GENERAL STRUCTURE

3-[4-(5-methyl-1,3,4-oxadiazol-2-yl)phenoxy]-5-[[(3R)-1-methyl-2-oxo-3-pyrrolidinyl]oxy]-N-2-thiazolyl- Benzamide
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide……S CONF…..WO2011013141A2
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide…..R CONF…..WO2011013141A2
CAS 1263402-84-1 R CONF
CAS 1263402-76-1 S CONF
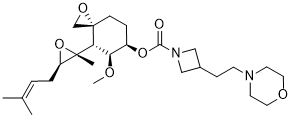
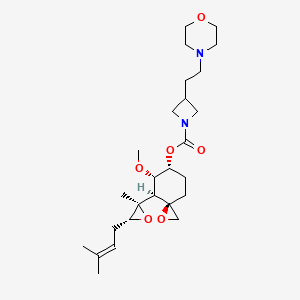
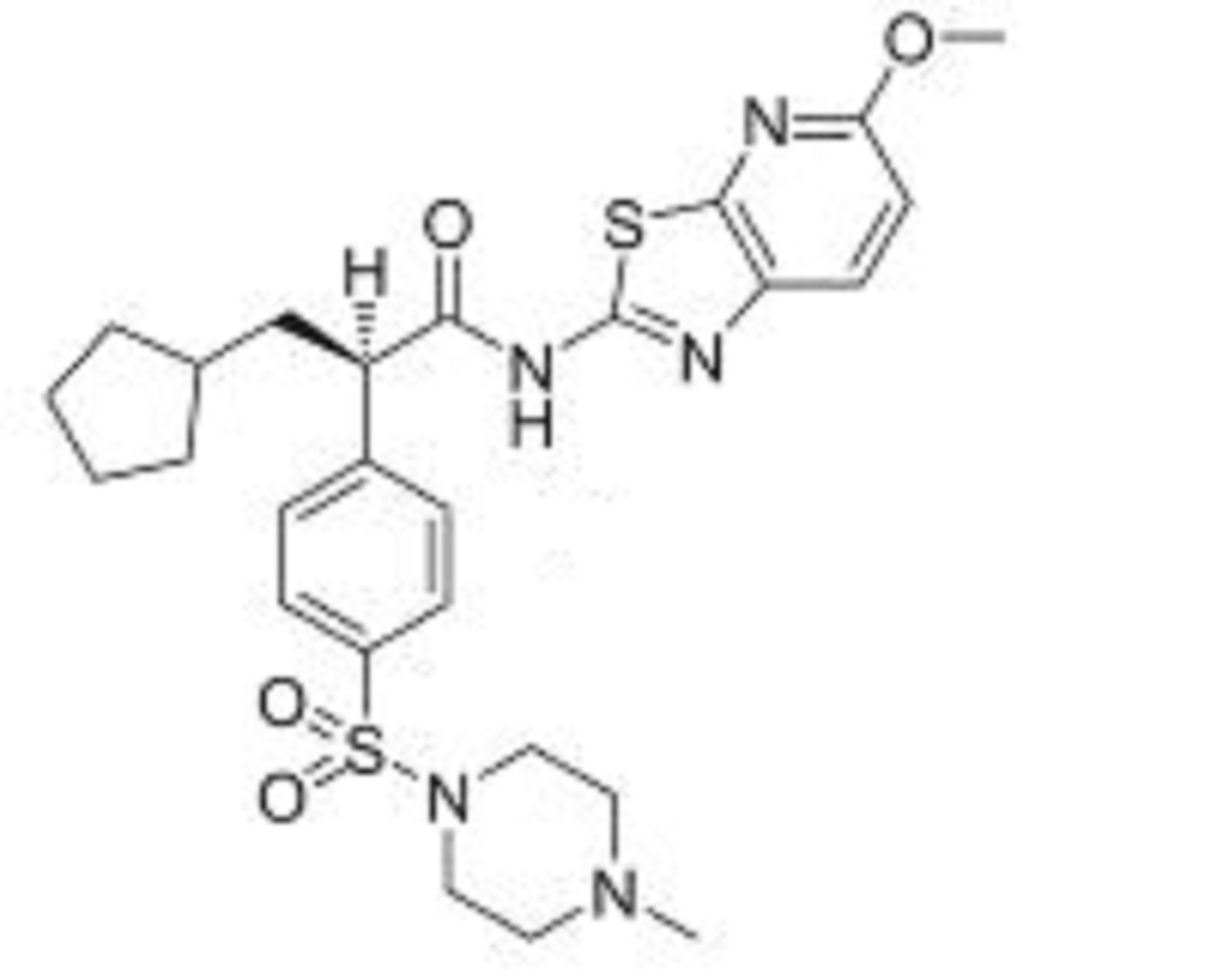
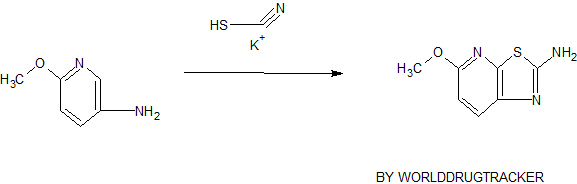
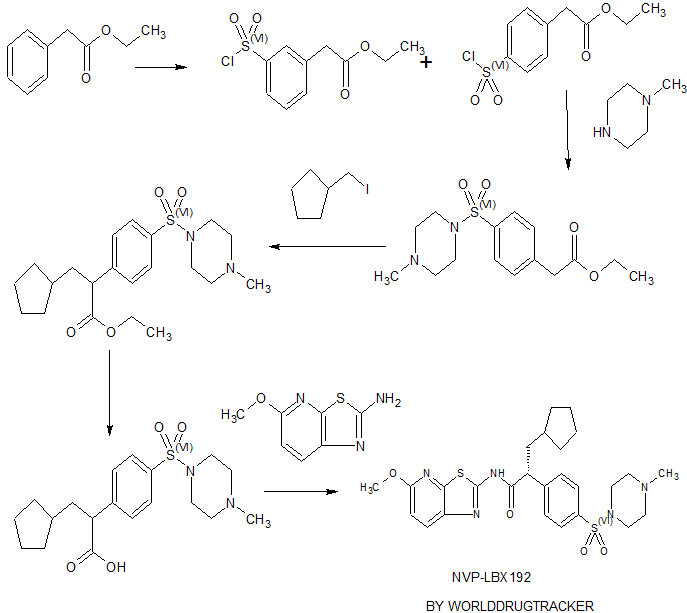
ZYD 1/ZYDPLA 1……….Probable Representative structure only, I will modify it as per available info
Watch out on this post as I get to correct structure………..

Cadila Healthcare Limited

ZYDPLA1 is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre, the NCE research wing of Zydus. ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents. It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1.
By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
In October 2013, Zydus received IND approval from the US FDA to initiate a phase I trial in type II diabetes
Clinical trials..Type 2 Diabetes Mellitus
NCT01972893; ZYD1/1001;
CTRI/2011/04/001684;
ZYD1
ZYD1/1001
ZYD1 is a novel GLP-1 receptor agonist. The ZYD1 exhibits increased stability to proteolytic cleavage, especially against dipeptidyl peptidase-4 (DPP-IV).ZYD1 is a potent antidiabetic agent without gastrointestinal side-effects. A first in human (FIH) Phase I study intends to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of ZYD1 in normal healthy adult volunteers……..https://clinicaltrials.gov/show/NCT01972893
