
Home » Posts tagged 'APPROVALS 2025' (Page 4)
Tag Archives: APPROVALS 2025
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MIRDAMETINIB
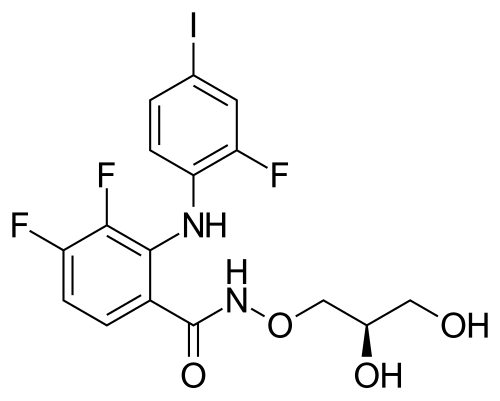

MIRDAMETINIB
391210-10-9
Chemical Formula: C16H14F3IN2O4
Molecular Weight: 482.19
PD0325901; PD 0325901; PD-325901; mirdametinib
FDA APPROVED 2/11/2025, Gomekli, To treat neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection
IUPAC/Chemical Name: (R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-((2-fluoro-4-iodophenyl)amino)benzamide
SpringWorks Therapeutics (a spin out of Pfizer ) is developing mirdametinib, a second-generation, non-ATP competitive, allosteric MEK1 and MEK2 inhibitor derived from CI-1040, for treating type 1 neurofibromatosis (NF1) and advanced solid tumors. In June 2021, a phase I/II trial was initiated in patients with low grade glioma.
- OriginatorPfizer
- DeveloperAstraZeneca; BeiGene; BIOENSIS; Pfizer; SpringWorks Therapeutics; St. Jude Childrens Research Hospital; University of Oxford
- ClassAniline compounds; Anti-inflammatories; Antineoplastics; Benzamides; Immunotherapies; Small molecules
- Mechanism of ActionMAP kinase kinase 1 inhibitors; MAP kinase kinase 2 inhibitors
- Orphan Drug StatusYes – Neurofibromatosis 1
- Phase IINeurofibromatosis 1
- Phase I/IIGlioma
- Phase ISolid tumours
- PreclinicalChronic obstructive pulmonary disease
- No development reportedCervical cancer
- DiscontinuedBreast cancer; Cancer; Colorectal cancer; Malignant melanoma; Non-small cell lung cancer
- 22 Jul 2021SpringWorks Therapeutics receives patent allowance for mirdametinib from the US Patent and Trademark Office for the treatment of Neurofibromatosis type 1-associated plexiform neurofibromas
- 16 Jun 2021SpringWorks Therapeutics and St. Jude Children’s Research Hospital agree to develop mirdametinib in USA for glioma
- 15 Jun 2021Efficacy and safety data from the phase IIb RENEU trial for Neurofibromatosis type 1-associated plexiform neurofibromas released by SpringWorks Therapeutics
Mirdametinib, sold under the brand name Gomekli, is a medication used for the treatment of people with neurofibromatosis type 1.[1] Mirdametinib is a kinase inhibitor.[1][2] It is taken by mouth.[1]
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib was approved for medical use in the United States in February 2025.[1][3]
SCHEME
SIDE CHAIN

MAIN
Medical uses
Mirdametinib is indicated for the treatment of people with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection.[1]
Adverse effects
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib can cause left ventricular dysfunction and ocular toxicity including retinal vein occlusion, retinal pigment epithelial detachment, and blurred vision.[3]
History
The efficacy of mirdametinib was evaluated in ReNeu (NCT03962543), a multicenter, single-arm trial in 114 participants aged two years of age and older (58 adults, 56 pediatric participants) with symptomatic, inoperable NF1-associated plexiform neurofibromas causing significant morbidity.[3] An inoperable plexiform neurofibromas was defined as a plexiform neurofibromas that could not be completely surgically removed without risk for substantial morbidity due to encasement or close proximity to vital structures, invasiveness, or high vascularity.[3]
The US Food and Drug Administration (FDA) granted the application for mirdametinib priority review, fast track, and orphan drug designations along with a priority review voucher.[3]
Society and culture
Legal status
Mirdametinib was approved for medical use in the United States in February 2025.[3][4][5]
PATENT
US-11066358
On July 20, 2021, SpringWorks Therapeutics announced that the United States Patent and Trademark Office (USPTO) has issued US11066358 , directed to mirdametinib , the Company’s product candidate in development for several oncology indications, including as a monotherapy for patients with neurofibromatosis type 1-associated plexiform neurofibromas (NF1-PN) and was assigned to Warner-Lambert Company (a subsidiary of Pfizer ).This patent was granted on July 20, 2021, and expires on Feb 17, 2041. Novel crystalline forms of mirdametinib and compositions comprising them are claimed.
| N—((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (“mirdametinib”, or “PD-0325901”) is a small molecule drug which has been designed to inhibit mitogen-activated protein kinase kinase 1 (“MEK1”) and mitogen-activated protein kinase kinase 2 (“MEK2”). MEK1 and MEK2 are proteins that play key roles in the mitogen-activated protein kinase (“MAPK”) signaling pathway. The MAPK pathway is critical for cell survival and proliferation, and overactivation of this pathway has been shown to lead to tumor development and growth. Mirdametinib is a highly potent and specific allosteric non-ATP-competitive inhibitor of MEK1 and MEK2. By virtue of its mechanism of action, mirdametinib leads to significantly inhibited phosphorylation of the extracellular regulated MAP kinases ERK1 and ERK2, thereby leading to impaired growth of tumor cells both in vitro and in vivo. In addition, evidence indicates that inflammatory cytokine-induced increases in MEK/ERK activity contribute to the inflammation, pain, and tissue destruction associated with rheumatoid arthritis and other inflammatory diseases. |
Example 1: Production of Essentially Pure Form IV
Lab Scale Production of Essentially Pure Form IV
| All reactions were performed in toluene other than otherwise stated. Triflic anhydride gave the best yield. |
[TABLE-US-00002]TABLE 1 Coupling Agents for Step 1Entry No.Coupling AgentYieldNotes 1Mesyl Chloridedid not react 2Benzyl chloride27Had to heat 70° C. for 166 hr34-fluorobenzensulfonylchloride27Ran 93 hrs. at 70° C.44-chlorobenzensulfonylchloride35Complete after 68 hrs. 50° C.5Tosyl Chloride36Had to heat to 70° C. for 164 hrs6Benzyl chloride52study solvent effects: DMF, DMSO, NMP – all similar DMSO fastest all complete after 110 hrs., heated to 70° C. after 66 hrs.7Triflic anhydride91Cooled to −74° C. |
| [TABLE-US-00004]TABLE 3 Yields for base deprotection ReagentYield* Methyl hydrazine85-95% Anhydrous NH3 (sparged)78-90% Anhydrous NH3 (50 psi)80-92% Aqueous NH390-97% *from PD-0333760 |
Step 2: Fluoride Displacement
Pilot Plant Preparation of Essentially Pure Form IV
Step 1: Preparation of “Side Chain”, PD-0337792
Step 2: Preparation of PD-0315209
Step 3: Preparation of PD-0325901
Polymorph Transformation
| 21.4 kg PD-0315209, 9.7 kg CDI (1.05 equiv.), 91 kg solution of 9.7% PD-0337792 in Toluene (1.1 equiv.) were used and resulted in 12.74 kg of PD-0325901 (assay 99.4%, 100% Form IV, Yield 48%). |
PATENT
WO2006134469 , claiming methods of preparing MEK inhibitor, assigned to Warner-Lambert Co .
https://patents.google.com/patent/WO2006134469A1/enThe compound Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide represented by formula 1

i is a highly specific non-ATP-competitive inhibitor of MEK1 and MEK2. The compound of formula ± (Compound I) is also known as the compound PD 0325901. Compound I is disclosed in WO 02/06213; WO 04/045617; WO 2005/040098; EP 1262176; U.S. Patent Application Pub. No. 2003/0055095 A1 ; U.S. Patent Application Pub. No. 2004/0054172 A1; U.S. Patent Application Pub. No. 2004/0147478 A1 ; and U.S. Patent Application No. 10/969,681, the disclosures of which are incorporated herein by reference in their entireties.Numerous mitogen-activated protein kinase (MAPK) signaling cascades are involved in controlling cellular processes including proliferation, differentiation, apoptosis, and stress responses. Each MAPK module consists of 3 cytoplasmic kinases: a mitogen-activated protein kinase (MAPK), a mitogen-activated protein kinase kinase (MAPKK), and a mitogen-activated protein kinase kinase kinase (MAPKKK). MEK occupies a strategic downstream position in this intracellular signaling cascade catalyzing the phosphorylation of its MAP kinase substrates, ERK1 and ERK2. Anderson et al. “Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase.” Nature 1990, v.343, pp. 651-653. In the ERK pathway, MAPKK corresponds with MEK (MAP kinase ERK Kinase) and the MAPK corresponds with ERK (Extracellular Regulated Kinase). No substrates for MEK have been identified other than ERK1 and ERK2. Seger et al. “Purification and characterization of mitogen-activated protein kinase activator(s) from epidermal growth factor-stimulated A431 cells.” J. Biol. Chem., 1992, v. 267, pp. 14373-14381. This tight selectivity in addition to the unique ability to act as a dual-specificity kinase is consistent with MEK’s central role in integration of signals into the MAPK pathway. The RAF-MEK-ERK pathway mediates proliferative and anti-apoptotic signaling from growth factors and oncogenic factors such as Ras and Raf mutant phenotypes that promote tumor growth, progression, and metastasis. By virtue of its central role in mediating the transmission of growth- promoting signals from multiple growth factor receptors, the Ras-MAP kinase cascade provides molecular targets with potentially broad therapeutic applications.One method of synthesizing Compound I is disclosed in the above-referenced WO 02/06213 andU.S. Patent Application Pub. No. 2004/0054172 A1. This method begins with the reaction of 2-fluoro-4- iodo-phenylamine and 2,3,4-trifluoro-benzoic acid in the presence of an organic base, such as lithium diisopropylamide, to form 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzoic acid, which is then reacted with (R)-0-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of a peptide coupling agent (e.g., diphenylphosphinic chloride) and a tertiary amine base (e.g., diisopropylethylamine). The resulting product is hydrolyzed under standard acidic hydrolysis conditions (e.g., p-TsOH in MeOH) to provide Compound 1. (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine is prepared by reaction of [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol with N-hydroxyphthalimide in the presence of Ph3P and diethyl azodicarboxylate.Another method of synthesizing Compound I, which is disclosed in the above-referenced U.S.Patent Application No. 10/969,681, comprises reaction of 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzoic acid with (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of N1N1– carbonyldiimidazole. The resulting product is hydrolyzed with aqueous acid and crystallized to provide polymorphic form IV of Compound I.Although the described methods are effective synthetic routes for small-scale synthesis of Compound I, there remains a need in the art for new synthetic routes that are safe, efficient and cost effective when carried out on a commercial scale.The present invention provides a new synthetic route including Steps I through Step III to the MEK inhibitor Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I).Step I: Preparation of 0-{r(4RV2.2-dimethyl-1.3-dioxolan-4-ynmethyl}hydroxylanπine (6) The method of the present invention comprises a novel Step I of preparing of 0-{[(4R)-2,2- dimethyl-1 ,3-dioxolan-4-yl]methyl}hydroxylamine (6) from [(4S)-2,2-dimethyl-1 ,3-dioxoIan-4-yl]methanol (1) through the formation of [(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methyl trifluoromethanesulfonate (3) and its coupling with N-hydroxyphthalimide (4) to afford 2-{[(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methoxy}-1 H- isoindole-1 ,3(2H)-dione (5), which is subsequently de-protected to give 6 as shown in Scheme 1.Scheme 1



The reaction of compound (1) with trifluoromethanesulfonic anhydride (2) is carried out in the presence of a non-nucleophilic base, such as, for example, a tertiary organic amine, in an aprotic solvent at a temperature of from -5O0C to 50C, preferably, at a temperature less than -150C, to form triflate (3). A preferred tertiary organic amine is triethylamine, and a preferred solvent is toluene. Treatment of triflate (3) with N-hydroxyphthalimide (4) furnishes phthalimide (5), which can be isolated if desired. However, in order to minimize processing time and increase overall yield, 0-{[(4R)- 2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) can be prepared in a one-pot process with no phthalimide (S) isolation. Cleavage of the phthalimide function could be achieved by methods known in the art, for example, by hydrazinolysis. However, the use of less hazardous aqueous or anhydrous ammonia instead of methyl hydrazine (CH3NHNH2) is preferred.Step II: Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) As shown in Scheme 2, Step Il of the method of the present invention provides 3,4-difluoro-2-(2- fluoro-4-iodophenylamino)-benzoic acid (9).Scheme 2

Preparation of compound (9) can be carried out by reacting compound (7), wherein X is halogen, or O-SC^R^ or 0-P(3O)(OR^, wherein R^ is alkyl or aryl, with compound (8) optionally in a solvent, and in the presence of from about 1 mol equivalent to about 10 mol equivalents of at least one base, wherein the base is selected from: a Group I metal cation hydride or a Group 2 metal cation hydride, including lithium hydride, sodium hydride, potassium hydride, and calcium hydride, a Group I metal cation dialkylamide or a Group 2 metal cation dialkylamide, including lithium diisopropylamide, a Group I metal cation amide or a Group 2 metal cation amide, including lithium amide, sodium amide, potassium amide, a Group I metal cation alkoxide or a Group 2 metal cation alkoxide, including sodium ethoxide, potassium terf-butoxide, and magnesium ethoxide, and a Group I metal cation hexamethyldisilazide, including lithium hexamethyldisilazide; for a time, and at a temperature, sufficient to yield compound (9).Preferably, preparation of compound (9) is carried out by reacting compound (7), wherein X is halogen, more preferably, X is fluorine, in an aprotic solvent with compound (8) in the presence of from about 3 mol equivalents to about 5 mol equivalents of a Group I metal cation amide at a temperature of from 2O C to 55°C, more preferably, at a temperature from 45°C to 55°C. A catalytic amount of Group I metal cation dialkylamide can be added if necessary. A preferred Group I metal cation amide is lithium amide, a preferred Group I metal cation dialkylamide is lithium diisopropylamide, and a preferred solvent is tetrahydrofuran. Preferably, the reaction is performed by adding a small amount of compound (7) and compound (8) to lithium amide in tetrahydrofuran followed by slow continuous addition of the remaining portion. This procedure minimizes the risk of reactor over-pressurization due to gas side product (ammonia) generation.Step III: Preparation of N-((RV2.3-dihydroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I)Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using a carboxylic acid activating reagent such as, for example, COCI2, S(O)C^, S(O)2Cl2, P(O)Cl3, triphenylphosphine/diethylazodicarboxylate, diphenylphosphinic chloride, N, N’-dicyclohexylcarbodiimide, (benzotriazol-1 -yloxy)tripyrolidinophosphonium hexafluorophosphate, (benzotriazol-1 – yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, N-ethyl-N’-(3- dimethylaminopropyl)carbodiimide hydrochloride, or 1,1′-carbonyldiimidazole (CDI).A preferred carboxylic acid activating reagent is 1,1′-carbonyldimidazole (CDI) shown in Scheme 3. Preparation of the desirable polymorphic Form IV of Compound I using CDI is described in the above- referenced U.S. Patent Application No. 10/969,681.Scheme 3

10

10 11 Compound IIn according to the present invention, the method was modified to include the advantageous procedure for product purification and isolation, which procedure is performed in single-phase systems such as, for example, toluene/acetonitrile for the first isolation/crystallization and ethanol/toluene for the second recrystallization. Water addition, implemented in the previous procedure, was omitted to avoid the two-phase crystallization from the immiscible water-toluene system that caused inconsistent product purity. The one-phase procedure of the present invention provides consistent control and removal of un- reacted starting material and side products. Alternatively, Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using thionyl chloride (SOCI2) as shown in Scheme 4.Scheme 4


Compound IExamplesThe reagents and conditions of the reactions described herein are merely illustrative of the wide variety of starting materials, their amounts and conditions which may be suitably employed in the present invention as would be appreciated by those skilled in the art, and are not intended to be limiting in any way.HPLC (Conditions A): 10 μL injection volume onto Agilent Zorbax RX-C18 150 mm x 4.6 mm x 3.5 μm column at 30°C column temperature, 1.0 mL/min flow rate and detection at 246 nm. Mobile phase A (v/v): 25 mM Acetate Buffer, pH 6.0; Mobile phase B (v/v): Acetonitrile, and Linear Gradient Table:

Sample Preparation: Dilute 100 μL reaction mixture to 10 mL with acetonitrile. Mix in a vial 200 μL of this sample solution with 300 μL carbonate buffer pH 10.0 and 300 μL solution of 2-mercaptopyridine in acetonitrile (18 mM), heat the vial for 10 minutes at 500C and dilute to 1:1 ratio in mobile phase A.GC (Conditions B): 1 μL injection onto an RTX-5 column (30 m x 0.25 mm x 0.25 μm) with initial oven temperature of 120°C for 2 min. to final temperature of 250°C in 15°C/minute ramping and a final time of 2.33 min; Flow rate: 1 mL/min.HPLC (Conditions C): 5 μL injection onto Phenomenex Luna C18(2) 150 mm x 4.6 mm x 3μm column ; flow rate : 1.0 mL/min; detection at 225 nm; mobile phase A: 95/5 v/v Water/Acetonitrile with 0.1% Trifluoroacetic acid (TFA), mobile phase B: 5/95 v/v Water/Acetonitriie with 0.1% TFA; Linear Gradient Table:

Sample preparation: Dilute 1 ml_ reaction mixture to 100 mL with acetonitrile and dilute 1 mL of this solution to 10 mL with 50:50 Water/Acetonitrile.HPLC (Conditions D): 5 μL injection onto Waters SymmetryShield RP 18, 150 mm x 4.6 mm x 3.5 μm column; flow rate: 1.0 mL/min; detection at 235 nm; mobile phase A: 25 mM Acetate Buffer adjusted to pH 5.5, mobile phase B: Acetonitrile; Linear Gradient Table:

Sample preparation: Dilute 40 μL of reaction mixture in 20 mL acetonitrile.HPLC (Conditions E): 10 μL sample injection onto YMC ODS-AQ 5 μm, 250 mm x 4.6 mm column; flow rate: 1.0 ml_/min; detection at 280 nm; temperature 30°C; mobile phase : 75/25 v/v Acetonitrile/Water with 0.1% Formic acid.Sample preparation: Quench reaction mixture sample with dipropylamine and stir for about 5 minutes before further dilution with mobile phase.DSC measurement was performed using a Mettler-Toledo DSC 822, temperature range 25° to 150°C with 5°C/min heating rate in a 40 μL aluminum pan. Experimental Conditions for Powder X-Rav Diffraction (XRD):A Rigaku Miniflex+ X-ray diffractometer was used for the acquisition of the powder XRD patterns. The instrument operates using the Cu Ka1 emission with a nickel filter at 1.50451 units. The major instrumental parameters are set or fixed at:X-ray: Cu / 30 kV (fixed) / 15 mA (fixed)Divergence Slit: Variable Scattering Slit: 4.2° (fixed) Receiving Slit: 0.3 mm (fixed) Scan Mode: FT Preset Time: 2.0 s Scan Width: 0.050° Scan Axis: 2Theta/Theta Scan Range: 3.000° to 40.000°Jade Software Version: 5.0.36(SP1) 01/05/01 (Materials Data, Inc.) Rigaku Software: Rigaku Standard Measurement for Windows 3.1 Version 3.6(1994-1995) Example 1. Preparation of 0-ffl4R)-2.2-dimethyl-1.3-dioxolan-4-vπmethyl}hvdroxylamine (6)A solution containing [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol (1) (13.54 ml_, 0.109 mol) (DAISO Co., Ltd., CAS# 22323-82-6) and triethylamine (18.2 ml_, 0.131 mol) in 115 mL toluene was cooled to -15 C, then trifluoromethanesulfonic anhydride (2) (18.34 mL, 30.75 g, 0.109 mol) (Aldrich, Catalog # 17,617-6 ) was added drop wise while maintaining the temperature at less than -15°C. The mixture was then stirred for 2 hours, and transferred to a separate flask containing a mixture (slurry) of N- hydroxyphthalimide (4) (18.99 g, 0.116 mol) (Aldrich, Catalog # H5.370-4) and 18.2 mL (0.13 mol) triethylamine in 95 mL toluene. The resulting mixture was warmed to 20-25°C and stirred for at least 5 hours or until reaction completion (determined by HPLC (Conditions A)). Water (93 mL) was then added to quench the reaction mixture, the phases were separated, and the bottom aqueous layer was discarded. The water quench was repeated two more times resulting in a pale yellow organic layer. The organic layer was heated to 35 C and treated with 36.7 mL ammonium hydroxide solution (contains about 28-29% wt/wt ammonia). The mixture was stirred for at least 12 hours or until the reaction was deemed complete as determined by GC (Conditions B). The water was then removed under reduced pressure by co- distilling it with toluene to about half of the original volume at temperatures around 35-45 C. Toluene (170 mL) was added to the concentrated solution and the distillation was repeated. A sample was drawn for water content determination by Karl Fisher method (using EM Science Aquastar AQV-2000 Titrator with a sample injected to a pot containing methanol and salicylic acid). The distillation was repeated ifl water content was more than 0.1%. The concentrated solution was filtered to remove the white solid side product, and the filtrate was stored as 112mL (98 g) product solution containing 9.7% w/w compound 6 in toluene. This solution was ready for use in the final coupling step (Example 3). Overall chemical yield was 59%. A small sample was evaporated to yield a sample for NMR identification.1H NMR (400 MHz, CDCI3): δ 5.5 (bs, 2H), 4.35 (m, 1H), 4.07 (dd, 1H), 3.77 (m, 2H), 3.69 (dd, 1H), 1.44 (s, 3H), 1.37 (s, 3H).Example 2. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9)A solution of 2-fluoro-4-iodoaniline (8) (16.4 g, 0.069 mol) (Aldrich, Catalog # 30,660-6) and 2,3,4- trifluorobenzoic acid (7) (11.98 g, 0.068 mol) (Aldrich, Cat # 33,382-4) in 38 mL tetrahydrofuran (THF) was prepared and a portion (about 5%) of this solution was added to a stirring slurry of lithium amide (5 g, 0.22 mol) in 40 mL THF at 50-55 C. After about 15-30 min. an exotherm followed by gas release and color change are observed. The remaining portion of the (8) and (7) solution was added slowly over 1-2 hr while maintaining temperatures within 45-55°C. The mixture was stirred until the reaction was deemed complete (by HPLC (Conditions C). The final mixture was then cooled to 20-25°C and transferred to another reactor containing 6 N hydrochloric acid (47 mL) followed by 25 mL acetonitrile, stirred, and the bottom aqueous phase was discarded after treatment with 40 mL 50% sodium hydroxide solution. The organic phase was concentrated under reduced pressure and 57 mL acetone was added. The mixture was heated to 50°C, stirred, and added with 25 mL warm (40-50°C) water and cooled to 25-30°C to allow crystallization to occur (within 1-4 hours). Once the crystallization occurred, the mixture was further cooled to 0 to -5°C and stirred for about 2 hours. The solid product was filtered and the wet cake was dried in vacuum oven at about 55°C. Overall chemical yield was 21.4 g, 80%. 1H NMR (400 MHz, (CD3)2SO): δ 13.74 (bs, 1H), 9.15 (m, 1 H), 7.80 (dd, 1H), 7.62 (d, 1H), 7.41 (d, 1H), 7.10 (q, 1H), 6.81 (m, 1H).Example 2B. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) by the solid addition of lithium amide methodTo a stirring solution of 2,3,4-trifluorobenzoic acid (13) (5.0 g, 28.4 mmol) and 2-fluoro-4- iodoaniline (14) (6.73 g, 28.4 mmol) in MeCN (100 mL), under N2 atmosphere was added lithium amide (2.61 g, 113.6 mmol) in small portions. The reaction mixture was heated to reflux for 45 minutes, cooled to ambient temperature and quenched with 1 N HCI and then water. The yellowish white precipitate was filtered, washed with water. The solid was triturated in CH2CI2 (30 mL) for 1h, filtered and dried in a vacuum oven at 45°C for 14 hours to give 8.Og (72%) of compound (9) as an off-white solid, mp 201.5-203 °C.Example 3. Preparation of N-((R)-2.3-dihvdroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound \)3,4-Difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (20 g, 0.051 mol) in 100 mL acetonitrile was treated with 1,1′-carbonyldiimidazole (CDI) (8.66 g, 0.053 mol) (Aldrich, Cat # 11,553-3) and stirred for about 2 hours at 20-25°C until the reaction was deemed complete by HPLC (Conditions D). 94 mL (84.9 g) of 9.7% w/w solution of O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) in toluene was then added and stirred for about 4 hours or until the reaction was deemed complete by HPLC (Conditions D). To this mixture was added 66 mL of 5.6 % hydrochloric acid solution, and after stirring, the bottom aqueous phase was discarded. Again 66 mL of 5.6 % hydrochloric acid solution was added to the organic phase and stirred at 20-25°C for 12-18 hours or until the reaction was deemed complete by HPLC (Conditions D). The bottom layer was then discarded and the remaining organic layer was concentrated under reduced pressure to remove about 10-20% solvent, and the volume was adjusted to about 9-11 mL/g with toluene (80 mL). Crude product was then crystallized at 10-15°C. The slurry was allowed to stir for about 2 hours and the crude solid product was filtered, and dried. The dried crude product was recharged to the reactor and dissolved into 150 mL of 5% v/v ethanol/toluene mixture at 55- 67°C. The solution was then clarified at this temperature through filter (line filter) to remove any remaining particulate matter. The solution was then cooled slowly to 5°C to crystallize and stirred for at least 2 h, filtered and dried. The dried solid product was redissolved in EtOH (60 mL) at 35°C, and product was precipitated out by adding water (300 mL) at 35°C followed by cooling to 200C. The slurry was stirred for at least 2 hours to transform the crystals to the desired polymorphic Form IV as determined by DSC and Powder X-ray Diffraction pattern (PXRD). The slurry was filtered and dried under vacuum oven at 70- 90°C to yield the final N-((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I) product. Overall chemical yield was 13 g, 53%. Melting point (DSC): 112+1° C. Appearance: White to off-white crystals.Shown in Figure 1, PXRD conforms to polymorphic crystal Form IV disclosed in the above mentioned U.S. Patent Application No. 10/969,681 1H NMR (400 MHz, (CD3)2SO): δ 11.89 (bs, 1H), 8.71 (bs, 1H), 7.57 (d, 1H), 7.37 (m, 2H), 7.20 (q, 1H), 6.67 (m, 1H), 4.84 (bs, 1H), 4.60 (m, 1H), 3.87 (m, 1 H), 3.7 (m, 2H), 3.34 (m, 2H).Example 4. Preparation of N-((R)-2.3-dihydroxypropoxyV3.4-difluoro-2-(2-fluoro-4-iodo-phenylanrιinoV benzamide (Compound \)To a stirring solution of 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (120 g, 0.30 mol) in a mixture of 1 mL N,N-dimethylformamide and 1000 mL toluene was added thionyl chloride (55 g, 0.462 mol). The mixture was heated to 50-65 C and stirred for 2 hours or until reaction completion as determined by HPLC (Conditions E). The final reaction mixture was then cooled and concentrated under reduced pressure to a slurry keeping the temperature below 35°C. Toluene (600 mL) was added to dissolve the slurry and vacuum distillation was repeated. Additional toluene (600 mL) was added to the slurry dissolving all solids and the solution was then cooled to 5° -10°C. The solution was then treated with O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) (63 g, 0.43 mol) solution in 207 mL toluene followed by potassium carbonate (65 g) and water (200 mL), stirred for at least 2 hours at 20- 25°C. The stirring was stopped to allow phase separation and the bottom phase was discarded. The remaining organic layer was treated with hydrochloric acid solution (7.4%, 240 mL) until pH was less than 1 and stirred for 2 hours. The final reaction mixture was slightly concentrated under vacuum collecting about 100 mL distillate and the resulting organic solution was cooled to 5°C to crystallize the product and filtered. The filter cake was washed with toluene (1000 mL) followed by water (100 mL) and the wet cake (crude product Compound I) was charged back to the flask. Toluene (100 mL), ethanol (100 mL) and water (100 mL) are then added, stirred at 30-35°C for about 15 min, and the bottom aqueous phase was discarded. Water (200 mL) was then added to the organic solution and the mixture was stirred at about 3O C to allow for crystallization. The stirring was continued for 2 hours after product crystallized, then it was further cooled to about 0°C and stirred for at least 2 hours. The slurry was filtered and wet cake was dried under reduced pressure at 55-85°C to yield the final product N-((R)-2,3-dihydroxypropoxy)-3,4- difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I) product. Overall chemical yield was 86 g, 58%.
PATENT
WO2002/006213 describes crystalline Forms I and II. U.S. Pat. No. 7,060,856 (“the ‘856 patent”)
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2002006213
| Clinical data | |
|---|---|
| Trade names | Gomekli |
| Other names | PD-0325901 |
| AHFS/Drugs.com | Gomekli |
| License data | US DailyMed: Mirdametinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | L01EE05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 391210-10-9 |
| PubChem CID | 9826528 |
| IUPHAR/BPS | 7935 |
| DrugBank | DB07101 |
| ChemSpider | 10814340 |
| UNII | 86K0J5AK6M |
| KEGG | D11675 |
| ChEBI | CHEBI:9826528 |
| ChEMBL | ChEMBL507361 |
| PDB ligand | 4BM (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C16H14F3IN2O4 |
| Molar mass | 482.198 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f “Gomekli- mirdametinib capsule; Gomekli- mirdametinib tablet, for suspension”. DailyMed. 27 February 2025. Retrieved 2 April 2025.
- ^ Armstrong AE, Belzberg AJ, Crawford JR, Hirbe AC, Wang ZJ (June 2023). “Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas”. BMC Cancer. 23 (1): 553. doi:10.1186/s12885-023-10996-y. PMC 10273716. PMID 37328781.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection”. U.S. Food and Drug Administration (FDA). 11 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “UPDATE: SpringWorks Therapeutics Announces FDA Approval of Gomekli (mirdametinib) for the Treatment of Adult and Pediatric Patients with NF1-PN” (Press release). SpringWorks Therapeutics. 12 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025 – via GlobeNewswire News Room.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 14 February 2025. Retrieved 16 February 2025.
External links
- “Mirdametinib (Code C52195)”. NCI Thesaurus.
- Clinical trial number NCT03962543 for “MEK Inhibitor Mirdametinib (PD-0325901) in Patients With Neurofibromatosis Type 1 Associated Plexiform Neurofibromas (ReNeu)” at ClinicalTrials.gov
- Moertel CL, Hirbe AC, Shuhaiber HH, Bielamowicz K, Sidhu A, Viskochil D, Weber MD, Lokku A, Smith LM, Foreman NK, Hajjar FM, McNall-Knapp RY, Weintraub L, Antony R, Franson AT, Meade J, Schiff D, Walbert T, Ambady P, Bota DA, Campen CJ, Kaur G, Klesse LJ, Maraka S, Moots PL, Nevel K, Bornhorst M, Aguilar-Bonilla A, Chagnon S, Dalvi N, Gupta P, Khatib Z, Metrock LK, Nghiemphu PL, Roberts RD, Robison NJ, Sadighi Z, Stapleton S, Babovic-Vuksanovic D, Gershon TR: ReNeu: A Pivotal, Phase IIb Trial of Mirdametinib in Adults and Children With Symptomatic Neurofibromatosis Type 1-Associated Plexiform Neurofibroma. J Clin Oncol. 2025 Feb 20;43(6):716-729. doi: 10.1200/JCO.24.01034. Epub 2024 Nov 8. [Article]
- Weiss BD, Wolters PL, Plotkin SR, Widemann BC, Tonsgard JH, Blakeley J, Allen JC, Schorry E, Korf B, Robison NJ, Goldman S, Vinks AA, Emoto C, Fukuda T, Robinson CT, Cutter G, Edwards L, Dombi E, Ratner N, Packer R, Fisher MJ: NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor Mirdametinib (PD-0325901) in Adolescents and Adults With NF1-Related Plexiform Neurofibromas. J Clin Oncol. 2021 Mar 1;39(7):797-806. doi: 10.1200/JCO.20.02220. Epub 2021 Jan 28. [Article]
- Ioannou M, Lalwani K, Ayanlaja AA, Chinnasamy V, Pratilas CA, Schreck KC: MEK Inhibition Enhances the Antitumor Effect of Radiotherapy in NF1-Deficient Glioblastoma. Mol Cancer Ther. 2024 Sep 4;23(9):1261-1272. doi: 10.1158/1535-7163.MCT-23-0510. [Article]
- FDA Approved Drug Products: GOMEKLI (mirdametinib) capsules and tablets for oral and oral suspension use (Feb 2024) [Link]
- FDA News: FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection [Link]
////////MIRDAMETINIB, Orphan Drug Status, Neurofibromatosis 1, PHASE 2, PD0325901, PD 0325901, PD-325901, FDA 2025, GOMEKLI, APPROVALS 2025
O=C(NOC[C@H](O)CO)C1=CC=C(F)C(F)=C1NC2=CC=C(I)C=C2F
Rilzabrutinib
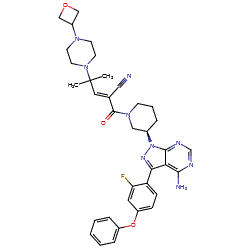
![(R)-2-(3-(4-Amino-3-(2-fluoro-4-phenoxyphenyl)-1H-pyrazolo[3,4-d]-pyrimidin-1-yl)piperidine-1-carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin-1-yl)pent-2-enenitrile.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118325989&t=l)


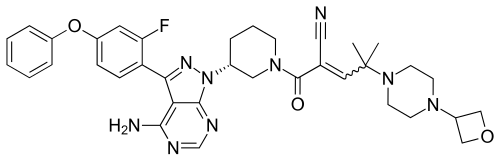
PRN 1008, Rilzabrutinib
CAS 1575591-66-0
| リルザブルチニブ; |
C36H40FN9O3,
| MW 665.7597 |
2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidine-1-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-1-yl]pent-2-enenitrile
Anti-inflammatory disease, Autoimmune disease treatment
Fda 2025, approvals 2025 8/29/2025, Wayrilz, To treat persistent or chronic immune thrombocytopenia that has not sufficiently responded to immunoglobulins, anti-D therapy, or corticosteroids
- OriginatorPrincipia Biopharma
- Class2 ring heterocyclic compounds; Amines; Anti-inflammatories; Fluorobenzenes; Nitriles; Phenyl ethers; Piperazines; Piperidines; Pyrazoles; Pyrimidines; Skin disorder therapies; Small molecules
- Mechanism of ActionAgammaglobulinaemia tyrosine kinase inhibitors
- Orphan Drug StatusYes – Idiopathic thrombocytopenic purpura; Pemphigus vulgaris
- Phase IIIIdiopathic thrombocytopenic purpura; Pemphigus vulgaris
- Phase IIAutoimmune disorders
- 02 Jun 2021Efficacy data from a phase IIa trial in Ankylosing spondylitis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
- 07 Apr 2021Sanofi initiates enrollment in a phase I pharmacokinetics trial in healthy volunteers in Australia (PO, Tablet, Capsule) (NCT04748926)
- 31 Mar 2021Sanofi announces intention to seek regulatory approval for Idiopathic thrombocytopenic purpura in 2023 (Sanofi pipeline, May 2021)
Rilzabrutinib, sold under the brand name Wayrilz, is an anti-cancer medication used for the treatment of immune thrombocytopenia.[1] Rilzabrutinib is a tyrosine kinase inhibitor.[1] It is taken by mouth.[1]
Rilzabrutinib may increase the risk of serious infections (including bacterial, viral, or fungal).[2] The most common side effects include diarrhea, nausea, headache, abdominal pain, and COVID-19.[2]
Rilzabrutinib was approved for medical use in the United States in August 2025.[2]
CLIP
Sanofi to acquire BTK inhibitor firm Principia for $3.7 billion
Principia is testing its small-molecule compounds in multiple sclerosis and immune system diseases
Sanofi will pay $3.7 billion to acquire Principia Biopharma, a San Francisco-based biotech firm developing small molecules that inhibit Bruton tyrosine kinase (BTK). The price represents about a 75% premium over Principia’s stock market value in early July, before reports surfaced that Sanofi was interested in buying the firm.
BTK is a protein important for both normal B cell development and the proliferation of lymphomas, which are B cell cancers. AbbVie, AstraZeneca, and BeiGene all market BTK inhibitors for treating specific kinds of lymphomas. Sales of AbbVie’s inhibitor, Imbruvica, approached $4.7 billion in 2019.
Other drug firms have been eager to get in on the action as well. In January, Merck & Co. spent $2.7 billion to acquire ArQule, whose experimental noncovalent BTK inhibitor is designed to overcome resistance that some cancers develop after treatment with current covalent BTK inhibitors. Eli Lilly and Company’s $8 billion acquisition of Loxo Oncology in 2019 also included a noncovalent BTK inhibitor.
BTK is also linked to inflammation, and Principia focuses on developing BTK inhibitors for immune system diseases and multiple sclerosis. Its compound rilzabrutinib is currently in clinical trials for pemphigus and immune thrombocytopenia. In 2017, Sanofi struck a deal to develop Principia’s brain-penetrant BTK inhibitor, SAR442168, for multiple sclerosis.
Sanofi announced in April of this year that the inhibitor reduced formation of new lesions—the scarred nervous tissue that gives multiple sclerosis its name—by 85% in a Phase II clinical trial. A Phase III trial of the compound began in June.
Upon announcing its deal to acquire Principia, Sanofi said that both rilzabrutinib and SAR442168 have the potential to become a “pipeline in a product,” indicating they can be used for many immune-related and neurological diseases, respectively.
The anti-inflammatory effects of BTK inhibitors have raised interest in the drugs as treatments for people hospitalized with COVID-19. Notably, the US National Cancer Institute conducted a small study suggesting acalabrutinib may help reduce the respiratory distress and inflammation in people with COVID-19. Based on that preliminary study, AstraZeneca—which markets acalabrutinib as Calquence—is conducting a 60-person randomized trial of the drug for COVID-19.
Sanofi has not indicated interest in investigating Principia’s BTK inhibitors as COVID-19 treatments.Chemical & Engineering NewsISSN 0009-2347
PATENT
WO 2021127231https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021127231&tab=PCTDESCRIPTION&_cid=P20-KRA0I9-18818-1
SOLID FORMS OF 2-[3-[4-AMTNO-3-(2-FT,TTORO-4-PHENOXY- PHEN¥L)PYRAZOLO[3,4 D]PYRIMIDIN l~YL]PIPERIDINE~l~CARBON¥L] 4~
METHYL-4-[4-(OXETAN-3-YL)PIPERAZIN-l-YLjPENT-2-ENENITRILE
[11 This application claims the benefit of priority to U.S. Provisional Application
No 62/951,958, filed December 20, 2019, and U.S Provisional Application No. 63/122,309, filed December 7, 2020, the contents of each of which are incorporated by reference herein in their entirety.
[2] Disclosed herein are solid forms of 2-[3-[4~amino-3~(2~fluoro-4-phenoxy-plienyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l Carbonyl]~4-nietliyl-4~[4-(oxetaii~3-yl)piperazin-!~yi]pent-2~enenitriie (Compound (I)), methods of using the same, and processes for making Compound (I), including its solid forms. The solid forms of Compound (I) may be inhibitors of Bruton’s tyrosine kinase (BTK) comprising low residual solvent content.
[3| The enzyme BTK is a member of the Tec family non-receptor tyrosine kinases.
BTK is expressed in most hematopoietic cells, including B cells, mast cells, and macrophages BTK plays a role in the development and activation of B cells. BTK activity has been implicated in the pathogenesis of several disorders and conditions, such as B cell-related hematological cancers (e.g., non-Hodgkin lymphoma and B cell chronic lymphocytic leukemia) and autoimmune diseases (e.g., rheumatoid arthritis, Sjogren’s syndrome, pemphigus, IBD, lupus, and asthma).
[4] Compound (I), pharmaceutically acceptable salts thereof, and solid forms of any of the foregoing may inhibit BTK and be useful in the treatment of disorders and conditions mediated by BTK activity. Compound (I) is disclosed in Example 31 of WO 2014/039899 and has the following structure:
where *C is a stereochemical center. An alternative procedure for producing Compound (!) is described in Example 1 of WO 2015/127310.
[5] Compound (I) obtained by the procedures described in WO 2014/039899 and WO 2015/127310 comprises residual solvent levels well above the limits described in the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (“ICH”) guidelines. In general, manufacturing processes producing residual solvent levels near or above the ICH limits are not desirable for preparing active pharmaceutical ingredients (APIs).
Example 1: Spray Drying Process A
[311] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was washed with pH 3 phosphate buffer to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. The dichloromethane solution was then washed with pH 7 buffer and solvent exchanged into isopropyl acetate. The isopropyl acetate solution was then washed with pH 3 phosphate buffer, bringing Compound (I) into the aqueous layer and removing non-basic impurities. The pH of the aqueous layer was adjusted to pH 9 with 10% sodium hydroxide, and the aqueous layer was extracted with isopropyl acetate. Upon concentration under vacuum, Compound (I) was precipitated from heptane at 0 °C, filtered and dried to give a white amorphous solid as a mixture of the (E) and (Z) isomers, as wet Compound (I). Wet Compound (I) was dissolved in methanol and spray dried at dryer inlet temperature of 125 °C to 155 °C and dryer outlet temperature of 48 to 58 °C to obtain the stable amorphous Compound (I) free base with levels of isopropyl acetate and heptane below 0.5% and 0.05%, respectively.
Example 2: Spray Drying Process B
intermediate A
Compound (!)
[241] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe, and recirculating fluid chiller/heater was charged with Intermediate A (20.2 kg) and Intermediate B (13.6 kg, 1.5 equiv). DCM (361.3 kg, 14.5 vol) was charged to the reactor. The mixture was agitated, and the batch cooled to 0 °C to 5 °C. The reactor was charged with pyrrolidine (18.3 kg, 6 equiv) and then charged with TMSC1 (18.6 kg, 4 eq). Stirring was continued at 0 °C to 5 °C for 0.5 to 1 hour
[242] At 0 °C to 5 °C, acetic acid (2.0 equiv) was charged to the reactor followed by water (5 equiv). Stirring was continued at 0 °C to 5 °C for 1 to 1.5 hours. Water (10 equiv) was charged to the reactor, and the solution was adjusted to 20 °C to 25 °C. The internal temperature was adjusted to 20 °C to 25 °C and the biphasic mixture was stirred for 15 to 20 mins. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed.
[243] Water (7 vol) was charged to the reactor. The pH was adjusted to 2.8-3.3 with a 10 wt. % solution of citric acid. Stirring was continued at 0 to 5 °C for 1 to 1.5 hours. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed.
[244] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe, and recirculating fluid chiller/heater was charged with an approximately 9% solution of NaHCCri (1 vol) and the organic layer. The internal temperature was adjusted to 20 °C to 25 °C, and the biphasic mixture was stirred for 15 to 20 mins. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed. The aqueous layer was measured to have a pH greater than 7.
[245] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe and recirculating fluid chiller/heater was charged with the organic layer. The organic phase ¾s distilled under vacuum at less than 25 °C to 4 total volumes. IP AC (15 vol) was charged to the reactor. The organic phase was distilled under vacuum at less than 25 °C to 10 total volumes. Water (15 vol) followed by pH 2.3 phosphate buffer were charged to the reactor at an internal temperature of 20 °C to 25 °C. The pH adjusted to 3 Stirring was stopped and phases allowed to separate for at least 0.5 h. The organic phase was removed.
[246] The following steps were repeated twice: IP AC (5 vol) was charged to the reactor containing the aqueous layer. Stirring was continued for 0.25 to 0.5 hours. Stirring was stopped and phases allowed to separate for at least 0.5 h. The organic phase was removed. [247] IP AC (15 vol) was charged to the reactor containing the aqueous layer. A pH 10 phosphate buffer was charged to the reactor and the pH adjusted to 10 with 14% NaOH solution. Stirring was continued for 1.5 to 2 hours. Stirring was stopped and phases allowed to separate for at. least 0.5 h. The aqueous layer was discarded. The organic layer was dried over brine.
[248] The organic solution was distilled under vacuum at less than 25 °C to 5 total volumes.
[249] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe and recirculating fluid chiller/heater was charged with n-heptane (20 vol). The internal temperature was adjusted to 0 to 5 °C, and the IP AC solution was added.
[250] The suspension was filtered. The filter cake was washed with n-heptane and the tray was dried at 35 °C. Compound (I) (24.6 kg) was isolated in 86% yield.
[251] Compound (1) was dissolved in methanol (6 kg) and spray dried to remove residual IP AC and n-heptane.
Example 3: Precipitation Process A
[252] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing with pH 3 aqueous solution to remove basic impurities that are more soluble than Compound (1) in the aqueous layer. Washing was repeated as needed to reduce impurities. Methanesulfonic acid was added to the dichloromethane solution, and the dichloromethane solution was concentrated by distillation under reduced pressure, followed by addition of 1% NaCi aqueous solution and isopropyl acetate before adjustment of pH to approximately 3 with potassium hydroxide. The isopropyl acetate layer was removed and discarded. The aqueous layer containing Compound (I) was washed with isopropyl acetate to remove hydrophobic impurities. Washing was repeated as needed to reduce related substance impurities. Residual isopropyl acetate was removed by distillation under reduced pressure. The aqueous solution containing Compound (I) was cooled to 0 to 5°C before adjusting the pH to approximately 9 with potassium hydroxide. The free base of Compound (I) was allowed to precipitate and maturate at 20 °C for 20 hours. The mixture temperature was then adjusted to 20 °C to 25 °C, and the hydrate impurity was verified to be less than 0.3% (< 0.3%). The cake of the free base of Compound (I) was filtered and washed as needed to reduce conductivity. The cake was then allowed to dry on the filter under vacuum and nitrogen swept to reduce water content by Karl-Fischer (KF < 50%) before transferring to the oven for drying. The wet cake of the free base of Compound (1) was dried under vacuum at 25 °C until water content by Karl -Fischer was less than 1.5% (KF < 1.5%), and then dehmiped by milling to yield a uniform white amorphous solid as a mixture of the (E) and (Z) isomers, with no detectible levels of isopropyl acetate or heptane.
Example 4: Precipitation Process 3B
[253] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing with pH 3 aqueous solution to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. The washing was repeated as needed to reduce residual solvents and impurities. The dichloromethane solution was then washed with saturated sodium bicarbonate (pH > 7). Dichloromethane was removed by distillation under reduced pressure, followed by addition of water and isopropyl acetate. The pH of the aqueous layer was adjusted to pH to 2.8 – 3.3 with 2 M aqueous sulfuric acid (H2SQ4) at 0 – 5 °C, and the mixture rvas stirred and settled. After phase separation removal of the organic layer, the aqueous layer was washed with isopropyl acetate three times and the residual isopropyl acetate in aqueous layer was distilled out under vacuum at a temperature below 25 °C and the solution was basitied with 5% aqueous KOFI to pH 9 – 10 to a slurry . The resulting suspension was stirred and warmed up to 20 °C to 25 °C and aged for 20 h. The product was filtered and washed with water and dried to give white solid in 86% yield.
Example 5: Precipitation Process C
[254] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. Washing was repeated as needed to reduce impurities. Methanesulfonic acid was added to the d chloromethane solution, and the dichloromethane solution was concentrated under reduced pressure to obtain a thin oil. The concentrated oil was cooled to approximately 5°C before washing with an aqueous solution of sodium chloride. The organic phase was discarded. Washing of the aqueous layer was repeated as needed with dichloromethane to remove low level impurities. The pH of the aqueous solution was adjusted to approximately 3 with an aqueous solution of potassium hydroxide. Residual dichloromethane was removed
under reduced pressure. The level of residual acetic acid was determined by, for example, titration. The aqueous solution containing Compound (I) was cooled to a temperature between 0°C and 5°C. Acetic acid was present at 0 wt % to 8 wt. %. Acetic acid level was 0 wt % if the aqueous acid solution was washed with aqueous sodium bicarbonate or another aqueous inorganic base. Optionally, additional acetic acid was added to achieve a 0 wt.% to 8 wt. % acetic acid level. An aqueous solution of potassium hydroxide was constantly charged to the aqueous solution to obtain a pH to approximately 9.5. The free base of Compound (I) was allowed to precipitate and maturate at approximately 20 °C for least 3 hours. The cake (wet solid) of the free base of Compound (I) was filtered and washed with water. The wet cake was then dried under reduced vacuum with slight heat. Alternatively, instead of washing the wet cake with water, the wet cake was reslurried with water at approximately 15 °C for at least 1 hour before filtering. The free base of Compound (I) in the fomi of a wet cake was dried under vacuum with slight heat at 25°C.
[255] FIGs. 12-15 are example SEM images showing the variable morphologies of particles of Compound (I) during the filtration step to isolate Compound (I) based on the amount acetic acid added during the initial step in the precipitation of Compound (Ϊ) (FIG. 12: at 0 wt. % acetic acid; FIG 13: at 3 wt. % acetic acid; FIG. 14: at 5 wt. % acetic acid; FIG 15: at 8 wt. % acetic acid). Filtration speed depended on the morphology and was the fastest for 0 wt. % acetic acid. At 1 wt. % acetic acid, the filtration speed diminished considerably, improving at 2 wt. % to 3 wt. % acetic acid. Morphologies with more open holes (such as, e.g., more porous particles) resulted in improved filtration speeds, whereas more compact particles resulted in decreased filtration speed.
Example 6: Conversion of a Crystalline Form of Compound (Ϊ) to an Amorphous Form
[256] 9.8 grams of a crystalline form of Compound (I) were dissolved in approximately 20 mL of dichloromethane and approximately 120 ml. of brine solution. Then, approximately 1 equivalent of methanesulfonic acid was added. The pH w¾s approximately 2. The layers were separated. The aqueous layer was concentrated at a temperature between 0°C and 5°C to remove residual dichloromethane before slowly adding aqueous KOI I solution (approximately 5%) to adjust the pH to a value between 9 and 10. During aqueous KOH addition, an amorphous form of Compound (I) precipitated out. The slurry was slowly warmed to room temperature and then was stirred for approximately 24 hours before filtering and rinsing the wet cake with water. The wet cake was dried under vacuum with slight heat at approximately 30°C to provide 7 grams of a white to an off-white solid (87% yield and 98 4% purity). XRPD showed that the product was an amorphous solid form of Compound (I).
Example 7: Micronization of Compound (I) Particles Obtained by Precipitation Processes
[257] A fluid jet mill equipment was used during lab scale jet milling trials. The fluid jet mill equipment includes a flat cylindrical chamber with 1.5” diameter, fitted with four symmetric jet nozzles winch are tangentially positioned in the inner wall. Prior to feeding material to the fluid jet mill in each trial, the material was sieved in a 355 iim screen to remove any agglomerates and avoid blocking of the nozzles during the feed of material to the micronization chamber. The material to be processed was drawn into the grinding chamber through a vacuum created by the venturi (P vent ~ 0 5 – 1 0 bar above P grind). The feed flow rate of solids (F_feed) was controlled by a manual valve and an infinite screw volumetric feeder. Compressed nitrogen was used to inject the feed material; compressed nitrogen was also used for the jet nozzles in the walls of the milling chamber. Compressed fluid issuing from the nozzles expands from P grind and imparts very’ high rotational speeds in the chamber. Accordingly, material is accelerated by rotating and expanding gases and subjected to centrifugal forces. Particles move outward and are impacted by high velocity jets, directing the particles radially inward at very high speeds. Rapidly moving particles impact the slower moving path of particles circulating near the periphery of the chamber. Attrition takes place due to the violent impacts of particles against each other. Particles with reduced size resulting from this sequence of impacts are entrained in the circulating stream of gas and swept against the action of centrifugal force toward the outlet at the center. Larger particles in the gas stream are subjected to a centrifugal force and returned to the grinding zone. Fine particles are carried by the exhaust gas to the outlet and pass from the grinding chamber into a collector.
[258] The feeder has continuous feed rate control; however, to more precisely control the feed rate, the full scale of feed rates was arbitrary divided in 10 positions. To calibrate F feed, the feeder was disconnected from milling chamber and 10 g of Compound (I) powder was fed through the feeder operating at various feed rate positions. The mass of powder flowing through the feeder over 6 minutes was marked. The resulting feed rate was directly proportional to feeder position. After processing each of the four trials, the jet mill was stopped, micronized product removed from the container, and the milling chamber checked for any powder accumulation.
Variables/Parameters
F_feed Feed flow rate of solids [kg/h]
P grind Grinding pressure inside the
drying chamber [bar]
P vent Feed pressure in the venturi [bar]
Example 8: Residual Solvent Levels
[251] Retention of process solvents (/.<?., res dual solvents) depends on van der Waal s’ forces that are unique to and an inherent property of each molecule. Additionally, solvent retention depends how the API solid is formed, isolated, washed, and dried (i.e., during the manufacturing process). Because residual solvents may pose safety risks, pharmaceutical processes should be designed to minimize residual solvent levels (e.g , to result in residual solvent levels below the limits established in the ICH guidelines).
[252] Residual solvent analysis was performed using gas chromatography-mass spectrometry. The residual solvent levels in solid forms of Compound (I) prepared by spray drying processes described herein and precipitation processes described herein are provided in Table 2. The residual solvent levels in crude Compound (I) listed in Table 2 are comparable to the residual solvent levels in crude Compound (I) prepared according to the procedures detailed in Example 31 of WO 2014/039899 and Example 1 of WO 2015/127310.
Table 2: Residual solvent levels in solid forms of Compound (I)
PATENT
WO 2015127310
https://patents.google.com/patent/WO2015127310A1/enExample 1Synthesis of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l- yl]-piperidine-l-carbonyl]-4-m iperazin-l-yl]pent-2-enenitrile

