
Home » Posts tagged 'drugs' (Page 10)
Tag Archives: drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
VINCRISTINE……..Chemistry, Isolation

VINCRISTINE
(3aR,3a1R,4R,5S,5aR,10bR)-methyl 4-acetoxy-3a-ethyl-9-((5S,7S,9S)-5-ethyl-5-hydroxy-9-(methoxycarbonyl)-2,4,5,6,7,8,9,10-octahydro-1H-3,7-methano[1]azacycloundecino[5,4-b]indol-9-yl)-6-formyl-5-hydroxy-8-methoxy-3a,3a1,4,5,5a,6,11,12-octahydro-1H-indolizino[8,1-cd]carbazole-5-carboxylate
…………………………………………………………..
………………………………………………………………….


Vincristine (brand name, Oncovin), formally known as leurocristine, sometimes abbreviated “VCR”, is a vinca alkaloid from the Catharanthus roseus (Madagascar periwinkle), formerly Vinca rosea and hence its name. It is amitotic inhibitor, and is used in cancer chemotherapy. Vincristine is created by the coupling of indole alkaloids vindoline and catharanthine in the vinca plant.[1]
Mechanism
Tubulin is a structural protein that polymerizes to microtubules. The cell cytoskeleton and mitotic spindle, among other things, are made of microtubules. Vincristine binds to tubulin dimers, inhibiting assembly of microtubule structures. Disruption of the microtubules arrests mitosis in metaphase. Therefore, the vinca alkaloids affect all rapidly dividing cell types including cancer cells, but also those of intestinal epithelium and bone marrow.

Uses
Vincristine is delivered via intravenous infusion for use in various types of chemotherapy regimens. Its main uses are in non-Hodgkin’s lymphoma as part of the chemotherapy regimen CHOP, Hodgkin’s lymphoma as part of MOPP, COPP, BEACOPP, or the less popular Stanford V chemotherapy regimen, in acute lymphoblastic leukemia, and in treatment for nephroblastoma (Wilms tumor, a kidney tumor most common in young children). It is also used to induce remission in ALL with Dexamethasone and L-Asparaginase. Vincristine is occasionally used as an immunosuppressant, for example, in treating thrombotic thrombocytopenic purpura (TTP) or chronic idiopathic thrombocytopenic purpura (ITP). It is used in combination with prednisone to treat childhood leukemia.
The main side-effects of vincristine are peripheral neuropathy, hyponatremia, constipation, and hair loss.
Peripheral neuropathy can be severe, and hence a reason to avoid, reduce, or stop the use of vincristine. One of the first symptoms of peripheral neuropathy is foot drop: A person with a family history of foot drop and/or Charcot-Marie-Tooth disease (CMT) should avoid the taking of vincristine.[2]
Accidental injection of vinca alkaloids into the spinal canal (intrathecal administration) is highly dangerous, with a mortality rate approaching 100 percent. The medical literature documents cases of ascending paralysis due to massive encephalopathy and spinal nerve demyelination, accompanied by intractable pain, almost uniformly leading to death; a handful of survivors were left with devastating neurological damage with no hope of recovery. Rescue treatments consist of washout of the cerebrospinal fluid and administration of protective medications.[3] A significant series of inadvertent intrathecal vincristine administration occurred in China in 2007 when batches of cytarabine andmethotrexate (both often used intrathecally) manufactured by the company Shanghai Hualian were found to be contaminated with vincristine.[4]

Having been used as a folk remedy for centuries, studies in the 1950s revealed that C. roseus contained 70 alkaloids, many of which are biologically active. While initial studies for its use in diabetes mellitus were disappointing, the discovery that it caused myelosuppression (decreased activity of the bone marrow) led to its study in mice withleukemia, whose lifespan was prolonged by the use of a vinca preparation. Treatment of the ground plant with Skelly-B defatting agent and an acid benzene extract led to a fraction termed “fraction A”. This fraction was further treated withaluminium oxide, chromatography, trichloromethane, benz-dichloromethane, and separation by pH to yield vincristine.[5]
Vincristine was approved by the United States Food and Drug Administration (FDA) in July 1963 as Oncovin. The drug was initially discovered by a team led by Dr. J.G. Armstrong, then marketed by Eli Lilly and Company.
Like LSD, the microtubule toxin vincristine allegedly causes not-unpleasant visual hallucinations in humans. Other side-effects of vincristine include depression, agitation, and insomnia. Very small doses are needed for the effects of LSD or vincristine, for example, these drugs are active at concentrations of 4.3E-7 M-1 vincristine and 1.0E-8 M-1 LSD.
Many researchers have favored the drug-receptor theory to explain drug-induced hallucinations, usually at the 5-HT2A receptor. In the drug-receptor theory, signal amplification takes place when one molecule of drug binds to a receptor, which activates G-proteins, which affects more proteins, thus signaling cascades explain how a small amount of LSD can lead to widespread changes in the cell.
Van Woerkom suggests instead that LSD binds an element of the cytoskeleton, in a fashion similar to colchicine or vinblastine, which directly bind tubulin. The amount of LSD needed to produce hallucinations is so vanishly small, that it seems hard to believe that a submicromolar dosage of LSD could act on a substrate as vast as the cytoskeleton. However, some microtubule inhibitors such as vincristine are effective at very low dosages. The potency of vincristine may partly explain the success of this drug as a chemotherapeutic drug.
Three generic drug makers supply vincristine in the United States – APP, Mayne, and Sicor (Teva).
- ^ “Pharmacognosy of Vinca Alkaloids”.
- Graf, W. D.; Chance, P. F.; Lensch, M. W.; Eng, L. J.; Lipe, H. P.; Bird, T. D. (1996). “Severe Vincristine Neuropathy in Charcot-Marie-Tooth Disease Type 1A”. Cancer 77 (7): 1356–1362. doi:10.1002/(SICI)1097-0142(19960401)77:7<1356::AID-CNCR20>3.0.CO;2-#. PMID 8608515.
- Qweider, M.; Gilsbach, J. M.; Rohde, V. (2007). “Inadvertent Intrathecal Vincristine Administration: A Neurosurgical Emergency. Case Report”. Journal of Neurosurgery: Spine 6 (3): 280–283. doi:10.3171/spi.2007.6.3.280. PMID 17355029.
- Jake Hooker and Walt Bogdanich (January 31, 2008). “Tainted Drugs Tied to Maker of Abortion Pill”. New York Times.
- Johnson, I. S.; Armstrong, J. G.; Gorman, M.; Burnett, J. P. (1963). “The Vinca Alkaloids: A New Class of Oncolytic Agents” (pdf). Cancer Research 23 (8 Part 1): 1390–1427.PMID 14070392.
External links
- Vincristine chemotherapy
- Vincristine and vinblastine
- Description and Natural History of the Periwinkle
- The Boger Route to (-)-Vindoline
- U.S. National Library of Medicine: Drug Information Portal – Vincristine
-
Cytostatic Vinca alkaloids rosea L. Catharanthus roseus G.Don) are now well known anticancer and particularly useful. Given the small amount of vincristine in Catharanthus present, quite a number of ways of preparation have been proposed by chemists. Thus FR-A-2296418 describes the synthesis of vincristine by coupling Catha-ranthine and vindoline. Other laboratories have achieved the transformation of vinblastine vincristine oxidation under controlled conditions, very strict.
-
FR-A-2210393 and US-A-3899493 perform the oxidation by chromic acid at -30, -90 ° C in a mixture of acetic acid-acetone or chloroform-acetic acid at -55 ° C.
-
In U.S. 4,375,432, chromic compound is also used in acid medium at -65 ° C, -50 ° C in a medium based solvent THF. In addition, EP-A-37289 boasts an oxidation mixture ferrous salt, hydrogen peroxide, perchlorate in acetonitrile. ZA-A-82 08939 discloses a method with chromic acid and an ether-chloroform.
-
HU-A-23638 offers diterbutylchromate in pelargonic acid, and finally EP-A-117861 gets vinblastinel transformation vincristine oxidant potassium permanganate in acetic acid medium. It is clear that these dimeric alkaloids are a valuable material because of their low levels in vegetable raw materials, and therefore the processes of synthesis or semi-synthesis performance are of extreme interest.
-
Vincristine is used in cancer chemotherapy, particularly for the treatment of certain acute leukemias.
-
This alkaloid is obtained mainly by extraction from leaves of Catharanthus Ro-seus (U.S. Patent No. 3,205,220) where it is accompanied by other alkaloids bis-Indo-holic, especially vinblastine.Vinblastine (I, R = CH 3), however, is present at a concentration much higher than that of vincristine and is therefore a precursor of choice for the semisynthesis of the latter.
-
Several processes of vincristine from vinblastine were disclosed. We note in particular patents or patent applications include:
- a) Belgian Patent 739,337 (Gedeon Richter) which describes a method for the oxidation of vinblastine vincristine in a mixture chromic acid, acetic acid and acetone.
- b) Belgian Patent 823560 (Gedeon Richter) the oxidation is performed with oxygen in the presence of formic acid and of a catalyst based on platinum at room temperature.
- c) European Patent Application 18231 (Gedeon Richter): is carried out by oxidation with chromic acid or an alkali metal dichromate in the presence of acetic anhydride and, optionally, of ethanol and an organic solvent immis target with water.
- d) European Patent Application 37289 (Eli Lil-ly): the oxidation is effected by the perchlorate of iron (II) in the presence of hydrogen peroxide and acetonitrile.
-
In addition, the European patent application 37. 290 discloses a process for the oxidation of vinblastine base with Na 2 Cr 2 O 7 in the presence of sulfuric acid in tetrahydrofuran. This reaction led to -50 ° C, is achieved with a yield of 80-92% calculated for each estimation.
-
Observed yields or purity of the products obtained characterizing the processes described above are, however, significant disadvantages.
-
Frequently a secondary product formed is N-demethyl vinblastine need then reformulate for vincristine.
Thus Potier and Kutney obtained products with the C18’S-C2’R absolute configuration, which is critical for anti-tumor activity, by a coupling reaction of the N.sup.b -oxide of catharanthine, or its derivatives, with vindoline, in the presence of trifluoroacetic anhydride, followed by a reduction reaction. [See Potier et. al. J. Am. Chem. Soc. 98. 7017 (1976) and Kutney et. al. Helv. Chim. Acta, 59, 2858 (1976)].
The Potier and Kutney coupling process has disadvantages. The yields are not satisfactory except for the coupling of catharanthine N-oxide with vindoline and even there the preparative yield is low. While vindoline is the most abundant alkaloid of Vinca rosea and is thus readily available, the other possible components of the Potier-Kutney coupling process (catharanthine, allocatharanthine, voacangine,) are relatively inaccessible, costly, and they do not allow a wide range of structural variation of that component of the coupling process.
- …………………………………………………………………………………………………………………………………………………………………………………………………………..
-
EP 0117861 B1
-
clips
-
The process of the present invention produces a simple vincristine, in quantity and purity requiring little or no additional purification by recrystallization or chromatography.
-
[0009]The reagent used is oxidation permanganate ion dissolved in toluene or dichloromethane as solvent. An alternative consists in immobilizing the resin on a permanganate anion, for example a polymer such as polystyrene comprising ammonium groups. Solubilization can be achieved by the action of a complexing agent crown ether (“crown-ether”) of potassium permanganate.
-
[0010]The permanganate anion can also be solubilized by preparing an ammonium salt or quaternary phosphonium corresponding which is soluble in methylene chloride or toluene. For this purpose, it is preferable to use potassium permanganate benzyltriethylammonium.
-
[0011]Obtaining from vincristine vinblastine using a permanganate salt is unexpected since the potassium permanganate used in some acetone oxide derivatives of vinblastine at the portion of the molecule velbanamine (Kutney, Balsevich and Worth, Heterocycles, 11, 69, 1978). The N-methyl group of the vindoline part intact.
-
[0012]The formation of N-CHO indoline skeleton on a bis-indole group vinblastine using a permanganate salt has never been reported.
-
[0013]According to one embodiment of the method of the present invention, vinblastine, preferably in the form of sulphate, is treated in the presence of an organic acid such as acetic acid, with an excess of potassium permanganate dissolved in dichloromethane or toluene in the presence of “18-crown-6” or ether derivatives dibenzo-or di-cyclohexylcorrespondants. The reaction is conducted at a temperature between -40 ° C and -75 ° C and is preferably followed by thin layer chromatography. The reaction time generally ranges from 5 minutes to 3 hours.
-
[0014]Potassium permanganate is preferably dissolved in dichloromethane and the oxidation reaction is then carried out at -70 ° C.
-
[0015]The solubility of potassium permanganate is indeed substantially increased in the presence of a macrocyclic polyether as the “18-crown-6” ether (1, 4, 7, 10, 13, 16-hexaoxacy-clooctadécane) or derivative dibenzo – or corresponding dicyclohexyl-hexyl.
-
[0016]The reaction mixture is then treated simultaneously by a mild reducing and alkaline. For this purpose, use is preferably an aqueous solution of bisulfite, disulfite or sodium metabisulfite and ammonia.
-
[0017]The organic phase was separated and the aqueous phase is extracted several times with methylene chloride. The combined organic phases were concentrated in vacuo to give a residue containing 80-85% of base vincristine, a 90-95% yield.
-
[0018]Alternatively, you can proceed with the extraction of the reaction mixture after reduction without conducting a simultaneous alkalinization. The acidic aqueous solution was then extracted with dichloromethane. This route is a novel process for purification of vincristine formed in the reaction medium.
-
[0019]According to another embodiment of the present invention, vincristine is obtained by oxidation of vinblastine by reacting a quaternary ammonium permanganate. The ammonium cation is preferably benzyltriethylammonium group or benzyl trimethyl ammonium (see eg Angew. Chem., Intern. Ed. 13, 170, 1974). The reaction is carried out in 2 to 6 hours at -60 ° C in an inert solvent wherein the ammonium salt is soluble, and an acid, preferably an organic acid of low molecular weight. A mixture of dichloromethane and glacial acetic acid can be used. After treatment with a mild reducing agent in aqueous medium, the resulting acidic solution is extracted with dichloromethane, and the organic phase is made alkaline by washing with a basic aqueous solution and concentrated. Vincristine solvate is isolated with a yield higher than 90%.
-
[0020]The latest variant of the method of the invention is particularly advantageous in terms of economic and technical.
-
[0021]Purification or separation may be effected by crystallization and chromatography using techniques well known this from the crude product of the reaction. The product can also be lyophilized.
-
[0022]In most cases, vincristine thus obtained can be converted directly into an addition salt with an organic or inorganic acid, preferably pharmaceutically acceptable. This salt is preferably a sulfate that may arise in a more or less solvated or hydrated.
-
[0023]We can also prepare vincristine dissolved in a physiologically acceptable solvent and ready to be injected.
-
[0024]In particular, vincristine sulfate is obtained by addition of H 2 S0 4 to a solution of vincristine gross or recrystallized from ethanol, dissolved in a mixture of methylene chloride and anhydrous ethanol, partial removal in vacuo chloride methylene and crystallization.
-
[0025]Vincristine sulfate thus obtained has a purity sufficient for use as a medicament, particularly in the form of injectable solutions.