A randomized, double blind, placebo controlled Phase I clinical study to evaluate the safety, tolerability and pharmacokinetics of ZYD1, a selective GLP-1 agonist, following the subcutaneous administrations in healthy volunteers …………http://www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=2263&EncHid=&modid=&compid=%27,%272263det%27
Some clippings I found

ONE MORE……………


Zydus announces data presentations on ZYDPLA1 “A once-weekly small molecule DPP-IV inhibitor for treating diabetes”, at the ENDO conference in Chicago, Illinois, USA. Ahmedabad, India June 9, 2014 The Zydus group will be presenting data on its molecule ZYDPLA1 a novel compound in the Gliptin class of anti-diabetic agents during the joint meeting of the International Society of Endocrinology and the Endocrine Society: ICE/ENDO 2014 to be held from June 21-24, 2014 in Chicago, Illinois.
ZYDPLA1, currently in Phase I clinical evaluation in USA, is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre. ZYDPLA1 works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP- 1 levels, ZYDPLA1 glucose-dependently increases insulin secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
The Chairman & Managing Director of Zydus, Mr. Pankaj R. Patel said, “Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.”
The abstract of Poster Number: LB-PP02-4 can also be viewed on the ENDO web program at https://endo.confex.com/endo/2014endo/webprogram/authora.html. The Poster Preview is scheduled on Sunday, June 22, 2014 at McCormick Place West.