Step 1To a solution of 3-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin-l -yl]-l-piperidyl]-3-oxo-propanenitrile (15 g, 3.12mmol), 2-methyl-2-[4- (oxetan-3-yl)piperazin-l-yl]propanal (794.25mg, 3.74mmol) in DCM (40mL), pyrrolidine (1.54mL,18.71mmol) at 0-5 °C was added, which is followed by TMS-Cl (1.58mL,12.47mmol). The reaction mixture was stirred at 0-5 °C for 3 h and was quenched with 1 M potassium phosphate buffer (pH 3). Layers were separated and the organic layer was washed once more with 1 M potassium phosphate buffer (pH 3). The organic layer was extracted withl M potassium Phosphate buffer at pH 1.5. Layers were separated. The aqueous phase contained the desired product while the impurities stayed in the organic phase. The aqueous phase was neutralized with 1 M potassium phosphate (pH 7) and was extracted with isopropylacetate (10 volumes). Upon concentration 2-[(3R)-3-[4-amino-3-(2-fluoro-4- phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2-enenitrile was obtained as a foam having >99% HPLC purity. MS (pos. ion) m/z: 666 (M+l ).The foam containing high levels of residual solvent was dissolved in 2 M HC1 and the resulting solution was placed under vacuum to remove residual organic solvents. pH of the solution was then adjusted to ~ 7 and the resulting paste was filtered and dried in vacuum without heat. This resulted in isolation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3- yl)piperazin- l-yl]pent-2-enenitrile containing residual water up to 10%. Drying under vacuum without heat reduces the water level but lead to generation of impurities.Step 1AAlternatively, the isopropylacetate solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4- phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4- (oxetan-3-yl)piperazin-l -yl]pent-2-enenitrile can be concentrated to 4 vol and added to heptane (20 volume) at 0 °C. The resulting suspension was stirred at 0 °C overnight and the product was filtered, washed twice with heptane and dried at 45 °C for 2 days under vacuum to give 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l – yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2-enenitrile in 85 – 90 % yield as a free flowing solid. However, the solids obtained by this method contained high residual solvents (3.9 wt% isopropylacetate and 1.7 wt% heptane). In addition, the free base form was not very stable as degradation products were observed during the drying process at less than 45 °C.Salt formationExample 2Preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4-d]pyrimidin- l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2-enenitrile hemisulfate and sulfate saltHemisulfate: To the solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin-l-yl]-piperidine -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2- enenitrile (4.2 g) in EtOAc (60 mL, 15 vol) was added sulfuric acid (0.31 g, 0.17 mL, 0.5 eq) in EtOAc (20 mL, 5 vol) at ambient temperature. The suspension was stirred at ambient temperature for ~ 2 hr and then 40 °C for 4 hr and then at ambient temperature for at least 1 hr. After filtration and drying at ambient temperature under vacuum, 1.5 g of white powder was obtained. Solubility of the hemi-sulfate at ambient temperature was > 100 mg/mL in water.Sulfate saltTo the solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2- enenitrile (810 mg) in EtOAc (8 mL, 10 vol) was added sulfuric acid (0.06 mL, 1.0 equiv.) in EtOAc (2.5 mL, 5 vol) at ambient temperature. The resulting suspension was stirred at 40 °C for 2 hr and then cooled to ambient temperature for at least 1 hr. After filtration, solids were dried by suction under Argon for 1 h to give a white powder (0.68 g) in 69% yield.
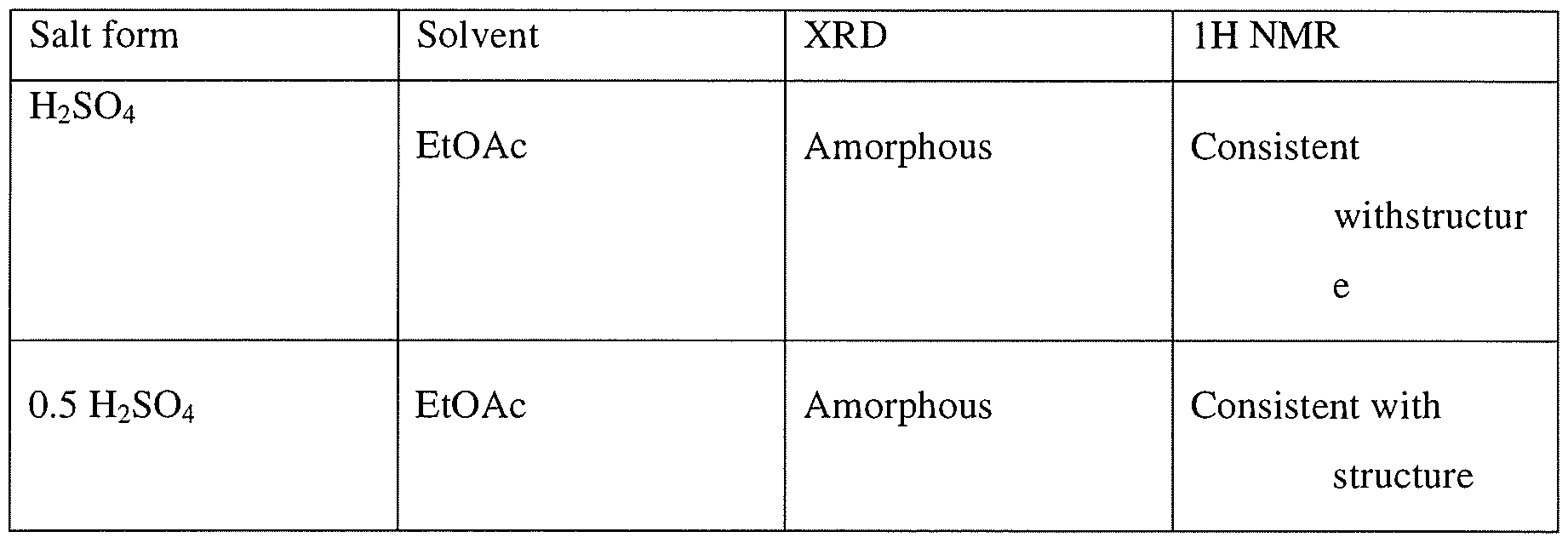
Example 3Preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin- 1 -yl]-piperidine- 1 -carbonyl] -4-methyl-4-[4-(oxetan-3-yl)-piperazin- 1 -yl]pent-2- enenitrile hydrochlorideTo a solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile (100 mg, 0.15 mmol) in CH2CI2 (1ml) at ambient temperature was added 2 equivalent of HC1 (0.3 mmol, 0.15 ml of 2M HC1 in 1 : 1 dioaxane:CH2Cl2). The resulting homogeneous solution was stirred at ambient temperature for 1 h and was added dropwise to 15 volumes of ethylacetate (as compared to CH2C12) resulting in formation of a white solid. The mixtures was aged at ambient temperature for lh and placed at 2-8 C for 19 h. Upon filtration and washing of the filter cake with ethylacetate and drying a white solid was obtained. Analysis by XRPD indicated formation of an amorphous solid. Both Ή-NMR and IC analysis indicated formation of the salt. IC indicated formation mono-HCl salt.

Example 4General procedure for preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)- piperazin-l-yl]pent-2-enenitrile mono- and di-mesylate saltsTo a solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2- enenitrile (100 mg, 0.15 mmol) in CH2C12 (1 ml) at ambient temperature was added either 1 equivalent of methanesulfonic acid (0.15 mmol, 0.2 ml of 74 mg/ml solution in CH2C12) or 2 equivalent of methanesulfonic acid (0.3 mmol, 0.4 ml of 74 mg/ml solution in CH2C12). The resulting homogeneous solution was stirred at ambient temperature for 1 h and was added dropwise to 10 volumes of antisolvents (ethylacetate, methyl tert-butylether (MTBE), or cyclohexane) (10 ml as compared to CH2C12) resulting in formation of a white solid. The mixture was aged at ambient temperature for lh and placed at 2-8 °C for 19 h. Upon filtration and washing of the filter cake with the antisolvent and drying, a white solid was obtained. Analysis by XRPD indicated formation of an amorphous solid. Both Ή-NMR and IC analysis indicated formation of the salt as well as counterion ratio.Alternatively 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]- pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile can be dissolved in 4 volumes of isopropylacetate and added to 2 equivalent of methanesulfonic acid in 6 volumes of isopropylacetate at 0 °C to generate the dimesylate salt.

1. Theoretical mesylate content, monomesylate=12.6% and dimesylate=22.4%, NO- not determinedExample 5 General procedure for the preparation of carboxylate salt Approximately 20 mg of the compound (I) was dissolved in minimum amount of the allocated solvent system. These were then mixed with the appropriate number of equivalents of counterion dissolved or slurried in the allocated solvent.If compound (I) was insoluble in the selected solvent, slurry of the sample was used after adding 300 μί.If the acid was insoluble in the selected solvent, slurry of the acid was used after adding 300 xL.If the acid was a liquid, the acid was added to the dissolved/slurried compound (I) from a stock solution in the allocated solvent.The suspensions/ precipitates resulting from the mixtures of compound (I) were temperature cycled between ambient (ca. 22°C) and 40°C in 4 hour cycles for ca. 48 hrs (the cooling/heating rate after each 4 hour period was ca. 1 °C/min). The mixtures were visually checked and any solids present were isolated and allowed to dry at ambient conditions prior to analysis. Where no solid was present, samples were allowed to evaporate at ambient. Samples which produced amorphous material, after the treatment outlined above, were re- dissolved and precipitated using anti-solvent (ter/-butylmethylether) addition methods at ambient conditions (ca. 22°C). i.e. the selected anti-solvent was added to each solution, until no further precipitation could be observed visually or until no more anti-solvent could be added. The solvents used in this preparation were acetonitrile, acetone, isopropyl acetate, THF and MTBE. The acid used were oxalic acid, L-aspartic acid, maleic acid, malonic acid, L-tartaric acid, and fumaric acid.Example 6General procedure for preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)- piperazin-l-yl]pent-2-enenitrile hemicitrate saltTo a solution 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]- pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile (5 g, 7.5 mmol) in ethanol (50 ml) was added citric acid (720.5 mg, 3.76 mmol) dissolved in 2 ml of water. Mixture was stirred at ambient temperature for 15 min, additional 0.5 ml of water was added and the mixture was stirred for 1 h, concentrated in vacuo to a gum. Ethanol was added and the mixture was concentrated. This process was repeated twice more and then CH2CI2 was added to the mixture. Upon concentration a white solid was obtained which was tumble dried under reduced pressure at 40 C for 4 h, then in a vacuum oven for 19h to give 5.4 g of a solid. Analysis by XRD indicated formation of an amorphous solid
PATENT
WO2014039899, Example 31
Rilzabrutinib (PRN1008) is an oral, reversible covalent inhibitor of Bruton’s tyrosine kinase (BTK) [1].
https://patents.google.com/patent/WO2014039899A1/enExample 31Synthesis of (R)-2-(3-(4-amino-3-(2-fluoro-4-phenoxyphenyl)- 1 H-pyrazolo[3,4-d]pyrimidin- 1 -yl)piperidine- 1 -carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin- 1 -yl)pent-2-enenitrile

Step 1A solution of 2-bromo-2-methyl-propanal (696.6 mg, 4.61 mmol) in DCM (10 mL) was cooled with an ice bath and l -(oxetan-3-yl)piperazine (328 mg, 2.31 mmol), diluted with 5-10 mL of DCM, was slowly added via addition funnel over a 15 min period. Next, Hunig’s base (0.4 mL, 2.31 mmol) was added and then the cooling bath was removed. The reaction mixture was stirred at room temperature overnight and the DCM layer was washed three times with 0.5N HC1. The combined aqueous layer was neutralized with NaOH to pH 10-11 and extracted with DCM. The combined organic layer was washed with brine and dried over Na?S04. Filtration and removal of solvent afforded 2-methyl-2-[4-(oxetan-3-yl)piperazin-l- yl]propanal as a light yellow liquid, which was used directly in the next step without further purification.Step 2To a cooled (0 °C) solution of 3-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)- pyrazolo[3,4-d]pyrimidin-l-yl]-l-piperidyl]-3-oxo-propanenitrile (80 mg, 0.17 mmol), was added 2-methyl-2-[4-(oxetan-3-yl)piperazin-l-yl]propanal (-108 mg, 0.51 mmol) in DCM (10 mL) followed by pyrrolidine (0.08 mL, 1.02 mmol) and TMS-C1 (0.09 raL, 0.68 mmol.) The ice bath was removed, and the reaction stirred 1 hour. Most of the solvent was removed and the residues were purified by chromatography, using 95:5 CH2Cl2:MeOH to obtain 79 mg of (R)-2-(3-(4-amino-3-(2-fluoro-4-phenoxyphenyl)-lH-pyrazolo[3,4-d]-pyrimidin-l- yl)piperidine- 1 -carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin- 1 -yl)pent-2-enenitrile as a white solid. MS (pos. ion) m/z: 666 (M+l).
PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0223523421001781?dgcid=rss_sd_all
Therapy based on Bruton’s tyrosine kinase (BTK) inhibitors one of the major treatment options currently recommended for lymphoma patients. The first generation of BTK inhibitor, Ibrutinib, achieved remarkable progress in the treatment of B-cell malignancies, but still has problems with drug-resistance or off-target induced serious side effects. Therefore, numerous new BTK inhibitors were developed to address this unmet medical need. In parallel, the effect of BTK inhibitors against immune-related diseases has been evaluated in clinical trials. This review summarizes recent progress in the research and development of BTK inhibitors, with a focus on structural characteristics and structure-activity relationships. The structure-refinement process of representative pharmacophores as well as their effects on binding affinity, biological activity and pharmacokinetics profiles were analyzed. The advantages and disadvantages of reversible/irreversible BTK inhibitors and their potential implications were discussed to provide a reference for the rational design and development of novel potent BTK inhibitors.