Madagascar Periwinkle: Public Domain Illustration by Sydenham Edwards
The Madagascar periwinkle, an attractive flowering plant, contains the powerful anti-cancer chemicals vinblastine and vincristine. Velvet beans, which are named from the covering of soft hairs on the young plant, contain L-dopa, a very helpful chemical in the treatment of Parkinson’s disease. The Madagascar periwinkle and the velvet bean are just two of the large number of plants that have been found to contain medicinal chemicals. There are almost certainly many more plants that have undiscovered health benefits.
The Madagascar Periwinkle
The Madagascar periwinkle is native to Madagascar and India, but is now grown in many countries as a garden plant. It has also escaped from gardens and grows as a weed. The red, purple, pink or white flowers often have a center which is a different color from the rest of the flower. Madagascar periwinkles may grow up to one meter tall and have glossy green leaves.
The sap of the Madagascar periwinkle, which has a milky appearance and is poisonous, contains vinblastine, vincristine and many other alkaloids. Researchers are discovering that many of these alkaloids are biologically active inside the human body.
Vinblastine and Vincristine
Vinblastine and vincristine have very similar chemical structures, but their effects on the body are not the same. Vinblastine is used to treat specific types of cancer, such as Hodgkin’s disease, breast cancer, testicular cancer and non-small cell lung cancer. Vincristine is used in the treatment of acute lymphoblastic leukemia (ALL) and has provided a great breakthrough in successful treatment of this disease in children. When vincristine is added to the treatment regimen for children suffering from ALL, the survival rate reaches eighty percent. Vincristine is not so impressive in the treatment of ALL in adults.
Cells contain a supporting network of protein tubules, which are known as microtubules. Microtubules also play a vital role in the process of cell division. Before a cell divides, each chromosome in the cell is replicated. The replicated chromosomes are separated from their partners and pulled to opposite ends of the cell by microtubules during a process called mitosis. The cell then divides down the middle.
Vinblastine and vincristine stop microtubule formation during mitosis and therefore prevent cells from reproducing. This effect is strongest in cells that have a high rate of division, such as cancer cells. However, vinblastine and vincristine also affect cells lining the intestine, the cells in the bone marrow that produce blood cells, and the cells in the hair follicles, since these too have a high rate of cell division.
Possible vinblastine or vincristine side effects include constipation, hair loss, a low platelet count, which can cause increased bleeding, a low white blood cell count, which can lead to increased infections, or a low red blood cell count, resulting in anemia. There may occasionally be nerve damage, possibly due to the effect of the medicines on the microctubules in the nerve cells. Vincristine is more likely to cause nerve damage than vinblastine.

,,,,,,,,,,,,,


Total synthesis of (+)-vincristine (2). TFA, trifluoroacetic acid or trifluoroacetyl; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene.
Stereocontrolled total synthesis of (+)-vincristine

………………….
see docstoc presentation
click below
Vincristine
var docstoc_docid=”51697405″;var docstoc_title=”Vincristine”;var docstoc_urltitle=”Vincristine”;
…………………….
isolation
Kumar A, Patil D, Rajamohanan PR, Ahmad A (2013)
Isolation, Purification and Characterization of Vinblastine and Vincristine from Endophytic Fungus Fusarium oxysporumIsolated from Catharanthus roseus. PLoS ONE 8(9): e71805. doi:10.1371/journal.pone.0071805
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0071805
Isolation, purification and characterization of vinblastine and vincristine from the endophytic fungus Fusarium oxysporum
A two stage fermentation procedure was employed for the isolation of vinblastine and vincristine by Fusarium oxysporum. In the first stage, 500 ml Erlenmeyer flasks containing 100 ml medium (MGYP, (0.3%) malt extract, (1.0%) glucose, (0.3%) yeast extract and (0.5%) peptone) were inoculated with 7 days old culture and incubated at 28°C on a rotary shaker (240 rpm) for 4–5 days, which was used as seed culture (I stage). Later, 10 ml seed culture was transferred to 500 ml Erlenmeyer flask containing 100 ml production medium called as vinca medium-1 (Glucose: 3%, Succinic acid: 1%, Sodium benzoate: 100 mg, Peptone: 1%, Magnesium sulphate: 3.6 mg, Biotin: 1 mg, Thiamine: 1 mg, Pyridoxal: 1 mg, Calcium pentothenate: 1 mg, Phosphate buffer: 1 ml (pH 6.8), L-Tryptophan: 0.1%, Geranium oil: 0.05%.) which were incubated at 28°C for 20 days as shake culture (II stage), after which it was harvested and used for further study. Culture filtrates and mycelia were separated with the help of muslin cloth and then lyophilized. Lyophilized culture filtrate was extracted using ethyl acetate as a solvent system. The organic layer was separated from the aqueous layer using separating funnel. The extraction was repeated thrice and the solvent was dried using anhydrous sodium sulphate and concentrated under vacuum using rotavapour at 40°C in order to get crude extract. A small amount of crude extract was dissolved in ethyl acetate and subjected to thin layer chromatography (TLC) on silica gel-G (0.5 mm thickness) using chloroform:methanol (8:2) as a solvent system. The TLC plates were sprayed with ceric ammonium sulphate reagent. Vinca alkaloids spots produced brilliant violet color as well as purple color with above spraying reagent. Purification of fungal vinblastine and vincristine were done by silica gel column chromatography. The crude extract was loaded on silica gel column (60–120 mesh size, 40 cm×2 cm length width) pre-equilibrated with chloroform and eluted with a gradient of chloroform:methanol (100% chloroform, 9:1, 8:2, 7:3, 1:1 and 3:7 and 100% methanol). Fractions containing compounds with Rf values similar to that of the standard vinblastine and vincristine were pooled and subjected to preparative TLC on a 0.5 mm thick (20 cm×20 cm) silica plate and developed in chloroform:methanol (8:2) solvent system. The putative bands of fungal vinblastine and vincristine were scraped and eluted out with methanol. Purity of the isolated compounds was checked on TLC in the solvent systems such as (a) chloroform:methanol (8:2) (b) chloroform:methanol (9:1) and (c) ethyl acetate: acetonitrile (8:2).
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0071805
see also
http://www.ncbi.nlm.nih.gov/pubmed/20209002
ALSO
large-scale isolation of native catharantine, vindoline and 3′,4′-anhydrovinblastine whereby the isolation of vincristine, vinblastine, leurosine and the corresponding desacetoxy, desacetyl and N-desmethyl derivatives in a manner known per se can also be accomplished.
For the isolation of the two monoindole alkaloids: vindoline and catharantine from the dried plant Vinca rosea L. Svoboda [J. Am. Pharm. Assoc. 48, (11), 659 (1959)] described a method, which can be accomplished only with a very modest yield. From 1 kg. of the dried plant–subjecting the whole plant to a suitable treatment–approximately 0.6 g. of vindoline and 0.05 g. of catharantine were obtained.
3′,4′-ANHYDROVINBLASTINE UNTIL NOW HAS NEITHER BEEN ISOLATED FROM THE PLANT Vinca rosea L. nor identified in it.
For the preparation of the diindole alkaloid components starting from the leaves of Vinca rosea L. there are more methods known in the art (U.S. Pat. nos. 3,097,137; 3,205,220; 3,225,030 and Hungarian Pat. Nos. 153,200; 154,715; 160,967 and 164,958 as well as Austrian Pat. Nos. 313,435, 313,485, Australian pat. No. 458,629 and Swiss Pat. No. 572,488 and British Pat Nos. 1,412,932, 1,382,460 corresponding to the preceding two patents). According to these known processes from 1 kg. of the dried leaves of Vinca rosea L. about 0.1 to 0.2 g. of leurosine can be obtained and vinblastine, vincristine and optionally the corresponding N-desmethyl, desacetyl and desacetoxy derivatives are also simultaneously isolated.
Further on it is well known that the synthetic catharantine and vindoline may be coupled by the Polonovszky reaction to give 3′,4′-anhydrovinblastine which can thereafter be epoxidized to leurosine [Potier et al. Tetrahedron Letters 3945 (1976); DT-OS 25 58,124; Helv. Chim. Acta 59, 2858 (1976); Heterocycles 4, 997 (1976), Belgian patent specification No. 842,200 equivalent to U.S. patent application Ser. No. 582,372]. Leurosine itself has a valuable tumour growth inhibiting activity and the N-desmethyl-N-formyl derivative thereof is the most promising substance against leukemia (Hungarian Pat. No. 165,986 equivalent to U.S. patent application Ser. No. 422,100, and Austrian Pat. No. 332,566 which has issued as British Pat. No. 1,412,932).
EMA Accepts AstraZeneca’s Naloxegol Application