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPP IV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
About Zydus
Headquartered in Ahmedabad, India, Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 16,000 people worldwide including over 1100 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global researchbased pharmaceutical company by 2020. The group has a strong research pipeline of NCEs, biologics and vaccines which are in various stages of clinical trials including late stage.
About Zydus Research Centre
The Zydus Research Centre has over 20 discovery programmes in the areas of cardio-metabolic disorders, pain, inflammation and oncology. Zydus has in-house capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. The Zydus Research group had identified and developed Lipaglyn™ (Saroglitazar) which has now become India’s first NCE to reach the market. Lipaglyn™ is a breakthrough therapy in the treatment of diabetic dyslipidemia and Hypertriglyceridemia. The company recently announced the commencement of Phase III trials of LipaglynTM (Saroglitazar) in patients suffering from Lipodystrophy.
PATENT
http://www.google.com/patents/WO2011013141A2?cl=en
Rajendra Kharul, Mukul R. Jain, Pankaj R. Patel
Substituted benzamide derivatives as glucokinase (gk) activators

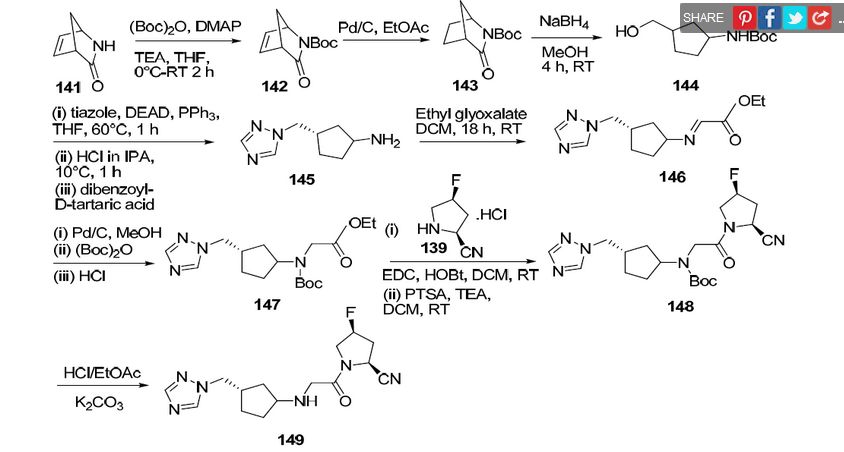
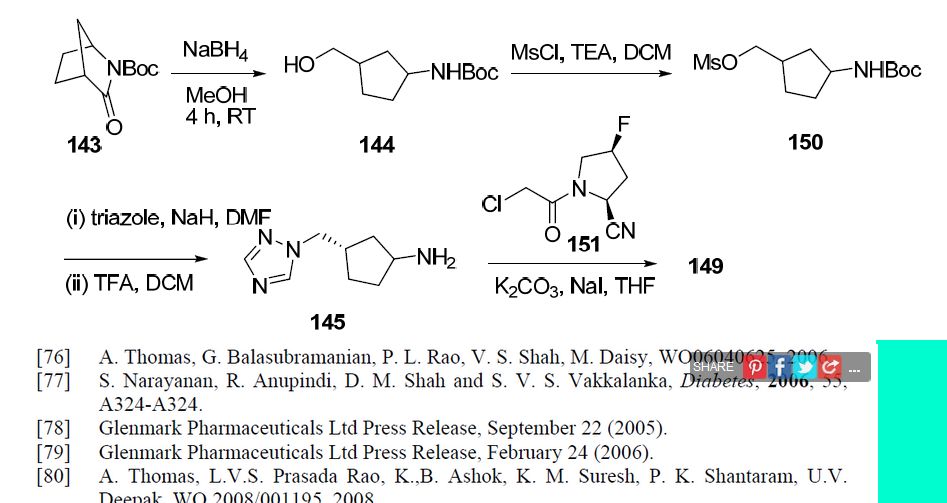
Scheme 2:
Scheme 3:
Scheme 4A:

Scheme 4B.
] Scheme 5 A:
Scheme 5B:
Scheme 6:

Example 1
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
4-(Dimethylamino)pyridine (DMAP) (0.149 g), N-(3-Dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDCI.HC1) (0.524 g) were added to a solution of 3-
( 1 -Methoxypropan-2-yloxy)-5-(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl) phenoxy) benzoic acid (0.5 g) (Intermediate 1) in dry DCM under nitrogen at 0-5 0C. 2-Aminothiazole (0.134 g) was added and the mixture was stirred for 16 h at room temperature. It was diluted with commercially available DCM. Organic phase was washed with dil HCl, saturated solution of NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude residue. The residue was chromatographed using silica gel as stationary phase and MeOH: CHCl3 gradient as mobile phase up to yield the product (0.3 g) as a white solid.
