
Home » Posts tagged 'BIOMARIN'
Tag Archives: BIOMARIN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pegvaliase
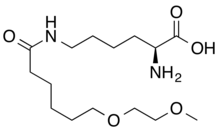
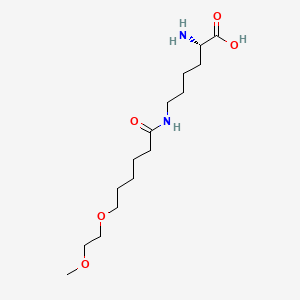

Pegvaliase
(2S)-2-amino-6-[6-(2-methoxyethoxy)hexanoylamino]hexanoic acid
CAS 1585984-95-7
- Molecular FormulaC15H30N2O5
- Average mass318.409 Da
BMN-165
Palynziq
pegvaliase
pegvaliase-pqpz
L-Lysine, N6-[6-(2-methoxyethoxy)-1-oxohexyl]-
N6-[6-(2-Methoxyethoxy)hexanoyl]-L-lysine
AUSTRALIA APPROVAL 2021
PALYNZIQ![]()
Evaluation commenced: 30 Sep 2020
Registration decision: 6 Jul 2021
Date registered: 14 Jul 2021
Approval time: 166 (175 working days)
pegvaliase
BioMarin Pharmaceutical Australia Pty Ltd
PALYNZIQ (solution for injection, pre-filled syringe) is indicated for the treatment of patients with phenylketonuria (PKU) aged 16 years and older who have inadequate blood phenylalanine control despite prior management with available treatment options.
Pegvaliase, sold under the brand name Palynziq, is a medication for the treatment of the genetic disease phenylketonuria.[2][3] Chemically, it is a pegylated derivative of the enzyme phenylalanine ammonia-lyase that metabolizes phenylalanine to reduce its blood levels.[4]
It was approved by the Food and Drug Administration for use in the United States in 2018.[2] The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[5]
Pegvaliase is a recombinant phenylalanine ammonia lyase (PAL) enzyme derived from Anabaena variabilis that converts phenylalanine to ammonia and trans-cinnamic acid. Both the U.S. Food and Drug Administration and European Medicines Agency approved pegvaliase-pqpz in May 2018 for the treatment of adult patients with phenylketonuria (PKU). Phenylketonuria is a rare autosomal recessive disorder that is characterized by deficiency of the enzyme phenylalanine hydroxylase (PAH) and affects about 1 in 10,000 to 15,000 people in the United States. PAH deficiency and inability to break down an amino acid phenylalanine (Phe) leads to elevated blood phenylalanine concentrations and accumulation of neurotoxic Phe in the brain, causing chronic intellectual, neurodevelopmental and psychiatric disabilities if untreated. Individuals with PKU also need to be under a strictly restricted diet as Phe is present in foods and products with high-intensity sweeteners. The primary goal of lifelong treatment of PKU, as recommended by the American College of Medical Genetics and Genomics (ACMG) guidelines, is to maintain blood Phe concentration in the range of 120 µmol/L to 3690 µmol/L. Pegvaliase-pqpz, or PEGylated pegvaliase, is used as a novel enzyme substitution therapy and is marketed as Palynziq for subcutanoues injection. It is advantageous over currently available management therapies for PKU, such as [DB00360], that are ineffective to many patients due to long-term adherence issues or inadequate Phe-lowering effects. The presence of a PEG moiety in pegvaliase-pqpz allows a reduced immune response and improved pharmacodynamic stability.
References
- ^ Jump up to:a b “Palynziq”. Therapeutic Goods Administration (TGA). 23 July 2021. Retrieved 5 September 2021.
- ^ Jump up to:a b “FDA approves a new treatment for PKU, a rare and serious genetic disease” (Press release). Food and Drug Administration. May 24, 2018.
- ^ Mahan KC, Gandhi MA, Anand S (April 2019). “Pegvaliase: a novel treatment option for adults with phenylketonuria”. Current Medical Research and Opinion. 35 (4): 647–651. doi:10.1080/03007995.2018.1528215. PMID 30247930.
- ^ “Palynziq”. BioMarin Pharmaceutica.
- ^ New Drug Therapy Approvals 2018 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2019. Retrieved 16 September 2020.