Research
Rilzabrutinib is an oral, reversible covalent inhibitor of Bruton’s tyrosine kinase, that may increase platelet counts in people with immune thrombocytopenia by means of dual mechanisms of action: decreased macrophage (Fcγ receptor)–mediated platelet destruction and reduced production of pathogenic autoantibodies.[5]
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219685s000lbl.pdf
- “FDA Approves Drug to Treat Adults with Persistent or Chronic Immune Thrombocytopenia”. U.S. Food and Drug Administration. 2 September 2025. Retrieved 5 September 2025. This article incorporates text from this source, which is in the public domain.
- “Press Release: Sanofi’s Wayrilz approved in US as first BTK inhibitor for immune thrombocytopenia” (Press release). Sanofi. 29 August 2025. Retrieved 5 September 2025 – via GlobeNewswire.
- World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 83”. WHO Drug Information. 34 (1). hdl:10665/339768.
- Kuter DJ, Efraim M, Mayer J, Trněný M, McDonald V, Bird R, et al. (April 2022). “Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia”. The New England Journal of Medicine. 386 (15): 1421–1431. doi:10.1056/NEJMoa2110297. PMID 35417637.
External links
- “Rilzabrutinib ( Code – C174769 )”. EVS Explore.
- Clinical trial number NCT04562766 for “Study to Evaluate Rilzabrutinib in Adults and Adolescents With Persistent or Chronic Immune Thrombocytopenia (ITP) (LUNA 3)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Wayrilz |
| Other names | PRN-1008 |
| AHFS/Drugs.com | Wayrilz |
| License data | US DailyMed: Rilzabrutinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1575591-66-0 |
| PubChem CID | 73388818 |
| DrugBank | DB17709 |
| ChemSpider | 58893525 |
| UNII | NWN58M4F5T |
| KEGG | D11873 |
| ChEMBL | ChEMBL3702854 |
| Chemical and physical data | |
| Formula | C36H40FN9O3 |
| Molar mass | 665.774 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////////PRN-1008, PRN 1008, Rilzabrutinib, リルザブルチニブ, Fda 2025, approvals 2025 8/29/2025, Wayrilz,
N#CC(=CC(N(C1COC1)C)(C)C)C(=O)N1CCCC1Cn1nc(c2c1ncnc2N)c1ccc(cc1F)Oc1ccccc1
PAT
PAT



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Delgocitinib

Delgocitinib
デルゴシチニブ
3-[(3S,4R)-3-methyl-7-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,7-diazaspiro[3.4]octan-1-yl]-3-oxopropanenitrile
1,6-Diazaspiro(3.4)octane-1-propanenitrile, 3-methyl-beta-oxo-6-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-, (3S,4R)-
3-((3S,4R)-3-methyl-6-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-1,6-diazaspiro(3.4)octan-1-yl)-3-oxopropanenitrile
| Formula |
C16H18N6O
|
|---|---|
| CAS |
1263774-59-9
|
| Mol weight |
310.3537
|
Approved, Japan 2020, Corectim, 2020/1/23, atopic dermatitis, Japan Tobacco (JT)
Torii
7/23/2025 fda approved, Anzupgo
| To treat moderate-to-severe chronic hand eczema when topical corticosteroids are not advisable or produce an inadequate response |
UNII-9L0Q8KK220, JTE-052, LP-0133, ROH-201, 9L0Q8KK220, LEO 124249A, LEO 124249, HY-109053
CS-0031558, D11046, GTPL9619, JTE-052A, JTE052
Delgocitinib, also known as LEO-124249 and JTE052, is a potent and selective JAK inhibitor. JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation.
Delgocitinib is a JAK inhibitor first approved in Japan for the treatment of atopic dermatitis in patients 16 years of age or older. Japan Tobacco is conducting phase III clinical trials for the treatment of atopic dermatitis in pediatric patients. Leo is developing the drug in phase II clinical trials for the treatment of inflammatory skin diseases, such as atopic dermatitis, and chronic hand eczema and for the treatment of discoid lupus erythematosus. Rohto is evaluating the product in early clinical development for ophthalmologic indications.
In 2014, the drug was licensed to Leo by Japan Tobacco for the development, registration and marketing worldwide excluding Japan for treatment of inflammatory skin conditions. In 2016, Japan Tobacco licensed the rights of co-development and commercialization in Japan to Torii. In 2018, Japan Tobacco licensed the Japanese rights of development and commercialization to Rohto for the treatment of ophthalmologic diseases.
Delgocitinib, sold under the brand name Corectim among others, is a medication used for the treatment of autoimmune disorders and hypersensitivity, including inflammatory skin conditions.[3] Delgocitinib was developed by Japan Tobacco and approved in Japan for the treatment of atopic dermatitis.[3] In the United States, delgocitinib is in Phase III clinical trials and the Food and Drug Administration has granted delgocitinib fast track designation for topical treatment of adults with moderate to severe chronic hand eczema.[4]
Delgocitinib works by blocking activation of the JAK-STAT signaling pathway which contributes to the pathogenesis of chronic inflammatory skin diseases.[5]
PATENTS
WO 2018117151
IN 201917029002
IN 201917029003
IN 201917029000
PATENTS
WO 2011013785
https://patents.google.com/patent/WO2011013785A1/en
[Production Example 6]: Synthesis of Compound 6

(1) Optically active substance of 2-benzylaminopropan-1-ol

To a solution of (S)-(+)-2-aminopropan-1-ol (50.0 g) and benzaldehyde (74 ml) in ethanol (500 ml) was added 5% palladium carbon (5.0 g) at room temperature and normal pressure. Hydrogenated for 8 hours. The reaction mixture was filtered through celite and concentrated under reduced pressure to give the title compound (111.2 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.23-7.18 (1H, m), 4.53-4.47 (1H, m), 3.76 (1H, d, J = 13.5 Hz) , 3.66 (1H, d, J = 13.5 Hz), 3.29-3.24 (2H, m), 2.65-2.55 (1H, m), 1.99 (1H, br s), 0.93 (3H, d, J = 6.4 Hz) .
(2) Optically active substance of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester

To a mixture of optically active 2-benzylaminopropan-1-ol (111.2 g), potassium carbonate (111.6 g) and N, N-dimethylformamide (556 ml) cooled to 0 ° C., tert-butyl bromoacetate was added. Ester (109 ml) was added dropwise over 20 minutes and stirred at room temperature for 19.5 hours. The mixture was acidified to pH 2 by adding 2M aqueous hydrochloric acid and 6M aqueous hydrochloric acid, and washed with toluene (1000 ml). The separated organic layer was extracted with 0.1 M aqueous hydrochloric acid (300 ml). The combined aqueous layer was adjusted to pH 10 with 4M aqueous sodium hydroxide solution and extracted with ethyl acetate (700 ml). The organic layer was washed successively with water (900 ml) and saturated aqueous sodium chloride solution (500 ml). The separated aqueous layer was extracted again with ethyl acetate (400 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the title compound (160.0 g).
1 H-NMR (DMSO-D 6 ) δ: 7.37-7.26 (4H, m), 7.24-7.19 (1H, m), 4.26 (1H, dd, J = 6.9, 3.9 Hz), 3.76 (1H, d, J = 14.1 Hz), 3.68 (1H, d, J = 13.9 Hz), 3.45-3.39 (1H, m), 3.29-3.20 (1H, m), 3.24 (1H, d, J = 17.2 Hz), 3.13 ( 1H, d, J = 17.0 Hz), 2.84-2.74 (1H, m), 1.37 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
(3) Optically active substance of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester

(3)-(1) Optically active form of [benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester

To a solution of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (160.0 g) cooled to 0 ° C. in chloroform (640 ml) was added thionyl chloride (50.0 ml). Was added dropwise and stirred at 60 ° C. for 2 hours. The reaction mixture was cooled to 0 ° C., saturated aqueous sodium hydrogen carbonate solution (1000 ml) and chloroform (100 ml) were added and stirred. The separated organic layer was washed with a saturated aqueous sodium chloride solution (500 ml), and the aqueous layer was extracted again with chloroform (450 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain the title compound (172.9 g).
1 H-NMR (CDCl 3 ) δ: 7.40-7.22 (5H, m), 4.05-3.97 (0.4H, m), 3.93-3.81 (2H, m), 3.70-3.65 (0.6H, m), 3.44- 3.38 (0.6H, m), 3.29 (0.8H, s), 3.27 (1.2H, d, J = 2.4 Hz), 3.24-3.15 (0.6H, m), 3.05-2.99 (0.4H, m), 2.94 -2.88 (0.4H, m), 1.50 (1.2H, d, J = 6.4 Hz), 1.48 (3.6H, s), 1.45 (5.4H, s), 1.23 (1.8H, d, J = 6.8 Hz) .
(3)-(2) Optically active form of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester

[Benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (172.9 g) was dissolved in N, N-dimethylformamide (520 ml) and stirred at 80 ° C. for 140 minutes. did. The reaction mixture was cooled to 0 ° C., water (1200 ml) was added, and the mixture was extracted with n-hexane / ethyl acetate (2/1, 1000 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (400 ml), and the separated aqueous layer was extracted again with n-hexane / ethyl acetate (2/1, 600 ml). The combined organic layers were concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 50/1 to 40/1) to give the title compound (127.0 g )
1 H-NMR (CDCl 3 ) δ: 7.37-7.29 (4H, m), 7.28-7.23 (1H, m), 4.05-3.97 (1H, m), 3.91 (1H, d, J = 13.5 Hz), 3.86 (1H, d, J = 13.7 Hz), 3.29 (2H, s), 3.03 (1H, dd, J = 13.9, 6.6 Hz), 2.91 (1H, dd, J = 13.9, 6.8 Hz), 1.50 (3H, d, J = 6.4 Hz), 1.48 (9H, s).
(4) Optically active substance of 1-benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester

To a solution of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester optically active substance (60.0 g) cooled to −72 ° C. and hexamethylphosphoramide (36.0 ml) in tetrahydrofuran (360 ml), Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 242 ml) was added dropwise over 18 minutes, and the temperature was raised to 0 ° C. over 80 minutes. A saturated aqueous ammonium chloride solution (300 ml) and water (400 ml) were sequentially added to the reaction mixture, and the mixture was extracted with ethyl acetate (500 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (500 ml), and the separated aqueous layer was extracted again with ethyl acetate (300 ml). The combined organic layers were dried over anhydrous sodium sulfate, concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: n-hexane / ethyl acetate = 50/1 to 4/1). To give the title compound (50.9 g).
1 H-NMR (CDCl 3 ) δ: 7.34-7.21 (5H, m), 3.75 (1H, d, J = 12.6 Hz), 3.70-3.67 (1H, m), 3.58 (1H, d, J = 12.6 Hz ), 3.05-3.01 (1H, m), 2.99-2.95 (1H, m), 2.70-2.59 (1H, m), 1.41 (9H, s), 1.24 (3H, d, J = 7.1 Hz).
(5) Optically active substance of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester

1-Benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester optically active substance (43.5 g) and di-tert-butyl dicarbonate (38.2 g) in tetrahydrofuran / methanol (130 ml / 130 ml) solution 20% Palladium hydroxide carbon (3.5 g) was added thereto, and hydrogenated at 4 atm for 2 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (48.0 g).
1 H-NMR (DMSO-D 6 ) δ: 4.44 (1H, d, J = 8.8 Hz), 3.99-3.77 (1H, m), 3.45-3.37 (1H, m), 3.00-2.88 (1H, m) , 1.45 (9H, s), 1.40-1.30 (9H, m), 1.02 (3H, d, J = 7.2 Hz).
(6) Optically active substance of 3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester

Optically active substance (48.0 g) of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester cooled to -69 ° C. and 1-bromo-3-methyl-2-butene (25.4 ml) Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 200 ml) was added to a tetrahydrofuran solution (380 ml). The reaction mixture was warmed to −20 ° C. in 40 minutes and further stirred at the same temperature for 20 minutes. A saturated aqueous ammonium chloride solution (200 ml) and water (300 ml) were successively added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1 / 1,500 ml). The separated organic layer was washed successively with water (200 ml) and saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 15/1 to 8/1) to give the titled compound (44.5 g).
1 H-NMR (CDCl 3 ) δ: 5.29-5.21 (1H, m), 3.77-3.72 (1H, m), 3.49-3.44 (1H, m), 2.73-2.52 (3H, m), 1.76-1.74 ( 3H, m), 1.66-1.65 (3H, m), 1.51 (9H, s), 1.43 (9H, s), 1.05 (3H, d, J = 7.3 Hz).
(7) Optically active substance of 3-methyl-2- (2-oxoethyl) azetidine-1,2-dicarboxylic acid di-tert-butyl ester

3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (44.5 g) in chloroform / cooled to −70 ° C. An ozone stream was passed through the methanol solution (310 ml / 310 ml) for 1 hour. To this reaction mixture, a solution of triphenylphosphine (44.7 g) in chloroform (45 ml) was added little by little, and then the mixture was warmed to room temperature. To this mixture were added saturated aqueous sodium thiosulfate solution (200 ml) and water (300 ml), and the mixture was extracted with chloroform (500 ml). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to obtain the title compound (95.0 g). This product was subjected to the next step without further purification.
1 H-NMR (DMSO-D 6 ) δ: 9.65 (1H, t, J = 2.6 Hz), 3.79-3.74 (1H, m), 3.45-3.40 (1H, m), 2.99-2.80 (3H, m) , 1.46 (9H, s), 1.34 (9H, s), 1.06 (3H, d, J = 7.2 Hz).
(8) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester

To a solution of the residue (95.0 g) obtained in (7) in tetrahydrofuran (300 ml) was added benzylamine (34 ml) at room temperature, and the mixture was stirred for 2 hours. The mixture was cooled to 0 ° C., sodium triacetoxyborohydride (83.3 g) was added, and the mixture was stirred at room temperature for 1.5 hours. Water (300 ml) was added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1/3, 600 ml). The separated organic layer was washed with water (300 ml) and saturated aqueous sodium chloride solution (200 ml), and then extracted twice with 5% aqueous citric acid solution (300 ml, 200 ml) and three times with 10% aqueous citric acid solution (250 ml × 3). . The combined aqueous layers were basified to pH 10 with 4M aqueous sodium hydroxide solution and extracted with chloroform (300 ml). The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (46.9 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.26 (4H, m), 7.22-7.17 (1H, m), 3.74-3.65 (2H, m), 3.61 (1H, t, J = 7.8 Hz) , 3.28 (1H, t, J = 7.5 Hz), 2.76-2.66 (2H, m), 2.57-2.45 (1H, m), 2.15 (1H, br s), 2.05-1.89 (2H, m), 1.42 ( 9H, s), 1.27 (9H, s), 0.96 (3H, d, J = 7.1 Hz).
(9) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride

2- (2-Benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (46.5 g), 4M hydrochloric acid 1,4-dioxane (230 ml) and water (4.1 ml) was mixed and stirred at 80 ° C. for 2 hours. The mixture was concentrated under reduced pressure, azeotroped with toluene, and then slurry washed with n-hexane / ethyl acetate (1/1, 440 ml) to give the title compound (30.1 g).
1 H-NMR (DMSO-D 6 ) δ: 10.24 (1H, br s), 9.64 (2H, br s), 8.90 (1H, br s), 7.58-7.53 (2H, m), 7.47-7.41 (3H , m), 4.21-4.10 (2H, m), 4.02-3.94 (1H, m), 3.46-3.37 (1H, m), 3.20-3.10 (1H, m), 2.99-2.85 (2H, m), 2.69 -2.54 (2H, m), 1.10 (3H, d, J = 7.2 Hz).
(10) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one

To a solution of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride optically active substance (29.1 g) and N, N-diisopropylethylamine (65 ml) in chloroform (290 ml), At room temperature, O- (7-azabenzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (41.3 g) was added and stirred for 4 hours. To this reaction mixture were added saturated aqueous sodium hydrogen carbonate solution (200 ml) and water (100 ml), and the mixture was extracted with chloroform (200 ml). The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 20/1 to 10/1) to give the titled compound (21.3 g).
1 H-NMR (DMSO-D 6 ) δ: 7.38-7.31 (2H, m), 7.30-7.22 (3H, m), 4.52 (1H, d, J = 14.8 Hz), 4.29 (1H, d, J = 14.8 Hz), 3.35-3.27 (2H, m), 3.22-3.17 (1H, m), 3.05 (2H, dd, J = 9.5, 4.0 Hz), 2.77-2.66 (1H, m), 2.16-2.10 (1H , m), 1.96-1.87 (1H, m), 0.94 (3H, d, J = 7.1 Hz).
(11) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester

Concentrated sulfuric acid (4.8 ml) was slowly added dropwise to a suspension of lithium aluminum hydride (6.8 g) in tetrahydrofuran (300 ml) under ice cooling, and the mixture was stirred for 30 minutes. To this mixture was added dropwise a solution of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one optically active substance (21.3 g) in tetrahydrofuran (100 ml) at the same temperature. Stir for 45 minutes. Water (7.0 ml), 4M aqueous sodium hydroxide solution (7.0 ml) and water (14.0 ml) were sequentially added to the reaction mixture, and the mixture was stirred as it was for 30 minutes. To this mixture was added anhydrous magnesium sulfate and ethyl acetate (100 ml), and the mixture was stirred and filtered through celite. Di-tert-butyl dicarbonate (23.4 g) was added to the filtrate at room temperature and stirred for 3 hours. The mixture was concentrated under reduced pressure to a half volume and washed twice with a saturated aqueous ammonium chloride solution (200 ml × 2). N-Hexane (200 ml) was added to the separated organic layer, and the mixture was extracted 5 times with a 10% aqueous citric acid solution. The separated aqueous layer was basified with 4M aqueous sodium hydroxide solution and extracted with chloroform. The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 40/1 to 20/1) to give the titled compound (15.6 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.26-7.21 (1H, m), 3.84-3.69 (1H, m), 3.62-3.47 (2H, m), 3.19- 3.05 (1H, m), 3.02-2.92 (1H, m), 2.76-2.69 (1H, m), 2.47-2.24 (4H, m), 1.95-1.77 (1H, m), 1.36 (9H, s), 1.03 (3H, d, J = 7.0 Hz).
(12) Optically active substance of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester

20% of optically active form of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (10.0 g) in tetrahydrofuran / methanol (50 ml / 50 ml) solution Palladium hydroxide on carbon (2.0 g) was added and hydrogenated at 4 atm for 24 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (7.3 g).
1 H-NMR (DMSO-D 6 ) δ: 3.88-3.71 (1H, m), 3.44-3.06 (2H, m), 3.02-2.64 (4H, m), 2.55-2.38 (1H, m), 2.31- 2.15 (1H, m), 1.81-1.72 (1H, m), 1.37 (9H, s), 1.07 (3H, d, J = 7.0 Hz).
(13) Optical activity of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester body

The optically active substance (6.9 g) of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester was converted into 4-chloro-7H-pyrrolo [2,3-d] pyrimidine ( 4.3 g), potassium carbonate (7.7 g) and water (65 ml) and stirred for 4 hours at reflux. The mixture was cooled to room temperature, water (60 ml) was added, and the mixture was extracted with chloroform / methanol (10/1, 120 ml). The organic layer was washed successively with water, saturated aqueous ammonium chloride solution and saturated aqueous sodium chloride solution, and dried over anhydrous sodium sulfate. To this mixture, silica gel (4 g) was added, stirred for 10 minutes, filtered through celite, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / ethyl acetate = 1/1, then chloroform / methanol = 50/1 to 20/1) to give the title compound (10.0 g). Obtained.
1 H-NMR (DMSO-D 6 ) δ: 11.59 (1H, br s), 8.09 (1H, s), 7.12-7.09 (1H, m), 6.64-6.59 (1H, m), 4.09-3.66 (5H , m), 3.39-3.21 (1H, m), 2.64-2.44 (2H, m), 2.27-2.06 (1H, m), 1.36 (3H, s), 1.21 (6H, s), 1.11 (3H, d , J = 6.5 Hz).
(14) Optically active form of 4- (3-methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride

Optically active form of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (9 0.5 g), 4M hydrochloric acid 1,4-dioxane (50 ml), chloroform (50 ml) and methanol (100 ml) were mixed and stirred at 60 ° C. for 30 minutes. The mixture was concentrated under reduced pressure and azeotroped with toluene to give the title compound (9.3 g).
1 H-NMR (DMSO-D 6 ) δ: 12.91 (1H, br s), 9.97-9.64 (2H, m), 8.45-8.35 (1H, m), 7.58-7.47 (1H, m), 7.04-6.92 (1H, m), 4.99-4.65 (1H, m), 4.32-3.21 (7H, m), 3.04-2.90 (1H, m), 2.46-2.31 (1H, m), 1.27 (3H, d, J = 6.0 Hz).
(15) 3- [3-Methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] oct-1-yl] -3-oxo Optically active form of propionitrile