1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.92-2.01 (m, 1 H), 2.59 (s, 3 H), 2.60-2.65 (m,
I H), 2.79 (s, 3 H), 3.31-3.34 (m, 1 H), 3.36-3.44 (m ,1 H), 5.15 (t, J = 7.6 Hz, 1 H),
7.08 (s, 1 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.27-7.29 (m, 1 H), 7.40 (s, 1 H), 7.54 (s, 1 H),
7.62 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H), 12.60 (bs, 1 H); ESI-MS mix (relative intensities): 492.03 (M+H)+ (100 %), 514.02 (M+Na)+(15 %); UPLC Purity: 93.59 %, Rettime: 3.59 min.
Intermediate 1: 3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxo pyrrolidin -3-yloxy)benzoic acid
A solution of Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl- 2-oxopyrrolidin-3-yloxy)benzoate (7 g) (Intermediate 2) in a mixture of THF and methanol (1 :1 ratio) was treated with a solution of sodium hydroxide (2 g) in water and the reaction mixture was stirred for 1 h at room temperature. The resulting solution was concentrated under vacuum to remove THF and methanol, diluted with water, and washed with EtOAc. The aqueous phase was cooled and acidified with 0.1 N HCl and extracted with DCM, combined organic extracts washed with brine, dried over Na2SO4 and concentrated in vacuo to give the product (3.5 g) as white solid.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.20-2.27 (m, 1 H), 2.59-2.67 (m, 1 H), 2.77 (s, 3 H), 2.95 (s, 3 H), 3.38-3.44 (m, 1 H), 3.49-3.54 (m, 1 H), 4.96 (t, J = 7.2 Hz, 1 H), 6.93-6.95 (m, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.32-7.34 (m, 1 H), 7.52 (d, J= 8.8 Hz, 2 H), 9.96-9.98 (m, 2 H); ESI-MS (relative intensities): 431.9 (M+ Na)+ (70%).
Intermediate 2: Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2- oxo- pyrrolidin-3-yloxy)benzoate
To a stirred mixture of Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy) benzoate (15 g) (Intermediate 3), N,N-dimethylglycine hydrochloride (2.3 g), copper (II) iodide (1 g) in dry 1,4-dioxane was added 2-(4-iodophenyl)-5 -methyl- 1,3,4- oxadiazole (15.4 g) (Intermediate 4) under nitrogen. The reaction mixture was refluxed for 24 h. The reaction mixture was cooled, quenched with water and extracted with DCM. Combined organic washings were washed with water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude product. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether (9:1) as mobile phase to give the product (7 g) as thick liquid. 1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.91-1.98 (m, 1 H), 2.49-2.54 (m, 1 H), 2.56 (s, 3 H), 2.77 (s, 3 H), 3.34-3.41 (m, 2 H), 3.81 (s, 3 H), 5.12 (t, J= 7.6 Hz, 1 H), 7.13- 7.15 (m, 2 H), 7.22 (d, J = 8.8 Hz, 2 H), 7.42 (s, 1 H), 7.97 (d, J = 8.8 Hz, 2 H); ESI- MS (relative intensities): 423.9 (M+H)+ (100%), 446.2 (M+ Na)+ (30%).
Intermediate 3: Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy)benzoate
To a stirred solution of Methyl 3, 5-dihydroxybenzoate (20 g) [CAS No. 2150- 44-9] in dry DMF was added potassium carbonate (48 g) and the suspension stirred at ambient temperature under nitrogen. To this 3-Bromo-l-methyl-pyrrolidin-2-one (4Og) (Intermediate 5) [J. Med. Chem., 1987, 30, 1995-98] was added in three equal portions in 4 h intervals at room temperature and stirred overnight at ambient temperature. It was then quenched with water. The aqueous suspension was extracted with DCM. The combined extracts were washed with water, brine, dried over Na2SO4, and filtered, concentrated under reduced pressure to get the thick liquid residue. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether as a mobile phase to yield the product as white solid (15 g).1H NMR (CDCl3, 400 MHz) δ ppm: 2.08-2.10 (m, 1 H), 2.60-2.67 (m, 1 H), 3.04 (s, 3 H), 3.40-
3.43 (m, 1 H), 3.48-3.51 (m, 1 H), 3.87 (s, 3 H), 4.91 (t, J = 7.2 Hz, 1 H), 6.59- 6.61 (m, 1 H), 7.07-7.09 (m, 1 H), 7.09-7.13 (m, 1 H), 8.02 (s, 1 H); ESI-MS (relative intensities): 287.9 (M+ Na)+ (30%).