External links
- “Pegvaliase”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | peg val’ i ase |
| Trade names | Palynziq |
| Other names | Pegvaliase-pqpz; PEG-PAL; RAvPAL-PEG |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a618057 |
| License data | US DailyMed: Pegvaliase |
| Pregnancy category | AU: D[1] |
| Routes of administration | Subcutaneous |
| ATC code | A16AB19 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-onlyEU: Rx-only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1585984-95-7 |
| PubChem CID | 86278362 |
| DrugBank | DB12839 |
| ChemSpider | 58172730 |
| UNII | N6UAH27EUV |
| KEGG | D11077 |
| Chemical and physical data | |
| Formula | C15H30N2O5 |
| Molar mass | 318.414 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////pegvaliase, PALYNZIQ, AUSTRALIA 2021, APPROVALS 2021, BioMarin, BMN 165, Palynziq, pegvaliase, pegvaliase-pqpz
COCCOCCCCCC(=O)NCCCCC(C(=O)O)N


NEW DRUG APPROVALS
ONE TIME
$10.00
FDA Approves Vimizim to Treat Mucopolysaccharidosis Type IVA
STRUCTURAL FORMULA
Monomer
APQPPNILLL LMDDMGWGDL GVYGEPSRET PNLDRMAAEG LLFPNFYSAN 50
PLCSPSRAAL LTGRLPIRNG FYTTNAHARN AYTPQEIVGG IPDSEQLLPE 100
LLKKAGYVSK IVGKWHLGHR PQFHPLKHGF DEWFGSPNCH FGPYDNKARP 150
NIPVYRDWEM VGRYYEEFPI NLKTGEANLT QIYLQEALDF IKRQARHHPF 200
FLYWAVDATH APVYASKPFL GTSQRGRYGD AVREIDDSIG KILELLQDLH 250
VADNTFVFFT SDNGAALISA PEQGGSNGPF LCGKQTTFEG GMREPALAWW 300
PGHVTAGQVS HQLGSIMDLF TTSLALAGLT PPSDRAIDGL NLLPTLLQGR 350
LMDRPIFYYR GDTLMAATLG QHKAHFWTWT NSWENFRQGI DFCPGQNVSG 400
VTTHNLEDHT KLPLIFHLGR DPGERFPLSF ASAEYQEALS RITSVVQQHQ 450
EALVPAQPQL NVCNWAVMNW APPGCEKLGK CLTPPESIPK KCLWSH 496
Disulfide bridges
139-139′ 282-393 282′-393′ 463-492 463′-492′ 475-481 475′-481′
Modified residues
C
53 , 53′
3-oxoAla
O
CO2H
H NH2
Glycosylation sites (N)
Asn-178 Asn-178′ Asn-397 Asn-397′
Vimizim (elosufase alfa)
Elosulfase alfa nonproprietary drug name GET STRUCTURE
MOLECULAR FORMULA C5020H7588N1364O1418S34
MOLECULAR WEIGHT 110.8 kDa (peptide)
SPONSOR BioMarin Pharmaceutical Inc.
CODE DESIGNATION BMN 110, rhGALNS
CAS REGISTRY NUMBER 9025-60-9
THERAPEUTIC CLAIM Treatment of Morquio Syndrome
CHEMICAL NAMES
1. Sulfatase, chondroitin
2. Human N-acetylgalactosamine-6-sulfatase (chondroitinsulfatase, galactose-6-sulfate
sulfatase, EC=3.1.6.4) dimer (139-139′)-disulfide glycosylated (produced by CHO cells)
Company: BioMarin Pharmaceutical Inc.
Date of FDA Approval: February 14, 2014
Treatment for: Mucopolysaccharidosis Type IVA
- BMN 110
- Chondroitin 6-sulfatase
- Chondroitin sulfatase
- Chondroitin sulfate sulfatase
- Chondroitinase
- Chondrosulfatase
- E.C. 3.1.6.4
- Elosulfase alfa
- rhGALNS
- UNII-ODJ69JZG85
- Vimizim
CLINICAL….http://clinicaltrials.gov/search/intervention=Elosulfase%20alfa%20OR%20bmn%20110
Vimizim (elosufase alfa) is an enzyme replacement therapy for patients with Mucopolysaccharidosis Type IVA (Morquio A syndrome).
- FDA Advisory Committee Recommends Approval for BioMarin’s Vimizim for the Treatment of Patients With Morquio A Syndrome – November 20, 2013
Feb 16, 2014 Approval FDA Approves Vimizim to Treat Mucopolysaccharidosis Type IVA

The U.S. Food and Drug Administration today approved Vimizim (elosulfase alfa), the first FDA-approved treatment for Mucopolysaccharidosis Type IVA (Morquio A syndrome). Morquio A syndrome is a rare, autosomal recessive lysosomal storage disease caused by a deficiency in N-acetylgalactosamine-6-sulfate sulfatase (GALNS). Vimizim is intended to replace the missing GALNS enzyme involved in an important metabolic pathway. Absence of this enzyme leads to problems with bone development, growth and mobility. There are approximately 800 patients with Morquio A syndrome in the United States.