naloxegol
http://www.ama-assn.org/resources/doc/usan/naloxegol.pdf
Morphinan-3,14-diol, 4,5-epoxy-6-(3,6,9,12,15,18,21-heptaoxadocos-1-yloxy)-17-(2-
propen-1-yl)-, (5α,6α)-
4,5α-epoxy-6α-[(3,6,9,12,15,18,21-heptaoxadocosan-1-yl)oxy]-17-(prop-2-en-1-
yl)morphinan-3,14-diol
MOLECULAR FORMULA C34H53NO11
MOLECULAR WEIGHT 651.8
SPONSOR AstraZeneca
CODE DESIGNATION NKTR-118
CAS REGISTRY NUMBER 854601-70-0
WHO NUMBER 9434
Marketing Authorisation Application for naloxegol accepted by European Medicines Agency
Friday, 27 September 2013
AstraZeneca today announced that the European Medicines Agency (EMA) has accepted the Marketing Authorisation Application (MAA) for naloxegol, an investigational peripherally-acting mu-opioid receptor antagonist, which has been specifically designed for the treatment of opioid-induced constipation (OIC) for adult patients 18 years and older, including patients with inadequate response to laxatives.
read more
http://www.pharmalive.com/ema-accepts-astrazeneca-s-naloxegol-application
Naloxegol (INN; NKTR-118), or PEGylated naloxol,[1] is a peripherally–selective opioid antagonist under development byAstraZeneca, licensed from Nektar, for the treatment of opioid-induced constipation.[2]
- ^ Roland Seifert; Thomas Wieland; Raimund Mannhold; Hugo Kubinyi, Gerd Folkers (17 July 2006). G Protein-Coupled Receptors as Drug Targets: Analysis of Activation and Constitutive Activity. John Wiley & Sons. p. 227. ISBN 978-3-527-60695-5. Retrieved 14 May 2012.
- ^ “Nektar | R&D Pipeline | Products in Development | CNS/Pain | Oral Naloxegol (NKTR-118) and Oral NKTR-119”. Retrieved 2012-05-14.
Naloxegol (NKTR-118) is an investigational drug candidate in Phase 3 studies being developed as a once-daily oral tablet for the treatment of opioid-induced constipation. Naloxegol (NKTR-118) was designed using Nektar’s proprietary small molecule polymer conjugate technology. Results of the Phase 2 study of naloxegol (NKTR-118) were presented in October 2009 at the American College of Gastroenterology Annual Clinical Meeting and the American Academy of Pain Management. NKTR-119 is an early stage drug development program that is intended to combine oral naloxegol (NKTR-118) with selected opioids, with the goal of treating pain without the side effect of constipation traditionally associated with opioid therapy.
Nektar and AstraZeneca have a global agreement for both naloxegol (NKTR-118) and NKTR-119. Under the agreement, AstraZeneca has responsibility for the development, global manufacturing and marketing of both naloxegol (NKTR-118) and NKTR-119. For naloxegol (NKTR-118), Nektar is eligible to receive up to $235 million in aggregate payments upon the achievement of certain regulatory milestones, as well as additional tiered sales milestone payments of up to $375 million if the product achieves considerable levels of commercial success. Nektar will also be eligible to receive significant double-digit royalty payments on net sales of naloxegol (NKTR-118) worldwide. For NKTR-119, Nektar would receive development milestone payments as well as tiered sales milestone payments. Nektar will also receive significant double-digit royalty payments on NKTR-119 net sales worldwide.
oxalate derivative
http://www.ama-assn.org/resources/doc/usan/naloxegol-oxalate.pdf
Morphinan-3,14-diol, 4,5-epoxy-6-(3,6,9,12,15,18,21-heptaoxadocos-1-yloxy)-
17-(2-propen-1-yl)-, (5α,6α)-, ethanedioate (1:1)
4,5α-epoxy-6α-[(3,6,7,12,15,18,21-heptaoxadocosyl)oxy]-17-(prop-2-
enyl)morphinan-3,14-diol hydrogen ethanedioate
MOLECULAR FORMULA C34H53NO11 . C2H2O4
MOLECULAR WEIGHT 741.8
TRADEMARK None as yet
SPONSOR AstraZeneca
CODE DESIGNATIONS NKTR-118 oxalate, AZ13337019 oxalate
CAS REGISTRY NUMBER 1354744-91-4
Drugs for Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
<div style=”margin-bottom:5px”> <strong> <a href=”https://www.slideshare.net/CTEPH/drugs-for-cteph-studi-farmacologici” title=”Drugs for CTEPH – studi farmacologici” target=”_blank”>Drugs for CTEPH – studi farmacologici</a> </strong> from <strong><a href=”http://www.slideshare.net/CTEPH” target=”_blank”>CTEPH</a></strong> </div>
Glaxo Gets EU OK for New Revolade Indication
GSK receives marketing authorisation from the European Commission for additional Revolade™ (eltrombopag) indication as the first approved treatment for chronic hepatitis C-associated thrombocytopenia
GlaxoSmithKline plc announced today that the European Commission has granted an additional indication for Revolade™ (eltrombopag) as a treatment for low platelet counts (thrombocytopenia) in adult patients with chronic hepatitis C infection, where the degree of thrombocytopenia is the main factor preventing the initiation or limiting the ability to maintain optimal interferon (IFN)-based therapy
read all at
http://www.pharmalive.com/glaxo-gets-eu-ok-for-new-revolade-indication
Purdue Pharma L.P. Receives FDA Approval For 15 mcg/hour Dosage Strength Of Butrans (buprenorphine) Transdermal System CIII

buprenorphine
STAMFORD, Conn., Sept. 24, 2013 /PRNewswire/ — Purdue Pharma L.P. announced that the U.S. Food and Drug Administration (FDA) approved a new 15 mcg/hour dosage strength of Butrans® (buprenorphine) Transdermal System CIII, which will provide an additional titration option for healthcare professionals. Four strengths of Butrans will now be available: 5, 10, 15 and 20 mcg/hour. Purdue expects to launch Butrans 15 mcg/hour commercially in the U.S. in October 2013.
read all at

Buprenorphine is a semi-synthetic opioid that is used to treat opioid addiction in higher dosages (>2 mg), to control moderate acute pain in non-opioid-tolerant individuals in lower dosages (~200 µg), and to control moderate chronic pain in dosages ranging from 20–70 µg/hour. It is available in a variety of formulations: Subutex, Suboxone, Zubsolv (buprenorphine HCl and naloxone HCl; typically used for opioid addiction), Temgesic (sublingual tablets for moderate to severe pain), Buprenex (solutions for injection often used for acute pain in primary-care settings), Norspan and Butrans (transdermal preparations used for chronic pain).
-
The treatment of opiate abuse and dependence by substitution of the abused opiate with a safer, longer-acting opioid is often a successful pharmacotherapeutic intervention strategy. Heroin, a widely abused opiate, acts as an agonist for the mu-opioid receptor (MOR). Heroin is often abused using intravenous injection, often resulting in needle-sharing among addicts, which is often responsible for the spread of life-threatening infections such as hepatitis C and HIV/AIDS. Methadone has been used as a substitute MOR agonist. Methadone is orally active, and has sufficient duration of action to enable it to be given as a single daily dose. More recently, buprenorphine 1, 21-(cyclopropyl-7α-[(S)-1-hydroxy-1,2,2-trimethylpropyl]-6,14-endo-ethano-6,7,8,14-tetrahydro-oripavine, a MOR partial agonist, has been used as a pharmacotherapy (see, e.g., U.S. Pat. No. 4,935,428 ). As a partial MOR agonist, it has a lower ceiling to its MOR-mediated effects than a full MOR agonist (e.g., methadone). As a result, buprenorphine has a greater margin of safety than full MOR agonists. In addition, buprenorphine also has a long duration of action. Buprenorphine’s enhanced safety, coupled with its extended duration, enables a relatively long dosing interval, typically every 24 hours, but this can be extended to every 72 hours or more.

Buprenorphine’s favorable safety profile compared to methadone has allowed it to be prescribed by office-based physicians, which has substantially decreased the cost of treatment, and increased the number of addicts in pharmacotherapy treatment.
-
For the treatment of opiate abuse and dependence, buprenorphine is available as tablets formulated for sublingual administration, and is sold under the trademark Subutex®. The daily maintenance dose for Subutex® is in the range 4-16 mg. Subutex®is readily soluble in aqueous media, making it possible for addicts to misuse the formulation by dissolving the tablets in water, and then injecting the resulting solution. To counter this misuse, buprenorphine has been formulated as a mixture with the MOR antagonist naloxone in a 4:1 ratio (Suboxone®).
-
Sublingual administration of buprenorphine has several drawbacks, notably the need to avoid swallowing the tablet because of buprenorphine’s low bioavailability (∼5%) when taken orally. In comparison, buprenorphine’s bioavailability is approximately fifty percent when absorbed sublingually (see, e.g., Jasinski and Preston, Buprenorphine, Ed. A Cowan, JW Lewis, Wiley-Lis, NY pp. 189-211).
-
Several buprenorphine ester derivatives are described by Stinchcomb et al. in Pharm. Res (1995), 12, 1526-1529. The physiochemical properties of the esters are described, and compared with those of buprenorphine hydrochloride and its free base. Stinchcomb et al. also describe transdermal absorption of these esters in Biol. Pharm. Bull. (1996), 19, 263-267 and Pharm. Res. (1996), 13, 1519-1523. Wang, Published U.S. Patent Application No. 2005/0075361 , also describes some buprenorphine derivatives, which are apparently useful for pain relief when delivered intramuscularly or subcutaneously. EP 1 422 230 discloses buprenorphine monocarboxylic ester derivatives and dibuprenorphine dicarboxylic ester derivatives which exert a longer analgesic effect as compared to buprenorphine hydrochloride.
Buprenorphine hydrochloride was first marketed in the 1980s by Reckitt & Colman (now Reckitt Benckiser) as an analgesic, generally available as Temgesic 0.2 mg sublingual tablets, and as Buprenex in a 0.3 mg/mL injectable formulation. In October 2002, the Food and Drug Administration (FDA) of the United States also approved Suboxone and Subutex, buprenorphine’s high-dose sublingual tablet preparations indicated for detoxification and long-term replacement therapy in opioid dependency, and the drug is now used predominantly for this purpose.
In the European Union, Suboxone and Subutex, buprenorphine’s high-dose sublingual tablet preparations, were approved for opioid addiction treatment in September 2006.[3] In the Netherlands, buprenorphine is a List II drug of the Opium Law, though special rules and guidelines apply to its prescription and dispensation. In the United States, it was rescheduled to Schedule III drug from Schedule V just before FDA approval of Suboxone and Subutex.[4] In recent years, buprenorphine has been introduced in most European countries as a transdermal formulation for the treatment of chronic pain.
Commercial preparations
British firm Reckitt & Colman (now Reckitt Benckiser) first marketed buprenorphine under the trade names Temgesic (sublingual/parenteral preparations) and Buprenex (parenteral). Subsequently, two more formulations were released: Subutex (white, oval-shaped, bitter, no active additives) and Suboxone (white color [orange in the U.S.], hexagonal tablet, lemon-lime-flavored, one part naloxone for every four parts buprenorphine). The orange film strips form of Suboxone are lemon flavor. More than 71% of patients gave Suboxone film a favorable taste rating.[5]