Example 68…. S CONFIGURATION
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of S-(-)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5- [(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (3.5 g) (Intermediate 13) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, 4- (dimethylamino)pyridine (2.24 g) followed by N-(3-Dimethy lam inopropy I)-N5– ethylcarbodiimide hydrochloride (EDCI. HCl) (3.3 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.94 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (200 mL), washed with dil HCl (20 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (3.5 g). The crude brown solid was purified by solvent trituration.
1H ΝMR (CDCl3, 400 MHz) δ ppm: 2.13-2.22 (m, 1 H), 2.62 (s, 3 H), 2.56-2.64 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.92 (t, J= 7.2 Hz, 1 H), 7.01 (s, 1 H), 7.04 (t, J= 2 Hz, 1 H), 7.21 (d, J = 8.8 Hz, 2 H), 7.26 (s, 1 H), 7.36 (s, 1 H), 7.44 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 513.8 (M+Νa)+ (10 %); UPLC Purity: 98.13 %, Ret. time: 3.577 min. Chiral Purity by HPLC: 97.31 %, Ret. time: 22.93 min. % ee: 94.62 %
Intermediate 13: S-(-)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2- oxo-pyrro- lidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 1.5 g) was added to a stirring mixture of (.S)-(-)-Methyl 3- [4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy] benzoate (5.3g) (Intermediate 14) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (3.5 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.17-2.22 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.99 (t, J= 7.2 Hz, 1 H), 6.89 (t, J = 2.4 Hz, 1 H), 7.07 (d, J = 8.8 Hz, 2 H), 7.28 (s, 1 H), 7.53 (s, 1 H), 7.95 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410 (M+H)+ (100 %); UPLC Purity: 97.85 %, Ret. time: 3.136 min. Chiral Purity by HPLC: 99.59 %, Ret. Time: 57.46 min. % ee: 99.18 %
Intermediate 14: (S) -(-) -Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate
Sodium hydride suspension (0.71 g, 50 %) was added to a stirring solution of (£)-(-)- methyl 3 -(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (5.5 g) (Intermediate 15) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.91 mL) was added and stirred till the reaction completion. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vaccuo to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product (5.3 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.14-2.21 (m, 1 H), 2.58-2.63 (m, 1 H), 2.64 (s, 3 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m , 1 H), 3.89 (s, 3 H), 4.99 (t, J = 7.2 Hz, 1 H), 6.99 (t, J = 2 Hz, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.35 (s, 1 H), 7.53 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 424.1 (M+H)+ (100 %); UPLC Purity: 96.1 1 %, Ret. time: 3.68 min. Chiral Purity by HPLC: 92.05 %, Ret. Time: 39.33 min.
Intermediate 15: (S) -(-) -Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxo pyrrolidin-3-yl)oxy) benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (7 g) (Intermediate 7) and (/?)-(+)-3-hydroxy-2-pyrrolidinone (Intermediate 16) (2.4g) in dry THF (200 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (1 1.3 g) was added. Diisopropyl azodicarboxylate (DIAD) (6.2 mL) in dry THF (10 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (6 g). 1H NMR (CDCl3, 400 MHz) δ ppm: 2.26-2.33 (m, 1 H), 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.47 (m, 1 H), 3.51-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.07 (bs, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (100 %); UPLC Purity: 98.35 %, Ret. time: 3.47 min. Chiral Purity by HPLC: 95.31 %, Ret. Time: 47.97 min. ee: 90.62 %.
Intermediate 16: (R)-(+)-3-Hydroxy-2-pyrrolidinone
To a stirring mixture of 4-Nitrobenzoic acid (21.5 g) and (5)-(-)-3-hydroxy-2- pyrrolidinone (11.8 g) (Intermediate 17) in dry THF (360 mL) taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (61.2 g) was added. To this reaction mixture, diisopropyl diazodicarboxylate (DIAD) (34 mL) was added drop wise in three portions at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was concentrated under vacuum to obtain residue. Methanol (360 mL) was added to the residue followed by potassium carbonate (10 g) at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was diluted with CHCl3 and filtered through celite. Celite bed was successively washed with 1 % MeOH:CHCl3. The filtrates were combined and concentrated to dryness to remove solvents. The residues were partitioned between EtOAc: dil. HCl (200 mL, 9:1) and stirred for 15 min. Layers were separated, aq. layer was washed with EtOAc thrice until all organic impurities were washed out. The aq. Layer was concentrated to dryness to remove the water and solid residues were obtained. The residues obtained were washed with 1-2 % MeOH: CHCl3 (3 x 100 mL), dried over sodium sulfate, filtered trough cotton, concentrated to get brown thick liquid product.