Vimizim was granted priority review. An FDA priority review provides for an expedited review of drugs for serious diseases or conditions that may offer major advances in treatment. Vimizim is also the first drug to receive the Rare Pediatric Disease Priority Review Voucher – a provision that aims to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases.
“This approval and rare pediatric disease priority review voucher underscores the agency’s commitment to making treatments available to patients with rare diseases,” said Andrew E. Mulberg, M.D., deputy director, Division of Gastroenterology and Inborn Errors Products in the FDA’s Center for Drug Evaluation and Research (CDER). “Prior to today’s approval, patients with this rare disease have had no approved drug treatment options.”
The safety and effectiveness of Vimizim were established in a clinical trial involving 176 participants with Morquio A syndrome, ranging in age from 5 to 57 years. Participants treated with Vimizim showed greater improvement in a 6-minute walk test than participants treated with placebo. On average, patients treated with Vimizim in the trial walked 22.5 meters farther in 6 minutes compared to the patients who received placebo.
The most common side effects in patients treated with Vimizim during clinical trials included fever, vomiting, headache, nausea, abdominal pain, chills and fatigue. The safety and effectiveness of Vimizim have not been established in pediatric patients less than 5 years of age. Vimizim is being approved with a boxed warning to include the risk of anaphylaxis. During clinical trials, life-threatening anaphylactic reactions occurred in some patients during Vimizim infusions.
Vimizim is marketed by Novato, Calif.-based BioMarin Pharmaceutical Inc.
Elosulfase alfa (GALNS), a proposed treatment for Morqio A syndrome. Morquio A syndrome is an inherited, autosomal recessive disease caused by a deficiency of a particular lysosomal enzyme, N- acetylgalactosamine- 6 sulfatase. BioMarin’s experimental drug for Morquio A syndrome is an enzyme replacement of elosulfase alfa (called BMN 110), which is designed to clear keratan sulfate from the lysosome. BMN 110 is being studied to determine if it is safe, if it will slow the progression of the disease and if it will improve some of the symptoms.
BioMarin started BMN 110 clinical studies in humans in 2009 to evaluate safety and efficacy. In a phase III Multicenter, Multinational, Extension Studythe Long-Term Efficacy and Safety of BMN 110 in Patients With Mucopolysaccharidosis IVA (Morquio A Syndrome) MOR-005 was evaluated. Participants will receive 2 mg/kg weekly or every other weekly dosing of study drug via infusion until the MOR- 004 study is unblinded and the optimal dose is selected. All subjects will then be treated with the optimal dose for up to approximately 5 years or until the drug is approved.
BioMarin gets US priority review for rare disease drug
![]()
31 may 2013
BioMarin will be celebrating news that regulators on both sides of the Atlantic have agreed to review Vimzin under accelerated review processes.
Vimzin is an enzyme replacement therapy for which the drugmaker is seeking approval as a treatment for the rare lysosomal storage disorder Mucopolysaccharidosis Type IVA (MPS IVA), also known as Morquio A Syndrome.
During an initial review of the Biologics License Application the US Food and Drug Administration requested extra Chemistry, Manufacturing and Controls information, and following receipt of this from BioMarin it has extended the PDUFA action date by three months to February 28 next year.
In the EU, assuming the marketing application remains on the accelerated assessment timeline, a CHMP opinion is anticipated in December this year, which means a decision from the European Commission could be handed down in the first quarter of 2014, the firm said.
The estimated prevalence of MPS IVA is around 3,000 patients in the developed world, and it is believed that around 20% or patients are in North America and 50% in Europe.