8mg Suboxone film strip
Subutex and Suboxone are available in 2 mg and 8 mg sublingual dosages. (Suboxone Film is also available in doses of 4 mg/1 mg & 12 mg/3 mg buprenorphine/naloxone respectively). On October 8, 2009, Roxane Laboratories of Columbus, Ohio, United States won FDA approval for a generic preparation of Subutex[6] and as of October 23, 2009, announced that it is ready for distribution nationwide in 2 mg and 8 mg sublingual dosages. The demand for this generic was so high that Roxane did not produce enough to meet market demand, resulting in pharmacies running out and being unable to order more.[7] Teva Pharmaceutical Laboratories of Tel Aviv, Israel also received approval (as of April 1, 2010) for a generic formulation of Subutex sublingual tablets in 2 mg and 8 mg dosages that are currently available in limited distribution in America as of June 20, 2010. In 2013, Reckitt Benckiser voluntarily discontinued the sale of Suboxone tablets in the United States based on data from Poison control centers that consistently found significantly higher rates (7.8–8.5 times greater) of accidental pediatric exposure with Suboxone tablets as compared with Suboxone Film.[8]
Since 2001, buprenorphine is also available transdermally as 35, 52.5, and 70 µg/h transdermal patches that deliver the dose over 96 hours. This dosage form is marketed as Transtec in most European countries by Grunenthal (Napp Pharmaceuticals in the UK,[9][10] Norpharma in Denmark) for the treatment of moderate to severe cancer pain and severe non-cancer pain not responding to non-opioids.
Other available buprenorphine formulations include a 5, 10, and 20 µg/h, 7-day patch, marketed as Butrans in the U.S. by Purdue Pharma (and by Napp Pharmaceuticals in the UK) indicated for the management of moderate to severe chronic pain in patients requiring a continuous, around-the-clock opioid analgesic for an extended period of time.[11] A similar transdermal system is marketed by a collaboration between Mundipharma and Grunenthal in Australia under the name Norspan, with indications for moderate chronic pain not responding to non-opioids, dosed in 5, 10, or 20 µg/h patches.[12]
In India: Addnok 0.4, 2 & 8 Mg Sublingual Tablets by Rusan Pharma Ltd.,[13] Tidigesic 0.2 mg (slow release) or 0.3 mg/mL injectable by Sun Pharmaceuticals;[14] Buprigesic (0.3 mg/mL) by Neon Laboratories;[15] Morgesic (0.3 mg/mL) by Samarth Pharma; Norphin (0.3 mg/mL) Unichem Laboratories.
A novel implantable formulation of buprenorphine (Probuphine), using a polymer matrix sustained-release technology, has been developed to offer treatment for opioid dependence while minimizing risks of patient noncompliance and illicit diversion. FDA requested additional information about Probuphine on April 30, 2013, in a complete response letter to Titan Pharmaceuticals pending NDA.[16]
In addition to the sublingual tablet, Suboxone is now marketed in the form of a sublingual film, available in the 2 mg/0.5 mg, 4 mg/1 mg, 8 mg/2 mg, and recently 12 mg/3 mg dosages; the film is not available in Canada or the United Kingdom (where it was discovered). The makers of Suboxone, Reckitt Benckiser, claim that the film has some advantages over the traditional tablet in that it dissolves faster and, unlike the tablet, adheres to the oral mucosa under the tongue, preventing it from being swallowed or falling out; that patients favor its taste over the tablet, stating that “more than 71% of patients scored the taste as neutral or better”; that each film strip is individually wrapped in a compact unit-dose pouch that is child-resistant and easy to carry; and that it is clinically interchangeable with the Suboxone tablet and can also be dosed once daily.[17] Reckitt Benckiser also states that the film discourages misuse and abuse, as the paper-thin film is more difficult to crush and snort. Also, a ten-digit code is printed on each pouch, which helps facilitate medication counts and, therefore, serves to deter diversion into the illegal drug market. Although Suboxone film may deter snorting the drug it makes injecting the drug much easier as the films are extremely easy to dissolve in water making for easy injection and the fact that the naloxone in suboxone is ineffective at blocking the effects of buprenorphine when injected by addicts not dependent on another opioid.
Physicochemical properties
Buprenorphine is a semi-synthetic derivative of thebaine, one of the most chemically reactive opium alkaloids. Buprenorphine has a molecular weight of 467 and its structure is typically opioid with the inclusion of a C-7 side-chain containing a t-butyl group. This group confers overall lipophilicity on the molecule which has an important influence on its pharmacology.
Opioids exert their pharmacological effects by binding to opioid receptors. The pharmacological effects are determined by the nature of opioid-receptor interaction. Some of these effects such as analgesia, mediated by an agonistic action at the μ-opioid receptor are desirable, whereas others such as nausea, sedation, or constipation can be considered as unwanted adverse effects. Buprenorphine is a μ-opioid receptor agonist with high affinity, but low intrinsic activity. Compared with morphine (a full μ-opioid agonist) buprenorphine is considered a partial μ-opioid agonist displaying high affinity for and slow dissociation from the μ-opioid receptor. A full dose-dependent effect on analgesia has been seen within the clinically relevant dose range (up to 10 mg), but no respiratory depression which levels off at higher doses (Dahan et al. 2005). Clinically, there is also a less marked effect of buprenorphine-binding to μ-opioid receptors on gastrointestinal transit times, and indeed constipation seen in the clinic is remarkably low (Griessinger et al. 2005). Buprenorphine also shows partial agonistic activity at the opioid receptor-like receptor 1 (ORL1)-receptors which are (at least at supraspinal receptors) postulated to induce a pronociceptive effect. A study by Lutfy et al. (2003) reported that co-activation of ORL1-receptors compromises the antinociception induced by activation of the μ-opioid receptor. ORL1-activation has also an effect on hyperalgesia. It might be that buprenorphine’s partial agonism reduces this effect compared with full ORL1-agonists such as morphine or fentanyl. Buprenorphine’s antagonistic action at the δ-receptors which have a marked anti-opioid action and seem to negatively modulate central analgesia seems further to contribute to its clinically seen analgesic effect. Its likewise antagonistic activity at the κ-opioid receptors might explain the fact that it induces much less sedation and psychotomimetic effects than morphine or fentanyl (Lewis 1985; Leander 1988). Animal studies have shown that buprenorphine has a 20–40 times higher potency than morphine (Martin et al. 1976).
The strong binding of buprenorphine to the μ-opioid receptor has several consequences. Initial binding is relatively slow compared with other opioids such as fentanyl (Boas and Villiger 1985). However, the onset of analgesia is not dissimilar, since buprenorphine achieves effective analgesia at relatively low receptor occupancy (5%–10%) (Tyers 1980) and thus relatively low plasma concentrations of buprenorphine are sufficient to provide effective pain relief. The slow dissociation of buprenorphine from the receptor results in a long duration of effect and also confers another advantage in that when the drug is withdrawn an abstinence syndrome is rarely seen because of the long time taken for the drug to come off the receptor (Bickel et al. 1988).
1 Weinberg, D. S.; Inturrisi, C. E.; Reidenberg, B.; Moulin, D. E.; Nip, T. J.; Wallenstein, S.; Houde, R. W.; Foley, K. M. (1988). “Sublingual absorption of selected opioid analgesics”. Clinical pharmacology and therapeutics 44 (3): 335–342. PMID 2458208.
2. Eriksen, J.; Jensen, N. H.; Kamp-Jensen, M.; Bjarnø, H.; Friis, P.; Brewster, D. (1989). “The systemic availability of buprenorphine administered by nasal spray”. The Journal of pharmacy and pharmacology 41 (11): 803–805. PMID 2576057.
3. Suboxone EU Approval
4.DEA Rescheduling
5.What flavor do suboxone come in?. Kgbanswers.com (2012-09-06). Retrieved on 2013-05-19.
6.FDA Approval Letter to Roxane
7.Generic buprenorphine shortage
8.Reckitt Benckiser Announcement of Suboxone tablets withdrawal
9.Napp Pharmaceuticals
10.electronic Medicines Compendium (eMC) of UK medicines, Transtec product characteristics. Medicines.org.uk (2010-10-21). Retrieved on 2013-05-19.
11. “Butrans”, accessed January 23, 2011.
12. “Norspan Buprenorphine Drug/Medicine information”. news-medical.net
13.Addnok information
14. Tidigesic in India
15. Buprigesic in India
16.Probuphine complete response letter
17. Suboxone film patient information
18 Likar, R. (2006). “Transdermal buprenorphine in the management of persistent pain – safety aspects”. Therapeutics and clinical risk management 2 (1): 115–125. PMC 1661652. PMID 18360586.
- U.S. National Library of Medicine: Buprenorphine information portal
- U.S. Federal government buprenorphine program for opioid addiction
- U.S. Federal Government listing of doctors who can prescribe buprenorphine for opioid addiction
- U.S. Non-government listing of doctors who can prescribe buprenorphine for opioid addiction
- NAABT: Non-profit buprenorphine advocate
- Australian national buprenorphine policy
- The bitter pill: A Wired Magazine article on Suboxone
- Subu Must Die – How a nation of junkies went cold turkey: A New Republic article on Subutex abuse in the nation of Georgia
- Erowid.org buprenorphine page
- Methadone support.org: A methadone anonymous site
- Methdone.US: A resource center on opioid addiction treatment

-
Buprenorphine acts as a mixed agonist / antagonist and it is an important treatment option for opiate addiction and analgesia.
-
-
Opiate compounds such as (-)-naltrexone, (-)-naloxone, (-)-nalbuphene, (-)-nalmefene, and (-)-buprenorphine have been used for addiction therapy. (-)-Buprenorphine, in particular, is increasingly being used for the treatment of heroin addiction. Recently, the (+)-opiate enantiomers have been shown to have important bioactivities that differ from their (-) counter parts. Because of the exceptional opiate medicinal activity of (-)-buprenorphine, there is great interest in the therapeutic efficacy of (+)-buprenorphine. In order to explore the possible benefits of this compound, there is a need in the art for synthetic routes to produce (+)-buprenorphine or its derivatives in an efficient and cost effective manner that generates a high yield of product having a high degree of purity.
-
The following documents disclose processes for the preparation of buprenorphine:
- D1: WO 2007/081506 A (MALLINCKRODT INC [US]) 19 July 2007 (2007-07-19)
- D2: J. MARTON ET AL: “Herstellung von 6,14-Ethenomorphinan-Derivaten” LIEBIGS ANNALEN DER CHEMIE, 1993, pages 915-919, XP002519987
- D1 discloses a process for the preparation of buprenorphine from oripavine.
- D2 discloses a process
-
-
[0002]The conventional synthetic route used world-wide to prepare buprenorphine utilizes thebaine as the starting material.

-
[0003]Through a series of chemical reactions, thebaine is converted into nor-buprenorphine, the immediate precursor to buprenorphine. The final step adds a cyclopropyl methyl group to the nitrogen to form buprenorphine from nor-buprenorphine.
-
[0004]An outline of the conventional series of reactions from thebaine to buprenorphine follows:
- 1. Reaction of thebaine with methyl vinyl ketone to form the 4 + 2 reaction product.
- 2. Hydrogenation of the carbon-carbon double bond.
- 3. Addition of a tertiary butyl group via a Grignard Reaction.
- 4. An N-demethylation, via a two step reaction sequence.
- 5. An O-demethylation reaction and an N-cyano hydrolysis.
- 6. Addition of the cyclopropyl methyl group to form buprenorphine.
-
[0005]A drawback of this conventional production scheme is that the O-demethylation step is considered a low to moderate yield transformation. There is therefore a need for a norbuprenorphine/buprenorphine production scheme that does not include an O-demethylation step.
-
[0006]

-
[0007]An aspect of the present invention is to provide a method for producing norbuprenorphine utilizing oripavine as the starting material. The method comprises:
- reacting oripavine according to Formula I with methyl vinyl ketone to form a compound according to Formula II;

- hydrogenating the compound according to Formula II to form a compound according to Formula III;

- adding a t-butyl group to the compound according to Formula III to form a compound according to Formula X; and

- demethylating the nitrogen of the compound according to Formula X to form norbuprenorphine, Formula VIII.

- reacting oripavine according to Formula I with methyl vinyl ketone to form a compound according to Formula II;
-
[0008]Another aspect of the present invention is to provide a method of making buprenorphine utilizing oripavine as the starting material.




-
[0009]There is provided a method utilizing oripavine as the preferred starting material for the synthesis of nor-buprenorphine and optionally buprenorphine. Oripavine is a naturally occurring alkaloid of Papaver somniferum. The key difference between the conventional technology and the present use of oripavine as a starting material is that the O-demethylation step, typically a low to moderate yield transformation, is not needed since the oripavine molecule lacks an O-3 methyl group. In a synthesis involving several steps, it is advantageous to have only high yield reactions, in order for the overall transformation to be economical. Since the present oripavine based synthesis does not require the O-3 demethylation step, the overall yield from oripavine provides an improved yield over that traditionally achieved when thebaine is used as the starting material. The conversion route from oripavine to produce buprenorphine is convenient and more straightforward as compared to other synthetic routes.
-
[0010]An illustrative embodiment of the steps for converting oripavine into norbuprenorphine, and optionally buprenorphine, is as follows:


-
[0011]The sequence outlined above is an illustrative embodiment presented to show the transformations required, but is not limited as to the order in which the transformations may be employed. In an alternative embodiment, the hydrogenation of the Diels-Alder double bond can also be accomplished as part of step 7 when the removal of the Y protecting group is through catalytic hydrogenation.