1U NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.46-2.54 (m, 1 H), 3.28-3.35 (m, IH), 3.38-3.48 (m, 1 H), 4.50 (t, J = 8.4 Hz, 1 H), 4.55 (bs, 1 H), 7.02 (bs, 1 H); [α]D25: + 68, c = l, CHCl3
Intermediate 17: (S)-(-)-3-hydroxy-2-pyrrolidinone
Cone. H2SO4 (14.8 g, 8 mL) was added drop wise over 5 min to the stirring solution of (5)-(-)-4-Amino-2-hydroxybutyric acid (15 g) [CAS No. 40371-51-5] in MeOH (95 rnL) under dry conditions using anhydrous CaCl2 guard tube. After refluxing for 4 h, the reaction mixture was allowed to cool to room temperature and diluted with water (15 mL). Potassium carbonate (24 g) was added in portions to the reaction mixture and stirred overnight (20 h). Reaction mixture was diluted with CHCl3, filtered through celite. Celite bed was thoroughly washed with 1 % MeOHiCHCl3. The filtrates were combined and evaporated to dryness to obtain thick liquid residue. The residue was subjected to aging using 1-2 % MeOHiCHCl3 and then filtered. Organic layers were combined, dried over anhydrous sodium sulphate, filtered and concentrated to obtain the white solid. (1 1.8 g)
1H NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.48-2.55 (m, 1 H), 3.30-3.35
(m, IH), 3.36-3.50 (m, 1 H), 4.34 (t, J = 8.4 Hz, 1 H), 6.51 (bs, 1 H); [α]D25: + 98, c =
1, CHCl3
Following examples (Example 70-76) were prepared by using similar procedure as that of example lwith suitable modifications as are well within the scope of a skilled person
Example 77 R CONFIGURATION
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide
CORRECTED AS (R)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of (/?j-(+)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-
[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (0.2 g) (Intermediate 18) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, N.ΛP-dimethylamino pyridine (0.060 g) followed by EDCI. HCl (0.23 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.054 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (20 mL), washed with dil HCl (5 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (0.080 g). The crude brown solid was purified by solvent trituration.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.15-2.20 (m, 1 H), 2.55-2.60 (m, 1 H), 2.62 (s, 3 H), 2.93 (s, 3 H), 3.38-3.43 (m, 1 H), 3.47-3.53 (m, 1 H), 4.91 (t, J= 6.8 Hz, 1 H), 6.99 (d, J= 8.8 Hz, 2 H), 7.10-7.14 (m, 2 H), 7.23-7.26 (m, 1 H), 7.36 (s, 1 H), 7.43 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H), 10.75 (bs, 1 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 514.0 (M+Na)+ (20 %); UPLC Purity: 95.25 %, Ret.time: 3.578 min. Chiral Purity by HPLC: 95.93 %, Ret.time: 14.17min. % ee: 91.86 %
Intermediate 18: (R)-(+)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl- 2-oxo- pyrrolidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 0.35 g) was added To a stirring mixture of (/?)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate (1.1 g) (Intermediate 19) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (0.76 g).
1H NMR (DMSO-J6, 400 MHz) δ ppm: 1.92-1.99 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 3.31 (s, 3 H), 3.32-3.40 (m, 2 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.08 (s, 1 H), 7.14 (s, 1 H), 7.23 (d, J= 8.8 Hz, 2 H), 7.40 (s, 1 H), 7.99 (d, J= 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (65 %), 410.1 (M+H)+ (100 %); UPLC Purity: 96.95 %, Ret. time: 3.12 min. Chiral Purity by HPLC: 89.04 %, Ret. Time: 48.15 min. Intermediate 19: (R)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate:
Sodium hydride suspension (0.16 g, 50 %) was added to a stirring solution of (R)- (+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (1.5 g) (Intermediate 20) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.20 mL) was added and stirred till the reaction completed. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vacuum to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product
(1.2 g).