4- (3-Methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride optically active substance (8.8 g) was converted to 1- The mixture was mixed with cyanoacetyl-3,5-dimethylpyrazole (6.8 g), N, N-diisopropylethylamine (20 ml) and 1,4-dioxane (100 ml) and stirred at 100 ° C. for 1 hour. The mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform / methanol (10/1). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 30/1 to 9/1). The residue obtained by concentration under reduced pressure was slurry washed with n-heptane / ethanol (2/1, 90 ml) to obtain a solid (7.3 g). The solid was slurried again with n-heptane / ethanol (5/1, 90 ml) to give the title compound as crystals 1 (6.1 g).
1 H-NMR (DMSO-D 6 ) δ: 11.60 (1H, br s), 8.08 (1H, s), 7.11 (1H, dd, J = 3.5, 2.4 Hz), 6.58 (1H, dd, J = 3.4 , 1.9 Hz), 4.18-4.14 (1H, m), 4.09-3.93 (3H, m), 3.84-3.73 (1H, m), 3.71 (1H, d, J = 19.0 Hz), 3.66 (1H, d, J = 18.7 Hz), 3.58 (1H, dd, J = 8.2, 6.0 Hz), 2.70-2.58 (2H, m), 2.24-2.12 (1H, m), 1.12 (3H, d, J = 7.1 Hz).
[Α] D = + 47.09 ° (25 ° C., c = 0.55, methanol)
1-Butanol (39 ml) was added to the obtained crystal 1 (2.6 g), and the mixture was heated and stirred at 100 ° C. After complete dissolution, the solution was cooled to room temperature by 10 ° C. every 30 minutes and further stirred at room temperature overnight. The produced crystals were collected by filtration, washed with 1-butanol (6.2 ml), and dried under reduced pressure to give crystals 2 (2.1 g) of the title compound.
PATENTS
WO 2017006968
WO 2018117152
WO 2018117151
PATENT
WO 2018117153
https://patentscope.wipo.int/search/zh/detail.jsf?docId=WO2018117153&tab=FULLTEXT
Janus kinase (JAK) inhibitors are of current interest for the treatment of various diseases including autoimmune diseases, inflammatory diseases, and cancer. To date, two JAK inhibitors have been approved by the U.S. Food & Drug Administration (FDA). Ruxolitinib has been approved for the treatment of primary myelofibrosis and polycythemia vera (PV), and tofacitinib has been approved for the treatment of rheumatoid arthritis. Other JAK inhibitors are in the literature. The compound 3-((3S,4R)-3-methyl-6-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,6-diazaspiro[3.4]octan-1-yl)-3-oxopropanenitrile (Compound A) (see structure below) is an example of a spirocyclic JAK inhibitor reported in U.S. Pat. Pub. Nos. 2011/0136778 and International Pat. Pub. No. PCT/JP2016/070046.
[Chem. 1]
[Chem. 2]
Step 1
[Chem. 3]
A crude product of S-BBMO which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.36-7.13 (5H, m), 4.26 (1H, dd, J = 6.8, 3.9 Hz), 3.72 (2H, dd, J = 14.2, 6.8 Hz), 3.47-3.38 (1H, m), 3.30-3.08 (3H, m), 2.79 (1H, sext, J = 6.8 Hz), 1.35 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
MS: m/z = 280 [M+H] +
[Chem. 4]
A crude product of R-BCAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.11 (5H, m), 4.24-4.11 (1H, m), 3.80 (2H, d, J = 3.6 Hz), 3.24 (2H, d, J = 3.6 Hz), 2.98-2.78 (2H, m), 1.46-1.37 (12H, m).
MS: m/z = 298 [M+H] +
[Chem. 5]
A crude product of S-MABB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.25 (10H, m), 3.75 (1H, d, J = 12.7 Hz), 3.68 (1H, d, J = 1.4 Hz), 3.66 (1H, d, J = 6.7 Hz), 3.46 (2H, d, J = 12.7 Hz), 3.30-3.17 (2H, m), 2.95 (1H, dd, J = 6.2, 1.2 Hz), 2.77 (1H, dd, J = 6.1, 2.2 Hz), 2.65-2.55 (1H, m), 2.48-2.40 (2H, m), 1.35 (9H, s), 1.35 (9H, s), 1.12 (3H, d, J = 7.2 Hz), 1.09 (3H, d, J = 6.2 Hz).
MS: m/z = 262 [M+H] +
[Chem. 6]
S-MABB-HC which was prepared by the same process was measured about NMR, MS, and Cl-content.
1H-NMR (DMSO-d 6) δ: 11.08 (1H, br s), 10.94 (1H, br s), 7.52-7.42 (10H, m), 5.34 (1H, t, J = 8.4 Hz), 4.90 (1H, br s), 4.45-4.10 (5H, m), 3.92-3.49 (3H, br m), 3.10-2.73 (2H, br m), 1.35 (9H, s), 1.29 (9H, s), 1.24 (3H, d, J = 6.7 Hz), 1.17 (3H, d, J = 7.4 Hz).
MS: m/z = 262 [M+H-HCl] +
Cl content (ion chromatography): 11.9 % (in theory: 11.9 %).
[Chem. 7]
A crude product of S-MACB-HC which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 9.60 (br s, 1H), 4.97 (d, 1H, J = 9.2 Hz), 4.61 (d, 1H, J = 8.4 Hz), 4.01 (dd, 1H, J = 10.0, 8.4 Hz), 3.78-3.74 (m, 1H), 3.54 (dd, 1H, J = 9.6, 8.4 Hz), 3.35 (dd, 1H, J = 10.0, 6.0 Hz), 3.15-3.03 (m, 1H), 3.00-2.88 (m, 1H), 1.49 (s, 9H), 1.47 (s, 9H), 1.22 (d, 3H, J = 6.8 Hz), 1.14 (d, 3H, J = 7.2 Hz).
MS: m/z = 172 [M+H] + (free form)
[Chem. 8]
A crude product of S-ZMAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.38-7.28 (m, 10H), 5.16-5.04 (m, 4H), 4.60 (d, 1H, J = 9.2 Hz), 4.18-4.12 (m, 2H), 4.04 (t, 1H, J = 8.6 Hz), 3.66 (dd, 1H, J = 7.6, 7.2 Hz), 3.50 (dd, 1H, J = 8.0, 5.2 Hz), 3.05-2.94 (m, 1H), 2.60-2.50 (m, 1H), 1.43 (br s, 18H), 1.33 (d, 3H, J = 6.5 Hz), 1.15 (d, 3H, J = 7.2 Hz).
MS: m/z = 328 [M+Na] +.
[Chem. 9]
A crude product of RS-ZMBB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.38-7.29 (m, 5H), 5.09-4.96 (m, 2H), 3.91 (t, 0.4H, J = 8.0 Hz), 3.79 (t, 0.6H, J = 8.0 Hz), 3.55 (t, 0.4H, J = 7.2 Hz), 3.46 (t, 0.6H, J = 7.5 Hz), 3.14-3.04 (m, 1H), 2.83-2.72 (m, 2H), 1.38 (br s, 9H), 1.37 (br s, 3.6H), 1.34 (br s, 5.4H), 1.12-1.09 (m, 3H).
MS: m/z = 420 [M+H] +.
[Chem. 10]
RS-ZMAA-DN .2H 2O which was prepared by the same process was measured about NMR, MS, Na-content, and water-content.
1H-NMR (DMSO-d 6) δ: 7.32-7.22 (m, 5H), 4.97 (d, 1H, J = 12.7 Hz), 4.84 (d, 1H, J = 12.7 Hz), 3.79 (t, 1H, J = 8.0 Hz), 3.29 (d, 1H, J = 14.8 Hz), 3.16-3.12 (m, 1H), 2.17-2.09 (m, 2H), 1.07 (d, 3H, J = 6.9 Hz).
MS: m/z = 352 [M+H] + (anhydrate)
Na content (ion chromatography): 13.3 % (after correction of water content)(13.1 % in theory)
Water content (Karl Fischer’s method): 9.8 % (9.3 % in theory)
[Chem. 11]
RS-ZMAA which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.35-7.28 (m, 5H), 5.06-4.94 (m, 2H), 3.86 (dt, 1H, J = 48.4, 7.9 Hz), 3.50 (dt, 1H, J = 37.9, 7.4 Hz), 3.16-3.02 (br m, 1H), 2.91-2.77 (br m, 2H), 1.08 (d, 3H, J = 6.9 Hz)
MS: m/z = 308 [M+H] +.
[Chem. 12]
RS-ZMOO which was prepared by the same process was measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.39-7.30 (m, 5H), 5.10 (s, 2H), 4.15-4.01 (br m, 2H), 3.83-3.73 (br m, 3H), 3.48 (dd, 1H, J = 8.3, 6.4 Hz), 2.59-2.50 (br m, 1H), 2.46-2.40 (br m, 1H), 2.07-1.99 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 280 [M+H]+.
[Chem. 13]
RS-ZMSS which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.37-7.27 (br m, 5H), 5.10-4.98 (m, 2H), 4.58-4.22 (br m, 4H), 3.84 (dt, 1H, J = 45.6, 8.1 Hz), 3.48-3.33 (br m, 1H), 3.17-3.10 (m, 6H), 2.81-2.74 (br m, 1H), 2.22-2.12 (m, 2H)
MS: m/z = 436 [M+H] +.
[Chem. 14]
1H-NMR (CDCl 3) δ: 7.35-7.20 (m, 10H), 5.08 (d, 2H, J = 23.6 Hz), 3.94 (q, 1H, J = 7.9 Hz), 3.73-3.42 (br m, 2H), 3.30-3.23 (m, 1H), 3.05 (dd, 1H, J = 19.7, 9.5 Hz), 2.79 (dt, 1H, J = 69.6, 6.1 Hz), 2.57-2.32 (br m, 4H), 1.96-1.89 (m, 1H), 1.09 (d, 3H, J = 6.9 Hz)
MS: m/z = 351 [M+H] +.
[Chem. 15]
SR-MDOZ which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.37-7.28 (m, 5H), 5.08 (dd, 2H, J = 16.8, 12.8 Hz), 4.00 (dd, 1H, J = 17.1, 8.3 Hz), 3.40-3.31 (m, 1H), 3.24 (d, 1H, J = 12.7 Hz), 3.00 (dd, 1H, J = 54.9, 12.4 Hz), 2.87-2.57 (m, 3H), 2.47-2.27 (m, 1H), 1.91-1.80 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 261 [M+H] +.
[Chem. 16]
SR-MDOZ-OX which was prepared by the same process was measured about NMR, MS, and elementary analysis.
1H-NMR (DMSO-D 6) δ: 7.37-7.30 (m, 5H), 5.15-5.01 (m, 2H), 3.92 (dt, 1H, J = 43.5, 8.4 Hz), 3.48-3.12 (br m, 5H), 2.67-2.56 (m, 1H), 2.46-2.35 (m, 1H), 2.12-2.05 (m, 1H), 1.13 (d, 3H, J = 6.9 Hz)
MS: m/z = 261 [M+H] +
elementary analysis: C 58.4wt % , H 6.4wt % , N 7.9 % wt % (theoretically, C 58.3wt % , H 6.3wt % , N 8.0wt % )
[Chem. 17]
SR-MDPZ which was prepared by the same process was isolated as a solid from a mixture of ethyl acetate and n-heptane, and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.41-7.26 (br m, 3H), 7.22-7.08 (br m, 3H), 6.64-6.51 (br m, 1H), 5.07-4.91 (br m, 2H), 4.09-3.67 (br m, 5H), 3.47-3.32 (br m, 1H), 2.67-2.55 (br m, 2H), 2.21-2.15 (br m, 1H), 1.11 (d, 3H, J = 6.9 Hz).
MS: m/z = 378 [M+H] +
[Chem. 18]
SR-MDOP which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.57 (br s, 1H), 8.07 (s, 1H), 7.10 (d, 1H, J = 3.2 Hz), 6.58 (d, 1H, J = 3.2 Hz), 3.92-3.59 (br m, 4H), 3.49 (dd, 1H, J = 8.3, 7.2 Hz), 2.93 (dd, 1H, J = 7.2, 6.1 Hz), 2.61-2.53 (m, 2H), 2.12-2.01 (br m, 2H), 1.10 (d, 3H, J = 6.9 Hz).
MS: m/z = 244 [M+H] +.
[Chem. 19]
Compound A mono-ethanolate which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.34 (t, 1H, J = 5.1 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.92 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 3.44 (dq, 2H, J = 6.7, 5.1 Hz), 2.69-2.60 (m, 2H), 2.23-2.13 (br m, 1H), 1.12 (d, 3H, J = 7.1 Hz), 1.06 (t, 3H, J = 6.7 Hz).
MS: m/z = 311 [M+H] +
[Chem. 20]
Compound A which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.5 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.69-2.59 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H] +
(1) Preparation of Single crystal
To 10 mg of Compound A in a LaPha ROBO Vial(R) 2.0 mL wide-mouthed vial was added 0.5 mL of chloroform. The vial was covered with a cap, in which Compound A was completely dissolved. In order to evaporate the solvent slowly, a hole was made on the septum attached in the cap with a needle of a TERUMO(R) syringe, and the vial was still stood at room temperature. The resulting single crystal was used in the structural analysis.
(2) Measuring instrument
Beam line: SPring-8 BL32B2
Detector: Rigaku R-AXIS V diffractometer
(3) Measuring method
The radiant light of 0.71068Å was irradiated to the single crystal to measure X-ray diffraction data.
(4) Assay method
Using the X-ray anomalous scattering effect of the chlorine atom in the resulting Compound A chloroform-solvate, the absolute configuration of Compound A was identified as (3S,4R). Based on the obtained absolute configuration of Compound A, the absolute configurations of each process intermediate were identified.
REFERENCES
1: Nakagawa H, Nemoto O, Yamada H, Nagata T, Ninomiya N. Phase 1 studies to assess the safety, tolerability and pharmacokinetics of JTE-052 (a novel Janus kinase inhibitor) ointment in Japanese healthy volunteers and patients with atopic dermatitis. J Dermatol. 2018 Jun;45(6):701-709. doi: 10.1111/1346-8138.14322. Epub 2018 Apr 17. PubMed PMID: 29665062; PubMed Central PMCID: PMC6001687.
2: Nakagawa H, Nemoto O, Igarashi A, Nagata T. Efficacy and safety of topical JTE-052, a Janus kinase inhibitor, in Japanese adult patients with moderate-to-severe atopic dermatitis: a phase II, multicentre, randomized, vehicle-controlled clinical study. Br J Dermatol. 2018 Feb;178(2):424-432. doi: 10.1111/bjd.16014. Epub 2018 Jan 15. PubMed PMID: 28960254.
3: Tanimoto A, Shinozaki Y, Yamamoto Y, Katsuda Y, Taniai-Riya E, Toyoda K, Kakimoto K, Kimoto Y, Amano W, Konishi N, Hayashi M. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. Exp Dermatol. 2018 Jan;27(1):22-29. doi: 10.1111/exd.13370. Epub 2017 Jul 3. PubMed PMID: 28423239.
4: Nomura T, Kabashima K. Advances in atopic dermatitis in 2015. J Allergy Clin Immunol. 2016 Dec;138(6):1548-1555. doi: 10.1016/j.jaci.2016.10.004. Review. PubMed PMID: 27931536.
5: Amano W, Nakajima S, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. JAK inhibitor JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation. J Dermatol Sci. 2016 Dec;84(3):258-265. doi: 10.1016/j.jdermsci.2016.09.007. Epub 2016 Sep 13. PubMed PMID: 27665390.
6: Tanimoto A, Shinozaki Y, Nozawa K, Kimoto Y, Amano W, Matsuo A, Yamaguchi T, Matsushita M. Improvement of spontaneous locomotor activity with JAK inhibition by JTE-052 in rat adjuvant-induced arthritis. BMC Musculoskelet Disord. 2015 Nov 6;16:339. doi: 10.1186/s12891-015-0802-0. PubMed PMID: 26546348; PubMed Central PMCID: PMC4636776.
7: Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, Dainichi T, Honda T, Otsuka A, Kimoto Y, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. 2015 Sep;136(3):667-677.e7. doi: 10.1016/j.jaci.2015.03.051. Epub 2015 Jun 24. PubMed PMID: 26115905.
8: Tanimoto A, Ogawa Y, Oki C, Kimoto Y, Nozawa K, Amano W, Noji S, Shiozaki M, Matsuo A, Shinozaki Y, Matsushita M. Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflamm Res. 2015 Jan;64(1):41-51. doi: 10.1007/s00011-014-0782-9. Epub 2014 Nov 12. PubMed PMID: 25387665; PubMed Central PMCID: PMC4286029.
References
- “Anzupgo EPAR”. European Medicines Agency. 25 July 2024. Retrieved 25 July 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Anzupgo PI”. Union Register of medicinal products. 23 September 2024. Retrieved 27 September 2024.
- Dhillon S (April 2020). “Delgocitinib: First Approval”. Drugs. 80 (6): 609–615. doi:10.1007/s40265-020-01291-2. PMID 32166597. S2CID 212681247.
- Park B (5 August 2020). “Delgocitinib Cream Gets Fast Track Status for Chronic Hand Eczema”. empr.com.
- Szalus K, Trzeciak M, Nowicki RJ (November 2020). “JAK-STAT Inhibitors in Atopic Dermatitis from Pathogenesis to Clinical Trials Results”. Microorganisms. 8 (11): 1743. doi:10.3390/microorganisms8111743. PMC 7694787. PMID 33172122.
- “Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 22-25 July 2024”. European Medicines Agency (Press release). 25 July 2024. Retrieved 29 July 2024.
/////////Delgocitinib, デルゴシチニブ , JAPAN 2020, 2020 APPROVALS, Corectim, UNII-9L0Q8KK220, JTE-052, 9L0Q8KK220, LEO 124249A, LEO 124249, HY-109053, CS-0031558, D11046, GTPL9619, JTE-052A, JTE052, LP-0133 , ROH-201, atopic dermatitis
CC1CN(C12CCN(C2)C3=NC=NC4=C3C=CN4)C(=O)CC#N

 |
|
| Clinical data | |
|---|---|
| Trade names | Corectim, others |
| Other names | JTE-052; JTE-052A |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C16H18N6O |
| Molar mass | 310.361 g·mol−1 |
| 3D model (JSmol) | |
https://pubs.acs.org/doi/10.1021/acs.oprd.1c00031
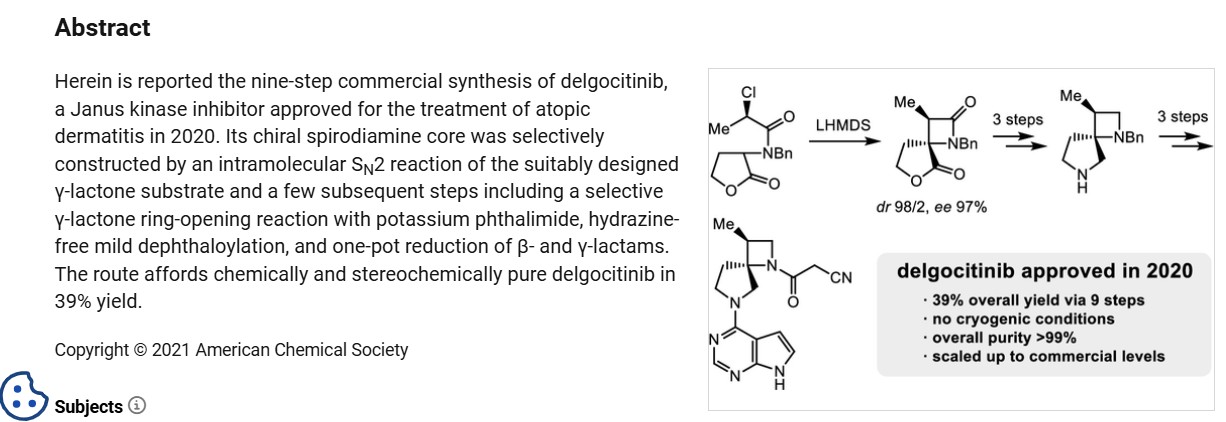
https://www.chemicalbook.com/article/synthesis-of-delgocitinib.htm
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

Synthesis of Delgocitinib
Dec 26,2023
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

A stereocontrolled kilogram scale synthesis of delgocitinib has been disclosed, beginning with an SN2 reaction involving bromolactone 128 and benzyl amine to provide α-amino lactone 129, which was isolated as the HCl salt after precipitation from hydrochloric acid in ethyl acetate. Amine 129 was then acylated with enantiomerically pure acid chloride 131 (prepared by thionyl chloride treatment of commercial acid 130) to furnish lactone 132. In the crucial spirocyclic ring ringforming sequence of the synthesis, lactone 132 was treated with LHMDS to form an enolate that underwent SN2 displacement of the chloride, forming the spirolactone 133 and establishing both stereocenters with 98:2 dr and 96% ee.
The lactone ring of 133 was then opened by an attack of potassium phthalimide on the γ- carbon, and the resulting carboxylic acid was converted to the ethyl ester by treatment with ethyl iodide. Finally, treatment with diethylenetriamine released phthalimide, providing a free amine for subsequent cyclization to spirolactam 134 via the corresponding ethyl ester intermediate. This sequence took place in 80% yield over four steps and provided the spirolactam in >99% de after recrystallization.
The carbonyl groups within spirolactam 134 were then reduced with lithium aluminum hydride and aluminum chloride in THF, and the resulting diamine 135 was crystallized as a succinic acid salt in 86% yield. The SNAr reaction of 135 with chloropyrrolopyrimidine 136 followed by hydrogenative removal of the benzyl protecting group provided amine 137 in 92% yield over 2 steps. Finally, amine 137 was acylated with cyanoacetyl pyrazole 138 and recrystallized from n-butanol with 3 wt % BHT to provide delgocitinib in 86% yield, >99% ee, and >99% de.
Tradipitant, традипитант , تراديبيتانت , 曲地匹坦 ,

Tradipitant
VLY-686, LY686017
- Molecular Formula C28H16ClF6N5O
- Average mass 587.903 Da
PHASE 2, Gastroparesis; Pruritus
FDA 2025, APPROVALS 2025, 12/30/2025, To treat vomiting associated with motion
pyridine-containing NK-1 receptor antagonist ie tradipitant, useful for treating anxiety, pruritus and alcoholism.
Vanda Pharmaceuticals, under license from Eli Lilly, was developing tradipitant, a NK1 antagonist, for treating anxiety disorder, pruritus and alcohol dependence. The company was also investigating the drug for treating gastroparesis. In February 2017, tradipitant was reported to be in phase 2 clinical development for treating anxiety and pruritus.
- Originator Eli Lilly
- Developer Eli Lilly; National Institute on Alcohol Abuse and Alcoholism; Vanda Pharmaceuticals
- Class Antipruritics; Anxiolytics; Chlorobenzenes; Pyridines; Small molecules; Triazoles
- Mechanism of Action Neurokinin 1 receptor antagonists; Substance P inhibitors
Highest Development Phases
- Phase II Gastroparesis; Pruritus
- Discontinued Alcoholism; Social phobia
- The drug had been in phase II clinical trials at Lilly and the National Institute on Alcohol Abuse and Alcoholism for the treatment of alcoholism; however, no recent development has been reported for this research.
- A phase II clinical trial for the treatment of social phobia has been completed by Lilly.
PATENT WO 2003091226
SYNTHESIS