BioMarin Submits Vimizim BLA to the U.S. FDA for the Treatment of MPS IVA
![]()
Elosulfase alfa, BMN-110
STRUCTURAL FORMULA
Monomer
APQPPNILLL LMDDMGWGDL GVYGEPSRET PNLDRMAAEG LLFPNFYSAN 50
PLCSPSRAAL LTGRLPIRNG FYTTNAHARN AYTPQEIVGG IPDSEQLLPE 100
LLKKAGYVSK IVGKWHLGHR PQFHPLKHGF DEWFGSPNCH FGPYDNKARP 150
NIPVYRDWEM VGRYYEEFPI NLKTGEANLT QIYLQEALDF IKRQARHHPF 200
FLYWAVDATH APVYASKPFL GTSQRGRYGD AVREIDDSIG KILELLQDLH 250
VADNTFVFFT SDNGAALISA PEQGGSNGPF LCGKQTTFEG GMREPALAWW 300
PGHVTAGQVS HQLGSIMDLF TTSLALAGLT PPSDRAIDGL NLLPTLLQGR 350
LMDRPIFYYR GDTLMAATLG QHKAHFWTWT NSWENFRQGI DFCPGQNVSG 400
VTTHNLEDHT KLPLIFHLGR DPGERFPLSF ASAEYQEALS RITSVVQQHQ 450
EALVPAQPQL NVCNWAVMNW APPGCEKLGK CLTPPESIPK KCLWSH 496
Disulfide bridges
139-139′ 282-393 282′-393′ 463-492 463′-492′ 475-481 475′-481′
Sulfatase, chondroitin,
structure
http://www.ama-assn.org/resources/doc/usan/elosulfase-alfa.pdf
1 APRIL, 2013
BioMarin Pharmaceutical Inc. has submitted a Biologics License Application (BLA) to the U.S. Food and Drug Administration (FDA) for Vimizim (elosulfase alfa), an enzyme replacement therapy under evaluation for the treatment of patients with the rare lysosomal storage disorder mucopolysaccharidosis type IVA (MPS IVA), also called Morquio A syndrome. The company intends to submit an application for registration in the European Union (EU) by the end of April 2013.
“Based on the positive results from our Phase 3 pivotal study, we believe that Vimizim offers a substantial benefit to patients with MPS IVA, a severely debilitating and progressive disease for which there is no current treatment,” said Hank Fuchs, M.D., Chief Medical Officer of BioMarin. “The submission of the BLA represents a significant milestone for BioMarin and is the result of the strong, collaborative effort of many hard working employees, investigators, patients, and their families. With this application, BioMarin continues in its long-standing tradition of developing important therapies for those who are most in need. We look forward to working with the U.S. regulatory authorities to bring this treatment to patients.”
About MPS IVA
Mucopolysaccharidosis IVA (MPS IVA, also known as Morquio A Syndrome) is a disease characterized by deficient activity of N-acetylgalactosamine-6-sulfatase (GALNS) causing excessive lysosomal storage of glycosaminoglycans such as keratan sulfate and chondroitin sulfate. This excessive storage causes a systemic skeletal dysplasia, short stature, and joint abnormalities, which limit mobility and endurance. Malformation of the chest impairs respiratory function, and looseness of joints in the neck cause spinal instability and potentially spinal cord compression. Other symptoms may include hearing loss, corneal clouding, and heart disease. Initial symptoms often become evident in the first five years of life. The disease substantially limits both the quality and length of life of those affected.
The rate of incidence of MPS IVA is as yet unconfirmed and varies among different populations but estimates vary between 1 in 200,000 live births and 1 in 250,000 live births. The estimated prevalence is between 1,000 and 1,500 patients in the U.S., EU and Japan and between 1,500 to 2,000 patients in the rest of the world for a total of 2,500 to 3,000 patients.
About BioMarin
BioMarin develops and commercializes innovative biopharmaceuticals for serious diseases and medical conditions. The company’s product portfolio comprises four approved products and multiple clinical and pre-clinical product candidates. Approved products include Naglazyme® (galsulfase) for mucopolysaccharidosis VI (MPS VI), a product wholly developed and commercialized by BioMarin; Aldurazyme® (laronidase) for mucopolysaccharidosis I (MPS I), a product which BioMarin developed through a 50/50 joint venture with Genzyme Corporation; Kuvan® (sapropterin dihydrochloride) Tablets, for phenylketonuria (PKU), developed in partnership with Merck Serono, a division of Merck KGaA of Darmstadt, Germany; and Firdapse™ (amifampridine), which has been approved by the European Commission for the treatment of Lambert Eaton Myasthenic Syndrome (LEMS). Product candidates include BMN-110 (elosulfase alfa), formally referred to as GALNS, which successfully completed Phase III clinical development for the treatment of MPS IVA, PEG-PAL (PEGylated recombinant phenylalanine ammonia lyase), which is currently in Phase II clinical development for the treatment of PKU, BMN-701, a novel fusion protein of insulin-like growth factor 2 and acid alpha glucosidase (IGF2-GAA), which is currently in Phase I/II clinical development for the treatment of Pompe disease, BMN-673, a poly ADP-ribose polymerase (PARP) inhibitor, which is currently in Phase I/II clinical development for the treatment of genetically-defined cancers, and BMN-111, a modified C-natriuretic peptide, which is currently in Phase I clinical development for the treatment of achondroplasia. For additional information, please visit www.BMRN.com. Information on BioMarin’s website is not incorporated by reference into this press release.

{kind=link}