1U NMR (DMSO-J6, 400 MHz) δ ppm: 1.95-1.98 (m, 1 H), 2.51-2.55 (m, 1 H), 2.56 (s, 3 H), 2.88 (s, 3 H), 3.29-3.34 (m, 1 H), 3.37-3.40 (m ,1 H), 3.81 (s, 3 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.13-7.17 (m, 2 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.41 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 423.9 (M+H)+ (100 %); UPLC Purity: 90.38 %, Ret. time: 3.68 min.
Intermediate 20: (R)-(+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxopyrrolidin -3-yl)oxy)benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (2.5 g) (Intermediate 7) and (5)-(-)-3-hydroxy-2-pyrrolidinone (Intermediate 17) (0.8 g) in dry THF (70 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (3.77 g) was added. Diisopropyl azodicarboxylate (DIAD) (2.1 mL) in dry THF (2 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (2 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.23-2.30 (m, 1 H); 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.46 (m, 1 H), 3.50-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (45 %); UPLC Purity: 96.40 %, Ret. time: 3.48 min. Chiral Purity by HPLC: 90.92 %, Ret. Time: 48.36 min.
Zydus announces US FDA approval for initiating Phase I clinical trials of ‘ZYDPLA1’ – a novel next generation orally active, small molecule DPP-4 inhibitor to treat Type 2 Diabetes Ahmedabad, October 23, 2013
• Zydus strengthens its cardiometabolic pipeline with the addition of ZYDPLA1
• Novel next generation New Chemical Entity (NCE) would offer once-a-week oral treatment option, a significant benefit to Type-2 diabetic patients
Close on the heels of launching Lipaglyn, the breakthrough therapy to treat diabetic dyslipidemia and India’s first NCE to reach the market, the Zydus group announced the Phase I clinical trial approval from the USFDA for ZYDPLA1 – a Next Generation, long-acting DPP-4 Inhibitor.
ZYDPLA1 is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre, the NCE research wing of Zydus. ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents.
It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen, would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.
Speaking on the new development, Mr. Pankaj R. Patel, Chairman and Managing Director, Zydus Group, said, “After a promising start with Lipaglyn, we take another big leap forward in the area of diabetic research and long term management of Type 2 diabetes. The IND approval by USFDA is another major regulatory milestone for us. We believe that ZYDPLA1 holds promise and would take us closer to our mission of reducing the burden of chronic diseases and addressing unmet medical needs in the treatment of diabetes.”
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPPIV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
About Zydus Zydus
Cadila is an innovative, global pharmaceutical company that discovers, develops, manufactures and markets a broad range of healthcare therapies. The group employs over 15,000 people worldwide and is dedicated to creating healthier communities globally. Zydus is the only Indian pharma company to launch its own patented NCE – Lipaglyn™, the world’s first drug to be approved for the treatment of diabetic dyslipidemia. It aims to be a leading global healthcare provider with a robust product pipeline, achieve sales of over $3 billion by 2015 and be a research-based pharmaceutical company by 2020.
About Zydus Research Centre
The Zydus Research Centre has over 20 discovery programmes ongoing with several candidates in the pre-clinical development stage focused on metabolic, cardiovascular, pain, inflammation and oncology therapeutic areas. With over 400 research professionals spearheading its research programme, Zydus has inhouse capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. ZYDPLA1 is the latest addition to the group’s strong research pipeline of 6 NCEs which are in various stages of clinical trials. For more information, please visit:
http://www.zyduscadila.comREFERENCES
International Society of Endocrinology and the Endocrine Society: ICE/ENDO 2014 to be held from June 21-24, 2014 in Chicago, Illinois.