Condensation of 2-chloropyridine with thiophenol in the presence of K2CO3 in DMF at 110ºC yields sulfide intermediate,
which is then oxidized by means of NaOCl in AcOH to give 2-(benzenesulfonyl)pyridine.
This is treated with (iPr)2NH and n-BuLi in THF at -60 to -70°C and subsequently couples with 2-chlorobenzaldehyde in THF at -60 to -70°C to furnish (2-(phenylsulfonyl)pyridin-3-yl)-(2-chlorophenyl)methanone.
Ketone couples with the enolate of 4-acetylpyridine (formed by treating 4-acetylpyridine (VII) with t-BuOK in DMSO) in the presence of LiOH in DMSO and subsequently is treated with PhCOOH in iPrOAc to give rise to pyridine benzoate derivative.
This finally couples with 1-azidomethyl-3,5-bistrifluoromethylbenzene (obtained by treating 3,5-bis(trifluoromethyl)benzylchloride with NaN3 ini DMSO) in the presence of K2CO3 in t-BuOH to afford the title compound Tradipitant.
Tradipitant (VLY-686 or LY686017) is an experimental drug that is a neurokinin 1 antagonist. It works by blocking substance P, a small signaling molecule. Originally, this compound was owned by Eli Lilly and named LY686017. VLY-686 was purchased by Vanda Pharmaceuticals from Eli Lilly and Company in 2012.[1] Vanda Pharmaceuticals is a U.S. pharmaceutical company that as of November 2015 only has 3 drugs in their product pipeline: tasimelteon, VLY-686, and iloperidone.[2]
Tachykinins are a family of peptides that are widely distributed in both the central and peripheral nervous systems. These peptides exert a number of biological effects through actions at tachykinin receptors. To date, three such receptors have been characterized, including the NK-1 , NK-2, and NK-3 subtypes of tachykinin receptor.
The role of the NK-1 receptor subtype in numerous disorders of the central nervous system and the periphery has been thoroughly demonstrated in the art. For instance, NK-1 receptors are believed to play a role in depression, anxiety, and central regulation of various autonomic, as well as cardiovascular and respiratory functions. NK- 1 receptors in the spinal cord are believed to play a role in pain transmission, especially the pain associated with migraine and arthritis. In the periphery, NK-1 receptor activation has been implicated in numerous disorders, including various inflammatory disorders, asthma, and disorders of the gastrointestinal and genitourinary tract.
There is an increasingly wide recognition that selective NK-1 receptor antagonists would prove useful in the treatment of many diseases of the central nervous system and the periphery. While many of these disorders are being treated by new medicines, there are still many shortcomings associated with existing treatments. For example, the newest class of anti-depressants, selective serotonin reuptake inhibitors (SSRIs), are increasingly prescribed for the treatment of depression; however, SSRIs have numerous side effects, including nausea, insomnia, anxiety, and sexual dysfunction. This could significantly affect patient compliance rate. As another example, current treatments for chemotherapy- induced nausea and emesis, such as the 5-HT3receptor antagonists, are ineffective in managing delayed emesis. The development of NK-1 receptor antagonists will therefore greatly enhance the ability to treat such disorders more effectively. Thus, the present invention provides a class of potent, non-peptide NK-1 receptor antagonists, compositions comprising these compounds, and methods of using the compounds.
Indications
Pruritus
It is being investigated by Vanda Pharmaceuticals for chronic pruritus (itchiness) in atopic dermatitis. In March 2015, Vanda announced positive results from a Phase II proof of concept study.[3] A proof of concept study is done in early stage clinical trials after there have been promising preclinical results. It provides preliminary evidence that the drug is active in humans and has some efficacy.[4]
Alcoholism
VLY-686 reduced alcohol craving in recently detoxified alcoholic patients as measured by the Alcohol Urge Questionnaire.[5] In a placebo controlled clinical trial of recently detoxified alcoholic patients, VLY-686 significantly reduced alcohol craving as measured by the Alcohol Urge Questionnaire. It also reduced the cortisol increase seen after a stress test compared to placebo. The dose given was 50 mg per day.
Social anxiety disorder
In a 12-week randomized trial of LY68017 in 189 patients with social anxiety disorder, 50 mg of LY68017 did not provide any statistically significant improvement over placebo.[6]
PATENT
WO03091226,
https://www.google.com/patents/WO2003091226A1?cl=en
PATENT
The compound {2-[l-(3,5-bis-trifluoromethyl-benzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]- pyridin-3-yl}-(2-chlorophenyl)-methanone, depicted below as the compound of Formula I, was first described in PCT published application WO2003/091226.

(I)
Because the compound of Formula I is an antagonist of the NK-I subtype of tachykinin receptor, it is useful for the treatment of disorders associated with an excess of tachykinins. Such disorders include depression, including major depressive disorder; anxiety, including generalized anxiety disorder, panic disorder, obsessive compulsive disorder, and social phobia or social anxiety disorder; schizophrenia and other psychotic disorders, including bipolar disorder; neurodegenerative disorders such as dementia, including senile dementia of the Alzheimer’s type or Alzheimer’s disease; disorders of bladder function such as bladder detrusor hyper-reflexia and incontinence, including urge incontinence; emesis, including chemotherapy-induced nausea and acute or delayed emesis; pain or nociception; disorders associated with blood pressure, such as hypertension; disorders of blood flow caused by vasodilation and vasospastic diseases, such as angina, migraine, and Reynaud’s disease; hot flushes; acute and chronic obstructive airway diseases such as adult respiratory distress syndrome, bronchopneumonia, bronchospasm, chronic bronchitis, drivercough, and asthma; inflammatory diseases such as inflammatory bowel disease; gastrointestinal disorders or diseases associated with the neuronal control of viscera such as ulcerative colitis, Crohn’s disease, functional dyspepsia, and irritable bowel syndrome (including constipation-predominant, diarrhea- -?-
predominant, and mixed irritable bowel syndrome); and cutaneous diseases such as contact dermatitis, atopic dermatitis, urticaria, and other eczematoid dermatitis.
In PCT published application, WO2005/042515, novel crystalline forms of the compound of Formula I, identified as Form IV and Form V, are identified. Also described in WO2005/042515 is a process for preparation of the compound of Formula I, comprising reacting (2-chlorophenyl)-[2-(2- hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone or a phosphate salt thereof with l-azidomethyl-3,5- bistrifluoromethylbenzene in the presence of a suitable base and a solvent. Use of this procedure results in several shortcomings for synthesis on a commercial scale. For example, use of the solvent DMSO, with (2- chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone phosphate, requires a complex work-up that has a propensity to emulsify. This process also requires extraction with CH2CI2, the use of which is discouraged due to its potential as an occupational carcinogen, as well as the use of MgSC>4 and acid-washed carbon, which can generate large volumes of waste on a commercial scale. Conducting the reaction with (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone in isopropyl alcohol, as also described in WO2005/042515, is also undesirable due to the need to incorporate a free base step. Furthermore, variable levels of residual l-azidomethyl-3,5-bistrifluoromethylbenzene, a known mutagen, are obtained from use of the procedures described in WO2005/042515.
An improved process for preparing the compound of Formula I would control the level of 1- azidomethyl-3,5-bistrifluoromethylbenzene impurity, and improve the yield. We have discovered that use of the novel salt, (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate, as well as use of tert-butanol as the reaction solvent, improves reaction times and final yield, and decreases impurities in the final product. In addition, a novel process for the preparation of (2-chlorophenyl)- [2-(2- hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate, in which a pre-formed enolate of 4-acetyl pyridine is added to (2-phenylsulfonyl-pyridine-3-yl)-(2-chlorophenyl)methanone, results in an overall improved yield and improved purity, and is useful on a commercial scale.
EXAMPLES
Example 1 {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)- methanone (Form IV)

Suspend (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl] methanone benzoate (204.7 g; 1.04 equiv; 445 mmoles) in t-butanol (614 mL) and treat the slurry with potassium carbonate (124.2 g; 898.6 mmoles). Heat to 7O0C with mechanical stirring for 1 hour. Add l-azidomethyl-3,5- bistrifluoromethylbenzene (115.6 g; 1.00 equiv; 429.4 mmoles) in a single portion, then heat the mixture to reflux. A circulating bath is used to maintain a condenser temperature of 3O0C. After 18 hours at reflux, HPLC reveals that the reaction is complete (<2% l-azidomethyl-3,5-bistrifluoromethylbenzene remaining). The mixture is cooled to 7O0C, isopropanol (818 mL) is added, then the mixture is stirred at 7O0C for 1 hour. The mixture is filtered, and the waste filter cake is rinsed with isopropanol (409 mL). The combined filtrate and washes are transferred to a reactor, and the mechanically stirred contents are heated to 7O0C. To the dark purple solution, water (1.84 L) is added slowly over 35 minutes. The solution is cooled to 6O0C, then stirred for 1 hour, during which time a thin precipitate forms. The mixture is slowly cooled to RT, then the solid is filtered, washed with 1 : 1 isopropanol/water (614 mL), subsequently washed with isopropanol (410 mL), then dried in vacuo at 450C to produce 200.3 g of crude {2-[l-(3,5- bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)-methanone as a white solid. Crude {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin- 3-yl}-(2-chlorophenyl)-methanone (200.3 g) and isopropyl acetate (600 mL) are charged to a 5L 3-neck jacketed flask, then the contents heated to 750C. After dissolution is achieved, the vessel contents are cooled to 550C, then the solution polish filtered through a 5 micron filter, and the filter rinsed with a volume of isopropyl acetate (200 mL). After the polish filtration operation is complete, the filtrates are combined, and the vessel contents are adjusted to 5O0C. After stirring for at least 15 minutes at 5O0C, 0.21 grams of {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2- chlorophenyl)-methanone Form IV seed (d90 = 40 microns) is added, and the mixture stirred at 5O0C for at least 2 h. Heptanes (1.90 L) are then added over at least 2 h. After the heptanes addition is completed, the slurry is stirred for an hour at 5O0C, cooled to 230C at a rate less then 2O0C per hour, then aged at 230C for an hour prior to isolation. The mixture is then filtered in portions through the bottom outlet valve in the reactor into a 600 mL filter. The resulting wetcake is washed portionwise with a solution containing heptanes (420 mL) and isopropyl acetate (180 mL), which is passed directly through the 5L crystallization vessel. The wetcake is blown dry for 5 minutes with nitrogen, then transferred to a 500 mL plastic bottle. The product is dried at 5O0C for 4 h. to produce 190.3g of pure {2-[l-(3,5- bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)- methanone, Form IV in 75.0% yield with 100% purity, as determined by HPLC analysis. Particle size is reduced via pin or jet mill. 1H NMR (400 MHz, CDCl3): 5.46 (s, 2H); 7.19 (m, 5H); 7.36 (dd, IH, J = 4.9, 7.8); 7.45 (s, 2H); 7.59 (m, IH); 7.83 (s, IH); 7.93 (dd, IH, J = 1.5, 7.8); 8.56 (dd, IH, J= 1.5, 4.9); 8.70 (d, 2H, J= 5.9).
Preparation 1-A (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate Charge powdered KOfBu (221.1 g, 1.93 moles, 1.40 eq.) to Reactor A, then charge DMSO (2 L) at
250C over 10 min. The KOfBu/DMSO solution is stirred for 30 min at 230C, then a solution of 4-acetyl pyridine (92 mL, 2.07 moles, 1.50 eq) in DMSO (250 mL) is prepared in reactor B. The contents of reactor B are added to Reactor A over 10 minutes, then the Reactor A enolate solution is stirred at 230C for Ih. In a separate 12-L flask (Reactor C), solid LiOH (84.26 g, 3.45 moles, 2.0 eq) is poured into a mixture of (2- phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone (500.0 g, 1.34 moles, 1.0 eq) and DMSO (2L), with stirring, at 230C. The enolate solution in reactor A is then added to Reactor C over a period of at least 15 minutes, and the red suspension warmed to 4O0C. The reaction is stirred for 3h, after which time HPLC analysis reveals less than 2% (2-phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone. Toluene (2.5 L) is charged, and the reactor temperature cooled to 3O0C. The mixture is quenched by addition of glacial acetic acid (316 mL, 5.52 moles, 4.0 eq), followed by 10 % NaCl (2.5 L). The biphasic mixture is transferred to a 22-L bottom-outlet Morton flask, and the aqueous layer is removed. The aqueous layer is then extracted with toluene (750 mL). The combined organic layers are washed with 10 % NaCl (750 mL), then concentrated to 4 volumes and transferred to a 12-L Morton flask and rinsed with isopropyl acetate (4 vol, 2 L). The opaque amber solution is warmed to 75 degrees to 750C over 40 min. Benzoic acid (171. Ig, 1.34 moles, 1.0 eq) is dissolved in hot isopropyl acetate (1.5 L), and charged to the crude free base solution over at least 30 min. The crude solution containing benzoate salt is stirred for 0.5 h at 750C then cooled to 23 0C. When solids are first observed, the cooling is stopped and the mixture is aged for an hour at the temperature at which crystals are first observed. Alternatively, if seed crystal is available, the mixture may be seeded with (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate (2.25g) at 750C, followed by stirring for 0.5 h at 750C, then cooling to 230C over at least 1.5 h. The mixture is then cooled to <5 0C, then filtered through paper on a 24cm single-plate filter. The filtercake is then rinsed with cold z‘-PrOAc (750 mL) to produce granular crystals of bright orange-red color. The wet solid is dried at 550C to produce 527.3 g (83% yield) with 99.9% purity. (2-chlorophenyl)-[2-(2-hydroxy-2- pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate. Anal. Calcd. for C26Hi9N2ClO4: C, 68.05; H, 4.17; N, 7.13. Found: C, 67.89; H, 4.15; N 6.05. HRMS: calcd for C19H13ClN2O2, 336.0666; found 336.0673.
The synthesis of(2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate proceeds optimally when the potassium enolate of 4-acetyl pyridine is pre-formed using KOfBu in DMSO. Pre-formation of the enolate allows the SNAR (nucleophilic aromatic substitution) reaction to be performed between room temperature and 4O0C, which minimizes the amount of degradation. Under these conditions, the SNAR is highly regioselective, resulting in a ratio of approximately 95:5 preferential C – acylation. In all cases, less polar solvents such as THF or toluene, or co-solvents of these solvents mixed with DMSO, results in a substantial increase of acylation at the oxygen in the SNAR, and leads to a lower yield of product. This is a substantial improvement over the procedures described in WO2005/042515 for synthesis of the free base or the phosphate salt, in which the SNAR is performed at 60-700C, resulting in a substantial increase in chemical impurity. Using the conditions described in WO2005/042515, when scaled to 2kg, results in maximum yields of 55%, with sub-optimal potency. In comparison, the improved conditions described herein can be run reproducibly from 0.4 to 2kg scale to give yields of 77-83%, with >99% purity. In addition, the reaction can be held overnight at 4O0C with minimal degradation, whereas holding the reaction for 1 h past completion at 60-70°C results in substantial aromatized impurity. The reaction may also be performed using sodium tert-amylate as the base, in combination with an aprotic solvent, such as DMSO or DMF.
The title compound exists as a mixture of tautomers and geometric isomers. It is understood that each of these forms is encompassed within the scope of the invention.

Preparation 1-B
(2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone toluate The procedure described in Preparation 1-A is followed, with the following exception. Solid toluic acid (1.0 eq) is added to the crude free base solution at 550C, then the solution cooled to 45 0C. The solution is stirred for one hour at 45 0C, then slowly cooled to 23 0C. When solids are first observed, the cooling is stopped and the mixture is aged for an hour at the temperature at which crystals are first observed. Alternatively, if seed crystal is available, the mixture may be seeded, aged for 3 h at 450C , then cooled to O0C over 4 h. The isolation slurry is filtered, and the wetcake washed with MeOH (3 volumes). The wetcake is dried at 5O0C to provide 14.0 g (76.4%) of (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl- vinyl)pyridin-3-yl]methanone toluate as a light red powder.
As with the benzoate salt, the toluate salt can also exist as a mixture of tautomers and geometric isomers, each of which is encompassed within the scope of the invention. (2-chlorophenyl)-[2-(2-hydroxy- 2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone toluate . 13C NMR (125 MHz,DMS0-d6) δ 194.5, 167.8, 167.4, 155.5, 150.7 (2C), 147.4, 144.0, 143.4, 142.7, 138.6, 133.0, 130.8, 130.7, 130.5, 129.8(2C), 129.5(2C), 128.5, 128.0, 127.9, 119.9 (2C), 118.6, 92.6, 21.5.
Preparation 1-C
(2-phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone
A solution of 1.3 eq of diisopropylamine (based on 2-benzenesulfonyl pyridine) in 5 volumes of THF in a mechanically stirred 3 -necked flask is cooled to -70 to -75 0C. To this solution is added 1.05 eq of w-butyllithium (1.6M in hexanes) at such a rate as to maintain the temperature below -6O0C. The light yellow solution is stirred at -60 to -70 0C for 30 minutes. Once the temperature has cooled back down to – 60 to -650C, 1.0 eq of 2-benzene-sulfonyl pyridine, as a solution in 3 volumes of THF, is added at the fastest rate that will maintain the reaction temperature under -6O0C. A yellow suspension forms during the addition that becomes yellow-orange upon longer stirring. This mixture is stirred for 3 hours at -60 to – 750C, and then 1.06 eq of 2-chlorobenzaldehyde, as a solution in 1 volume of THF, is added dropwise at a sufficient rate to keep the temperature under -55 0C. The suspension gradually turns orange-red, thins out, and then becomes a clear red solution. The reaction mixture is allowed to stir at -60 to -7O0C for 1 hour, 3N aqueous HCl (7 volumes) is added over 20-30 minutes, and the temperature is allowed to exotherm to 0-100C. The color largely disappears, leaving a biphasic yellow solution. The solution is warmed to at least 1O0C, the layers are separated, and the aqueous layer is back-extracted with 10 volumes of ethyl acetate. The combined organic layers are washed with 10 volumes of saturated sodium bicarbonate solution and concentrated to about 2 volumes. Ethyl acetate (10 volumes) is added, and the solution is once again concentrated to 2 volumes. The thick solution is allowed to stand overnight and is taken to the next step with no purification of the crude alcohol intermediate. The crude alcohol intermediate is transferred to a 3 -necked flask with enough ethyl acetate to make the total solution about 10 volumes. The yellow solution is treated with 3.2 volumes of 10% aqueous (w/w) potassium bromide, followed by 0.07 eq of 2,2,6,6-Tetramethylpiperidine-N-oxide (TEMPO). The orange mixture is cooled to 0-50C and treated with a solution of 1.25 eq of sodium bicarbonate in 12% w/w sodium hypochlorite (9 volumes) and 5 volumes of water over 30-60 minutes while allowing the temperature to exotherm to a maximum of 2O0C. The mixture turns dark brown during the addition, but becomes yellow, and a thick precipitate forms. The biphasic light yellow mixture is allowed to stir at ambient temperature for 1-3 hours, at which time the reaction is generally completed. The biphasic mixture is cooled to 0-50C and stirred for 3 hours at that temperature. The solid is filtered off, washed with 4 volumes of cold ethyl acetate, followed by 4 volumes of water, and dried in vacuo at 450C to constant weight. Typical yield is 80-83% with a purity of greater than 98%. 1H NMR (600 MHz, CDCl3-^) δ ppm 7.38 (td, ./=7.52, 1.28 Hz, 1 H) 7.47 (dd, ./=7.80, 1.30 Hz, 1 H) 7.51 (td, ./=7.79, 1.60 Hz, 1 H) 7.51 (t, ./=7.89 Hz, 2 H) 7.50 – 7.54 (m, J=7.75, 4.63 Hz, 1 H) 7.60 (t, J=7.43 Hz, 1 H) 7.73 (dd, J=7.75, 1.60 Hz, 1 H) 7.81 (dd, J=7.79, 1.56 Hz, 1 H) 8.00 (dd, ./=8.44, 1.10 Hz, 2 H) 8.76 (dd, ./=4.63, 1.61 Hz, 1 H).
Preparation 1-D 1 -azidomethyl-3,5-bistrifluoromethyl-benzene
Sodium azide (74.3 g, 1.14 mol) is suspended in water (125 mL), then DMSO (625 mL) is added. After stirring for 30 minutes, a solution consisting of 3,5-Bis(trifluoromethyl)benzyl chloride (255.3 g, 0.97 moles) and DMSO (500 mL) is added over 30 minutes. (The 3,5-Bis(trifluoromethyl)benzyl chloride is heated to 350C to liquefy prior to dispensing (MP = 30-320C)). The benzyl chloride feed vessel is rinsed with DMSO (50 mL) into the sodium azide solution, the mixture is heated to 4O0C, and then maintained for an hour at 4O0C, then cooled to 230C.
In Process Analysis: A drop of the reaction mixture is dissolved in d6-DMSO and the relative intensities of the methylene signals are integrated (NMR verified as a 0.35% limit test for 3,5- Bis(trifluoromethyl)benzyl Chloride). Work-up: After the mixture reaches 230C , it is diluted with heptanes (1500 mL), then water (1000 mL) is added, and the mixture exotherms to 350C against a jacket setpoint of 230C. The aqueous layer is removed (-2200 mL), then the organic layer (approximately 1700 mL) is washed with water (2 X 750 mL). The combined aqueous layers (-3700 mL) are analyzed and discarded.
The solvent is then partially removed via vacuum distillation with a jacket set point of 850C, pot temperature of 60-650C and distillate head temperature of 50-550C to produce 485g (94.5% yield) of 51 Wt% solution title compound as a clear liquid. Heptanes can be either further removed by vacuum distillation or wiped film evaporation technology. 1H NMR (400 MHz, CDCl3): 4.58 (s, 2H); 7.81 (s, 2H); 7.90 (s, IH).
Preparation 1-E 2-benzene-sulfonyl pyridine Charge 2-chloropyridine (75 mL, 790 mmol), thiophenol (90 mL, 852 mmol), and DMF (450 mL) to a 2L flask. Add K2CO3 (134.6 g, 962 mmol), then heat to HO0C and stir for 18 hours. Filter the mixture, then rinse the waste cake with DMF (195 mL). The combined crude sulfide solution and rinses are transferred to a 5-L flask, and the waste filtercake is discarded. Glacial acetic acid (57 mL, 995 mmol) is added to the filtrate, then the solution is heated to 4O0C, and 13 wt % NaOCl solution (850 mL, 1.7 mol) is added over 2 hours. After the reaction is complete, water (150 mL) is added, then the pH of the mixture adjusted to 9 with 20 % (w/v) NaOH solution (250 mL). The resulting slurry is cooled to <5 0C, stirred for 1.5 h, then filtered, and the cake washed with water (3 x 200 mL). The product wetcake is dried in a 550C vacuum oven to provide 2-benzene-sulfonyl pyridine (149 g, 676 mmol) in 86 % yield: 1H NMR (500 MHz, CDCl3) δ 8.66 (d, J = 5.5 Hz, IH), 8.19 (d, J = 1.1 Hz, IH), 8.05 (m, 2H), 7.92 (ddd, J= 9.3, 7.7, 1.6 Hz, IH), 7.60 (m, IH), 7.54 (m, 2H), 7.44 (m, IH); IR (KBr) 788, 984, 1124, 1166, 1306, 1424, 1446, 1575, 3085 cm“1; MS (TOF) mlz 220.0439 (220.0427 calcd for C11H10NO2S, MH); Anal, calcd for C11H9NO2S: C, 60.26; H, 4.14; N, 6.39; S, 14.62. Found: C, 60.40; H, 4.02; N, 6.40; S, 14.76.
As noted above, use of the improved process of the present invention results in an improved habit of the crystalline Form IV compound of Formula I. The improved habit reduces surface area of the crystal, improves the filtration, and washing, and improves the efficiency of azide mutagen rejection. These improvements are described in greater detail below.
In patent application WO2005/042515, the polish filtration is carried out in 7 volumes (L/kg) of isopropanol near its boiling point (65-83 0C), a process that is difficult and hazardous to execute in commercial manufacturing because of the high risk of crystallization on the filter and/or vessel transfer lines due to supersaturation. In the preferred crystallization solvent, isopropyl acetate, the polish filtration is conducted in four volumes of isopropyl acetate at temperatures from 45 to 55 0C. This temperature range is 35 to 45 0C lower than the boiling point of isopropyl acetate, which provides a key safety advantage.
PATENT
PATENT
WO 2017031215
EXAMPLES
Example 1: Preparation of Compound (I) via Negishi Coupling Route
Example 1 provides a scheme including preparations 1A-1D, described below, for the synthesis of the compound of Formula (I) and intermediates used in the route. An overview of the scheme is as follows:

80 on ma s ale
Example 1A: Preparation of Compound (I)

Zinc dust (200 mg, 3.06 mmol) combined with 2.0 mL of dimethylformamide was treated with 0.010 mL of 1,2-dibromoethane and heated to 65°C for 3 minutes. The mixture was cooled to ambient temperature and treated with 0.010 mL of trimethylsilyl chloride. After 5 minutes, 1.26 mL of 1M zinc chloride in diethyl ether was added to the mixture followed by Compound (Ila) (600 mg, 1.20 mmol). The mixture was heated to 65°C and further treated with 0.020 mL each of 1,2-dibromoethane and trimethylsilyl chloride. After 2.5 hours, via HPLC chromatogram, the reaction showed some formation of the zincate and was allowed to stir at ambient temperature for 16 hours. At this time
tetrakis(triphenylphosphine)palladium(0) (70 mg, 0.06 mmol), Compound (Ilia) (357 mg, 1.20 mmol) were added to the reaction and the mixture heated to 65°C. HPLC analysis showed the formation of Compound (I) in the reaction.
IB: Preparation of Comp

To a solution of Compound (IV) (8.00 g, 18 mmol) in 40 mL of 1,2-dichloroethane was added a solution of iodine monochloride (10.7 g, 65.9 mmol) in 40 mL of 1,2-dichloroethane resulting in a slurry. The slurry was heated to 75°C for 4 hours then cooled to ambient temperature. The solids were collected by filtration, washed with heptane, then combined with 90 mL of ethyl acetate and 80 mL of saturated sodium thiosulfate solution. The organic phase was washed with saturated sodium chloride solution and dried with sodium sulfate. The mixture was concentrated to yield 7.80 g (87%) of Compound (Ila) as a yellow solid. The product could be further purified by silica gel chromatography. Thus 2.0 g of yellow solid was dissolved in dichloromethane and charged onto a silica gel column. The product was eluted using tert-butyl methyl ether to provide 1.87 g (93% recovery) of Compound (Ila) as a white powder. Analytical data: Iodine monochloride complex: ¾ NMR (500 MHz, DMSO-de) δ 8.80 (2 H), 8.05 (1 H), 7.77 (2 H), 7.59 (2 H), 5.86 (2 H).
Uncomplexed: ¾ NMR (500 MHz, DMSO-de) δ 8.71 (2 H), 8.03 (1 H), 7.74 (2 H), 7.44 (2 H), 5.86 (2 H).
It was observed that the iodination proceeded smoothly as a suspension in 1,2-dichloroethane with IC1 (4.0 equiv) at 75°C. An ICl-Compound (Ila) complex was initially isolated by filtration. Compound (Ila) was then obtained in approximately 85% yield by treatment of the ICl-Compound (Ila) complex with sodium thiosulfate. This protocol provided a viable means of isolation of Compound (Ila) without the use of DMF.
Example 1C: Preparation of silyl substituted triazole (Compound IV)

A mixture of Compound (V) (8.07 g, 30.0 mmol) and Compound (VI) (5.12 g, 29.2 mmol) was heated to 100°C for 18 hours. To the mixture was added 40 mL of heptane and the reaction was allowed to cool with rapid stirring. After 1 hour the solids were collected by filtration and washed with heptane then dried to 9.30 g (72%) of Compound (IV) as a tan solid. Analytical data: ¾ NMR (500 MHz, DMSO-de) δ 8.66 (2 H), 8.04 (1 H), 7.67 (2 H), 7.32 (2 H), 5.72 (2 H), 0.08 (9 H).
It was further found that combining Compound (V) and Compound (VI) (neat) and heating at 95 – 105°C afforded a 92: 8 mixture of regioisomers as shown below:

Crystallization of the mixture from heptane afforded Compound (IV) in 62-72% yield, thus obviating the need for chromatography to isolate Compound (IV).
Example ID: Preparation of starting material Compound (VI)

Zinc bromide (502 g, 2.23 mole) was added in approximately 100 g portions to 2.0 L of tetrahydrofuran cooled to between 0 and 10°C. To this cooled solution was added 4-bromopyridine hydrochloride (200 g, 1.02 mol), triphenylphosphine (54 g, 0.206 mol), and palladium (II) chloride (9.00 g, 0.0508 mol). Triethylamine (813 g, 8.03 mol) was then added at a rate to maintain the reaction temperature at less than 10°C, and finally
trimethylsilylacetylene (202 g, 2.05 mol) was added. The mixture was heated to 60°C for 4.5 hours. The reaction was cooled to -5°C and combined with 2.0 L of hexanes and treated with 2 L of 7.4 M NH4OH. Some solids were formed and were removed as much as possible with the aqueous phase. The organic phase was again washed with 2.0 L of 7.4 M NH4OH, followed by 2 washes with 500 mL of water, neutralized with 1.7 L of 3 M hydrochloric acid, dried with sodium sulfate, and concentrate to a thick slurry. The slurry was combined with 1.0 L of hexanes to give a precipitate. The precipitate was removed by filtration and the filtrate was concentrated to 209 g of dark oil. The product was purified by distillation (0.2 torr, 68°C) to give 172 g (96%) of Compound (VI) as colorless oil. Analytical data: ¾ NMR (500 MHz, DMDO-de) δ 8.57 (2 H), 7.40 (2 H), 0.23 (9 H).
EXAMPLE 2 – Preparation of Compound (Ilia)
Example 2 provides a morpholine amide route for the synthesis of Compound (Ilia). In this approach, morpholine amide (Compound VII) was prepared from 2-chlorobenzoyl chloride (Preparation 2A). Metallation of 2-bromopyridine with LDA (1.09 equiv.) in THF at -70°C followed by addition of (Compound VII) afforded Compound (Ilia) in 37% yield after crystallization from IP A/heptane (Preparation 2B). This sequence provides a direct route to Compound (Ilia), and a means to isolate Compound (Ilia) without the use of
chromatography. Compound (Ilia) may then be used to form Compound (I) as shown in Example 1A above (Preparation 2C).
Preparation 2A: Preparation of Compound (VII)

Toluene (1.5 L) was added to Compound (IX) (150 g, 0.86 mol) and cooled to 10°C. Morpholine (82 mL, 0.94 mol) was added to the clear solution over 10 minutes. The resulting white slurry was stirred for 20 minutes then pyridine (92 mL, 1.2 mol) was added dropwise over 20 minutes. The cloudy white mixture was stirred in a cold bath for 1 hour. Water (600 mL) was added in a single portion and the cold bath removed. The mixture was stirred for 20 minutes and the layers are separated. The organic layer was washed with a mixture of 1 N HC1 and water (2: 1, 500 mL:250 mL). The pH of the aqueous layer was ~ 2. The organic layer was washed with a mixture of saturated NaHCCb and water (1 : 1, 100 mL: 100 mL). The pH of the aqueous layer was ~ 9. The layers were separated. The organic layer was concentrated in vacuo to an oil. The oil was dissolved in IPA (70 mL) and heated at 60°C for 30 min. The clear solution was allowed to cool to 30°C, then heptane (700 mL, 4.7 v) was added dropwise. The resulting slurry was stirred at RT for 2 hours then cooled to 0°C for 1 hour. The slurry was filtered at RT, washed with heptane then dried under vacuum at 30°C overnight. Compound (VII) (156.2 g, 81%) was obtained as a white solid. Analytical data: ¾ NMR (500 MHz, CDCh) δ 7.42-7.40 (m, 1 H), 7.35-7.29 (m, 3 H), 3.91-3.87 (m, 1 H), 3.80-3.76 (m, 3 H), 3.71 (ddd, J= 11.5, 6.8, 3.3 Hz, 1 H), 3.60 (ddd, J = 11.2, 6.4, 3.4 Hz, 1 H), 3.28 (ddd, J= 13.4, 6.3, 3.2 Hz, 1 H), 3.22 (ddd, J= 13.7, 6.8, 3.3 Hz, 1 H); LRMS (ES+) calcd for CnHi3F6ClN02 (M+H)+ 226.1, found 225.9 m/z.
Preparation 2B: Preparation of Compound (Ilia)

THF (75 mL) was added to diisopropyl amine (4.9 mL, 34.8 mmol) and cooled to a
temperature of -70°C under N2 atmosphere. 2.5 M w-BuLi in hexanes (13.9 mL, 34.8 mmol) was added in a single portion (a 30-40°C exotherm) to the clear solution and cooled back to -70°C. Compound (VIII) (5.0 g, 31.6 mmol) was added neat to the LDA solution (a 2 to 5°C exotherm) followed by a THF (10 mL) rinse, keeping T< -65°C. This clear yellow solution was stirred at -70°C for 15 min. Compound (VII) (7.1 g, 31.6 mmol) in THF (30 mL) was added keeping T< -65°C. The resulting clear orange solution was stirred at -70°C for 3 hours. MeOH (3 mL) was added to quench reaction mixture and the cold bath was removed. 5 N HC1 (25 mL) was added to the reaction solution. MTBE (25 mL) was added, and the layers were separated. The organic layer was washed with water (25 mL X 2). The organic layer was dried over MgS04 and filtered. The organic layer was concentrated in vacuo to an orange oil. The oil was dissolved in IPA (15 mL, 3 vol) at ambient temperature. Heptane (25 mL) was added dropwise and the resulting slurry was stirred at RT for 1 hour. The slurry was cooled to 0°C for 1 hour and filtered. The filter cake was rinsed with chilled heptane (20 mL) and dried under vacuum at 30°C overnight. Compound (Ilia) (4.25 g, 45%) was obtained as a yellow solid.
Several reactions were run at different temperatures and with different addition rates of Compound (VII). If the reaction temperature was maintained below -65°C and Compound (VII) was added in <5 min, it was found that the reaction worked well. If the temperature was increased and/or the addition time of Compound (VII) was increased, then yields suffered, and the work-up was complicated by emulsions.
Preparation 2C: Preparation of Compound (I)
Compound (Ilia) may then reacted with Compound (Ila) to produce Compound (I) as shown in Preparation 1A.
EXAMPLE 3
Example 3 describes a new route for the synthesis of an intermediate free base, which may be used to form Compound (I) as described further below.
Example 3A: Preparation of starting material (Compound X) from 2-Chloronicotinonitrile

A mixture of NaH (40.0 g, 1 mol, 60% dispersion in mineral oil) and 2-chloronicotinonitrile (69.3 g, 500 mmol) in THF (1 L) was heated to reflux. A solution of 4-acetylpyridine (60.6 g, 500 mmol) in THF (400 mL) was added over a period of 40 min. The resulting dark brown mixture was stirred at reflux for ~ 2 h. The heating mantle was then removed, and AcOH (58 mL, 1 mol) was added. EtOAc (1 L) and H2O (1 L) were then added, and the layers were separated. The organic layer was concentrated to afford an oily solid. CH3CN (500 mL) was added, and the mixture was stirred for 30 min. H2O (1 L) was then added. The mixture was stirred for 1 h then filtered. The solid was rinsed with 2: 1
CH3CN-H2O (900 mL) and hexanes (400 mL) then dried under vacuum at 45°C overnight to afford 61.4 g (55% yield) of Compound (X) as yellow solid. Compound (X) exists as an approximate 95:5 enol-ketone mixture in CDCI3. Analytical data for enol: IR (CHCI3): 3024, 2973, 2229, 1631, 1597, 1579, 1550, 1497; ¾ NMR (500 MHz, CDCI3) δ 8.69 (dd, J= 4.4,
1.7 Hz, 2H), 8.55 (dd, J = 5.2, 1.8 Hz, 1H), 7.97 (dd, J= 7.9, 1.8 Hz, 1H), 7.70 (dd, J= 4.6, 1.5 Hz, 2H, 7.17 (dd, J = 7.8, 5.0 Hz, 1H), 6.59 (s, 1H); LRMS (ES+) calcd for C13H10N3O (M+H)+ 224.1, found 224.0 m/z.
Preparation 3B: Preparation of Compound (XI)
Preparation 3B(1):

(X) (XI)
Compound (XI) may be prepared using Compound (X).
Preparation 3B(2):
Alternatively, the following procedure for the conversion of nitrile into an acid which may also yield compound (XI). A mixture of Compound (X) (1 eq) and NaOH (1.5 eq) in 1 : 1 fhO-EtOH (3.5 mL/g of Compound (X)) was heated at 65°C overnight. The reaction mixture was cooled to RT then added to CH2C12 (12.5 mL/g of Compound (X)) and H20 (12.5 mL/g of Compound (X)). Cone. HC1 (2.5 mL/g of Compound (X)) was then added, and the layers were separated. The aqueous layer was extracted with CH2CI2 (10 mL/g of Compound (X)). The combined organic extracts were washed with H2O (12.5 ml/g of Compound (X)), dried (MgS04), filtered and concentrated to afford Compound (XI).
Preparation 3C
Compound Compound (XI) may then be converted into a Stage C intermediate free base, with observed 87% conversion in Grignard reaction as shown above. A complete synthesis route for Com ound (I) starting from compound Compound (XI) is depicted below.

Detailed experimental procedures for the synthesis of benzoate salt and final step are given in
International Patent Application Publication WO 2008/079600 Al .
References
- “Company Overview of Eli Lilly & Co., Worldwide License to Develop and Commercialize VLY-686”. Bloomberg Business. Retrieved 16 November 2015.
- [1]
- “Vanda Pharmaceuticals Announces Tradipitant Phase II Proof of Concept Study Results for Chronic Pruritus in Atopic Dermatitis”. PR Newswire. Retrieved 16 November 2015.
- Schmidt, B (2006). “Proof of principle studies”. Epilepsy Res. 68 (1): 48–52. doi:10.1016/j.eplepsyres.2005.09.019. PMID 16377153.
- George, DT; Gilman, J; Hersh, J; et al. (2008). “Neurokinin 1 receptor antagonism as a possible therapy for alcoholism.”. Science. 6: 1536–1539. doi:10.2147/SAR.S70350. PMC 4567173
 . PMID 26379454.
. PMID 26379454. - Tauscher, J; Kielbasa, W; Iyengar, S; et al. (2010). “Development of the 2nd generation neurokinin-1 receptor antagonist LY686017 for social anxiety disorder”. European Neuropsychopharmacology. 20 (2): 80–87. doi:10.1016/j.euroneuro.2009.10.005. PMID 20018493.
George, D.T.; Gilman, J.; Hersh, J.; Thorsell, A.; Herion, D.; Geyer, C.; Peng, X.; Kielbasa, W.; Rawlings, R.; Brandt, J.E.; Gehlert, D.R.; Tauscher, J.T.; Hunt, S.P.; Hommer, D.; Heilig, M. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism, Science 2008, 319(5869): 1536
Gackenheimer, S.L.; Gehlert, D.R.In vitro and in vivo autoradiography of the NK-1 antagonist (3H)-LY686017 in guinea pig brain39th Annu Meet Soc Neurosci (October 17-21, Chicago) 2009, Abst 418.16
Tonnoscj, K.; Zopey, R.; Labus, J.S.; Naliboff, B.D.; Mayer, E.A.
The effect of chronic neurokinin-1 receptor antagonism on sympathetic nervous system activity in irritable bowel syndrome (IBS) Dig Dis Week (DDW) (May 30-June 4, Chicago) 2009, Abst T1261
Kopach, M.E.; Kobierski, M.E.; Coffey, D.S.; et al.
Process development and pilot-plant synthesis of (2-chlorophenyl)[2-(phenylsulfonyl)pyridin-3-yl]methanone
Org Process Res Dev 2010, 14(5): 1229
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016060250 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2015-11-10 | 2016-03-03 |
| US2015320866 | PHARMACEUTICAL COMPOSITION COMPRISING ANTIEMETIC COMPOUNDS AND POLYORTHOESTER | 2013-12-13 | 2015-11-12 |
| US2014206877 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2014-03-27 | 2014-07-24 |
| US2012225904 | New 7-Phenyl-[1, 2, 4]triazolo[4, 3-a]Pyridin-3(2H)-One Derivatives | 2010-11-09 | 2012-09-06 |
| US2010056795 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2010-03-04 | |
| US7381826 | Crystalline forms of {2-[1-(3, 5-bis-trifluoromethyl-benzyl)-5-pyridin-4-yl-1H-[1, 2, 3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)-methanone | 2007-04-05 | 2008-06-03 |
| US7320994 | Triazole derivatives as tachykinin receptor antagonists | 2005-10-27 | 2008-01-22 |
 |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| Chemical and physical data | |
| Formula | C28H16ClF6N5O |
| Molar mass | 587.90 g/mol |
| 3D model (Jmol) | |
Tradipitant
Tradipitant is being evaluated in a Phase II study in treatment resistant pruritus in atopic dermatitis.
Tradipitant is an NK-1 receptor antagonist licensed from Eli Lilly in 2012. Tradipitant has demonstrated proof-of-concept in alcohol dependence in a study published by the NIH1. In that study tradipitant was shown to reduce alcohol cravings and voluntary alcohol consumption among patients with alcohol dependence. NK-1R antagonists have been evaluated in a number of indications including chemotherapy-induced nausea and vomiting (CINV), post-operative nausea and vomiting (PONV), alcohol dependence, anxiety, depression, and pruritus.
The NK-1R is expressed throughout different tissues of the body, with major activity found in neuronal tissue. Substance P (SP) and NK-1R interactions in neuronal tissue regulate neurogenic inflammation locally and the pain perception pathway through the central nervous system. Other tissues, including endothelial cells and immune cells, have also exhibited SP and NK-1R activity2. The activation of NK-1R by the natural ligand SP is involved in numerous physiological processes, including the perception of pain, behavioral stressors, cravings, and the processes of nausea and vomiting1,2,3. An inappropriate over-expression of SP either in nervous tissue or peripherally could result in pathological conditions such as substance dependence, anxiety, nausea/vomiting, and pruritus1,2,3,4. An NK-1R antagonist may possess the ability to reduce this over-stimulation of the NK-1R, and as a result address the underlying pathophysiology of the symptoms in these conditions.
References
- George DT, Gilman J, Hersh J, Thorsell A, Herion D, Geyer C, Peng X, Keilbasa W, Rawlings R, Brandt JE, Gehlert DR, Tauscher JT, Hunt SP, Hommer D, Heilig M. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008; 319(5869):1536-9
- Almeida TA, Rojo J, Nieto PM, Pinto FM, Hernandez M, et al. Tachykinins and tachykinin receptors: structure and activity relationships. Current Medicinal Chemistry. 2004;11:2045-2081.
- Hargreaves R, Ferreira JC, Hughes D, Brands J, Hale J, Mattson B, Mill S. Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Annals of the New York Academy of Sciences. 2011; 1222:40-48.
- Stander S, Weisshaar E, Luger A. Neurophysiological and neurochemical basis of modern pruritus treatment. Experimental Dermatology. 2007;17:161-69.
///////////////////tradipitant, PHASE 2, VLY-686, LY686017, традипитант , تراديبيتانت , 曲地匹坦 , VANDA, ELI LILLY, Gastroparesis Pruritus, FDA 2025, APPROVALS 2025, vomiting associated with motion
Atrasentan