Step 1:
-
[0012]The first step involves reaction of the oripavine with methyl vinyl ketone. This addition reaction may be accomplished by any conventional method known in the art. An illustrative embodiment is a Diels-Alder reaction in which the oripavine and methyl vinyl ketone are dissolved in a solvent and refluxed until the reaction is substantially complete. Illustrative suitable solvents include isopropyl alcohol, methanol, ethanol, toluene and mixtures thereof. The reaction mixture is then filtered to isolate the Diels-Alder adduct solids. Typical reactions result in at least about an 85% yield of at least about 98% purity.
Step 2:
-
[0013]The second step involves the hydrogenation of the C-C double bond. In an illustrative embodiment, the Diels-Alder adduct formed in step 1 was charged to a reaction vessel with Pd/ Carbon catalyst, then dissolved in a solvent. A presently preferred solvent is methanol, but any suitable solvent may be used, including methanol, ethanol, isopropyl alcohol, acetic acid and mixtures thereof. The hydrogenation takes place under nitrogen at an elevated pressure and temperature. The temperature and pressure are selected to insure substantial completion of the reaction, as is well known in the art. An illustrative temperature range typical of this reaction is about 50-90°C, with about 60°C being preferred and an illustrative pressure range typical of this reaction is about 20-60 psi, with about 35 psi being preferred. The reaction mixture is filtered to remove the catalyst and the resulting filtrate reduced under vacuum to yield the product according to Formula III.
Step 3:
-
[0014]Optional step 3 discloses the addition of a protecting group Y to form a compound according to Formula IV. The preferred method using oripavine as the starting material for norbuprenorphine and then buprenorphine utilizes an O-3 protecting group. However, the reaction can be accomplished without the use of the protection group, although the overall yield may be compromised. Further, the protecting group may be removed simultaneously with another step thereby eliminating one chemical step. Addition of an O-3 protecting group may minimize unwanted chemical reactions involving the unprotected phenol function at the 3-position. Illustrative suitable protecting groups include benzyl, O-t-butyl and silyl groups.
-
[0015]In an illustrative embodiment, the reduced Diels-Alder adduct according to Formula III and ground K2CO3 are added (K2CO3 not soluble) in chloroform and benzyl bromide, heated and refluxed. After cooling to room temperature, the reaction mixture is filtered to remove the K2CO3. The filtrate is then reduced under vacuum and azeo dried in toluene.
Step 4:
-
[0016]The fourth step utilizes the crude material according to Formula IV, formed in step 3. in a Grignard reaction. Under moisture free conditions and further under an inert atmosphere, t-BuMgCl is added, followed by anhydrous toluene. The solution is distilled until a pot temperature of about 100 °C is achieved, and the compound according the Formula IV is added. The reaction is quenched, and the temperature of the reaction mixture lowered. The organic and aqueous layers are separated, and the organic layer is concentrated under vacuum yielding an oily residue. The oily residue is then purified resulting in a compound according to Formula V, of up to about 93% purity.
-
[0017]In an alternate embodiment, the t-butyl group is added using a t-butyl lithium reagent, as is well known in the art.
-
[0018]The N-demethylation reaction may be accomplished by any suitable method known in the art. In the illustrative embodiment shown in steps 5 and 6, the methyl group is first converted into a nitrite in step 5, followed by reduction of the nitrile group in step 6.
Step 5:
-
[0019]In an illustrative embodiment of step 5, the tertiary alcohol starting material according to Formula V is dissolved in a solvent, flushed with an inert atmosphere, and then K2CO3 and cyanogen bromide are added. This reaction mixture is then refluxed until the reaction is substantially complete, cooled to room temperature and filtered to remove the K2CO3. The reaction mixture is then extracted and the organic layers reduced and dried under vacuum. The resulting solid is purified yielding up to about 93% clean material after drying.
Step 6:
-
[0020]In an illustrative embodiment, potassium hydroxide is dissolved in diethylene glycol and heated. The N-CN compound according to Formula VII is added and the reaction mixture heated until the reaction is substantially complete. After cooling to room temperature, distilled water is added and the resulting solid collected and dried, with up to about 100% yield.
-
[0021]The N-demethylation may be accomplished by any method know to those skilled in the art without departing from the instant method.
Step 7:
-
[0022]The seventh step involves the removal of the optional protecting group. In the illustrative embodiment, the Y protecting group added in step 3 is removed. In this embodiment, the secondary amine starting material may be catalytically removed by Pd/Carbon in a suitable , solvent. Suitable solvents include methanol, ethanol, isopropyl acetate and mixtures thereof. The resulting filtrate is dried under vacuum to yield norbuprenorphine. In another embodiment, the Y protecting group may be removed with an acid, such as HCl, HOAc, HF or an F anion.
Step 8:
-
[0023]Finally the norbuprenorphine is optionally converted to buprenorphine as illustrated in step 8. In an illustrative embodiment, the norbuprenorphine is converted to buprenorphine.
-
[0024]In an illustrative embodiment, a mixture of norbuprenorphine, a mild base, and cyclopropylmethyl bromide are heated in an oilbath at about 80-100°C until the reaction is substantially complete. The reaction mixture is then added over 5 minutes to 160ml of water, with mechanical stirring, yielding a gum. The mixture is stirred and filtered, and the filter cake is washed with water. The HPLC will show about 90% by area of desired product, and 0.2-0.5% of an N-butenyl substituted impurity. The resulting product is dried, and then boiled in alcohol, cooled, and filtered to yield buprenorphine.
-
[0025]In the alternative, norbuprenorphine can be converted to buprenorphine by reductive amination, or by acylation followed by reduction of the amide.
-
[0026]In an alternative embodiment, the hydrogenation step 2 is performed on the crude reaction mixture formed in step 1, thereby providing a one-pot reaction scheme for forming a compound according to Formula III.

-
[0027]The oripavine, methyl vinyl ketone and isopropyl alcohol are heated under pressure. Upon cooling, a Pd-C catalyst is added, and the reaction mixture is heated under pressure until the reaction is substantially complete. The product is then solubilized and the catalyst removed by filtration. The filtrate is then concentrated under vacuum.
-
[0028]In another alternative embodiment, illustrated below, a method for producing norbuprenorphine, and optionally buprenorphine, from oripavine, without the use of a protecting group on O-3. The individual reactions are as discussed in more detail above.
-
[0029]The method comprises:
- a) reacting the oripavine according to Formula I with methyl vinyl ketone to form a compound according to Formula II;

- b) hydrogenating the compound according to Formula II to form a compound according to Formula III;

- c) adding a t-butyl group to the compound according to Formula III to form a compound according to Formula X; and

- d) demethylating the nitrogen of the compound according to Formula X to form norbuprenorphine, Formula VIII.

………………………………………………………………………………………………………………………………………………………………………………………………………………………………………………………………….
WO 2009078986 A1
Th invention provides processes and intermediate compounds for producing buprenorphine. In particular, the process encompasses synthetic routes for the production of buprenorphine or derivatives of buprenorphine from norhydromorphone or derivatives of norhydromorphone. While it is envisioned that the synthetic routes described herein may be utilized to produce (+/-)-buprenorphine, in an exemplary aspect of the invention, the process encompasses the production of (+)-buprenorphine or derivatives of (+)-buprenorphine.
For purposes of illustration, Reaction Scheme 1 depicts the production of compound 8 from compound 1 in accordance with one aspect of the present invention:


………………………………………………………………………………………………………………………………………………………
- a) reacting the oripavine according to Formula I with methyl vinyl ketone to form a compound according to Formula II;





Glenmark receives final ANDA approval for Desoximetasone Ointment USP, 0.25%

Desoximetasone 382-67-2 cas

September 23, 2013: Glenmark Generics Inc., USA the subsidiary of Glenmark Generics Limited has been granted final abbreviated new drug approval (ANDA) from the United States Food
and Drug Administration (U.S. FDA) for Desoximetasone Ointment USP, 0.25% their generic version of Topicort® by Taro Pharmaceuticals USA Inc and shipping will commence immediately.
Desoximetasone Ointment is indicated for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses. According to IMS Health sales data for the
12 month period ending June 2013, Desoximetasone Ointment garnered annual sales of approximately USD 40 million.
more info
Desoximetasone is a medication belonging to the family of medications known as topical corticosteroids. It is used for the relief of various skin conditions, including rashes. It helps to reduce redness, itching, and irritation. Desoximetasone is a synthetic corticosteroid, a class of primarily synthetic steroids used as anti-inflammatory and anti-pruritic agents.
There are two brand name products:
- Topicort Emollient Cream (0.25% desoximetasone)
- Topicort LP Emollient Cream (0.05% desoximetasone)
When using desoximetasone, some of the medication may be absorbed through the skin and into the bloodstream. Too much absorption can lead to unwanted side effects elsewhere in the body. To keep this problem to a minimum, avoid using large amounts of desoximetasone over large areas, do not use it for extended periods of time, and do not cover it with airtight dressings such as plastic wrap or adhesive bandages unless specifically told to by your doctor. Children may absorb more medication than adults do. Desoximetasone is for use only on the skin and should be kept out of the eyes.
Desoximetasone can also be used to treat some types of psoriasis.


Stelara (ustekinumab) Receives FDA Approval to Treat Active Psoriatic Arthritis

Ustekinumab
| CAS No: | 815610-63-0 |
|---|---|
| Molecular Weight: | 145.64 g/mol |
| Chemical Formula: | C9H18N2O2 |
| IUPAC Name: | Immunoglobulin G1, anti-(human interleukin 12 p40 subunit) (human monoclonal CNTO 1275 gamma1-chain), disulfide with human monoclonal CNTO 1275 kappa-chain, dimer |
HORSHAM, Pa., Sept. 23, 2013 /PRNewswire/ — Janssen Biotech, Inc., announced today that the U.S. Food and Drug Administration (FDA) has approved Stelara (ustekinumab) alone or in combination with methotrexate for the treatment of adult patients (18 years or older) with active psoriatic arthritis. It is estimated that more than two million people in the U.S. are living with psoriatic arthritis, a chronic autoimmune disease characterized by both joint inflammation and psoriasis skin lesions
read all at
Ustekinumab (INN, experimental name CNTO 1275, proprietary commercial name Stelara, Centocor) is a human monoclonal antibody. It is directed against interleukin 12 and interleukin 23, naturally occurring proteins that regulate the immune system and immune-mediated inflammatory disorders.
Ustekinumab is a fully human monoclonal antibody (mAb) targeting the interleukin (IL)-12/23p40 subunit.
Interleukins are small soluble proteins that communicate between white blood cells (leukocytes), such as T cells. Interleukins mediate the differentiation, proliferation and many other processes of these cells. IL-12 and IL-23 are involved in the differentiation of naive T cells into T helper (Th) 1 and Th17 cells respectively.
Th1 and Th17 cells have been implicated in several autoimmune disorders, such as psoriasis. Ustekinumab targets the common p40 subunit of IL-12 and IL-23 to stop these cytokines from binding to their receptors and consequently preventing the development of Th1 and Th17 cells in an immune response.
In two Phase III trials for moderate to severe psoriasis, the longest >76 weeks, ustekinumab was safe and effective.
A third Phase III trial, ACCEPT, compared the efficacy and safety of ustekinumab with etanercept in the treatment of moderate to severe plaque psoriasis. This trial found a significantly higher clinical response with ustekinumab over the 12-week study period compared to high-dose etanercept. It also demonstrated the clinical benefit of ustekinumab among patients who failed to respond to etanercept.
Ustekinumab is approved in Canada, Europe and the United States to treat moderate to severe plaque psoriasis.
As of November 2009, the drug is being investigated for the treatment of psoriatic arthritis. It has also been tested in Phase II studies for multiple sclerosis and sarcoidosis, the latter versus golimumab (Simponi).
The US Food and Drug Administration (FDA) and European Union (EU) have approved the interleukin (IL) 12/23 inhibitor ustekinumab (Stelara, Janssen Biotech) for adults with active psoriatic arthritis who have not responded adequately to previous nonbiological disease-modifying antirheumatic drug therapy, the company announced today.
Approval of ustekinumab for psoriatic arthritis is “significant for patients and physicians as it marks the first treatment approved for this devastating and complex disease since the introduction of anti-TNF biologic medicines more than a decade ago,” Jerome A. Boscia, MD, vice president and head of immunology development, Janssen Research & Development, LLC, said in a statement.
The European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) recommended approval of ustekinumab for active psoriatic arthritis in June, as reported by Medscape Medical News.
Ustekinumab is already approved in the US and EU for treatment of moderate to severe psoriatic plaques in adults. The drug, which can be used alone or in combination with methotrexate, is novel in that it targets both IL-12 and IL-23.
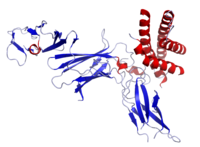
Image source: Crystal structure of human IL-12, Wikipedia, public domain

Ustekinumab binding to IL-12/23p40
EFAVIRENZ – Huahai Pharma China-Approved to Produce AIDS Treatment