Mukul R Jain, PhD1, Amit Arvind Joharapurkar, PhD1, Rajesh Bahekar, PhD2, Harilal Patel, MSc3, Samadhan Kshirsagar, MPharm1, Pradip Jadav, MSc2, Vishal Patel, MPharm1, Kartikkumar Patel, MPharm1, Vikram K Ramanathan, PhD3, Pankaj R Patel, MPharm4 and Ranjit Desai, PhD2, (1)Pharmacology and Toxicology, Zydus Research Centre, Ahmedabad, India
(2)Medicinal Chemistry, Zydus Research Centre, Ahmedabad, India
(3)Drug Metabolism and Pharmacokinetics, Zydus Research Centre, Ahmedabad, India
(4)Cadila Healthcare Limited, Ahmedabad, India
Poster Board Number: LBSU-1075
http://zyduscadila.com/wp-content/uploads/2015/09/ZYDPLA1-a-Novel-LongActing-DPP-4-Inhibitor.pdf
http://zyduscadila.com/wp-content/uploads/2015/05/PressNote23-10-13.pdf
http://zyduscadila.com/wp-content/uploads/2015/07/annual_report_14-15.pdf
http://pharmaxchange.info/press/2012/08/glucokinase-activators-gkas-in-diabetes-management/
LB-PP02-4 ZYDPLA1, a novel long-acting DPP-4 inhibitor
Jt Int Congr Endocrinol Annu Meet Endocr Soc (ICE/ENDO) (June 21-24, Chicago) 2014, Abst LBSU-1075
LB-PP02-4 ZYDPLA1, a Novel Long-Acting DPP-4 Inhibitor
Late-Breaking Abstracts
LBSU 1074-1087-Diabetes & Obesity
Translational
Hall F (McCormick Place West Building)
Poster Board LBSU-1075
Mukul R Jain, PhD1, Amit Arvind Joharapurkar, PhD1, Rajesh Bahekar, PhD1, Harilal Patel, MSc1, Samadhan Kshirsagar, MPharm1, Pradip Jadav, MSc1, Vishal Patel, MPharm1, Kartikkumar Patel, MPharm1, Vikram K Ramanathan, PhD1, Pankaj R Patel, MPharm2 and Ranjit Desai, PhD1
1Zydus Research Centre, Ahmedabad, India, 2Cadila Healthcare Limited, Ahmedabad, India
DPP-4 inhibitors inhibit degradation of glucagon like peptide-1 (GLP-1) and GIP, the endogenous incretin hormones responsible for stimulating glucose-dependent insulin secretion. ZYDPLA1 is a novel and potent DPP-4 inhibitor under clinical development for the treatment of type 2 diabetes and has shown potential for once a week administration in humans. The in vitro effect of ZYDPLA1 was assessed using recombinant DPP-4 enzyme. ZYDPLA1 competitively inhibited DPP-4 activity in vitro with an IC50 of 2.99 nM, and Ki of 9.3 nM. The calculated Koff rate for ZYDPLA1 was 5.12 × 10–5S-1. ZYDPLA1 was more than 8000 fold selective for DPP-4 relative to DPP-8, and DPP-9, and was more than 10000 fold selective relative to fibroblast activation protein in vitro. The potency of ZYDPLA1 for DPP-4 inhibition was similar across the species. In C57BL/6J mice ZYDPLA1 administration showed a potent antihyperglycemic effect in oral glucose tolerance test. This effect was mediated through elevated circulating levels of GLP-1 and insulin. Potent antihyperglycemic effect was also observed in Zucker fatty rats following meal tolerance test. Significant DPP-4 inhibition was observed for more than 48 hours in mice and rats and up to 168 hours in dogs and non-human primates. In conclusion, ZYDPLA1 is a potent, selective inhibitor of DPPP-4 that has the potential to become once a week therapy for treatment of type 2 diabetes.
Disclosure: MRJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. AAJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. RB: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. HP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. SK: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. KP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VKR: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PRP: Chairman, Cadila Healthcare Limited, Ahmedabad, India. RD: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India.

http://www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=2263&EncHid=&modid=&compid=%27,%272263det%27
////////Dipeptidyl Peptidase IV, CD26, DPP-IV, DP-IV, Inhibitors

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






























 Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb
Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb




















 Born in the year 1950, Dr. Chaitanya Dutt holds an MD in Medicine. He practiced as a consulting physician before joining the company in 1982. Since then he has been associated with the Company. His rich experience spans in the areas of Pharma R&D, clinical research, manufacturing, quality assurance, etc. He is one of the key professionals in the top management team of the Company. He has been instrumental in setting up the Torrent Research Centre (TRC), the research wing of the Company. Under his prudent guidance and leadership, TRC has achieved tremendous progress in the areas of discovery research as well as development work on formulations. He does not hold any directorship in any other company.
Born in the year 1950, Dr. Chaitanya Dutt holds an MD in Medicine. He practiced as a consulting physician before joining the company in 1982. Since then he has been associated with the Company. His rich experience spans in the areas of Pharma R&D, clinical research, manufacturing, quality assurance, etc. He is one of the key professionals in the top management team of the Company. He has been instrumental in setting up the Torrent Research Centre (TRC), the research wing of the Company. Under his prudent guidance and leadership, TRC has achieved tremendous progress in the areas of discovery research as well as development work on formulations. He does not hold any directorship in any other company.