Atrasentan
- 173937-91-2

- as HCl: 195733-43-8
A-147627, (+)-A-127722, ABT-627,173937-91-2,
(2R,3R,4S)-4-(1,3-benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid
Endothelin ET-A antagonist
Diabetic nephropathy; End stage renal disease; Renal disease
FDA APPROVED 4/02/2025, Vanrafia, To reduce proteinuria in adults with primary immunoglobulin A nephropathy at risk of rapid disease progression
- (2R,3R,4S)-4-(Benzo[d][1,3]dioxol-5-yl)-1-(2-(dibutylamino)-2-oxoethyl)-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid
- (2R,3R,4S)-4-Benzo[1,3]dioxol-5-yl-1-dibutylcarbamoylmethyl-2-(4-methoxy-phenyl)-pyrrolidine-3-carboxylic acid
- 3-Pyrrolidinecarboxylic acid, 4-(1,3-benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)-, (2alpha,3beta,4alpha)-
- 3-Pyrrolidinecarboxylic acid, 4-(1,3-benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)-, (2R,3R,4S)-rel-
- 4-(1,3-Benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)-3-pyrrolidinecarboxylic acid, (2R,3R,4S)-rel-
- rel-(2R,3R,4S)-4-(Benzo[d][1,3]dioxol-5-yl)-1-(2-(dibutylamino)-2-oxoethyl)-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Atrasentan hydrochloride | E4G31X93ZA | 195733-43-8 | IJFUJIFSUKPWCZ-SQMFDTLJSA-N |
Atrasentan is an experimental drug that is being studied for the treatment of various types of cancer,[1] including non-small cell lung cancer.[2] It is also being investigated as a therapy for diabetic kidney disease.
Atrasentan failed a phase 3 trial for prostate cancer in patients unresponsive to hormone therapy.[3] A second trial confirmed this finding.[4]
It is an endothelin receptor antagonist selective for subtype A (ETA). While other drugs of this type (sitaxentan, ambrisentan) exploit the vasoconstrictive properties of endothelin and are mainly used for the treatment of pulmonary arterial hypertension, atrasentan blocks endothelin induced cell proliferation.
In April 2014, de Zeeuw et al. showed that 0.5 mg and 1.25 mg of atrasentan reduced urinary albumin by 35 and 38% respectively with modest side effects. Patients also had decreased home blood pressures (but no change in office readings) decrease total cholesterol and LDL. Patients in the 1.25 mg dose group had increased weight gain which was presumably due to increased edema and had to withdraw from the study more than the placebo or 0.5 mg dose group.[5] Reductions in proteinuria have been associated with beneficial patient outcomes in diabetic kidney disease with other interventions but is not an accepted end-point by the FDA.
The recently initiated SONAR trial[6] will determine if atrasentan reduces kidney failure in diabetic kidney disease.
Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes. For a prior filing see WO2015006219 , claiming the stable solid composition in the form of a tablet comprising atrasentan and an anti-oxidant. AbbVie (following its spin-out from Abbott), is developing atrasentan (phase III; February 2015) for treating chronic kidney disease, including diabetic nephropathy.
PAPER
European Journal of Organic Chemistry
Enantioselective Synthesis of the Pyrrolidine Core of Endothelin Antagonist ABT-627 (Atrasentan) via 1,2-Oxazines
Year:2003
Volume:2003
Issue:18
page:3524-3533
PATENT
http://www.google.com/patents/US20080132710
EXAMPLE 1
A mixture of bromoacetyl bromide (72.3 mL) in toluene (500 mL) at 0° C. was treated with dibutylamine (280 mL) in toluene (220 mL) while keeping the solution temperature below 10° C., stirred at 0° C. for 15 minutes, treated with 2.5% aqueous phosphoric acid (500 mL) and warmed to 25° C. The organic layer was isolated, washed with water (500 mL) and concentrated to provide the product as a solution in toluene.
EXAMPLE 25-((E)-2-nitroethenyl)-1,3-benzodioxole
3,4-methylenedioxybenzaldehyde (15.55 Kg) was treated sequentially with ammonium acetate (13.4 Kg,), acetic acid (45.2 Kg) and nitromethane (18.4 Kg), warmed to 70° C., stirred for 30 minutes, warmed to 80° C., stirred for 10 hours, cooled to 10° C. and filtered. The filtrant was washed with acetic acid (2×8 Kg) and water (2×90 Kg) and dried under a nitrogen stream then in under vacuum at 50° C. for 2 days.
EXAMPLE 3ethyl 3-(4-methoxyphenyl)-3-oxopropanoate
A mixture of potassium tert-amylate (50.8 Kg) in toluene (15.2 Kg) at 5° C. was treated with 4-methoxyacetophenone (6.755 Kg) and diethyl carbonate (6.4 Kg) in toluene over 1 hour while keeping the solution temperature below 10° C., warmed to 60° C. for 8 hours, cooled to 20° C. and treated with acetic acid (8 Kg) and water (90 Kg) over 30 minutes while keeping the solution temperature below 20° C. The organic layer was isolated, washed with 5% aqueous sodium bicarbonate (41 Kg) and concentrated at 50° C. to 14.65 Kg.
EXAMPLE 4ethyl 2-(4-methoxybenzoyl)-4-nitromethyl-3-(1,3-benzodioxol-5-yl)butyrate
A mixture of EXAMPLE 3 (7.5 Kg) in THF (56 Kg) was treated with EXAMPLE 3 (8.4 Kg), cooled to 17° C., treated with sodium ethoxide (6.4 g), stirred for 30 minutes, treated with more sodium ethoxide (6.4 g), stirred at 25° C. until HPLC shows less than 1 area % ketoester remaining and concentrated to 32.2 Kg.
EXAMPLE 5ethyl cis,cis-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate
Raney nickel (20 g), from which the water had been decanted, was treated sequentially with THF (20 mL), EXAMPLE 4 (40.82 g), and acetic acid (2.75 mL). The mixture was stirred under hydrogen (60 psi) until hydrogen uptake slowed, treated with trifluoroacetic acid, stirred under hydrogen (200 psi) until HPLC shows no residual imine and less than 2% nitrone and filtered with a methanol (100 mL) wash. The filtrate, which contained 13.3 g of EXAMPLE 5, was concentrated with THF (200 mL) addition to 100 mL, neutralized with 2N aqueous NaOH (50 mL), diluted with water (200 mL), and extracted with ethyl acetate (2×100 mL). The extract was used in the next step.
EXAMPLE 6ethyl trans,trans-2-(4-methoxyphenyl)-4-(1,3 -benzodioxol-5 -yl)pyrrolidine-3-carboxylate
Example 501E (38.1 g) was concentrated with ethanol (200 mL) addition to 100 mL, treated with sodium ethoxide (3.4 g), heated to 75° C., cooled to 25° C. when HPLC showed less than 3% of EXAMPLE 1E and concentrated. The concentrate was mixed with isopropyl acetate (400 mL), washed with water (2×150 mL) and extracted with 0.25 M phosphoric acid (2×400 mL). The extract was mixed with ethyl acetate (200 mL) and neutralized to pH 7 with sodium bicarbonate (21 g), and the organic layer was isolated.
EXAMPLE 7ethyl (2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate, (S)-(+) mandelate
EXAMPLE 501F was concentrated with acetonitrile (100 mL) addition to 50 mL, treated with (S)-(+)-mandelic acid (2.06 g), stirred until a solution formed, stirred for 16 hours, cooled to 0° C., stirred for 5 hours and filtered. The filtrant was dried at 50° C. under a nitrogen stream for 1 day. The purity of the product was determined by chiral HPLC using Chiralpak AS with 95:5:0.05 hexane/ethanol/diethylamine, a flow rate of 1 mL/min. and UV detection at 227 nm. Retention times were 15.5 minutes for the (+)-enantiomer and 21.0 minutes for the (−)-enantiomer.
EXAMPLE 8(2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)pyrrolidine-3-carboxylic acid
A mixture of EXAMPLE 7 (20 g) in ethyl acetate (150 mL) and 5% aqueous sodium bicarbonate was stirred at 25° C. until the salt dissolved and gas evolution stopped. The organic layer was isolated and concentrated. The concentrate was treated with acetonitrile (200 mL), concentrated to 100 mL, cooled to 10° C., treated with diisopropylethylamine (11.8 mL) and EXAMPLE 1 (10.5 g), stirred for 12 hours and concentrated. The concentrate was treated with ethanol (200 mL), concentrated to 100 mL, treated with 40% aqueous NaOH (20 mL), stirred at 60° C. for 4 hours, cooled, poured into water (400 mL), washed with hexanes (2×50 mL then 2×20 mL), treated with ethyl acetate (400 mL) and adjusted to pH 5 with concentrated HCl (12 mL). The organic layer was isolated and concentrated.
………………….



SYN
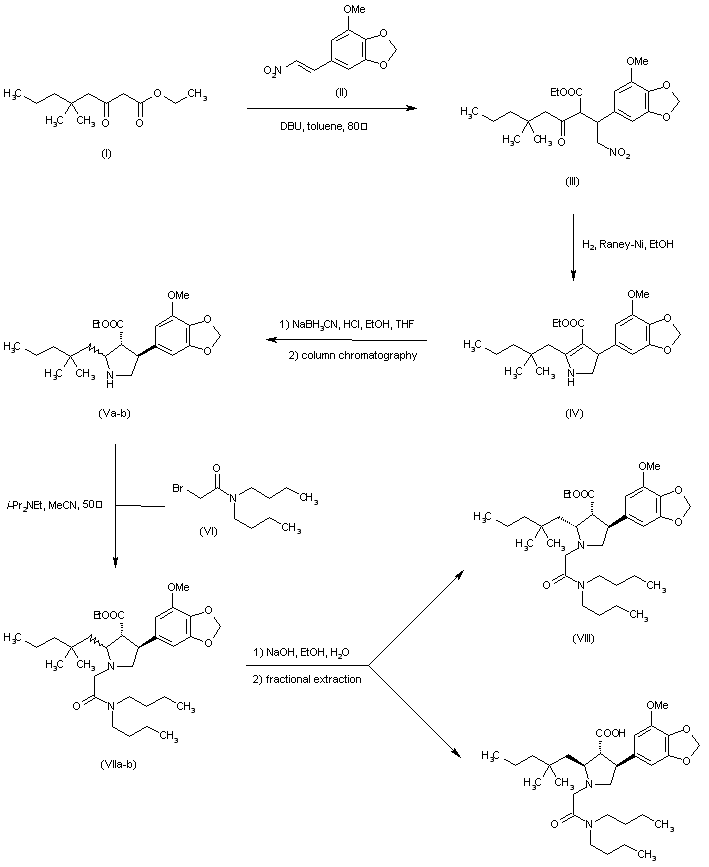
Condensation of ketoester (I) with nitrovinyl benzodioxole (II) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene gave adduct (III). Hydrogenation of the nitro group of (III) over Raney Nickel with concomitant cyclization yielded dihydropyrrole (IV). Further reduction of (IV) with sodium cyanoborohydride provided a mixture of diastereomeric pyrrolidines. Chromatographic separation removed the cis,cis isomer, affording a mixture of trans,trans and cis,trans products (V). N-Alkylation of the pyrrolidine (V) with N,N-dibutyl bromoacetamide (VI) furnished (VIIa-b). Finally, selective hydrolysis of the ester group from the trans,trans isomer produced a mixture of cis,trans ester (VIII) and the target trans,trans acid, which were readily separated by fractional extraction.
SYN
SYN
J Med Chem 1996,39(5),1039
The Michael reaction between 3,4-(methylenedioxy)-beta-nitrostyrene (I) and ethyl (4-methoxybenzoyl)acetate (II) in the presence of DBU gave adduct (III) as a mixture of isomers. Hydrogenation of this nitro ketone over Raney-Ni afforded, after spontaneous cyclization of the resulting amino ketone, the pyrroline (IV). Further reduction of the imine with NaBH3CN yielded a mixture of three pyrrolidine isomers. The desired trans-trans isomer (VI) could not be separated from the cis-trans isomer by column chromatography. However, the pure cis-cis compound (V) was isomerized to (VI) with NaOEt in refluxing EtOH. The protection of the amine as the tert-butyl carbamate with Boc2O, and saponification of the ester function provided the racemic acid (VII). Resolution of (VII) was achieved by conversion to the mixed anhydride (VIII) with pivaloyl chloride, followed by condensation with the lithium salt of (S)-4-benzyl-2-oxazolidinone (IX), and chromatographic separation of the resulting diastereomeric imides. Alternatively, racemic (VII) could be resolved by crystallization of its salt with (R)-a-methylbenzylamine. Removal of the Boc group from the appropriate isomer (X) with HCl in dioxan, followed by alkylation with N,N-dibutylbromoacetamide (XI) in the presence of i-Pr2NEt furnished the pyrrolidinylacetamide (XII). Finally, hydrolysis of the imide with lithium hydroperoxide provided the target acid.
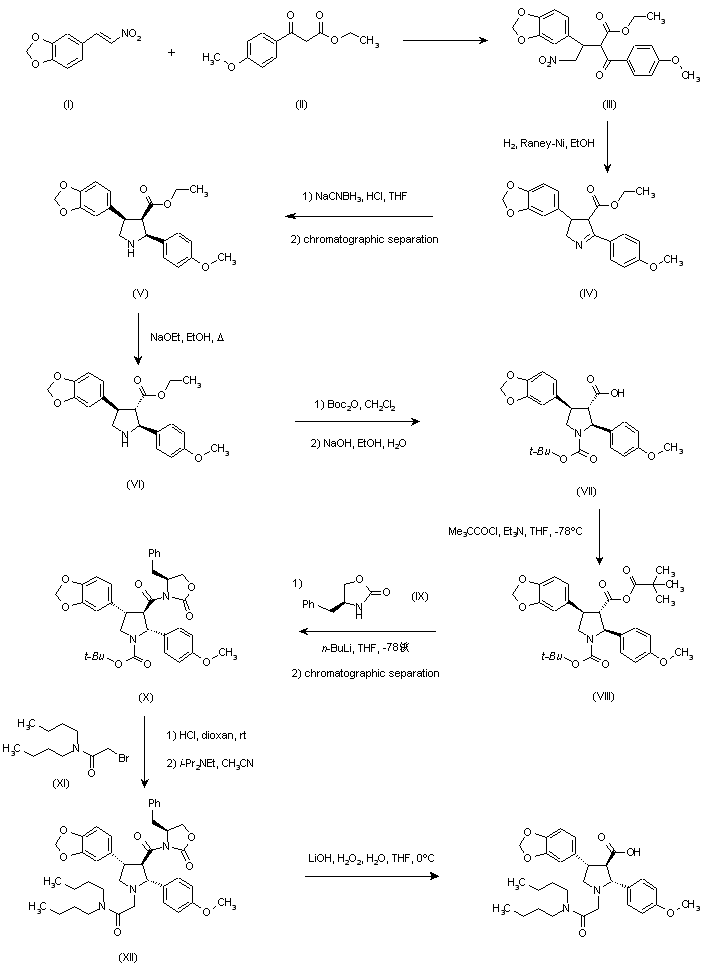
SYN
Reaction of 2-(1,3-dioxol-5-yl)acetic acid (XXI) with pivaloyl chloride and TEA gives the corresponding anhydride (XXII), which is condensed with the chiral oxazolidinone (XXIII) by means of n-BuLi in THF to yield the amide (XXIV). Condensation of (XXIV) with 2-bromoacetic acid tert-butyl ester (XXV) by means of NaHMDS in THF affords the adduct (XXVI). Elimination of the chiral auxiliary of (XXVI) by means of LiOOH in THF/water provides the chiral succinic acid hemiester (XXVII) (93% ee), which is selectively reduced with BH3璗HF complex to give the 4-hydroxysuccinate (XXVIII). Reaction of succinate (XXVIII) with 4-chlorophenylsulfonyl chloride, TEA and DMAP in dichloromethane yields the sulfonate (XXIX), which is condensed with 4-methoxybenzaldoxime (XXX) by means of Cs2CO3 in hot acetonitrile to afford the oxime ether (XXXI). Transesterification of the tert-butyl ester of (XXXI) with trimethyl orthoformate and p-toluenesulfonic acid in hot methanol provides the methyl ester (XXXII), which is cyclized by means of trimethylsilyl triflate and tributylamine in dichloroethane to afford a 9:1 diastereomeric mixture of perhydro-1,2-oxazines (XXXIII) and (XXXIV) which is easily separated. The reductive N-O-bond cleavage of the major oxazine diastereomer (XXXIII) by means of Zn/HOAc or H2 over Pd/C gives the trisubstituted 4-aminobutanol (XXXV), which is cyclized by means of CBr4, PPh3 and TEA to yield chiral pyrrolidine (XXXVI) (4). Finally, pyrrolidine (XXXVI) is alkylated with N,N-dibutyl-2-bromoacetamide (XIII) followed by ester hydrolysis as before.
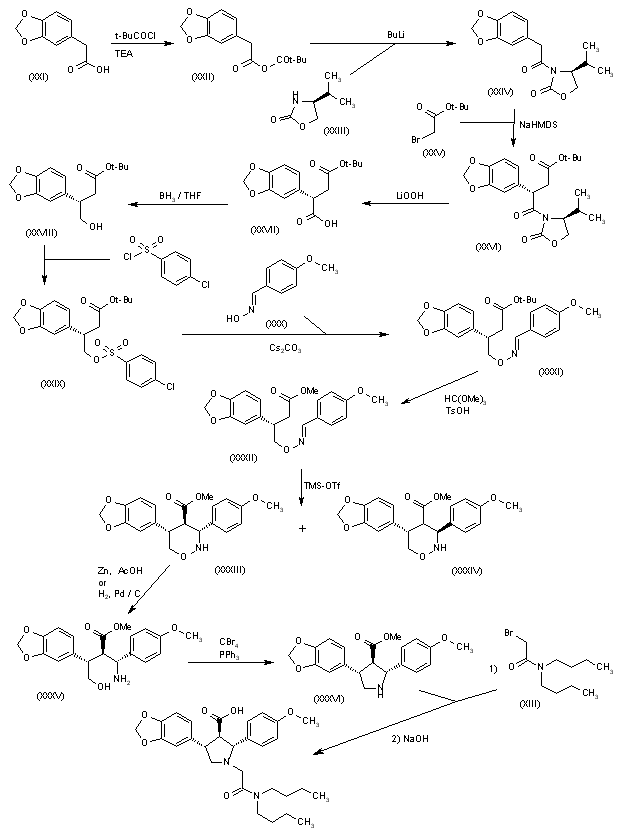
References
1
- “Atrasentan”. NCI Dictionary of Cancer Terms. National Institute of Cancer.
- 2
- Chiappori, Alberto A.; Haura, Eric; Rodriguez, Francisco A.; Boulware, David; Kapoor, Rachna; Neuger, Anthony M.; Lush, Richard; Padilla, Barbara; Burton, Michelle; Williams, Charles; Simon, George; Antonia, Scott; Sullivan, Daniel M.; Bepler, Gerold (March 2008). “Phase I/II Study of Atrasentan, an Endothelin A Receptor Antagonist, in Combination with Paclitaxel and Carboplatin as First-Line Therapy in Advanced Non–Small Cell Lung Cancer”. Clinical Cancer Research 14 (5): 1464–9. doi:10.1158/1078-0432.CCR-07-1508. PMID 18316570.
- 3
- “Addition of experimental drug to standard chemotherapy for advanced prostate cancer shows no benefit in phase 3 clinical trial” (Press release). National Cancer Institute. April 21, 2011. Retrieved October 18, 2014.
- 4
- Quinn, David I; Tangen, Catherine M; Hussain, Maha; Lara, Primo N; Goldkorn, Amir; Moinpour, Carol M; Garzotto, Mark G; Mack, Philip C; Carducci, Michael A; Monk, J Paul; Twardowski, Przemyslaw W; Van Veldhuizen, Peter J; Agarwal, Neeraj; Higano, Celestia S; Vogelzang, Nicholas J; Thompson, Ian M (August 2013). “Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (SWOG S0421): a randomised phase 3 trial”. The Lancet Oncology 14 (9): 893–900. doi:10.1016/S1470-2045(13)70294-8. PMID 23871417.
- 5
- de Zeeuw, Dick; Coll, Blai; Andress, Dennis; Brennan, John J.; Tang, Hui; Houser, Mark; Correa-Rotter, Ricardo; Kohan, Donald; Lambers Heerspink, Hiddo J.; Makino, Hirofumi; Perkovic, Vlado; Pritchett, Yili; Remuzzi, Giuseppe; Tobe, Sheldon W.; Toto, Robert; Viberti, Giancarlo; Parving, Hans-Henrik (May 2014). “The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy”. Journal of the American Society of Nephrology 25 (5): 1083–93. doi:10.1681/ASN.2013080830. PMID 24722445.
- 6
Clinical trial number NCT01858532 for “Study Of Diabetic Nephropathy With Atrasentan (SONAR)” at ClinicalTrials.gov
Granted in February 2015, this patent claims novel crystalline anhydrous S-mandelate salt of atrasentan. Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2R,3R,4S)-4-(1,3-Benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid | |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 173937-91-2 |
| ATC code | None |
| PubChem | CID 159594 |
| ChemSpider | 140321 |
| UNII | V6D7VK2215 |
| ChEMBL | CHEMBL9194 |
| Chemical data | |
| Formula | C29H38N2O6 |
| Molecular mass | 510.621 g/mol |
READ MORE ON SENTAN SERIES………..http://medcheminternational.blogspot.in/p/sentan-series.html
- Szczepankiewicz BG, Bal RB, von Geldern TW, Wu-Wong JR, Chiou WJ, Dixon DB, Opgenorth TJ, Hoffman DJ, Borre AJ, Marsh KC, Nguyen BN: The effects of diminishing albumin binding to some Endothelin receptor antagonists. Life Sci. 1998;63(21):1905-12. doi: 10.1016/s0024-3205(98)00466-4. [Article]
- Rajasekaran A, Julian BA, Rizk DV: IgA Nephropathy: An Interesting Autoimmune Kidney Disease. Am J Med Sci. 2021 Feb;361(2):176-194. doi: 10.1016/j.amjms.2020.10.003. Epub 2020 Oct 8. [Article]
- FDA Approved Drug Products: Vanrafia (atrasentan) tablets for oral use (April 2025) [Link]
- Novartis Media Release: Novartis receives FDA accelerated approval for Vanrafia® (atrasentan), the first and only selective endothelin A receptor antagonist for proteinuria reduction in primary IgA nephropathy (IgAN) [Link]
- StatPearls [Internet]: IgA Nephropathy (Berger Disease) [Link]
- ResearchGate: Total Synthesis of Atrasentan (Craig S. Harris, Reims Symposium, October 2002) [Link]
//////////ATRASENTAN, FDA 2025, APPROVALS 2025, Vanrafia, A 147627, (+)-A-127722, ABT 627, UNII-V6D7VK2215

{kind=link}
{kind=link}