Efavirenz
DMP 266
- Sustiva (USA, Bristol-Myers Squibb)
- Stocrin (EU, MSD)
- Aspen Efavirenz (Sub-Saharan Africa, Aspen Pharmacare)
- E.F (McNeil & Argus)
- Efavir (Cipla)
- Efcure (Emcure Pharmaceuticals)
- Efferven (Ranbaxy Laboratories)
- Estiva (Hetero)
- Evirenz (Alkem Laboratories)
- Viranz (Aurobindo Pharma)
Zhejiang Huahai Pharma received CFDA approval to produce efavirenz, an oral non-nucleoside reverse transcriptase inhibitor (NNRTI) used to control the symptoms of AIDS. Huahai is the first China drugmaker approved to make the drug. Huahai produced efavirenz API for Merck, which marketed the drug under the name Stocrin
read at
http://www.sinocast.com/readbeatarticle.do?id=99634
Efavirenz (EFV), sold under the brand names Sustiva among others, is a non-nucleoside reverse transcriptase inhibitor (NNRTI). It is used as part of highly active antiretroviral therapy (HAART) for the treatment of a human immunodeficiency virus (HIV) type 1. For HIV infection that has not previously been treated, the United States Department of Health and Human Services Panel on Antiretroviral Guidelines currently recommends the use of efavirenz in combination with tenofovir/emtricitabine (Truvada) as one of the preferred NNRTI-based regimens in adults and adolescents.[1] Efavirenz is also used in combination with other antiretroviral agents as part of an expanded postexposure prophylaxis regimen to reduce the risk of HIV infection in people exposed to a significant risk (e.g. needlestick injuries, certain types of unprotected sex etc.).
It is usually taken on an empty stomach at bedtime to reduce neurological and psychiatric adverse effects.
Efavirenz was combined with the HIV medications tenofovir and emtricitabine, all of which are reverse transcriptase inhibitors. This combination of three medications under the brand name Atripla, provides HAART in a single tablet taken once a day.
Efavirenz was discovered at Merck Research Laboratories. It is on the WHO Model List of Essential Medicines, the most important medication needed in a basic health system.[2] As of 2015 the cost for a typical month of medication in the United States is more than 200 USD.[3]
Efavirenz (EFV, brand names Sustiva, Stocrin, Efavir etc.) is a non-nucleoside reverse transcriptase inhibitor (NNRTI) and is used as part of highly active antiretroviral therapy(HAART) for the treatment of a human immunodeficiency virus (HIV) type 1.
For HIV infection that has not previously been treated, the United States Department of Health and Human Services Panel on Antiretroviral Guidelines currently recommends the use of efavirenz in combination with tenofovir/emtricitabine (Truvada) as one of the preferred NNRTI-based regimens in adults and adolescents.
Efavirenz is also used in combination with other antiretroviral agents as part of an expanded postexposure prophylaxis regimen to reduce the risk of HIV infection in people exposed to a significant risk (e.g. needlestick injuries, certain types of unprotected sex etc.).
The usual adult dose is 600 mg once a day. It is usually taken on an empty stomach at bedtime to reduce neurological and psychiatric adverse effects.
Efavirenz was combined with the popular HIV medication Truvada, which consists oftenofovir and emtricitabine, all of which are reverse transcriptase inhibitors. This combination of three medications approved by the U.S. Food and Drug Administration(FDA) in July 2006 under the brand name Atripla, provides HAART in a single tablet taken once a day. It results in a simplified drug regimen for many patients.

doi:10.1016/0040-4039(95)01955-H

Merck synthesis of Efavirenz
History
Efavirenz was approved by the FDA on September 21, 1998, making it the 14th approved antiretroviral drug.
-
Efavirenz is a non-nucleoside reverse trancriptase inhibitor being studied clinically for use in the treatment of HIV infections and AIDS.
- Efavirenz chemically known as (-) 6-Chloro-4-cyclopropylethynyl-4-trifluoromethyl- 1 , 4- dihydro-2H-3, 1-benzoxa zin-2-one, is a highly potent non-nucleoside reverse transcriptase inhibitor (NNRTI).A number of compounds are effective in the treatment of the human immunodeficiency virus (HIV) which is the retrovirus that causes progressive destruction of the human immune system. Effective treatment through inhibition of HIV reverse transcriptase is known for non- nucleoside based inhibitors. Benzoxazinones have been found to be useful non-nucleoside based inhibitors of HIV reverse transcriptase.(-) β-chloro^-cyclopropylethynyM-trifluoromethyl-l ,4-dihydro-2H-3,l -ben zoxazin-2-one (Efavirenz) is efficacious against HIV reverse transcriptase resistance. Due to the importance of (-)6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-l,4-dihydro-2H-3,l-ben zoxazin-2- one, economical and efficient synthetic processes for its production needs to be developed.The product patent US5519021. discloses the preparation of Efavirenz, in Example-6, column-29, involving cyclisation of racemic mixture of 2-(2-amino-5-chlorophenyl)-4- cyclopropyl-l,l,l-trifluoro-3-butyn-2-ol using l ,l ‘-carbonyldiimidazole as carbonyl delivering agent to give racemic Efavirenz. Further, resolution of the racemic Efavirenz is carried out using (-) camphanic acid chloride to yield optically pure Efavirenz. However, research article published in the Drugs of the future, 1998, 23(2), 133-141 discloses process for manufacture of optically pure Efavirenz. The process involves cyclisation of racemic 2-(2-amino-5-chlorophenyl)-4-cyclopropyl-l, 1, l-trifluoro-3-butyn-2- ol using 1, 1-carbonyldiimidazole as carbonyl delivering agent to give racemic Efavirenz and further resolution by (-) camphanic acid chloride.Similarly research article published in Synthesis 2000, No. 4, 479-495 discloses stereoselective synthesis of Efavirenz (95%yield, 99.5%ee), as shown below

Even though many prior art processes report method for the preparation of Efavirenz, each process has some limitations with respect to yield, purity, plant feasibility etc. Hence in view of the commercial importance of Efavirenz there remains need for an improved process.
- US 6 028 237 discloses a process for the manufacture of optically pure Efavirenz.
-
The synthesis of efavirenz and structurally similar reverse transcriptase inhibitors are disclosed in US Patents 5,519,021, 5,663,169, 5,665,720 and the corresponding PCT International Patent Application WO 95/20389, which published on August 3, 1995. Additionally, the asymmetric synthesis of an enantiomeric benzoxazinone by a highly enantioselective acetylide addition and cyclization sequence has been described by Thompson, et al., Tetrahedron Letters 1995, 36, 8937-8940, as well as the PCT publication, WO 96/37457, which published on November 28, 1996.
-
Additionally, several applications have been filed which disclose various aspects of the synthesis of(-)-6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-1,4-dihydro-2H-3,1-benzoxazin-2-one including: 1) a process for making the chiral alcohol, U.S.S.N. 60/035,462, filed 14 January 1997; 2) the chiral additive, U.S.S.N. 60/034,926, filed 10 January 1997; 3) the cyclization reaction, U.S.S.N. 60/037,059, filed 12 February 1997; and the anti-solvent crystallization procedure, U.S.S.N. 60/037,385 filed 5 February 1997 and U.S.S.N. 60/042,807 filed 8 April 1997.





Syntheses of EFV API; different routes of manufacturingAPI, active pharmaceutical ingredient; EFV efavirenz. BELOW
Related substances and degradants (partial listing) in EFVAPI, active pharmaceutical ingredient; CPA, cyclopropylacetylene; EFV, efavirenz
Syntheses of EFV API; different routes of manufacturingAPI, active pharmaceutical ingredient; EFV efavirenz.
Chemical properties
Efavirenz is chemically described as (S)-6-chloro-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one. Its empirical formula is C14H9ClF3NO2. Efavirenz is a white to slightly pink crystalline powder with a molecular mass of 315.68 g/mol. It is practically insoluble in water (<10 µg/mL).
History
Efavirenz was approved by the FDA on September 21, 1998, making it the 14th approved antiretroviral drug.
Society and culture
Pricing information
A one-month supply of 600 mg tablets cost approximately $550 in April 2008.[16] Merck provides efavirenz in certain developing countries at cost, currently about $0.65 per day.[17] Some emerging countries have opted to purchase Indian generics[18] such as Efavir by Cipla Ltd.[19] In Thailand, one month supply of efavirenz + truvada, as of June 2012, costs THB 2900 ($90), there’s also a social program for poorer patients who can’t afford even this price. In South Africa, a license has been granted to generics giant Aspen Pharmacare to manufacture, and distribute to Sub-Saharan Africa, a cost-effective antiretroviral drug.[20]
PATENT
http://www.google.com/patents/WO1999061026A1?cl=en
EXAMPLE 1
Cl

1a

To a solution of trifluoroethanol and (IR, 2S)-N-pyrrolidinyl norephedrine in THF (9 L) under nitrogen is added a solution of diethylzinc in hexane at 0 °C slowly enough to keep the temperature below 30 °C. The mixture is stirred at room temperature for 0.5 ~ 1 h. In another dry flask a solution of chloromagnesium cyclopropyl acetylide is prepared as follows: To neat cyclopropyl acetylene at 0 °C is added a solution of rc-butylmagnesium chloride slowly enough to keep the internal temperature < 30 °C. The solution is stirred at 0 °C for ~ 40 min and transfered to the zinc reagent via cannula with 0.36 L of THF as a wash. The mixture is cooled to -10 °C and ketoaniline la is added. The mixture is stirred at -2 to -8 °C for 35 h, warmed to room temperature, stirred for 3 h, and quenched with 30% potassium carbonate over 1.5 h. The mixture is stirred for 4 h and the solid is removed by filtration and washed with THF (2 cake volume). The wet solid still contains -18 wt% of pyrrolidinyl norephedrine and is saved for further study. The filtrate and wash are combined and treated with 30% citric acid. The two layers are separated. The organic layer is washed with water (1.5 L). The combined aqueous layers are extracted with 2.5 L of toluene and saved for norephedrine recovery. The toluene extract is combined with the organic solution and is concentrated to ~ 2.5 L. Toluene is continuously feeded and distilled till THF is not detectable by GC. The final volume is controlled at 3.9 L. Heptane (5.2 L) is added over 1 h. The slurry is cooled to 0 °C, aged for 1 h, and filtered. The solid is washed with heptane (2 cake volume) and dried to give 1.234 Kg (95.2% yield) of amino alcohol 3 as a white crystalline. The material is 99.8 A% pure and 99.3% ee.
EXAMPLE 2


To a three necked round bottom flask, equipped with a mechanical stirrer, nitrogen line, and thermocouple, was charged the solid amino alcohol 3, MTBE (500 L), and aqueous KHCO3 (45 g in 654 mL H2O). Solid 4-nitrophenyl chloroformate was added, in 4 batches, at 25°C. During the addition the solution pH was monitored. The pH was maintained between 8.5 and 4 during the reaction and ended up at 8.0. The mixture was stirred at 20-25°C for two hours. Aqueous KOH (2N) was added over 20 minutes, until the pH of the aqueous layer reached 11.0.
The layers were separated and 500 mL brine was added to the MTBE layer. 0.1 N Acetic acid was added until the pH was 6-7. The layers were separated and the organic phase was washed with brine (500 mL). At this point the mixture was solvent switched to EtOH/IPA and crystallized as recited in Examples 5 and 6.
EXAMPLE 3


To a three necked round bottom flask, equipped with a mechanical stirrer, nitrogen line, and thermocouple, was charged the solid amino alcohol 3a, toulene (500 mL), and aqueous KHCO3 (86.5 g in 500 L H2O). Phosgene solution in toulene was added at 25°C, and the mixture was stirred at 20-25°C for two hours.
The layers were separated and the organic phase was washed with brine (500 mL). At this point the mixture was solvent switched to EtOH/IPA and crystallized as recited in Examples 5 and 6.
EXAMPLE 4


