx (information provided by manufacturer)
- “Melphalan: profile and news”. Archived from the original on 2007-09-28. (on cytostatic combination therapy)
- Beşişik SK, Oztürk GB, Calişkan Y, Sargin D (March 2005). “Complete resolution of transplantation-associated thrombotic microangiopathy and hepatic veno-occlusive disease by defibrotide and plasma exchange”. Turk J Gastroenterol 16 (1): 34–7. PMID 16252186.
| WO2003101468A1 * | Jun 2, 2003 | Dec 11, 2003 | Guenther Eissner | Method for the protection of endothelial and epithelial cells during chemotherapy |
| US4985552 | Jul 5, 1989 | Jan 15, 1991 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| US5223609 | May 26, 1992 | Jun 29, 1993 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999026639A1 * | 24 Nov 1998 | 3 Jun 1999 | Allegheny University Of The He | Methods for mobilizing hematopoietic facilitating cells and hematopoietic stem cells into the peripheral blood |
| EP0317766A1 * | 20 Oct 1988 | 31 May 1989 | Crinos Industria Farmacobiologica S.p.A. | A method for preventing blood coaguli from being formed in the extra-body circuit of dialysis apparatus and composition useful thereof |
| EP0416678A1 * | 10 Aug 1990 | 13 Mar 1991 | Crinos Industria Farmacobiologica S.p.A. | Topical compositions containing Defibrotide |
| US5199942 * | 26 Sep 1991 | 6 Apr 1993 | Immunex Corporation | Method for improving autologous transplantation |
| US5977083 * | 5 Jun 1995 | 2 Nov 1999 | Burcoglu; Arsinur | Method for using polynucleotides, oligonucleotides and derivatives thereof to treat various disease states |
| Reference | ||
|---|---|---|
| 1 | * | CARLO-STELLA, C. (1) ET AL: “Defibrotide significantly enhances peripheral blood progenitor cell mobilization induced by recombinant human granulocyte colony – stimulating factor ( rhG – CSF.” BLOOD, ( NOVEMBER 16, 2000 ) VOL. 96, NO. 11 PART 1, PP. 553A. PRINT. MEETING INFO.: 42ND ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY SAN FRANCISCO, CALIFORNIA, USA DECEMBER 01-05, 2000 AMERICAN SOCIETY OF HEMATOLOGY. , XP002176349 |
| 2 | * | GURSOY A: “PREPARATION, CHARACTERIZATION AND ANTI-INFLAMMATORY EFFECT OF DEFIBROTIDE LIPOSOMES” PHARMAZIE,DD,VEB VERLAG VOLK UND GESUNDHEIT. BERLIN, vol. 48, no. 7, 1 July 1993 (1993-07-01), pages 549-550, XP000372658 ISSN: 0031-7144 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2005017160A2 * | 12 Aug 2004 | 24 Feb 2005 | Childrens Hosp Medical Center | Mobilization of hematopoietic cells |
| WO2009115465A1 * | 13 Mar 2009 | 24 Sep 2009 | Gentium Spa | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| EP2103689A1 * | 19 Mar 2008 | 23 Sep 2009 | Gentium S.p.A. | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| US7417026 | 12 Aug 2004 | 26 Aug 2008 | Children’s Hospital Medical Center | Mobilization of hematopoietic cells |
| US7915384 | 5 Jan 2009 | 29 Mar 2011 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8242246 | 28 Feb 2011 | 14 Aug 2012 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8674075 | 13 Aug 2012 | 18 Mar 2014 | Children’s Medical Center Corporation | Chimeric peptides for the regulation of GTPases |
| US8980862 | 12 Nov 2010 | 17 Mar 2015 | Gentium S.P.A. | Defibrotide for use in prophylaxis and/or treatment of Graft versus Host Disease (GVHD) |
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
oral, i.m., i.v. |
| Pharmacokinetic data | |
| Bioavailability | 58 – 70% orally (i.v. and i.m. = 100%) |
| Biological half-life | t1/2-alpha = minutes; t1/2-beta = a few hours |
| Identifiers | |
| CAS Registry Number | 83712-60-1 |
| ATC code | B01AX01 |
| DrugBank | DB04932 |
| UNII | 438HCF2X0M |
| KEGG | D07423 |
///////////Approved, 3/30/3016, US FDA, defibrotide sodium, NDA 208114, FDA 2016
Updates……….
FDA approves first treatment for rare disease in patients who receive stem cell transplant from blood or bone marrow
For Immediate Release
March 30, 2016
Release
The U.S. Food and Drug Administration today approved Defitelio (defibrotide sodium) to treat adults and children who develop hepatic veno-occlusive disease (VOD) with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation (HSCT). This is the first FDA-approved therapy for treatment of severe hepatic VOD, a rare and life-threatening liver condition.
HSCT is a procedure performed in some patients to treat certain blood or bone marrow cancers. Immediately before an HSCT procedure, a patient receives chemotherapy. Hepatic VOD can occur in patients who receive chemotherapy and HSCT. Hepatic VOD is a condition in which some of the veins in the liver become blocked, causing swelling and a decrease in blood flow inside the liver, which may lead to liver damage. In the most severe form of hepatic VOD, the patient may also develop failure of the kidneys and lungs. Fewer than 2 percent of patients develop severe hepatic VOD after HSCT, but as many as 80 percent of patients who develop severe hepatic VOD do not survive.
“The approval of Defitelio fills a significant need in the transplantation community to treat this rare but frequently fatal complication in patients who receive chemotherapy and HSCT,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
The efficacy of Defitelio was investigated in 528 patients treated in three studies: two prospective clinical trials and an expanded access study. The patients enrolled in all three studies had a diagnosis of hepatic VOD with liver or kidney abnormalities after HSCT. The studies measured the percentage of patients who were still alive 100 days after HSCT (overall survival). In the three studies, 38 to 45 percent of patients treated with Defitelio were alive 100 days after HSCT. Based on published reports and analyses of patient-level data, the expected survival rates 100 days after HSCT would be 21 to 31 percent for patients with severe hepatic VOD who received only supportive care or interventions other than Defitelio.
The most common side effects of Defitelio include abnormally low blood pressure (hypotension), diarrhea, vomiting, nausea and nosebleeds (epistaxis). Serious potential side effects of Defitelio that were identified include bleeding (hemorrhage) and allergic reactions. Defitelio should not be used in patients who are having bleeding complications or who are taking blood thinners or other medicines that reduce the body’s ability to form clots.
The FDA granted the Defitelio application priority review status, which facilitates and expedites the development and review of certain drugs in light of their potential to benefit patients with serious or life-threatening conditions. Defitelio also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Defitelio is marketed by Jazz Pharmaceuticals based in Palo Alto, California

{kind=link}