To a three necked round bottom flask, equipped with a mechanical stirrer, nitrogen line, and thermocouple, was charged the solid amino alcohol 3a, MTBE (500 mL), and aqueous KHCO3 (86.5 g in 500 mL H2O). Phosgene gas was slowly passed into the solution at 25°C, until the reaction was complete.
The layers were separated and the organic phase was washed with brine (500 mL). At this point the mixture was solvent switched to EtOH/IPA and crystallized as recited in Examples 5 and 6.
EXAMPLE 5
Crystallization of efavirenz from 30% 2-Propanol in Water using a ratio of 15 ml solvent per gram efavirenz Using Controlled Anti-Solvent Addition on a 400 g Scale.
400 g. of efavirenz starting material is dissolved in 1.8 L of 2- propanol. The solution is filtered to remove extraneous matter. 1.95 L of deionized (DI) water is added to the solution over 30 to 60 minutes. 10 g. to 20 g. of efavirenz seed (Form II wetcake) is added to the solution. The seed bed is aged for 1 hour. The use of Intermig agitators is preferred to mix the slurry. If required (by the presence of extremely long crystals or a thick slurry), the slurry is wet-milled for 15 – 60 seconds. 2.25 L of DI water is added to the slurry over 4 to 6 hours. If required (by the presence of extremely long crystals or a thick slurry), the slurry is wet- milled for 15 – 60 seconds during the addition. The slurry is aged for 2 to 16 hours until the product concentration in the supernatant remains constant. The slurry is filtered to isolate a crystalline wet cake. The wet cake is washed with 1 to 2 bed volumes of 30 % 2-propanol in water and then twice with 1 bed volume of DI water each. The washed wet cake is dried under vacuum at 50°C.
EXAMPLE 6
Crystallization of efavirenz from 30% 2-Propanol in Water using a ratio of 15 ml solvent per gram efavirenz Using a Semi-Continuous Process on a 400 g Scale.
400 g. of efavirenz starting material is dissolved in 1.8 L of 2- propanol. A heel slurry is produced by mixing 20 g. of Form II efavirenz in 0.3 L of 30 % (v/v) 2-propanol in water or retaining part of a slurry froma previous crystallization in the crystallizer. The dissolved batch and 4.2 L of DI water are simultaneously charged to the heel slurry at constant rates over 6 hours to maintain a constant solvent composition in the crystallizer. Use of Intermig agitators during the crystallization is preferred. During this addition the slurry is wet-milled when the crystal lengths become excessively long or the slurry becomes too thick. The slurry is aged for 2 to 16 hours until the product concentration in the supernatant remains constant. The slurry is filtered to isolate a crystalline wet cake. The wet cake is washed with 1 to 2 bed volumes of 30 % 2-propanol in water and then twice with 1 bed volume of DI water each. The washed wet cake is dried under vacuum at 50°C.
EXAMPLE 7 Preparation of Amino Alcohol 3 and ee Upgrading— Through Process

1a

A solution of diethyl zinc in hexane was added to a solution of trifluoroethanol (429.5 g, 4.29’mol) and (IR, 2S)-N-pyrrolidinyl norephedrine (1.35 kg, 6.58 mol) in THF (9 L), under nitrogen, at 0 °C. The resulting mixture was stirred at room temperature for approx. 30 min. In another dry flask a solution of chloromagnesium- cyclopropylacetylide was prepared as follows. To a solution of n- butylmagnesium chloride in THF (2 M, 2.68 L, 5.37 mol) was added neat cyclopropylacetylene at 0 °C keeping the temperature < 25 °C. The solution was stirred at 0 °C for 1 ~ 2 h. The solution of chloromagnesiumcyclopropylacetylide was then warmed to room temperature and was transferred into the zinc reagent via cannula over 5 min followed by vessel rinse with 0.36 L of THF. The resulting mixture was aged at ~ 30 °C for 0.5 h and was then cooled to 20 °C. The ketoaniline 1 (1.00 kg, 4.47 mol) was added in one portion as a solid, and the resulting mixture was stirred at 20-28 °C for 3 h.
The reaction was quenched with 30% aq. potassium carbonate (1.2 L) and aged for 1 h. The solid waste was filtered and the cake was washed with THF (3 cake volumes). The filtrate and wash were combined and solvent switched to IP Ac.
The IPAc solution of product 3 and pyrrolidinyl norephedrine was washed with citric acid (3.5 L) and with water (1.5 L). The combined aqueous layers were extracted with IPAc (2 L) and saved for norephedrine recovery. To the combined organic layers was added
12N HC1 (405 mL, 4.88 mol), to form a thin slurry of the amino alcohol-
HC1 salt. The mixture was aged for 30 min at 25 °C and was then dried azeotropically. The slurry was aged at 25 °C for 30 min and filtered. The cake was washed with 2.5 L of IPAc and dried at 25 °C under vacuum/nitrogen for 24 h to give 1.76 kg of the wet HC1 salt.
The salt was dissolved in a mixture of MTBE (6 L) and aq Na2Cθ3 (1.18 kg in 6.25 L water). The layers were separated and the organic layer was washed with 1.25 L of water. The organic layer was then solvent switched into toluene.
Heptane (5 L) was added over 1 h at 25 °C. The slurry was cooled to 0 °C, aged for 1 h, and filtered. The solid was washed with heptane (2 cake volumes) and was dried to give 1.166 kg (90% overall yield) of amino alcohol 3 as a white crystalline solid. Norephedrine recovery
The aqueous solution was basified to pH13 using 50% aq NaOH, and extracted with heptane (2 L). The heptane solution was washed with water (1 L) and concentrated to remove residual IPAc and water. The final volume was adjusted to about 3 L. The heptane solution was cooled to -20 °C, aged for 2 h, and filtered. The solid was washed with cold heptane (1 cake volume) and dried to give 1.269 kg solid (94% recovery)




CLIPS
http://www.mdpi.com/1420-3049/21/2/221/htm



| WO2007013047A2 * | Jul 31, 2006 | Feb 1, 2007 | Ranbaxy Lab Ltd | Water-dispersible anti-retroviral pharmaceutical compositions |
| WO2007013047A3 * | Jul 31, 2006 | May 31, 2007 | Ranbaxy Lab Ltd | Water-dispersible anti-retroviral pharmaceutical compositions |
| WO2007052289A2 * | Jul 24, 2006 | May 10, 2007 | Rubicon Res Pvt Ltd | Novel dispersible tablet composition |
| WO2007052289A3 * | Jul 24, 2006 | Dec 27, 2007 | Rubicon Res Pvt Ltd | Novel dispersible tablet composition |
| WO2011131943A2 | Apr 20, 2011 | Oct 27, 2011 | Cipla Limited | Pharmaceutical compositions |
| WO2012048884A1 | Oct 14, 2011 | Apr 19, 2012 | Lonza Ltd | Process for the synthesis of cyclic carbamates |
| WO2012048886A1 | Oct 14, 2011 | Apr 19, 2012 | Lonza Ltd | Process for the synthesis of cyclic carbamates |
| WO2015059466A1 | Oct 22, 2014 | Apr 30, 2015 | Cipla Limited | Pharmaceutical compositions comprising efavirenz |
| EP1448170A2 * | Nov 26, 2002 | Aug 25, 2004 | Bristol-Myers Squibb Company | Efavirenz tablet formulation having unique biopharmaceutical characteristics |
| EP2441759A1 | Oct 14, 2010 | Apr 18, 2012 | Lonza Ltd. | Process for the synthesis of cyclic carbamates |
| EP2447255A1 | Oct 14, 2010 | May 2, 2012 | Lonza Ltd. | Process for the synthesis of cyclic carbamates |
| US6238695 | Apr 6, 1999 | May 29, 2001 | Dupont Pharmaceuticals Company | Formulation of fast-dissolving efavirenz capsules or tablets using super-disintegrants |
| US6555133 | Apr 2, 2001 | Apr 29, 2003 | Bristol-Myers Squibb Company | Formulation of fast-dissolving efavirenz capsules or tablets using super-disintegrants |
| US8871271 | Jul 29, 2013 | Oct 28, 2014 | Gilead Sciences, Inc. | Method and composition for pharmaceutical product |
| US8957204 | Oct 14, 2011 | Feb 17, 2015 | Lonza Ltd. | Process for the synthesis of cyclic carbamates |
| US8969550 | Oct 14, 2011 | Mar 3, 2015 | Lonza Ltd. | Process for the synthesis of cyclic carbamates |
| US9018192 | Oct 10, 2013 | Apr 28, 2015 | Bristol-Myers Squibb & Gilead Sciences, Inc. | Unitary pharmaceutical dosage form |
| US9198862 | Jul 24, 2006 | Dec 1, 2015 | Rubicon Research Private Limited | Dispersible tablet composition |
| WO1995020389A1 * | Jan 24, 1995 | Aug 3, 1995 | Merck & Co Inc | Benzoxazinones as inhibitors of hiv reverse transcriptase |
| WO1996037457A1 * | May 21, 1996 | Nov 28, 1996 | Merck & Co Inc | Asymmetric synthesis of (-) 6-chloro-4-cyclopropyl-ethynyl-4-trifluoromethyl-1,4-dihydro-2h-3,1-benzoxazin-2-one |
| WO1998052570A1 * | May 14, 1998 | Nov 26, 1998 | David Walter Barry | Antiviral combinations containing the carbocyclic nucleoside 1592u89 |
References
- 1 “Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents”. Retrieved 10 May 2013.
- 2
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- 3
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 62. ISBN 9781284057560.
- 4
- Cespedes, MS; Aberg, JA (2006). “Neuropsychiatric complications of antiretroviral therapy.”. Drug safety : an international journal of medical toxicology and drug experience 29 (10): 865–74. doi:10.2165/00002018-200629100-00004. PMID 16970510.
- 5
- “www.accessdata.fda.gov” (PDF).
- 6
- DHHS panel. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents (October 10, 2006). (Available for download from AIDSInfo)
- 7
- Ford, N.; Mofenson, L.; Kranzer, K.; Medu, L.; Frigati, L.; Mills, E. J.; Calmy, A. (2010). “Safety of efavirenz in first-trimester of pregnancy: A systematic review and meta-analysis of outcomes from observational cohorts”. AIDS 24 (10): 1461–1470. doi:10.1097/QAD.0b013e32833a2a14. PMID 20479637.
- 8
- Rossi, S; Yaksh, T; Bentley, H; Van Den Brande, G; Grant, I; Ellis, R (2006). “Characterization of interference with 6 commercial delta9-tetrahydrocannabinol immunoassays by efavirenz (glucuronide) in urine”. Clinical Chemistry 52 (5): 896–7. doi:10.1373/clinchem.2006.067058. PMID 16638958.
- 9
- Röder, CS; Heinrich, T; Gehrig, AK; Mikus, G (2007). “Misleading results of screening for illicit drugs during efavirenz treatment”. AIDS (London, England) 21 (10): 1390–1. doi:10.1097/QAD.0b013e32814e6b3e. PMID 17545727.
- 10
- Ren J, Bird LE, Chamberlain PP; et al. (2002). “Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors”. Proc Natl Acad Sci USA 99 (22): 14410–15. doi:10.1073/pnas.222366699. PMC 137897. PMID 12386343.
- 11
- Sustiva (efavirenz) capsules and tablets. Product information (April 2005)
- 12
- Simen AA, Ma J, Svetnik V, Mayleben D, Maynard J, Roth A, Mixson L, Mogg R, Shera D, George L, Mast TC, Beals C, Stoch A, Struyk A, Shire N, Fraser I (2014). “Efavirenz modulation of sleep spindles and sleep spectral profile”. J Sleep Res 24: 66–73. doi:10.1111/jsr.12196. PMID 25113527.
- 13
- Gatch MB, Kozlenkov A, Huang RQ, Yang W, Nguyen JD, González-Maeso J, Rice KC, France CP, Dillon GH, Forster MJ, Schetz JA (2013). “The HIV antiretroviral drug efavirenz has LSD-like properties”. Neuropsychopharmacology 38 (12): 2373–84. doi:10.1038/npp.2013.135. PMC 3799056. PMID 23702798.
- 14
- Dabaghzadeh F, Ghaeli P, Khalili H, Alimadadi A, Jafari S, Akhondzadeh S, Khazaeipour Z (2013). “Cyproheptadine for prevention of neuropsychiatric adverse effects of efavirenz: a randomized clinical trial”. AIDS Patient Care STDS 27 (3): 146–54. doi:10.1089/apc.2012.0410. PMID 23442031.
- 15
- Dabaghzadeh F, Khalili H, Ghaeli P, Dashti-Khavidaki S (2012). “Potential benefits of cyproheptadine in HIV-positive patients under treatment with antiretroviral drugs including efavirenz”. Expert Opin Pharmacother 13 (18): 2613–24. doi:10.1517/14656566.2012.742887. PMID 23140169.
- 16
- Price listed on http://drugstore.com website, 4/20/2008
- 17
- “Merck & Co., Inc., Again Reduces Price of Stocrin (efavirenz) for Patients in Least Developed Countries and Countries Hardest Hit by Epidemic – Drugs.com MedNews”.
- 18
- IndiaDaily – A new trend in emerging nations – Brazil opts for Indian generic drug ignoring US pharmaceutical giant Merck’s patent on AIDS drug Efavirenz
- 19
- http://www.cipla.com
- 20
- Patrick Lumumba Osewe; Yvonne Korkoi Nkrumah; Emmanuel K. Sackey (15 June 2008). Improving Access to HIV/AIDS Medicines in Africa: Trade-Related Aspects of Intellectual Property Rights (TRIPS) Flexibilities Utilization. World Bank Publications. pp. 35–39. ISBN 978-0-8213-7544-0. Retrieved 30 June 2012.
- 21
- http://www.sustiva.com/
- 22
- http://www.medsafe.govt.nz/consumers/cmi/s/stocrin.pdf
- 23
- Drugsupdate.com generic brands list: http://www.drugsupdate.com/brand/generic/Efavirenz/87
- 24
- http://mcneilargusindia.com/
- 25
- http://www.alkemlabs.com/
- 26
- “Regast® (efavirenz) film-coated tablets.”. http://www.pharmasyntez.com (in Russian). Pharmasyntez, 2011. Retrieved 28 June 2015. External link in
|website=(help) - 27
- IOL: Thugs get high on stolen Aids drugs IOL News May 12, 2007
- 28
- Getting high on HIV drugs in S Africa. BBC News, 8 December 2008.
- 29
- ‘No Turning Back’: Teens Abuse HIV Drugs. ABC News, April 6, 2009.
- 30
- Getting High On HIV Medication Vice 7.04.2014.
- 31
- Gatch, M. B.; Kozlenkov, A.; Huang, R. Q.; Yang, W.; Nguyen, J. D.; González-Maeso, J.; Rice, K. C.; France, C. P.; Dillon, G. H.; Forster, M. J.; Schetz, J. A. (2013). “The HIV Antiretroviral Drug Efavirenz has LSD-Like Properties”. Neuropsychopharmacology 38 (12): 2373–84. doi:10.1038/npp.2013.135. PMC 3799056. PMID 23702798.
- Sütterlin, S.; Vögele, C.; Gauggel, S. (2010). “Neuropsychiatric complications of Efavirenz therapy: suggestions for a new research paradigm”. The Journal of Neuropsychiatry and Clinical Neurosciences 22 (4): 361–369. doi:10.1176/jnp.2010.22.4.361.
External links


|
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(4S)-6-chloro-4-(2-cyclopropylethynyl)-4-(trifluoromethyl)-2,4-dihydro-1H-3,1-benzoxazin-2-one
|
|
| Clinical data | |
| Trade names | Sustiva, Stocrin, others |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699004 |
| Pregnancy category |
|
| Routes of administration |
By mouth (capsules, tablets) |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 40–45% (under fasting conditions) |
| Protein binding | 99.5–99.75% |
| Metabolism | Hepatic (CYP2A6 and CYP2B6-mediated) |
| Onset of action | 3–5 hours |
| Biological half-life | 40–55 hours |
| Excretion | Urine (14–34%) and feces (16–61%) |
| Identifiers | |
| CAS Number | 154598-52-4 |
| ATC code | J05AG03 (WHO) |
| PubChem | CID 64139 |
| DrugBank | DB00625 |
| ChemSpider | 57715 |
| UNII | JE6H2O27P8 |
| KEGG | D00896 |
| ChEBI | CHEBI:119486 |
| ChEMBL | CHEMBL223228 |
| NIAID ChemDB | 032934 |
| PDB ligand ID | EFZ (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C14H9ClF3NO2 |
| Molar mass | 315.675 g/mol |



VITAMINS, COMMON INFORMATION

A vitamin (US /ˈvaɪtəmɪn/ or UK /ˈvɪtəmɪn/) is an organic compound required by an organism as a vital nutrient in limited amounts. An organic chemical compound (or related set of compounds) is called a vitamin when it cannot be synthesized in sufficient quantities by an organism, and must be obtained from the diet. Thus, the term is conditional both on the circumstances and on the particular organism. For example, ascorbic acid (vitamin C) is a vitamin for humans, but not for most other animals, and biotin (vitamin H) and vitamin D are required in the human diet only in certain circumstances.







|
Vitamin A – discovered in 1913 What it does:
Foods that have vitamin A:
Deficiency problems:
|
 |
|
What it does:
Foods that have vitamin D:
Deficiency problems:
|
 |
|
What it does:
Foods that have vitamin E:
Deficiency problems:
|
 |
|
Vitamin K – made by bacteria in our intestines What it does:
Foods that have vitamin K:
Deficiency problems:
|
 |
……….
By convention, the term vitamin includes neither other essential nutrients, such as dietary minerals, essential fatty acids, or essential amino acids (which are needed in larger amounts than vitamins) nor the large number of other nutrients that promote health but are otherwise required less often. Thirteen vitamins are universally recognized at present.
Vitamins are classified by their biological and chemical activity, not their structure. Thus, each “vitamin” refers to a number of vitamer compounds that all show the biological activity associated with a particular vitamin. Such a set of chemicals is grouped under an alphabetized vitamin “generic descriptor” title, such as “vitamin A“, which includes the compounds retinal, retinol, and four known carotenoids. Vitamers by definition are convertible to the active form of the vitamin in the body, and are sometimes inter-convertible to one another, as well.
itamins have diverse biochemical functions. Some, such as vitamin D, have hormone-like functions as regulators of mineral metabolism, or regulators of cell and tissue growth and differentiation (such as some forms of vitamin A). Others function as antioxidants (e.g., vitamin E and sometimesvitamin C). The largest number of vitamins, the B complex vitamins, function as precursors for enzyme cofactors, that help enzymes in their work as catalysts in metabolism. In this role, vitamins may be tightly bound to enzymes as part of prosthetic groups: For example, biotin is part of enzymes involved in making fatty acids. They may also be less tightly bound to enzyme catalysts as coenzymes, detachable molecules that function to carry chemical groups or electrons between molecules. For example, folic acid may carry methyl, formyl, and methylene groups in the cell. Although these roles in assisting enzyme-substrate reactions are vitamins’ best-known function, the other vitamin functions are equally important.

Until the mid-1930s, when the first commercial yeast-extract vitamin B complex and semi-synthetic vitamin C supplement tablets were sold, vitamins were obtained solely through food intake, and changes in diet (which, for example, could occur during a particular growing season) usually greatly altered the types and amounts of vitamins ingested. However, vitamins have been produced as commodity chemicals and made widely available as inexpensive semisynthetic and synthetic-source multivitamin dietary and food supplements and additives, since the middle of the 20th century.,,,,,,,

List of vitamins
Each vitamin is typically used in multiple reactions, and, therefore, most have multiple functions.
| Vitamin generic
descriptor name |
Vitamerchemical name(s) (list not complete) | Solubility | Recommended dietary allowances
(male, age 19–70)[6] |
Deficiency disease | Upper Intake Level
(UL/day)[6] |
Overdose disease | Food sources |
|---|---|---|---|---|---|---|---|
| Vitamin A | Retinol, retinal, and
four carotenoids including beta carotene |
Fat | 900 µg | Night-blindness,Hyperkeratosis, andKeratomalacia[7] | 3,000 µg | Hypervitaminosis A | Orange, ripe yellow fruits, leafy vegetables, carrots, pumpkin, squash, spinach, liver, soy milk, milk |
| Vitamin B1 | Thiamine | Water | 1.2 mg | Beriberi, Wernicke-Korsakoff syndrome | N/D[8] | Drowsiness or muscle relaxation with large doses.[9] | Pork, oatmeal, brown rice, vegetables, potatoes, liver, eggs |
| Vitamin B2 | Riboflavin | Water | 1.3 mg | Ariboflavinosis | N/D | Dairy products, bananas, popcorn, green beans, asparagus | |
| Vitamin B3 | Niacin, niacinamide | Water | 16.0 mg | Pellagra | 35.0 mg | Liver damage (doses > 2g/day)[10] and other problems | Meat, fish, eggs, many vegetables, mushrooms, tree nuts |
| Vitamin B5 | Pantothenic acid | Water | 5.0 mg[11] | Paresthesia | N/D | Diarrhea; possibly nausea and heartburn.[12] | Meat, broccoli, avocados |
| Vitamin B6 | Pyridoxine,pyridoxamine,pyridoxal | Water | 1.3–1.7 mg | Anemia[13] peripheral neuropathy. | 100 mg | Impairment ofproprioception, nerve damage (doses > 100 mg/day) | Meat, vegetables, tree nuts, bananas |
| Vitamin B7 | Biotin | Water | 30.0 µg | Dermatitis, enteritis | N/D | Raw egg yolk, liver, peanuts, certain vegetables | |
| Vitamin B9 | Folic acid, folinic acid | Water | 400 µg | Megaloblastic anemiaand Deficiency during pregnancy is associated with birth defects, such as neural tube defects | 1,000 µg | May mask symptoms of vitamin B12 deficiency;other effects. | Leafy vegetables, pasta, bread, cereal, liver |
| Vitamin B12 | Cyanocobalamin,hydroxycobalamin,methylcobalamin | Water | 2.4 µg | Megaloblastic anemia[14] | N/D | Acne-like rash [causality is not conclusively established]. | Meat and other animal products |
| Vitamin C | Ascorbic acid | Water | 90.0 mg | Scurvy | 2,000 mg | Vitamin C megadosage | Many fruits and vegetables, liver |
| Vitamin D | Cholecalciferol | Fat | 10 µg[15] | Rickets andOsteomalacia | 50 µg | Hypervitaminosis D | Fish, eggs, liver, mushrooms |
| Vitamin E | Tocopherols,tocotrienols | Fat | 15.0 mg | Deficiency is very rare; mild hemolytic anemiain newborn infants.[16] | 1,000 mg | Increased congestive heart failure seen in one large randomized study.[17] | Many fruits and vegetables, nuts and seeds |
| Vitamin K | phylloquinone,menaquinones | Fat | 120 µg | Bleeding diathesis | N/D | Increases coagulation in patients taking warfarin.[18] | Leafy green vegetables such as spinach, egg yolks, liver |

Watson Files ANDA for Ranbaxy’s Absorica

isotretinoin
RANBAXY RECEIVES PARAGRAPH IV CERTIFICATION
Gurgaon, India, Sept. 19, 2013 – Ranbaxy Laboratories Inc. (RLI), a wholly owned subsidiary of Ranbaxy Laboratories Limited, today announced that the company has received a Paragraph IV Certification Notice of filing from Watson Laboratories Inc. of an Abbreviated New Drug Application (“ANDA”) to the U.S. Food and Drug Administration (“FDA”) for a generic version of Absorica™ (isotretinoin capsules), a product that is licensed from Cipher Pharmaceuticals Inc. (TSX: DND) (”Cipher”) of Mississauga, Ontario. read all a thttp://www.pharmalive.com/watson-files-anda-for-ranbaxys-absorica
Isotretinoin, INN, /ˌaɪsoʊtrɨˈtɪnoʊ.ɨn/, first marketed as Accutane by Hoffmann-La Roche, is a medication primarily to curecystic acne. Rarely, it is also used to prevent certain skin cancers (squamous-cell carcinoma), and can be used in the treatment of brain, pancreatic and other cancers. It is used to treat harlequin-type ichthyosis, a usually lethal skin disease, and lamellar ichthyosis. It is a retinoid, meaning it is related to vitamin A, and is found in small quantities naturally in the body.
Isotretinoin is currently the standard of care for treatment of severe, scarring cystic acne. The most common adverse effects are a transient worsening of acne (lasting 2–3 weeks), dry lips (cheilitis), dry skin, and a propensity to sunburn easily. Other side effects are rare but do include: muscle aches and pains (myalgias), headaches. Isotretinoin is known to cause birth defectsdue to in utero exposure because of the molecule’s close resemblance to retinoic acid, a natural vitamin A derivative which controls normal embryonic development.
In the United States a special procedure is required to obtain the pharmaceutical. In most other countries a consent form is required which explains these risks. Women taking isotretinoin must not get pregnant during, and for 1 month after isotretinoin therapy. Sexual abstinence, or effective contraception is mandatory during this period. Barrier methods by themselves (such as condoms) are not considered adequate due to the unacceptable failure rates of approximately 3%. Women who fall pregnant whilst on isotretinoin therapy are generally counselled to have a termination. Isotretionin has no effect on male reproduction.
There is little evidence in the medical literature linking isotretinoin use with depression and suicide. Despite this, there exists a popular misconception amongst the public that isotretinoin use commonly causes depression.
In 2009, Roche decided to remove Accutane from the US market after juries had awarded millions of dollars in damages to former Accutane users over inflammatory bowel disease claims. Other common brands are Roaccutane (Hoffman-La Roche, known as Accutane in the United States before July 2009), Amnesteem (Mylan), Claravis (Barr), Isotroin (Cipla) or Sotret(Ranbaxy).

{kind=link}